Ubiquitin and ATP-Dependent Proteolysis: From Nobel Discovery to Targeted Protein Degradation Therapeutics
This article explores the groundbreaking discovery of ATP-dependent proteolysis by Avram Hershko, Aaron Ciechanover, and Irwin Rose, which earned them the 2004 Nobel Prize in Chemistry.
Ubiquitin and ATP-Dependent Proteolysis: From Nobel Discovery to Targeted Protein Degradation Therapeutics
Abstract
This article explores the groundbreaking discovery of ATP-dependent proteolysis by Avram Hershko, Aaron Ciechanover, and Irwin Rose, which earned them the 2004 Nobel Prize in Chemistry. We trace the foundational research that identified the ubiquitin-proteasome system as a crucial regulatory mechanism for intracellular protein degradation, moving beyond the lysosomal paradigm. The content examines the methodological breakthroughs that enabled this discovery and their direct application in modern drug development, particularly in PROTACs (Proteolysis Targeting Chimeras) and other targeted protein degradation technologies. For researchers and drug development professionals, we analyze current challenges in optimizing these therapeutic strategies and compare the ubiquitin system with other ATP-dependent proteases. The synthesis provides a comprehensive resource for understanding both the historical significance and future directions of this transformative field in biomedical research.
The Paradigm Shift: Discovering the Ubiquitin-Proteasome System
The fundamental understanding of cellular protein degradation was revolutionized in the late 20th century by the groundbreaking work of Avram Hershko, Aaron Ciechanover, and Irwin Rose, who discovered the ubiquitin-mediated ATP-dependent proteolytic system. Their research addressed a central biochemical paradox: why would the hydrolysis of peptide bonds, an exergonic process, require energy input in the form of adenosine triphosphate (ATP)? This energy requirement suggested the existence of a complex regulatory mechanism far beyond simple enzymatic cleavage [1].
Before this discovery, intracellular proteolysis was poorly understood, with many scientists believing it occurred primarily through lysosomal action or via bacterial Lon protease. The key breakthrough came from the convergence of intellectual curiosity and rigorous biochemical experimentation, which revealed that ATP-dependent proteolysis involved a sophisticated system of covalent protein modification [1]. This review examines the foundational experiments that uncovered the ubiquitin system, the methodological approaches used, and the implications of these discoveries for understanding cellular regulation.
Historical and Theoretical Background
The Energy Paradox in Cellular Protein Turnover
The energy paradox of intracellular proteolysis was first highlighted by Melvin Simpson in 1953 through isotopic labeling studies, which demonstrated that protein degradation in mammalian cells required energy [1]. For the next 25 years, this observation remained largely unexplained, as the hydrolysis of peptide bonds is thermodynamically favorable and thus should not require energy input. The scientific community recognized that this energy requirement must indicate unknown regulatory complexity in cellular proteolytic systems [1].
Concurrently, research by Goldberg's group revealed that damaged or abnormal proteins were rapidly cleared from cells in an energy-dependent manner [1]. They also observed that rate-limiting enzymes in metabolic pathways were often short-lived, with their concentrations responsive to metabolic conditions. These findings suggested that energy-dependent proteolysis served regulatory functions beyond simple protein disposal [1].
Prelude to Discovery: Key Observations
Several critical observations set the stage for the discovery of the ubiquitin system:
- ATP dependence: Reticulocyte lysates, which lack lysosomes, exhibited ATP-dependent proteolysis of denatured proteins
- Fractionation potential: The proteolytic system could be separated into biochemical fractions that required recombination for activity
- Heat-stable factors: Initial fractionation studies revealed the existence of heat-stable components essential for proteolysis [1]
These observations suggested that ATP-dependent proteolysis involved multiple protein components rather than a single protease, pointing to a more complex system than previously imagined.
Experimental Breakthroughs: Methodology and Key Findings
The Reticulocyte Lysate System and Biochemical Fractionation
The Hershko and Ciechanover laboratory developed a reticulocyte lysate system that reproduced ATP-dependent proteolysis, enabling biochemical dissection of the process. Their experimental approach involved several key steps [1]:
Table 1: Key Experimental Steps in the Discovery of Ubiquitin-Mediated Proteolysis
| Experimental Step | Methodological Approach | Key Outcome |
|---|---|---|
| System Development | Use of reticulocyte lysates lacking lysosomes | Established ATP-dependent proteolysis in cell-free system |
| Biochemical Fractionation | Separation of lysate into Fractions I and II | Identified requirement for multiple components |
| Factor Identification | Heat stability and chromatographic purification | Isolated APF-1 (later identified as ubiquitin) |
| Conjugation Analysis | Use of ¹²⁵I-labeled APF-1 and SDS-PAGE | Demonstrated covalent attachment to multiple proteins |
The experimental workflow began with separating reticulocyte lysates into two essential fractions: Fraction I contained a heat-stable protein component, while Fraction II contained higher molecular weight components. Both fractions were required to reconstitute ATP-dependent proteolysis [1].
Identification of APF-1 and the Covalent Modification System
The critical breakthrough came from the identification and characterization of ATP-dependent Proteolysis Factor 1 (APF-1), a small, heat-stable protein that was absolutely required for ATP-dependent proteolysis. The key experiments included [1]:
- Covalent linkage demonstration: Incubation of ¹²⁵I-labeled APF-1 with Fraction II and ATP resulted in the formation of high molecular weight complexes
- Stability analysis: The linkage between APF-1 and target proteins was stable to NaOH treatment, indicating a covalent bond
- Specificity controls: The conjugation required ATP and specific metal ions, with requirements matching those for proteolysis
Art Haas, a postdoctoral fellow in Rose's laboratory, made the crucial observation that the association survived high pH treatment, demonstrating its covalent nature [1]. This finding was particularly surprising because it revealed that proteolytic targeting involved covalent modification of substrates rather than simple receptor-ligand interactions.
Identification of APF-1 as Ubiquitin
The discovery that APF-1 was identical to the previously known protein ubiquitin (first discovered by Gideon Goldstein in his search for thymopoietin) connected the ATP-dependent proteolysis system to known biology [1]. This identification came through:
- Biochemical comparison: Direct comparison of APF-1 with authentic ubiquitin samples
- Functional analysis: Demonstration that ubiquitin could replace APF-1 in supporting ATP-dependent proteolysis
- Historical connection: Recognition that histone H2A was known to be modified by ubiquitin, suggesting broader biological significance
This connection revealed that the covalent attachment of proteins to other proteins served as a regulatory mechanism, analogous to phosphorylation or acetylation [1].
Key Research Reagents and Experimental Tools
Table 2: Essential Research Reagents in the Discovery of Ubiquitin-Mediated Proteolysis
| Reagent/Resource | Function/Role in Discovery | Key Experimental Application |
|---|---|---|
| Reticulocyte Lysate | ATP-dependent proteolysis system | Cell-free model for biochemical analysis |
| Fraction I | Source of APF-1/ubiquitin | Reconstitution of proteolytic activity |
| Fraction II | High molecular weight components | Contains conjugation enzymes and proteolytic machinery |
| ¹²⁵I-labeled APF-1 | Radiolabeled tracer | Tracking covalent conjugation to target proteins |
| ATP and ATP-regenerating System | Energy source | Driving conjugation and proteolytic reactions |
| Ion-exchange Chromatography | Protein separation and purification | Isolation of APF-1 and other essential factors |
The experimental system relied heavily on classical biochemical techniques, including fractionation, ion-exchange chromatography, and radiolabeling. The reticulocyte lysate system was particularly valuable because it contained minimal lysosomal contamination, allowing clear observation of the ATP-dependent cytoplasmic proteolytic system [1].
Mechanistic Insights: From Covalent Modification to Proteolysis
The seminal PNAS papers from 1980 outlined the essential mechanism of ubiquitin-mediated proteolysis, which can be summarized in several key steps:
- Activation of ubiquitin: ATP-dependent activation of ubiquitin for conjugation
- Target recognition: Identification of protein substrates for modification
- Covalent conjugation: Formation of isopeptide bonds between ubiquitin and substrate proteins
- Polyubiquitin chain formation: Processive addition of multiple ubiquitin molecules
- Proteolytic targeting: Recognition of polyubiquitinated substrates by the proteasome
- ATP-dependent degradation: Unfolding and proteolysis of targeted substrates
The researchers demonstrated that authentic substrates of the system were heavily modified, with multiple molecules of APF-1/ubiquitin attached to each molecule of substrate [1]. This polyubiquitination was later shown to involve chains linked through K48 of one ubiquitin and the C-terminus of the next, forming the specific signal for proteasomal recognition [1].
Comparative Analysis: Bacterial vs. Eukaryotic ATP-Dependent Proteolysis
The discovery of ubiquitin-mediated proteolysis highlighted fundamental differences between eukaryotic and bacterial protein degradation systems, though both utilize AAA+ proteases (ATPases Associated with diverse cellular Activities) [2].
Table 3: Comparison of ATP-Dependent Proteolytic Systems in Eukaryotes and Bacteria
| Feature | Eukaryotic Ubiquitin-Proteasome System | Bacterial AAA+ Protease Systems |
|---|---|---|
| Targeting Mechanism | Covalent ubiquitin modification | Direct recognition of degron sequences |
| Key Proteases | 26S Proteasome | Lon, ClpXP, ClpAP, FtsH, HslUV |
| ATPase Components | AAA-ATPases in regulatory particle | Integral AAA+ domains in proteases |
| Specificity Control | Multiple E3 ubiquitin ligases | Adaptor proteins (e.g., SspB) |
| Energy Requirements | ATP for ubiquitination and degradation | ATP primarily for unfolding/degradation |
| Quality Control Role | Misfolded protein clearance | Misfolded protein clearance |
While eukaryotic cells use the ubiquitin tagging system, bacteria employ AAA+ proteases that recognize specific degradation signals (degrons) directly or through adaptor proteins. For example, the bacterial ClpXP protease recognizes the ssrA tag added during trans-translation, with enhanced recognition through the SspB adaptor protein [2]. Similarly, Lon protease recognizes misfolded proteins through exposed hydrophobic patches or specific degrons without requiring covalent modification [2].
Impact and Implications of the Discovery
The discovery of ubiquitin-mediated proteolysis had far-reaching implications for understanding cellular regulation:
- New regulatory paradigm: The system established protein degradation as a regulated process rather than simply a disposal mechanism
- Connection to key cellular processes: Ubiquitin-mediated proteolysis was quickly connected to cell cycle regulation through cyclin degradation
- Therapeutic targeting: The system has become a major target for drug development, particularly in cancer therapy
- Disease connections: Dysregulation of the ubiquitin-proteasome system has been implicated in numerous diseases, including neurodegenerative disorders
The discovery exemplified how fundamental biochemical curiosity could unravel complex regulatory mechanisms with broad biological significance. The convergence of intellectual curiosity, collaboration, and rigorous biochemistry enabled this paradigm-shifting discovery that ultimately earned Hershko, Ciechanover, and Rose the Nobel Prize in Chemistry in 2004 [1].
The discovery of the ubiquitin-proteasome system, a cornerstone of modern cell biology, was propelled by the innovative use of an unassuming biochemical tool: the rabbit reticulocyte lysate. While the scientific community of the 1970s was largely focused on protein synthesis and lysosomal degradation, the pioneering work of Avram Hershko and Aaron Ciechanover leveraged this cell-free system to uncover a fundamental, ATP-dependent proteolytic pathway. This technical guide explores the pivotal role of reticulocyte lysates as a model system that enabled the dissection of ubiquitin-mediated protein degradation. We detail the experimental methodologies, key findings, and contemporary applications of this powerful in vitro platform, framing its development within the broader context of Hershko and Ciechanover's seminal research on regulated proteolysis.
Prior to the groundbreaking work of Hershko, Ciechanover, and Rose, intracellular protein degradation was considered a largely unregulated process occurring primarily within the lysosome, a membrane-bound organelle containing digestive enzymes [3]. However, several observations contradicted this view, including the finding that ATP was required for the degradation of many cellular proteins—a thermodynamic paradox given that protein breakdown is an energy-liberating process [3] [4].
Hershko encountered this paradox during his postdoctoral research and, upon establishing his own laboratory at the Technion-Israel Institute of Technology, sought to unravel its mechanism. He and his graduate student, Aaron Ciechanover, made a strategic decision to use a cell-free system derived from rabbit reticulocytes (immature red blood cells) for several key reasons [4]:
- Absence of Lysosomes: Reticulocytes naturally lose their lysosomes during maturation, allowing study of non-lysosomal proteolytic pathways without contamination.
- High Degradation Activity: As reticulocytes mature into erythrocytes, they extensively degrade their internal organelles and proteins, making them a rich source of the relevant proteolytic machinery.
- Biochemical Tractability: A cell-free extract could be fractionated, manipulated, and studied in a controlled manner, unlike intact cells.
This experimental choice was instrumental in leading to the discovery of the ubiquitin-proteasome system, for which Hershko, Ciechanover, and Rose were awarded the 2004 Nobel Prize in Chemistry [5]. The reticulocyte lysate system provided the essential platform for identifying the key components and biochemical steps of this pathway.
The Reticulocyte Lysate System: Fundamental Principles and Advantages
Reticulocyte lysates are translationally active cytoplasmic extracts that retain the core machinery for protein synthesis, modification, and degradation. Their utility extends far beyond their initial application, serving as a foundational tool for biochemical reconstitution.
Table: Key Characteristics of Rabbit Reticulocyte Lysates in Proteolysis Research
| Characteristic | Description | Experimental Implication |
|---|---|---|
| Lysosome-free | Naturally lacking lysosomal compartments [4] | Enables isolated study of non-lysosomal, ATP-dependent proteolysis. |
| ATP-dependent | Contains functional machinery for energy-dependent ubiquitination and proteasomal degradation [6] | Allows investigation of the energy requirement for targeted protein degradation. |
| Reconstitution Capability | Can be separated into complementary fractions that regain activity upon mixing [3] | Permits fractionation, identification, and purification of individual system components (E1, E2, E3, proteasome). |
| Heat-Stable Components | Contains heat-stable factors (e.g., Ubiquitin/APF-1) that remain active after boiling [3] [4] | Enabled the critical purification and identification of ubiquitin. |
The system's power lies in its biochemical flexibility. Researchers can supplement lysates with radiolabeled substrates, energy-regenerating systems, specific inhibitors, or purified recombinant proteins to dissect complex processes [6] [7]. Furthermore, the ability to create a "dialyzed" lysate—depleted of endogenous small molecules like ATP and ubiquitin—allows for precise control over reaction conditions and the addition of defined components [6].
Historical Breakthrough: The Discovery of Ubiquitin-Mediated Proteolysis
The elucidation of the ubiquitin pathway serves as a masterclass in biochemical discovery using the reticulocyte lysate model.
Key Experimental Workflow
The initial experiments followed a logical progression of fractionation and reconstitution, as visualized below.
The Discovery of APF-1/Ubiquitin and the Conjugation Mechanism
A critical breakthrough came when the researchers, struggling to separate the active component in Fraction I from hemoglobin, decided to boil the fraction. Unlike most proteins, the active factor remained soluble, allowing them to purify it and name it ATP-dependent proteolysis factor 1 (APF-1) [3] [4].
Subsequent experiments revealed APF-1's unique behavior: in the presence of ATP, it formed covalent conjugates with a wide range of proteins in the lysate. This observation, supported by crucial input from Irwin Rose, led to the then-heretical hypothesis that proteins are marked for degradation by the covalent attachment of a label, not by direct protease recognition [3]. APF-1 was later identified by others as ubiquitin, a previously known but functionally enigmatic small protein [5].
Further work in the reticulocyte lysate system elucidated the enzymatic cascade responsible for ubiquitin conjugation:
- E1 (Ubiquitin-activating enzyme): Activates ubiquitin in an ATP-dependent reaction.
- E2 (Ubiquitin-conjugating enzyme): Accepts ubiquitin from E1.
- E3 (Ubiquitin ligase): Recognizes specific protein substrates and facilitates the transfer of ubiquitin from E2 to the target, forming an isopeptide bond [3].
The power of multi-ubiquitin chains as the definitive degradation signal was also demonstrated using this system [3].
Contemporary Applications and Methodologies
Reticulocyte lysates remain a vital tool for studying protein degradation and other complex cellular processes.
Studying Endoplasmic Reticulum-Associated Degradation (ERAD)
Reticulocyte lysates, when supplemented with canine pancreas microsomal membranes, provide a powerful system to reconstitute ERAD. This combined system supports:
- ER targeting, translocation, and glycosylation of newly synthesized proteins.
- Generation of ³⁵S-labeled, membrane-integrated substrates with defined topology.
- ATP-dependent degradation of misfolded ER proteins via the ubiquitin-proteasome pathway [6].
The degradation process can be quantitatively tracked by monitoring the conversion of radiolabeled, full-length substrate into trichloroacetic acid (TCA)-soluble peptides [6].
Analysis of Translation Mechanisms
Nuclease-treated reticulocyte lysates are a cornerstone for in vitro translation studies. They support various initiation mechanisms (cap-dependent, IRES-mediated) and allow for the analysis of ribosomal complex formation via sucrose density gradients. A key advantage is that reactions in lysates occur at or near the in vivo rate, unlike systems reconstituted from purified factors [7].
Table: Essential Research Reagents for Reticulocyte Lysate Experiments
| Research Reagent | Function/Description | Key Application |
|---|---|---|
| Nuclease-Treated Lysate | Lysate treated with micrococcal nuclease to degrade endogenous mRNA. | Ensures that translation of added reporter mRNA can be monitored without background [7]. |
| Energy Mix (ATP, GTP) | Provides essential energy for both translation and ubiquitination. | Required for efficient protein synthesis and for the activity of the ubiquitin-proteasome pathway. |
| ³⁵S-Methionine/Cysteine | Radiolabeled amino acids incorporated into newly synthesized proteins. | Allows sensitive detection and quantification of synthesized or degraded proteins via SDS-PAGE and autoradiography [7]. |
| Canine Microsomal Membranes | Membrane vesicles derived from the endoplasmic reticulum. | Used to study co-translational translocation, glycosylation, and ERAD of membrane proteins [6]. |
| Proteasome Inhibitors (e.g., MG132) | Specific inhibitors of the 26S proteasome's proteolytic activity. | Used to confirm ubiquitin-proteasome-dependent degradation and to accumulate ubiquitinated substrates [6]. |
A standard translation reaction in a 50 µl volume typically contains 35 µl of lysate, a mixture of amino acids (minus methionine), ²⁵S-methionine, RNase inhibitor, and the experimental mRNA [7]. Optimization of mRNA and salt concentrations (particularly Mg²⁺ and K⁺) is critical for maximal efficiency.
The Scientist's Toolkit: Core Methodologies
This section provides a concise protocol for key experiments enabled by the reticulocyte lysate system.
Protocol: Reconstituting ATP-Dependent Proteolysis
This foundational protocol is adapted from the original approaches used to discover ubiquitination [3] [4].
- Lysate Preparation: Use fresh or commercially available rabbit reticulocyte lysate. For degradation assays, the lysate may be dialyzed to remove endogenous amino acids and small molecules.
- Reaction Setup:
- Test Sample: Combine lysate (e.g., 25 µl), ²⁵S-labeled substrate (e.g., 1 µl), and an energy-regenerating system (e.g., 2 µl of 10x ATP/GTP mix).
- Negative Control: Set up an identical reaction without ATP or with a non-hydrolyzable ATP analog.
- Incubation: Incubate reactions at 30-37°C for 0-120 minutes.
- Termination and Analysis:
- Stop reactions at desired time points by adding an equal volume of 2x SDS-PAGE sample buffer (for visualizing ubiquitin conjugates) or by transferring to ice.
- For degradation quantification, precipitate proteins with 10% Trichloroacetic Acid (TCA). The amount of TCA-soluble radioactivity (degraded peptides) in the supernatant, measured by scintillation counting, is directly proportional to proteolytic activity.
Protocol: In Vitro Translation for Degradation Substrate Generation
This protocol is used to generate a radiolabeled protein substrate for subsequent degradation assays [7].
- mRNA Template: Prepare the mRNA of interest via in vitro transcription. A 5' cap structure (m7G) is recommended for efficient initiation.
- Translation Reaction:
- Assemble a 50 µl reaction containing:
- 35 µl Nuclease-Treated Reticulocyte Lysate
- 2 µl 1mM Amino Acid Mixture (minus Methionine)
- 2 µl ¹⁵S-Methionine (10 mCi/ml)
- 1 µl RNasin Ribonuclease Inhibitor
- 0.5-1 µg of mRNA template
- Nuclease-free water to 50 µl.
- Incubate at 30°C for 60-90 minutes.
- Assemble a 50 µl reaction containing:
- Product Verification: Analyze 2-5 µl of the reaction by SDS-PAGE and autoradiography to confirm synthesis of the full-length, radiolabeled protein.
The relationship between these core applications is summarized below.
The rabbit reticulocyte lysate system stands as a testament to the power of a well-chosen model system in driving scientific revolution. It was the indispensable biochemical canvas upon which Hershko, Ciechanover, and Rose painted their picture of the ubiquitin-proteasome system, fundamentally altering our understanding of how cells control their protein repertoire. From its historic role in discovering fundamental principles to its continued utility in modeling complex processes like ERAD and translational regulation, the reticulocyte lysate remains a versatile and powerful platform for mechanistic cell biology. Its legacy underscores the importance of methodological innovation in enabling the discovery of profound biological truths.
Identifying APF-1: The Heat-Stable Factor Later Recognized as Ubiquitin
This whitepaper delineates the seminal discovery of ATP-dependent Proteolysis Factor 1 (APF-1) and its subsequent identification as the protein ubiquitin, the central component of a novel proteolytic system. The research, pioneered by Avram Hershko, Aaron Ciechanover, and Irwin Rose, fundamentally reconfigured our understanding of intracellular protein degradation, moving from a model of nonspecific lysosomal digestion to one of a highly specific, energy-dependent, and enzymatically controlled process [1] [8]. This document provides a comprehensive technical account of the key experiments, methodologies, and biochemical evidence that established APF-1/ubiquitin's role in targeted protein degradation—a pathway now recognized as critical in regulating cell cycle, DNA repair, and numerous other cellular functions, with profound implications for therapeutic drug development [8] [3].
Prior to the 1980s, intracellular protein degradation was largely considered an unregulated, bulk process occurring within lysosomes. A key biochemical paradox challenged this view: the hydrolysis of peptide bonds is an exergonic process, yet the degradation of intracellular proteins was demonstrated to require metabolic energy in the form of adenosine triphosphate (ATP) [1] [8]. This ATP requirement suggested the existence of a previously unrecognized, non-lysosomal proteolytic pathway characterized by high specificity and complex regulation.
The collaborative work of Hershko, Ciechanover, and Rose was instrumental in resolving this paradox. They utilized a cell-free extract derived from rabbit reticulocytes, which, due to their lack of lysosomes, provided an ideal model system for studying ATP-dependent, non-lysosomal proteolysis [9] [10]. Through biochemical fractionation of this system, they identified an essential, heat-stable polypeptide they designated APF-1, which would later be recognized as the previously described but functionally enigmatic protein, ubiquitin [1] [11].
Experimental Breakthroughs: The APF-1 Discovery Pipeline
The identification and characterization of APF-1 involved a series of critical experiments that systematically uncovered its unique properties and central role.
Initial Fractionation and the Discovery of a Heat-Stable Factor
The initial breakthrough came from the fractionation of the reticulocyte lysate using anion-exchange chromatography (DEAE-cellulose). This process separated the lysate into two complementary fractions:
- Fraction I: Contained proteins not adsorbed to the resin, including the bulk of hemoglobin.
- Fraction II: Contained proteins adsorbed to and eluted from the resin [9] [10].
Critically, neither fraction alone could support ATP-dependent proteolysis; activity was only restored upon their recombination. The key finding was that the essential component in Fraction I was a low-molecular-weight (~8.5 kDa), heat-stable protein that retained its activity even after being boiled, a treatment that denatures most cellular proteins. This factor was named APF-1 [12] [10].
Table 1: Key Properties of the Reticulocyte Lysate Fractions and APF-1
| Component | Key Characteristics | Role in Proteolysis |
|---|---|---|
| Reticulocyte Lysate | Cell-free system from immature red blood cells; lacks lysosomes. | Source of ATP-dependent proteolytic activity [9]. |
| Fraction I | Not adsorbed to DEAE-cellulose; contained hemoglobin and heat-stable proteins. | Provided an essential factor (APF-1) for reconstituting activity [10]. |
| Fraction II | Adsorbed to and eluted from DEAE-cellulose. | Contained additional essential enzymes and the proteolytic machinery [10]. |
| APF-1 | Heat-stable polypeptide; molecular weight ~8.5 kDa. | Essential stimulating factor; later identified as ubiquitin [12]. |
The Covalent Conjugation Hypothesis
A pivotal experiment involved incubating iodine-125-labeled APF-1 (¹²⁵I-APF-1) with Fraction II and ATP. Instead of observing free APF-1, researchers discovered the label was incorporated into multiple high-molecular-weight complexes [1]. This association was:
- ATP-dependent: Required the presence of ATP.
- Covalent: The complexes were stable to treatment with sodium hydroxide (NaOH) and sodium dodecyl sulfate (SDS), indicating a strong, covalent linkage [1] [9].
- Reversible: The process could be reversed upon ATP depletion.
This led to the then-revolutionary hypothesis that APF-1 was not an enzyme activator but was itself covalently conjugated to protein substrates,
marking them for degradation [1] [8]. Further work demonstrated that a single target protein could be modified by multiple molecules of APF-1, a process termed polyubiquitination [8].
Diagram 1: Experimental Workflow for APF-1 Isolation.
The Critical Identification: APF-1 is Ubiquitin
The discovery of covalent protein conjugation prompted a search for precedents. Researchers noted that a known protein, ubiquitin, had been previously found conjugated to histone H2A in a similar isopeptide linkage [1]. This prompted a direct comparison, which confirmed APF-1 and ubiquitin were identical through several lines of evidence [11]:
Table 2: Experimental Evidence Identifying APF-1 as Ubiquitin
| Assay Type | Observation | Interpretation |
|---|---|---|
| Gel Electrophoresis & Isoelectric Focusing | APF-1 and authentic ubiquitin co-migrated identically across five different polyacrylamide gel systems and in isoelectric focusing [11]. | The two proteins share identical molecular weight and isoelectric point. |
| Amino Acid Analysis | The amino acid composition of APF-1 showed excellent agreement with that of ubiquitin [11]. | The two proteins share an identical primary structure. |
| Functional Reconstitution | Purified ubiquitin could replace APF-1 in activating the ATP-dependent proteolytic system from reticulocytes [11]. | The two proteins are functionally interchangeable. |
| Covalent Conjugation | ¹²⁵I-ubiquitin formed electrophoretically identical covalent conjugates with reticulocyte proteins as ¹²⁵I-APF-1 [11]. | The conjugation mechanism and targets are identical. |
This conclusive evidence, published in 1980, established that the ATP-dependent proteolysis factor, APF-1, was the protein ubiquitin [11].
The Ubiquitin-Proteasome System: A Modern Technical Perspective
The initial discoveries around APF-1/ubiquitin laid the foundation for the elaborate ubiquitin-proteasome system (UPS) as it is understood today. The process involves a tightly regulated enzymatic cascade.
The Enzymatic Cascade
The attachment of ubiquitin to a substrate protein is a multi-step process mediated by three key enzyme classes:
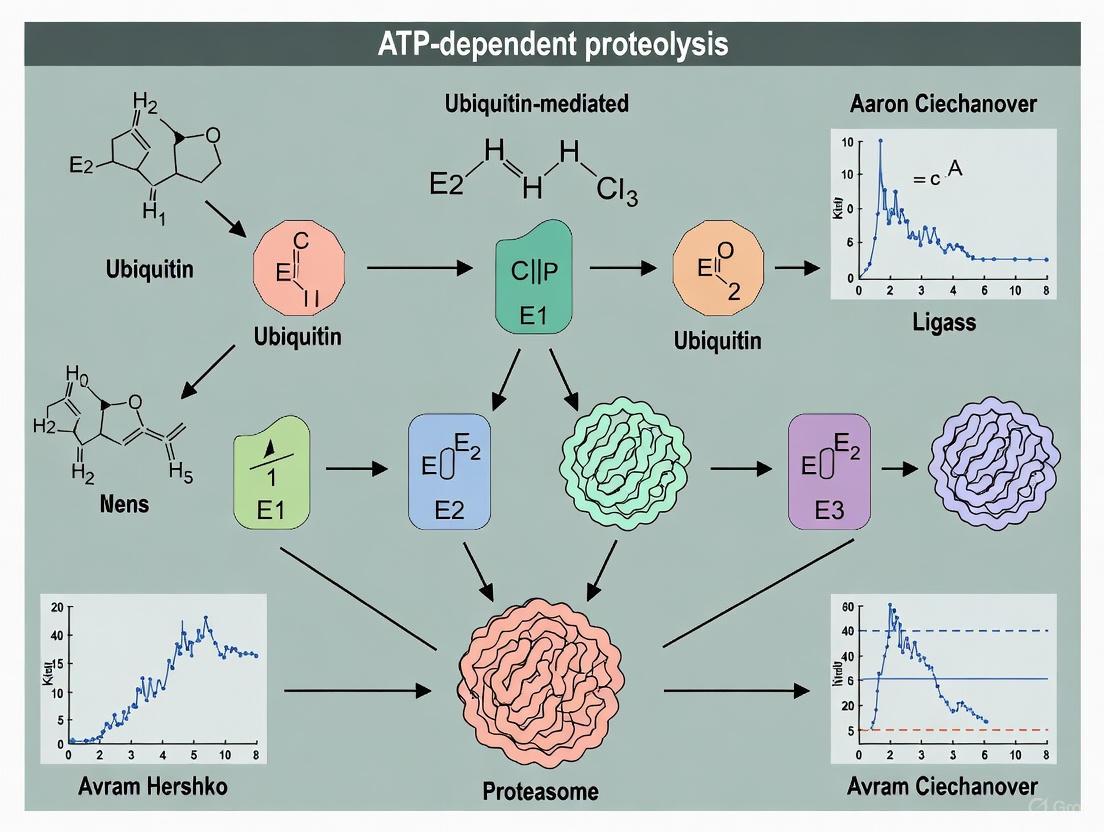
- E1 (Ubiquitin-Activating Enzyme): Activates ubiquitin in an ATP-dependent reaction, forming a high-energy thioester bond with the C-terminal glycine of ubiquitin.
- E2 (Ubiquitin-Conjugating Enzyme): Accepts the activated ubiquitin from E1 via a trans-thioesterification reaction.
- E3 (Ubiquitin Ligase): Recognizes specific protein substrates and facilitates the transfer of ubiquitin from E2 to a lysine residue on the substrate, forming an isopeptide bond [8] [3].
A polyubiquitin chain is built by reiterating this process, where additional ubiquitin molecules are attached to a lysine residue (typically Lys48) on the previously conjugated ubiquitin. This K48-linked polyubiquitin chain serves as the definitive recognition signal for the 26S proteasome [1] [8].
Diagram 2: The Ubiquitin-Proteasome System Pathway.
The Scientist's Toolkit: Key Research Reagent Solutions
The discovery of the UPS was enabled by specific reagents and model systems. The following toolkit details resources critical for both foundational and contemporary research in this field.
Table 3: Research Reagent Solutions for Ubiquitin System Studies
| Reagent / Model System | Function in Research | Key Application in Discovery |
|---|---|---|
| Reticulocyte Lysate | A cell-free system capable of protein synthesis and degradation. | Served as the source for fractionating and identifying the essential components of the UPS, including APF-1/ubiquitin [9] [10]. |
| DEAE-Cellulose Resin | An anion-exchange chromatography medium for separating protein mixtures. | Used to fractionate the reticulocyte lysate into complementary Fractions I and II, enabling the identification of APF-1 [9] [10]. |
| Heat-Stable Protein Fractions | A preparation enriched for proteins that resist denaturation at high temperatures (e.g., 100°C). | The key step that allowed for the separation of APF-1/ubiquitin from the bulk of hemoglobin and other denatured proteins in Fraction I [3] [10]. |
| ATPγS (ATP analog) | A non-hydrolyzable analog of ATP. | Used in modern studies to probe ATP-dependent steps; its failure to support conjugation would confirm an ATP hydrolysis requirement [1]. |
| ts20 Cell Line | A temperature-sensitive mutant mouse cell line with a thermolabile E1 enzyme. | Provided crucial genetic evidence in intact cells that a functional ubiquitin system is essential for cell cycle progression and degradation of short-lived proteins [8] [3]. |
Research Applications and Drug Development Implications
The elucidation of the ubiquitin-proteasome system has created a vast field of therapeutic opportunity. By understanding the rules of targeted protein degradation, researchers can now design strategies to eliminate specific disease-causing proteins.
- Targeted Protein Degradation (TPD): Modern modalities like PROTACs (Proteolysis-Targeting Chimeras) are heterobifunctional molecules that recruit a target protein to an E3 ubiquitin ligase, inducing its ubiquitination and degradation by the proteasome [13]. This approach can target proteins previously considered "undruggable."
- Cancer Therapeutics: The UPS is crucial for regulating cell cycle proteins (e.g., cyclins) and tumor suppressors (e.g., p53). Bortezomib, a proteasome inhibitor, is a validated therapy for multiple myeloma, proving the clinical relevance of this pathway [8].
- Quality Control and Disease: The UPS is the primary system for eliminating misfolded and damaged proteins. Defects in this system are implicated in neurodegenerative diseases like Alzheimer's and Parkinson's, making UPS components attractive targets for therapeutic intervention [8] [9].
The journey from an unknown, heat-stable factor dubbed APF-1 to its identification as the pivotal regulatory protein ubiquitin represents a paradigm shift in cell biology. The rigorous biochemical approaches of Hershko, Ciechanover, and Rose—centered on fractionation, functional reconstitution, and covalent linkage analysis—uncovered a previously unimaginable layer of cellular regulation. Their work demonstrated that cells employ a highly specific, energy-dependent system to label and destroy individual proteins, a process as sophisticated and vital as protein synthesis itself. This discovery of the ubiquitin-proteasome system has not only illuminated fundamental biological processes but has also forged a new and expanding frontier in drug development, enabling innovative strategies to treat cancer, neurodegenerative disorders, and other human diseases.
The concept of deliberately tagging proteins for destruction represents a paradigm shift in chemical biology and therapeutic development. This approach, known as targeted protein degradation, has evolved from a fundamental understanding of cellular protein turnover into a powerful strategy for eliminating disease-causing proteins. At its core lies the revolutionary discovery of the ubiquitin-proteasome system by Avram Hershko, Aaron Ciechanover, and their colleagues, who elucidated the ATP-dependent proteolytic machinery that cells use to selectively mark and destroy proteins [3]. Their pioneering work in the late 1970s and 1980s identified the crucial role of a small, heat-stable protein they initially termed APF-1 (later identified as ubiquitin), which serves as a molecular "death tag" for proteins [14] [3]. This foundational research uncovered the enzymatic cascade (E1, E2, E3) that conjugates ubiquitin to target proteins, marking them for destruction by the proteasome [3]. The conceptual breakthrough that proteins must be covalently modified to be recognized for degradation paved the way for developing intentional protein degradation strategies that now dominate modern drug discovery efforts.
Historical Context: Hershko and Ciechanover's ATP-Dependent Proteolysis
The intellectual foundation for contemporary protein degradation technologies stems directly from the groundbreaking work on ATP-dependent proteolysis. Hershko's initial curiosity was sparked by an apparent paradox: why would protein degradation, an inherently energy-liberating process, require ATP hydrolysis? [3] This question led to a series of elegant experiments using reticulocyte (immature red blood cell) lysates, which provided a cell-free system for studying non-lysosomal protein degradation.
Key Experimental Breakthroughs and Methodologies
Table: Seminal Experiments in the Discovery of the Ubiquitin System
| Year | Experimental Approach | Key Finding | Significance |
|---|---|---|---|
| 1977-1978 | Fractionation of reticulocyte lysates followed by boiling | Identification of heat-stable APF-1 (ubiquitin) | Discovered the central component of the tagging system [3] |
| 1978-1980 | Radiolabeling of APF-1 with ATP addition | Demonstration of covalent APF-1-protein conjugates | Established the tagging mechanism [3] |
| 1980s | Biochemical reconstitution | Identification of E1, E2, E3 enzymatic cascade | Elucidated the complete ubiquitination machinery [3] |
| 1985 | Characterization of multi-ubiquitin chains | Proteins with ubiquitin chains are better degradation substrates | Revealed the polyubiquitin signal requirement [3] |
The critical methodological breakthrough came when Hershko and Ciechanover separated reticulocyte lysates into two fractions (I and II) and discovered that neither could promote ATP-dependent proteolysis alone [14] [3]. The active component in fraction I displayed remarkable heat stability—unusual for a biological effector—remaining active after boiling while hemoglobin and other proteins denatured and precipitated [3]. This observation allowed the purification and characterization of APF-1, later identified as ubiquitin.
The experimental protocol that revealed the tagging mechanism involved radioactively labeling APF-1 and incubating it with cellular fractions. In the absence of ATP, APF-1 migrated as a single small protein, but with ATP added, multiple radioactive protein bands appeared, suggesting covalent attachment of APF-1 to various target proteins [3]. Further experiments confirmed that this conjugation occurred through stable, peptide-like bonds, and that proteins destined for degradation were marked with multiple ubiquitin molecules, creating a recognizable signal for the proteolytic machinery.
The Covalent Conjugation Machinery
The Ubiquitin-Proteasome System
The ubiquitin-proteasome system represents nature's primary covalent conjugation machinery for targeted protein degradation in eukaryotic cells. This system operates through a precise enzymatic cascade that tags proteins for destruction with high specificity.
The ubiquitination pathway involves three key enzymatic components that work in concert [15] [3]:
- E1 (Ubiquitin-activating enzyme): Activates ubiquitin in an ATP-dependent manner through the formation of a thioester bond
- E2 (Ubiquitin-conjugating enzyme): Receives activated ubiquitin from E1
- E3 (Ubiquitin ligase): Recognizes specific protein substrates and facilitates ubiquitin transfer from E2 to the target protein
The result is a polyubiquitin chain (typically K48-linked) attached to the target protein, which serves as a recognition signal for the proteasome, a large multi-subunit protease complex that degrades the tagged protein into small peptides [15] [3].
Covalent Warheads in Targeted Therapeutics
The understanding of natural ubiquitination inspired the development of synthetic covalent warheads that can deliberately mark proteins for degradation. These warheads form the business end of targeted degradation technologies.
Table: Covalent Warheads in Protein Degradation Technologies
| Warhead Type | Reaction Mechanism | Target Residue | Key Characteristics | Applications |
|---|---|---|---|---|
| α-Cyanoacrylamides | Reversible Michael addition | Cysteine | Tunable reactivity, reversible binding [16] | Reversible covalent inhibitors |
| Aldehydes | Reversible hemithioacetal formation | Cysteine | Moderate electrophilicity [16] | TCIs (e.g., FGF401) [16] |
| Acrylamides | Irreversible Michael addition | Cysteine | Irreversible modification [16] | Covalent kinase inhibitors (e.g., ibrutinib) [16] |
| Boronic Acids | Reversible ester formation | Serine | Electrophilic character [16] | Proteasome inhibitors |
| Sulfonyl Fluorides | Irreversible SuFEx chemistry | Serine/Lysine/Tyrosine | High chemoselectivity [16] | Chemoproteomic probes |
These warheads can be classified as either irreversible or reversible covalent modifiers. Irreversible warheads (e.g., acrylamides) form permanent bonds with their target proteins, providing sustained inhibition but potentially increasing off-target risks [16]. Reversible warheads (e.g., α-cyanoacrylamides, aldehydes) form transient covalent bonds, offering a balance between sustained engagement and reduced potential for idiosyncratic toxicity [16].
Modern Covalent Protein Degradation Technologies
Proteolysis-Targeting Chimeras (PROTACs)
PROTACs represent the most advanced and widely adopted technology for targeted protein degradation. These heterobifunctional molecules consist of three key elements [15]:
- A warhead that binds to the protein of interest (POI)
- A ligand that recruits an E3 ubiquitin ligase
- A chemical linker that connects these two elements
The mechanism of PROTACs embodies the covalent conjugation principle by hijacking the natural ubiquitination machinery. PROTACs do not themselves covalently modify the target protein, but instead induce proximity-induced ubiquitination, leading to covalent tagging of the POI with ubiquitin chains [16] [15].
The development of PROTAC technology has followed an evolutionary path from peptide-based early constructs to fully small-molecule degraders [15]. The first PROTAC, reported in 2001 by Crews and Deshaies, used a phosphopeptide to recruit the SCF ubiquitin ligase complex and ovalicin to target methionine aminopeptidase-2 (MetAP-2) [15]. This was followed in 2008 by the first fully small-molecule PROTAC, which used non-steroidal androgen receptor ligands and MDM2 E3 ligase ligands to degrade the androgen receptor [15].
Covalent Molecular Glue Degraders
Molecular glue degraders represent a more recent innovation in covalent protein degradation. Unlike the heterobifunctional design of PROTACs, molecular glues are typically monovalent small molecules that induce or stabilize interactions between an E3 ubiquitin ligase and a target protein [15] [17].
A notable advance in this field is the discovery of covalent molecular glue degraders such as EN450, identified through chemoproteomic screening [17]. EN450 covalently targets cysteine 111 (C111) in the E2 ubiquitin-conjugating enzyme UBE2D, acting as a molecular glue that induces proximity between UBE2D and the transcription factor NF-κB (NFKB1), leading to its degradation [17]. This mechanism represents a unique approach where the covalent modification occurs not on the target protein, but on a component of the ubiquitination machinery itself, effectively repurposing it for targeted degradation.
Targeted Covalent Inhibitors (TCIs)
Targeted Covalent Inhibitors represent another application of covalent conjugation in protein targeting. TCIs consist of a reversible binding element and an electrophilic warhead that covalently modifies a nucleophilic residue (typically cysteine) on the target protein [16]. The mechanism follows a two-step process:
- Reversible binding that positions the warhead near the target residue
- Covalent bond formation that irreversibly or reversibly inactivates the protein [16]
The kinetic profile of TCIs is described by the equation:
[E + I \rightleftharpoons{k{off}}^{k{on}} E \cdot I \rightarrow{k_{inact}} E-I]
Where the overall potency is determined by the second-order rate constant of target inactivation (k{inact}/Ki) [16]. This approach has been successfully applied to previously "undruggable" targets like KRAS G12C, with covalent inhibitors such as sotorasib (AMG 510) achieving clinical success [16].
The Scientist's Toolkit: Research Reagent Solutions
Table: Essential Research Reagents for Protein Degradation Studies
| Reagent/Category | Function/Application | Key Examples | Experimental Considerations |
|---|---|---|---|
| E3 Ligase Ligands | Recruit specific E3 ubiquitin ligases | VHL ligands (e.g., VH032), CRBN ligands (e.g., lenalidomide/pomalidomide), MDM2 ligands [15] | Ligand choice influences degradation efficiency and tissue specificity |
| Covalent Warheads | Covalently bind target proteins | α-Cyanoacrylamides, acrylamides, aldehydes, boronic acids [16] | Warhead reactivity must be balanced between efficacy and selectivity |
| Linker Chemistry | Connect warheads to E3 ligands | PEG chains, alkyl chains, heterocyclic linkers [15] [18] | Linker length and composition affect ternary complex formation and degradation efficiency |
| Proteasome Inhibitors | Validate proteasome-dependent degradation | Bortezomib, MG132, carfilzomib [15] | Essential control experiments to confirm UPS mechanism |
| NEDDylation Inhibitors | Disrupt cullin-RING ligase activity | MLN4924 [17] | Control for E3 ligase functionality in degradation assays |
| Chemoproteomic Platforms | Identify covalent binding sites and mechanisms | Activity-based protein profiling (ABPP) [17] | Enables discovery of novel covalent interactions and mechanisms |
This toolkit enables researchers to design, optimize, and validate covalent protein degradation systems. Recent advances include innovative warhead designs, optimized linker strategies, and expanded E3 ligase ligands that collectively enhance the potency, selectivity, and scope of targeted degradation [18].
Experimental Protocols for Covalent Degradation Studies
Assessing Target Engagement and Degradation
A standard protocol for evaluating covalent protein degraders involves multiple steps to confirm mechanism of action:
Cellular Viability Assays:
- Treat cells with degrader compounds across a concentration range (typically 0.1 nM - 10 μM)
- Assess viability using ATP-based (CellTiter-Glo) or metabolic (MTT) assays after 72-96 hours
- Confirm NEDDylation and proteasome dependence using inhibitors like MLN4924 and bortezomib [17]
Protein Degradation Analysis:
Ternary Complex Assessment:
- Use techniques like cellular thermal shift assays (CETSA) or proximity-based methods (BRET, FRET)
- Confirm formation of POI-PROTAC-E3 ternary complex
- Evaluate cooperative binding behavior [15]
Chemoproteomic Profiling for Covalent Degrader Discovery
The discovery of covalent molecular glue degraders like EN450 employed sophisticated chemoproteomic approaches [17]:
Phenotypic Screening:
- Screen covalent ligand libraries against disease-relevant cell models
- Identify hits that impair cell viability in a proteasome-dependent manner
Activity-Based Protein Profiling (ABPP):
- Use probe-based platforms to map cysteine reactivity across the proteome
- Identify specific covalent binding sites (e.g., C111 of UBE2D for EN450)
Quantitative Proteomics:
- Employ multiplexed quantitative mass spectrometry (e.g., TMT, SILAC)
- Monitor global protein abundance changes following degrader treatment
- Identify putative degradation targets (e.g., NF-κB identification for EN450) [17]
This integrated approach enables the rapid discovery of covalent degradation mechanisms directly in native cellular environments.
The field of targeted protein degradation through covalent conjugation has evolved dramatically from the foundational discoveries of Hershko and Ciechanover. What began as fundamental research into ATP-dependent proteolysis has transformed into a sophisticated therapeutic modality with immense potential. Current research focuses on expanding the scope of degradable targets, improving tissue specificity through advanced delivery systems, and developing technologies for spatiotemporal control of degradation activity [18]. The integration of covalent chemistry with targeted degradation represents one of the most promising frontiers in chemical biology, offering potential solutions to previously intractable therapeutic challenges. As the field continues to mature, the covalent conjugation breakthrough promises to revolutionize both biological understanding and therapeutic intervention across a broad spectrum of diseases.
This technical review delineates the seminal discovery of ATP-dependent proteolysis, wherein the initial identification of an ATP-dependent proteolysis factor (APF-1) was subsequently revealed to be the well-known protein ubiquitin. The elucidation of this connection, primarily by Avram Hershko, Aaron Ciechanover, and Irwin A. Rose, established the foundational principles of the ubiquitin-proteasome system (UPS). This review provides an in-depth analysis of the key experimental protocols that uncovered APF-1's identity and function, summarizes the core components of the UPS, and explores the modern therapeutic applications, such as PROTACs, that have emerged from this fundamental research. Framed within the broader context of Hershko and Ciechanover's work, this article serves as a comprehensive guide for researchers and drug development professionals, connecting a pivotal historical discovery to its extensive implications in contemporary cellular biology and targeted protein degradation.
For decades, the metabolic logic behind ATP-dependent intracellular proteolysis was a biochemical curiosity. As the hydrolysis of peptide bonds is an exergonic process, there was no apparent thermodynamic reason for an energy requirement [1]. The collaboration between Avram Hershko, Aaron Ciechanover, and Irwin A. Rose was uniquely positioned to solve this mystery. Their work, which would later earn them the Nobel Prize in Chemistry in 2004, shifted the paradigm from the view of proteolysis as a simple, unregulated digestive process to the understanding of a highly complex, targeted, and energy-dependent regulatory mechanism [19] [1].
The initial research model used reticulocyte lysates, which are devoid of lysosomes, thus allowing the team to isolate a non-lysosomal ATP-dependent proteolytic system. Fractionation of this lysate revealed that the system could be separated into two essential fractions: Fraction I and Fraction II [1]. Fraction I was found to contain a single, heat-stable component that was crucial for the reaction, which the researchers termed APF-1 (ATP-dependent Proteolysis Factor 1). The journey to connect this unknown factor to a known protein would redefine the landscape of cell biology.
Experimental Breakthrough: The Discovery of APF-1 and its Covalent Attachment
Key Experimental Protocol: Identifying the Covalent Link
The following workflow summarizes the pivotal experiment that demonstrated the covalent nature of APF-1 attachment.
Title: Experimental Workflow for APF-1 Conjugation
Detailed Methodology:
- System Reconstitution: The ATP-dependent proteolytic system was reconstituted by combining Fraction I (containing APF-1) and Fraction II [1].
- Radiolabeling: APF-1 was tagged with iodine-125 (¹²⁵I) to allow for sensitive tracking [1].
- Incubation with ATP: The ¹²⁵I-labeled APF-1 was incubated with Fraction II in the presence of ATP.
- Analysis: The reaction mixture was analyzed using SDS-PAGE (Sodium Dodecyl Sulfate–Polyacrylamide Gel Electrophoresis), a technique that separates proteins by molecular weight. The covalent nature of the bond was confirmed by the stability of the complexes to high pH (NaOH) treatment [1].
Interpretation of Results: The appearance of radiolabeled APF-1 in high molecular weight complexes on SDS-PAGE, even under denaturing conditions, provided definitive evidence for the covalent attachment of APF-1 to multiple proteins in Fraction II. This conjugation was reversible upon ATP removal and its requirements for ATP and Fraction II mirrored those of ATP-dependent proteolysis, strongly suggesting a functional link between conjugation and protein degradation [1].
The Critical Connection: APF-1 is Ubiquitin
The discovery of covalent attachment prompted the critical question: What was the identity of APF-1? The observation that a small protein was covalently linked to cellular proteins recalled a previous finding: that histone H2A was modified by a small, ubiquitous protein named ubiquitin [1]. This similarity was pursued, and through direct comparison, it was confirmed that APF-1 was identical to the previously characterized protein, ubiquitin [1] [20]. This connection merged two separate lines of inquiry and revealed the physiological role of ubiquitin.
Table 1: From APF-1 to Ubiquitin - Key Characteristics
| Feature | APF-1 (Initial Discovery) | Ubiquitin (Later Identification) |
|---|---|---|
| Identity | ATP-dependent Proteolysis Factor 1 | Ubiquitous Immunopoietic Polypeptide (UBIP) [21] [20] |
| Known Role | Unknown | Covalent modifier of histone H2A [1] |
| Primary Function | Mediate ATP-dependent proteolysis | Target proteins for degradation via the UPS [19] [20] |
| Size | Small, heat-stable protein | 76 amino acids, 8.6 kDa [21] [20] |
| Conservation | Not initially known | Highly conserved in eukaryotes [21] |
The Ubiquitin-Proteasome System: From Discovery to Core Mechanism
The connection of APF-1 to ubiquitin unlocked the understanding of a major cellular pathway. The ubiquitin-proteasome system (UPS) is responsible for the degradation of over 80% of cellular proteins, including damaged, misfolded, and short-lived regulatory proteins [20] [15].
The Enzymatic Cascade and the Ubiquitin Code
The process of ubiquitination involves a sequential enzymatic cascade:
- Activation: A ubiquitin-activating enzyme (E1) activates ubiquitin in an ATP-dependent manner [15].
- Conjugation: The activated ubiquitin is transferred to a ubiquitin-conjugating enzyme (E2) [15].
- Ligation: A ubiquitin ligase (E3) recognizes the specific substrate protein and facilitates the transfer of ubiquitin from E2 to a lysine residue on the substrate, forming an isopeptide bond [15].
A critical feature of ubiquitin is that it itself contains seven lysine residues (Lys6, Lys11, Lys27, Lys29, Lys33, Lys48, Lys63). These residues can themselves be ubiquitinated, leading to the formation of polyubiquitin chains. The type of linkage determines the fate of the modified protein, creating a complex "ubiquitin code" [21] [20].
Table 2: The Ubiquitin Code: Linkage and Functional Consequences
| Ubiquitin Linkage | Primary Function | Key Examples / Notes |
|---|---|---|
| K48-linked | Proteasomal degradation [20] [15] | The most abundant degradation signal; targets proteins like p53 and HIF1α [20]. |
| K63-linked | Non-proteolytic signaling | Regulates lysosome functions, endocytosis, DNA repair, and inflammatory response [20] [15]. |
| Met1-linked (Linear) | NF-κB activation | Assembled by the LUBAC complex [20]. |
| K11-linked | Proteasomal degradation | Works with K48; key in cell cycle regulation by APC/C [20]. |
| K27-linked | Mitophagy, Immune signaling | Associated with Parkin-mediated mitophagy [20]. |
| K29-linked | Lysosomal degradation? | Implicated in some lysosomal degradation pathways [20]. |
| K33-linked | Non-degradative signaling | Inhibits necroptosis by modulating RIPK3 [20]. |
| K6-linked | DNA repair | Associated with BRCA1 auto-ubiquitination [20]. |
The following diagram illustrates the core ubiquitin-proteasome pathway and the diversity of the ubiquitin code.
Title: The Ubiquitin-Proteasome System and Signaling Outcomes
The Scientist's Toolkit: Key Research Reagents and Applications
The research into the UPS relies on a suite of essential reagents and molecular tools.
Table 3: Research Reagent Solutions in Ubiquitin and Protein Degradation Research
| Reagent / Tool | Function in Research | Application Example |
|---|---|---|
| Reticulocyte Lysate | A cell-free system for biochemical fractionation and reconstitution of ATP-dependent proteolysis. | Used in the original discovery to isolate Fractions I and II, identifying APF-1/Ubiquitin [1]. |
| E1, E2, E3 Enzymes | Recombinant enzymes used to reconstitute specific ubiquitination cascades in vitro. | Studying the activity and specificity of particular E3 ligases for their target substrates. |
| Proteasome Inhibitors | Small molecules (e.g., Bortezomib, MG132) that selectively inhibit the proteasome's proteolytic activity. | Validating UPS-dependent degradation; used clinically to treat multiple myeloma [20]. |
| Ubiquitin Binding Domains (UBDs) | Protein domains that non-covalently recognize and bind specific ubiquitin chains. | As tools to purify, detect, and characterize the type of ubiquitin linkage on modified proteins [21]. |
| PROTACs (Proteolysis Targeting Chimeras) | Heterobifunctional molecules with a warhead for a target protein, a ligand for an E3 ligase, and a linker. | Inducing targeted degradation of disease-causing proteins, a novel therapeutic strategy [15]. |
| Molecular Glue Degraders | Small molecules that induce proximity between an E3 ligase and a target protein, leading to its ubiquitination. | Examples include thalidomide and its analogs, which redirect CRBN E3 ligase activity [15]. |
Modern Applications: Targeted Protein Degradation in Drug Discovery
The profound understanding of the UPS has directly fueled the emergence of a revolutionary therapeutic strategy: Targeted Protein Degradation (TPD). Unlike traditional small-molecule inhibitors that merely block a protein's activity, TPD aims to completely remove the disease-causing protein from the cell [15].
The most prominent TPD technology is PROteolysis TArgeting Chimeras (PROTACs). These are heterobifunctional molecules consisting of three elements:
- A warhead that binds to the Protein of Interest (POI).
- A ligand that recruits a specific E3 ubiquitin ligase.
- A linker connecting the two [15].
PROTACs act as a catalytic "matchmaker," bringing the E3 ligase into proximity with the POI, leading to its ubiquitination and subsequent degradation by the proteasome. This approach has key advantages, including the ability to target proteins previously considered "undruggable" and its event-driven, sub-stoichiometric mechanism of action [15].
The journey from the enigmatic APF-1 to the universally recognized ubiquitin represents one of the most elegant stories of scientific discovery in modern biology. The meticulous biochemical fractionation and reconstitution experiments conducted by Hershko, Ciechanover, and Rose not only solved the long-standing puzzle of ATP-dependent proteolysis but also unveiled a universal regulatory mechanism of unparalleled complexity. The connection established that a known protein, ubiquitin, was repurposed by the cell as a precise molecular tag for destruction, a finding that fundamentally altered our understanding of cellular regulation. Today, the legacy of this discovery extends far beyond basic science, directly inspiring a new generation of therapeutic modalities like PROTACs that leverage the cell's own degradation machinery to treat disease. The field continues to evolve, promising further insights into the ubiquitin code and novel applications in research and medicine.
The period from 1978 to 1980 marked a paradigm shift in our understanding of cellular proteolysis. Prior to this groundbreaking work, intracellular protein degradation was largely attributed to lysosomal function or ATP-dependent proteases like bacterial Lon. The collaborative research of Avram Hershko, Aaron Ciechanover, and Irwin Rose revealed a far more sophisticated system—ubiquitin-dependent proteolysis—that would fundamentally alter cell biology and earn them the 2004 Nobel Prize in Chemistry [1] [13]. Their discovery that eukaryotic cells covalently attach a small protein (ubiquitin) to target proteins to mark them for degradation established a new regulatory mechanism now recognized as being as crucial as phosphorylation or acetylation [1].
This research was framed by a fundamental biochemical curiosity: why did intracellular proteolysis require energy? The hydrolysis of peptide bonds is exergonic, presenting no thermodynamic requirement for ATP. Simpson's 1953 observation of this energy dependence had remained unexplained for 25 years, suggesting unknown regulatory mechanisms [1]. The Hershko-Ciechanover-Rose collaboration, forged through international scientific partnership and sabbatical work at Fox Chase Cancer Center, was uniquely positioned to address this mystery using biochemical approaches that would redefine protein turnover regulation [1] [22].
Experimental Foundations and Key Reagents
The Reticulocyte Lysate System and Initial Fractionation
The research program employed rabbit reticulocyte lysates as a model system, building on seminal observations by Etlinger and Goldberg that these lysates (which lack lysosomes) exhibited ATP-dependent proteolysis of denatured proteins [1] [13]. This system proved amenable to biochemical fractionation, allowing the team to separate the lysate into functionally distinct components.
Table 1: Key Research Reagent Solutions in Ubiquitin-Proteasome Research
| Research Reagent | Function in Experimental System | Experimental Role |
|---|---|---|
| Reticulocyte Lysate | ATP-dependent proteolytic extract lacking lysosomes | Source of ubiquitin-system components; foundational experimental system |
| APF-1 (Ubiquitin) | Heat-stable polypeptide; ATP-dependent proteolysis factor 1 | Covalent tag for protein targeting; later identified as ubiquitin |
| Fraction I | Contains APF-1/ubiquitin | Required component for reconstituting ATP-dependent proteolysis |
| Fraction II | High molecular weight fraction containing conjugating enzymes and proteasome | Contains E1, E2, E3 enzymes and 26S proteasome (APF-2) |
| ATP-regenerating System | Maintains constant ATP levels during incubations | Prevents ATP depletion during proteolysis assays |
The initial fractionation experiments demonstrated that the reticulocyte lysate could be separated into two fractions (I and II), both required to reconstitute ATP-dependent proteolysis [1] [13]. Fraction I contained a single essential heat-stable component termed APF-1 (ATP-dependent Proteolysis Factor 1), which would later be identified as ubiquitin [1]. Fraction II contained a high molecular weight component (APF-2) that was stabilized by ATP and later recognized as the 26S proteasome [1].
The Discovery of Covalent Protein Modification
The pivotal experimental breakthrough came from studies examining the association between (^{125})I-labeled APF-1 and components in Fraction II [1]. Surprisingly, the association persisted under denaturing conditions (high pH), suggesting a covalent linkage rather than non-covalent binding [1]. This covalent modification exhibited striking characteristics:
- ATP dependence: The conjugation required low ATP concentrations and was reversed upon ATP removal [1]
- Multi-substrate targeting: APF-1 was bound to many different proteins in Fraction II as judged by SDS-PAGE [1]
- Stability: The bond was stable to NaOH treatment, suggesting an isopeptide linkage rather than ester or thioester bond [1]
The connection to the previously known protein ubiquitin came through collaborative discussions and experimental verification. Art Haas, Keith Wilkinson, and Michael Urban recognized the similarity between APF-1 and ubiquitin, a protein previously identified by Gideon Goldstein and known to conjugate with histone H2A [1]. This critical insight connected the ATP-dependent proteolysis system with a known protein modifier.
Key Publications and Experimental Workflows (1978-1980)
The 1978 Biochemical Characterization
The initial report in 1978 (Ciechanover, Hod, and Hershko) established the heat-stable polypeptide component (APF-1) and its essential role in the ATP-dependent proteolytic system [13]. This work provided the foundational biochemical characterization of the system.
Table 2: Key Experimental Publications in the Discovery of Ubiquitin-Dependent Proteolysis
| Publication Year & Authors | Journal | Key Findings | Experimental Methods |
|---|---|---|---|
| 1978: Ciechanover, Hod, Hershko | Biochemical and Biophysical Research Communications | Identification of heat-stable polypeptide component (APF-1) essential for ATP-dependent proteolysis | Biochemical fractionation of reticulocyte lysates; ATP-depletion studies; reconstitution assays |
| 1980: Ciechanover, Heller, Elias, Haas, Hershko | Proceedings of the National Academy of Sciences | Demonstration of covalent conjugation of APF-1/ubiquitin to multiple proteins in ATP-dependent manner | (^{125})I-labeled APF-1 tracking; SDS-PAGE analysis; ATP dependence studies |
| 1980: Hershko, Ciechanover, Heller, Haas, Rose | Proceedings of the National Academy of Sciences | Evidence that protein substrates are multi-ubiquitinated; relationship between conjugation and proteolysis | Use of natural protein substrates; quantification of ubiquitin conjugates per substrate molecule |
Diagram 1: Experimental Workflow Leading to Covalent Modification Discovery
The 1980 PNAS Papers: Mechanistic Insights
Two seminal 1980 PNAS papers established the core mechanism of ubiquitin-dependent proteolysis. The first paper (Ciechanover et al.) systematically characterized the covalent conjugation process [1] [13], while the second (Hershko et al.) demonstrated that authentic protein substrates were heavily modified with multiple ubiquitin molecules [1].
The experimental protocols employed in these studies included:
- ATP-depletion and reconstitution: Preparing Fraction II under ATP-depleted conditions was essential for demonstrating the APF-1 requirement, as endogenous ATP led to pre-formed conjugates [1]
- Radiolabel tracing: (^{125})I-labeled APF-1 allowed visualization and quantification of conjugation events [1]
- Native substrate analysis: Using known proteolytic substrates to demonstrate physiological relevance [1]
- Kinetic correlation: Showing parallel nucleotide and metal ion requirements for conjugation and proteolysis [1]
A critical insight from the second paper was that multiple ubiquitin molecules were attached to each substrate molecule, suggesting a cooperative recognition mechanism [1]. The conjugation system exhibited processivity, preferring to add additional ubiquitin molecules to existing conjugates rather than initiating new conjugation events on free substrates [1].
Diagram 2: Ubiquitin-Proteasome Pathway Conceptual Relationship
Technical Challenges and Methodological Innovations
The research team encountered and overcame several significant technical challenges:
The ATP Depletion Artifact
A particularly insightful finding emerged from understanding why some laboratories failed to demonstrate APF-1 dependence. When Fraction II was prepared without ATP depletion, most APF-1 existed in pre-formed high molecular weight conjugates that were subsequently disassembled by amidases in the fraction, liberating sufficient free APF-1 to support proteolysis without supplementation [1]. This explained discrepancies between laboratories and highlighted the importance of ATP-depletion protocols.
Identification of the Central Components
The terminology used in the original papers reflected the functional characterization approach:
- APF-1: The heat-stable, essential factor in Fraction I, later identified as ubiquitin [1]
- APF-2: The high molecular weight, ATP-stabilized component in Fraction II, later recognized as the 26S proteasome [1]
The connection to ubiquitin came through collaborative discussions recognizing the similarity between APF-1 and the previously known ubiquitin protein [1]. This connection was experimentally verified by showing that authentic ubiquitin could replace APF-1 in supporting ATP-dependent proteolysis [1].
Impact and Evolution of the Model
The period from 1978-1980 established the fundamental framework for understanding ubiquitin-dependent proteolysis, but subsequent research would reveal additional complexity:
- Enzyme cascade: The conjugation system involves E1 (activating), E2 (conjugating), and E3 (ligase) enzymes [1]
- Polyubiquitin signaling: Chau et al. later showed that proteolytic targeting requires polyubiquitin chains linked through Lys48 of ubiquitin [1]
- Proteasome structure: The 26S proteasome consists of 20S catalytic core and 19S regulatory particles [1] [23]
The model system established in this work—the fractionated reticulocyte lysate—remained the foundation for purifying and characterizing the individual components of the ubiquitin-proteasome system over the following decades.
The 1978-1980 work established not only a new biochemical pathway but also a novel regulatory paradigm in cell biology: targeted protein destruction through covalent protein tagging. This system would later be recognized as controlling virtually all major cellular processes, from cell cycle progression to signal transduction [1].
The therapeutic implications emerged decades later with the development of proteasome inhibitors such as bortezomib (Velcade) for multiple myeloma and mantle cell lymphoma [22] [24]. By 2018, these drugs had treated over half a million patients worldwide [22], demonstrating how fundamental biochemical research into basic cellular mechanisms can yield transformative clinical applications.
The collaboration between Hershko, Ciechanover, and Rose—supported by international scientific partnerships and sabbatical opportunities—exemplifies how intellectual curiosity, biochemical rigor, and collaborative spirit can unravel nature's complexities and ultimately benefit human health.
Mechanistic Insights and Therapeutic Translation: From Ubiquitination to PROTACs
The discovery of a non-lysosomal, ATP-dependent proteolytic pathway by Avram Hershko, Aaron Ciechanover, and Irwin Rose fundamentally reshaped our understanding of intracellular protein degradation and earned them the 2004 Nobel Prize in Chemistry [13]. Their pioneering work in the late 1970s and early 1980s revealed the existence of a biochemical marker that labels doomed proteins for destruction—a molecule they termed ubiquitin [12] [13]. They demonstrated that ubiquitin is activated in an ATP-consuming process and forms a covalent conjugate with target proteins, marking them for degradation by a large protease complex [13]. This process, now known as the ubiquitin-proteasome system (UPS), involves a precise enzymatic cascade wherein E1 (activating), E2 (conjugating), and E3 (ligating) enzymes work in sequence to transfer ubiquitin onto specific substrate proteins [25] [26]. The UPS has since been recognized as a master regulator of countless cellular processes, governing the controlled destruction of proteins involved in cell cycle progression, signal transduction, and stress response [13] [26]. This technical guide explores the intricate mechanisms of this enzymatic cascade, framed within the revolutionary context of the Hershko and Ciechanover research that laid its foundation.
The Ubiquitination Cascade: A Three-Enzyme Mechanism
The conjugation of ubiquitin to substrate proteins follows a conserved, three-step mechanism that ensures precision and specificity in targeting proteins for degradation.
Step 1: Ubiquitin Activation by E1
The process initiates with the E1 ubiquitin-activating enzyme. This step is ATP-dependent, wherein the E1 enzyme catalyzes the formation of a high-energy thioester bond between its active-site cysteine residue and the C-terminal glycine (Gly76) of ubiquitin [27] [26]. Prior to this, ubiquitin is adenylated, forming a UB-AMP intermediate. The human genome encodes two E1 enzymes, Ube1 and Uba6, which launch the ubiquitin transfer cascade [27].
Step 2: Ubiquitin Conjugation by E2
The activated ubiquitin is subsequently transferred from E1 to a cysteine residue of an E2 ubiquitin-conjugating enzyme (Ubc) via a trans-thioesterification reaction. This results in the formation of a E2~Ub thioester conjugate (where "~" denotes the thioester bond) [27] [26]. The human genome encodes approximately 50 E2 enzymes, which function as central hubs in the cascade [27] [28].
Step 3: Ubiquitin Ligation by E3
The final step is catalyzed by an E3 ubiquitin ligase, which is responsible for the specific recognition of substrate proteins. The E3 facilitates the transfer of ubiquitin from the E2~Ub complex to a lysine residue on the substrate protein, forming an isopeptide bond [26]. In some cases, E3 ligases first form a thioester intermediate with ubiquitin before transferring it to the substrate. E3s are the most diverse components of the cascade, with over 600 E3 ligases identified in humans, providing immense substrate specificity [29] [30]. The final product can be a mono-ubiquitinated protein or a polyubiquitin chain, where subsequent ubiquitin molecules are attached to a lysine residue on the preceding ubiquitin [30].
Table 1: Core Enzymes of the Ubiquitin Transfer Cascade
| Enzyme | Role | Human Genes | Key Functional Domain | Output |
|---|---|---|---|---|
| E1 (Activating) | Activates Ub via ATP hydrolysis | 2 (Ube1, Uba6) | Active-site Cysteine | E1~Ub thioester |
| E2 (Conjugating) | Accepts and carries activated Ub | ~50 | Active-site Cysteine | E2~Ub thioester |
| E3 (Ligating) | Recognizes substrate and catalyzes Ub transfer | >600 | RING, HECT, or RBR | Ubiquitinated substrate |
Figure 1: The E1-E2-E3 Enzymatic Cascade. Ubiquitin is activated by E1 in an ATP-dependent step, transferred to E2, and finally ligated to a protein substrate by an E3 ligase.
Historical and Structural Basis of E1-Ubiquitin Recognition
The C-terminal sequence of ubiquitin is critical for its recognition and activation by E1 enzymes. Early research by Hershko, Ciechanover, and Rose established that a heat-stable polypeptide was essential for the ATP-dependent proteolytic system [13]. Subsequent studies revealed that this polypeptide was ubiquitin and that its C-terminal glycine was indispensable for conjugation [27] [13].
Structural Insights from Phage Display
Phage display profiling of the ubiquitin C-terminal sequence (ending with 71LRLRGG76) has provided deep insights into E1 specificity. Key findings include [27]:
- Arg72 is absolutely required for E1 recognition.
- Residues at positions 71, 73, and 74 can be replaced with bulky aromatic side chains while maintaining E1 reactivity.
- Gly75 can be mutated to Ser, Asp, or Asn, and UB variants can still be efficiently activated by E1 and transferred to E2 enzymes.
- However, these UB mutants are often blocked from further transfer to E3 enzymes, indicating that the C-terminal sequence is also crucial for discharge from E2.
Table 2: Ubiquitin C-Terminal Residue Specificity for E1 Recognition
| Ubiquitin Residue | Wild-Type Amino Acid | Mutant Compatibility | Functional Importance |
|---|---|---|---|
| 71 | Leucine (L) | Bulky aromatic side chains | Tolerates mutation; not absolute |
| 72 | Arginine (R) | None (Absolute requirement) | Essential for E1 binding; 58-fold ↑ Kd with Leu mutant |
| 73 | Leucine (L) | Bulky aromatic side chains | Tolerates mutation; not absolute |
| 74 | Arginine (R) | Bulky aromatic side chains | Tolerates mutation; not absolute |
| 75 | Glycine (G) | Serine, Aspartate, Asparagine | Required for E1 activation but tolerates some mutation |
| 76 | Glycine (G) | None (Absolute requirement) | Indispensable for thioester formation with E1 |
E3 Ubiquitin Ligases: Architects of Specificity
E3 ubiquitin ligases serve as the primary determinants of substrate specificity within the ubiquitin cascade. They are broadly classified into three major families based on their structural domains and mechanisms of action [30].
RING Domain Family
RING-type E3s (e.g., MDM2, cIAP) contain a Really Interesting New Gene domain. They act as scaffolds that simultaneously bind to both the E2~Ub complex and the substrate protein, facilitating the direct transfer of ubiquitin from E2 to the substrate without forming a covalent E3-Ub intermediate. This is the largest family of E3 ligases [26] [30].
HECT Domain Family
HECT-type E3s (e.g., NEDD4, E6AP) contain a Homologous to E6-AP C-Terminus domain. Unlike RING E3s, they form a covalent thioester intermediate with ubiquitin transferred from the E2~Ub complex before ultimately transferring it to the substrate protein [27] [26].
RBR Type Family
RBR-type E3s (e.g., Parkin, HOIP) possess two RING domains connected by an In-Between-RING domain. These hybrid enzymes employ a two-step mechanism, combining features of both RING and HECT families. They first bind the E2~Ub non-covalently (RING-like) and then form a transient thioester intermediate with ubiquitin (HECT-like) before substrate modification [30].
Experimental Methodologies for Profiling the Ubiquitin Cascade
Phage Display for E1-E2-UB Specificity Profiling
This methodology identifies ubiquitin variants reactive with E1 enzymes from a randomized library [27].
Protocol:
- Library Construction: Create a UB phage display library with randomized C-terminal residues (positions 71-75), leaving Gly76 unchanged. A library size of 1×10^8 clones ensures coverage of sequence diversity.
- Enzyme Immobilization: Express E1 enzymes as fusions with a peptidyl carrier protein (PCP) domain. Label the PCP domain with biotin using Sfp phosphopantetheinyl transferase and biotin-CoA. Immobilize the biotin-labeled PCP-E1 fusions on a streptavidin-coated plate.
- Selection Reaction: Incubate the phage-displayed UB library with immobilized E1 in the presence of 1 mM Mg-ATP to catalyze the formation of UB~E1 thioester conjugates. Covalently bound phage are selected.
- Elution and Amplification: Release bound phage particles by cleaving the thioester linkage with Dithiothreitol (DTT). Amplify the eluted phage for subsequent rounds of selection.
- Stringency Optimization: Over iterative selection rounds, decrease the amount of phage input, E1 enzyme, and reaction time to enrich for the most catalytically active UB variants. For example, in the 8th round, use 1×10^10 phage reacted with 1 pmol E1 for 10 minutes.
- Validation: Confirm enrichment by comparing phage output from the selection reaction (with E1 and Mg-ATP) versus controls lacking E1 or Mg-ATP. A 350-fold enrichment indicates successful selection of catalytically active clones.
Orthogonal Ubiquitin Transfer (OUT) Cascade Engineering
This approach engineers an orthogonal UB transfer cascade to map the substrate specificity of individual E3s [28].
Protocol:
- Component Engineering: Use phage display and mutagenesis to engineer orthogonal pairs of UB and E1 (xUB-xE1), and E1 and E2 (xE1-xE2), that do not cross-react with their native counterparts.
- Cascade Assembly: Combine the xUB, xE1, xE2, and a specific E3 of interest (xE3) to form a self-contained OUT cascade.
- Substrate Identification: Use the OUT cascade to ubiquitinate the cellular substrate proteins of the specific E3, enabling the mapping of E3-specific signaling networks without interference from the native UPS.
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Research Reagents for Ubiquitin Cascade Studies
| Reagent / Solution | Function / Application | Example Use-Case |
|---|---|---|
| Sfp Phosphopantetheinyl Transferase | Catalyzes site-specific biotinylation of PCP-tagged proteins | Labeling PCP-E1 fusions for immobilization in phage display [27] |
| Biotin-CoA Conjugate | Donor substrate for Sfp-mediated biotinylation | Labeling PCP-tagged E1 enzymes for streptavidin plate capture [27] |
| Streptavidin-Coated Plates | Solid support for immobilizing biotinylated proteins | Capturing biotinylated PCP-E1 for phage selection [27] |
| Dithiothreitol (DTT) | Reducing agent that cleaves thioester bonds | Releasing phage covalently bound via UB~E1 thioester [27] |
| PROTAC Molecules | Bifunctional degraders recruiting E3 ligases to target proteins | Inducing targeted degradation of disease-associated proteins [31] |
| E3 Ligase Inhibitors | Small molecules inhibiting specific E3 ligases | Probing E3 function; therapeutic development (e.g., Nutlins for MDM2) [26] |
Therapeutic Applications and Future Directions
The understanding of the ubiquitin cascade, built upon the foundation of Hershko and Ciechanover's work, has opened revolutionary therapeutic avenues, most notably in Targeted Protein Degradation (TPD) [31].
Proteolysis-Targeting Chimeras (PROTACs)
PROTACs are heterobifunctional molecules that consist of:
- A ligand that binds a Protein of Interest (POI).
- A linker.
- A second ligand that recruits an E3 ubiquitin ligase.
PROTACs induce the formation of a ternary complex (POI-PROTAC-E3), leading to the ubiquitination and subsequent proteasomal degradation of the POI [31]. This event-driven pharmacology allows for the degradation of targets previously considered "undruggable," such as transcription factors and scaffolding proteins [29] [31]. Clinical candidates like ARV-110 (for prostate cancer) and ARV-471 (for breast cancer) exemplify the rapid translation of this technology [31].
E3 Ligases in Disease and Drug Discovery
Dysregulation of E3 ligases is implicated in numerous diseases, making them prime therapeutic targets [29] [30].
- Cancer: E3s like MDM2 (regulator of p53) are targeted for inhibition, while others like CRBN and VHL are hijacked by PROTACs [26] [31].
- Neurodegenerative Disorders: E3s are involved in clearing misfolded proteins; their dysfunction or modulation is linked to diseases like Parkinson's and Alzheimer's [29].
- Bone Disorders: E3s such as RNF185 and Trim21 act as negative regulators of osteogenic differentiation, presenting potential targets for bone metabolic diseases [30].
Figure 2: PROTAC Mechanism of Action. A heterobifunctional PROTAC molecule brings an E3 ligase into proximity with a target protein, leading to its ubiquitination and degradation by the proteasome.
The E1-E2-E3 enzymatic cascade for ubiquitin transfer represents a masterfully orchestrated system for controlling protein fate, a process whose fundamental principles were unveiled by the groundbreaking work of Hershko, Ciechanover, and Rose. From the initial activation of ubiquitin by E1 to the precise substrate selection dictated by E3s, each step is critical for maintaining cellular homeostasis. The legacy of the Nobel-winning discovery of ATP-dependent ubiquitin-mediated proteolysis continues to expand, driving a paradigm shift in drug discovery. As research delves deeper into the specificity and regulatory mechanisms of these enzymes, and as technologies like PROTACs mature in the clinic, the potential to develop powerful, precise therapies for cancer, neurodegenerative disorders, and other diseases grows ever more tangible.
Polyubiquitin Chains as the Proteasomal Degradation Signal
The regulated degradation of intracellular proteins is a fundamental biological process essential for cellular homeostasis, and the ubiquitin-proteasome system (UPS) stands as its primary executor. At the heart of this system lies a elegant signaling mechanism: the covalent attachment of a polyubiquitin chain to a target protein, which serves as a potent signal for its degradation by the 26S proteasome. This review is framed within the groundbreaking work of Avram Hershko, Aaron Ciechanover, and Irwin Rose, who were awarded the Nobel Prize in Chemistry in 2004 for their discovery of ubiquitin-mediated protein degradation [32]. Their seminal research in the early 1980s, using biochemical fractionation of reticulocyte lysates, revealed that ATP-dependent proteolysis required a small, heat-stable protein they termed APF-1 (ATP-dependent Proteolysis Factor 1), later identified as ubiquitin [1] [19]. They made the astoundingly prescient observation that this factor was covalently conjugated to target proteins in an ATP-dependent manner, a modification that was necessary for their degradation [1]. This foundational discovery unveiled an entirely new paradigm for post-translational regulation and established the core principle that a polyubiquitin chain is the canonical degradation signal.
The Ubiquitin Conjugation Machinery and Degradation Signal Diversity
Ubiquitination is a highly enzymatic process mediated by a cascade of E1 (ubiquitin-activating), E2 (ubiquitin-conjugating), and E3 (ubiquitin-ligase) enzymes [33]. The E3 ligase, of which there are hundreds in humans, provides substrate specificity and is the most important factor in determining which proteins are targeted for degradation [33]. A fundamental property of ubiquitin is its ability to form polymers (polyubiquitin chains) through its internal lysine residues or N-terminal methionine. The type of linkage within the chain creates a unique "ubiquitin code" that determines the fate of the modified protein [34].
While several chain types exist (e.g., K63, M1-linear), the K48-linked polyubiquitin chain is the archetypal and most extensively studied signal for proteasomal degradation [35] [36]. Early work established that a chain of at least four ubiquitin molecules (tetra-ubiquitin) linked through K48 is the minimal efficient signal for targeting substrates to the 26S proteasome [35] [33]. The proteasome recognizes this signal through intrinsic ubiquitin receptors in its 19S regulatory particle, leading to substrate unfolding, deubiquitination, and translocation into the proteolytic core for digestion into short peptides [33].
However, the ubiquitin code is not limited to homotypic K48 chains. The system exhibits remarkable diversity, and recent evidence underscores the importance of heterotypic and branched ubiquitin chains in regulating degradation. For instance, branched chains containing K48 linkages, such as K11/K48 or K48/K63 hybrids, can also target proteins for proteasomal destruction, often with enhanced efficiency or under specific regulatory conditions [37]. This complexity allows for a vast expansion of the informational content of the ubiquitin code, enabling nuanced control over protein stability.
Table 1: Major Types of Polyubiquitin Chains and Their Primary Functions
| Linkage Type | Structure | Primary Function | Proteasomal Degradation Role |
|---|---|---|---|
| K48 | Homotypic | Classical proteasomal targeting [36] | Primary signal; requires chain of ≥4 ubiquitins [35] [33] |
| K11 | Homotypic / Branched (with K48) | Cell cycle regulation; proteasomal degradation [37] | Contributes to degradative signal, often in conjunction with K48 [37] |
| K63 | Homotypic | Signal transduction, DNA repair, endocytosis [36] | Generally non-degradative; can be converted to degradative signal when branched with K48 [37] [36] |
| M1 (Linear) | Homotypic | NF-κB signaling, inflammation [34] | Non-degradative [34] |
| K48/K63 | Branched | NF-κB signaling, apoptosis [37] | Enhances degradation; converts non-proteolytic K63 signal into degradative signal [37] |
| K29/K48 | Branched | Ubiquitin Fusion Degradation (UFD) pathway [37] | Targets specific substrates for proteasomal degradation [37] |
Structural Mechanisms of Polyubiquitin Chain Recognition
The precise molecular recognition of K48-linked polyubiquitin chains by the proteasome and associated factors is critical for ensuring fidelity in protein degradation. Recent structural biology studies have provided deep insights into this process.
A key mechanism involves the recognition of the hydrophobic surface patch on ubiquitin, centered on residues L8, I44, and V70 [35]. This patch is a common hot spot for interaction with many ubiquitin-binding domains (UBDs). In the proteasome, intrinsic ubiquitin receptors like Rpn10 and Rpn13 bind to ubiquitin chains via their own UBDs [33]. Furthermore, specific adaptor proteins, which contain both a polyubiquitin-binding domain and a proteasome-binding domain, can shuttle ubiquitinated substrates to the proteasome, adding another layer of regulation [33].
Groundbreaking research on macrocyclic peptide inhibitors, such as Ub4a, has illuminated a novel mechanism of chain recognition. These peptides, which selectively bind K48-linked tetra-ubiquitin with nanomolar affinity, were found to engage three consecutive ubiquitin units in a trimeric moiety within the chain. The cyclic peptide sits in a central hole lined by the hydrophobic patches of all three ubiquitins, effectively being encircled by the K48-linked trimer [35]. Intriguingly, the peptide exhibits a strong preference for binding the proximal trimer moiety (the end with the unanchored C-terminus) of a tetra-ubiquitin chain. This selectivity is governed by interactions with the flexible C-terminal tail (residues L73-R74-G75-G76) of the proximal ubiquitin itself. Truncation of this tail redirects the peptide's binding site on the chain, demonstrating its role as a determinant for proximal-end recognition [35].
Diagram 1: Mechanism of Selective K48-linked Tetra-Ubiquitin Recognition. The macrocyclic peptide Ub4a selectively engages the proximal trimer moiety (Ub units A-C), with interactions mediated by the hydrophobic patches and the C-terminal tail of the proximal ubiquitin.
Advanced Methodologies for Studying Ubiquitination
The complexity of the ubiquitin code demands sophisticated tools for its study. Below are detailed protocols and reagents for investigating polyubiquitin chains and their role in degradation.
Experimental Protocol: Analysis of Linkage-Specific Ubiquitination using TUBEs
Tandem Ubiquitin Binding Entities (TUBEs) are engineered protein reagents containing multiple ubiquitin-binding domains in tandem, which confer high-affinity, linkage-specific recognition of polyubiquitin chains. This protocol, adapted from recent research, details their use in a plate-based assay to capture endogenous ubiquitinated proteins [36].
Table 2: Key Research Reagents for Ubiquitination Studies
| Research Reagent | Composition / Type | Primary Function in Experiments |
|---|---|---|
| TUBEs (Tandem Ubiquitin Binding Entities) | Engineered proteins with multiple UBDs (e.g., UBA, UBAN domains) [36] | High-affinity capture and protection of polyubiquitin chains from deubiquitinases (DUBs) during cell lysis [36]. |
| Linkage-Specific TUBEs | TUBEs engineered for selective binding (e.g., K48-TUBE, K63-TUBE) [36] | Selective enrichment and analysis of specific polyubiquitin chain linkages (e.g., K48 for degradation) from complex cell lysates [36]. |
| Deubiquitinase (DUB) Inhibitors | Small molecules (e.g., PR-619) or ubiquitin variants [35] | Preserve labile ubiquitin signals during sample preparation by inhibiting endogenous DUBs. Often used in conjunction with TUBEs. |
| Linkage-Specific Antibodies | Monoclonal antibodies (e.g., K48-linkage specific antibody) [38] | Detect specific chain types in immunoblotting or immunofluorescence. Useful for validating TUBE capture or visualizing endogenous chains. |
| Macrocyclic Peptide Inhibitors (e.g., Ub4a) | De novo synthesized cyclic peptides [35] | Function as specific antagonists of K48-chain recognition, useful for probing the functional consequences of blocking this interaction in vitro and in vivo. |
Procedure:
- Plate Coating: Coat the wells of a 96-well plate with chain-selective TUBEs (e.g., K48-TUBE for degradative signals, K63-TUBE for non-degradative signals) or Pan-TUBE (for total ubiquitin capture) in phosphate-buffered saline (PBS) overnight at 4°C [36].
- Cell Lysis and Treatment: Prepare cell lysates (e.g., from THP-1 cells) under denaturing conditions or using a specialized lysis buffer optimized to preserve polyubiquitination. This buffer should contain a cocktail of deubiquitinase (DUB) inhibitors and proteasome inhibitors to prevent the loss of ubiquitin signals during extraction [36].
- Capture: Apply the clarified cell lysates to the TUBE-coated wells and incubate for 2-4 hours at 4°C with gentle agitation to allow for linkage-specific binding [36].
- Washing: Wash the wells extensively with a mild wash buffer to remove non-specifically bound proteins.
- Elution and Detection: Elute the captured ubiquitinated proteins using Laemmli sample buffer containing SDS and DTT. Analyze the eluates by SDS-PAGE and immunoblotting using an antibody against the protein of interest (e.g., anti-RIPK2) to detect its ubiquitinated forms [36]. Alternatively, for a higher-throughput quantitative readout, detect the captured proteins using an antibody conjugated to a fluorescent or luminescent tag in the well.
Application: This assay was used to demonstrate that an inflammatory agent (L18-MDP) induces K63-linked ubiquitination of RIPK2, which is captured by K63-TUBEs but not K48-TUBEs. Conversely, a PROTAC designed to degrade RIPK2 induced its K48-linked ubiquitination, which was specifically captured by K48-TUBEs [36].
Experimental Protocol: Structural Analysis of Peptide-Ubiquitin Complexes
Understanding the atomic-level interactions of ubiquitin chain recognition, as with the macrocyclic peptide Ub4a, requires a combination of structural biology techniques [35].
Procedure:
- Sample Preparation: Produce and purify isotopically labeled (15N, 13C) ubiquitin and the K48-linked polyubiquitin chains (e.g., tri-Ub, tetra-Ub) using recombinant expression and enzymatic assembly. Synthesize the macrocyclic peptide (Ub4a) using solid-phase peptide synthesis.
- Complex Formation: Form the complex by titrating the peptide into the solution of labeled polyubiquitin chain.
- X-ray Crystallography:
- Crystallize the purified peptide-polyubiquitin complex.
- Collect X-ray diffraction data at a synchrotron source.
- Solve the structure by molecular replacement using a known ubiquitin structure as a search model.
- Outcome: This reveals how three consecutive ubiquitins form a ring around the cyclic peptide, with the peptide nestled in the central hole lined by the hydrophobic patches [35].
- Solution NMR Spectroscopy:
- Acquire a series of 2D and 3D NMR spectra (e.g., 1H-15N HSQC) of the free 15N-labeled polyubiquitin chain and the chain in complex with the peptide.
- Monitor chemical shift perturbations (CSPs) and signal broadening to map the binding interface and determine the binding affinity.
- Outcome: NMR confirmed the engagement of the hydrophobic patch (I44) and, crucially, identified the critical role of the flexible C-terminal tail of the proximal ubiquitin in directing binding selectivity [35]. Truncation of this tail (e.g., ΔLRGG) altered the binding mode, as evidenced by changes in the NMR spectra of the distal ubiquitin unit.
- Biochemical Validation: Use deubiquitinase (DUB) assays to confirm the functional consequence of binding, such as protection of the polyubiquitin chain from disassembly [35].
Implications for Drug Discovery and Therapeutic Development
The understanding of polyubiquitin chains as degradation signals has directly catalyzed novel therapeutic paradigms. The most prominent example is the development of PROteolysis TArgeting Chimeras (PROTACs). These heterobifunctional molecules consist of a ligand that binds a target protein of interest, linked to another ligand that recruits an E3 ubiquitin ligase [36]. By bringing the E3 ligase into proximity with the target, PROTACs induce its site-specific polyubiquitination, primarily with K48-linked chains, leading to its degradation by the proteasome [36]. This technology has opened the door to degrading proteins previously considered "undruggable."
Furthermore, directly targeting the ubiquitin signal itself is an emerging strategy. The macrocyclic peptide inhibitors like Ub4a, which selectively bind K48-linked tetra-ubiquitin, represent a novel class of potential anti-cancer therapeutics. By shielding the degradation signal from recognition by the proteasome and deubiquitinases, they cause the accumulation of polyubiquitinated proteins, leading to proteotoxic stress and apoptosis in cancer cells, and have been shown to attenuate tumor growth in vivo [35]. The structural insights from these studies are now guiding the development of next-generation compounds with enhanced potency and selectivity [35].
Diagram 2: Therapeutic Strategies Targeting the Polyubiquitin Degradation Signal. Two main approaches are shown: PROTACs hijack the system to degrade specific proteins, while direct inhibitors like Ub4a block the recognition of K48 chains by the proteasome.
The elegant work initiated by Hershko, Ciechanover, and Rose revealed that the covalent attachment of a protein, ubiquitin, could serve as a molecular "kiss of death." We now understand that this signal is not a simple mark but a sophisticated polyubiquitin code, with the K48-linked chain as its premier degradation signal. The structural mechanisms governing the recognition of this signal by receptors and inhibitors, involving specific ubiquitin surfaces and chain topology, are remarkably complex. Modern tools like TUBEs and structural biology techniques continue to decode this complexity, revealing new layers of regulation such as branched chains. This deep knowledge is no longer purely academic; it is the foundation for a new class of medicines, including PROTACs and signal-binding peptides, that leverage the cell's own degradation machinery to treat disease, fully realizing the therapeutic potential of the foundational discovery of ubiquitin-dependent proteolysis.
The 26S proteasome serves as the degrading arm of the ubiquitin system, a major pathway for regulated protein degradation in eukaryotic cells. The groundbreaking discovery of this ATP-dependent proteolytic system by Avram Hershko, Aaron Ciechanover, and Irwin Rose, for which they were awarded the 2004 Nobel Prize in Chemistry, revealed a sophisticated mechanism far more complex than previously imagined [1] [19]. Their seminal work in the late 1970s and early 1980s identified ATP-dependent proteolysis factor 1 (APF-1), later recognized as ubiquitin, and established the fundamental principle of covalent protein modification as a targeting signal for degradation [1]. This discovery laid the foundation for understanding the 26S proteasome as the central protease responsible for the selective, ATP-dependent degradation of polyubiquitinated cellular proteins, governing essential processes from cell cycle progression to stress response [39] [40].
Architectural Organization of the 26S Proteasome
The 26S proteasome is a ~2.5 MDa multi-subunit complex consisting of a 20S core particle (CP) capped by one or two 19S regulatory particles (RP) [40] [41]. This structural organization enables the complex to recognize, prepare, and degrade a diverse array of protein substrates with remarkable selectivity.
The 20S Core Particle
The 20S core particle forms the proteolytic heart of the proteasome and exhibits a conserved barrel-shaped structure across species [41]. It is composed of four stacked heptameric rings forming a central pore [41]. The two outer rings consist of seven distinct α-subunits (α1-α7) that function as a tightly regulated "gate" controlling access to the interior degradation chamber; the N-terminal tails of these α-subunits block unregulated entry of substrates [40]. The two inner rings contain seven β-subunits (β1-β7), three of which (β1, β2, and β5) contain proteolytic active sites with different cleavage specificities: caspase-like, trypsin-like, and chymotrypsin-like activities, respectively [40]. These catalytic subunits utilize an N-terminal threonine residue as the catalytic nucleophile [40].
Table 1: Proteolytic Activities of the 20S Core Particle β-Subunits
| β-Subunit | Proteolytic Activity | Catalytic Residue | Cleavage Preference |
|---|---|---|---|
| β1 | Caspase-like | N-terminal Thr | Acidic residues |
| β2 | Trypsin-like | N-terminal Thr | Basic residues |
| β5 | Chymotrypsin-like | N-terminal Thr | Hydrophobic residues |
| β1i (Immuno) | Caspase-like | N-terminal Thr | Altered specificity |
| β2i (Immuno) | Trypsin-like | N-terminal Thr | Altered specificity |
| β5i (Immuno) | Chymotrypsin-like | N-terminal Thr | Altered specificity |
| β5t (Thymo) | Chymotrypsin-like | N-terminal Thr | Reduced activity |
Note: Alternative "immuno" and "thymo" subunits are expressed in specialized tissues and cell types, modifying cleavage preferences to support immune function and thymic selection [40].
The 19S Regulatory Particle
The 19S regulatory particle is a multifunctional complex that identifies, binds, deubiquitinates, unfolds, and translocates substrates into the proteolytic chamber of the CP [40]. It can be further divided into two subcomplexes: the base and the lid [39].
The base subcomplex contains six AAA+ ATPase subunits (Rpt1-Rpt6) that form a heterohexameric ring, serving as the mechanical motor for substrate unfolding and translocation [39] [40]. The base also includes four non-ATPase subunits (Rpn1, Rpn2, Rpn10, and Rpn13) that function as ubiquitin receptors, recognizing substrates targeted to the proteasome [39] [40].
The lid subcomplex consists of nine subunits (Rpn3, Rpn5-Rpn9, Rpn11, Rpn12, and Rpn15/Sem1) that form a horseshoe-shaped structure [40] [42]. A primary function of the lid is deubiquitination of incoming substrates, carried out by the deubiquitinating enzyme Rpn11 [39] [42]. The lid undergoes substantial conformational rearrangements during incorporation into the 26S holoenzyme, which activates Rpn11 for substrate processing [42].
Table 2: Subunit Composition of the 19S Regulatory Particle
| Subcomplex | S. cerevisiae | H. sapiens | Function |
|---|---|---|---|
| Base | Rpn1 | PSMD2/S2 | Ubiquitin/UBL binding |
| Base | Rpn2 | PSMD1/S1 | Structural |
| Base | Rpn13 | ADRM1 | Ubiquitin binding |
| Base | Rpt1-Rpt6 | PSMC1-6 | ATPase motor |
| Lid | Rpn3 | PSMD3/S3 | Structural, PCI domain |
| Lid | Rpn5 | PSMD12 | Structural, PCI domain |
| Lid | Rpn6 | PSMD11/S9 | Structural, PCI domain |
| Lid | Rpn7 | PSMD6/S10 | Structural, PCI domain |
| Lid | Rpn8 | PSMD7/S12 | MPN domain, Rpn11 partner |
| Lid | Rpn9 | PSMD13/S11 | Structural, PCI domain |
| Lid | Rpn11 | PSMD14/Poh1 | Deubiquitinase (JAMM/MPN) |
| Lid | Rpn12 | PSMD8/S14 | Structural, PCI domain |
| Lid | Sem1 | PSMD9/Dss1 | Structural |
| Bridge | Rpn10 | PSMD4/S5a | Ubiquitin binding |
| Associated DUB | Ubp6 | Usp14 | Deubiquitinase |
| Associated DUB | - | Uch37 | Deubiquitinase |
Note: The table summarizes the conserved subunits of the 19S regulatory particle and their functions across species [39] [40].
The ATP-Dependent Proteolytic Mechanism
The degradation of ubiquitinated proteins by the 26S proteasome involves a highly coordinated, multi-step process that consumes ATP at several stages. This mechanism ensures proper substrate selection before the proteasome commits to processive degradation.
Diagram Title: ATP-Dependent Proteolytic Mechanism of the 26S Proteasome
Substrate Recognition and Initial Binding
Proteins destined for degradation are first tagged with polyubiquitin chains through a cascade of E1, E2, and E3 enzymes [1] [41]. These ubiquitin modifications are recognized by multiple ubiquitin receptors on the proteasome, primarily Rpn10, Rpn13, and Rpn1 [39] [40]. Initial binding of ubiquitin conjugates to the 26S proteasome is stimulated 2- to 4-fold by ATP or its non-hydrolyzable analog ATPγS, indicating that nucleotide binding (but not hydrolysis) enhances this initial interaction [43]. This step is reversible and represents the first checkpoint in substrate selection.
The Commitment Step
Following initial binding, substrates undergo a critical commitment step that requires ATP hydrolysis and the presence of a loosely folded or unstructured region of approximately 25 amino acids in the substrate [43] [41]. This step involves a major conformational change in the proteasome that transitions the regulatory particle from a substrate-accommodating state to a substrate-engaging state, tightly gripping the substrate and committing it to degradation [39] [43]. The requirement for an unstructured region ensures that only properly engaged substrates proceed to unfolding and degradation, preventing futile ATP hydrolysis cycles [43].
Deubiquitination and Substrate Engagement
During substrate engagement, the deubiquitinase Rpn11 removes the polyubiquitin chain from the substrate in a translocation-coupled manner [39] [42]. Rpn11 is a Zn²⁺-dependent metalloprotease of the JAMM/MPN family that cleaves at the base of the ubiquitin chain, releasing it for recycling [39] [42]. In the isolated lid complex, Rpn11 is inhibited through interactions with Rpn5, but upon incorporation into the 26S holoenzyme, conformational rearrangements activate the deubiquitinase, ensuring it only acts on properly engaged substrates [42].
Unfolding and Translocation
The six AAA+ ATPase subunits (Rpt1-Rpt6) of the base form a ring-shaped motor that uses conserved pore loops to engage the unstructured region of the substrate and apply mechanical force to unfold it [39]. ATP hydrolysis drives conformational changes in the ATPase ring that generate a pulling force, unraveling the native structure of the substrate and translocating the unfolded polypeptide through the gated channel into the degradation chamber of the 20S core particle [39] [40]. The opening of the gate in the α-ring is coordinated with the ATPase activity, ensuring substrates only enter when the proteasome is committed to degradation [40].
Proteolytic Degradation
Once inside the central chamber of the 20S core particle, the unfolded polypeptide is cleaved by the proteolytic active sites of the β-subunits [40] [41]. The resulting peptides are typically 7-8 amino acids long and are released from the proteasome for further degradation by cellular peptidases or for presentation by major histocompatibility complex (MHC) class I molecules in immune cells [41].
Conformational Dynamics and Allosteric Regulation
Recent cryo-electron microscopy studies have revealed that the 26S proteasome exhibits complex conformational dynamics that coordinate the various steps of substrate processing [39]. The regulatory particle exists in multiple distinct states, primarily classified as substrate-free (s1) and substrate-processing (s3) states, with several intermediate conformations [39]. These conformational states are influenced by nucleotide binding and hydrolysis, substrate engagement, and the occupancy of ubiquitin receptors and deubiquitinating enzymes [39]. The transitions between these states ensure proper coordination of substrate recognition, deubiquitination, unfolding, and degradation, allowing the proteasome to combine high promiscuity with exceptional substrate selectivity [39].
Experimental Analysis of Proteasome Function
Key Methodologies
The molecular mechanisms of the 26S proteasome have been elucidated through a combination of biochemical, structural, and biophysical approaches. Rapid kinetic assays at reduced temperatures (4°C) have been instrumental in separating the initial binding of ubiquitin conjugates from subsequent commitment steps, revealing that ATP binding alone stimulates initial conjugate binding, while ATP hydrolysis is required for the subsequent tighter binding [43]. Cryo-electron microscopy has provided atomic-resolution structures of the proteasome in multiple conformational states, revealing the intricate architectural changes that occur during substrate processing [39] [42]. Additionally, mutagenesis studies of specific residues in the lid complex have identified the network of interactions that maintain Rpn11 in an inhibited state prior to proteasome assembly [42].
Table 3: Essential Research Reagents and Methodologies
| Reagent/Method | Function/Application | Key Findings Enabled |
|---|---|---|
| ATPγS (non-hydrolyzable ATP analog) | Distinguishing nucleotide binding vs. hydrolysis effects | Demonstrated ATP binding alone stimulates initial ubiquitin conjugate binding [43] |
| Cryo-electron microscopy | High-resolution structural analysis | Revealed multiple conformational states of the 26S proteasome [39] [42] |
| Rapid kinetic assays at 4°C | Separation of binding and commitment steps | Identified two-step binding mechanism with distinct ATP requirements [43] |
| Site-directed mutagenesis | Functional analysis of specific residues | Identified Rpn5 interactions that inhibit Rpn11 in isolated lid [42] |
| 125I-labeled APF-1/ubiquitin | Tracing ubiquitin conjugation | Established covalent attachment of ubiquitin to substrate proteins [1] |
| Reticulocyte lysate fractionation | Biochemical reconstitution of ubiquitin-proteasome system | Enabled identification of essential components (APF-1/ubiquitin, APF-2/proteasome) [1] |
Experimental Workflow
A representative experimental workflow for analyzing ATP-dependent steps in ubiquitin conjugate binding to the 26S proteasome involves several key stages, as illustrated below.
Diagram Title: Experimental Workflow for ATP-Dependent Binding Assays
The 26S proteasome represents a sophisticated molecular machine that integrates multiple regulatory steps to achieve selective protein degradation. The architectural features of this complex—including multiple ubiquitin receptors, a regulated deubiquitination enzyme, and a powerful ATP-dependent unfoldase—work in concert to process a diverse array of substrates while maintaining cellular protein homeostasis. The conformational plasticity of the proteasome enables it to coordinate substrate recognition, commitment, deubiquitination, unfolding, and degradation through a series of carefully orchestrated steps. The pioneering work of Hershko, Ciechanover, and Rose on ATP-dependent proteolysis not only revealed the ubiquitin-proteasome system but also established a new paradigm in cell biology: that covalent protein modification can serve as a specific targeting signal for regulated proteolysis. Ongoing structural and biochemical studies continue to refine our understanding of this essential proteolytic complex, providing insights that may inform therapeutic strategies for diseases linked to proteasome dysfunction.
The conceptual foundation of Proteolysis-Targeting Chimeras (PROTACs) is rooted in the pioneering work of Avram Hershko, Aaron Ciechanover, and Irwin Rose on ATP-dependent proteolysis. Their research in the late 1970s and 1980s solved a fundamental biological paradox: why would the inherently exergonic process of protein breakdown require energy input from ATP hydrolysis? This question led to the discovery of the ubiquitin-proteasome system (UPS), a landmark achievement recognized with the 2004 Nobel Prize in Chemistry [1] [3].
Through meticulous biochemical fractionation of reticulocyte lysates, the researchers identified APF-1 (ATP-dependent Proteolysis Factor 1), later recognized as the protein ubiquitin [10]. They demonstrated that this small, heat-stable protein was covalently attached to target proteins in an ATP-dependent manner, marking them for destruction [1] [3]. This tagging mechanism represented a revolutionary departure from the prevailing lysosomal hypothesis of protein degradation and revealed an entirely new layer of cellular regulation. The subsequent elucidation of the three-enzyme cascade (E1 activating, E2 conjugating, and E3 ligase enzymes) established the core machinery that PROTAC technology would later co-opt for therapeutic purposes [3].
The Ubiquitin-Proteasome System: Core Mechanism
The ubiquitin-proteasome system is the primary quality control and regulatory proteolytic machinery in eukaryotic cells, responsible for degrading short-lived proteins and soluble misfolded proteins [15]. This sophisticated system operates through a well-orchestrated enzymatic cascade:
- Ubiquitin Activation: The 76-amino acid protein ubiquitin is first activated in an ATP-dependent manner by the E1 ubiquitin-activating enzyme, forming a high-energy thioester bond [15] [44].
- Ubiquitin Conjugation: The activated ubiquitin is transferred to an E2 ubiquitin-conjugating enzyme.
- Ubiquitin Ligation: An E3 ubiquitin ligase recognizes specific substrate proteins and catalyzes the transfer of ubiquitin from E2 to a lysine residue on the substrate, forming an isopeptide bond [44].
- Polyubiquitination: Repeated cycles attach additional ubiquitin molecules to Lys48 of the previously conjugated ubiquitin, forming a polyubiquitin chain [15] [1].
- Proteasomal Degradation: The K48-linked polyubiquitin chain serves as a recognition signal for the 26S proteasome, which unfolds the tagged protein, degrades it into small peptides, and recycles ubiquitin molecules [15] [44].
The specificity of this system is largely determined by the E3 ubiquitin ligases, of which humans possess approximately 600 varieties, allowing precise recognition of diverse substrates [15] [44].
PROTAC Technology: Mechanism and Design
PROTAC technology represents a therapeutic strategy to hijack the endogenous ubiquitin-proteasome system for targeted protein degradation. The first PROTAC molecule was conceptualized and developed by Crews and Deshaies in 2001 [15] [44].
Core Components and Mechanism of Action
A PROTAC molecule is a heterobifunctional complex consisting of three essential elements:
- A warhead that binds to the Protein of Interest (POI)
- A ligand that recruits an E3 ubiquitin ligase
- A chemical linker connecting these two moieties [15] [45] [44]
The mechanism of action occurs through a catalytic cycle:
- The PROTAC molecule simultaneously engages both the target protein and an E3 ubiquitin ligase, forming a ternary complex.
- This complex positions the E2-charged ubiquitin in close proximity to lysine residues on the target protein surface.
- The E3 ligase facilitates the transfer of ubiquitin from E2 to the target protein.
- Polyubiquitination marks the target protein for recognition and degradation by the 26S proteasome.
- The PROTAC molecule is released and can catalyze additional rounds of degradation [15] [44].
Table 1: Key E3 Ligases Utilized in PROTAC Development
| E3 Ligase | Full Name | Ligand Examples | Applications |
|---|---|---|---|
| CRBN | Cereblon | Thalidomide, Lenalidomide, Pomalidomide | Hematological malignancies, immune disorders |
| VHL | Von Hippel-Lindau | VHL ligand 1/2 | Hypoxia-related pathways, diverse oncology targets |
| MDM2 | Murine Double Minute 2 | Nutlin, RG7112 | p53-related pathways, oncology |
| cIAP1 | Cellular Inhibitor of Apoptosis Protein 1 | Bestatin analogs | Apoptosis regulation, oncology |
Advantages Over Traditional Therapeutic Modalities
PROTAC technology offers several distinct advantages compared to conventional small molecule inhibitors:
- Event-Driven Catalysis: Unlike traditional inhibitors that require sustained binding for functional inhibition, PROTACs operate catalytically, degrading targets after ubiquitination and recycling for multiple rounds of activity [15] [44].
- Expanded Target Scope: PROTACs can degrade proteins without defined active sites (e.g., scaffolding proteins), potentially addressing "undruggable" targets [46] [44].
- Overcoming Resistance: By eliminating the entire protein, PROTACs can circumvent resistance mechanisms arising from point mutations or protein overexpression that often plague conventional inhibitors [45] [44].
- Reduced Dosing Requirements: The catalytic mechanism may enable lower therapeutic doses, potentially improving safety profiles [15].
Current Clinical Landscape and Applications
The translational potential of PROTAC technology is evidenced by the rapidly expanding clinical pipeline. As of 2025, over 40 PROTAC candidates are in various stages of clinical development, targeting diverse proteins including androgen receptor (AR), estrogen receptor (ER), Bruton's tyrosine kinase (BTK), and interleukin-1 receptor-associated kinase 4 (IRAK4) [47].
Table 2: Selected PROTAC Degraders in Advanced Clinical Development (2025 Update)
| Drug Candidate | Company/Institution | Target | Indication | Development Phase |
|---|---|---|---|---|
| Vepdegestran (ARV-471) | Arvinas/Pfizer | ER | ER+/HER2- breast cancer | Phase III |
| BMS-986365 (CC-94676) | Bristol Myers Squibb | AR | Metastatic castration-resistant prostate cancer (mCRPC) | Phase III |
| BGB-16673 | BeiGene | BTK | Relapsed/refractory B-cell malignancies | Phase III |
| ARV-110 | Arvinas | AR | mCRPC | Phase II |
| KT-474 (SAR444656) | Kymera | IRAK4 | Hidradenitis suppurativa and atopic dermatitis | Phase II |
| DT-2216 | Dialectic Therapeutics | BCL-XL | Liquid and solid tumors | Phase I |
Promising clinical results have emerged from several trials. Vepdegestran (ARV-471) demonstrated a statistically significant improvement in progression-free survival compared to fulvestrant in patients with ESR1 mutations in the Phase III VERITAC-2 trial for ER+/HER2- advanced breast cancer [47]. Similarly, BMS-986365 showed dose-dependent activity in metastatic castration-resistant prostate cancer, with 55% of patients receiving the 900 mg twice-daily dose achieving a ≥30% decline in PSA levels [47].
Experimental Design and Methodologies
Key In Vitro Assays for PROTAC Development
Ternary Complex Formation Assays:
- Surface Plasmon Resonance (SPR): Measures real-time binding kinetics and affinity between PROTAC, POI, and E3 ligase.
- Isothermal Titration Calorimetry (ITC): Quantifies binding thermodynamics of ternary complex formation.
- Time-Resolved Fluorescence Energy Transfer (TR-FRET): High-throughput screening for ternary complex stabilization.
Degradation Efficacy Assessment:
- Western Blotting: Standard method to quantify target protein levels over time following PROTAC treatment.
- Cellular Thermal Shift Assay (CETSA): Confirms target engagement by measuring thermal stability changes.
- Immunofluorescence Microscopy: Visualizes subcellular localization and depletion of target proteins.
Ubiquitination Detection:
- Tandem Ubiquitin Binding Entity (TUBE) Assay: Pull-down of ubiquitinated proteins using ubiquitin-binding domains.
- Ubiquitin Remnant Profiling: Mass spectrometry-based identification of ubiquitination sites on target proteins.
In Vivo Evaluation Protocols
Pharmacokinetic/Pharmacodynamic Studies:
- Administration routes: Oral gavage, intravenous injection, subcutaneous delivery
- Blood collection at predetermined time points for plasma concentration analysis
- Tissue harvest (e.g., tumor biopsies) for target engagement and degradation assessment
- Dose-ranging studies to establish exposure-response relationships
Efficacy Models:
- Cell Line-Derived Xenografts (CDX): Standard tumor models using established cancer cell lines.
- Patient-Derived Xenografts (PDX): Models that better recapitulate human tumor heterogeneity.
- Genetically Engineered Mouse Models (GEMM): Syngeneic models with intact immune systems.
Research Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for PROTAC Development
| Reagent/Material | Function/Application | Examples/Specifications |
|---|---|---|
| E3 Ligase Ligands | Recruit specific E3 ligases to ternary complex | CRBN: Thalidomide derivatives; VHL: VH032/VH098 derivatives |
| Target Protein Ligands | Bind protein of interest with high affinity | Kinase inhibitors, receptor antagonists, epigenetic reader inhibitors |
| Linker Libraries | Connect warhead and E3 ligand with optimal geometry | PEG-based chains, alkyl chains, rigid aromatic spacers (varying lengths: 5-20 atoms) |
| Proteasome Inhibitors | Confirm UPS-dependent mechanism of action | MG132, Bortezomib, Carfilzomib (used as controls) |
| Ubiquitination Assay Kits | Detect and quantify target ubiquitination | Commercial kits with ubiquitin-binding matrices, anti-ubiquitin antibodies |
| CRISPR-Modified Cell Lines | Validate E3 ligase specificity | E3 ligase knockout lines, endogenous tagging of target proteins |
Emerging Innovations and Future Perspectives
Next-Generation PROTAC Technologies
The field has rapidly evolved beyond conventional PROTACs to address limitations and expand applications:
- Molecular Glues: Monovalent small molecules that induce neo-interactions between E3 ligases and target proteins, often with improved pharmacological properties [15].
- LYTACs (Lysosome-Targeting Chimeras): Degrade extracellular and membrane proteins by recruiting lysosomal trafficking receptors [15] [45].
- AbTACs (Antibody-based PROTACs): Fully recombinant bispecific antibodies that engage cell surface E3 ligases and target proteins [15].
- Photoactivatable PROTACs: Opto-PROTACs with photolabile caging groups (e.g., DMNB, DEACM) enable spatiotemporal control of protein degradation with light activation [45] [48].
- Dual-Degrading PROTACs: Simultaneously target multiple disease-relevant proteins or protein complexes [48].
Artificial Intelligence in PROTAC Design
The increasing complexity of PROTAC development has driven integration of computational approaches:
- Predictive Modeling: AI platforms (e.g., AIMLinker, ShapeLinker, DeepPROTAC) generate novel linker designs and predict degradation efficacy [45] [49].
- Patent-Based Datasets: Large-scale compound collections (e.g., 63,136 unique PROTACs from patent literature) provide training data for machine learning models [49].
- Ternary Complex Prediction: Molecular dynamics simulations guide rational design of optimized ternary complex formation.
Visualization of Core Concepts
The Ubiquitin-Proteasome System Pathway
Ubiquitin-Proteasome System Mechanism
PROTAC Mechanism of Action
PROTAC-Mediated Targeted Degradation
Historical Development Timeline
Key Milestones in Targeted Protein Degradation
PROTAC technology represents a paradigm shift in therapeutic intervention, building upon the foundational discoveries of ATP-dependent proteolysis by Hershko, Ciechanover, and Rose. By hijacking the endogenous ubiquitin-proteasome system, this approach transcends the limitations of traditional occupancy-driven pharmacology, offering catalytic, event-driven protein elimination. The rapid clinical advancement of PROTAC degraders, with multiple candidates now in late-stage trials for oncology and inflammatory indications, underscores the translational potential of this platform. As the field evolves to address challenges in tissue-specific delivery and expand the scope of degradable targets, PROTAC technology continues to validate the therapeutic strategy of targeted protein degradation, fundamentally expanding the druggable proteome and offering new hope for addressing previously untreatable diseases.
The groundbreaking work of Avram Hershko, Aaron Ciechanover, and Irwin Rose in the early 1980s fundamentally reshaped our understanding of cellular protein degradation [19] [13]. They discovered a non-lysosomal, ATP-dependent proteolytic pathway, revealing that cells employ a sophisticated tagging system to mark proteins for destruction [13]. Their research established that a small protein, ubiquitin, is covalently attached to target proteins through a sequential enzymatic cascade involving E1 (activating), E2 (conjugating), and E3 (ligating) enzymes [50] [13]. This polyubiquitination acts as a molecular signal, directing tagged proteins to the 26S proteasome for degradation [50]. This Nobel Prize-winning discovery of the ubiquitin-proteasome system (UPS) not only explained a crucial biological mechanism but also unveiled a entirely new therapeutic paradigm [13].
The field of targeted protein degradation (TPD), particularly the use of molecular glues, is a direct intellectual descendant of this foundational work. These small molecules harness the cellular machinery detailed by Hershko and Ciechanover to achieve selective degradation of disease-causing proteins [51] [52]. By reprogramming the cell's natural "trash disposal" system, molecular glues offer a powerful strategy to eliminate proteins previously considered "undruggable" due to the lack of conventional binding pockets [52] [53] [50]. This technical guide explores the mechanisms, experimental characterization, and therapeutic applications of these transformative molecules.
Molecular Glues: Mechanisms and Comparative Analysis
Fundamental Mechanisms of Action
Molecular glues (MGs) are small, often monovalent molecules that induce or stabilize interactions between two proteins that otherwise would not associate or would do so only weakly [52] [50]. In the context of protein degradation, they typically function by binding to an E3 ubiquitin ligase or a target protein (POI), creating a novel protein-protein interaction (PPI) interface that facilitates the formation of a productive ternary complex [51] [54]. This induced proximity leads to the polyubiquitination of the target protein and its subsequent degradation by the proteasome [52] [50].
A key advantage of molecular glues is their catalytic mode of action. They operate in a substoichiometric manner, meaning a single degrader molecule can facilitate the ubiquitination and degradation of multiple copies of the target protein. This allows for efficient and sustained activity at very low concentrations, resulting in greater therapeutic efficacy with reduced dosing requirements [52].
Molecular Glues vs. PROTACs: A Technical Comparison
While both Molecular Glues and PROteolysis TArgeting Chimeras (PROTACs) achieve targeted protein degradation via the UPS, they differ significantly in structure, mechanism, and discovery.
Table 1: Comparative Analysis of Molecular Glues and PROTACs
| Feature | Molecular Glues | PROTACs |
|---|---|---|
| Molecular Structure | Monovalent, small molecules (typically <500 Da) [50] | Heterobifunctional molecules with two ligands connected by a linker [51] [53] |
| Mechanism of Action | Induce novel PPIs by binding to one protein (E3 or POI), altering its surface [52] [54] | Simultaneously engage E3 ligase and POI with two separate warheads [51] |
| Molecular Weight | Lower, more "drug-like" [53] [50] | Higher, often beyond the Rule of 5 [53] [54] |
| Hook Effect | Not observed, as they bind predominantly to one protein [54] | Can occur at high concentrations, saturating binding sites and reducing ternary complex formation [54] |
| Discovery Paradigm | Historically serendipitous; rational discovery is challenging [52] [50] | More amenable to rational design by linking known binders [51] |
| Pharmacological Properties | Generally better membrane permeability, cellular uptake, and potential for blood-brain barrier penetration [53] | Can face bioavailability challenges due to higher molecular weight [54] |
The following diagram illustrates the fundamental mechanistic differences between PROTACs and the two primary binding modes of molecular glues:
Experimental Characterization of Molecular Glues
Biochemical Assays and Workflows
Characterizing molecular glues presents unique challenges, as activity depends not only on affinity for a single protein but also on the induced cooperativity (KD shift) between the protein partners [54]. A high-throughput compatible workflow has been developed to derive key parameters from concentration-response experiments.
The assay often uses Time-Resolved Fluorescence Resonance Energy Transfer (TR-FRET) to monitor the enhancement of the PPI. In a characterized model system, a molecular glue (NRX-252262) enhanced the interaction between the E3 ligase β-TrCP1 and a β-catenin peptide. The affinity (KD) improved from a basal level of 430-570 nM to a much tighter bond in the presence of the glue, demonstrating a massive KD shift [54].
The core of this methodology involves performing titrations of the molecular glue at a fixed concentration of one protein (e.g., the E3 ligase) and varying concentrations of the other protein (the target), expressed as a fraction (fKD) of the basal KD. The normalized span (Sn) from the concentration-response curve relates to the cooperativity (α) via the following derived equation, which allows for the calculation of the glue-induced KD shift [54]:
This workflow, summarized below, enables the quantification of the key parameter for molecular glue optimization—the induced cooperativity—from standard concentration-response data, making it suitable for high-throughput screening and structure-activity relationship (SAR) assessment [54].
The Scientist's Toolkit: Essential Research Reagents
Research into molecular glues relies on a suite of specialized reagents and methodologies to discover, validate, and characterize these compounds.
Table 2: Key Research Reagent Solutions for Molecular Glue Studies
| Reagent / Technology | Function & Application |
|---|---|
| E3 Ligase Components (e.g., CRBN, VHL, MDM2) | The most popular E3 ligases used as recruitable components for TPD; their ligands are starting points for degrader design [53] [55]. |
| TR-FRET Assay Kits | Enable high-throughput biochemical characterization of molecular glue-induced PPIs by measuring energy transfer between tagged proteins [54]. |
| Non-Canonical Amino Acids (e.g., AHA) | Used in pulse-chase assays like QUAD to label newly synthesized proteins and measure global protein stability rates in tissues via mass spectrometry [56]. |
| Spectral Shift Technology | A high-throughput biophysical method to directly identify molecular glues by characterizing ternary complex formation in a label-free manner [55]. |
| Targeted Proteomics | Utilizes high-throughput mass spectrometry to systematically discover novel degraders and their targets across the native cellular proteome [55]. |
| Cryo-Electron Microscopy (Cryo-EM) | Provides high-resolution structural insights into the ternary complexes formed by molecular glues, guiding rational design [55]. |
Applications and Clinical Outlook
Therapeutic Applications
Molecular glues hold significant promise across a wide spectrum of diseases, primarily by targeting pathological proteins for degradation.
- Oncology: This is the most prominent application area. Drugs like lenalidomide and pomalidomide are FDA-approved molecular glues that bind to the E3 ligase cereblon (CRBN), leading to the degradation of transcription factors IKFZ1 and IKFZ3, which are critical for multiple myeloma and myelodysplastic syndromes [52] [53]. Degraders targeting Bruton's Tyrosine Kinase (BTK), WEE1, and CDK8/19 are in development for various cancers [55].
- Neurodegenerative Disorders: Strategies are being explored to employ molecular glues that target mutant huntingtin protein in Huntington's disease for degradation via the autophagosome, not the proteasome [50].
- Other Diseases: Applications are also being investigated in inflammatory, autoimmune, and infectious diseases, leveraging the ability to degrade key immune or pathogen-derived proteins [53].
Current Challenges and Future Directions
Despite the excitement, the field faces several hurdles. A primary challenge is specificity and selectivity, as off-target effects can occur from engaging multiple proteins or E3 ligases [52]. Furthermore, rational design remains difficult; discovery has often been serendipitous because predicting productive ternary complex formation is non-trivial [52] [50].
Future progress is closely tied to overcoming these challenges. This includes the development of better predictive computational models, high-throughput screening methods, and a deeper mechanistic understanding of E3 ligase biology. The clinical pipeline is expanding, with companies like Monte Rosa Therapeutics, Ranok Therapeutics, and Neomorph advancing molecular glue candidates into and through preclinical development [53]. As of 2025, the field is experiencing explosive growth, indicating that molecular glues are poised to become a major therapeutic modality in the coming years [51] [53].
The conceptual foundation for PROteolysis TArgeting Chimeras (PROTACs) was laid by the pioneering work of Avram Hershko, Aaron Ciechanover, and Irwin Rose on ATP-dependent proteolysis. Their research, for which they received the 2004 Nobel Prize in Chemistry, unraveled the ubiquitin-proteasome system (UPS), the cell's intrinsic machinery for selective protein degradation [1] [3]. They discovered that a small, heat-stable protein they termed APF-1 (later identified as ubiquitin) was covalently attached to target proteins in an ATP-dependent manner, marking them for destruction by a large, multi-subunit protease complex [10] [3]. This seminal work resolved the long-standing paradox of why intracellular proteolysis required energy—an exergonic process—by revealing the complex enzymatic cascade involved in the "death tag" of ubiquitination [1] [3].
PROTAC technology, first introduced in 2001, represents a direct therapeutic application of this foundational knowledge [57] [15]. It harnesses the UPS to achieve targeted degradation of disease-causing proteins. Unlike traditional small-molecule inhibitors that merely block protein function and require sustained target occupancy, PROTACs act catalytically to eliminate the target protein entirely, offering a novel event-driven mechanism of action [57] [31]. This paradigm shift has not only expanded the druggable proteome to include proteins previously considered "undruggable," such as transcription factors and scaffold proteins, but also provides a promising strategy to overcome drug resistance in cancer therapy [57] [31] [46].
PROTAC Mechanism and Design: Harnessing the Ubiquitin-Proteasome System
Molecular Architecture and Mechanism of Action
A PROTAC is a heterobifunctional molecule composed of three distinct elements:
- A ligand that binds to the Protein of Interest (POI).
- A ligand that recruits an E3 ubiquitin ligase.
- A linker that covalently connects the two ligands [31] [46] [15].
The mechanism is illustrated in the diagram below, which synthesizes the core process described across multiple sources.
The formation of this ternary complex brings the E3 ligase into close proximity with the POI. The E3 ligase then mediates the transfer of ubiquitin chains from an E2 conjugating enzyme (activated by an E1 enzyme) onto lysine residues of the POI [15]. Once polyubiquitinated, primarily with K48-linked chains, the POI is recognized and degraded by the 26S proteasome [1] [15]. The PROTAC molecule is not consumed in this process and can be released to catalyze multiple rounds of degradation, operating in a sub-stoichiometric, catalytic manner [57] [31].
Key Research Reagents and Their Functions
The following table details essential reagents and components used in PROTAC research and development, as derived from the literature.
Table 1: Key Research Reagent Solutions in PROTAC Development
| Reagent / Component | Function in PROTAC Research |
|---|---|
| E3 Ligase Ligands (e.g., for VHL, CRBN) | Recruit the cell's endogenous ubiquitin ligase machinery to the ternary complex. These are often small molecules like immunomodulatory imide drugs (IMiDs) for CRBN or specific inhibitors for VHL [57] [31]. |
| POI-Targeting Ligands | Bind with high affinity to the target protein intended for degradation. These can be derived from known enzyme inhibitors, receptor antagonists, or other small-molecule binders [31] [15]. |
| Chemical Linkers | Spatially connect the E3 and POI ligands. Linker chemistry (length, flexibility, polarity) is systematically varied to optimize ternary complex formation and degradation efficacy [31] [58]. |
| Ubiquitin-Proteasome System | A cellular extract or living cell system containing functional E1, E2, E3 enzymes, ubiquitin, and the 26S proteasome. This is the essential endogenous machinery required for the degradation process [1] [3]. |
| Proteasome Inhibitors (e.g., Bortezomib) | Used as experimental controls to confirm that observed protein loss is mediated by the proteasome, as they should block PROTAC-induced degradation [59]. |
PROTACs in Clinical Trials: A 2025 Landscape
The clinical translation of PROTACs has progressed rapidly, with numerous candidates now being evaluated across a spectrum of cancers. Their ability to degrade key oncogenic drivers has shown particular promise in overcoming resistance to standard therapies.
The following table summarizes key PROTAC candidates in clinical development, highlighting their targets, indications, and current status.
Table 2: Select PROTACs in Clinical Trials (2025 Update)
| Drug Candidate | Company/Sponsor | Target | Indication(s) | Latest Reported Status |
|---|---|---|---|---|
| Vepdegestrant (ARV-471) | Arvinas / Pfizer | Estrogen Receptor (ER) | ER+/HER2- Advanced or Metastatic Breast Cancer | Phase III (NDA preparation following mixed Phase III results) [47] |
| BMS-986365 (CC-94676) | Bristol Myers Squibb | Androgen Receptor (AR) | Metastatic Castration-Resistant Prostate Cancer (mCRPC) | Phase III [47] |
| BGB-16673 | BeiGene | Bruton's Tyrosine Kinase (BTK) | Relapsed/Refractory B-cell Malignancies | Phase III [47] |
| Bavdegalutamide (ARV-110) | Arvinas | Androgen Receptor (AR) | mCRPC | Phase II [57] [47] |
| ARV-766 | Arvinas / Novartis | Androgen Receptor (AR) | mCRPC | Phase II [57] [47] |
| KT-474 (SAR444656) | Kymera | IRAK4 | Hidradenitis Suppurativa & Atopic Dermatitis | Phase II (showing expansion beyond oncology) [47] |
| DT-2216 | Dialectic Therapeutics | BCL-XL | Liquid and Solid Tumors | Phase I [47] |
Analysis of Key Clinical Programs
- Vepdegestrant (ARV-471) in Breast Cancer: As the first oral PROTAC to reach Phase III trials, Vepdegestrant targets the estrogen receptor for degradation. Recent results from the VERITAC-2 Phase III trial demonstrated a statistically significant improvement in progression-free survival (PFS) compared to fulvestrant in patients with ESR1 mutations [47]. While it did not meet the primary endpoint in the overall intent-to-treat population, this highlights the potential of PROTACs in genetically defined patient subgroups. Based on these data, Arvinas and Pfizer plan to submit Vepdegestrant for regulatory approval as a second-line monotherapy [47].
- Androgen Receptor Degraders in Prostate Cancer: Multiple AR-targeting PROTACs are in development for mCRPC, a setting plagued by resistance to conventional anti-androgens. Bavdegalutamide (ARV-110) has shown antitumor activity in patients who progressed on standard therapies, including those with specific AR mutations [57]. BMS-986365 is another potent AR degrader that has demonstrated up to 100 times greater potency in suppressing AR-driven transcription compared to the antagonist enzalutamide in preclinical models [47].
- Targeting BTK and Beyond: The advancement of BGB-16673 for B-cell malignancies underscores the expansion of TPD beyond nuclear hormone receptors. Targeting BTK via degradation, rather than inhibition, presents a promising strategy to overcome common resistance mutations seen with covalent BTK inhibitors like ibrutinib [31] [47].
The clinical development workflow for these agents is complex, as visualized below.
Experimental Protocols for PROTAC Evaluation
The development and validation of PROTACs require a multi-faceted experimental approach to confirm mechanism of action and therapeutic potential.
In Vitro Degradation and Mechanism Studies
Objective: To demonstrate and quantify target protein degradation and confirm UPS dependence in cell-based models. Workflow:
- Cell Culture & Treatment: Culture appropriate cell lines endogenously expressing or engineered to express the POI. Treat cells with a range of PROTAC concentrations and time points (e.g., 0.1 nM - 10 µM, over 3-24 hours) [31].
- Confirmation of Degradation: Lyse cells and analyze lysates by Western Blot to detect changes in POI protein levels. Quantification of band intensity normalized to loading controls (e.g., GAPDH, Actin) determines DC₅₀ (concentration for 50% degradation) and Dmax (maximum degradation) [31].
- Mechanism Validation:
- Proteasome Inhibition: Co-treat cells with a proteasome inhibitor (e.g., MG132, Bortezomib). Blockade of degradation confirms proteasome dependence [59].
- Ligase Dependence: Use a matched negative control PROTAC with an inactive E3 ligand, or employ CRISPR/Cas9 to knockout the relevant E3 ligase (e.g., CRBN, VHL). Loss of activity confirms E3 ligase dependence [31].
- Ubiquitination Assay: Perform immunoprecipitation of the POI from PROTAC-treated cells, followed by Western Blot with an anti-ubiquitin antibody to detect increased polyubiquitination of the POI [1] [15].
Ternary Complex Stability and Cooperativity Assays
Objective: To biophysically characterize the formation and stability of the POI-PROTAC-E3 ligase complex, a key determinant of degradation efficiency. Methodologies:
- Surface Plasmon Resonance (SPR) & Biolayer Interferometry (BLI): These label-free techniques can quantify binding kinetics (on-rate, off-rate) and affinity of the PROTAC for the POI and E3 ligase individually, as well as the stability of the ternary complex [31].
- Isothermal Titration Calorimetry (ITC): Measures the heat change associated with binding, providing information on binding affinity, stoichiometry, and thermodynamic parameters, which can help assess cooperativity—the enhanced binding affinity in the ternary complex versus the binary complexes [31] [58].
- X-ray Crystallography & Cryo-Electron Microscopy (Cryo-EM): Used to solve high-resolution structures of the ternary complex. This provides atomic-level insights into the protein-protein interface and informs rational design of improved PROTACs by guiding linker optimization [58].
Challenges and Future Directions
Despite the remarkable progress, the clinical advancement of PROTACs faces several hurdles. Their typically high molecular weight can lead to poor oral bioavailability and cellular permeability [31] [58]. The "hook effect," where degradation efficiency decreases at high concentrations due to the formation of non-productive binary complexes, complicates dose optimization [31]. Furthermore, the field is still expanding its repertoire of E3 ligase ligands beyond the most commonly used CRBN and VHL recruiters to improve tissue selectivity and overcome potential resistance [31] [58].
Future development will focus on optimizing the drug-like properties of PROTACs, exploring novel E3 ligases, and combining PROTACs with other therapeutic agents. The integration of Artificial Intelligence (AI) is poised to accelerate this process by predicting ternary complex structures, optimizing linker design, and forecasting pharmacokinetic properties, thereby creating a more rational and efficient framework for the next generation of targeted protein degraders [58].
Overcoming Challenges in Targeted Protein Degradation Systems
Substrate Recognition and Selection Mechanisms
The fundamental understanding of substrate recognition and selection mechanisms in intracellular proteolysis was revolutionized by the pioneering research of Avram Hershko, Aaron Ciechanover, and Irwin Rose. Their discovery of the ubiquitin-proteasome system revealed that cells employ sophisticated biochemical pathways to identify and mark proteins for degradation with extraordinary specificity, contrary to previous assumptions of non-specific proteolysis. This ATP-dependent process represents one of the most precisely regulated substrate selection systems in eukaryotic cells, where protein destruction is controlled through a complex tagging mechanism that serves as a molecular "kiss of death" for targeted substrates [1] [3].
The historical context of this discovery emerged from a puzzling biochemical paradox: while peptide bond hydrolysis is inherently exergonic, intracellular proteolysis required ATP hydrolysis [1]. This energy requirement suggested the existence of previously unrecognized regulatory steps preceding proteolysis itself. By the late 1970s, researchers had established that reticulocyte lysates (which lack lysosomes) exhibited ATP-dependent proteolysis of abnormal proteins, providing an experimental system amenable to biochemical fractionation [1] [10]. It was through the meticulous analysis of this system that the key components of the ubiquitin-mediated substrate recognition pathway were identified.
Historical Foundations: The Discovery of Ubiquitin-Mediated Substrate Selection
Key Experimental Breakthroughs
The elucidation of the substrate recognition mechanism began with critical fractionation experiments conducted by Hershko and Ciechanover. They demonstrated that ATP-dependent proteolysis in reticulocyte lysates required two complementary fractions [1] [10]:
- Fraction I: Contained a small, heat-stable protein they termed APF-1 (ATP-dependent Proteolysis Factor 1)
- Fraction II: Contained higher molecular weight components, including the proteolytic machinery
The key insight emerged when researchers radiolabeled APF-1 and discovered that it formed covalent conjugates with multiple proteins in Fraction II in an ATP-dependent manner [1]. This conjugation was unexpectedly stable to harsh treatments, including high pH, suggesting a covalent linkage rather than a non-covalent association. Subsequent experiments demonstrated that authentic proteolytic substrates became heavily modified with multiple molecules of APF-1, leading to the conclusion that conjugation served as a targeting signal for proteolysis [1].
Table 1: Historical Experimental Evidence for Ubiquitin-Mediated Substrate Recognition
| Experimental Approach | Key Finding | Technical Innovation | Citation |
|---|---|---|---|
| Reticulocyte lysate fractionation | Identification of two required fractions (I and II) | Biochemical complementation assay | [1] [10] |
| Heat stability analysis | APF-1 remained active after boiling | Unconventional protein purification | [3] [10] |
| Radiolabeled APF-1 conjugation | Covalent attachment to multiple proteins | Radioiodination tracking | [1] |
| Substrate modification analysis | Multiple APF-1 molecules per substrate | Quantitative conjugation assessment | [1] |
| Comparative biochemistry | Identification of APF-1 as ubiquitin | Cross-reactivity with known ubiquitin | [1] |
The Ubiquitin Tagging Mechanism
The discovery that APF-1 was identical to the previously known protein ubiquitin connected these findings to a broader biological context [1]. The ubiquitin tagging mechanism explained several critical aspects of substrate recognition:
- Specificity: Ubiquitin conjugation provided a specific recognition signal that could explain why certain proteins were degraded while otherwise stable proteins remained intact
- Energy Requirement: The ATP dependence reflected the enzymatic cascade required to activate and transfer ubiquitin to substrate proteins
- Regulation: The reversible nature of ubiquitination allowed for potential regulation of the degradation signal
Researchers subsequently identified the enzymatic cascade responsible for ubiquitin conjugation: E1 (ubiquitin-activating enzyme), E2 (ubiquitin-conjugating enzyme), and E3 (ubiquitin ligase) enzymes [3]. This three-tiered enzymatic system provided the molecular basis for both the energy requirement and substrate specificity of the recognition process.
Molecular Machinery of Substrate Recognition
The Ubiquitin-Proteasome System
The complete ubiquitin-proteasome pathway for substrate recognition and degradation involves a highly coordinated sequence of events:
- Substrate Identification: E3 ubiquitin ligases recognize specific degradation signals (degrons) on target proteins
- Ubiquitin Activation: E1 enzyme activates ubiquitin in an ATP-dependent reaction
- Ubiquitin Transfer: Activated ubiquitin is transferred to E2 enzyme
- Substrate Tagging: E3 facilitates transfer of ubiquitin from E2 to substrate protein
- Polyubiquitin Chain Formation: Repeated cycles create a polyubiquitin chain on substrate
- Proteasomal Recognition: Polyubiquitinated substrates are recognized by the 26S proteasome
- Degradation: Substrate is unfolded, translocated into proteolytic chamber, and degraded
- Ubiquitin Recycling: Ubiquitin is cleaved and recycled for further use
The discovery that substrates are modified with multiple ubiquitin molecules rather than single ubiquitin moieties was crucial for understanding the specificity of recognition [1] [3]. Later work would establish that K48-linked polyubiquitin chains serve as the principal signal for proteasomal degradation.
Diagram 1: Ubiquitin-Proteasome Substrate Recognition Pathway. This diagram illustrates the sequential enzymatic cascade that identifies, tags, and directs protein substrates to the proteasome for degradation.
AAA+ Protease Systems in Substrate Recognition
Beyond the ubiquitin-proteasome system, cells employ additional ATP-dependent proteases with distinct substrate recognition mechanisms. AAA+ ATPases represent a family of ring-shaped hexameric complexes that function as sophisticated molecular motors, converting chemical energy from ATP hydrolysis into mechanical work for substrate unfolding and translocation [23].
The ClpXP system serves as a paradigm for AAA+ protease substrate recognition:
- ClpX: A AAA+ ATPase unfoldase that recognizes specific degradation tags ("degrons") on substrate proteins
- ClpP: A tetradecameric proteolytic chamber that degrades unfolded polypeptides
ClpX recognizes client proteins bearing unstructured N- or C-terminal or specific degron motifs, using ATP hydrolysis cycles to unfold and translocate these substrates through its central pore into the ClpP proteolytic barrel [23]. This architecture ensures that only recognized substrates are unfolded and degraded, while preventing uncontrolled proteolysis.
Table 2: Comparative Substrate Recognition Mechanisms in ATP-Dependent Proteolytic Systems
| Proteolytic System | Recognition Mechanism | Energy Coupling | Key Recognition Elements | Specificity Determinants |
|---|---|---|---|---|
| Ubiquitin-Proteasome | Polyubiquitin tagging | ATP-dependent conjugation | E3 ubiquitin ligases | Degron motifs, Polyubiquitin chains |
| ClpXP | Degron recognition | ATP-dependent unfolding | ClpX pore loops | N/C-terminal degrons, ssrA tags |
| Other AAA+ Proteases | Specific tag recognition | ATP-dependent translocation | Pore-1 loops | Varied degradation signals |
Structural Basis of Substrate Recognition
Proteasome Architecture and Substrate Engagement
Structural studies have revealed how the 26S proteasome recognizes polyubiquitinated substrates. The proteasome consists of:
- 20S Core Particle: Contains the proteolytic active sites
- 19S Regulatory Particle: Recognizes ubiquitinated substrates, removes ubiquitin chains, unfolds substrates, and translocates them into the core particle
The regulatory particle contains ubiquitin receptors that specifically recognize polyubiquitin chains, while specialized ATPases in the base of the regulatory particle unfold substrates and translocate them into the proteolytic chamber [1].
Recent cryo-EM studies of AAA+ proteases like ClpXP have provided unprecedented insights into the mechanical aspects of substrate recognition and processing. These structures reveal how conserved pore loops within the central channel of the ATPase ring engage substrate proteins and exert mechanical force during ATP hydrolysis cycles to unfold them [23].
Principles of Degron Recognition
Research across different proteolytic systems has revealed common principles of degron recognition:
- Structural Accessibility: Degrons must be accessible to the recognition machinery
- Sequence Specificity: Specific amino acid sequences or motifs are recognized by particular E3 ligases or AAA+ proteases
- Context Dependence: Cellular context, post-translational modifications, and protein oligomerization state influence degron recognition
In the ubiquitin system, the enormous diversity of E3 ubiquitin ligases (numbering in the hundreds in humans) provides the molecular basis for recognizing the vast array of potential substrates [1].
Experimental Approaches for Studying Substrate Recognition
Key Methodologies and Techniques
The understanding of substrate recognition mechanisms has been advanced through diverse experimental approaches:
Diagram 2: Experimental Approaches for Studying Substrate Recognition. This workflow illustrates how complementary methodologies have advanced the understanding of substrate recognition mechanisms.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Studying Substrate Recognition Mechanisms
| Reagent/Tool | Composition/Type | Primary Function | Application Examples |
|---|---|---|---|
| Reticulocyte Lysate | Cell extract from immature red blood cells | ATP-dependent proteolysis system | Initial fractionation studies [1] [3] |
| Heat-stable Protein Fractions | Thermostable cellular components | Source of ubiquitin/APF-1 | Identification of ubiquitin [3] [10] |
| ATPγS (ATP analog) | Non-hydrolyzable ATP analog | Distinguish ATP-dependent steps | Demonstration of energy requirements [1] |
| Radioiodinated APF-1 | 125I-labeled ubiquitin | Tracking conjugation | Covalent attachment studies [1] |
| Proteasome Inhibitors (e.g., MG132) | Peptide aldehydes/boronic acids | Proteasome activity inhibition | Validation of proteasome dependence [1] |
| E1/E2/E3 Enzyme Systems | Recombinant enzymes | Reconstitution of ubiquitination | Mechanism of hierarchical recognition [3] |
| Cryo-EM Grids | UltrAuFoil or Quantifoil | High-resolution structure determination | Structural studies of AAA+ proteases [23] |
| Defined Protein Substrates | Engineered substrate proteins | Recognition specificity analysis | Degron mapping studies [23] |
Contemporary Research and Therapeutic Applications
Modern Structural Insights
Recent advances in structural biology, particularly cryo-electron microscopy (cryo-EM), have revolutionized our understanding of substrate recognition at near-atomic resolution. Studies of AAA+ protease complexes like ClpXP have captured:
- Substrate-engaged intermediates: Structures showing how unfolded polypeptides are translocated through the central pore
- Nucleotide-dependent conformational changes: Different states corresponding to ATP binding, hydrolysis, and product release
- Mechanical unfolding mechanisms: How conserved pore loops apply force to unfold stable protein domains [23]
These structural insights reveal a conserved core mechanism among AAA+ proteases, where sequential ATP hydrolysis around the ring drives coordinated conformational changes that exert mechanical force on substrate proteins.
Therapeutic Targeting of Substrate Recognition
The understanding of substrate recognition mechanisms has enabled novel therapeutic strategies:
- PROTACs (Proteolysis-Targeting Chimeras): Bifunctional molecules that recruit E3 ubiquitin ligases to target proteins of interest, inducing their degradation [13]
- Antifungal therapies: Targeting ClpX in Cryptococcus neoformans reverses fluconazole resistance, demonstrating the clinical relevance of these mechanisms [60]
- Cancer therapeutics: Inhibitors of specific E3 ligases or components of the ubiquitin-proteasome system
Recent research has shown that disruption of ClpX, an ATP-dependent unfoldase, can reverse antifungal resistance in Cryptococcus neoformans, restoring fluconazole susceptibility in resistant strains [60]. This approach demonstrates how targeting substrate recognition machinery can have significant therapeutic implications.
The investigation of substrate recognition and selection mechanisms represents a cornerstone of molecular cell biology, with roots in the pioneering work of Hershko, Ciechanover, and Rose on ATP-dependent proteolysis. From the initial discovery of ubiquitin-mediated tagging to the contemporary structural understanding of AAA+ protease mechanisms, this field has continually revealed the exquisite precision with which cells identify proteins for destruction. The fundamental principles emerging from these studies—specific recognition through defined degrons, energy-dependent unfolding, and regulated access to proteolytic chambers—continue to guide both basic research and therapeutic development. As structural biology techniques advance and our understanding of these systems deepens, new opportunities for manipulating substrate recognition for therapeutic benefit will undoubtedly emerge, continuing the legacy of this transformative research.
Optimizing Ternary Complex Formation for Efficient Degradation
The groundbreaking work of Avram Hershko, Aaron Ciechanover, and Irwin Rose in the late 1970s and early 1980s fundamentally reshaped our understanding of intracellular protein degradation [1] [13]. Their discovery of the ubiquitin-proteasome system revealed that ATP-dependent proteolysis was far more complex and regulated than previously thought, moving beyond simple lysosomal degradation or ATP-dependent protease action [1]. This paradigm shift began with their identification of a heat-stable protein called APF-1 (later identified as ubiquitin) that formed covalent conjugates with target proteins in an ATP-dependent manner [1] [19]. This conjugation system essentially labels proteins for destruction, representing a targeting mechanism every bit as important as phosphorylation or acetylation [1].
The researchers demonstrated that this system required multiple components: APF-1 (ubiquitin), which serves as the degradation tag; fraction II containing the proteolytic machinery; and ATP as an energy source [1]. Their key insight was recognizing that multiple ubiquitin molecules were attached to protein substrates in a process that preceded degradation [1]. This ubiquitin system has since been recognized as a central regulatory mechanism controlling diverse cellular processes including cell cycle progression, signal transduction, and quality control [13].
The contemporary field of targeted protein degradation directly builds upon these foundational discoveries. Modern approaches now deliberately exploit this natural system by designing molecules that specifically recruit target proteins to E3 ubiquitin ligases, forming a ternary complex that triggers ubiquitination and subsequent proteasomal degradation [61] [62]. This whitepaper examines current methodologies for optimizing this crucial ternary complex formation to achieve efficient and specific protein degradation.
Fundamental Principles of Ternary Complex Formation
Thermodynamic and Mathematical Framework
Ternary complex formation follows defined thermodynamic principles governed by three independent equilibrium constants [61]. For a heterobifunctional ligand (PROTAC) engaging a target protein (P) and an E3 ubiquitin ligase (E), the system can be completely described by:
- K~P1~: Binary equilibrium dissociation constant for target protein-PROTAC interaction
- K~E1~: Binary equilibrium dissociation constant for E3 ligase-PROTAC interaction
- α: Cooperativity factor representing the ratio between ternary and binary equilibrium dissociation constants [61]
The cooperativity factor (α) is particularly crucial as it quantifies the effect of simultaneous binding events. When α > 1, positive cooperativity enhances ternary complex stability through favorable protein-protein interactions at the interface. When α < 1, negative cooperativity destabilizes the ternary complex due to unfavorable interactions [61] [62]. The mathematical relationship describing ternary complex concentration [PLE] at equilibrium as a function of free ligand concentration [L] has been solved exactly for the first time recently, providing a universal framework for analyzing these systems [61].
Key Structural Determinants of Efficient Complex Formation
Several structural factors critically influence ternary complex formation:
Linker Architecture: The nature of the linker—its length, composition, and attachment points to the two ligand moieties—significantly impacts the overall affinity and efficiency of ternary complex formation [61] [62]. Although heterobifunctional ligands often feature flexible linkers in solution, crystal structures of ternary complexes frequently reveal tight linker folding that accommodates protein-protein interactions [61].
Molecular Geometry: The relative orientation of the two proteins in the ternary complex affects the efficiency of ubiquitin transfer. Even with high-affinity ternary complex formation, ubiquitination may fail if surface lysine residues on the target protein are inaccessible to ubiquitin-loaded E2 enzymes [62].
Binding Interface: The total buried surface area at the protein-protein interface correlates with measured ternary complex binding affinity, providing a predictive parameter for degrader optimization [62].
Quantitative Analysis of Ternary Complex Parameters
Correlation Between Binding Parameters and Degradation Efficiency
Recent systematic studies have quantified the relationship between ternary complex binding parameters and degradation efficiency. The data reveal clear correlations that guide degrader optimization.
Table 1: Correlation Between Ternary Complex Parameters and Degradation Activity
| Ternary Complex Parameter | Impact on Degradation Potency | Impact on Degradation Rate | Experimental Evidence |
|---|---|---|---|
| Binding Affinity (K~LPT~) | Strong positive correlation | Strong positive correlation | SPR measurements show tighter binding correlates with increased cellular potency and faster degradation [62] |
| Cooperativity (α) | Positive correlation | Positive correlation | Higher cooperativity enhances both degradation potency and initial degradation rates [62] |
| Buried Surface Area | Positive correlation | Not directly measured | Computational modeling shows larger interface BSA correlates with higher binding affinity [62] |
| Hook Effect Concentration | Critical for dosing | Affects maximal degradation | Bell-shaped dose-response requires careful titration to avoid complex dissociation [61] [63] |
Mathematical Modeling of Ternary Complex Formation
The formation of the ternary complex follows a predictable mathematical model that explains the characteristic "hook effect" observed in dose-response curves [61]. Unlike traditional binary interactions that follow a simple sigmoidal curve, ternary complex formation displays a bell-shaped curve where excessive PROTAC concentrations dissociate the complex by forming binary complexes with each protein separately [61] [63].
The exact mathematical equation describing ternary complex concentration at equilibrium as a function of free ligand concentration [L] is:
Where f(L) represents a function of the free ligand concentration and the relevant equilibrium constants [61]. This equation provides a universal mathematical tool for analyzing ternary complex systems and predicting their behavior under different concentration conditions.
Experimental Methods for Assessing Ternary Complex Formation
Biochemical and Biophysical Assays
Multiple experimental approaches have been developed to quantitatively measure ternary complex formation:
Surface Plasmon Resonance (SPR): This label-free technique directly measures binding affinity of PROTAC-mediated ternary complex formation by flowing a preformed binary complex (PROTAC + target protein) over a surface-immobilized E3 ligase [62]. SPR provides quantitative data on binding affinity (K~LPT~) and cooperativity (α) [62].
Isothermal Titration Calorimetry (ITC): Similar to SPR, ITC offers label-free measurement of ternary complex thermodynamics, providing information on binding constants, stoichiometry, and enthalpy changes [62].
Lumit Biochemical Assays: This homogeneous immunoassay approach uses anti-tag antibodies conjugated to complementary fragments of a luciferase enzyme to detect ternary complex formation [63]. When the ternary complex brings the tags into proximity, the luciferase fragments complement to produce a luminescent signal [63].
Cellular Assays
NanoBRET Technology: This live-cell assay monitors ternary complex formation in physiologically relevant environments [63]. The target protein is tagged with a luciferase donor (NanoLuc or HiBiT), while the E3 ligase is tagged with a fluorescent acceptor (HaloTag). PROTAC-induced proximity generates a BRET signal measurable in real-time [63].
Cellular Degradation Assays: Western blotting or immunofluorescence methods measure downstream target protein depletion, providing functional validation of ternary complex activity [62].
Structural and Computational Approaches
X-ray Crystallography and Cryo-EM: These techniques provide high-resolution structural information about ternary complex architecture, revealing molecular details of protein-protein interfaces and linker conformation [62].
Computational Modeling: Recent advances enable prediction of ternary complex structures and calculation of buried surface area at interfaces, which correlates with measured binding affinity [62].
Figure 1: Experimental workflows for assessing ternary complex formation and their measured parameters.
The Scientist's Toolkit: Essential Research Reagents and Methods
Table 2: Key Research Reagent Solutions for Ternary Complex Studies
| Reagent/Method | Function | Application Context | Key Features |
|---|---|---|---|
| NanoBRET Ternary Complex Assay | Live-cell detection of ternary complex formation | Cellular studies, kinetic analysis | Endpoint or kinetic measurement in physiologically relevant environments [63] |
| Lumit Anti-Tag Protein Interaction Reagents | Biochemical detection of ternary complexes | In vitro screening, relative potency determination | Homogeneous assay format, no washing steps [63] |
| SPR with Preformed Binary Complex | Direct measurement of binding affinity | Biophysical characterization | Label-free, quantitative K~LPT~ and cooperativity data [62] |
| HiBiT Tagging with CRISPR/Cas9 | Endogenous tagging of target proteins | Cellular studies in native context | Preserves endogenous regulation and expression levels [63] |
| HaloTag-E3 Ligase Fusions | Acceptor for proximity assays | Cellular and biochemical studies | Compatible with multiple detection modalities [63] |
Optimization Strategies for Enhanced Ternary Complex Formation
Linker Optimization Strategies
The linker component of heterobifunctional degraders represents a critical optimization parameter that significantly influences ternary complex formation:
Length Optimization: Systematic variation of linker length can identify the optimal distance that allows simultaneous engagement of both target protein and E3 ligase without introducing strain [61] [62]. Both excessively short and excessively long linkers typically impair ternary complex formation.
Composition and Rigidity: Linker composition (flexible PEG chains, rigid aromatics, or aliphatic chains) affects the conformational entropy cost upon binding [61]. Optimal linkers often balance flexibility for initial engagement with sufficient rigidity to position proteins correctly.
Attachment Points: The specific atoms on each ligand where linkers are attached significantly influence the overall topology of the ternary complex [62]. Even small changes in attachment points can dramatically alter cooperativity and degradation efficiency.
Leveraging Cooperativity Effects
Positive cooperativity (α > 1) emerges from favorable protein-protein interactions at the ternary complex interface and significantly enhances degradation efficiency [62]. Strategies to enhance cooperativity include:
Interface Engineering: Designing PROTACs that promote complementary interactions between the target protein and E3 ligase [62].
Structural Informatics: Using computational modeling to predict ternary complex structures and interface buried surface area, which correlates with binding affinity [62].
Ligand Selection: Choosing warheads that orient proteins to create favorable interaction surfaces rather than just high-affinity binders [62].
Balancing Binary Affinities
Optimal degraders require careful balancing of the individual binary affinities (K~P1~ and K~E1~) rather than simply maximizing both [61] [62]. The relationship between these affinities follows:
This equation reveals that the maximal ternary complex formation depends on both cooperativity (α) and the ratio of binary dissociation constants [62]. In practice, this means that extremely high affinity for one partner may require compensation in the other binding event to maintain optimal ternary complex formation.
The optimization of ternary complex formation represents the crucial first step in achieving efficient targeted protein degradation, directly building upon the foundational discoveries of Hershko, Ciechanover, and Rose in ATP-dependent proteolysis [1]. Through quantitative assessment of binding parameters, systematic optimization of linker architecture, and strategic enhancement of cooperativity, researchers can now design degraders with improved efficiency and predictability. The mathematical frameworks and experimental methods reviewed here provide a roadmap for advancing this promising therapeutic modality, continuing the legacy of the ubiquitin-proteasome system discoveries that revealed the intricate regulation of cellular protein degradation.
Addressing Specificity and Off-Target Effects in PROTAC Design
The groundbreaking work of Avram Hershko, Aaron Ciechanover, and Irwin Rose in the late 1970s and early 1980s unveiled the ubiquitin-proteasome system (UPS), the fundamental ATP-dependent proteolytic machinery that would ultimately inspire targeted protein degradation therapeutics [1] [10]. Their discovery that a small, heat-stable protein (initially termed APF-1, later identified as ubiquitin) mediated ATP-dependent proteolysis through covalent tagging of substrate proteins established the biochemical framework for today's proteolysis-targeting chimera (PROTAC) technology [1] [10]. This foundational research revealed that intracellular protein degradation is not a nonspecific process but rather a highly regulated system employing covalent protein modification to mark specific substrates for destruction—a principle that directly enables the targeted degradation approach of PROTACs.
PROTACs represent a revolutionary approach in drug discovery that moves beyond traditional occupancy-based inhibition toward induced protein degradation [31]. These heterobifunctional molecules harness the natural UPS to selectively degrade disease-driving proteins, offering potential solutions for previously "undruggable" targets [31]. However, as the field advances, addressing specificity and off-target effects remains a central challenge in PROTAC design and development. This technical guide examines current strategies and methodologies to enhance PROTAC precision, framed within the historical context of ATP-dependent proteolysis research.
PROTAC Mechanism and Specificity Challenges
Molecular Architecture and Degradation Mechanism
PROTACs consist of three fundamental components: a ligand that binds to the protein of interest (POI), an E3 ubiquitin ligase-recruiting ligand, and a chemical linker connecting these two moieties [45] [64] [31]. The molecular mechanism proceeds through several well-defined steps:
- Ternary Complex Formation: The PROTAC simultaneously engages both the POI and an E3 ubiquitin ligase, forming a POI-PROTAC-E3 ternary complex [64] [31].
- Ubiquitin Transfer: The E3 ligase transfers ubiquitin chains to lysine residues on the POI surface [45] [31].
- Proteasomal Degradation: Polyubiquitinated POI is recognized and degraded by the 26S proteasome [23] [31].
- PROTAC Recycling: The PROTAC molecule is released and can catalyze additional degradation cycles [31].
Table 1: Core Components of PROTAC Design and Their Influence on Specificity
| PROTAC Component | Specificity Considerations | Key Design Principles |
|---|---|---|
| POI Ligand | Binding affinity, selectivity for target versus related proteins | Optimize for target engagement without inhibiting native function; suboptimal ligands can still produce effective degraders |
| E3 Ligand | E3 ligase expression patterns across tissues/cell types, intrinsic specificity | Match E3 expression to target tissue; consider tissue-specific E3 ligases to reduce off-target effects |
| Linker | Length, composition, flexibility/rigidity | Enable optimal ternary complex geometry; influence degradation efficiency and selectivity through spatial orientation |
The following diagram illustrates the complete PROTAC mechanism, from ternary complex formation through proteasomal degradation:
Fundamental Specificity Challenges in PROTAC Design
Despite their therapeutic potential, PROTACs face several specificity challenges that can limit clinical application:
- Binary versus Ternary Complex Specificity: A ligand with high specificity for its target in binary binding may demonstrate different specificity when incorporated into a PROTAC due to altered ternary complex dynamics [64] [31].
- Hook Effect: At high concentrations, PROTACs can form non-productive binary complexes (POI-PROTAC and E3-PROTAC) instead of productive ternary complexes, reducing degradation efficiency and potentially increasing off-target effects [31].
- Tissue-Specific E3 Ligase Expression: The restricted repertoire of currently utilized E3 ligases (only ~13 of approximately 600 human E3s) means that degraders may act in tissues where they are not needed, potentially causing toxicity [65].
- Linker-Dependent Off-Target Effects: Optimized linkers can improve selectivity, but suboptimal linkers may facilitate degradation of structurally similar off-target proteins [64].
Strategic Approaches to Enhance PROTAC Specificity
Ternary Complex Optimization
The formation of a productive ternary complex is the central determinant of PROTAC efficacy and specificity. Several factors influence ternary complex quality:
- Cooperativity: Positive cooperativity occurs when binding of the PROTAC to one protein enhances its affinity for the second protein, improving degradation efficiency and potentially specificity [31].
- Linker Optimization: Linker length and composition directly influence the geometry of the ternary complex, with even small modifications significantly impacting degradation efficiency and selectivity [64] [31].
Table 2: Experimental Approaches for Ternary Complex Characterization
| Method | Application in PROTAC Development | Key Output Parameters |
|---|---|---|
| Surface Plasmon Resonance (SPR) | Measures binding kinetics and affinity in binary and ternary complexes | Binding affinity (KD), association/dissociation rates, cooperative effects |
| Cryo-Electron Microscopy (Cryo-EM) | High-resolution structural analysis of ternary complexes | Ternary complex architecture, protein-protein interfaces, spatial orientation |
| Isothermal Titration Calorimetry (ITC) | Quantifies thermodynamic parameters of binding | Binding stoichiometry, enthalpy (ΔH), entropy (ΔS), cooperativity factors |
| Cellular Thermal Shift Assay (CETSA) | Evaluves target engagement in cellular contexts | Thermal stability shifts, confirmation of intracellular ternary complex formation |
Pro-PROTAC and Conditional Activation Strategies
PROTAC prodrugs (pro-PROTACs) represent an emerging strategy to enhance specificity through controlled activation:
- Photocaged PROTACs (opto-PROTACs): Incorporation of photolabile groups (e.g., DMNB, DEACM, NPOM) that prevent E3 ligase or POI binding until removed by light exposure enables spatiotemporal control of protein degradation [45].
- Bio-removable Protecting Groups: Installation of enzymatically cleavable moieties that are selectively removed in target tissues or disease environments [45].
- Dual-Latent PROTACs: Simultaneous caging of both E3 and POI binding motifs to minimize off-target activity before activation [45].
The following diagram illustrates the conditional activation mechanism of pro-PROTACs:
Expanding the E3 Ligase Toolkit
The current reliance on a limited set of E3 ligases (predominantly CRBN and VHL) represents both a specificity challenge and opportunity:
- Tissue-Restricted E3 Ligases: Identification and utilization of E3 ligases with restricted expression patterns could enable tissue-specific degradation [65].
- Disease-Associated E3 Ligases: Leveraging E3 ligases upregulated in specific disease states could enhance therapeutic windows [65].
- Engineered E3 Ligases: Computational design of E3 ligases with altered specificity profiles for customized degradation applications [64].
Experimental Protocols for Specificity Assessment
Comprehensive Specificity Profiling
Rigorous experimental validation is essential for characterizing PROTAC specificity:
Protocol 1: Global Proteomics Analysis for Off-Target Degradation
- Objective: Identify unintended protein degradation events across the proteome.
- Methodology:
- Treat cells with PROTAC at multiple concentrations and time points.
- Perform quantitative mass spectrometry-based proteomics (e.g., TMT or label-free approaches).
- Analyze significantly downregulated proteins beyond the intended target.
- Validate putative off-targets using orthogonal methods (e.g., Western blot).
- Key Parameters: Fold-change threshold (typically ≥70% degradation), statistical significance (p-value < 0.05), concentration dependence.
Protocol 2: Ternary Complex Cooperativity Measurements
- Objective: Quantitatively assess cooperative effects in ternary complex formation.
- Methodology:
- Measure PROTAC binding affinity to POI and E3 ligase individually using SPR.
- Employ analytical ultracentrifugation or SPR to assess ternary complex formation.
- Calculate cooperativity factor (α) from binding isotherms.
- Correlate cooperativity with degradation efficiency in cellular models.
- Interpretation: α > 1 indicates positive cooperativity; α < 1 indicates negative cooperativity.
Functional Validation of Target Specificity
Protocol 3: Resistance Mutation Profiling
- Objective: Confirm on-target degradation through mutation-based validation.
- Methodology:
- Introduce point mutations in the POI at the PROTAC binding interface.
- Express mutant proteins in genetically null backgrounds.
- Assess degradation resistance compared to wild-type protein.
- Evaluate rescue of phenotypic effects in PROTAC-treated cells.
- Key Controls: Include wild-type rescue controls and monitor mutation effects on protein stability.
Protocol 4: Kinase Selectivity Profiling
- Objective: Comprehensive assessment of kinase degradation specificity (for kinase-targeting PROTACs).
- Methodology:
- Utilize kinase-conjugated beads for affinity purification from PROTAC-treated lysates.
- Perform quantitative mass spectrometry to assess kinase abundance.
- Compare degradation patterns across the kinome.
- Validate hits using cellular assays monitoring pathway activation.
- Applications: Particularly valuable for PROTACs derived from multi-kinase inhibitors.
Research Reagent Solutions for PROTAC Development
Table 3: Essential Research Tools for Specificity Optimization
| Reagent Category | Specific Products/Assays | Research Application |
|---|---|---|
| E3 Ligase Recruitment Assays | HTRF-based ubiquitination assays, AlphaLigase | Quantify E3 ligase engagement and ubiquitination efficiency |
| Ternary Complex Characterization | Cryo-EM structural analysis, SPR co-binding experiments | Elucidate ternary complex structure and binding dynamics |
| Proteomic Profiling Platforms | TMT-based global proteomics, phosphoproteomics | Systematically identify degradation events and signaling consequences |
| Cellular Model Systems | CRISPR-engineered E3 knockout cells, resistance mutation models | Validate mechanism of action and target specificity |
| Computational Tools | AIMLinker, ShapeLinker, DeepPROTAC | Predict linker structures and optimize ternary complex formation |
The quest for enhanced specificity in PROTAC design continues to build upon the foundational discoveries of ATP-dependent proteolysis research initiated by Hershko, Ciechanover, and Rose [1] [10]. As the field advances, several promising approaches are emerging:
- Integration of Artificial Intelligence: Machine learning models like AIMLinker and DeepPROTACs are revolutionizing linker design and ternary complex prediction, enabling more rational and specific PROTAC development [45].
- Nano-Based Delivery Systems: Advanced delivery platforms including nano-PROTACs and biomacromolecule conjugates are being developed to improve tissue targeting and overcome bioavailability limitations [65] [66].
- Multi-Omics Characterization: Comprehensive proteomic, phosphoproteomic, and metabolomic analyses enable systems-level understanding of PROTAC effects, revealing both on-target and off-target consequences [65].
As PROTAC technology continues its clinical advancement, with multiple candidates now in Phase III trials [47], addressing specificity challenges remains paramount for realizing the full therapeutic potential of targeted protein degradation. The ongoing expansion of E3 ligase tools, refinement of conditional activation strategies, and development of sophisticated characterization methods promise to deliver increasingly precise degraders for clinical application.
The Role of Deubiquitinating Enzymes (DUBs) in Regulation
The discovery of ubiquitin-dependent proteolysis by Avram Hershko, Aaron Ciechanover, and Irwin Rose fundamentally reshaped our understanding of cellular protein regulation. Their seminal 1980 studies revealed that ATP-dependent intracellular proteolysis involved a complex enzymatic cascade, centered on a small protein they termed APF-1, later identified as ubiquitin [1] [19]. This research established the foundational principle that covalent attachment of ubiquitin to target proteins serves as a critical regulatory mechanism, particularly for marking proteins for degradation by the proteasome. This reversible modification is now recognized as being every bit as important as phosphorylation or acetylation for eukaryotic cell regulation [19].
Deubiquitinating enzymes (DUBs) constitute the essential regulatory counterpoint to the ubiquitination machinery. As proteases that cleave the isopeptide or peptide bonds between ubiquitin and substrate proteins, DUBs provide dynamic reversibility to ubiquitin signaling, ensuring appropriate biological responses to cellular cues [67]. This review examines the sophisticated mechanisms governing DUB activity and specificity, exploring how these regulatory principles enable DUBs to control diverse physiological processes and contribute to human disease pathogenesis.
DUB Classification and Fundamental Functions
The human genome encodes approximately 100 DUBs, which are categorized into two main classes based on their catalytic mechanisms: cysteine proteases and metalloproteases [67] [68]. The cysteine proteases represent the majority of DUBs and are further subdivided into four primary families: ubiquitin-specific proteases (USPs), ubiquitin C-terminal hydrolases (UCHs), ovarian tumor proteases (OTUs), and Machado-Josphin domain proteases (MJDs) [67] [68]. The metalloprotease class contains only the JAMM domain proteases [68].
Table 1: Major Deubiquitinating Enzyme (DUB) Families
| DUB Family | Catalytic Mechanism | Representative Members | Key Features |
|---|---|---|---|
| USP | Cysteine protease | USP7, USP15, USP28 | Largest family (~58 members); diverse domain architecture; regulates multiple cellular processes |
| OTU | Cysteine protease | OTUB1, OTULIN, A20 | High linkage specificity for polyubiquitin chains; regulated by oxidation |
| UCH | Cysteine protease | UCH-L1, UCH-L3 | Primarily processes ubiquitin precursors and small adducts |
| MJD | Cysteine protease | Ataxin-3, JOSD1 | Josephin domain; involved in protein aggregation diseases |
| JAMM | Zinc metalloprotease | AMSH, RPN11 | Requires zinc for catalysis; often part of macromolecular complexes |
DUBs perform several essential functions in maintaining ubiquitin homeostasis [68]:
- Processing ubiquitin precursors: DUBs cleave inactive ubiquitin polymers or ubiquitin-ribosomal fusion proteins to generate active monoubiquitin.
- Reversing ubiquitin signals: By removing ubiquitin from modified proteins, DUBs rescue substrates from proteasomal degradation or alter their localization and activity.
- Maintaining free ubiquitin pools: DUBs recycle ubiquitin from proteasome-bound chains and dismantle unanchored polyubiquitin.
- Proofreading ubiquitin conjugates: DUBs correct erroneous ubiquitination events to ensure signaling fidelity.
Molecular Mechanisms of DUB Regulation
Active Site Rearrangements and Substrate-Induced Activation
Structural studies have revealed that many DUBs exist in latent states with misaligned catalytic residues, requiring activation through specific molecular events. USP7 (HAUSP), which regulates p53 and other critical factors, exemplifies this regulatory mechanism [67]. In its apo state, USP7's catalytic cysteine and histidine residues are positioned in a non-productive conformation. Ubiquitin binding triggers a substantial conformational rearrangement that properly aligns the catalytic triad for efficient isopeptide bond hydrolysis [67].
This activation mechanism is further modulated by USP7's C-terminal ubiquitin-like (UBL) domains. These UBL domains interact with the catalytic domain and enhance ubiquitin binding affinity. GMP synthase promotes the activated conformation of USP7 by facilitating association between the UBL domains and the catalytic domain, thereby stabilizing the active site in a catalytically competent configuration [67]. This requirement for substrate-induced alignment may also protect the reactive cysteine from oxidative inactivation, adding another layer of regulation [67].
OTU family DUBs employ similar activation mechanisms but achieve remarkable linkage specificity through distinct molecular strategies. OTULIN, which specifically cleaves linear (M1-linked) ubiquitin chains, demonstrates exquisite mechanism for substrate discrimination [67]. In the apoenzyme, OTULIN's catalytic histidine is misaligned for catalysis. The backbone carbonyl oxygen of the N-terminal methionine in the proximal ubiquitin mechanically coaxes this histidine into proper position—an interaction unique to peptide bonds that is absent in isopeptide linkages [67]. Additionally, Glu16 in the proximal ubiquitin further promotes the active conformation by displacing an inhibitory acidic side chain and orienting the catalytic asparagine [67]. This dual requirement for structural elements found only in linear ubiquitin chains ensures OTULIN's remarkable specificity.
Regulation by Partner Protein Interactions
Many DUBs require incorporation into macromolecular complexes for full enzymatic activity and proper substrate targeting. The yeast USP enzyme Ubp8 provides a compelling example of this regulatory principle [67]. Ubp8 possesses weak intrinsic isopeptidase activity that is dramatically enhanced upon association with the SAGA (Spt-Ada-Gcn5-acetyltransferase) histone modification complex. Within this complex, other subunits both activate Ubp8's catalytic function and target it to specific chromatin regions where it deubiquitinates histone H2B [67].
Proteomic studies have revealed that DUBs frequently form complexes with E3 ubiquitin ligases, creating functional modules that potentially allow for precise coordination between ubiquitination and deubiquitination activities [67]. These associations can have dual regulatory consequences: they may activate or inhibit the DUB's catalytic function, and they can simultaneously recruit the DUB to specific subcellular locations or substrate populations. This partnership principle represents a sophisticated mechanism for ensuring spatial and temporal control of deubiquitination activity.
Post-Translational Modification and Allosteric Regulation
DUB activity is frequently modulated by post-translational modifications that alter catalytic efficiency, substrate specificity, or subcellular localization. Several USP-class DUBs, including USP1, undergo reversible oxidation of the active site cysteine to sulphenic acid, effectively inactivating the enzyme under conditions of oxidative stress [67]. This regulatory mechanism directly couples DUB activity to cellular redox state and may contribute to signaling pathways involving reactive oxygen species.
Phosphorylation has also emerged as an important regulatory modification for multiple DUBs. While the search results do not provide specific examples, this mechanism can influence DUB stability, enzymatic activity, or interactions with binding partners. The presence of additional protein domains in many DUBs, including ubiquitin-associated (UBA), ubiquitin-interacting motif (UIM), and zinc finger domains, provides platforms for allosteric regulation and integration into diverse signaling networks.
Experimental Approaches for DUB Research
Structural and Biophysical Methodologies
Understanding DUB regulatory mechanisms has relied heavily on structural biology approaches. The following experimental workflow has been instrumental in elucidating mechanisms of active site regulation:
Title: Structural Biology Workflow for DUB Regulation Studies
Key methodologies include:
- X-ray crystallography: Provides high-resolution structures of DUBs in apo states and in complex with ubiquitin or substrate analogs, revealing active site organization and conformational changes [67].
- Small-angle X-ray scattering (SAXS): Offers solution-based structural information about conformational changes in multi-domain DUBs, such as those involving UBL domains [67].
- Site-directed mutagenesis: Validates functional importance of specific residues identified through structural analysis, including catalytic triad residues and regulatory elements [67].
- Hydrolysis kinetics: Measures enzymatic activity of wild-type and mutant DUBs using ubiquitin-AMC or diubiquitin substrates to quantify catalytic efficiency and linkage specificity [67].
Chemical Tools and Research Reagents
The development of selective chemical probes has been essential for dissecting DUB functions in physiological contexts.
Table 2: Key Research Reagents for DUB Investigation
| Reagent Category | Specific Examples | Research Applications | Mechanistic Insights |
|---|---|---|---|
| Activity-Based Probes | Ubiquitin vinyl sulfone (UbVS), Ubiquitin aldehyde (Ubal) | Profiling active DUBs in complex mixtures, structural studies | Covalently traps catalytic cysteine, allowing identification and purification |
| Small Molecule Inhibitors | P22077 (USP7 inhibitor), IU1 (USP14 inhibitor) | Functional validation in cellular and animal models | Targets regulatory domains or active sites to inhibit DUB function |
| Genetic Tools | siRNA/shRNA, CRISPR-Cas9 knockout, Dominant-negative mutants | Loss-of-function studies in cells and animal models | Reveals physiological consequences of DUB inhibition or deletion |
| Ubiquitin Variants | Linkage-specific diubiquitins, Mutant ubiquitins | Enzymatic characterization of specificity | Determines linkage preference and cleavage efficiency |
Activity-based probes like ubiquitin vinyl sulfone (UbVS) function as mechanism-based inhibitors that covalently modify the active site cysteine of cysteine protease DUBs, enabling identification of catalytically active enzymes in complex biological samples [69]. These tools have been particularly valuable for profiling DUB activity states under different physiological conditions and for validating target engagement of small molecule inhibitors.
DUBs in Disease and Therapeutic Targeting
Pathological Roles in Human Diseases
Dysregulation of DUB activity contributes to numerous human diseases, particularly cancer, where DUBs can function as either oncogenes or tumor suppressors depending on cellular context. USP28 is overexpressed in multiple cancer types, including colon and lung carcinomas, where it stabilizes oncogenic transcription factors like c-Myc, Notch1, and c-Jun by preventing their proteasomal degradation [68]. In squamous tumors, USP28 promotes chemotherapy resistance by regulating DNA repair through the ΔNp63-Fanconia anemia pathway axis [68].
The contextual duality of DUB functions is exemplified by USP9X in pancreatic ductal adenocarcinoma (PDAC). In human PDAC models, USP9X promotes tumor cell survival and malignancy, whereas in KPC (KrasLSL-G12D/+; Trp53LSL-R172H/+; Pdx1-Cre) mouse models, it acts as a tumor suppressor by regulating the Hippo pathway and cooperating with LATS kinase to inhibit YAP/TAZ activity [70]. Transposon-mediated mutagenesis screens identified USP9X as having the highest mutation frequency in PDAC tumors, highlighting its significance as both a prognostic marker and therapeutic target [70].
Beyond oncology, DUBs play critical roles in other pathological conditions. In osteoarthritis, USP7 stabilizes NOX4 to amplify ROS–NLRP3-dependent pyroptosis and cartilage catabolism, while USP15 deubiquitinates ERK2 and SMAD2 to enhance TGF-β signaling and chondrocyte anabolism [71]. In diabetic nephropathy, specific DUBs dynamically regulate glycolipid metabolism, oxidative stress, and fibrotic processes, presenting novel therapeutic opportunities for this condition [72].
Emerging Therapeutic Strategies
Several innovative approaches are being developed to target DUBs therapeutically:
Small molecule inhibitors: Multiple selective DUB inhibitors have entered preclinical development, with some advancing to clinical trials. VLX1570, an inhibitor of USP14 and UCHL5, reached Phase I/II clinical trials for multiple myeloma but was terminated due to toxicity concerns [69]. Structure-based drug design and advanced delivery technologies are being employed to improve the selectivity and therapeutic indices of next-generation DUB inhibitors.
DUBTACs (Deubiquitinase-Targeting Chimeras): This emerging therapeutic modality employs heterobifunctional molecules that recruit DUBs to specific target proteins, promoting their stabilization by removing ubiquitin chains [73]. DUBTACs consist of a DUB-binding ligand connected via a linker to a protein-of-interest (POI) targeting ligand, enabling selective stabilization of disease-relevant proteins that are aberrantly degraded.
Targeted DUB degraders: Similar to PROTACs (Proteolysis-Targeting Chimeras), these molecules facilitate the degradation of specific DUBs by recruiting them to E3 ubiquitin ligases, offering an alternative strategy for modulating DUB activity [69].
The following diagram illustrates the molecular mechanism of DUBTACs:
Title: DUBTAC Mechanism for Targeted Protein Stabilization
The regulatory mechanisms governing deubiquitinating enzymes represent a sophisticated control system that ensures precision in ubiquitin-dependent signaling. Through active site rearrangements, partnership with protein complexes, post-translational modifications, and subcellular targeting, DUB activity is finely tuned to maintain cellular homeostasis. The foundational research on ATP-dependent proteolysis by Hershko, Ciechanover, and Rose not only revealed the ubiquitin system but also implicitly highlighted the importance of its reversal through deubiquitination.
As our understanding of DUB regulatory mechanisms deepens, new therapeutic opportunities continue to emerge across diverse disease contexts. The development of selective chemical probes, innovative therapeutic modalities like DUBTACs, and improved structural and functional characterization methods will accelerate the translation of DUB biology into clinical applications. Future research will undoubtedly uncover additional layers of regulation and expand the therapeutic potential of targeting this fascinating enzyme class.
Linker Optimization in PROTAC Molecule Design
The groundbreaking work of Avram Hershko, Aaron Ciechanover, and Irwin Rose in elucidating the ATP-dependent ubiquitin-proteasome system (UPS) laid the fundamental mechanistic foundation for the development of Proteolysis Targeting Chimeras (PROTACs) [1] [3]. Their discovery that covalent attachment of ubiquitin (originally termed APF-1) serves as an energy-dependent "death tag" for proteins marked for proteasomal degradation solved the long-standing paradox of ATP requirement in intracellular proteolysis [1] [3]. This foundational knowledge directly enabled the conceptualization of PROTACs as heterobifunctional molecules that hijack this natural protein degradation machinery [74]. PROTACs consist of three key components: a target protein-binding warhead, an E3 ubiquitin ligase-binding anchor, and a connecting linker that enables the formation of a productive ternary complex [45] [64]. While early PROTAC development focused predominantly on warhead and anchor optimization, the critical role of the linker in determining degradation efficacy has become increasingly apparent [64] [75]. This technical guide examines linker optimization strategies within the historical context of ATP-dependent proteolysis research, providing researchers with experimentally-validated methodologies for rational PROTAC design.
The PROTAC Linker: Function and Strategic Importance
The linker in PROTAC design serves not merely as a spatial connector but as a critical determinant of ternary complex formation, degradation efficiency, and selectivity [64] [75]. Unlike traditional small molecules where optimization focuses primarily on target binding affinity, PROTACs operate through an event-driven catalytic mechanism where the linker governs the geometry and stability of the POI-PROTAC-E3 ligase ternary complex [64] [58]. The strategic importance of linkers stems from their influence on multiple pharmacological parameters:
- Ternary Complex Formation: Optimal linker length and composition enable proper spatial orientation between the POI and E3 ligase, facilitating effective ubiquitin transfer [64].
- Degradation Efficiency: Linker properties directly impact the cooperativity and stability of the ternary complex, which correlates with degradation potency [64] [75].
- Selectivity Profiling: Linker optimization can impart selectivity between homologous protein targets by exploiting subtle differences in ternary complex formation [64].
- Pharmacokinetic Properties: Linker composition significantly influences PROTAC solubility, membrane permeability, and metabolic stability [58] [75].
The critical nature of linker optimization becomes evident when considering that PROTACs often violate Lipinski's rules due to their high molecular weight (700-1,100 Da) and flexibility, making linker design essential for maintaining drug-like properties [58] [76].
Table 1: Key Linker Characteristics and Their Impact on PROTAC Efficacy
| Linker Characteristic | Structural Impact | Functional Consequences | Experimental Evidence |
|---|---|---|---|
| Length | Determines spatial distance between warhead and anchor | Optimal length enables productive ternary complex formation; too short or too long linkers reduce degradation efficiency [75] | BRD4 degraders show 2-3 orders of magnitude potency difference based on linker length optimization [64] |
| Flexibility | Governs conformational freedom and entropy | Balanced flexibility allows adaptation to protein surfaces while maintaining optimal orientation [75] | Comparison of alkyl (flexible) vs. aryl (rigid) linkers in BET degraders demonstrates distinct degradation profiles [64] |
| Composition | Affects physicochemical properties and protein interactions | Hydrophilic linkers (e.g., PEG) improve solubility; hydrophobic linkers may enhance membrane permeability [64] [75] | MZ1 utilizes a triazole-containing linker that contributes to selective BRD4 over BRD2/3 degradation [64] |
| Attachment Points | Influences vector orientation of binding motifs | Proper alignment of warhead and anchor binding vectors enhances ternary complex stability [75] | VHL-based PROTACs show dramatically different efficacy based on hydroxyproline attachment chemistry [64] |
Linker Design and Optimization Strategies
Structural Classes and Chemical Motifs
PROTAC linkers encompass diverse structural classes, each imparting distinct physicochemical properties and conformational preferences. The evolution from simple alkyl and polyethylene glycol (PEG) linkers to more sophisticated functionalized linkers represents a significant advancement in PROTAC design [64]. Common linker classes include:
- Alkyl Chain Linkers: Comprised of saturated carbon chains, these offer flexibility but may adopt collapsed conformations that reduce degradation efficiency [75].
- PEG-Based Linkers: Ethylene glycol repeats provide enhanced solubility and reduced aggregation propensity, though excessive flexibility can be detrimental to ternary complex formation [64] [75].
- Aromatic/Rigid Linkers: Incorporating phenyl or other aromatic rings reduces conformational flexibility, potentially improving selectivity by enforcing specific binding orientations [75].
- Heterocyclic Linkers: Triazole rings (from click chemistry) and other heterocycles can provide balanced rigidity and polarity while enabling efficient synthetic assembly [64].
The choice of linker class should be guided by the structural characteristics of both the target protein and E3 ligase, with particular attention to the spatial arrangement required for productive ubiquitin transfer [75].
Key Optimization Parameters
Length Optimization
Systematic variation of linker length represents a fundamental optimization strategy, as even small changes can dramatically alter degradation potency [75]. The optimal length is typically determined empirically through the synthesis of analogous series with incremental increases in chain length:
Table 2: Experimental Length Optimization Guidelines
| Linker Type | Optimal Length Range | Structural Considerations | Representative Examples |
|---|---|---|---|
| Alkyl Chains | 8-16 atoms | Sufficient length to span protein surfaces without excessive flexibility | BET degraders show optimal activity with 11-atom alkyl chains [64] |
| PEG Linkers | 4-12 ethylene glycol units | Balance between solubility and conformational entropy | VHL-recruiting PROTACs often utilize PEG6-8 linkers [64] |
| Aromatic-Aliphatic Hybrids | 10-14 atoms | Rigid segments maintain orientation while aliphatic spacers provide adaptability | CRBN-based PROTACs with alternating rigid-flexible motifs show enhanced nuclear localization [64] |
Flexibility and Rigidity Balance
The optimal balance between linker flexibility and rigidity depends on the structural compatibility between the POI and E3 ligase [75]. Highly flexible linkers allow adaptation to protein surface contours but may incur entropic penalties upon ternary complex formation. Conversely, rigid linkers enforce specific orientations but may prevent productive complex formation if the geometry is suboptimal [64]. Strategic incorporation of conformational constraints through ring structures or stereochemical control can improve degradation efficiency by reducing the entropic cost of ternary complex formation [75].
Attachment Point Optimization
The specific sites where linkers connect to warhead and anchor ligands significantly influence PROTAC efficacy [75]. For instance, in VHL ligands, linker attachment to the hydroxyproline moiety requires careful consideration of stereochemistry and bonding geometry to maintain high-affinity ligase binding [64]. Similarly, for CRBN ligands, attachment points on the glutarimide ring must preserve critical hydrogen-bonding interactions [45]. Molecular modeling and structural analysis of binary complexes can inform optimal attachment vector selection before synthetic investment.
Experimental Methodologies for Linker Evaluation
In Vitro Assessment Protocols
Ternary Complex Formation Assays
Surface Plasmon Resonance (SPR)
- Protocol: Immobilize either POI or E3 ligase on sensor chip surface; inject PROTAC analogs followed by the binding partner; measure response units reflecting ternary complex formation [64].
- Data Interpretation: Enhanced binding responses (cooperativity) indicate productive ternary complex formation; calculate α-values to quantify cooperative effects.
- Application: Direct comparison of linker variants' ability to facilitate ternary complex formation independent of cellular permeability.
Cellular Thermal Shift Assay (CETSA)
- Protocol: Treat cells with PROTAC analogs; heat cell lysates to denature proteins; quantify remaining soluble POI and E3 ligase via immunoblotting [74].
- Data Interpretation: Thermal stabilization indicates intracellular target engagement and ternary complex formation.
- Advantage: Assesses PROTAC activity in physiological cellular environment while accounting for membrane permeability.
Degradation Efficiency Profiling
Western Blot Analysis
- Protocol: Treat cells with serial dilutions of PROTAC analogs for 4-24 hours; lyse cells; separate proteins via SDS-PAGE; immunoblot for POI and loading controls [74].
- Quantification: Determine DC50 (half-maximal degradation concentration) and Dmax (maximal degradation) values through densitometric analysis.
- Time-Course: Include multiple time points to assess kinetics of degradation and potential rebound effects.
Cellular Viability and Proliferation Assays
- Protocol: Treat susceptible cell lines with PROTAC analogs for 72-96 hours; measure viability via ATP content (CellTiter-Glo) or metabolic activity (MTT) [74].
- Correlation: Compare IC50 values with DC50 values to establish relationship between degradation and functional effects.
Structural Characterization Techniques
X-ray Crystallography of Ternary Complexes
Protocol Overview:
- Purify POI and E3 ligase to homogeneity
- Form ternary complex with PROTAC in vitro
- Crystallize complex using vapor diffusion methods
- Collect diffraction data and solve structure
Application: The seminal structure of BRD4-MZ1-VHL ternary complex revealed how linker composition influences cooperative interactions between proteins, explaining the selectivity of MZ1 for BRD4 over BRD2/3 [64]. This structural insight guides rational linker design by identifying optimal spatial arrangements.
Computational Modeling and Molecular Dynamics
Protocol:
- Generate homology models of POI and E3 ligase if crystal structures unavailable
- Dock warhead and anchor fragments to respective binding sites
- Model linker conformations using flexible sampling methods
- Perform molecular dynamics simulations to assess ternary complex stability
Application: Predict optimal linker lengths and identify potential protein-protein interfaces that stabilize ternary complexes [58].
Visualization of PROTAC Mechanism and Linker Optimization
PROTAC Mechanism and ATP-Dependent Degradation Pathway
Diagram 1: PROTAC Mechanism and ATP-Dependent Degradation. This visualization illustrates the complete PROTAC mechanism, from ternary complex formation through ATP-dependent ubiquitination to final proteasomal degradation, highlighting the historical connection to Hershko and Ciechanover's discovery of the ubiquitin-proteasome system.
Linker Optimization Experimental Workflow
Diagram 2: Linker Optimization Workflow. This diagram outlines the iterative process of linker optimization, from initial PROTAC synthesis through ternary complex assessment to degradation profiling and structure-activity relationship analysis.
Research Reagent Solutions for Linker Optimization
Table 3: Essential Research Reagents for PROTAC Linker Optimization
| Reagent Category | Specific Examples | Function in Linker Optimization | Commercial Sources |
|---|---|---|---|
| Functionalized PEG Linkers | NH₂-PEG₄-OH, Boc-NH-PEG₃-Tosylate, NH₂-PEG₃-OH | Provide variable length spacers with improved solubility; enable modular assembly [76] | Biopharma PEG, Sigma-Aldrich |
| Click Chemistry Reagents | Boc-NH-PEG₁₁-N₃, DBCO-PEG4-NHS | Enable efficient, modular coupling of warhead and anchor modules; facilitate library synthesis [64] | Click Chemistry Tools, Thermo Fisher |
| E3 Ligase Ligands | VHL ligands (VH032), CRBN ligands (Pomalidomide), MDM2 ligands (Nutlin) | Serve as anchor components for ternary complex formation; determine compatibility with linker motifs [64] | MedChemExpress, Cayman Chemical |
| Characterized Warheads | BET inhibitors (JQ1), kinase inhibitors, AR/AR antagonists | Provide target protein-binding modules for degradation studies; establish structure-degradation relationships [74] | Tocris Bioscience, Selleck Chemicals |
| Proteasome Inhibitors | Bortezomib, MG132, Carfilzomib | Confirm proteasome-dependent degradation mechanism; validate on-target PROTAC activity [74] | Cell Signaling Technology, MilliporeSigma |
Emerging Trends and Future Perspectives
The field of linker optimization is rapidly evolving beyond traditional alkyl and PEG-based spacers toward more sophisticated design strategies [64]. Several emerging trends show particular promise:
Functionalized and Cleavable Linkers
Recent advances include the development of PROTAC prodrugs (pro-PROTACs) where linkers incorporate labile groups that can be selectively removed under specific physiological or experimental conditions [45]. Photocaged PROTACs (opto-PROTACs) utilize photolabile groups such as 4,5-dimethoxy-2-nitrobenzyl (DMNB) moieties installed on critical functional groups of E3 ligase ligands, enabling spatiotemporal control of PROTAC activation through light exposure [45]. This strategy addresses potential on-target toxicity by restricting degradation activity to specific tissues or timepoints.
AI-Guided Linker Design
Artificial intelligence is revolutionizing linker optimization through predictive modeling and generative design [58]. Machine learning approaches such as AIMLinker employ deep encoder-decoder neural networks to generate novel linker moieties by extracting structural fragment information and filtering non-druggable candidates [45]. Similarly, ShapeLinker implements fragment-linking using reinforcement learning on SMILES generators to design PROTAC linkers with optimized properties [45]. These computational methods address the traditional trial-and-error approach that has hampered rapid PROTAC development.
Expanding the E3 Ligase Toolkit
While current PROTAC designs predominantly utilize CRBN and VHL ligases, linker optimization strategies must adapt as the E3 ligase toolkit expands [64] [58]. Emerging E3 ligases such as KEAP1, DCAF16, RNF114, and FEM1B present new opportunities for tissue-specific degradation but will require customized linker optimization approaches tailored to their unique structural characteristics [58]. The limited ligand availability for most of the 600+ human E3 ligases represents both a challenge and opportunity for future PROTAC development [58].
Linker optimization represents a critical determinant of PROTAC efficacy, bridging the historical discoveries of ATP-dependent proteolysis by Hershko, Ciechanover, and Rose with modern targeted protein degradation therapeutics [1] [3]. The multifaceted role of linkers in governing ternary complex formation, degradation efficiency, selectivity, and drug-like properties necessitates systematic optimization approaches combining empirical screening and rational design. As PROTAC technology advances toward clinical application, with compounds such as ARV-110 and ARV-471 demonstrating promising results in trials, linker optimization strategies will continue to evolve in sophistication [45] [74]. Emerging technologies including AI-guided design, functionalized cleavable linkers, and expanded E3 ligase utilization promise to address current challenges in PROTAC development, potentially unlocking new therapeutic opportunities for previously undruggable targets [58] [75]. The continued integration of structural biology, computational modeling, and synthetic chemistry will further advance rational linker design, fulfilling the therapeutic potential inherent in the fundamental discoveries of the ubiquitin-proteasome system.
Navigating Cellular Resistance to Protein Degradation Therapies
The pioneering work of Avram Hershko, Aaron Ciechanover, and Irwin Rose on ATP-dependent proteolysis in the late 1970s and 1980s unveiled what seemed paradoxical at the time: cellular protein degradation requires metabolic energy [1] [3] [10]. Their discovery of the ubiquitin-proteasome system (UPS) revealed an elegant enzymatic cascade where target proteins are marked for destruction by covalent attachment of ubiquitin molecules, culminating in their recognition and degradation by the 26S proteasome [1] [3]. This foundational knowledge has catalyzed a revolutionary approach to therapeutics—targeted protein degradation (TPD), most notably through proteolysis-targeting chimeras (PROTACs) [31] [77]. However, as with any transformative therapy, the emergence of resistance mechanisms threatens their long-term efficacy. This guide examines the cellular resistance landscape against protein degradation therapies, offering strategic navigation for researchers and drug development professionals.
The Ubiquitin-Proteasome System: From Fundamental Mechanism to Therapeutic Exploitation
Historical Foundation and Core Mechanism
The ubiquitin-proteasome system operates through a precise enzymatic cascade. The E1 ubiquitin-activating enzyme activates ubiquitin in an ATP-dependent manner. The E2 ubiquitin-conjugating enzyme then carries the activated ubiquitin. Finally, an E3 ubiquitin ligase facilitates the transfer of ubiquitin to specific substrate proteins [3] [15]. Polyubiquitinated substrates are subsequently recognized and degraded by the 26S proteasome into small peptides [78] [15]. The specificity of this system is largely determined by the approximately 600 E3 ligases in the human genome, which recognize distinct sets of substrates [78] [77].
PROTACs: Hijacking the Natural Destruction Machinery
PROTACs are heterobifunctional molecules that consist of three elements: a ligand that binds a protein of interest (POI), a ligand that recruits an E3 ubiquitin ligase, and a linker connecting these two moieties [31] [79]. By inducing proximity between an E3 ligase and a POI, PROTACs facilitate ubiquitination and subsequent degradation of the target [31] [15]. Unlike traditional inhibitors that merely block protein function, PROTACs catalytically eliminate the entire protein, potentially addressing challenges posed by non-enzymatic functions, scaffold properties, and adaptive resistance mechanisms [31] [77].
Table 1: Key E3 Ligases Utilized in PROTAC Technology
| E3 Ligase | Native Biological Function | Common Ligands Used in PROTACs | Notable Features |
|---|---|---|---|
| CRBN (Cereblon) | Substrate receptor for CRL4 E3 ubiquitin ligase complex | Thalidomide, Lenalidomide, Pomalidomide derivatives | Binds immunomodulatory drugs (IMiDs); extensive clinical validation |
| VHL (Von Hippel-Lindau) | Recognition component for hypoxia-inducible factor (HIF-1α) | Hydroxyproline-containing peptides and mimetics | Well-defined binding pocket; high specificity designs |
| MDM2 | Negative regulator of p53 tumor suppressor | Nutlin-3 derivatives | Potential for p53 stabilization in wild-type tumors |
| IAP (Inhibitor of Apoptosis Protein) | Regulation of apoptosis and inflammatory signaling | LCL-161, MV1, Bestatin derivatives | Can induce auto-ubiquitination and degradation of IAPs themselves |
Mechanisms of Resistance to Protein Degradation Therapies
Target-Related Resistance Mechanisms
Target Mutations: Point mutations in the POI can disrupt PROTAC binding while preserving protein function. For instance, mutations in the binding domain of Bruton's tyrosine kinase (BTK) have been observed following treatment with BTK-directed degraders, analogous to resistance seen with traditional inhibitors but requiring different mutational profiles [79]. These mutations may affect either the warhead binding site or regions critical for ternary complex formation.
Target Overexpression: Cancer cells may amplify gene copy numbers or increase transcription/translation of oncoproteins targeted by PROTACs, effectively overwhelming the degradation capacity [77]. This mechanism is particularly relevant for proteins with short half-lives that can rapidly reappear after degrader removal.
UPS Component Alterations
E3 Ligase Downregulation: Maladaptive changes in E3 ligase expression represent a common resistance mechanism. Chronic engagement of specific E3 ligases by PROTACs can lead to their downregulation through auto-ubiquitination or transcriptional repression, limiting ternary complex formation [77]. For example, CRBN levels have been shown to decrease in response to continuous CRBN-recruiting PROTAC exposure.
Proteasome Capacity Limitations: The catalytic nature of PROTACs depends on available proteasome capacity. Resistance can emerge through reduced proteasome subunit expression or impaired proteasome function, particularly in cells stressed by the accumulation of misfolded proteins or under oxidative stress conditions [15].
Compensatory Pathway Activation and Cellular Adaptation
Bypass Signaling Pathways: Tumor cells activate alternative signaling pathways to compensate for degraded oncoproteins, maintaining proliferative and survival signals despite effective target degradation [77]. This adaptive resistance mirrors mechanisms seen with targeted kinase inhibitors but may involve different pathway preferences.
Efflux Pump Upregulation: The relatively large molecular weight and hydrophobic nature of many PROTACs make them susceptible to recognition by multidrug resistance transporters, particularly P-glycoprotein (P-gp) [31] [79]. Upregulation of these efflux pumps reduces intracellular PROTAC concentrations below therapeutic thresholds.
Table 2: Documented Resistance Mechanisms to Protein Degradation Therapies
| Resistance Category | Specific Mechanism | Experimental Evidence | Potential Workarounds |
|---|---|---|---|
| Target-Related | Point mutations in warhead-binding domain | BTK mutations in lymphoma models following BTK degrader treatment | Develop PROTACs targeting alternative protein domains |
| Target-Related | Target protein overexpression | Gene amplification in oncogenic transcription factors | Higher degrader doses or combination with transcriptional inhibitors |
| UPS Component | E3 ligase downregulation | CRBN depletion in myeloma cells after IMiD-based PROTAC exposure | Rotate E3 ligases or use dual-PROTAC approaches |
| UPS Component | Proteasome insufficiency | Reduced proteasome subunit expression in solid tumors | Episodic dosing schedules to allow proteasome recovery |
| Cellular Adaptation | Upregulation of efflux pumps | P-gp-mediated export of PROTACs in multidrug-resistant cell lines | Develop less lipophilic PROTACs or use pump inhibitors |
| Cellular Adaptation | Activation of compensatory pathways | Alternative kinase signaling in RTK-degraded cells | Rational combination therapies targeting escape pathways |
Methodologies for Investigating Resistance Mechanisms
Protocol: Continuous Dose Escalation for Resistance Selection
Principle: Mimicking clinical resistance development through prolonged exposure to increasing PROTAC concentrations enables identification of adaptive cellular mechanisms [77].
Procedure:
- Initiate culture with low-dose PROTAC (typically 10-50 nM) corresponding to ~IC20 concentration
- Maintain cells in logarithmic growth phase with medium refreshment every 48-72 hours
- Gradually increase PROTAC concentration by 1.5-2 fold increments upon observed recovery of proliferation
- Continue escalation over 4-6 months until reaching micromolar resistance levels
- Clone isolated populations for mechanistic studies
- Validate resistance through comparative viability assays and Western blot analysis of target protein levels
Protocol: Genome-Wide CRISPR Screening for Resistance Modifiers
Principle: Systematic identification of genetic determinants of PROTAC sensitivity through loss-of-function screening [77].
Procedure:
- Transduce target cells with genome-wide CRISPR knockout library (e.g., Brunello or GeCKO v2)
- Select transduced cells with puromycin for 5-7 days
- Split cells into PROTAC-treated and vehicle-control groups at high coverage (>500x)
- Maintain cultures for 14-21 population doublings under PROTAC selection pressure
- Harvest genomic DNA and amplify integrated sgRNA sequences by PCR
- Sequence amplicons by next-generation sequencing to quantify sgRNA abundance
- Identify enriched/depleted sgRNAs using specialized algorithms (MAGeCK, BAGEL)
- Validate candidate genes through individual sgRNA knockout studies
Protocol: Ternary Complex Stability Assessment
Principle: Evaluate resistance mechanisms related to impaired protein-protein interactions in the POI-PROTAC-E3 complex [31].
Procedure:
- Express and purify recombinant POI and E3 ligase components
- Develop fluorescence polarization (FP) or surface plasmon resonance (SPR) assays measuring ternary complex formation
- Titrate PROTAC concentrations while maintaining constant POI and E3 concentrations
- Determine binding affinity (Kd) and cooperativity factors (α) for ternary complex
- Compare complex stability between wild-type and resistant cell-derived protein variants
- Correlate biophysical parameters with cellular degradation efficacy (DC50, Dmax)
Strategic Approaches to Overcome Resistance
E3 Ligase Diversification and Engineering
The current heavy reliance on CRBN and VHL ligands in PROTAC design creates vulnerability to resistance through E3 downregulation [77] [15]. Expanding the E3 ligase repertoire represents a critical strategy:
- Exploration of alternative E3s: IAP, MDM2, DCAF, and RNF family E3 ligases offer complementary degradation capacities
- Tissue-specific E3 utilization: Leveraging E3s with restricted expression patterns may enhance therapeutic index
- Dual-PROTACs: Simultaneously engaging two E3 ligases may reduce the likelihood of resistance through E3 loss
Structure-Guided PROTAC Optimization
Rational design based on ternary complex structural biology can address specific resistance mechanisms:
- Warhead optimization: Developing ligands targeting alternative binding sites less prone to resistance mutations
- Linker engineering: Adjusting length, composition, and rigidity to optimize ternary complex geometry despite target variations
- Bivalent degraders: Targeting multiple domains on the same protein to require simultaneous mutations for resistance
Combination Therapy Strategies
Integrating PROTACs with complementary therapeutic modalities provides multidimensional attack:
- Traditional inhibitors: Concurrently blocking the target's enzymatic function while degrading it
- Epigenetic modulators: Preventing compensatory pathway upregulation at the transcriptional level
- Efflux pump inhibitors: Co-administration to maintain intracellular PROTAC concentrations
Diagram: Strategic Framework for Addressing PROTAC Resistance
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Investigating Resistance Mechanisms
| Reagent Category | Specific Examples | Research Application | Commercial Sources |
|---|---|---|---|
| E3 Ligase Ligands | Pomalidomide (CRBN), VHL ligand VH032, MDM2 ligand Nutlin-3 | PROTAC construction and E3 specificity studies | MedChemExpress, Cayman Chemical, Tocris |
| Proteasome Inhibitors | Bortezomib, MG132, Carfilzomib | Validation of ubiquitin-proteasome system dependence | Selleck Chemicals, Sigma-Aldrich |
| Ubiquitination Assays | Ubiquitin aldehyde, TUBE reagents (Tandem Ubiquitin Binding Entities) | Monitoring substrate ubiquitination status | LifeSensors, Enzo Life Sciences |
| CRISPR Libraries | Brunello whole-genome knockout library, custom E3/TPD-focused libraries | Genome-wide resistance modifier screening | Addgene, Dharmacon |
| Protein Degradation Assays | HTRF ubiquitin assay kits, proteasome activity probes | Quantifying degradation efficiency and kinetics | Cisbio Bioassays, Boston Biochem |
| Ternary Complex Assays | Time-resolved FRET kits, SPR chips with immobilized E3/POI | Evaluating ternary complex formation and stability | Cisbio Bioassays, Cytiva |
Future Directions and Clinical Outlook
The field of targeted protein degradation continues to evolve rapidly beyond traditional PROTACs. Emerging technologies including lysosome-targeting chimeras (LYTACs), antibody-based PROTACs (AbTACs), and molecular glues offer alternative degradation routes that may bypass established resistance mechanisms [15]. The clinical success of ARV-471 (for breast cancer) and ARV-110 (for prostate cancer) in advanced trials demonstrates the transformative potential of protein degradation therapies, while simultaneously highlighting the inevitability of resistance development [31] [79]. Future research directions should prioritize proactive resistance management through rational degrader design, predictive biomarkers for resistance susceptibility, and adaptive treatment strategies that anticipate and counter resistance evolution.
The journey from the fundamental discovery of ATP-dependent proteolysis to modern targeted degradation therapeutics exemplifies how deep biological understanding enables therapeutic innovation. By applying this same rigorous mechanistic approach to understanding and addressing resistance, the promise of protein degradation therapies can be fully realized across a broad spectrum of diseases.
Ubiquitin System in Context: Validation and Comparison with Other Proteolytic Pathways
- Protein degradation pathways: Introduces cellular protein degradation mechanisms and historical context.
- Ubiquitin-proteasome system: Details UPS mechanisms, components, and experimental analysis.
- Lysosomal degradation pathway: Explores lysosomal mechanisms, pathways, and research methods.
- Comparative analysis: Presents side-by-side comparison of key characteristics and physiological roles.
- Therapeutic applications: Examines targeted protein degradation technologies and research tools.
Comparative Analysis: Ubiquitin-Proteasome vs. Lysosomal Degradation
Protein homeostasis (proteostasis) represents a fundamental biological process through which cells maintain optimal concentrations, conformations, and subcellular localization of proteins. Eukaryotic cells have evolved two primary protein degradation systems that operate in a complementary manner to regulate protein turnover: the ubiquitin-proteasome system (UPS) and the lysosomal degradation pathway. The discovery of ATP-dependent proteolysis by Avram Hershko, Aaron Ciechanover, and Irwin Rose in the late 1970s and early 1980s revolutionized our understanding of intracellular protein degradation [1]. Their pioneering research identified a heat-stable polypeptide initially called ATP-dependent proteolysis factor 1 (APF-1), later recognized as ubiquitin, which forms covalent conjugates with target proteins in an ATP-requiring reaction [80] [14] [81]. This breakthrough revealed that intracellular proteolysis is far more complex and regulated than previously imagined, establishing the conceptual foundation for understanding both major degradation pathways.
The biological significance of these degradation systems extends beyond simple protein disposal. They collectively regulate diverse cellular processes including cell cycle progression, stress response, apoptosis, autophagy, signal transduction, and DNA repair [15] [82]. The proper functioning of both pathways is crucial for neuronal health, with dysfunction implicated in various neurodegenerative diseases including Alzheimer's disease (AD), Parkinson's disease, and other tauopathies [83] [84] [85]. Understanding the distinct mechanisms, substrates, and regulatory aspects of the UPS and lysosomal pathways provides critical insights for developing novel therapeutic strategies for cancer, neurodegenerative disorders, and other proteinopathies.
The Ubiquitin-Proteasome System (UPS)
Mechanism and Components
The ubiquitin-proteasome system represents the primary mechanism for targeted degradation of intracellular proteins in eukaryotic cells. This sophisticated system employs a three-enzyme cascade to mark proteins for destruction: ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3) [15] [84] [82]. The process begins with E1 activating ubiquitin in an ATP-dependent manner, followed by transfer to E2, and finally E3 facilitates the covalent attachment of ubiquitin to lysine residues on target proteins [15] [84]. Repeated cycles lead to the formation of polyubiquitin chains, with specific chain types determining the fate of modified proteins [15].
The 26S proteasome is the macromolecular protease complex responsible for degrading ubiquitinated proteins. It consists of two primary subcomplexes: the 20S core particle (CP) that contains the proteolytic active sites, and the 19S regulatory particle (RP) that recognizes ubiquitinated substrates, unfolds them, and translocates them into the catalytic chamber [84]. The 20S CP is composed of 28 subunits arranged in four stacked heptameric rings (α7β7β7α7), with three different β-subunits (β1, β2, and β5) possessing caspase-like, trypsin-like, and chymotrypsin-like proteolytic activities, respectively [84]. The 19S RP contains six different ATPases that facilitate substrate unfolding and multiple ubiquitin receptors (Rpn1, Rpn10, and Rpn13) that recognize polyubiquitin chains [84].
Table 1: Key Components of the Ubiquitin-Proteasome System
| Component | Structure | Function |
|---|---|---|
| Ubiquitin | 76-amino acid protein (8.5 kDa) | Tags proteins for degradation via covalent attachment |
| E1 Enzyme | Single component in humans | Activates ubiquitin in ATP-dependent manner |
| E2 Enzyme | ~37 varieties in humans | Accepts ubiquitin from E1 and transfers to E3 |
| E3 Ligase | ~1000 varieties in humans | Confers substrate specificity for ubiquitination |
| 26S Proteasome | 2.5 MDa complex | Recognizes and degrades ubiquitinated proteins |
| 20S Core Particle | 28 subunits in 4 rings | Catalytic core with multiple proteolytic activities |
| 19S Regulatory Particle | 19 subunits in base and lid | Recognizes ubiquitinated proteins, unfolds substrates |
Experimental Analysis of UPS
The foundational experiments elucidating the UPS mechanism stem from the work of Hershko, Ciechanover, and Rose using reticulocyte lysates as a model system [1] [80]. Their experimental approach involved fractionating reticulocyte lysates by DEAE-cellulose chromatography into two essential components: Fraction I (containing APF-1/ubiquitin) and Fraction II (containing the proteolytic machinery) [80] [14]. The key methodology involved:
- ATP-dependent conjugate formation: Incubating ¹²⁵I-labeled APF-1 with Fraction II in the presence of ATP and Mg²⁺ resulted in covalent conjugates of APF-1 with endogenous proteins, demonstrated by SDS-PAGE autoradiography [80].
- Characterization of conjugates: The APF-1-protein conjugates were stable to SDS, heat, acid, alkali, and reduction, indicating covalent isopeptide bonds between APF-1 and substrate proteins [80] [81].
- Functional correlation: Conjugate formation required identical conditions (ATP, Mg²⁺) as ATP-dependent proteolysis, and both processes were inhibited by N-ethylmaleimide, suggesting conjugates were intermediates in proteolysis [80].
- Polyubiquitination: Analysis of conjugates with protein substrates like lysozyme revealed multiple bands on SDS-PAGE, representing proteins with increasing numbers of APF-1 molecules attached [81].
The following diagram illustrates the experimental workflow used in these foundational studies:
The Lysosomal Degradation Pathway
Mechanism and Pathways
The lysosomal degradation pathway represents the cell's primary system for degrading extracellular proteins, cell-surface receptors, and intracellular organelles. Lysosomes are acidic, membrane-bound organelles containing numerous hydrolases including lipases, phosphatases, glycosidases, peptidases, and nucleosidases that function optimally at low pH [82]. Unlike the UPS, which primarily degrades soluble intracellular proteins, the lysosomal system processes materials through multiple distinct mechanisms:
- Receptor-mediated endocytosis: Specific ligands bind cell-surface receptors, leading to clathrin-coated pit formation, internalization, and trafficking to lysosomes [82]. The acidic pH of endosomes triggers ligand-receptor dissociation, directing receptors for recycling and ligands for degradation.
- Phagocytosis: Specialized cells (e.g., macrophages) engulf large particles (>0.5 μm) such as microorganisms or cellular debris, forming phagosomes that fuse with lysosomes for content degradation [15] [82].
- Autophagy: A conserved process for degrading cytoplasmic components, including organelles and long-lived proteins, through formation of double-membrane autophagosomes that deliver contents to lysosomes [15] [83]. Autophagy includes three subtypes: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) [85].
The autophagy-lysosomal pathway (ALP) has received particular attention for its role in quality control and neurodegeneration. During macroautophagy, cytoplasmic components are sequestered within phagophores that mature into autophagosomes, which subsequently fuse with lysosomes to form autolysosomes where degradation occurs [83] [85]. Recent research has revealed that autophagy can be highly selective through receptors that recognize ubiquitinated cargo, blurring the distinction between UPS and lysosomal degradation [83] [84].
Experimental Analysis of Lysosomal Degradation
Research methodologies for studying lysosomal degradation pathways employ various biochemical, cell biological, and imaging approaches:
- Lysosome isolation and characterization: Differential centrifugation allows isolation of lysosome-enriched fractions from tissue homogenates, with characterization based on acid phosphatase activity and presence of marker proteins (LAMP1, LAMP2).
- Tracking endocytic pathways: Fluorescent membrane stains, pH-sensitive dyes (pHrodo), and fluorescent dextran conjugates enable visualization and quantification of endocytic processes [82]. pHrodo indicators exhibit increased fluorescence in acidic environments, allowing monitoring of endosomal maturation.
- Autophagy flux assays: Monitoring LC3-I to LC3-II conversion by immunoblotting and tracking p62/SQSTM1 degradation provide quantitative measures of autophagic activity [82]. Fluorescent protein-tagged LC3 allows visualization of autophagosome formation by microscopy.
- Phagocytosis assays: Fluorescent BioParticles (bacteria or yeast labeled with pH-sensitive dyes) enable quantification of phagocytic uptake and lysosomal degradation in immune cells [82].
The following diagram illustrates the primary lysosomal degradation pathways:
Comparative Analysis: Key Distinctions and Interdependence
Characteristic Features
The ubiquitin-proteasome and lysosomal degradation pathways represent complementary systems with distinct characteristics, substrates, and cellular functions. The following table provides a comprehensive comparison of their key features:
Table 2: Comparative Analysis of Ubiquitin-Proteasome and Lysosomal Degradation Pathways
| Characteristic | Ubiquitin-Proteasome System (UPS) | Lysosomal Degradation Pathway |
|---|---|---|
| Primary substrates | Short-lived intracellular proteins, soluble misfolded proteins [15] [85] | Long-lived proteins, extracellular proteins, protein aggregates, organelles [15] [85] |
| Degradation mechanism | 26S proteasome complex with multiple proteolytic activities [84] | Acidic hydrolases in lysosomal compartment [82] |
| Targeting signal | K48-linked polyubiquitin chains (primarily) [15] [84] | K63-linked ubiquitin chains, receptor-mediated targeting [15] [84] |
| Energy requirement | ATP-dependent (ubiquitination and proteasomal degradation) [80] | ATP-dependent (acidification, vesicle trafficking) [15] |
| Degradation rate | Rapid (minutes to hours) | Slower (hours to days) |
| Selectivity | High (E3 ligase-specific) [15] | Variable (non-selective to highly selective) |
| Cellular location | Cytosol, nucleus [15] | Endosomal-lysosomal compartment |
| Key components | E1/E2/E3 enzymes, 26S proteasome [84] [82] | Lysosomal hydrolases, LAMP proteins, autophagy-related proteins |
| Inhibitors | MG132, bortezomib, lactacystin | Chloroquine, bafilomycin A1, leupeptin |
| Primary functions | Protein quality control, cell cycle regulation, signal transduction [82] | Nutrient sensing, organelle turnover, pathogen defense [15] [82] |
Physiological Roles and Interdependence
Both degradation pathways play crucial roles in protein quality control and cellular homeostasis, with their relative importance varying by cell type and physiological context. In neurons, both systems are essential for maintaining neuronal health, and their dysfunction is implicated in Alzheimer's disease pathogenesis [83] [84]. The UPS is particularly important for degrading soluble, misfolded proteins and regulating key neuronal proteins including tau, while the lysosomal system clears protein aggregates and damaged organelles [85].
Despite their distinct mechanisms, the UPS and lysosomal pathways exhibit significant interdependence and cross-talk:
- Ubiquitin as a common signal: Both systems recognize ubiquitinated proteins, though they typically prefer different ubiquitin chain linkages (K48 for UPS, K63 for lysosomal degradation) [84].
- Compensatory activation: Inhibition of one system often upregulates the other; proteasome inhibition induces autophagy activation, while autophagy impairment can increase ubiquitinated protein accumulation [83] [85].
- Shared regulatory components: Proteins such as p62/SQSTM1 and HDAC6 function as shuttle factors that can direct ubiquitinated proteins to both proteasomal and autophagic degradation [85].
- Collaborative substrate processing: Some substrates, including tau, can be degraded by both systems depending on their aggregation state—soluble tau primarily by UPS, while aggregated tau is cleared by autophagy [85].
The following diagram illustrates the collaborative relationship between these degradation systems:
Therapeutic Applications and Research Tools
Targeted Protein Degradation Technologies
The understanding of cellular degradation mechanisms has inspired novel therapeutic approaches, particularly in targeted protein degradation (TPD). These technologies harness the cell's natural degradation machinery to eliminate disease-causing proteins:
- PROTACs (PROteolysis TArgeting Chimeras): Heterobifunctional molecules with ligands for both a target protein and an E3 ubiquitin ligase, enabling targeted ubiquitination and proteasomal degradation [15] [82]. PROTACs offer advantages over traditional inhibitors, including catalytic activity, ability to target "undruggable" proteins, and potential reduced drug resistance [15].
- Molecular glues: Monovalent small molecules that induce or stabilize interactions between E3 ligases and target proteins, leading to ubiquitination and degradation [15]. Examples include thalidomide, lenalidomide, and pomalidomide, which recruit neosubstrates to the CRL4CRBN E3 ligase [15].
- LYTACs (LYsosome-TArgeting Chimeras): Bispecific molecules that bind both extracellular proteins and cell-surface lysosome-targeting receptors, directing extracellular and membrane proteins for lysosomal degradation [15] [82].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Protein Degradation Studies
| Reagent Category | Specific Examples | Research Applications |
|---|---|---|
| UPS Inhibitors | MG132, bortezomib, lactacystin | Proteasome function studies, accumulation of ubiquitinated proteins |
| Lysosomal Inhibitors | Chloroquine, bafilomycin A1, leupeptin | Lysosomal function studies, autophagy flux measurements |
| Ubiquitination Tools | Ubiquitin-activating enzyme (E1) inhibitors, TAK-243 | Investigation of ubiquitination cascade |
| Autophagy Modulators | Rapamycin (inducer), 3-MA (inhibitor) | Regulation of autophagic activity |
| Fluorescent Reporters | pHrodo dyes, LC3-GFP constructs, ubiquitin probes | Visualization and tracking of degradation pathways |
| Metabolic Labeling | L-azidohomoalanine (AHA), Click-iT reagents | Protein synthesis and degradation measurements |
| Antibodies | Anti-ubiquitin, anti-LC3, anti-p62, anti-K48/K63 ubiquitin | Detection of degradation intermediates and pathway activity |
These research tools enable scientists to dissect the complexities of protein degradation pathways and develop novel therapeutic strategies for diseases characterized by proteostasis dysfunction, including neurodegenerative disorders, cancer, and infectious diseases [82]. The growing interest in targeted protein degradation technologies highlights the translational potential of understanding these fundamental biological processes.
The comparative analysis of the ubiquitin-proteasome and lysosomal degradation pathways reveals two evolutionarily conserved systems with distinct yet complementary roles in cellular homeostasis. The UPS provides rapid, selective degradation of short-lived and soluble misfolded proteins through an ATP-dependent mechanism centered on the 26S proteasome. In contrast, the lysosomal system offers broader degradation capacity for extracellular proteins, protein aggregates, and organelles through multiple mechanisms including endocytosis, phagocytosis, and autophagy. The pioneering work of Hershko, Ciechanover, and Rose on ATP-dependent proteolysis and ubiquitination laid the foundation for understanding these essential pathways, earning them the Nobel Prize in Chemistry in 2004 [1].
The interdependence between these systems, particularly through shared recognition of ubiquitinated substrates and compensatory activation, highlights the complexity of cellular proteostasis regulation. In neurodegenerative diseases like Alzheimer's, both systems contribute to the clearance of pathogenic proteins such as tau, with the UPS predominantly handling soluble forms and autophagy managing aggregated species [85]. The emerging field of targeted protein degradation harnesses this mechanistic understanding to develop novel therapeutic modalities like PROTACs and LYTACs that can address previously "undruggable" targets [15] [82]. Continuing research on these degradation pathways promises not only fundamental biological insights but also innovative treatments for human diseases characterized by proteostasis dysfunction.
The seminal work of Avram Hershko, Aaron Ciechanover, and Irwin Rose, awarded the Nobel Prize in Chemistry in 2004, established the paradigm of ATP-dependent ubiquitin-mediated proteolysis in eukaryotes [32] [13]. Their discovery revealed that targeted protein degradation is not a passive process but an energy-dependent, highly regulated circuit essential for cellular homeostasis. This foundational principle extends beyond the ubiquitin-proteasome system. In prokaryotes, mitochondria, and chloroplasts, a parallel paradigm operates through sophisticated proteolytic machines including Lon, FtsH, and Clp proteases [86]. These enzymes are central to protein quality control, regulatory circuit regulation, and stress responses, functioning as self-compartmentalized proteases where the proteolytic active sites are sequestered within internal chambers, and access is governed by ATP-fueled chaperone modules [87] [86]. This review provides an in-depth analysis of the structure, mechanism, and function of these critical proteolytic systems.
Protease System Architectures and Mechanisms
The Lon Protease Family
Domain Organization and Subfamilies: Lon proteases are conserved from bacteria to eukaryotic organelles and are characterized by the fusion of ATPase and proteolytic activities into a single polypeptide chain [87] [88]. They are categorized into subfamilies based on domain architecture:
- LonA: Found in bacteria and eukaryotic organelles, LonA possesses a large N-terminal domain (N-domain), an AAA+ ATPase domain (A-domain), and a proteolytic domain (P-domain) [87] [89]. The N-domain is involved in substrate recognition and binding [87].
- LonB: Present in archaea, LonB proteases lack the N-terminal domain but contain a membrane-anchoring domain within their AAA+ module, tethering them to the cytoplasmic side of the membrane [87] [89].
- LonC: A more recently identified subfamily, primarily in bacteria, which lacks the N-terminal domain but features a large helical insert (HHE/LID) in its AAA+ module and has degenerated ATPase motifs that may abolish nucleotide hydrolysis [89].
Oligomeric State and Catalytic Mechanism: Lon proteases function as homohexameric assemblies, forming a ring-shaped structure with a central chamber [87] [89]. The proteolytic active site is unusual, employing a serine-lysine catalytic dyad (Ser-Lys) instead of the classic serine protease triad [87] [89]. The consensus sequences around the catalytic residues differ between subfamilies, contributing to their functional specialization [89].
ATP binding and hydrolysis by the AAA+ module drive conformational changes that power the unfolding of target proteins and their translocation through a narrow axial channel into the internal proteolytic chamber, where processive degradation occurs [87].
The FtsH Protease Family
Domain Organization and Membrane Integration: FtsH is the only known membrane-anchored AAA+ protease and is essential in prokaryotes and eukaryotic organelles [90] [91]. Its primary structure includes:
- An N-terminal domain with one or two transmembrane helices.
- A highly conserved AAA+ ATPase domain featuring Walker A and B motifs.
- A C-terminal zinc metalloprotease domain (M41 peptidase) with a characteristic HEXXH zinc-binding motif [90].
Complex Assembly and Molecular Mechanism: FtsH forms hexameric complexes that can be homomeric or, in organisms with multiple FtsH homologs (like cyanobacteria and plant chloroplasts), heteromeric [90]. The soluble ATPase and protease domains assemble into a cytoplasmic double-ring structure, while the transmembrane domains anchor the complex [91].
The current model suggests that FtsH recognizes membrane-bound and soluble substrates. A flexible linker between the transmembrane and ATPase domains provides space for substrate access [90]. A key phenylalanine residue in the FVG motif on the ATPase domain surface is critical for substrate binding [90]. ATP hydrolysis induces a "spiral staircase" rotation of the ATPase domains, unfolding the substrate and translocating it through the central pore to the proteolytic site for degradation into peptides approximately 12 amino acids long [90] [91]. Studies suggest around 8 ATP molecules are consumed per peptide bond cleavage [90].
The Clp Protease System
Modular Composition and Assembly: The Clp system is unique for its modular nature, comprising separate proteolytic core and ATPase regulatory particles that assemble into a functional holoenzyme [92] [86].
- Proteolytic Core (ClpP): The ClpP subunit forms a stable tetradecamer (two stacked heptameric rings) that assembles into a barrel-shaped structure with an internal proteolytic chamber [92] [86]. Its active site contains a canonical catalytic triad (Asp-His-Ser) [92].
- Regulatory ATPase Partners: AAA+ proteins such as ClpA, ClpX, or ClpC form homohexameric rings that associate with one or both ends of the ClpP core, forming complexes like ClpAP or ClpXP [92]. These ATPase partners are responsible for substrate recognition, unfolding, and translocation.
Functional Versatility: Different ATPase partners confer distinct substrate specificities to the Clp complex. For example, in E. coli, proteins tagged with the SsrA peptide are degraded by ClpXP and ClpAP [92]. Some bacteria, like Pseudomonas aeruginosa, possess multiple ClpP isoforms (ClpP1 and ClpP2) that form mixed complexes with different cleavage specificities, adding a layer of regulatory complexity linked to processes like biofilm formation [92].
Table 1: Comparative Overview of ATP-Dependent Protease Systems
| Feature | Lon Protease | FtsH Protease | Clp Protease |
|---|---|---|---|
| Domain Organization | Single polypeptide (N, AAA+, Protease) | Single polypeptide (TM, AAA+, Protease) | Separate subunits (ClpP core + AAA+ partner) |
| Protease Type | Serine protease (Ser-Lys dyad) | Zinc metalloprotease | Serine protease (Asp-His-Ser triad) |
| Oligomeric State | Homohexamer | Homo-/Heterohexamer | ClpP: Tetradecamer; AAA+: Hexamer |
| Membrane Association | LonB subfamily only | Yes (integral membrane) | Soluble (some mitochondrial membrane association) |
| Key Structural Features | Central chamber, axial channel | Spiral staircase ATPase, membrane anchor | Barrel-shaped core, axial channels |
| ATPase Partner | Covalently fused AAA+ domain | Covalently fused AAA+ domain | Separate AAA+ proteins (ClpA, ClpX, ClpC) |
| Core Biological Functions | Protein quality control, regulatory protein turnover | Membrane protein quality control, stress response | Protein quality control, regulation, virulence |
Experimental Analysis of ATP-Dependent Proteases
Structural Methodologies
Understanding the mechanistic details of these proteases has been revolutionized by structural biology. The following workflow is typical for determining high-resolution structures using cryo-electron microscopy (cryo-EM), as applied to FtsH [91].
Diagram 1: Cryo-EM Structure Determination Workflow
Detailed Protocol:
- Protein Expression and Purification: The gene for the protease (e.g., full-length FtsH) is cloned and overexpressed in a system like E. coli. The protein is purified using affinity and size-exclusion chromatography, often in detergents to maintain solubility of membrane proteins [91].
- Membrane Protein Reconstitution (if applicable): For membrane proteins like FtsH, the purified protein is reconstituted into lipid nanodiscs using membrane scaffold proteins (e.g., MSP1D1). This step stabilizes the protein in a near-native lipid environment, which is crucial for obtaining high-quality particles for cryo-EM [91].
- Cryo-EM Grid Preparation and Data Collection: The sample is applied to cryo-EM grids, vitrified in liquid ethane, and imaged in an electron microscope. For difficult samples, additives like CHAPS detergent or ultra-thin carbon support films may be used to improve particle distribution and stability [91].
- Image Processing and Reconstruction: Thousands of particle images are picked and classified using software like RELION or cryoSPARC. 2D class averages are generated to assess particle quality. 3D reconstruction with or without imposed symmetry (e.g., C6 for hexamers) is performed to generate an electron density map [91].
- Model Building and Refinement: An atomic model is built into the density map using programs like Coot and refined against the map. The presence of endogenous ligands like ADP can be identified from distinct densities within the map [91].
Functional and Genetic Analyses
Functional studies often involve genetic and biochemical assays to link protease activity to cellular phenotypes. A recent study on Mycobacterium tuberculosis exemplifies a protocol for identifying protease mutations linked to antibiotic resistance [93].
Protocol for Identifying Resistance Mutations in Clinical Isolates:
- Drug Susceptibility Testing (DST): Clinical bacterial isolates are tested for resistance to drugs like pyrazinamide (PZA) using systems like the BACTEC MGIT 960. Phenotypic resistance is confirmed by comparing growth in drug-containing versus drug-free media [93].
- Enzyme Activity Assay: For PZA, the enzymatic activity of pyrazinamidase (PZase) is assessed using a modified Wayne test. Bacterial colonies are incubated with PZA, and a colorimetric change after adding ferrous ammonium sulfate indicates PZase activity [93].
- Targeted Gene Sequencing: Genomic DNA is extracted from resistant and susceptible isolates. Customary resistance-associated genes (e.g., pncA for PZA) are amplified by PCR and sequenced. Sequences are compared to a reference genome to identify mutations [93].
- Whole-Genome Sequencing (WGS): For resistant strains lacking mutations in known genes, WGS is performed. This unbiased approach can reveal novel mutations in other genes, such as clpC1, a gene encoding an AAA+ ATPase component of the Clp protease system [93].
- Functional Validation: The impact of identified mutations is validated. For example, the ATP-dependent proteolytic activity of complexes like ClpC1P1P2 can be measured in vitro using degradation assays to confirm that the mutation impairs function [93].
Table 2: Essential Research Reagents and Their Applications
| Reagent / Material | Function in Research | Specific Application Example |
|---|---|---|
| MSP1D1 Nanodiscs | Provides a native-like lipid bilayer environment for stabilizing membrane proteins for structural studies. | Reconstitution of full-length FtsH for cryo-EM analysis [91]. |
| BACTEC MGIT 960 System | Automated liquid culture system for rapid drug susceptibility testing (DST) of bacterial isolates. | Phenotypic screening of M. tuberculosis for pyrazinamide resistance [93]. |
| Cross-linking Reagents | Stabilize weak protein-protein interactions and transient complexes for biochemical analysis. | Stabilizing oligomeric states of Lon protease for structural characterization [89]. |
| Active-site Mutants | Catalytically inactive mutants used to trap substrates and reaction intermediates. | Lon S679A mutant to stabilize the protease domain for crystallization [89]. |
| ATPγS (ATP-gamma-S) | A poorly hydrolyzable ATP analog used to trap the ATP-bound state of AAA+ proteins. | Used in crystallography/cryo-EM to study the ATP-state of FtsH and other AAA+ proteases. |
Connection to the Hershko-Ciechanover Paradigm and Therapeutic Implications
The discoveries of Hershko, Ciechanover, and Rose established that selective, energy-dependent proteolysis is a central regulatory mechanism in eukaryotic cells, governed by the ubiquitin-proteasome system [32] [13]. The Lon, FtsH, and Clp systems represent the evolutionary and functional parallels to this paradigm in prokaryotes and organelles. They share the core principle: ATP hydrolysis is used to mechanically unfold protein substrates and translocate them into a sequestered proteolytic chamber for destruction [86]. This mechanistic conservation underscores the fundamental importance of compartmentalized, energy-dependent proteolysis in all domains of life.
The therapeutic implications are substantial. As essential virulence factors in many pathogenic bacteria, these proteases are promising targets for novel antimicrobial agents [92] [93]. For instance, inhibiting ClpP or its ATPase partners can disrupt protein homeostasis and attenuate virulence in pathogens like P. aeruginosa and M. tuberculosis [92] [93]. Furthermore, in humans, the mitochondrial versions of these proteases are critical for organellar function. Mutations in human ClpP are linked to Perrault syndrome, characterized by hearing loss and ovarian failure, while upregulation of ClpP has been implicated in cancer cell survival [92]. Understanding the detailed structure and mechanism of these molecular machines, as illuminated by the research methodologies detailed herein, paves the way for developing targeted therapies against infectious diseases and certain genetic disorders.
Structural and Functional Conservation Across Evolution
The discovery of ATP-dependent proteolysis by Avram Hershko, Aaron Ciechanover, and Irwin Rose in the late 1970s and early 1980s revolutionized our understanding of cellular protein degradation [1]. Their pioneering research revealed a complex, energy-dependent system far removed from the previously accepted models of lysosomal proteolysis or simple ATP-dependent proteases. This work, which earned them the Nobel Prize in Chemistry in 2004, began with a simple biological curiosity: the paradoxical observation that intracellular proteolysis requires energy despite being thermodynamically exergonic [1] [10]. Their initial experiments using reticulocyte lysates led to the identification of a heat-stable polypeptide component they termed APF-1 (ATP-dependent Proteolysis Factor 1), later identified as ubiquitin [1] [10]. This discovery unveiled an entirely novel proteolytic paradigm where proteins are marked for degradation through covalent attachment of a small protein tag, establishing the foundation for what we now recognize as the ubiquitin-proteasome system (UPS).
The significance of this discovery extends beyond the mechanism of protein degradation. It revealed that the covalent attachment of small proteins as targeting signals represents a regulatory mechanism as crucial to eukaryotic cells as phosphorylation or acetylation [1]. The ubiquitin system has since been implicated in the regulation of diverse cellular processes including nuclear localization, chromatin structure, genetic integrity, protein quality control, and signaling [1]. This article explores how the structural and functional principles discovered in this system are conserved across evolution, from prokaryotic protease complexes to eukaryotic proteasomes, and how this conservation informs modern drug discovery approaches.
Quantitative Measures of Structural Conservation
The conservation of protein structures across evolution provides critical insights into functional relationships that may be obscured at the sequence level. Recent proteome-wide structural analyses using both experimental and AlphaFold2-predicted structures have quantified this conservation across the evolutionary tree.
Structural Similarity in the "Twilight Zone"
The "twilight zone" of protein evolution refers to sequence identities of 20-25%, where evolutionary relationships become difficult to detect through sequence alignment alone [94]. In this range, structural similarity often reveals evolutionary relationships that sequences cannot. Comparative analyses of Homo sapiens, Escherichia coli, and Methanocaldococcus jannaschii proteomes demonstrate that structurally similar homologous protein pairs in the twilight zone account for approximately 0.004%-0.021% of all possible protein pair combinations, which translates to 8%-32% of protein-coding genes depending on the species compared [94]. This indicates that a significant proportion of proteomes maintain structural conservation despite extensive sequence divergence.
Table 1: Proteome-Wide Structural Conservation Analysis
| Metric | H. sapiens | E. coli | M. jannaschii |
|---|---|---|---|
| Proteins without folded domains (>50 aa) | 11.5% | 7.6% | 4.2% |
| Average number of domains per protein | 1.58 | 1.16 | 1.15 |
| Proteins with structural homologs in twilight zone | 8-32% (depending on comparison) | 8-32% (depending on comparison) | 8-32% (depending on comparison) |
| Structural homologs as percentage of all possible pairs | 0.004-0.021% | 0.004-0.021% | 0.004-0.021% |
Evolutionary Origins of Human Protein Pathways
Structural comparisons reveal distinct evolutionary origins for different functional categories of human proteins. Human proteins involved in energy supply show greater structural similarity to their E. coli homologs, while proteins relating to the central dogma (DNA replication, transcription, translation) are more similar to their M. jannaschii homologs [94]. This pattern supports the hypothesis that eukaryotic cells originated from endosymbiotic events between ancestral bacteria and archaea, with different cellular processes retaining structural features from different prokaryotic ancestors.
Functional Conservation in ATP-Dependent Proteolytic Systems
The structural conservation observed across evolution is mirrored by remarkable functional conservation in ATP-dependent proteolytic systems. The core architecture and mechanism of these molecular machines remain consistent from prokaryotes to eukaryotes.
Conserved Architecture of AAA+ Proteases
AAA+ ATPases (ATPases Associated with diverse cellular Activities) represent a family of ring-shaped hexameric protein complexes that function as elaborate macromolecular motors [23]. These complexes share a common overall architecture despite limited sequence conservation outside their ATP-binding domains [95]. They all form oligomeric, barrel-shaped complexes composed of one or more rings with active proteolytic sites sequestered inside a central degradation chamber [95]. Narrow access channels control entry to these sites, requiring substrate proteins to be unfolded before degradation [95]. Regulatory particles belonging to the AAA+ family assemble on either end of the proteolytic chamber, recognizing substrates, unfolding them, and translocating the unfolded polypeptide through a central channel to the proteolytic chamber using energy derived from ATP hydrolysis [95] [23].
Table 2: Comparative Analysis of ATP-Dependent Proteolytic Systems
| Protease System | Organism Type | Architecture | Unfolding Ability | Primary Targeting Mechanism |
|---|---|---|---|---|
| ClpXP | Bacteria, Mitochondria | ClpX hexamer + ClpP heptamer (x2) | Moderate | Recognition of degron sequences |
| ClpAP | Bacteria, Mitochondria | ClpA hexamer + ClpP heptamer (x2) | High | Recognition of degron sequences |
| 26S Proteasome | Eukaryotes | 19S regulatory particle + 20S core particle | High | Polyubiquitin conjugation |
| HslUV (ClpYQ) | Bacteria | HslU hexamer + HslV hexamer (x2) | Moderate | Recognition of degron sequences |
| Lon | Bacteria, Mitochondria | Homooligomeric complex | Low | Recognition of unstructured regions |
| FtsH | Bacteria, Mitochondria | Membrane-bound hexamer | Low | Recognition of unstructured regions |
Functional Conservation from Prokaryotic Proteases to Eukaryotic Proteasomes
The ClpXP protease serves as an exemplary model for illustrating functional conservation across evolution. ClpXP consists of two components: ClpX, an AAA+ ATPase that functions as an unfoldase, and ClpP, the proteolytic component [23] [96]. ClpX recognizes client proteins bearing specific degradation signals ("degrons"), uses ATP hydrolysis to unfold these substrates, and translocates them through its central pore into the proteolytic chamber of ClpP [23]. This functional architecture is conserved in the eukaryotic 26S proteasome, where the 19S regulatory particle (functionally analogous to ClpX) recognizes ubiquitinated substrates, unfolds them, and translocates them into the 20S core particle (functionally analogous to ClpP) for degradation [95] [97].
This conservation extends to specific functional domains. ClpP proteases form a tetradecameric structure consisting of two stacked heptameric rings, with 14 canonical catalytic domains based on a Ser-His-Asp catalytic triad [23]. Similarly, the 20S proteasome core particle forms a barrel-like structure with multiple proteolytic active sites facing an interior degradation chamber [97]. In both systems, the proteolytic components alone can only degrade small peptides, preventing uncontrolled proteolysis of native proteins - access to the degradation chamber is strictly controlled by the regulatory components [23].
Experimental Analysis of Structural and Functional Conservation
Quantitative Protease Unfolding Assays
Comparative studies of ATP-dependent proteases have quantified their unfolding abilities, revealing more than 100-fold variation across different protease classes [95]. These differences in unfolding capability represent an additional layer of substrate selectivity beyond recognition of specific degradation signals.
Experimental Protocol:
- Substrate Design: Engineer model substrate proteins containing folded domains (e.g., barnase, barstar, DHFR) fused to specific protease recognition signals (e.g., ssrA peptide, λO fragment, cytochrome b2 presequence) [95].
- Radioactive Labeling: Express substrates using in vitro transcription/translation systems supplemented with [³⁵S]methionine for detection [95].
- Protease Purification: Purify proteases of interest using multi-step chromatographic methods (e.g., ion exchange, hydrophobic interaction, size exclusion) [95].
- Degradation Assays: Incubate radiolabeled substrates with proteases and ATP regeneration systems at defined temperatures [95].
- Analysis: Resolve reaction products by SDS-PAGE, visualize and quantify using phosphorimaging, and calculate degradation rates [95].
This methodology revealed that different classes of ATP-dependent proteases exhibit distinct unfolding capabilities. For example, ClpAP demonstrates high unfolding ability, while Lon and FtsH have relatively weak unfolding abilities, making them selective for damaged or unfolded polypeptides [95]. These differences contribute to substrate selectivity independent of recognition sequences.
Structural Dynamics Analysis
The internal dynamics of proteins can be conserved even when structural similarity becomes marginal. Several methodological approaches enable quantitative comparison of protein dynamics:
Essential Dynamics Analysis Protocol:
- Molecular Dynamics Simulations: Perform atomic-level simulations of protein structures under physiological conditions [98].
- Coarse-Grained Modeling: Apply elastic network models to characterize large-scale motions [98].
- Collective Coordinate Identification: Use principal component analysis to identify dominant modes of motion [98].
- Comparative Dynamics Alignment: Quantify similarity in functional dynamics across protein families using specialized algorithms [98].
These approaches have revealed that aspects of protein flexibility and dynamics that impact biological function are often subject to evolutionary conservation, sometimes even when structural similarity becomes limited [98]. For example, comparative analyses have identified conserved dynamic features across structurally heterogeneous members of protein families and even across different protein families with similar functional requirements [98].
Visualization of Conserved Systems and Experimental Approaches
The Ubiquitin-Proteasome Pathway
Ubiquitin-Proteasome Pathway
ATP-Dependent Protease Experimental Workflow
Protease Assay Workflow
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Research Reagents for Studying ATP-Dependent Proteolysis
| Reagent/Category | Function/Application | Specific Examples |
|---|---|---|
| ATP-Regeneration Systems | Maintains constant ATP levels during assays | Creatine phosphate + creatine kinase [95] |
| Protease Inhibitors | Characterize catalytic mechanisms | Z-Leu-Tyr-ChloroMethylKetone (Z-LY-CMK) for serine proteases [23] |
| Tagged Substrates | Model substrates for degradation assays | Barnase, barstar, DHFR domains with targeting signals [95] |
| Radiolabeled Amino Acids | Detect and quantify protein degradation | [³⁵S]Methionine for in vitro translation [95] |
| Ubiquitination System Components | Study eukaryotic protein tagging | E1 (activating), E2 (conjugating), E3 (ligating) enzymes [95] |
| Chromatography Resins | Purify protease components | Ion-exchange, hydrophobic interaction, affinity resins [95] |
| Thermostable Proteases | Study evolutionary adaptations | Proteases from thermophilic organisms (M. jannaschii) [94] |
Implications for Drug Discovery and Therapeutic Development
The conserved nature of ATP-dependent proteolytic systems has profound implications for drug discovery, particularly in the development of targeted protein degradation (TPD) technologies [97]. These approaches leverage the cell's natural degradation machinery to eliminate disease-causing proteins.
Proteolysis-Targeting Chimeras (PROTACs)
PROTACs are heterobifunctional molecules that simultaneously bind to a target protein and an E3 ubiquitin ligase, bringing them into proximity and leading to target ubiquitination and degradation by the proteasome [97]. This approach offers advantages over traditional inhibitors, including the ability to target "undruggable" proteins, achieve catalytic activity (degraders can act multiple times), and potentially overcome drug resistance [97]. The conservation of the ubiquitin-proteasome system across eukaryotes makes PROTACs particularly valuable for therapeutic development.
Targeting Conserved Bacterial Proteases
The functional conservation of proteolytic systems also enables targeting of bacterial proteases for antimicrobial development. Recent research has demonstrated that disruption of the ATP-dependent unfoldase ClpX can reverse antifungal resistance in Cryptococcus neoformans, restoring susceptibility to fluconazole in resistant strains [60]. This approach highlights how understanding the conserved mechanisms of proteolytic systems can inform strategies to combat drug resistance.
The structural and functional conservation of ATP-dependent proteolytic systems, from the initial discoveries of Hershko, Ciechanover, and Rose to contemporary structural biology, reveals fundamental principles of cellular organization and regulation. The conserved architecture of AAA+ proteases, the mechanistic similarities between prokaryotic protease complexes and eukaryotic proteasomes, and the evolutionary persistence of specific structural domains all testify to the optimization of these essential cellular machines through evolution. Quantitative analyses of structural conservation, particularly in the "twilight zone" where sequence similarity becomes marginal, continue to reveal unexpected evolutionary relationships and functional similarities. As drug discovery increasingly focuses on targeted protein degradation technologies, this evolutionary conservation provides both opportunities for broad therapeutic applications and challenges for achieving specificity. The continued investigation of these conserved systems will undoubtedly yield further insights into cellular function and new avenues for therapeutic intervention.
Validation Through Genetic and Cellular Models
The seminal research of Avram Hershko, Aaron Ciechanover, and Irwin Rose fundamentally reshaped our understanding of intracellular protein degradation. Their discovery of the ubiquitin-proteasome system revealed a sophisticated, energy-dependent mechanism that diverged radically from the previously accepted model of lysosomal proteolysis [1] [10]. This ATP-dependent proteolytic system employs a covalent tagging mechanism, whereby a small protein called ubiquitin (initially identified as ATP-dependent proteolysis factor 1, or APF-1) is attached to substrate proteins, marking them for degradation by a large protease complex [1]. This foundational principle of targeted, energy-dependent proteolysis extends beyond the ubiquitin-proteasome pathway to include other ATP-dependent proteases such as Lon, FtsH, and ClpXP, which are crucial for protein quality control and regulatory processes across all domains of life [99] [23] [86]. This whitepaper details the genetic and cellular models that have been instrumental in validating the core mechanisms of ATP-dependent proteolysis, providing researchers with established methodologies to further explore this essential biological system.
Historical Context and Foundational Discoveries
The intellectual journey to this discovery began with the puzzling observation that intracellular proteolysis requires metabolic energy, a thermodynamic paradox since peptide bond hydrolysis is itself an exergonic process [1] [10]. For decades, the lysosome was presumed to be the site of cellular protein turnover. However, experimental evidence, including the use of lysosomal inhibitors that failed to block the degradation of most intracellular proteins, suggested the existence of a non-lysosomal pathway [10].
The critical breakthrough came from a series of experiments using a cell-free reticulocyte lysate system, which lacks lysosomes yet exhibits robust ATP-dependent proteolysis [1]. The key experimental strategy involved biochemical fractionation:
- Fractionation and Reconstitution: The reticulocyte lysate was separated into two essential fractions (I and II). ATP-dependent proteolytic activity was only observed upon recombining these fractions [1].
- Identification of APF-1: Fraction I was found to contain a single, heat-stable component termed APF-1. This factor was later identified as the previously known protein, ubiquitin [1].
- Covalent Conjugation: The critical mechanistic insight was that APF-1/ubiquitin was covalently attached to a wide array of proteins in Fraction II in an ATP-dependent manner. This conjugation was reversible upon ATP removal, and substrates destined for degradation were found to be modified by multiple molecules of ubiquitin [1].
This pioneering work established a new paradigm where proteolysis is not catalyzed by a single enzyme but by a complex system involving a tagging mechanism that provides specificity and explains the energy requirement [10].
Classification and Structures of ATP-Dependent Proteases
The discovery of the ubiquitin system paved the way for recognizing a broader family of ATP-dependent proteases. These enzymes can be classified based on their architectural principles, primarily into two categories: those with proteolytic and ATPase activities on a single polypeptide chain and those with these functions on separate subunits that assemble into a complex.
Table 1: Major Families of ATP-Dependent Proteases
| Protease Family | Architectural Principle | ATPase Component | Protease Component | Typical Structure | Key Characteristics |
|---|---|---|---|---|---|
| Lon | Single polypeptide | Integrated AAA+ domain | Integrated Serine protease domain | Homo-hexameric or heptameric ring [99] | Self-compartmentalized; each subunit contains N-domain, ATPase A-domain, sensor SSD, and proteolytic P-domain [99]. |
| FtsH | Single polypeptide | Integrated AAA+ domain | Integrated Zinc metalloprotease domain | Membrane-anchored hexamer [100] [86] | Degrades misassembled membrane proteins; cytosolic region forms symmetric protease domains with asymmetric AAA+ domains [100]. |
| ClpXP | Separate subunits | ClpX (AAA+ unfoldase) | ClpP (protease) | Double-heptameric ClpP barrel capped by single or double hexameric ClpX rings [23] | ClpP is a serine protease with a Ser-His-Asp catalytic triad; ClpX recognizes degrons and translocates unfolded substrates [23]. |
| Proteasome | Separate subunits | Regulatory Particle (RP) | Core Particle (CP) | Cylindrical CP of 4 stacked heptameric rings capped by one or two RPs [86] | CP is a threonine protease; in eukaryotes, substrates are typically tagged with polyubiquitin for recognition by the RP [1] [86]. |
| HslUV | Separate subunits | HslU (AAA+ unfoldase) | HslV (protease) | HslV forms a twin-hexameric proteolytic chamber [86] | HslV is a threonine protease and considered a prokaryotic proteasome counterpart [86]. |
The following diagram illustrates the core architecture and functional workflow shared by these diverse ATP-dependent protease complexes.
Diagram 1: Generalized mechanism of AAA+ proteases. The substrate is recognized and unfolded by the AAA+ ATPase component, then translocated into the sequestered proteolytic chamber for degradation.
Quantitative Analysis of Protease Mechanisms
Understanding the kinetics and energy requirements of these proteases is critical for validating their mechanisms. Pre-steady-state kinetic studies, particularly of the Lon protease, have provided deep insights into the coupling between ATP hydrolysis and proteolysis.
Table 2: Kinetic Parameters for E. coli Lon Protease Mutants
| Parameter | S679A Lon Mutant | S679W Lon Mutant | Interpretation & Biological Significance |
|---|---|---|---|
| Rate of Conformational Change (Step 1) | 0.74 ± 0.10 s⁻¹ | 0.57 ± 0.10 s⁻¹ | Reflects a peptide-induced conformational change; rate is comparable to the lag phase rate constant (k~lag~ ~1 s⁻¹) observed in peptide hydrolysis, suggesting it is a rate-limiting step prior to peptide bond cleavage [101]. |
| Rate of Conformational Change (Step 2) | Not detected | 7.6 ± 1.0 s⁻¹ | An ATP-dependent conformational change within the proteolytic site; rate is essentially identical to the burst rate constant for ATP hydrolysis, indicating this step is coupled to ATP hydrolysis and likely activates the proteolytic site [101]. |
| ATP Hydrolysis Burst Rate | ~7.6 s⁻¹ (Wild-type) | ~7.6 s⁻¹ (Inferred) | Occurs prior to peptide bond cleavage, providing energy for the conformational changes that facilitate substrate translocation and proteolytic site activation [101]. |
| Mutation Impact | Abolishes proteolysis, does not affect ATPase activity [101]. | Abolishes proteolysis, does not affect intrinsic ATPase activity [101]. | Confirms that the catalytic Ser-679 is essential for proteolysis but not for ATP binding or hydrolysis, demonstrating functional separation of the active sites. |
Experimental Protocol: Stopped-Flow Kinetics to Monitor Conformational Changes
This protocol is used to detect rapid, nucleotide-dependent conformational changes in proteases like Lon [101].
- Protein Preparation: Generate and purify a proteolytically inactive Lon mutant (e.g., S679A or S679W) where the active-site serine is replaced. The S679W mutation introduces a tryptophan fluorophore directly into the proteolytic site.
- Substrate Design: Use a synthetic peptide substrate based on a known endogenous target (e.g., the λN protein). A dansylated version (e.g., S4 peptide) allows for fluorescence detection.
- Stopped-Flow Experiment:
- Load one syringe with the Lon mutant and ATP (or a non-hydrolyzable analog like AMPPNP).
- Load the second syringe with the dansylated peptide substrate.
- Rapidly mix the solutions and monitor fluorescence over time.
- Excitation: 290 nm (to excite intrinsic Trp residues in Lon).
- Emission Detection: > 450 nm (to capture emission from the dansyl group).
- Data Analysis: The increase in dansyl fluorescence upon binding and energy transfer from Trp residues (a FRET-based signal) is tracked. The resulting fluorescence time courses are fitted to kinetic models to determine the rate constants of binding and conformational changes.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Studying ATP-Dependent Proteolysis
| Reagent / Tool | Function / Description | Example Application |
|---|---|---|
| Reticulocyte Lysate | A cell-free extract from rabbit reticulocytes, rich in the ubiquitin-proteasome system components [1] [10]. | Foundational system for fractionating and reconstituting ATP-dependent proteolysis; identifying essential factors like ubiquitin [1]. |
| Proteolytically Inactive Mutants | Mutant proteases where catalytic residues are mutated (e.g., Lon S679A, ClpP S97A). Abolishes proteolysis while preserving ATPase and binding functions [101]. | Trapping enzyme-substrate complexes for structural studies; dissecting the steps of ATP hydrolysis and substrate translocation independently of degradation [101]. |
| Non-hydrolyzable ATP Analogs | Nucleotide analogs that bind but cannot be hydrolyzed (e.g., AMPPNP, ATPγS). | Decoupling the effects of ATP binding from ATP hydrolysis; used to demonstrate that ATP binding alone can activate proteolysis of certain substrates in Lon [101]. |
| Defined Peptide Substrates | Short, fluorogenic, or fluorophore-labeled peptides based on native protease targets (e.g., dansylated S4 peptide for Lon) [101]. | High-throughput kinetic assays; pre-steady-state stopped-flow experiments to measure binding and conformational changes [101]. |
| Limited Proteolysis-Mass Spectrometry (LiP-MS) | A proteome-wide technique that uses partial proteolysis to detect protein structural changes [102]. | Identifying structural changes in proteins upon drug binding, mutations, or in different cellular states; drug-target deconvolution [102]. |
Advanced Model: The ClpXP Protease
The ClpXP system serves as a premier model for understanding the mechanism of AAA+ proteases. The unfoldase (ClpX) and protease (ClpP) are separate entities that assemble into a functional complex, making it ideal for biochemical and structural studies [23].
Experimental Protocol: Analyzing ClpXP Protease Activity
This protocol outlines how to assay the degradation of a protein substrate by the ClpXP complex.
- Reconstitution:
- Purify ClpX and ClpP separately. ClpP from E. coli forms a tetradecameric (two heptameric rings) core [23].
- Combine ClpX and ClpP in a molar ratio (typically 2:1 for ClpX hexamer to ClpP tetradecamer) in an appropriate buffer containing Mg²⁺.
- Substrate Design:
- Use a model substrate protein fused to a specific degradation tag ("degron") recognized by ClpX (e.g., the ssrA tag) [23].
- The substrate can be labeled with a fluorophore for sensitive detection.
- Degradation Assay:
- In an assay buffer (e.g., 50 mM HEPES, pH 8.0, 5 mM Mg(OAc)₂, 5 mM DTT, 100 mM KCl), add the reconstituted ClpXP complex.
- Initiate the reaction by adding ATP (typically 2-5 mM) and the tagged substrate.
- Incubate at 37°C.
- Detection and Analysis:
- Time-point Assay: Remove aliquots at regular intervals, quench with SDS-PAGE loading buffer, and analyze by gel electrophoresis to visualize substrate disappearance.
- Continuous Assay: If using a fluorescently labeled substrate, monitor fluorescence loss (e.g., due to fluorescence quenching upon degradation) in real-time using a fluorometer.
The architecture and functional cycle of the ClpXP complex are summarized in the following diagram.
Diagram 2: The ClpXP degradation cycle. The ClpX hexamer recognizes a degradation-tagged substrate, uses ATP hydrolysis to unfold it, and translocates the unfolded polypeptide into the ClpP proteolytic chamber for digestion into small peptides.
The validation of ATP-dependent proteolysis through genetic, biochemical, and cellular models has been a cornerstone of modern cell biology. The pioneering work of Hershko, Ciechanover, and Rose on the ubiquitin system unveiled a new principle of cellular regulation. Subsequent research on proteases like Lon, FtsH, and ClpXP has shown that the coupling of ATP hydrolysis to targeted protein degradation is a conserved and essential mechanism from bacteria to humans. The experimental frameworks and tools detailed herein provide a roadmap for continued investigation into these complex molecular machines, with significant implications for understanding disease mechanisms and developing novel therapeutic strategies, particularly in areas such as cancer and neurodegenerative disorders where proteostasis is disrupted.
Therapeutic Advantages of PROTACs vs. Traditional Inhibitors
The discovery of the ubiquitin-proteasome system (UPS) by Avram Hershko, Aaron Ciechanover, and Irwin Rose, which earned them the Nobel Prize in Chemistry in 2004, unveiled a fundamental biological process: ATP-dependent intracellular proteolysis [1] [10]. Their pioneering work in the late 1970s and 1980s demonstrated that protein degradation is not a passive process but a highly specific, energy-dependent mechanism central to cellular regulation [1] [10]. This foundational research identified ubiquitin as a critical tag for protein destruction and laid the groundwork for understanding the enzymatic cascade (E1-E2-E3) that governs cellular protein homeostasis [1] [15].
Building directly upon this fundamental knowledge, PROteolysis TArgeting Chimeras (PROTACs) have emerged as a revolutionary therapeutic modality [103] [15]. First conceptualized in 2001, PROTAC technology hijacks the body's natural protein degradation machinery to target disease-causing proteins for destruction [45] [104]. This review delineates the considerable therapeutic advantages of the PROTAC platform over traditional small-molecule inhibitors, underscoring how a deep understanding of basic biological mechanisms can directly inform transformative drug discovery efforts.
Fundamental Mechanisms: A Paradigm Shift in Pharmacology
Traditional Small-Molecule Inhibitors: Occupancy-Driven Inhibition
Traditional inhibitors operate on the receptor occupancy model, where a drug's effect is proportional to the number of target binding sites it occupies [105]. These molecules typically function by binding to an enzyme's active site or a receptor's ligand-binding domain, thereby blocking its activity [106]. This approach requires maintaining a high, sustained systemic drug concentration to ensure sufficient target occupancy, often leading to dose-limiting toxicity and off-target effects [76] [105]. Furthermore, this strategy only inhibits specific functions of a protein (e.g., enzymatic activity), leaving its structural, scaffolding, or other non-enzymatic roles unaffected, which can contribute to compensatory signaling and drug resistance [15] [104].
PROTACs: Event-Driven Catalytic Degradation
In contrast, PROTACs employ an event-driven, catalytic mechanism that fundamentally differs from occupancy-driven inhibition [103] [106]. As heterobifunctional molecules, PROTACs consist of three key components:
- A ligand that binds to the Protein of Interest (POI)
- An E3 ubiquitin ligase-recruiting ligand
- A linker connecting the two moieties [45] [76]
The mechanism of action is illustrated in the following diagram and involves a cascade of events:
- Ternary Complex Formation: The PROTAC molecule simultaneously engages the target protein and an E3 ubiquitin ligase, forming a productive ternary complex [103] [104].
- Ubiquitination: The recruited E3 ligase catalyzes the transfer of ubiquitin chains from an E2 conjugating enzyme to lysine residues on the target protein's surface [45] [15].
- Degradation: The polyubiquitinated protein is recognized and degraded by the 26S proteasome into small peptides [23] [15].
- PROTAC Recycling: The PROTAC molecule is released unchanged and can initiate further rounds of degradation, functioning catalytically [105] [106].
This event-driven mechanism means that PROTACs need only bind transiently to their targets to induce degradation, eliminating the need for sustained high occupancy [106].
Table 1: Fundamental Mechanistic Differences Between PROTACs and Traditional Inhibitors
| Feature | Traditional Small-Molecule Inhibitors | PROTAC Degraders |
|---|---|---|
| Mode of Action | Occupancy-driven | Event-driven, catalytic |
| Effect on POI | Inhibits function (often reversible) | Eliminates protein entirely |
| Pharmacology | Stoichiometric | Sub-stoichiometric/Catalytic |
| Dosing Requirement | High, sustained concentrations for efficacy | Lower, catalytic doses often sufficient |
| Target Scope | Proteins with defined functional pockets | Any protein with a bindable site |
| Impact on Protein Functions | Selective inhibition (e.g., enzymatic activity only) | Removal of all functions (enzymatic, scaffolding, structural) |
Key Therapeutic Advantages of PROTACs
Overcoming Drug Resistance
A significant challenge with traditional targeted therapies is the emergence of resistance, often through target protein overexpression or mutations that reduce drug-binding affinity [104]. PROTACs can overcome several common resistance mechanisms:
- Addressing Target Overexpression: Since PROTACs catalytically degrade their targets, they can manage situations where cancer cells survive by producing more of the target protein, a common resistance mechanism against inhibitors [104].
- Bypassing Mutations: Resistance to kinase inhibitors frequently arises from mutations in the kinase's active site. As PROTACs require only transient binding to any accessible surface on the target protein, they can often effectively degrade mutated proteins even when traditional inhibitors fail [15] [104]. For example, PROTACs targeting the androgen receptor (AR) have shown efficacy in degrading enzalutamide-resistant mutants in prostate cancer models [45].
Enhanced Selectivity and Targeting "Undruggable" Proteins
PROTACs can achieve remarkable selectivity, sometimes surpassing that of the warhead ligand used in their construction.
- Isoform Selectivity: PROTACs have demonstrated the ability to achieve isoform-selective degradation, even among highly similar protein family members. For instance, researchers have developed p38α- and p38δ-selective PROTACs (SJFα and SJFδ) from the non-selective kinase inhibitor foretinib by optimizing linker length and attachment points [103]. This selectivity arises from the formation of the ternary complex, where degradation efficiency depends on optimal spatial positioning of the entire complex, not just binding affinity [103].
- Expanding the Druggable Proteome: Traditional small-molecule drugs are largely restricted to proteins with well-defined, druggable pockets (e.g., kinases, GPCRs). This leaves many pathogenic proteins, such as transcription factors, scaffolding proteins, and non-enzymatic regulators, classified as "undruggable" [15] [104]. PROTACs require only a bindable site on the target protein, not a functional pocket, opening the door to therapeutic targeting of these previously inaccessible proteins [105] [104].
Catalytic Activity and Sustained Pharmacological Effects
The catalytic nature of PROTACs is a fundamental advantage. A single PROTAC molecule can theoretically mediate the degradation of multiple copies of the target protein, enabling potent effects at low concentrations and reducing the potential for off-target toxicity [105] [106]. Furthermore, because the protein must be resynthesized to regain function, the pharmacological effect of a successful degradation event persists long after the PROTAC is cleared from the system. This can lead to a more sustained duration of action, potentially allowing for less frequent dosing compared to traditional inhibitors that require constant occupancy [76].
Experimental Evaluation of PROTACs
Key Methodologies and Protocols
Evaluating PROTAC efficacy and mechanism requires a combination of biochemical, cellular, and biophysical techniques.
Ternary Complex Formation Analysis:
- Surface Plasmon Resonance (SPR): Used to characterize the affinity and kinetics of ternary complex formation, which is a critical determinant of degradation efficiency [103].
- Isothermal Titration Calorimetry (ITC): Measures the binding thermodynamics of the POI-PROTAC-E3 ligase complex.
- X-ray Crystallography/Cryo-Electron Microscopy (Cryo-EM): Provides high-resolution structural insights into the ternary complex, guiding rational design and explaining selectivity [23].
Cellular Degradation Assessment:
- Western Blotting/Immunoblotting: The standard method for quantifying target protein levels in cells following PROTAC treatment. Cells are typically treated with a range of PROTAC concentrations and for various durations, followed by lysis and protein level analysis [45].
- Cellular Thermal Shift Assay (CETSA): Can confirm target engagement by measuring the thermal stabilization of the POI upon PROTAC binding.
- Anti-proliferative and Apoptosis Assays: Functional assays (e.g., MTS, colony formation, caspase activation) to determine the downstream biological consequences of target degradation [104].
Hook Effect Analysis: A critical experiment in PROTAC characterization is testing a wide concentration range (e.g., from nM to µM). At excessively high concentrations, PROTACs can saturate both the POI and E3 ligase independently, preventing productive ternary complex formation and leading to a decrease in degradation efficiency—a phenomenon known as the "hook effect." This is typically observed as a bell-shaped dose-response curve in degradation assays [105].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagents and Solutions for PROTAC Development and Analysis
| Research Reagent / Tool | Function & Application in PROTAC Research |
|---|---|
| E3 Ligase Ligands | Recruit specific E3 ligases (e.g., CRBN, VHL, MDM2). These are core components of the PROTAC molecule. [76] [106] |
| PROTAC Linkers (e.g., PEG-based, alkyl chains) | Connect the POI ligand and E3 ligand. Their length, composition, and rigidity are systematically optimized to facilitate efficient ternary complex formation. [45] [76] |
| Proteasome Inhibitors (e.g., MG132, Bortezomib) | Used as control reagents to confirm that PROTAC-induced degradation is proteasome-dependent. Treatment should block degradation. [15] |
| Lysosome Inhibitors (e.g., Chloroquine, Bafilomycin A1) | Used to rule out lysosomal involvement in the degradation mechanism, confirming UPS dependence. [15] |
| Selective POI Ligands (Warheads) | Bind the target protein. These can be inhibitors, agonists, or simply binders, and often do not require high inhibitory potency. [106] |
| Caged/Opto-PROTACs (e.g., with DMNB group) | Research tools where PROTAC activity is controlled by light (e.g., UV). Allows for precise spatiotemporal control of protein degradation in experimental models. [45] [105] |
Clinical Translation and Future Perspectives
The PROTAC field has progressed rapidly from a scientific concept to clinical-stage investigation. As of the date of this review, there are over 30 PROTAC candidates in clinical trials, with therapies targeting the androgen receptor (ARV-110) and estrogen receptor (ARV-471) being the most advanced, demonstrating promising early results in prostate and breast cancer, respectively [45] [103] [106].
Future developments focus on addressing remaining challenges and expanding the technology's reach:
- Overcoming Pharmacokinetic Limitations: The high molecular weight of PROTACs can pose challenges for oral bioavailability. Strategies like nanotechnology-based delivery systems (Nano-PROTACs) and the development of macrocyclic PROTACs (MacroPROTACs) to constrain the molecule into a more bioactive conformation are being explored to improve drug-like properties [105].
- Improving Tissue and Cell Selectivity: Novel approaches such as Antibody-conjugated PROTACs (DAC-PROTACs) and activatable pro-PROTACs are in development. These are designed to be inert until they reach a specific tissue or encounter a unique cellular trigger (e.g., hypoxia, enzyme activity), thereby minimizing on-target/off-tumor effects [105].
- Expanding the E3 Ligase Repertoire: Currently, most clinical-stage PROTACs recruit a limited set of E3 ligases (CRBN, VHL). Research is actively exploring ligands for other E3 ligases, particularly those with tissue-restricted expression, to enhance tumor selectivity and reduce potential toxicity in normal tissues [103] [106].
PROTAC technology represents a paradigm shift in therapeutic intervention, moving beyond simple inhibition to the complete elimination of pathogenic proteins. Its core advantages—the ability to overcome drug resistance, achieve high selectivity, target previously "undruggable" proteins, and function catalytically with sustained effects—are direct consequences of its event-driven mechanism of action, which itself is built upon the foundational ubiquitin-proteasome research of Hershko, Ciechanover, and Rose. While challenges in drug delivery and pharmacokinetics remain, the rapid clinical progression of PROTACs and the development of next-generation technologies signal a transformative era in drug discovery, offering new hope for targeting a wide array of debilitating diseases.
The groundbreaking work of Avram Hershko, Aaron Ciechanover, and Irwin Rose in the late 1970s and early 1980s fundamentally reshaped our understanding of intracellular protein degradation [13] [10]. They discovered a non-lysosomal, ATP-dependent proteolytic system where a small, heat-stable protein—ubiquitin—marks target proteins for destruction [19] [10]. This ubiquitin-proteasome system (UPS) explained the high specificity and metabolic energy requirement of intracellular proteolysis, a finding that earned them the 2004 Nobel Prize in Chemistry. For two decades, the lysosome was considered the primary site for protein degradation; however, their research demonstrated that the bulk of cellular proteins under basal conditions are degraded by this novel, non-lysosomal pathway [10]. The discovery that ubiquitin conjugation serves as a specific tagging mechanism for proteasomal degradation laid the essential foundation for all subsequent targeted protein degradation (TPD) technologies [15] [107].
While revolutionary, the initial application of this discovery was largely confined to intracellular proteins. The emerging TPD strategies, exemplified by PROteolysis TArgeting Chimeras (PROTACs), have revolutionized drug discovery by targeting proteins previously categorized as "undruggable" [108] [15]. However, a significant limitation remained: these strategies were ineffective against extracellular and membrane proteins, which constitute over 40% of the human proteome [108] [109]. This review focuses on the latest expansion of the TPD toolkit—technologies like LYTACs and AbTACs that hijack distinct cellular machinery to degrade this previously inaccessible class of proteins, thereby fulfilling huge unmet medical needs [108].
Core Protein Degradation Pathways in the Cell
Eukaryotic cells maintain protein homeostasis, or proteostasis, through two primary degradation pathways: the ubiquitin-proteasome system (UPS) and the lysosomal pathway [15]. These systems are independent yet interconnected, handling different types of substrates and fulfilling complementary roles [107].
- The Ubiquitin-Proteasome System (UPS): This system is primarily responsible for degrading short-lived proteins and soluble misfolded proteins [15]. It involves a cascade where a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2), and a ubiquitin ligase (E3) work together to tag target proteins with a polyubiquitin chain [15] [107]. Proteins marked with a K48-linked ubiquitin chain are recognized and degraded by the 26S proteasome in an ATP-dependent process [15]. Most early TPD technologies, such as PROTACs and molecular glues, exploit this pathway [108] [15].
- The Lysosomal Pathway: Lysosomes are the primary degradative compartments for long-lived proteins, insoluble protein aggregates, and entire organelles [15]. They receive cargo for degradation through three main routes: endocytosis (engulfing extracellular material and cell-surface proteins), phagocytosis (engulfing large particles like pathogens), and autophagy (sequestering intracellular components into autophagosomes that fuse with lysosomes) [15]. Novel degrader technologies like LYTACs co-opt the endocytic pathway to target extracellular and membrane proteins for lysosomal degradation [108] [109].
The following diagram illustrates the core cellular protein degradation pathways and how different TPD technologies hijack them.
Diagram 1: Core Cellular Protein Degradation Pathways Hijacked by TPD Technologies. The Ubiquitin-Proteasome System (UPS) is exploited by PROTACs and AbTACs, while the Lysosomal Pathway is leveraged by LYTACs.
Expanding the Scope: Degradation of Extracellular and Membrane Proteins
The limitations of UPS-based technologies prompted the development of novel strategies capable of targeting proteins outside the cell or on its surface. The following table summarizes the key features of these emerging technologies.
Table 1: Comparison of Novel Targeted Protein Degradation Technologies
| Technology | Key Components | Mechanism of Action | Primary Pathway | Target Class | Notable Features |
|---|---|---|---|---|---|
| LYTAC [108] [110] | Bispecific conjugate with a POI-binding moiety and a ligand for a lysosome-targeting receptor (e.g., CI-M6PR, ASGPR) | Engages cell-surface receptor to shuttle POI into endocytosis-lysosome pathway | Lysosomal | Extracellular & membrane proteins | First technology to systematically target extracellular proteins; can be cell-type specific (ASGPR-LYTAC) |
| AbTAC [108] [110] | Bispecific IgG antibody binding a membrane POI and a transmembrane E3 ligase (e.g., RNF43) | Recruits membrane-bound E3 ligase to induce ubiquitination and lysosomal degradation | Lysosomal (via UPS initiation) | Membrane proteins | Fully recombinant antibody; exploits transmembrane E3 ligases |
| KineTAC [110] | Bispecific antibody with a cytokine-binding arm and a target-binding arm | Uses cytokine receptor (e.g., CXCR7) to internalize and traffic POI to lysosomes | Lysosomal | Membrane & extracellular proteins | Genetically encoded; modular platform using endogenous cytokine pathways |
| PROTAB [110] | Bispecific antibody tethering transmembrane POI to transmembrane E3 ligase (e.g., ZNRF3) | Induces degradation via proximity-driven recruitment of specific E3 ligases | Lysosomal | Membrane proteins | Antibody-driven; demonstrated in vivo efficacy in colorectal cancer models |
| REULR [110] | Nanobody-based bifunctional tool recruiting transmembrane E3 ligase to membrane receptor | Promotes receptor degradation via ubiquitin ligase recruitment | Lysosomal | Membrane receptors (e.g., EGFR, PD-1) | Nanobody-based; can be designed for "fratricide" degradation of E3 ligases themselves |
LYTAC (Lysosome-Targeting Chimaera)
Mechanism and Design: LYTAC is a bifunctional conjugate composed of a targeting moiety (e.g., an antibody or small molecule) specific to an extracellular or membrane protein of interest (POI), linked to a ligand that binds a cell-surface lysosome-targeting receptor (LTR) [108] [109]. The first-generation LYTAC, termed M6Pn-LYTAC, utilized a ligand for the cation-independent mannose-6-phosphate receptor (CI-M6PR), a receptor widely expressed and known to shuttle enzymes to the lysosome [108] [110]. A second-generation system, GalNAc-LYTAC, engages the liver-specific asialoglycoprotein receptor (ASGPR), enabling tissue-specific degradation of extracellular proteins in hepatocytes [108] [110].
Experimental Workflow:
- LYTAC Synthesis: Conjugate a POI-binding antibody or small molecule to a CI-M6PR or ASGPR ligand (e.g., poly-M6Pn polymers or GalNAc) via a chemical linker [108] [110].
- Cell Treatment: Incubate cells expressing both the POI and the relevant LTR with the synthesized LYTAC molecule.
- Complex Formation: The LYTAC simultaneously binds the POI and the LTR, forming a ternary complex on the cell surface [108].
- Internalization and Degradation: The complex undergoes clathrin-mediated endocytosis. It traffics through early and late endosomes, where the acidic pH causes dissociation. The POI is delivered to the lysosome for degradation, while the LTR is recycled back to the membrane [108].
- Validation: Degradation efficiency is typically measured using Western blot to assess POI levels, flow cytometry for surface protein quantification, and immunofluorescence microscopy to visualize protein internalization and co-localization with lysosomal markers (e.g., LAMP1) [108].
AbTAC (Antibody-based PROTAC) and PROTAB (Protein-Targeting Antibodies)
Mechanism and Design: AbTACs are fully recombinant bispecific IgG antibodies [108] [110]. One arm binds a membrane-bound POI (e.g., PD-L1), while the other arm recruits a transmembrane E3 ubiquitin ligase, such as RNF43 [108] [110]. The formation of the RNF43-AbTAC-POI complex leads to ubiquitination of the POI, its internalization, and subsequent lysosomal degradation [108] [19]. The exact mechanism, including whether RNF43 directly ubiquitinates the POI's intracellular domain, is an area of active investigation [108]. PROTABs represent a closely related approach, also using bispecific antibodies to tether a membrane POI to a transmembrane E3 ligase (e.g., ZNRF3), inducing potent and specific degradation [110].
Experimental Workflow:
- Antibody Generation: Generate bispecific antibodies (e.g., using recombinant DNA technology) against the extracellular domains of the POI and the transmembrane E3 ligase [108] [110].
- Cell-Based Assay: Treat cells expressing the target POI and the E3 ligase with the AbTAC/PROTAB.
- Complex Formation and Internalization: The bispecific antibody bridges the POI and the E3 ligase, leading to complex internalization.
- Degradation Analysis: Evaluate POI degradation via Western blot and flow cytometry. Confirm the role of the specific E3 ligase using knockout cell lines or siRNA-mediated knockdown [108] [19]. Perform whole-cell proteomics to assess selectivity and off-target effects [108].
The diagram below illustrates the distinct mechanistic pathways of LYTAC and AbTAC technologies.
Diagram 2: Mechanisms of LYTAC and AbTAC. LYTAC engages lysosome-targeting receptors to internalize proteins, while AbTAC recruits transmembrane E3 ligases to initiate ubiquitination and lysosomal degradation.
The Scientist's Toolkit: Essential Research Reagents and Methodologies
Successful research in this field relies on a suite of specialized reagents and methodologies. The following table details key resources for developing and characterizing novel degraders.
Table 2: Key Research Reagent Solutions for TPD Development
| Reagent / Material | Function and Application | Examples / Specifications |
|---|---|---|
| CI-M6PR Ligand [108] | A key component of 1st-gen LYTACs; binds CI-M6PR to hijack lysosomal trafficking. | Synthetic poly(M6Pn) polymers conjugated to antibodies or small molecules via linkers. |
| GalNAc Ligand [108] [110] | A key component of 2nd-gen LYTACs; binds ASGPR for liver-specific degradation. | N-Acetylgalactosamine (GalNAc) conjugated to the POI binder. |
| Bispecific Antibody Platforms [108] [110] | Technological backbone for generating AbTACs and PROTABs. | Recombinant IgG formats (e.g., knob-into-hole) for simultaneous binding to POI and transmembrane E3 ligase. |
| Transmembrane E3 Ligase Constructs [108] [110] | Essential tools for studying and developing AbTACs/PROTABs. | Plasmids or cell lines expressing E3 ligases like RNF43 and ZNRF3. |
| Lysosomal Markers [108] | Critical for validating the lysosomal degradation pathway via imaging. | Antibodies against LAMP1, LAMP2; fluorescent dyes (e.g., LysoTracker). |
| BACTEC MGIT 960 System [93] | An example of a standardized, automated system for phenotypic drug susceptibility testing. | Used in microbiological contexts (e.g., for Pyrazinamide resistance testing) as a model for rigorous degradation validation. |
The field of targeted protein degradation has expanded dramatically from its origins in the UPS. The foundational discovery of ATP-dependent, ubiquitin-mediated proteolysis by Hershko, Ciechanover, and Rose provided the conceptual framework that underpins not only PROTACs but also the latest extracellular and membrane protein degraders [10]. Technologies like LYTACs and AbTACs represent a logical and powerful extension of this framework, leveraging either endocytic receptors or transmembrane E3 ligases to exploit the lysosomal degradation pathway [108] [109].
Despite the promising preclinical data, these novel technologies face significant challenges on their path to the clinic. For LYTACs, the pharmacokinetics and immunogenicity of typically large, antibody-based constructs need careful evaluation [108]. For AbTACs and PROTABs, the limited current knowledge of membrane E3 ligase biology and the potential for on-target, off-tissue toxicity require further investigation [108] [110]. Furthermore, the optimal design of linkers (for LYTACs) and the identification of additional tissue-specific LTRs or degradative receptors remain active areas of research [108] [110] [109].
The ongoing evolution of TPD, from a basic scientific discovery to a broad therapeutic platform, highlights the power of fundamental biological research. As the toolkit continues to expand with KineTACs, REULRs, and other emerging technologies, the potential to target previously "undruggable" proteins across virtually every disease area becomes increasingly tangible, heralding a new era in drug discovery [108] [110].
Conclusion
The discovery of ATP-dependent proteolysis and the ubiquitin-proteasome system by Hershko, Ciechanover, and Rose fundamentally transformed our understanding of cellular regulation, moving protein degradation from a simple waste disposal process to a sophisticated regulatory mechanism. This paradigm shift has opened entirely new avenues for therapeutic intervention, exemplified by the rapid development of PROTACs and other targeted protein degradation technologies that are now showing promise in clinical trials. The foundational principles of ubiquitin tagging continue to inspire innovative approaches to degrade disease-causing proteins previously considered 'undruggable.' Future directions will likely focus on expanding the repertoire of E3 ligases for therapeutic use, improving tissue specificity, and combining protein degradation strategies with other treatment modalities. As we continue to unravel the complexities of the ubiquitin system and other ATP-dependent proteolytic pathways, we can anticipate further transformative advances in treating cancer, neurodegenerative diseases, and other conditions characterized by protein dysregulation.
References
- 1. The discovery of ubiquitin- dependent - PMC proteolysis [https://pmc.ncbi.nlm.nih.gov/articles/PMC1266097/]
- 2. Regulated Proteolysis in Bacteria - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6013389/]
- 3. Ubiquitin system for regulated protein degradation [https://laskerfoundation.org/winners/ubiquitin-system-for-regulated-protein-degradation/]
- 4. A conversation with Aaron Ciechanover - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3784551/]
- 5. A prize for protein degradation | Nature Cell Biology [https://www.nature.com/articles/ncb1104-1011]
- 6. Reticulocyte lysate as a model system to study ... [https://pubmed.ncbi.nlm.nih.gov/15917633/]
- 7. Use of Reticulocyte Lysates for Mechanistic Studies ... [https://www.sciencedirect.com/science/article/abs/pii/S0076687907290019#!]
- 8. The Nobel Prize in Chemistry 2004 - Popular information [https://www.nobelprize.org/prizes/chemistry/2004/popular-information/]
- 9. Research Profile - Avram Hershko - Lindau Mediatheque [https://mediatheque.lindau-nobel.org/laureates/hershko/research-profile]
- 10. Tracing the history of the ubiquitin proteolytic system [https://www.sciencedirect.com/science/article/abs/pii/S0006291X09011887]
- 11. Ubiquitin is the ATP-dependent proteolysis factor I of rabbit ... [https://pubmed.ncbi.nlm.nih.gov/6249803/]
- 12. Useful Links / Further Reading [https://www.nobelprize.org/prizes/chemistry/2004/8689-useful-links-further-reading/]
- 13. Ubiquitin and its role in proteolisis: the 2004 Nobel prize in ... [http://ukrbiochemjournal.org/2022/12/ubiquitin-and-its-role-in-proteolisis-the-2004-nobel-prize-in-chemistry.html]
- 14. A heat-stable polypeptide component of an ATP ... [https://www.sciencedirect.com/science/article/abs/pii/0006291X78912494]
- 15. Targeted protein degradation: mechanisms, strategies and ... [https://www.nature.com/articles/s41392-022-00966-4]
- 16. Applications of Covalent Chemistry in Targeted Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9669245/]
- 17. Chemoproteomics-enabled discovery of a covalent ... [https://www.sciencedirect.com/science/article/pii/S2451945623000570]
- 18. Recent breakthroughs in innovative elements ... [https://www.sciencedirect.com/science/article/pii/S0753332224014707]
- 19. The discovery of ubiquitin-dependent proteolysis [https://pubmed.ncbi.nlm.nih.gov/16230621/]
- 20. Targeted Protein Degradation: Principles and Applications ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10378610/]
- 21. Ubiquitin: Structure and Function [https://www.intechopen.com/chapters/87831]
- 22. The Chance Meeting Behind a Nobel Discovery [https://www.foxchase.org/blog/2018-02-12-Forward-friendship-best-medicine]
- 23. Recent Advances in the Structural Studies of the Proteolytic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12383707/]
- 24. Israel and the U.S: Partners in Science [https://www.jewishpolicycenter.org/2018/04/11/israel-and-the-u-s-partners-in-science/]
- 25. ubiquitin/proteasome system (UPS) - Qian Lab @ Cornell [http://qian.human.cornell.edu/Research-UPS.htm]
- 26. E1, E2, E3 Enzyme and Their Regulators [https://www.bocsci.com/resources/e1-e2-e3-enzyme-and-their-regulators.html?srsltid=AfmBOooPY2McG-nYXMGw2nCnmNd-vycfla3GGuNNGND-lfZHLPeB6g6d]
- 27. Specificity of the E1-E2-E3 Enzymatic Cascade for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4652587/]
- 28. Orthogonal Ubiquitin Transfer through Engineered E1-E2 ... [https://www.sciencedirect.com/science/article/pii/S1074552112002888]
- 29. E3 Ubiquitin assisted technologies ligases ... in protein degradation [https://pubmed.ncbi.nlm.nih.gov/38521359/]
- 30. Frontiers | E3 ubiquitin : key regulators of osteogenesis and... ligases [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2024.1447093/full]
- 31. Proteolysis‐Targeting Chimera (PROTAC) - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC12495453/]
- 32. The Nobel Prize in Chemistry 2004 - NobelPrize.org [https://www.nobelprize.org/prizes/chemistry/2004/summary/]
- 33. Degradation signal diversity in the ubiquitin-proteasome system [https://pmc.ncbi.nlm.nih.gov/articles/PMC2606094/]
- 34. Visualizing ubiquitination in mammalian cells - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6362358/]
- 35. Mechanism of selective recognition of Lys48-linked ... [https://www.nature.com/articles/s41467-023-43025-4]
- 36. Unraveling chain specific ubiquitination in cells using tandem ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12217038/]
- 37. Emerging functions of branched ubiquitin chains [https://www.nature.com/articles/s41421-020-00237-y]
- 38. Polyamines sustain epithelial regeneration in aged ... [https://www.nature.com/articles/s41556-025-01804-9]
- 39. Structure and Function of the 26S Proteasome - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6422034/]
- 40. The life cycle of the 26S proteasome: from birth, through ... [https://www.nature.com/articles/cr201686]
- 41. Proteasome [https://en.wikipedia.org/wiki/Proteasome]
- 42. Atomic structure of the 26S proteasome lid reveals ... [https://elifesciences.org/articles/13027]
- 43. ATP-Dependent Steps in the Binding of Ubiquitin ... [https://www.sciencedirect.com/science/article/pii/S1097276510008403]
- 44. PROTAC: An Effective Targeted Protein Degradation ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2021.692574/full]
- 45. Recent Advances in the Development of Pro-PROTAC for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12473374/]
- 46. Design and development of PROTACs: A new paradigm in ... [https://www.sciencedirect.com/science/article/pii/S2590098625000181]
- 47. PROTAC Degraders in Clinical Trails: 2025 Update [https://www.biochempeg.com/article/434.html]
- 48. PROTAC 2.0: Expanding the frontiers of targeted protein ... [https://pubmed.ncbi.nlm.nih.gov/40348076/]
- 49. A PROTAC Patent Compound Dataset | Scientific Data [https://www.nature.com/articles/s41597-025-06136-9]
- 50. : enhanced Molecular - glues interactions and cell... protein protein [https://link.springer.com/article/10.1007/s00044-022-02882-2]
- 51. and PROTACs in targeted Molecular ... glues protein degradation [https://pubmed.ncbi.nlm.nih.gov/40907798/]
- 52. Molecular glues: A new path to the world of the unattainable [https://www.sciencedirect.com/science/article/abs/pii/S0141813025094188]
- 53. tackle undruggable targets | CAS Molecular glues [https://www.cas.org/ja/resources/cas-insights/molecular-glues-protein-degraders]
- 54. A high-throughput compatible workflow for the biochemical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12167809/]
- 55. Degraders & Molecular Glues Part 1 | April 15-16, 2025 [https://www.drugdiscoverychemistry.com/25/protein-degradation-molecular-glues]
- 56. Quantitative analysis of global protein stability rates in tissues [https://www.nature.com/articles/s41598-020-72410-y]
- 57. Proteolysis‐targeting chimeras in cancer therapy [https://pmc.ncbi.nlm.nih.gov/articles/PMC12535346/]
- 58. Advancing PROTAC Discovery Through Artificial Intelligence [https://www.mdpi.com/1424-8247/18/12/1793]
- 59. A Critical Appraisal of Quantitative Studies of Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3483835/]
- 60. Disruption of the ATP-dependent unfoldase ClpX reverses ... [https://www.nature.com/articles/s41467-025-61412-x]
- 61. A suite of mathematical solutions to describe ternary complex ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7650257/]
- 62. Affinity and cooperativity modulate ternary complex ... [https://www.nature.com/articles/s41467-023-39904-5]
- 63. Ternary Complex Formation [https://www.promega.com/applications/small-molecule-drug-discovery/protein-degradation-drug-discovery/e3-ternary-complex/]
- 64. Current strategies for the design of PROTAC linkers [https://pmc.ncbi.nlm.nih.gov/articles/PMC9400730/]
- 65. The Future of PROTAC Research: Emerging Opportunities ... [https://panomebio.com/blog/the-future-of-protac-research-emerging-opportunities-and-challenges/]
- 66. Targeted protein degradation technologies: Emerging ... [https://www.sciopen.com/article/10.26599/NR.2025.94907983]
- 67. Mechanisms for regulating deubiquitinating enzymes - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3970886/]
- 68. Deubiquitinating enzyme [https://en.wikipedia.org/wiki/Deubiquitinating_enzyme]
- 69. Recent advances in the development of deubiquitinases ... [https://www.sciencedirect.com/science/article/abs/pii/S0223523424000412]
- 70. The multifaceted roles of deubiquitinating enzymes (DUBs) ... [https://www.nature.com/articles/s41419-025-08052-7]
- 71. Deubiquitinating Enzymes in Osteoarthritis [https://orthopedicreviews.openmedicalpublishing.org/article/144733]
- 72. Deubiquitinating enzymes as therapeutic targets in diabetic ... [https://www.sciencedirect.com/science/article/pii/S0165614725001580]
- 73. Deubiquitinase-Targeting Chimeras (DUBTACs) as a ... [https://pubmed.ncbi.nlm.nih.gov/40135978/]
- 74. An overview of PROTACs: a promising drug discovery ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9763089/]
- 75. Characteristic roadmap of linker governs the rational ... [https://www.sciencedirect.com/science/article/pii/S2211383524001357]
- 76. PROTACs VS. Traditional Small Molecule Inhibitors [https://www.biochempeg.com/article/233.html]
- 77. Targeted protein degradation: advances in drug discovery ... [https://www.nature.com/articles/s41392-024-02004-x]
- 78. Chemically Induced Cellular Proteolysis: An Emerging ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6277563/]
- 79. Proteolysis-targeting chimeras in biotherapeutics [https://www.sciencedirect.com/science/article/abs/pii/S0223523423004130]
- 80. ATP-dependent conjugation of reticulocyte proteins with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC348495/]
- 81. Proposed role of ATP in protein breakdown: conjugation ... [https://snu.elsevierpure.com/en/publications/proposed-role-of-atp-in-protein-breakdown-conjugation-of-protein-/]
- 82. Mechanisms of Protein Degradation [https://www.thermofisher.com/us/en/home/life-science/cell-analysis/cell-analysis-learning-center/protein-degradation-resource-center/mechanisms-protein-degradation.html]
- 83. The Ubiquitin–Proteasome System and the Autophagic– ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3405832/]
- 84. Interplay Between the Autophagy-Lysosomal Pathway and ... [https://www.frontiersin.org/journals/cellular-neuroscience/articles/10.3389/fncel.2018.00126/full]
- 85. Tau degradation: The ubiquitin–proteasome system versus ... [https://www.sciencedirect.com/science/article/abs/pii/S0301008213000154]
- 86. Structure and mechanism of ATP-dependent proteases [https://www.sciencedirect.com/science/article/abs/pii/S1367593199000137]
- 87. Slicing a protease: Structural features of the ATP-dependent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2242575/]
- 88. Lon protease family [https://en.wikipedia.org/wiki/Lon_protease_family]
- 89. Structure and the mode of activity of Lon proteases from ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9013511/]
- 90. Recent Advances in Understanding the Structural and ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2022.837528/full]
- 91. Cryo-EM structure of transmembrane AAA+ protease FtsH ... [https://www.nature.com/articles/s42003-022-03213-2]
- 92. ATP-dependent Clp protease proteolytic subunit [https://en.wikipedia.org/wiki/ATP-dependent_Clp_protease_proteolytic_subunit]
- 93. The Identification of Novel Mutations in ATP-Dependent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12196252/]
- 94. Proteome-wide structural analysis quantifies structural ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10760455/]
- 95. ATP-dependent Proteases Differ Substantially in Their Ability ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2707231/]
- 96. Sequence and structure of Clp P, the proteolytic ... [https://www.sciencedirect.com/science/article/pii/S0021925819383784]
- 97. An updated review on the role of small molecules in ... [https://www.sciencedirect.com/science/article/abs/pii/S0223523425001357]
- 98. Comparing proteins by their internal dynamics [https://www.sciencedirect.com/science/article/abs/pii/S1571064512001327]
- 99. Functional mechanics of the ATP-dependent Lon protease [https://pmc.ncbi.nlm.nih.gov/articles/PMC2443057/]
- 100. Structure of the Whole Cytosolic Region of ATP-Dependent ... [https://www.sciencedirect.com/science/article/pii/S1097276506002681]
- 101. Detection and characterization of two ATP-dependent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2537469/]
- 102. Benchmarking of Quantitative Proteomics Workflows for ... [https://www.sciencedirect.com/science/article/pii/S153594762500043X]
- 103. PROTACs: An Emerging Therapeutic Modality in Precision ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9424844/]
- 104. PROTACs to Address the Challenges Facing Small Molecule ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7779748/]
- 105. PROTAC 2.0: Expanding the frontiers of targeted protein ... [https://www.sciencedirect.com/science/article/abs/pii/S1359644625000893]
- 106. PROTAC Technology as a New Tool for Modern ... [https://www.mdpi.com/1420-3049/30/10/2123]
- 107. Synthesis, biological evaluation and clinical trials of ... [https://www.nature.com/articles/s42004-025-01598-9]
- 108. Emerging protein degradation strategies: expanding the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8344007/]
- 109. Degradation from the outside in: Targeting extracellular ... [https://www.sciencedirect.com/science/article/pii/S2451945621001094]
- 110. Emerging Targeted Protein Degradation Technologies [https://www.bocsci.com/blog/overview-expanding-the-landscape-of-targeted-protein-degradation-tpd/?srsltid=AfmBOoo--dd-6mIlwTo6rVQPzNaruZru18TwjUCdlcW7apwxXZkFOcLU]