The ATP-Dependent Proteolysis System in Rabbit Reticulocyte Lysate: From Foundational Mechanisms to Modern Research Applications
This article provides a comprehensive examination of the ATP-dependent proteolysis system in rabbit reticulocyte lysate (RRL), a foundational in vitro tool for studying intracellular protein degradation.
The ATP-Dependent Proteolysis System in Rabbit Reticulocyte Lysate: From Foundational Mechanisms to Modern Research Applications
Abstract
This article provides a comprehensive examination of the ATP-dependent proteolysis system in rabbit reticulocyte lysate (RRL), a foundational in vitro tool for studying intracellular protein degradation. We explore the historical discovery of this soluble, non-lysosomal system and its core mechanism involving ubiquitin conjugation and ATP hydrolysis. The content details methodological applications in protein degradation assays and drug discovery, addresses common troubleshooting and optimization challenges, and presents validation data through comparative analyses with other proteolytic systems. Tailored for researchers, scientists, and drug development professionals, this review synthesizes decades of research to guide effective experimental utilization and contextualize the continued relevance of RRL in biomedical research.
Discovering the RRL Proteolysis System: Historical Foundations and Core Mechanisms
In 1977, a landmark study by Etlinger and Goldberg established the existence of a soluble, non-lysosomal, ATP-dependent proteolytic system in rabbit reticulocytes, fundamentally reshaping our understanding of intracellular protein degradation. This pioneering work, published in Proceedings of the National Academy of Sciences, demonstrated for the first time that the energy-dependent degradation of abnormal proteins could be reconstituted in a cell-free extract, providing a crucial experimental system for dissecting the biochemical machinery governing regulated proteolysis in eukaryotic cells. This discovery laid the essential groundwork for the subsequent identification of the ubiquitin-proteasome system and other ATP-dependent proteases, forming the foundation of a research field of immense importance to cellular physiology and drug development.
Prior to the 1970s, the prevailing paradigm attributed intracellular protein degradation primarily to the lysosome, an organelle discovered by Christian de Duve and known to contain a battery of acid hydrolases [1]. However, several lines of evidence were inconsistent with this view. Notably, the degradation of many abnormal and short-lived regulatory proteins was observed to be energy-dependent, occurring under conditions where lysosomal function was impaired [1]. A key experimental model for studying this phenomenon was the rabbit reticulocyte, which actively degrades abnormal proteins, such as globin synthesized in the presence of the valine analog 2-amino-3-chlorobutyric acid (ClAbu) [2] [3]. This analog-containing globin was degraded with a half-life of approximately 15 minutes, while normal hemoglobin was stable, highlighting the selectivity and regulatory potential of the process [3]. The central enigma was the biochemical mechanism responsible for this ATP-dependent, non-lysosomal proteolysis.
The Pioneering Experiment: Key Methodology and Findings
The seminal 1977 paper by Etlinger and Goldberg provided the first direct biochemical characterization of this system in a cell-free environment [2] [3].
Experimental Workflow and Setup
The researchers established a robust assay to probe the proteolytic machinery directly. The core experimental workflow is summarized below.
Key Experimental Steps [2] [3]:
- Reticulocyte Isolation: Reticulocytes were isolated from phenylhydrazine-induced anemic rabbits.
- Synthesis of Abnormal Protein Substrate: Cells were incubated with the amino acid analog 2-amino-3-chlorobutyric acid (ClAbu) to produce a population of abnormal, rapidly degraded globin.
- Preparation of Cell-Free Extract: A soluble cytosolic fraction (S100) was prepared by lysing the cells and centrifuging the lysate at 100,000 × g for 90 minutes. This step deliberately removed membranes and organelles, including lysosomes.
- Proteolysis Assay: The cell-free extract was incubated with the radioactively labeled abnormal globin under various conditions. Proteolysis was quantified by measuring the release of acid-soluble radioactivity, indicating the hydrolysis of the protein substrate into small peptides and amino acids.
Critical Results and Quantitative Data
The experimental results provided unambiguous evidence for a novel proteolytic system. The key quantitative findings are consolidated in the table below.
Table 1: Key Experimental Findings from the Etlinger & Goldberg (1977) Study [2] [3]
| Experimental Condition | Effect on Proteolysis of Abnormal Globin | Key Implication |
|---|---|---|
| ATP Addition | Stimulated degradation several-fold | Process is energy-dependent |
| ADP Addition | Slight stimulation | Specific requirement for nucleotide triphosphates |
| AMP / cAMP Addition | No significant effect | Specific requirement for nucleotide triphosphates |
| Lysosomal Inhibitors | No inhibition reported | System is non-lysosomal in origin |
| Serine Protease Inhibitors (TPCK, TLCK) | Inhibited proteolysis | Involves serine protease catalytic activity |
| Sulfhydryl Reagents (NEM, Iodoacetamide) | Inhibited proteolysis | Essential cysteine residues are required |
| Metal Chelator (o-Phenanthroline) | Inhibited proteolysis | Dependence on metal ions (e.g., Zn²⁺) |
The correlation between inhibitor sensitivities in the cell-free system and in intact reticulocytes confirmed the physiological relevance of the reconstituted system [3]. Furthermore, the system exhibited a pH optimum of 7.8, distinct from the acidic pH optimum of lysosomal proteases, providing additional evidence for its unique identity [2].
The Scientist's Toolkit: Key Research Reagents and Materials
The investigation of the ATP-dependent proteolytic system relied on a specific set of biochemical reagents and materials.
Table 2: Essential Research Reagents for Studying ATP-Dependent Proteolysis in Reticulocytes
| Reagent / Material | Function in the Experimental Context |
|---|---|
| Rabbit Reticulocytes | Biological source of the ATP-dependent proteolytic machinery; ideal model due to high proteolytic activity and lack of lysosomes. |
| Amino Acid Analog (ClAbu) | Incorporated into proteins during synthesis to generate defined "abnormal" protein substrates (e.g., globin) that are rapidly recognized and degraded. |
| Adenosine Triphosphate (ATP) | The key nucleotide used to stimulate the proteolytic system; its non-hydrolyzable analogs helped define the energy requirement. |
| Protease Inhibitors (TPCK, TLCK) | Serine protease inhibitors used to characterize the catalytic class of the protease(s) involved and confirm the proteolytic nature of the activity. |
| Sulfhydryl Reagents (NEM, Iodoacetamide) | Used to alkylate free cysteine thiol groups, demonstrating the essential role of cysteine residues in the proteolytic pathway. |
| Metal Chelator (o-Phenanthroline) | Chelator of divalent cations (e.g., Zn²⁺, Fe²⁺); its inhibitory effect suggested the involvement of a metalloprotease or a metal-dependent step. |
Mechanistic Insights and the Path Forward
The 1977 discovery was a breakthrough that opened the door to mechanistic dissection. The core finding was that ATP serves two distinct roles in the degradation pathway, a concept later clarified by subsequent studies.
The Dual Role of ATP
Following the initial discovery, further research, including a key 1983 study, demonstrated that ATP hydrolysis is required for two distinct steps in the pathway [4]:
- Conjugation to Ubiquitin: The covalent attachment of the small protein ubiquitin to substrate proteins, marking them for degradation.
- Proteolysis per se: A step independent of ubiquitin conjugation, later understood to involve the unfolding of protein substrates and their translocation into the proteolytic core of the proteasome.
This dual requirement is illustrated in the following mechanism diagram.
The Etlinger and Goldberg system was thus the starting point for separating these two energy-dependent functions. Experiments with amino-blocked proteins that could not be ubiquitinated showed they were still degraded in an ATP-stimulated manner, proving the existence of the second, conjugation-independent role for ATP [4].
Legacy and Evolution of the Model
The cell-free system developed from rabbit reticulocytes was the direct experimental foundation for the next decade of Nobel Prize-winning work. It enabled the fractionation of the proteolytic machinery, leading directly to:
- The identification of ubiquitin as the ATP-dependent proteolysis factor (APF-1) [1].
- The purification and characterization of the 26S proteasome, the large proteolytic complex that degrades ubiquitin-tagged proteins [5] [6].
- The elucidation of the complex enzymatic cascade (E1, E2, E3) responsible for ubiquitin conjugation [6] [1].
The discovery also had conceptual breadth, as it paralleled findings in bacteria where ATP-dependent proteases like Lon (protease La) were being identified as key regulators of protein quality control and the degradation of specific regulatory proteins [7] [4].
The initial identification of a soluble ATP-dependent proteolytic system in rabbit reticulocytes was a transformative event in cell biology. By providing a well-defined and reproducible cell-free system, Etlinger and Goldberg enabled the reductionist biochemical dissection of a process fundamental to cellular regulation. This pioneering work moved the field beyond the lysosomal paradigm and laid the indispensable groundwork for discovering the ubiquitin-proteasome system, a discovery that has profoundly impacted our understanding of human disease mechanisms and created entirely new avenues for therapeutic intervention, particularly in cancer and neurodegenerative disorders. The reticulocyte lysate system remains a vital tool in biochemical research, a testament to the power of its initial discovery.
Within the broader context of research on the rabbit reticulocyte lysate ATP-dependent proteolysis system, the subcellular localization and origin of the proteolytic machinery are fundamental characteristics. The discovery that an ATP-dependent proteolytic system was present in the soluble fraction of reticulocytes and was non-lysosomal marked a significant departure from the then-prevailing understanding of cellular protein degradation [3]. This characteristic is essential for understanding the system's mechanism, its role in cellular physiology, and its distinction from other degradative pathways. This guide details the experimental evidence and methodologies that established these core system characteristics, providing a technical resource for researchers aiming to apply or build upon this knowledge.
Core Experimental Findings
The definitive characterization of the ATP-dependent proteolytic system in rabbit reticulocytes was established through a series of key experiments. The foundational study by Etlinger and Goldberg (1977) provided direct evidence for its soluble and non-lysosomal nature [3].
Table 1: Key Experimental Findings on System Localization and Origin
| Experimental Characteristic | Finding | Interpretation |
|---|---|---|
| Subcellular Fractionation | Proteolytic activity was recovered in the 100,000 x g supernatant fraction [3]. | The active components are soluble cytosolic proteins, not associated with heavy organelles or membranes. |
| Sedimentation Analysis | The activity did not sediment under high-speed centrifugation [3]. | Confirms the system is not part of large, sedimentable structures like lysosomes, mitochondria, or the cytoskeleton. |
| pH Optimum | The system exhibited a pH optimum of 7.8 [3]. | This alkaline pH preference is distinct from the acidic pH optimum (∼4.5-5.0) characteristic of lysosomal proteases. |
| Comparative Cell Biology | A sedimentable, ATP-dependent proteolytic activity was found in less mature erythroleukemia cells [8]. | Suggests the system's form may change with cellular maturation, becoming soluble in terminally differentiated reticulocytes. |
These findings collectively demonstrated the existence of a novel, energy-dependent proteolytic pathway operating in the cytosol, separate from the lysosomal system.
Detailed Experimental Protocols
Subcellular Fractionation of Reticulocytes
This protocol is used to isolate the soluble cytosolic fraction containing the ATP-dependent proteolytic activity.
Principle: Differential centrifugation separates cellular components based on size and density, allowing for the isolation of the 100,000 x g supernatant (S-100) as the soluble cytosolic fraction.
Procedure:
- Lysate Preparation: Isolate rabbit reticulocytes from phenylhydrazine-treated animals. Wash the cells and lyse them in a hypotonic buffer (e.g., 1-2 mM MgCl₂, 1 mM dithiothreitol).
- Low-Speed Centrifugation: Remove cell membranes and any unlysed cells by centrifuging the lysate at 10,000 x g for 20 minutes. Retain the supernatant.
- High-Speed Centrifugation: Transfer the 10,000 x g supernatant to ultracentrifuge tubes. Centrifuge at 100,000 x g for 1-2 hours at 4°C. This step pellets ribosomes, large organelles, and any remaining particulate matter.
- Fraction Collection: The resulting supernatant (S-100 fraction) contains the soluble cytosolic proteins, including the ATP-dependent proteolytic activity. The pellet can be resuspended in buffer for comparison.
- Activity Assay: Assay both the S-100 supernatant and the resuspended pellet for ATP-dependent proteolytic activity using a labeled substrate like analog-containing globin [3].
Assaying ATP-Dependent Proteolytic Activity
This protocol measures the degradation of abnormal proteins in the isolated fractions.
Principle: The degradation of a radiolabeled or otherwise tagged abnormal protein substrate is measured by the release of acid-soluble fragments in an ATP-dependent manner.
Procedure:
- Reaction Setup: Prepare a reaction mixture containing:
- Buffer: 50 mM Tris-HCl, pH 7.8.
- Cofactors: 5 mM MgCl₂, 2 mM dithiothreitol.
- Energy Source: 1-2 mM ATP (an ATP-regenerating system, e.g., creatine phosphate and creatine kinase, is often included).
- Substrate: ³H-labeled abnormal protein (e.g., globin containing the valine analog 2-amino-3chlorobutyric acid) [3].
- Enzyme Source: The S-100 fraction or other subcellular fraction.
- Incubation: Incubate the reaction at 37°C for 30-60 minutes.
- Reaction Termination: Stop the reaction by adding an equal volume of cold 10% (w/v) trichloroacetic acid (TCA).
- Quantification: Keep the samples on ice for 30 minutes to precipitate intact protein, then centrifuge. Measure the radioactivity in the TCA-soluble supernatant, which represents the peptides and amino acids released by proteolysis [3].
- Controls: Include control reactions without ATP and without the enzyme source to determine background and ATP-stimulated activity.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for Studying the ATP-Dependent Proteolytic System
| Reagent | Function in the Experimental Context |
|---|---|
| Rabbit Reticulocyte Lysate | The biological source material for preparing the S-100 fraction and studying the native proteolytic system [3]. |
| Abnormal Protein Substrate (e.g., ClAbu-globin) | A selectively degraded model substrate that demonstrates the system's specificity for misfolded or abnormal proteins [3]. |
| Adenosine Triphosphate (ATP) | The essential energy source required to stimulate proteolysis; its hydrolysis is a key characteristic of the system [3]. |
| ATP-regenerating System | Prevents the depletion of ATP during the assay, ensuring sustained proteolytic activity. |
| Protease Inhibitors (TPCK, TLCK) | Serine protease inhibitors used to characterize the protease components and distinguish the system from other proteases [3]. |
| Sulfhydryl Reagents (N-ethylmaleimide, Iodoacetamide) | Inhibitors that target cysteine residues, demonstrating the essential role of cysteine proteases in the system [3]. |
| Metal Chelator (o-phenanthroline) | Inhibits metalloproteases; its inhibitory effect helps characterize the type of proteases involved [3]. |
Experimental and Conceptual Workflows
The following diagrams illustrate the key experimental and conceptual frameworks for establishing the system's characteristics.
Reticulocyte Fractionation Workflow
Non-Lysosomal Evidence Synthesis
Discussion and Research Context
The localization of the ATP-dependent proteolytic system in the 100,000 x g supernatant firmly established its identity as a soluble, cytosolic pathway. This characteristic differentiates it from the lysosomal/vacuolar system and from other particulate-associated ATP-dependent activities found in less mature erythroid cells, such as erythroleukemia cells [8]. The non-lysosomal origin was further supported by the system's alkaline pH optimum and its specific inhibitor profile, which did not align with known lysosomal protease inhibitors. Subsequent research built upon this foundational characteristic, revealing that ATP plays two distinct roles in the pathway: one in the ubiquitin conjugation pathway and another, independent of ubiquitination, in the proteolytic process itself [4]. Understanding these system characteristics is crucial for drug development professionals targeting specific intracellular proteolytic pathways, as it allows for the rational design of inhibitors that avoid off-target effects on the lysosomal system.
The ATP-dependent proteolysis system in rabbit reticulocytes represents a fundamental biological pathway responsible for the selective degradation of abnormal proteins. Initial investigations into this system revealed a surprising requirement for metabolic energy in a process that is thermodynamically favorable, suggesting the involvement of sophisticated regulatory mechanisms [3] [1]. This energy-dependent pathway was subsequently shown to be non-lysosomal and present in the cytosol, representing a major breakthrough in understanding cellular protein quality control [3] [5]. The discovery of this system laid the foundation for identifying the ubiquitin-proteasome pathway, which has since been recognized as a critical regulator of diverse cellular processes, with aberrations leading to various human diseases [1]. This technical guide examines the precise roles of ATP binding and hydrolysis in proteolytic activation, focusing specifically on insights gained from the rabbit reticulocyte lysate model system.
Historical Context and Fundamental Discoveries
The investigation of ATP-dependent proteolysis began with the observation that abnormal proteins in reticulocytes are rapidly degraded through an energy-requiring process. Seminal work in 1977 demonstrated that cell-free extracts from rabbit reticulocytes could hydrolyze abnormal proteins in the 100,000 × g supernatant fraction, exhibiting a pH optimum of 7.8 and showing no evidence of lysosomal involvement [3]. This soluble proteolytic system was stimulated several-fold by ATP, with ADP providing slight stimulation, while AMP and cyclic AMP had no significant effect [3]. The energy requirement was particularly puzzling because peptide bond hydrolysis is thermodynamically favorable, suggesting that ATP hydrolysis must be fulfilling other functions beyond driving the proteolytic reaction itself.
Further fractionation studies revealed that the ATP-dependent proteolytic system could be separated into distinct functional components. Researchers demonstrated that rabbit reticulocytes contain two distinct high molecular weight proteases: one approximately 1500 kDa enzyme that degrades proteins only when ATP and conjugating fractions are added, and a smaller 670 kDa protease that does not require ATP or ubiquitin [5]. The larger protease, described as the ubiquitin-conjugate degrading enzyme (UCDEN), was found to be labile in the absence of nucleotides and was strongly inhibited by various compounds including heparin, hemin, and N-ethylmaleimide [5]. This fractionation approach enabled researchers to dissect the individual steps in the ATP-dependent proteolytic pathway.
The 26S Proteasome: Architecture and ATP-Dependent Assembly
The 26S proteasome represents the central proteolytic machine in eukaryotic cells, comprising a 20S core particle (CP) and one or two 19S regulatory particles (RP). The 20S proteasome is a barrel-shaped structure composed of four stacked heptameric rings, with the catalytic sites located in an interior chamber [9]. Access to these catalytic sites is restricted by narrow pores (approximately 13 Å) at either end of the cylinder, which are further occluded by N-terminal peptide extensions of the outer ring subunits [9]. The 19S regulatory particle contains six homologous AAA ATPase subunits arranged in a ring that interfaces with the 20S proteasome, positioning them to mediate various ATP-dependent functions [9].
Table 1: Components of the 26S Proteasome Complex
| Component | Structure | Molecular Weight | Function |
|---|---|---|---|
| 20S Core Particle (CP) | 4 stacked heptameric rings | 700,000 Da | Catalytic core; contains proteolytic active sites |
| 19S Regulatory Particle (RP) | 18 different subunits including 6 AAA ATPases | 700,000 Da | Substrate recognition, unfolding, translocation |
| 26S Proteasome (singly-capped) | 1 RP + 1 CP | 1,400,000 Da | Primary functional form in reticulocytes |
| 26S Proteasome (doubly-capped) | 2 RP + 1 CP | 2,400,000 Da | Enhanced degradation capacity |
Research using purified 26S proteasome from rabbit reticulocytes has demonstrated that ATP binding is necessary and sufficient for assembly of the 26S proteasome from 20S core and 19S regulatory subcomplexes [9]. This assembly is accompanied by a 20- to 50-fold increase in hydrolysis of peptide substrates, resulting from the opening of the gated pores at the ends of the 20S proteasome [9]. The stability of the assembled 26S proteasome is also maintained by ATP binding, as nucleotide depletion promotes dissociation into subcomplexes [9].
Distinct Roles of ATP Binding Versus ATP Hydrolysis
A critical advancement in understanding ATP-dependent proteolysis came from studies delineating the separate functions of ATP binding and ATP hydrolysis. Investigations with non-hydrolyzable ATP analogs revealed that ATP binding alone is sufficient for proteasome assembly and activation, though hydrolysis is required for complete proteolytic function [9].
Table 2: Nucleotide Requirements for 26S Proteasome Functions
| Proteasome Function | ATP Requirement | Nucleotide Specificity | Magnitude of Effect |
|---|---|---|---|
| Assembly | Binding only | ATPγS, AMP-PNP effective | Half-maximal at ~40 μM ATP |
| Peptidase Activation | Binding only | ATPγS, AMP-PNP effective | 20-50 fold activation |
| Unstructured Protein Degradation | None | Not nucleotide dependent | Similar rates regardless of ATP |
| Folded Protein Degradation | Hydrolysis required | ATP specific | No degradation with analogs |
| Polyubiquitinated Protein Degradation | Hydrolysis required | ATP specific | Coupled to deubiquitination |
Experimental evidence demonstrated that half-maximal proteasome activation occurs at approximately 40 μM ATP, while non-hydrolyzable analogs ATPγS and AMP-PNP promoted assembly and activation at similar or lower concentrations (1 μM for ATPγS and 20 μM for AMP-PNP) [9]. Neither the 26S proteasome nor isolated PA700/19S complex hydrolyzed ATPγS at detectable rates, confirming that hydrolysis is not required for assembly [9].
The energy requirement for degradation depends significantly on substrate characteristics. The 26S proteasome can degrade non-ubiquitinated, unstructured proteins without ATP hydrolysis, indicating that substrate translocation through the opened pore does not inherently require energy [9]. However, both folded proteins and polyubiquitinated proteins require ATP hydrolysis for degradation, suggesting that the energy requirement is imposed by the mechanistic coupling of multiple processes rather than any single proteolytic step [9].
Diagram 1: ATP utilization in proteasome functions. ATP binding (green) is sufficient for assembly and pore opening, while ATP hydrolysis (red) is required for substrate unfolding and coupled processes.
Experimental Approaches and Methodologies
Proteasome Purification and Assembly Assays
Purification of intact 26S proteasome from rabbit reticulocytes requires ATP in all buffers to maintain complex stability [9]. In contrast, the 20S proteasome and PA700/19S subcomplexes can be purified separately in ATP-free buffers [9]. This differential stability provides a key methodological advantage for studying assembly mechanisms.
Native Gel Electrophoresis for Assembly Monitoring: The assembly of 26S proteasome from 20S core and PA700/19S subcomplexes can be monitored by native polyacrylamide gel electrophoresis [9]. Assembly is accompanied by a dramatic increase (20-50 fold) in hydrolysis of fluorogenic peptide substrates like Suc-Leu-Leu-Val-Tyr-AMC, which can be quantified using solution assays or substrate overlay assays in native gels [9].
Nucleotide Dependency Experiments: To test the specific roles of ATP binding versus hydrolysis, researchers employ non-hydrolyzable ATP analogs including ATPγS and AMP-PNP [9]. These experiments require careful depletion of ATP from proteasome preparations using apyrase pretreatment, followed by repletion with the nucleotide of choice [9]. Such approaches demonstrated that ATPγS supports proteasome assembly despite being completely resistant to hydrolysis by the proteasomal ATPases [9].
Substrate-Specific Degradation Assays
Different classes of substrates reveal distinct ATP requirements:
Unstructured Protein Degradation: Non-ubiquitinated, unstructured proteins can be degraded without ATP hydrolysis, requiring only initial ATP-dependent proteasome activation [9].
Folded Protein Degradation: Folded proteins and certain polyubiquitinated folded proteins require ATP hydrolysis for degradation [9]. Those that are refractory to proteolysis may be deubiquitinated through an ATP-independent mechanism [9].
Deubiquitination Activity: The 26S proteasome contains deubiquitinating activity that can function without ATP, though for some substrates, ATP hydrolysis is required for both degradation and deubiquitination [9].
The Researcher's Toolkit: Essential Reagents and Methods
Table 3: Key Research Reagents for Studying ATP-Dependent Proteolysis
| Reagent/Condition | Function/Application | Experimental Notes |
|---|---|---|
| ATPγS | Non-hydrolyzable ATP analog | Distinguishes binding vs hydrolysis requirements; 1 μM for half-maximal effect |
| AMP-PNP | Non-hydrolyzable ATP analog | Confirms binding sufficiency; 20 μM for half-maximal effect |
| Apyrase | ATP depletion enzyme | Used to remove endogenous ATP before nucleotide repletion studies |
| Suc-LLVY-AMC | Fluorogenic peptide substrate | Standard activity assay; 20-50 fold activation upon assembly |
| Mg²⁺ | Essential cofactor | Required for all nucleotide effects on assembly and activation |
| Potassium Chloride (KCl) | Ionic activator | Stimulates peptidase activity with AMP-PNP; eliminates time lag |
| Ubiquitin-Conjugating System | Substrate targeting | Required for ubiquitin-dependent degradation pathways |
Regulatory Mechanisms and Physiological Significance
The ATP-dependent proteolytic system displays sophisticated regulation that maintains cellular protein homeostasis. The intracellular ATP concentration serves as a key regulator of proteolytic activity, particularly during nutrient stress. Research in Salmonella Typhimurium has demonstrated that reduced ATP concentrations during stationary phase decrease proteolysis by AAA+ proteases, thereby stabilizing functional proteins and facilitating rapid recovery when nutrients become available [10]. This mechanism appears to be conserved across species, including yeast [10].
The ATP dependence also provides a crucial control mechanism that prevents inappropriate degradation of normal cellular proteins. The 19S regulatory particle contains six AAA ATPase subunits that undergo conformational changes during ATP binding and hydrolysis, driving mechanical unfolding of substrate proteins [9] [11]. This unfolding activity is essential for translocation of folded substrates through the narrow proteasome pore, explaining why ATP hydrolysis is required for degradation of structured proteins but not unstructured polypeptides [9].
Comparative Analysis with Related ATP-Dependent Proteases
The principles discovered in the rabbit reticulocyte system extend to other AAA+ proteases across evolution. The bacterial protease HslVU exhibits similar uncoupling of ATP binding and hydrolysis functions. Notably, AMP-PNP together with K⁺ can induce a form of HslVU that degrades proteins without energy consumption [12]. Similarly, ClpXP complexes in bacteria use cycles of ATP hydrolysis to unfold and translocate substrates through the central pore of the complex [11]. These conserved mechanisms highlight the fundamental nature of ATP utilization in cellular proteolysis.
Diagram 2: Substrate processing workflow. ATP binding initiates recognition and unfolding, while hydrolysis powers mechanical unfolding and translocation before degradation.
The rabbit reticulocyte lysate system has provided profound insights into the critical role of ATP hydrolysis in proteolytic activation. The separation of ATP binding and hydrolysis functions represents a sophisticated regulatory mechanism that enables controlled protein degradation without compromising cellular protein homeostasis. The 26S proteasome utilizes ATP binding for complex assembly and gated pore opening, while reserving ATP hydrolysis for the mechanistically demanding processes of substrate unfolding and translocation. These fundamental principles, first discovered in the reticulocyte system, have broad relevance to AAA+ proteases throughout biology and continue to inform drug discovery efforts targeting the ubiquitin-proteasome pathway in human diseases.
The controlled degradation of proteins is a fundamental cellular process, essential for maintaining homeostasis, regulating critical signaling pathways, and disposing of damaged or misfolded proteins. The rabbit reticulocyte lysate system has been a cornerstone model for elucidating the biochemical mechanisms of ATP-dependent proteolysis [13]. Within this framework, two distinct yet complementary pathways have been characterized: the well-established ubiquitin-dependent pathway and the increasingly recognized ubiquitin-independent pathway. The ubiquitin-proteasome system (UPS) is responsible for the degradation of most intracellular proteins, where a polyubiquitin chain acts as the primary signal for proteasomal recognition and degradation [14] [15] [16]. Conversely, ubiquitin-independent proteasomal degradation (UbInPD) allows for the direct recognition and processing of specific substrates by the proteasome without the need for a ubiquitin tag [17] [18] [19]. This article delves into the mechanisms, key players, and experimental dissection of these dual degradation pathways, framed within the context of seminal research on rabbit reticulocyte lysate.
The Ubiquitin-Proteasome System (UPS)
The ubiquitin-dependent pathway is a multi-step enzymatic cascade that results in the tagging of a protein substrate with a polyubiquitin chain, marking it for degradation by the 26S proteasome.
The Ubiquitination Cascade
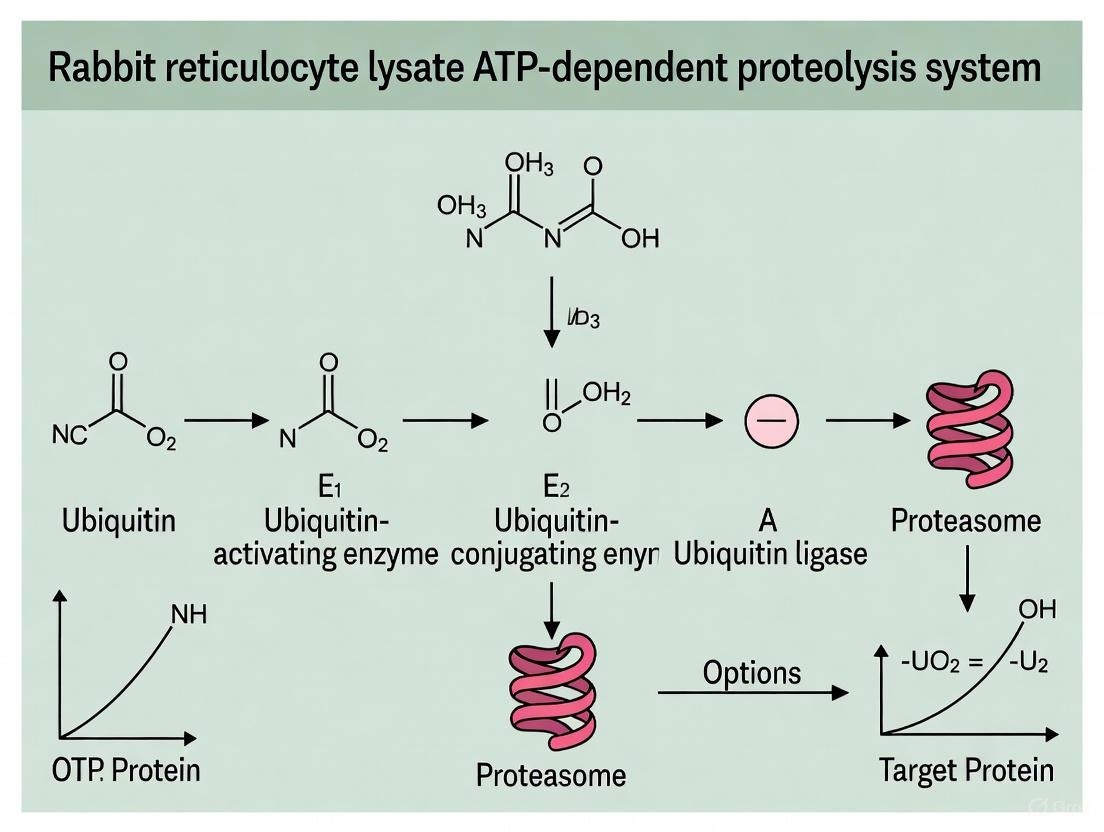
The process of ubiquitination involves a sequential action of three key enzymes [15] [16]:
- E1 (Ubiquitin-Activating Enzyme): This enzyme initiates the pathway by activating ubiquitin in an ATP-dependent reaction, forming a high-energy thioester bond.
- E2 (Ubiquitin-Conjugating Enzyme): The activated ubiquitin is then transferred from E1 to the catalytic cysteine residue of an E2 enzyme.
- E3 (Ubiquitin Ligase): Finally, an E3 ligase facilitates the transfer of ubiquitin from E2 to a lysine residue on the target protein, forming an isopeptide bond. The E3 ligase is primarily responsible for substrate recognition, providing specificity to the system.
Repeated cycles of this cascade lead to the formation of a polyubiquitin chain on the substrate. The type of ubiquitin chain linkage determines the fate of the modified protein. While all eight polyubiquitin chain types (K6, K11, K27, K29, K33, K48, and K63) exist, K48-linked chains are the principal signal for proteasomal degradation [14] [20].
The 26S Proteasome and Substrate Processing
The 26S proteasome is the executive component of the UPS and is a multi-subunit complex composed of two primary entities [14] [19]:
- The 20S Core Particle (20S): This barrel-shaped structure contains the proteolytically active sites (β1, β2, β5 subunits) within its inner chamber, sequestered from the cellular environment.
- The 19S Regulatory Particle (19S): This cap structure recognizes polyubiquitinated substrates, unfolds them, and translocates the unfolded polypeptide into the 20S core for degradation.
The 19S regulatory particle contains several key components that facilitate degradation [14]:
- Ubiquitin Receptors (e.g., Rpn1, Rpn10, Rpn13): These subunits recognize and bind the polyubiquitin chain on the substrate.
- Deubiquitinating Enzymes (DUBs, e.g., Rpn11): These enzymes remove the ubiquitin chain from the substrate prior to its entry into the 20S core, allowing for ubiquitin recycling.
- AAA-ATPase Motor: This hexameric ring utilizes ATP hydrolysis to unfold the target protein and thread it into the degradation chamber of the 20S core.
For some substrates, particularly those embedded in complexes or membranes, an additional ATPase complex, p97/VCP (Cdc48 in yeast), is required to extract and unfold the ubiquitinated protein before delivery to the proteasome [14].
dot-1-Biochemical Pathway of Ubiquitin Dependent Degradation
Ubiquitin-Independent Proteasomal Degradation (UbInPD)
Contrary to long-held beliefs, ubiquitin is not an obligatory tag for proteasomal degradation. A subset of cellular proteins can be degraded in a ubiquitin-independent manner.
Mechanisms and Key Players
Ubiquitin-independent degradation is mediated primarily by the 20S core proteasome alone, often facilitated by alternative regulatory complexes instead of the 19S cap [18] [19]. The 20S proteasome can directly recognize and degrade specific substrate proteins that possess inherent structural features making them susceptible to degradation.
Key aspects of UbInPD include [17] [18] [19]:
- C-degron Pathways: Specific amino acid sequences at the C-terminus of a protein (C-degrons) can act as potent signals for ubiquitin-independent degradation.
- Intrinsically Disordered Regions (IDRs): Proteins or protein regions that lack a stable three-dimensional structure can directly access the proteolytic chamber of the 20S proteasome without requiring ATP-dependent unfolding.
- Oxidized/Damaged Proteins: The 20S proteasome serves as a major defender against oxidative stress by selectively degrading proteins that have been damaged and partially unfolded by reactive oxygen species.
- Alternative Proteasome Activators: Complexes such as PA28αβ (11S regulator) and PA200 can bind to the 20S core and activate its peptidase activity in an ATP- and ubiquitin-independent manner, facilitating the degradation of specific substrates like acetylated histones or damaged proteins.
Recent systematic studies using GPS-peptidome technology have revealed that UbInPD is far more prevalent than previously appreciated, identifying thousands of potential degron sequences and dozens of full-length human proteins subject to this pathway, including regulatory proteins like REC8 and CDCA4 [17]. Furthermore, certain Ubiquilin family proteins have been identified as adaptors that can mediate the proteasomal targeting of a subset of UbInPD substrates, bridging the gap between the two pathways [17].
dot-2-Ubiquitin Independent Degradation Pathways
Comparative Analysis of Degradation Pathways
The table below summarizes the key characteristics of ubiquitin-dependent and ubiquitin-independent degradation pathways.
Table 1: Comparative Analysis of Ubiquitin-Dependent and Ubiquitin-Independent Degradation Pathways
| Feature | Ubiquitin-Dependent Pathway | Ubiquitin-Independent Pathway |
|---|---|---|
| Key Signal | Polyubiquitin chain (primarily K48-linked) [14] [20] | C-degrons, Intrinsically Disordered Regions, oxidative damage [17] [18] [19] |
| Primary Proteasome Complex | 26S (20S core + 19S cap) [14] [15] | 20S core alone or with alternative activators (PA28, PA200) [18] [13] [19] |
| ATP Requirement | Yes (for ubiquitination & 19S cap function) [14] [16] | No (for 20S core degradation) [13] |
| Energy of Activation (Ea) | ~27 kcal/mol (in rabbit reticulocyte lysate) [21] | Not explicitly defined, but lower due to lack of ubiquitination and unfolding requirements |
| Primary Function | Regulated turnover of normal proteins; quality control [15] [16] | Quality control (oxidized/misfolded proteins); rapid regulation of specific proteins [17] [19] |
| Example Substrates | Cyclins, transcription factors, misfolded proteins [14] [16] | Ornithine decarboxylase, thymidylate synthase, p21, oxidized proteins [17] [18] |
Experimental Analysis in Reticulocyte Lysate Systems
The rabbit reticulocyte lysate has been an indispensable model system for reconstituting and studying both ubiquitin-dependent and independent degradation.
Key Experimental Workflow
The foundational experiments involved fractionating the lysate to isolate and characterize the proteolytic machinery. The following workflow outlines the key steps for analyzing ATP-dependent and independent degradation:
dot-3-Experimental Workflow for Analyzing Degradation Pathways
Key Research Reagents and Tools
Table 2: Essential Research Reagents for Studying Protein Degradation Pathways
| Reagent/Tool | Primary Function | Key Insights Enabled |
|---|---|---|
| Rabbit Reticulocyte Lysate | A cell-free system rich in proteasomes, ubiquitin, and associated enzymes [13] | Enabled initial fractionation and identification of 20S and 26S proteasomes; demonstration of ATP-dependent and independent proteolysis [21] [13] |
| Proteasome Inhibitors (e.g., MG132) | Reversibly inhibits the proteolytic activity of the 20S core particle [15] | Confirms proteasome involvement in a degradation process; leads to accumulation of polyubiquitinated proteins in UPS studies [15] |
| ATPγS (Non-hydrolyzable ATP analog) | Inhibits ATP-dependent processes by replacing ATP without supporting hydrolysis [14] | Differentiates between ATP-dependent (26S/UPS) and ATP-independent (20S/UbInPD) degradation pathways [14] [13] |
| Ubiquitin Enrichment Kits | Isolate polyubiquitinated proteins from cell or tissue lysates using high-affinity resins [15] | Allows for specific detection and analysis of proteins targeted for ubiquitin-dependent degradation via Western blot or mass spectrometry |
| E1 Inhibitor (e.g., PYR-41) | Specifically inhibits the E1 ubiquitin-activating enzyme, blocking the entire ubiquitination cascade [15] | Used to distinguish ubiquitin-dependent from ubiquitin-independent degradation of a specific substrate |
| GPS-Peptidome Screening | A systematic method for high-throughput discovery of degron sequences [17] | Identified thousands of sequences, including C-degrons, that promote ubiquitin-independent degradation, revealing its prevalence |
Detailed Experimental Protocol: Assessing Pathway Dependence
To determine whether a protein of interest is degraded via a ubiquitin-dependent or independent pathway in a reticulocyte lysate system, the following experimental approach can be employed, based on classical and modern methodologies [17] [15] [13]:
- Reaction Setup: Set up a series of degradation reactions containing rabbit reticulocyte lysate and the substrate protein (radiolabeled for sensitive detection).
- Inhibitor/Addback Conditions:
- Condition A (Control): Lysate + ATP-regenerating system.
- Condition B (ATP-depletion): Lysate + ATPase (e.g., Apyrase) or non-hydrolyzable ATP analog (ATPγS).
- Condition C (Proteasome Inhibition): Lysate + ATP-regenerating system + MG132 (or other proteasome inhibitor).
- Condition D (E1 Inhibition): Lysate + ATP-regenerating system + E1 inhibitor (e.g., PYR-41).
- Condition E (UbInPD Promotion): Lysate where 26S is dissociated (e.g., by mild oxidative stress) and 20S activity is assessed.
- Incubation and Analysis:
- Incubate reactions at a defined temperature (e.g., 37°C). The degradation of proteins in this system exhibits a high Energy of Activation (Ea) of ~27 kcal/mol, a characteristic feature of the rate-limiting step in this pathway [21].
- At timed intervals, stop the reactions.
- Analyze the remaining substrate by SDS-PAGE and autoradiography (for radiolabeled substrates) or immunoblotting.
- Interpretation of Results:
- Degradation that is inhibited in Conditions B, C, and D indicates a Ubiquitin-Dependent process.
- Degradation that occurs in Conditions B and D but is blocked in Condition C indicates a Ubiquitin-Independent, 20S-mediated process.
- Degradation that is enhanced in Condition E further supports a UbInPD mechanism.
Discussion and Pathological Significance
The coexistence of ubiquitin-dependent and independent pathways provides the cell with a versatile and layered system for protein degradation. The UPS is optimized for the highly specific, regulated turnover of normal cellular proteins, such as cyclins and transcription factors [14] [16]. In contrast, the UbInPD pathway, particularly via the 20S proteasome, acts as a first line of defense against proteotoxic stress by rapidly degrading damaged, misfolded, or intrinsically disordered proteins that might otherwise aggregate [18] [19].
The importance of these pathways is starkly evident in neurodegenerative diseases such as Alzheimer's, Parkinson's, and Huntington's disease. These conditions are characterized by the accumulation of toxic protein aggregates like tau, α-synuclein, and huntingtin. Evidence suggests that many of these aggregation-prone proteins are substrates for both degradation pathways, and a decline in proteasomal activity with age contributes to pathology [22] [19]. Notably, some of these pathogenic proteins, especially in their native or modified states, can be degraded directly by the 20S proteasome in a ubiquitin-independent manner, highlighting this pathway's role in neuroprotective protein quality control [19].
From a therapeutic perspective, the ubiquitin-dependent pathway is being successfully targeted by PROTACs (PROteolysis TArgeting Chimeras) and molecular glues, which are bifunctional molecules that recruit E3 ubiquitin ligases to neosubstrates, inducing their degradation [20]. A deeper understanding of the UbInPD pathway, including its C-degron signals and associated adaptors like Ubiquilins, may open new avenues for drug development, potentially allowing for the targeted degradation of proteins currently considered "undruggable" [17].
Research originating from the rabbit reticulocyte lysate system was instrumental in uncovering the complexity of intracellular proteolysis, revealing not one but two major pathways for proteasomal degradation. The ubiquitin-dependent pathway represents a sophisticated, energy-dependent mechanism for precise regulatory control, while the ubiquitin-independent pathway provides an efficient, streamlined system for protein quality control and the rapid degradation of specific substrates. Rather than operating in isolation, these pathways likely form an integrated network, with cross-talk facilitated by shared components like the proteasome core and adaptor proteins. Future research will continue to elucidate the full scope of UbInPD substrates, the detailed mechanisms of degron recognition, and the therapeutic potential of harnessing both pathways to combat disease.
The ATP-dependent proteolytic system in rabbit reticulocyte lysate serves as a foundational model for understanding intracellular protein degradation. This in-depth technical guide characterizes three critical chemical inhibitors—hemin, vanadate, and N-ethylmaleimide—that have been instrumental in mapping the components and mechanistic principles of the ubiquitin-proteasome system. Through their distinct and specific inhibitory profiles, these compounds have enabled researchers to dissect the multi-step proteolytic pathway, from ubiquitin conjugation to final substrate hydrolysis by the 26S proteasome. This whitepaper synthesizes historical and current research findings into structured quantitative data, detailed experimental protocols, and visual workflows, providing life science researchers and drug development professionals with essential tools for investigating proteasome function and developing targeted therapeutic interventions.
Rabbit reticulocyte lysate has served as a premier cell-free system for elucidating the biochemical mechanisms of ATP-dependent intracellular protein degradation. This system is uniquely capable of selectively degrading abnormal or damaged proteins while sparing normal cellular proteins, mirroring a critical quality control process in living cells. Early seminal work established that reticulocytes contain a soluble, nonlysosomal proteolytic pathway that requires adenosine triphosphate (ATP) for function [3]. This system operates through a consecutive biochemical cascade: first, the ATP-dependent covalent attachment of ubiquitin to protein substrates, and second, the ATP-dependent degradation of these ubiquitin-protein conjugates by a high-molecular-weight protease complex [5].
The discovery and characterization of specific inhibitors have been paramount to disentangling this complex proteolytic machinery. Hemin, vanadate, and N-ethylmaleimide each inhibit distinct steps in the pathway through different mechanisms, enabling researchers to isolate and study individual components. Their use has been fundamental in identifying the ubiquitin-proteasome system (UPS) as the primary executor of energy-dependent proteolysis in eukaryotic cells—a discovery with profound implications for understanding cellular regulation and human disease pathogenesis [1]. This whitepaper provides a comprehensive technical resource on these essential inhibitors, framing their mechanisms within the broader context of UPS research.
Inhibitor Profiles and Mechanisms of Action
Hemin: Regulator of Ubiquitin Conjugate Degradation
Hemin acts as a specific allosteric inhibitor of the final degradation step in the UPS pathway. Research demonstrates that hemin exclusively targets the ATP-dependent ubiquitin-dependent proteolytic system without affecting basal proteolysis that occurs independently of ATP or ubiquitin [23] [24].
Quantitative Inhibition Data
Table 1: Quantitative Characterization of Hemin Inhibition
| Parameter | Value | Experimental Context |
|---|---|---|
| IC₅₀ | ~25 µM | 50% inhibition of ¹²⁵I-BSA degradation [23] [24] |
| Conjugate Formation Impact | ~50% of control | ATP-dependent ubiquitin conjugation at fully inhibitory hemin concentrations [23] [24] |
| Conjugate Degradation Impact | Complete blockade | Full inhibition at concentrations that completely block overall proteolysis [23] [24] |
| Specificity | Ubiquitin-dependent pathway exclusive | No effect on basal ATP-independent or ubiquitin-independent proteolysis [23] [24] |
Mechanistic Insights
Hemin functions as a negative allosteric effector specifically targeting the initial step in the degradation of ubiquitin-protein conjugates. At concentrations that completely inhibit proteolysis (approximately 25-50 µM), hemin still permits continued ATP-dependent formation of ubiquitin conjugates at approximately half the control rate, but completely blocks their subsequent breakdown [23] [24]. This results in the accumulation of higher molecular weight ubiquitin conjugates that rapidly disappear when hemin is removed, indicating the reversibility of its inhibitory effect. The specific molecular target is the ubiquitin conjugate-degrading enzyme (UCDEN), an approximately 1500 kDa protease essential for ATP-dependent proteolysis [5]. This specificity has made hemin an invaluable tool for distinguishing the conjugation and degradation phases of the UPS.
Vanadate: Inhibitor of ATP-Stimulated Proteolysis
Vanadate represents a distinct class of inhibitor that blocks ATP-stimulated proteolysis without affecting the ubiquitin conjugation system, providing crucial evidence for the multi-component nature of the UPS.
Quantitative Inhibition Data
Table 2: Quantitative Characterization of Vanadate Inhibition
| Parameter | Value | Experimental Context |
|---|---|---|
| Kᵢ | 50 µM | Inhibition of ATP-dependent [³H]methylcasein and denatured ¹²⁵I-BSA degradation [25] |
| Specificity | ATP-stimulated step | Does not affect ubiquitin conjugation to protein substrates [25] |
| Reversibility | Yes | Inhibition is reversed by dialysis [25] |
| Specificity Marker | Not mimicked by molybdate | Distinguishes from lysosomal protease inhibition [25] |
Mechanistic Insights
Vanadate inhibits an ATP-stimulated proteolytic step that is functionally distinct from the ATP requirement for ubiquitin conjugation. This was demonstrated through experiments using proteins with covalently modified amino groups that prevent ubiquitin conjugation—these derivatized proteins still undergo ATP-stimulated degradation that remains sensitive to vanadate inhibition [25]. Additionally, vanadate does not reduce the ATP-dependent conjugation of ¹²⁵I-ubiquitin to endogenous reticulocyte proteins, despite markedly inhibiting their degradation. This clear dissociation between conjugation and degradation established that these are functionally separable processes with distinct ATP requirements. In intact reticulocytes, vanadate also inhibits endogenous protein degradation, though secondary effects on protein synthesis and ATP levels complicate interpretation in cellular contexts [25].
N-Ethylmaleimide: Differential Effects on Proteolytic Components
N-Ethylmaleimide (NEM) exhibits complex, differential effects on the various components of the UPS, highlighting the biochemical diversity within this proteolytic system.
Quantitative Inhibition Data
Table 3: Effects of N-Ethylmaleimide on Proteolytic Components
| Protease Component | NEM Effect | Functional Implications |
|---|---|---|
| UCDEN (Ubiquitin-Conjugate Degrading Enzyme) | Inhibition | Essential for ATP-dependent proteolysis; contains sulfhydryl groups critical for activity [5] |
| 670 kDa Protease (Non-essential) | Stimulation | Does not require ATP or ubiquitin; not required for ATP-dependent proteolysis [5] |
| Overall ATP-Dependent Proteolysis | Inhibition | Consistent with blockade of the essential UCDEN component [3] |
Mechanistic Insights
The differential effects of NEM on distinct protease components revealed the existence of multiple high molecular weight proteases in reticulocyte cytosol. The essential ubiquitin conjugate-degrading enzyme (UCDEN) is inhibited by NEM, consistent with its classification as a cysteine protease or a protease requiring reduced sulfhydryl groups for activity [5]. In contrast, the smaller 670 kDa protease, which is not required for ATP-dependent proteolysis and hydrolyzes proteins that cannot be conjugated to ubiquitin, is actually stimulated by NEM treatment. This contrasting sensitivity highlights fundamental biochemical differences between these co-existing proteolytic complexes and underscores the value of NEM as a tool for distinguishing their individual contributions to overall proteolysis.
Experimental Protocols & Methodologies
Reticulocyte Lysate Preparation and Fractionation
The standard methodology for investigating ATP-dependent proteolysis begins with preparation of rabbit reticulocyte lysate and its subsequent fractionation into functionally distinct biochemical components.
Protocol Steps:
- Lysate Preparation: Collect rabbit reticulocytes and lyse cells in low-ionic-strength buffer (e.g., 1-5 mM MgCl₂, 1 mM DTT, pH 7.5-8.0) followed by centrifugation at 100,000 × g for 60 minutes to obtain the soluble cytosolic fraction [5] [3].
DEAE Chromatography: Apply the cytosolic fraction to a DEAE-cellulose column equilibrated with low-salt buffer. Elute with a linear NaCl gradient (0-0.5 M) to separate the ubiquitin conjugation system (eluting at approximately 0.15-0.25 M NaCl) from the proteolytic components [5].
Ammonium Sulfate Fractionation: Precipitate proteolytic activities by sequential ammonium sulfate fractionation. The UCDEN activity precipitates at 0-38% saturation, while the non-essential 670 kDa protease precipitates at 40-80% saturation [5].
Gel Filtration Chromatography: Further purify precipitated fractions by size exclusion chromatography (e.g., Sephacryl S-300 or S-400) to isolate high molecular weight protease complexes (>450 kDa) [5].
Standard ATP-Dependent Proteolysis Assay
The foundational assay for monitoring ATP-dependent proteolysis utilizes radiolabeled protein substrates to quantify degradation rates.
Protocol Steps:
- Substrate Preparation: Prepare ¹²⁵I-labeled bovine serum albumin (BSA) or incorporate amino acid analogs (e.g., 2-amino-3-chlorobutyric acid) into proteins to generate abnormal substrates recognized by the UPS [23] [3].
Reaction Conditions: In a standard 100-250 µL reaction volume, combine reticulocyte fraction(s), 1-2 mg/mL substrate protein, 2 mM ATP, 10 mM MgCl₂, 50 mM Tris-HCl (pH 7.8), 1 mM DTT, and an ATP-regenerating system (e.g., 10 mM creatine phosphate, 0.1 mg/mL creatine phosphokinase) [5] [3].
Inhibitor Addition: Add inhibitors at specified concentrations: hemin (25-50 µM), vanadate (50-100 µM), or N-ethylmaleimide (0.1-1 mM). Include appropriate solvent controls.
Incubation and Measurement: Incubate at 37°C for 30-120 minutes. Terminate reactions by adding trichloroacetic acid (TCA) to 10% final concentration. Measure TCA-soluble radioactivity by gamma counting as an index of protein degradation [23] [3].
Data Analysis: Calculate ATP-dependent proteolysis by subtracting values from parallel reactions without ATP. Express inhibitor effects as percentage inhibition relative to uninhibited controls.
Ubiquitin Conjugation Assay
This specialized assay specifically monitors the first step in the UPS pathway independently of downstream degradation.
Protocol Steps:
- Reaction Conditions: Combine the conjugation fraction (DEAE-purified), ¹²⁵I-ubiquitin, substrate protein, 2 mM ATP, 10 mM MgCl₂, and 50 mM Tris-HCl (pH 7.8) in a total volume of 50-100 µL [23].
Inhibitor Testing: Include hemin (25-50 µM) or other inhibitors to assess their specific effects on conjugation separate from degradation.
Analysis: Terminate reactions with SDS-PAGE sample buffer. Separate proteins by SDS-PAGE and visualize high molecular weight ubiquitin conjugates by autoradiography or Western blotting [23].
Interpretation: Hemin typically reduces conjugation by approximately 50% while completely blocking degradation, confirming its primary site of action at the degradation step [23] [24].
Pathway Diagrams and Experimental Workflows
Ubiquitin-Proteasome System with Inhibitor Targets
The following diagram illustrates the sequential steps of the ATP-dependent ubiquitin-proteasome system in reticulocyte lysate, with specific inhibitor targets indicated:
Experimental Workflow for Inhibitor Characterization
This workflow outlines the standard experimental approach for characterizing inhibitors in the reticulocyte lysate system:
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents for UPS Inhibition Studies
| Reagent | Function/Application | Key Characteristics |
|---|---|---|
| Rabbit Reticulocyte Lysate | Source of ATP-dependent proteolytic system | Contains complete ubiquitin conjugation and degradation machinery; can be fractionated [5] |
| ATP-Regenerating System | Maintains constant ATP levels during assays | Typically includes creatine phosphate and creatine phosphokinase [3] |
| ¹²⁵I-Labeled Substrate Proteins | Proteolysis quantification | BSA or abnormal proteins containing amino acid analogs; TCA-soluble radioactivity measures degradation [23] [3] |
| ¹²⁵I-Ubiquitin | Conjugation assay substrate | Tracks ubiquitin conjugation to proteins independently of degradation [23] |
| DEAE-Cellulose | Chromatographic separation | Separates conjugation factors from proteolytic components [5] |
| Ammonium Sulfate | Protease fractionation | Precipitates UCDEN at 0-38% saturation; 670 kDa protease at 40-80% saturation [5] |
Research Implications and Future Directions
The characterization of hemin, vanadate, and N-ethylmaleimide has provided fundamental insights into the UPS that continue to resonate through modern biomedical research. The differential inhibition patterns established that ubiquitin conjugation and substrate degradation are biochemically separable processes with distinct enzymatic requirements and regulatory mechanisms [5] [23] [25]. This conceptual framework directly enabled the subsequent identification and purification of the 26S proteasome as the molecular machine responsible for ubiquitin-conjugate degradation [1] [26].
Contemporary research continues to build upon these foundational inhibitor studies. The recognition that UPS dysregulation contributes to cancer, neurodegenerative diseases, and cardiovascular disorders has stimulated intensive efforts to develop targeted UPS modulators [1]. Current investigations are exploring natural metabolites, including circulating polyphenol-derived compounds such as valerolactones and urolithins, as potential proteasome modulators with therapeutic applications [27]. These modern approaches benefit greatly from the established experimental paradigms and mechanistic insights generated through earlier inhibitor studies using the reticulocyte lysate system.
Future research directions include developing increasingly specific inhibitors that target distinct proteasome subpopulations or regulatory particles, building upon the recognition that multiple forms of both 20S multicatalytic complexes and 26S ubiquitin/ATP-dependent proteases exist in rabbit reticulocyte lysate [26]. The continued integration of classic biochemical approaches with modern structural biology and chemical biology techniques promises to yield new generations of UPS-targeted therapeutics with enhanced specificity and reduced off-target effects.
Practical Implementation: RRL Proteolysis Assays in Contemporary Research
Rabbit Reticulocyte Lysate (RRL) is a premier cell-free protein expression system derived from the lysates of immature red blood cells from rabbits, which are naturally engaged in high rates of hemoglobin synthesis [28]. These systems are a cornerstone of molecular biology, providing a versatile platform for in vitro translation of a wide array of viral, prokaryotic, and eukaryotic mRNAs [28]. A critical distinction in these systems lies in their pre-treatment: untreated RRL contains endogenous mRNAs, while nuclease-treated RRL has been processed with micrococcal nuclease to degrade these endogenous mRNAs, thereby reducing background translation and favoring the synthesis of proteins from exogenously added templates [29] [28]. The choice between these systems is fundamental, as it significantly influences the physiological relevance of the experimental environment, particularly for research focused on ATP-dependent proteolysis and the intricate regulation of protein degradation pathways.
Comparative Analysis: Untreated vs. Nuclease-Treated RRL
The decision to use an untreated or nuclease-treated RRL system hinges on the specific scientific question. The table below summarizes the core characteristics, advantages, and optimal applications of each system.
Table 1: Direct Comparison of Untreated and Nuclease-Treated RRL Systems
| Feature | Untreated RRL | Nuclease-Treated RRL |
|---|---|---|
| Endogenous mRNA | Present (e.g., globin, lipoxygenase) [29] | Degraded via micrococcal nuclease [28] |
| Background Translation | Higher, due to endogenous mRNAs [29] | Very low, enabling high signal-to-noise for exogenous templates [28] |
| Key Advantage | Recapitulates a 'near to physiological' environment; supports cap/poly(A) tail synergy [29] | Ideal for controlled expression of a single exogenous protein [28] |
| Primary Application | Studying regulated translation, mRNA circularization, and competitive translation [29] | Standard protein expression, identification of mRNA products, and functional proteomics [28] |
| Proteolysis Research Context | Superior for studying endogenous ubiquitin-dependent proteolysis in a more native context [30] | Useful for controlled studies on the degradation of specific, in vitro synthesized substrate proteins [30] |
Essential Supplementations for RRL Systems
To function effectively in vitro, both untreated and nuclease-treated RRL require supplementation with key components that mimic the intracellular environment. These additions provide energy, building blocks, and regulatory factors necessary for efficient translation and proteolysis.
Table 2: Essential Supplementations for RRL Systems
| Supplementation | Function | Typical Concentration | Notes |
|---|---|---|---|
| Energy System | |||
| Creatine Phosphate | ATP regeneration substrate [29] [28] | 5 mg/mL [29] | |
| Creatine Phosphokinase | Enzyme for ATP regeneration from creatine phosphate [29] [28] | 25 µg/mL [29] | |
| Cofactors & Regulators | |||
| Hemin | Prevents activation of heme-regulated eIF-2α kinase (HRI), avoiding translation inhibition [29] [28] | 25 µM [29] | Critical for maintaining high translation efficiency |
| Building Blocks | |||
| Amino Acid Mix | Provides substrates for protein synthesis [29] | 20 µM (minus methionine) [29] | Often used with labeled methionine for detection |
| Bovine Liver tRNAs | Balances tRNA populations, optimizing codon usage for a wider range of mRNAs [29] [28] | 50 µg/mL [29] | |
| Salts | |||
| KCl & MgCl₂ | Optimizes ionic conditions for translation initiation and elongation [29] | 75 mM and 0.5 mM, respectively [29] | Concentrations may require optimization |
RRL in ATP-Dependent Proteolysis Research
The RRL system is not only a powerful tool for protein synthesis but also an established model for studying ATP-dependent proteolysis, the primary pathway for targeted protein degradation in eukaryotic cells. This system contains the necessary machinery, including the ubiquitin-proteasome system, which recognizes and degrades proteins tagged with polyubiquitin chains [6]. The proteasome and other ATP-dependent proteases are complex molecular machines that unfold and translocate substrate proteins into an internal chamber for hydrolysis, a process fueled by ATP [6].
Research has demonstrated key differences in proteolytic function between RRL and other systems. For instance, a comparative study found that a retinal pigment epithelial (RPE) cell line supernatant showed a more pronounced ubiquitin-dependent enhancement in degrading rod outer segment proteins compared to RRL [30]. This highlights the value of selecting the appropriate in vitro system for specific proteolysis questions. The untreated RRL, with its more physiological milieu, may provide a more accurate picture of how ubiquitin-dependent degradation occurs in a competitive cellular environment.
Diagram 1: Ubiquitin-proteasome pathway.
Detailed Experimental Protocols
Preparation of a Functional Untreated RRL System
The following protocol is adapted from studies utilizing untreated RRL to recapitulate physiological translation and proteolysis [29].
- Supplement Preparation: Prepare a master mix containing the following components to be added to 1 mL of commercial untreated RRL (e.g., from Promega):
- Lysate Supplementation: Thaw the untreated RRL on ice. Gently add the master mix to the lysate and mix by pipetting slowly. Avoid introducing air bubbles.
- Aliquoting and Storage: Dispense the supplemented lysate into small, single-use aliquots. Flash-freeze the aliquots in liquid nitrogen and store them at -70°C or below to preserve activity.
Protocol for ATP-Dependent Proteolysis Assay
This protocol outlines a method for studying ATP- and ubiquitin-dependent degradation of substrates in the RRL system, based on established comparative methodologies [30].
- Reaction Setup: For a standard 50 µL degradation reaction, combine the following on ice:
- 35 µL of supplemented untreated or nuclease-treated RRL.
- 1-2 µL of your substrate (e.g., in vitro translated, radiolabeled protein or purified protein).
- An ATP-regenerating system (if not already present in the supplementation). This typically includes 2 mM ATP, 10 mM creatine phosphate, and 0.1 mg/mL creatine phosphokinase.
- Optionally, include 12-120 µM ubiquitin to stimulate ubiquitin-dependent pathways [30].
- Controls: Prepare control reactions containing:
- An ATP-depleting system (e.g., apyrase or hexokinase/glucose) to confirm ATP dependence.
- Specific inhibitors of the ubiquitin-proteasome pathway, such as hemin or vanadate, to confirm the mechanism [30].
- Incubation: Transfer the reaction tubes to a 30°C or 37°C water bath and incubate for 30-120 minutes.
- Termination and Analysis: Stop the reactions by adding 2x SDS-PAGE loading buffer and heating to 95°C for 5 minutes. Analyze the degradation of the substrate by:
- SDS-PAGE and Autoradiography: If the substrate is radiolabeled, run the samples on a gel, dry it, and expose it to a phosphorimager screen or X-ray film. Quantify the band intensity to determine the rate of substrate degradation.
- Western Blotting: For non-radiolabeled substrates, use specific antibodies to detect the full-length substrate and its degradation products.
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for RRL and Proteolysis Research
| Reagent / Material | Function in Research | Example in Context |
|---|---|---|
| Untreated RRL | Creates a competitive, physiological environment for studying translation and endogenous proteolysis [29]. | Used to demonstrate cap/poly(A) tail synergy in translation initiation [29]. |
| Nuclease-Treated RRL | Enables high-efficiency, specific expression of a single exogenous protein with low background [28]. | Used to express and then degrade a specific rod outer segment protein in proteolysis assays [30]. |
| Ubiquitin | Key regulator of proteasome targeting; its addition stimulates ATP-dependent degradation of specific substrates [30] [6]. | Addition of 12 µM ubiquitin to RPE supernatant stimulated degradation of multiple substrates by ≥3-fold [30]. |
| ATPγS (ATP analog) | A non-hydrolyzable ATP analog used to probe the energy dependence of a process. | Expected to inhibit both the ubiquitination cascade and proteasome activity, serving as a critical control. |
| Hemin | An inhibitor of the heme-regulated eIF-2α kinase and of ATP-/ubiquitin-dependent proteolysis [30] [28]. | Completely inhibited ATP-dependent degradation of ROS proteins in reticulocyte lysate [30]. |
| Proteasome Inhibitors (e.g., MG132) | Specific inhibitors of the proteasome's proteolytic activity, used to confirm proteasome involvement. | Useful for distinguishing proteasome-dependent degradation from other protease activities in the lysate. |
Proteolysis, the enzymatic cleavage of peptide bonds, is a critical biochemical process for cellular function, regulating everything from cell proliferation and death to immune response and digestion [31] [32]. The rabbit reticulocyte lysate system has been a foundational model for studying one specialized class of these events: ATP-dependent proteolysis [3]. This system is responsible for the selective degradation of abnormal proteins, a crucial quality-control mechanism in cells [3]. Early work demonstrated that reticulocyte lysates contain a soluble, non-lysosomal proteolytic system that requires ATP and can be inhibited by agents like N-ethylmaleimide and iodoacetamide [3]. Subsequent research revealed that this system comprises multiple high molecular weight proteases, including a ~1500 kDa enzyme essential for degrading ubiquitin-protein conjugates [5]. Mastery of standard proteolysis protocols is therefore indispensable for researchers investigating protein turnover, protease function, and drug mechanisms. This guide provides an in-depth technical framework for setting up proteolysis experiments, optimizing incubation parameters, and selecting detection methods, with specific emphasis on the context of ATP-dependent proteolysis research.
Core Principles of Proteolytic Systems
The Rabbit Reticulocyte Lysate System
The ATP-dependent proteolytic system in rabbit reticulocytes has served as a key model for understanding intracellular protein degradation. This system is characterized by several defining features:
- Location and ATP Dependence: It is a soluble, non-lysosomal system located in the 100,000 × g supernatant fraction of the lysate. Its activity is stimulated several-fold by ATP, while ADP provides a slight stimulation, and AMP and cAMP have no significant effect [3].
- Inhibitor Profile: The proteolysis in this system is inhibited by agents such as N-ethylmaleimide, iodoacetamide, and L-1-tosylamido-2-phenylethylchloromethyl ketone (TPCK), indicating a reliance on specific catalytic residues and a distinct mechanism from lysosomal degradation [3].
- Enzymatic Complexity: The system can be fractionated into multiple components. A "Conjugation Fraction" (30-300 kDa) handles the ATP-dependent conjugation of ubiquitin to substrates, while a high mass fraction (>450 kDa) is necessary for the hydrolysis of the conjugated proteins [5]. This high mass fraction contains at least two distinct proteases: a ~1500 kDa protease that degrades ubiquitin conjugates (UCDEN) and requires ATP, and a ~670 kDa protease that does not require ATP or ubiquitin [5].
Signaling Pathway of ATP-Dependent Proteolysis
The following diagram illustrates the core biochemical pathway of the ATP-dependent ubiquitin-proteasome system as characterized in rabbit reticulocytes.
Figure 1: ATP-dependent ubiquitin-proteasome pathway in rabbit reticulocytes. The pathway involves ubiquitin conjugation followed by degradation via a high-molecular-weight protease complex.
Experimental Setup and Protocol
Reagent Preparation
A successful proteolysis experiment begins with the careful preparation of reagents. The following buffers are commonly used in proteolysis studies, including those involving reticulocyte lysates.
Table 1: Essential Buffers for Proteolysis Studies
| Buffer Name | pH | Key Components | Primary Function |
|---|---|---|---|
| Spin Buffer [33] | 6.8 | 50 mM MOPS, 25 mM NaCl | Provides a stable ionic environment for spin-labeling and EPR studies. |
| HEPES Buffer [33] | 7.4 | 50 mM HEPES, 50 mM NaCl | Maintains physiological pH for a wide range of proteolytic reactions. |
| Lysis Buffer [34] | 7.4 | 50 mM HEPES, 150 mM NaCl, 1% Triton X-100 (EDTA-free protease inhibitors optional) | Efficiently extracts proteins while preserving native conformations and ligand-binding capabilities. |
Sample Preparation
The initial quality of the biological sample is paramount. For cell-based systems like reticulocyte lysates, the protocol involves:
- Harvesting and Lysis: Cells should be harvested and rinsed with cold PBS. Lysis is then performed using an ice-cold, appropriate buffer (e.g., Table 1). For tissues, mechanical homogenization under cold conditions is required [34].
- Clarification: The lysate is centrifuged at 14,000 × g for 15 minutes at 4°C to remove insoluble debris. The clear supernatant is carefully collected [34].
- Quantification: The total protein concentration of the lysate should be determined using an assay like the BCA assay. Lysates are typically normalized to a consistent concentration (e.g., 1–3 mg/mL) for reproducible experimental conditions [34].
Protease Immobilization (Optional)
A powerful technique to simplify downstream analysis is protease immobilization. Metal-Organic Materials (MOMs) like Ca-BPDC can be used for this purpose via co-crystallization [33].
- Preparation of BPDC-Na₂: Biphenyl-4,4′-dicarboxylic acid (BPDC-COOH) is dissolved in a 1:2 molar ratio with NaOH in water. After vigorous stirring for 2 hours, the soluble BPDC-Na₂ is precipitated with cold 2-isopropanol, collected by filtration, and dried [33].
- Immobilization: Stock solutions of CaCl₂ and BPDC-Na₂ are mixed in the presence of the target protease, allowing the enzyme to be trapped within the growing MOM crystals. This enables the physical separation of the protease from the reaction mixture by centrifugation, preventing self-degradation and simplifying the mass spectrometric analysis of substrate cleavage products [33].
Incubation Parameters and Optimization
The conditions under which proteolysis occurs are critical for the efficiency and specificity of the reaction. The key parameters to control and optimize are summarized in the table below.
Table 2: Key Incubation Parameters for Proteolysis Experiments
| Parameter | Typical Range | Impact on Proteolysis | Considerations for Reticulocyte System |
|---|---|---|---|
| Temperature | 37°C (for most assays); 4°C for unstable ligands [34] | Higher temperatures increase reaction rates but may denature proteins. | The native ATP-dependent system operates at physiological temperature [3]. |
| Time | 10–30 min (kinetic); Overnight (for complete digestion) [35] [34] | Duration must be optimized to balance completeness of digestion with protease stability. | Time courses can reveal early vs. late cleavage events during processes like apoptosis [32]. |
| ATP | 1–5 mM [3] | Essential for the activity of the ubiquitin-proteasome system. | ATP concentration is a critical variable; the system shows maximal stimulation with ATP over ADP or AMP [3]. |
| Protease:Substrate Ratio | 1:20 to 1:100 (w/w) for trypsin [35] | Higher ratios accelerate digestion but may increase non-specific cleavage. | The endogenous protease concentration is fixed in lysates; activity is modulated by ATP and inhibitors. |
| pH | 7.4–7.8 (physiological); specific proteases have unique optima [3] [36] | Drastically affects protease activity and protein conformation. | The ATP-dependent system in reticulocytes has a pH optimum of 7.8 [3]. |
Compound Incubation for Binding Studies
When studying the effect of small molecules (e.g., potential drugs or inhibitors) on proteolysis:
- The lysate is split into compound-treated and vehicle-treated control groups.
- The small molecule is added to the desired final concentration (commonly 1–10 μM), ensuring the solvent concentration (e.g., DMSO) is matched and does not exceed 1% (v/v) in all samples [34].
- Incubation is typically performed at room temperature for 15 minutes to 2 hours to allow for ligand-target engagement [34].
Detection and Analysis Methods
A variety of methods exist to monitor proteolytic activity and identify cleavage products. The choice of method depends on the specific research question, whether it is measuring general activity, identifying specific cleavage sites, or probing structural changes.
Table 3: Comparison of Proteolysis Detection and Analysis Methods
| Method | Principle | Key Applications | Throughput |
|---|---|---|---|
| Fluorometric Assays (e.g., Red Protease Kit) [36] | Protease cleaves quenched substrate, releasing a fluorescent signal. | Screening protease activity/inhibitors; quantifying contamination. | High |
| Limited Proteolysis-Coupled MS (LiP-MS) [37] | Non-specific protease under native conditions cleaves accessible regions; peptides analyzed by MS. | Identifying protein structural changes; drug target discovery. | Medium-High |
| Gel-Based Methods (e.g., PROTOMAP) [32] | Proteins separated by SDS-PAGE; cleavage observed by band shifts or disappearance. | Identifying protein substrates and approximating cleavage sites. | Low-Medium |
| N-Terminal Enrichment (e.g., COFRADIC, Subtiligase) [32] | Isolation and MS-identification of native or neo-N-terminal peptides. | Unbiased discovery of cleavage sites and substrates. | Medium |
| EPR/SDSL with MS [33] | Site-directed spin labeling tracks local molecular motion via EPR; MS confirms sequences. | Residue-level, time-resolved analysis of proteolytic kinetics and cleavage sites. | Low-Medium |
Detailed Protocol: Limited Proteolysis-Coupled Mass Spectrometry (LiP-MS)
LiP-MS is a powerful method for detecting protein structural changes on a proteome-wide scale [37]. The workflow is illustrated below and involves the following steps:
Figure 2: Limited Proteolysis-Coupled Mass Spectrometry (LiP-MS) workflow for detecting protein structural changes.
- Perturbation and Lysis: Subject cells or tissues to the condition of interest (e.g., drug treatment, pH change). Prepare the proteome extract via lysis [37].
- Limited Proteolysis: Subject the lysate to a non-specific protease (e.g., pronase) under native conditions. This step is critical, as the protease will only cleave in solvent-accessible and flexible regions of proteins. The digestion time and protease concentration must be carefully optimized to achieve "limited" cleavage [37].
- Denaturation and Complete Digestion: Halt the limited proteolysis and denature the proteins. Then, perform a complete digestion with a sequence-specific protease like trypsin under denaturing conditions. This second step generates a complex mixture of peptides suitable for MS analysis [37].
- MS Analysis and Quantification: Analyze the peptides using liquid chromatography-tandem mass spectrometry (LC-MS/MS). Use label-free quantification to compare samples from different perturbations. Peptides whose abundance changes between conditions represent protein regions that have undergone structural alterations, as their accessibility to the non-specific protease was affected [37].
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Research Reagents for Proteolysis Studies
| Reagent / Tool | Function / Principle | Example Use Case |
|---|---|---|
| Trypsin, Mass Spectrometry Grade [35] | Highly specific serine protease; cleaves C-terminal to Arg/Lys. Gold standard for bottom-up proteomics. | Digestion of proteins into peptides for LC-MS/MS identification and quantification. |
| Trypsin/Lys-C Mix [35] | Combination of trypsin and Lys-C; reduces missed cleavages, especially in folded proteins. | Two-step digestion of difficult-to-digest proteins; improved sequence coverage. |
| Red Protease Detection Kit [36] | Uses quenched TAMRA-labeled casein; proteolysis generates fluorescent signal. | High-throughput screening of protease activity or inhibitor potency. |
| Pronase [34] [37] | A mixture of broad-specificity proteases. | Used in DARTS and LiP-MS assays to probe global protein structure and stability. |
| Sequencing Grade Modified Trypsin [35] | Reductive methylation suppresses autolysis, ensuring high cleavage specificity. | Applications requiring stringent specificity over long incubation periods. |
| Metal-Organic Materials (MOMs) [33] | Platform for protease immobilization via co-crystallization. | Enables easy separation of protease from reaction products, simplifying MS analysis. |
Troubleshooting and Data Interpretation
A common challenge in quantitative proteomics is incomplete proteolysis, which can lead to inaccurate quantification. For example, when using the QconCAT method for absolute quantification, different peptides from the same protein can yield different concentration values if digestion is not complete and the standard and analyte peptides are in different sequence contexts [38]. It is essential to optimize digestion conditions (time, protease ratio, denaturants) and to assess the completeness of digestion by monitoring the presence of mis-cleaved peptides.
When working with the ATP-dependent reticulocyte system, consistency in ATP regeneration and the use of appropriate inhibitors are critical. The system is sensitive to N-ethylmaleimide, which can be used to confirm the ATP-dependent component of proteolysis [3] [5]. Data interpretation should always account for the complex nature of the lysate, where multiple proteolytic activities coexist.
The ATP-dependent proteolytic system in rabbit reticulocyte lysate represents a foundational model for understanding the controlled degradation of cellular proteins. This system is responsible for the selective turnover of abnormal proteins, a process essential for maintaining cellular homeostasis and protein quality control [3]. The seminal discovery of a soluble, ATP-dependent proteolytic complex in reticulocytes established a non-lysosomal pathway for the selective recognition and degradation of damaged or misfolded polypeptides [3]. This article examines the molecular mechanisms governing substrate specificity within this system, focusing on its capacity to identify aberrant proteins, process structurally unique substrates like rod outer segment proteins, and hydrolyze well-characterized model substrates. We will explore the quantitative parameters of these degradation processes, detail critical experimental methodologies for their study, and visualize the core mechanisms, all within the context of ongoing research utilizing rabbit reticulocyte lysate.
Core System and Quantitative Parameters
The ATP-dependent proteolytic machinery in rabbit reticulocyte lysate comprises high molecular weight complexes with distinct biochemical properties. The key proteases identified include a 26 S protease (~1,000,000 Da) that requires ATP, and a 20 S protease (~700,000 Da) which is ATP-independent [13]. The following table summarizes the core quantitative data characterizing this system and its substrate degradation profiles.
Table 1: Key Quantitative Parameters of the Reticulocyte ATP-Dependent Proteolytic System
| Parameter | 26 S Protease | 20 S Protease (Multicatalytic Proteinase Complex) | Experimental Context |
|---|---|---|---|
| Molecular Weight | 1,000,000 ± 100,000 Da [13] | 700,000 ± 20,000 Da [13] | Sedimentation, gel filtration |
| Subunit Composition | Multisubunit complex [13] | 8-10 subunits (Mr 21,000-32,000) [13] | Nondenaturing PAGE, gel filtration |
| ATP Dependence | Required for protein degradation [13] | Not required [13] | Degradation of 125I-lysozyme-ubiquitin conjugates |
| Specific Activity (Model Substrate) | Stimulated degradation of Suc-LLVY-MCA [13] | Degrades Suc-LLVY-MCA [13] | Peptide hydrolysis (4-methylcoumaryl-7-amide peptides) |
| Inhibitor Profile | Inhibited by hemin, thiol reagents, chymostatin, leupeptin [13] | Inhibited by hemin, thiol reagents, chymostatin, leupeptin [13] | In vitro protease assays |
| Degradation Half-Life (Abnormal Protein) | ~15 minutes (ClAbu-globin) [3] | Not specifically determined | In intact reticulocytes and cell-free extracts |
Table 2: Substrate Specificity and Degradation Triggers
| Substrate Class | Recognition Signal / Degron | Key Degradation Stimulus | Protease System |
|---|---|---|---|
| Abnormal Proteins | Exposed unstructured regions, hydrophobic patches [6] | Amino acid analog incorporation (e.g., 2-amino-3chlorobutyric acid) [3] | ATP-dependent 26S Proteasome [3] |
| Ubiquitinated Model Substrates | K48-linked polyubiquitin chain (≥4 ubiquitins) [6] | Covalent ubiquitination by E1-E2-E3 enzyme cascade [6] | 26S Proteasome (19S RP recognizes ubiquitin) [6] |
| Native Regulatory Proteins | Specific degron sequences + unstructured initiation site [6] | Allosteric activation, post-translational modification | ATP-dependent Proteases |
| Rod Outer Segment Proteins (e.g., Rhodopsin) | Not fully elucidated; part of daily phagocytic turnover [39] | Phagocytosis of OS tips by RPE, followed by lysosomal degradation [39] | Phagolysosomal degradation (ATP-dependent in RPE) [39] |
Mechanisms of Substrate Recognition and Degradation
The specificity of ATP-dependent proteolysis is not a function of the proteolytic active sites themselves, which show little intrinsic sequence preference, but is instead conferred by sophisticated targeting mechanisms that deliver specific substrates to the protease [6]. In the ubiquitin-proteasome pathway, which operates in the reticulocyte system, substrate specificity is primarily achieved through the covalent attachment of a polyubiquitin tag. This process involves a cascade of E1 (activating), E2 (conjugating), and E3 (ligating) enzymes that collectively recognize degradation signals, or degrons, on the substrate protein and assemble a chain of ubiquitin molecules linked through lysine 48 (K48) [6]. A chain of at least four ubiquitin moieties constitutes the minimal signal for efficient targeting to the 26S proteasome, where it is recognized by ubiquitin receptors such as Rpn10 and Rpt5 in the 19S regulatory particle [6].
A critical second step in degradation is the initiation of unfolding. Even a ubiquitinated protein must possess an unstructured region, often at its N- or C-terminus, to engage the ATP-dependent unfolding machinery of the proteasome's regulatory subunit [6]. This requirement ensures that the degradation of folded proteins begins at a specific, accessible point. The protease then processively unfolds the substrate and translocates the polypeptide chain into the internal proteolytic chamber.
This mechanism is highly conserved. In prokaryotes and organelles, AAA+ proteases like ClpXP perform an analogous function. ClpX, a AAA+ ATPase, recognizes specific degradation tags ("degrons") on substrates and uses ATP hydrolysis to mechanically unfold and translocate them into the ClpP peptidase for degradation [11]. The mechanical unfolding mechanism involves powerful conformational changes in the ATPase ring that pull on the substrate polypeptide, translocating it through a narrow central pore [11].
For the degradation of abnormal proteins, as studied in the classic reticulocyte model, recognition often occurs via exposed hydrophobic patches or unstructured regions that are buried in properly folded native proteins. The incorporation of amino acid analogs, such as 2-amino-3chlorobutyric acid (ClAbu) in place of valine, leads to the production of misfolded proteins that are rapidly flagged for degradation, with a half-life of approximately 15 minutes, in contrast to stable native proteins like hemoglobin [3].
It is important to distinguish this cytosolic, proteasomal degradation from the turnover of highly organized structures like rod outer segment (OS) proteins. The daily renewal of rod OS involves the phagocytosis of the OS tips by the retinal pigment epithelium (RPE) [39]. Within the RPE phagolysosomes, proteins like rhodopsin are degraded in an ATP-dependent manner, but this process is part of an organellar clearance pathway distinct from the cytosolic ubiquitin-proteasome system [39].
Experimental Protocols
Protocol 1: Purification of ATP-Dependent Proteases from Rabbit Reticulocyte Lysate
This protocol is adapted from the seminal work that first isolated the high molecular weight proteases from rabbit reticulocyte lysate [13].
Key Reagents:
- Source: Fresh or frozen rabbit reticulocyte lysate.
- Assay Substrates: 125I-alpha-casein and fluorogenic peptides like Suc-Leu-Leu-Val-Tyr-AMC (Suc-LLVY-MCA).
- Inhibitors: Hemin, N-ethylmaleimide (NEM), chymostatin, leupeptin.
- Buffers: Standard phosphate or Tris-based buffers, pH 7.0-7.8.
Methodology:
- Lysate Preparation: Start with post-ribosomal supernatant from rabbit reticulocytes, obtained by centrifugation at 100,000 × g [3] [13].
- Initial Fractionation: Subject the 100,000 × g supernatant to ammonium sulfate precipitation. The proteolytic activity of interest is typically found in the 40-70% saturation fraction.
- Size-Exclusion Chromatography: Resuspend the ammonium sulfate pellet and load onto a Sepharose 6B or similar gel filtration column. Elute with a suitable buffer (e.g., 20 mM Tris-HCl, pH 7.5). Collect fractions and monitor for protein content and ATP-stimulated protease activity.
- Ion-Exchange Chromatography: Pool active fractions from size exclusion and apply to an anion-exchange column (e.g., DEAE-Sephacel). Elute with a linear NaCl gradient (e.g., 0-0.5 M).
- Activity Assay: Monitor purification success by assaying fractions for:
- ATP-dependent degradation of 125I-labeled proteins (e.g., abnormal globin or 125I-lysozyme-ubiquitin conjugates).
- Hydrolysis of fluorogenic peptides like Suc-LLVY-AMC, which is stimulated by ATP for the 26S protease.
- Purity Assessment: Analyze final fractions by nondenaturing polyacrylamide gel electrophoresis and confirm molecular weight via sedimentation analysis (∼20 S for the core particle, ∼26 S for the ATP-dependent complex) [13].
Protocol 2: Assessing Degradation of Abnormal Proteins in Cell-Free Extracts
This protocol outlines the method for studying the degradation of experimentally-induced abnormal proteins, as described in the foundational 1977 study [3].
Key Reagents:
- Abnormal Protein Substrate: Reticulocytes that have incorporated the valine analog 2-amino-3chlorobutyric acid (ClAbu) to produce abnormal globin.
- Inhibitors: TPCK, TLCK, N-ethylmaleimide, iodoacetamide, o-phenanthroline.
- Nucleotides: ATP, ADP, AMP, cAMP.
Methodology:
- Substrate Generation: Incubate intact rabbit reticulocytes with ClAbu to generate abnormal, radiolabeled hemoglobin as the degradation substrate.
- Extract Preparation: Prepare a cell-free extract from control or analog-treated reticulocytes by lysing cells and centrifuging at 100,000 × g to obtain the soluble supernatant.
- Degradation Assay: Incubate the cell-free extract with the radiolabeled abnormal protein substrate.
- Energy Dependence: Supplement reactions with 2-5 mM ATP or other nucleotides (ADP, AMP) to test energy stimulation.
- Inhibitor Studies: Pre-incubate extracts with class-specific protease inhibitors (e.g., serine protease inhibitors like TPCK/TLCK, thiol reagents like NEM, metalloprotease inhibitors like o-phenanthroline) to characterize the protease involved.
- Quantification: Measure the release of acid-soluble radioactivity over time to quantify protein degradation. The ATP-dependent degradation of ClAbu-globin has a half-life of approximately 15 minutes in this system [3].
Pathway and Mechanism Visualization
Ubiquitin-Proteasome Pathway
The following diagram illustrates the primary pathway for targeted protein degradation in the rabbit reticulocyte lysate system.
Diagram Title: Ubiquitin-Proteasome Degradation Pathway
ATP-Dependent Protease Mechanism
This diagram depicts the conserved functional mechanism of AAA+ ATP-dependent proteases, such as the 26S proteasome and bacterial ClpXP.
Diagram Title: ATP-Dependent Protease Mechanism
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for Studying ATP-Dependent Proteolysis
| Reagent / Material | Function in Research | Specific Examples & Notes |
|---|---|---|
| Rabbit Reticulocyte Lysate | Source of ATP-dependent proteolytic activity; provides the native system for assay development and purification. | Prepared from phenylhydrazine-treated rabbits; used as crude lysate or fractionated (100,000 x g supernatant) [3] [13]. |
| Amino Acid Analogs | Induce production of abnormal protein substrates within cells or extracts to study recognition and degradation. | 2-amino-3chlorobutyric acid (ClAbu, a valine analog) used to generate abnormal globin [3]. |
| Defined Model Substrates | Quantitative assay of protease activity; fluorogenic peptides measure specific catalytic activities. | Suc-LLVY-AMC: Chymotrypsin-like activity of proteasome [13]. 125I-alpha-casein: General proteolysis [13]. 125I-lysozyme-ubiquitin conjugates: ATP-stimulated degradation [13]. |
| Nucleotides | To probe the energy dependence of the proteolytic system and characterize ATPase components. | ATP (required), ADP (weak stimulation), AMP/cAMP (no effect) [3]. |
| Class-Specific Protease Inhibitors | To pharmacologically characterize the class of protease and identify catalytic mechanisms. | Serine/Cysteine Protease Inhibitors: TPCK, TLCK, chymostatin, leupeptin [3] [13]. Thiol Reagents: N-ethylmaleimide, iodoacetamide [3]. Metalloprotease Inhibitor: o-phenanthroline [3]. |
| Ubiquitination System Components | To reconstitute the substrate targeting pathway for mechanistic studies. | Purified E1, E2, E3 enzymes, ubiquitin, and ATP for in vitro ubiquitination of substrates [6]. |
The rabbit reticulocyte lysate (RRL) has long been recognized as a foundational in vitro tool for studying ATP-dependent proteolytic pathways [4]. However, its utility extends far beyond degradation, serving as a powerful physiological environment for investigating the complex process of protein folding assisted by molecular chaperones. This technical guide focuses on leveraging the RRL system, particularly the untreated, non-nuclease-treated lysate, to study the function of the essential eukaryotic chaperonin CCT (chaperonin containing TCP-1) or TRiC (TCP-1 ring complex) [29] [40]. Unlike simplified systems, untreated RRL maintains the endogenous macromolecular complexity, energy regeneration components, and cofactor requirements that recapitulate a near-physiological environment for protein folding, making it an indispensable tool for researchers investigating proteostasis mechanisms relevant to health and disease [29].
The chaperonin CCT/TRiC represents a sophisticated folding machinery that is indispensable for cellular proteostasis. As an essential eukaryotic molecular chaperone, CCT is a multi-subunit oligomer consisting of two stacked rings, each composed of eight different but related protein subunits that form a defined cylindrical structure with a central folding chamber [40] [41]. This complex is responsible for the ATP-dependent folding of approximately 5-10% of newly synthesized cytosolic proteins, with its most prominent substrates being the abundant structural proteins actin and tubulin [40] [41]. The folding cycle involves substrate recognition, ATP-regulated conformational changes, and encapsulation within the central cavity, providing a protected environment for proper tertiary structure acquisition [42].
Theoretical Background: CCT/TRiC Structure and Function
Architectural Complexity of the CCT/TRiC Complex
CCT/TRiC belongs to the group II chaperonins and exhibits remarkable structural sophistication. Each of the eight subunits (CCTα/β/γ/δ/ε/ζ/η/θ, or Cct1p-8p in yeast) occupies a specific position within the chaperonin ring, creating a geometrically defined binding interface formed from divergent sequences within the substrate binding domains [41]. This subunit arrangement is not random; the subunits are organized in a specific clockwise orientation: Cct1p→Cct5p→Cct6p→Cct2p→Cct3p→Cct8p→Cct4p→Cct7p [40]. This precise arrangement suggests functional specialization, where different subunits may recognize distinct structural features or motifs in substrate proteins, significantly expanding the repertoire of proteins that CCT can assist [40] [41].
Similar to other chaperonins, each CCT subunit contains three principal domains: an equatorial domain that contains the nucleotide-binding pocket and facilitates most intra- and inter-subunit interactions; an apical domain that encompasses the entrance of the central cavity and contains hydrophobic residues responsible for substrate binding; and an intermediate domain that connects both and acts as a hinge for ATP-induced conformational changes [40]. Unlike the group I chaperonin GroEL, which requires the separate co-chaperonin GroES, CCT/TRiC has built-in flexible protrusions in its apical domains that function as an intrinsic lid, opening and closing the central cavity in an ATP-dependent manner [40] [42].
Functional Mechanism and Substrate Diversity
The CCT-mediated folding cycle begins with the recognition and binding of non-native substrate proteins, typically through hydrophobic and electrostatic interactions [40]. Following ATP binding and hydrolysis, the chaperonin undergoes substantial conformational changes that close the lid and encapsulate the substrate within the central cavity. This encapsulated environment serves as an "Anfinsen cage" that prevents aggregation and allows the protein to fold efficiently without interference from the crowded cellular environment [40]. After folding is complete, another ATP hydrolysis event triggers the release of the properly folded protein.
While actin and tubulin are considered obligate folding substrates that absolutely require CCT for their proper folding, the chaperonin also assists a diverse array of other proteins, including cell cycle regulators, signaling molecules, and proteins involved in complex assembly [40] [41]. The ability of CCT to facilitate the folding of such structurally diverse proteins stems from the combinatorial substrate recognition potential afforded by its hetero-oligomeric nature, where different subunit combinations can interact with distinct structural features or motifs in substrate proteins [40].
Table 1: CCT/TRiC Subunit Composition and Characteristics
| Subunit Name | Alternative Name | Molecular Weight (kDa) | Notable Features |
|---|---|---|---|
| CCTα | Cct1p | 52-65 | Apical domain can suppress huntingtin aggregation |
| CCTβ | Cct2p | 52-65 | Essential for complex assembly |
| CCTγ | Cct3p | 52-65 | Positioned between Cct2p and Cct8p in ring |
| CCTδ | Cct4p | 52-65 | Binds to dynactin complex; affects aggregate morphology |
| CCTε | Cct5p | 52-65 | Component of SRF signaling pathway; not involved in Htt aggregation |
| CCTζ | Cct6p | 52-65 | Located between Cct5p and Cct2p in ring |
| CCTη | Cct7p | 52-65 | Final subunit in clockwise arrangement |
| CCTθ | Cct8p | 52-65 | Positioned between Cct3p and Cct4p |
Experimental System: Preparing the Untreated RRL Platform
Advantages of Untreated RRL for Folding Studies
The conventional nuclease-treated RRL has been widely used for in vitro translation but fails to recapitulate important physiological aspects of translation and folding, particularly the synergistic interaction between the 5' cap structure and 3' poly(A) tail that promotes efficient translation initiation [29]. The untreated RRL preserves endogenous globins and other components that create a more competitive, physiologically relevant environment. Research has demonstrated that the untreated RRL system faithfully recreates cap/poly(A) tail synergy and is able to reproduce biological phenomena such as the selective advantage of the EMCV IRES under conditions of cap-dependent translation inhibition, which is not observed in nuclease-treated systems [29].
The preservation of endogenous components in untreated RRL extends beyond mRNA to include full complements of chaperones, co-chaperones, and folding factors that create a proteostasis network capable of assisting proper protein folding. This makes it particularly valuable for studying complex chaperone systems like CCT/TRiC that may require coordinated action with other chaperones such as Hsp70 and prefoldin for optimal function [40] [42].
Preparation of Supplemented Untreated RRL
To maximize the translational efficiency and folding capacity of untreated RRL, proper supplementation is essential. The following protocol has been established for preparing functional untreated RRL for folding studies [29]:
- Start with 1 ml of commercial untreated RRL (e.g., from Promega).
- Supplement with:
- 25 μM haemin (from Fluka)
- 25 μg/ml creatine phosphokinase (Sigma-Aldrich)
- 5 mg/ml creatine phosphate (Fluka)
- 50 μg/ml bovine liver tRNAs (Sigma-Aldrich)
- 3 mM d-glucose (Sigma-Aldrich)
The supplemented lysate should be aliquoted and stored at -80°C to preserve activity. Each batch should be quality-controlled for translational efficiency and folding capacity using standard substrates.
Table 2: Research Reagent Solutions for RRL-Based Folding Studies
| Reagent | Function | Working Concentration | Key Characteristics |
|---|---|---|---|
| Untreated RRL | Provides chaperone system and folding environment | 50-80% of reaction volume | Contains endogenous CCT, Hsp70, and other chaperones |
| Haemin | Regulates eIF2α phosphorylation and protein synthesis | 25 μM | Prevents inhibition of translation initiation |
| Creatine Phosphate/Creatine Phosphokinase | ATP regeneration system | 5 mg/ml / 25 μg/ml | Maintains constant ATP levels for chaperone function |
| Bovine Liver tRNAs | Enhances translation efficiency | 50 μg/ml | Supplements tRNA pool for diverse codon usage |
| Amino Acid Mix (minus methionine) | Provides building blocks for translation | 20 μM each | Can be supplemented with labeled amino acids for detection |
| ATP/Mg²⁺ | Energy source for chaperones | 1-3 mM ATP, 0.5-2 mM Mg²⁺ | Essential for CCT ATPase activity and folding cycle |
Methodologies for Investigating CCT/TRiC Function in RRL
Analyzing Folding of Specific Substrates
To monitor CCT/TRiC-mediated folding of specific substrates in the RRL system, researchers can employ several complementary approaches:
Radiolabeled Folding Assays: Substrate proteins can be synthesized directly in the RRL system in the presence of [³⁵S]-methionine or translated exogenously and then introduced into the folding-competent RRL. The folding status can be assessed by native gel electrophoresis, limited proteolysis, or immunoprecipitation with conformation-specific antibodies. For actin and tubulin, proper folding can be further verified by their ability to polymerize into filaments [40] [41].
Functional Folding Assays: For enzymatic substrates, folding efficiency can be quantified by measuring the acquisition of catalytic activity over time. For example, when studying the folding of firefly luciferase or other reporter proteins, the appearance of enzymatic activity provides a direct measure of successful folding [29].
Co-immunoprecipitation of CCT-Substrate Complexes: To capture transient interactions between CCT and its folding substrates, researchers can perform co-immunoprecipitation using antibodies against specific CCT subunits at different time points during folding reactions. This approach can reveal the kinetics of substrate binding and release throughout the folding cycle [41].
Perturbation Strategies for Functional Analysis
Several specific perturbation approaches can be employed in the RRL system to establish CCT's role in folding particular substrates:
ATP Depletion and Regeneration Systems: Since CCT function is ATP-dependent, manipulating ATP levels through apyrase treatment or using non-hydrolyzable ATP analogs can specifically inhibit CCT-mediated folding. Conversely, optimizing ATP regeneration systems can enhance folding efficiency [4].
Targeted Protein Depletion: Immunodepletion of specific CCT subunits or associated co-chaperones (e.g., prefoldin) from the RRL before conducting folding assays can establish requirements for these components in the folding of specific substrates [41].
Chemical Inhibitors: While no highly specific CCT inhibitors are available, general chaperone inhibitors such as geldanamycin (targeting Hsp90) can be used to investigate the cooperation between CCT and other chaperone systems in the RRL [40].
CCT/TRiC in Disease and Therapeutic Applications
Role in Protein Aggregation Disorders
Compelling evidence has established connections between CCT/TRiC dysfunction and protein aggregation diseases. RNAi screens in C. elegans identified several CCT subunits as modulators of polyglutamine tract aggregation, and subsequent studies have confirmed CCT's role in suppressing the aggregation of huntingtin protein containing expanded polyglutamine tracts [41]. The mechanistic basis for this protection appears to involve direct binding of the CCT apical domain, particularly from CCTα and CCTδ subunits, to helical segments in huntingtin that act as switches for initiating aggregation [41].
The therapeutic potential of CCT subunits has been demonstrated in cellular models, where application of the recombinant CCT1(α) apical domain reduced inclusion body formation in PC12 cells expressing aggregating huntingtin variants and improved respiratory function in affected striatal cells [41]. Notably, the CCT1(α) apical domain was able to cross the plasma membrane, suggesting potential for therapeutic development [41].
Implications for Drug Development
The ability to study CCT function in the context of untreated RRL provides valuable opportunities for drug discovery, particularly for neurodegenerative diseases characterized by protein aggregation. The RRL system can serve as a platform for:
Screening Small Molecule Modulators: Identifying compounds that enhance CCT-mediated folding or reduce aggregation of disease-related proteins.
Testing Chaperone-Based Therapeutics: Evaluating the efficacy of chaperone subunits or mimetics in preventing aggregation in a physiologically relevant environment.
Investigating Combination Therapies: Exploring synergistic effects between CCT and other proteostasis network components that could be targeted therapeutically.
Diagram 1: CCT/TRiC-Mediated Folding vs. Aggregation Pathway. This diagram illustrates the competitive pathways between productive protein folding assisted by CCT/TRiC and the formation of toxic aggregates linked to neurodegenerative diseases.
Advanced Applications and Future Directions
Integration with Modern Biopharmaceutical Trends
The use of physiologically relevant systems like untreated RRL for studying protein folding aligns with several advancing trends in biopharmaceutical research. The industry is increasingly emphasizing physiological relevance in early discovery to improve translational success rates [43]. Furthermore, the integration of artificial intelligence and machine learning in structural biology, exemplified by tools like AlphaFold for protein structure prediction, creates opportunities for combining computational approaches with experimental validation in systems like RRL [43].
The growing focus on personalized therapeutics for neurodegenerative disorders also underscores the importance of understanding patient-specific variations in chaperone function and proteostasis network efficiency that could be modeled and studied using ex vivo systems [43].
Technical Advancements and Methodological Innovations
Future applications of the RRL system for studying CCT/TRiC and other chaperones will likely benefit from several technical developments:
Single-Molecule Visualization: Advanced microscopy techniques could be adapted to monitor real-time chaperone-substrate interactions in the RRL environment.
Multi-Omics Integration: Combining the RRL system with proteomic, transcriptomic, and structural approaches could provide comprehensive insights into chaperone networks.
Microfluidics and High-Throughput Screening: Miniaturization and automation of RRL-based folding assays would enable larger-scale investigations of chaperone function and modulator screening.
The untreated rabbit reticulocyte lysate represents a sophisticated experimental platform that extends far beyond its traditional use in degradation studies, offering a physiologically relevant environment for investigating complex chaperone systems like CCT/TRiC. By maintaining endogenous chaperones, cofactors, and energy systems, untreated RRL provides a unique window into the mechanisms of protein folding and proteostasis maintenance. As research continues to illuminate the critical role of CCT/TRiC in health and disease, particularly in protein aggregation disorders, the utilization of untreated RRL as an experimental system will remain an essential approach for basic mechanistic studies and therapeutic development alike.
The rabbit reticulocyte lysate (RRL) system, a foundational in vitro model for studying intracellular protein degradation, has been instrumental in elucidating the ubiquitin-proteasome system (UPS). This ATP-dependent proteolytic pathway is crucial for maintaining cellular homeostasis by degrading abnormal proteins and regulating key cellular processes. The RRL system provides a biologically relevant environment for drug discovery, enabling the identification and characterization of modulators that can either enhance or inhibit targeted proteolysis. This guide details the application of the RRL ATP-dependent proteolysis system in screening for novel therapeutic agents, with a focus on experimental protocols, key reagents, and data interpretation.
The RRL ATP-Dependent Proteolysis System: A Historical and Mechanistic Foundation
Discovery and Significance
The discovery of a soluble, ATP-dependent proteolytic system in rabbit reticulocytes in 1977 marked a paradigm shift in the understanding of intracellular protein degradation. This system was identified as the primary mechanism for the selective breakdown of abnormal proteins, such as analog-containing globin, within the cell cytosol [3]. Subsequent research confirmed that this system was non-lysosomal and responsible for the energy-dependent degradation of a wide range of intracellular proteins [1]. The RRL system served as the essential experimental foundation for the stepwise reconstitution and biochemical characterization of the UPS, which is now recognized as a central regulator in cell cycle, transcription, and quality control [1].
Mechanistic Insights and Key Enzymes
Fractionation of the RRL system revealed it comprises multiple essential components. Research demonstrated that the ATP-dependent system could be separated into a conjugation fraction responsible for ubiquitinating protein substrates and a high molecular mass protease fraction necessary for the hydrolysis of the conjugated proteins [5].
Further purification efforts identified two distinct high molecular weight proteases in RRL:
- A ~1000 kDa (26S) protease whose activity is stimulated by ATP. This multisubunit complex is essential for ATP-dependent proteolysis and is responsible for degrading ubiquitinated proteins [13].
- A ~700 kDa (20S) protease that does not require nucleotides for its activity. This complex, similar to the multicatalytic proteinase complex, hydrolyzes peptide substrates and certain proteins independently of ubiquitin [13].
The degradation of ubiquitin conjugates is carried out by the ubiquitin-conjugate degrading enzyme (UCDEN), while the function of the smaller ATP-independent protease in the context of the UPS remains less certain [5]. The high activation energy (Ea ~27 kcal/mol) observed for protein degradation in cell-free RRL systems and intact cells suggests that neither simple protein unfolding nor proteolysis alone is rate-limiting, pointing to a more complex, coordinated biochemical process [21].
Figure 1: Mechanism of the Ubiquitin-Proteasome System and PROTAC-Induced Targeted Degradation. PROTAC molecules act as bifunctional linkers, bringing the target protein and E3 ubiquitin ligase into proximity to facilitate ubiquitination and subsequent degradation by the 26S proteasome.
Modern Drug Discovery: Proteolysis-Targeting Chimeras (PROTACs)
The PROTAC Paradigm
PROTACs are heterobifunctional small molecules that represent a revolutionary approach in drug discovery. They consist of three elements: a ligand that binds to the protein of interest (POI), a ligand that recruits an E3 ubiquitin ligase, and a linker connecting these two moieties [44]. By simultaneously engaging both the target protein and an E3 ligase, PROTACs induce the ubiquitination of the POI, leading to its recognition and destruction by the 26S proteasome [45]. This technology has expanded the druggable proteome, allowing targeting of proteins previously considered "undruggable," such as transcription factors, scaffolding proteins, and non-enzymatic regulators [46] [44].
Key E3 Ligases in PROTAC Design
The efficacy of a PROTAC is heavily dependent on the E3 ligase it recruits. Several E3 ligases have been successfully harnessed:
Table 1: Key E3 Ubiquitin Ligases Utilized in PROTAC Technology
| E3 Ligase | PROTAC Ligand | Significance and Applications |
|---|---|---|
| Cereblon (CRBN) | Thalidomide and derivatives (e.g., Pomalidomide) | Most widely used; successful in degrading transcription factors like BRD4 and IKZF1/3 [44] [47]. |
| von Hippel-Lindau (VHL) | VH032 and related compounds | High specificity; used in degraders for BRD4, BCR-ABL, and ALK [44] [47]. |
| MDM2 | Nutlins | Key regulator of p53; explored for degrading oncoproteins [47]. |
| cIAP1 | MeBS | Early use in PROTACs; induces auto-ubiquitination and degradation of IAPs [46]. |
Experimental Protocols for Screening Proteolysis Modulators
Core ATP-Dependent Degradation Assay in RRL
This protocol forms the basis for evaluating proteolysis modulation.
- Principle: Measure the ATP-dependent degradation of a radiolabeled or otherwise detectable substrate protein in a cell-free RRL extract.
- Reagents:
- RRL Preparation: Lysate from rabbit reticulocytes, prepared by ammonium sulfate precipitation or commercial source.
- Substrate: ^125I-α-casein, ^125I-lysozyme, or abnormal proteins like ClAbu-globin [3] [5] [13].
- Energy-Regenerating System: ATP (2-5 mM), Mg²⁺, and creatine phosphate/creatine phosphokinase.
- Test Compounds: Small molecules, fragments, or PROTAC candidates.
- Proteasome Inhibitors: MG132, lactacystin (for control).
- Ubiquitination Inhibitors: E1 inhibitor (PYR-41) (for control).
- Procedure:
- Set up reaction mixtures containing RRL, energy-regenerating system, and the substrate.
- Pre-incubate test compounds with the RRL for 15-30 minutes.
- Initiate the reaction by adding substrate. Incubate at 37°C for 1-2 hours.
- Terminate the reaction by adding trichloroacetic acid (TCA) to 10%.
- Measure TCA-soluble counts (degradation products) by scintillation counting or TCA-precipitable protein by SDS-PAGE/immunoblot.
- Data Analysis: Calculate the percentage of substrate degraded relative to controls (no ATP, with inhibitor). Compounds showing significant stimulation or inhibition of degradation relative to vehicle control are considered hits.
Specialized Assays for PROTAC Characterization
For targeted degraders like PROTACs, more specific assays are required.
- Ternary Complex Assay (e.g., AlphaLISA): Quantifies the formation of the POI-PROTAC-E3 ligase complex. A key predictor of PROTAC efficacy [47].
- Cycloheximide Chase Assay: Measures the half-life of the target protein in cells following PROTAC treatment. Cells are treated with PROTAC, then with cycloheximide to block new protein synthesis, and target protein levels are monitored over time by immunoblotting [47].
- Ubiquitination Assay: Confirms the PROTAC-induced ubiquitination of the target protein, often using purified components or cell-based assays with epitope-tagged ubiquitin [47].
- Cellular Thermal Shift Assay (CETSA): Assesss target engagement by the PROTAC within cells, based on ligand-induced thermal stabilization of the target protein [47].
Figure 2: Experimental Workflow for Screening Proteolysis Modulators in a Rabbit Reticulocyte Lysate (RRL) System. This flowchart outlines the key steps and decision points in identifying compounds that either stimulate or inhibit ATP-dependent proteolysis.
The Scientist's Toolkit: Key Research Reagents
Successful screening and characterization of proteolysis modulators rely on a defined set of reagents and tools.
Table 2: Essential Research Reagents for Proteolysis Modulator Screening
| Reagent / Tool | Function and Utility | Specific Examples |
|---|---|---|
| Rabbit Reticulocyte Lysate (RRL) | Source of the ATP-dependent ubiquitin-proteasome system for in vitro assays; provides physiological context [3] [5]. | Freshly prepared or commercial lysates. |
| Defined Protein Substrates | Act as reporters for proteolytic activity in assays [5] [13]. | ^125I-α-casein, ^125I-lysozyme, abnormal ClAbu-globin [3]. |
| Ubiquitin System Enzymes | For reconstitution studies and specific ubiquitination assays. | Purified E1, E2s, E3s (e.g., CRBN, VHL). |
| E3 Ligase Ligands | Critical components for designing and testing PROTACs [44] [47]. | Pomalidomide (CRBN), VH032 (VHL), Nutlin (MDM2). |
| Specific Inhibitors | Essential control compounds for pathway validation and mechanism determination. | MG132 (proteasome), Hemin (global inhibitor), PYR-41 (E1 inhibitor) [5] [13]. |
| Energetic Cofactors | Fuel the enzymatic reactions of the UPS. | ATP, Mg²⁺, and an ATP-regenerating system (creatine phosphate/kinase). |
Quantitative Data and Clinical Translation
Characterizing PROTAC Potency
The activity of PROTACs is quantified using specific metrics:
- DC₅₀: The concentration at which 50% of the target protein is degraded [47].
- Dmax: The maximal degradation observed, expressed as a percentage of protein remaining [47].
Table 3: Representative Profile of Clinical-Stage PROTACs
| PROTAC (Company) | Target | E3 Ligase | Indication | Clinical Status (as of 2022) |
|---|---|---|---|---|
| ARV-110 (Bavdegalutamide) | Androgen Receptor (AR) | CRBN | Prostate Cancer | Phase II [44] |
| ARV-471 | Estrogen Receptor (ER) | CRBN | Breast Cancer | Phase II [44] |
| KT-474 | IRAK4 | IAP | Autoimmune Diseases | Phase I |
| CFT7455 | IKZF1/3 | CRBN | Multiple Myeloma | Phase I/II |
Challenges and Delivery Strategies
Despite their promise, PROTACs face challenges related to their large molecular weight and high polarity, which can lead to poor solubility, low cell permeability, and suboptimal pharmacokinetics [45]. Advanced delivery strategies are being explored to overcome these hurdles, including:
- Nanocarrier Systems: Lipid nanoparticles and polymeric micelles can encapsulate PROTACs, improving solubility and bioavailability [45].
- Targeted Delivery: Conjugating PROTACs to antibodies or ligands can enhance tissue-specific delivery and mitigate off-target effects [45].
- Oral Formulation Optimization: Salt formation, co-crystals, and solid dispersions are being investigated to improve the oral bioavailability of PROTAC drugs [45].
Enhancing Experimental Outcomes: Troubleshooting and System Optimization Strategies
The rabbit reticulocyte lysate (RRL) system represents a foundational model for elucidating the mechanisms of ATP-dependent intracellular proteolysis. This in-depth technical guide synthesizes classic and contemporary research to provide a detailed framework for optimizing the core components—ATP, ubiquitin, and essential cofactors—within this system. We present summarized quantitative data, detailed experimental protocols, and clear visualizations of the proteolytic machinery. By outlining precise methodological approaches and key reagent solutions, this review serves as an essential resource for researchers aiming to maximize the efficiency of the RRL system for studying protein turnover, with direct implications for biochemical research and drug development targeting the ubiquitin-proteasome system.
The discovery of an ATP-dependent proteolytic system in rabbit reticulocytes marked a pivotal advancement in cell biology, fundamentally shaping our understanding of controlled protein turnover [3]. This system, now recognized as the ubiquitin-proteasome system (UPS), is responsible for the selective degradation of abnormal and short-lived regulatory proteins. The RRL system is a cell-free extract that faithfully recapitulates this energy-dependent process, making it an indispensable tool for in vitro biochemical studies. Its historical and continued relevance lies in its ability to support the degradation of abnormal proteins, such as analog-containing globin, with kinetics that mirror intracellular proteolysis, providing a well-characterized and controllable experimental platform [3].
Early characterizations established that the soluble, non-lysosomal proteolytic activity in RRL has a pH optimum of 7.8 and is localized in the 100,000 × g supernatant fraction [3]. The system's core machinery was later resolved through the purification of two high molecular weight proteases from RRL: a massive ~1 MDa ATP-dependent 26S proteasome complex and a smaller ~700 kDa ATP-independent 20S protease, often called the multicatalytic proteinase complex [13]. The degradation of ubiquitin-conjugated proteins is specifically catalyzed by the 26S proteasome, which requires ATP for its function [13]. This technical guide is framed within this broader thesis of RRL research, providing a contemporary manual for optimizing the critical components that drive the efficiency of this sophisticated proteolytic system.
Core System Components and Their Functions
The efficient operation of ATP-dependent proteolysis in RRL relies on the coordinated activity of three fundamental classes of components, each with a distinct and essential role.
ATP: Serves as the fundamental energy source for the system. Its hydrolysis is required for multiple steps, including the activation of ubiquitin, the assembly and stability of the 26S proteasome complex, and the mechanical unfolding and translocation of substrate proteins into the proteasome's catalytic core [3] [48]. An ATP-regenerating system is often employed in vitro to maintain a constant supply.
Ubiquitin: Functions as the primary degradation signal. Through an ATP-dependent enzymatic cascade, ubiquitin is covalently conjugated to substrate proteins, often forming a polyubiquitin chain [49]. This chain is then recognized by specific receptors on the 26S proteasome, marking the substrate for degradation.
Cofactors: A diverse set of proteins that confer specificity and efficiency to the system. This category includes:
- E1, E2, E3 Enzymes: The activating (E1), conjugating (E2), and ligating (E3) enzymes that work in cascade to tag substrate proteins with ubiquitin [49].
- Cdc48/p97 and Cofactors (e.g., Ufd1/Npl4): A specialized AAA+ ATPase complex that assists in the extraction of ubiquitylated proteins from membranes or macromolecular complexes, preparing them for proteasomal degradation [50] [51]. Recent studies highlight the role of accessory cofactors like VCF1, which binds p97 with high affinity and promotes the recognition of ubiquitylated substrates by the Ufd1/Npl4 complex [51].
The interplay between these components is summarized in the diagram below, which outlines the logical workflow of substrate processing.
Quantitative Optimization Guidelines
Optimizing the concentrations of ATP, ubiquitin, and cofactors is critical for achieving maximal proteolytic efficiency. The tables below consolidate key quantitative data from foundational and modern studies to serve as a reference for experimental design.
Table 1: Optimization of ATP and Ubiquitin Concentrations
| Component | Optimal Concentration Range | Functional Role | Experimental Notes |
|---|---|---|---|
| ATP | 2-5 mM [3] | Energy source for ubiquitin activation and 26S proteasome function. | Stimulates proteolysis several-fold over basal levels; ADP provides slight stimulation; AMP and cAMP are ineffective [3]. Use an ATP-regenerating system for prolonged incubations. |
| Ubiquitin | Not fully quantified in early RRL studies | Signal for proteasomal degradation; conjugated to substrates via E1-E2-E3 cascade. | System has an absolute requirement for ubiquitin [49]. Endogenous levels in RRL may be sufficient for some substrates, but supplementation can enhance degradation of challenging targets. |
Table 2: Key Cofactors and Their Roles in the RRL System
| Cofactor / Complex | Key Functional Role | Optimization Insight |
|---|---|---|
| Ufd1/Npl4 | Primary cofactor for Cdc48/p97; binds K48-linked polyubiquitin chains and initiates substrate unfolding [50]. | Preferentially processes substrates with K48-linked chains of 4-6 ubiquitins [50]. The UT3 domain of Ufd1 is critical for binding a ubiquitin moiety proximal to the initiator. |
| VCF1 (FAM104A) | High-affinity p97 cofactor that promotes p97-Ufd1/Npl4 recruitment to ubiquitylated substrates [51]. | Binds the p97 N-domain with very high affinity (Kd ~10 nM), exceeding that of many other cofactors. Enhances substrate turnover without direct ubiquitin binding [51]. |
| Proteasome Inhibitors | Used to confirm UPS-dependent degradation. | In RRL, proteolysis is inhibited by TPCK, TLCK, N-ethylmaleimide, iodoacetamide, and o-phenanthroline [3]. Hemin, thiol reagents, chymostatin, and leupeptin also inhibit the purified 26S and 20S proteases [13]. |
Detailed Experimental Protocols
Protocol 1: ATP-Dependence and Ubiquitin Requirement in RRL
This foundational protocol is adapted from the seminal work that first characterized the soluble ATP-dependent proteolytic system in reticulocytes [3].
- Reticulocyte Lysate Preparation: Isulate reticulocytes from phenylhydrazine-treated rabbits. Lysate cells in a hypotonic buffer (e.g., 1 mM DTT, 1 mM MgCl2). Centrifuge the lysate at 100,000 × g for 1 hour to obtain the soluble S100 supernatant fraction, which contains the ATP-dependent proteolytic activity.
- Reaction Setup: Assemble degradation reactions containing:
- S100 lysate fraction (e.g., 50-100 µg of protein).
- A radiolabeled or otherwise detectable abnormal protein substrate (e.g., globin containing the valine analog 2-amino-3chlorobutyric acid).
- A buffer system with a pH of 7.8 (e.g., Tris-HCl).
- Experimental Variables:
- +ATP: Add ATP to a final concentration of 2-5 mM.
- -ATP Control: Omit ATP and/or include a non-hydrolyzable ATP analog like ATPγS.
- +Ubiquitin: In some cases, supplement with additional ubiquitin (exact concentration to be optimized, as endogenous levels are present).
- Incubation and Analysis:
- Incubate reactions at 37°C for a defined period (e.g., 15-60 minutes).
- Stop the reaction by adding trichloroacetic acid (TCA) to a final concentration of 10%.
- Measure the release of acid-soluble radioactivity or peptides by scintillation counting or fluorescence to quantify substrate degradation.
- Expected Outcome: Degradation of the abnormal globin substrate should be stimulated several-fold in the presence of ATP compared to the no-ATP control, with a half-life of approximately 15 minutes [3].
Protocol 2: Analyzing Cofactor-Driven Substrate Engagement
This protocol, based on modern structural and biochemical studies, outlines how to investigate the role of specific cofactors like Ufd1/Npl4 in substrate processing, which can be reconstituted in RRL or similar extracts [50].
- Substrate Design: Prepare a substrate protein conjugated to a defined polyubiquitin chain. For studying Cdc48-UN (Ufd1/Npl4), K48-linked chains of 4-6 ubiquitins are optimal. For FRET-based assays, use chains with fluorophores (e.g., Cy3Ub-Cy5Ubn) to monitor conformational changes during engagement.
- Reconstitution:
- Purify complexes of Cdc48/p97 with wild-type or mutant Ufd1/Npl4 cofactor. Critical mutants include those in the ubiquitin-binding groove of Npl4 (e.g., W252A/R253E, D460K/Y461A) which abrogate initiator ubiquitin unfolding [50].
- In RRL or a defined buffer, mix the ubiquitylated substrate with the Cdc48-UN complex in the presence of a non-hydrolyzable ATP analog (e.g., ATPγS) to trap the engagement state.
- Measurement:
- For FRET assays, monitor the decrease in FRET efficiency (increase in donor fluorescence) as an indicator of initiator ubiquitin unfolding and engagement by Npl4.
- Use stopped-flow instruments for kinetic analysis, which typically reveals a double-exponential unfolding process [50].
- Expected Outcome: Wild-type Cdc48-UN will cause a rapid decrease in FRET, while mutant complexes will show significantly reduced activity, confirming the specific role of the cofactor in substrate engagement.
The Scientist's Toolkit: Key Research Reagent Solutions
Successful experimentation in the RRL system requires a suite of specific reagents. The following table details essential materials and their functions.
Table 3: Essential Reagents for RRL ATP-Dependent Proteolysis Research
| Reagent | Function & Application | Specific Examples |
|---|---|---|
| ATP & Analogs | To energize the system and probe mechanism. | ATP (2-5 mM, for activity); ATPγS (non-hydrolyzable, for trapping intermediate states); ADP/AMP (to test nucleotide specificity) [3]. |
| Proteasome & Protease Inhibitors | To confirm the involvement of specific enzymatic activities. | N-ethylmaleimide, iodoacetamide (thiol reagent inhibitors); o-phenanthroline (metalloprotease inhibitor); TPCK, TLCK (serine protease inhibitors); Hemin, Chymostatin, Leupeptin (inhibitors of the purified 26S/20S proteases) [3] [13]. |
| Ubiquitin System Reagents | To manipulate the ubiquitination status of substrates. | Ubiquitin (wild-type for supplementation); E1 enzyme (for reconstituting ubiquitination); Methylated Ubiquitin (to inhibit polyubiquitin chain formation). |
| Cofactor Complexes | To study specific pathways of substrate recognition and processing. | Purified Cdc48/p97, Ufd1/Npl4 heterodimer; Mutant Cofactors (e.g., Npl4 groove mutants, Ufd1 UT3 domain mutants) [50]; Recombinant VCF1 (to study its augmenting effect on p97-UN) [51]. |
| Stabilizing Additives | To preserve labile protein complexes during extraction. | Proteasome inhibitors (e.g., MG132); Phosphatase inhibitors; ATP cofactor; Reductants (e.g., DTT) – critical for stabilizing unstable proteins like PPR proteins during extraction [52]. |
The rabbit reticulocyte lysate system remains a powerful and versatile platform for dissecting the intricate mechanics of the ubiquitin-proteasome system. Maximizing its efficiency hinges on the precise optimization of ATP, ubiquitin, and a growing list of regulatory cofactors, as detailed in this guide. By applying the summarized quantitative data, detailed protocols, and reagent knowledge, researchers can design robust experiments to not only answer fundamental biological questions but also to screen and characterize novel therapeutics targeting protein homeostasis. The continued integration of foundational RRL biochemistry with modern structural and mechanistic insights, particularly regarding cofactor networks, promises to further unlock the potential of this classic experimental system.
The rabbit reticulocyte lysate (RRL) system is a cornerstone for studying intracellular protein degradation, particularly ATP-dependent proteolysis. This non-lysosomal process is essential for understanding cellular protein quality control, yet researchers frequently encounter two persistent challenges: low degradation activity and high background noise. These issues can compromise data interpretation and hinder experimental progress. This technical guide examines the underlying mechanisms of these pitfalls within the RRL ATP-dependent proteolysis system and provides evidence-based strategies to overcome them, leveraging foundational research that continues to inform current practices.
Understanding ATP-Dependent Proteolysis in RRL Systems
Rabbit reticulocyte lysate contains a sophisticated proteolytic system responsible for the selective degradation of abnormal and short-lived proteins. Early groundbreaking work established that this process is both soluble and ATP-dependent, contrasting with lysosomal degradation pathways [3]. The system demonstrates a pH optimum of 7.8 and requires magnesium ions for maximal activity [4] [3].
A critical breakthrough in understanding this system came with the discovery of ATP's dual roles in protein degradation. Seminal research demonstrated that ATP stimulates proteolysis through both ubiquitin-dependent and ubiquitin-independent mechanisms [4]. When amino groups of protein substrates (such as methyl-casein and denatured bovine serum albumin) were chemically blocked—preventing ubiquitin conjugation—ATP still stimulated degradation approximately two-fold, though the magnitude of stimulation was reduced compared to unmodified substrates [4]. This fundamental finding explains why both pathways must be considered when troubleshooting degradation efficiency issues.
Table 1: Key Characteristics of ATP-Dependent Proteolysis in RRL
| Parameter | Characteristics | Experimental Evidence |
|---|---|---|
| Cellular Location | Soluble, non-lysosomal, 100,000 × g supernatant | [3] |
| ATP Stimulation | 2-4 fold stimulation for unmodified proteins; 2-fold for amino-blocked proteins | [4] |
| Cofactor Requirements | Requires Mg²⁺ and ATP hydrolysis | [4] |
| pH Optimum | 7.8 | [3] |
| Inhibitors | Hemin, N-ethylmaleimide, iodoacetamide, o-phenanthroline | [4] [3] |
Troubleshooting Low Degradation Activity
Biochemical Optimization Strategies
Low degradation activity typically stems from suboptimal biochemical conditions or inadequate proteolytic components. The following approaches can significantly enhance activity:
ATP Regeneration Systems: Maintain constant ATP levels (typically 1-2 mM) using creatine phosphate and creatine phosphokinase to prevent depletion during extended incubations. Early studies established that ATP hydrolysis is essential for proteolytic activity, with the terminal high-energy phosphate being particularly important [3].
Divalent Cation Optimization: Magnesium is an absolute requirement for both ubiquitin-dependent and ubiquitin-independent pathways. Optimize Mg²⁺ concentrations between 2-5 mM, as excess can promote non-specific degradation while insufficient amounts impair both proteolysis and ubiquitination [4].
Inhibitor Management: Hemin powerfully inhibits ATP-dependent proteolysis in reticulocytes, while protoporphyrin IX does not [4]. Avoid hemin contamination from lysate preparation. Additionally, serine protease inhibitors like TPCK and TLCR can reduce degradation, as can sulfhydryl reagents including N-ethylmaleimide and iodoacetamide [3].
Substrate Preparation: Denatured proteins typically serve as better substrates than native structures. Ensure proper substrate denaturation when studying abnormal protein degradation. Interestingly, studies with extensively modified proteins (93-99% amino group blockage) revealed that ubiquitin conjugation, while important for certain substrates like BSA, is not absolutely essential for all ATP-stimulated proteolysis [4].
Experimental Protocols: Assessing ATP-Dependence
To diagnose the specific cause of low degradation activity:
Set up reaction mixtures containing: 50-100 μg RRL protein, 1-2 mM ATP, 5-10 mM creatine phosphate, 0.1-0.2 U creatine phosphokinase, 2-5 mM MgCl₂, and 50-100 mM Tris-HCl (pH 7.8) in a total volume of 50-100 μL [3].
Include controls without ATP and without energy-regenerating system to establish baseline proteolysis.
Incubate at 37°C for 60-120 minutes, terminating reactions with SDS-PAGE sample buffer or trichloroacetic acid for precipitation.
Analyze degradation by SDS-PAGE followed by staining/densitometry or by measuring trichloroacetic acid-soluble radioactivity for labeled substrates.
Managing High Background Noise
Source Identification and Reduction Strategies
High background noise often arises from non-specific proteolysis, incomplete inhibition of non-target pathways, or substrate aggregation. Implement these evidence-based reduction strategies:
Ubiquitin System Modulation: For studies focusing on ubiquitin-independent degradation, remove endogenous ubiquitin using ion exchange chromatography [4]. This approach demonstrated that ATP still stimulated casein breakdown two-fold even after ubiquitin removal [4]. Conversely, when studying ubiquitin-dependent pathways, ensure adequate ubiquitin levels.
Protease Inhibition Profiling: Utilize class-specific protease inhibitors to identify noise sources. The RRL system is sensitive to sulfhydryl reagents and metalloprotease inhibitors like o-phenanthroline [3]. However, note that some inhibitors affect both ubiquitin-dependent and ubiquitin-independent pathways equally, suggesting shared mechanistic features [4].
Substrate Quality Control: Address protein aggregation through proper substrate handling. Recent research highlights that limited accessibility of peptide bonds due to protein structure significantly impacts proteolytic efficiency [53]. Consider mild denaturation or reduction of disulfide bonds where appropriate for your experimental goals.
Quantitative Assessment Protocols
To quantify and minimize background signal:
Establish baseline proteolysis using no-ATP controls and heat-inactivated RRL to measure ATP-independent and non-enzymatic substrate breakdown.
Implement time-course experiments with multiple time points to distinguish linear degradation from early plateauing, which may indicate substrate limitations or inhibitor accumulation.
Utilize substrate controls with known degradation profiles (e.g., normal hemoglobin vs. analog-containing globin) to validate system functionality [3].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagent Solutions for RRL ATP-Dependent Proteolysis Studies
| Reagent | Function | Application Notes |
|---|---|---|
| ATP Regeneration System (ATP, creatine phosphate, creatine phosphokinase) | Maintains constant ATP levels during prolonged incubations | Essential for observing maximum degradation; prevents false negatives [4] [3] |
| MgCl₂ | Cofactor for ATP hydrolysis and proteolytic machinery | Optimize between 2-5 mM; required for both ubiquitin-dependent and independent pathways [4] |
| Hemín | Regulatory inhibitor of proteolysis | Use as experimental control (50-100 μM); avoid contamination during lysate preparation [4] |
| N-Ethylmaleimide (NEM) | Sulfhydryl alkylating agent | Inhibits multiple proteolytic components; use for system characterization (1-5 mM) [3] |
| Ion Exchange Resins (DEAE-cellulose, etc.) | Remove endogenous ubiquitin | Isolate ubiquitin-independent degradation; demonstrated to retain ATP-stimulated activity [4] |
| Analog-Containing Proteins (e.g., ClAbu-globin) | Defined proteolysis substrates | Provide consistent, rapidly-degraded substrates for system optimization [3] |
Visualization of Proteolytic Pathways and Experimental Workflows
Pathways of ATP-Dependent Protein Degradation
Experimental Troubleshooting Workflow
Advanced Applications and Contemporary Relevance
While foundational RRL research established core principles of ATP-dependent proteolysis, these findings continue to inform modern protein quality control research. Contemporary studies have revealed that orphaned proteins from multiprotein complexes are eliminated through quality control pathways involving sequential ubiquitination, where priming E3 ligases like HERC1/HERC2 first mono-ubiquitinate substrates before chain-elongating enzymes like UBR4-KCMF1 build poly-ubiquitin degradation signals [54].
Furthermore, current research increasingly recognizes the importance of substrate accessibility in proteolytic efficiency, with kinetic models of proteolysis accounting for the limited accessibility of some peptide bonds due to protein structure [53]. This modern work builds upon early observations that protein modifications altering structure can significantly impact degradation rates [4].
The rabbit reticulocyte lysate system remains an invaluable tool for studying ATP-dependent proteolysis decades after its initial characterization. By understanding the dual roles of ATP in ubiquitin-dependent and independent pathways, researchers can systematically address low degradation activity and high background noise. Methodical optimization of ATP and magnesium levels, careful management of inhibitors like hemin, strategic use of ubiquitin depletion, and proper substrate preparation collectively enable robust experimental outcomes. The enduring principles established in early RRL research continue to provide fundamental insights into cellular protein quality control mechanisms that remain relevant to contemporary studies of protein homeostasis.
The rabbit reticulocyte lysate system represents a foundational model for understanding ATP-dependent intracellular protein degradation. This soluble, non-lysosomal proteolytic pathway is responsible for the selective breakdown of abnormal and short-lived proteins within eukaryotic cells [3]. The discovery that proteolysis in reticulocyte extracts requires adenosine triphosphate (ATP) hydrolysis represented a paradigm shift in understanding cellular protein turnover mechanisms [3]. This system has since been characterized as comprising multiple high molecular weight proteases, including a ~1500 kDa enzyme essential for degrading ubiquitin-conjugated proteins (UCDEN) and a ~670 kDa protease that functions independently of ATP and ubiquitin [5]. Optimizing the pH and ionic environment for these enzymatic complexes is crucial for maintaining their activity and elucidating their physiological functions, particularly in the context of drug development targeting proteostatic pathways.
Fundamental Components of the Reticulocyte Proteolytic System
Key Proteolytic Complexes and Their Characteristics
Table 1: High Molecular Weight Proteases in Rabbit Reticulocytes
| Protease | Molecular Weight | ATP Dependence | Ubiquitin Dependence | Sensitivity to Inhibitors |
|---|---|---|---|---|
| UCDEN (Ubiquitin-Conjugate Degrading Enzyme) | ~1500 kDa | Required | Required | Inhibited by hemin, dichloroisocoumarin, N-ethylmaleimide, peptide chloromethyl ketones [5] |
| ATP-Independent Protease | ~670 kDa | Not required | Not required | Stimulated by N-ethylmaleimide and 3,4-dichloroisocoumarin [5] |
Research Reagent Solutions for ATP-Dependent Proteolysis Studies
Table 2: Essential Research Reagents for Reticulocyte Proteolysis Experiments
| Reagent | Function/Application | Experimental Context |
|---|---|---|
| ATP, ADP, ATPγS | Nucleotide effectors; ATP required, ADP slight stimulation, ATPγS for trapping complexes [3] [55] | Fundamental energy source and allosteric regulator |
| N-ethylmaleimide, Iodoacetamide | Sulfhydryl-reactive inhibitors; block essential cysteine residues [3] [5] | Probing cysteine-dependent mechanisms |
| Hemin | Natural inhibitor; regulates proteolytic activity [5] | Physiological regulation studies |
| 3,4-Dichloroisocoumarin | Serine protease inhibitor [5] | Characterizing serine protease components |
| Leupeptin, TLCK, TPCK | Protease inhibitors; specificity profiling [3] | Enzyme classification and mechanism studies |
| o-Phenanthroline | Metalloprotease inhibitor [3] | Assessing metalloprotease contributions |
| Ubiquitin | Protein co-factor; essential for conjugate formation [5] | Ubiquitin-dependent degradation pathways |
pH Optimization in ATP-Dependent Proteolysis
pH Profile of Reticulocyte Proteases
The ATP-dependent proteolytic system in rabbit reticulocytes displays a distinct pH optimum of 7.8, as established in the seminal characterization of this system [3]. This alkaline pH preference distinguishes it from lysosomal proteases, which typically function optimally at acidic pH, and aligns with its cytosolic localization. The pH sensitivity reflects the dependence of catalytic residues on proper protonation states and influences substrate binding affinity. Recent structural studies on related AAA+ proteases like ClpXP have revealed that catalytic triads (Ser-His-Asp in serine proteases) require specific protonation states for efficient function [11] [56]. The observation that the reticulocyte system maintains near-maximal activity within the physiological pH range (7.0-8.0) underscores its adaptation to cytoplasmic conditions.
Experimental Protocol: Determining pH Optima for ATP-Dependent Proteolysis
Materials:
- Rabbit reticulocyte lysate (prepared from phenylhydrazine-treated rabbits)
- ATP regeneration system (2 mM ATP, 10 mM creatine phosphate, 0.2 mg/mL creatine phosphokinase)
- Radioactively labeled substrate protein (e.g., [³H]- or ¹⁴C]-labeled abnormal globin)
- Buffer systems spanning pH 4.0-10.0 (acetate, phosphate, Tris-HCl, glycine-NaOH)
- Precipitation reagents (10% trichloroacetic acid)
- Liquid scintillation counter
Method:
- Prepare reaction mixtures containing 50 μL reticulocyte lysate, 2 mM ATP regeneration system, and labeled substrate (50,000 cpm) in a total volume of 100 μL.
- Adjust reactions to different pH values using appropriate buffering systems (pH 4.0-6.0: acetate; pH 6.0-8.0: phosphate; pH 8.0-10.0: Tris-glycine).
- Incubate at 37°C for 30 minutes.
- Terminate reactions by adding 500 μL of 10% trichloroacetic acid.
- Remove precipitate by centrifugation at 10,000 × g for 10 minutes.
- Measure acid-soluble radioactivity in the supernatant by liquid scintillation counting.
- Calculate proteolytic rates as pmol of substrate degraded per minute per mg of lysate protein.
Interpretation: The pH profile establishes the ionization requirements of catalytic residues and informs buffer selection for subsequent assays. Deviation from the established optimum may indicate protease dysfunction or the presence of inhibitory factors.
Ionic Environment Effects on Proteolytic Activity
Hofmeister Series and Ion Specificity
The ionic environment significantly influences protease activity through specific ion effects that follow the Hofmeister series [57]. This empirical classification arranges ions based on their ability to stabilize or destabilize protein structure:
- Kosmotropic anions: SO₄²⁻ > HPO₄²⁻ > F⁻ > CH₃COO⁻ > Cl⁻ > NO₃⁻ > Br⁻ > I⁻ > SCN⁻ (stabilizing)
- Chaotropic cations: NH₄⁺ > Rb⁺ > K⁺ > Na⁺ > Li⁺ > Mg²⁺ > Ca²⁺ > Ba²⁺ (destabilizing)
Kosmotropic anions and chaotropic cations generally stabilize enzymes and enhance activity, while chaotropic anions and kosmotropic cations typically destabilize protein structure [57]. These effects are mediated through alterations in water structure and direct ion-protein interactions that modulate enzyme stability and catalytic efficiency.
Divalent Cation Effects
Divalent cations exhibit complex effects on proteolytic systems, with both inhibitory and activating roles depending on the specific protease:
Table 3: Effects of Divalent Cations on Protease Activity
| Cation | Effect on Reticulocyte System | Effect on Marine Bacterial ClpP |
|---|---|---|
| Mg²⁺ | Required for ATP hydrolysis [3] | Inhibitory at 2 mM concentration [58] |
| Ca²⁺ | Information missing | Inhibitory at 2 mM concentration [58] |
| Zn²⁺ | Inhibitory [3] | Inhibitory at 2 mM concentration [58] |
| Co²⁺ | Information missing | Marked activation in absence of ATP [58] |
| Mn²⁺ | Information missing | Inhibitory at 2 mM concentration [58] |
The marine bacterial ClpP protease from Cobetia amphilecti demonstrates distinctive ion sensitivity, with Co²⁺ causing marked activation in the absence of ATP, while Mg²⁺, Ca²⁺, Ni²⁺, Mn²⁺, Li⁺, and Zn²⁺ at 2 mM concentration significantly inhibit activity [58]. This contrasting behavior highlights the diversity of ion effects across different proteolytic systems.
Experimental Protocol: Ionic Optimization Using the Hofmeister Series
Materials:
- Dialyzed reticulocyte lysate (to remove endogenous ions)
- ATP regeneration system
- Labeled substrate protein
- Salts representing kosmotropic and chaotropic ions (Na₂SO₄, NaCl, NaBr, NaI, NaClO₄, (NH₄)₂SO₄, NH₄Cl, etc.)
- Precipitation and scintillation counting reagents
Method:
- Dialyze reticulocyte lysate overnight against 20 mM HEPES, pH 7.8, 1 mM DTT to remove small molecules and ions.
- Prepare reaction mixtures with dialyzed lysate, ATP regeneration system, and labeled substrate.
- Add test salts at concentrations ranging from 0-200 mM.
- Incubate at 37°C for 30 minutes.
- Measure proteolytic activity as described in Section 3.2.
- Plot activity versus ion concentration and classify ions according to their effects.
Interpretation: Optimal ionic conditions can be determined by identifying salts that maximize proteolytic rate. Kosmotropic anions paired with chaotropic cations typically yield maximal activity, providing a rational basis for buffer optimization.
Structural Basis for pH and Ion Sensitivity
AAA+ Protease Architecture and Catalytic Mechanisms
Recent cryo-EM and crystallographic studies of AAA+ proteases such as ClpXP have revealed the structural features governing pH and ion sensitivity [11] [55]. These large macromolecular machines consist of hexameric AAA+ ATPase rings (ClpX) stacked upon tetradecameric proteolytic chambers (ClpP). The catalytic sites within ClpP contain Ser-His-Asp triads that require specific protonation states for nucleophilic attack on substrate peptides [11]. The aspartic acid residues must be deprotonated to properly orient the catalytic histidine, which acts as a general base to activate the serine nucleophile. This precise arrangement is highly pH-dependent and explains the sharp pH optimum observed in reticulocyte proteases.
Visualizing the ATP-Dependent Proteolysis Workflow
Ion Binding Sites in Protease Structures
Structural analyses have identified specific ion binding sites that modulate protease function. In metalloproteases, zinc ions are coordinated by histidine and glutamate residues to activate a catalytic water molecule [56]. The ClpP proteolytic chamber contains intersubunit contacts that are stabilized by ions, with the marine bacterial CamClpP exhibiting optimal activity at 0.3 M NaCl or 0.2 M KCl [58]. These ions likely shield negative charges at subunit interfaces, promoting proper oligomerization. The AAA+ ATPase components contain conserved magnesium-binding motifs essential for ATP hydrolysis, explaining the Mg²⁺ requirement in reticulocyte systems [3].
Advanced Optimization Strategies
Integrated pH and Ionic Optimization
For precise control of proteolytic activity, simultaneous optimization of pH and ionic conditions is essential. This approach acknowledges the interdependence of these parameters, as ion binding can alter the pKₐ of catalytic residues, and pH can influence ion coordination geometry. The reticulocyte system operates optimally in phosphate buffer at pH 7.8, which provides both buffering capacity and kosmotropic phosphate anions that stabilize protease structure [57].
Macromolecular Crowding and Compatible Solutes
Intracellular environments feature macromolecular crowding that significantly influences proteolytic efficiency. Compatible solutes such as glycerol, proline, and glycine betaine can stabilize proteases under suboptimal conditions, though their effects vary. Glycerol generally stabilizes protein structure but may inhibit certain proteases like CamClpP [58]. Optimization should consider these physiological context factors when designing in vitro assays.
Technical Applications in Drug Development
High-Throughput Screening Assay Design
The principles of pH and ionic optimization directly inform drug discovery efforts targeting proteolytic systems. Luminescence-based high-throughput screening assays for enzyme activity employ optimized buffer conditions to maximize signal-to-noise ratios [59]. For ATP-dependent proteases, this includes maintaining pH at 7.8-8.0, Mg²⁺ at 2-5 mM for ATP hydrolysis, and kosmotropic salts (e.g., phosphate or sulfate) to stabilize enzyme structure during screening campaigns.
Visualizing Protease Structure and Mechanism
Pathological Implications and Therapeutic Targeting
Dysregulation of ATP-dependent proteolysis contributes to various diseases, including neurodegeneration, cancer, and infectious diseases [60] [56]. The mitochondrial AAA+ protease YME1L regulates proteostasis at the inner membrane, with mutations linked to neurological disorders [60]. Rhomboid intramembrane proteases are implicated in Parkinson's disease and parasitic infections [61]. Pharmaceutical targeting of these enzymes requires thorough understanding of their pH and ionic requirements, as these factors influence inhibitor binding and specificity. Structure-based drug design utilizing QM/MM computational approaches has successfully developed inhibitors for therapeutic targets like HIV protease by modeling catalytic mechanisms under physiological ionic conditions [56].
The ATP-dependent proteolytic system in rabbit reticulocyte lysate provides a fundamental model for understanding regulated intracellular protein degradation. Optimal activity requires precise control of pH at 7.8 and specific ionic conditions that follow Hofmeister series principles. Kosmotropic anions and chaotropic cations generally stabilize the proteolytic complexes, while Mg²⁺ is essential for ATP hydrolysis. These biochemical parameters reflect the structural features of AAA+ proteases, with their catalytic triads and ion coordination sites requiring specific protonation states and ionic environments for efficient function. As drug development increasingly targets proteostatic pathways, rigorous optimization of pH and ionic conditions remains essential for accurate in vitro modeling of physiological and pathological processes.
The ATP-dependent proteolytic system in rabbit reticulocyte lysate represents a fundamental non-lysosomal pathway responsible for the selective degradation of abnormal and short-lived proteins within eukaryotic cells [3]. This system is characterized by its location in the 100,000 × g supernatant, a pH optimum of 7.8, and a functional requirement for adenosine triphosphate (ATP) as a critical energy source [3]. Originally characterized in the 1970s, this system provided the first evidence that cells possess a soluble, energy-dependent mechanism for protein quality control, distinguishing it markedly from lysosomal degradation pathways. The system demonstrates remarkable substrate selectivity, efficiently degrading abnormal proteins such as analog-containing globin (e.g., incorporating 2-amino-3chlorobutyric acid) with a half-life of approximately 15 minutes, while sparing normal hemoglobin from significant breakdown [3]. This discriminative capability makes it an invaluable model system for studying targeted proteolysis and for testing inhibitors that modulate this crucial cellular process.
Core Mechanisms of ATP-Dependent Proteolysis
The rabbit reticulocyte system primarily operates through the ubiquitin-proteasome pathway, wherein proteins destined for degradation are first tagged with ubiquitin chains before being recognized and processed by the 26S proteasome complex. The 26S proteasome itself consists of a 20S core particle that carries out proteolytic activity, capped by 19S regulatory particles that recognize ubiquitinated substrates, unfold them, and translocate them into the catalytic core in an ATP-dependent manner. This system represents a soluble non-lysosomal proteolytic pathway that demonstrates specificity for particular protein substrates based on their structural abnormalities or regulatory signals [3].
Table 1: Key Characteristics of the Rabbit Reticulocyte ATP-Dependent Proteolytic System
| Characteristic | Description | Experimental Basis |
|---|---|---|
| Cellular Location | 100,000 × g supernatant (soluble, non-lysosomal) | Centrifugation fractionation studies [3] |
| pH Optimum | 7.8 | pH-dependent activity assays [3] |
| Energy Requirement | ATP-dependent (stimulated several-fold by ATP, slightly by ADP) | Nucleotide dependence experiments [3] |
| Primary Function | Selective degradation of abnormal proteins | Comparative degradation of normal vs. analog-containing globin [3] |
| Inhibition Profile | Sensitive to TPCK, TLCK, N-ethylmaleimide, iodoacetamide, o-phenanthroline | Inhibitor screening in cell-free systems and intact reticulocytes [3] |
Established Inhibitors and Their Mechanisms of Action
Classical Protease Inhibitors
The foundational study of the ATP-dependent proteolytic system in reticulocytes identified several chemical inhibitors that effectively block proteolytic activity, providing crucial insights into the mechanistic requirements of this pathway [3]. These inhibitors include L-1-tosylamido-2-phenyl-ethylchloromethyl ketone (TPCK), N-alpha-p-tosyl-L-lysine chloromethyl ketone (TLCK), N-ethylmaleimide, iodoacetamide, and o-phenanthroline. The effectiveness of these inhibitors, which target various protease classes and functional groups, indicates the involvement of multiple enzyme types and essential cysteine residues in the proteolytic cascade. Particularly noteworthy is the consistent inhibition pattern observed between intact reticulocytes and cell-free extracts, validating the physiological relevance of the in vitro system for inhibitor studies [3].
Physiological Modulators: Polyamines
Physiological concentrations of polyamines serve as endogenous regulators of the ubiquitin-dependent proteolytic pathway in reticulocyte lysates [62]. These naturally occurring cations exert concentration-dependent inhibition, with spermine demonstrating the most potent effect (71-96% inhibition), followed by spermidine (26-72% inhibition) and putrescine (26-72% inhibition) [62]. Through systematic fractionation of the ATP-dependent system, researchers determined that polyamines primarily inhibit the ATP-dependent degradation of ubiquitin-protein conjugates rather than affecting the ATP-independent breakdown of oxidant-damaged proteins [62]. This specific action point underscores the regulatory potential of polyamines in modulating ubiquitin-mediated protein turnover under physiological conditions.
Table 2: Quantitative Inhibition of ATP-Dependent Proteolysis by Polyamines
| Polyamine | Inhibition Range | Specificity | Proposed Mechanism |
|---|---|---|---|
| Spermine | 71-96% | Inhibits ATP-dependent degradation of ubiquitin-protein conjugates | Interference with ubiquitin-conjugate degradation step [62] |
| Spermidine | 26-72% | Inhibits ATP-dependent degradation of ubiquitin-protein conjugates | Interference with ubiquitin-conjugate degradation step [62] |
| Putrescine | 26-72% | Inhibits ATP-dependent degradation of ubiquitin-protein conjugates | Interference with ubiquitin-conjugate degradation step [62] |
Synthetic Peptide Inhibitors
Synthetic peptides mimicking viral inhibitory sequences represent a sophisticated approach to targeting specific aspects of the ubiquitin-proteasome pathway. A prime example is the glycine-alanine repeat (GAr) peptide derived from the Epstein-Barr virus nuclear antigen-1, which effectively inhibits ubiquitin/proteasome-dependent proteolysis in rabbit reticulocyte lysates at micromolar concentrations [63]. This 20-mer synthetic GAr peptide inhibits lysozyme degradation without significantly affecting its ubiquitination or the hydrolytic activity of purified proteasomes against fluorogenic substrates [63]. Mechanistic studies indicate that the GAr does not compete with tetra-ubiquitin chains for binding to the S5a ubiquitin-recognition subunit of the 19S proteasomal regulator [63]. Instead, evidence suggests that the peptide may act by destabilizing the interaction between ubiquitinated substrates and the proteasome, potentially promoting premature substrate release prior to degradation [63].
Experimental Approaches for Inhibitor Characterization
Core Assay Protocol for Inhibitor Testing
The standard methodology for evaluating proteolysis inhibitors in the reticulocyte system involves the following detailed protocol adapted from foundational studies [3] [62]:
Lysate Preparation: Isolate rabbit reticulocytes and lyse them in hypotonic buffer (e.g., 10 mM Tris-HCl, pH 7.8). Centrifuge at 100,000 × g for 60 minutes at 4°C to obtain the soluble fraction containing the ATP-dependent proteolytic activity [3].
Substrate Labeling: Prepare radiolabeled or otherwise detectable substrate proteins. Common substrates include:
Reaction Setup: In a typical assay, combine:
- 25-50 μL reticulocyte lysate (supernatant fraction)
- ATP-regenerating system (2 mM ATP, 10 mM phosphocreatine, 0.1 mg/mL creatine phosphokinase)
- Substrate protein (50-100 μg/mL)
- Test inhibitor at varying concentrations
- Buffer components (50 mM Tris-HCl, pH 7.8, 5 mM MgCl₂, 1 mM DTT)
- Incubate at 37°C for 30-120 minutes [3] [62] [63]
Proteolysis Quantification:
Diagram Title: Experimental Workflow for Proteolysis Inhibitor Characterization
Specialized Methodologies
Ubiquitin-Conjugate Analysis
For inhibitors potentially affecting the ubiquitination phase, additional protocols are necessary:
- Monitor formation of high-molecular-weight ubiquitin-protein conjugates by SDS-PAGE and immunoblotting with anti-ubiquitin antibodies [30] [63]
- Assess ubiquitin-conjugating enzyme (E2) and ubiquitin-ligase (E3) activities in the presence of inhibitors
- Evaluate the requirement for ubiquitin in inhibitor action by testing in ubiquitin-depleted lysates [30]
Proteasome Interaction Studies
To determine whether inhibitors directly affect proteasome function:
- Test inhibitors against purified 20S and 26S proteasomes using fluorogenic substrates (e.g., Suc-LLVY-AMC)
- Perform competition assays with known proteasome inhibitors (e.g., MG132, lactacystin)
- Analyze substrate binding to the proteasome using native gels or co-sedimentation assays [63]
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Proteolysis Inhibition Studies
| Reagent Category | Specific Examples | Function/Application | Experimental Notes |
|---|---|---|---|
| ATP-System Components | ATP, phosphocreatine, creatine phosphokinase | Maintain ATP levels during prolonged incubations | ATP analogs help determine energy requirement specificity [3] |
| Protease Inhibitors | TPCK, TLCK, N-ethylmaleimide, iodoacetamide | Characterize protease classes involved | Different inhibitors help distinguish between cysteine vs. serine proteases [3] |
| Physiological Inhibitors | Spermine, spermidine, putrescine | Study endogenous regulation of proteolysis | Concentration-dependent effects; spermine most potent (71-96% inhibition) [62] |
| Synthetic Peptide Inhibitors | GAr (glycine-alanine repeat) peptide | Probe substrate-proteasome interactions | Does not affect ubiquitination or proteasome active sites [63] |
| Ubiquitin System Reagents | Ubiquitin, methylated ubiquitin, E1/E2/E3 enzymes | Dissect ubiquitination vs. degradation steps | Methylated ubiquitin acts as dominant-negative inhibitor [30] |
| Specialized Substrates | Analog-containing globin, iodinated lysozyme, rod outer segment proteins | Monitor proteolysis efficiency | Analog-containing proteins show rapid degradation (t½ = 15 min) [3] [30] |
Interpretation Framework for Inhibition Scenarios
Mechanistic Inference from Inhibition Patterns
Proper interpretation of inhibitor effects requires systematic analysis of the inhibition pattern across different aspects of the proteolytic pathway:
Diagram Title: Decision Framework for Inferring Inhibitor Mechanisms
Contextual Factors in Data Interpretation
When analyzing inhibition results, researchers must consider several contextual factors:
Cellular Environment Effects: Inhibitor efficacy may vary between purified systems and complex cellular environments. For example, the affinity between α-1-antitrypsin Z-variant and neutrophil elastase was more than two times better in plasma from individuals with high lung function compared to those with impaired function, demonstrating how environmental components modulate protein interactions [64].
Temporal Aspects of Inhibition: The timing of inhibitor addition relative to substrate introduction can reveal mechanistic information. Adding inhibitors at different time points in the proteolysis reaction helps distinguish between effects on initial recognition, ubiquitin conjugation, or degradation phases.
Concentration-Response Relationships: Quantitative analysis of inhibition potency should include IC₅₀ determinations and assessment of whether complete inhibition is achievable, suggesting irreversibility or allosteric effects.
Specificity Validation: Use complementary assays to verify inhibitor specificity, such as testing against unrelated enzymatic systems or using genetic approaches when available.
The rabbit reticulocyte ATP-dependent proteolytic system continues to serve as a robust platform for characterizing proteolysis inhibitors, from classical chemical inhibitors to physiological modulators and engineered peptide inhibitors. The well-defined nature of this system, combined with the comprehensive methodological framework presented herein, enables researchers to systematically classify inhibition mechanisms and translate these findings to more complex physiological and pathological contexts. As research advances, particularly in structural biology of AAA+ proteases [11] and the exploration of metabolite-based inhibitors [27], this foundational system will undoubtedly continue to yield critical insights into targeted proteolysis and its therapeutic manipulation.
The rabbit reticulocyte lysate (RRL) system represents a foundational in vitro tool for studying ATP-dependent intracellular protein degradation. First characterized as a soluble, non-lysosomal proteolytic system responsible for the energy-dependent degradation of abnormal proteins [3], this system has been instrumental in revealing the core components and mechanisms of the ubiquitin-proteasome pathway. Research using RRL has demonstrated that the system contains all necessary enzymatic machinery for the ubiquitination and degradation of target proteins, including two distinct high molecular weight proteases, one of which specifically degrades ubiquitin conjugates (UCDEN) [5]. This technical guide explores how this classical system can be adapted for contemporary research challenges, leveraging structural insights and innovative methodologies to study novel substrates and achieve complex experimental goals in drug discovery and basic research.
Structural and Mechanistic Basis for System Adaptation
Conserved Architecture of ATP-Dependent Proteases
A comprehensive understanding of ATP-dependent protease architecture provides the foundation for rational system adaptation. These proteases share a common structural organization despite limited sequence identity:
- Multisubunit Assemblies: ATP-dependent proteases form large multisubunit complexes ranging from approximately 425 kDa (FtsH) to 2 MDa (proteasome) [6].
- Compartmentalized Design: Proteolytic active sites are sequestered deep inside cylindrical structures, accessible only through narrow channels that exclude folded proteins [6].
- AAA+ ATPase Modules: ATPase subunits positioned at the entrance to proteolytic chambers perform dual functions of gating access and selecting/unfolding substrates through ATP hydrolysis [6].
Table 1: Major ATP-Dependent Protease Families and Their Characteristics
| Protease Family | Organismic Distribution | Architecture | Catalytic Mechanism | Key Features |
|---|---|---|---|---|
| Proteasome | Eukaryotes | 20S core + 19S regulatory particles | Threonine protease | Recognizes polyubiquitinated substrates [6] |
| Lon | Prokaryotes, eukaryotic organelles | Homohexameric rings | Ser-Lys catalytic dyad [65] | Contains AAA+ and proteolytic domains in single polypeptide [65] |
| ClpXP | Eubacteria, mitochondria | ClpX hexamer + ClpP heptameric rings | Ser-His-Asp catalytic triad [11] | Symmetry mismatch enables rotary mechanism [66] |
| ClpAP/ClpCP | Eubacteria, eukaryotes | Separate AAA+ (ClpA/C) + protease (ClpP) | Serine protease | Modular assembly with adaptor proteins [67] |
| FtsH | Prokaryotes, organelles | Homohexameric complex | Zinc metalloprotease | Membrane-associated [6] |
Structural Insights from Cryo-EM and Crystallography
Recent advances in structural biology, particularly cryo-electron microscopy (cryo-EM), have revolutionized our understanding of ATP-dependent proteases at near-atomic resolution:
- ClpXP Rotary Mechanism: Cryo-EM structures of substrate-bound ClpXP from Neisseria meningitidis at 2.3-3.3 Å resolution reveal how the sequential hydrolysis of ATP couples to motions of ClpX pore loops, leading to directional substrate translocation and rotation of ClpX relative to ClpP [66].
- Symmetry Mismatch Adaptation: The structural mismatch between hexameric ClpX and heptameric ClpP rings is accommodated through dynamic interactions that enable continuous degradation as ClpX rotates relative to ClpP [66].
- Pore Loop Mechanisms: Conserved aromatic and aliphatic residues in pore-1 (GYVG) and pore-2 (RDV) loops transmit mechanical force to substrates, facilitating unfolding through a processive rotary mechanism [66].
Substrate Targeting and Recognition Mechanisms
Ubiquitin-Dependent Targeting
The ubiquitin-proteasome system represents the primary degradation pathway in eukaryotic cells, with the RRL system containing all necessary components for reconstituting this pathway:
- Polyubiquitin Chain Assembly: A cascade of E1 (activating), E2 (conjugating), and E3 (ligating) enzymes attaches polyubiquitin chains to substrate proteins, typically through Lys48 linkages [6].
- Proteasome Recognition: The minimal proteasome targeting signal consists of four ubiquitin moieties, recognized by the 19S regulatory particle through surfaces formed by Rpn10 and Rpt5 subunits [6].
- Dynamic Tag Regulation: Deubiquitinating enzymes (DUBs) like Rpn11 and Ubp6, along with ubiquitin ligases such as Hul5, create a dynamic equilibrium that fine-tunes substrate commitment to degradation [6].
Degron Recognition and Initiation Mechanisms
Beyond ubiquitination, specific degradation signals (degrons) direct substrates to ATP-dependent proteases:
- Two-Step Targeting Model: Effective proteolysis requires both substrate recognition (e.g., through ubiquitination) and the presence of an unstructured region that serves as a degradation initiation site [6].
- Initiation Site Requirements: Unstructured regions, either at polypeptide termini or internally, engage the unfolding machinery of the protease and are indispensable for degradation of folded proteins [6].
- Local Stability Determinants: Susceptibility to unfolding depends more on the stability of local structures first encountered by the protease than on global protein stability [6].
Table 2: Quantitative Parameters for Substrate Degradation in ATP-Dependent Proteolytic Systems
| Parameter | Typical Range | Influencing Factors | Experimental Measurement Approaches |
|---|---|---|---|
| Degradation Rate (Kcat) | 0.1-10 min⁻¹ | Substrate stability, degron strength, protease concentration | Fluorescence depletion, radiolabel release [6] [3] |
| Unfolding Susceptibility | Varies >10 orders of magnitude [68] | Local structural stability, secondary structure content | cDNA display proteolysis, yeast display proteolysis [68] |
| Ubiquitin Chain Optimal Length | ≥4 ubiquitins [6] | E3 ligase processivity, DUB activity | Immunoblotting, mass spectrometry [6] |
| Peptide Product Size | 3-30 amino acids [6] | Protease specificity, processivity | HPLC, mass spectrometry [6] |
| Protease Processivity | Hundreds of ATP hydrolysis events/substrate [66] | Substrate stability, protease efficiency | Single-turnover assays, fluorescence monitoring [66] |
Experimental Methodologies for System Adaptation
Limited Proteolysis Mass Spectrometry (LiP-MS)
LiP-MS has emerged as a powerful technique for detecting protein structural changes and interactions on a proteome-wide scale:
- Workflow Optimization: Comprehensive benchmarking of 12 quantitative workflows demonstrates that both data-independent acquisition (DIA) and tandem mass tag (TMT) labeling effectively quantify LiP-MS experiments, with DIA-MS exhibiting greater accuracy in identifying true drug targets [69].
- Structural Proteomics Applications: LiP-MS can detect drug-induced structural changes, map binding sites, and identify allosteric effects without requiring specific reagents or labels [69].
- Protocol Implementation:
- Limited Proteolysis: Treat native protein samples with low protease concentrations under non-denaturing conditions.
- Proteolysis Quenching: Acidify samples to terminate proteolytic reactions.
- Peptide Digestion: Completely digest samples with a high-activity protease (e.g., trypsin).
- MS Analysis: Utilize either DIA or TMT approaches for peptide quantification.
- Data Analysis: Identify structural changes through protection/differential accessibility patterns [69].
cDNA Display Proteolysis for High-Throughput Stability Mapping
The recently developed cDNA display proteolysis method enables massive parallel analysis of protein folding stability:
- Unprecedented Throughput: Measurement of up to 900,000 protein domains in a single one-week experiment at minimal cost [68].
- Dual Protease Strategy: Incorporation of both trypsin and chymotrypsin proteases accounts for sequence-specific cleavage biases and improves stability inference accuracy [68].
- Quantitative Model: Bayesian inference of protease stability parameters (K50) enables calculation of thermodynamic folding stabilities (ΔG) [68].
- Validation: High consistency (R > 0.75) with traditional purified protein measurements establishes method reliability [68].
Cryo-EM Structural Analysis of Proteolytic Complexes
Recent technical advances enable high-resolution structural analysis of ATP-dependent proteases during substrate processing:
- Substrate-Bound Complex Trapping: Use of Walker B mutants (e.g., ClpX E185Q) slows ATP hydrolysis, prolonging complex lifetime for structural analysis [66].
- Symmetry Mismatch Resolution: Focused classification and refinement strategies overcome challenges posed by symmetry mismatches in complexes like ClpXP [66].
- Integrative Structural Biology: Combination of cryo-EM with solution-state NMR and computational methods provides comprehensive views of conformational dynamics [11].
Adaptation Strategies for Novel Substrates and Experimental Goals
Targeting Non-Natural Substrates
Adapting the RRL system for novel substrates requires strategic engineering of recognition elements:
- Degron Tagging: Fusion of natural degrons (e.g., SsrA tag) to proteins of interest directs them to specific ATP-dependent proteases [66].
- Ubiquitination Machinery Engineering: Co-expression of specific E3 ubiquitin ligases with substrates enables targeted ubiquitination and degradation in the RRL system [6].
- Unstructured Region Introduction: Incorporation of flexible linkers or destabilizing N-terminal provides initiation sites for protease engagement [6].
Modifying Proteolytic Specificity
The proteolytic specificity of the system can be tuned through multiple approaches:
- Protease Inhibitor Profiling: Use class-specific inhibitors (3,4-dichloroisocoumarin, N-ethylmaleimide, hemin) to characterize and modulate proteolytic activities [5].
- Subunit Composition Engineering: Recombinant expression of modified protease subunits alters cleavage preferences and regulatory properties [67].
- Allosteric Modulation: Small molecules (e.g., ADEPs for ClpP) can gate proteolytic chamber access and modulate degradation rates [66].
Quantitative Analysis Frameworks
Implementation of robust quantitative models enhances data interpretation from adapted systems:
- Two-Step Proteolysis Model: Accounts for demasking of peptide bonds through protein structure opening followed by bond hydrolysis [53].
- Selectivity Parameter Determination: Analysis of individual peptide bond cleavage rates relative to all potential cleavage sites [53].
- Kinetic Parameter Extraction: Determination of kcat and KM values for specific proteolytic events under defined conditions [53].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for ATP-Dependent Proteolysis Studies
| Reagent/Material | Function/Application | Example Specifications | Key References |
|---|---|---|---|
| Rabbit Reticulocyte Lysate | Source of ATP-dependent proteolytic machinery | Commercially prepared, nuclease-treated | [3] [5] |
| ATP Regeneration System | Maintains ATP levels during prolonged incubations | Creatine phosphate/creatine phosphokinase | [3] |
| Protease Inhibitors | Class-specific protease inhibition | 3,4-Dichloroisocoumarin (serine proteases), N-ethylmaleimide (cysteine proteases) | [5] |
| Ubiquitination System Components | Reconstitution of ubiquitin-dependent degradation | E1, E2, E3 enzymes, ubiquitin | [6] |
| cDNA Display Proteolysis Kit | High-throughput stability measurements | Includes transcription/translation and pull-down reagents | [68] |
| LiP-MS Workflow Solutions | Structural proteomics analysis | Optimized protease panels, TMT or DIA mass spectrometry | [69] |
| Cryo-EM Grids and Vitrification | Structural analysis of proteolytic complexes | UltraAuFoil grids, vitrification devices | [11] [66] |
| Walker B Mutant Constructs | Trapping transient proteolytic states | ClpX E185Q, other hydrolysis-deficient mutants | [66] |
The rabbit reticulocyte lysate ATP-dependent proteolysis system continues to evolve from a basic biochemical tool to a sophisticated platform for addressing complex research questions. By integrating structural insights from cryo-EM with innovative methodologies like cDNA display proteolysis and LiP-MS, researchers can now adapt this system to study increasingly diverse substrates and biological questions. The future of this field lies in further refining these adaptation strategies, particularly through engineered components with tailored specificities and enhanced computational integration of multi-dimensional data. As these tools become more accessible and sophisticated, they will undoubtedly accelerate both basic understanding of protein homeostasis and applied drug discovery efforts targeting proteolytic pathways.
Contextual Validation: Comparative Analysis with Other Biological Systems
The study of intracellular protein degradation is fundamental to understanding cellular homeostasis, quality control, and the pathogenesis of numerous diseases. Within this broad field, two experimental systems have provided particularly invaluable insights: the rabbit reticulocyte lysate (RRL) and retinal pigment epithelial (RPE) cell systems. The RRL represents a foundational in vitro model that was instrumental in discovering the core mechanisms of the ubiquitin-proteasome system (UPS). In contrast, RPE cells constitute a complex, physiologically relevant in vivo and ex vivo model for investigating how multiple proteolytic pathways, including the UPS, autophagy, and phagocytosis, integrate to maintain tissue function. This whitepaper provides a direct technical comparison of these two systems, delineating their distinct advantages, methodological applications, and relevance for researchers and drug development professionals working on proteostasis, neurodegenerative diseases, and ocular therapeutics.
System Fundamentals and Biological Context
Understanding the origin and inherent complexity of each system is crucial for experimental design and data interpretation.
Table 1: Fundamental Characteristics of RRL and RPE Systems
| Characteristic | Rabbit Reticulocyte Lysate (RRL) System | Retinal Pigment Epithelial (RPE) Cell System |
|---|---|---|
| System Type | Cell-free, biochemical extract [3] | Cellular, either primary, immortalized, or stem-cell derived [70] [71] |
| Biological Context | Simplified model for cytoplasmic proteolysis during red blood cell maturation [1] | Complex, polarized epithelial monolayer in a specific tissue (retina) [72] [70] |
| Key Primary Function | Degradation of abnormal proteins during terminal differentiation [3] [4] | Maintenance of retinal health via phagocytosis of photoreceptor outer segments (POS), visual cycle, and blood-retinal barrier formation [72] [70] [73] |
| Native Proteolytic Challenges | Clearance of misfolded globins and other unwanted proteins [3] | Daily phagocytosis and degradation of POS; clearance of damaged organelles and proteins in a high-oxidative-stress environment [72] [74] |
Mechanisms of Proteolysis: A Comparative Analysis
The core distinction between these systems lies in the diversity and regulation of their proteolytic pathways.
The Ubiquitin-Proteasome System in RRL
The RRL system was the experimental basis for the seminal discovery of the ATP-dependent ubiquitin-proteasome system. This pathway involves a cascade of enzymes (E1, E2, E3) that conjugate the small protein ubiquitin to substrate proteins, marking them for degradation by the 26S proteasome [1]. A key breakthrough from RRL studies was the identification of ATP's dual role: it is required both for ubiquitin conjugation and for a distinct, ubiquitin-independent step in the proteolytic process itself [4].
Multiple Degradative Pathways in RPE Cells
RPE cells are professional phagocytes that utilize a more complex network of degradative systems to maintain retinal health:
- Phagocytosis: RPE cells ingest the shed tips of photoreceptor outer segments (POS) in a daily, circadian rhythm-driven process. The ingested POS are packaged into phagosomes, which mature and fuse with lysosomes for degradation [72].
- Autophagy: This pathway rids the cell of damaged organelles, such as mitochondria (mitophagy), and toxic protein aggregates. A decline in autophagic flux is implicated in RPE pathology and age-related macular degeneration (AMD) [72] [74].
- The Ubiquitin-Proteasome System (UPS): Functions as a crucial quality control system for cytosolic proteins in RPE, similar to its role in other cells. Evidence suggests increased oxidative stress in AMD leads to protein damage and increased UPS component content in the retina [72].
The following diagram illustrates the convergence of these three major degradation pathways in the RPE cell.
Quantitative Functional Parameters
The functional output of these systems can be quantified through key biochemical and cellular metrics, as summarized in the table below.
Table 2: Quantitative Functional Parameters
| Parameter | Rabbit Reticulocyte Lysate (RRL) System | Retinal Pigment Epithelial (RPE) Cell System |
|---|---|---|
| ATP Dependence | Absolute requirement for ATP; stimulates degradation 2- to 4-fold [3] [4] | ATP required for UPS and autophagic flux; phagocytosis is also energy-dependent [72] |
| Degradation Rate of Model Substrates | Abnormal globin (t½ ~15 min) [3] | Phagocytosed POS are largely degraded within a few hours [72] |
| pH Optimum | ~7.8 [3] | Lysosomal degradation occurs at acidic pH (~4.5-5.0) [72] |
| Key Inhibitors | N-ethylmaleimide, iodoacetamide, o-phenanthroline, hemin [3] [4] | Proteasomal inhibitors (e.g., MG132); lysosomal inhibitors (e.g., chloroquine); cytoskeletal disruptors [72] |
| Pathological Hallmarks of Dysfunction | N/A (Acellular system) | Accumulation of lipofuscin and drusen; mitochondrial dysfunction; geographic atrophy in AMD [72] [74] [73] |
Detailed Experimental Protocols
Protocol 1: Assessing ATP-Dependent Proteolysis in RRL
This protocol is adapted from the pioneering work of Etlinger & Goldberg (1977) [3].
- RRL Preparation: Induce reticulocytosis in rabbits with phenylhydrazine. Draw blood via cardiac puncture and purify reticulocytes by centrifugation. Lysate cells in a low-ionic-strength buffer and centrifuge at 100,000 x g to obtain a soluble supernatant [3].
- Reaction Setup:
- Standard Reaction Mixture: Combine 25 µL of RRL, 5 µL of an ATP-regenerating system (2 mM ATP, 10 mM Creatine Phosphate, 0.1 mg/mL Creatine Phosphokinase), and 1 µL of radiolabeled abnormal protein substrate (e.g., ²H-methyl-casein or analog-containing globin).
- Controls: Include reactions with a non-hydrolyzable ATP analog (e.g., AMP-PNP) or an ATP-depleting system (e.g., Apyrase) [4].
- Incubation: Incubate the reactions at 37°C for 60 minutes.
- Termination and Measurement: Stop the reaction by adding trichloroacetic acid (TCA) to a final concentration of 10%. Precipitate on ice, then centrifuge. Measure the radioactivity in the TCA-soluble supernatant (degraded peptides/amino acids) via scintillation counting [3].
- Data Analysis: Calculate the rate of proteolysis as the pmol of amino acids released per mg of lysate protein per hour. ATP-dependent degradation is calculated as the difference between rates in the presence of ATP and in its absence.
Protocol 2: Measuring Photoreceptor Outer Segment (POS) Phagocytosis in RPE Cells
This protocol models the critical in vivo function of RPE cells [72].
- Cell Culture: Culture polarized human RPE cells (e.g., ARPE-19, hTERT-RPE, or stem-cell derived RPE) on Transwell filters for at least 4-6 weeks to establish mature, polarized monolayers with tight junctions [71].
- POS Isolation: Isolate POS from fresh bovine or porcine retinas by sucrose density gradient centrifugation. Label POS with a fluorescent dye (e.g., FITC or pHrodo) [72].
- Phagocytosis Assay: Add the labeled POS particles to the apical medium of the RPE monolayer. To synchronize uptake, a common method is to briefly spin the POS onto the cell surface. Incubate at 37°C for a defined pulse (e.g., 3-5 hours).
- Quenching and Washing: After the pulse, remove non-internalized POS by extensively washing the cells. Treat with Trypan Blue to quench the fluorescence of any surface-bound, non-internalized POS.
- Analysis:
- Flow Cytometry: Trypsinize the cells and analyze by flow cytometry to quantify the mean fluorescence intensity per cell, representing internalized POS.
- Immunofluorescence: Fix cells and visualize internalized POS via confocal microscopy. Co-staining with tight junction markers (e.g., ZO-1) confirms monolayer integrity.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents and Their Applications
| Reagent / Solution | Function in RRL System | Function in RPE Cell System |
|---|---|---|
| ATP-regenerating System | Maintains constant ATP levels crucial for ubiquitination and proteasomal degradation [3]. | Used in in vitro assays to sustain energy-dependent processes like phagocytosis and autophagy. |
| Ubiquitin | The central tagging polypeptide; conjugation to substrates is a key step in the degradative pathway [1] [4]. | Studied in the context of cytosolic protein quality control; alterations linked to RPE stress in disease. |
| Proteasome Inhibitors (e.g., MG132) | Used to confirm proteasome-dependent degradation and to isolate ubiquitination steps [72]. | Tool to induce proteostatic stress and model UPS impairment in RPE pathology. |
| Lysosomal Inhibitors (e.g., Chloroquine, Bafilomycin A1) | Largely ineffective, confirming the non-lysosomal nature of RRL proteolysis [3]. | Used to inhibit autophagic and phagocytic flux, leading to accumulation of substrates (e.g., lipofuscin) [72]. |
| Hemin | A potent inhibitor of ATP-dependent proteolysis in RRL; used to probe regulatory mechanisms [4]. | Not a standard reagent; iron homeostasis is studied in the context of oxidative stress and ferroptosis in RPE. |
| Reconstituted POS | Not applicable. | The physiological substrate for RPE phagocytosis; essential for functional assays [72]. |
| Oxidative Stress Inducers (e.g., tBHP) | Can be used to generate oxidized protein substrates for degradation studies. | Used to model AMD-related stress; induces protein damage, alters antioxidant protein expression, and disrupts RPE function [74] [75]. |
Research Applications and Therapeutic Relevance
The choice between RRL and RPE systems is dictated by the research question, balancing reductionist power against physiological complexity.
RRL System Applications
- Mechanistic Biochemistry: Ideal for dissecting the step-by-step molecular mechanics of the UPS, including ubiquitin conjugation and proteasomal gating/processing [1] [4].
- Drug Screening: Can be used for high-throughput screening of compounds that modulate UPS activity, with relevance to cancer and neurodegenerative diseases.
- Study of Mutant Proteins: Serves as a platform to investigate the degradation kinetics and mechanisms of disease-associated mutant proteins.
RPE System Applications
- Disease Modeling: Essential for studying the pathophysiology of retinal degenerative diseases like AMD, Stargardt disease, and retinitis pigmentosa, where RPE dysfunction is primary [72] [74] [73].
- Therapeutic Development: Critical for evaluating the efficacy and mechanisms of action of new therapeutics, including antioxidants, visual cycle modulators, and complement inhibitors [74] [75].
- Cell and Tissue Therapy: Serves as the target for regenerative medicine approaches, including stem cell-derived RPE transplantation and tissue-engineered RPE patches [70] [73].
The RRL and RPE systems represent two complementary yet distinct pillars in proteolysis research. The RRL system is a powerful, reductionist tool for elucidating fundamental biochemical principles of the UPS with unparalleled clarity. In contrast, RPE cell systems offer a holistic model to investigate the intricate crosstalk between multiple proteolytic pathways within a highly specialized, disease-relevant cellular context. For researchers aiming to bridge fundamental mechanisms with therapeutic applications, a synergistic approach that leverages the strengths of both systems will be most fruitful. Understanding the specific proteolytic failures in RPE cells, informed by foundational knowledge from RRL studies, is key to developing novel treatments for the millions of patients affected by retinal degenerative diseases.
The rabbit reticulocyte lysate (RRL) system stands as a foundational discovery in the understanding of intracellular protein degradation, providing the first evidence of an ATP-dependent proteolytic pathway soluble in the cytosol. This seminal model revealed that the degradation of abnormal proteins is an energy-dependent process, a characteristic that extends to the highly specialized environment of the cardiac muscle. Subsequent research has delineated a complex spectrum of proteolytic control, encompassing ubiquitin-dependent degradation via the 26S proteasome and ubiquitin-independent pathways facilitated by the 20S proteasome and other ATP-dependent proteases. This whitepaper synthesizes quantitative data and experimental protocols from key studies, framing them within the context of RRL research to explore how the heart muscle adeptly utilizes both arms of this proteolytic system to maintain proteostasis. The intricate balance between these pathways holds significant implications for therapeutic interventions in cardiac disease and drug development.
The discovery of an ATP-dependent proteolytic system in rabbit reticulocyte lysates (RRL) marked a paradigm shift in cell biology. Prior to this, protein degradation was largely considered a passive, lysosomal process. The RRL system demonstrated that the selective degradation of abnormal proteins—such as globin incorporating the valine analog 2-amino-3-chlorobutyric acid (ClAbu)—was carried out by a soluble, non-lysosomal machinery that required metabolic energy [3]. This ATP requirement was a critical clue pointing to the existence of novel biochemical mechanisms, fundamentally altering the pursuit of the responsible enzymes.
The RRL model established several core characteristics of what would later be identified as the ubiquitin-proteasome system (UPS):
- Energy Dependence: Proteolysis was stimulated several-fold by ATP, and slightly by ADP, while AMP and cyclic AMP had no significant effect [3].
- Cytosolic Localization: The activity was found in the 100,000 x g supernatant, indicating a non-lysosomal origin [3].
- Specificity for Abnormal Proteins: The system selectively degraded analog-containing abnormal globin (half-life ~15 minutes) while leaving normal hemoglobin largely intact [3].
- Biochemical Sensitivity: Proteolysis in the cell-free RRL system was inhibited by agents like N-ethylmaleimide, iodoacetamide, and serine protease inhibitors, a profile that paralleled inhibition within intact cells [3].
This foundational work in RRL provided the essential biochemical framework for the subsequent discovery of ubiquitin and its central role in the majority of ATP-dependent cytoplasmic proteolysis. It established a baseline from which the full spectrum of proteolytic mechanisms, including ubiquitin-independent pathways, could be explored.
The Ubiquitin Dependence Spectrum in Proteolysis
The proteolytic landscape is not a simple binary but a spectrum of mechanisms defined by their reliance on ubiquitin. The RRL system was instrumental in initially characterizing the ubiquitin-dependent branch, which is responsible for degrading the majority of short-lived and misfolded proteins in the cytosol and nucleus. However, parallel and complementary ubiquitin-independent pathways also exist, providing the cell with a versatile and layered proteostatic network.
Ubiquitin-Dependent Proteolysis
This is the predominant pathway for targeted protein degradation in eukaryotic cells. It involves the covalent attachment of a ubiquitin chain to a substrate protein, which acts as a molecular tag for recognition and degradation by the 26S proteasome.
- The Enzymatic Cascade: Ubiquitination is mediated by a sequential cascade involving E1 (activating), E2 (conjugating), and E3 (ligating) enzymes. The E3 ubiquitin ligases, of which there are hundreds, provide substrate specificity [76] [77].
- The Proteasome: The 26S proteasome is a massive multi-subunit complex comprising a 20S core particle (where proteolysis occurs) and one or two 19S regulatory particles that recognize ubiquitinated substrates, remove the ubiquitin chain, and unfold the substrate for translocation into the core [76].
- Chain Topology Dictates Fate: The fate of the ubiquitinated protein is determined by the topology of the ubiquitin chain. K48-linked and K11-linked polyubiquitin chains are primary signals for proteasomal degradation, whereas K63-linked and linear chains typically serve non-proteolytic roles in signaling and trafficking [78] [77].
Ubiquitin-Independent Proteolysis
Certain proteins can be degraded by the proteasome without prior ubiquitination. This pathway often handles proteins that have become structurally disordered or damaged.
- The 20S Core Proteasome: The 20S proteasome itself can function independently, degrading unfolded proteins without the need for the 19S regulator or ubiquitin tagging. This is considered a primary defense against oxidative stress, where damaged proteins often expose hydrophobic regions that can be directly recognized by the 20S core [79].
- Alternative Activators: The 20S proteasome can also be activated by other regulatory complexes, such as the PA28αβ (11S) regulator, which facilitates the degradation of small, unfolded peptides in an ATP- and ubiquitin-independent manner [79].
- Direct Proteasome Binding: Some proteins contain intrinsic, unstructured regions that allow them to bypass the ubiquitin system and bind directly to the proteasome for degradation. For instance, studies on the BAF57 subunit of the SWI/SNF complex revealed a ubiquitin-independent degradation mechanism that is blocked when the subunit is incorporated into the larger complex [80].
Table 1: Comparative Features of Ubiquitin-Dependent and Ubiquitin-Independent Proteolytic Pathways
| Feature | Ubiquitin-Dependent Pathway | Ubiquitin-Independent Pathway |
|---|---|---|
| Key Signal | K48/K11-linked polyubiquitin chain | Unfolded, oxidized, or intrinsically disordered regions |
| Protease Complex | 26S Proteasome (20S core + 19S cap) | 20S Core Proteasome (alone or with PA28) |
| ATP Requirement | Yes (for ubiquitination & substrate unfolding) | Not strictly required for core proteolysis |
| Primary Function | Targeted degradation of specific substrates | Bulk degradation of damaged/unstructured proteins |
| Historical Model | Rabbit Reticulocyte Lysate (RRL) | Bacterial Protease La / Mitochondrial Protease |
Experimental Protocols: From Foundational RRL to Modern Cardiac Studies
Core Protocol: ATP-Dependent Proteolysis in Rabbit Reticulocyte Lysate
The foundational experiment that established the energy dependence of proteolysis can be reconstructed as follows [3]:
Objective: To measure the ATP-dependent degradation of abnormal proteins in a cell-free system derived from rabbit reticulocytes.
Key Reagents & Solutions:
- Reticulocyte Lysate: Prepared from phenylhydrazine-treated anemic rabbits. Reticulocytes are lysed in a low-ionic-strength buffer and centrifuged at 100,000 x g to obtain a clear supernatant (S100).
- Abnormal Protein Substrate: [^{3}\text{H}]- or [^{14}\text{C}]-leucine-labeled globin synthesized in the presence of the amino acid analog 2-amino-3-chlorobutyric acid (ClAbu). The analog-containing globin is isolated to high specific activity.
- ATP-Regenerating System: 2 mM ATP, 10 mM phosphocreatine, 50 µg/mL creatine phosphokinase, 5 mM MgCl₂.
- Inhibitor Controls: 0.5 mM N-ethylmaleimide (NEM) or 10 mM iodoacetamide.
Procedure:
- Reaction Setup: In a final volume of 100 µL, combine 50 µL of S100 lysate, 10 µg of labeled abnormal globin, and the ATP-regenerating system. Control reactions replace ATP with an equal volume of water or include NEM/iodoacetamide.
- Incubation: Incubate the reactions at 37°C for 60 minutes.
- Reaction Termination: Stop the reaction by adding 100 µL of 10% (w/v) trichloroacetic acid (TCA).
- Measurement of Degradation: Centrifuge the TCA-precipitated samples. Measure the radioactivity in the TCA-soluble supernatant (containing amino acids and small peptides) using a liquid scintillation counter. This represents the degraded material.
- Data Analysis: Calculate the rate of proteolysis as the percentage of TCA-precipitable counts converted to TCA-soluble counts per hour. The ATP-dependent component is determined by subtracting the degradation rate in the absence of ATP from the rate in its presence.
Protocol: Assessing Proteasome Activity in Cardiac Muscle
Modern studies on cardiac proteostasis often measure proteasome activity directly, building upon the principles established in the RRL system [81].
Objective: To measure the chymotrypsin-like activity of the proteasome in cardiac tissue lysates.
Key Reagents & Solutions:
- Cardiac Tissue Homogenate: Fresh or snap-frozen heart tissue homogenized in a buffer containing 50 mM Tris-HCl (pH 7.5), 1 mM DTT, 5 mM MgCl₂, 2 mM ATP, and 10% glycerol. The homogenate is centrifuged at 10,000 x g, and the supernatant is used for the assay.
- Fluorogenic Proteasome Substrate: Suc-LLVY-AMC (for chymotrypsin-like activity). The proteasome cleaves the peptide bond, releasing fluorescent AMC (7-amino-4-methylcoumarin).
- Proteasome Inhibitor Control: 20 µM MG132 or 10 µM lactacystin.
- Assay Buffer: 50 mM HEPES (pH 7.5), 5 mM MgCl₂, 1 mM DTT, 2 mM ATP.
Procedure:
- Reaction Setup: In a black 96-well plate, combine 50 µg of cardiac tissue supernatant, 100 µM Suc-LLVY-AMC, and assay buffer to a final volume of 200 µL. Include control wells with tissue supernatant pre-incubated with MG132 for 30 minutes to confirm specificity.
- Incubation and Measurement: Incubate the plate at 37°C and measure the fluorescence (excitation ~380 nm, emission ~460 nm) kinetically every 5 minutes for 60-90 minutes using a plate reader.
- Data Analysis: Calculate the rate of AMC release (relative fluorescence units per minute, RFU/min). The proteasome-specific activity is the MG132-inhibitable portion of this rate. Data is often normalized to total protein concentration.
Table 2: Key Research Reagent Solutions for ATP-Dependent Proteolysis Studies
| Research Reagent | Function in Experiment | Example Application |
|---|---|---|
| Rabbit Reticulocyte Lysate (S100) | Source of cytosolic ATP-dependent proteolytic machinery (E1, E2, E3, proteasome). | Foundational assay for ubiquitin-dependent proteolysis [3]. |
| Amino Acid Analogs (e.g., ClAbu) | Incorporated into nascent proteins during synthesis to generate misfolded, degradation-prone substrates. | Creating defined abnormal protein substrates for degradation assays [3]. |
| ATP-Regenerating System (ATP, CP, CPK) | Maintains a constant, high level of ATP in cell-free reactions, crucial for energy-dependent processes. | Essential for sustaining ubiquitination and proteasomal degradation in vitro [3]. |
| Proteasome Inhibitors (MG132, Lactacystin, Bortezomib) | Specifically and reversibly/irreversibly inhibit the proteolytic activity of the 20S proteasome. | Validating proteasome involvement in a degradation process; cancer therapy [81] [77]. |
| Fluorogenic Peptide Substrates (e.g., Suc-LLVY-AMC) | Proteasome-specific peptides linked to a fluorophore. Cleavage releases a measurable fluorescent signal. | Quantifying proteasome activity in tissue lysates or purified systems [81]. |
| NEDD8-Activating Enzyme (NAE) Inhibitor (MLN4924) | Inhibits neddylation of cullins, thereby inactivating Cullin-RING Ligases (CRLs), a major class of E3s. | Probing the role of CRLs in processes like cardiac development and cell cycle [77] [82]. |
Cardiac Muscle: A Paradigm of Specialized Proteostasis
The heart, as a post-mitotic organ with limited regenerative capacity, is critically dependent on efficient protein quality control systems to maintain function over a lifetime. Cardiomyocytes utilize both ubiquitin-dependent and independent pathways to manage proteostatic stress, with the balance often determining pathological outcomes.
The Ubiquitin-Proteasome System in Cardiac Health and Disease
The UPS is the primary system for targeted protein turnover in the heart. Its role is multifaceted, ranging from routine protein quality control to adaptive and maladaptive remodeling in disease.
- Evidence of UPS Insufficiency in Failure: Human failing hearts from various etiologies consistently show elevated levels of total ubiquitinated proteins and ubiquitin-positive protein aggregates. This suggests that the demand for ubiquitin-mediated proteolysis may exceed the system's capacity during pathological stress [76].
- Regulation of Key Processes: The UPS controls the stability of transcription factors, ion channels, and sarcomeric proteins, thereby influencing cardiac hypertrophy, metabolism, and contractility. For example, specific E3 ligases like certain Cullin-RING ligases (CRLs) are essential for cardiac development, with their disruption leading to defects such as ventricular non-compaction [82].
- Therapeutic Targeting: The proteasome inhibitor Bortezomib, used to treat multiple myeloma, can cause cardiac dysfunction, highlighting the heart's sensitivity to UPS perturbation [76]. Conversely, targeting upstream components like the NEDD8-activating enzyme (with MLN4924) is being explored for its therapeutic potential, though it also demonstrates cardiotoxicity in developmental contexts [77] [82].
Ubiquitin-Independent and Complementary Pathways in the Heart
While the UPS is dominant, the heart also leverages ubiquitin-independent mechanisms and closely linked systems like autophagy to manage proteostasis.
- Autophagy as a Key Partner: Autophagy is a lysosomal degradation pathway that clears damaged organelles (e.g., mitochondria), protein aggregates, and long-lived proteins. It is critically regulated by ubiquitin through receptors like p62/SQSTM1, which bind ubiquitinated cargo and target them to autophagosomes [83]. This creates a functional interface where ubiquitin signals can direct cargo to the proteasome or the autophagosome.
- Metabolic Adaptation: Autophagy is rapidly upregulated in the heart during starvation, providing amino acids, lipids, and nucleic acids to support energy production and cellular homeostasis. Blocking autophagy accelerates energy depletion and cell death under such stress [83].
- Context-Dependent Role in Disease: In ischemic heart disease, autophagy is generally considered cardioprotective during ischemia (by providing energy), but its role during reperfusion is more complex and can be maladaptive if overactivated [83]. This illustrates the nuanced balance of proteolytic pathways in cardiac pathology.
The journey from the initial characterization of ATP-dependent proteolysis in rabbit reticulocyte lysates to the current understanding of the heart's sophisticated proteostatic network underscores a fundamental biological principle: cellular protein degradation is an active, regulated, and energetically costly process. The spectrum of ubiquitin dependence—from the highly specific, ubiquitin-dependent degradation of regulatory proteins to the more general, ubiquitin-independent clearance of damaged proteins—provides the cardiac muscle with the versatility needed to adapt to constant physiological and pathological challenges.
Future research and therapeutic development will continue to be informed by this spectrum. Key frontiers include:
- Defining the Interactome: Systematically mapping the interactions between E3 ubiquitin ligases and their specific substrates in the heart under different conditions will reveal novel regulatory nodes.
- Targeting Deubiquitinases (DUBs): As counter-regulatory enzymes to E3 ligases, DUBs like the Ubiquitin-Specific Proteases (USPs) are emerging as critical players in cardiomyopathy [84]. Developing specific pharmacological modulators for DUBs represents a promising therapeutic strategy.
- Harnessing Ubiquitin-Independence: A deeper understanding of how the 20S proteasome is activated and regulated could lead to strategies for enhancing the clearance of oxidized and misfolded proteins in conditions of oxidative stress, such as ischemia-reperfusion injury, without overburdening the ubiquitin system.
The RRL system remains a powerful historical and experimental paradigm. Its legacy is a framework that continues to guide the exploration of proteolytic mechanisms in specialized tissues like the heart, driving the development of next-generation therapies aimed at preserving cardiac function by maintaining the delicate balance of protein synthesis and degradation.
The ATP-dependent proteolytic system, first extensively characterized in rabbit reticulocyte lysates, represents a fundamental biological pathway responsible for the selective degradation of abnormal and short-lived proteins in eukaryotic cells [85] [3]. This proteolytic pathway exhibits distinct tissue-specific variations in activity, regulation, and molecular composition, leading to divergent responses to pharmacological inhibitors across different tissue extracts. Understanding these differential inhibitor profiles is crucial for both basic biochemical research and drug development, particularly in the context of targeted therapeutic interventions.
Research spanning several decades has established that the ATP-dependent proteolytic system is highly conserved yet demonstrates remarkable functional specialization across tissues. Early investigations revealed that reticulocytes possess a soluble, non-lysosomal proteolytic system that requires ATP for the degradation of abnormal proteins [3]. Subsequent studies identified that this system consists of multiple components, including a ubiquitin-conjugating apparatus and high molecular weight proteases that degrade ubiquitinated substrates [5]. The tissue-specific nature of this system became apparent when comparative studies demonstrated significant variations in ATP-dependent proteolytic activity across different tissue types, with adrenal cortex mitochondria, for instance, exhibiting particularly high activity [86].
The investigation of inhibitor sensitivities across tissue extracts provides critical insights into the molecular diversity of proteolytic complexes and their regulatory mechanisms. This technical guide synthesizes current understanding of comparative inhibitor profiles, experimental methodologies for assessing proteolytic activity, and practical applications for research and drug discovery within the broader context of rabbit reticulocyte lysate ATP-dependent proteolysis system research.
The ATP-Dependent Proteolytic System in Reticulocytes: Core Components
The ATP-dependent proteolytic system in rabbit reticulocytes comprises several functionally distinct components that work in concert to identify, tag, and degrade target proteins. The system fundamentally consists of two major parts: the ubiquitin conjugation machinery that marks proteins for degradation, and the proteasome complex that executes the actual proteolysis.
Historical Foundation and Key Discoveries
The foundational discovery of ATP-dependent proteolysis in reticulocytes emerged in the 1970s, when Etlinger and Goldberg identified a soluble, non-lysosomal system responsible for degrading abnormal proteins in rabbit reticulocytes [3]. This system was found to be localized in the 100,000 × g supernatant, have a pH optimum of 7.8, and require ATP for maximal activity. Subsequent research by Hershko, Ciechanover, and Rose led to the discovery of the ubiquitin-dependent pathway, which revolutionized understanding of intracellular protein degradation [85] [1].
The ubiquitin-mediated proteolytic pathway involves a cascade of enzymes (E1, E2, E3) that conjugate ubiquitin to target proteins, marking them for destruction by the 26S proteasome. The 26S proteasome itself consists of a 20S core particle (CP) that carries the catalytic activity and a 19S regulatory particle (RP) that recognizes ubiquitinated substrates [1]. The discovery of these components explained the previously observed ATP dependence of proteolysis, as both the ubiquitination process and proteasomal degradation require energy.
Molecular Composition and Functional Specialization
Fractionation studies of rabbit reticulocytes revealed two distinct high molecular weight proteases with different functions [5]. The first is an approximately 1500 kDa protease that degrades proteins only when ATP and the conjugating fractions are added. This enzyme, essential for ATP-dependent proteolysis, is labile in the absence of nucleotides and is inhibited by various compounds including heparin, poly(Glu-Ala-Tyr), 3,4-dichloroisocoumarin, hemin, decavanadate, N-ethylmaleimide, and peptide chloromethyl ketones. The second protease, approximately 670 kDa, does not require ATP or ubiquitin and is therefore not required for ATP-dependent proteolysis [5].
The ATP-dependent protease from bovine adrenal cortex mitochondria shares some characteristics with the reticulocyte system but also exhibits distinct properties, including stimulation by ATP plus MgCl₂ at alkaline pH (8.2), and inhibition by vanadate derivatives and quercetin but not by oligomycin and ouabain [86]. This tissue-specific variation in inhibitor sensitivity forms the basis for comparative studies of inhibitor profiles across different tissue extracts.
Tissue-Specific Variations in ATP-Dependent Proteolysis
Comparative analyses of ATP-dependent proteolytic activity across tissues reveal significant variations in expression levels, functional properties, and inhibitor sensitivities. These differences reflect tissue-specific adaptations to metabolic requirements, protein quality control needs, and specialized regulatory mechanisms.
Quantitative Variations in Proteolytic Activity
Studies measuring ATP-dependent proteolytic activities in mitochondrial fractions from several tissues in rats and bovines demonstrated that the adrenal cortex exhibits one of the highest activities among tissues examined [86]. This tissue-specific pattern of activity correlates with specialized physiological functions, suggesting that tissues with high protein turnover or complex secretory functions have evolved enhanced proteolytic capacity.
The molecular basis for these tissue-specific variations includes differences in the expression levels of proteasome subunits, regulatory particles, and ubiquitin-conjugating enzymes. Additionally, tissue-specific proteasome activators and regulators may contribute to the functional specialization of the proteolytic system across different cell types.
Tissue-Specific Inhibitor Sensitivities
The differential sensitivity of ATP-dependent proteolytic systems to various inhibitors across tissues provides crucial insights into their structural and functional diversity. The following table summarizes documented inhibitor sensitivities in different tissue extracts:
Table 1: Comparative Inhibitor Sensitivities of ATP-Dependent Proteolytic Systems Across Tissues
| Inhibitor | Reticulocyte System | Adrenal Cortex Mitochondria | General Sensitivity |
|---|---|---|---|
| N-Ethylmaleimide | Sensitive [5] | Sensitive [86] | Sulfhydryl group modifier |
| Hemin | Sensitive [85] [5] | Not reported | Heme-regulated inhibitor |
| Vanadate derivatives | Sensitive (decavanadate) [5] | Sensitive (o-vanadate, m-vanadate, vanadyl sulfate) [86] | Phosphate analog |
| Chloromethyl ketones | Sensitive [5] | Not reported | Serine protease inhibitor |
| Heparin | Sensitive [5] | Not reported | Glycosaminoglycan |
| 3,4-Dichloroisocoumarin | Sensitive [5] | Not reported | Serine protease inhibitor |
| Phenylmethylsulfonyl fluoride | Not reported | Sensitive [86] | Serine protease inhibitor |
| Quercetin | Not reported | Sensitive [86] | Flavonoid |
| Mersalyl acid | Not reported | Sensitive [86] | Organomercurial |
The molecular basis for these differential inhibitor profiles includes variations in proteasome subunit composition, differences in the regulatory particles, tissue-specific post-translational modifications, and the presence of tissue-specific proteasome activators and inhibitors.
Experimental Protocols for Assessing Inhibitor Sensitivity
Comprehensive evaluation of inhibitor profiles across tissue extracts requires standardized experimental approaches that enable direct comparison of proteolytic activities and inhibitor sensitivities. The following section details essential methodologies for such comparative analyses.
Preparation of Tissue Extracts
Reticulocyte Lysate Preparation:
- Induce reticulocytosis in rabbits through phenylhydrazine treatment (40 mg/kg for 3-5 days)
- Collect blood via cardiac puncture and isolate reticulocytes by centrifugation
- Wash cells repeatedly in isotonic saline solution
- Lyse cells by osmotic shock in hypotonic buffer (5 mM Tris-HCl, pH 7.5, 2 mM DTT)
- Centrifuge at 100,000 × g for 60 minutes to obtain soluble fraction
- Aliquot and store at -80°C until use [3]
Mitochondrial Extract Preparation from Adrenal Cortex:
- Homogenize fresh adrenal cortex tissue in ice-cold isolation buffer (0.25 M sucrose, 10 mM Tris-HCl, pH 7.4)
- Centrifuge homogenate at 600 × g for 10 minutes to remove nuclei and debris
- Collect supernatant and centrifuge at 8,000 × g for 15 minutes to pellet mitochondria
- Wash mitochondrial pellet twice in isolation buffer
- Solubilize mitochondria in extraction buffer (20 mM Tris-HCl, pH 7.5, 5 mM MgCl₂, 1 mM DTT, 0.5% Triton X-100)
- Centrifuge at 100,000 × g for 60 minutes and collect supernatant as mitochondrial extract [86]
Proteolytic Activity Assay
Standard Reaction Conditions:
- Substrate: ¹⁴C-methyl-casein (50,000-100,000 cpm/mg)
- Reaction buffer: 50 mM Tris-HCl (pH 7.5-8.2), 5 mM MgCl₂, 1 mM DTT
- ATP regenerating system: 2 mM ATP, 10 mM phosphocreatine, 50 μg/mL creatine phosphokinase
- Tissue extract: 50-100 μg protein
- Total reaction volume: 100-250 μL
- Incubation: 37°C for 60-120 minutes [86]
Termination and Measurement:
- Stop reactions by adding 1 mL of 10% trichloroacetic acid (TCA)
- Incubate on ice for 30 minutes to precipitate undegraded protein
- Centrifuge at 10,000 × g for 10 minutes
- Measure radioactivity in TCA-soluble supernatant by liquid scintillation counting
- Calculate proteolytic activity as TCA-soluble radioactivity released per mg protein per hour [3] [86]
Inhibitor Sensitivity Profiling
Comprehensive Inhibitor Screening:
- Prepare inhibitor stocks at 100-1000× final concentration in appropriate solvents
- Pre-incubate tissue extracts with inhibitors for 10 minutes at 25°C before adding substrate
- Include solvent controls for each inhibitor concentration
- Test a range of concentrations (typically 0.1-100 μM) to establish dose-response relationships
- Normalize activities to uninhibited controls (100% activity)
- Calculate IC₅₀ values from inhibition curves [5] [87]
Ubiquitin-Dependence Assessment:
- Compare proteolytic activity in the presence and absence of added ubiquitin (0.1-1 mg/mL)
- Assess ATP dependence by omitting ATP regenerating system
- Include negative controls with heat-denatured tissue extracts
- Use specific ubiquitin pathway inhibitors (e.g., ubiquitin-aldehyde) to confirm ubiquitin involvement [5]
Data Analysis and Interpretation
Quantitative Comparison of Inhibitor Profiles
Systematic analysis of inhibitor sensitivity data enables comprehensive comparison of proteolytic systems across tissue extracts. The following table presents a template for organizing and comparing inhibitor efficacy data:
Table 2: Template for Comparative Inhibitor Profile Analysis Across Tissue Extracts
| Inhibitor | Mechanism of Action | IC₅₀ Reticulocyte (μM) | IC₅₀ Adrenal Cortex (μM) | IC₅₀ Liver (μM) | IC₅₀ Muscle (μM) | Selectivity Index |
|---|---|---|---|---|---|---|
| N-Ethylmaleimide | Sulfhydryl alkylation | [Value] | [Value] | [Value] | [Value] | [Value] |
| Hemin | Heme-regulated inhibitor | [Value] | [Value] | [Value] | [Value] | [Value] |
| Decavanadate | Phosphate analog | [Value] | [Value] | [Value] | [Value] | [Value] |
| 3,4-Dichloroisocoumarin | Serine protease inhibitor | [Value] | [Value] | [Value] | [Value] | [Value] |
| Quercetin | Flavonoid, inhibits ATPases | [Value] | [Value] | [Value] | [Value] | [Value] |
| Lactacystin | Specific proteasome inhibitor | [Value] | [Value] | [Value] | [Value] | [Value] |
| MG132 | Peptide aldehyde proteasome inhibitor | [Value] | [Value] | [Value] | [Value] | [Value] |
The selectivity index can be calculated as the ratio of IC₅₀ values between different tissue extracts, highlighting inhibitors with tissue-specific potency that may reflect structural or functional differences in proteolytic complexes.
Statistical Analysis and Validation
Robust statistical analysis is essential for establishing significant differences in inhibitor sensitivity:
- Perform triplicate measurements for each inhibitor concentration
- Calculate mean ± standard deviation for normalized proteolytic activities
- Use nonlinear regression to determine IC₅₀ values with 95% confidence intervals
- Apply two-way ANOVA to assess significant differences between tissue extracts and inhibitor concentrations
- Calculate selectivity ratios with statistical significance testing
Validation of inhibitor specificity should include counter-screening against unrelated enzymatic systems to exclude non-specific effects and confirmation using multiple substrate types to verify mechanism-based inhibition.
Research Reagent Solutions
Comprehensive profiling of inhibitor sensitivities requires a carefully selected toolkit of research reagents. The following table details essential materials for investigating ATP-dependent proteolysis across tissue extracts:
Table 3: Essential Research Reagents for ATP-Dependent Proteolysis Studies
| Reagent/Category | Specific Examples | Function/Application | Key Characteristics |
|---|---|---|---|
| Protease Inhibitor Cocktails | Commercial mammalian protease inhibitor cocktail (e.g., Sigma P8340) [87] | Broad-spectrum inhibition of serine, cysteine, acid proteases, and aminopeptidases | Contains AEBSF, aprotinin, E-64, bestatin, leupeptin, pepstatin A; soluble in DMSO |
| Specific Proteasome Inhibitors | Lactacystin, MG132, Epoxomicin, Bortezomib | Selective inhibition of proteasome catalytic activities | Varying specificity for different proteasome active sites; essential for mechanism determination |
| ATP System Components | ATP, ATPγS, AMP-PNP, Creatine phosphate/creatine phosphokinase system | Energy source and ATP analog studies | Distinguish ATP-dependent and independent proteolysis; identify energy requirements |
| Ubiquitin System Reagents | Ubiquitin, Methylated ubiquitin, Ubiquitin aldehyde, E1/E2/E3 enzymes | Ubiquitin-dependent pathway modulation | Determine ubiquitin dependence; investigate conjugation machinery |
| Substrate Panel | ¹⁴C-methyl-casein, Fluorescent peptides, Ubiquitin-¹²⁵I-lysozyme conjugates | Proteolytic activity measurements | Different substrates may show varying degradation rates and inhibitor sensitivities |
| Specialized Inhibitors | Hemin, N-Ethylmaleimide, Vanadate derivatives, 3,4-Dichloroisocoumarin | Investigation of specific inhibitory mechanisms | Target distinct regulatory components; reveal tissue-specific variations |
Signaling Pathways and Experimental Workflows
The ATP-dependent proteolytic pathway involves a complex sequence of molecular events that can be systematically investigated through structured experimental approaches. The following diagrams illustrate key pathways and workflows relevant to comparative inhibitor profiling.
ATP-Dependent Ubiquitin-Proteasome Pathway
Diagram Title: ATP-Dependent Ubiquitin-Proteasome Pathway with Inhibition Points
Experimental Workflow for Comparative Inhibitor Profiling
Diagram Title: Experimental Workflow for Inhibitor Profiling
Applications in Drug Discovery and Development
The comparative analysis of inhibitor profiles across tissue extracts has significant implications for pharmaceutical research, particularly in the development of targeted therapeutic agents.
Mechanism-Based Drug Discovery
Understanding tissue-specific variations in proteasome structure and function enables the rational design of selective inhibitors with improved therapeutic indices. Contemporary drug discovery efforts increasingly incorporate tissue-specific profiling early in development pipelines. For instance, growth rate inhibition (GR) metrics combined with intracellular drug exposure measurements provide robust evaluation of cellular drug sensitivity [88]. This approach quantifies drug sensitivity on a per-cell division basis, generating parameters such as GR₅₀ (concentration at which growth rate is inhibited by 50%) and GRmax (maximal effect of the drug) that are more informative than traditional IC₅₀ values [88].
The application of these advanced metrics to proteasome inhibitors could reveal tissue-specific vulnerabilities not apparent from conventional inhibition assays. Similarly, comprehensive evaluation of cellular sensitivities to targeted therapies like CDK4/6 inhibitors in breast cancer has demonstrated the importance of baseline cell cycle and immune profiles in predicting treatment response [89], establishing a precedent for mechanism-based patient stratification that could be extended to proteasome-targeting agents.
Tissue-Specific Toxicity Assessment
Comparative inhibitor profiling across tissue extracts provides valuable insights for predicting and understanding tissue-specific toxicities of proteasome-targeting drugs. The differential expression of proteasome subunits and regulatory components across tissues may explain the varied toxicity profiles observed with clinically used proteasome inhibitors such as bortezomib, carfilzomib, and ixazomib.
The integration of tissue extract profiling with advanced cell culture models, including 3D spheroids that better replicate in vivo microenvironments [90], enhances the predictive power of preclinical toxicity assessment. This approach is particularly relevant for evaluating organ-specific toxicities and identifying potential off-target effects early in drug development.
The comparative analysis of inhibitor profiles across tissue extracts reveals fundamental aspects of the structural and functional diversity of ATP-dependent proteolytic systems. The documented variations in inhibitor sensitivity between reticulocyte lysates, adrenal cortex mitochondria, and other tissue extracts highlight the molecular specialization of proteolytic complexes to meet tissue-specific physiological requirements.
Standardized experimental approaches for assessing inhibitor sensitivity, coupled with robust data analysis frameworks, enable systematic characterization of these differential profiles. The integration of this information with contemporary drug discovery methodologies, including growth rate inhibition metrics and advanced cell culture models, provides powerful approaches for developing more selective therapeutic agents with improved safety profiles.
As research continues to elucidate the molecular basis of tissue-specific variations in proteasome structure and function, the strategic application of comparative inhibitor profiling will remain essential for advancing both basic biochemical knowledge and targeted therapeutic development in the context of the rabbit reticulocyte lysate ATP-dependent proteolysis system and its tissue-specific counterparts.
The rabbit reticulocyte lysate (RRL) system has served as a foundational in vitro model for deciphering the mechanisms of ATP-dependent proteolysis in eukaryotic cells. Early research using RRL was instrumental in identifying and characterizing two central components of the cellular protein quality control machinery: the ubiquitin-proteasome system and the chaperonin TRiC/CCT. This whitepaper delineates the evolutionary and functional connections between the proteolytic pathways first discovered in RRL and the conserved ATP-dependent proteases and chaperonins central to cellular homeostasis. We provide a detailed analysis of the core machinery, summarize key quantitative data, and present standardized protocols for studying these systems, offering a comprehensive resource for researchers in protein biochemistry and drug development.
Rabbit reticulocyte lysate (RRL) has been an indispensable tool for biochemical research due to its concentrated cytoplasmic content and high metabolic activity, which is specialized for hemoglobin synthesis. Historically, it was in the RRL system that researchers first identified and fractionated ATP-dependent proteolytic activities [13]. These pioneering studies led to the purification of two high molecular-weight proteases: a 700 kDa multicatalytic complex (the 20S proteasome) and a larger ~1,000 kDa ATP-dependent complex (the 26S proteasome) [13]. A parallel line of investigation using RRL revealed a soluble, ATP-dependent proteolytic system responsible for the selective degradation of abnormal proteins, which was non-lysosomal, had a pH optimum of 7.8, and was inhibited by agents like N-ethylmaleimide and iodoacetamide [3]. This system was notably implicated in the degradation of abnormal globin produced in the presence of valine analogs [3].
The RRL system also proved vital for chaperonin research. It provided a cell-free environment to demonstrate that the eukaryotic chaperonin TRiC/CCT (TCP-1 Ring Complex) interacts with its substrates, such as the signal transducer and activator of transcription 3 (Stat3), during co-translational folding [91]. This review synthesizes these discoveries, placing the RRL system within the broader evolutionary context of conserved ATP-dependent protein homeostasis machines.
Core Components and Evolutionary Relationships
The ATP-dependent machinery identified in RRL represents two major classes of conserved protein quality control systems: energy-dependent proteases and ring-shaped chaperonins.
ATP-Dependent Proteases: From RRL to AAA+ Proteases
The early characterizations from RRL described a high molecular weight protease that degraded proteins and peptides in an ATP-stimulated manner [13]. We now recognize this as the 26S proteasome, an archetypal member of the AAA+ protease superfamily. This superfamily includes bacterial counterparts like ClpXP and ClpAP [92]. These proteases share a common architecture:
- A hollow compartmentalized protease core (e.g., the 20S proteasome, ClpP) that sequesters proteolytic active sites.
- A regulatory AAA+ ATPase "unfoldase" (e.g., the 19S proteasomal cap, ClpX, ClpA) that recognizes substrates, uses ATP hydrolysis to unfold them, and translocates the unfolded polypeptide into the protease chamber [11] [92].
Table 1: Key Characteristics of ATP-Dependent Proteolytic Systems
| Feature | RRL 26S Proteasome [13] | Bacterial ClpXP [92] | Mitochondrial ClpXP [11] |
|---|---|---|---|
| Molecular Weight | ~1,000 kDa | ~450 kDa (ClpX₆ + ClpP₁₄) | Similar to bacterial |
| ATP Dependence | Required for protein degradation | Required for unfolding/translocation | Required for unfolding/translocation |
| Core Protease | 20S Multicatalytic Complex | ClpP Tetradecamer | ClpP Tetradecamer |
| Unfoldase | 19S Regulatory Particle (ATPases) | ClpX Hexamer | ClpX Hexamer |
| Primary Role | Protein turnover, quality control | Protein turnover, stress response, regulation | Protein turnover, respiratory chain maintenance |
The TRiC/CCT Chaperonin
TRiC is an essential, hetero-oligomeric chaperonin that facilitates the folding of approximately 10% of the eukaryotic proteome. It is a ~1 MDa complex composed of two stacked rings, each containing eight different subunits (CCT1-8), forming an enclosed chamber for folding [93] [42]. Unlike the proteasome, TRiC is not a protease; it is a folding nanomachine. Its function is coupled to ATP hydrolysis, which drives conformational changes necessary for substrate encapsulation, folding, and release [42]. Substrates include cytoskeletal proteins like actin and tubulin, as well as complex proteins with β-strand-rich domains, such as STAT3 and the von Hippel-Lindau protein (pVHL) [93] [91].
The evolutionary link is underscored by the use of RRL in identifying TRiC substrates. Pulse-chase analyses in RRL showed that newly synthesized polypeptides interact with TRiC before being released upon folding, confirming its role in de novo protein folding [93] [91].
Quantitative Data and Functional Comparisons
The following tables consolidate key functional data for these systems, highlighting their operational parameters and substrate profiles.
Table 2: Functional Properties of Degradation and Folding Machines
| Parameter | RRL ATP-Dependent Proteolysis [3] | TRiC/CCT [93] [91] | Bacterial ClpXP [92] |
|---|---|---|---|
| pH Optimum | 7.8 | Not Specified | Not Specified |
| Stimulated by ATP | Several-fold | Required for folding cycle | Required for unfolding/translocation |
| Inhibitors | TPCK, TLCK, N-ethylmaleimide, Iodoacetamide, o-phenanthroline | Not Specified | Specific peptide inhibitors (e.g., Z-LY-CMK for ClpP) [11] |
| Estimated Substrates | Selective for abnormal proteins | ~10% of cytosolic proteome (~300 proteins) | Specific tagged proteins (e.g., ssrA-tagged) |
| Key Substrates | Abnormal globin (e.g., ClAbu-containing) | Actin, Tubulin, STAT3, pVHL | ssrA-tagged proteins, RecN, RseA |
Detailed Experimental Protocols
The following protocols are adapted from seminal studies that utilized RRL to investigate these systems.
Protocol 1: Assessing ATP-Dependent Proteolysis in RRL
This protocol is based on the classic study that identified a soluble, ATP-dependent proteolytic system in reticulocytes [3].
Materials:
- Reticulocyte Lysate: Prepare from phenylhydrazine-treated rabbits.
- Substrate: ²⁵I-labeled abnormal globin (e.g., from reticulocytes incorporating 2-amino-3chlorobutyric acid (ClAbu)).
- ATP-Regenerating System: 2 mM ATP, 10 mM Creatine Phosphate, 50 μg/mL Creatine Phosphokinase.
- Inhibitors: 1 mM TPCK, 1 mM N-ethylmaleimide (NEM), 5 mM o-phenanthroline.
- Buffers: Tris-HCl buffer (pH 7.8), Stop Solution (10% TCA).
Procedure:
- Reaction Setup: In a final volume of 100 μL, combine:
- 50 μL RRL (100,000 x g supernatant).
- 5 μg of ²⁵I-labeled abnormal globin substrate.
- 1x ATP-regenerating system.
- Incubate at 37°C for 30-60 minutes.
- Control Reactions: Set up parallel reactions without ATP or with non-hydrolyzable ATP analogs.
- Inhibition Studies: Pre-incubate RRL with inhibitors (TPCK, NEM, o-phenanthroline) for 10 minutes on ice before adding substrate and ATP.
- Termination and Quantification:
- Stop the reaction by adding 500 μL of 10% TCA.
- Incubate on ice for 30 minutes to precipitate undegraded protein.
- Centrifuge at 15,000 x g for 15 minutes.
- Measure the radioactivity in the TCA-soluble supernatant (degraded peptides) using a gamma counter.
- Data Analysis: Calculate the rate of proteolysis as the pmol of TCA-soluble radioactivity released per minute per mg of lysate protein.
Protocol 2: Co-Immunoprecipitation of TRiC-Substrate Complexes from RRL
This protocol is used to identify novel TRiC substrates during their biogenesis, as demonstrated for STAT3 [91].
Materials:
- Nuclease-Treated RRL: For cell-free translation.
- cDNA: Encoding the protein of interest (e.g., STAT3, pVHL).
- ³⁵S-Methionine: For radiolabeling newly synthesized proteins.
- Antibodies: Anti-TRiC antibodies (e.g., against CCT2 and CCT5 subunits).
- Control IgG: Non-specific species-matched antibody.
- Protein A-Agarose Beads.
- Lysis/Wash Buffer: 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, supplemented with protease inhibitors.
Procedure:
- In Vitro Translation:
- Program the RRL with the cDNA of interest in the presence of ³⁵S-methionine according to the manufacturer's instructions.
- Incubate at 30°C for 90 minutes to allow for protein synthesis and TRiC interaction.
- Immunoprecipitation:
- Dilute the translation reaction 1:5 with Lysis/Wash Buffer.
- Pre-clear the lysate by incubating with Protein A-Agarose beads for 30 minutes at 4°C.
- Incubate the pre-cleared lysate with 2-5 μg of anti-TRiC antibody or control IgG overnight at 4°C with gentle rotation.
- Add Protein A-Agarose beads and incubate for an additional 2 hours.
- Washing and Elution:
- Pellet beads and wash 3-4 times with Lysis/Wash Buffer.
- Elute bound proteins by boiling the beads in 1x SDS-PAGE sample buffer.
- Analysis:
- Resolve the eluted proteins by SDS-PAGE.
- Dry the gel and visualize the radiolabeled proteins that co-precipitated with TRiC by autoradiography or phosphorimaging.
Visualizing Pathways and Relationships
Functional and Evolutionary Relationships of RRL Systems
This diagram illustrates the evolutionary conservation of the core AAA+ module and the distinct functional specialization between the degradation and folding machines studied in RRL.
TRiC-Mediated Protein Folding Workflow
This diagram outlines the key steps in the TRiC-assisted folding of a substrate like STAT3, as delineated using RRL experiments.
The Scientist's Toolkit: Essential Research Reagents
The following table lists key reagents used in the foundational RRL experiments, which remain crucial for researchers exploring these systems today.
Table 3: Key Research Reagents for RRL-Based Studies of ATP-Dependent Systems
| Reagent/Solution | Function in Experiment | Example from Literature |
|---|---|---|
| ATP-Regenerating System | Maintains constant, high levels of ATP during prolonged incubations to support energy-dependent processes. | Used in proteolysis assays to demonstrate ATP stimulation [3]. |
| N-Ethylmaleimide (NEM) | Thiol-reactive compound that inhibits ATP-dependent proteolysis by alkylating cysteine residues in critical enzymes. | Validated the presence of a essential cysteine in the RRL proteolytic system [3]. |
| ²⁵I- or ³⁵S-Labeled Protein Substrates | Provides a highly sensitive, quantitative readout for proteolysis (TCA-soluble counts) or synthesis/immunoprecipitation (SDS-PAGE). | ²⁵I-labeled casein/lysozyme for proteolysis [13]; ³⁵S-methionine for TRiC co-IP [91]. |
| Anti-TRiC Antibodies | Specific immunoprecipitation of the chaperonin complex to identify associated substrate proteins during or after synthesis. | Antibodies against CCT2/CCT5 used to confirm STAT3 as a TRiC substrate in RRL [91]. |
| SsrA-Degron Tagged Proteins | A well-defined degradation signal used to target substrates for degradation by AAA+ proteases like ClpXP, useful for comparative studies. | While bacterial, illustrates the principle of degron recognition conserved in the 26S proteasome [92]. |
| Amino Acid Analogs (e.g., ClAbu) | Incorporated into proteins during synthesis to produce misfolded or abnormal proteins, which are then recognized by quality control systems. | Used to generate abnormal globin, a model substrate for the RRL ATP-dependent proteolytic system [3]. |
The rabbit reticulocyte lysate system has provided an unparalleled window into the evolutionarily conserved world of ATP-dependent protein homeostasis. The foundational work conducted in RRL established direct links between a soluble, energy-dependent proteolytic activity and the modern 26S proteasome, and simultaneously served as a versatile platform for defining the substrate repertoire and mechanism of the TRiC chaperonin. The experimental paradigms and core discoveries originating from RRL studies continue to inform our understanding of the complex chaperone and protease networks that maintain proteome integrity across all domains of life. For drug development professionals, these conserved pathways—particularly the proteasome and chaperonins—represent critical targets for therapeutic intervention in cancer, neurodegenerative diseases, and other protein misfolding disorders.
The rabbit reticulocyte lysate (RRL) system has established itself as a foundational in vitro translation tool in biochemical research, particularly for studying ATP-dependent cellular processes. First developed decades ago, this system continues to provide invaluable insights into protein synthesis and degradation mechanisms. The RRL's commercial availability and high translational capacity have made it an attractive platform for fundamental discoveries, including the components of the ATP-dependent proteolytic system [94] [95]. However, the translational gap between these reductionist biochemical findings and their relevance in complex living systems represents a significant challenge in therapeutic development. This whitepaper outlines a comprehensive framework for validating RRL-derived findings through increasingly complex cellular and physiological models, thereby strengthening their relevance to human biology and disease.
A critical barrier in utilizing conventional RRL for investigating specialized machinery like ribosomes has been the technical difficulty of depleting specific proteins. As noted in recent methodology studies, "RRL struggles in depleting proteins by gene knockout or knockdown," which limits its utility for studying modified ribosomes with potentially specialized functions [95]. To overcome this limitation, researchers have developed the hybrid translation system, which combines ribosome-depleted RRL with purified human ribosomes, creating a more physiologically relevant experimental platform [95]. This technical evolution exemplifies the ongoing innovation necessary to enhance the translational value of the RRL platform.
The RRL ATP-Dependent Proteolysis System: Foundational Insights
Historical Context and Core Discoveries
Seminal research using the RRL system resolved the ATP-dependent proteolytic machinery into essential components, revealing fundamental mechanisms that underpin protein quality control in cells. Early work demonstrated that this system requires three absolutely required components for acid solubilization of protein substrates: (1) a heat-stable polypeptide, (2) a high molecular weight protein (~450,000) remarkably stabilized by ATP, and (3) a third protein component separable from the heat-labile species by salt precipitation [94]. These foundational discoveries using RRL established the core principles of ATP-dependent proteolysis that continue to inform our understanding of cellular protein turnover.
Further research illuminated the mechanistic basis of ATP-dependent proteases, revealing that these complexes "degrade their substrates by processively unraveling them from the degradation signal" [96]. This unfolding capability represents a crucial activity shared by evolutionarily conserved proteases including ClpAP and the proteasome. The RRL system was instrumental in demonstrating that "protein unfolding is a key step" in degradation and that "in multidomain proteins, independently stable domains are unfolded sequentially" [96]. These findings established a mechanistic framework for understanding how proteases achieve controlled degradation of structured cellular proteins.
Quantitative Characterization of RRL System Components
Table 1: Essential Components of the RRL ATP-Dependent Proteolytic System
| Component | Key Properties | Role in Proteolysis | ATP Interaction |
|---|---|---|---|
| Heat-stable Polypeptide | Previously reported factor | Required for substrate degradation | Not specified |
| High MW Factor (~450kDa) | Labile at 42°C; stabilized by ATP | Essential for proteolytic activity | Direct interaction demonstrated |
| Third Protein Component | Stable at 42°C; separable by salt | Required alongside other factors | Not determined |
| ATP | Nucleotide triphosphate | Activates proteolysis; stabilizes components | Hydrolyzed for energy requirement |
| ADP/ATP analogs | Non-hydrolyzable variants | Cause stabilization but not activation | Bind without supporting proteolysis |
Principles of Validation for Translational Research
Establishing Validation Frameworks
The movement of findings from reductionist systems like RRL to clinically relevant models requires robust validation frameworks. In physiological modeling and simulation, validation credibility is paramount, particularly when findings may affect "real-world decision-making, including within the clinic, in regulatory science, or in the design and engineering of novel therapeutics" [97]. Without proper validation, the usefulness of models remains impaired, casting shadows over their credibility across domains. The fundamental question in validation revolves around establishing what constitutes sufficient evidence that a model or finding adequately represents the real-world phenomenon it purports to explain.
A systematic review of AI system validation methods reveals that while "various validation strategies have all been relatively widely applied, only few studies report on continuous validation" [98]. This insight applies equally to biological model validation, where ongoing assessment strengthens translational relevance. The taxonomy of validation approaches includes trials, simulations, model-centred validation, and expert opinion, with continuous validation methods including "failure monitors, safety channels, redundancy, voting, and input and output restrictions" [98]. These structured approaches provide methodological rigor for establishing confidence in research findings across experimental platforms.
Accuracy Assessment in Physiological Models
For physiological models specifically, accuracy assessment requires specialized methodologies. Recent research has developed "tools for evaluating the accuracy of physiological models and establishes fundamental measures for predictive capability assessment across different physiological models" [99]. These include comparing calibration performance using metrics like root-mean-squared error (RMSE), Akaike information criterion (AIC), and multi-dimensional approaches utilizing normalized features extracted from fitting error. Enhanced predictive capability is demonstrated when a significantly larger proportion of measurements fall within the prediction envelope in both transient and leave-one-out prediction scenarios [99].
Methodological Framework for Validation
The Hybrid Translation System: Bridging RRL and Cellular Physiology
The human-rabbit hybrid translation system represents a powerful methodological advance for validating RRL findings in more physiologically relevant contexts. This system "is based on the supplementation of purified human ribosomes into ribosome-depleted RRL," creating an experimental platform that combines the biochemical tractability of RRL with human-specific components [95]. The protocol involves four major steps: (1) preparation of ribosome-depleted RRL, (2) purification of ribosomes from human cells (e.g., HEK293), (3) preparation of reporter mRNAs, and (4) the translation reaction itself.
Table 2: Hybrid Translation System Components and Applications
| Component | Source | Preparation Method | Function in System |
|---|---|---|---|
| Ribosome-depleted RRL | Commercial RRL | Ultracentrifugation | Provides translation-competent lysate without endogenous ribosomes |
| Ribosomes | HEK293 cells | Purification by sedimentation | Supplies human ribosomes for physiological relevance |
| Reporter mRNA | In vitro transcription | Plasmid-based with luciferase genes | Measures translation elongation efficacy |
| Supplemental Factors | RRL inherent | N/A | Provides tRNAs, amino acids, initiation/elongation factors |
This hybrid approach enables researchers to "study the function of ribosomes in the context of active protein synthesis," particularly specialized ribosomes with diverse functions proposed based on growing "evidence of modifications of ribosomal RNA, ribosomal proteins, and the heterologous composition of the constituents" [95]. When combined with a dual-luciferase reporter system that measures ribosome processivity through the synthesis rate difference between upstream Renilla and downstream firefly luciferases, this platform enables detailed investigation of elongation efficacy on specific codon or RNA sequences [95].
Workflow for Validating RRL Findings Across Model Systems
Advanced Transcriptomic and Single-Cell Validation Approaches
Cutting-edge transcriptomic technologies provide powerful validation pathways for RRL-derived findings. Single-cell RNA sequencing (scRNA-seq) analyzes gene expression profiles of individual cells from both homogeneous and heterogeneous populations, enabling researchers to "identify and characterize different cell types, states, and subpopulations" with unprecedented resolution [100]. Unlike bulk sequencing that provides population-averaged data, scRNA-seq can "detect cell subtypes or gene expression variations that would otherwise be overlooked" [100], making it ideal for validating mechanisms discovered in reductionist systems.
Table 3: Comparison of Bulk vs Single-Cell RNA Sequencing for Validation
| Characteristic | Bulk RNA-seq | Single-Cell RNA-seq |
|---|---|---|
| Resolution | Average gene expression across heterogeneous cells | Gene expression profiles of individual cells |
| Cell Type Detection | Masks cellular heterogeneity | Reveals rare cell subtypes and continuous transitions |
| Applications | Population-level analysis | Cellular diversity, lineage tracing, developmental trajectories |
| Limitations | Obscures cell-to-cell variation | Higher cost, technical noise, computational complexity |
| Validation Power | Moderate for population mechanisms | High for cell-type specific mechanisms |
Spatial transcriptomics represents a further advancement by facilitating "the identification of molecules such as RNA in their original spatial context within tissue sections at the single-cell level" [100]. This capability offers a substantial advantage over traditional single-cell sequencing techniques by preserving architectural information, thus providing critical validation evidence for physiological relevance of RRL-derived mechanisms in structured tissues.
Case Study: Implementing a Validation Workflow
Experimental Protocol: Hybrid System with Processivity Reporter
The following detailed protocol exemplifies a robust approach for validating RRL findings regarding translation elongation mechanisms:
Step 1: Ribosome-depleted RRL Preparation
- Start with commercial nuclease-treated RRL.
- Subject to ultracentrifugation at high speed (e.g., 100,000 × g for 2-4 hours) to pellet endogenous ribosomes.
- Collect the ribosome-free supernatant, which retains translation factors, tRNAs, and enzymes.
- Confirm ribosome depletion by absorbance measurement at 260nm.
Step 2: Human Ribosome Purification
- Culture HEK293T cells (wild-type or genetically modified) under standard conditions.
- Harvest cells and prepare cytoplasmic extract by gentle lysis.
- Separate ribosomes through sucrose density gradient centrifugation.
- Confirm ribosome integrity and purity by appropriate analytical methods.
Step 3: Reporter mRNA Construction
- Design plasmid with T7 promoter followed by Renilla luciferase, multiple cloning site (for inserting sequences of interest), and firefly luciferase in sequence.
- In vitro transcribe mRNA using T7 RNA polymerase with proper 5' capping and 3' polyadenylation.
- Validate mRNA integrity by gel electrophoresis and quantify by spectrophotometry.
Step 4: Translation Reaction and Analysis
- Combine ribosome-depleted RRL, purified human ribosomes, reporter mRNA, and energy regeneration system.
- Incubate at 32°C for appropriate time periods (typically 60-90 minutes).
- Measure Renilla and firefly luciferase activities sequentially using dual-luciferase assay reagents.
- Calculate elongation efficiency as the ratio of firefly to Renilla luciferase activity, normalized to appropriate controls.
This protocol "could be used to study the potency of heterologous ribosomes" and specifically "to study the effect of the histidine methylation of ribosomal protein uL3 (or RPL3)" as discovered in initial RRL screens [95]. The system enables direct comparison between ribosomes purified from naïve HEK293T cells and those from genetically modified cells (e.g., METTL18 deficient), assessing functional consequences of ribosomal modifications on translation elongation.
Workflow Visualization: Hybrid Translation System
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagent Solutions for RRL Validation Studies
| Reagent/Category | Specific Examples | Function/Application | Validation Role |
|---|---|---|---|
| RRL Systems | Commercial nuclease-treated RRL (e.g., Promega L4960) | Base in vitro translation system | Initial discovery platform |
| Cell Lines | HEK293T, METTL18 KO HEK293T | Source of human ribosomes and cellular validation | Provides human physiological context |
| Ribosome Purification Tools | Sucrose gradients, ultracentrifugation equipment | Isolation of intact ribosomal complexes | Enables hybrid system creation |
| Reporter Plasmids | Dual-luciferase constructs with MCS | Assessment of translation elongation efficacy | Quantifies functional outcomes |
| scRNA-seq Platforms | 10x Genomics, Smart-seq2 | Single-cell resolution transcriptomics | Validates cell-type specific mechanisms |
| Spatial Transcriptomics | 10x Visium, Slide-seq | Tissue context preservation | Bridges cellular and physiological relevance |
| Validation Software | Root-mean-squared error (RMSE), Akaike information criterion (AIC) tools | Model accuracy assessment | Quantifies predictive capability |
Concluding Perspective: Strengthening the Translational Pipeline
The journey from RRL-based discoveries to clinically relevant insights requires methodical validation across increasingly complex experimental systems. The framework outlined in this whitepaper emphasizes structured approaches that progressively build evidence for physiological relevance, from hybrid translation systems to single-cell genomics and physiological modeling. This multi-layered strategy addresses the fundamental challenge that "high-throughput technologies may not always provide clear insights into the regulatory relationships between molecules" [101], while leveraging the complementary strengths of reductionist and systems-level approaches.
For drug development professionals, this validation continuum represents a critical pathway for derisking therapeutic programs based on initial RRL findings. By implementing robust validation workflows that include "trials, simulations, model-centred validation, and expert opinion" [98], researchers can progressively strengthen the evidence supporting the translational relevance of mechanistic discoveries. In an era of increasingly sophisticated experimental tools, establishing these methodological bridges between foundational biochemistry and human physiology remains essential for advancing both scientific knowledge and therapeutic innovation.
Conclusion
The rabbit reticulocyte lysate ATP-dependent proteolysis system remains an indispensable and robust model for dissecting the biochemical principles of intracellular protein degradation. Its well-characterized mechanism, encompassing both ubiquitin-dependent and independent pathways, provides a simplified yet physiologically relevant platform that has yielded fundamental insights into proteostasis. The comparative analyses with systems like RPE cell extracts and cardiac muscle highlight both the conserved features and specialized adaptations of proteolytic pathways across tissues. For future research, leveraging the RRL system will continue to be critical for elucidating the role of proteostasis in disease mechanisms, including neurodegeneration and cancer, and for screening novel therapeutic compounds that modulate protein degradation pathways. Its integration with modern techniques ensures its continued value in advancing biomedical knowledge and drug development.
References
- 1. Intracellular protein degradation: from a vague idea thru ... [https://www.nature.com/articles/4401692]
- 2. A soluble ATP-dependent proteolytic system responsible ... [https://pubmed.ncbi.nlm.nih.gov/264694/]
- 3. A soluble ATP-dependent proteolytic system responsible ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC393195/]
- 4. ATP serves two distinct roles in protein degradation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2112434/]
- 5. Demonstration of two distinct high molecular weight ... [https://www.sciencedirect.com/science/article/pii/S002192581861525X]
- 6. Protein targeting to ATP-dependent proteases - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC2346608/]
- 7. Functional mechanics of the ATP-dependent Lon protease [https://pmc.ncbi.nlm.nih.gov/articles/PMC2443057/]
- 8. A particle-associated ATP-dependent proteolytic activity in ... [https://www.sciencedirect.com/science/article/pii/S0021925818895060]
- 9. ATP Binding and ATP Hydrolysis Play Distinct Roles in the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3951175/]
- 10. Reduced ATP-dependent proteolysis during stationary ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8378506/]
- 11. Recent Advances in the Structural Studies of the Proteolytic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12383707/]
- 12. Proteolytic Activity of the ATP-dependent Protease HslVU ... [https://www.sciencedirect.com/science/article/pii/S0021925819657650]
- 13. Purification of two high molecular weight proteases from ... [https://www.sciencedirect.com/science/article/pii/S0021925818475643]
- 14. Ubiquitin-Dependent and Independent Proteasomal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10536765/]
- 15. Protein Degradation using the Ubiquitin-Proteasome ... [https://www.thermofisher.com/us/en/home/life-science/cell-analysis/cell-analysis-learning-center/protein-degradation-resource-center/protein-degradation-using-ubiquitin-proteasome-pathway.html]
- 16. Protein Degradation - The Cell - NCBI Bookshelf - NIH [https://www.ncbi.nlm.nih.gov/books/NBK9957/]
- 17. Ubiquitin-independent proteasomal degradation driven by C ... [https://pubmed.ncbi.nlm.nih.gov/37201526/]
- 18. Ubiquitin-independent proteasomal degradation [https://www.sciencedirect.com/science/article/pii/S0167488913001882]
- 19. Mechanisms of ubiquitin-independent proteasomal ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2024.1531797/full]
- 20. Targeted protein degradation: mechanisms, strategies and ... [https://www.nature.com/articles/s41392-022-00966-4]
- 21. Effects of temperature on the degradation of proteins in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC344616/]
- 22. Ubiquitin, the proteasome and protein degradation in ... [https://www.nature.com/articles/nrn2499]
- 23. role of hemin in regulating ubiquitin conjugate degradation [https://pmc.ncbi.nlm.nih.gov/articles/PMC349148/]
- 24. role of hemin in regulating ubiquitin conjugate degradation [https://pubmed.ncbi.nlm.nih.gov/6273891/]
- 25. Vanadate inhibits the ATP-dependent degradation of ... [https://www.osti.gov/biblio/6444140]
- 26. Multiple forms of the 20 S multicatalytic and the 26 S ... [https://www.sciencedirect.com/science/article/pii/S0021925818416808]
- 27. Modulation of the Ubiquitin-Proteasome System by ... [https://www.sciencedirect.com/science/article/pii/S095528632500289X]
- 28. Protein Expression Guide I An Introduction to ... [https://worldwide.promega.com/resources/guides/protein-analysis/protein-expression-methods/]
- 29. Back to basics: the untreated rabbit reticulocyte lysate as a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2094066/]
- 30. A comparison of ubiquitin- dependent of rod outer... proteolysis [https://pubmed.ncbi.nlm.nih.gov/8529413/]
- 31. Monitoring Proteolytic Activity in Real Time: A New World of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7199732/]
- 32. Methods for the proteomic identification of protease ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2787889/]
- 33. A Protocol to Depict the Proteolytic Processes Using a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10777052/]
- 34. Step-by-Step Protocol: How to Perform a DARTS Assay [https://www.creative-proteomics.com/resource/step-by-step-protocol-performing-darts-assay-target-identification.htm]
- 35. Protease Digestion for Mass Spectrometry [https://www.promega.com/resources/guides/protein-analysis/protease-digestion-for-mass-spec/]
- 36. Better Protease Detection | High-Sensitivity ... [https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/protein-biology/enzyme-activity-assays/high-sensitivity-protease-detection-assay?srsltid=AfmBOoqGaWuWl1Js3ZrJ3ExR6tZAjCtuLhR6Lu1ijGo3HcsfyUR4CcbC]
- 37. Measuring protein structural changes on a proteome-wide ... [https://www.nature.com/articles/nprot.2017.100]
- 38. Proteolysis and absolute quantification in proteomics [https://www.sciencedirect.com/science/article/abs/pii/S1046202311000971]
- 39. Quantitative modeling of rod outer segment phagocytosis ... [https://www.nature.com/articles/s41598-025-06356-4]
- 40. Functional Subunits of Eukaryotic Chaperonin CCT/TRiC in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3268035/]
- 41. The Molecular Chaperone CCT/TRiC: An Essential ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2020.00172/full]
- 42. The structural basis of eukaryotic chaperonin TRiC/CCT [https://www.sciencedirect.com/science/article/pii/S1016847824000050]
- 43. The Future of Biopharmaceutical Research: Trends to ... [https://www.genemod.net/blog/the-future-of-biopharmaceutical-research-trends-to-watch-in-2025]
- 44. An overview of PROTACs: a promising drug discovery ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9763089/]
- 45. Advancements in delivery Systems for Proteolysis ... [https://www.sciencedirect.com/science/article/abs/pii/S0168365925003396]
- 46. Chemically Induced Cellular Proteolysis: An Emerging ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6277563/]
- 47. Modulators of Proteolysis for the Treatment of Carcinomas [https://pmc.ncbi.nlm.nih.gov/articles/PMC6378659/]
- 48. Proteasome dynamics in response to metabolic changes [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2025.1523382/full]
- 49. Ubiquitin-activating enzyme. Mechanism and role in protein ... [https://pubmed.ncbi.nlm.nih.gov/6277905/]
- 50. The Ufd1 cofactor determines the linkage specificity of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9992269/]
- 51. VCF1 is a p97/VCP cofactor promoting recognition of ... [https://www.nature.com/articles/s41467-024-46760-4]
- 52. Optimization of non-denaturing protein extraction conditions ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0187753]
- 53. Towards a Quantitative Description of Proteolysis [https://pmc.ncbi.nlm.nih.gov/articles/PMC11719913/]
- 54. Convergence of orphan quality control pathways at a ... [https://www.sciencedirect.com/science/article/pii/S1097276525000024]
- 55. Structures of the ATP-fueled ClpXP proteolytic machine ... [https://elifesciences.org/articles/52774]
- 56. Mechanisms of Proteolytic Enzymes and Their Inhibition in QM ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8004986/]
- 57. Effect of ions and other compatible solutes on enzyme ... [https://www.sciencedirect.com/science/article/abs/pii/S138111770500130X]
- 58. Production, Purification, and Biochemical Characterization ... [https://www.mdpi.com/2076-2607/13/2/307]
- 59. Optimization of a Luminescence-Based High-Throughput ... [https://www.sciencedirect.com/science/article/pii/S2472555222069398]
- 60. Engineered AAA+ proteases reveal principles of ... [https://www.nature.com/articles/ncomms13301]
- 61. Proteolysis inside the membrane is a rate-governed reaction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3917317/]
- 62. Polyamines inhibit the ATP-dependent proteolytic pathway ... [https://www.sciencedirect.com/science/article/pii/0014579389801164]
- 63. Inhibition of ubiquitin-dependent proteolysis by a synthetic ... [https://www.sciencedirect.com/science/article/pii/S0014579302028971]
- 64. Quantitative analysis of protease recognition by inhibitors ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5064372/]
- 65. Slicing a protease: Structural features of the ATP-dependent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2242575/]
- 66. A processive rotary mechanism couples substrate ... [https://elifesciences.org/articles/52158]
- 67. Structure and Function of a Novel Type of ATP-dependent ... [https://www.sciencedirect.com/science/article/pii/S0021925820582070]
- 68. Mega-scale experimental analysis of protein folding ... [https://www.nature.com/articles/s41586-023-06328-6]
- 69. Benchmarking of Quantitative Proteomics Workflows for ... [https://www.sciencedirect.com/science/article/pii/S153594762500043X]
- 70. Advances in retinal pigment epithelial cell transplantation for ... [https://stemcellres.biomedcentral.com/articles/10.1186/s13287-024-04007-5]
- 71. Two new retinal pigment epithelial cell models developed [https://www.drugtargetreview.com/news/52794/two-new-retinal-pigment-epithelial-cell-models-developed/]
- 72. Defects in Retinal Pigment Epithelial Cell Proteolysis and the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4769684/]
- 73. Advances in the study of tissue-engineered retinal pigment ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11062139/]
- 74. Dysfunctional Autophagy, Proteostasis, and Mitochondria ... [https://www.mdpi.com/1422-0067/24/10/8763]
- 75. Proteome changes in a human retinal pigment epithelial ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2023.1138519/full]
- 76. Ubiquitin and ubiquitin-like proteins in cardiac disease ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4899309/]
- 77. exploring the ubiquitin system for drug development [https://www.nature.com/articles/cr201631]
- 78. Ubiquitin‐dependent regulation of transcription in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8025022/]
- 79. ATP-Requiring Proteolytic Pathways in Bacterial and ... [https://www.fao.org/4/x5550e/x5550e0d.htm]
- 80. Ubiquitin-dependent and Ubiquitin-independent Control of ... [https://www.sciencedirect.com/science/article/pii/S0021925820469078]
- 81. Activation of ATP-ubiquitin-dependent Proteolysis in ... [https://pubmed.ncbi.nlm.nih.gov/11461093/]
- 82. Emerging Roles of Cullin-RING Ubiquitin Ligases in ... [https://www.mdpi.com/2073-4409/13/3/235]
- 83. Cardiac Ubiquitin Ligases: Their Role in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5159290/]
- 84. Ubiquitin-specific protease: an emerging key player in ... [https://biosignaling.biomedcentral.com/articles/10.1186/s12964-025-02123-0]
- 85. ATP stimulates proteolysis in reticulocyte extracts by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC394092/]
- 86. ATP-dependent protease in bovine adrenal cortex. Tissue ... [https://www.sciencedirect.com/science/article/pii/S0021925818890524]
- 87. Protease Inhibitor Cocktail for Mammalian Cell and Tissue Extracts in DMSO Solution Broad-Spectrum Inhibition Stable Storage Sigma-Aldrich [https://www.sigmaaldrich.com/US/en/product/sigma/p8340]
- 88. Application of drug-induced growth rate inhibition and intracellular drug exposures for comprehensive evaluation of cellular drug sensitivity | Scientific Reports [https://www.nature.com/articles/s41598-025-86919-7]
- 89. Baseline cell cycle and immune profiles indicate CDK4/6 inhibitor response in metastatic HR + /HER2- breast cancer | npj Breast Cancer [https://www.nature.com/articles/s41523-025-00767-2]
- 90. Comparative Analysis of the Effect of the BRAF Inhibitor Dabrafenib in 2D and 3D Cell Culture Models of Human Metastatic Melanoma Cells | In Vivo [https://iv.iiarjournals.org/content/38/4/1579]
- 91. Modulation of STAT3 Folding and Function by TRiC/CCT ... [https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.1001844]
- 92. ATP-dependent proteases of bacteria: recognition logic and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2717004/]
- 93. Defining the TRiC/CCT interactome links chaperonin function ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2658641/]
- 94. Resolution of the ATP-dependent proteolytic system from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC383772/]
- 95. Human-rabbit Hybrid Translation System to Explore ... [https://en.bio-protocol.org/en/bpdetail?id=4714&type=0]
- 96. ATP-Dependent Proteases Degrade Their Substrates by ... [https://www.sciencedirect.com/science/article/pii/S109727650100209X]
- 97. Physiological Modeling and Simulation-Validation, Credibility ... [https://pubmed.ncbi.nlm.nih.gov/32501771/]
- 98. Systematic literature review of validation methods for AI ... [https://www.sciencedirect.com/science/article/pii/S0164121221001473]
- 99. Accuracy assessment methods for physiological model ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0251001]
- 100. Advancements in single-cell RNA sequencing and spatial ... [https://www.frontierspartnerships.org/journals/acta-biochimica-polonica/articles/10.3389/abp.2025.13922/full]
- 101. Development and Validation of an AI‐Driven System for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11600262/]