The Ubiquitin-Proteasome System: From Molecular Mechanism to Therapeutic Application in Disease and Drug Development
This article provides a comprehensive exploration of the ubiquitin-proteasome system (UPS), a crucial pathway for intracellular protein degradation and regulation.
The Ubiquitin-Proteasome System: From Molecular Mechanism to Therapeutic Application in Disease and Drug Development
Abstract
This article provides a comprehensive exploration of the ubiquitin-proteasome system (UPS), a crucial pathway for intracellular protein degradation and regulation. We examine the foundational biochemistry of the UPS, including the E1-E2-E3 enzymatic cascade and proteasome structure. The review highlights cutting-edge methodological applications, particularly targeted protein degradation technologies like PROTACs and molecular glues, with updates on their clinical progression. We address system crosstalk with autophagy and common research challenges. Finally, we evaluate the UPS as a therapeutic target across cancer, neurodegenerative diseases, and other pathologies, offering a vital resource for researchers and drug development professionals navigating this dynamic field.
Decoding the UPS: Architecture, Mechanism, and Cellular Functions
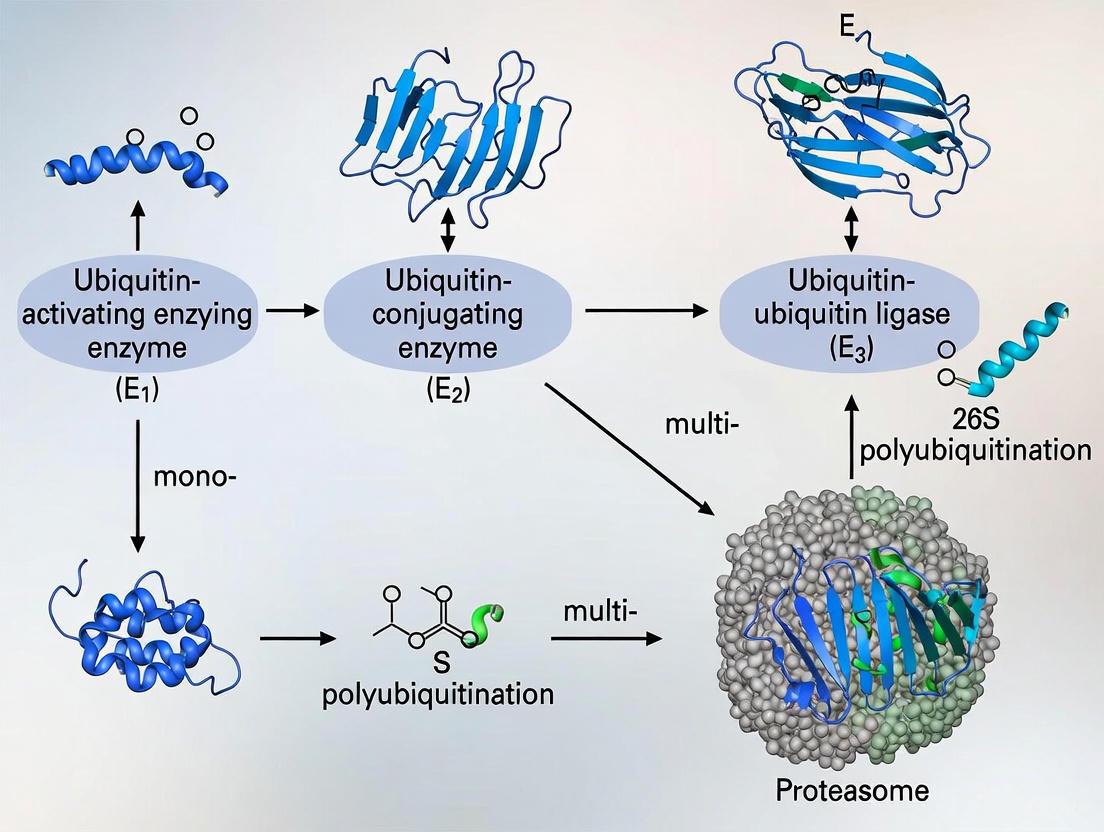
The ubiquitin-proteasome system (UPS) represents a highly conserved and selective mechanism for protein degradation and signaling, playing an indispensable role in virtually all aspects of eukaryotic cell biology [1]. This sophisticated system regulates protein turnover through a cascade of enzymatic reactions that culminate in the covalent attachment of ubiquitin, a 76-amino acid protein, to target substrates [2]. The process of ubiquitination serves as a critical post-translational modification (PTM) that influences diverse cellular processes including cell cycle progression, DNA repair, immune signaling, and apoptosis [2] [1]. Dysregulation of the ubiquitination cascade contributes to numerous pathological conditions, making its components attractive therapeutic targets for cancer, neurodegenerative disorders, and immune diseases [3] [1]. This technical guide examines the core enzymatic machinery—E1, E2, and E3 enzymes—that executes and regulates the ubiquitination cascade, with particular emphasis on recent mechanistic insights and experimental approaches relevant to researchers and drug development professionals.
The Ubiquitination Enzymatic Cascade
The ubiquitination process proceeds through a three-step enzymatic cascade involving E1 (ubiquitin-activating), E2 (ubiquitin-conjugating), and E3 (ubiquitin-ligase) enzymes [2]. This coordinated mechanism ensures precise targeting and modification of substrate proteins.
E1 Ubiquitin-Activating Enzymes
The initial step in ubiquitination involves E1 enzymes, which activate ubiquitin in an ATP-dependent manner [2]. The human genome encodes only two E1 enzymes, making this the most limited component of the cascade [4]. The activation mechanism proceeds as follows:
- ATP-dependent activation: E1 catalyzes the formation of a thioester bond between its active-site cysteine residue and the C-terminal glycine of ubiquitin, with concomitant generation of AMP [2].
- E2 transfer: The activated ubiquitin is subsequently transferred to the catalytic cysteine of an E2 conjugating enzyme via a trans-thiolation reaction [2] [5].
Table 1: Key Characteristics of E1 Ubiquitin-Activating Enzymes
| Feature | Description | Research Significance |
|---|---|---|
| Number in Humans | 2 genes | Limited diversity facilitates broad inhibition strategies |
| Reaction Mechanism | ATP-dependent thioester formation with ubiquitin | Requires Mg²⁺ and ATP for in vitro reconstitution |
| Primary Function | Initiate ubiquitination cascade by activating ubiquitin | Essential for all downstream ubiquitination events |
| Key Structural Features | Ubiquitin-fold domain, active site cysteine, adenylation domain | Target for structural biology and inhibitor design |
E2 Ubiquitin-Conjugating Enzymes
E2 enzymes serve as central intermediaries in the ubiquitination cascade, receiving activated ubiquitin from E1 and cooperating with E3 ligases to modify specific substrates [2]. The human genome encodes approximately 40 E2 enzymes, each exhibiting distinct specificities for particular E3s and substrate types [6] [4]. Key functional aspects include:
- Ubiquitin charging: E2 enzymes form a thioester bond with ubiquitin transferred from E1 [2].
- Chain-type specificity: Different E2s determine the topology of polyubiquitin chains formed on substrates [6]. For instance, UBE2G2 primarily generates Lys48-linked chains targeting proteins for proteasomal degradation, while UBE2J2 can modify serine and threonine residues in addition to lysines [6].
- Regulatory sensing: Recent research reveals that certain E2s, such as the membrane-anchored UBE2J2, can sense lipid packing density in endoplasmic reticulum membranes, directly linking ubiquitination to organelle homeostasis [6].
Table 2: Selected E2 Ubiquitin-Conjugating Enzymes and Their Functions
| E2 Enzyme | Ubiquitin Linkage Preference | Cellular Functions | Experimental Considerations |
|---|---|---|---|
| UBE2J2 | K48, K63, serine/threonine | ER-associated degradation (ERAD), lipid sensing | Membrane reconstitution required for functional studies |
| UBE2G2 | Primarily K48 | Proteasomal degradation, ERAD | Requires AUP1 for membrane association |
| UBE2J1 | K48, K63 | ERAD, protein quality control | Functional in ER-like membranes without additional activation |
| Ubc13 (Yeast) | K63 | DNA damage response, NF-κB signaling | Often paired with E2 variants for K63 chain formation |
E3 Ubiquitin Ligases
E3 ubiquitin ligases represent the most diverse and specialized components of the ubiquitination cascade, with over 600 members in the human genome [5] [4]. These enzymes confer substrate specificity by simultaneously recognizing target proteins and E2-ubiquitin conjugates, thereby catalyzing ubiquitin transfer to specific substrates [5] [3]. E3 ligases are categorized into several structural families based on their mechanism of action:
- RING (Really Interesting New Gene) E3 ligases: The largest E3 family, characterized by a RING domain that directly recruits E2-ubiquitin conjugates and facilitates direct ubiquitin transfer without forming an E3-ubiquitin intermediate [5] [4]. RING E3s can function as single polypeptides (e.g., MDM2, CBL) or as multi-subunit complexes (e.g., Cullin-RING ligases/CRLs) [5] [4].
- HECT (Homologous to E6AP C-Terminus) E3 ligases: Utilize a conserved HECT domain that forms a thioester intermediate with ubiquitin before transferring it to substrates [5] [4]. This family includes the NEDD4 subfamily, HERC subfamily, and other HECT ligases such as E6AP and HUWE1 [4].
- RBR (RING-Between-RING) E3 ligases: Hybrid enzymes that employ a RING domain for E2 binding and a catalytic domain that forms a thioester intermediate with ubiquitin, similar to HECT ligases [7] [4]. Key examples include Parkin and HOIP (component of the LUBAC complex) [7] [4].
Table 3: Major E3 Ubiquitin Ligase Families and Their Characteristics
| E3 Family | Catalytic Mechanism | Representative Members | Key Structural Domains |
|---|---|---|---|
| RING | Direct transfer from E2 to substrate | MDM2, CBL, BRCA1, APC/C | RING domain, substrate recognition domains |
| HECT | E3-ubiquitin thioester intermediate | NEDD4, HERC, HUWE1, E6AP | HECT domain, C2 domain, WW domains |
| RBR | Hybrid RING-HECT mechanism | Parkin, HOIP, HOIL-1 | RING1, IBR, RING2 domains |
| U-box | Structurally similar to RING | CHIP, UFD2 | U-box domain, tetratricopeptide repeats |
Ubiquitin Signaling Diversity and Functional Outcomes
The ubiquitination code extends beyond simple monoubiquitination to include diverse polyubiquitin chains that dictate distinct functional outcomes for modified substrates.
Ubiquitin Chain Linkages and Functions
Ubiquitin contains seven lysine residues (K6, K11, K27, K29, K33, K48, K63) and an N-terminal methionine (M1) that serve as linkage points for polyubiquitin chain formation [1] [4]. The specific topology of these chains determines the fate and function of the modified protein:
- K48-linked chains: The most abundant linkage type, primarily targeting substrates for proteasomal degradation [2] [4].
- K63-linked chains: Mainly involved in non-proteolytic signaling processes including DNA damage repair, kinase activation, and intracellular trafficking [2] [4].
- M1-linked (linear) chains: Assembled by the LUBAC complex (HOIP, HOIL-1), these regulate inflammatory signaling pathways, particularly NF-κB activation [7] [1].
- Atypical linkages (K6, K11, K27, K29, K33): Participate in diverse processes including DNA damage response, cell cycle regulation, and innate immunity [4].
Recent research has revealed additional complexity through heterotypic and branched ubiquitin chains, which may integrate multiple signals to fine-tune cellular responses [3] [1].
Diagram 1: The three-step ubiquitination cascade. This diagram illustrates the sequential action of E1 (activation), E2 (conjugation), and E3 (ligation) enzymes in mediating ubiquitin transfer to substrate proteins.
Experimental Approaches for Studying the Ubiquitination Cascade
In Vitro Reconstitution of Ubiquitination
Recent advances in biochemical reconstitution have enabled detailed mechanistic studies of ubiquitination components. The following protocol, adapted from research on UBE2J2 regulation, demonstrates methodology for analyzing lipid-dependent E2 activity [6]:
Objective: Assess ubiquitin loading of membrane-anchored E2 enzymes in liposomes of defined lipid composition.
Reagents and Equipment:
- Purified E1, E2 (e.g., UBE2J2, UBE2J1), ubiquitin
- Phospholipids: phosphatidylcholine (PC), phosphatidylethanolamine (PE)
- ATP regeneration system
- Detergents for solubilization (e.g., n-dodecyl-β-D-maltopyranoside)
- Fluorescent labeling reagents (optional)
- Equipment: Fast protein liquid chromatography (FPLC), SDS-PAGE, immunoblotting apparatus
Procedure:
- Liposome Preparation: Form liposomes with defined lipid compositions (e.g., ER-like membranes: 80:20 PC:PE molar ratio with 33% saturated fatty acyl chains) using extrusion or dialysis methods [6].
- E2 Reconstitution: Incorporate purified E2 enzymes into pre-formed liposomes via detergent dilution and removal.
- Ubiquitin Loading Assay: Incubate E2-containing proteoliposomes with E1 (100 nM), ubiquitin (5 μM), and ATP (2 mM) in reaction buffer at 30°C.
- Time-course Sampling: Remove aliquots at specified time points (0, 30s, 1, 2, 5, 10, 30 min) and immediately mix with non-reducing SDS sample buffer to preserve thioester linkages.
- Analysis: Resolve proteins by non-reducing SDS-PAGE, followed by immunoblotting with anti-ubiquitin or anti-E2 antibodies.
Key Controls:
- Include detergent-solubilized E2 as an activation-positive control
- Verify E2 orientation in liposomes via protease protection assays
- Test lipid packing effects using liposomes with varying saturation levels (10% vs. 50% SFAs)
Identification of E3 Substrates
Determining physiological substrates for E3 ligases remains a central challenge in ubiquitin research. The Global Protein Stability (GPS) profiling system represents a powerful approach for substrate identification [2]:
Methodology:
- Library Construction: Generate a comprehensive library of reporter proteins fused to potential substrate candidates.
- E3 Perturbation: Employ CRISPR-Cas9-mediated knockout or RNA interference to deplete specific E3 ligases.
- Stability Monitoring: Quantify reporter accumulation resulting from reduced ubiquitination and degradation.
- Validation: Confirm direct ubiquitination through in vitro assays with purified components.
Technical Considerations:
- Genome-wide screening enables discovery of novel E3-substrate relationships
- Requires rigorous validation to distinguish direct from indirect effects
- Complementary approaches include ubiquitin remnant profiling and BioID proximity labeling
Research Reagent Solutions for Ubiquitination Studies
Table 4: Essential Research Reagents for Ubiquitination Cascade Investigations
| Reagent Category | Specific Examples | Research Applications | Technical Considerations |
|---|---|---|---|
| E1 Inhibitors | PYR-41, TAK-243 | Pan-inhibition of ubiquitination cascade | High toxicity in live cells; use for acute inhibition |
| E2 Tools | UBE2J2, UBE2G2, UBE2J1 purified proteins | In vitro ubiquitination, chain linkage specificity | Require co-expression with E1 for functional assays |
| Proteasome Inhibitors | Bortezomib, MG132, Carfilzomib | Block substrate degradation, stabilize ubiquitinated proteins | Non-specific effects on overall protein turnover |
| DUB Inhibitors | PR-619 (broad-specificity), VLX1570 (USP14) | Stabilize ubiquitin signals, study deubiquitination | Varying specificity profiles require careful controls |
| Linkage-Specific Antibodies | Anti-K48, Anti-K63, Anti-M1 ubiquitin | Detection of specific ubiquitin chain types | Cross-reactivity validation essential |
| Reconstitution Systems | Purified E1-E2-E3 components, liposome kits | Mechanistic studies of ubiquitination | Lipid composition critically affects membrane-associated E2s |
| Activity Reporters | Ubiquitin vinyl sulfones, diGly remnant antibodies | Proteomic identification of ubiquitination sites | Require specialized mass spectrometry expertise |
Therapeutic Targeting of the Ubiquitination Cascade
Components of the ubiquitination cascade represent promising targets for therapeutic intervention, particularly through strategies that exploit E3 ligases for targeted protein degradation [3]. Key approaches include:
- Proteolysis-Targeting Chimeras (PROTACs): Bifunctional molecules that simultaneously bind E3 ligases and target proteins, inducing proximity and ubiquitination of disease-relevant proteins [3].
- Molecular Glues: Small molecules that enhance or induce interactions between E3 ligases and specific substrates [3].
- Ubiquitin Variants (UbVs): Engineered ubiquitin mutants that selectively inhibit or modulate specific E2-E3 interactions.
Recent structural studies have revealed new mechanistic classes of E3 ligases, including RING-Cys-Relay and RZ finger ligases, expanding the potential toolbox for therapeutic development [3]. Additionally, research into branched and mixed-linkage ubiquitin chains has uncovered complex regulatory signals that integrate cellular stress pathways, offering new opportunities for intervention in cancer and neurodegenerative diseases [3].
The ubiquitination cascade, comprising the coordinated action of E1, E2, and E3 enzymes, represents a sophisticated regulatory system that controls protein fate and function in eukaryotic cells. Continued elucidation of the mechanisms underlying ubiquitin signal specification, including the expanding roles of atypical ubiquitin linkages and the regulatory potential of E2 enzymes as environmental sensors, promises to unlock new therapeutic strategies for human diseases. For researchers and drug development professionals, leveraging advanced tools such as in vitro reconstitution systems, substrate identification platforms, and targeted degradation technologies will be essential for translating fundamental insights into clinical applications.
The ubiquitin-proteasome system (UPS) represents the primary pathway for selective intracellular protein degradation in eukaryotic cells, responsible for degrading over 80% of cellular proteins [8]. This system maintains protein homeostasis, regulates critical cellular processes including cell cycle progression, DNA repair, signal transduction, and eliminates damaged or misfolded proteins [8] [9]. At the heart of the UPS lies the 26S proteasome, a massive 2.6 MDa multi-subunit complex that degrades ubiquitin-tagged proteins into small peptides [10]. The 26S proteasome comprises two main subcomplexes: the 20S core particle (CP) that executes proteolysis, and the 19S regulatory particle (RP) that recognizes ubiquitinated substrates, prepares them for degradation, and regulates access to the core [10]. Understanding the intricate structure and functional mechanics of these components is fundamental to biomedical research, particularly in drug development for cancer and neurodegenerative diseases where proteasomal function is frequently impaired [8] [11].
Architectural Organization of the 26S Proteasome
The 20S Core Particle: Proteolytic Chamber
The 20S core particle forms the catalytic heart of the proteasome, organized as a hollow cylinder composed of four stacked heptameric rings [8]. The two outer rings consist of seven distinct α-subunits (α1-α7) that form a gated channel, while the two inner rings contain seven different β-subunits (β1-β7), three of which (β1, β2, and β5) harbor the proteolytic active sites [8] [10]. The organization creates an enclosed degradation chamber where substrates are sequestered to prevent uncontrolled protein destruction.
Table 1: 20S Core Particle Subunits and Their Functions
| Subunit Type | Yeast Gene | Human Gene | Proteolytic Activity/Function |
|---|---|---|---|
| α-subunits | PRE9 | PSMA4 | N-terminal tail constitutes major component of 20S gate |
| PRE6 | PSMA7 | Structural component of outer ring | |
| PRE5 | PSMA1 | Structural component of outer ring | |
| β-subunits | PRE3 | PSMB6 | Caspase-like (C-L) activity |
| PUP1 | PSMB7 | Trypsin-like (T-L) activity | |
| PRE2 | PSMB5 | Chymotrypsin-like (CT-L) activity; primary target of proteasome inhibitors |
The 20S core particle maintains a gated channel that restricts access to the proteolytic chamber, requiring regulatory particles to open for substrate entry [10]. Recent research has demonstrated that the 20S proteasome can also function independently of the 19S regulator, providing a specialized pathway for degrading intrinsically disordered proteins (IDPs) and oxidatively damaged proteins without ubiquitination [12].
The 19S Regulatory Particle: Substrate Recognition and Processing
The 19S regulatory particle recognizes ubiquitinated substrates, removes ubiquitin chains, unfolds target proteins, and translocates them into the 20S core particle [10]. This complex can be structurally and functionally divided into two subcomplexes: the base and the lid.
Table 2: Major 19S Regulatory Particle Subunits and Functions
| Subcomplex | Subunit | Human Gene | Function |
|---|---|---|---|
| Base | Rpt1-Rpt6 | PSMC2-PSMC6 | AAA-ATPase unfoldase; substrate translocation |
| Rpn1 | PSMD2 | Scaffold; ubiquitin receptor docking | |
| Rpn2 | PSMD1 | Scaffold; Rpn13 docking site | |
| Rpn10 | PSMD4 | Ubiquitin receptor (UIM domain) | |
| Rpn13 | ADRM1 | Ubiquitin receptor (PRU domain) | |
| Lid | Rpn11 | PSMD14 | Deubiquitinase (MPN+ domain) |
| Rpn3,5,6,7,8,9,12 | PSMD3,12,11,6,7,13,8 | PCI domain proteins; structural scaffold |
The base contains six AAA-ATPase subunits (Rpt1-Rpt6) that form a heterohexameric ring, which uses ATP hydrolysis to unfold substrates and translocate them into the 20S core [10]. The base also contains three ubiquitin receptors (Rpn1, Rpn10, and Rpn13) that recognize polyubiquitin chains on substrates [13]. The lid consists of nine non-ATPase subunits (Rpn3, Rpn5-Rpn9, Rpn11, Rpn12, and Sem1) and contains the deubiquitinating enzyme Rpn11, which removes ubiquitin chains from substrates prior to degradation [9] [10].
Functional Mechanics of Proteasomal Degradation
Substrate Recognition Mechanisms
The proteasome employs multiple ubiquitin receptors to recognize substrates with diverse ubiquitin chain configurations. Rpn10 serves as the primary receptor for proteins with single chains of K48-linked ubiquitin, while Rpn1 can act as a co-receptor with Rpn10 for K63 chains and other chain types [13]. Surprisingly, Rpn13 appears to retard degradation of various single-chain substrates in steady-state assays, suggesting complex regulatory roles for different receptors [13]. Substrates with multiple short ubiquitin chains can be presented for degradation through any of the known receptors, indicating remarkable versatility in recognition mechanisms [13].
Diagram 1: Proteasomal Degradation Pathway
Ubiquitin-Independent Degradation Mechanisms
While ubiquitin-dependent degradation represents the canonical pathway, recent research has revealed significant ubiquitin-independent proteasomal degradation mechanisms. The 20S core particle can directly degrade intrinsically disordered proteins (IDPs) and oxidatively damaged proteins without 19S regulation [12]. A 2025 study demonstrated that a hyperactive 20S proteasome (α3ΔN) engineered in C. elegans markedly enhanced IDP and misfolded protein degradation, reduced oxidative damage, and improved endoplasmic reticulum-associated degradation (ERAD) [12].
Furthermore, research published in 2025 revealed that depletion of 19S PSMD lid proteins causes aberrant ubiquitin-independent degradation of the kinesin motor protein KIF11 by the 20S core, leading to defects in bipolar spindle assembly during mitosis [14]. This demonstrates that the 19S particle not only facilitates ubiquitin-dependent degradation but also restrains inappropriate ubiquitin-independent degradation, highlighting a dual regulatory function.
Experimental Approaches for Proteasome Research
Structural Analysis Methods
Structural elucidation of proteasome complexes has advanced significantly through cryo-electron microscopy (cryo-EM). Recent technical breakthroughs have enabled near-atomic resolution views of the 26S proteasome, revealing conformational states during substrate processing [10]. For example, a 2025 cryo-EM study of human thioredoxin-like protein 1 (TXNL1) bound to the 19S regulatory particle revealed key interaction interfaces with PSMD1 (Rpn2), PSMD4 (Rpn10), and PSMD14 (Rpn11), establishing structural requirements for stress-induced ubiquitin-independent degradation [15].
Table 3: Key Experimental Methods in Proteasome Research
| Method | Application | Key Insights Generated |
|---|---|---|
| Cryo-EM | Structural analysis of proteasome complexes | Conformational states during substrate processing; ubiquitin receptor organization |
| Reconstituted proteasomes with mutated subunits | Functional analysis of specific ubiquitin receptors | Role of Rpn10, Rpn13, and Rpn1 in different substrate degradation pathways |
| Genetic engineering (e.g., CRISPR-Cas9) | Generation of hyperactive proteasome mutants | Mechanism of ubiquitin-independent degradation; proteostasis regulation |
| Affinity purification + Mass spectrometry | Identification of proteasome-interacting proteins | TXNL1-proteasome interactions; stress-induced degradation pathways |
| Tandem Mass Tag Mass Spectrometry (TMT-MS) | Proteomic analysis of proteasome function | Global proteome changes in response to proteasome hyperactivation |
Functional Assays and Genetic Approaches
Genetic manipulation combined with biochemical assays has been instrumental in deciphering proteasome function. Site-directed mutagenesis of ubiquitin receptors in yeast proteasomes has revealed specialized functions: Rpn10 primarily mediates degradation of K48-linked ubiquitin chains, while Rpn1 acts as a co-receptor for K63 chains and other chain types [13]. The development of hyperactive 20S proteasome models using CRISPR-Cas9 to induce N-terminal truncation of the α3 subunit (α3ΔN) has enabled research into ubiquitin-independent degradation pathways and their role in proteostasis [12].
RNA interference (RNAi) approaches have demonstrated the essential nature of 20S proteasome subunits across species. In Locusta migratoria, RNAi-mediated knockdown of 20S proteasome subunits resulted in complete mortality, midgut and gastric cecum atrophy, and significant reductions in body length and weight [16]. Similarly, silencing of proteasome subunits impaired ovarian growth, underscoring the crucial role of proteasomal function in development and tissue homeostasis [16].
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Research Reagents for Proteasome Studies
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Proteasome Inhibitors | MG132, Bortezomib, Carfilzomib | Inhibit proteolytic activity of 20S core particle; study substrate accumulation |
| E1 Inhibitors | PYR-41, PYZD-4409 | Block ubiquitin activation; investigate upstream ubiquitination |
| NEDD8-Activating Enzyme (NAE) Inhibitor | MLN4924 | Disrupts cullin neddylation and SCF E3 ligase function; in clinical trials |
| E2 Inhibitors | CC0651, NSC697923, BAY 11-7082 | Allosteric inhibition of specific E2 enzymes; study chain assembly |
| Genetic Tools | CRISPR-Cas9 (α3ΔN mutant), RNAi constructs | Generate hyperactive proteasomes; study subunit-specific functions |
| Affinity Tags | 3xFLAG tags on Rpn11, Pre1 | Purify proteasome subcomplexes; study assembly and interactions |
| Ubiquitin Chain Linkage-Specific Reagents | K48-only, K63-only ubiquitin mutants | Study substrate targeting specificity; receptor preferences |
Implications for Therapeutic Development
The intricate mechanics of the 20S core and 19S regulatory particles present multiple therapeutic intervention points. Cancer cells exhibit heightened dependence on proteasomal function, making proteasome inhibitors valuable therapeutics [8] [17]. Bortezomib, carfilzomib, and ixazomib target the 20S core particle and have been approved for treating multiple myeloma and other hematological malignancies [8].
Emerging strategies focus on more specific targeting of ubiquitin system components to enhance therapeutic efficacy while reducing side effects. The NEDD8-activating enzyme inhibitor MLN4924 is currently in phase II clinical trials, demonstrating promising results in disrupting cullin-RING ligase function [11]. Research into 20S proteasome hyperactivation presents a novel approach for treating neurodegenerative diseases characterized by protein aggregation, with studies showing that enhanced 20S activity reduces toxic protein accumulation in models of Alzheimer's and Parkinson's disease [12].
The structural and mechanistic insights into proteasome function continue to reveal new therapeutic opportunities. Understanding the specialized roles of ubiquitin receptors, the regulation of ubiquitin-independent degradation, and the assembly pathways of proteasome subcomplexes provides a foundation for developing next-generation therapeutics targeting the ubiquitin-proteasome system in cancer, neurodegenerative disorders, and other human diseases.
Ubiquitination is a crucial post-translational modification wherein a small, 76-amino acid protein, ubiquitin, is covalently attached to substrate proteins. The versatility of this signal arises from ubiquitin's ability to form polymers, or chains, through its seven lysine (K) residues (K6, K11, K27, K29, K33, K48, K63) or its N-terminal methionine (M1) [18] [19]. The type of linkage, the length of the chain, and its architecture (homotypic, mixed-linkage, or branched) constitute a sophisticated "ubiquitin code" that determines the fate and function of the modified substrate [20]. This code is decoded by ubiquitin-binding proteins (UBPs) containing ubiquitin-binding domains (UBDs), which direct downstream cellular processes [20]. Among the different linkage types, K48 and K63 represent the most abundant and well-studied chain types, with K48-linked chains being the classical signal for proteasomal degradation and K63-linked chains playing key roles in non-proteolytic signaling pathways [21] [20]. This review delves into the diversity of the ubiquitin code, with a specific focus on the biological roles, recognition, and experimental dissection of K48, K63, and other key linkages.
The Functional Spectrum of Ubiquitin Linkages
Different ubiquitin chain linkages create a functional spectrum of cellular signals. The table below summarizes the key characteristics and functions of the major linkage types.
Table 1: Diversity of Ubiquitin Chain Linkages and Their Functions
| Linkage Type | Primary Functions | Key E2 Enzymes / E3 Complexes | Representative Decoders / Effectors |
|---|---|---|---|
| K48-linked | Proteasomal degradation [21] [20] | CDC34 [20] | Proteasome (RPN10, RPN13) [22], RAD23B [20] |
| K63-linked | DNA repair, endocytosis, NF-κB signaling, inflammation, kinase activation [18] [21] [20] | Ubc13/Uev1a (Mms2) heterodimer [18] [20] | EPN2 [20], USP53/USP54 (DUBs) [23] |
| K11-linked | Proteasomal degradation (cell cycle regulators, ERAD) [24] | UbcH10 (with APC/C) [24] | Proteasome (RPN1, RPN10) [22] |
| K11/K48-branched | Priority signal for proteasomal degradation [22] | Not specified in search results | Proteasome (RPN1, RPN2, RPN10) [22], UCHL5 (DUB) [22] |
| Linear (M1-linked) | Innate immune response, NF-κB signaling [18] | LUBAC (HOIP/HOIL-1/SHARPIN) [18] | NF-κB pathway components [18] |
K48-Linked Ubiquitin Chains: The Canonical Degradation Signal
Discovery and Core Function
The discovery of K48-linked polyubiquitin chains by Chau et al. was a landmark event that established the paradigm of ubiquitin as a signal for protein degradation [18]. This linkage is the most abundant in the cell and serves as the primary signal for targeting substrates to the 26S proteasome for degradation [20]. The modification of a substrate with a chain of four or more K48-linked ubiquitins is conventionally considered the signal for efficient proteasomal recognition and degradation [20].
Mechanism of Proteasomal Recognition
The 26S proteasome recognizes K48-linked chains through its intrinsic ubiquitin receptors, including RPN10 and RPN13 [22]. Structural studies have revealed that these receptors bind to the hydrophobic patch centered around Ile44 on ubiquitin, facilitating substrate engagement and translocation into the proteolytic core particle.
K63-Linked Ubiquitin Chains: Masters of Non-Proteolytic Signaling
A Paradigm-Shifting Discovery
The discovery of K63-linked ubiquitin chains fundamentally changed the perception of ubiquitin's role in cell signaling. In 1999, Hofmann and Pickart found that a K63R ubiquitin mutation in yeast caused defects in DNA repair, a process independent of the proteasome [18]. They subsequently identified the Ubc13/Mms2 heterodimer as the specific E2 complex responsible for synthesizing K63-linked chains [18]. This revealed that ubiquitin functions as a signaling molecule beyond protein degradation.
Structural Basis of K63-Linked Chain Assembly
The structural basis for K63-chain specificity was elucidated through a collaboration that determined the crystal structure of Ubc13/Mms2. The structure revealed that Mms2, a catalytically inactive ubiquitin E2 variant (UEV), acts as a scaffold. It positions the acceptor ubiquitin molecule so that its K63 residue is oriented towards the active site cysteine of Ubc11 [18]. A key hydrophobic residue in Mms2 engages the Ile44 patch of the acceptor ubiquitin, ensuring linkage specificity [18].
Diverse Cellular Roles
K63-linked ubiquitin chains are involved in a wide array of non-degradative processes.
- Intracellular Trafficking and MVB Sorting: K63-linked chains act as a specific signal for sorting membrane proteins, like the Gap1 permease and carboxypeptidase S (CPS), into the multivesicular body (MVB) pathway for vacuolar/lysosomal degradation [21]. In contrast, monoubiquitination is often sufficient for the initial internalization of these proteins from the plasma membrane [21].
- DNA Damage Response: K63 chains play a critical role in recruiting DNA repair proteins to sites of damage [18] [20].
- Innate Immunity and Inflammation: The K63 linkage is essential for activating NF-κB signaling pathways in response to inflammatory stimuli [20].
- Kinase Activation: K63 ubiquitination can directly activate protein kinases, such as the nerve growth factor receptor TrkA [21].
Beyond K48 and K63: K11-Linked and Branched Ubiquitin Chains
K11-Linked Chains in Cell Cycle and Degradation
The Anaphase-Promoting Complex/Cyclosome (APC/C), a key regulator of the cell cycle, preferentially assembles K11-linked ubiquitin chains to trigger the degradation of mitotic regulators like cyclin B1 and securin [24]. The E2 enzyme UbcH10 provides specificity for K11-chain assembly. A unique surface on ubiquitin, the TEK-box, is critical for the elongation of K11-linked chains by facilitating the interaction between the E2 and the acceptor ubiquitin [24]. Strikingly, similar TEK-box motifs are found in APC/C substrates, enabling the ligase to switch from modifying substrate lysines to elongating chains on ubiquitin itself [24]. K11-linked chains are also recognized as efficient proteasomal targeting signals [24].
The Complexity of Branched Ubiquitin Chains
Branched ubiquitin chains, where a single ubiquitin molecule has more than one ubiquitin attached to it, represent a higher level of complexity in the ubiquitin code. Among these, K48/K63-branched chains are the best characterized, accounting for a significant portion of cellular K63 linkages [20]. Recent cryo-EM structures have revealed how the human 26S proteasome recognizes K11/K48-branched chains. The proteasome uses a multivalent recognition mechanism involving a novel K11-linked Ub binding site in a groove formed by RPN2 and RPN10, in addition to the canonical K48-linkage binding site [22]. This branched architecture acts as a "priority signal" for the proteasomal degradation of substrates during cell cycle progression and proteotoxic stress [22].
Experimental Toolkit for Decoding the Ubiquitin Code
Studying specific ubiquitin linkages requires specialized reagents and methodologies. The following table outlines key tools used in this field.
Table 2: Key Research Reagents and Methodologies for Ubiquitin Research
| Research Tool / Reagent | Function and Specificity | Key Application Examples |
|---|---|---|
| Linkage-Specific TUBEs (Tandem Ubiquitin Binding Entities) | High-affinity reagents (nanomolar Kd for tetra-ubiquitin) with specificity for K48 or K63 linkages. Protect polyubiquitinated proteins from deubiquitination and degradation [19]. | Immunoprecipitation of endogenous K48- or K63-polyubiquitinated proteins from cell lysates without cross-reactivity [19]. |
| Linkage-Specific DUBs | Deubiquitinases that cleave specific ubiquitin linkages. Used for linkage validation (UbiCRest assay) [20]. | OTUB1 (K48-specific) and AMSH (K63-specific) can be used to confirm chain linkage in pulldown experiments [20]. |
| Ubiquitin Mutants | Ubiquitin where all lysines are mutated to arginine except one (e.g., ubi-K48 only, ubi-K63 only), or single lysine-to-arginine mutants (e.g., ubi-K63R) [24]. | Determining if a specific linkage is required or sufficient for a biological process in in vitro assays or in cells [24]. |
| DUB Inhibitors (CAA, NEM) | Cysteine alkylators used in lysate-based pulldowns to inhibit endogenous DUBs and prevent disassembly of ubiquitin chains on bait proteins [20]. | Stabilizing immobilized Ub chains during ubiquitin interactor pulldown screens from cell lysates. Choice of inhibitor (CAA vs. NEM) can affect results and must be considered [20]. |
Detailed Experimental Protocol: Ubiquitin Interactor Pulldown Screen
This protocol, adapted from [20], is used to identify proteins that bind to specific ubiquitin chain types.
- Synthesis and Immobilization of Ubiquitin Chains: Enzymatically synthesize the desired homotypic or branched ubiquitin chains (e.g., K48-Ub3, K63-Ub3, Br-Ub3) using linkage-specific E2 enzymes (e.g., CDC34 for K48, Ubc13/Uev1a for K63). Purify the chains and immobilize them on streptavidin resin via a C-terminal biotin tag.
- Cell Lysate Preparation and DUB Inhibition: Prepare lysates from the cell line of interest (e.g., HeLa cells). Divide the lysate and treat with either Chloroacetamide (CAA) or N-Ethylmaleimide (NEM) to inhibit deubiquitinases. Note that the choice of inhibitor can impact the stability of the bait chains and the set of interactors identified.
- Pulldown Incubation: Incubate the immobilized ubiquitin chains with the inhibited cell lysates to allow ubiquitin-binding proteins (UBPs) to bind.
- Wash and Elution: Wash the resin thoroughly to remove non-specifically bound proteins. Elute the bound proteins.
- Identification by Mass Spectrometry: Analyze the eluted proteins using liquid chromatography-mass spectrometry (LC-MS).
- Data Analysis: Statistically compare the enrichment of proteins across the different chain types to identify linkage-specific, chain length-specific, or branch-specific interactors.
The workflow for this screen is visualized below.
Advanced Concepts and Recent Discoveries
Linkage-Specific Deubiquitinases (DUBs)
DUBs are essential for editing the ubiquitin code. A landmark 2025 study revised the annotation of USP53 and USP54, previously thought to be inactive, as highly specific K63-linkage-directed DUBs [23]. Disease-associated mutations in USP53 abrogate this activity, linking loss of K63-deubiquitination to pediatric cholestasis [23]. The study also revealed distinct mechanisms: USP54 cleaves within K63-linked chains, while USP53 performs "en bloc" deubiquitination of substrate proteins in a K63-specific manner, a previously unobserved activity [23]. Structural analysis identified cryptic S2 ubiquitin-binding sites within their catalytic domains that underpin this specificity [23].
The Role of Chain Length
Beyond linkage type, the length of a ubiquitin chain contributes to the specificity of its decoding. Some UBPs and DUBs display a clear preference for longer chains. For instance, the proteasome may require a chain of at least four ubiquitins (K48 ≥Ub4) for efficient substrate degradation, although this is debated [20]. Recent interactome screens have identified proteins like CCDC50, FAF1, and DDI2 that prefer Ub3 over Ub2 chains, highlighting the importance of chain length as a parameter in the ubiquitin code [20].
Concluding Perspectives
The exploration of the ubiquitin code has evolved from a simple dichotomy of K48 (degradation) versus K63 (signaling) to an appreciation of a complex language comprising multiple linkage types, chain lengths, and branched architectures. Advanced structural biology techniques, such as cryo-EM, are revealing how cellular machinery like the proteasome multivalently recognizes complex signals like K11/K48-branched chains [22]. Simultaneously, the development of more sophisticated tools, such as linkage-specific TUBEs and quantitative interactome screens, is enabling researchers to decode this language with ever-greater precision [20] [19]. Understanding the nuances of the ubiquitin code is not only fundamental to cell biology but also holds immense therapeutic potential, as dysregulation of ubiquitin signaling is implicated in cancer, neurodegenerative diseases, and other disorders [25] [26]. Future research will continue to unravel how the combinatorial complexity of ubiquitin modifications is integrated to control cellular homeostasis.
Cellular Roles in Protein Homeostasis and Quality Control
The ubiquitin-proteasome system (UPS) represents a cornerstone of cellular protein homeostasis, orchestrating the precise, selective degradation of myriad regulatory, damaged, and misfolded proteins. This targeted degradation is fundamental to critical physiological processes, including cell cycle progression, signal transduction, immune responses, and synaptic plasticity. Dysregulation of the UPS is mechanistically linked to a spectrum of human diseases, most notably cancer, neurodegenerative proteinopathies, and renal disorders, positioning it as a prime target for therapeutic intervention. This whitepaper provides an in-depth technical analysis of the UPS pathway, detailing its molecular mechanisms, exploring its multifaceted roles in disease pathogenesis, and highlighting cutting-edge research methodologies and reagent toolkits that are propelling both fundamental discovery and drug development in this field. The content is framed within the context of advancing UPS pathway research to unravel novel biological insights and therapeutic opportunities.
The ubiquitin-proteasome system is a highly conserved and regulated cascade responsible for the majority of selective intracellular protein degradation in eukaryotic cells. It governs the turnover of approximately 80% of cellular proteins, particularly short-lived, regulatory, damaged, or misfolded proteins, thereby maintaining protein homeostasis [27]. The process involves two major coordinated steps: first, the covalent attachment of a ubiquitin chain to a target protein, and second, the recognition and degradation of that tagged protein by the proteasome [28]. The exquisite specificity of the UPS is largely conferred by a vast array of E3 ubiquitin ligases, which recognize specific substrates, while the proteasome provides the proteolytic core. Beyond its fundamental housekeeping role, the UPS is a rapid and potent regulator of key signaling pathways, dynamically controlling the levels of proteins critical for processes as diverse as photomorphogenesis in plants [29] and neuronal plasticity in the mammalian brain [30]. The system's centrality to cellular function means that even minor perturbations can contribute to disease pathogenesis, making its components attractive diagnostic and therapeutic targets.
Core Mechanism of the Ubiquitin-Proteasome Pathway
The UPS functions through a precise, ATP-dependent enzymatic cascade that results in the targeted degradation of substrate proteins.
The Ubiquitination Cascade
Ubiquitination is a multi-step process mediated by a sequential enzyme cascade:
- E1 (Ubiquitin-Activating Enzyme): This initiation step involves a single or few E1 enzymes that activate ubiquitin in an ATP-dependent reaction, forming a high-energy thiol-ester bond between the C-terminal glycine of ubiquitin and a cysteine residue in the E1 active site [28] [30].
- E2 (Ubiquitin-Conjugating Enzyme): The activated ubiquitin is then transferred to the active site cysteine of an E2 enzyme. Humans possess approximately 25-30 different E2s, which begin to impart specificity to the process [30].
- E3 (Ubiquitin Ligase): This final and most diverse group of enzymes (numbering over 600 in humans) functions as a scaffold, simultaneously binding the E2~ubiquitin complex and the specific protein substrate. The E3 catalyzes the transfer of ubiquitin from E2 to a lysine residue on the substrate protein [28] [30]. Successive rounds of this process build a polyubiquitin chain on the substrate.
Table 1: Key Enzymatic Components of the Ubiquitination Cascade
| Component | Estimated Number in Humans | Primary Function | Families/Examples |
|---|---|---|---|
| E1 Enzyme | ~2 | Ubiquitin activation via ATP hydrolysis | UBA1, UBA6 |
| E2 Enzyme | 25-30 | Accepts activated ubiquitin from E1 | UbcH5, UbcH7 |
| E3 Ligase | >600 | Substrate recognition & ubiquitin transfer | RING, HECT, RBR [30] |
E3 ligases are primarily categorized into two major families:
- RING Finger E3s: These facilitate the direct transfer of ubiquitin from the E2 enzyme to the substrate. They can function as single subunits (e.g., Mdm2) or as multi-subunit complexes (e.g., CRL4COP1-SPA in plants) [29] [30].
- HECT Domain E3s: These form a thioester intermediate with ubiquitin before transferring it to the substrate. A well-characterized example is E6-AP (Ube3a) [30].
The nature of the polyubiquitin chain linkage determines the fate of the modified protein. A chain linked through lysine 48 (K48) of ubiquitin is the canonical signal for proteasomal degradation, whereas other linkages, such as K63, often play roles in non-proteolytic signaling pathways [30] [27].
The Proteasome and Degradation
The 26S proteasome is the macromolecular machine that recognizes and degrades polyubiquitinated proteins. It is composed of two primary complexes:
- 20S Core Particle (CP): A barrel-shaped structure comprising four stacked heptameric rings (α7β7β7α7) that contains the proteolytically active sites (housed in the β-subunits) facing the inner chamber. The activities include caspase-like, trypsin-like, and chymotrypsin-like hydrolysis [28] [27].
- 19S Regulatory Particle (RP): One or two RP "caps" attach to the 20S CP to form the 26S or 30S proteasome, respectively. The RP is responsible for recognizing ubiquitinated substrates, deubiquitinating them, unfolding the polypeptide, and translocating it into the 20S core for degradation in an ATP-dependent manner [28] [27].
Specialized proteasome variants, such as the immunoproteasome, incorporate alternative catalytic subunits and play specific roles in processes like antigen presentation [31].
Quantitative Data on UPS Composition and Function
The functionality of the UPS can be quantified by measuring the activity of its enzymatic components and the proteasome itself. The following table summarizes key quantitative assays and typical findings in disease contexts.
Table 2: Quantitative Profiling of UPS Components and Activity
| Assay Target | Experimental Method | Exemplary Finding / Relevance |
|---|---|---|
| E1 Activating Activity | Thioester assay, ATP/AMP quantification | A single E1 enzyme activates ubiquitin in most mammalian cells [27]. |
| E2 Conjugating Diversity | MS-based proteomics, yeast two-hybrid | ~25-30 E2 enzymes in humans interact selectively with specific E3s [30]. |
| E3 Ligase Specificity | IP-MS, protein microarrays, CRISPR screens | >600 E3 ligases provide substrate specificity; e.g., CRL1EBF1/2 degrades PIFs in light-grown plants [29]. |
| Proteasome Hydrolytic Activity | Fluorogenic peptide substrates (e.g., Suc-LLVY-AMC) | Cancer cells (e.g., multiple myeloma) show elevated chymotrypsin-like activity, targeted by drugs like Bortezomib [28]. |
| Global Ubiquitination Levels | Anti-Ub Western Blot, Ubiquitinome MS | Viral infection in maize significantly increases total protein ubiquitination levels [32]. |
| Proteasome Interactome | Proximity Labeling (ProteasomeID), APEX-MS | ProteasomeID identified novel interacting proteins and substrates across mouse organs [31]. |
Detailed Experimental Protocol: ProteasomeID for Mapping Interactomes
The following methodology details the ProteasomeID approach, a state-of-the-art technique for quantitatively mapping proteasome interactomes and substrates in vivo [31].
Background and Principle
Traditional methods like co-immunoprecipitation often fail to capture the dynamic and transient interactions of the proteasome. ProteasomeID utilizes proximity-dependent biotinylation (e.g., using the promiscuous biotin ligase BirA*) fused to specific proteasome subunits. This allows for the labeling of proteins within a ~10 nm radius, enabling subsequent streptavidin-based capture and mass spectrometric identification of interactors and induced substrates in their native cellular context.
Required Materials and Reagents
- Cell Line: HEK293 FlpIn TREx (HEK293T) cells or similar.
- Expression Constructs: Plasmids encoding BirA* fusions to proteasome subunits (e.g., PSMA4-BirA, PSMC2-BirA, BirA*-PSMD3), under a tetracycline-inducible promoter with an N- or C-terminal FLAG tag.
- Control: A construct expressing BirA* alone.
- Chemicals: Biotin, Tetracycline, Proteasome inhibitor (e.g., MG132), Streptavidin-coated magnetic beads.
- Buffers: Lysis buffer (e.g., RIPA with protease inhibitors), Wash buffers.
- Instrumentation: Mass spectrometer capable of Data Independent Acquisition (DIA).
Step-by-Step Procedure
Cell Line Generation & Induction:
- Generate stable HEK293T cell lines expressing the BirA* fusion proteins and the BirA*-only control.
- Induce expression with Tetracycline and supplement the culture medium with biotin for a defined period (typically 18-24 hours) to allow for biotinylation.
Cell Lysis and Protein Extraction:
- Harvest cells and lyse using an optimized lysis buffer.
- Clarify the lysate by centrifugation to remove insoluble debris.
Streptavidin Affinity Purification:
- Incubate the clarified lysate with chemically modified streptavidin magnetic beads to capture biotinylated proteins.
- Wash the beads stringently with a series of buffers to remove non-specifically bound proteins.
On-Bead Digestion and Peptide Preparation:
- Subject the beads to on-bead digestion with a protease (e.g., trypsin) to release peptides from the captured proteins.
- Use an optimized protocol to minimize co-elution of streptavidin-derived peptides, which can interfere with MS analysis.
Mass Spectrometric Analysis and Data Processing:
- Analyze the resulting peptides by Liquid Chromatography tandem Mass Spectrometry (LC-MS/MS) using a DIA method for deep, quantitative profiling.
- Process the raw data using a dedicated bioinformatics pipeline to identify and quantify biotinylated peptides and their corresponding proteins.
Key Applications and Validation
- Identification of Novel Interactors: ProteasomeID has been used to chart tissue-specific proteasome interactomes across different mouse organs, revealing previously unknown regulatory associations [31].
- Substrate Identification: When combined with proteasome inhibition (e.g., with MG132), the method enables the identification of endogenous proteasome substrates, including low-abundance transcription factors, by capturing proteins that accumulate near the proteasome upon inhibition.
- Validation: Findings should be validated using orthogonal methods such as immunofluorescence co-localization, co-immunoprecipitation, or functional assays measuring protein half-life.
The Scientist's Toolkit: Key Research Reagents and Solutions
Advanced research into the UPS relies on a suite of specialized reagents and molecular tools. The following table catalogs essential materials for probing UPS function.
Table 3: Essential Research Reagents for UPS Investigation
| Reagent / Tool | Category | Primary Function in Research | Specific Examples |
|---|---|---|---|
| Proteasome Inhibitors | Small Molecule Inhibitors | Inhibit proteolytic activity of the 20S core particle; used for mechanistic studies and cancer therapy. | Bortezomib, Carfilzomib, MG132 [28] [32] |
| E1/E2/E3 Inhibitors/Modulators | Small Molecule Inhibitors | Target specific steps in the ubiquitin cascade to dissect pathway mechanics and for therapeutic targeting. | PYR-41 (E1 inhibitor), Lenalidomide (Cereblon E3 modulator) [28] |
| Ubiquitin-Activating Enzyme (E1) | Recombinant Protein | For in vitro ubiquitination assays to study the enzymatic cascade and screen for inhibitors. | Recombinant UBA1 [27] |
| Ubiquitin-Conjugating Enzyme (E2) | Recombinant Protein | For in vitro ubiquitination assays to study specific E2-E3-substrate relationships. | Recombinant UbcH5, UbcH7 [30] |
| E3 Ligase Expression Constructs | Plasmid DNA/CRISPR Tools | To overexpress or knockout specific E3s for functional studies of substrate recognition and degradation. | Plasmids for CRL4COP1-SPA, Mdm2 [29] [30] |
| Anti-Ubiquitin Antibodies | Immunological Reagent | Detect mono- and polyubiquitinated proteins via Western Blot or Immunoprecipitation. | Anti-K48-linkage, Anti-K63-linkage specific antibodies [32] [27] |
| Anti-Proteasome Subunit Antibodies | Immunological Reagent | Detect proteasome composition, assembly, and localization. | Anti-PSMA4, Anti-PSMC2 [31] |
| Fluorogenic Proteasome Substrates | Activity Probe | Quantitatively measure the chymotrypsin-like, trypsin-like, and caspase-like activities of the proteasome. | Suc-LLVY-AMC [28] |
| BirA* Proximity Labeling System | Molecular Biology Tool | To map proteasome interactomes and proximal substrates in live cells and in vivo models. | PSMA4-BirA* knock-in mouse model [31] |
| PROTACs (Proteolysis-Targeting Chimeras) | Bifunctional Molecules | Induce targeted degradation of specific proteins of interest by recruiting them to an E3 ligase. | ARV-471 (targets ER for degradation) [28] |
UPS Dysregulation in Disease and Therapeutic Targeting
The critical role of the UPS in maintaining cellular homeostasis means its dysregulation is a contributory factor in numerous pathologies.
Cancer: Many oncoproteins (e.g., cyclins) are short-lived and controlled by the UPS, while tumor suppressors (e.g., p53) are often inactivated via ubiquitin-mediated degradation. Proteasome inhibitors, such as Bortezomib, are first-line therapies for blood cancers like multiple myeloma, inducing apoptosis by disrupting protein homeostasis in malignant cells [28]. Furthermore, targeted protein degradation strategies, including PROTACs, are a revolutionary class of therapeutics that hijack the UPS to degrade previously "undruggable" oncogenic targets [28].
Neurodegenerative Diseases: Conditions like Alzheimer's, Parkinson's, and Huntington's disease are characterized by the accumulation of toxic protein aggregates (e.g., Aβ, α-synuclein, huntingtin). This is frequently associated with an age-related decline in UPS activity and a failure in protein quality control [30] [33]. Neurons are particularly vulnerable to UPS impairment due to their post-mitotic nature and high metabolic demands [33].
Renal Diseases: The UPS is implicated in acute kidney injury (AKI), diabetic kidney disease, and renal fibrosis. For example, the E3 ligase RBBP6 promotes K48-linked ubiquitination and degradation of ERRα, exacerbating mitochondrial damage in tubular cells in diabetic kidney disease [27]. The balance between ubiquitination and deubiquitination is crucial for renal health.
Infection and Immunity: Viruses can manipulate the host UPS to promote their own replication. For instance, maize chlorotic mottle virus (MCMV) and sugarcane mosaic virus (SCMV) infection significantly alter the host ubiquitinome, and inhibition of the proteasome with MG132 enhances viral accumulation, indicating a role for the UPS in antiviral defense [32]. Specialized immunoproteasomes are also critical for generating peptides for antigen presentation [31].
Once considered exclusively a marker for proteasomal degradation, ubiquitination is now recognized as a versatile post-translational modification regulating diverse non-proteolytic cellular processes. This technical review examines the mechanisms by which non-degradative ubiquitination controls intracellular signaling cascades and membrane trafficking pathways. We detail how specific ubiquitin chain linkages and attachment sites directly modulate protein function, complex assembly, and subcellular localization through structural and conformational changes. The review synthesizes current experimental approaches for investigating these processes and discusses the implications for therapeutic intervention in human diseases characterized by disrupted ubiquitin signaling.
The ubiquitin-proteasome system (UPS) represents a crucial pathway for maintaining cellular proteostasis through targeted protein degradation. However, contemporary research has revealed that ubiquitination serves functions far beyond mere protein destruction [34]. The ubiquitin code—comprising monoubiquitination, multiple monoubiquitination, and various polyubiquitin chain linkages—generates tremendous functional diversity that regulates nearly all cellular processes [34] [35].
Non-degradative ubiquitination operates through distinct mechanisms that differ fundamentally from proteasome-targeting signals. Whereas K48-linked polyubiquitin chains typically target substrates for proteasomal degradation, K63-linked chains, monoubiquitination, and other atypical linkages (K6, K11, K27, K29, K33, M1) mediate regulatory functions including signal transduction, protein trafficking, DNA repair, and inflammatory responses [34] [36]. This functional divergence stems from both structural differences in chain conformation and the specific recognition of ubiquitin signals by proteins containing ubiquitin-binding domains (UBDs) [37].
The importance of non-degradative ubiquitination is particularly evident in immune signaling, where components like TRAF6 and TAK1 undergo K63-linked ubiquitination to activate NF-κB signaling independently of degradation [36]. Similarly, monoubiquitination regulates membrane trafficking by controlling the endocytosis of surface receptors [35]. This review systematically examines the mechanisms, experimental approaches, and pathophysiological significance of non-degradative ubiquitination in cellular signaling and trafficking regulation.
Mechanisms of Non-degradative Ubiquitin Signaling
Ubiquitin Chain Linkages and Their Functional Consequences
The structural basis for non-degradative ubiquitin signaling lies in the diverse topologies of ubiquitin chains, which determine specific interactions with ubiquitin-binding proteins.
Table 1: Non-degradative Ubiquitin Linkages and Their Cellular Functions
| Linkage Type | Structural Features | Primary Functions | Key Examples |
|---|---|---|---|
| K63-linked | Extended, open conformation [37] | DNA repair, endocytosis, NF-κB signaling, inflammation [37] [34] | TAK1 activation, histone ubiquitylation in DDR [34] |
| K6-linked | Not well characterized | Mitophagy, protein stabilization [34] | Parkin-mediated mitophagy [34] |
| K11-linked | Mixed features | DNA damage response [34] | Not specified |
| K27-linked | Not well characterized | Innate immunity, DDR recruitment [34] | RNF168-mediated histone marking [34] |
| K29-linked | Not well characterized | Wnt signaling, neurodegenerative disorders [34] | SPOP-mediated 53BP1 regulation [34] |
| K33-linked | Extended structure [37] | Protein trafficking, TCR signaling [34] | TCR-zeta regulation [37] |
| M1-linear | Linear structure | Immune signaling, cell death [34] | LUBAC in NF-κB activation [38] |
| Monoubiquitination | Single ubiquitin moiety | Endocytosis, chromatin regulation, protein activation [37] [35] | Histone H2B, Ras activation [37] |
Molecular Mechanisms of Action
Non-degradative ubiquitination regulates cellular processes through several distinct mechanisms:
Steric Regulation and Conformational Changes
Ubiquitin moieties can directly alter protein conformation and function. Molecular dynamics simulations of ZAP-70 kinase revealed that monoubiquitination at specific sites induces distinct conformational shifts—ubiquitination at K377 disrupted the active conformation, while modification at K476 stabilized an active-like state [37] [39]. This demonstrates how ubiquitin can allosterically regulate enzyme activity independent of degradation.
Platform for Complex Assembly
K63-linked and linear polyubiquitin chains serve as scaffolds for protein complex assembly. In NF-κB signaling, K63-linked polyubiquitin chains generated by TRAF6 create binding platforms that recruit and activate the TAK1 kinase complex through proteins with ubiquitin-binding domains like TAB2 [36]. Similarly, during Salmonella infection, linear ubiquitination promotes complex formation necessary for NF-κB activation [38].
Regulation of Protein-Protein Interactions
Monoubiquitination can either promote or inhibit specific protein interactions. For example, monoubiquitination of PCNA at K164 creates a binding site for specialized polymerases that facilitate translational DNA synthesis [37]. Conversely, monoubiquitination can sterically hinder interactions, as demonstrated in histone H2B, where ubiquitination prevents chromatin compaction [37].
Methodologies for Studying Non-degradative Ubiquitination
Proteomic Identification of Ubiquitination Sites
Mass spectrometry-based proteomics has revolutionized the identification of ubiquitination sites. The following workflow represents a standard approach for ubiquitin remnant profiling:
Key Experimental Details:
- Cell Lysis: Use denaturing conditions (e.g., 8M urea, 1% SDS) to preserve modifications and inhibit deubiquitinases [37].
- Digestion: Trypsin digestion generates di-glycine remnants (K-ε-GG) on modified lysines, serving as mass tags (+114.0429 Da) [37].
- Enrichment: Immunoaffinity purification with K-ε-GG-specific antibodies significantly enhances detection sensitivity [37] [38].
- Quantification: Stable Isotope Labeling with Amino acids in Cell culture (SILAC) or tandem mass tag (TMT) approaches enable comparative analysis between conditions [37].
- Proteasome Inhibition: Treatment with MG132 or bortezomib helps distinguish degradative from non-degradative ubiquitination by revealing sites unaffected by proteasomal blockade [37].
Functional Validation Approaches
Molecular Dynamics Simulations
Computational approaches provide mechanistic insights into how ubiquitin modifications affect protein dynamics. For ZAP-70 kinase studies:
- System Setup: Generate ubiquitinated protein structures using native chemical ligation or in silico modeling [37].
- Simulation Parameters: Run all-atom simulations in explicit solvent for hundreds of nanoseconds to capture conformational sampling [39].
- Analysis: Measure metrics like root-mean-square deviation (RMSD), radius of gyration, and inter-residue distances to quantify structural changes [39].
Functional Assays
- Kinase Activity Assays: Measure activity of ubiquitinated versus non-ubiquitinated kinases using radioactive or fluorescence-based phosphorylation assays [39].
- Co-immunoprecipitation: Assess protein-protein interactions affected by ubiquitination status [37].
- Pulldown Assays: Use ubiquitin-binding domains to detect specific chain linkages [34].
Signaling Pathways Regulated by Non-degradative Ubiquitination
Immune and Inflammatory Signaling
The NF-κB pathway represents a paradigm for non-degradative ubiquitin signaling, employing multiple chain types for precise regulation:
Key Mechanisms:
- K63-linked Ubiquitination: TRAF6 auto-ubiquitination creates K63 chains that recruit TAK1 through TAB2/3 ubiquitin-binding domains, facilitating IKK phosphorylation and activation [36].
- Linear Ubiquitination: The LUBAC complex (HOIP, HOIL-1L, SHARPIN) generates M1-linked chains on NEMO/IKKγ, reinforcing NF-κB signaling [38] [34].
- Negative Regulation: Deubiquitinating enzymes like A20/TNFAIP3 terminate signaling by removing K63 chains from key signaling intermediates [36].
During bacterial infection, Salmonella Typhimurium induces extensive rewiring of the host ubiquitinome, promoting CDC42 activity and linear ubiquitination to activate NF-κB [38]. Pathogens have evolved effector proteins that manipulate host ubiquitination to promote survival, highlighting the critical role of ubiquitin signaling in host-pathogen interactions.
DNA Damage Response
The DNA damage response employs a sophisticated ubiquitin signaling system for repair protein recruitment:
Table 2: Ubiquitin Ligases and Linkages in DNA Damage Response
| E3 Ligase | Ubiquitin Linkage | Substrate | Functional Outcome |
|---|---|---|---|
| RNF168 | K27-linked [34] | Histones H2A/H2A.X [34] | Recruitment of 53BP1 and BRCA1 to damage sites |
| RNF8 | K63-linked [34] | H1 histones [34] | Initial ubiquitin platform for RNF168 recruitment |
| RNF8 | K63-linked [34] | Akt [34] | Facilitates Akt membrane translocation and activation |
| SPOP | K27-linked [34] | Geminin [34] | Prevents DNA re-replication during S phase |
| SPOP | K29-linked [34] | 53BP1 [34] | Excludes 53BP1 from chromatin during S phase |
The sequential action of RNF8 and RNF168 establishes a ubiquitin-dependent recruitment platform that amplifies the DNA damage signal and facilitates the assembly of repair complexes at damage sites [34]. This exemplifies how different ubiquitin linkages create a sophisticated signaling code that coordinates the temporal and spatial organization of DNA repair.
Kinase Regulation and Cell Signaling
Protein kinases represent prominent targets for regulatory ubiquitination. Global ubiquitinome analyses reveal that kinases are frequently ubiquitinated within structured domains critical for catalytic activity and regulation [37] [39]. Unlike phosphorylation, which predominantly occurs in disordered regions, ubiquitination sites cluster in regions governing conformational stability and substrate access.
The TGF-β signaling pathway demonstrates how non-degradative ubiquitination both positively and negatively regulates signaling. Smad proteins undergo mono- and polyubiquitination that modulates their activity and complex formation without targeting them for degradation [37] [39]. Similarly, TAK1 activation requires K63-linked polyubiquitination, which facilitates its association with upstream regulators [37].
Membrane Trafficking and Protein Localization
Monoubiquitination serves as a versatile signal for controlling membrane trafficking processes:
Endocytic Trafficking
Monoubiquitination of cell surface receptors targets them for internalization and endosomal sorting [35]. The ubiquitin signal is recognized by endocytic proteins containing UBDs, such as epsins and Hrs, which facilitate cargo selection and vesicle formation.
Inflammatory Signaling and Trafficking
During Salmonella infection, host cells remodel their ubiquitinome to regulate actin cytoskeleton components and small GTPases like CDC42, linking membrane trafficking to inflammatory responses [38]. This coordination ensures precise spatiotemporal control of immune signaling.
Research Tools and Reagent Solutions
Table 3: Essential Research Reagents for Studying Non-degradative Ubiquitination
| Reagent Category | Specific Examples | Research Applications | Key Features |
|---|---|---|---|
| Ubiquitin Antibodies | K-ε-GG monoclonal antibodies [37] | Ubiquitin remnant immunoaffinity enrichment | Enrichment of ubiquitinated peptides for MS |
| Linkage-specific Antibodies | K63-linkage specific, K48-linkage specific, M1-linear specific antibodies [34] | Immunoblotting, immunofluorescence | Detection of specific chain types |
| Proteasome Inhibitors | Bortezomib, MG132, Carfilzomib [40] [41] | Distinguishing degradative vs non-degradative ubiquitination | Selective inhibition of proteasomal activity |
| Activity-Based Probes | Ubiquitin vinyl sulfones, HA-Ub-VS [40] | DUB activity profiling | Identification of active deubiquitinating enzymes |
| Cell Lines | HEK293, Jurkat T-cells [37] [39] | Ubiquitin proteomics, signaling studies | Well-characterized models for ubiquitin research |
| Expression Plasmids | Wild-type ubiquitin, ubiquitin mutants (K63R, K48R, K63-only, K48-only) [37] | Mechanistic studies in cell culture | Linkage-specific ubiquitin signaling |
Therapeutic Implications and Future Perspectives
The regulatory functions of non-degradative ubiquitination have profound implications for human disease and therapeutic development. In Alzheimer's disease, UPS proteins are elevated in cerebrospinal fluid decades before symptom onset, with increases correlating with tau pathology [42]. This suggests UPS dysregulation contributes to neurodegeneration through both degradative and non-degradative mechanisms.
In cancer, multiple components of non-degradative ubiquitination pathways are dysregulated. The success of proteasome inhibitors like bortezomib in multiple myeloma validates the UPS as a therapeutic target, though these agents broadly affect both degradative and non-degradative functions [40] [41]. More selective targeting of specific E3 ligases or deubiquitinases represents an emerging therapeutic strategy.
Viral myocarditis progression to dilated cardiomyopathy involves UPS-mediated regulation of inflammatory signaling, particularly through modulation of NF-κB and interferon responses [43]. Targeting specific ubiquitin pathways in inflammatory heart disease may offer therapeutic opportunities while minimizing global proteostatic disruption.
Future research directions include:
- Developing linkage-specific ubiquitin probes to precisely manipulate non-degradative signaling
- Elucidating the structural basis for ubiquitin-induced conformational changes
- Exploring crosstalk between ubiquitination and other post-translational modifications
- Investigating tissue-specific functions of non-degradative ubiquitination in disease contexts
Non-degradative ubiquitination has emerged as a crucial regulatory mechanism that parallels phosphorylation in its complexity and functional significance. Through specific chain linkages and attachment sites, ubiquitin modifications directly control protein function, complex assembly, and subcellular localization across diverse cellular processes. The continued development of experimental tools and analytical approaches will further illuminate the intricacies of the ubiquitin code and its therapeutic potential in human disease.
UPS in Neurodevelopment and Synaptic Plasticity
The ubiquitin-proteasome system (UPS) represents a crucial regulatory pathway for protein degradation in eukaryotic cells, functioning as a master coordinator of neurodevelopment and synaptic plasticity. This highly conserved system targets proteins for degradation through a coordinated enzymatic cascade involving E1 (activating), E2 (conjugating), and E3 (ligating) enzymes that tag substrates with ubiquitin chains, leading to their recognition and processing by the 26S proteasome [44]. In neurons, the UPS maintains proteostatic balance—a particularly challenging task given their extreme polarization, complex subcellular compartmentalization, and the need for rapid, localized responses to synaptic activity [45]. Emerging research has firmly established that beyond its housekeeping functions, the UPS actively participates in shaping neuronal connectivity and information processing through precise control of protein abundance at critical locations and times [45] [44].
The importance of the UPS in nervous system function is underscored by the growing number of neurodevelopmental disorders (NDDs) linked to genetic lesions in UPS components. Strikingly, genomic alterations in genes encoding various UPS elements—including ubiquitin-activating (E1), -conjugating (E2) enzymes, ubiquitin ligases (E3), ubiquitin hydrolases, and proteasome subunits—have been identified as causative factors in monogenic forms of NDDs [44]. This connection highlights the non-redundant roles that specific UPS components play during brain development and function, positioning the UPS as a central pathway in both neurodevelopmental and neurodegenerative conditions.
Fundamental Mechanisms of the UPS
The UPS Enzymatic Cascade
The ubiquitination process begins with E1 ubiquitin-activating enzymes, which activate ubiquitin in an ATP-dependent reaction. The activated ubiquitin is then transferred to an E2 ubiquitin-conjugating enzyme, which cooperates with an E3 ubiquitin ligase to catalyze the covalent attachment of ubiquitin to specific substrate proteins [44]. E3 ligases provide substrate specificity and can be divided into three major classes based on their mechanism of action: RING (Really Interesting New Gene), HECT (Homologous to E6AP C-Terminus), and RBR (RING-Between-RING) types [44].
The fate of ubiquitinated proteins depends on the topology of the ubiquitin modification. Monoubiquitination (single ubiquitin moiety) or multiple monoubiquitination (single ubiquitin on multiple sites) typically regulates subcellular localization, endocytosis, and protein trafficking [44]. Alternatively, ubiquitin itself can be modified on any of its eight acceptor sites (K6, K11, K27, K29, K33, K48, K63, and M1), generating polyubiquitin chains with distinct biological functions. Whereas K48-linked chains predominantly target substrates for proteasomal degradation, other linkage types (e.g., K63-linked) often serve non-proteolytic functions in signaling and trafficking [44].
The 26S Proteasome Complex
The 26S proteasome is a massive multi-subunit complex comprising a 20S core particle capped by one or two 19S regulatory particles. The 20S core particle contains the proteolytic active sites within its hollow cylindrical structure, while the 19S regulatory particle recognizes ubiquitinated substrates, removes ubiquitin chains, unfolds target proteins, and translocates them into the proteolytic chamber [44]. The regulatory particle contains ubiquitin receptors such as PSMD4 and ADRM1 that recognize K48-linked polyubiquitin chains, facilitating substrate engagement and processing [44].
Table 1: Major Components of the Ubiquitin-Proteasome System
| Component Type | Key Subtypes/Families | Primary Functions |
|---|---|---|
| E1 Enzymes | UBA1-UBA6 | Ubiquitin activation via ATP hydrolysis |
| E2 Enzymes | ~40 members in humans | Ubiquitin conjugation; determines chain topology |
| E3 Ligases | RING, HECT, RBR | Substrate recognition; specific ubiquitin transfer |
| E3 Complexes | Cullin-RING ligases (CRLs) | Multi-subunit complexes; cullins scaffold substrate receptors to RING proteins |
| Deubiquitinases | ~100 members in humans | Ubiquitin removal; proteasome processing; chain editing |
| Proteasome | 20S core, 19S regulatory cap | Protein degradation; substrate recognition & unfolding |
Recent research has revealed additional complexity in proteasomal regulation, including the existence of ubiquitin-independent proteasomal protein degradation (UIPP) pathways that can degrade intrinsically unstructured proteins without prior ubiquitination [46]. Furthermore, proteasome activators such as PA28γ (REGγ) can modulate proteasomal function in ways that are still being elucidated, particularly in the context of chromatin remodeling and DNA repair [46].
UPS in Neurodevelopmental Processes
Regulation of Neural Progenitor Fate and Cortical Development
During embryonic brain development, the UPS plays instrumental roles in determining neuronal cell fate through precise control of key developmental regulators. Recent research using ribosome profiling and RNA sequencing in mouse embryos has revealed that mRNA translation is dynamically regulated during cortical development, with the UPS contributing to the degradation of specific factors that must be eliminated at precise developmental transitions [45]. This regulated protein degradation is particularly important for the maintenance of neural progenitor pools and the timing of their differentiation into specific neuronal subtypes.
Studies have demonstrated that the protein translation rate itself serves as a critical determinant of neocortical neuron fate, with the UPS potentially contributing to the degradation of factors that maintain progenitor identity [45]. The development of advanced technologies allowing examination of protein synthesis and degradation at cellular resolution has revealed that proteostatic mechanisms are actively regulated during neurogenesis, rather than simply maintaining static protein levels [45]. This dynamic regulation ensures the proper progression of developmental programs and the emergence of appropriate neuronal diversity in the developing cortex.
Axonal and Dendritic Development
The UPS plays a particularly important role in shaping neuronal morphology by regulating the stability of proteins involved in axon guidance, dendritic arborization, and synapse formation. During axon development, the UPS facilitates the remodeling of the axonal proteome in response to guidance cues, eliminating proteins that are no longer required while allowing the accumulation of those needed for the next developmental phase [45]. This localized proteostatic control is essential for the proper pathfinding of growing axons and the establishment of functional neural circuits.
In dendritic development, the UPS regulates the stability of cytoskeletal components, adhesion molecules, and signaling proteins that collectively determine dendritic complexity and targeting specificity. The importance of this regulation is highlighted by the identification of several UPS components, including ubiquitin ligases and deubiquitinating enzymes, that when mutated cause neurodevelopmental disorders characterized by altered dendritic morphology and connectivity [44]. These findings position the UPS as a central regulator of the structural development of both pre- and postsynaptic compartments.
UPS in Synaptic Plasticity
Presynaptic Mechanisms
At the presynaptic terminal, the UPS regulates neurotransmitter release, synaptic vesicle cycling, and active zone organization through the controlled degradation of key presynaptic proteins. Recent research has revealed an impressive amount of ongoing translation in presynaptic terminals, with the UPS providing a complementary mechanism for rapidly adjusting the presynaptic proteome in response to activity [45]. This localized protein synthesis and degradation allows presynaptic terminals to autonomously control their protein composition without relying on somatic supply.
Studies examining presynaptic protein synthesis have shown that it supports structural and functional plasticity of glutamatergic axon terminals [45]. The UPS interacts with these local translation mechanisms to maintain presynaptic function, with disruption of either system leading to impaired neurotransmission. For instance, inhibition of presynaptic protein synthesis alters transmitter release, while proteasome inhibition similarly disrupts presynaptic function, indicating that both systems must be coordinately regulated to maintain synaptic efficacy [45].
Postsynaptic Mechanisms and Density Regulation
In the postsynaptic compartment, the UPS controls the abundance of neurotransmitter receptors, scaffolding proteins, and signaling molecules that collectively determine synaptic strength. The postsynaptic density (PSD) contains numerous UPS components, including ubiquitin ligases and proteasomal subunits, allowing for localized protein degradation in response to synaptic activity [45]. This postsynaptic UPS activity is particularly important for long-term plasticity processes such as long-term potentiation (LTP) and long-term depression (LTD).
Recent technological advances have enabled the examination of protein synthesis and degradation at subcellular resolution, revealing ongoing translation and UPS activity in dendritic spines [45]. What has emerged is a model in which local protein synthesis provides new proteins for plasticity-related changes, while the UPS removes proteins that must be eliminated to enable structural and functional reorganization of the synapse. The balance between these opposing processes allows for the precise control of synaptic protein composition that underlies information storage in neural circuits.
Table 2: Quantitative Assessments of UPS Activity in Neuronal Compartments
| Experimental System | Measurement Type | Key Finding | Reference Technique |
|---|---|---|---|
| Drosophila oocytes (quiescent) | Proteasome activity | 3-fold increase during quiescence | AMC peptide-substrate assay [47] |
| Mitochondrial fractions (quiescent) | Mitochondria-associated proteasome activity | 12-fold increase vs. growing follicles | Proteasome activity probe [47] |
| Neuronal processes | Local proteasome abundance | Proteasomes present in axons & dendrites | Subcellular fractionation [45] |
| Synaptogenesis | Ribosome levels in axons | Stimulation reduces ribosomal proteins | Proteomic analysis [45] |
| GSK3-RNAi oocytes | Mitochondrial proteasome activity | Reduced activity & 26S proteasome content | Proteasome activity assays [47] |
Methodologies for Studying Neuronal UPS
Proteasome Activity Assays
Multiple approaches have been developed to quantitatively assess UPS function in neuronal contexts. The AMC peptide-substrate proteasome activity assay utilizes fluorogenic peptides that release aminomethylcoumarin (AMC) upon proteasomal cleavage, providing a sensitive measure of proteasomal chymotrypsin-like, trypsin-like, and caspase-like activities [47]. This approach can be applied to tissue homogenates, subcellular fractions, or intact cells and has revealed a 3-fold increase in overall UPS activity as Drosophila oocytes enter cellular quiescence [47].
More recently, activity-based probes such as Me4BodipyFL-Ahx3Leu3VS (UbiQ-018) have been developed that covalently label active proteasomal subunits, allowing both quantification and visualization of proteasome activity [47]. These probes can be used to examine all three proteasomal activities simultaneously and have confirmed elevations in proteasome function during quiescence entry. When combined with subcellular fractionation protocols, these probes have demonstrated a striking 12-fold increase in proteasome activity associated with mitochondrial fractions from quiescent oocytes compared to growing follicles [47].
Genetic and Pharmacological Manipulation
The functional significance of UPS components in neurodevelopment and plasticity has been extensively investigated using both genetic and pharmacological approaches. RNA interference (RNAi) techniques have been employed to knock down specific UPS components, such as GSK3, resulting in reduced mitochondrial proteasome activity and impaired mitochondrial respiratory quiescence [47]. Similarly, pharmacological inhibitors including MG132 (a proteasome inhibitor) and Bortezomib have been used to acutely inhibit proteasomal function, leading to increased mitochondrial membrane potential and disrupted synaptic function [47] [48].
The combination of cell type-specific genetic manipulations with electrophysiological and behavioral assessments has been particularly powerful in establishing causal relationships between UPS function and neuronal phenotypes. For example, deletion of Fmr1 in parvalbumin-expressing neurons results in dysregulated translation and selective behavioral deficits associated with fragile X syndrome, illustrating how cell type-specific UPS-related manipulations can produce distinct functional outcomes [45].
Diagram 1: UPS in Activity-Dependent Synaptic Plasticity. Neuronal activity triggers postsynaptic signaling that modulates UPS function, leading to degradation of specific substrates and structural/functional plasticity changes.
Research Reagent Solutions
Table 3: Essential Research Reagents for Investigating Neuronal UPS
| Reagent Category | Specific Examples | Key Applications | Functional Outcome |
|---|---|---|---|
| Proteasome Inhibitors | MG132, Bortezomib | Acute proteasome inhibition | Increased ubiquitinated proteins; disrupted synaptic function [47] [48] |
| Activity Probes | Me4BodipyFL-Ahx3Leu3VS (UbiQ-018) | Labeling active proteasomes | Quantification of proteasome activity & localization [47] |
| Fluorogenic Substrates | AMC-conjugated peptides | Proteasome activity assays | Measurement of chymotrypsin-/trypsin-/caspase-like activities [47] |
| Genetic Tools | RNAi constructs, Cre-lox system | Cell type-specific manipulation | Defined UPS component functions [45] [47] |
| Ubiquitin Linkage Tools | Linkage-specific antibodies | Ubiquitin chain characterization | Differentiation of degradative vs signaling ubiquitination [44] |
UPS Dysfunction in Neurodevelopmental Disorders
Genomic alterations in UPS components are increasingly recognized as causative factors in monogenic forms of neurodevelopmental disorders (NDDs) [44]. These include mutations in genes encoding E3 ubiquitin ligases such as UBE3A (associated with Angelman syndrome), HUWE1 (associated with X-linked intellectual disability), and CUL3 (implicated in autism spectrum disorder) [45] [44]. Interestingly, a comprehensive analysis of NDDs caused by UPS genomic alterations reveals that a majority of the affected proteins have described functions in the negative regulation of innate immune response, suggesting a possible involvement of autoinflammation in NDD pathogenesis [44].
The mechanisms through which UPS mutations disrupt neurodevelopment are diverse and include altered degradation of specific substrates critical for neuronal migration, synapse formation, or circuit refinement. For example, increased rates of cerebral protein synthesis have been observed in Shank3 knockout mice, suggesting a link between synaptic protein deficit and dysregulated protein synthesis in autism spectrum disorder and intellectual disability [45]. Similarly, excessive proteostasis has been shown to contribute to pathology in fragile X syndrome, indicating that both increased and decreased UPS activity can be detrimental to neuronal function [45].
Diagram 2: Pathological Cascades from UPS Dysfunction to Neurodevelopmental Disorders. Multiple UPS impairment types converge on cellular dysfunction, leading to structural and functional neuronal deficits.
Emerging Research Directions and Therapeutic Implications
Recent research has revealed unexpected aspects of UPS function in neurons, including the discovery that proteasomes are recruited to mitochondria during quiescence to support mitochondrial respiratory quiescence (MRQ) [47]. This recruitment, which is regulated by GSK3 phosphorylation of mitochondrial outer membrane proteins like VDAC, represents a conserved mechanism for coordinating metabolic shifts with proteostatic regulation across fungi, Drosophila, and mammals [47]. The finding that proteasome inhibition increases mitochondrial membrane potential underscores the intimate relationship between UPS function and metabolic regulation in neurons.
Another emerging area concerns the crosstalk between the UPS and ubiquitin-independent proteasomal protein degradation (UIPP) pathways [46]. Intrinsically unstructured proteins (IUPs) can be degraded by 20S stand-alone proteasomes without prior ubiquitination, with proteasome activators such as PA28γ playing important regulatory roles [46]. Understanding how these alternative degradation pathways contribute to neuronal proteostasis, particularly under conditions of oxidative stress, represents an important frontier with potential therapeutic implications.
From a therapeutic perspective, the UPS represents a promising target for treating neurodevelopmental disorders, though manipulating this system presents significant challenges given its broad functions. Strategies that target specific E3 ligases or regulatory complexes rather than global proteasome activity may offer better therapeutic windows. Additionally, understanding the parallels between immune dysregulation and neurodevelopment in UPS-related NDDs may reveal novel treatment approaches that address both neurological and inflammatory aspects of these conditions [44].
Harnessing the UPS: Targeted Degradation Technologies and Clinical Translation
Targeted protein degradation via PROteolysis TArgeting Chimeras (PROTACs) represents a revolutionary therapeutic strategy that harnesses the body's natural protein disposal machinery. This technology marks a paradigm shift from traditional inhibition to complete elimination of disease-causing proteins, offering unique advantages for targeting proteins previously considered "undruggable" [49] [50]. The approach is firmly grounded in the biology of the ubiquitin-proteasome system (UPS), the primary pathway for regulated intracellular protein degradation in eukaryotic cells [50].
The UPS maintains cellular proteostasis by selectively degrading damaged, misfolded, or short-lived regulatory proteins, thereby controlling critical processes including cell cycle progression, DNA repair, and stress response [50]. This system functions through a coordinated enzymatic cascade: ubiquitin-activating enzymes (E1) activate ubiquitin in an ATP-dependent manner, ubiquitin-conjugating enzymes (E2) accept the activated ubiquitin, and ubiquitin ligases (E3) transfer ubiquitin to specific substrate proteins [49] [50]. Repeated cycles result in polyubiquitin chain formation on substrates, with K48-linked chains primarily targeting proteins for degradation by the 26S proteasome [49]. The proteasome then recognizes, unfolds, and proteolytically cleaves ubiquitinated proteins into small peptides, while ubiquitin molecules are recycled for future use [50]. PROTAC technology strategically co-opts this sophisticated endogenous quality control system for therapeutic purposes.
The Molecular Mechanism of PROTACs
Core Components and Degradation Cycle
A PROTAC molecule is a heterobifunctional complex comprising three essential elements: a ligand that binds the protein of interest (POI), a ligand that recruits an E3 ubiquitin ligase, and a linker connecting these two moieties [50] [51]. Mechanistically, the PROTAC functions as a molecular bridge that induces proximity between a target protein and an E3 ubiquitin ligase [52]. This forced association facilitates the formation of a POI-PROTAC-E3 ligase ternary complex, positioning the POI for ubiquitin transfer from the E2-loaded ubiquitin conjugating enzyme [50] [52].
Following successful ubiquitination, the polyubiquitinated target protein is recognized and degraded by the 26S proteasome [50]. Crucially, the PROTAC molecule is not consumed in this process; it dissociates from the degradation complex upon completion and can subsequently initiate additional rounds of ubiquitination and degradation [50] [51]. This catalytic mechanism enables sustained protein degradation at low compound concentrations, offering a significant advantage over traditional occupancy-driven inhibitors [51].
Visualization of the PROTAC Mechanism
The following diagram illustrates the sequential mechanism of action of PROTAC-induced targeted protein degradation.
Design and Evolution of PROTAC Molecules
Generations of PROTAC Development
PROTAC technology has evolved significantly since its inception, progressing through distinct generations characterized by key innovations in molecular design and functionality.
Table 1: Generations of PROTAC Development
| Generation | Time Period | Key Characteristics | Representative Achievements | Limitations |
|---|---|---|---|---|
| First Generation | Early 2000s | Peptide-based E3 ligase ligands [50] | PROTAC-1 targeting MetAP-2 using IκBα-derived peptide to recruit SCF complex [50] | Poor cell permeability and metabolic stability [50] |
| Second Generation | Late 2000s onward | Small molecule-based ligands for both POI and E3 ligase [50] | Use of nutlin-3a (MDM2 ligand) with androgen receptor ligand for AR degradation [50] | Improved pharmacokinetics but still substantial molecular weight |
| Third Generation | Present & Future | Advanced modalities including reversible covalent, dual-target, and tissue-specific PROTACs [51] | Pro-PROTACs (prodrugs), dual-PROTACs targeting multiple POIs, Nano-PROTACs for improved delivery [53] [51] | Addressing selectivity, delivery, and resistance challenges |
Key E3 Ligases in PROTAC Design
The selection of appropriate E3 ubiquitin ligases is critical for PROTAC design, as different E3 ligases exhibit distinct tissue expression patterns, subcellular localization, and substrate preferences. Although over 600 E3 ligases exist in humans, current PROTAC designs primarily utilize a limited subset with well-characterized ligands and suitable biological properties [54].
Table 2: Major E3 Ubiquitin Ligases Utilized in PROTAC Technology
| E3 Ligase | Ligand | Key Characteristics | Common Applications |
|---|---|---|---|
| Cereblon (CRBN) | Thalidomide, Lenalidomide, Pomalidomide [49] [50] | Component of CRL4^CRBN complex; targets transcription factors [50] | Hematological malignancies; BRD4 degraders (e.g., dBET1) [50] |
| Von Hippel-Lindau (VHL) | VHL ligand derived from hydroxyproline [50] | Component of CRL2^VHL complex; oxygen-sensing pathway [50] | Solid tumors; HIF-1α related pathologies [50] |
| MDM2 | Nutlin-3a [50] | Regulates p53 tumor suppressor; natural inhibitor of p53 [50] | Cancers with wild-type p53; first small molecule PROTAC [50] |
| cIAP1 | Methyl bestatin [50] | Apoptosis regulator; uses dimerization-dependent mechanism [49] | Apoptosis-resistant cancers; early-stage PROTAC development [50] |
Advantages of PROTACs over Traditional Inhibitors
PROTAC technology offers several fundamental advantages over traditional small molecule inhibitors, deriving from its unique event-driven mechanism of action rather than occupancy-based inhibition.
Table 3: Comparative Analysis: PROTACs vs. Small Molecule Inhibitors
| Parameter | PROTACs | Small Molecule Inhibitors |
|---|---|---|
| Mechanism of Action | Event-driven, catalytic protein degradation [52] [51] | Occupancy-driven, direct inhibition [52] |
| Active Site Requirement | Not required; can target proteins without functional binding pockets [52] [54] | Essential; requires accessible, druggable active site [52] |
| Occupancy Model | Sub-stoichiometric; single molecule degrades multiple POI copies [52] [51] | Stoichiometric; continuous exposure required for inhibition [52] |
| Druggable Target Space | Expanded scope including scaffolding proteins and non-enzymatic functions [52] [54] | Limited to proteins with suitable binding pockets (~15% of proteome) [52] |
| Selectivity | Enhanced through ternary complex formation and cooperative binding [52] | Primarily determined by binary ligand-target interactions [52] |
| Resistance Potential | Lower; effective against mutant and overexpressed targets [50] [51] | Higher; susceptible to mutations and overexpression [50] |
| Target Fate | Complete destruction; sustained effect requiring new protein synthesis [52] | Target remains intact; rapid recovery after drug removal [52] |
The catalytic nature of PROTACs enables sustained pharmacological effects even after drug clearance, as protein function can only be restored through new protein synthesis [52]. This technology particularly excels in addressing drug resistance mechanisms common with traditional inhibitors, including target overexpression, mutations, and compensatory pathway activation [50] [51]. Furthermore, PROTACs can target "undruggable" proteins such as transcription factors, scaffolding proteins, and non-enzymatic regulators that lack conventional binding pockets for inhibitor development [54] [51].
Experimental Approaches in PROTAC Development and Validation
Quantitative Measurement of Intracellular Target Engagement
Successful PROTAC development requires rigorous assessment of cellular penetration, target engagement, and degradation efficiency. The NanoBRET (Bioluminescence Resonance Energy Transfer) platform provides a robust method for monitoring these parameters in live cells [52]. This system utilizes a Nano-luciferase (nLuc) tagged target protein (e.g., BTK-nLuc) and a cell-permeable fluorescent tracer that competes with the PROTAC for binding to the target. As PROTACs displace the tracer, decreased BRET signals quantitatively reflect intracellular target engagement in real-time [52].
Complementary cellular permeability and accumulation assays are essential for prioritizing PROTAC candidates. Quantitative measurements of intracellular drug concentrations and target binding constants (Kd) enable calculation of the Relative Intracellular Accumulation Coefficient, a critical parameter for optimizing PROTAC efficacy [52]. For covalent PROTACs, additional kinetic studies of bond formation and reversal rates provide insights into their unique mechanism of action [52].
Experimental Workflow for PROTAC Evaluation
The following diagram outlines a comprehensive experimental workflow for evaluating PROTAC efficacy, from initial design to functional validation.
Essential Research Reagents and Methodologies
Table 4: Research Reagent Solutions for PROTAC Development
| Reagent/Method | Function | Application Examples |
|---|---|---|
| NanoBRET Target Engagement Assay | Quantitative measurement of intracellular target binding [52] | Real-time monitoring of POI-PROTAC interactions in live cells [52] |
| ATP Site-Dependent Competition Binding Assay | Determination of binding affinity (Kd) between target and degraders [52] | Screening PROTAC libraries for optimal POI binding moieties [52] |
| HaloTag/Degradation Tag (dTAG) Systems | Controlled degradation of engineered fusion proteins [52] | Validation of degradation-specific phenotypes; tool compound development [52] |
| Phage-Based Binding Assays | High-throughput screening of binding constants [52] | Ranking PROTAC candidates based on target affinity [52] |
| Photo-activatable PROTACs | Spatiotemporal control of PROTAC activity [53] | Precise mechanistic studies; reducing off-target effects in validation [53] |
| Cellular Viability & Proliferation Assays | Assessment of functional consequences of protein degradation [55] | Determining anticancer efficacy in relevant disease models [55] |
Current Clinical Landscape and Emerging Frontiers
PROTACs in Clinical Development
The PROTAC clinical pipeline has expanded rapidly, with over 40 candidates currently in various stages of clinical trials as of 2025 [55]. Several programs have advanced to pivotal Phase III studies, demonstrating the translational potential of this technology.
Table 5: Selected PROTACs in Advanced Clinical Development (2025)
| PROTAC Candidate | Company | Target | Indication | Development Phase |
|---|---|---|---|---|
| Vepdegestrant (ARV-471) | Arvinas/Pfizer | Estrogen Receptor (ER) | ER+/HER2- Breast Cancer | Phase III [55] |
| BMS-986365 (CC-94676) | Bristol Myers Squibb | Androgen Receptor (AR) | Metastatic Castration-Resistant Prostate Cancer | Phase III [55] |
| BGB-16673 | BeiGene | BTK | B-cell Malignancies | Phase III [55] |
| ARV-110 | Arvinas | Androgen Receptor (AR) | Prostate Cancer | Phase II [55] |
| KT-474 (SAR444656) | Kymera | IRAK4 | Hidradenitis Suppurativa & Atopic Dermatitis | Phase II [55] |
Recent clinical data has demonstrated both promise and challenges. Vepdegestrant showed statistically significant improvement in progression-free survival in patients with ESR1 mutations in the VERITAC-2 trial, though it did not reach significance in the overall intent-to-treat population [55]. This highlights the importance of patient stratification strategies in PROTAC development.
Emerging Innovations and Future Directions
The PROTAC field continues to evolve with several next-generation technologies addressing current limitations:
- PROTAC 2.0 Modalities: Advanced designs including antibody-PROTAC conjugates (DAC-PROTACs), macrocyclic PROTACs, and trivalent degraders that enhance selectivity and tissue targeting [51].
- Alternative Degradation Pathways: Lysosome-Targeting Chimeras (LYTACs) for extracellular and membrane proteins, and Autophagy-Targeting Chimeras (AUTACs) leveraging autophagy pathways [53].
- Ubiquitin-Independent Degradation: Exploiting bacterial effector proteins like SAP05 that mediate ubiquitin-independent proteasomal degradation by directly bridging targets to proteasomal subunits [54].
- Spatiotemporal Control: Photo-activatable PROTACs (opto-PROTACs) with caging groups (e.g., DMNB) that enable precise light-controlled activation for research and potential therapeutic applications [53].
These innovations collectively address key challenges in PROTAC technology, including tissue specificity, expansion of targetable proteins, and overcoming potential resistance mechanisms related to E3 ligase function [54] [51].
PROTAC technology represents a transformative approach in therapeutic development, fundamentally shifting the paradigm from protein inhibition to targeted elimination. By harnessing the ubiquitin-proteasome system, PROTACs overcome significant limitations of traditional small molecule inhibitors, particularly for targets previously considered undruggable. The modular design and catalytic mechanism of PROTACs provide unique pharmacokinetic and pharmacodynamic advantages, including sustained target suppression and potential application against resistant disease variants.
As the field advances, next-generation PROTACs with enhanced selectivity, novel degradation mechanisms, and improved delivery systems promise to expand the clinical utility of this technology. With multiple candidates in advanced clinical trials and a robust pipeline of innovative approaches, targeted protein degradation continues to establish itself as a cornerstone of modern drug discovery, offering new hope for addressing challenging disease targets across oncology, neurodegeneration, and inflammatory disorders.
Targeted protein degradation (TPD) represents a paradigm shift in therapeutic intervention, moving beyond simple inhibition to the complete removal of disease-causing proteins from cells [49]. This approach primarily harnesses the body's natural protein quality-control machinery, most notably the ubiquitin-proteasome system (UPS) [49] [56]. The UPS serves as a critical regulatory pathway for maintaining cellular protein homeostasis (proteostasis) through a highly orchestrated enzymatic cascade [49]. Central to this process are E3 ubiquitin ligases, which confer substrate specificity by recognizing target proteins and facilitating their tagging with ubiquitin chains [49] [56]. This ubiquitination marks proteins for destruction by the proteasome, a multi-subunit complex that degrades proteins into small peptides [49].
Molecular glue degraders (MGDs) constitute an emerging class of monovalent small molecules that exploit the UPS by inducing novel protein-protein interactions (PPIs) between E3 ubiquitin ligases and target proteins [57] [58]. Unlike traditional inhibitors that merely block protein activity, MGDs promote the ubiquitination and subsequent degradation of target proteins, offering a catalytic mechanism of action that can address previously "undruggable" targets [58] [59]. These compounds typically exhibit favorable drug-like properties, including lower molecular weights and improved bioavailability compared to other TPD modalities such as proteolysis-targeting chimeras (PROTACs) [60] [57]. The therapeutic potential of MGDs spans oncology, neurodegenerative diseases, and other therapeutic areas where eliminating pathogenic proteins could transform treatment paradigms [61] [58].
Molecular Mechanisms of Glue Degraders
Fundamental Mechanistic Principles
Molecular glue degraders function through induced proximity, remodeling protein surfaces to facilitate interactions between E3 ubiquitin ligases and target proteins that would not normally occur [56] [58]. This mechanism stands in contrast to heterobifunctional degraders like PROTACs, which physically bridge two proteins using separate binding moieties connected by a linker [49] [60]. MGDs are typically monovalent, binding to a single protein—most commonly an E3 ubiquitin ligase—and altering its surface conformation to create a novel interface that recognizes specific target proteins [56] [59]. This induced complementarity enables the formation of a productive ternary complex wherein the target protein is positioned for ubiquitin transfer [56].
A key advantage of MGDs is their catalytic mode of action [58]. Once the ternary complex forms and ubiquitination occurs, the MGD dissociates and can facilitate additional rounds of degradation, enabling sustained pharmacological effects at low doses [56] [58]. This substoichiometric activity contrasts with traditional occupancy-driven inhibitors that require continuous target engagement [56]. The degradation mechanism follows the natural ubiquitin-proteasome pathway: after polyubiquitination (typically via K48-linked chains), the target protein is recognized and unfolded by the proteasome, then degraded into small peptide fragments [49].
Structural Basis of Molecular Glue Action
The structural biology of MGD-mediated ternary complexes reveals diverse mechanisms of action. Some MGDs, like the immunomodulatory imide drugs (IMiDs), bind deeply within the tri-tryptophan pocket of cereblon (CRBN), a substrate receptor of the CRL4 E3 ubiquitin ligase complex [62] [63]. This binding event alters the CRBN surface, creating a neo-interface that recognizes specific protein motifs such as the G-loop degron found in transcription factors like IKZF1 and IKZF3 [62]. Recent structural insights from cryo-EM studies have illuminated even more sophisticated mechanisms, as demonstrated by MRT-31619, which drives CRBN homodimerization by forming a helix-like structure that mimics a native degron [62].
The cooperativity of ternary complex formation represents a critical parameter determining MGD efficacy [56]. Positive cooperativity occurs when binding of the MGD to one protein enhances its affinity for the second protein, leading to more stable ternary complexes and efficient degradation [56]. This cooperativity arises from extensive interfacial contacts between the E3 ligase and target protein, with the MGD stabilizing these interactions through shape complementarity and specific molecular contacts [62] [56]. The resulting protein-protein interface varies considerably across different MGD systems, with some creating extensive contact surfaces (>1500 Ų) that ensure specificity and efficient ubiquitin transfer [62].
Figure 1: Molecular Glue Degrader Mechanism. MGDs bind to E3 ubiquitin ligases, inducing conformational changes that create novel surfaces for target protein recognition, leading to ternary complex formation, ubiquitination, and proteasomal degradation.
Key Experimental Methods for Molecular Glue Research
Target Identification and Validation
The discovery and characterization of molecular glue degraders relies on multidisciplinary approaches that integrate biophysical, biochemical, and cellular techniques. High-throughput screening (HTS) platforms employ various detection methods to identify compounds that induce protein-protein interactions between E3 ligases and potential target proteins [57]. These include protein-fragment complementation assays, fluorescence polarization, and time-resolved fluorescence resonance energy transfer (TR-FRET) [57]. For known protein targets, cellular thermal shift assays (CETSA) can demonstrate compound engagement by measuring protein stabilization against thermal denaturation [63].
Proteomic profiling represents a crucial unbiased approach for identifying novel MGDs and their mechanisms. Techniques like thermal protein profiling (TPP) coupled with mass spectrometry can reveal global changes in protein stability and abundance following MGD treatment [62]. As demonstrated in the discovery of MRT-31619, quantitative proteomics enables researchers to monitor degradation kinetics and specificity across thousands of proteins simultaneously, distinguishing true substrates from nonspecific effects [62]. For target validation, RNA interference and CRISPR-Cas9 screening validate the essentiality of identified E3 ligases in the degradation process [59].
Mechanistic Characterization Techniques
Structural biology methods provide atomic-level insights into MGD mechanisms. Cryogenic electron microscopy (cryo-EM) has proven particularly valuable for visualizing ternary complexes, as evidenced by the structural determination of CRBN homodimers induced by MRT-31619 [62]. This technique can capture conformational flexibility in complexes that might be challenging for crystallography [62]. X-ray crystallography continues to contribute high-resolution structures of MGDs bound to individual proteins, revealing detailed interaction networks as seen in CRBN-IMiD structures [63].
Biophysical assays quantitatively characterize MGD-induced interactions. Surface plasmon resonance (SPR) and isothermal titration calorimetry (ITC) measure binding affinities and thermodynamics [56]. NanoBRET and NanoLuc complementation assays enable real-time monitoring of protein-protein interactions in live cells, as utilized in confirming MRT-31619-induced CRBN homodimerization [62]. These cellular proximity assays provide critical functional validation of ternary complex formation under physiologically relevant conditions.
Biochemical validation of degradation mechanisms typically involves demonstrating UPS dependence through pharmacological inhibition using proteasome inhibitors (e.g., bortezomib) or neddylation inhibitors (e.g., MLN4924) that disrupt cullin-RING ligase activity [62]. Western blotting quantitatively assesses target protein depletion over time, while cycloheximide chase experiments can distinguish reduced synthesis from enhanced degradation [63].
Figure 2: Experimental Workflow for Molecular Glue Degrader Research. A multi-phase approach integrating discovery, validation, and characterization methods identifies and confirms MGD activity and mechanism.
Clinical Examples and Applications
Approved Molecular Glue Therapies
Several molecular glue degraders have received regulatory approval, predominantly in hematologic malignancies. Thalidomide and its analogs lenalidomide and pomalidomide (collectively known as immunomodulatory imide drugs, or IMiDs) were the first clinically approved MGDs, though their mechanism was only elucidated years after their approval [49] [63]. These compounds recruit the E3 ligase CRBN to promote the degradation of transcription factors IKZF1 and IKZF3, leading to complex immunomodulatory and antitumor effects [63]. Their approval for multiple myeloma and other hematologic malignancies established the clinical proof-of-concept for targeted protein degradation [63].
The discovery that IMiDs function as molecular glues emerged from retrospective mechanistic studies, highlighting the serendipitous nature of early MGD development [49] [56]. Structural analyses revealed that these compounds bind the tri-tryptophan pocket of CRBN, creating a neomorphic surface that recognizes specific zinc finger domains in IKZF1 and IKZF3 [63]. This degradation underlies their therapeutic efficacy in multiple myeloma, where loss of IKZF1 and IKZF3 disrupts oncogenic transcriptional programs in malignant plasma cells [63].
Investigational Molecular Glue Degraders
The clinical pipeline for molecular glue degraders has expanded beyond IMiDs, with several innovative candidates entering clinical trials. CFT7455 is a next-generation CRBN-based MGD designed for enhanced potency and specificity against IKZF1/3 in multiple myeloma and non-Hodgkin lymphoma (NCT04756726) [60] [63]. Preclinical data demonstrates superior degradation efficiency and antitumor activity compared to earlier IMiDs [63]. E7820 represents a distinct mechanistic class, recruiting the DCAF15 E3 ligase to degrade the RNA-binding protein RBM39 [60]. This compound is being evaluated in phase II trials for hematologic malignancies (NCT05024994) [60].
Recent discoveries have revealed MGDs with novel mechanisms, such as MRT-31619, which promotes CRBN homodimerization and self-degradation [62]. This "chemical knockout" approach provides a tool for studying CRBN biology and represents a unique degradation strategy. Another emerging candidate, BI-3802, promotes the polymerization and degradation of the oncogenic transcription factor BCL6, demonstrating the structural diversity of MGD mechanisms [60].
Table 1: Clinically Approved Molecular Glue Degraders
| Drug Name | E3 Ligase | Primary Target(s) | Clinical Indications | Key Characteristics |
|---|---|---|---|---|
| Thalidomide | CRBN | IKZF1/IKZF3 | Multiple myeloma, leprosy | First discovered MGD; teratogenic effects |
| Lenalidomide | CRBN | IKZF1/IKZF3, CK1α | Multiple myeloma, MDS | Enhanced potency over thalidomide |
| Pomalidomide | CRBN | IKZF1/IKZF3 | Multiple myeloma | Activity in lenalidomide-resistant disease |
Table 2: Selected Investigational Molecular Glue Degraders in Clinical Development
| Drug Name | E3 Ligase | Primary Target | Clinical Stage | NCT Identifier |
|---|---|---|---|---|
| CFT7455 | CRBN | IKZF1/IKZF3 | Phase I/II | NCT04756726 |
| E7820 | DCAF15 | RBM39 | Phase II | NCT05024994 |
| CC-90009 | CRBN | GSPT1 | Phase I | (Various trials) |
| ARV-471* | CRBN | Estrogen receptor | Phase III | NCT04072952 |
Note: ARV-471 is a PROTAC, not a molecular glue, included for comparison of advanced TPD clinical candidates.
Research Reagent Solutions
Table 3: Essential Research Reagents for Molecular Glue Degrader Studies
| Reagent Category | Specific Examples | Research Applications |
|---|---|---|
| E3 Ligase Ligands | Thalidomide, Lenalidomide, Pomalidomide, CC-885 | Positive controls; CRBN-directed degradation studies |
| Proteasome Inhibitors | Bortezomib, Carfilzomib, MG-132 | Validation of UPS-dependent degradation |
| Neddylation Inhibitors | MLN4924 | Disruption of cullin-RING ligase activity |
| Ubiquitin Assay Reagents | Ubiquitin E1, E2 enzymes, ATP, Fluorogenic ubiquitin substrates | In vitro ubiquitination assays |
| Protein Interaction Assays | HTRF, AlphaScreen, TR-FRET kits | Quantification of ternary complex formation |
| Cellular Degradation Reporters | HaloTag, NanoLuc, GFP-tagged substrates | Real-time monitoring of protein degradation |
| CRISPR/Cas9 Screening Libraries | E3 ligase-focused knockout/activation libraries | Identification of essential E3 ligases |
| Proteomic Analysis Kits | TMT/Isobaric labeling, Protein stability kits | Global profiling of degradation effects |
Molecular glue degraders represent a transformative approach in chemical biology and therapeutic development, harnessing the ubiquitin-proteasome system to eliminate pathogenic proteins [58] [59]. The field has evolved from serendipitous discoveries to increasingly rational design strategies, aided by structural biology insights and sophisticated screening platforms [57] [59]. Current research focuses on expanding the repertoire of E3 ligases beyond the well-characterized CRBN and VHL systems, developing predictive algorithms for ternary complex formation, and addressing challenges such as tissue-specific delivery and resistance mechanisms [57] [64].
The therapeutic potential of MGDs extends far beyond their current applications in hematology-oncology [61] [58]. Their favorable drug-like properties, including lower molecular weight and improved bioavailability compared to PROTACs, position them favorably for central nervous system applications and chronic diseases requiring long-term dosing [57]. As discovery platforms like GLUESEEKER demonstrate the feasibility of systematic MGD identification, the clinical pipeline will likely diversify to include degraders for neurodegenerative disorders, autoimmune conditions, and infectious diseases [59]. The ongoing elucidation of fundamental mechanisms, coupled with advances in rational design, promises to accelerate the development of this innovative therapeutic modality toward its full potential [57] [58].
Proteolysis Targeting Chimeras (PROTACs) represent a revolutionary therapeutic paradigm in targeted protein degradation (TPD), harnessing the body's natural ubiquitin-proteasome system (UPS) to eliminate disease-causing proteins. Unlike traditional small-molecule inhibitors that merely block protein function, PROTACs catalytically degrade target proteins, offering a promising strategy for addressing previously "undruggable" targets and overcoming drug resistance [65] [66]. This whitepaper provides an in-depth analysis of the three most advanced PROTAC candidates currently in Phase III clinical trials, framing their development within the broader context of ubiquitin-proteasome pathway research.
A typical PROTAC molecule is a heterobifunctional structure comprising three components: a ligand that binds the protein of interest, a ligand that recruits an E3 ubiquitin ligase, and a linker connecting the two [55] [67]. This design enables the PROTAC to form a ternary complex, bringing the target protein into proximity with an E3 ubiquitin ligase. This proximity facilitates the transfer of ubiquitin chains onto the target protein, marking it for recognition and degradation by the 26S proteasome [54] [65]. The catalytic nature of PROTACs allows for sub-stoichiometric activity, meaning a single degrader molecule can facilitate the destruction of multiple target protein molecules, providing a significant pharmacological advantage [51].
PROTACs in Phase III Clinical Trials: 2025 Status
As of 2025, the PROTAC clinical landscape has matured significantly, with three candidates advancing to Phase III trials, demonstrating the translational potential of this technology. Vepdegestrant (ARV-471) targets the estrogen receptor for breast cancer, BMS-986365 (CC-94676) targets the androgen receptor for prostate cancer, and BGB-16673 targets Bruton's tyrosine kinase for B-cell malignancies [55] [68]. The following section provides a detailed comparison of these candidates.
Table 1: PROTACs in Phase III Clinical Trials (2025)
| Drug Candidate (Company) | Target | Indication | Key Clinical Trial Findings & Status |
|---|---|---|---|
| Vepdegestrant (ARV-471)(Arvinas/Pfizer) | Estrogen Receptor (ER) | ER+/HER2- Advanced or Metastatic Breast Cancer | VERITAC-2 Phase III Trial: Met primary endpoint in patients with ESR1 mutations, showing statistically significant and clinically meaningful improvement in PFS vs. fulvestrant. Did not reach statistical significance in overall ITT population. NDA submitted; PDUFA date: June 5, 2026 [55] [69]. |
| BMS-986365 (CC-94676)(Bristol Myers Squibb) | Androgen Receptor (AR) | Metastatic Castration-Resistant Prostate Cancer (mCRPC) | Phase I data showed dose-dependent PSA reductions. At 900 mg twice daily, 55% of patients achieved ≥30% PSA decline (PSA30). First AR-targeting PROTAC to reach Phase III [55] [70]. |
| BGB-16673(BeiGene) | Bruton's Tyrosine Kinase (BTK) | Relapsed/Refractory B-cell Malignancies | First BTK-targeting PROTAC to advance to Phase III trials, aiming to overcome resistance to conventional BTK inhibitors [55] [68]. |
Table 2: Mechanistic and Pharmacological Profile of Phase III PROTACs
| Drug Candidate | E3 Ligase Recruited | Mechanistic Highlights | Reported Pharmacological Advantages |
|---|---|---|---|
| Vepdegestrant (ARV-471) | Not Specified in Sources | Novel oral PROTAC ER degrader; designed to degrade wild-type and mutant ER [69]. | Fast Track designation by FDA; potential in patients with ESR1 mutations post-CDK4/6 inhibitor therapy [55] [69]. |
| BMS-986365 | CRL4CRBN [55] | Dual-functioning: acts as a ligand-directed degrader (LDD) and a competitive antagonist of AR [70]. | Degrades both wild-type and clinically relevant mutant AR. Preclinically, ~100x more potent than enzalutamide in suppressing AR-driven transcription [55] [70]. |
| BGB-16673 | Not Specified in Sources | Targets BTK for degradation in B-cell malignancies [55] [68]. | Aims to overcome resistance to traditional BTK inhibitors by removing the BTK protein entirely [68]. |
The Ubiquitin-Proteasome System: Mechanism and PROTAC Hijacking
The Canonical Ubiquitin-Proteasome Pathway
The ubiquitin-proteasome system is the primary pathway for targeted protein degradation in eukaryotic cells, a highly conserved process fundamental to cellular homeostasis [54] [71]. The process occurs through a well-orchestrated enzymatic cascade:
- Activation: A ubiquitin-activating enzyme (E1) activates ubiquitin in an ATP-dependent manner.
- Conjugation: The activated ubiquitin is transferred to a ubiquitin-conjugating enzyme (E2).
- Ligation: A ubiquitin ligase (E3) recognizes a specific substrate and facilitates the transfer of ubiquitin from the E2 to a lysine residue on the target protein.
- Polyubiquitination: Repetition of this process forms a polyubiquitin chain on the substrate.
- Degradation: The polyubiquitinated protein is recognized by the 26S proteasome and degraded into small peptides [54] [67] [65].
E3 ubiquitin ligases provide substrate specificity to this system. Humans possess over 600 E3 ligases, but current PROTAC designs primarily leverage a small subset, most commonly Cereblon (CRBN) and Von Hippel-Lindau (VHL) [54] [67] [65]. The dependency on a limited set of E3 ligases presents both a current limitation and a significant opportunity for future expansion of the PROTAC field [54] [68].
PROTAC-Mediated Hijacking of the UPS
PROTACs are event-driven catalysts that co-opt the UPS by artificially creating a ternary complex (POI-PROTAC-E3 Ligase). The stability and geometry of this ternary complex are critical for efficient ubiquitination and degradation, influenced more by cooperative interactions than just the individual binding affinities of the ligands [65]. The subsequent degradation of the target protein leads to a more sustained and profound pharmacological effect compared to mere inhibition.
Experimental Protocols for PROTAC Development and Validation
The transition of PROTACs from concept to clinic relies on a suite of specialized experimental protocols designed to validate their efficacy, mechanism, and specificity.
In Vitro Degradation and Potency Assays
Objective: To quantify the ability of a PROTAC to degrade the target protein and inhibit cell proliferation in relevant cellular models. Detailed Methodology:
- Cell Culture: Maintain appropriate cell lines (e.g., breast cancer lines for ER degraders, prostate cancer lines for AR degraders, B-cell malignancy lines for BTK degraders) under standard conditions.
- PROTAC Treatment: Treat cells with a concentration gradient of the PROTAC candidate (e.g., ranging from 1 nM to 10 µM) for a predetermined time (typically 16-24 hours). Include controls (vehicle) and positive controls if available.
- Protein Lysate Preparation: Lyse cells using RIPA buffer supplemented with protease and phosphatase inhibitors.
- Western Blot Analysis:
- Separate proteins by SDS-PAGE and transfer to a PVDF membrane.
- Probe with antibodies against the target protein (e.g., anti-AR, anti-ER, anti-BTK).
- Use antibodies against a housekeeping protein (e.g., GAPDH, β-Actin) for normalization.
- Quantify band intensity using densitometry software to determine the percentage of protein degradation relative to control and calculate the half-maximal degradation concentration (DC₅₀).
- Proliferation Assays: In parallel, assess the impact on cell viability using assays like CellTiter-Glo after 72-120 hours of PROTAC treatment to determine the half-maximal inhibitory concentration (IC₅₀) [70].
Ternary Complex Stability and Cooperativity Analysis
Objective: To evaluate the formation and stability of the POI-PROTAC-E3 ligase complex, a critical determinant of PROTAC efficiency. Detailed Methodology:
- Surface Plasmon Resonance (SPR) or Bio-Layer Interferometry (BLI): Immobilize the E3 ligase complex (e.g., VHL/Elongin B/C) or the target protein onto a sensor chip.
- Complex Formation: Pre-incubate the PROTAC with one binding partner (e.g., POI) and then analyze its binding kinetics to the immobilized partner (e.g., E3 ligase).
- Data Analysis: Measure the binding response (RU for SPR, nm shift for BLI). A positive binding response upon injection of the pre-formed binary complex, compared to minimal binding of the individual components, indicates ternary complex formation.
- Cooperativity Calculation: The cooperative factor (α) can be determined by comparing the dissociation constant (Kd) of the ternary complex to the Kd values of the individual binary interactions. An α > 1 indicates positive cooperativity, which is highly desirable for PROTAC function [67] [65].
In Vivo Efficacy Studies
Objective: To evaluate the anti-tumor activity and pharmacodynamics of the PROTAC in animal models of human disease. Detailed Methodology:
- Model Generation: Establish cell line-derived xenograft (CDX) or patient-derived xenograft (PDX) models in immunocompromised mice (e.g., NSG mice). For example, implant AR-positive VCaP prostate cancer cells for AR degraders [70].
- Dosing Regimen: Once tumors reach a predefined volume (e.g., 150-200 mm³), randomize mice into groups (n=8-10) for treatment with vehicle, the PROTAC at various doses, and a standard-of-care control (e.g., enzalutamide for prostate cancer).
- Monitoring: Measure tumor volumes and animal body weights 2-3 times per week. Calculate tumor growth inhibition (TGI).
- Pharmacodynamic Analysis: At the end of the study, harvest tumors and key tissues. Analyze protein lysates by Western blot to confirm target degradation (e.g., AR protein levels) in the tumor tissue [70].
The Scientist's Toolkit: Key Research Reagent Solutions
Advancing PROTAC research requires a specialized set of tools and reagents to dissect their mechanism and efficacy.
Table 3: Essential Research Reagents for PROTAC Development
| Reagent / Tool | Function in PROTAC R&D | Specific Application Example |
|---|---|---|
| Recombinant E3 Ligase Complexes (e.g., VHL, CRBN) | In vitro characterization of binding affinity and ternary complex formation. | Used in SPR/BLI assays to measure PROTAC-E3 binding kinetics and ternary complex stability [67]. |
| Target-Specific Cell Lines (Wild-type & Mutant) | Cellular validation of degradation potency and selectivity. | Using engineered cell lines expressing mutant AR to demonstrate degradation of resistant variants by BMS-986365 [55] [70]. |
| Ubiquitination Assay Kits | Direct measurement of PROTAC-induced ubiquitination of the target protein. | In vitro reconstitution of the ubiquitination cascade with E1, E2, E3, ubiquitin, and the target protein to confirm PROTAC functionality [65]. |
| Proteasome Inhibitors (e.g., MG-132, Bortezomib) | Mechanistic validation of UPS-dependent degradation. | Used as a control in cellular degradation assays; pre-treatment with MG-132 should block PROTAC-induced protein loss, confirming proteasomal dependence [67]. |
| CRISPR/Cas9 Knockout Tools | To confirm E3 ligase specificity and identify resistance mechanisms. | Generating E3 ligase (e.g., CRBN, VHL) knockout cell lines. Loss of degradation in knockout cells confirms the specific E3 pathway utilized by the PROTAC [54]. |
The advancement of ARV-471, BMS-986365, and BGB-16673 into Phase III trials marks a critical inflection point for PROTAC technology, demonstrating its transition from an innovative concept to a viable therapeutic strategy firmly grounded in the principles of the ubiquitin-proteasome system. These clinical-stage assets validate the core hypothesis that targeted protein degradation offers a unique therapeutic advantage, particularly for tackling resistance mechanisms in oncology, as seen with mutant AR and ER [55] [70].
Future development will focus on overcoming current challenges, such as the "hook effect" and limited oral bioavailability [65] [51]. Key frontiers include expanding the repertoire of utilizable E3 ligases beyond CRBN and VHL to achieve tissue specificity and degrade a wider range of targets, and exploring ubiquitin-independent pathways for proteasomal degradation as an alternative mechanism [54]. Furthermore, novel modalities such as activatable PROTACs, nanotechnology-based delivery systems, and antibody-conjugated degraders are being actively researched to improve the pharmacokinetics and selectivity of future degraders [51].
In conclusion, the clinical progress of the PROTAC pipeline underscores the successful hijacking of the ubiquitin-proteasome system for therapeutic purposes. As the field matures, the integration of deeper mechanistic insights with innovative chemical and biological strategies will undoubtedly unlock the full potential of targeted protein degradation, expanding its utility beyond oncology and solidifying its role in the future of drug discovery.
Targeted protein degradation (TPD) has emerged as a transformative therapeutic strategy, moving beyond traditional inhibition to the complete removal of disease-causing proteins. While the ubiquitin-proteasome system (UPS) has been the foundation for pioneering TPD platforms like PROTACs, it inherently limits targets to intracellular proteins [72]. The UPS relies on E1, E2, and E3 enzymes to tag proteins with ubiquitin chains, primarily K48-linked polyubiquitination, marking them for degradation by the 26S proteasome [28] [72]. This pathway is optimal for degrading soluble, cytosolic proteins but cannot access extracellular or membrane-bound proteins, which constitute a significant portion of the proteome and include high-value therapeutic targets such as immune checkpoints, receptor tyrosine kinases, and cytokine receptors [73] [74].
The limitations of the UPS and proteasome have catalyzed the development of novel degrader platforms that hijack alternative cellular machinery, notably the lysosomal degradation pathway. Lysosome-Targeting Chimeras (LYTACs), Antibody-Based PROTACs (AbTACs), and other related technologies represent a paradigm shift by enabling the degradation of proteins previously considered "undruggable" by proteasome-based approaches [75] [74]. These platforms leverage the cell's endogenous lysosomal trafficking receptors, expanding the TPD landscape to encompass extracellular, membrane-bound, and even aggregated proteins, thereby opening new frontiers in drug discovery for oncology, neurodegenerative diseases, and beyond [76] [74]. This review provides an in-depth technical guide to the mechanisms, applications, and experimental methodologies of these next-generation degraders, framed within the context of UPS pathway research.
The lysosome serves as the primary degradative compartment of the cell, capable of breaking down a diverse array of materials, including long-lived proteins, insoluble protein aggregates, and entire organelles [72]. Cargo is delivered to the lysosome through several distinct but interconnected pathways:
- Endocytosis: The process by which cells internalize cell surface proteins, lipids, and extracellular material. Following internalization, some receptors are recycled back to the plasma membrane, while others are sorted into intraluminal vesicles of multivesicular bodies (MVBs) by the Endosomal Sorting Complex Required for Transport (ESCRT) machinery and ultimately degraded upon MVB-lysosome fusion [72].
- Autophagy: An evolutionarily conserved process where cytoplasmic components are sequestered within double-membrane vesicles called autophagosomes, which subsequently fuse with lysosomes for content degradation. This pathway is particularly important for clearing protein aggregates and damaged organelles [74] [72].
- Phagocytosis: A specialized form of endocytosis used by immune cells to engulf large particles such as microbial pathogens [72].
Unlike the proteasome, which requires substrates to be unfolded, the lysosome can degrade large macromolecular complexes and membrane-bound structures, making it an ideal machinery for degrading cell surface proteins when the appropriate targeting mechanisms are engaged [74].
Platform Mechanisms and Technological Profiles
LYTAC (Lysosome-Targeting Chimera)
Mechanism of Action: LYTACs are bifunctional molecules designed to bridge a target protein on the cell surface to a lysosome-trafficking receptor [73]. One moiety, typically an antibody or a high-affinity peptide, binds with high specificity to the protein of interest (POI). The other moiety is a ligand that engages a lysosomal shuttle receptor, such as the cation-independent mannose-6-phosphate receptor (CI-M6PR) or the liver-specific asialoglycoprotein receptor (ASGPR) [75] [77]. The formation of this ternary complex co-opts the receptor's natural endocytic cycle. The entire complex is internalized via clathrin-mediated endocytosis, trafficked through the endosomal system, and ultimately delivered to the lysosome, where the acidic environment and hydrolytic enzymes degrade the target protein. The lysosomal receptor can then be recycled back to the cell membrane [77] [76].
Key Receptors and Technologies:
- CI-M6PR LYTACs: The first-generation LYTACs utilized synthetic glycopeptides containing M6P-mimetics (M6Pn) to engage the ubiquitously expressed CI-M6PR [77] [73]. This platform has been demonstrated to degrade a range of therapeutically relevant targets, including EGFR and PD-L1 [75].
- GalNAc-LYTACs: To achieve tissue-specific degradation, later-generation LYTACs were engineered to engage ASGPR, a receptor highly expressed on hepatocytes [77]. These LYTACs are conjugated to a triantennary N-acetylgalactosamine (tri-GalNAc) ligand, which binds ASGPR with low nanomolar affinity. This not only restricts degradation activity to the liver but also demonstrates superior internalization efficiency compared to CI-M6PR engagers in hepatocyte-derived cell lines [77].
- Pep-TACs: A recent innovation to overcome the synthetic challenges and poor solid tumor penetration of antibody-based LYTACs. Pep-TACs are covalent chimeric peptides that engage the transferrin receptor (TFRC), a highly recyclable lysosomal targeting receptor overexpressed on many tumor cells [76]. A proof-of-concept study designed a Pep-TAC by conjugating a TFRC-targeting peptide (DT7) to a PD-L1-targeting peptide. To enhance binding stability, the PD-L1-binding peptide was modified with a flexible aryl sulfonyl fluoride (k-ASF) group, enabling covalent cross-linking to PD-L1 upon binding. This design led to significant and sustained degradation of PD-L1 in tumor models, and demonstrated an ability to cross the blood-brain barrier [76].
The following diagram illustrates the core mechanism of LYTAC-mediated protein degradation.
LYTAC Mechanism: A LYTAC molecule binds a cell-surface POI and a lysosomal trafficking receptor, triggering internalization and lysosomal degradation of the POI.
AbTAC (Antibody-Based PROTAC)
Mechanism of Action: AbTACs are fully recombinant bispecific antibodies designed to degrade cell-surface proteins [75] [73]. Unlike LYTACs, which use chemical conjugates, AbTACs are genetically encoded. One arm of the AbTAC binds to the target membrane protein, while the other arm engages a specific cell-surface E3 ubiquitin ligase, such as RNF43 [75]. The simultaneous engagement induces the formation of a complex that leads to the internalization of the target protein. Although the precise mechanism is still under investigation, it is believed that the complex is trafficked to the lysosome for degradation [75]. AbTACs are particularly well-suited for targeting membrane proteins, including immune checkpoint proteins like PD-L1, and represent a convergence of biologic therapeutics with targeted protein degradation principles [75].
AUTAC (AUTophagy-Targeting Chimera)
Mechanism of Action: While LYTACs and AbTACs target extracellular and membrane proteins, AUTACs represent a complementary strategy for degrading intracellular targets that are inaccessible to the proteasome, such as protein aggregates and damaged organelles [74]. An AUTAC molecule consists of a target-binding ligand linked to a degradation tag, often a guanine derivative that mimics S-guanylation, a natural post-translational modification associated with cellular clearance during stress [74]. This tag is recognized by the autophagy machinery, leading to the engulfment of the tagged cargo into an autophagosome, which subsequently fuses with the lysosome for degradation. AUTACs have been shown to clear dysfunctional mitochondria (mitophagy) and aggregate-prone proteins implicated in neurodegenerative diseases, thereby expanding the functional scope of TPD [74].
Comparative Analysis of TPD Platforms
The table below provides a structured, quantitative comparison of the key features of LYTACs, AbTACs, and the related AUTAC platform against the established PROTAC technology.
Table 1: Comparative Analysis of Targeted Protein Degradation Platforms
| Technology | Mechanism of Action | Delivery Strategy | Target Type | Key Advantages & Limitations |
|---|---|---|---|---|
| PROTAC [75] [72] | Recruits an intracellular E3 ligase (e.g., VHL, CRBN) to the POI, inducing ubiquitination and proteasomal degradation. | Oral or injectable; good cell permeability. | Intracellular proteins (e.g., BRD4, estrogen receptors). | Advantages: Broad applications in cancer; targets "undruggable" intracellular proteins. Limitations: Limited to cytosolic/nuclear targets; potential resistance. |
| LYTAC [75] [77] [76] | Recruits a lysosomal trafficking receptor (e.g., CI-M6PR, ASGPR, TFRC) to a cell-surface POI, inducing endocytosis and lysosomal degradation. | Intravenous injection; chemical conjugation of ligand to antibody or peptide. | Extracellular and membrane-bound proteins (e.g., EGFR, PD-L1, apolipoproteins). | Advantages: Expands TPD to extracellular space; tissue-specificity possible (e.g., ASGPR for liver). Limitations: Synthesis can be challenging; potential immunogenicity; larger size may limit tissue penetration. |
| AbTAC [75] [73] | A bispecific antibody that binds a cell-surface E3 ligase (e.g., RNF43) and a membrane POI, leading to internalization and lysosomal degradation. | Intravenous injection; recombinant antibody format. | Primarily cell surface proteins (e.g., PD-L1). | Advantages: Fully recombinant; excellent specificity and stability. Limitations: High manufacturing cost; risks of immune responses; limited tissue penetration. |
| AUTAC [74] | Tags the POI with an S-guanylation mimic, recruiting the autophagy machinery for lysosomal degradation. | N/A (Information not specified in search results) | Intracellular proteins, protein aggregates, and organelles (e.g., mitochondria). | Advantages: Degrades large aggregates and organelles; potential for neurodegenerative diseases. Limitations: Degradation is slower than proteasomal pathways; complex trafficking. |
Experimental Protocols and Validation
This section outlines key methodologies for designing and validating novel degrader molecules, with a focus on LYTAC development and mechanism confirmation.
Protocol: Development and Validation of GalNAc-LYTACs
The following workflow, derived from a study on ASGPR-engaging LYTACs, details the steps from synthesis to functional validation [77].
- Synthesis of Tri-GalNAc Ligand: Synthesize the tri-GalNAc-DBCO ligand (e.g., over 8 steps from peracetylated GalNAc and a dendrimer scaffold). The DBCO group enables copper-free "click" chemistry conjugation [77].
- Antibody Functionalization: Introduce azide groups onto the target antibody (e.g., Cetuximab for EGFR) via non-specific labeling chemistries.
- Conjugation: Conjugate the tri-GalNAc-DBCO ligand to the azide-functionalized antibody using strain-promoted azide-alkyne cycloaddition (SPAAC). Monitor the reaction and conjugate formation using native gel electrophoresis, which will show a mobility shift due to increased hydrodynamic size [77].
- Characterization: Determine the average number of tri-GalNAc moieties conjugated per antibody using mass spectrometry (e.g., MALDI-MS) [77].
Functional Validation Assays:
- Internalization Assay: Treat ASGPR-expressing cells (e.g., HEPG2) with the GalNAc-LYTAC and a fluorescently labeled target antibody. Quantify cellular internalization using flow cytometry and confirm subcellular localization (e.g., co-localization with Lysotracker dye) via confocal microscopy [77].
- Degradation Efficiency:
- Cell-Surface Levels: Treat target cells (e.g., HEP3B for EGFR) with the LYTAC. Measure the remaining cell-surface levels of the POI using flow cytometry with an orthogonal, non-competing detection antibody [77].
- Total Protein Levels: Analyze whole cell lysates by Western blot to quantify the reduction in total POI protein levels. A concentration-dependent (e.g., 1-100 nM) and time-dependent (e.g., over 48 hours) degradation should be demonstrated [77].
- Mechanistic Validation:
- Receptor Dependency: Knock down the target receptor (e.g., ASGPR) using siRNA. Ablation of degradation upon receptor knockdown confirms mechanism dependency [77].
- Ligand Competition: Co-incubate the LYTAC with a large excess of free tri-GalNAc ligand. Competitive inhibition of degradation confirms the role of receptor-ligand engagement [77].
- Pathway Inhibition: Treat cells with endo-lysosomal pathway inhibitors such as Bafilomycin A1 (a v-ATPase inhibitor) or Chloroquine. Abrogation of degradation confirms the dependence on endosomal acidification and a functional lysosomal pathway [77].
The workflow for such an experimental process is summarized below.
LYTAC Experimental Workflow: Key steps for developing and validating a novel LYTAC molecule, from synthesis to functional and mechanistic studies.
Researcher's Toolkit: Key Reagents and Assays
Table 2: Essential Research Reagent Solutions for Novel Degrader Development
| Item | Function/Application | Example(s) |
|---|---|---|
| Tri-GalNAc-DBCO [77] | A homogeneous, high-affinity ligand for engaging the liver-specific ASGPR in GalNAc-LYTAC synthesis. | Synthesized in 8 steps from peracetylated GalNAc [77]. |
| M6Pn (Mannose-6-Phosphonate) [75] | A synthetic ligand for engaging the cation-independent mannose-6-phosphate receptor (CI-M6PR) in first-generation LYTACs. | N/A |
| k-ASF (Aryl Sulfonyl Fluoride) [76] | A flexible covalent warhead based on SuFEx chemistry; enables stable, covalent binding to target proteins (e.g., PD-L1) in Pep-TAC platforms. | Modified D-lysine with a long side chain (k-ASF) for improved reaction radius [76]. |
| DT7 Peptide [76] | A D-configuration peptide with high affinity and stability that specifically targets the Transferrin Receptor (TFRC). | Used as the lysosomal shuttle component in TFRC-based Pep-TACs [76]. |
| Pathway Inhibitors | Tool compounds for validating the mechanism of action and the specific degradation pathway utilized. | Bafilomycin A1 (v-ATPase inhibitor), Chloroquine (lysosomal function), MLN4924 (NEDD8 activation inhibitor for Cullin-RING ligases), MG-132 (proteasome inhibitor) [77] [78]. |
| HiBiT System [79] | A quantitative luminescence-based system for measuring real-time changes in target protein levels in live cells, ideal for degrader potency and kinetics studies. | Used for measuring level of target rescue after treatment with control compounds [79]. |
| SPR (Surface Plasmon Resonance) [78] | A biophysical technique used to measure the binding affinity and kinetics between a degrader warhead and its target (e.g., E3 ligase or POI). | Used to characterize binders to novel E3 ligases like KLHDC2 [78]. |
Future Directions and Therapeutic Applications
The expansion of the TPD toolkit is paving the way for novel therapeutic strategies across diverse disease areas. In oncology, LYTACs and AbTACs are being explored to degrade immune checkpoint proteins like PD-L1 directly from the surface of tumor cells, potentially overcoming resistance to simple blockade and reinvigorating anti-tumor immunity [75] [76]. The ability to degrade receptor tyrosine kinases (e.g., EGFR) from the cell membrane offers a more complete inhibition of oncogenic signaling compared to traditional inhibitors [77]. In neurodegenerative diseases like Parkinson's and Alzheimer's, AUTACs hold promise for clearing toxic, aggregated proteins such as α-synuclein and tau, which are refractory to proteasomal degradation [74]. Furthermore, Pep-TACs have demonstrated the ability to cross the blood-brain barrier, opening a potential avenue for treating brain tumors and other CNS disorders [76].
Future innovation will focus on several key areas:
- Expanding the Lysosomal Receptor Toolkit: Discovery and validation of new lysosomal trafficking receptors will enable degradation in specific tissues and cell types, improving therapeutic windows [76].
- Combinatorial Therapies: The synergistic combination of different degrader platforms (e.g., a PROTAC for an intracellular oncoprotein and a LYTAC for a surface receptor) or degraders with other therapeutic agents (e.g., checkpoint inhibitors or chemotherapy) represents a powerful strategy to enhance efficacy and overcome resistance [75].
- Beyond Cereblon and VHL: For all TPD modalities, the heavy reliance on a small subset of E3 ligases (e.g., CRBN, VHL) poses a risk of resistance and limits the scope of actionable targets. Research is actively focused on hijacking novel E3 ligases, such as Ligase X (functioning through CUL1/SKP1 SCF complex) and KLHDC2 (a CUL2 complex member), to develop next-generation degraders with novel mechanisms and potentially improved safety profiles [78].
The advent of LYTACs, AbTACs, AUTACs, and related platforms marks a significant evolution in the field of targeted protein degradation, moving beyond the constraints of the ubiquitin-proteasome system. By co-opting the lysosomal and autophagy pathways, these technologies have vastly expanded the druggable proteome to include extracellular, membrane-bound, and aggregated proteins. As the underlying mechanisms are further elucidated and the toolkit of lysosomal receptors and covalent warheads grows, these novel degraders are poised to unlock new therapeutic frontiers. Their successful integration into the drug development pipeline, potentially in combination with UPS-based approaches, promises to deliver transformative treatments for a wide spectrum of diseases, from cancer to neurodegenerative disorders.
The ubiquitin-proteasome system (UPS) serves as a critical regulatory mechanism for intracellular protein degradation in eukaryotic cells, maintaining protein homeostasis through the precise, ATP-dependent breakdown of ubiquitin-tagged proteins [28]. This system regulates a vast array of cellular processes, including cell cycle progression, transcriptional regulation, apoptosis, and signal transduction [28] [80]. The UPS operates through a coordinated enzymatic cascade: ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3) work sequentially to tag target proteins with ubiquitin molecules, marking them for recognition and degradation by the 26S proteasome [28] [81]. The 26S proteasome itself is a multi-subunit complex comprising a 20S core particle (CP) that contains the proteolytic active sites, capped by one or two 19S regulatory particles (RP) that recognize ubiquitinated substrates, remove ubiquitin chains, and unfold proteins for degradation within the core [28] [82].
In cancer biology, UPS dysregulation represents a fundamental pathogenic mechanism. Malignant cells frequently exhibit aberrant expression of E3 ubiquitin ligases and ubiquitin-binding enzymes, leading to abnormal accumulation of ubiquitinated proteins that drive tumor initiation, progression, and therapeutic resistance [28]. Cancer cells develop a heightened dependence on UPS function to eliminate misfolded proteins resulting from genomic instability and high protein synthesis rates, creating a therapeutic vulnerability that can be exploited pharmacologically [83] [84]. This dependency is particularly pronounced in hematological malignancies like multiple myeloma (MM), where malignant plasma cells produce enormous quantities of immunoglobulins, creating exceptional proteostatic stress that renders them uniquely sensitive to proteasome inhibition [83]. The clinical validation of this approach came with the development of proteasome inhibitors (PIs), which have revolutionized treatment paradigms for MM and mantle cell lymphoma, establishing the UPS as a legitimate target for cancer therapy [28] [85] [83].
Fundamental Mechanisms of the Ubiquitin-Proteasome System
The Ubiquitination Cascade
Protein degradation via the UPS follows a meticulously regulated three-step enzymatic process that tags substrates for proteasomal destruction:
- Step 1: Ubiquitin Activation - An E1 ubiquitin-activating enzyme activates ubiquitin in an ATP-dependent reaction, forming a thioester bond between a cysteine residue in E1's active site and the C-terminal glycine of ubiquitin [28].
- Step 2: Ubiquitin Conjugation - The activated ubiquitin is transferred to a cysteine residue of an E2 ubiquitin-conjugating enzyme via trans-esterification [28] [81].
- Step 3: Ubiquitin Ligation - An E3 ubiquitin ligase facilitates the transfer of ubiquitin from E2 to a lysine residue on the target protein, forming an isopeptide bond [28]. E3 ligases provide substrate specificity, recognizing particular target proteins through specialized binding domains [80].
Following the initial monoubiquitination, additional ubiquitin molecules are attached to previously conjugated ubiquitins, forming polyubiquitin chains. The Lys48-linked polyubiquitin chains primarily signal proteasomal degradation, while other linkage types (e.g., Lys63-linked) regulate non-proteolytic processes including DNA repair, immune signaling, and inflammation [81].
Figure 1: The Ubiquitination Cascade. This process involves sequential action of E1, E2, and E3 enzymes, culminating in polyubiquitination and proteasomal degradation.
The Proteasome: Architecture and Function
The 26S proteasome serves as the executioner of the UPS pathway, with its structure precisely engineered for selective protein degradation:
Figure 2: Proteasome Structure. The 26S proteasome comprises 19S regulatory particles and a 20S core particle.
The 19S regulatory particle performs multiple functions: it recognizes polyubiquitinated protein substrates, removes the ubiquitin tags (deubiquitination) for recycling, unfolds the target protein using ATP-dependent mechanisms, and gates the entry channel to the 20S core particle [28] [82]. The 20S core particle contains three primary proteolytic activities: chymotrypsin-like, trypsin-like, and caspase-like, which cleave after hydrophobic, basic, and acidic amino acid residues, respectively [85] [84]. These collaborative activities degrade proteins into short peptide fragments (typically 3-25 amino acids in length) that are subsequently released into the cytoplasm for further processing by cellular peptidases or for antigen presentation [28].
Current UPS-Targeted Therapeutics in Oncology
Proteasome Inhibitors: Mechanisms and Clinical Applications
Proteasome inhibitors represent the most clinically validated approach to UPS modulation in cancer therapy. These compounds primarily target the catalytic activities of the 20S proteasome core particle, with particular emphasis on the chymotrypsin-like site due to its critical role in processive protein degradation [85].
Table 1: FDA-Approved Proteasome Inhibitors in Clinical Oncology
| Inhibitor | Chemical Class | Binding Mechanism | Primary Cancer Indications | Key Clinical Features |
|---|---|---|---|---|
| Bortezomib | Peptide boronate | Reversible covalent binding to chymotrypsin-like site | Multiple Myeloma (MM), Mantle Cell Lymphoma | First-in-class PI; IV/SC administration; Peripheral neuropathy risk |
| Carfilzomib | Peptide epoxyketone | Irreversible covalent binding to chymotrypsin-like site | Relapsed/Refractory MM | Reduced neurotoxicity; Cardiotoxicity risk |
| Ixazomib | Peptide boronate | Reversible covalent binding to chymotrypsin-like site | Relapsed/Refractory MM | First oral PI; Improved convenience |
The therapeutic efficacy of proteasome inhibitors in multiple myeloma stems from the unique biology of plasma cells, which produce massive amounts of immunoglobulins and consequently experience high proteostatic stress [83]. Proteasome inhibition disrupts the degradation of misfolded proteins, leading to the accumulation of polyubiquitinated proteins and proteotoxic stress that triggers apoptosis via the unfolded protein response (UPR) and ER stress pathways [83] [84]. Additionally, PIs prevent the degradation of pro-apoptotic factors such as NOXA, BIM, and p53, while simultaneously stabilizing tumor suppressors, thereby shifting the cellular balance toward apoptosis [80].
Experimental Methodologies for Proteasome Inhibitor Evaluation
Proteasome Activity Assays
Fluorogenic substrate-based assays represent the gold standard for measuring proteasome activity in response to inhibitor treatment. These assays utilize short peptides conjugated to fluorescent reporters (e.g., AMC, AFC) that emit fluorescence upon proteolytic cleavage:
- Protocol for Chymotrypsin-like Activity Assessment:
- Prepare cell lysates from treated samples or purified 20S proteasomes in assay buffer (50 mM HEPES, pH 7.5, 5 mM EDTA, 0.05% NP-40).
- Add fluorogenic substrate Suc-LLVY-AMC (for chymotrypsin-like activity) at 50-100 μM final concentration.
- Incubate at 37°C for 30-120 minutes protected from light.
- Measure fluorescence (excitation 380 nm, emission 460 nm) using a plate reader.
- Calculate percentage inhibition relative to DMSO-treated controls [85].
Parallel assays should be conducted for caspase-like (Z-LLE-AMC substrate) and trypsin-like (Boc-LRR-AMC substrate) activities to determine inhibitor specificity. For cellular studies, include the non-specific protease inhibitor MG132 as a control to confirm signal specificity.
Assessment of Apoptotic Response
Proteasome inhibitor-induced apoptosis can be quantified through multiple complementary approaches:
Western Blot Analysis of Apoptotic Markers:
- Extract proteins from PI-treated cells at various time points (e.g., 6, 12, 24 hours)
- Probe for cleaved caspase-3, caspase-9, PARP cleavage, and accumulation of polyubiquitinated proteins
- Evaluate Bcl-2 family protein dynamics (stabilization of BIM, NOXA; changes in Mcl-1, Bcl-2) [80]
Flow Cytometric Analysis of Apoptosis:
- Stain cells with Annexin V-FITC and propidium iodide after 24-48 hours of PI treatment
- Quantify percentage of early apoptotic (Annexin V+/PI-) and late apoptotic/necrotic (Annexin V+/PI+) populations
- Compare dose-response curves across different cancer cell types [83]
Resistance Mechanisms and Novel Therapeutic Approaches
Molecular Mechanisms of Resistance to Proteasome Inhibition
Despite the clinical success of PIs, resistance remains a substantial challenge in oncology practice, driven by multiple adaptive cellular responses:
- Proteasome Bounce-Back Response: Inhibited proteasome activity triggers activation of the transcription factor NRF1 (NFE2L1), which is normally retained in the ER membrane and continuously degraded. Proteasome impairment stabilizes NRF1, which undergoes DDI2-mediated cleavage and translocation to the nucleus, where it transcriptionally upregulates all proteasome subunit genes, restoring proteasome capacity [83].
- Activation of Compensatory Degradation Pathways: PI-resistant cells frequently exhibit enhanced autophagy, as evidenced by increased LC3-I to LC3-II conversion and elevated SQSTM1/p62 degradation. This provides an alternative route for protein clearance that bypasses proteasome inhibition [83] [84].
- Reduced Immunoglobulin Synthesis: MM cells may undergo dedifferentiation to a pre-plasma cell state with reduced immunoglobulin production, thereby decreasing the proteotoxic burden and dependency on proteasome function [83].
- Proteasome Subunit Mutations: Mutations in the PSMB5 gene encoding the β5 subunit (primary target of bortezomib and carfilzomib) can alter inhibitor binding affinity, particularly in long-term treated patients [85] [83].
Figure 3: Proteasome Inhibitor Resistance Mechanisms. Multiple adaptive responses contribute to clinical resistance.
Emerging UPS-Targeting Strategies
Immunoproteasome-Selective Inhibitors
The immunoproteasome, containing alternative catalytic subunits (LMP2, LMP7, MECL-1), is expressed in hematopoietic cells and plays specialized roles in antigen presentation. Selective immunoproteasome inhibitors (e.g., KZR-616) demonstrate enhanced efficacy in hematologic malignancies while potentially reducing off-target effects in non-malignant tissues [85]. These agents are being explored for both oncologic and autoimmune applications.
PROTACs and Targeted Protein Degradation
Proteolysis-Targeting Chimeras (PROTACs) represent a revolutionary approach that hijacks the UPS for targeted protein degradation. These heterobifunctional molecules consist of three elements: a warhead that binds the protein of interest (POI), an E3 ligase recruiter, and a linker connecting these domains. PROTACs facilitate the formation of a ternary complex between the POI and E3 ligase, leading to polyubiquitination and proteasomal degradation of the target [28]. This technology enables the degradation of historically "undruggable" targets, including transcription factors and scaffold proteins.
Combination Therapies Overcoming Resistance
Rational combination strategies represent the most promising approach to overcome PI resistance:
- PI + Bcl-2 Inhibitors: Co-targeting the proteasome and anti-apoptotic Bcl-2 family members (e.g., using venetoclax) synergistically induces apoptosis, particularly in double-hit myeloma models [80].
- PI + HDAC Inhibitors: Histone deacetylase inhibitors further disrupt protein homeostasis by inhibiting aggresome formation, creating complementary proteotoxic stress [83].
- PI + NRF1 Pathway Inhibitors: Suppressing the bounce-back response through DDI2 or NRF1 inhibition prevents compensatory proteasome upregulation, enhancing PI sensitivity [83].
The Scientist's Toolkit: Essential Research Reagents and Methodologies
Table 2: Key Research Reagents for UPS Investigation
| Reagent Category | Specific Examples | Research Applications | Key Considerations |
|---|---|---|---|
| Proteasome Inhibitors | MG132, Bortezomib, Carfilzomib, Epoxomicin | Mechanistic studies, apoptosis induction, protein stabilization | Varying specificity profiles; differential cytotoxicity |
| Activity Assay Substrates | Suc-LLVY-AMC, Z-LLE-AMC, Boc-LRR-AMC | Proteasome activity profiling, inhibitor screening | Substrate specificity for different catalytic sites |
| UPS-Related Antibodies | Anti-polyubiquitin, anti-K48 ubiquitin, anti-20S/19S subunits, anti-NRF1 | Western blot, immunohistochemistry, monitoring UPS inhibition | Linkage-specific ubiquitin antibodies available |
| E3 Ligase Modulators | MLN4924 (NEDD8-activating enzyme inhibitor), JNJ-165 | E3 ligase function studies, substrate identification | Impacts multiple cullin-RING ligase family members |
| DUB Inhibitors | b-AP15, PR-619 | Deubiquitinating enzyme function, protein stabilization studies | Varying specificity across DUB families |
| Cell Lines | MM.1S (PI-sensitive), ANBL-6 (PI-resistant), HCT-116 p53+/+ | Resistance mechanism studies, combination screening | Isogenic pairs valuable for specific pathway analysis |
The therapeutic targeting of the ubiquitin-proteasome system has evolved substantially from the initial development of broad-spectrum proteasome inhibitors to increasingly sophisticated approaches including immunoproteasome-selective inhibitors, PROTAC technology, and rational combination strategies. The continued elucidation of resistance mechanisms, particularly the NRF1-mediated bounce-back response and the role of compensatory protein degradation pathways, provides critical insights for next-generation therapeutic development [83].
Future directions in UPS-targeted cancer therapy will likely focus on several key areas: First, the development of tissue-specific and subunit-selective proteasome inhibitors may enhance therapeutic efficacy while reducing off-target toxicities [85]. Second, the advancement of PROTAC technology continues to expand the druggable proteome, enabling targeted degradation of oncoproteins previously considered undruggable [28]. Third, biomarker-driven patient selection strategies are needed to identify tumors with inherent UPS vulnerabilities, potentially through assessment of proteasome workload, immunoglobulin production, or NRF1 pathway activation status [83] [84]. Finally, innovative combination approaches that simultaneously target multiple nodes of the proteostasis network—including the UPS, autophagy, heat shock response, and apoptotic machinery—hold promise for overcoming resistance and improving outcomes across diverse cancer types [80] [84].
As our understanding of UPS biology in malignancy continues to mature, so too will our ability to therapeutically exploit this fundamental cellular pathway for cancer therapy, moving beyond proteasome inhibitors toward increasingly precise and effective UPS modulation strategies.
Emerging Applications in Neurodegenerative and Inflammatory Diseases
The ubiquitin-proteasome system (UPS) represents the primary pathway for targeted intracellular protein degradation in eukaryotic cells, an essential process for maintaining cellular homeostasis. This sophisticated system orchestrates the precise regulation of protein abundance through a hierarchical enzymatic cascade, ultimately directing substrate proteins to the 26S proteasome for degradation [86] [87]. The UPS's fundamental role extends to virtually all cellular processes, including cell cycle progression, signal transduction, stress response, and immune activation [86]. Dysregulation of this system underpins a diverse spectrum of human pathologies, positioning the UPS as a critical frontier for therapeutic intervention. This technical review examines the emerging applications of UPS-targeted strategies in two major disease domains: neurodegenerative disorders and inflammatory/autoimmune conditions. We explore the molecular mechanisms, preclinical evidence, and clinical translation of therapies designed to modulate this sophisticated proteolytic machinery, providing researchers and drug development professionals with a comprehensive framework for understanding this rapidly evolving field.
Molecular Machinery of the Ubiquitin-Proteasome System
The Enzymatic Cascade
The UPS operates through a precise, multi-step enzymatic cascade that covalently tags substrate proteins with ubiquitin molecules, marking them for proteasomal degradation:
- Ubiquitin Activation: The process initiates with an ATP-dependent step where a ubiquitin-activating enzyme (E1) forms a high-energy thioester bond with the C-terminal glycine of ubiquitin. The human genome encodes only one or two E1 enzymes, representing an upstream regulatory node [88].
- Ubiquitin Conjugation: Activated ubiquitin is subsequently transferred to approximately 38 ubiquitin-conjugating enzymes (E2s), which maintain the ubiquitin via a similar thioester linkage [88].
- Ubiquitin Ligation: The final and most specific step involves ubiquitin-protein ligases (E3s), which number in the hundreds. E3s recognize specific substrate proteins and facilitate the transfer of ubiquitin from E2 to a lysine residue on the substrate, forming an isopeptide bond [88] [86]. This hierarchical system (E1→E2→E3) ensures precise targeting of thousands of distinct protein substrates.
Ubiquitin Chain Topologies and Signaling Outcomes
Ubiquitination is not a uniform modification but creates diverse topological signals through different chain structures:
Table: Ubiquitin Chain Linkages and Their Functional Consequences
| Linkage Type | Primary Function | Cellular Processes | Proteasomal Degradation |
|---|---|---|---|
| K48-linked | Primary degradation signal | Cell cycle, stress response | Yes |
| K11-linked | Degradation signal | ER-associated degradation, cell cycle | Yes |
| K63-linked | Non-degradative signaling | DNA repair, inflammation, endocytosis | No |
| Linear (M1-linked) | Inflammatory signaling | NF-κB activation, immunity | No |
| K29-linked | Degradation signal | Protein quality control | Yes |
| K6, K27, K33-linked | Specialized signaling | Mitophagy, trafficking | Context-dependent |
The 26S proteasome recognizes primarily K48- and K11-linked polyubiquitin chains, typically requiring a chain of four or more ubiquitin molecules for efficient substrate recognition [88]. The proteasome itself is a massive multiprotein complex comprising a cylindrical 20S core particle capped by 19S regulatory particles. The 20S core contains three catalytic subunits (β1, β2, β5) with caspase-like, trypsin-like, and chymotrypsin-like activities, respectively, which collectively degrade target proteins into small peptides [88].
Regulatory Components and Specialized Adaptations
The system incorporates several regulatory layers that refine its specificity:
- Deubiquitinating Enzymes (DUBs): Approximately 79 human DUBs counteract ubiquitination by removing ubiquitin chains from substrates, providing a proofreading mechanism that can rescue proteins from degradation or edit ubiquitin signals [88].
- Immunoproteasome: Under inflammatory conditions, cytokine exposure (IFN-γ, TNF-α) induces replacement of standard catalytic subunits with inducible counterparts (β1i/LMP2, β2i/MECL-1, β5i/LMP7), enhancing production of antigenic peptides for MHC class I presentation [86].
Diagram: The Ubiquitin-Proteasome System Enzymatic Cascade. This diagram illustrates the sequential E1-E2-E3 enzymatic cascade that culminates in substrate polyubiquitination and subsequent recognition and degradation by the 26S proteasome. Deubiquitinating enzymes (DUBs) provide regulatory counterbalance by removing ubiquitin chains.
UPS Dysfunction in Neurodegenerative Diseases
Pathogenic Mechanisms in Proteinopathies
Neurodegenerative diseases are characterized by the accumulation of misfolded, aggregation-prone proteins that ultimately form pathogenic inclusions. The UPS plays a central role in both the genesis and potential resolution of these proteinopathies through several interconnected mechanisms:
- Proteostatic Failure: In Alzheimer's disease (Aβ, tau), Parkinson's disease (α-synuclein), and Huntington's disease (mHtt), pathogenic proteins either directly impair proteasome function or overwhelm its degradative capacity, creating a vicious cycle of proteostatic collapse [26] [89]. The initial hypothesis that UPS dysfunction primarily initiates neurodegeneration has evolved to recognize that the UPS remains at least partially functional in many disease models, offering therapeutic opportunities [89].
- Mitochondrial Impairment: UPS components, particularly the E3 ligase Parkin, regulate mitophagy—the selective clearance of damaged mitochondria. Mutations in PARK2 (encoding Parkin) disrupt this quality control mechanism, permitting the accumulation of dysfunctional mitochondria with elevated oxidative stress, a key driver of neuronal vulnerability in Parkinson's disease [26].
- Synaptic Dysfunction: The specialized morphology and extreme polarization of neurons create unique challenges for protein quality control. The UPS regulates presynaptic vesicle release and postsynaptic receptor composition, with demonstrated roles in long-term potentiation and synaptic plasticity [90]. UPS dysfunction at synapses contributes to the early connectivity deficits observed across neurodegenerative conditions.
Genetic Evidence Linking UPS Components to Neurodegeneration
Strong genetic evidence supports the causal role of UPS components in familial neurodegenerative syndromes:
Table: UPS Components Implicated in Neurodegenerative Diseases
| UPS Component | Gene | Neurodegenerative Disease | Molecular Consequence |
|---|---|---|---|
| E3 Ubiquitin Ligase | PARK2 (Parkin) | Early-onset Parkinson's disease | Impaired mitophagy, mitochondrial dysfunction |
| E3 Ubiquitin Ligase | CHIP | Various proteinopathies | Reduced clearance of toxic protein aggregates |
| Deubiquitinase | USP14 | Ataxia, neurodegeneration | Altered synaptic protein turnover |
| Deubiquitinase | UCHL1 | Parkinson's disease | Disrupted ubiquitin recycling |
| E3 Ubiquitin Ligase | UBR1-4 | Neurodevelopmental disorders | Impaired degradation of misfolded proteins |
Mutations in these components disrupt the precise regulation of neuronal proteostasis, leading to the accumulation of neurotoxic proteins and ultimately neuronal death [26]. The vulnerability of post-mitotic neurons to proteostatic stress is particularly acute due to their limited capacity for protein dilution through cell division [90].
UPS Dysregulation in Inflammatory and Autoimmune Diseases
Regulation of Innate and Adaptive Immunity
The UPS exerts sophisticated control over immune signaling pathways at multiple levels, with dysregulation contributing to various inflammatory and autoimmune conditions:
- NF-κB Pathway Regulation: The NF-κB pathway serves as a master regulator of inflammation and is extensively controlled by ubiquitination. The linear ubiquitin chain assembly complex (LUBAC) generates M1-linked ubiquitin chains on NF-κB essential modulator (NEMO), facilitating IKK complex activation and subsequent NF-κB translocation [81]. Conversely, deubiquitinating enzymes including A20, CYLD, and OTULIN negatively regulate this pathway by removing activating ubiquitin signals [81].
- Inflammasome Activation: UPS components regulate the stability and activity of inflammasome complexes, which control the maturation of pro-inflammatory cytokines IL-1β and IL-18. Both E3 ligases and DUBs fine-tune this process, with implications for sterile inflammation and autoinflammatory diseases [87].
- Interferon Pathway Modulation: Type I interferon production in response to viral infection requires non-proteolytic K63-linked ubiquitination of signaling intermediates including TRAF3, TBK1, and IKKε. E3 ligases like TRAF3 and TRIM27, along with DUBs such as USP21 and OTUD5, provide balancing regulation to prevent excessive interferon responses that could drive autoimmunity [87].
Pathogenic Role in Specific Autoimmune Conditions
UPS dysregulation manifests in distinct autoimmune syndromes through cell-type-specific mechanisms:
- Antiphospholipid Syndrome (APS): In APS, UPS dysregulation promotes a prothrombotic phenotype in endothelial cells and activates monocytes/neutrophils, leading to tissue factor expression and inflammatory cytokine production. The UPS mediates critical signaling events downstream of antiphospholipid antibody engagement with cell surface receptors including TLR2/4 and apoER2 [81].
- Myocarditis and Inflammatory Cardiomyopathy: Viral myocarditis progression to dilated cardiomyopathy involves UPS-mediated regulation of viral replication, pattern recognition receptor signaling, and innate/adaptive immune responses. The immunoproteasome is particularly important in shaping the antigenic landscape presented to infiltrating T cells [86].
- Autoimmune Lymphoproliferative Conditions: E3 ligases including Cbl-b, Itch, and GRAIL function as critical gatekeepers of T cell activation thresholds. Their deficiency or dysfunction lowers the threshold for T cell activation, breaking tolerance and promoting autoimmunity [87].
Diagram: UPS Regulation of Inflammatory Signaling. This diagram illustrates how E3 ligases and DUBs reciprocally regulate key inflammatory signaling pathways (particularly NF-κB) downstream of various inflammatory stimuli, including those relevant to antiphospholipid syndrome and viral myocarditis.
Emerging Therapeutic Strategies and Experimental Approaches
Targeted Molecular Interventions
Therapeutic targeting of the UPS has evolved from broad proteasome inhibition to highly specific molecular interventions:
- Proteasome Inhibitors: First-generation proteasome inhibitors like bortezomib have demonstrated efficacy in multiple myeloma by inducing endoplasmic reticulum stress and apoptosis in malignant plasma cells [88] [91]. Dose-limiting peripheral neuropathy remains a significant challenge, driving development of next-generation inhibitors with improved therapeutic indices.
- E3 Ligase Modulators: Tissue-specific E3 ligases represent attractive targets for precision therapeutics. Small molecule inhibitors of E3s including MDM2, IAPs, and others are in clinical development for cancer, with potential applications in inflammatory conditions where these ligases regulate cell survival and inflammatory signaling [88] [87].
- DUB Inhibitors: Selective DUB inhibition offers an alternative approach to modulate ubiquitin signaling. Preclinical studies demonstrate efficacy of DUB inhibitors in models of cancer and inflammation, though achieving sufficient selectivity among the 79 human DUBs remains challenging [88].
- Targeted Protein Degradation (TPD): Bifunctional molecules including PROTACs (PROteolysis TArgeting Chimeras) and molecular glues represent a paradigm shift in UPS therapeutics. These compounds recruit E3 ligases to neosubstrates, enabling targeted degradation of disease-driving proteins that were previously considered "undruggable" [92] [87].
Table: Emerging UPS-Targeted Therapeutic Agents
| Therapeutic Class | Representative Agents | Molecular Target | Development Stage | Primary Indications |
|---|---|---|---|---|
| Proteasome Inhibitors | Bortezomib, Carfilzomib | 20S proteasome core | FDA-approved | Multiple myeloma |
| Immunoproteasome Inhibitors | KZR-504, PR-957 | Immunoproteasome subunits | Preclinical/Phase I | Autoimmune diseases |
| E1 Inhibitors | PYR-41, TAK-243 | UBA1 (E1 enzyme) | Preclinical/Phase I | Cancer, inflammation |
| E3 Ligase Modulators | MLN4924 (Pevonedistat) | NEDD8-activating enzyme | Phase III | Cancer, immune disorders |
| DUB Inhibitors | b-AP15, VLX1570 | Proteasomal DUBs | Preclinical/Phase I | Cancer, autoimmunity |
| PROTACs | ARV-110, ARV-471 | E3 ligase recruiter | Phase II | Cancer, neurodegenerative |
Non-Pharmacological and Adjunctive Approaches
Complementary strategies that modulate UPS function indirectly offer additional therapeutic avenues:
- Natural Compounds: Curcumin and resveratrol demonstrate UPS-modulating activity in preclinical models, potentially contributing to their neuroprotective and anti-inflammatory effects [26].
- Dietary Interventions: Mediterranean and ketogenic diets may enhance proteasome function through metabolic reprogramming, representing low-risk adjunctive approaches for neurodegenerative and inflammatory conditions [26].
- RNA-Based Therapies: microRNAs (e.g., miR-101) and circular RNAs (e.g., circHIPK3) that regulate expression of UPS components represent emerging therapeutic modalities with potential for high specificity [26].
Experimental Methodologies for UPS Research
Ubiquitinome Profiling and Proteomic Approaches
Comprehensive analysis of the ubiquitinome requires specialized methodologies to capture the dynamics and complexity of ubiquitin signaling:
- diGly Antibody Enrichment: Antibodies specifically recognizing the diglycine remnant left on tryptic peptides after ubiquitin modification enable enrichment of ubiquitinated peptides from complex protein digests. This approach forms the foundation for most mass spectrometry-based ubiquitinome studies [92].
- Data-Independent Acquisition (DIA) Mass Spectrometry: DIA methods provide superior reproducibility and quantitative precision compared to data-dependent acquisition for ubiquitinome profiling. Optimized DIA workflows can identify over 40,000 diGly precursors corresponding to more than 7,000 ubiquitinated proteins in a single measurement from proteasome inhibitor-treated cells [92].
- Tandem Ubiquitin Binding Entities (TUBEs): engineered ubiquitin-binding domains with enhanced affinity for polyubiquitin chains enable purification of ubiquitinated proteins from cell lysates, preserving labile ubiquitin modifications that might be lost during standard processing.
Functional Assays for UPS Activity
- Reporter-Based Assays: Fluorescent (GFU) or luciferase-based UPS substrates enable real-time monitoring of proteasome activity in living cells and in vivo. These reporters typically consist of a degradation signal (degron) fused to the reporter protein, producing a fluorescent or luminescent signal that increases when proteasome function is impaired [89].
- Activity-Based Probes: Chemical probes that covalently modify active proteasome subunits allow direct assessment of proteasome composition and activity, including discrimination between standard proteasome and immunoproteasome activities [86].
- In Vitro Degradation Assays: Reconstituted systems with purified 26S proteasomes, ubiquitin, E1, E2, E3 enzymes, and candidate substrates enable detailed mechanistic studies of degradation kinetics and requirements [89].
The Scientist's Toolkit: Essential Research Reagents
Table: Key Research Reagents for UPS Investigation
| Reagent Category | Specific Examples | Research Application | Key Utility |
|---|---|---|---|
| Proteasome Inhibitors | MG132, Bortezomib, Epoxomicin | Inducing ubiquitinated protein accumulation | Experimental UPS impairment |
| E1 Inhibitors | PYR-41, TAK-243 | Blocking ubiquitin activation | Upstream pathway inhibition |
| E2 Inhibitors | NSC697923 | Targeting specific E2 enzymes | Selective pathway disruption |
| E3 Ligase Modulators | MLN4924, JNJ-165 | Inhibiting specific E3 ligase families | Precision targeting of ubiquitination |
| DUB Inhibitors | b-AP15, PR-619 | Pan-deubiquitinase inhibition | Studying deubiquitination effects |
| Ubiquitin-Activating Enzyme | Recombinant UBA1 (E1) | In vitro ubiquitination assays | Reconstitution experiments |
| diGly-Specific Antibodies | K-ε-GG antibodies | Ubiquitinome enrichment | Mass spectrometry sample prep |
| Ubiquitin Binding Domains | TUBEs, UIMs, UBA domains | Affinity purification | Isolation of ubiquitinated proteins |
| Activity-Based Probes | MV151, Ub-AMC | Profiling DUB activities | Enzyme activity measurement |
The ubiquitin-proteasome system represents a remarkably versatile regulatory network whose therapeutic potential is only beginning to be realized. The ongoing paradigm shift from viewing the UPS solely as a pathogenic mechanism to harnessing it as a therapeutic target reflects the growing sophistication of our molecular understanding. Future directions in this field will likely include the development of tissue-specific UPS modulators, enhanced targeted protein degradation technologies with improved pharmacological properties, and combination approaches that address the multifactorial nature of both neurodegenerative and inflammatory diseases. As our tools for investigating and manipulating this system continue to advance, particularly in the areas of structural biology, chemoproteomics, and gene editing, the therapeutic landscape for UPS-targeted interventions will expand substantially. The integration of UPS-focused strategies with complementary approaches targeting protein aggregation, immune dysregulation, and cellular metabolism holds particular promise for addressing these complex and devastating human diseases.
Navigating UPS Complexity: System Crosstalk, Challenges, and Research Solutions
Within eukaryotic cells, protein homeostasis (proteostasis) is governed by two principal degradation systems: the ubiquitin-proteasome system (UPS) and autophagy. Historically viewed as independent pathways, emerging research reveals extensive compensatory crosstalk wherein inhibition of one system prompts functional upregulation of the other. This interplay ensures robustness of cellular protein quality control, a feature critical for survival under proteotoxic stress. This review synthesizes current mechanistic understanding of how autophagy and the UPS functionally compensate for one another, with a particular focus on the role of nuclear proteasomes as a backup for impaired autophagy. The implications for disease pathogenesis, particularly in neurodegenerative contexts such as Huntington's and Parkinson's disease, and for therapeutic development are discussed.
Cellular protein homeostasis, or proteostasis, is a dynamic balance maintained by integrated pathways controlling protein synthesis, folding, trafficking, and degradation [93] [94]. The ubiquitin-proteasome system (UPS) and autophagy are the two major degradation systems responsible for the clearance of the majority of cellular proteins [95] [96]. The UPS primarily degrades short-lived soluble proteins, often marked for destruction by K48-linked ubiquitin chains, in a rapid, processive manner [97] [98]. In contrast, autophagy, particularly macroautophagy, is responsible for the turnover of long-lived proteins, insoluble protein aggregates, and damaged organelles via the lysosome [95] [96]. A common feature of both pathways is the utilization of ubiquitin signaling as a degradation signal, providing a molecular platform for their functional interconnection [98] [99]. The emerging paradigm posits that these systems do not operate in isolation but form a single, collaborative proteostatic network, capable of compensatory activation to maintain cellular viability under stress [93] [100] [98].
Mechanisms of Compensatory Crosstalk
Functional Compensation and Synthetic Lethality
A cornerstone finding in the field is the phenomenon of synthetic lethality observed when both the UPS and autophagy are genetically or pharmacologically inhibited. A seminal 2024 study by Park et al. demonstrated that autophagy-deficient cells (e.g., lacking ATG9A or ATG16L1) exhibit severely compromised viability when proteasome function (e.g., knockout of PSMD7) or nuclear pore complex (NPC) components (e.g., NUP98, NUP133) are concurrently disrupted [93]. This synthetic lethal interaction indicates that proteasome activity and nucleoporin function are essential backup mechanisms that support cell survival when autophagy is compromised.
The mechanistic basis for this compensation involves the nuclear shuttling of cytoplasmic proteins for degradation. In autophagy-impaired cells, cytoplasmic proteins, including misfolded species and autophagy substrates like the A53T mutant of SNCA/α-synuclein (associated with Parkinson's disease), are actively transported into the nucleus in an NPC-dependent manner [93]. Once in the nucleus, these proteins are degraded by nuclear proteasomes. Inhibition of the proteasome in this context leads to marked accumulation of aberrant proteins within the nucleus, while inhibition of the nuclear pore complex prevents their nuclear accumulation, confirming the pathway's sequence [93].
Table 1: Key Experimental Findings on UPS-Autophagy Compensation
| Experimental Finding | System/Method | Interpretation |
|---|---|---|
| Synthetic lethality between ATG9A/ATG16L1 KO and PSMD7/NUP98/NUP133 KO [93] | CRISPR-Cas9 knockout in HeLa cells | Proteasome and nucleoporin activity are essential for survival in autophagy-deficient cells. |
| Increased nuclear translocation of cytoplasmic proteins upon autophagy inhibition [93] | Analysis of bulk proteins, misfolded proteins, and A53T SNCA mutant | Autophagy compromise triggers an alternative degradation pathway via the nucleus. |
| Accumulation of nuclear proteins upon additional proteasome inhibition [93] | Protein quantification and imaging | Nuclear proteasomes are responsible for degrading the translocated cytoplasmic cargo. |
| Impaired compensation in Huntington disease models [93] | Patient-derived iPSC neurons, primary fibroblasts, and mouse striatal neurons | Co-existing defects in autophagy and nuclear pore function (as in HD) create a vulnerable state. |
Molecular Mediators of Crosstalk
The compensatory dialogue between the UPS and autophagy is facilitated by shared molecular players and regulatory nodes.
Ubiquitin as a Universal Degron: Ubiquitin is the common language of both systems. While K48-linked chains are the canonical proteasomal degron, other linkages, including K63, K11, and K29, can also target substrates for proteasomal or autophagic degradation [97] [98]. Furthermore, autophagy receptors like p62/SQSTM1 and NBR1 contain ubiquitin-associated domains (UBA) that bind ubiquitinated cargo and LC3-interacting regions (LIR) that tether them to the growing autophagosome, thereby enabling selective autophagy of ubiquitinated substrates [98].
HDAC6 in Aggresome Clearance: The histone deacetylase HDAC6 is a key sensor of proteasome impairment. It binds ubiquitinated proteins and, together with dynein, facilitates their transport along microtubules to form an aggresome [94] [95]. The aggresome is then targeted for clearance by autophagy, providing a compensatory route for aggregated proteins that resist proteasomal degradation [94] [95].
The EI24 Bridge: The transmembrane autophagy-related protein EI24 (also known as PIG8) has been identified as a bridging molecule that regulates the stability of several RING-domain E3 ubiquitin ligases, thereby directly linking autophagic activity to the regulation of the UPS [101].
Transcription Factor NRF1: Upon severe proteasome impairment, the transcription factor NRF1 is activated and upregulates the expression of both proteasome subunits and p62, thereby coordinately enhancing the capacity of both degradation arms [99].
The following diagram illustrates the core compensatory mechanism identified in recent research, where nuclear proteasomes serve as a backup for impaired autophagy.
Experimental Evidence and Methodologies
A Workflow for Identifying Compensatory Pathways
The discovery of the nuclear proteasome backup pathway exemplifies a multi-faceted experimental approach. The following workflow, based on Park et al. (2024), outlines key steps for investigating compensatory mechanisms between autophagy and the UPS [93].
Detailed Experimental Protocols
Protocol 1: Genetic Interaction Screen for Synthetic Lethality (adapted from [93])
- Objective: To identify genes that are essential for viability in autophagy-deficient backgrounds.
- Procedure:
- Cell Line Generation: Establish isogenic autophagy-competent and autophagy-deficient cell lines (e.g., ATG9A or ATG16L1 knockout in HeLa cells) using CRISPR-Cas9.
- Candidate Gene Knockout: Transduce the above cell lines with a library of gRNAs targeting a curated list of candidate genes (e.g., proteasome subunits, nuclear pore components).
- Viability Assessment: Monitor cell viability and proliferation over several days using assays like Cell Titer-Glo.
- Data Analysis: Identify gRNAs that are depleted specifically in the autophagy-deficient lines, indicating a synthetic lethal interaction. Key hits from the Park et al. study included proteasome subunit gene PSMD7 and nucleoporin genes NUP98 and NUP133.
Protocol 2: Assessing Nuclear-Cytoplasmic Shuttling of Autophagy Substrates
- Objective: To validate the translocation of cytoplasmic proteins to the nucleus upon autophagy inhibition.
- Procedure:
- Treatment: Treat cells with autophagy inhibitors (e.g., chloroquine to block lysosomal degradation or siRNA against core ATG genes).
- Fractionation & Imaging: Perform cellular fractionation to separate nuclear and cytoplasmic components, followed by immunoblotting for candidate proteins (e.g., mutant SNCA, bulk misfolded proteins). Alternatively, use live-cell imaging of fluorescently tagged proteins (e.g., GFP-A53T SNCA).
- Pharmacological/Gentic Perturbation: Co-inhibit the proteasome (e.g., with MG132) to observe nuclear accumulation, or inhibit the nuclear pore complex (e.g., with exportin inhibitors or siRNA against NUP98/NUP133) to observe a block in nuclear accumulation.
Table 2: The Scientist's Toolkit: Key Research Reagents
| Reagent / Tool | Function / Application | Example Use in Context |
|---|---|---|
| CRISPR-Cas9 KO Systems | Targeted gene knockout to establish genetic models. | Generation of ATG9A/ATG16L1 null cells to study autophagy deficiency [93]. |
| Proteasome Inhibitors (e.g., MG132, Bortezomib) | Pharmacological inhibition of proteasome activity. | Testing synthetic lethality with autophagy defects; confirming proteasome's role in degrading nuclear cargo [93] [94]. |
| Autophagy Inhibitors (e.g., Chloroquine, 3-MA) | Pharmacological blockade of autophagic flux at various stages. | Inducing autophagy compromise to trigger compensatory pathways [93] [102]. |
| Surrogate UPS Substrates (e.g., GFPdgn, GFPu) | Fluorescent reporters for monitoring UPS function in live cells. | Measuring UPS capacity and its functional coupling to ubiquitination under proteotoxic stress [94]. |
| LC3-II Antibodies & Turnover Assays | Marker for autophagosome number and autophagic flux. | Quantifying autophagy induction or blockade in response to UPS impairment [95]. |
| HDAC6 Inhibitors (e.g., Tubastatin A) | Selective inhibition of HDAC6 activity. | Probing the role of the aggresome pathway in clearing ubiquitinated aggregates when the UPS is impaired [94] [95]. |
Implications for Disease and Therapeutics
The failure of compensatory mechanisms between autophagy and the UPS is increasingly implicated in pathogenesis, especially in neurodegenerative diseases.
Huntington's Disease (HD): The vulnerability of proteostasis in HD is a prime example. Research shows that in HD models (e.g., patient-derived neurons, HTT knock-in cells), both autophagy and nuclear pore complex function are compromised [93]. This dual defect prevents the activation of the nuclear proteasome backup pathway. Consequently, HD cells are exquisitely sensitive to even mild perturbations of the remaining proteasomal activity in the cytosol, as they cannot effectively shunt cargo to the nucleus for degradation [93]. This creates a "perfect storm" of proteostatic failure.
Therapeutic Strategies: Understanding this crosstalk opens new therapeutic avenues. Strategies aimed at enhancing the capacity of the compensatory pathway could be beneficial. For instance, in conditions where autophagy is deficient, boosting nuclear proteasome activity or facilitating nuclear import of toxic cytoplasmic proteins could be explored. Conversely, in diseases linked to proteasome dysfunction, pharmacological induction of autophagy via HDAC6 inhibitors or mTOR modulators may help clear accumulating aggregates [94] [97]. The critical consideration is that therapeutic modulation must be context-dependent, as the effectiveness of targeting one pathway depends on the functional integrity of the other.
The intricate interplay between autophagy and the UPS represents a fundamental aspect of the cellular proteostasis network. The discovery of mechanisms such as the nuclear proteasome backup pathway underscores the remarkable adaptability of this network in responding to functional deficits in one degradation arm. This crosstalk, mediated by shared signals like ubiquitin and specialized adaptors, ensures a robust defense against proteotoxic stress. Its failure is a key contributor to the pathology of several age-related and neurodegenerative diseases. Future research leveraging integrative omics and sophisticated disease models will continue to unravel the complexity of this interplay, paving the way for novel therapeutic strategies that harness the compensatory potential of the proteostasis network to treat human disease.
Nuclear Proteasomes as Autophagy Backup in Proteostasis
Protein homeostasis (proteostasis) is fundamental to cellular health, governed primarily by the ubiquitin-proteasome system (UPS) and autophagy. Emerging research reveals these are not isolated pathways but function as an interconnected network. Recent studies demonstrate that under autophagy-deficient conditions, cytoplasmic proteins are transported to the nucleus for degradation by nuclear proteasomes. This compensatory mechanism ensures proteostasis maintenance when one system is compromised. This whitepaper examines the mechanistic basis of this backup system, its implications for neurodegenerative diseases and cancer, and its potential as a therapeutic target within the broader context of ubiquitin-proteasome pathway research.
Cellular protein homeostasis (proteostasis) represents the dynamic balance between protein synthesis, folding, trafficking, and degradation, essential for proper cellular function and survival [103] [104]. The proteostasis network comprises highly conserved pathways including the ubiquitin-proteasome system (UPS), autophagy, the unfolded protein response, and stress response pathways [104]. The UPS serves as the primary degradation route for short-lived, misfolded, and regulated proteins, degrading up to 80-90% of cellular proteins [105] [106]. Autophagy, particularly macroautophagy, primarily degrades long-lived proteins, aggregated proteins, and damaged organelles [107] [106]. While historically investigated as separate systems, recent advances reveal extensive cross-talk and functional coordination between these degradation pathways, highlighting the robustness of cellular proteostasis maintenance [103] [106] [108].
The Ubiquitin-Proteasome System: Core Mechanisms
Biochemical Cascade of Ubiquitination
The UPS employs a sophisticated enzymatic cascade to target proteins for degradation:
- Activation: Ubiquitin is activated by E1 ubiquitin-activating enzyme in an ATP-dependent manner [105] [106].
- Conjugation: Activated ubiquitin is transferred to E2 ubiquitin-conjugating enzyme [105] [106].
- Ligation: E3 ubiquitin ligase recognizes specific substrates and facilitates ubiquitin transfer from E2 to the target protein [105] [106].
- Polyubiquitination: Repeated cycles add polyubiquitin chains, typically via lysine-48 (K48) linkages, serving as the primary proteasomal degradation signal [106] [108].
The Proteasome Complex
The 26S proteasome comprises:
- 20S Core Particle: Barrel-shaped complex with three catalytic activities: chymotrypsin-like (CT-L), trypsin-like (T-L), and caspase-like (C-L) [108].
- 19S Regulatory Particle: Recognizes ubiquitinated substrates, removes ubiquitin chains, unfolds proteins, and translocates them into the 20S core [108].
Table 1: Major Ubiquitin Chain Linkages and Their Primary Functions
| Linkage Type | Primary Function | Recognition Features |
|---|---|---|
| K48 | Canonical proteasomal degradation signal | High affinity for proteasomal recognition |
| K63 | Endocytosis, DNA repair, kinase activation; autophagy signal | Preferred by autophagy receptors like p62 |
| K11 | Cell cycle regulation, ER-associated degradation | Recognized by specific proteasomal receptors |
| K29 | Proteasomal degradation (alternative pathway) | Less characterized degradation signal |
| K33 | Non-degradative functions | Immune signaling, T-cell regulation |
| K6 | DNA damage response, mitophagy | Parkin-mediated mitophagy |
| Linear | NF-κB pathway activation | Generated by LUBAC complex |
Autophagy: Complementary Degradation Pathway
Autophagy Machinery and Process
Autophagy proceeds through distinct stages:
- Initiation: ULK1 complex activation under stress conditions (nutrient deprivation, oxidative stress) [107] [109].
- Phagophore Formation: Double-membrane structure nucleation requiring Class III PI3K complex (VPS34, Beclin-1) [109].
- Elongation and Cargo Sequestration: LC3/GABARAP conjugation systems mediate autophagosome expansion and selective cargo recruitment via adaptors like p62 [109] [108].
- Fusion and Degradation: Autophagosomes fuse with lysosomes forming autolysosomes where cargo is degraded and recycled [107] [109].
Selective Autophagy and Cargo Recognition
Autophagy adaptor proteins enable selective degradation:
- p62/SQSTM1: Multidomain protein containing PB1, ZZ, LIR, and UBA domains that bridge ubiquitinated cargo to LC3 on autophagosomes [108].
- NBR1: Shares architectural similarity with p62 and collaborates in aggregate clearance [108].
- Optineurin, NDP52: Additional receptors for specialized selective autophagy processes [108].
Nuclear Proteasomes as Autophagy Backup: Mechanistic Insights
Synthetic Lethality Screens Reveal Functional Interconnection
Genetic interaction studies demonstrate compensatory relationships between degradation systems:
- Synthetic Lethality: Loss of both autophagy (ATG16L1 or ATG9) and proteasome components (PSMD7) or nuclear pore complex (NUP98, NUP133) causes synergistic viability defects [110].
- Conserved Mechanism: These genetic interactions are evolutionarily conserved from yeast to human cells, indicating fundamental biological importance [110].
- Vulnerability in Disease: Cells with compromised autophagy and impaired nucleocytoplasmic transport (as in Huntington's disease) show heightened sensitivity to proteostasis perturbation [103] [110].
Nucleocytoplasmic Transport During Autophagy Compromise
Mechanistic studies reveal redirected protein flux when autophagy is impaired:
- Altered Localization: Autophagy-deficient cells show increased nuclear accumulation of cytoplasmic proteins like A53T α-synuclein, particularly under proteasome inhibition [110].
- Enhanced Nuclear Import: Fluorescence recovery after photobleaching (FRAP) demonstrates accelerated nuclear transport of autophagy substrates in ATG16L1-null cells [110].
- Bulk Protein Redirection: Metabolic labeling with AHA (L-azidohomoalanine) confirms increased nuclear localization of newly synthesized proteins upon autophagy inhibition [110].
Experimental Evidence for Compensatory Degradation
Key findings establishing the backup mechanism:
- Nuclear Aggresome Formation: Proteasome inhibition in autophagy-null cells promotes nuclear accumulation of ubiquitinated protein aggregates [110].
- Importin-Dependent Transport: Nuclear import of autophagy substrates like α-synuclein depends on importin α, inhibited by ivermectin treatment [110].
- Proteasome Dependency: The viability of autophagy-deficient cells relies on functional nuclear proteasomes and intact nucleocytoplasmic transport [103] [110].
Figure 1: Proteostasis Network and Nuclear Compensation Mechanism. Under normal conditions (top), the ubiquitin-proteasome system and autophagy maintain protein homeostasis. During autophagy impairment (bottom), cytoplasmic proteins accumulate and are redirected to the nucleus via importin-dependent transport for degradation by nuclear proteasomes.
Quantitative Experimental Data
Table 2: Key Quantitative Findings from Genetic and Pharmacological Studies
| Experimental Manipulation | Cell Model | Key Measured Outcome | Quantitative Result |
|---|---|---|---|
| PSMD7 knockout + Autophagy deficiency | ATG16L1-/-, ATG9-/- HeLa | Cell survival reduction | Synthetic lethality ( synergistic viability loss) |
| NUP98/NUP133 knockout + Autophagy deficiency | ATG16L1-/-, ATG9-/- HeLa | Cell survival reduction | Synthetic lethality ( synergistic viability loss) |
| Proteasome inhibition (MG132, bortezomib) + Autophagy deficiency | ATG16L1-/- vs WT | Cell death enhancement | Significant increase in autophagy-null vs WT |
| Nuclear import inhibition (ivermectin) + Autophagy inhibition (SBI-0206965) | Wild-type cells | Cell death induction | Dramatic increase vs single treatments |
| Proteasome inhibition + Autophagy deficiency | ATG16L1-/- vs WT | Nuclear A53T α-synuclein accumulation | Significant increase in autophagy-null cells |
| Autophagy inhibition (SBI-0206965) | Wild-type cells | Nuclear AHA-labeled proteins | Increased nuclear intensity |
Detailed Experimental Protocols
Genetic Interaction Screening Protocol
Objective: Identify synthetic lethal interactions between autophagy and proteasome/nuclear pore components [110].
Methodology:
- Cell Line Generation:
- Create isogenic ATG16L1 knockout (ATG16-), ATG9 knockout (ATG9-), and corresponding wild-type (ATG16+, ATG9+) HeLa cells using CRISPR/Cas9.
- Validate autophagy deficiency by LC3-I/II conversion and p62 accumulation assays.
CRISPR Screening:
- Target 30 human orthologues of yeast genes with synthetic lethal interactions with core autophagy genes.
- Use at least two distinct guide RNAs per gene to minimize off-target effects.
- Transduce autophagy-null and wild-type cells with lentiviral CRISPR constructs.
Viability Assessment:
- Monitor cell numbers over time using fluorescence-activated cell sorting (FACS)-based assays.
- Calculate relative survival compared to wild-type controls.
- Validate hits using siRNA approaches as orthogonal validation.
Hit Validation:
- Select candidates showing consistent synthetic lethality with multiple guides in both ATG16L1-null and ATG9-null backgrounds.
- Confirm with complementary approaches (pharmacological inhibition, siRNA).
Protein Localization and Transport Assays
Objective: Quantify nuclear translocation of cytoplasmic proteins during autophagy compromise [110].
Methodology:
- Fluorescence Recovery After Photobleaching (FRAP):
- Express GFP-tagged autophagy substrates (GFP-A53T α-synuclein) in ATG16L1-null and wild-type cells.
- Photobleach nuclear GFP signal using high-intensity laser.
- Monitor fluorescence recovery every 30 seconds for 15 minutes.
- Calculate recovery halftime and mobile fraction.
- Normalize to cytoplasmic fluorescence in unbleached regions.
Subcellular Fractionation and Immunoblotting:
- Separate nuclear and cytoplasmic fractions using differential centrifugation.
- Treat cells with proteasome inhibitors (MG132, 10μM, 6 hours) or vehicle control.
- Quantify protein distribution by Western blotting using nuclear (lamin A/C) and cytoplasmic (GAPDH) markers.
- Calculate nuclear-to-cytoplasmic ratios for proteins of interest.
Metabolic Labeling and Click Chemistry:
- Starve cells of methionine for 1 hour in DMEM without methionine.
- Label newly synthesized proteins with AHA (L-azidohomoalanine, 50μM) for 2 hours.
- Fix cells and perform copper-catalyzed azide-alkyne cycloaddition with fluorescent azides.
- Quantify nuclear vs. cytoplasmic fluorescence intensity by confocal microscopy.
Aggregate Formation and Clearance Assays
Objective: Monitor protein aggregation and clearance under combinatorial stress [110].
Methodology:
- Proteostat Aggresome Detection:
- Treat ATG16L1-null and wild-type cells with proteasome inhibitors (MG132, 5μM, 12 hours).
- Stain with Proteostat dye (1:2000 dilution) according to manufacturer's protocol.
- Counterstain nuclei with Hoechst 33342.
- Quantify nuclear and cytoplasmic aggresomes by high-content imaging.
- Time-Course Combination Treatments:
- Treat cells with autophagy inhibitors (SBI-0206965, 10μM), proteasome inhibitors (bortezomib, 100nM), and nuclear import inhibitors (importazole, 25μM) individually and in combination.
- Monitor cell viability every 12 hours using real-time cell analysis or MTT assays.
- Assess cell death by annexin V/propidium iodide staining and flow cytometry.
Figure 2: Experimental Workflow for Studying Nuclear Proteasome Backup Mechanism. The comprehensive approach combines genetic screening with mechanistic validation to establish functional interconnection between degradation pathways.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Tools for Investigating Nuclear Proteasome-Autophagy Crosstalk
| Reagent/Category | Specific Examples | Primary Function/Application | Key Experimental Use |
|---|---|---|---|
| Autophagy Modulators | SBI-0206965 (ULK1 inhibitor), Chloroquine, Bafilomycin A1 | Inhibit autophagy at distinct steps | ULK1 inhibition, lysosomal acidification blockade |
| Proteasome Inhibitors | MG132, Bortezomib, Carfilzomib | Reversible/irreversible proteasome inhibition | Induce proteostatic stress, measure compensatory mechanisms |
| Nuclear Transport Inhibitors | Ivermectin (importin α/β), Importazole (importin β) | Block nucleocytoplasmic transport | Test nuclear import dependency of compensation |
| Genetic Tools | CRISPR/Cas9 (ATG16L1, ATG9), siRNA (NUP98, NUP133) | Targeted gene knockout/knockdown | Establish synthetic lethality, validate genetic interactions |
| Tracking and Labeling | AHA, GFP-tagged substrates, Proteostat dye | Monitor protein synthesis, localization, aggregation | Visualize protein flux, aggregate formation, compartmentalization |
| Cell Lines | ATG16L1-/-, ATG9-/- HeLa, Huntington's disease models | Autophagy-deficient backgrounds | Study compensation in genetic deficiency contexts |
| Analytical Methods | FRAP, Subcellular fractionation, FACS viability assays | Quantitative measurement of protein dynamics | Measure transport kinetics, compartment-specific degradation |
Implications for Disease and Therapeutics
Neurodegenerative Diseases
The nuclear proteasome backup system has particular relevance for neurodegenerative disorders:
- Huntington's Disease: Both autophagy and nucleocytoplasmic transport are impaired, explaining enhanced vulnerability to proteostatic stress [103] [110].
- Parkinson's Disease: α-synuclein nuclear translocation observed during autophagy compromise may contribute to pathology [110].
- Therapeutic Opportunity: Enhancing nuclear proteasome function or facilitating cytoplasmic-nuclear transport could compensate for autophagy defects [103].
Cancer and Therapeutic Resistance
Dual targeting of degradation pathways represents emerging anticancer strategy:
- Synthetic Lethality Applications: Simultaneous inhibition of autophagy and proteasome/nuclear transport selectively targets cancer cells [107] [110].
- Pancreatic Cancer Context: Autophagy plays dual roles in PDAC—tumor-suppressive early and pro-survival later—influencing therapeutic approach [111].
- Combination Therapy Rationale: Proteasome inhibitors with autophagy blockers show enhanced cytotoxicity in preclinical models [107] [110].
Pharmacological Considerations
Therapeutic targeting requires precise pathway modulation:
- Sequential Inhibition: Autophagy inhibition followed by proteasome targeting maximizes synthetic lethality [110].
- Tissue-Specific Effects: Proteostasis network components show tissue-specific expression and function, impacting therapeutic index [104].
- Compensatory Activation: Inhibition of one degradation pathway may upregulate the other, requiring monitoring of adaptive responses [103] [106].
The emerging paradigm of nuclear proteasomes serving as backup for autophagy underscores the remarkable plasticity of cellular proteostasis networks. This compensatory mechanism represents a fundamental adaptive response that maintains protein homeostasis when primary degradation pathways are compromised. From a therapeutic perspective, this interconnection offers both challenges and opportunities—while complicating single-pathway targeting, it reveals novel synthetic lethal approaches for selective cell elimination, particularly relevant in cancer and neurodegenerative diseases. Future research should focus on elucidating the precise signals that trigger nuclear redistribution of cytoplasmic proteins, identifying additional components of this backup system, and developing strategies to therapeutically modulate this compensatory pathway for disease treatment.
Addressing Specificity Challenges in E3 Ligase Targeting
The ubiquitin-proteasome system (UPS) represents one of the most sophisticated and selective protein degradation pathways in eukaryotic cells, with E3 ubiquitin ligases serving as the central determinants of specificity. These enzymes facilitate the final step in the ubiquitination cascade, recognizing specific substrate proteins and mediating the transfer of ubiquitin from E2 conjugating enzymes to target substrates [4]. The human genome encodes over 600 E3 ligases, yet current targeted protein degradation (TPD) approaches, particularly proteolysis-targeting chimeras (PROTACs), heavily rely on only a small fraction of these enzymes, primarily von Hippel-Lindau tumor suppressor (VHL) and Cereblon (CRBN) [112] [113]. This limited utilization creates significant specificity challenges and therapeutic constraints, including potential on-target toxicities in healthy tissues, acquired resistance mechanisms, and restricted substrate targeting capabilities [112] [3].
The specificity challenge extends beyond mere E3 ligase selection to encompass complex regulatory mechanisms within the UPS. Recent research has revealed that E3 ligases exhibit remarkable diversity in their structural classifications, activation mechanisms, and substrate recognition patterns [4] [3]. Furthermore, the UPS incorporates multiple layers of regulation, including crosstalk between ubiquitin-dependent and ubiquitin-independent proteasomal degradation pathways, specialized proteasome isoforms such as the immunoproteasome (i20S) and constitutive proteasome (c20S), and context-dependent activity modulation by diverse proteasome activators and inhibitors [114]. Understanding these intricate regulatory networks is essential for developing next-generation TPD strategies with enhanced specificity and therapeutic windows.
E3 Ligase Diversity and Current Utilization Landscape
Structural and Functional Classification of E3 Ligases
E3 ubiquitin ligases demonstrate remarkable structural diversity, which directly influences their substrate recognition patterns and catalytic mechanisms. The major E3 ligase families include:
- RING-finger Type E3 Ligases: Characterized by a Really Interesting New Gene (RING) domain that directly binds E2 conjugating enzymes and facilitates ubiquitin transfer without forming an E3-ubiquitin thioester intermediate. This largest E3 family includes both monomeric forms (MDM2, TRAF6) and multi-subunit complexes like cullin-RING ligases (CRLs) [4].
- HECT Type E3 Ligases: Contain a Homologous to E6AP C-Terminus (HECT) domain that forms a thioester intermediate with ubiquitin before transferring it to substrates. This family includes the Nedd4 subfamily (with WW and C2 domains), HERC subfamily (characterized by RCC-like domains), and other HECT ligases such as E6AP and HUWE1 [4].
- RBR Type E3 Ligases: Feature a RING-between-RING architecture that combines aspects of both RING and HECT mechanisms, functioning as RING-HECT hybrids [3].
- U-box Type E3 Ligases: Utilize a U-box domain that is structurally similar to RING domains but stabilized by salt bridges rather than zinc coordination [4].
Table 1: Major E3 Ligase Classes and Their Characteristics
| E3 Class | Catalytic Mechanism | Representative Members | Key Structural Features |
|---|---|---|---|
| RING-finger | Direct transfer from E2 to substrate | MDM2, BRCA1, CBL | RING domain, often multi-subunit complexes |
| HECT | E3-ubiquitin thioester intermediate | NEDD4, HERC, HUWE1 | HECT domain, substrate-binding N-terminal domains |
| RBR | Hybrid RING-HECT mechanism | Parkin, HOIP | RING1, in-between-RING, RING2 domains |
| U-box | RING-like without metal coordination | CHIP, UFD2 | U-box domain, tetratricopeptide repeats |
Quantitative Analysis of E3 Ligase Utilization in TPD
Despite the extensive repertoire of E3 ligases in the human genome, current TPD platforms remain heavily dependent on a minimal subset of well-characterized ligases. Systematic analysis reveals that of 1,075 unique E3 ligases compiled from major databases including Ge et al., UbiHub, and UbiBrowser2.0, only 12 (approximately 1.1%) have been successfully co-opted for PROTAC design [112]. Even more strikingly, clinical-stage PROTACs exclusively recruit either VHL or CRBN, highlighting a critical limitation in current TPD approaches [112]. This constrained utilization creates significant bottlenecks in therapeutic development, particularly given that 275 E3 ligases (25.6%) have confidence scores of 5 or 6 (on a 1-6 scale), indicating sufficient characterization and validation for potential inclusion in TPD strategies [112].
Table 2: Experimentally Validated E3 Ligases with High Potential for TPD Applications
| E3 Ligase | Confidence Score | Known Substrates | Current TPD Status | Therapeutic Potential |
|---|---|---|---|---|
| VHL | 6 | HIF-1α | Clinically validated | Oncology |
| CRBN | 6 | IKZF1/3 | Clinically validated | Oncology, Immunology |
| MDM2 | 5 | p53 | Preclinical development | Oncology |
| DCAF16 | 5 | Undefined | Experimental | Oncology |
| KEAP1 | 5 | NRF2 | Experimental | Oncology, Neurodegeneration |
| RNF4 | 5 | Multiple oncoproteins | Experimental | Oncology |
| HUWE1 | 5 | MCL1, MFN1 | Not yet co-opted | Oncology |
| FBXO7 | 5 | Mitofusin 1 | Not yet co-opted | Neurodegeneration |
Methodologies for Systematic E3 Ligase Characterization and Validation
Multi-Parameter Assessment Framework for E3 Ligase Prioritization
Comprehensive characterization of E3 ligases requires a systematic framework evaluating multiple essential parameters that influence their suitability for TPD applications. Recent research has established a seven-dimensional assessment strategy that integrates diverse experimental and computational approaches [112]:
- Chemical Ligandability: Evaluation of existing small-molecule binders, drugs, or covalent fragments that can serve as starting points for degrader development. Current data indicates that 686 E3 ligases (63.8%) interact with known ligands from at least one category of ligand sources, with 127 (11.8%) having evidence of targeting by approved or investigational drugs [112].
- Expression Patterns: Analysis of E3 ligase expression across normal and diseased tissues at both bulk and single-cell resolution to identify tissue-restricted expression profiles that could enable selective targeting.
- Protein-Protein Interactions (PPI): Mapping of endogenous E3-substrate interactions to understand natural substrate recognition patterns and identify potential compensatory mechanisms.
- Structural Characterization: Assessment of available high-resolution structures (X-ray crystallography, cryo-EM) to enable structure-based degrader design.
- Functional Essentiality: Evaluation of E3 ligase essentiality using CRISPR knockout screens across hundreds of cell lines to predict potential on-target toxicities.
- Subcellular Localization: Determination of compartment-specific E3 localization to match with target protein distribution.
- PPI Interface Analysis: Characterization of protein-protein interaction interfaces to identify potential binding pockets for molecular glues.
Figure 1: Systematic Framework for E3 Ligase Characterization and Prioritization
Experimental Protocols for E3 Ligase Validation
Fragment-Based Ligand Discovery Using Protein-Observed NMR
Objective: Identify fragment-sized ligands for E3 ligases with restricted expression patterns using protein-observed NMR screening.
Methodology Details:
- Protein Expression and Purification: Express E3 ligases (e.g., CBL-c, TRAF-4) in E. coli with (^{15})N isotopic labeling for NMR detection. Purify using affinity (Ni-NTA for His-tagged constructs) and size-exclusion chromatography to ensure proper folding [113].
- Fragment Library Preparation: Curate a diverse fragment library (500-1,000 compounds) with molecular weight <300 Da and high structural diversity. Prepare stock solutions in DMSO-d6 [113].
- NMR Screening: Collect (^{1})H-(^{15})N HSQC spectra of 50-100 µM (^{15})N-labeled E3 ligase in the absence and presence of fragments (0.5-2 mM). Monitor chemical shift perturbations (CSPs) indicative of binding [113].
- Hit Validation: Conduct titration experiments with confirmed hits to determine binding affinity (K(_d)). Validate binding specificity through competition experiments with known ligands or site-directed mutagenesis [113].
- Structural Characterization: Determine X-ray co-crystal structures of E3 ligase-fragment complexes to elucidate binding modes and inform medicinal chemistry optimization [113].
Key Advantages: This approach is particularly valuable for E3 ligases with no known ligands, as it requires minimal prior structural knowledge and can identify weak binders (mM-µM K(_d)) suitable for subsequent optimization.
In-Cell E3 Ligase Evaluation Using Genetic Code Expansion
Objective: Evaluate E3 ligase suitability for TPD without requiring pre-existing ligands through genetic code expansion and click chemistry.
Methodology Details:
- Tetrazine-incorporated E3 Expression: Utilize amber codon suppression to site-specifically incorporate tetrazine-containing non-canonical amino acids (Tet-ncAA) at various surface-exposed positions on the E3 ligase (e.g., CRBN, SPOP) in living cells [115].
- Binder Conjugation: Conjugate the incorporated Tet with a strained trans-cyclooctene (sTCO) tethered to a neosubstrate protein binder (e.g., JQ1 for BRD2/4) via inverse electron demand Diels-Alder click chemistry [115].
- Degradation Efficiency Assessment: Measure degradation of the neosubstrate (e.g., BRD2/4) using Western blotting or targeted proteomics to evaluate E3 ligase functionality and identify optimal TPD interfaces [115].
- Systematic Interface Mapping: Screen multiple Tet-ncAA incorporation sites across the E3 surface with varying linker lengths to map functional degron sites and optimize degrader geometry [115].
Key Advantages: This "E3-ligand-free degrader" (ELF degrader) platform preserves native E3 conformation in live cells and enables direct functional assessment without requiring known ligands, significantly expanding the range of evaluable E3 ligases [115].
Table 3: Essential Research Reagents for E3 Ligase Characterization and Validation
| Reagent/Category | Specific Examples | Experimental Function | Key Applications |
|---|---|---|---|
| Fragment Libraries | Diverse chemical fragments (MW <300 Da) | Identify initial ligand binding sites | NMR-based screening, X-ray crystallography |
| Isotopically Labeled Proteins | (^{15})N-labeled E3 ligases | Protein-observed NMR spectroscopy | Binding site mapping, binding affinity determination |
| Genetic Code Expansion System | Tetrazine-ncAA, pyrrolysyl-tRNA synthetase | Site-specific non-canonical amino acid incorporation | In-cell E3 surface functionalization |
| Click Chemistry Reagents | sTCO-linker-binder conjugates | Covalent E3-neosubstrate linkage | ELF degrader assembly, interface mapping |
| CRISPR Screening Libraries | E3-focused sgRNA libraries | Functional essentiality assessment | Toxicity prediction, resistance mechanism identification |
| Proteasome Activity Reporters | Fluorogenic peptide substrates (LLVY-AMC) | Proteasome function assessment | Degradation efficiency validation |
Advanced Computational and AI-Driven Approaches
Machine Learning for Molecular Glue Prediction
The identification of molecular glues—small molecules that induce novel interactions between E3 ligases and target proteins—represents a promising strategy for expanding E3 ligase utilization. Recent advances in machine learning (ML) have demonstrated significant potential for in silico prediction of molecular glue candidates:
- Dataset Curation: Comprehensive datasets of known molecular glues (2,091 compounds) and non-molecular glues (196 compounds) compiled from literature and patent sources enable robust model training [116].
- Feature Engineering: Extensive molecular descriptors (1,500+ features) encompassing 2D and 3D structural properties, electronic features, and physicochemical parameters are calculated using RDKit, Mordred, PaDEL, Dragon, alvaDesc, and PyDisciptors [116].
- Model Development: Multiple ML algorithms, including XGBoost, Random Forest (RF), Support Vector Machines (SVM), Naïve Bayes, and k-Nearest Neighbors (k-NN), are employed with feature selection methods (RFE, RFECV, Boruta) to identify optimal predictive features [116].
- Performance Metrics: Top-performing models achieve ROC-AUC values exceeding 0.95, with XGBoost and RF consistently demonstrating high predictive accuracy. Deep learning-based D-MPNN architectures show slightly superior performance, suggesting enhanced capacity for capturing complex molecular patterns [116].
- Structural Alert Identification: Analysis of molecular glue datasets has identified 54 significant structural alerts specific to molecular glues, with the top 10 alerts demonstrating high accuracy and low false positive rates [116].
E3 Ligase Atlas and Web-Based Resource Platforms
To accelerate the development of PROTACs utilizing under-explored E3 ligases, researchers have developed comprehensive web resources that systematically characterize E3 ligases across multiple dimensions. The E3 Atlas (https://hanlaboratory.com/E3Atlas/) represents one such platform, integrating analysis of 30 large-scale datasets to provide researchers with a flexible tool for rapidly identifying E3 ligases with promising TPD activities against specific targets of interest [112]. This resource enables:
- Multi-dimensional E3 Ligase Scoring: Integration of confidence scores, ligandability assessments, expression patterns, and protein-protein interaction data to prioritize E3 ligases for specific therapeutic contexts.
- Tissue-Specific Expression Filtering: Identification of E3 ligases with restricted expression patterns in target tissues (e.g., tumor-selective expression) to enable tissue-specific degradation strategies.
- Ligandability Assessment: Evaluation of existing chemical starting points for E3 ligase targeting, including drugs, small-molecule ligands, and covalent binders from databases including DrugBank, DGIdb, ChEMBL, and SLCABPP [112].
- Essentiality Analysis: Integration of CRISPR knockout data from DepMap to identify E3 ligases whose inhibition may cause minimal toxicity in normal tissues.
Emerging Strategies for Enhancing Targeting Specificity
Tissue-Restricted E3 Ligase Exploitation
A particularly promising approach for enhancing E3 ligase specificity involves the strategic selection of ligases with naturally restricted expression patterns. This strategy leverages differential E3 expression between diseased and normal tissues to minimize on-target toxicities in healthy cells. Systematic analysis of RNA-seq data from 11,057 tumors (TCGA) and 17,382 normal samples (GTEx) has identified multiple E3 ligases with tumor-enriched expression patterns, including CBL-c and TRAF-4 [113]. These E3 ligases demonstrate significantly elevated expression in various cancer types compared to normal tissues, creating potential therapeutic windows for tumor-selective protein degradation.
The therapeutic potential of this approach has been validated preclinically with the PROTAC DT2216, which recruits VHL to degrade Bcl-xL. Since VHL expression is naturally low in platelets, DT2216 effectively spares Bcl-xL function in these cells, mitigating the thrombocytopenia typically associated with Bcl-xL inhibition [113]. This proof-of-concept demonstrates how strategic E3 ligase selection based on expression patterns can significantly improve therapeutic indices.
Figure 2: Workflow for Developing Tissue-Restricted E3 Ligase-Based Degraders
Structural Diversification and Ternary Complex Engineering
Beyond expression-based strategies, structural approaches to E3 ligase engineering offer additional avenues for enhancing degradation specificity. Recent research has revealed that E3 ligases exhibit remarkable plasticity in their capacity to form productive ternary complexes with diverse target proteins. The in-cell genetic code expansion platform has demonstrated that multiple surface regions on E3 ligases can support targeted degradation when properly engaged, with degradation efficiency depending on both the spatial placement of the engagement site and the geometry of the connecting linker [115].
This structural plasticity enables rational engineering approaches to optimize degradation specificity:
- Interface Mapping: Systematic screening of E3 surface positions identifies optimal engagement sites that maximize degradation efficiency for specific target classes.
- Linker Optimization: Variation of linker length and composition fine-tunes the geometry of the E3-PROTAC-target ternary complex to enhance specificity and potency.
- Selectivity Engineering: Strategic engagement of E3 surfaces that are structurally distinct from endogenous substrate binding interfaces may reduce competition with natural substrates and improve degradation selectivity.
The systematic expansion of targetable E3 ligases represents one of the most critical challenges and opportunities in the continued development of targeted protein degradation therapeutics. Current overreliance on VHL and CRBN creates fundamental limitations in therapeutic scope, specificity, and durability. The integrated experimental and computational frameworks outlined in this review provide a roadmap for unlocking the substantial untapped potential within the E3 ligase family.
Future progress in E3 ligase targeting will likely emerge from several key directions: First, continued development of fragment-based screening platforms and AI-driven ligand prediction will expand the repertoire of chemically tractable E3 ligases. Second, advanced understanding of tissue-specific E3 expression patterns and functions will enable more precise therapeutic targeting with reduced off-tissue toxicities. Third, structural characterization of ternary complex formation will facilitate rational design of degraders with enhanced specificity and efficiency. Finally, the integration of these approaches through comprehensive resources like the E3 Atlas will accelerate the translation of E3 biology into novel therapeutic strategies across diverse disease contexts, particularly in oncology, neurodegeneration, and immune disorders where targeted protein degradation holds exceptional promise.
As these efforts mature, the strategic diversification of utilized E3 ligases will fundamentally transform the TPD landscape, enabling precision degradation strategies with enhanced therapeutic windows and reduced susceptibility to resistance mechanisms. The systematic addressing of E3 ligase specificity challenges outlined herein represents a critical step toward realizing the full potential of the ubiquitin-proteasome system as a therapeutic platform.
Overcoming Drug Resistance in UPS-Targeted Therapies
The ubiquitin-proteasome system (UPS) represents a pivotal therapeutic target for combating drug resistance in cancer and other diseases. As the primary pathway for intracellular protein degradation, the UPS regulates countless cellular processes through post-translational modification and controlled proteolysis. Despite the initial success of UPS-targeting therapies, the emergence of resistance mechanisms continues to challenge clinical efficacy. This technical review examines the molecular foundations of resistance in UPS-targeted therapies, delineates experimental approaches for its investigation, and explores innovative strategies to overcome these limitations through emerging technologies and combination approaches.
The ubiquitin-proteasome system (UPS) constitutes a sophisticated enzymatic cascade responsible for controlled protein degradation and maintenance of cellular homeostasis. This system coordinates a hierarchical process involving E1 activating enzymes, E2 conjugating enzymes, and E3 ligase enzymes that collectively tag target proteins with ubiquitin chains for recognition and degradation by the 26S proteasome [117]. The specificity of this process is largely determined by E3 ubiquitin ligases, with over 600 encoded in the human genome, though only a subset has been utilized therapeutically [118]. The proteasome itself is a multi-subunit complex comprising a 20S core particle (CP) capped by 19S regulatory particles (RP) that recognize ubiquitinated substrates, remove ubiquitin chains, and unfold proteins for degradation [117].
UPS-targeted therapies have evolved along two primary axes: direct proteasome inhibition and targeted protein degradation (TPD). Proteasome inhibitors such as bortezomib represent the first generation of UPS-targeted drugs, functioning through direct inhibition of proteasomal proteolytic activity [26]. More recently, TPD strategies including proteolysis-targeting chimeras (PROTACs) and molecular glues have emerged as revolutionary approaches that hijack the UPS to selectively degrade disease-causing proteins [118]. These technologies have expanded the druggable proteome to include traditionally "undruggable" targets lacking conventional binding pockets. However, the clinical application of these therapies faces significant challenges from both innate and acquired resistance mechanisms that limit their long-term efficacy.
Molecular Mechanisms of Resistance in UPS-Targeted Therapies
Resistance to Conventional Proteasome Inhibitors
Resistance to proteasome inhibitors involves multifaceted adaptations within cancer cells. The table below summarizes the primary resistance mechanisms observed in response to proteasome inhibitor treatment.
Table 1: Mechanisms of Resistance to Proteasome Inhibitors
| Resistance Mechanism | Molecular Components | Functional Consequences |
|---|---|---|
| Proteasome subunit mutations | β5 subunit (PSMB5) | Reduced drug binding affinity while maintaining catalytic activity |
| Proteasome subunit overexpression | Immunoproteasome subunits | Increased capacity for protein degradation via alternative complexes |
| Efflux pump upregulation | P-glycoprotein (P-gp) | Enhanced drug export reducing intracellular concentrations |
| UPS compensatory activation | NRF2 pathway, E1/E2/E3 enzymes | Enhanced ubiquitination capacity and substrate processing |
| Aggresome formation | HDAC6, vimentin, dynein | Alternative protein clearance pathway activation |
Resistance in Targeted Protein Degradation Approaches
Targeted protein degradation technologies, particularly PROTACs and molecular glues, face distinct resistance challenges. These include:
E3 Ligase Alterations: Downregulation or mutations in E3 ligase components (e.g., VHL, CRBN) compromise ternary complex formation and degradation efficiency. For instance, mutations in the CRBN substrate-binding domain can disrupt molecular glue binding without affecting E3 ligase activity [118].
Ternary Complex Disruption: Impaired formation or stability of the POI-PROTAC-E3 ligase complex significantly reduces degradation efficacy. The "hook effect" observed at high PROTAC concentrations further complicates dosing strategies [118].
Ubiquitination Pathway Adaptations: Alterations in E2 conjugating enzymes or ubiquitin availability can limit ubiquitin chain formation on target proteins, even with successful ternary complex formation.
Deubiquitinating Enzyme (DUB) Activity: Upregulation of DUBs, particularly ubiquitin-specific proteases (USPs), can reverse ubiquitination before substrates reach the proteasome. USP14 and UCHL1 overexpression has been documented as a resistance mechanism to multiple PROTACs [119].
UPS-Mediated Regulation of Immune Checkpoints and Therapeutic Implications
The UPS plays a crucial role in modulating immune checkpoint protein stability, significantly impacting cancer immunotherapy resistance. PD-L1, the primary ligand for PD-1, is regulated by multiple E3 ubiquitin ligases that determine its turnover rate and cell surface expression [117] [120].
Table 2: E3 Ubiquitin Ligases Regulating PD-L1 Stability and Cancer Immunity
| E3 Ligase | Cancer Type | Regulatory Mechanism | Therapeutic Implications |
|---|---|---|---|
| SPOP | Colorectal cancer | Promotes K48-linked ubiquitination and degradation of PD-L1 | SPOP agonists may enhance anti-tumor immunity |
| TRIM21 | Non-small cell lung cancer | Ubiquitinates PD-L1 leading to proteasomal degradation | TRIM21 activators overcome immunotherapy resistance |
| ARIH1 | Lymphosarcoma, NSCLC | GSK3α-mediated phosphorylation enhances PD-L1 recognition | EGFR inhibition stabilizes GSK3α activity |
The intricate regulation of PD-L1 by SPOP demonstrates how cancer cells exploit the UPS for immune evasion. In colorectal cancer, SPOP normally promotes PD-L1 ubiquitination and degradation. However, elevated ALDH2 expression competitively binds PD-L1, preventing SPOP-mediated ubiquitination and increasing PD-L1 stability [117]. Similarly, in hepatocellular carcinoma, BCLAF1 binds and inactivates SPOP, thereby stabilizing PD-L1 [117]. These mechanisms highlight the potential of targeting UPS components to overcome resistance to immune checkpoint blockade therapy.
The following diagram illustrates the complex regulatory network controlling PD-L1 stability through ubiquitination:
Experimental Approaches for Investigating UPS-Mediated Resistance
Protocol for Assessing UPS Component Expression in Resistant Cells
Objective: Quantify changes in UPS component expression in therapy-resistant versus sensitive cell lines.
Methodology:
- Establish resistant cell lines through gradual exposure to increasing concentrations of UPS-targeting agents over 3-6 months
- Perform RNA sequencing to identify transcriptional alterations in E3 ligases, DUBs, and proteasome subunits
- Validate findings using Western blot analysis for protein expression levels
- Conduct co-immunoprecipitation assays to evaluate protein-protein interactions in ubiquitination pathways
- Implement proteasome activity assays using fluorogenic substrates (LLVY-AMC for chymotrypsin-like activity)
Key Reagents:
- Fluorogenic proteasome substrates (LLVY-AMC, LRR-AMC, nLPnLD-AMC)
- E1 inhibitor (TAK-243) as positive control
- Proteasome inhibitors (MG132, bortezomib) for control conditions
- Antibodies against UPS components (E3 ligases, DUBs, ubiquitin chains)
Protocol for Evaluating Ternary Complex Formation in PROTAC Resistance
Objective: Assess efficiency of ternary complex formation in PROTAC-resistant cells.
Methodology:
- Generate CRISPR-modified cell lines with tagged E3 ligases (VHL, CRBN) and target proteins
- Treat cells with PROTACs at varying concentrations (1nM-10μM) for time course experiments
- Perform proximity ligation assays (PLA) to visualize and quantify ternary complex formation
- Conduct cellular thermal shift assays (CETSA) to evaluate target engagement
- Implement ubiquitination assays by immunoprecipitating target proteins and probing with ubiquitin-specific antibodies
Expected Outcomes: Resistant cells typically show reduced ternary complex formation and decreased target protein ubiquitination despite adequate PROTAC concentrations.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Investigating UPS-Mediated Resistance
| Reagent Category | Specific Examples | Research Application | Resistance Insights |
|---|---|---|---|
| E3 Ligase Inhibitors | Nutlin-3 (MDM2), LCL-161 (IAP) | Modulate specific ubiquitination pathways | Identify compensatory E3 ligases in resistant cells |
| DUB Inhibitors | IU1-47 (USP14), b-AP15 (UCHL1) | Probe deubiquitination mechanisms | Assess DUB upregulation as resistance mechanism |
| PROTAC Molecules | ARV-471 (ER degrader), KT-253 (MDM2-recruiting) | Study targeted protein degradation | Evaluate ternary complex stability in resistance |
| Ubiquitin Variants | TUBE reagents (Tandem Ubiquitin Binding Entities) | Enrich and detect ubiquitinated proteins | Quantify changes in global ubiquitination patterns |
| Activity-Based Probes | Ub-AMC, proteasome substrates | Measure enzymatic activity in live cells | Detect proteasome adaptation in resistant lines |
| CRISPR Libraries | E3 ligase, DUB-focused libraries | High-throughput genetic screening | Identify novel resistance mediators |
Emerging Strategies to Overcome UPS-Targeted Therapy Resistance
Next-Generation Targeted Protein Degradation Technologies
Novel TPD approaches are being developed to circumvent existing resistance mechanisms:
Heterobifunctional Degraders Beyond PROTACs: Lysosome-targeting chimeras (LYTACs) and autophagy-targeting chimeras (AUTACs) exploit alternative degradation pathways, bypassing proteasome-dependent resistance [118].
Molecular Glue Optimization: Through systematic screening approaches, molecular glues with enhanced E3 ligase binding or the ability to recruit novel E3 ligases are being developed to overcome ligase-specific resistance [118].
Dual-Targeting Degraders: These compounds simultaneously engage two E3 ligases or target multiple pathogenic proteins, reducing the probability of resistance emergence through single pathway alterations.
Combination Therapies to Overcome Resistance
Rational combination strategies represent a promising approach to overcome resistance:
PROTACs with E1 Inhibitors: Combining PROTACs with E1 ubiquitin-activating enzyme inhibitors (e.g., TAK-243) can create synthetic lethality in resistant cells [119].
DUB Inhibitors with PROTACs: Simultaneous inhibition of compensatory DUBs enhances degradation efficacy by prolonging ubiquitin chain residence on target proteins [119].
PROTACs with Immune Checkpoint Inhibitors: Coordinated targeting of oncoproteins and immune checkpoints addresses both cell-autonomous and microenvironmental resistance mechanisms [117] [120].
The following diagram illustrates a comprehensive experimental workflow for investigating UPS-mediated drug resistance:
Overcoming resistance in UPS-targeted therapies requires a multidimensional approach that addresses the dynamic adaptability of the ubiquitin-proteasome system. The future of this field lies in developing more sophisticated degradation technologies that engage multiple E3 ligases simultaneously, creating resistance-resistant therapeutic modalities. Additionally, advanced biomarker development for predicting resistance mechanisms before treatment initiation will enable more personalized application of UPS-targeted therapies.
The integration of artificial intelligence and machine learning approaches to model ternary complex formation and predict resistance mutations will accelerate the design of next-generation degraders. Furthermore, combining UPS-targeted therapies with complementary modalities, such as epigenetic regulators and immune checkpoint inhibitors, presents a promising strategy to overcome the heterogeneity of resistance mechanisms. As our understanding of UPS biology deepens, so too will our ability to develop increasingly effective strategies to circumvent therapeutic resistance, ultimately improving outcomes for patients with cancer and other diseases driven by protein homeostasis dysregulation.
Technical Considerations in UPS Activity Measurement and Validation
The ubiquitin-proteasome system (UPS) represents a highly conserved protein degradation pathway that plays an essential role in maintaining cellular protein homeostasis [29]. This system ensures precise regulation of key cellular regulators, with its dysregulation implicated in the pathogenesis of numerous chronic diseases, including neurodegenerative, cardiovascular, and oncological disorders [25]. For researchers and drug development professionals investigating the UPS pathway, accurate and validated measurement of proteasome activity is paramount. This technical guide provides an in-depth examination of current methodologies, validation frameworks, and experimental protocols for reliable UPS activity assessment, supporting the broader research objective of understanding UPS pathway dynamics in both physiological and disease contexts.
Core Principles of the Ubiquitin-Proteasome System
The UPS is a fundamental cellular mechanism responsible for the controlled degradation of intracellular proteins. This system ensures precise regulation of key regulators within various signaling pathways, enabling dynamic cellular responses to changing conditions [29]. In practical terms, the UPS pathway consists of two main sequential processes:
- Ubiquitination: Target proteins are marked for degradation through the covalent attachment of ubiquitin molecules. This process involves a cascade of E1 (activating), E2 (conjugating), and E3 (ligating) enzymes, with E3 ligases providing substrate specificity [29].
- Degradation by the Proteasome: Polyubiquitinated proteins are recognized and degraded by the 26S proteasome complex, a multi-subunit protease that breaks down proteins into small peptides.
Table 1: Core Components of the Ubiquitin-Proteasome System
| Component | Function | Research Significance |
|---|---|---|
| Ubiquitin | Small regulatory protein that tags substrates for degradation | Conserved across eukaryotes; can be fluorescently tagged for tracking |
| E1 Enzymes | Activate ubiquitin in an ATP-dependent manner | Initial step in ubiquitination cascade; few genes encode E1 enzymes |
| E2 Enzymes | Carry activated ubiquitin and collaborate with E3 ligases | ~40 genes in humans; determine ubiquitin chain topology |
| E3 Ligases | Recognize specific substrates and facilitate ubiquitin transfer | >600 genes in humans; provide substrate specificity; potential drug targets |
| 26S Proteasome | Multi-protein complex that degrades ubiquitinated substrates | Composed of 20S core and 19S regulatory particles; primary activity measurement target |
Understanding these core principles is essential for designing appropriate experimental approaches for UPS activity measurement, as each component presents potential intervention points or measurement targets for research and therapeutic development.
Quantitative UPS Activity Assays
Accurate measurement of proteasome activity relies primarily on monitoring the degradation of specific fluorogenic substrates. The following table summarizes the core assay types and their respective applications in UPS research.
Table 2: Quantitative Assays for UPS Activity Measurement
| Assay Type | Principle | Measured Parameters | Dynamic Range | Advantages | Limitations |
|---|---|---|---|---|---|
| Fluorogenic Substrate-Based | Proteasome cleaves peptide-fluorophore bonds, releasing detectable fluorescence | Chymotrypsin-like, trypsin-like, and caspase-like activity | 5-1000 μM substrate concentration | High sensitivity, real-time kinetics, adaptable to high-throughput | Potential interference from other proteases |
| Luminescence-Based Assays | Luciferase fusion proteins report degradation kinetics in live cells | UPS-dependent protein turnover | 2-3 log linear range | Live-cell monitoring, temporal resolution | Requires genetic manipulation, potential artifacts |
| Immunoblot-Based Methods | Detection of ubiquitin conjugates or proteasome subunits | Polyubiquitinated proteins, subunit expression | Semi-quantitative, dependent on antibody affinity | Specific component analysis, widely accessible | Low throughput, semi-quantitative at best |
| Activity-Based Probes | Irreversible binding to active proteasome sites | Active proteasome quantification, subunit profiling | Probe concentration-dependent | Specific for active proteasomes, visualization possible | Not a functional activity measure |
Fluorogenic Substrate Assay Protocol
Title: Determination of 26S Proteasome Chymotrypsin-like Activity in Cell Lysates
Principle: The proteasome's chymotrypsin-like activity cleaves the peptide bond after hydrophobic residues in the substrate Suc-LLVY-AMC, releasing the fluorescent 7-amino-4-methylcoumarin (AMC) group.
Materials:
- Assay Buffer: 50 mM HEPES, pH 7.5, 5 mM MgCl₂, 1 mM DTT, 1 mM ATP
- Substrate: Suc-LLVY-AMC (250 μM stock in DMSO)
- Proteasome Inhibitor: MG-132 (10 mM stock in DMSO)
- Cell Lysis Buffer: 50 mM Tris-HCl, pH 7.5, 250 mM sucrose, 5 mM MgCl₂, 1 mM DTT, 0.5% NP-40, 1 mM ATP
- Fluorescence Plate Reader (Excitation: 380 nm, Emission: 460 nm)
Procedure:
- Sample Preparation: Harvest cells and wash with ice-cold PBS. Lyse cells in lysis buffer (100 μL per 10⁶ cells) for 30 minutes on ice. Centrifuge at 16,000 × g for 20 minutes at 4°C. Collect supernatant and determine protein concentration.
- Assay Setup: In a 96-well plate, add 90 μL assay buffer per well. Add 10 μL cell lysate (10-20 μg protein) to sample wells. For inhibitor controls, pre-incubate lysate with 10 μM MG-132 for 15 minutes on ice.
- Reaction Initiation: Add 10 μL substrate solution (final concentration: 25 μM) to all wells. Mix gently by pipetting.
- Kinetic Measurement: Immediately place plate in pre-warmed plate reader (37°C) and measure fluorescence every 5 minutes for 60-90 minutes.
- Data Analysis: Calculate reaction velocity from the linear portion of the fluorescence increase. Subtract background activity from inhibitor-treated controls. Express activity as pmol AMC released/min/mg protein using an AMC standard curve.
Data Validation Techniques
Robust validation of UPS activity data requires a multi-layered approach to ensure accuracy, specificity, and reproducibility. The following techniques should be systematically implemented.
Specificity Controls
Inhibitor Profiling: Use specific proteasome inhibitors (e.g., MG-132, bortezomib, epoxomicin) at multiple concentrations to confirm that measured activity is proteasome-specific. Include negative controls with inhibitors of other protease classes (e.g., E-64 for cysteine proteases, PMSF for serine proteases) to rule out non-specific contributions.
Substrate Specificity: Validate activity measurements with multiple substrate types (Suc-LLVY-AMC for chymotrypsin-like, Z-LLE-AMC for caspase-like, Boc-LRR-AMC for trypsin-like activity) to ensure consistent patterns of inhibition and activation.
Range Validation
Establish acceptable activity ranges for different sample types (e.g., cell lines, tissues) through repeated measurements of control samples. Define minimum and maximum constraints based on biological plausibility to flag potentially erroneous data [121]. For mammalian cell lysates, typical chymotrypsin-like activity ranges from 50-500 pmol/min/mg protein, depending on cell type and growth conditions.
Constraint Validation
Implement complex business rules for data integrity, such as enforcing expected ratios between different proteasome activities (e.g., chymotrypsin-like activity is typically 3-5 times higher than trypsin-like activity in most mammalian cells) [121]. Reject datasets that violate these predefined constraints to maintain data quality.
Advanced Methodological Approaches
Live-Cell UPS Monitoring
For dynamic assessment of UPS function in intact cells, implement reporter systems based on UPS-dependent degradation of fluorescent proteins (e.g., GFP-Ubiquitin fusion constructs). The critical protocol parameters include:
GFP-Ubiquitin Degradation Assay:
- Transfect cells with GFP-ubiquitin plasmid or use stable cell lines
- Treat with proteasome inhibitors (10 μM MG-132) or activators as experimental conditions
- Monitor GFP fluorescence intensity by flow cytometry or live-cell imaging over 24-48 hours
- Calculate protein half-life from fluorescence decay curves
- Normalize data to protein synthesis rates using cycloheximide chase controls
Tissue-Specific Activity Profiling
Measuring UPS activity in tissue samples presents unique challenges related to heterogeneity and sample integrity. The modified protocol includes:
Tissue Processing:
- Homogenize fresh or snap-frozen tissue in hypotonic buffer (10 mM Tris, pH 7.5) using Dounce homogenizer
- Centrifuge at 3,000 × g for 10 minutes to remove nuclei and debris
- Use supernatant for immediate assay or aliquot and store at -80°C
- Include protease inhibitor cocktails excluding proteasome inhibitors
- Normalize activity to both total protein and tissue weight
Single-Cell Proteasome Activity
Advanced flow cytometry approaches enable UPS activity measurement at single-cell resolution:
Flow Cytometry Protocol:
- Load cells with fluorogenic proteasome substrates (5 μM final concentration)
- Incubate for 30-60 minutes at 37°C in serum-free media
- Include control samples with proteasome inhibitors
- Analyze by flow cytometry using appropriate laser/filter combinations
- Gate on live cells using viability dyes
- Report geometric mean fluorescence intensity of specific activity
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for UPS Activity Studies
| Reagent Category | Specific Examples | Function/Application | Key Considerations |
|---|---|---|---|
| Proteasome Inhibitors | MG-132, Bortezomib, Epoxomicin, Lactacystin | Specific inhibition of proteasome activity; validation controls | Varying specificity, cell permeability, and reversibility profiles |
| Activity-Based Probes | MV151, BodipyFL-Ahx3L3VS | Labeling active proteasomes; visualization and quantification | Require click chemistry for detection; specific for active sites |
| Fluorogenic Substrates | Suc-LLVY-AMC, Z-LLE-AMC, Boc-LRR-AMC | Specific measurement of proteasome catalytic activities | Different cleavage specificities; potential non-proteasome hydrolysis |
| Antibodies for UPS | Anti-ubiquitin, anti-K48-ubiquitin, anti-proteasome subunits | Immunodetection of UPS components and ubiquitination | Specificity validation critical; chain linkage specificity important |
| UPS Reporter Systems | GFP-Ubiquitin, ZsProSensor-1, Ub-G76V-GFP | Live-cell monitoring of UPS function | May overload endogenous system; requires controlled expression |
| E3 Ligase Modulators | Small molecule inhibitors/activators of specific E3s | Pathway-specific UPS modulation | Increasing availability for specific E3 ligases (e.g., MDM2, CRL4) |
Troubleshooting and Data Interpretation
Common Technical Challenges
High Background Activity:
- Cause: Contamination from other cellular proteases
- Solution: Include appropriate inhibitor controls (e.g., E-64, pepstatin, PMSF) and optimize lysis conditions
- Validation: Compare activity with and without specific proteasome inhibitors
Low Signal-to-Noise Ratio:
- Cause: Suboptimal substrate concentration or poor cell lysis
- Solution: Perform substrate titration (typically 10-100 μM) and verify lysis efficiency
- Validation: Include positive control (commercial proteasome) to establish maximum expected signal
Inconsistent Replicates:
- Cause: Variable protein quantification or temperature fluctuations during assay
- Solution: Standardize protein assay method and use pre-warmed plates with temperature control
- Validation: Include internal control samples on every plate
Data Normalization Strategies
Normalize proteasome activity data using multiple approaches to ensure robust interpretation:
- Total Protein Normalization: Most common method using BCA or Bradford assay
- Cellular Protein Normalization: Per million cells for cell-based assays
- Proteasome Content Normalization: Using immunoblotting for specific subunits
- Reference Activity Normalization: To housekeeping protease activities
Robust measurement and validation of UPS activity requires meticulous attention to methodological details, appropriate controls, and multi-layered validation approaches. The techniques outlined in this guide provide a framework for generating reliable, reproducible data on proteasome function in diverse experimental systems. As research continues to uncover the UPS's complex roles in cellular physiology and disease, these standardized approaches will facilitate comparison across studies and support the development of UPS-targeted therapeutics. Future methodological developments will likely focus on single-cell resolution, real-time monitoring in intact organisms, and integration with other omics approaches for systems-level understanding of proteostasis networks.
Managing UPS Dysfunction in Disease Models
The ubiquitin-proteasome system (UPS) serves as the primary pathway for regulated intracellular protein degradation, playing an indispensable role in maintaining cellular proteostasis. This sophisticated system employs a hierarchical enzymatic cascade to tag proteins with ubiquitin for recognition and degradation by the 26S proteasome, a multi-subunit protease complex [122] [123]. The UPS governs numerous critical cellular processes beyond protein quality control, including cell cycle progression, signal transduction, DNA repair, and immune responses [123]. Given its central regulatory role, dysfunction of the UPS has been implicated in the pathogenesis of a wide spectrum of human diseases. Neurodegenerative disorders such as Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis (ALS) are characterized by accumulation of toxic protein aggregates, often linked to impaired UPS function [122] [124] [125]. Conversely, many cancers exhibit upregulated proteasome activity that supports rapid proliferation and evasion of cell death [126] [127]. Additionally, UPS dysfunction contributes to cardiac pathologies [128], skeletal muscle disorders [129], and metabolic diseases [123].
Understanding and managing UPS dysfunction requires a multifaceted approach that encompasses accurate disease modeling, precise monitoring of UPS activity, and targeted therapeutic interventions. This technical guide synthesizes current methodologies for investigating UPS dysfunction across disease contexts, providing researchers with standardized protocols, quantitative benchmarks, and visualization tools to advance both basic research and drug development efforts. The complex interplay between the UPS and other proteostatic pathways, particularly autophagy, further necessitates integrated experimental strategies to capture the system's dynamics comprehensively [122] [123].
Molecular Basis of UPS Dysfunction
Key Components and Dysregulation Mechanisms
The molecular architecture of the UPS consists of sequential enzymatic components that coordinate substrate recognition and degradation. The system begins with E1 ubiquitin-activating enzymes that initiate ubiquitin transfer, followed by E2 ubiquitin-conjugating enzymes, and culminating with E3 ubiquitin ligases that confer substrate specificity through recognition of degrons on target proteins [123]. The human genome encodes over 600 E3 ligases, enabling precise recognition of a vast array of substrates [123]. Polyubiquitinated proteins are subsequently degraded by the 26S proteasome, which consists of a 20S core particle capped by 19S regulatory particles that recognize ubiquitin signals, unfold substrates, and facilitate translocation into the proteolytic chamber [122] [123].
Dysfunction can occur at multiple levels within this pathway. Genetic mutations in UPS components represent a primary mechanism of dysfunction, as exemplified by mutations in the E3 ligase Parkin that cause early-onset Parkinson's disease [122]. Similarly, mutations in KLHL40, a CUL3 E3 ligase adapter, result in severe congenital nemaline myopathy due to disrupted sarcomeric protein turnover [129]. Beyond genetic lesions, oxidative damage can impair proteasomal function, particularly following ischemia-reperfusion injury or in neurodegenerative contexts [128] [125]. The accumulation of aggregation-prone proteins such as α-synuclein, β-amyloid, and polyglutamine-expanded huntingtin can physically obstruct the proteasome, creating a self-reinforcing cycle of proteostatic collapse [122]. Additionally, altered expression of deubiquitinases (DUBs) that normally recycle ubiquitin and edit ubiquitin chains can disrupt the delicate balance of ubiquitin signaling, contributing to pathogenesis [123].
Compensatory Pathways and Adaptive Responses
Cells deploy multiple adaptive responses to mitigate UPS dysfunction. The transcription factor Nrf1 (NFE2L1) serves as a master regulator of proteostatic stress responses. Under normal conditions, Nrf1 is retained in the endoplasmic reticulum membrane and continuously degraded via ER-associated degradation (ERAD) [130]. When proteasomal activity is impaired, Nrf1 escapes degradation and is processed by the protease DDI2 into its active form, which translocates to the nucleus and activates transcription of proteasome subunit genes and autophagy-lysosomal pathway genes [130]. This "bounce-back response" represents a critical adaptive mechanism that allows cells to restore proteolytic capacity. However, in disease states, this compensatory response may become overwhelmed or maladaptive. The interplay between the UPS and autophagy is particularly important, as autophagy can partially compensate for impaired proteasomal function by clearing protein aggregates and damaged organelles [122] [123]. Understanding these compensatory mechanisms is essential for developing holistic therapeutic strategies that target the broader proteostatic network rather than isolated components.
Quantitative Assessment of UPS Function
Proteasomal Activity Assays
Accurate quantification of proteasomal activity is fundamental to evaluating UPS function in disease models. The chymotrypsin-like, trypsin-like, and caspase-like activities of the proteasome can be measured using fluorogenic substrates that emit fluorescence upon cleavage (Table 1). Standardized protocols typically employ cell lysates or tissue homogenates incubated with specific substrates such as Suc-LLVY-AMC for chymotrypsin-like activity [128] [127]. Activity measurements should be normalized to total protein content and performed in the presence and absence of specific proteasome inhibitors like MG-132 or lactacystin to confirm signal specificity [128]. For in situ assessment, cell-permeable fluorescent substrates can be applied to live cells, though permeability variations must be controlled for. Recent advances in ubiquitinomics approaches enable system-wide quantification of ubiquitin conjugates, providing comprehensive insights into UPS substrate flux under pathological conditions [129] [123].
Table 1: Quantitative Parameters of Proteasome Inhibition in Disease Models
| Disease Model | Intervention | Proteasome Activity Reduction | Key Functional Outcomes | Reference |
|---|---|---|---|---|
| Cardiac I/R (High Glucose) | 5 µM Lactacystin | ~40% (Chymotrypsin-like) | Improved cardiac function recovery; Reduced infarct size | [128] |
| Cardiac I/R (High Glucose) | 10 µM MG-132 | ~50% (Chymotrypsin-like) | Enhanced anti-oxidant defenses; Attenuated inflammation | [128] |
| Melanoma (A375 cells) | 1.258 µM MG-132 (IC50) | 50% (Cell viability) | 85.5% apoptosis induction; Suppressed migration | [127] |
| Motor Neuron (Psmc4 CKO) | Genetic disruption | ~70% (Proteasome function) | Progressive motor neuron loss; TDP-43 aggregation | [125] |
Markers of Proteostatic Stress
Beyond direct activity measurements, molecular markers provide valuable indirect assessments of UPS dysfunction. Immunoblotting for polyubiquitinated proteins reveals overall accumulation of UPS substrates, though this approach cannot distinguish between adaptive ubiquitination signaling and pathological impairment [125]. More specific markers include transcription factors that respond to proteostatic stress, such as Nrf1 and its target genes [130]. In motor neurons with UPS dysfunction, elevated expression of N-myc downstream regulated 1 (NDRG1) has been identified as a sensitive biomarker that correlates with apoptotic activation [125]. Additionally, the accumulation of characteristic misfolded proteins associated with specific diseases (e.g., TDP-43 in ALS, α-synuclein in Parkinson's) provides disease-relevant readouts of UPS impairment [122] [125]. For comprehensive assessment, multi-parametric approaches that combine activity assays, ubiquitin conjugate profiling, and stress marker quantification offer the most robust evaluation of UPS status in disease models.
Experimental Models of UPS Dysfunction
Pharmacological Inhibition Models
Pharmacological proteasome inhibitors provide a tractable approach for inducing controlled UPS dysfunction in cellular and animal models (Table 2). These compounds can be applied at specific concentrations and durations to achieve partial or complete proteasome inhibition, mimicking various degrees of UPS impairment observed in human diseases. MG-132, a reversible aldehyde inhibitor, primarily targets the chymotrypsin-like activity of the proteasome's β5 subunit and is widely used for in vitro studies due to its cell permeability and rapid action [128] [127]. Lactacystin, a natural product isolated from Streptomyces, irreversibly inhibits primarily the chymotrypsin-like activity with lesser effects on trypsin-like and caspase-like activities [128]. For in vivo applications and clinical translation, FDA-approved inhibitors including bortezomib, carfilzomib, and ixazomib offer more specific pharmacological profiles, though their use in modeling neurodegenerative diseases requires careful dose optimization to achieve partial inhibition without excessive toxicity [130].
Table 2: Research Reagent Solutions for UPS Dysfunction Studies
| Reagent/Category | Specific Examples | Function/Application | Key Considerations |
|---|---|---|---|
| Proteasome Inhibitors | MG-132, Lactacystin, Bortezomib, Carfilzomib | Induce acute UPS dysfunction; Study consequences and compensatory mechanisms | Dose-dependent effects; Reversible vs. irreversible inhibition; Subunit specificity |
| Genetic Models | Psmc4 (Rpt3) conditional KO, KLHL40 mutants, Nrf1 KO | Tissue-specific UPS dysfunction; Study developmental and chronic effects | Compensation by other systems; Developmental lethality in full KOs |
| Cell Lines | A375 melanoma, NIH-3T3 fibroblasts, MDA-MB-231/468 TNBC | High-throughput screening; Mechanistic studies | Cell-type specific responses; Variable basal UPS activity |
| Activity Reporters | Fluorogenic substrates (Suc-LLVY-AMC), Ubiquitinomics, Ub-GFP reporters | Quantify proteasome activity; Monitor protein degradation | Signal specificity; Substrate permeability; Normalization methods |
| Pathway Modulators | NMS-873 (p97 inhibitor), DDI2 inhibitors, Anthracyclines | Target specific UPS regulatory nodes; Block adaptive responses | Off-target effects; Specificity validation required |
The concentration and duration of inhibitor treatment critically influence experimental outcomes and biological interpretations. High-dose exposure (e.g., 10-20 µM MG-132 for 24 hours) typically induces rapid apoptosis through stabilization of pro-death regulators, mimicking acute proteostatic collapse [127]. In contrast, low-dose, chronic inhibition (e.g., 0.1-1 µM for several days) better models the gradual UPS decline observed in aging and neurodegeneration, enabling study of adaptive responses including Nrf1 activation and autophagy induction [130]. In cardiac ischemia-reperfusion models under high glucose conditions, partial proteasome inhibition (approximately 40-50% reduction in chymotrypsin-like activity) during early reperfusion paradoxically protected against injury by enhancing antioxidant defenses and attenuating inflammatory responses [128]. This demonstrates the complex, context-dependent consequences of proteasome modulation and highlights the importance of carefully calibrated inhibition protocols.
Genetic Models of UPS Dysfunction
Genetic models provide powerful tools for investigating cell-type-specific consequences of chronic UPS impairment. Conditional knockout mice with motor neuron-specific deletion of Psmc4 (Rpt3), an essential ATPase subunit of the 19S regulatory particle, recapitulate key features of ALS including progressive motor neuron loss, TDP-43 protein aggregation, and glial activation [125]. Similarly, KLHL40 mutations in zebrafish and mice disrupt sarcomeric protein turnover, leading to nemaline myopathy with structural defects in myofibers and aberrant protein trafficking [129]. These models enable investigation of UPS dysfunction during development and in post-mitotic tissues that are particularly vulnerable to proteostatic stress.
For mechanistic dissection of specific UPS components, CRISPR-Cas9-mediated gene editing allows targeted disruption of E1, E2, E3, or proteasomal subunits in cultured cells [130] [129]. Knockout of Nrf1, for instance, abrogates the bounce-back response to proteasome inhibition, sensitizing cells to proteotoxic stress and enhancing the efficacy of proteasome inhibitors in cancer models [130]. Genetic models are particularly valuable for distinguishing primary UPS defects from secondary consequences observed in protein aggregation diseases, where aggregates may physically impair proteasome function rather than intrinsic UPS components being compromised.
Pathway Visualization: Nrf1-Mediated Bounce-Back Response
Nrf1 Bounce-Back Response to Proteasome Inhibition
Methodologies for Monitoring UPS Dysfunction
Protocol: Quantitative Assessment of Proteasomal Activity in Tissue Homogenates
Principle: This protocol measures the chymotrypsin-like activity of the proteasome in cardiac tissue following ischemia-reperfusion injury, adapted from established methodologies [128]. The assay utilizes the fluorogenic substrate Suc-LLVY-AMC, which emits fluorescence upon cleavage by the proteasome's β5 subunit.
Reagents:
- Homogenization buffer: 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 5 mM MgCl₂, 1 mM DTT, 1 mM ATP, 0.5% Triton X-100
- Assay buffer: 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 5 mM MgCl₂, 1 mM DTT, 1 mM ATP
- Substrate solution: 100 µM Suc-LLVY-AMC in DMSO
- Inhibitor control: 10 µM MG-132 in DMSO
- Protein quantification reagent (e.g., BCA assay)
Procedure:
- Tissue Homogenization: Excise approximately 20 mg of cardiac tissue and homogenize in 200 µL ice-cold homogenization buffer using a motorized homogenizer. Centrifuge at 12,000 × g for 15 minutes at 4°C and collect the supernatant.
- Protein Quantification: Determine protein concentration of the supernatant using BCA assay. Dilute samples to a uniform concentration of 2 mg/mL with assay buffer.
- Reaction Setup: In a black 96-well plate, add 90 µL of diluted sample per well. Include technical triplicates for each biological sample. Set up control wells containing 10 µL of inhibitor solution for background subtraction.
- Activity Measurement: Add 10 µL of substrate solution to each well (final concentration: 10 µM). Immediately measure fluorescence (excitation: 380 nm, emission: 460 nm) every 5 minutes for 60 minutes using a plate reader maintained at 37°C.
- Data Analysis: Calculate the rate of fluorescence increase (RFU/min) during the linear phase of the reaction. Subtract the rate from inhibitor-treated controls to determine specific proteasomal activity. Normalize activities to protein content and express as percentage of sham-operated controls.
Technical Notes: Maintain samples on ice throughout preparation to prevent activity loss. Include positive controls (commercial proteasome) to validate assay performance. For comprehensive assessment, parallel measurements of trypsin-like and caspase-like activities can be performed using appropriate substrates (Z-ARR-AMC and Z-LLE-AMC, respectively).
Protocol: Monitoring the Nrf1-Mediated Bounce-Back Response
Principle: This protocol assesses the transcriptional bounce-back response to proteasome inhibition using luciferase reporter assays and qRT-PCR analysis, based on established screening approaches [130]. The assay quantifies Nrf1-dependent transcription following proteasome inhibition and its modulation by anthracyclines.
Reagents:
- NIH-3T3 cells stably expressing 8xARE-firefly luciferase reporter
- Renilla luciferase control vector (transfection control)
- Proteasome inhibitor: 0.5 µM carfilzomib (CFZ)
- Anthracycline: 1 µM doxorubicin (DOX)
- Dual-luciferase reporter assay system
- RNA extraction kit and qRT-PCR reagents
- Primers for proteasome subunit genes (PSMB5, PSMB6) and autophagy genes (GABARAPL1, SQSTM1)
Procedure:
- Cell Seeding: Plate NIH-3T3 reporter cells in 24-well plates at 1×10⁵ cells/well and culture for 24 hours.
- Treatment: Pre-treat cells with 1 µM DOX or vehicle for 1 hour, then add 0.5 µM CFZ or vehicle. Incubate for 16 hours.
- Luciferase Assay: Lyse cells and measure firefly and Renilla luciferase activities using dual-luciferase reporter system according to manufacturer's instructions. Normalize firefly luminescence to Renilla values.
- Gene Expression Analysis: Extract total RNA from parallel samples. Perform reverse transcription and qRT-PCR using primers for proteasome and autophagy genes. Calculate fold changes using the 2^(-ΔΔCt) method with GAPDH as reference.
- Functional Validation: For proteasome activity recovery assays, treat cells with 0.5 µM CFZ for 3 hours, wash twice with PBS, then culture in fresh medium with or without 1 µM DOX. Measure proteasome activity at 0, 6, 12, and 24 hours post-wash using fluorogenic substrates as described in Section 5.1.
Technical Notes: The anthracycline doxorubicin disrupts Nrf1 binding to antioxidant response elements (AREs) without affecting Nrf1 processing or nuclear localization, providing a specific means to inhibit the bounce-back response [130]. Aclarubicin, a non-DNA-damaging anthracycline, can be used as a negative control to confirm that observed effects are DNA damage-independent.
Experimental Workflow: Integrated UPS Dysfunction Analysis
Integrated Workflow for UPS Dysfunction Analysis
Therapeutic Strategies for Managing UPS Dysfunction
Proteasome Inhibition in Hyperglycemic Ischemia-Reperfusion
Cardiac ischemia-reperfusion injury under hyperglycemic conditions presents a unique therapeutic scenario where partial UPS inhibition confers significant protection. The experimental approach involves isolated rat heart perfusions with Krebs-Henseleit buffer containing 33 mM glucose (simulating acute hyperglycemia) versus controls (11 mM glucose) for 60 minutes stabilization, followed by 20 minutes global ischemia and 60 minutes reperfusion [128]. Proteasome inhibitors (5 µM lactacystin or 10 µM MG-132) are added during the first 20 minutes of the reperfusion phase. This specific timing is critical for cardioprotection, which manifests as improved recovery of left ventricular developed pressure, reduced infarct size, and attenuated oxidative stress. The protective mechanisms involve enhanced superoxide dismutase protein levels (SOD1, SOD2), diminished pro-inflammatory responses, and upregulation of autophagic markers that collectively mitigate reperfusion injury [128]. This model demonstrates the context-dependent nature of UPS modulation, where the same system that requires support in neurodegenerative diseases may benefit from selective inhibition in other pathological conditions.
Targeting Adaptive Resistance in Cancer Therapy
In multiple myeloma and other malignancies, resistance to proteasome inhibitors frequently develops through activation of the Nrf1-mediated bounce-back response, which increases de novo proteasome synthesis to counteract therapeutic inhibition [130]. Combination approaches that simultaneously inhibit proteasome activity and block this adaptive response have demonstrated enhanced efficacy. Anthracyclines, including doxorubicin, disrupt Nrf1 binding to antioxidant response elements (AREs) without affecting Nrf1 processing or nuclear localization, thereby attenuating transcriptions of proteasome subunit genes and autophagy-lysosomal pathway genes [130]. The experimental protocol involves co-treatment of cancer cells with carfilzomib (0.5 µM) and doxorubicin (1 µM) for 16-24 hours, resulting in impaired proteasome recovery and restored sensitivity in resistant cell lines. Importantly, the non-DNA-damaging anthracycline aclarubicin produces similar effects, confirming that DNA damage is not required for Nrf1 inhibition [130]. This approach exemplifies the strategic targeting of compensatory pathways to enhance primary therapeutic efficacy.
Emerging Directions and Combination Therapies
Future therapeutic strategies for managing UPS dysfunction increasingly focus on system-level approaches that acknowledge the interconnected nature of proteostatic pathways. As UPS impairment often triggers compensatory autophagy induction, combined modulation of both systems may yield synergistic benefits [122] [123]. In neurodegenerative models, enhancing autophagy when the UPS is compromised can alleviate proteotoxic stress by clearing aggregation-prone proteins that would otherwise overwhelm proteasomal capacity. Conversely, in cancers with elevated proteasome activity, simultaneous inhibition of both degradation pathways can induce synergistic proteotoxic stress [127].
Advanced therapeutic platforms including proteolysis-targeting chimeras (PROTACs) that harness endogenous E3 ligases to degrade specific disease-causing proteins represent a promising direction that shifts the strategy from inhibiting the UPS to redirecting its activity against pathological targets [123]. Similarly, molecular tweakers that modulate E3 ligase specificity or activity offer precision approaches for restoring UPS function in specific disease contexts without global proteasome inhibition. The integration of ubiquitinomics profiling with functional screening approaches will continue to identify novel nodes for therapeutic intervention in the complex network of UPS regulation [129] [123].
The strategic management of UPS dysfunction in disease models requires sophisticated experimental approaches that account for the system's complexity, contextual dependencies, and adaptive capacity. This technical guide has outlined standardized methodologies for modeling UPS impairment, quantifying its functional consequences, and targeting its pathological manifestations across diverse disease contexts. The paradoxical nature of UPS modulation—where inhibition may be therapeutic in some contexts while detrimental in others—underscores the importance of precise, context-aware interventions. The continuing development of targeted protein degradation technologies and combinatorial approaches that address the broader proteostatic network will undoubtedly yield novel therapeutic strategies for diseases characterized by UPS dysfunction. Researchers should implement the protocols and visualization tools presented here to advance our understanding of UPS biology and translate these insights into effective treatments for human diseases.
UPS in Context: Disease Validation, Pathway Comparison, and Therapeutic Assessment
The ubiquitin-proteasome system (UPS) represents a crucial regulatory pathway for intracellular protein degradation, maintaining cellular homeostasis through precise control of protein turnover. In cancer, dysregulation of UPS components leads to aberrant accumulation of oncoproteins and enhanced degradation of tumor suppressors, driving tumor initiation, progression, and therapeutic resistance. This comprehensive review examines the molecular mechanisms of UPS dysregulation in cancer pathogenesis, highlighting the therapeutic validation of UPS-targeting strategies. We explore how cancer cells exploit specific UPS components—including E3 ubiquitin ligases, deubiquitinating enzymes (DUBs), and the proteasome—to support survival, proliferation, and immune evasion. The clinical advances in proteasome inhibitors, emerging targeted protein degradation technologies, and novel small molecule inhibitors are critically evaluated, providing a framework for developing next-generation anti-cancer therapies centered on UPS manipulation.
The ubiquitin-proteasome system (UPS) is the primary selective pathway for intracellular protein degradation in eukaryotic cells, responsible for regulating approximately 80-90% of cellular proteins [28]. This sophisticated system controls protein stability through a hierarchical enzymatic cascade that tags target proteins with ubiquitin chains for recognition and degradation by the proteasome. The UPS maintains precise regulation of critical cellular processes including cell cycle progression, transcription, DNA repair, apoptosis, and immune responses [28] [131]. The foundational discovery of the UPS dates to the 1980s, when scientists first identified ubiquitination as a novel protein modification mechanism. By the 1990s, the association between UPS dysfunction and human diseases, particularly cancer, became increasingly evident [28]. The system's importance was further validated when the clinical application of proteasome inhibitors demonstrated significant efficacy in hematological malignancies, establishing the UPS as a legitimate therapeutic target in oncology [28] [85].
Molecular Mechanisms of the UPS
The Ubiquitination Cascade
Ubiquitination involves a coordinated three-step enzymatic cascade that conjugates the small, highly conserved ubiquitin protein to specific substrate proteins:
- Activation (E1): A single ubiquitin-activating enzyme (E1) activates ubiquitin in an ATP-dependent reaction, forming a thioester bond between a cysteine residue in E1's active site and the C-terminal glycine of ubiquitin [28].
- Conjugation (E2): The activated ubiquitin is transferred to one of approximately 40 ubiquitin-conjugating enzymes (E2) through transesterification [28].
- Ligation (E3): One of over 600 ubiquitin ligases (E3) facilitates the transfer of ubiquitin from E2 to a lysine residue on the target protein, forming an isopeptide bond [28] [132].
E3 ubiquitin ligases provide substrate specificity by recognizing target proteins and facilitating polyubiquitin chain assembly. The major classes of E3 ligases—RING, HECT, and RBR—employ distinct mechanisms for ubiquitin transfer [80]. Polyubiquitin chains linked through lysine 48 (K48) or lysine 11 (K11) typically target substrates for proteasomal degradation, while K63-linked chains generally regulate proteasome-independent signaling processes [131] [132].
Figure 1: The Ubiquitination Cascade. This diagram illustrates the sequential enzymatic process of protein ubiquitination, culminating in the tagging of substrate proteins with polyubiquitin chains that signal for proteasomal degradation.
The Proteasome and Protein Degradation
The 26S proteasome is a massive, multi-subunit proteolytic complex that recognizes and degrades ubiquitinated proteins. It consists of:
- 20S Core Particle (CP): A barrel-shaped structure containing proteolytic active sites that degrade target proteins into small peptides [28] [132].
- 19S Regulatory Particle (RP): Caps on one or both ends of the 20S core that recognize ubiquitinated substrates, remove ubiquitin chains, unfold target proteins, and facilitate their translocation into the catalytic core [28] [132].
The degradation process is ATP-dependent and results in the release of short peptide fragments (typically 3-25 amino acids in length) that are further degraded to amino acids for recycling or immune presentation [28]. Deubiquitinating enzymes (DUBs) counterbalance ubiquitination by removing ubiquitin chains, thereby rescuing substrates from degradation or modulating their signaling functions [131] [133]. The coordinated action of ubiquitination and deubiquitination allows dynamic, precise control of protein half-lives essential for cellular homeostasis.
Mechanisms of UPS Dysregulation in Cancer
Cancer cells systematically dysregulate specific UPS components to create a microenvironment conducive to tumor survival, proliferation, and metastasis. The table below summarizes key UPS components frequently dysregulated in human cancers.
Table 1: Key UPS Components Dysregulated in Cancer
| UPS Component | Type | Cancer-Associated Alteration | Oncogenic Effect |
|---|---|---|---|
| MDM2 | E3 Ligase | Overexpression | Degrades tumor suppressor p53, promoting uncontrolled growth [134] |
| VHL | E3 Ligase | Inactivating mutations | Stabilizes HIF-1α, driving angiogenesis and metabolic reprogramming [135] |
| SPOP | E3 Ligase | Mutations/Competitive binding | Stabilizes PD-L1, facilitating immune evasion [132] |
| c-CBL | E3 Ligase | Loss-of-function mutations | Enhances receptor tyrosine kinase signaling [131] |
| USP21 | DUB | Overexpression | Stabilizes oncoproteins (NF-κB, β-catenin) [133] |
| USP7 | DUB | Overexpression | Stabilizes MDM2, suppressing p53 function [133] |
| Immunoproteasome | Proteasome variant | Overexpression in hematologic cancers | Enhanced survival, drug resistance [85] |
Dysregulation of Tumor Suppressors and Oncogenes
UPS-mediated control of critical tumor suppressors and oncogenes represents a fundamental mechanism in carcinogenesis:
p53 Regulation: MDM2, an E3 ubiquitin ligase frequently overexpressed in cancers, targets the p53 tumor suppressor for degradation. This leads to genomic instability, impaired DNA damage response, and uncontrolled cell cycle progression [134]. Mutations in the MDM2-p53 axis occur in over 50% of human cancers.
HIF-1α Stabilization: Under normoxic conditions, the von Hippel-Lindau (VHL) E3 ligase targets hypoxia-inducible factor 1α (HIF-1α) for proteasomal degradation. In many cancers, VHL loss or mutation results in HIF-1α stabilization even under oxygen-rich conditions, driving angiogenesis, metabolic reprogramming, and metastasis [135].
Transcription Factor Control: Numerous transcription factors critical for cancer progression are regulated by ubiquitination. For example, the ubiquitination of NF-κB subunits, STAT proteins, and β-catenin significantly impacts their stability and transcriptional activity in cancer cells [135].
Apoptosis Evasion via Bcl-2 Regulation
The UPS precisely regulates the balance between pro-apoptotic and anti-apoptotic Bcl-2 family proteins. Cancer cells exploit this regulation to resist programmed cell death:
Anti-apoptotic Protein Stabilization: Overexpression of anti-apoptotic proteins like Bcl-2, Bcl-xL, and Mcl-1 is common in many cancers. These proteins are frequently stabilized through reduced ubiquitination or increased deubiquitination, enhancing cell survival [80].
Pro-apoptotic Protein Degradation: Conversely, pro-apoptotic proteins such as Bax, Bak, and various BH3-only proteins may be targeted for accelerated degradation in cancer cells, further tilting the balance toward survival [80].
Therapeutic Implications: The proteasome-Bcl-2 axis represents a promising therapeutic target. Proteasome inhibitors can indirectly modulate this balance by preventing the degradation of pro-apoptotic factors, thereby promoting cancer cell death [80].
Immune Evasion Through PD-1/PD-L1 Regulation
Tumor cells manipulate the UPS to evade anti-tumor immunity by regulating immune checkpoint proteins:
PD-L1 Stabilization: The E3 ubiquitin ligase SPOP normally targets programmed death-ligand 1 (PD-L1) for ubiquitination and degradation. In various cancers, including colorectal and hepatocellular carcinomas, competitive binding by proteins like ALDH2 or BCLAF1 disrupts SPOP-mediated PD-L1 degradation, leading to PD-L1 accumulation on tumor cells and subsequent T-cell inhibition [132].
Metabolic Regulation of PD-L1: SGLT2, a glucose transporter upregulated in some cancers, competitively binds PD-L1 and prevents its SPOP-mediated ubiquitination. SGLT2 inhibitors like canagliflozin can disrupt this interaction, restoring PD-L1 degradation and enhancing T-cell-mediated tumor killing [132].
UPS Modulation in Immune Cells: Beyond directly regulating checkpoint proteins, the UPS controls T-cell activation, differentiation, and function through ubiquitination of key signaling molecules, further influencing anti-tumor immunity [131].
Therapeutic Targeting of the UPS in Cancer
Proteasome Inhibitors
Proteasome inhibitors were the first class of UPS-targeting agents to achieve clinical success, particularly for hematological malignancies:
Table 2: Clinically Approved Proteasome Inhibitors in Cancer Therapy
| Drug Name | Chemical Class | Mechanism of Action | Primary Indications | Key Limitations |
|---|---|---|---|---|
| Bortezomib | Peptide boronate | Reversible proteasome inhibition | Multiple myeloma, mantle cell lymphoma [28] [85] | Peripheral neuropathy, drug resistance |
| Carfilzomib | Epoxyketone | Irreversible proteasome inhibition | Relapsed/refractory multiple myeloma [28] [85] | Cardiotoxicity, limited efficacy in solid tumors |
| Ixazomib | Peptide boronate | Oral reversible inhibitor | Multiple myeloma [28] [85] | Gastrointestinal toxicity |
| Marizomib | β-lactone | Pan-proteasome inhibitor | Clinical trials for multiple myeloma and glioblastoma [85] | CNS-related adverse effects |
The mechanism of action of proteasome inhibitors involves disrupting protein homeostasis, leading to the accumulation of pro-apoptotic proteins, cell cycle regulators, and unfolded proteins. This ultimately induces endoplasmic reticulum stress and activates apoptotic pathways in cancer cells, which are particularly vulnerable to proteotoxic stress due to their high protein synthesis rates [28] [85]. While highly effective in hematological malignancies, proteasome inhibitors have demonstrated limited efficacy in solid tumors, prompting research into combination therapies and next-generation agents.
Targeted Protein Degradation: PROTACs and Molecular Glues
Targeted protein degradation represents a paradigm shift in UPS-focused cancer therapy, enabling precise targeting of specific oncoproteins:
PROTAC Technology: Proteolysis-Targeting Chimeras (PROTACs) are heterobifunctional molecules consisting of three elements: a warhead that binds the target protein, a linker, and an E3 ligase recruiter. PROTACs hijack endogenous E3 ligases (such as cereblon or VHL) to selectively ubiquitinate and degrade target oncoproteins [28] [136].
Molecular Glues: These small molecules induce or stabilize interactions between E3 ubiquitin ligases and target proteins, leading to selective ubiquitination and degradation. Unlike PROTACs, molecular glues are typically smaller, monomeric compounds that don't require a linker [131].
Advantages over Traditional Inhibitors: Targeted protein degradation offers several advantages, including the ability to target "undruggable" proteins, catalytic activity (single degrader molecules can facilitate multiple degradation cycles), and potential overcoming of resistance mutations [28] [136].
E3 Ligase and DUB Inhibitors
The development of specific inhibitors targeting individual UPS components represents a promising therapeutic approach:
E3 Ligase Inhibitors: Compounds targeting specific E3 ligases, such as MDM2 inhibitors (nutlins), can reactivate p53 signaling in tumors with wild-type p53, promoting apoptosis [134] [135].
DUB Inhibitors: Small molecule inhibitors targeting deubiquitinating enzymes overexpressed in cancers can restore the degradation of oncoproteins. For example, USP21 inhibitors such as the phytoconstituents Ranmogenin A and Tokorogenin have shown promise in preclinical studies for promoting the degradation of oncoproteins like NF-κB and β-catenin [133].
Specificity Challenges: Achieving specificity remains a significant hurdle for E3 and DUB inhibitors due to the large number of structurally similar enzymes in these families. Advanced screening techniques and structural biology approaches are being employed to develop more selective agents [133].
Experimental Approaches for UPS Research
Research Reagent Solutions
Table 3: Essential Research Reagents for UPS Investigation
| Reagent Category | Specific Examples | Research Applications | Key Functions |
|---|---|---|---|
| Proteasome Inhibitors | MG132, Epoxomicin, Bortezomib | In vitro and cellular UPS inhibition studies | Block proteasomal activity, induce ER stress and apoptosis [85] |
| E3 Ligase Modulators | Nutlin-3 (MDM2 inhibitor), MLN4924 (NEDD8 inhibitor) | Target validation, mechanism studies | Modulate specific ubiquitination pathways [135] |
| DUB Inhibitors | P5091 (USP7 inhibitor), Ranmogenin A, Tokorogenin (USP21 inhibitors) | DUB functional studies, therapeutic development | Inhibit deubiquitination, promote substrate degradation [133] |
| Ubiquitination Assays | Ubiquitin conjugation kits, TUBE (Tandem Ubiquitin Binding Entity) reagents | In vitro and cellular ubiquitination detection | Detect, quantify, and characterize protein ubiquitination [133] |
| Activity Reporters | Ubiquitin-proteasome pathway reporters, GFPu degradation sensor | Real-time UPS function monitoring | Measure proteasome activity in live cells [133] |
Methodological Framework for USP21 Inhibitor Screening
Recent research has established comprehensive protocols for identifying and validating novel UPS-targeting compounds:
Figure 2: USP21 Inhibitor Screening Workflow. This diagram outlines the integrated virtual screening strategy for identifying bioactive phytoconstituents as USP21 inhibitors, combining computational and experimental approaches.
The screening methodology involves sequential filtering stages:
Virtual Screening: A library of approximately 18,000 phytochemicals from the IMPPAT 2.0 database is initially filtered using Lipinski's Rule of Five to identify compounds with favorable physicochemical properties [133].
Molecular Docking: Filtered compounds (approximately 12,000) undergo molecular docking against the target USP21 structure (PDB ID: 3I3T) using MGL AutoDock Tools and InstaDock v1.2 with a standardized grid configuration (79×65×85 Å) centered at coordinates X:14.508, Y:19.994, Z:-34.675 [133].
ADMET Prediction: Top-binding candidates are evaluated for absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties using tools like Deep-PK, with PAINS filtering to eliminate promiscuous binders [133].
PASS Analysis: The Prediction of Activity Spectra for Substances (PASS) algorithm predicts biological activity spectra based on structure-activity relationships, prioritizing compounds with Pa > 0.3 for predicted anticancer activity [133].
Molecular Dynamics Simulations: Selected compounds undergo 500 ns all-atom MD simulations using GROMACS with GROMOS 96 force field to evaluate complex stability, conformational flexibility, and binding thermodynamics [133].
This integrated approach successfully identified Ranmogenin A and Tokorogenin as promising USP21 inhibitors with favorable binding affinities and pharmacokinetic properties, demonstrating the utility of systematic screening methodologies for UPS drug discovery [133].
The ubiquitin-proteasome system represents a critically important pathway in cancer biology, with dysregulation of its components contributing fundamentally to tumor pathogenesis. Understanding the precise molecular mechanisms of UPS dysfunction in specific cancer types provides valuable insights for developing targeted therapeutic interventions. While proteasome inhibitors have established clinical utility, particularly in hematological malignancies, emerging strategies including PROTACs, molecular glues, and specific E3/DUB inhibitors offer promising avenues for enhancing therapeutic efficacy and overcoming resistance.
Future research directions should focus on developing isoform-selective proteasome inhibitors, expanding the repertoire of E3 ligases available for targeted protein degradation, and identifying optimal combination therapies that leverage UPS-targeting agents with conventional chemotherapy, radiation, or immunotherapy. Additionally, advancing our understanding of UPS modulation in the tumor microenvironment may reveal novel opportunities for manipulating anti-tumor immunity. As screening methodologies and structural biology techniques continue to evolve, the systematic targeting of specific UPS components holds significant potential for precision oncology approaches tailored to individual tumor vulnerabilities.
The ubiquitin-proteasome system (UPS) represents the primary intracellular pathway for targeted protein degradation in eukaryotic cells, serving as a critical mechanism for maintaining cellular proteostasis. This sophisticated system regulates the turnover of short-lived regulatory proteins, damaged polypeptides, and misfolded proteins, thereby influencing essential cellular processes including cell cycle progression, stress responses, and apoptotic signaling [137]. The UPS operates through a coordinated two-step process: (1) ubiquitination, which involves the covalent attachment of ubiquitin chains to target proteins, and (2) proteasomal degradation, where labeled proteins are recognized and processed into short peptides [137]. In the context of neuronal health and function, the UPS exhibits particular significance given the post-mitotic nature of neurons and their limited capacity for protein dilution through cell division. The system maintains normal central nervous system function by regulating protein degradation, and when compromised, cellular proteostasis becomes disrupted, accelerating neurodegenerative processes [138].
Neurodegenerative disorders (NDDs), including Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD), and amyotrophic lateral sclerosis (ALS), share a common pathological feature: the accumulation of ubiquitin-positive intracellular inclusions formed by aggregate-prone neurotoxic proteins [139]. This observation initially positioned UPS dysfunction as a prime suspect in the pathophysiology of neurodegeneration. However, over the past decade, a paradigm shift has occurred, repositioning the UPS from being merely a dysfunctional bystander to an attractive therapeutic target that can be harnessed to accelerate the clearance of disease-linked proteins [122]. This whitepaper comprehensively examines the role of UPS in neurodegenerative diseases, exploring both its pathological implications and emerging therapeutic opportunities for drug development professionals and researchers.
Molecular Architecture of the Ubiquitin-Proteasome System
The Ubiquitination Cascade
The ubiquitination process employs a sequential enzymatic cascade that confers specificity to protein targeting:
- E1 Ubiquitin-Activating Enzymes: Initiate ubiquitination by activating ubiquitin in an ATP-dependent manner through formation of a thioester bond with a catalytic cysteine residue [139].
- E2 Ubiquitin-Conjugating Enzymes: Receive activated ubiquitin from E1 enzymes. Mammalian systems express approximately 35 distinct E2 enzymes that contribute to substrate diversity [137].
- E3 Ubiquitin Ligases: Facilitate the transfer of ubiquitin from E2 enzymes to specific substrate proteins. With over 600 E3 ligases identified in mammalian cells, this enzyme class provides the system with remarkable substrate specificity [137]. E3 ligases are categorized into three major families based on their structural domains and mechanisms: Really Interesting New Gene (RING)/U-box domain-containing E3s, Homologous to E6AP C-terminus (HECT) domain-containing E3s, and RING-between-RING (RBR) domain-containing E3s [139].
The fate of ubiquitinated proteins is determined by the topology of the polyubiquitin chain. Lys48-linked chains typically target substrates for proteasomal degradation, while Lys63-linked chains are involved in non-proteolytic functions including signaling, endocytosis, and DNA repair [122]. Other chain topologies, including Lys11 and Lys29 linkages, can also serve as proteasomal targeting signals [122].
Proteasome Assemblies and Architecture
The 26S proteasome represents the fundamental functional unit of the UPS and consists of two primary structural components:
- 20S Core Particle (CP): A barrel-shaped complex composed of four stacked heptameric rings (arranged as α7β7β7α7) that houses the proteolytic active sites within its interior cavity. The β-subunits contain three principal proteolytic activities: caspase-like (β1), trypsin-like (β2), and chymotrypsin-like (β5) [137].
- 19S Regulatory Particle (RP/PA700): A multi-subunit complex that caps one or both ends of the 20S core particle. The 19S RP recognizes ubiquitinated substrates, removes ubiquitin chains, unfolds target proteins, and translocates them into the catalytic chamber of the 20S core in an ATP-dependent manner [137].
The following diagram illustrates the core components and degradation process of the UPS:
Figure 1: UPS Pathway Schematic. The ubiquitin-proteasome system involves sequential enzymatic activities (E1, E2, E3) that tag substrate proteins with ubiquitin chains, leading to recognition and degradation by the proteasome complex.
Beyond the constitutive proteasome, specialized variants exist with tissue-specific expression and functional specialization:
- Immunoproteasome: Induced by interferon-γ exposure, features alternative catalytic subunits (LMP2/β1i, MECL-1/β2i, LMP7/β5i) with enhanced cleavage activity after hydrophobic and basic residues, potentially improving generation of antigenic peptides [137].
- Hybrid Proteasomes: Contain different regulatory particles at each end of the 20S core (e.g., 19S-20S-11S configurations) that exhibit distinct substrate preferences and catalytic efficiencies [137].
Table 1: Proteasome Assemblies and Their Functional Characteristics
| Proteasome Type | Structural Composition | Activator Particles | Primary Functions | Distribution |
|---|---|---|---|---|
| Constitutive Proteasome | 20S core with standard β subunits | 19S (unidirectional or bidirectional) | Bulk protein degradation, cell cycle regulation | Ubiquitous |
| Immunoproteasome | 20S core with β1i, β2i, β5i subunits | 19S or 11S | Antigen processing, inflammatory responses | Immune cells, inflamed tissues |
| Hybrid Proteasome | 20S core | 19S at one end, 11S at other | Specialized substrate processing | Multiple tissues |
| Blm10/PA200 Complex | 20S core | Blm10/PA200 | DNA repair, spermatogenesis | Nucleus, testes |
UPS Dysregulation in Major Neurodegenerative Disorders
Alzheimer's Disease (AD)
Alzheimer's disease pathology is characterized by extracellular amyloid-β plaques and intracellular neurofibrillary tangles composed of hyperphosphorylated tau protein. The UPS intersects with AD pathogenesis through multiple mechanisms:
- Amyloid-β Clearance: The UPS participates in the degradation of amyloid precursor protein (APP) and amyloid-β peptides. UPS dysfunction promotes amyloidogenic processing of APP and accumulation of Aβ oligomers [140].
- Tau Metabolism: Normal tau is a UPS substrate, but hyperphosphorylated tau aggregates resist degradation and may directly impair proteasome function [140] [139].
- Ubiquitin-Positive Inclusions: Both amyloid plaques and neurofibrillary tangles contain ubiquitinated proteins, suggesting unsuccessful targeting of pathological aggregates to the UPS [139].
Recent evidence indicates that UPS dysfunction in AD may occur early in disease pathogenesis and contributes to the accumulation of multiple pathological proteins. The system offers a promising therapeutic target for AD prevention and treatment, with ongoing preclinical studies investigating UPS modulation strategies [140].
Parkinson's Disease (PD)
Parkinson's disease involves the degeneration of dopaminergic neurons in the substantia nigra and the presence of Lewy bodies containing aggregated α-synuclein:
- Genetic Evidence: Multiple familial PD-linked genes encode UPS components, including Parkin (an E3 ubiquitin ligase) and UCHL1 (a deubiquitinating enzyme) [139].
- α-Synuclein Degradation: Both the UPS and autophagy-lysosomal pathway participate in α-synuclein clearance. Misfolded α-synuclein species can directly inhibit proteasome function, creating a vicious cycle of impaired proteostasis [122].
- Mitophagy Connection: Parkin, together with PINK1, regulates mitochondrial quality control through ubiquitin-mediated targeting of damaged mitochondria to autophagic degradation, linking UPS dysfunction to mitochondrial impairment in PD [122].
Polyglutamine Disorders
Huntington's disease and other polyglutamine expansion diseases share common pathogenic features:
- Aggregation-Prone Proteins: Proteins with expanded polyQ tracts (e.g., mutant huntingtin) exhibit structural instability and resistance to UPS-mediated degradation [122].
- Proteasome Impairment: Early hypotheses suggested that polyQ aggregates physically obstruct the proteasome, but recent evidence indicates the UPS remains largely functional in many polyQ disease models [122].
- Selective Vulnerability: Distinct UPS components may be differentially affected in various brain regions, contributing to selective neuronal vulnerability despite widespread expression of mutant proteins [122].
Table 2: UPS-Related Abnormalities in Major Neurodegenerative Disorders
| Disease | Key Pathological Proteins | UPS-Related Genetic Factors | Primary UPS Defects | Characteristic Inclusions |
|---|---|---|---|---|
| Alzheimer's Disease | Aβ, hyperphosphorylated tau | Ubiquilin polymorphisms, PS1 mutations | Impaired proteasome activity, deficient ubiquitination | NFTs, amyloid plaques |
| Parkinson's Disease | α-Synuclein | Parkin, UCHL1, PINK1 mutations | E3 ligase dysfunction, impaired mitochondrial clearance | Lewy bodies |
| Huntington's Disease | mutant Huntingtin (polyQ) | - | Impaired substrate recognition, sequestration of UPS components | Nuclear inclusions |
| Amyotrophic Lateral Sclerosis | TDP-43, SOD1 | - | Proteasomal dysfunction, impaired protein targeting | Cytoplasmic inclusions |
Emerging Therapeutic Strategies Targeting the UPS
Proteasome Activation Approaches
Unlike oncological applications where proteasome inhibition is therapeutic, neurodegenerative diseases may benefit from enhanced UPS function:
- Small Molecule Activators: Compounds that increase proteasome assembly or enhance proteolytic activity offer potential for reducing toxic protein burden [138].
- Gene-Based Therapies: Modulation of UPS component expression through viral vector-mediated delivery of E3 ligases, deubiquitinating enzymes, or proteasome subunits [138].
- Hybrid Modulators: Molecules that simultaneously enhance proteasome function while activating complementary degradation pathways like autophagy [25].
Targeted Protein Degradation
Novel strategies leverage UPS components to selectively target disease proteins:
- PROTACs (Proteolysis-Targeting Chimeras): Bifunctional molecules that recruit E3 ubiquitin ligases to specific pathogenic proteins, promoting their ubiquitination and degradation [138].
- Molecular Triage Enhancement: Compounds that improve recognition of misfolded proteins by the UPS while sparing normally folded cellular proteins [122].
UPS Modulation by Natural Compounds
Circulating polyphenol-derived metabolites demonstrate UPS-modulating properties:
- Valerolactones (from flavan-3-ols) and Urolithins (from ellagitannins) exhibit tissue-specific effects on proteasome function [25].
- Hydroxycinnamic acids (ferulic and caffeic acids) can affect proteasome activity through diverse mechanisms, including autophagy induction and modulation of ubiquitination enzymes [25].
- Dose Considerations: Current evidence primarily derives from supraphysiological concentrations in cellular models, highlighting the need for physiologically relevant dosing studies [25].
Experimental Approaches for UPS Research
Methodologies for Assessing UPS Function
Research into UPS function employs multiple complementary approaches:
- Proteasome Activity Assays: Fluorogenic peptide substrates that measure chymotrypsin-like, trypsin-like, and caspase-like proteasome activities in cell lysates or tissue homogenates [122].
- Ubiquitin-Proteasome Reporter Systems: Fluorescent or luminescent protein substrates containing degradation signals that enable real-time monitoring of UPS function in living cells [122].
- Protein Turnover Measurements: Pulse-chase analyses combined with immunoprecipitation to determine degradation kinetics of specific UPS substrates [139].
- Ubiquitin Chain Topology Analysis: Mass spectrometry-based methods to characterize polyubiquitin chain linkages associated with specific substrates or pathological inclusions [122].
The following experimental workflow illustrates a comprehensive approach to investigating UPS function in neurodegenerative disease models:
Figure 2: UPS Research Workflow. Comprehensive experimental approach for investigating UPS function in neurodegenerative disease models, from baseline characterization to therapeutic intervention.
Case Study: OTULIN and Tau Regulation
A groundbreaking recent study revealed unexpected mechanisms by which UPS components influence neurodegenerative processes:
- Initial Hypothesis: Researchers hypothesized that inhibiting the deubiquitinase OTULIN would enhance tau clearance through ubiquitin-mediated degradation [141].
- Unexpected Discovery: Complete OTULIN knockout eliminated tau not through enhanced degradation, but by preventing its production at the transcriptional level [141].
- Mechanistic Insights: OTULIN deficiency caused dramatic changes in gene expression (13,341 genes downregulated) and RNA metabolism, positioning this deubiquitinase as a master regulator of neuronal gene expression [141].
- Therapeutic Translation: Partial pharmacological inhibition of OTULIN with small molecule inhibitor UC495 reduced pathological tau forms without complete elimination, suggesting a viable therapeutic window [141].
Table 3: Essential Research Reagents for UPS Investigation
| Reagent Category | Specific Examples | Research Applications | Technical Considerations |
|---|---|---|---|
| Proteasome Activity Reporters | Fluorogenic peptides (LLVY-AMC), GFP-based degradation reporters | Dynamic assessment of proteasome function in live cells and tissues | Substrate specificity (chymotrypsin-like vs caspase-like activities) |
| UPS Modulators | MG132 (inhibitor), Bortezomib, Betulinic acid (activator) | Experimental manipulation of UPS function | Dose-dependent effects, potential off-target activities |
| Genetic Tools | CRISPR/Cas9 constructs, siRNA libraries, Viral vectors for UPS component expression | Targeted manipulation of specific UPS pathway elements | Compensation by related genes, transduction efficiency |
| Ubiquitin Probes | Tandem ubiquitin-binding entities (TUBEs), Linkage-specific antibodies | Detection and isolation of ubiquitinated proteins | Affinity variations, chain linkage specificity |
| Patient-Derived Cells | iPSC-derived neurons from neurodegenerative disease patients | Human disease modeling, personalized therapeutic screening | Differentiation efficiency, phenotypic stability |
The ubiquitin-proteasome system represents both a vulnerable pathway in neurodegenerative pathogenesis and a promising therapeutic target. The historical view of UPS as a passive victim of protein aggregation has evolved to recognize its active role in disease progression and potential for therapeutic manipulation. Current evidence suggests that rather than global UPS impairment, more selective dysfunction affecting specific substrates or cellular compartments may drive neurodegeneration [122].
Future research directions should prioritize:
- Precision UPS Therapeutics: Developing strategies that enhance degradation of specific pathological proteins without disrupting overall proteostasis [138].
- Biomarker Development: Identifying UPS component alterations in accessible biofluids that reflect brain pathology for diagnostic and monitoring purposes [142].
- Combination Approaches: Integrating UPS-targeted therapies with complementary protein clearance mechanisms, such as autophagy enhancement [25].
- Temporal Considerations: Recognizing that UPS therapeutic interventions may have differing efficacy across disease stages, necessitating careful timing of interventions [140].
The expanding toolkit for investigating and modulating UPS function, coupled with growing understanding of its complex roles in neuronal health and disease, positions this pathway as a fertile ground for developing transformative therapies for neurodegenerative disorders. As research methodologies advance and large-scale collaborative efforts like the Global Neurodegeneration Proteomics Consortium generate comprehensive datasets [142], the integration of UPS-focused strategies into the neurodegenerative disease therapeutic pipeline holds significant promise for addressing these devastating conditions.
1. Introduction
Within eukaryotic cells, protein homeostasis is maintained by two primary degradation systems: the Ubiquitin-Proteasome System (UPS) and the Autophagic-Lysosomal Pathway (ALP). The UPS is a highly selective, rapid-response system for degrading short-lived proteins, while autophagy handles bulk cytoplasmic components, damaged organelles, and protein aggregates. Understanding their distinct mechanisms, regulatory cross-talk, and functional interplay is fundamental to cellular biology and therapeutic development, particularly for cancer and neurodegenerative diseases. This review provides a comparative analysis of these systems, detailing their molecular mechanisms, experimental methodologies, and emerging therapeutic applications.
2. System Overview and Key Characteristics
The UPS and ALP represent complementary yet interconnected proteolytic pathways. The UPS primarily degrades proteins marked by ubiquitin chains, providing precise temporal control over key regulatory proteins. In contrast, autophagy, particularly macroautophagy, engulfs large cytoplasmic structures within double-membraned vesicles for lysosomal degradation, acting as a bulk clearance mechanism during stress or for cellular renewal [100].
Table 1: Core Characteristics of UPS and Autophagic-Lysosomal Degradation
| Feature | Ubiquitin-Proteasome System (UPS) | Autophagic-Lysosomal Pathway (ALP) |
|---|---|---|
| Primary Function | Selective degradation of short-lived soluble proteins [143] | Bulk degradation of long-lived proteins, aggregates, and organelles (e.g., mitochondria, lysosomes) [143] [144] |
| Degradation Machinery | 26S proteasome complex | Lysosomal hydrolases [143] |
| Key Recognition Signal | K48-linked polyubiquitin chain (primarily for proteasomal degradation) | Ubiquitin-dependent (e.g., via p62) and ubiquitin-independent (e.g., via LC3 receptors) signals [144] |
| Cellular Role | Rapid regulation of cell cycle, transcription, signaling | Cellular quality control, nutrient recycling during stress, organelle turnover [144] [100] |
| Therapeutic Target Examples | Proteasome inhibitors (Bortezomib) [145] | AUTAC, ATTEC, AUTOTAC degraders [144] |
3. Molecular Mechanisms and Signaling Pathways
3.1. The Ubiquitin-Proteasome System (UPS) The UPS is a master regulator of intracellular protein turnover. Its operation involves a well-defined enzymatic cascade:
- Ubiquitination: A three-enzyme cascade (E1 activating, E2 conjugating, E3 ligating) tags target proteins with a polyubiquitin chain. K48-linked ubiquitination typically serves as the degradation signal [145].
- Recognition and Degradation: The ubiquitinated protein is recognized by the 26S proteasome, a multi-subunit complex comprising a regulatory particle and a core particle. The protein is unfolded, deubiquitinated, and degraded into short peptides within the proteasome's catalytic chamber [100].
Diagram 1: The Ubiquitin-Proteasome System (UPS) Pathway
3.2. The Autophagic-Lysosomal Pathway (ALP) The ALP, specifically macroautophagy, is a multi-step process for encapsulating and degrading cytoplasmic cargo:
- Phagophore Formation: Cellular stress signals (e.g., nutrient starvation) inhibit mTOR, initiating the formation of an isolation membrane (phagophore) [144].
- Cargo Recognition and Autophagosome Formation: The phagophore elongates and engulfs cargo. Selective autophagy relies on receptors like p62/SQSTM1, which binds both ubiquitin on cargo and LC3-II on the expanding phagophore membrane. LC3-II, a lipidated form of LC3, is a key marker embedded in the autophagosome membrane [144].
- Fusion and Degradation: The completed autophagosome fuses with a lysosome to form an autolysosome. The acidic environment and powerful hydrolases within the lysosome degrade the inner autophagosome membrane and its contents [143] [144].
Diagram 2: The Autophagic-Lysosomal Pathway (Macroautophagy)
3.3. Functional Crosstalk Between UPS and ALP The UPS and ALP are not isolated systems but engage in complex crosstalk. They can reciprocally regulate each other's components; for instance, autophagy can degrade proteasomes (a process known as "proteaphagy"), and the UPS can degrade key autophagy proteins [100]. The decision to degrade a substrate via one pathway or the other depends on factors like the substrate's aggregation status, ubiquitination code, and subcellular localization [100].
Diagram 3: UPS and ALP Crosstalk
4. Experimental Methodologies for Pathway Analysis
4.1. Analyzing UPS Function
- Western Blot for Ubiquitinated Proteins: Detect accumulation of polyubiquitinated proteins to infer UPS activity or inhibition. Treating cells with a proteasome inhibitor (e.g., MG132) serves as a positive control.
- Proteasome Activity Assay: Use fluorogenic peptides (e.g., Suc-LLVY-AMC) that, when cleaved by the proteasome's chymotrypsin-like activity, release a measurable fluorescent signal.
- Monitoring E3 Ligase-Specific Degradation: For a specific target like the transcription factor FOXP3, researchers can overexpress or knock down E3 ligases like STUB1 or CBLB and measure FOXP3 protein levels via Western blot to demonstrate UPS-mediated regulation [145].
4.2. Analyzing Autophagic-Lysosomal Function
- Western Blot for LC3 and p62: Monitor the conversion of LC3-I to LC3-II (indicating autophagosome formation) and the level of p62 (a substrate that decreases when autophagic flux is complete). Blocking lysosomal degradation with Bafilomycin A1 can confirm flux [144].
- Immunofluorescence Microscopy: Visualize autophagosome formation by tracking the punctate localization of GFP-LC3.
- Lysosomal Function Assays: Measure lysosomal enzyme activity (e.g., Cathepsin B/L) or use lysosomotropic dyes (e.g., LysoTracker) to assess lysosomal integrity and acidity, which are crucial for ALP function [143].
5. Quantitative Data and Substrate Specificity
Table 2: Substrate Specificity and Degradation Output
| Parameter | Ubiquitin-Proteasome System (UPS) | Autophagic-Lysosomal Pathway (ALP) |
|---|---|---|
| Primary Substrates | Short-lived regulatory proteins (e.g., cyclins, transcription factors), misfolded soluble proteins [143] | Protein aggregates, damaged mitochondria (mitophagy), damaged lysosomes (lysophagy), long-lived proteins [143] [144] |
| Degradation Rate | Rapid (minutes) | Slower (hours) |
| Energy Dependence | ATP-dependent (ubiquitination, proteasomal unfolding) | ATP-dependent (autophagosome formation, vesicle fusion) |
| Degradation Products | Short peptides (3-25 amino acids) | Amino acids, fatty acids, sugars |
6. Therapeutic Targeting and Technologies
Dysregulation of both pathways is implicated in numerous diseases, making them prime therapeutic targets. The UPS is a well-established target in oncology, while the ALP is increasingly targeted for neurodegenerative diseases.
Table 3: Emerging Therapeutic Technologies
| Technology | Targeted Pathway | Mechanism of Action | Application/Therapeutic Area |
|---|---|---|---|
| PROTACs [144] [145] | UPS | Bifunctional molecule binding E3 ubiquitin ligase and a target protein, leading to ubiquitination and proteasomal degradation. | Cancer (e.g., ARV-471 targeting ER for breast cancer) [144] |
| LYTACs [144] | Lysosome | Degrades extracellular and membrane proteins by recruiting them to lysosomal targeting receptors. | Cancer |
| ATTECs [144] | ALP | Bifunctional molecule binding both the target (e.g., lipid droplets, mutant protein) and LC3, tethering it to the autophagosome for degradation. | Neurodegenerative diseases |
| AUTACs [144] | ALP | Bifunctional molecule attaching a ubiquitin-like tag (guanine derivative) to the target, signaling it for selective autophagy. | Diseases with dysfunctional organelles |
7. The Scientist's Toolkit: Key Research Reagents
Table 4: Essential Reagents for UPS and ALP Research
| Research Reagent | Function/Application | Key Examples |
|---|---|---|
| Proteasome Inhibitors | Inhibit proteasomal activity to study UPS function or induce ER stress. | Bortezomib, MG132, Carfilzomib [145] |
| UPS Substrate Reporters | Monitor UPS-dependent degradation in live or fixed cells. | Ubiquitin-specific antibodies, GFP-based degradation reporters (e.g., GFPu) |
| Autophagy Inducers | Activate autophagy to study its effects or measure maximal flux. | Rapamycin (mTOR inhibitor), Starvation media |
| Autophagy Inhibitors | Block specific stages of autophagy to dissect the process. | 3-Methyladenine (early stage), Bafilomycin A1 (lysosomal fusion) [144] |
| LC3 Antibodies | Detect and quantify autophagosomes via Western blot or immunofluorescence. | Antibodies against LC3A/B |
| p62/SQSTM1 Antibodies | Monitor autophagic flux; levels inversely correlate with functional flux. | Anti-SQSTM1/p62 antibody [144] |
| Lysosomal Stains | Assess lysosomal mass, acidity, and integrity. | LysoTracker (acidity), LysoSensor, anti-LAMP1/2 antibodies [143] |
| E3 Ligase Modulators | Study the role of specific E3 ligases in target degradation. | Overexpression plasmids, siRNA/shRNA for knock-down (e.g., for STUB1, CBLB) [145] |
8. Conclusion
The Ubiquitin-Proteasome System and the Autophagic-Lysosomal Pathway represent two pillars of cellular proteostasis. The UPS offers speed and selectivity for targeted protein elimination, while the ALP provides capacity and range for large-scale clearance. Their intricate crosstalk ensures cellular adaptability. Current research is increasingly focused on exploiting this crosstalk and developing innovative technologies like PROTACs and ATTECs to target previously "undruggable" proteins. A deep understanding of both systems' mechanisms, their interactions, and the tools to study them is essential for advancing fundamental biology and developing next-generation therapeutics for cancer, neurodegenerative disorders, and other age-related diseases.
Targeted protein degradation via Proteolysis-Targeting Chimeras (PROTACs) represents a paradigm shift in therapeutic strategy, moving beyond the occupancy-driven model of traditional small-molecule inhibitors toward an event-driven catalytic approach. This whitepaper provides a comprehensive technical analysis comparing the efficacy and resistance profiles of PROTACs against conventional inhibitors within the framework of ubiquitin-proteasome system (UPS) pathway research. PROTAC technology harnesses the native UPS to achieve selective protein degradation, offering distinct advantages in targeting undruggable proteins, overcoming resistance mechanisms, and providing sustained pharmacological effects. We examine the mechanistic foundations, synthesize comparative efficacy data, and detail experimental protocols for evaluating degrader performance, providing researchers with essential methodologies for advancing this transformative therapeutic modality.
The ubiquitin-proteasome system (UPS) is the primary pathway for controlled intracellular protein degradation in eukaryotic cells, playing a crucial role in maintaining protein homeostasis by eliminating damaged, misfolded, or regulatory proteins [146] [49]. This highly regulated process involves a sequential enzymatic cascade: a ubiquitin-activating enzyme (E1) activates ubiquitin in an ATP-dependent manner; a ubiquitin-conjugating enzyme (E2) receives and transfers ubiquitin; and a ubiquitin ligase (E3) catalyzes the final attachment of ubiquitin to specific substrate proteins [147] [146]. Proteins tagged with K48-linked polyubiquitin chains are recognized and degraded by the 26S proteasome, a multi-subunit complex comprising a 20S core particle with proteolytic activity and 19S regulatory particles that recognize ubiquitinated substrates [146].
Targeted protein degradation (TPD) represents a revolutionary approach that exploits the natural protein quality control machinery to selectively eliminate disease-causing proteins [65] [49]. Unlike traditional inhibitors that merely block protein activity, TPD strategies aim to completely remove target proteins from cells. Among TPD technologies, PROteolysis TArgeting Chimeras (PROTACs) have emerged as the most advanced platform, with the first molecules entering clinical trials in 2019 and progressing to Phase III completion by 2024 [65]. This approach has unlocked therapeutic possibilities for previously "undruggable" targets, including transcription factors, mutant oncoproteins, and scaffolding proteins lacking conventional binding pockets [65] [67].
Mechanistic Foundations: PROTACs vs. Traditional Inhibitors
Traditional Small-Molecule Inhibitors
Conventional small-molecule therapeutics operate through an occupancy-driven mechanism of action [65] [148]. These inhibitors typically function by binding to active sites or allosteric pockets on target proteins, thereby blocking their biological function. This approach requires sustained high systemic drug concentrations to maintain target inhibition, as the pharmacological effect is directly proportional to the number of occupied receptors [51] [148]. Traditional inhibitors face fundamental limitations against proteins that lack well-defined binding pockets, exhibit high conformational plasticity, or display functional redundancy [65]. Current estimates suggest that only 10-15% of the human proteome is accessible to conventional small-molecule approaches, leaving vast territories of disease-relevant biology beyond therapeutic intervention [65].
PROTAC Mechanism of Action
PROTACs are heterobifunctional molecules consisting of three covalently linked components: a ligand that binds the protein of interest (POI), a ligand that recruits an E3 ubiquitin ligase, and a linker that bridges these two moieties [67] [148]. The molecular weight of PROTACs typically exceeds traditional drugs, often falling outside the scope of Lipinski's Rule of Five [148]. Unlike inhibitors, PROTACs do not require high-affinity binding to the POI's active site; even weak-affinity ligands can drive potent degradation if the linker supports favorable ternary complex geometry [65].
PROTACs operate through an event-driven, catalytic mechanism [65] [148]. The chimeric molecule facilitates the formation of a POI-PROTAC-E3 ternary complex, inducing spatial proximity between the target protein and the E3 ligase [67]. This proximity enables the transfer of ubiquitin chains from the E2 conjugating enzyme to lysine residues on the target protein [65]. The polyubiquitinated protein is then recognized and degraded by the 26S proteasome, while the PROTAC molecule is recycled to catalyze multiple rounds of degradation [65] [51]. This sub-stoichiometric mode of action differentiates PROTACs fundamentally from occupancy-driven inhibitors and can achieve more sustained effects [65].
Diagram 1: Comparative mechanisms of traditional inhibitors and PROTACs.
Comparative Efficacy Profiles
Quantitative Comparison of Pharmacological Properties
Table 1: Direct comparison of key characteristics between traditional inhibitors and PROTACs
| Feature/Capability | Small Molecule Inhibitors | Monoclonal Antibodies | PROTAC Protein Degraders |
|---|---|---|---|
| Mechanism of Action | Occupancy-driven (binds & inhibits) [65] | Occupancy-driven (blocks function or signaling) [65] | Event-driven, catalytic degradation [65] [148] |
| Target Scope | ~10-15% of proteome (requires functional pockets) [65] | Extracellular and membrane proteins [65] | Greatly expanded, including "undruggable" targets [65] [149] |
| Resistance Development | High (mutations affect binding) [149] | Moderate (mainly through antigen loss) [149] | Reduced (effective against some resistant mutants) [149] |
| Dosing Requirements | High, continuous (occupancy-driven) [51] | High, periodic [51] | Low, catalytic (sub-stoichiometric) [65] [51] |
| Effect Duration | Short (depends on PK half-life) [149] | Long (weeks) [149] | Extended (beyond PK, depends on protein resynthesis) [149] |
| Hook Effect | Not applicable | Not applicable | Present (high concentrations reduce efficacy) [65] [51] |
Efficacy Against Challenging Target Classes
PROTACs demonstrate particular efficacy against target classes that have proven resistant to traditional inhibition strategies. Transcription factors like STAT3, long considered among the most challenging cancer targets, are now tractable through systematic degradation [65]. Similarly, scaffolding proteins and regulatory molecules that lack conventional enzymatic activity can be effectively eliminated through PROTAC-mediated degradation, whereas traditional inhibitors would have limited efficacy against such targets [51] [149].
The catalytic nature of PROTACs enables potent effects even with lower target engagement. For example, BTK degraders have demonstrated superiority over ibrutinib in overcoming resistance mutations in hematological malignancies [65]. In prostate cancer, the PROTAC degrader ARV-110 has shown clinical efficacy in patients who developed resistance to enzalutamide and abiraterone, highlighting its potential against treatment-resistant disease [67] [150].
Resistance Profiles: Comparative Analysis
Resistance to Traditional Inhibitors
Traditional small-molecule inhibitors face several well-characterized resistance mechanisms [149]:
- Target mutations that reduce drug binding affinity (e.g., kinase gatekeeper mutations) [149]
- Target overexpression that requires higher drug concentrations to achieve inhibition [149]
- Activation of bypass signaling pathways that circumvent the blocked target [149]
- Increased drug efflux through transporters like P-glycoprotein [149]
- Drug inactivation through metabolic modifications [149]
In hematological malignancies, resistance to BCR-ABL inhibitors like imatinib commonly develops through point mutations in the kinase domain, particularly the T315I "gatekeeper" mutation that sterically hinders drug binding [149]. Similarly, resistance to androgen receptor antagonists in prostate cancer frequently occurs through AR amplification, mutations, or splice variants that render the receptor constitutively active [149].
Resistance to PROTACs
Emerging evidence suggests that resistance to PROTAC degraders may follow different patterns [149]:
- Reduced ternary complex formation due to mutations in either the POI or E3 ligase [149]
- Downregulation or mutation of the recruited E3 ligase [149]
- Compensatory upregulation of the target protein exceeding degradation capacity [149]
- Impaired ubiquitin-proteasome system function [149]
- Altered subcellular localization preventing ternary complex formation [149]
Notably, PROTACs can overcome certain resistance mechanisms that limit traditional inhibitors. For example, PROTACs targeting the androgen receptor (AR) remain effective against some mutant forms that drive resistance to standard antagonists [65]. Similarly, BCR-ABL targeted PROTACs have demonstrated efficacy against multiple imatinib-resistant mutants, with the exception of the T315I mutation when ATP-competitive warheads are used [149].
Experimental Protocols for Evaluating PROTAC Efficacy and Resistance
In Vitro Degradation Assays
Purpose: To quantify PROTAC-mediated degradation of the target protein and establish structure-activity relationships [67].
Materials:
- PROTAC compounds dissolved in DMSO at appropriate stock concentrations
- Cell lines expressing the target protein of interest
- Culture media and supplements for maintaining cell lines
- Lysis buffer (RIPA buffer with protease and phosphatase inhibitors)
- Antibodies for Western blot (target protein, loading control)
- Proteasome inhibitor (MG132) as control
- E3 ligase ligand as competition control
Procedure:
- Seed cells in appropriate multi-well plates and allow to adhere overnight.
- Treat cells with serially diluted PROTAC compounds for predetermined timepoints (typically 4-24 hours).
- Include controls: DMSO vehicle, proteasome inhibitor (MG132, 10 μM), and excess E3 ligand for competition.
- Harvest cells using ice-cold PBS and lyse in RIPA buffer.
- Quantify protein concentration using BCA assay.
- Perform Western blot analysis with antibodies against the target protein and loading control (e.g., GAPDH, β-actin).
- Quantify band intensities using densitometry software.
- Calculate DC₅₀ values (concentration causing 50% degradation) and Dmax (maximum degradation achieved) [67].
Troubleshooting: The "hook effect" may be observed at high PROTAC concentrations (>1 μM), where degradation efficiency decreases due to formation of binary complexes instead of productive ternary complexes [65] [51]. Always include a broad concentration range (nM to μM).
Ternary Complex Formation assays
Purpose: To evaluate the formation and stability of the POI-PROTAC-E3 ligase ternary complex, a critical determinant of degradation efficiency [65].
Materials:
- Purified proteins: POI and E3 ligase complex
- PROTAC compounds
- Surface Plasmon Resonance (SPR) equipment or Cellular Thermal Shift Assay (CETSA) reagents
- Size exclusion chromatography equipment
- X-ray crystallography or cryo-EM facilities for structural studies
Procedure - SPR Method:
- Immobilize the E3 ligase on SPR sensor chip.
- Pre-incubate POI with varying concentrations of PROTAC.
- Inject POI-PROTAC mixtures over E3-coated surface.
- Measure binding response compared to POI alone.
- Determine cooperative binding factors from binding kinetics.
Procedure - CETSA Method:
- Treat intact cells with PROTAC or vehicle control.
- Heat cells at different temperatures (37-65°C) to denature proteins.
- Separate soluble protein from precipitates.
- Detect target protein stability by Western blot.
- Shift in melting temperature indicates stabilization due to ternary complex formation.
Resistance Induction and Profiling assays
Purpose: To model and characterize potential resistance mechanisms to PROTAC treatment [149].
Materials:
- Parental cell lines sensitive to PROTAC treatment
- PROTAC compounds
- Culture flasks and media for long-term culture
- Genomic DNA extraction kit
- RNA sequencing and whole exome sequencing services
- CRISPR/Cas9 screening libraries
Procedure - Chronic Dosing:
- Treat cells with sub-lethal concentrations of PROTAC over multiple passages (3-6 months).
- Gradually increase PROTAC concentration as resistance develops.
- Isolate single-cell clones from resistant populations.
- Characterize degradation efficiency in resistant clones versus parental lines.
- Perform RNA-seq and whole exome sequencing to identify transcriptional and genomic alterations.
- Validate resistance mechanisms through CRISPR knockout or cDNA overexpression.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key research reagents for PROTAC development and evaluation
| Reagent Category | Specific Examples | Research Application |
|---|---|---|
| E3 Ligase Ligands | VHL ligand VH032, CRBN ligands (thalidomide, lenalidomide, pomalidomide), IAP antagonists [67] | Recruitment of specific E3 ligases for target ubiquitination |
| Proteasome Inhibitors | Bortezomib, Carfilzomib, MG132 [147] [146] | Validation of proteasome-dependent degradation mechanism |
| Ubiquitination Assay Reagents | Ubiquitin, E1 enzyme, E2 enzymes, ATP regeneration system [146] | In vitro ubiquitination assays to confirm E3 ligase engagement |
| Protein Stability Probes | Cycloheximide [149] | Measurement of protein half-life and degradation kinetics |
| Ternary Complex Assay Kits | SPR chips, CETSA reagents, FP-based ternary complex assays [65] | Evaluation of ternary complex formation efficiency and cooperativity |
| CRISPR Screening Libraries | Whole-genome KO libraries, E3 ligase-focused libraries [149] | Identification of resistance mechanisms and essential components for PROTAC activity |
Future Perspectives and Clinical Translation
The PROTAC field has advanced rapidly from concept to clinical validation. ARV-110 (for prostate cancer) and ARV-471 (for breast cancer) represent the most advanced PROTAC candidates, having demonstrated efficacy in clinical trials [65] [67]. These pioneers have established clinical proof-of-concept for the TPD modality and paved the way for next-generation degraders.
Future developments focus on expanding the E3 ligase toolbox beyond the commonly used CRBN and VHL ligases, developing tissue-specific degraders, and improving drug-like properties through advanced delivery systems [65] [51]. Innovations such as activatable PROTACs with spatiotemporal control, nanotechnology-based delivery, and antibody-PROTAC conjugates aim to enhance selectivity and overcome pharmacokinetic limitations [51] [151].
Diagram 2: Current challenges and innovative solutions in PROTAC technology.
PROTAC technology represents a fundamental paradigm shift in therapeutic intervention, moving beyond occupancy-driven inhibition to event-driven degradation. The unique mechanism of action of PROTACs confers distinct advantages in targeting challenging proteins, overcoming resistance, and achieving sustained pharmacological effects. While considerations such as the hook effect, molecular weight limitations, and emerging resistance mechanisms present ongoing challenges, continuous innovation in PROTAC design and delivery holds promise for expanding the therapeutic landscape. As research in the ubiquitin-proteasome system advances, PROTACs are poised to make increasingly significant contributions to precision medicine across oncology, neurodegenerative disorders, and other therapeutic areas.
Evaluating Therapeutic Index and Safety of UPS-Targeting Agents
The ubiquitin-proteasome system (UPS) represents a sophisticated target for therapeutic intervention in cancer, neurodegenerative disorders, and other diseases. This technical review provides a comprehensive assessment of strategies to enhance the therapeutic index of UPS-targeting agents, with particular focus on E3 ubiquitin ligase inhibition, proteasome modulation, and novel targeting technologies. We evaluate quantitative safety profiles, detailed experimental methodologies for profiling UPS agents, and the reagent toolkit required for preclinical development. The analysis synthesizes current evidence on component-specific toxicity relationships and mechanism-based safety considerations to inform the development of UPS-targeting therapies with optimized risk-benefit profiles for clinical application.
The ubiquitin-proteasome system (UPS) is a highly conserved protein degradation pathway essential for cellular homeostasis, regulating critical processes including cell cycle progression, gene expression, and stress responses [152] [153]. This sophisticated system operates through a sequential enzymatic cascade involving E1 (activating), E2 (conjugating), and E3 (ligase) enzymes that collectively tag target proteins with ubiquitin chains, primarily marking them for degradation by the 26S proteasome [153] [154]. The specificity of ubiquitination is largely determined by E3 ubiquitin ligases, with approximately 1000 members in the human genome enabling precise substrate recognition [154]. UPS dysfunction is implicated in multiple pathological states, most notably in oncogenesis where aberrant degradation of tumor suppressors occurs, and in neurodegenerative disorders characterized by pathological protein accumulation [155] [25]. The therapeutic targeting of this system presents unique challenges and opportunities for optimizing therapeutic index—the ratio between efficacious and toxic doses—which is paramount for clinical success.
Therapeutic Targeting Strategies for the UPS
E3 Ubiquitin Ligase Targeting
E3 ubiquitin ligases represent the most promising targets for therapeutic intervention within the UPS due to their substrate specificity and central role in determining degradation fate. The major E3 classes include RING-finger type (e.g., MDM2), HECT domain, and multi-subunit SCF complexes [154]. Among these, MDM2 has emerged as a validated cancer target through its regulation of the tumor suppressor p53. Inhibition of the MDM2-p53 interaction using small molecules such as Nutlin-3a stabilizes wild-type p53, activating apoptosis in cancer cells while theoretically sparing normal cells with minimal p53 dependence [155] [154]. This approach demonstrates the fundamental principle of enhancing therapeutic index through target specificity. Clinical development of E3 inhibitors faces the challenge of achieving sufficient selectivity while avoiding off-target effects on structurally similar E3 ligases with distinct physiological functions.
Proteasome Inhibitors and Emerging Modalities
Proteasome inhibitors such as bortezomib represent the first clinically validated UPS-targeting agents, demonstrating efficacy in hematological malignancies but exhibiting dose-limiting toxicities including peripheral neuropathy and hematological suppression [156]. Beyond direct inhibition, novel technologies including PROTACs (PROteolysis TArgeting Chimeras) represent a paradigm shift in UPS modulation by hijacking the ubiquitination machinery to target specific disease-driving proteins [153]. These heterobifunctional molecules simultaneously bind an E3 ligase and a target protein of interest, facilitating target ubiquitination and degradation. This approach offers potential advantages in dosing frequency and resistance mitigation compared to conventional occupancy-based inhibitors. Natural compound metabolites, including valerolactones and urolithins derived from dietary polyphenols, have also demonstrated UPS modulatory capacity, though their clinical translation requires further investigation into tissue distribution at physiologically relevant concentrations [25].
Table 1: Current UPS-Targeting Therapeutic Modalities
| Modality | Molecular Target | Therapeutic Index Considerations | Development Status |
|---|---|---|---|
| E3 Ligase Inhibitors (e.g., MDM2) | Specific E3-substrate interaction | High theoretical index in tumors with wild-type p53; potential on-target toxicity in normal tissues | Phase 1-3 clinical trials [155] |
| Proteasome Inhibitors (e.g., Bortezomib) | 20S proteasome catalytic subunits | Narrow therapeutic index; dose-limiting hematological and neurological toxicity | FDA-approved [156] |
| PROTACs | E3 ligase + target protein | Enhanced selectivity; catalytic mechanism may allow lower dosing | Preclinical/early clinical [153] |
| Natural UPS Modulators | Multiple UPS components | Favorable safety profile but uncertain tissue bioavailability | Preclinical investigation [25] |
Therapeutic Index and Safety Considerations
Component-Specific Toxicity Relationships
The safety profile of UPS-targeting agents is intrinsically linked to their specific molecular targets and structural components. For complex biologics such as antibody-drug conjugates (ADCs), which often utilize UPS-related payloads, systematic analyses reveal that specific toxicities correlate with distinct components [156]. Tubulin-binding payloads are significantly associated with peripheral neuropathy (OR 3.24, 95% CI 1.88-5.59), while non-cleavable linkers demonstrate strong associations with ocular toxicities (OR 4.71, 95% CI 2.33-9.53) and pulmonary events (OR 3.95, 95% CI 1.87-8.35) [156]. Similarly, drug-to-antibody ratio (DAR) emerges as a critical determinant of safety, with high-DAR ADCs (>4) showing increased hematological and pulmonary adverse events. These findings underscore the importance of component-level optimization in UPS-targeting strategies to maximize therapeutic index.
Mechanism-Based Safety Considerations
The fundamental role of the UPS in normal cellular physiology necessitates careful consideration of mechanism-based toxicities. As the UPS regulates approximately 80-90% of cellular proteins, non-selective inhibition inevitably disrupts essential processes including cell cycle regulation and stress response pathways [152] [153]. This is evidenced by the toxicity profile of broad-spectrum proteasome inhibitors, which cause dose-limiting hematological and gastrointestinal effects [156]. Strategies to enhance therapeutic index include tissue-selective targeting approaches and the development of agents targeting disease-specific E3 ligases with restricted expression patterns. Additionally, the temporal modulation of UPS activity—transient versus sustained inhibition—represents an important consideration for balancing efficacy and safety, particularly for targets involved in both pathological and physiological processes.
Table 2: Adverse Event Associations with UPS-Targeting Agent Components
| Component Class | Specific Component | Associated Adverse Events | Odds Ratio (95% CI) |
|---|---|---|---|
| Payload | Tubulin-binding agents | Peripheral neuropathy | 3.24 (1.88-5.59) [156] |
| Linker | Non-cleavable linkers | Ocular toxicity | 4.71 (2.33-9.53) [156] |
| Linker | Non-cleavable linkers | Pulmonary toxicity | 3.95 (1.87-8.35) [156] |
| Structural | DAR >4 | Hematological toxicity | Significant association [156] |
| Target | On-target, off-tumor | Tissue-specific toxicity | Case-dependent [156] |
Experimental Protocols for Profiling UPS-Targeting Agents
In Vitro Ubiquitination and Degradation Assays
Objective: To quantitatively assess the effect of candidate compounds on substrate-specific ubiquitination and degradation. Methodology: Recombinant E1, E2, E3 enzymes, ubiquitin, and ATP are incubated with the target substrate protein in the presence of test compounds. The reaction is terminated at various time points and analyzed via immunoblotting using ubiquitin- and substrate-specific antibodies [154]. For high-throughput screening, HTRF (Homogeneous Time-Resolved Fluorescence) or ALISA (Acceptor-Linked Immunosorbent Assay) platforms can be implemented to quantify polyubiquitin chain formation. Degradation kinetics are determined in cellular systems by treating engineered cell lines expressing tagged substrates with compounds, followed by cycloheximide chase and quantitative immunoblotting to measure substrate half-life extension [155] [154]. Key controls: Include reactions without ATP (negative control), with excess known inhibitor (positive control), and assessment of compound effects on individual E1/E2/E3 enzymes to identify specific inhibition points.
Computational and Multi-Omics Approaches for Biomarker Identification
Objective: To identify ubiquitin-related genetic signatures predictive of therapeutic response and toxicity. Methodology: Leverage transcriptomic data from public databases (TCGA, GEO) and implement Weighted Gene Co-expression Network Analysis (WGCNA) to identify gene modules correlated with ubiquitin scores calculated via single-sample gene set enrichment analysis (ssGSEA) [157]. Preprocess gene expression data by removing low-variance genes (median absolute deviation threshold of 0.01, retaining top 75% highest variance genes). Identify optimal soft threshold power (β) using the pickSoftThreshold function, then construct a weighted neighbor-joining matrix and calculate topological overlap measure (TOM) [157]. Identify gene modules with minimum size of 60 (deepSplit=2, mergeCutHeight=0.3) and correlate module eigengenes with ubiquitin scores. Intersect module genes with differentially expressed genes (DEGs) between disease and control samples (∣log~2~(FC)∣ > log~2~(1.5), p<0.05) to identify ubiquitin-associated candidate biomarkers [157]. Validate prognostic value through Cox regression and risk modeling.
Research Reagent Solutions
Table 3: Essential Research Tools for UPS-Targeting Agent Development
| Reagent Category | Specific Examples | Research Application | Key Features |
|---|---|---|---|
| E1 Activating Enzymes | UBE1 | Biochemical ubiquitination assays | Initiates ubiquitin activation; essential for in vitro reconstitution [153] |
| E2 Conjugating Enzymes | UBE2D2 | Enzyme cascade studies | Transfers ubiquitin from E1 to E3; determines chain topology [153] |
| E3 Ligase Assay Systems | MDM2, IAP, SCF complexes | Target engagement and inhibition screening | Provide substrate specificity; primary drug targets [154] |
| Tandem Ubiquitin-Binding Entities (TUBEs) | Pan-selective TUBEs | Polyubiquitin chain detection and purification | High-affinity ubiquitin chain sensors; protect chains from deubiquitination [153] |
| Proteasome Activity Probes | Fluorogenic substrates (LLVY-AMC) | Proteasome inhibition profiling | Measure chymotrypsin-like, trypsin-like, and caspase-like activities [25] |
| Ubiquitin Variants | Lys~48~- vs Lys~63~-linked ubiquitin chains | Specific pathway interrogation | Distinguish proteasomal vs non-proteasomal ubiquitination outcomes [153] |
The strategic targeting of specific UPS components, particularly E3 ubiquitin ligases, offers a promising path for enhancing therapeutic index compared to broad proteasome inhibition. The continued development of sophisticated experimental approaches for profiling UPS agent efficacy and safety, coupled with advanced reagent systems for delineating mechanism of action, will accelerate the rational design of next-generation UPS-targeting therapeutics. Future efforts should focus on leveraging tissue-specific delivery platforms, combination strategies that permit dose reduction, and comprehensive biomarker development to identify patient populations most likely to benefit from UPS-targeted interventions while minimizing toxicity risks.
Diagrams
Biomarkers for UPS Function and Therapeutic Response Monitoring
The ubiquitin-proteasome system (UPS) serves as the primary cellular machinery for regulated protein degradation, orchestrating the turnover of approximately 80% of intracellular proteins in eukaryotic cells [158]. This sophisticated system encompasses ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), ubiquitin ligases (E3), ubiquitin itself, and the 26S proteasome, which consists of a 20S core protease and 19S regulatory particles [158] [159]. The UPS governs fundamental cellular processes including cell cycle progression, apoptosis, differentiation, autophagy, epigenetic regulation, angiogenesis, drug resistance, signal transduction, inflammation, and immune responses [158]. Given its central role in cellular homeostasis, dysregulation of UPS components through functional mutations or abnormal expression contributes to various diseases, particularly neurodegenerative disorders and cancer [158]. The development of biomarkers for UPS function consequently provides critical tools for diagnosing UPS-related pathologies, monitoring disease progression, and evaluating therapeutic responses, especially for targeted therapies such as proteasome inhibitors and proteolysis-targeting chimeras (PROTACs) [158] [159].
Current UPS Biomarkers: Mechanisms and Measurement
Circulating Proteasomes as Direct UPS Biomarkers
The 20S proteasome exists not only intracellularly but also as extracellular "circulating proteasomes" detectable in plasma, serum, cerebrospinal fluid, and bronchoalveolar lavage fluids [159]. These circulating proteasomes represent quantifiable direct biomarkers of UPS presence, with levels fluctuating under various physiological and pathological conditions:
- Elevated Levels: Increased 20S proteasome levels occur in response to cellular trauma and disease states, including aggressive hematologic malignancies, inflammatory liver disorders, melanoma, sepsis, surgeries, burns, inhalation injuries, and critical illness [159]. The concentration of circulating proteasomes reflects the degree of cell damage irrespective of the specific cause, making them valuable nonspecific markers of tissue injury and inflammatory response.
- Diagnostic Utility: In neurological conditions, proteasome activity increases after various traumatic stressors including hyperoxia, radiation, or oxidative damage [159]. The removal of damaged proteins via UPS is essential for providing necessary conditions for cell repair, positioning proteasome levels as indicators of neuronal damage and recovery processes.
Table 1: Circulating Proteasome Biomarkers in Various Conditions
| Biomarker | Biological Sample | Associated Conditions | Direction of Change |
|---|---|---|---|
| 20S Proteasome | Plasma, Serum | Trauma, Neoplasms, Autoimmune Diseases | Increased [159] |
| 20S Proteasome | Cerebrospinal Fluid | Critical Illness, Inflammatory Disorders | Increased [159] |
| 20S Proteasome | Early-stage Chronic Lymphatic Leukemia | Early-stage cancer | Decreased [159] |
| 20S Proteasome | Advanced Hematologic Malignancies | Advanced cancer | Increased [159] |
Ubiquitin C-Terminal Hydrolase L1 (UCHL1) as a Neuronal Damage Biomarker
Ubiquitin C-terminal hydrolase L1 (UCHL1) represents another crucial UPS component serving as a specific biomarker:
- Tissue Specificity: UCHL1 demonstrates particular relevance for identifying brain and neuronal damage, advancing the diagnosis and prognosis of traumatic brain injury (TBI) and other neurological conditions [159].
- Functional Role: As a deubiquitinating enzyme (DUB), UCHL1 proof-reads ubiquitin-protein conjugates, removes ubiquitin, and maintains the 26S proteasome free of inhibitory ubiquitin chains [159] [160]. The human genome encodes approximately 100 distinct DUBs, with UCHL1 belonging to the ubiquitin C-terminal hydrolases subfamily [159].
Disease-Specific UPS Gene Signatures as Biomarkers
Recent bioinformatics approaches have identified specific UPS-related gene signatures serving as prognostic biomarkers across various diseases:
- Thyroid Carcinoma: A prognostic model comprising six UPS genes effectively forecasts patient prognosis in thyroid carcinoma (THCA) [158]. The ubiquitin-proteasome system prognostic model score (UPS-PMS) shows substantial correlation with immune microenvironmental factors, particularly demonstrating a significant association between high UPS-PMS and an immunosuppressive microenvironment [158].
- Type 2 Diabetes Mellitus: Integrated bioinformatics analysis identified four ubiquitin-pyroptosis-related biomarkers (ABCC8, RBP4, RASGRF1, and SLC34A2) in type 2 diabetes mellitus (T2DM) [161]. These biomarkers show enrichment in oxidative phosphorylation and mitogen-activated protein kinase signaling pathways relevant to diabetes pathogenesis and modulate immune cell infiltration [161].
- Synucleinopathies: In neurodegenerative disorders like Parkinson's disease (PD), dementia with Lewy bodies (DLB), and multiple system atrophy (MSA), quantitative measurements of α-synuclein in cerebrospinal fluid (CSF) have emerged as promising biomarkers [162]. The development of α-synuclein seed amplification assays (SAAs) and immunohistochemical detection from skin biopsies provides additional methods for detecting UPS-related pathology in synucleinopathies [162].
Methodologies for UPS Biomarker Analysis
Bioinformatics Approaches for UPS Biomarker Discovery
The discovery of UPS-related biomarkers increasingly employs sophisticated bioinformatics pipelines on large-scale genomic data:
Figure 1: Bioinformatics workflow for UPS biomarker discovery and validation [161] [158].
Detailed Experimental Protocol:
- Data Acquisition: Download gene expression datasets (e.g., from TCGA or GEO databases) containing disease samples and normal controls [161] [158].
- Differential Expression Analysis: Perform differential gene expression analysis using the limma package in R with criteria of |log2 fold change| ≥ 0.5 and adjusted p < 0.05 [161].
- Weighted Gene Co-expression Network Analysis (WGCNA): Identify clusters of highly correlated genes and evaluate associations with phenotypes using WGCNA to construct module-trait relationship networks [158].
- Functional Enrichment: Conduct Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis using clusterProfiler with false discovery rate < 0.05 [161] [158].
- Prognostic Model Construction: Employ LASSO regression analysis to build a risk score model (Risk Score = Σ(expi * betai)) and construct nomograms incorporating clinical pathological features and core UPS-related genes [158].
- Immune Infiltration Analysis: Use single-sample Gene Set Enrichment Analysis (ssGSEA) to assess enrichment scores for immune cell subtypes and the ESTIMATE algorithm to evaluate stromal and immune infiltration [161] [158].
- Drug Sensitivity Analysis: Utilize the R package "oncoPredict" to estimate half-maximal inhibitory concentration (IC50) values of anti-cancer drugs and identify potential therapeutic compounds [158].
- Experimental Validation: Validate expression trends of identified biomarkers through quantitative PCR (qPCR) and western blot analysis [161].
Analytical Techniques for Proteasome and Ubiquitination Assessment
Direct measurement of UPS components employs various biochemical and analytical techniques:
- Proteasome Activity Assays: Fluorogenic peptide substrates (e.g., Suc-LLVY-AMC) measure the chymotrypsin-like activity of the proteasome's β5 subunit; other substrates assess trypsin-like (β2) and caspase-like (β1) activities [159].
- Enzyme-Linked Immunosorbent Assay (ELISA): Quantifies concentrations of 20S proteasomes, UCHL1, and other UPS components in biological fluids including plasma, serum, and cerebrospinal fluid [159].
- Western Blotting: Detects protein expression levels and post-translational modifications of UPS components; for example, using rabbit anti-KCNA1 monoclonal antibody (1:1000; ab32433, Abcam) and anti-Tubulin monoclonal antibody as loading control [158].
- Mass Spectrometry-Based Proteomics: Identifies and quantifies ubiquitinated proteins, proteasome subunits, and interacting proteins in complex biological samples [162].
- Seed Amplification Assays (SAAs): Detect pathological protein aggregates in synucleinopathies by amplifying α-synuclein fibrils to measure UPS dysfunction in neurodegenerative diseases [162].
Table 2: Key Research Reagents for UPS Biomarker Analysis
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Proteasome Substrates | Suc-LLVY-AMC, Boc-LRR-AMC, Ac-nLPnLD-AMC | Fluorogenic peptides measuring proteasome catalytic activities (chymotrypsin-like, trypsin-like, caspase-like) [159] |
| Antibodies | Anti-KCNA1 (ab32433, Abcam), Anti-UCHL1, Anti-20S Proteasome | Detection and quantification of specific UPS proteins via Western blot, ELISA, immunohistochemistry [158] [159] |
| Bioinformatics Tools | limma R package, clusterProfiler, WGCNA, glmnet | Differential expression analysis, functional enrichment, co-expression network construction, LASSO regression [161] [158] |
| Proteasome Inhibitors | Bortezomib, MG132, Carfilzomib | Experimental controls for validating proteasome-dependent processes and therapeutic mechanisms [158] |
Regulatory Qualification of UPS Biomarkers
The regulatory acceptance of biomarkers, including those related to UPS function, follows structured pathways to ensure appropriate validation and context of use (COU) [163]. The FDA's Biomarker Qualification Program (BQP), formally established in 2016 under the 21st Century Cures Act, provides a key pathway for developing novel biomarkers for regulatory use [164] [160]. This program employs a three-phase process: (1) Letter of Intent (LOI), (2) Qualification Plan (QP), and (3) Full Qualification Package (FQP) [164].
Figure 2: FDA Biomarker Qualification Program (BQP) pathway [164] [163].
As of July 2025, 61 projects were accepted into the BQP, with safety (30%), diagnostic (21%), and pharmacodynamic response (20%) biomarkers being the most common categories [164] [160]. Molecular (46%) and radiologic/imaging (39%) methods represent the primary assessment approaches [164]. However, the program has seen limited use for biomarkers intended as surrogate endpoints, with only five such projects accepted [164]. The qualification process involves substantial timelines, with QP development taking a median of 32 months and LOI and QP reviews frequently exceeding FDA targets by three months and seven months, respectively [164].
For UPS-related biomarkers, the fit-for-purpose validation approach depends on the specific biomarker category and intended use [163]. Susceptibility/risk biomarkers require epidemiological evidence and biological plausibility, while diagnostic biomarkers prioritize sensitivity and specificity across diverse populations [163]. Prognostic biomarkers need robust clinical data showing consistent correlation with disease outcomes, and pharmacodynamic/response biomarkers require evidence of a direct relationship between drug action and biomarker changes [163].
Therapeutic Applications and Response Monitoring
UPS biomarkers play increasingly important roles in therapeutic development, particularly in oncology and neurodegenerative diseases:
- Proteasome Inhibitor Therapy: Biomarkers such as circulating proteasome levels may predict response to proteasome inhibitors like bortezomib in cancers including multiple myeloma and anaplastic thyroid carcinoma [158] [159]. Drug sensitivity analyses demonstrate that specific UPS gene expressions can influence therapeutic efficacy; for example, high KCNA1 expression promotes gemcitabine resistance in thyroid carcinoma patients, while KCNA1 knockdown increases sensitivity to gemcitabine [158].
- Immunotherapy Response: UPS-related gene signatures correlate with immune microenvironment characteristics, enabling stratification of patients for immunotherapy [158]. Specifically, high UPS-PMS associates with an immunosuppressive microenvironment in thyroid carcinoma, potentially influencing response to immune checkpoint inhibitors [158].
- Targeted Protein Degradation: For novel therapeutic approaches including PROTACs that harness E3 ubiquitin ligases for targeted protein degradation, UPS biomarkers provide crucial pharmacodynamic information on target engagement and degradation efficiency [159].
- Natural Compound Effects: Circulating polyphenol-derived metabolites (e.g., valerolactones, urolithins, hydroxycinnamic acids) demonstrate UPS modulatory activity, suggesting potential for therapeutic intervention in chronic diseases where UPS dysregulation occurs [25].
Biomarkers for UPS function and therapeutic response monitoring represent rapidly advancing tools with significant implications for disease diagnosis, prognosis, and treatment optimization. The integration of bioinformatics approaches with experimental validation has accelerated the discovery of UPS-related biomarkers across diverse pathological conditions. Current evidence supports the utility of circulating proteasomes, UCHL1, and disease-specific UPS gene signatures as valuable indicators of UPS status and therapeutic response. However, translation of these biomarkers into clinical and regulatory applications requires rigorous validation through structured pathways such as the FDA's Biomarker Qualification Program. As research continues to elucidate the complex roles of UPS in health and disease, the development and qualification of additional UPS biomarkers will enhance precision medicine approaches across therapeutic areas.
Conclusion
The ubiquitin-proteasome system represents a master regulatory network with profound implications for cellular homeostasis and disease pathology. Our understanding has evolved from basic protein degradation to recognizing its sophisticated roles in signaling, synaptic plasticity, and system-wide proteostasis. The emergence of targeted protein degradation technologies, particularly PROTACs advancing through clinical trials, marks a paradigm shift in therapeutic development, moving beyond inhibition to complete target elimination. Future directions include expanding the repertoire of addressable targets, developing novel E3 ligase recruiters, and precisely manipulating the ubiquitin code. The interconnected nature of protein degradation pathways necessitates integrated approaches considering UPS-autophagy crosstalk. As research continues to unravel the complexity of this system, the UPS promises to yield transformative therapies for cancer, neurodegenerative diseases, and other disorders with high unmet need, solidifying its position as a cornerstone of modern biomedical research and precision medicine.
References
- 1. The ubiquitin system: from cell signalling to disease ... [https://www.nature.com/articles/s41418-020-00703-w]
- 2. Biochemistry, Ubiquitination - StatPearls - NCBI Bookshelf - NIH [https://www.ncbi.nlm.nih.gov/books/NBK556052/]
- 3. E3 ubiquitin ligases in signaling, disease, and therapeutics [https://www.sciencedirect.com/science/article/pii/S0968000425001860]
- 4. E3 ubiquitin ligases: styles, structures and functions - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8607428/]
- 5. Ubiquitin ligase [https://en.wikipedia.org/wiki/Ubiquitin_ligase]
- 6. UBE2J2 sensitizes the ERAD ubiquitination cascade to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12511567/]
- 7. The Role of Protein Ubiquitination in the Onset and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12248740/]
- 8. Drug Development Targeting the Ubiquitin–Proteasome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7226376/]
- 9. The Assembly Pathway of the 19S Regulatory Particle ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1783769/]
- 10. Molecular Architecture and Assembly of the Eukaryotic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3827779/]
- 11. exploring the ubiquitin system for drug development [https://www.nature.com/articles/cr201631]
- 12. Hyperactive 20S proteasome enhances proteostasis and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12513426/]
- 13. The proteasome 19S cap and its ubiquitin receptors ... [https://www.nature.com/articles/s41467-019-13906-8]
- 14. 19S proteasome loss regulates mitotic spindle assembly ... [https://www.sciencedirect.com/science/article/pii/S2211124725008125]
- 15. Structure of the TXNL1-bound proteasome [https://www.nature.com/articles/s41594-025-01639-w]
- 16. Genome-wide investigation and functional analysis of 20S ... [https://www.sciencedirect.com/science/article/abs/pii/S0048357525002640]
- 17. From Discovery to Bedside: Targeting the Ubiquitin System [https://www.sciencedirect.com/science/article/pii/S2451945618303829]
- 18. Breaking the K48-Chain: Linking Ubiquitin Beyond Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11730971/]
- 19. Differentiate between K48- and K63-specific Ubiquitination [https://www.tebubio.com/en_fr_eur/content/post/how-to-differentiate-between-k48-and-k63-specific-ubiquitination]
- 20. K48- and K63-linked ubiquitin chain interactome reveals ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11109483/]
- 21. K63-linked ubiquitin chains as a specific signal for protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2700384/]
- 22. Structural basis of K11/K48-branched ubiquitin chain ... [https://www.nature.com/articles/s41467-025-64719-x]
- 23. Discovery and mechanism of K63-linkage-directed ... [https://www.nature.com/articles/s41589-024-01777-0]
- 24. Mechanism of Ubiquitin-Chain Formation by the human ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2696189/]
- 25. Modulation of the Ubiquitin-Proteasome System by ... [https://www.sciencedirect.com/science/article/pii/S095528632500289X]
- 26. The Ubiquitin-Proteasome System in Brain Disorders [https://pubmed.ncbi.nlm.nih.gov/41264052/?utm_source=FeedFetcher&utm_medium=rss&utm_campaign=None&utm_content=1x_POZXjwvkcsarQzozji89pJTuITeRBIY9P7KAo12saGGWkxF&fc=None&ff=20251120191010&v=2.18.0.post22+67771e2]
- 27. The roles of the ubiquitin-proteasome system in renal ... [https://www.medsci.org/v22p1791.htm]
- 28. Targeting the Ubiquitin–Proteasome System for Cancer - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12495026/]
- 29. The dual role of the ubiquitin-proteasome system in ... [https://pubmed.ncbi.nlm.nih.gov/41151053/]
- 30. Ubiquitin-Proteasome-Mediated Protein Degradation and ... [https://www.mdpi.com/1422-0067/26/3/966]
- 31. Quantitative mapping of proteasome interactomes and ... [https://elifesciences.org/articles/93256]
- 32. Proteome and Ubiquitinome Analyses Reveal the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12413316/]
- 33. The Proteostasis Network in Proteinopathies - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC12597693/]
- 34. Non-proteolytic ubiquitylation in cellular signaling and ... [https://www.nature.com/articles/s42003-022-03060-1]
- 35. Ubiquitin/Proteasome [https://www.cellsignal.com/pathways/ubiquitin-proteasome-pathway?srsltid=AfmBOoquvw5XkHChNk-LDedR-eL2b2TSkWGqvq3oPGDU5PnRxXOaFWgn]
- 36. Regulation of Signaling by Non-degradative Ubiquitination [https://pmc.ncbi.nlm.nih.gov/articles/PMC2659175/]
- 37. Non-degradative Ubiquitination of Protein Kinases - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4890936/]
- 38. Global Analysis of Host and Bacterial Ubiquitinome in ... [https://www.sciencedirect.com/science/article/pii/S109727651630065X]
- 39. Non-degradative Ubiquitination of Protein Kinases [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1004898]
- 40. Ubiquitin-like protein conjugation and the ubiquitin – proteasome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7097807/]
- 41. Drug discovery in the ubiquitin – proteasome system [https://www.nature.com/articles/nrd2056?error=cookies_not_supported]
- 42. Ubiquitin–proteasome system in the different stages of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12102666/]
- 43. The ubiquitin - proteasome system: A key regulatory hub in... [https://www.dovepress.com/the-ubiquitin-proteasome-system-a-key-regulatory-hub-in-myocarditis-le-peer-reviewed-fulltext-article-JIR]
- 44. Neurodevelopmental Disorders (NDD) Caused by ... [https://www.frontiersin.org/journals/molecular-neuroscience/articles/10.3389/fnmol.2021.733012/full]
- 45. Mechanisms of proteostasis in neuronal development and ... [https://www.sciencedirect.com/science/article/abs/pii/S0959438825001746]
- 46. Ubiquitin Proteasome System (UPS) and ... [https://www.frontiersin.org/research-topics/62657/ubiquitin-proteasome-system-ups-and-ubiquitin-independent-proteasome-mediated-proteolysis-uipp-crosstalk-in-development-and-diseaseundefined]
- 47. Highly conserved shifts in ubiquitin-proteasome system ... [https://www.nature.com/articles/s41467-022-32206-2]
- 48. Quantitative analysis of the ubiquitin-proteasome system ... [https://prelights.biologists.com/highlights/quantitative-analysis-of-the-ubiquitin-proteasome-system-under-proteolytic-and-folding-stressors/]
- 49. Targeted protein degradation: mechanisms, strategies and ... [https://www.nature.com/articles/s41392-022-00966-4]
- 50. Frontiers | PROTAC : a revolutionary technology propelling small... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2025.1676414/full]
- 51. PROTAC 2.0: Expanding the frontiers of targeted protein ... [https://www.sciencedirect.com/science/article/abs/pii/S1359644625000893]
- 52. Quantitative Measurement of PROTAC Intracellular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11102804/]
- 53. Recent Advances in the Development of Pro-PROTAC for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12473374/]
- 54. Ubiquitin‐Independent Degradation: An Emerging PROTAC ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11755708/]
- 55. Degraders in Clinical Trails: PROTAC Update | Biopharma PEG 2025 [https://www.biochempeg.com/article/434.html]
- 56. Molecular Glues: The Adhesive Connecting Targeted Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9910052/]
- 57. Rational discovery of molecular glue degraders based on ... [https://www.sciencedirect.com/science/article/abs/pii/S135964462500193X]
- 58. Molecular glues: A new path to the world of the unattainable [https://www.sciencedirect.com/science/article/abs/pii/S0141813025094188]
- 59. : Transforming Molecular Discovery | Technology Networks Glues Drug [https://www.technologynetworks.com/drug-discovery/articles/molecular-glues-the-next-frontier-in-targeted-protein-degradation-403331]
- 60. The Therapeutic Potential of Molecular and PROTAC Glue Degraders [https://www.biosynth.com/blog/the-therapeutic-potential-of-molecular-glue-and-protac-degraders]
- 61. : exciting opportunities for novel Molecular ... glue degraders drug [https://pubmed.ncbi.nlm.nih.gov/38240114/]
- 62. A degron-mimicking molecular glue drives CRBN homo- ... [https://www.nature.com/articles/s41467-025-65094-3]
- 63. Frontiers | Molecular for tumor treatment glue degrader [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2024.1512666/full]
- 64. Degraders & Molecular Glues Part 2 | April 16-17, 2025 [https://www.drugdiscoverychemistry.com/25/protein-degradation-molecular-glues-part2]
- 65. Proteolysis‐Targeting Chimera (PROTAC): Current Applications and Future Directions - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12495453/]
- 66. The mechanism of action and clinical value of PROTACs [https://www.sciencedirect.com/science/article/pii/S089865682200208X]
- 67. An overview of PROTACs: a promising drug discovery ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9763089/]
- 68. Global landscape of PROTAC: Perspectives from patents, drug pipelines, clinical trials, and licensing transactions - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S0223523425008207]
- 69. Press Release Details [https://ir.arvinas.com/news-releases/news-release-details/arvinas-announces-data-presentations-vepdegestrant-arv-471]
- 70. Discovery of BMS-986365, a first-in-class dual androgen ... [https://pubmed.ncbi.nlm.nih.gov/40788283/]
- 71. Tracking of Ubiquitin Signaling through 3.5 Billion Years of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11354881/]
- 72. Targeted protein degradation: mechanisms , strategies and application [https://www.nature.com/articles/s41392-022-00966-4?error=cookies_not_supported&code=37dfd49a-e0ed-4c8a-9a80-58e246d33432]
- 73. Degradation from the outside in: Targeting extracellular ... [https://www.sciencedirect.com/science/article/pii/S2451945621001094]
- 74. Unlocking New Frontiers with AUTACs and LYTACs [https://www.pharmasalmanac.com/articles/beyond-the-proteasome-unlocking-new-frontiers-with-autacs-and-lytacs]
- 75. vs. PROTAC vs. AbTAC Comparison - BOC Sciences LYTAC [https://ptc.bocsci.com/resource/abtacs-vs-protacs-vs-lytacs-a-comparative-guide.html]
- 76. A covalent peptide-based lysosome-targeting protein ... [https://www.nature.com/articles/s41467-025-56648-6]
- 77. LYTACs that engage the asialoglycoprotein receptor for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8387313/]
- 78. Unlocking new degradation mechanisms [https://www.news-medical.net/health/Unlocking-new-degradation-mechanisms-with-alternative-E3-ligases.aspx]
- 79. Functional Characterization of Pathway Inhibitors for the ... [https://pubmed.ncbi.nlm.nih.gov/39753207/]
- 80. A new era in cancer therapy: targeting the Proteasome-Bcl-2 ... [https://jeccr.biomedcentral.com/articles/10.1186/s13046-025-03505-5]
- 81. Frontiers | Ubiquitin - proteasome : a potential participant and... system [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1523799/full]
- 82. - Ubiquitin in Proteasome Neuroscience Pathway Systems [https://www.numberanalytics.com/blog/ubiquitin-proteasome-pathway-systems-neuroscience]
- 83. Impact of proteostasis workload on sensitivity to proteasome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12106538/]
- 84. Targeting proteostasis for cancer therapy: current advances ... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-025-02472-x]
- 85. The Evolution and Diversification of Proteasome Inhibitors ... [https://chemrxiv.org/engage/chemrxiv/article-details/6853081ac1cb1ecda0b1e33f]
- 86. The Ubiquitin-Proteasome System: A Key Regulatory Hub in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12497386/]
- 87. Ubiquitin proteasome system in immune regulation and ... [https://www.sciencedirect.com/science/article/abs/pii/S1471489222001370]
- 88. Emerging therapies targeting the ubiquitin proteasome system ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3871250/]
- 89. The ubiquitin - proteasome in system ... neurodegenerative diseases [https://pubmed.ncbi.nlm.nih.gov/25132814/]
- 90. , the Ubiquitin and... | Nature Reviews Neuroscience proteasome [https://www.nature.com/articles/nrn2499?error=cookies_not_supported&code=94be4525-993c-4afa-9089-c72344334498]
- 91. Emerging therapies targeting the ubiquitin proteasome system ... [https://pubmed.ncbi.nlm.nih.gov/24382383/]
- 92. Data-independent acquisition (DIA) approach for ... [https://sciety-discovery.elifesciences.org/articles/by?article_doi=10.1101/2025.05.27.656291]
- 93. Nuclear proteasomes as a backup for autophagy [https://pmc.ncbi.nlm.nih.gov/articles/PMC11702925/]
- 94. The interplay between autophagy and the ubiquitin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4277934/]
- 95. Autophagy and the ubiquitin-proteasome system [https://pmc.ncbi.nlm.nih.gov/articles/PMC2621359/]
- 96. Crosstalk Between Mammalian Autophagy and the ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2018.00128/full]
- 97. Interplay Between the Autophagy-Lysosomal Pathway and ... [https://www.frontiersin.org/journals/cellular-neuroscience/articles/10.3389/fncel.2018.00126/full]
- 98. Crosstalk and Interplay between the Ubiquitin-Proteasome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5547213/]
- 99. Ubiquitination-Proteasome System (UPS) and Autophagy ... [https://www.mdpi.com/2073-4409/11/5/851]
- 100. Interplay between autophagy and proteasome during ... [https://www.sciencedirect.com/science/article/pii/S1360138523000316]
- 101. Emerging Paradigm of Crosstalk between Autophagy and ... [https://www.sciencedirect.com/science/article/pii/S1016847823006817]
- 102. Review The interplay between autophagy and the ubiquitin ... [https://www.sciencedirect.com/science/article/pii/S092544391400249X]
- 103. as a Nuclear for proteasomes : interconnected... backup autophagy [https://pubmed.ncbi.nlm.nih.gov/39427251/]
- 104. The Proteostasis Network: A Global Therapeutic Target for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9658791/]
- 105. The Ubiquitin Proteasome Pathway (UPP) in the regulation of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3466981/]
- 106. Relationship between the proteasomal system and autophagy [https://pmc.ncbi.nlm.nih.gov/articles/PMC3627065/]
- 107. Autophagy as a potential therapeutic target in regulating ... [https://pubmed.ncbi.nlm.nih.gov/40444035/]
- 108. p62 links the autophagy pathway and the ubiqutin– proteasome ... [https://cmbl.biomedcentral.com/articles/10.1186/s11658-016-0031-z]
- 109. Autophagy as a potential therapeutic target in regulating ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1579183/full]
- 110. Nuclear proteasomes buffer cytoplasmic proteins during ... [https://www.nature.com/articles/s41556-024-01488-7]
- 111. Mechanistic insights and therapeutic strategies for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12018664/]
- 112. Expanding PROTACtable genome universe of E3 ligases [https://www.nature.com/articles/s41467-023-42233-2]
- 113. Identification of ligands for E3 ligases with restricted ... [https://pubs.rsc.org/en/content/articlehtml/2025/cb/d5cb00198f]
- 114. Multifaceted regulation of proteasomal activity [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2025.1748172/full]
- 115. In-Cell Approach to Evaluate E3 Ligases for Use ... [https://pubmed.ncbi.nlm.nih.gov/40493706/]
- 116. State of the art of Machine learning for Targeted Protein ... [https://chemrxiv.org/engage/chemrxiv/article-details/6903876fa482cba122f80125]
- 117. Molecular mechanisms and potential targeting strategies of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12511930/]
- 118. Targeted protein degradation: advances in drug discovery ... [https://www.nature.com/articles/s41392-024-02004-x]
- 119. Drug resistance mechanisms and treatment strategies ... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-024-02005-y]
- 120. Molecular mechanisms and potential targeting strategies of ... [https://pubmed.ncbi.nlm.nih.gov/41041864/]
- 121. 8 Essential Data Validation Techniques for 2025 [https://osher.com.au/blog/data-validation-techniques/]
- 122. The ubiquitin - proteasome in neurodegenerative system ... diseases [https://pmc.ncbi.nlm.nih.gov/articles/PMC4117186/]
- 123. Protein Degradation Through Ubiquitination : Insights... | Preprints.org [https://www.preprints.org/manuscript/202504.2585/v1]
- 124. of the Dysfunction – ubiquitin in multiple... | Scilit proteasome system [https://www.scilit.com/publications/21cd1d17a7a145f88ec0271462ec6421]
- 125. NDRG1 upregulation by ubiquitin proteasome system dysfunction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11515609/]
- 126. Papers - Academia.edu Ubiquitin Proteasome System Research [https://www.academia.edu/Documents/in/Ubiquitin_Proteasome_System]
- 127. Mechanistic insights into proteasome inhibitor MG132 ... [https://www.nature.com/articles/s41598-025-99151-0]
- 128. Partial inhibition of the ubiquitin–proteasome system ameliorates... [https://cardiab.biomedcentral.com/articles/10.1186/s12933-015-0258-4]
- 129. Dynamic regulation of inter-organelle communication by ubiquitylation... [https://elifesciences.org/articles/81966]
- 130. Anthracyclines attenuate Nrf1-dependent proteolytic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12574707/]
- 131. Ubiquitin proteasome system in immune regulation and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10163937/]
- 132. Molecular mechanisms and potential targeting strategies of ... [https://www.spandidos-publications.com/10.3892/or.2025.9000]
- 133. Exploring bioactive phytoconstituents as USP21 inhibitors ... [https://www.nature.com/articles/s41598-025-94825-1]
- 134. Study unravels the complex interplay of the UPS in cancer ... [https://www.news-medical.net/news/20250224/Study-unravels-the-complex-interplay-of-the-UPS-in-cancer-development-and-treatment.aspx]
- 135. Ubiquitination of transcription factors in cancer [https://pmc.ncbi.nlm.nih.gov/articles/PMC12330937/]
- 136. The Ubiquitin-Proteasome System and Cellular Signaling [https://www.frontiersin.org/research-topics/64761/the-ubiquitin-proteasome-system-and-cellular-signaling-mechanisms-and-regulatory-roles-in-cancer-and-infectious-diseasesundefined]
- 137. Ubiquitin-Proteasome System in Neurodegenerative Disorders [https://pmc.ncbi.nlm.nih.gov/articles/PMC6370320/]
- 138. Boosting Brain Clean-Up: Can Targeting UPS Genes Offer ... [https://pubmed.ncbi.nlm.nih.gov/40817956/]
- 139. Dysregulation of Ubiquitin-Proteasome System in ... [https://www.frontiersin.org/journals/aging-neuroscience/articles/10.3389/fnagi.2016.00303/full]
- 140. The ubiquitin-proteasome system in Alzheimer's disease ... [https://www.frontiersin.org/journals/aging-neuroscience/articles/10.3389/fnagi.2025.1730206/full]
- 141. Novel discovery reveals how brain protein OTULIN controls ... [https://www.eurekalert.org/news-releases/1107102]
- 142. The Global Neurodegeneration Proteomics Consortium [https://www.nature.com/articles/s41591-025-03834-0]
- 143. Integrated transcriptomic analysis identifies lysosomal ... [https://www.nature.com/articles/s41598-025-20595-5]
- 144. Targeted Degradation Technologies Utilizing Autophagy - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12295361/]
- 145. The ubiquitin-proteasome system in the tumor immune ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1522427/full]
- 146. Different Strategies to Overcome Resistance ... [https://www.mdpi.com/1422-0067/25/16/8949]
- 147. Repurposing old drugs as new inhibitors of the ubiquitin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7316602/]
- 148. PROTAC Technology as a New Tool for Modern ... [https://www.mdpi.com/1420-3049/30/10/2123]
- 149. Overcoming Cancer Drug Resistance Utilizing PROTAC ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2022.872729/full]
- 150. PROTAC targeted protein degraders: the past is prologue [https://www.nature.com/articles/s41573-021-00371-6]
- 151. Multitargeted protein degradation: Integrating small- ... [https://www.sciencedirect.com/science/article/pii/S259004982500089X]
- 152. The ubiquitin-proteasome system [https://pubmed.ncbi.nlm.nih.gov/16595883/]
- 153. Ubiquitin-Proteasome System [https://lifesensors.com/ubiquitinproteasomesystem/]
- 154. E3 Ubiquitin Ligases as Cancer Targets and Biomarkers - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC1601942/]
- 155. A Perspective on Therapeutic Targeting Against Ubiquitin ... [https://www.mdpi.com/2072-6694/17/4/626]
- 156. Treatment-related adverse events of antibody drug-conjugates ... [https://jhoonline.biomedcentral.com/articles/10.1186/s13045-025-01720-3]
- 157. Identification of biomarkers based on ubiquitin-correlated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12394886/]
- 158. A Molecular Signature of the Ubiquitin-Proteasome System for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11627108/]
- 159. Proteasomes and Ubiquitin C-Terminal Hydrolase L1 as ... [https://www.mdpi.com/2075-1729/15/3/413]
- 160. Timelines and Takeaways from the Biomarker Qualification ... [https://pubmed.ncbi.nlm.nih.gov/41139768/]
- 161. Identification and mechanistic insights of ubiquitin-proteasome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12362490/]
- 162. Current Status of α-Synuclein Biomarkers and the Need for α ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12384595/]
- 163. Use of Biomarkers in Drug Development for Regulatory ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12489174/]
- 164. Timelines and Takeaways from the Biomarker Qualification ... [https://link.springer.com/article/10.1007/s43441-025-00889-6]