Ubiquitin and Ubiquitin-like Signaling: Evolutionary Origins, Mechanisms, and Pathogen Interactions in Prokaryotes vs. Eukaryotes
This article provides a comprehensive comparison of ubiquitin and ubiquitin-like (Ubl) protein conjugation systems across eukaryotes and prokaryotes.
Ubiquitin and Ubiquitin-like Signaling: Evolutionary Origins, Mechanisms, and Pathogen Interactions in Prokaryotes vs. Eukaryotes
Abstract
This article provides a comprehensive comparison of ubiquitin and ubiquitin-like (Ubl) protein conjugation systems across eukaryotes and prokaryotes. For researchers and drug development professionals, we synthesize foundational knowledge on the eukaryotic ubiquitin-proteasome system (UPS) with groundbreaking discoveries of Ubl pathways in bacteria, such as Pup and newly identified bacterial E1-E2-E3 cascades. The scope spans evolutionary origins, structural and mechanistic parallels, methodological approaches for studying these systems, and how pathogens manipulate host ubiquitination. We highlight the implications of these fundamental differences and similarities for developing novel therapeutic strategies, including targeting bacterial virulence mechanisms and the bacterial ubiquitination machinery itself.
Ubiquitin and Ubls: From Eukaryotic Hallmark to Prokaryotic Ancestors
The ubiquitin-proteasome system (UPS) represents the primary pathway for targeted protein degradation in eukaryotic cells, operating as a sophisticated master regulator of cellular processes ranging from cell cycle progression to stress response [1] [2]. This system typically recognizes specific protein substrates and tags them with polyubiquitin chains, marking them for destruction by the proteasome—a massive proteolytic complex [3]. The UPS is remarkable not only for its precision in selecting targets among thousands of cellular proteins but also for its ability to regulate virtually all aspects of eukaryotic biology through both proteolytic and non-proteolytic mechanisms [2]. The discovery that bacteria possess ubiquitination-like pathways with striking mechanistic parallels to eukaryotic systems has revolutionized our understanding of the evolutionary origins of this complex machinery, suggesting these pathways arose first in bacteria before being adopted and refined by eukaryotes for expanded regulatory purposes [4] [5]. This comparison guide examines the canonical eukaryotic UPS, its core components, operational dogma, and experimental methodologies for studying its function, providing researchers with a comprehensive framework for understanding this essential biological system.
Core Components of the Eukaryotic Ubiquitin-Proteasome System
The Enzymatic Ubiquitination Cascade
The ubiquitination process employs a three-tiered enzymatic cascade comprising E1 (ubiquitin-activating), E2 (ubiquitin-conjugating), and E3 (ubiquitin-ligating) enzymes that work sequentially to tag substrate proteins [6] [2] [7].
E1 Activating Enzymes: The human genome encodes two ubiquitin E1 enzymes (UBA1 and UBA6) that initiate the ubiquitination cascade [6]. These enzymes use ATP to activate ubiquitin through the formation of a high-energy thioester bond between a catalytic cysteine residue in E1 and the C-terminal glycine of ubiquitin [6] [3]. This energy-dependent step makes the ubiquitin molecule competent for subsequent transfer reactions.
E2 Conjugating Enzymes: Approximately 38 E2 enzymes in humans receive the activated ubiquitin from E1 through transesterification, forming a similar thioester linkage [6]. E2 enzymes function not merely as passive carriers but play an active role in determining the topology of polyubiquitin chains, thereby influencing the fate of modified substrates [6].
E3 Ligating Enzymes: The human genome contains approximately 700 E3 ubiquitin ligases, which provide substrate specificity by recognizing target proteins and facilitating ubiquitin transfer from E2 to substrate lysine residues [6]. E3 ligases fall into three main subfamilies: RING E3s that act as scaffolding molecules to bring ubiquitin-charged E2 enzymes in close proximity to substrates; HECT E3s that form a transient thioester intermediate with ubiquitin before transfer; and RING-Between-RING (RBR) E3s that function as hybrids between RING and HECT mechanisms [6].
Table 1: Major E3 Ubiquitin Ligase Families and Their Characteristics
| E3 Family | Catalytic Mechanism | Representative Members | Key Features |
|---|---|---|---|
| RING E3s | Scaffold for direct ubiquitin transfer from E2 to substrate | SCF complexes, MDM2 | Largest E3 family; often multi-subunit complexes |
| HECT E3s | Forms E3~Ub thioester intermediate before substrate transfer | NEDD4, HACE1 | Catalytic cysteine residue; regulates diverse trafficking pathways |
| RBR E3s | Hybrid mechanism with RING and HECT-like features | PARKIN, ARIH1 | RING1 domain binds E2; RING2 domain has catalytic cysteine |
The Proteasome: Architecture and Degradation Machinery
The 26S proteasome is a 2.6 MDa molecular machine responsible for degrading ubiquitinated proteins [1]. It consists of two primary subcomplexes:
20S Core Particle (CP): This barrel-shaped structure contains the proteolytic active sites within its interior chamber, sequestered from the cellular environment to prevent uncontrolled protein degradation [1]. The CP is composed of four stacked heptameric rings: two identical outer α-rings that regulate gate opening, and two identical inner β-rings that contain the protease active sites [1].
19S Regulatory Particle (RP): This cap complex recognizes ubiquitinated substrates, removes ubiquitin chains, unfolds target proteins, and translocates them into the CP for degradation [1]. The RP contains at least 19 subunits organized into two subcomplexes: the "base" with six AAA+ ATPases (Rpt1-6) that unfold substrates, and the "lid" with ubiquitin receptors and deubiquitinating enzymes [1].
Table 2: Proteasome Subunits and Their Functions
| Subcomplex | Subunit Category | Representative Subunits | Primary Function |
|---|---|---|---|
| 20S Core Particle | α-subunits | PSMA1-PSMA7 (human) | Form gated channel for substrate entry |
| 20S Core Particle | β-subunits | PSMB1-PSMB7 (human) | Contain proteolytic active sites |
| 19S Regulatory Particle Base | AAA+ ATPases | Rpt1-Rpt6 (yeast) | Substrate unfolding and translocation |
| 19S Regulatory Particle Base | Non-ATPases | Rpn1, Rpn2, Rpn10, Rpn13 | Ubiquitin receptor docking |
| 19S Regulatory Particle Lid | PCI domain proteins | Rpn3, Rpn5-Rpn7, Rpn9, Rpn12 | Structural scaffolding |
| 19S Regulatory Particle Lid | MPN domain proteins | Rpn8, Rpn11 | Deubiquitination (Rpn11) |
The following diagram illustrates the complete ubiquitination and degradation pathway:
The Central Dogma: ubiquitin Code and Signal Interpretation
The Ubiquitin Code: A Complex Signaling Language
Ubiquitination creates a sophisticated post-translational modification code that extends far beyond a simple degradation signal [2] [7]. The "ubiquitin code" consists of:
Monoubiquitination: Single ubiquitin modifications that typically alter protein activity, localization, or interactions without triggering degradation [7].
Polyubiquitin Chains: Ubiquitin polymers formed through isopeptide bonds between the C-terminus of one ubiquitin and a lysine residue of another [2] [7]. With seven lysine residues in ubiquitin (Lys6, Lys11, Lys27, Lys29, Lys33, Lys48, Lys63) plus the N-terminal methionine (Met1), eight structurally and functionally distinct chain types can be formed [2] [7].
Chain Topology Diversity: Beyond homogeneous chains, cells generate heterogeneous (mixed linkage) and branched (multiple ubiquitination sites on a single ubiquitin) chains that expand the coding potential [7].
Table 3: Ubiquitin Chain Linkages and Their Primary Functions
| Linkage Type | Abundance in Cells | Primary Functions | Key Readers/Effectors |
|---|---|---|---|
| Lys48 | >50% of all linkages | Proteasomal degradation | Proteasome receptors |
| Lys63 | Second most abundant | NF-κB signaling, DNA repair, endocytosis | TAB2/TAB3, ESCRT components |
| Lys11 | Moderate | ER-associated degradation, cell cycle | Proteasome, Cdc48/p97 |
| Met1 (Linear) | Low | NF-κB activation, inflammation | NEMO/IKK complex |
| Lys29 | Low | Proteasomal degradation (with Lys48) | Proteasome receptors |
| Lys33 | Low | AKT signaling, trafficking | - |
| Lys6 | Low | DNA damage repair, mitophagy | - |
| Lys27 | Low | Immune signaling, autophagy | - |
Interpretation and Reversal of Ubiquitin Signals
The ubiquitin code is interpreted by ubiquitin-binding domains (UBDs) present in hundreds of cellular proteins [7]. These domains recognize specific ubiquitin modifications and translate them into appropriate cellular responses, such as proteasomal targeting, altered protein interactions, or changes in subcellular localization [7].
Ubiquitination is counterbalanced by deubiquitinating enzymes (DUBs) that remove ubiquitin modifications, providing dynamic regulation [6] [2]. The human genome encodes approximately 100 DUBs categorized into five major families: ubiquitin-specific proteases (USP), ubiquitin C-terminal hydrolases (UCH), ovarian tumor proteases (OTU), Machado-Joseph disease proteases (MJD), and JAMM/MPN metalloproteases [6]. DUBs not only reverse ubiquitination events but also process ubiquitin precursors and edit ubiquitin chains to refine signaling outcomes [6].
The following diagram illustrates the complexity of ubiquitin chain architecture and recognition:
Experimental Methods for UPS Analysis
Key Methodologies for Studying UPS Components
Research into the ubiquitin-proteasome system employs diverse experimental approaches to decipher its complexity:
Mass Spectrometry-Based Proteomics: Advanced quantitative mass spectrometry techniques, including SILAC (stable isotope labeling with amino acids in cell culture) and TMT (tandem mass tag) labeling, enable comprehensive identification and quantification of ubiquitination sites [7]. The AQUA (absolute quantification) method using labeled ubiquitin peptide standards provides precise measurement of specific ubiquitin chain linkages [7].
Linkage-Specific Reagents: Antibodies specific for Met1-, Lys11-, Lys48-, and Lys63-linked chains have been developed and validated, allowing immunohistochemical and immunoblotting applications [7]. Similarly, tandem ubiquitin-binding entities (TUBEs) serve as affinity reagents that protect polyubiquitinated proteins from deubiquitination during purification [8].
Structural Biology Approaches: Cryo-electron microscopy (cryo-EM) has been instrumental in determining the architecture of the 26S proteasome at near-atomic resolution [1]. X-ray crystallography continues to provide high-resolution structures of individual UPS components, such as the recent structural analysis of bacterial E1:E2:Ubl complexes that reveal evolutionary conservation [5].
Protein Engineering Techniques: Unnatural amino acid incorporation, expressed protein ligation (EPL), and phage/yeast display systems have generated specialized tools for probing UPS mechanisms, including ubiquitin variants (Ubvs) that selectively inhibit specific DUBs or E3 ligases [8].
Research Reagent Solutions for UPS Studies
Table 4: Essential Research Reagents for UPS Investigation
| Reagent Category | Specific Examples | Primary Applications | Key Features |
|---|---|---|---|
| E1 Inhibitors | PYR-41, PYZD-4409 | Block global ubiquitination | Irreversible covalent modification of E1 active site |
| E2 Inhibitors | CC0651, NSC697923 | Selective pathway inhibition | CC0651: allosteric inhibitor of CDC34; NSC697923: blocks UBE2N~Ub formation |
| Proteasome Inhibitors | Bortezomib, Carfilzomib | FDA-approved cancer therapeutics | Reversible vs. irreversible binding to proteolytic sites |
| Linkage-Specific Antibodies | Anti-K48, Anti-K63, Anti-M1 Ub | Chain linkage detection in cells | Enable monitoring of specific chain types in signaling |
| DUB Probes | Ubiquitin-based activity probes | DUB activity profiling | Covalent trapping of active DUBs for identification |
| Ubiquitin Variants (Ubvs) | Engineered Ub mutants | Specific pathway interruption | Phage display-derived inhibitors of E3s or DUBs |
| Tandem Ubiquitin-Binding Entities (TUBEs) | Multi-UBD fusion proteins | PolyUb protein enrichment | Protect Ub chains from DUBs during purification |
UPS-Targeted Therapeutic Applications
The ubiquitin-proteasome system presents numerous attractive targets for therapeutic intervention, particularly in oncology and neurodegenerative diseases [6] [2]. Several targeting strategies have shown clinical promise:
Proteasome Inhibitors: Drugs like bortezomib and carfilzomib inhibit the proteolytic activity of the 20S proteasome, leading to accumulation of polyubiquitinated proteins and apoptosis in rapidly dividing cells, particularly multiple myeloma cells [6].
E1 Enzyme Targeting: The NEDD8-activating enzyme (NAE) inhibitor MLN4924 (pevonedistat) has shown promise in clinical trials by blocking neddylation of cullins, thereby impairing the function of cullin-RING ligases (CRLs) and inducing DNA damage in cancer cells [6].
E3 Ligase Modulation: Molecular glues such as thalidomide and related immunomodulatory drugs (IMiDs) redirect the CRL4CRBN E3 ligase to target novel substrates for degradation, demonstrating the therapeutic potential of targeted protein degradation [8].
DUB Inhibition: Specific inhibitors of USP7, USP14, and other DUBs are under investigation for cancer therapy, aiming to stabilize tumor suppressors or disrupt oncogenic signaling pathways [6].
The following diagram illustrates major therapeutic targeting strategies within the UPS:
Evolutionary Perspective: Bacterial Antecedents of Eukaryotic UPS
Recent research has revealed that bacteria possess complete ubiquitination-like pathways with remarkable architectural and mechanistic parallels to eukaryotic systems [4] [5]. The discovery of Type II BilABCD operons in bacteria encoding E1, E2, Ubl, and DUB proteins demonstrates that these pathways likely originated in bacteria before being adopted by eukaryotes [5]. Key findings include:
Structural Conservation: Bacterial E1 proteins display the same domain architecture (IAD, AAD, and CYS domains) as eukaryotic E1s, despite limited sequence similarity [5].
Functional Parallels: Bacterial ubiquitination systems participate in antiviral defense, modifying virion structural proteins to provide immunity against phage infection—a functional analogy to eukaryotic immune signaling through ubiquitin [5].
Evolutionary Insight: These bacterial pathways challenge previous models that placed the origin of ubiquitination systems in archaea and suggest that eukaryotic ubiquitination machinery evolved from bacterial antecedents [5].
This evolutionary perspective provides researchers with a more comprehensive framework for understanding the fundamental principles of ubiquitin signaling across domains of life and may facilitate the development of novel experimental approaches based on these more ancient, simplified systems.
The canonical eukaryotic ubiquitin-proteasome system represents a sophisticated protein regulatory network that governs virtually all aspects of cell biology through both proteolytic and non-proteolytic mechanisms. Its core components—the E1-E2-E3 enzymatic cascade and the 26S proteasome—operate with remarkable specificity to control protein stability, activity, and interactions. The complexity of the ubiquitin code, with its diverse chain topologies and modifications, enables precise signal encoding that is interpreted by specialized receptors and reversed by deubiquitinating enzymes. From an evolutionary perspective, the discovery of analogous systems in bacteria highlights the ancient origins of this regulatory paradigm. For researchers and drug development professionals, understanding the detailed composition, operational dogma, and experimental methodologies of the UPS provides critical insights for developing novel therapeutic strategies that target this system in cancer, neurodegenerative disorders, and other human diseases.
Ubiquitin-like proteins (UBLs) are a family of small proteins involved in the post-translational modification of other proteins, thereby regulating an enormous range of physiological processes in eukaryotic cells [9] [10]. These modifiers share a common three-dimensional core structure known as the β-grasp fold and are conjugated to target proteins via a conserved enzymatic cascade [9] [11]. The first discovered and best-understood UBL is ubiquitin itself, best known for targeting proteins for degradation by the proteasome [9] [10]. Following the discovery of ubiquitin, many evolutionarily related UBLs were described, including SUMO (Small Ubiquitin-like Modifier), NEDD8 (Neural precursor cell-expressed, Developmentally down-regulated 8), and ISG15 (Interferon-Stimulated Gene 15) [9] [12].
These UBLs, while mechanistically parallel, are functionally distinct, controlling diverse processes such as transcription, the cell cycle, DNA repair, autophagy, immune responses, and inflammation [9] [12] [13]. Mounting evidence suggests that UBL-protein modification evolved from prokaryotic sulfurtransferase systems or related enzymes [10] [11]. Proteins similar to UBL-conjugating enzymes appear to have been present in the last universal common ancestor, indicating that UBL-protein conjugation is not a eukaryotic invention [10]. This review provides a comparative analysis of the major eukaryotic UBLs—SUMO, NEDD8, and ISG15—focusing on their distinct roles, regulatory mechanisms, and experimental approaches for their study, framed within the broader evolutionary context of ubiquitin-like systems.
The following table provides a systematic comparison of the core characteristics of SUMO, NEDD8, and ISG15, highlighting their structural and functional specialization.
Table 1: Core Characteristics of Major Eukaryotic Ubls
| Feature | SUMO | NEDD8 | ISG15 |
|---|---|---|---|
| Sequence Identity to Ubiquitin | ~18% (SUMO1) [10] | ~55% [10] | ~32-37% (per domain) [10] [12] |
| Protein Structure | Single UBL domain [14] | Single UBL domain [14] | Tandem UBL domains [12] [14] |
| Primary Known Functions | Transcription, DNA repair, nuclear transport, stress response [9] [13] | Activation of cullin-RING E3 ubiquitin ligases (CRLs) [9] [11] | Antiviral immunity, DNA damage response, autophagy, translation regulation [9] [12] |
| E1 Activating Enzyme | Heterodimer Aos1/Uba2 [11] | Heterodimer Uba3/NAE1 [11] | UBA7 (UBE1L) [12] |
| E2 Conjugating Enzyme | Ubc9 [11] [14] | Ubc12 [11] [14] | UBCH8 (UBE2L6) [12] [14] |
| Representative E3 Ligases | PIAS, RanBP2, ZNF451 [14] | RBX1/2, DCN1 [14] | HERC5, EFP (TRIM25), HHARI [12] [14] |
| Specific Proteases | SENPs, DeSI, USPL1 [14] | NEDP1, DEN1, SENP8 [14] | USP18 (UBP43) [12] [14] |
| Chain Formation | Yes (poly-SUMO) [9] [15] | Yes [9] | Yes (mixed Ub-ISG15 chains) [12] |
Ubl-Specific Conjugation Pathways and Cellular Functions
The SUMOylation Pathway
SUMOylation is crucial for nuclear processes, particularly transcription, DNA repair, and the maintenance of genome stability [13]. A unique feature of SUMOylation is the common use of a single E2 conjugating enzyme, Ubc9, which interacts directly with a consensus motif (ΨKxE/D, where Ψ is a hydrophobic residue) on the substrate [14]. SUMO can be conjugated as a monomer or form poly-SUMO chains, which act as scaffolds for the recruitment of proteins containing SUMO-interacting motifs (SIMs) [14] [15].
Figure 1: The SUMO Conjugation Pathway. SUMO is activated by the E1 heterodimer, transferred to the E2 enzyme Ubc9, and ligated to target substrates with the help of E3s, ultimately regulating key nuclear processes.
The NEDD8ylation Pathway
NEDD8, which shares the highest sequence identity with ubiquitin, primarily regulates the ubiquitin-proteasome system itself [15]. Its best-characterized function is the activation of cullin proteins, which are the core scaffolding components of Cullin-RING E3 ubiquitin ligases (CRLs) [9] [11]. NEDD8 is activated by its specific E1 (Uba3/NAE1) and E2 (Ubc12) enzymes. Conjugation of NEDD8 to a cullin, known as "neddylation," induces a conformational change that promotes the ubiquitin ligase activity of the CRL, thereby stimulating the polyubiquitination and degradation of proteins that control cell cycle progression and signaling [11].
Figure 2: The NEDD8 Conjugation Pathway. NEDD8 modification of cullins activates CRLs, which in turn drive the ubiquitination and degradation of a wide array of downstream proteins.
The ISGylation Pathway
ISG15 is a unique UBL composed of two ubiquitin-like domains [12] [14]. It is a central component of the innate immune response, strongly induced by interferons and viral infection [12]. Its conjugation cascade involves the E1 enzyme UBA7, the E2 enzyme UBCH8, and E3s such as HERC5. ISG15 can antagonize viral replication by conjugating to viral proteins or host factors, but it also has emerging roles in DNA damage response, autophagy, and cancer stem cell regulation [12] [16]. ISG15 can function both as a conjugated modifier and as a free cytokine, stimulating IFNγ secretion [12].
Figure 3: The ISG15 Conjugation Pathway. ISG15 is induced by interferon and conjugated to target proteins, playing key roles in antiviral defense and other cellular processes.
Experimental Data and Methodologies for Ubl Research
Key Experimental Protocols
Studying UBL modifications presents challenges, including the low abundance of endogenous conjugates and the activity of deconjugating enzymes. The following protocols are central to the field.
Table 2: Key Experimental Protocols in Ubl Research
| Method | Core Principle | Key Steps | Application & Insight |
|---|---|---|---|
| Denaturing Immuno-affinity Purification [15] | Use of epitope-tagged UBLs and purification under denaturing conditions. | 1. Express tagged UBL (e.g., HIS, FLAG, biotin) in cells.2. Lyse cells under denaturing conditions (e.g., SDS, Guanidine HCl).3. Purify conjugates with affinity resin (e.g., Ni-NTA, Streptavidin).4. Identify substrates by mass spectrometry. | Inactivates isopeptidases, reduces non-covalent interactors, and allows identification of low-abundance substrates. |
| In vivo Biotinylation (bioUbL) [15] | In vivo biotinylation of a short tag (AviTag) on the UBL by co-expressed BirA ligase. | 1. Co-express bio-tagged UBL and BirA from a multicistronic vector.2. Lyse under denaturing conditions.3. Capture biotinylated conjugates with streptavidin beads.4. Analyze by western blot or MS. | High-affinity streptavidin-biotin interaction enables stringent washes and minimal background. Ideal for proteomic studies in cells and model organisms. |
| Activity-Based Profiling with Chemical Synthesis [14] | Chemical synthesis of UBLs with tailored probes (e.g., fluorogenic, cross-linkers). | 1. Synthesize UBLs with C-terminal electrophilic traps or fluorogenic groups (e.g., AMC).2. Incubate with enzymes or cell lysates.3. Monitor activity via fluorescence or covalent capture. | Provides homogeneously modified substrates and probes to study enzyme kinetics, specificity, and inhibition. |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Reagents for Ubl Research
| Reagent / Tool | Function | Specific Examples |
|---|---|---|
| Epitope-Tagged Ubl Plasmids | Enables overexpression and purification of UBL conjugates. | HIS-SUMO, FLAG-NEDD8, Bio-ISG15 (for bioUbL system) [15]. |
| Protease Inhibitors | Protects UBL conjugates from deconjugating enzymes during lysis. | Inclusion of N-ethylmaleimide (NEM) or iodoacetamide to alkylate cysteine proteases [15]. |
| Activity-Based Probes | Monitors enzymatic activity and identifies interacting proteins. | UBL-AMC conjugates (e.g., Ub-AMC, ISG15-AMC) for kinetic assays; suicide inhibitors [17] [14]. |
| Linkage-Specific Antibodies | Detects endogenous UBL conjugates and specific chain linkages. | Antibodies against SUMO1, SUMO2/3, ISG15, or di-Gly remnant after tryptic digest (for MS) [15]. |
| Recombinant E1/E2/E3 Enzymes | Reconstitutes conjugation and deconjugation pathways in vitro. | Uba2/Aos1 (SUMO E1), Uba3/Nae1 (NEDD8 E1), Ubc9 (SUMO E2), USP18 (DeISGylase) [11] [14]. |
| Specific Small Molecule Inhibitors | Selectively inhibits UBL pathways for functional studies. | MLN4924 (NEDD8 E1 inhibitor) [11], GRL0617 (SARS-CoV-2 PLpro inhibitor affecting Ubl domain) [17]. |
Evolutionary Context: Prokaryotic Antecedents to Eukaryotic Ubls
The eukaryotic UBL conjugation system is believed to have evolved from ancient prokaryotic sulfur-transfer pathways [9] [10]. Key evidence for this includes:
- Structural and Mechanistic Homology: The bacterial sulfur carrier proteins ThiS and MoaD, involved in thiamine and molybdopterin biosynthesis, share the β-grasp fold and a similar activation mechanism involving adenylation and thiocarboxylate formation with eukaryotic UBLs [9] [10] [11].
- A Molecular Fossil: The eukaryotic UBL URM1 functions both as a protein modifier and a sulfur carrier, establishing a direct evolutionary link between these two systems [9] [10].
- Prokaryotic Ubl-like Systems: Some archaea and bacteria possess simplified UBL systems. For example, SAMPs in archaea and Pup in actinobacteria perform ubiquitin-like tagging for proteasomal degradation, though Pup is intrinsically disordered and lacks the β-grasp fold [9].
Figure 4: Evolutionary Trajectory of Ubl Systems. Eukaryotic UBL pathways evolved from prokaryotic sulfur-transfer systems involved in cofactor biosynthesis, through gene duplication and functional diversification.
SUMO, NEDD8, and ISG15 exemplify the functional diversification of ubiquitin-like proteins in eukaryotes. While they share a common structural fold and a conserved enzymatic cascade for conjugation, they regulate distinct cellular processes: SUMO in nuclear organization and stress, NEDD8 in ubiquitin ligase activation, and ISG15 in immunity and beyond. This specialization is enforced by dedicated sets of E1, E2, and E3 enzymes, as well as specific proteases. Research in this field relies on sophisticated methods like denaturing affinity purification and chemical biology tools to overcome the challenges of studying these dynamic modifications. The evolutionary perspective, which roots these complex eukaryotic systems in primordial prokaryotic sulfur metabolism, provides a unifying framework for understanding the biology of these critical regulatory proteins. Continued comparative research will deepen our understanding of their roles in health and disease and open new avenues for therapeutic intervention.
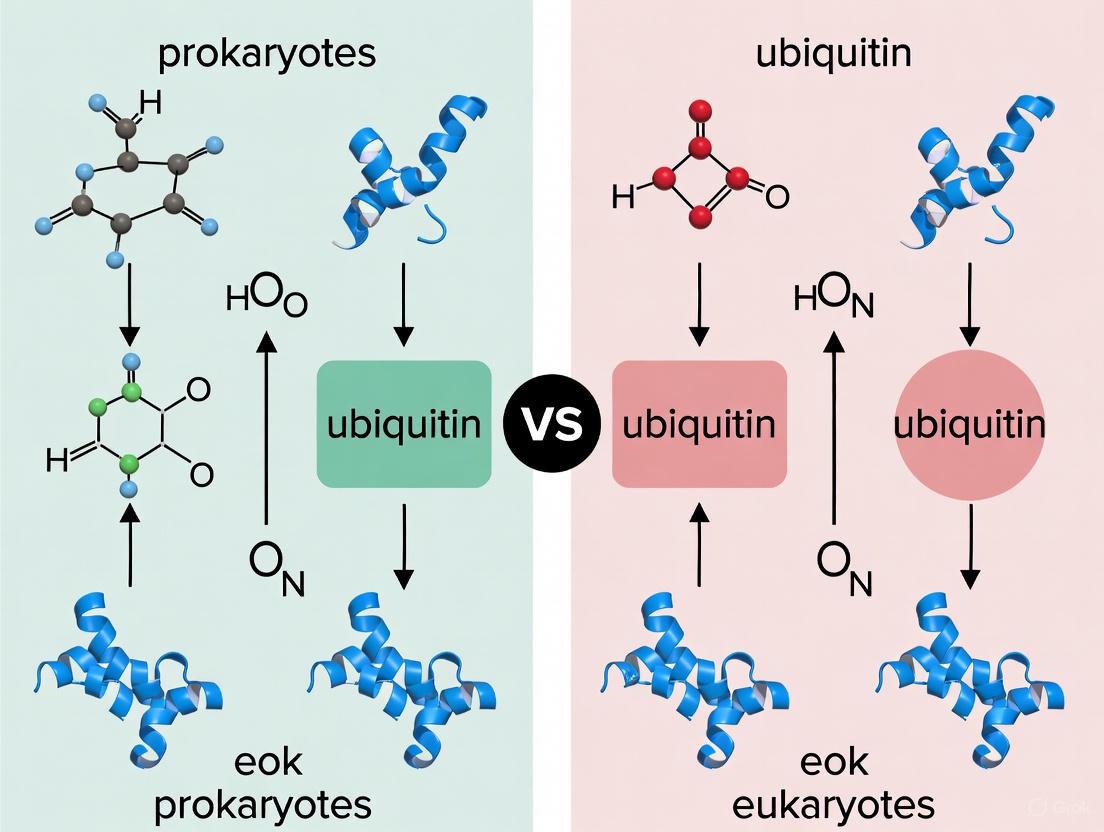
For decades, the ubiquitin-proteasome system was considered a hallmark of eukaryotic complexity, a sophisticated regulatory mechanism absent from prokaryotic organisms. This historical paradigm positioned ubiquitin as a eukaryote-specific modifier, fundamentally distinguishing eukaryotic cell biology from its supposedly simpler prokaryotic counterparts. The discovery of the ubiquitin-like protein (Pup) in prokaryotes has fundamentally challenged this long-standing assumption [18]. This revelation emerged from studies on Mycobacterium tuberculosis, where Pup was found to target proteins for proteolysis by the bacterial proteasome, representing a functional analogue to the eukaryotic ubiquitin system [18]. This discovery not only overturned the notion of ubiquitin as exclusively eukaryotic but also opened new avenues for understanding the evolution of post-translational modification systems and their implications for bacterial pathogenesis and drug development.
The subsequent identification of additional ubiquitin-like modifiers in archaea (SAMPs) and Thermus (TtuB), which share a common β-grasp fold with ubiquitin despite sequence differences, further eroded the eukaryotic monopoly on ubiquitin-like signaling [19] [20]. These findings suggest that the evolutionary origins of ubiquitin-like protein modifiers predate the divergence of life's domains, with prokaryotes possessing their own sophisticated protein-tagging systems that affect cellular processes, including targeted proteolysis [18]. This guide provides a comprehensive comparison between the eukaryotic ubiquitin system and its prokaryotic counterparts, examining their mechanisms, functions, and the experimental approaches used to characterize them.
Comparative Analysis of Ubiquitin and Prokaryotic Ubiquitin-Like Proteins
Core Structural and Functional Divergence
Table 1: Fundamental Comparison of Eukaryotic Ubiquitin and Prokaryotic Ubiquitin-like Proteins
| Feature | Eukaryotic Ubiquitin | Prokaryotic Pup (Mycobacteria) | Prokaryotic SAMPs/TtuB (Archaea/Thermus) |
|---|---|---|---|
| Protein Size & Structure | 76 amino acids; compact, globular β-grasp fold [21] | ~64 amino acids; intrinsically disordered [19] | β-grasp fold similar to Ub, but different sequence [19] |
| C-terminal Motif | Gly-Gly [21] | Gly-Gly-Gln (deamidated to Gly-Gly-Glu) [19] | Gly-Gly (in TtuB and some SAMPs) [19] |
| Conjugation Bond | Isopeptide bond (via C-terminal Gly α-carboxylate) [21] | Isopeptide bond (via C-terminal Glu γ-carboxylate) [19] | Isopeptide bond (presumably via C-terminal Gly) [19] |
| Enzymatic Cascade | E1-E2-E3 enzyme cascade (ATP-dependent) [22] | Deamidase (Dop) + Ligase (PafA) [19] | Appears streamlined; requires E1 but not E2/E3 [19] |
| Primary Function | Diverse: Proteasomal degradation, signaling, endocytosis [22] [23] | Targets proteins for bacterial proteasome degradation [18] | Protein modification & sulfur transfer for biosynthesis [19] |
| Proteasome Association | 26S proteasome (20S core + 19S regulatory cap) [18] | 20S core + ATPase (Mpa) regulator [18] | Not fully characterized |
Enzymatic Machinery and Conjugation Pathways
The fundamental distinction between these systems lies in their enzymatic machineries. The eukaryotic ubiquitination cascade is a three-step process involving E1 (activating), E2 (conjugating), and E3 (ligating) enzymes, which results in an isopeptide bond between the C-terminal glycine of ubiquitin and a lysine residue on the target protein [22] [21]. This system is highly complex, with humans encoding two E1s, ~35 E2s, and approximately 600-700 E3s, allowing for immense specificity and regulation [6] [24].
In contrast, the best-characterized prokaryotic system, pupylation, requires only two enzymes: the deamidase Dop (deamidase of Pup) and the ligase PafA (proteasome accessory factor A) [19]. Dop deamidates the C-terminal glutamine of Pup to glutamate in an ATP-dependent manner, and PafA then catalyzes the ATP-dependent formation of an isopeptide bond between the γ-carboxylate of this glutamate and the ε-amino group of a lysine residue on the target protein [19]. This mechanism is distinct from ubiquitylation in both enzymology and chemistry, despite the analogous functional outcome of targeting proteins for proteasomal degradation [19].
Table 2: Comparative Enzymology of Ubiquitin-like Modification Pathways
| Enzyme Type | Eukaryotic Ubiquitin System | Prokaryotic Pup System | Key Distinctions |
|---|---|---|---|
| Activating Enzyme | E1 (e.g., UBA1, UBA6); adenylates Ub, forms thioester [6] [21] | Dop; deamidates C-terminal Gln of Pup to Glu using ATP [19] | Dop does not form a thioester intermediate. |
| Conjugating Enzyme | E2 (e.g., CDC34, UBE2N); ~35 in humans, carries activated Ub [6] | Not present in pupylation pathway. | The E2 step is entirely absent in the pupylation cascade. |
| Ligating Enzyme | E3 (e.g., HECT, RING, RBR); ~600 in humans, provides substrate specificity [6] | PafA; directly ligates deamidated Pup to substrate lysine [19] | PafA is related to carboxylate-amine/ammonia ligases, not E3s. |
| Deconjugating Enzyme | Deubiquitinases (DUBs); ~100 in humans, remove Ub [23] [24] | Dop; also exhibits depupylase activity to reverse modification [19] | Dop is a bifunctional enzyme, unlike most DUBs. |
Experimental Protocols for Characterization
Key Methodologies for Studying Prokaryotic Ubiquitin-like Systems
1. Bacterial Two-Hybrid Screening for Protein Interaction Partners: The initial discovery of Pup relied on a bacterial two-hybrid screen using the proteasomal ATPase Mpa as bait to identify interacting partners [19]. This method involves co-expressing two putative interacting proteins fused to separate domains of a transcription factor in bacteria. If the proteins interact, they reconstitute the transcription factor and activate reporter genes, allowing for the selection and identification of novel binding partners like Pup.
2. Detection of Covalent Conjugates via Co-expression and Immunoblotting: To confirm the formation of covalent Pup-protein conjugates, researchers co-express Pup (or its mutants) with a candidate substrate protein (e.g., FabD, PanB) in mycobacteria or E. coli [19]. Cell lysates are then analyzed by SDS-PAGE and immunoblotting using an antibody specific for the substrate or Pup. A shift in the molecular weight of the substrate indicates the formation of a covalent complex, which can be further verified by mass spectrometry.
3. In Vitro Reconstitution of the Pupylation/Depupylation Cycle: This biochemical assay uses purified components to delineate the enzymatic pathway.
- Procedure:
- Purification: Recombinant Pup (GGQ form), Dop, PafA, and a candidate substrate protein (e.g., FabD) are purified.
- Deamidation Reaction: Pup is incubated with Dop and ATP (or a non-hydrolyzable analog). Deamidation to Pup-GGE can be monitored by gel shift or mass spectrometry.
- Ligation Reaction: Deamidated Pup is incubated with PafA, ATP, and the substrate. The formation of pupylated substrate is analyzed by SDS-PAGE and Coomassie staining or immunoblotting [19].
- Depupylation Assay: The pupylated conjugate is incubated with Dop, and the cleavage of the isopeptide bond is monitored over time, demonstrating the reversibility of the modification [19].
4. Tandem Mass Spectrometry (MS/MS) for Mapping Conjugation Sites: To definitively identify the specific lysine residue on a target protein that is modified by Pup, pupylated proteins are isolated and digested with a protease like trypsin. The resulting peptides are analyzed by tandem mass spectrometry. A diagnostic "di-glycine" remnant (or a modified mass for Glu) attached to a lysine residue identifies the precise site of modification, confirming an isopeptide linkage [19].
5. Genetic Deletion/Proteasome Inhibition to Identify Native Substrates: Steady-state levels of potential proteasomal substrates are analyzed in wild-type bacteria versus mutant strains lacking key components (e.g., ΔpafA, Δmpa, or Δproteasome) or treated with proteasome inhibitors [19]. Proteins that accumulate in the mutants or under inhibition are strong candidates for being native pupylation targets. These candidates can then be validated using the co-expression and in vitro reconstitution methods described above.
Diagram 1: Experimental Workflow for Characterizing Prokaryotic Ubiquitin-like Systems. This flowchart outlines the key methodological steps, from initial discovery to validation.
Signaling Pathways and Functional Outcomes
The functional consequences of ubiquitin and Pup modification reveal both convergent evolution and fundamental divergence. In eukaryotes, ubiquitination regulates a vast array of cellular processes. K48-linked polyubiquitin chains predominantly target proteins for degradation by the 26S proteasome, controlling the half-lives of key regulators like cyclins and transcription factors [22]. In contrast, K63-linked chains and monoubiquitination play critical non-proteolytic roles in signaling pathways, endocytosis, and DNA repair [22] [23]. A prime example is the NF-κB pathway, where K63-linked chains act as scaffolds to activate kinases, while K48-linked chains lead to the degradation of the inhibitor IκB, releasing NF-κB to initiate an inflammatory response [18] [22].
In prokaryotes such as mycobacteria, the current evidence indicates that pupylation primarily serves to target proteins for destruction by the proteasome, a function critical for virulence and resistance to nitric oxide stress from host immune cells [18] [19]. Substrates like FabD (malonyl Co-A acyl carrier protein transacylase) and PanB (ketopantoate hydroxymethyl-transferase) accumulate when the pupylation or proteasomal systems are disrupted, linking the pathway directly to metabolic regulation and stress adaptation [19]. Unlike the multifaceted ubiquitin code, evidence for a diverse "Pup code" with non-proteolytic functions is still emerging.
Diagram 2: Comparative Signaling Pathways: Ubiquitin vs. Pup. This diagram contrasts the multi-enzyme cascade in eukaryotes with the simpler, two-enzyme system in prokaryotes, highlighting different functional outcomes.
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Research Reagents for Investigating Prokaryotic Ubiquitin-like Systems
| Reagent / Material | Function in Research | Example Application |
|---|---|---|
| Anti-Pup Antibodies | Detect endogenous Pup and pupylated conjugates via immunoblotting. | Identify native pupylation substrates in mycobacterial lysates [19]. |
| ΔpafA / Δmpa / Δdop Mutant Strains | Genetically inactivate the pupylation system to study its physiological role. | Identify proteins that accumulate when pupylation is blocked, indicating potential substrates [19]. |
| Proteasome Inhibitors (e.g., MG132, Lactacystin) | Chemically inhibit the 20S proteasome to block degradation of pupylated targets. | Validate that a protein's turnover is dependent on both pupylation and the proteasome [19]. |
| Recombinant Dop, PafA, Pup (GGQ/GGE) | Purified components for in vitro biochemical reconstitution of the pathway. | Define the specific enzymatic steps of deamidation, ligation, and depupylation [19]. |
| Mycobacterial Protein Expression Systems | Express candidate substrate proteins and Pup variants in vivo. | Test for covalent conjugate formation via co-expression and pulldown assays [19]. |
| Pup-derived Peptides | Synthetic peptides spanning the C-terminus of Pup. | Use as interaction probes or competitors in binding assays with Mpa [18]. |
Implications for Drug Discovery and Therapeutic Development
The comparison between eukaryotic and prokaryotic ubiquitin-like systems has profound implications for drug development, particularly in targeting bacterial pathogens. The essential role of the pupylation-proteasome system in the virulence of M. tuberculosis makes its components attractive, pathogen-specific drug targets [18] [19]. Unlike the essential eukaryotic ubiquitin system, inhibiting the bacterial system could offer a therapeutic window with reduced host toxicity.
Drug discovery efforts targeting the ubiquitin system are advancing on multiple fronts. For eukaryotic targets, strategies include proteasome inhibitors (e.g., Bortezomib for multiple myeloma), NEDD8-activating enzyme (NAE) inhibitors (e.g., MLN4924 in clinical trials), and the development of E3 ligase and DUB inhibitors [6] [24]. Emerging technologies like Proteolysis-Targeting Chimeras (PROTACs) hijack the endogenous ubiquitin system to degrade specific disease-causing proteins, opening new therapeutic modalities [24]. Fragment-based drug discovery (FBDD), which uses small molecular fragments for efficient screening, is also being applied to target E1, E2, E3, and DUB enzymes with high specificity [25].
The simpler, two-enzyme pupylation pathway (Dop and PafA) in bacteria presents a unique opportunity for antibiotic development. The distinct mechanism of these enzymes from their eukaryotic counterparts suggests that specific inhibitors could be designed to disrupt bacterial proteostasis and virulence without affecting the human ubiquitin system, representing a promising avenue for novel anti-infectives.
The ubiquitin (Ub) signaling system, once considered a hallmark exclusive to eukaryotic cells, is a sophisticated protein modification machinery that regulates critical processes such as protein degradation, DNA repair, and stress response [26]. This system involves a cascade of E1 (activating), E2 (conjugating), and E3 (ligating) enzymes that attach ubiquitin to target proteins, marking them for various fates [26]. Surprisingly, the evolutionary origins of this complex system trace back to ancient prokaryotic sulfur carriers—ThiS and MoaD. These small proteins, involved in fundamental biosynthetic pathways, share striking structural and mechanistic similarities with eukaryotic ubiquitin and its activating enzymes [27] [28]. This article provides a comparative analysis of ThiS and MoaD, examining their roles as evolutionary precursors to the ubiquitin system. We explore their structural biology, functional mechanisms, and the experimental evidence linking them to eukaryotic ubiquitination, providing researchers with a foundational understanding of this conserved evolutionary module.
Structural and Functional Comparison of ThiS and MoaD
ThiS and MoaD are central components in the biosynthesis of essential cofactors in prokaryotes. Despite their lack of significant sequence similarity to ubiquitin, both proteins exhibit profound structural homology and operate through mechanistically similar activation pathways [27] [26]. The table below summarizes their core characteristics and functional roles.
Table 1: Comparative Overview of Prokaryotic Sulfur Carriers ThiS and MoaD
| Feature | ThiS | MoaD |
|---|---|---|
| Biological Function | Sulfur carrier in thiamine (vitamin B1) biosynthesis [26] | Sulfur carrier in molybdenum cofactor (Moco) biosynthesis [27] [26] |
| Activating Enzyme | ThiF [26] | MoeB [27] [26] |
| Final Sulfurated Form | ThiS-thiocarboxylate [26] | MoaD-thiocarboxylate [27] [26] |
| Structural Fold | β-grasp fold, identical to ubiquitin [26] | β-grasp fold, identical to ubiquitin [27] [26] |
| C-terminal Motif | Conserved Gly-Gly [26] | Conserved Gly-Gly [27] [26] |
| Activation Mechanism | Adenylation by ThiF → covalent acyl-adenylate → thiocarboxylate formation [26] | Adenylation by MoeB → covalent acyl-adenylate → thiocarboxylate formation [27] [26] |
| Covalent Intermediate with E1-like enzyme | Acyl-persulfide linkage with ThiF [26] | No direct covalent linkage with MoeB reported [26] |
The mechanistic similarities between these prokaryotic systems and eukaryotic ubiquitin activation are profound. Both ThiS and MoaD are activated by their respective E1-like enzymes (ThiF and MoeB) through adenylation of their C-terminus, consuming ATP to form a covalent acyl-adenylate intermediate [27] [26]. This activated form then receives sulfur from a sulfurtransferase to form a thiocarboxylate, which serves as the sulfur donor in the synthesis of thiamine and molybdenum cofactor [26]. The eukaryotic ubiquitin system follows an analogous initial step, where the E1 enzyme adenylates ubiquitin's C-terminal glycine before forming a thioester bond with a conserved cysteine residue [27] [26]. This conservation highlights the deep evolutionary connection between these distinct biochemical pathways.
Experimental Evidence and Key Methodologies
Structural Biology Techniques
The revelation that ThiS and MoaD are structural homologs of ubiquitin emerged primarily from X-ray crystallography studies. Seminal work on the MoeB-MoaD complex from Escherichia coli provided the first atomic-level view of this relationship [27] [29]. Researchers determined the crystal structures of the complex in three distinct states: apo (ligand-free), ATP-bound, and MoaD-adenylate form [27]. These structures revealed that despite minimal sequence similarity, MoaD possesses the characteristic β-grasp fold identical to ubiquitin, complete with the conserved C-terminal Gly-Gly motif essential for activation [27] [29]. The activation mechanism observed in the MoeB-MoaD complex provided a direct molecular framework for understanding the eukaryotic E1-ubiquitin interaction [27].
Table 2: Key Experimental Evidence Supporting the Ubiquitin-ThiS/MoaD Evolutionary Relationship
| Experimental Approach | Key Findings | Significance |
|---|---|---|
| X-ray Crystallography (MoeB-MoaD complex) [27] [29] | MoaD shares the β-grasp fold with ubiquitin; MoeB shares structural homology with E1 Uba domain; Revealed adenylate intermediate | Provided structural proof of evolutionary relationship; elucidated activation mechanism |
| Biochemical Analysis (ThiS-ThiF system) [26] | ThiF adenylates ThiS; ThiS forms acyl-persulfide with ThiF; ThiS-thiocarboxylate is sulfur donor | Demonstrated mechanistic parallels to ubiquitin activation cascade |
| Comparative Genomics [26] | Identified Ub-like β-grasp proteins in prokaryotes; discovered conserved gene neighborhoods linking Ub-like proteins, E1-like enzymes, JAB peptidases | Revealed functional associations in prokaryotes predating eukaryotic Ub-system |
| Functional Studies (Urm1 system) [28] | Urm1 acts as both sulfur carrier in tRNA thiolation and protein modifier | Identified molecular fossil linking prokaryotic sulfur carriers to eukaryotic Ub-like proteins |
Experimental Protocols
Protocol 1: Crystallographic Analysis of MoeB-MoaD Complex [27] [29]
- Protein Expression and Purification: Clone moeB and moaD genes into expression vectors. Express proteins in E. coli BL21. Purify using affinity and size-exclusion chromatography.
- Complex Formation: Mix purified MoeB and MoaD proteins in equimolar ratio.
- Crystallization: Use vapor-diffusion method to crystallize the complex. Obtain crystals in multiple states: apo, ATP-bound, and adenylate forms.
- Data Collection and Structure Determination: Collect X-ray diffraction data at synchrotron facilities. Solve structure using molecular replacement with known ubiquitin and E1 enzyme structures as search models.
- Structure Analysis: Compare molecular architectures with eukaryotic E1-ubiquitin complexes to identify conserved features.
Protocol 2: Analysis of ThiS Activation and Thiocarboxylate Formation [26]
- Adenylation Assay: Incubate ThiS with ThiF in reaction buffer containing ATP and Mg²⁺. Include [α-³²P]ATP to monitor adenylate formation.
- Sulfur Transfer Assay: After adenylation, add sulfur donor (cysteine desulfurase system) to reaction.
- Product Analysis: Resolve reaction products using non-reducing SDS-PAGE and autoradiography to detect thiocarboxylate formation.
- Functional Validation: Test thiocarboxylated ThiS in enzymatic assays for thiamine biosynthesis to confirm biological activity.
Visualization of Evolutionary and Mechanistic Relationships
Diagram 1: Evolutionary and mechanistic relationship between prokaryotic sulfur carriers and the eukaryotic ubiquitin system
This diagram illustrates the conserved module between prokaryotic sulfur carrier systems and eukaryotic ubiquitination. Both systems share the core mechanism of ATP-dependent adenylation by E1-like enzymes, followed by transfer to downstream components—sulfur carriers in prokaryotes and protein targets in eukaryotes via E2-E3 cascades. The structural conservation of the β-grasp fold in ThiS, MoaD, and ubiquitin, along with their similar activation mechanisms, provides compelling evidence for an evolutionary continuum [27] [26] [28].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Studying Prokaryotic Ubiquitin-like Systems
| Reagent / Resource | Function / Application | Example Use Case |
|---|---|---|
| Recombinant MoeB Protein [29] | E1-like enzyme for biochemical assays; protein crystallization | Structural studies of MoeB-MoaD complex; ATPase activity assays |
| Recombinant ThiS Protein [26] | Ubiquitin-like sulfur carrier for mechanistic studies | Analysis of adenylation and thiocarboxylation kinetics |
| MoeB-MoaD Crystallization Kits [27] [29] | Obtaining protein crystals for structural analysis | Determining atomic structures of complex in different catalytic states |
| Anti-ThiS / Anti-MoaD Antibodies | Detection and quantification of proteins in cellular extracts | Monitoring protein expression levels in genetic studies |
| ATP Analogues (e.g., ATPγS) | Probing enzyme mechanism and trapping intermediates | Characterizing adenylate formation in E1-like enzymes |
| E. coli Deletion Strains (ΔmoaD, ΔthiS) | Genetic analysis of pathway function | Phenotypic characterization of sulfur carrier mutants |
| pET Expression Vectors with MoaD/ThiS [30] | Heterologous protein expression in E. coli | Production of recombinant proteins for biochemical studies |
| Size-Exclusion Chromatography Matrix | Protein complex purification | Isolating native MoeB-MoaD complexes for biochemical analysis |
The structural and functional parallels between prokaryotic sulfur carriers ThiS/MoaD and the eukaryotic ubiquitin system represent a remarkable case of evolutionary conservation and adaptation. These proteins not only share the β-grasp structural fold but also operate through mechanistically similar activation pathways involving E1-like enzymes and ATP-dependent adenylation [27] [26]. The evolutionary journey from sulfur metabolism to protein modification highlights nature's capacity to repurpose successful molecular strategies for new biological functions.
Understanding these evolutionary relationships has practical significance beyond fundamental science. The conserved features of these systems offer opportunities for developing novel antimicrobial agents that target essential biosynthetic pathways in pathogenic bacteria [30]. Furthermore, the Urm1 system, which represents a "molecular fossil" bridging prokaryotic sulfur carriers and eukaryotic ubiquitin-like modifiers, provides insights into the evolutionary assembly of complex signaling networks [28]. Continued comparative studies of these systems will undoubtedly yield further surprises and deepen our understanding of how cells evolved sophisticated regulatory mechanisms from basic metabolic building blocks.
The discovery of a ubiquitin-like protein modification system in prokaryotes fundamentally expanded our understanding of protein homeostasis and degradation across life domains. In eukaryotes, the ubiquitin (Ub) system—comprising a conserved cascade of E1, E2, and E3 enzymes—tags proteins for proteasomal degradation and regulates numerous cellular processes [14] [31]. For decades, this system was considered a eukaryotic hallmark. However, the identification of prokaryotic ubiquitin-like protein (Pup) in Mycobacterium tuberculosis (Mtb) revealed that bacteria too employ small protein modifiers to target substrates for degradation [32] [19]. This Pup-proteasome system (PPS) is functionally analogous to eukaryotic ubiquitination yet distinct in its enzymology and evolutionary origin [19] [33]. The discovery of Pup has profound implications for understanding bacterial physiology and virulence, particularly in Mtb, where it is essential for surviving host immune responses [33]. This guide compares the core components, mechanisms, and experimental approaches defining the Pup system against eukaryotic ubiquitin signaling.
System Components and Comparative Mechanisms
The Pup-proteasome system and the eukaryotic ubiquitin system represent convergent evolutionary solutions for targeted protein degradation. The table below provides a structured comparison of their core components.
Table 1: Comparative Analysis: Prokaryotic Pup System vs. Eukaryotic Ubiquitin System
| Feature | Prokaryotic Pup System (PPS) | Eukaryotic Ubiquitin System |
|---|---|---|
| Key Modifier | Prokaryotic ubiquitin-like protein (Pup) [32] | Ubiquitin (Ub) [14] |
| Modifier Structure | Intrinsically disordered in free state; folds upon binding [32] | Stable β-grasp fold in solution [32] |
| Terminal Residue | C-terminal glutamate (or glutamine requiring deamidation) [34] [19] | C-terminal glycine with diglycine motif [14] |
| Conjugation Chemistry | Isopeptide bond via γ-carboxylate of Pup glutamate [19] | Isopeptide bond via α-carboxylate of Ub glycine [19] |
| Ligase Enzyme | PafA (Proteasome accessory factor A) [34] [33] | Cascade of E1, E2, and E3 enzymes [14] |
| Deconjugase Enzyme | Dop (Depupylase) [34] [19] | Deubiquitinating enzymes (DUBs) [14] |
| ATPase Unfoldase | Mpa (Mycobacterial proteasomal ATPase) [35] [33] | 19S Regulatory Particle (e.g., Rpt1-6 ATPases) [35] |
| Proteolytic Core | Bacterial 20S Core Particle (α7β7β7α7) [33] | Eukaryotic 20S Core Particle [33] |
| Primary Function | Target proteins for proteasomal degradation [19] | Target proteins for proteasomal degradation; diverse signaling [31] |
The PPS is phylogenetically restricted, found primarily in Actinobacteria (including mycobacteria) and Nitrospirae [19]. In Mtb, this system is a critical virulence factor, enabling the pathogen to resist nitric oxide stress from host macrophages [33]. The evolutionary origins of the enzymes are distinct; PafA and Dop are homologs derived from glutamine synthetase-like enzymes, not the E1/E2/E3 cascade used for ubiquitination [34] [19].
Key Experimental Methodologies and Findings
Structural Elucidation of the Pup-Dop Complex
Experimental Objective: To determine the high-resolution crystal structure of the depupylase Dop in complex with its substrate, Pup, to understand the mechanism of catalytic phosphate formation and substrate recognition [34].
Table 2: Key Reagents for Structural Studies of the Pup-Dop Complex
| Research Reagent | Function/Description in the Experiment |
|---|---|
| AcelDop Protein | Depupylase from Acidothermus cellulolyticus; the model enzyme for structural studies [34] |
| AcelPupQΔN43 | N-terminally truncated Pup fragment with C-terminal glutamine; used for co-crystallization [34] |
| AMP-PCP | A non-hydrolyzable ATP analog; used to trap the enzyme-substrate complex in a pre-hydrolysis state [34] |
| Crystallography | Method used to determine atomic structures at resolutions as high as 1.65 Å [34] |
Protocol Summary:
- Protein Purification and Complex Formation: Recombinantly express and purify AcelDop. Generate an N-terminally truncated Pup (PupQΔN43) to aid crystallization. Incubate Dop with Pup and AMP-PCP to form a stable complex [34].
- Crystallization and Data Collection: Crystallize the Dop-Pup-AMP-PCP complex using standard screening methods. Flash-cool crystals and collect X-ray diffraction data at a synchrotron source [34].
- Structure Determination and Refinement: Solve the crystal structure by molecular replacement using a known Dop structure as a model. Iteratively refine the atomic model to fit the electron density map [34].
Key Findings:
- Pup undergoes a disorder-to-order transition upon binding to Dop, forming two well-resolved helices that fit into a conserved binding groove on Dop [34].
- The C-terminal residue of Pup (glutamine in the structure) forms an extensive network of interactions within the Dop active site, more extensive than those observed in the homologous ligase, PafA [34].
- The structure revealed the role of the "Dop-loop," a conserved region that acts as an allosteric sensor for Pup binding and ATP cleavage [34].
Structural Analysis of the Mpa-Proteasome Complex
Experimental Objective: To visualize the structure of the mycobacterial proteasome ATPase (Mpa) engaged with the 20S core particle (CP) and a pupylated substrate during translocation, using single-particle cryo-electron microscopy (cryo-EM) [35].
Table 3: Key Reagents for Mpa-Proteasome Cryo-EM Studies
| Research Reagent | Function/Description in the Experiment |
|---|---|
| Mpa Hexamer | Mycobacterial proteasomal AAA+ ATPase unfoldase; forms a ring-shaped complex [35] |
| Δ7PrcA 20S CP | 20S core particle with N-terminal 7 residues deleted from α-subunits; promotes stable Mpa binding [35] |
| PupDHFR | Linear fusion protein model substrate; Pup domain recruits the complex, DHFR is the degradation target [35] |
| ATPγS | A slowly-hydrolyzable ATP analog; used to stall the translocation cycle for structural analysis [35] |
Protocol Summary:
- Complex Assembly and Substrate Engagement: Assemble the Mpa-20S CP complex in the presence of ATP. Add the PupDHFR substrate under ice-cold conditions to slow down the reaction and allow for initial engagement [35].
- Reaction Quenching and Vitrification: Add ATPγS to the reaction mixture to halt further ATP hydrolysis and stall the complex at a specific translocation step. Apply the sample to an EM grid and rapidly vitrify it by plunging into liquid ethane [35].
- Cryo-EM Data Collection and Processing: Collect micrographs of the frozen-hydrated particles. Use 2D and 3D classification to isolate homogeneous particle subsets. Reconstruct high-resolution density maps for the Mpa motor in different conformational states [35].
Key Findings:
- The cryo-EM structure revealed Mpa in a spiral-staircase arrangement around the substrate, a hallmark of AAA+ ATPase mechanism [35].
- Two distinct conformational states were resolved, corresponding to sequential steps in the ATP-hydrolysis cycle that drives substrate unfolding and translocation [35].
- The N-terminal coiled-coil domains of Mpa engage Pup in an antiparallel coiled-coil interaction, inducing Pup to form a single long helix [35].
Pathway and Mechanism Visualization
The following diagrams illustrate the core pathway of pupylation and the structural transitions of Pup during the process, synthesized from the experimental data.
Diagram 1: The Pup-Proteasome System Pathway. This diagram illustrates the complete cycle of pupylation, starting with the deamidation of Pup by Dop, ligation to a target protein by PafA, and culminating in recognition, unfolding, and degradation by the Mpa-20S proteasome complex. Dop also provides regulatory control through its depupylase activity [34] [19] [33].
Diagram 2: Induced Folding of Pup Upon Target Binding. Pup is intrinsically disordered in its free state but undergoes specific disorder-to-order transitions upon binding to different partners. When engaging the pupylation enzymes PafA and Dop, it folds into two orthogonal helices. In contrast, when bound to the unfoldase Mpa, it forms a single, long helix that engages in a coiled-coil interaction [35] [32].
The Scientist's Toolkit: Essential Research Reagents
The table below catalogues critical reagents and materials used to study the Pup-proteasome system, as identified from the cited experimental approaches.
Table 4: Essential Research Reagents for Investigating the Pup-Proteasome System
| Reagent / Material | Critical Function in Research |
|---|---|
| Pup (Wild-type & Mutants) | The central ubiquitin-like modifier; C-terminal (Gln/Glu) variants are essential for studying enzyme specificity and the pupylation cycle [34] [19]. |
| PafA (Ligase) | The enzyme responsible for covalently attaching deamidated Pup to target protein lysines [34] [33]. |
| Dop (Deamidase/Depupylase) | The dual-function enzyme that deamidates Pup-Gln and also reverses pupylation by cleaving the isopeptide bond [34] [19]. |
| Mpa (AAA+ ATPase) | The hexameric unfoldase that recognizes pupylated substrates, unfolds them, and translocates them into the proteasome [35] [33]. |
| 20S Core Particle (CP) | The proteolytic chamber where pupylated substrates are ultimately degraded [35] [33]. |
| Non-hydrolyzable ATP Analogs (e.g., AMP-PCP, ATPγS) | Used to trap and stabilize transient intermediate complexes for structural studies (e.g., X-ray crystallography, cryo-EM) [34] [35]. |
| Linear Pup-Substrate Fusions (e.g., PupDHFR) | Well-defined model substrates that facilitate in vitro degradation assays and structural studies of the complete machinery [35]. |
| pLink-UBL Software | A dedicated mass spectrometry search engine for the precise identification of UBL modification sites on protein substrates [36]. |
The β-grasp fold (β-GF) represents one of the most remarkable examples of evolutionary optimization in structural biology. Prototyped by the eukaryotic protein ubiquitin, this compact fold comprises a β-sheet with five anti-parallel strands that appears to "grasp" a single α-helical segment, creating a stable yet versatile structural platform [37] [38]. Initially discovered in ubiquitin and immunoglobulin-binding proteins of Gram-positive bacteria, subsequent research has revealed the β-grasp fold to be a widespread structural scaffold recruited for a strikingly diverse range of biochemical functions across all domains of life [37] [39]. The fold's extraordinary functional versatility encompasses sulfur transfer in metabolite biosynthesis, RNA and soluble ligand binding, enzymatic activities such as phosphohydrolase function, iron-sulfur cluster binding, adaptor functions in cellular signaling, and post-translational protein modification [37] [38]. This structural analysis will explore the genomic evidence demonstrating how this simple fold has been extensively deployed throughout evolution, with specific adaptations in prokaryotic versus eukaryotic systems, particularly focusing on its implications for ubiquitin-like signaling pathways.
Evolutionary reconstruction analyses indicate that the β-grasp fold had already differentiated into at least seven distinct lineages by the time of the last universal common ancestor (LUCA) of all extant organisms, encompassing much of the structural diversity observed in modern versions of the fold [37] [38]. The earliest β-grasp fold members were likely involved in RNA metabolism, subsequently radiating into various functional niches through extensive diversification in prokaryotes, while eukaryotic evolution was characterized by a specific expansion of ubiquitin-like β-grasp members [37]. This review will systematically compare the genomic distribution, structural variations, and functional adaptations of β-grasp fold domains across the tree of life, with particular emphasis on the evolutionary relationships between prokaryotic ubiquitin-like systems and the eukaryotic ubiquitin-signaling apparatus.
Comparative Genomic Analysis of β-Grasp Fold Domains
Phylogenetic Distribution and Diversity
Comprehensive sequence and structural analyses of β-grasp fold domains reveal their extensive presence across bacteria, archaea, and eukarya, demonstrating their ancient origin and adaptive radiation throughout evolutionary history. The fold has been recruited for an astonishing variety of molecular functions in different organisms, with specific lineages showing remarkable phylogenetic conservation patterns [37] [20].
Table 1: Major Lineages of β-Grasp Fold Domains and Their Distribution
| Lineage/Superfamily | Representative Members | Primary Functions | Distribution Across Domains |
|---|---|---|---|
| Ubiquitin-like | Ubiquitin, SUMO, NEDD8, ThiS, MoaD | Protein modification, Sulfur transfer | Eukaryotes (Ub/Ubls), Bacteria (ThiS/MoaD), Archaea |
| SLBB Superfamily | Transcobalamin, ComEA, Nqo1 | Soluble ligand binding, Vitamin B12 uptake, DNA uptake | Bacteria, Eukaryotes (animal-specific transcobalamin) |
| Enzymatic β-GF | NUDIX phosphohydrolases, Staphylokinases | Phosphohydrolase activity, Fibrinolysis | Widespread across all domains |
| Fe-S Cluster Binding | 2Fe-2S Ferredoxins | Electron transport | Bacteria, Archaea, Eukaryotes |
| Adaptor Domains | TGS, RA, PB1, FERM | RNA binding, Protein-protein interactions | Primarily eukaryotes, some bacterial versions |
Systematic genomic surveys indicate that most structural diversification of the β-grasp fold occurred in prokaryotes, with at least seven distinct lineages already established before the divergence of bacteria, archaea, and eukaryotes [37]. The eukaryotic phase of β-grasp evolution was predominantly marked by a specific expansion of ubiquitin-like family members, which diversified into at least 67 distinct families, with approximately 19-20 families already present in the eukaryotic common ancestor [37] [38]. Another key aspect of eukaryotic evolution was the dramatic increase in domain architectural complexity related to the expansion of Ub-like domains in numerous adaptor roles [37].
Ubiquitin-Like β-Grasp Domains: Prokaryotic Antecedents of Eukaryotic Systems
The evolutionary relationship between prokaryotic and eukaryotic ubiquitin-like systems provides compelling evidence for the deep conservation of the β-grasp fold. Originally considered a eukaryotic innovation, ubiquitin-mediated signaling is now understood to have profound connections to prokaryotic systems, particularly through the sulfur carrier proteins ThiS and MoaD, which are involved in thiamine and molybdenum cofactor biosynthesis, respectively [20]. These prokaryotic proteins share significant structural similarity with eukaryotic ubiquitin and even undergo analogous biochemical processes, including C-terminal adenylation and thiocarboxylate formation catalyzed by enzymes (ThiF and MoeB) that are structurally related to eukaryotic E1 ubiquitin-activating enzymes [37] [20].
Recent research has uncovered novel ubiquitin-like proteins in phylogenetically diverse bacteria that form functional associations with E1-like enzymes, JAB hydrolases, and E2-like enzymes, suggesting the existence of primitive ubiquitin-signaling systems in prokaryotes [20]. One significant discovery is the prokaryotic ubiquitin-like protein (Pup) in Mycobacterium tuberculosis, which targets proteins for degradation by the bacterial proteasome, demonstrating that post-translational protein tagging systems are not exclusive to eukaryotes [18]. This finding fundamentally challenges the paradigm that ubiquitin-like signaling is a uniquely eukaryotic feature and suggests that the eukaryotic ubiquitin-signaling apparatus was pieced together from prokaryotic antecedents [20].
Table 2: Comparison of Ubiquitin-Like Systems in Prokaryotes and Eukaryotes
| Feature | Prokaryotic Systems | Eukaryotic Systems |
|---|---|---|
| Representative β-GF | ThiS, MoaD, Pup, YukD | Ubiquitin, SUMO, NEDD8, URM1 |
| Modification Enzymes | ThiF, MoeB, PafA | E1, E2, E3 enzymes |
| Deconjugation Enzymes | JAB domain peptidases | Deubiquitinating enzymes (DUBs) |
| Primary Functions | Sulfur transfer, Cofactor biosynthesis, Proteasomal degradation | Protein degradation, Signaling, Trafficking |
| Structural Features | Core β-grasp fold with minimal inserts | Core β-grasp fold with family-specific modifications |
Methodologies for Identifying and Characterizing β-Grasp Fold Domains
Sequence and Structure Analysis Protocols
The identification and characterization of β-grasp fold domains across diverse organisms relies on sophisticated bioinformatic approaches that leverage both sequence and structural information. Due to the small size and extensive divergence of β-grasp domains, exhaustive identification requires multi-pronged computational strategies [37].
Protocol 1: Iterative Sequence Profile Searches
- Objective: Identify remote homologs with significant sequence similarity
- Methodology:
- Use known β-grasp structures from PDB as seeds for PSI-BLAST searches against NCBI NR database
- Iterate searches until convergence (e-value < 0.01 with statistical correction)
- Conduct transitive searches with newly detected members
- Evaluate sub-threshold hits for potential homologs
- Applications: Effective for identifying novel Ub-like domains and SLBB superfamily members [37] [39]
Protocol 2: Structural Similarity Searches
- Objective: Detect homologs with conserved structure but divergent sequence
- Methodology:
- Perform DALI structural comparisons with characterized β-grasp domains
- Analyze Z-scores for significant structural alignment
- Superimpose structures to identify core elements and variable inserts
- Map functional sites through conserved structural features
- Applications: Identification of SLBB domains through structural alignment with transcobalamin and Nqo1 [39]
Protocol 3: Genomic Context Analysis
- Objective: Infer functional associations through conserved gene neighborhoods
- Methodology:
- Identify conserved operons and gene clusters
- Analyze domain architectures in multidomain proteins
- Examine phylogenetic patterns of association
- Correlate with biochemical pathway information
- Applications: Revealed connections between Ub-like proteins, E1-like enzymes, and JAB peptidases in bacteria [20]
Experimental Validation Techniques
While bioinformatic analyses provide essential insights into the distribution and evolution of β-grasp domains, experimental validation is crucial for confirming functional predictions. Several key methodologies have been employed to characterize the biochemical and cellular functions of β-grasp proteins.
X-ray Crystallography and Structural Analysis: Determination of three-dimensional structures has been instrumental in characterizing the core β-grasp fold and its variations. For example, structural analysis of transcobalamin revealed how inserts in the SLBB superfamily create specialized ligand-binding sites for vitamin B12 [39]. Similarly, comparison of ubiquitin with ThiS and MoaD structures demonstrated conserved structural features despite divergent functions [20].
Enzymatic Assays: Biochemical characterization of enzymatic activities associated with β-grasp domains, such as the phosphohydrolase activity of NUDIX enzymes and the adenylation activity of E1-like enzymes, provides functional validation of predictions derived from sequence and structural analyses [37].
Interaction Studies: Investigation of protein-protein and protein-ligand interactions through methods such as co-immunoprecipitation, yeast two-hybrid screening, and surface plasmon resonance has been essential for understanding the diverse functional roles of β-grasp domains, particularly in ubiquitin-like signaling pathways and adaptor functions [37] [18].
Structural and Functional Adaptations Across Evolutionary Lineages
Key Structural Innovations in Major β-Grasp Lineages
The functional diversity of β-grasp domains arises from strategic structural elaborations on the conserved core fold. These innovations include distinctive insertions between core elements, modifications of surface properties, and development of specialized interaction interfaces [37].
The ubiquitin-like superfamily maintains the most conserved version of the fold, characterized by minimal inserts and a prominent β-sheet surface that facilitates diverse protein-protein interactions [37] [38]. In contrast, the SLBB (Soluble-Ligand-Binding β-grasp) superfamily exhibits two major structural variations: the transcobalamin-like clade features a β-hairpin insert after the helix that participates in ligand binding, while the Nqo1-like clade contains an insert between strands 4 and 5 of the core fold [39]. These inserts create specialized binding pockets for soluble ligands such as vitamin B12, with genomic context analyses supporting roles in B12 uptake, polysaccharide export, and DNA uptake in bacteria [39].
Enzymatic β-grasp domains like the NUDIX phosphohydrolases have evolved active sites on the common scaffold, demonstrating that the fold can support catalytic functions beyond binding and mediation of interactions [37]. Similarly, iron-sulfur cluster binding versions have developed unique cysteine-containing flaps that chelate iron atoms, representing one of the most striking functional adaptations of the fold [20].
Evolutionary Trajectory of Ubiquitin-Like Signaling Systems
The evolution of ubiquitin-like signaling systems from primitive sulfur transfer machinery illustrates how the β-grasp fold has been adapted for increasingly complex cellular functions throughout evolutionary history. Evolutionary reconstruction suggests that sulfur carrier proteins like ThiS and MoaD represent the ancestral state from which eukaryotic ubiquitin and ubiquitin-like modifiers descended [20].
The fundamental evolutionary transition involved a shift from sulfur transfer for cofactor biosynthesis to protein modification for regulatory purposes [20]. This transition was facilitated by the recruitment of additional enzymatic components, including E2-conjugating enzymes and E3 ligases, which increased the specificity and regulatory potential of the system. The discovery of Pup in mycobacteria demonstrates an independent evolutionary trajectory in which a β-grasp protein was recruited for proteasomal targeting, representing a convergent solution to the problem of substrate recognition for regulated proteolysis [18].
The eukaryotic ubiquitin system represents the most elaborate development of β-grasp-based signaling, with extensive diversification of E3 ligases that provide substrate specificity and the integration of deubiquitinating enzymes that add reversible control to the system [37] [20]. The expansion of ubiquitin-like modifiers with distinct functions (SUMO, NEDD8, etc.) further illustrates how gene duplication and functional specialization have built an intricate regulatory network on the versatile β-grasp scaffold.
Visualization of Evolutionary Relationships and Functional Diversity
Diagram Title: Evolutionary Radiation of β-Grasp Fold from LUCA
Essential Research Reagents and Computational Tools
Table 3: Research Toolkit for β-Grasp Fold Domain Analysis
| Tool/Reagent | Type | Primary Application | Key Features |
|---|---|---|---|
| PSI-BLAST | Algorithm | Remote homology detection | Position-specific scoring matrices, Iterative search |
| DALI | Algorithm | Structural similarity search | Pairwise structure alignment, Z-score significance |
| HMMER | Algorithm | Profile hidden Markov models | Sensitive domain detection, Multiple sequence alignment |
| PDB | Database | 3D structural data | Experimentally determined structures, Standardized formats |
| SCOP | Database | Structural classification | Evolutionary relationships, Structural hierarchy |
| ThiS/MoaD Proteins | Biochemical Reagents | Sulfur transfer studies | Prokaryotic Ub antecedents, C-terminal thiocarboxylate formation |
| E1-like Enzymes | Biochemical Reagents | Ub activation mechanism | Adenylation domain, Rossmann fold, Conserved cysteine |
| JAB Proteases | Biochemical Reagents | Deubiquitinating activity | Metalloprotease activity, Isopeptidase function |
The comprehensive genomic evidence presented in this analysis demonstrates that the β-grasp fold represents an extraordinary example of structural conservation coupled with functional diversification throughout the history of life. From its origins in the last universal common ancestor, this compact fold has been adapted for an astonishing array of molecular functions, with distinct evolutionary trajectories in prokaryotic and eukaryotic lineages [37] [38]. The fold's remarkable versatility stems from its stable core structure that can tolerate extensive surface modifications and strategic insertions, creating specialized binding sites and interaction surfaces while maintaining structural integrity [37].
The evolutionary relationship between prokaryotic sulfur carrier systems and the eukaryotic ubiquitin-signaling apparatus provides a compelling case study in molecular evolution, illustrating how complex regulatory systems can emerge through the elaboration and integration of simpler ancestral components [20]. The recent discovery of ubiquitin-like modification systems in diverse bacteria, including the Pup-proteasome system in Actinomycetes, further blurs the distinction between prokaryotic and eukaryotic post-translational regulatory mechanisms and suggests that the evolutionary history of ubiquitin-like signaling is more complex than previously appreciated [18] [4].
Future research directions should include more comprehensive surveys of β-grasp diversity in understudied bacterial and archaeal lineages, structural characterization of the novel β-grasp groups identified through bioinformatic analyses, and experimental investigation of the potential functional connections between prokaryotic ubiquitin-like proteins, JAB peptidases, and E2-like enzymes [20]. Such studies will not only provide deeper insights into the evolutionary history of this remarkable structural fold but may also reveal novel biological mechanisms with implications for understanding cellular regulation and developing therapeutic interventions.
Decoding Ubl Mechanisms: From Enzymatic Cascades to Pathogen Hijacking
The targeted degradation of proteins is a fundamental biological process conserved across all domains of life. While eukaryotes rely on a sophisticated ubiquitin-proteasome system (UPS) characterized by a multi-enzyme cascade, certain bacteria have evolved an analogous but mechanistically distinct system centered on prokaryotic ubiquitin-like protein (Pup) tagging, or pupylation [40]. This article provides a detailed comparative analysis of these two systems, focusing on their core enzymatic mechanisms. The eukaryotic pathway utilizes a trio of enzymes (E1, E2, E3) that function sequentially to attach ubiquitin to substrate proteins, often forming polyubiquitin chains that serve as a degradation signal [22] [21]. In contrast, the bacterial pupylation system, found primarily in Actinobacteria, employs a single ligase, PafA, to directly attach Pup to target proteins, enabling their recognition by the bacterial proteasome [41] [42] [40]. This comparison will explore the structural, mechanistic, and regulatory differences between these pathways, providing researchers with a foundation for understanding their biological implications and potential as therapeutic targets.
The eukaryotic ubiquitin and bacterial pupylation systems represent a fascinating case of convergent evolution, whereby biologically analogous solutions for targeted protein degradation evolved from unrelated ancestral proteins and enzymes [40]. The table below summarizes the core components and characteristics of each system.
Table 1: Core Components of Eukaryotic Ubiquitination and Bacterial Pupylation Systems
| Feature | Eukaryotic Ubiquitin System | Bacterial Pupylation System |
|---|---|---|
| Modifier Protein | Ubiquitin (Ub, 76 aa, structured) [21] | Prokaryotic Ubiquitin-like Protein (Pup, 64 aa in M. tuberculosis, intrinsically disordered) [40] |
| Final Bond | Isopeptide bond between Ub Gly76 and substrate Lys ε-amino group [21] | Isopeptide bond between Pup C-terminal Glu and substrate Lys ε-amino group [41] [40] |
| Activating Enzyme | E1 Ubiquitin-activating enzyme (ATP-dependent) [22] [21] | PafA (directly activates and ligates Pup) [41] [42] |
| Conjugating Enzyme | E2 Ubiquitin-conjugating enzyme [22] [21] | Not present |
| Ligase Enzyme | E3 Ubiquitin ligase (hundreds to thousands, e.g., RING, HECT) [22] [43] | PafA (sole ligase) [41] [42] [40] |
| Energy Source | ATP hydrolysis (by E1) [21] | ATP hydrolysis (by PafA, stoichiometric with ligation) [40] |
| Deconjugation Enzyme | Deubiquitinases (DUBs) [44] | Depupylase (Dop) [41] [40] |
| Proteasome Recruiter | Polyubiquitin chains (e.g., Lys48-linked) [22] [21] | Mono- or short poly-Pup chains [41] |
A critical distinction lies in the enzymatic architecture. The eukaryotic system is a multi-step cascade involving E1, E2, and E3 enzymes, which facilitates amplification and precise regulation [22] [21]. In contrast, the bacterial system is a single-enzyme pathway where PafA alone performs the roles of activation and ligation, making it a central regulatory hub [42] [40]. Furthermore, the modifier proteins themselves are structurally different; ubiquitin is a globular, stable protein, whereas Pup is intrinsically disordered, adopting a defined structure only upon binding to its interaction partners like the proteasomal ATPase Mpa [40].
Detailed Mechanistic Comparison
The Eukaryotic E1-E2-E3 Cascade
The ubiquitination pathway is a three-step enzymatic cascade that culminates in the covalent attachment of ubiquitin to a substrate protein.
- Step 1: Activation (E1). The process begins with the E1 ubiquitin-activating enzyme. In an ATP-dependent reaction, E1 catalyzes the adenylation of the C-terminal glycine (Gly76) of ubiquitin. This activated ubiquitin is then transferred to a catalytic cysteine residue within the E1 active site, forming a high-energy E1~Ub thioester intermediate and releasing AMP [22] [21] [44].
- Step 2: Conjugation (E2). The activated ubiquitin is subsequently transferred from E1 to a catalytic cysteine residue of an E2 ubiquitin-conjugating enzyme via a trans-thioesterification reaction. This results in an E2~Ub thioester intermediate [22] [21] [43].
- Step 3: Ligation (E3). The final step is mediated by an E3 ubiquitin ligase, which is responsible for substrate recognition. E3 enzymes bring the E2~Ub complex and the target protein into close proximity. For RING-type E3 ligases, the E3 acts as a scaffold to facilitate the direct transfer of ubiquitin from the E2 to a lysine residue on the substrate, forming an isopeptide bond [22] [43]. HECT-domain E3s, however, form a transient E3~Ub thioester intermediate before transferring ubiquitin to the substrate [21].
This cascade allows for immense specificity and regulation, with humans encoding two E1s, dozens of E2s, and hundreds to thousands of E3s [22] [43].
The following diagram illustrates this sequential cascade:
The Bacterial PafA-Mediated Pupylation
The bacterial pupylation pathway is a more streamlined process catalyzed by the single enzyme PafA.
- Step 1: Pup Activation. PafA first activates the C-terminal glutamate of Pup (PupE) using ATP. This reaction proceeds through the formation of a phospho-Pup mixed anhydride intermediate (γ-glutamyl phosphate) at the Pup C-terminus [40].
- Step 2: Isopeptide Bond Formation. The ε-amino group of a lysine residue on the target protein acts as a nucleophile, directly attacking the activated carbonyl carbon of the phospho-Pup intermediate. This leads to the formation of an isopeptide bond between the substrate lysine and the γ-carboxylate of Pup's C-terminal glutamate, with the release of inorganic phosphate (Pi) [41] [40].
Notably, PafA is an allosteric enzyme that functions as a dimer and exhibits positive cooperativity in substrate binding, allowing its activity to be regulated by the availability of target proteins [45]. Unlike the eukaryotic system, there is no stable covalent enzyme-Pup intermediate analogous to the E1~Ub or E2~Ub thioesters.
The following diagram illustrates the direct ligation mechanism of PafA:
Experimental Analysis of Mechanisms
Key Experimental Methodologies
Studying these conjugation mechanisms requires a combination of biochemical, structural, and genetic approaches. Key experimental protocols are summarized below.
Table 2: Key Experimental Methods for Studying Conjugation Mechanisms
| Method Category | Specific Technique | Application & Purpose | Key Experimental Details |
|---|---|---|---|
| In Vitro Reconstitution | Purified component assays [41] [46] | Define minimal requirements for conjugation; measure kinetics. | Incubate E1/E2/E3/Ub+ATP or PafA/Pup+ATP with substrate; analyze via SDS-PAGE/western blot for shifted bands [41]. |
| Enzyme Mutagenesis | Catalytic cysteine mutants (E1, E2, HECT E3) [21] | Trap thioester intermediates in ubiquitination. | Mutate E1 (Cys), E2 (Cys), or HECT E3 (Cys) to Ala/Ser; blocks transfer, isolating intermediates for study. |
| Site-Directed Mutagenesis | Pup lysine mutants (e.g., K7, K31, K61) [41] | Determine poly-Pup chain linkage topology. | Mutate Pup lysines; identify which are used for chain elongation in self-pupylation assays. |
| Structural Biology | X-ray crystallography, Cryo-EM [42] | Visualize enzyme-substrate complexes; identify interfaces. | Solve structures of PafA-Pup-substrate complexes to map electrostatic interaction motifs for substrate recognition [42]. |
| Mass Spectrometry | Tandem MS (LC-MS/MS) [41] [46] | Identify precise modification sites (lysine) on substrates. | Digest modified protein; identify peptides with Gly-Gly remnant (Ub) or Pup-derived peptides; confirm isopeptide linkage. |
Illustrative Experimental Findings
- PafA Self-Pupylation Mechanism: In vitro assays with M. smegmatis PafA revealed that its self-pupylation occurs via a specific mechanism. The attachment of the first Pup molecule is an intermolecular reaction catalyzed by one PafA molecule on another. However, the subsequent formation of a polymeric Pup chain proceeds in an intramolecular manner, catalyzed by the already mono-pupylated PafA molecule itself. The lysine residue K320 on PafA was identified as the primary pupylation site [41].
- Substrate Recognition by PafA: Contrary to initial assumptions that PafA non-specifically targets surface-accessible lysines, research has shown it recognizes a structural motif centered around the target lysine. Studies on the model substrate FabD demonstrated that PafA recognizes its targets via a compact interface mediated by a few key electrostatic interactions, allowing for selective substrate turnover despite the enzyme's broad target range [42].
- Functional Reconstruction of Archaeal Cascade: The evolutionary bridge between bacterial and eukaryotic systems was highlighted by the functional reconstruction of a minimized E1-E2-E3 cascade from the uncultured archaeon Caldiarchaeum subterraneum. This experiment demonstrated that a sequential ubiquitylation cascade, operationally similar to the eukaryotic one, can function with a minimal set of archaeal progenitor enzymes [46].
Essential Research Reagents and Tools
A successful research program in protein conjugation pathways requires a toolkit of well-characterized reagents. The following table lists key materials for investigating these systems.
Table 3: Essential Research Reagents for Studying Ubiquitination and Pupylation
| Reagent / Material | Function in Research | Specific Examples & Notes |
|---|---|---|
| Recombinant Enzymes | Core catalysts for in vitro assays and mechanistic studies. | Eukaryotic: Human E1 (UBA1), E2s (e.g., UbcH5b), E3s (e.g., MDM2, Skp2). Bacterial: M. tuberculosis or M. smegmatis PafA and Dop [41] [43]. |
| Modifier Proteins | The key signaling molecules conjugated to substrates. | Ubiquitin: Wild-type, mutant (e.g., K48R, K63R), fluorescently tagged. Pup: PupE (ligation-competent deamidated form) [41] [40]. |
| Energy Regeneration System | Fuel for the ATP-dependent activation steps. | ATP and MgCl₂; often included in reaction buffers (e.g., 5 mM ATP) [41]. |
| Affinity Purification Tags | For purifying proteins and pull-down assays to identify novel substrates. | His₆-tag, GST-tag, FLAG-tag. Used to tag Pup or Ub for pupylome/ubiquitome studies [41] [42]. |
| Proteasome Inhibitors | To stabilize poly-ubiquitinated/pupylated proteins in vivo by blocking their degradation. | Bortezomib (for eukaryotic proteasome), MG132 (less specific). Used to accumulate substrates for detection [22]. |
| Specific Antibodies | Detect modified proteins and endogenous components via western blot, immunofluorescence. | Anti-ubiquitin, anti-polyubiquitin (Lys48- or Lys63-linkage specific), anti-Pup [41]. |
The eukaryotic E1-E2-E3 cascade and the bacterial PafA-mediated pupylation system are two elegant yet mechanistically distinct solutions to the universal biological challenge of targeted protein degradation. The eukaryotic system leverages a multi-enzyme cascade for unparalleled specificity and regulatory complexity, with hundreds of E3 ligases enabling precise control over a vast array of substrates. In contrast, the bacterial system achieves its goals through a streamlined, single-ligase mechanism, where PafA combines activation and ligation functions and relies on allostery and electrostatic interactions for substrate selection [42] [45].
From an evolutionary perspective, these systems are a classic example of convergent evolution, having arisen independently from different ancestral proteins [40]. The recent discovery of minimized, functional ubiquitination cascades in archaea provides a tantalizing glimpse into a potential evolutionary missing link [46]. For drug development professionals, these mechanistic differences are of paramount importance. The eukaryotic UPS, particularly specific E3 ligases like MDM2, is a well-established target in oncology [22] [43]. Conversely, the Pup-proteasome system in pathogenic bacteria like Mycobacterium tuberculosis is a promising antibacterial target, as it is essential for virulence and persistence within host macrophages [41] [40]. The detailed mechanistic understanding of these divergent pathways, as outlined in this guide, provides the essential foundation for the rational design of specific inhibitors with potential therapeutic utility in both human disease and infectious disease.
The ubiquitin (Ub) signaling system, long considered a hallmark of eukaryotic cells, is a sophisticated protein modification pathway crucial for protein homeostasis, innate immunity, and cellular signaling. In eukaryotes, this system operates through a sequential enzymatic cascade involving E1 (activating), E2 (conjugating), and E3 (ligating) enzymes that conjugate ubiquitin or ubiquitin-like proteins (Ubls) to target proteins, ultimately determining their fate and function [47]. For decades, the evolutionary origins of this complex system remained enigmatic. While prokaryotes were known to encode distant relatives of E1 enzymes and Ubls involved in sulfur metabolism (ThiF/ThiS and MoeB/MoaD systems), these pathways lacked E2 enzymes and did not mediate Ubl conjugation to target proteins, suggesting bacteria had not evolved complete ubiquitination systems [5] [26]. This paradigm has been fundamentally challenged by groundbreaking research revealing that certain bacterial operons encode fully functional ubiquitination pathways with striking architectural and mechanistic parallels to their eukaryotic counterparts, revolutionizing our understanding of the evolutionary history of ubiquitin signaling and revealing ancient antecedents of eukaryotic immune mechanisms [47] [5] [4].
The recent discovery of complete eukaryotic-like E1-E2-Ubl pathways in bacterial antiviral defense represents a transformative shift in our understanding of ubiquitin signaling evolution. These findings emerged from investigations into bacterial immune systems, particularly those associated with phage defense islands and CBASS (cyclic oligonucleotide-based anti-phage signaling system) operons, which protect bacteria from bacteriophage infection [5] [48]. Structural and biochemical studies have now demonstrated that these bacterial systems possess bona fide ubiquitination machinery that parallels eukaryotic pathways in both complexity and function, suggesting these pathways arose first in bacteria rather than archaea or eukaryotes [47] [5] [4]. This article provides a comprehensive comparison between these newly discovered bacterial ubiquitination systems and canonical eukaryotic pathways, examining their structural relationships, enzymatic mechanisms, and functional roles in antiviral immunity.
Comparative Analysis of Bacterial and Eukaryotic Ubiquitination Systems
Table 1: Structural and Functional Comparison of Bacterial and Eukaryotic Ubiquitination System Components
| Component | Bacterial System | Eukaryotic System | Key Similarities | Notable Differences |
|---|---|---|---|---|
| E1 Enzyme | E1BilD (Type II BilABCD) | UBA6, NAE1-UBA3 | N-terminal inactive adenylation domain (IAD) + C-terminal active adenylation domain (AAD); Mobile CYS domain with catalytic cysteine; Structural zinc binding [5] | Bacterial E1 encoded as single polypeptide vs. heterodimeric NEDD8 E1; Different domain organization compared to some eukaryotic E1s [5] |
| E2 Enzyme | E2BilB (CEHH domain) | UBC-family E2s | Conserved catalytic cysteine; Forms thioester with E1~Ubl; Transfers Ubl to targets [5] | CEHH domain with unique Cys-Glu-His-His motif vs. canonical UBC fold [5] |
| Ubl Protein | UblBilA | Ubiquitin, Ubls | C-terminal glycine for adenylation; Processed by DUBs; Conjugated to target lysines [5] | Specific structural variations in β-grasp fold; Different target specificities [5] |
| DUB Enzyme | DUBBilC (JAB family) | JAB/MPN+ proteasomal DUBs | Pre-processes Ubl C-terminus; Cleaves Ubl-target isopeptide linkages [5] | Associated with specific bacterial operons vs. integrated into proteasomal system [5] |
| E3 Ligase | Not definitively identified | RING, HECT, RBR families | Potential E3-independent targeting in some bacterial systems [5] | Limited distribution in bacteria; Some pathogenic bacteria encode E3s that modulate host ubiquitination [5] |
Table 2: Functional Outcomes of Bacterial Ubiquitination Pathways in Antiviral Defense
| Pathway Type | Ubl Protein | Target Proteins | Antiviral Mechanism | Experimental Evidence |
|---|---|---|---|---|
| Type II BilABCD | UblBilA | Unknown phage/virion components [5] | Inhibition of phage replication through target protein modification [5] | Operon association with defense islands; Structural characterization of E1-E2-Ubl complex [5] |
| CBASS-Cap2 | cGAS (E. coli) | Unknown bacterial proteins [48] | Enhanced cGAMP production; Activation of phospholipase effectors (CapV) [48] | cGAS conjugation requires Cap2 E1/E2 activities; C-terminal glycine mutation abolishes anti-phage defense [48] |
| Other Ubl Pathways | Pup (Actinobacteria) | Proteosomal substrates | Target degradation via proteasome-like complexes [5] | Distinct from canonical E1-E2-E3 mechanism; Mycobacterial systems well-characterized [5] |
Architectural Parallels: Structural Biology of Bacterial Ubiquitination Complexes
Structural analyses of bacterial ubiquitination components have revealed remarkable architectural conservation with eukaryotic counterparts. The seminal work by Chambers et al. (2024) provided the first detailed structural insights into a bacterial E1-E2-Ubl complex from Ensifer aridi TW10 [47] [5]. Through X-ray crystallography determined to 2.5 Å and 2.7 Å resolution, they demonstrated that E1BilD exhibits the same domain organization as canonical eukaryotic E1 enzymes, featuring an N-terminal inactive adenylation domain (IAD), a C-terminal active adenylation domain (AAD), and a mobile α-helical CYS domain containing the catalytic cysteine [5]. This architecture closely mirrors eukaryotic E1 enzymes such as human UBA6 (ubiquitin/FAT10 E1) and the heterodimeric NEDD8 E1 (NAE1-UBA3), with structural alignment revealing conserved core elements despite sequence divergence [5].
The bacterial E1BilD also coordinates a structural zinc ion through four cysteine residues, a feature shared with eukaryotic SUMO and NEDD8 E1 proteins [5]. Two distinct structural states were captured: one representing a pre-reaction conformation with the UblBilA C-terminus positioned for adenylation, and a second mimicking the E1-to-E2 transthioesterification state with the CYS domain adjacent to the bound E2BilB [5]. These structures provide unprecedented mechanistic insights into the reaction cycle and confirm that bacterial systems undergo the same fundamental biochemical steps as eukaryotic ubiquitination. The E2BilB protein, though containing a distinct CEHH domain (named for conserved Cys-Glu-His-His residues) rather than the canonical UBC fold, nevertheless occupies the same structural position relative to E1 as observed in eukaryotic complexes and facilitates Ubl transfer to target proteins [5].
Diagram 1: Bacterial ubiquitination pathway. This diagram illustrates the complete enzymatic cascade in bacterial ubiquitination systems, from Ubl processing by DUB enzymes to target protein modification.
Detailed Experimental Protocols for Studying Bacterial Ubiquitination
Structural Characterization of E1-E2-Ubl Complexes
The elucidation of bacterial ubiquitination machinery has relied on sophisticated structural biological approaches. Chambers et al. employed a multi-step protocol to characterize the E1BilD-E2BilB-UblBilA complex [5]:
Complex Expression and Purification: Co-expression of E1BilD, E2BilB, and UblBilA in E. coli was essential, as the individual E1 and E2 components were insoluble when expressed alone. This suggests the complex forms a stable ternary structure in vivo.
Crystallization and Data Collection: The purified complex was crystallized using vapor diffusion methods. Two distinct crystal forms were obtained (Form 1 at 2.5 Å and Form 2 at 2.7 Å resolution), enabling the capture of different conformational states during the ubiquitination cycle.
Structure Determination: X-ray diffraction data were collected at synchrotron facilities and structures solved using molecular replacement with eukaryotic E1 structures as search models, highlighting the structural conservation between kingdoms.
Structural Analysis: Comparative analysis with eukaryotic E1 structures (UBA6, NAE1-UBA3) revealed conserved features including: the pseudo-dimeric IAD-AAD arrangement, the mobile CYS domain, catalytic residue positioning (including Arg9 from the IAD domain), and structural zinc coordination.
Biochemical Assays for Ubiquitination Activity
Functional validation of bacterial ubiquitination activity requires a series of complementary biochemical assays:
Thioester Formation Assays: Detection of covalent E1~Ubl and E2~Ubl intermediates using non-reducing SDS-PAGE that preserves thioester linkages. These bonds are sensitive to reducing agents (DTT) and hydroxylamine treatment, distinguishing them from stable isopeptide bonds [48].
Deubiquitinase Activity Assays: Recombinant DUBBilC protein is incubated with Ubl fusion substrates or native Ubl conjugates to demonstrate processing and deconjugation activity. Reaction products are analyzed by immunoblotting or mass spectrometry [5].
In Vitro Reconstitution assays: Purified E1, E2, Ubl, and ATP are combined to recapitulate conjugate formation independent of other cellular factors. ATP dependence confirms the enzymatic nature of the reaction [48].
Target Identification: Genetic screens and mass spectrometry-based proteomics identify physiological targets of bacterial ubiquitination systems, revealing their roles in antiviral defense [5] [48].
Diagram 2: Experimental workflow for characterizing bacterial ubiquitination systems. This diagram outlines the multidisciplinary approach required to identify and validate these pathways.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents for Studying Bacterial Ubiquitination Systems
| Reagent/Method | Specific Application | Function/Purpose | Key Considerations |
|---|---|---|---|
| Bacterial Strains | Ensifer aridi TW10 (Type II BilABCD); E. coli CBASS strains [5] [48] | Source of native ubiquitination genes; Heterologous expression host | Phylogenetic diversity important for comparative studies; Defense island association predicts immune function |
| Expression Vectors | Co-expression systems for E1-E2-Ubl complexes [5] | Enables production of soluble, functional complexes | Individual components often insoluble; Ternary complex formation essential for stability |
| Antibodies | Anti-Flag tags for immunodetection of conjugates [48] | Detection of Ubl conjugates and thioester intermediates | Critical for monitoring modification states; Commercial antibodies available for common tags |
| Phage Stocks | T4, T6, lambda phages for infection assays [48] | Functional assessment of antiviral defense | Phage sensitivity varies; Multiple phages needed for comprehensive analysis |
| Protease Inhibitors | Specific DUB inhibitors; Broad-spectrum protease cocktails | Preserve ubiquitination states during lysis | DUB activity can rapidly deconjugate products during preparation |
| Structural Tools | X-ray crystallography; AlphaFold predictions [5] | Determine atomic-level architecture of complexes | Eukaryotic structures useful as molecular replacement models; AF2 predictions guide experimental design |
| Biochemical Reagents | ATP, DTT, hydroxylamine [48] | Thioester assays and reaction requirements | ATP-dependence confirms enzymatic mechanism; DTT sensitivity distinguishes thioesters |
Implications and Future Directions in Ubiquitin Research
The discovery of complete eukaryotic-like E1-E2-Ubl pathways in bacteria represents a paradigm shift in our understanding of ubiquitin signaling evolution. These findings challenge previous models suggesting ubiquitination systems arose in archaea or eukaryotes and instead position bacteria as the originators of these sophisticated protein modification pathways [47] [5] [4]. The striking architectural and mechanistic conservation spanning billions of years of evolution highlights the fundamental efficiency of this signaling mechanism and suggests ancient bacterial immune pathways were repurposed or refined in eukaryotes for diverse regulatory functions beyond antiviral defense [5].
From a practical perspective, bacterial ubiquitination systems offer compelling advantages for structural and mechanistic studies. Their relative simplicity compared to eukaryotic counterparts—often comprising minimal operons with fewer components—makes them ideal model systems for understanding fundamental aspects of ubiquitination biochemistry [5]. Additionally, the association of these pathways with antiviral defense mechanisms opens new avenues for developing biotechnological tools to enhance bacterial resistance in industrial fermentation and phage therapy applications [5] [48]. Future research will likely focus on identifying the specific phage targets of these systems, elucidating the structural basis of target recognition, and exploring potential applications in synthetic biology and antimicrobial strategies.
The emerging paradigm of bacterial ubiquitination also raises intriguing questions about the evolutionary transition between bacterial and eukaryotic ubiquitination systems. How were these pathways horizontally transferred or maintained during eukaryotic origins? What selective pressures drove the expansion and diversification of ubiquitination components in eukaryotes? Continued investigation of bacterial ubiquitination systems will undoubtedly provide deeper insights into these fundamental questions while enhancing our understanding of ubiquitin signaling across all domains of life.
Ubiquitination, a crucial post-translational modification in eukaryotes, involves an enzymatic cascade of E1 (activating), E2 (conjugating), and E3 (ligating) proteins that conjugate ubiquitin or ubiquitin-like proteins (Ubls) to target lysine residues, thereby regulating protein homeostasis, signaling, and innate immunity [5] [47]. For decades, this sophisticated protein-tagging system was considered a hallmark of eukaryotic complexity, with bacteria known to encode only distant relatives of E1 and Ubl proteins involved primarily in sulfur metabolism rather than targeted protein conjugation [49]. However, groundbreaking research has fundamentally challenged this paradigm by revealing that certain bacterial operons associated with phage defense islands encode complete, functional ubiquitination pathways [5] [47] [50]. These bacterial systems exhibit striking architectural and mechanistic parallels with canonical eukaryotic ubiquitination machinery, suggesting these pathways may have originated in bacteria before being inherited and refined in eukaryotes [49] [47].
This comparison guide provides a structural biology-focused examination of eukaryotic and bacterial E1:E2:Ubl complexes, synthesizing recent high-resolution structural data to reveal both remarkable conservation and key distinctions. We present quantitative structural comparisons, detailed experimental methodologies, and evolutionary insights relevant to researchers investigating fundamental biological mechanisms and potential therapeutic applications. The emergence of bona fide ubiquitination systems in bacteria not only expands our understanding of this essential signaling pathway but also opens new avenues for exploring antibacterial strategies that target these systems.
Structural Architecture Comparison
Structural analyses reveal that bacterial E1:E2:Ubl complexes share profound architectural similarities with their eukaryotic counterparts despite billions of years of evolutionary divergence. The bacterial E1 (E1BilD) from Ensifer aridi displays the same domain organization characteristic of canonical eukaryotic E1 proteins, comprising an N-terminal inactive adenylation domain (IAD), a C-terminal active adenylation domain (AAD), and a mobile α-helical insertion containing the catalytic cysteine (CYS domain) [5]. This pseudo-dimeric arrangement mirrors that observed in eukaryotic E1s such as human UBA6 (ubiquitin/FAT10 E1) and the heterodimeric NEDD8 E1 NAE1-UBA3 [5]. The structural conservation extends to the coordination of zinc ions through cysteine residues in the AAD crossover loop and C-terminal region, a feature also present in SUMO and NEDD8 E1 proteins [5].
Table 1: Overall Architectural Features of E1:E2:Ubl Complexes
| Structural Feature | Eukaryotic Systems | Bacterial Systems | PDB References |
|---|---|---|---|
| E1 Domain Organization | IAD, AAD, CYS domain | IAD, AAD, CYS domain | 8TYZ [51] |
| E1 Quaternary Structure | Monomeric or heterodimeric | Monomeric | 8TYZ [51] |
| E2 Binding Interface | Conserved surface on E1 | Structurally similar interface | 8TYZ [51] [5] |
| Zinc Binding Sites | Present in SUMO, NEDD8 E1s | Present (4 cysteine residues) | 8TYZ [51] [5] |
| Ubl Processing | DUB-mediated C-terminal exposure | DUB (BilC)-mediated processing | [5] |
Domain-Specific Structural Comparisons
The core catalytic domains of ubiquitination machinery show exceptional conservation between kingdoms. In the bacterial E1BilD, the AAD contains a single active adenylation site, with a critical arginine residue (Arg9) supplied by the IAD, precisely as observed in eukaryotic E1s [5]. The CYS domain, connected to the AAD via flexible linkers, demonstrates significant mobility with high B-factors and adopts distinct positions in different structural forms—an open-like conformation with the catalytic cysteine positioned above the adenylation active site in Form 1, and a rotated upward orientation contacting the bound E2 in Form 2 [5]. This conformational flexibility mirrors the dynamic rearrangements observed in eukaryotic E1s during the catalytic cycle.
Table 2: Key Structural Parameters from Experimental Determinations
| Parameter | Eukaryotic UBL Domain Complex (2MWS) | Bacterial E1-E2-Ubl Complex (8TYZ) |
|---|---|---|
| PDB ID | 2MWS [52] | 8TYZ [51] |
| Resolution | Solution NMR | 2.68 Å [51] |
| Organism | Homo sapiens, Saccharomyces cerevisiae | Ensifer aridi [51] |
| Experimental Method | SOLUTION NMR | X-RAY DIFFRACTION [51] |
| Complex Stoichiometry | 1:1 ubiquitin:UBL domain | Multimeric E1:E2:Ubl |
| Key Structural Motifs | β-grasp domains | β-grasp domains, adenylation domains |
The Ubl components in both systems maintain the characteristic β-grasp fold, though bacterial Ubls display remarkable architectural diversity with some possessing up to three tandem β-grasp domains compared to the single domain typically found in eukaryotic ubiquitin or the two domains in ISG15 [50]. These multi-domain bacterial Ubls can form homodimers and helical filaments mediated by conserved Ca2+ ion binding sites, suggesting additional regulatory mechanisms not observed in eukaryotic systems [50]. The E2 proteins from bacteria, though highly diverged in sequence, maintain the canonical ubiquitin-conjugating (UBC) fold essential for transthioesterification activity [5].
Experimental Protocols and Methodologies
Structural Biology Approaches
The comparative insights between eukaryotic and bacterial E1:E2:Ubl complexes derive primarily from high-resolution structural techniques, particularly X-ray crystallography and complementary biophysical methods.
Protein Complex Production and Crystallization: For the bacterial E1:E2:Ubl complex, researchers cloned and expressed all four proteins (UblBilA, E2BilB, DUBBilC, and E1BilD) from the Type II BilABCD operon of Ensifer aridi TW10 [5]. While E1BilD and E2BilB were insoluble when expressed individually in E. coli, they formed a soluble complex when co-expressed with UblBilA, enabling structural studies. The complex was purified and crystallized using standard high-throughput screening approaches, yielding two distinct crystal forms (Form 1 and Form 2) that provided complementary structural information [5].
Data Collection and Structure Determination: X-ray diffraction data were collected for both crystal forms, with structures determined to 2.5 Å resolution (Form 1) and 2.7 Å resolution (Form 2) [5]. The structures were solved using molecular replacement or experimental phasing approaches, followed by iterative model building and refinement. The resulting structural models revealed distinct conformational states of the complex: Form 1 captures a pre-reaction state with the UblBilA C-terminus positioned for adenylation, while Form 2 mimics an E1-to-E2 transthioesterification state with the E1 CYS domain adjacent to the bound E2 [5].
Structure Analysis and Validation: Comprehensive structural comparisons were performed against eukaryotic E1:E2:Ub complexes using DALI and other structural alignment tools [5]. Validation followed standard crystallographic metrics with R-value free of approximately 0.245 for the 8TYZ deposition [51].
Functional Biochemistry Assays
Deubiquitinase Activity Assays: Researchers demonstrated that the bacterial deubiquitinase (DUBBilC) pre-processes the bacterial Ubl, exposing its C-terminal glycine for adenylation by E1BilD [5] [47]. This was established through in vitro cleavage assays using purified components and analysis by SDS-PAGE and mass spectrometry.
Conjugation Assays: The functionality of the bacterial ubiquitination system was confirmed through in vitro conjugation assays showing that E1BilD and E2BilB collaborate to conjugate Ubl to target protein lysine residues [5] [47]. These assays typically include ATP as an energy source and employ Western blotting with specific antibodies or tagged proteins to detect conjugation products.
Bioinformatic Analyses: Comprehensive sequence searches and phylogenetic analyses identified 136 complete nonredundant operons encoding these bacterial ubiquitination systems, with most instances found in the plant-associated Rhizobiaceae family [5]. AlphaFold structure predictions provided initial models that guided experimental design [5].
Mechanisms and Functional Implications
Catalytic Mechanisms and Conformational Dynamics
The catalytic mechanism of bacterial ubiquitination pathways mirrors the canonical eukaryotic process with remarkable fidelity. The reaction initiates with E1-mediated adenylation of the Ubl C-terminus, consuming ATP and forming a high-energy acyl adenylate intermediate [5]. Subsequently, the catalytic cysteine within the E1 CYS domain attacks this intermediate to form a thioester-linked E1~Ubl complex. Structural evidence shows the bacterial E1 CYS domain positioned directly above the adenylation active site with the conserved cysteine (Cys417 in E1BilD) approximately 14 Å from the C-terminus of bound UblBilA in the pre-reaction state [5]. Following this charging step, a cysteine residue on the E2 carrier protein attacks the E1~Ubl thioester to form an E2~Ubl thioester intermediate, a transthioesterification reaction captured in the Form 2 structure of the bacterial complex [5]. Finally, the E2~Ubl conjugate typically collaborates with an E3 ligase to mediate isopeptide bond formation between the Ubl C-terminus and a lysine side chain on target proteins.
Diagram 1: Ubiquitination Cascade. This diagram illustrates the conserved enzymatic cascade in both eukaryotic and bacterial systems, involving sequential activation and transfer of ubiquitin-like proteins.
The conformational dynamics observed in the bacterial structures directly parallel those seen in eukaryotic systems. The mobile CYS domain undergoes substantial rotation between the adenylation and transthioesterification states, precisely repositioning the charged Ubl from the E1 active site to the E2 binding interface [5]. This molecular gymnastics requires flexibility in the linker regions connecting the CYS domain to the AAD, a feature conserved across domains of life. The bacterial E1 also coordinates a structural zinc ion through four cysteine residues, maintaining structural integrity much like zinc-binding domains in eukaryotic SUMO and NEDD8 E1 proteins [5].
Biological Functions and Evolutionary Implications
While eukaryotic ubiquitination systems regulate diverse cellular processes including protein degradation, signaling transduction, and immune response, the bacterial counterparts appear primarily dedicated to antiviral defense, specifically protecting against bacteriophage infection [5] [47] [50]. In the Type II BilABCD operons, these systems confer immunity by specifically modifying virion structural proteins, thereby inhibiting phage assembly and infectivity [5]. The association of these operons with defense islands and their regulation by CapH/CapP transcriptional regulators that activate in response to DNA damage further supports their role in bacterial immunity [5].
The discovery of functionally competent ubiquitination pathways in bacteria has profound evolutionary implications, suggesting that these sophisticated protein modification systems arose first in bacteria before being horizontally transferred to or independently evolved in eukaryotes [49] [47]. The structural and mechanistic conservation despite minimal sequence similarity indicates strong functional constraints maintaining the core architecture through billions of years of evolution. The diversification of bacterial Ubls, with some possessing multiple β-grasp domains and the capacity for Ca2+-stimulated filament formation [50], suggests bacterial systems may have evolved additional regulatory layers not present in eukaryotes.
Diagram 2: Evolutionary Relationships. Proposed evolutionary pathway suggesting bacterial origins of ubiquitination systems later inherited or convergently evolved in eukaryotes.
Research Reagent Solutions
Table 3: Essential Research Reagents for E1:E2:Ubl Structural and Functional Studies
| Reagent/Category | Specific Examples | Function/Application | Experimental Context |
|---|---|---|---|
| Expression Systems | E. coli Rosetta2 pLysS [50] | Recombinant protein production | Bacterial protein expression |
| Proteases | TEV protease [50] | Fusion tag removal | Protein purification |
| Structural Biology Tools | X-ray crystallography [51], Solution NMR [52], Cryo-EM [50] | High-resolution structure determination | 3D architecture analysis |
| Bioinformatics Resources | AlphaFold [5], MAFFT [47], ColabFold [47] | Structure prediction, sequence analysis | Evolutionary studies, model building |
| Chromatography Media | Ni-NTA, ion-exchange, size-exclusion | Protein purification | Complex isolation |
| Crystallization Reagents | Commercial sparse matrix screens [5] | Crystal formation | Structure determination |
The structural comparison between eukaryotic and bacterial E1:E2:Ubl complexes reveals remarkable conservation in architectural organization and catalytic mechanisms, despite their evolutionary divergence. High-resolution structures demonstrate that bacterial systems maintain the core IAD-AAD-CYS domain organization characteristic of eukaryotic E1 enzymes, similar E2 binding interfaces, and conserved zinc coordination sites [51] [5]. The conformational dynamics essential for the ubiquitination cascade, particularly the mobility of the CYS domain during transthioesterification, are preserved in bacterial complexes [5].
Key distinctions emerge in biological function, with bacterial systems primarily dedicated to antiviral defense through modification of phage proteins [5] [47], while eukaryotic systems regulate diverse cellular processes. Additionally, bacterial Ubls display greater architectural diversity, with some containing multiple β-grasp domains capable of Ca2+-dependent oligomerization [50], suggesting regulatory mechanisms not observed in eukaryotes.
These findings fundamentally reshape our understanding of ubiquitination pathway evolution, suggesting an ancient bacterial origin for this sophisticated protein modification system [49] [47]. The structural insights provide frameworks for understanding the molecular mechanisms underlying bacterial immunity and highlight potential targets for developing antibacterial strategies that disrupt these essential defense pathways.
The ubiquitin-proteasome system (UPS) represents a crucial regulatory mechanism for protein degradation and modification in eukaryotic cells, influencing diverse processes from cell cycle control to stress responses [53]. This system relies on a sequential enzymatic cascade involving ubiquitin-activating (E1), ubiquitin-conjugating (E2), and ubiquitin-ligase (E3) enzymes that work in concert to tag substrate proteins with ubiquitin, typically marking them for degradation by the 26S proteasome [53]. While once considered exclusively eukaryotic, prokaryotic organisms have been found to possess simplified yet functionally relevant ubiquitin-like (Ubl) modification systems. The fundamental structural differences between prokaryotic and eukaryotic cells—notably the presence of membrane-bound organelles and a nucleus in eukaryotes—create distinct experimental challenges and considerations for studying Ubl pathways across these domains [54] [55]. This methodological guide provides a comprehensive comparison of the techniques available for identifying and characterizing Ubl substrates and conjugation pathways in both prokaryotic and eukaryotic systems, offering researchers a structured framework for cross-domain investigation.
Fundamental Biological Differences Informing Methodological Design
The strategic approach to studying Ubl pathways must account for profound structural and organizational differences between prokaryotic and eukaryotic cells. Eukaryotic cells possess membrane-bound organelles including a nucleus where genetic material is housed, while prokaryotic cells lack these compartmentalized structures, with their DNA bundled in the nucleoid region [54]. This fundamental distinction directly impacts methodological design, as eukaryotic Ubl pathways often involve complex subcellular trafficking and organelle-specific processes absent in prokaryotic systems.
Eukaryotic cells typically contain multiple linear DNA molecules organized into chromosomes within the nucleus, whereas prokaryotes generally possess a single, circular chromosome [54]. This genetic organization affects how Ubl genes are regulated and expressed across domains. Additionally, the larger size and greater structural complexity of eukaryotic cells (10-100 μm) compared to prokaryotes (0.1-5 μm) necessitate different approaches for cellular extraction and fractionation procedures [54]. The presence of specialized membrane-bound organelles in eukaryotes creates subcellular compartments that may harbor distinct Ubl pathway components, requiring more sophisticated subcellular fractionation techniques compared to prokaryotic studies.
Core Techniques for Ubl Pathway Component Identification
Genomic and Bioinformatic Identification Methods
Genome-wide identification of Ubl family genes represents the foundational first step in characterizing these pathways. This approach begins with database searches using conserved domain sequences, such as the UBC catalytic domain approximately 140-220 amino acids in length, followed by comprehensive bioinformatic analysis to characterize the identified genes [56] [57]. As demonstrated in studies of strawberry and wheat, this process typically involves phylogenetic construction, analysis of gene structure (exon-intron organization), motif identification, chromosomal location mapping, and examination of gene duplication events [56] [57]. The expansion of UBC genes in cultivated strawberry was found to be primarily driven by whole genome duplication (WGD) events, highlighting how evolutionary history shapes Ubl pathway complexity [56].
Table 1: Genomic Identification Workflow for Ubl Components
| Step | Methodology | Prokaryotic Considerations | Eukaryotic Considerations |
|---|---|---|---|
| Initial Identification | Database searches with conserved Ubl domains (e.g., HMMER, BLAST) | Simpler genomes with fewer Ubl components; potential horizontal transfer | Complex genomes with possible gene families; multiple subcellular localizations |
| Phylogenetic Analysis | Multiple sequence alignment and tree construction (e.g., MEGA, PhyML) | Broader evolutionary distance; distinct clustering from eukaryotic sequences | Detailed subgroup classification within species; orthology identification |
| Gene Structure Analysis | Exon-intron structure mapping using genome annotations | Fewer introns; more compact gene structure | Complex gene structures with varying exon numbers (e.g., 1-23 in strawberry FaUBCs) [56] |
| Cis-Element Analysis | Promoter region analysis for regulatory elements | Direct environmental response elements | Complex regulatory networks; hormone response elements [56] |
Expression Profiling Under Experimental Conditions
Quantitative expression analysis through methods like RNA-seq and quantitative RT-PCR (qRT-PCR) enables researchers to track Ubl gene expression patterns across different conditions, tissues, and developmental stages. In wheat studies, qRT-PCR has proven valuable for identifying UBC genes with significant expression changes following pathogen infection, with careful selection of time points (0, 3, 6, 12, 24, 48, and 72 hours post-inoculation) to capture the complete immune response trajectory [57]. This temporal profiling helps distinguish early from late responders in Ubl pathways. For qRT-PCR validation, researchers should employ the comparative 2–ΔΔCT method with appropriate internal controls (e.g., TaTubulin in wheat), include an initial denaturation at 95°C for 5 minutes followed by 40 cycles of denaturation at 95°C for 10s and extension at 60°C for 20s [57].
Functional Validation Through Genetic Manipulation
Functional characterization of putative Ubl pathway components requires genetic manipulation approaches tailored to the organismal system. Virus-induced gene silencing (VIGS) has emerged as a powerful technique for rapid functional assessment, particularly in plants like wheat where traditional genetic approaches are limited by long generation times and polyploidy [57]. The BSMV-VIGS protocol involves amplifying target gene fragments (typically 200-300bp) with specific primers, cloning them into appropriate viral vectors (e.g., BSMVγ), and infiltrating the constructs into plant tissues followed by phenotypic monitoring under controlled conditions [57]. In prokaryotic systems, targeted gene knockout or CRISPR-based approaches are typically employed, taking advantage of simpler genetic organization and transformation methods.
Analytical Techniques for Ubl Conjugation Mechanism Elucidation
Chemical Tools for Probing Conjugation Cascades
Advanced chemical tools have been developed to capture and characterize the transient intermediates and transition states within Ubl conjugation pathways [58]. These probes enable researchers to overcome the challenges posed by the labile nature of thioester-linked E1~Ub and E2~Ub complexes that are central to the ubiquitination cascade. The mechanism begins with E1-mediated ATP-dependent activation of the Ubl C-terminus, forming a thioester linkage with the E1 active site cysteine, followed by transference to the E2 active site cysteine, and culminates in E3-mediated substrate conjugation [53] [58]. These tools include activity-based probes that mimic native Ub/Ubl proteins, crosslinking agents that stabilize transient enzyme complexes, and stable isopeptide-linked mimics that resist hydrolysis for structural studies.
Activity and Interaction Assays
Enzymatic activity assays provide critical functional data on Ubl conjugation components. For E2 enzymes, these include autoubiquitination assays to assess intrinsic activity, ubiquitin discharge assays to measure thioester formation and transfer efficiency, and E3 collaboration assays to characterize specific E2-E3 partnerships. These assays typically employ Western blotting with ubiquitin-specific antibodies or epitope-tagged ubiquitin for detection. Protein interaction studies using yeast two-hybrid screening, co-immunoprecipitation (Co-IP), and pull-down assays help map the complex network of interactions between E1, E2, E3, and substrate proteins, revealing the combinatorial specificity that underlies Ubl signaling diversity.
Comparative Analysis of Ubiquitin Pathways: Prokaryotic vs. Eukaryotic Systems
The ubiquitination apparatus demonstrates remarkable divergence between prokaryotic and eukaryotic systems, reflecting their distinct cellular complexities and evolutionary histories. Eukaryotes typically possess expansive Ubl enzyme families, with Arabidopsis thaliana encoding 37 E2s and over 1400 potential E3 ligases, while prokaryotic systems operate with substantially reduced component sets [53]. This quantitative difference parallels functional specialization, with eukaryotic Ubl pathways coordinating intricate processes like hormone signaling, immune responses, and organelle-specific protein degradation that are largely absent in prokaryotes.
Table 2: Comparative Analysis of Ubl Pathways in Prokaryotes vs. Eukaryotes
| Feature | Prokaryotic Systems | Eukaryotic Systems |
|---|---|---|
| Cellular Context | Lack membrane-bound organelles; nucleoid region [54] | Membrane-bound organelles; defined nucleus [54] |
| Genetic Organization | Generally single, circular chromosome [54] | Multiple linear chromosomes in nucleus [54] |
| Component Complexity | Reduced Ubl enzyme sets; simpler pathways | Expanded families (e.g., 37 E2s in Arabidopsis) [53] |
| Ubiquitin Linkage Types | Limited linkage diversity | Extensive linkage diversity (K48, K63, K11, etc.) with functional specialization [53] |
| Functional Roles | Primarily stress adaptation, cell cycle regulation | Development, immunity, signaling, protein quality control [56] [57] |
| Experimental Challenges | Difficulty studying transient interactions; limited tools | Subcellular compartmentalization; functional redundancy |
The functional implications of these differences are substantial. Eukaryotic Ubl pathways have evolved to manage complex cellular compartmentalization, with specific E2s and E3s operating in distinct organelles. In contrast, prokaryotic Ubl systems function in a undivided cytoplasmic space, potentially enabling more direct substrate-enzyme interactions. The diversity of ubiquitin chain linkages in eukaryotes (K48, K63, K11, K6, K27, K29, K33) allows for sophisticated signaling coding, where different linkages can signal for proteasomal degradation, altered subcellular localization, or modified activity [53]. Prokaryotic Ubl systems appear to employ more limited linkage types, though the full repertoire remains an active research area.
Essential Research Reagents and Solutions
A standardized toolkit of research reagents is essential for experimental consistency and reproducibility in Ubl studies. The selection of appropriate reagents must account for the specific challenges posed by prokaryotic versus eukaryotic systems.
Table 3: Essential Research Reagent Solutions for Ubl Pathway Studies
| Reagent Category | Specific Examples | Function/Application | System Compatibility |
|---|---|---|---|
| Activity Assay Kits | Abcam CA Activity Assay Kit (ab284550) | Measures esterase activity of α-CAs via chromophore release [59] | Primarily eukaryotic |
| Enzymatic Assays | Wilbur-Anderson pH indicator assay | Quantifies CO₂ hydration kinetics using phenol red [59] | Adaptable to both systems |
| Detection Systems | Anti-Myc antibodies with colorimetric staining (Horseradish peroxidase + 4-Chloro-1-naphthol) | Protein detection in Western blotting [59] | Both systems |
| Genetic Tools | BSMV-VIGS constructs | Gene silencing in plants; 200-300bp target fragments [57] | Eukaryotic (plants) |
| Chemical Probes | Ub/Ubl active-site directed probes | Trapping transient enzyme intermediates [58] | Both systems (with modification) |
| Expression Vectors | Site-specific conjugation systems | Controlled E1/E2/E3 co-expression [60] | Both systems |
Integrated Workflow for Comprehensive Ubl Pathway Characterization
A robust methodological workflow for Ubl pathway characterization integrates multiple approaches to overcome the inherent limitations of individual techniques. The recommended workflow begins with genome-wide identification of Ubl components using conserved domain searches, proceeds through phylogenetic and structural analysis to classify components and predict relationships, then advances to expression profiling across relevant conditions and tissues to identify candidate genes for functional study [56] [57]. Selected candidates then undergo functional validation through genetic approaches like VIGS or gene knockout, followed by mechanistic biochemical studies using activity assays and interaction mapping to elucidate molecular functions [57].
This integrated approach enables researchers to move systematically from gene identification to mechanistic understanding, with each phase informing the next. The workflow should be adapted based on the specific biological system, with prokaryotic studies potentially emphasizing biochemical approaches due to limited genetic tools for some species, while eukaryotic studies might leverage more extensive genetic resources.
The methodological toolkit for studying Ubl substrates and conjugation pathways continues to evolve, with several emerging technologies promising to address current limitations. Cryo-electron microscopy is increasingly enabling visualization of transient E1-E2-E3 complexes at near-atomic resolution, while advanced mass spectrometry techniques offer unprecedented sensitivity for mapping ubiquitination sites and chain topology. The development of linkage-specific antibodies and activity-based probes continues to expand the toolbox for interrogating specific Ubl pathway aspects [58]. For prokaryotic systems, the application of CRISPR-based screening methods and bacterial two-hybrid systems adapted for Ubl studies shows particular promise. In eukaryotic research, organelle-specific profiling and single-cell analysis of Ubl pathways represent exciting frontiers. As these methods mature and integrate, they will undoubtedly reveal new insights into the remarkable conservation and divergence of Ubl signaling across the domains of life, with important implications for understanding fundamental biology and developing novel therapeutic strategies.
The ubiquitin-proteasome system (UPS), once considered an exclusive eukaryotic complex, is now recognized as a central battlefield in host-pathogen interactions. While prokaryotes lack a complete endogenous ubiquitin system, numerous bacterial pathogens have evolved sophisticated mechanisms to exploit the host's UPS for their survival and proliferation. This manipulation is primarily achieved through specialized virulence proteins, termed effectors, which are delivered into host cells via specialized secretion systems such as the Type III Secretion System (T3SS) [61] [62]. These effectors predominantly function as bacterial E3 ubiquitin ligases (BELs) and deubiquitinases (DUBs), mirroring and hijacking key components of eukaryotic ubiquitination machinery [63] [64]. The study of these bacterial tools not only reveals fundamental aspects of bacterial pathogenesis but also provides unique insights into the basic biology of the UPS itself, highlighting the evolutionary arms race between prokaryotic invaders and their eukaryotic hosts. This guide provides a comprehensive comparison of these bacterial tools, their mechanisms, and their experimental analysis.
Bacterial E3 Ubiquitin Ligases (BELs): Structure, Function, and Targets
Bacterial E3 ligases have evolved to mimic the core structural and functional features of their eukaryotic counterparts, yet they often possess unique characteristics that make them particularly potent virulence factors.
Major Classes of Bacterial E3 Ligases
Table 1: Comparison of Major Bacterial E3 Ubiquitin Ligase (BEL) Families
| BEL Family | Eukaryotic Mimic | Catalytic Mechanism | Key Structural Features | Representative Examples & Pathogens | Primary Functions in Pathogenesis |
|---|---|---|---|---|---|
| RING-type | RING E3 Ligases [65] | Direct transfer from E2 to substrate; acts as a scaffold [63] | RING or U-box domain for E2 binding [63] | AvrPtoB (Pseudomonas syringae) [63]; NleG family (EHEC/EPEC) [63] | Suppression of plant immunity (AvrPtoB) [62]; Virulence (NleG) |
| HECT-like | HECT E3 Ligases [65] | E3-Ub thioester intermediate via catalytic Cysteine [63] | Catalytic Cys residue; E2 binding site adjacent to substrate-binding domain [63] | SopA (Salmonella), NleL (EPEC) [63] | Dampening host inflammation [63] |
| NEL (Novel E3 Ligase) | None known (Bacterial-specific) [63] | E3-Ub thioester intermediate; "see-saw" mechanism for Ub transfer [63] | N-terminal LRR domain (substrate recognition), C-terminal NEL catalytic domain [63] [61] | IpaH family (Shigella, Salmonella) [63] [61] | Suppression of inflammatory response, xenophagy evasion; targeting host kinases and NF-κB pathway [63] [62] |
| XL-box | RING-type (mechanistically) [63] | Likely direct transfer, similar to RING [63] | N-terminal LRR domain, C-terminal XL-box domain (lacks Cys) [63] | XopL (Xanthomonas campestris) [63] | Suppression of ETI, causing disease in plants [63] |
Regulatory Mechanisms and Substrate Targeting
BEL activity is often tightly regulated to prevent premature action against bacterial proteins or inappropriate host targeting. A key regulatory model, elucidated for NEL ligases like SspH1 from Salmonella, involves auto-inhibition. In the absence of its specific substrate (e.g., PKN1), the leucine-rich repeat (LRR) domain of SspH1 interacts with and inhibits its own NEL catalytic domain. Upon substrate binding, a conformational change disrupts this interaction, relieving inhibition and activating ubiquitin ligase activity [63]. This precise control ensures that BELs are only deployed when they can productively engage their intended host targets, which include central immune signaling molecules such as the MAPK kinase Ste7 (targeted by IpaH9.8) and the kinase PKN1 (targeted by SspH1) [63].
Bacterial Deubiquitinases (DUBs): Reversing Host Defense Signals
To complement their arsenal of E3 ligases, bacterial pathogens also deploy effector DUBs to counteract ubiquitin-mediated host defenses. These enzymes hydrolyze the isopeptide bonds between ubiquitin molecules or between ubiquitin and substrate proteins, effectively erasing ubiquitin signals [64] [66].
Diversity and Specificity of Bacterial DUBs
Table 2: Comparison of Selected Bacterial Deubiquitinases (DUBs)
| DUB Effector | Pathogen | Reported Ubiquitin Chain Specificity | Cellular Targets / Functions |
|---|---|---|---|
| SseL | Salmonella Typhimurium | K63-linked chains [64] | Macrophage toxicity; manipulation of host trafficking and vacuole maintenance [64] |
| LotB | Legionella pneumophila | K63-linked chains [64] | Manipulates v-SNARE complex (deubiquitinates Sec22b) to aid vacuole formation [64] |
| LotA | Legionella pneumophila | K6-linked chains [64] | Vacuole maintenance; potential role in countering ubiquitin-mediated autophagy [64] |
| RavD | Legionella pneumophila | M1-linked (linear) chains [64] | Cleaves linear ubiquitin chains to inhibit NF-κB signaling and inflammation [64] |
| YopJ/P | Yersinia spp. | Not specified (broad activity implied) [66] | Suppression of MAPK and NF-κB pathways, induction of apoptosis [66] |
| Cdu1 | Chlamydia trachomatis | K48 and K63-linked chains [64] | Stabilizes host anti-apoptotic protein Mcl-1 by deubiquitination, promoting host cell survival [64] |
Bacterial DUBs exhibit remarkable specificity for different types of ubiquitin chain linkages, which dictates their functional roles. For instance, K63-linked and linear (M1) ubiquitin chains are often involved in innate immune signaling and inflammation [64] [65]. DUBs like RavD, which specifically cleave M1-linked chains, directly inhibit NF-κB signaling [64]. Conversely, K48-linked chains primarily target proteins for proteasomal degradation. By cleaving these chains, bacterial DUBs can stabilize host proteins that are beneficial for bacterial survival, as seen with Cdu1 stabilizing Mcl-1 [64].
Experimental Analysis: Validating BEL and DUB Activity
The discovery and characterization of bacterial E3s and DUBs rely on a combination of genetic, biochemical, and cellular techniques. Below are core experimental protocols used in the field.
Core Experimental Workflow
The following diagram outlines a generalized workflow for identifying and validating novel bacterial effector functions.
Key Experimental Protocols
Protocol 1: In Vitro Ubiquitination Assay for E3 Ligase Activity
This assay reconstitutes the ubiquitination cascade to test if a bacterial effector possesses E3 ligase activity [63].
- Recombinant Protein Purification: Express and purify the putative bacterial E3, along with necessary eukaryotic components: E1 enzyme, E2 enzyme, ubiquitin, and a suspected substrate protein.
- Reaction Setup: Combine the following in a reaction buffer:
- ATP (1-5 mM)
- E1 enzyme (50-100 nM)
- E2 enzyme (1-5 µM)
- Ubiquitin (10-50 µM)
- Bacterial E3 (1-5 µM)
- Substrate protein (1-5 µM)
- Incubation: Incubate the reaction at 30°C for 1-2 hours.
- Analysis: Terminate the reaction and analyze by SDS-PAGE and Western blotting. Probe for:
- Polyubiquitination: Smearing of high-molecular-weight ubiquitin signals on anti-Ub blot.
- Substrate Ubiquitination: Shift in substrate molecular weight or detection of ubiquitin on substrate immunoblot.
- Thioester Intermediate: For HECT/NEL-type E3s, a Ub-E3 intermediate can be detected under non-reducing conditions [63].
Protocol 2: In Vitro Deubiquitination Assay for DUB Activity
This assay determines if a bacterial effector can cleave ubiquitin chains or ubiquitin-protein conjugates [64] [66].
- Substrate Preparation: Generate ubiquitin substrates. Common options include:
- Di-ubiquitin Chains: Commercially available or purified chains of specific linkages (K48, K63, M1, etc.).
- Ubiquitinated Protein: In vitro ubiquitinated protein or purified ubiquitin-AMC (a fluorogenic substrate).
- Reaction Setup: Mix the bacterial DUB (10-500 nM) with the chosen ubiquitin substrate (1-5 µM) in an appropriate reaction buffer.
- Incubation and Detection: Incubate at 37°C and monitor cleavage over time (minutes to hours). Detection methods include:
- SDS-PAGE/Western Blot: Visualize the disappearance of the substrate (e.g., di-ubiquitin) and appearance of cleaved products (mono-ubiquitin).
- Fluorometry: If using Ub-AMC, measure the increase in fluorescence as AMC is released.
- Linkage Specificity: Repeat the assay with di-ubiquitin chains of different linkages (K6, K11, K48, K63, M1) to determine specificity [64].
The Scientist's Toolkit: Essential Research Reagents
Research in this field relies on a core set of reagents and tools to dissect the function of bacterial UPS manipulators.
Table 3: Key Research Reagent Solutions for Studying Bacterial UPS Effectors
| Reagent / Tool | Function & Application | Key Characteristics |
|---|---|---|
| Linkage-Specific Di-Ubiquitin Chains | Defining DUB specificity; probing E3-generated chain type [64] [65] | Recombinant K48, K63, K6, K11, M1-linked di-ubiquitin. Purity and defined linkage are critical. |
| Ubiquitin-Aldehyde (Ub-al) | A general, mechanism-based DUB inhibitor. Used in activity assays and to trap DUBs in complex with Ub for structural studies. | Irreversibly binds the active site cysteine of cysteine protease DUBs. |
| Active-Site Mutant Effectors | Essential negative controls (e.g., Cys to Ala for HECT/NEL E3s and many DUBs) to confirm catalytic dependence [63]. | Generated by site-directed mutagenesis; used in both in vitro and cellular assays. |
| T3SS-Deficient Bacterial Strains | Controls for confirming effector delivery and function is secretion-dependent [61] [62]. | Mutants in key structural components of the secretion apparatus (e.g., invA in Salmonella). |
| HEK293T Cells | A workhorse cell line for transient overexpression of effectors, protein-protein interaction studies (Co-IP), and assessing substrate ubiquitination/signaling. | Highly transferable, robust protein expression. |
Bacterial E3 ligases and DUBs represent a powerful toolkit evolved to precisely re-wire host ubiquitin signaling. Their study not only uncovers fundamental pathogenic strategies but also provides refined molecular tools for manipulating the UPS in basic research. The continued discovery and characterization of these effectors, supported by the experimental frameworks outlined here, will undoubtedly yield new insights into both bacterial pathogenesis and eukaryotic cell biology, potentially revealing novel targets for future anti-infective therapies.
The ubiquitin-proteasome system (UPS) is a master regulator of cellular protein homeostasis, controlling the degradation of nearly 80-90% of intracellular proteins in eukaryotic cells [67]. This system orchestrates vital processes including cell cycle progression, apoptosis, and immune responses by selectively tagging proteins with ubiquitin chains for destruction by the proteasome [67]. The recent discovery of ubiquitin-like machinery in bacteria has fundamentally reshaped our understanding of UPS evolution, revealing that these sophisticated degradation pathways originated in prokaryotic systems before being adopted and refined by eukaryotes [4] [5]. This evolutionary conservation underscores the fundamental biological importance of targeted protein degradation while highlighting key distinctions between prokaryotic and eukaryotic systems that inform therapeutic targeting.
Cancer cells exhibit a particular dependency on UPS function for their survival and proliferation, making this system an attractive therapeutic target [68]. The clinical validation of this approach came with the development of proteasome inhibitors for multiple myeloma, which revolutionized treatment paradigms for this malignancy [67] [69]. This article provides a comprehensive comparison of established and emerging UPS-targeting therapies, examining their mechanisms, clinical efficacy, and experimental approaches for evaluating novel candidates in cancer drug development.
Established UPS-Targeting Therapies: Proteasome Inhibitors
Mechanism of Action and Clinical Utility
Proteasome inhibitors represent the first clinically successful class of UPS-targeting anticancer agents. These compounds function by directly binding to and inhibiting the proteolytic activity of the 26S proteasome, a multi-subunit complex consisting of a 20S catalytic core particle and 19S regulatory particles [67]. The 20S core particle contains three primary proteolytic activities: chymotrypsin-like (β5 subunit), caspase-like (β1 subunit), and trypsin-like (β2 subunit) [69]. Most proteasome inhibitors preferentially target the chymotrypsin-like activity, leading to accumulation of polyubiquitinated proteins, disruption of protein homeostasis, and induction of apoptosis in malignant cells [68].
The heightened susceptibility of cancer cells to proteasome inhibition stems from their increased reliance on UPS function to degrade misfolded proteins resulting from rapid proliferation and genomic instability, as well as the specific need to regulate levels of oncoproteins and tumor suppressors [67] [68]. Multiple myeloma cells are particularly vulnerable due to their high protein synthetic burden for antibody production [69].
Comparative Analysis of Approved Proteasome Inhibitors
Table 1: Clinically Approved Proteasome Inhibitors for Cancer Therapy
| Drug Name | Molecular Target | Binding Mechanism | Clinical Indications | Key Adverse Effects |
|---|---|---|---|---|
| Bortezomib | 20S proteasome (β5 subunit) | Reversible covalent binding | Multiple myeloma, mantle cell lymphoma | Peripheral neuropathy, thrombocytopenia, gastrointestinal toxicity |
| Carfilzomib | 20S proteasome (β5 subunit) | Irreversible covalent binding | Relapsed/refractory multiple myeloma | Cardiotoxicity, renal dysfunction, thrombocytopenia |
| Ixazomib | 20S proteasome (β5 subunit) | Reversible covalent binding | Relapsed/refractory multiple myeloma | Gastrointestinal toxicity, thrombocytopenia, rash |
Table 2: Efficacy Profiles of Proteasome Inhibitors in Multiple Myeloma
| Drug | Response Rate in Relapsed/Refractory MM | Overall Survival Benefit | Administration Route | Common Combination Regimens |
|---|---|---|---|---|
| Bortezomib | 43% (single agent); higher in combinations | Significant improvement established | IV/Subcutaneous | Dexamethasone, lenalidomide, cyclophosphamide |
| Carfilzomib | 23-54% (varying by patient population) | Demonstrated in clinical trials | IV infusion | Dexamethasone, lenalidomide, daratumumab |
| Ixazomib | 15% (single agent); higher in combinations | Non-inferior to standard care | Oral | Lenalidomide, dexamethasone |
The efficacy of proteasome inhibitors extends beyond direct cytotoxicity to include disruption of key survival pathways in cancer cells. They inhibit nuclear factor kappa-B (NF-κB) signaling by preventing degradation of its inhibitor IκB, sensitize cells to apoptosis by affecting the balance of Bcl-2 family proteins, and induce endoplasmic reticulum stress through accumulation of misfolded proteins [70] [68]. The differential mechanisms of action—reversible versus irreversible binding—contribute to distinct resistance patterns and toxicity profiles among these agents [68].
Emerging UPS-Targeting Therapeutic Strategies
PROTACs and Molecular Glues
While proteasome inhibitors broadly disrupt proteasome function, newer technologies aim to achieve targeted protein degradation with greater precision. Proteolysis-Targeting Chimeras (PROTACs) are heterobifunctional molecules that consist of one ligand that binds to a target protein of interest, another ligand that recruits an E3 ubiquitin ligase, and a linker connecting these two moieties [67] [71]. By bringing the target protein into proximity with an E3 ligase, PROTACs facilitate target ubiquitination and subsequent degradation by the proteasome [72]. This approach offers several advantages over traditional inhibition: it catalytically degrades the entire protein rather than merely inhibiting its activity, can target proteins without classical active sites, and may overcome resistance mechanisms that develop against inhibitors [71].
Molecular glue degraders represent a related strategy wherein a single small molecule induces or stabilizes an interaction between a target protein and an E3 ligase, leading to ubiquitination and degradation [71]. Notably, immunomodulatory drugs (IMiDs) like thalidomide, lenalidomide, and pomalidomide, initially developed for other purposes, were later discovered to function as molecular glues that redirect the CRL4CRBN E3 ligase to degrade specific transcription factors [69].
Expanding the E3 Ligase Landscape for Targeted Degradation
The human genome encodes over 600 E3 ubiquitin ligases, yet most current targeted protein degradation approaches rely heavily on just a few, particularly cereblon (CRBN) and von Hippel-Lindau (VHL) [72]. This limitation has prompted efforts to identify and characterize novel E3 ligases to expand the therapeutic toolbox. Recent research has uncovered several promising alternative E3 ligases:
Ligase X: Functions through the CUL1/SKP1 SCF complex and demonstrates significant potential for degrading various oncology targets, including BRD4. This ligase is notably upregulated in many cancers, raising the possibility of cancer-selective degradation [72].
KLHDC2: A component of the CUL2 complex that can be recruited to degrade diverse proteins, including the inflammation target TYK2 and transcription factor FOXP3. Its wide tissue expression suggests potential for broad substrate scope [72] [73].
N-degron pathway ligases: An underutilized class of E3 ligases that recognize amino-terminal residues of proteins as degradation signals, offering novel degradation mechanisms [72].
Table 3: Novel E3 Ligases for Targeted Protein Degradation
| E3 Ligase | Complex | Targets Demonstrated | Therapeutic Areas | Key Advantages |
|---|---|---|---|---|
| Ligase X | CUL1/SKP1 SCF | BRD4, BRD9 | Oncology | Cancer-selective expression; broad substrate scope |
| KLHDC2 | CUL2 complex | TYK2, FOXP3, K-RAS, β-catenin | Oncology, Inflammation | Wide tissue expression; diverse substrate range |
| N-degron Ligase Y | Not specified | Under investigation | Multiple potential applications | Novel recognition mechanism; ubiquitous expression |
The diversification of recruitable E3 ligases addresses several limitations of current targeted protein degradation approaches, including resistance development from E3 ligase mutations, tissue-specific toxicity, and restricted substrate scope [72]. Furthermore, different E3 ligases exhibit varying expression patterns across tissues and cancer types, enabling development of context-specific degraders [73].
Experimental Approaches for Evaluating UPS-Targeting Therapies
Methodologies for Assessing Degradation Efficiency
Rigorous evaluation of UPS-targeting therapies requires standardized experimental protocols to quantify degradation efficiency, kinetics, and specificity. Key methodologies include:
Western Blot Analysis for Protein Level Quantification
- Protocol: Cells treated with degraders are lysed at multiple time points (e.g., 1, 6, 24 hours post-treatment). Proteins are separated by SDS-PAGE, transferred to membranes, and probed with target-specific antibodies. Band intensity is quantified using imaging software and normalized to loading controls.
- Data Interpretation: Calculate percentage degradation relative to DMSO-treated controls. Assess degradation kinetics by plotting protein levels over time. Evaluate specificity by probing for unrelated proteins.
- Optimization Notes: Include proteasome (MG-132) and neddylation (MLN-4924) inhibitors to confirm UPS-dependent degradation [72] [74].
Cellular Viability and Proliferation Assays
- Protocol: Seed cancer cells in 96-well plates and treat with serial dilutions of degraders. After 72-120 hours, measure viability using ATP-based (CellTiter-Glo) or metabolic activity (MTT) assays. Generate dose-response curves to calculate IC50 values.
- Controls: Include non-targeted cell lines to assess selectivity. Compare to conventional inhibitors of the same target.
- Considerations: Some E3 ligase recruiters (dTAG13, HaloPROTAC3) can independently affect proliferation, necessitating careful interpretation [74].
Ternary Complex Formation assays
- Protocol: Utilize techniques like HTRF (Homogeneous Time-Resolved Fluorescence) to detect proximity between target protein, degrader, and E3 ligase. Purified components are incubated with degraders, and energy transfer signals indicate complex formation.
- Applications: Validate mechanistic engagement and optimize linker length/chemistry in PROTAC design.
- Advantages: Provides direct evidence of targeted engagement rather than downstream degradation [72].
In Vivo Efficacy and Therapeutic Index Assessment
Translating UPS-targeting therapies from cellular models to preclinical in vivo systems requires specialized approaches:
Xenograft Mouse Models
- Protocol: Implant human cancer cells or patient-derived xenografts immunodeficient mice. Once tumors reach 100-200 mm³, randomize animals into treatment groups. Administer degraders via appropriate routes (oral, subcutaneous, intravenous) at predetermined schedules.
- Endpoint Measurements: Monitor tumor volume regularly by caliper measurements. At study termination, harvest tumors for pharmacodynamic analysis of target degradation and pathway modulation.
- Key Findings: Ligase X-based BRD4 degraders demonstrated complete loss of tumor-associated BRD4 levels with BID dosing in MV4.11 xenograft models [72].
Pharmacodynamic Biomarker Assessment
- Protocol: Collect tumor tissues at multiple time points post-dose. Analyze target protein levels by Western blot or immunohistochemistry. Assess downstream pathway modulation (e.g., c-MYC reduction for BRD4 degraders).
- Advanced Applications: Utilize reporter cell lines expressing luciferase-tagged targets for real-time degradation monitoring in vivo.
The UPS in Tumor Immunity and Combination Therapies
Beyond direct anticancer effects, UPS modulation significantly impacts antitumor immunity, creating opportunities for combination strategies. The UPS regulates key immune checkpoints through E3 ligase-mediated degradation of PD-L1 and controls the stability of critical transcription factors in immune cells [73]. For instance, FOXP3 stability in regulatory T cells (Tregs) is controlled by multiple E3 ligases, including STUB1, CBLB, and KLHDC2, which promote its degradation through K48-linked ubiquitination, while other ligases like Itch and RNF31 enhance FOXP3 stability through non-degradative ubiquitination [73]. This intricate regulation suggests that selective modulation of specific E3 ligases could reprogram the tumor immune microenvironment.
The combination of PROTACs with immune checkpoint inhibitors represents a promising frontier. Preclinical data demonstrate that USP5 inhibition synergizes with PD-(L)1 blockade, suggesting similar potential for targeted degraders [73]. Furthermore, the discovery that certain E3 ligases like SPOP and FBXO22 mediate PD-L1 degradation provides opportunities to enhance immune recognition by stabilizing this checkpoint protein through ligase inhibition [73].
Research Tools and Reagent Solutions
Table 4: Essential Research Tools for Investigating UPS-Targeted Therapies
| Reagent/Category | Specific Examples | Research Applications | Key Features & Considerations |
|---|---|---|---|
| Inducible Degron Systems | dTAG, HaloPROTAC, AID 2.0/2.1, IKZF3-based degrons | Rapid protein depletion studies; essential gene functional analysis | Varying kinetics, basal degradation, and reversibility profiles [74] |
| Proteasome Inhibitors | MG-132, bortezomib, epoxomicin | Validation of UPS-dependent degradation; control experiments | Different selectivity profiles (β5 vs. broad specificity) |
| E3 Ligase Modulators | MLN4924 (neddylation inhibitor), P5091 (USP7 inhibitor) | Study of cullin-RING ligase function; DUB inhibition experiments | MLN4924 blocks activity of cullin-based E3 ligases |
| Ubiquitination Assays | TUBE (Tandem Ubiquitin Binding Entity), linkage-specific antibodies | Detection and characterization of ubiquitin chains | Distinguish degradative (K48) vs. signaling (K63, M1) ubiquitination |
| Base Editing Systems | Cytosine base editors, adenine base editors | Directed evolution of E3 ligases; degron system optimization | Enables creation of gain-of-function variants (e.g., AID 2.1) [74] |
Visualizing Key UPS Pathways and Experimental Approaches
The Ubiquitin-Proteasome System Pathway
PROTAC Mechanism of Action
The therapeutic targeting of the ubiquitin-proteasome system has evolved dramatically from broad proteasome inhibitors to highly precise targeted protein degradation approaches. The expanding repertoire of recruitable E3 ligases, combined with advanced technologies like PROTACs and molecular glues, offers unprecedented opportunities for intervening in cancer pathways previously considered undruggable. Future directions include developing tissue-selective degraders, overcoming resistance mechanisms, and optimizing degradation kinetics for improved therapeutic indices. The continued exploration of UPS biology, informed by its evolutionary origins in prokaryotic systems, will undoubtedly yield further innovations in cancer therapy.
Navigating Complexity: Challenges in Studying and Targeting Prokaryotic Ubl Systems
The ubiquitin (Ub) signaling system, long considered a hallmark of eukaryotic cells, is a sophisticated protein modification machinery that regulates critical processes such as protein degradation and cellular signaling. Historically, prokaryotes were thought to lack such complex systems, but emerging research has fundamentally challenged this paradigm. We now understand that prokaryotes possess ancestral versions of ubiquitin-like (Ubl) signaling systems that represent evolutionary precursors to the eukaryotic ubiquitin apparatus [26] [20]. These prokaryotic Ubl systems utilize β-grasp fold proteins—ThiS, MoaD, TGS, and YukD—that are structurally and functionally related to eukaryotic ubiquitin [26] [20]. However, the identification and characterization of transient modifications and low-abundance conjugates in prokaryotic systems present unique experimental challenges that require specialized methodologies and tools.
The recent discovery of a ubiquitin-like machinery in bacteria by Chambers et al. underscores the prevalence of these systems in prokaryotes and their ancestral relationship to eukaryotic ubiquitin systems [4]. This breakthrough finding confirms that bacteria possess a functional ubiquitin-like system that was later adopted and refined by eukaryotes for diverse cellular purposes, including targeted protein degradation. Understanding these prokaryotic systems not only sheds light on evolutionary biology but also opens new avenues for therapeutic intervention, particularly for drug development professionals targeting bacterial pathogens.
Comparative Analysis of Ubiquitin-like Systems
Table 1: Comparison of Ubiquitin-like System Components Across Domains of Life
| Component | Eukaryotic Systems | Prokaryotic Systems | Key Differences |
|---|---|---|---|
| Ubiquitin-like Proteins | Ubiquitin, SUMO, Nedd8, URM1, Apg8/Apg12, ISG15 | ThiS, MoaD, TGS, YukD, and novel β-grasp fold proteins [26] [20] | Prokaryotic versions often specialized for sulfur transfer; lack extensive conjugation systems |
| Adenylating Enzymes (E1-like) | E1 ubiquitin-activating enzymes | ThiF, MoeB, and other E1-like adenylating enzymes [26] [20] | Similar catalytic mechanism but different substrate specificity |
| Conjugating Enzymes (E2-like) | E2 ubiquitin-conjugating enzymes (UBC) | UBC/E2-related proteins found in conserved gene neighborhoods [26] [20] | Less diverse in prokaryotes; often associated with specific metabolic pathways |
| Deconjugating Enzymes | Deubiquitinases (DUBs) of JAB, papain-like, and metalloprotease superfamilies | JAB domain peptidases and other isopeptidases [26] [20] | JAB domains in prokaryotes associated with diverse Ubl proteins |
| Functional Associations | Form strong functional associations between E1, E2, E3, and DUBs | Conserved gene neighborhoods combine JAB, E1-like, and Ubl proteins [26] [20] | Prokaryotic systems show precursor-level integration rather than comprehensive networks |
Table 2: Experimental Challenges in Prokaryotic Ubl Research
| Challenge | Impact on Research | Eukaryotic Counterpart |
|---|---|---|
| Transient Modification Detection | Difficult to capture short-lived Ubl conjugates due to rapid modification and reversal [26] [75] | Better established methodologies for capturing transient ubiquitination |
| Low-Abundance Conjugate Identification | Limited by sensitivity of detection methods and low expression of Ubl components [76] | Advanced enrichment strategies and sensitive mass spectrometry available |
| Dynamic Range Issues | High-abundance proteins obscure detection of rare Ubl conjugates [76] | Extensive fractionation and enrichment methods developed |
| Modification Heterogeneity | Multiple modification types and sites complicate analysis [76] | Well-characterized in eukaryotes but still challenging |
| Context-Dependent Modifications | Ubl modifications often condition-specific and growth-phase dependent [76] | Similar challenges but better tools for context preservation |
Key Experimental Hurdles and Methodological Approaches
Capturing Transient Protein-Protein Interactions
The transient nature of prokaryotic Ubl interactions represents a significant experimental hurdle. Bacterial effector proteins often engage in brief, "touch-and-go" interactions with host targets that are difficult to capture using conventional methods [75]. These interactions are typically short-lived and not sustained for prolonged periods, making them elusive targets for identification and characterization. The kinetic instability of these complexes means they may dissociate during cell lysis or purification procedures, leading to false negatives in interaction studies.
Traditional methods like yeast two-hybrid (Y2H) screening and affinity purification-mass spectrometry (AP-MS) have limitations in capturing these transient interactions. Y2H is particularly constrained by its requirement for interactions to occur in the non-native environment of yeast cells and its inability to detect interactions that depend on specific cellular contexts or larger protein scaffolds [75]. Similarly, AP-MS often struggles to capture weak, transient interactions or low-abundance proteins, especially in complex cellular environments, and is susceptible to false-positive protein-protein interactions (PPIs) occurring in non-native contexts following cell lysis [75].
Detecting Low-Abundance Conjugates
The identification of low-abundance Ubl conjugates in prokaryotes is complicated by several factors. These conjugates are often present in minute quantities compared to the overall cellular proteome, creating significant dynamic range challenges. Additionally, bacterial cultures in laboratory settings may not accurately reflect the conditions under which these Ubl systems are active in natural environments [76]. The standard practice of using batch cultures grown for short periods in rich media provides a poor reflection of the high-stress, nutrient-starved conditions in which bacteria typically spend most of their time in the wild, potentially leading to underestimation of Ubl conjugation events.
Mass spectrometry-based proteomics has revolutionized the detection of post-translational modifications, but its application to prokaryotic Ubl systems faces specific obstacles. The low stoichiometry of Ubl modifications means that modified peptides are often overshadowed by their unmodified counterparts in mass spectrometry analyses. Furthermore, the inherent heterogeneity of Ubl modifications—with variations in chain length, linkage type, and attachment sites—further complicates their comprehensive identification and quantification [76].
Advanced Methodologies for Overcoming Detection Challenges
Proximity-Dependent Labeling Techniques
Proximity labeling (PL) has emerged as a powerful strategy for overcoming the challenges associated with detecting transient interactions and low-abundance conjugates. This technique involves fusing a labeling enzyme to a protein of interest ("bait"), which then catalyzes the covalent attachment of small molecular tags to nearby proteins ("preys") [77] [75]. These labeled proteins can be purified, enriched, and analyzed through liquid chromatography tandem mass spectrometry (LC-MS/MS).
Table 3: Comparison of Proximity Labeling Methods
| Method | Enzyme | Size (kDa) | Labeling Time | Labeling Radius | Key Applications in Prokaryotic Research |
|---|---|---|---|---|---|
| BioID | BirA∗ | 35 | ~24 hours | 10-20 nm | First-generation biotin ligase-based PL [77] |
| BioID2 | Biotin ligase (A. aeolicus) | 27 | ~18 hours | ~10 nm | Smaller size reduces bait interference [77] |
| TurboID | Engineered biotin ligase | 35 | ≥10 minutes | 5-10 nm | Rapid labeling for capturing transient interactions [77] |
| miniTurbo | Engineered biotin ligase | 28 | ~10 minutes | 5-10 nm | Balanced size and activity advantages [77] |
| APEX/APEX2 | Engineered peroxidases | 28 | 1 minute | ~20 nm | Ultra-rapid labeling for highly dynamic processes [77] |
The implementation of PL techniques in prokaryotic research has already yielded significant insights. For example, Khan et al. (2018) first demonstrated the successful application of BioID in leaf tissues of the model plant Arabidopsis thaliana, showcasing its potential as an addition to the proteomic toolbox for plant research [77]. Subsequent work by Mair et al. showed that both TurboID and miniTurboID provided higher sensitivity than BioID in infiltrated Nicotiana benthamiana leaves and transformed A. thaliana seedlings [77]. These advanced PL methods have been used to profile various plant interactomes, including Arabidopsis meiotic chromosome axes, BRASSINOSTEROID-INSENSITIVE 2 (BIN2) kinase, and the target of rapamycin (TOR) complex [77].
Enrichment and Proteomic Strategies
Deep sampling across multiple growth conditions and timepoints is critical for comprehensive mapping of prokaryotic Ubl modifications. Research on Escherichia coli has demonstrated that post-translational modifications exhibit dynamic changes throughout the bacterial growth cycle, from early exponential phase to extended late stationary phase [76]. This temporal dimension is essential for capturing condition-specific modifications that might be absent in standard laboratory growth conditions.
Unbiased detection approaches for post-translational modifications in MS/MS proteomics data have facilitated the discovery of novel modification types and previously unobserved dynamic changes. Tools like MODa, which uses a combination of de novo sequence-tag matching and spectral alignment, enable researchers to conduct unrestricted analyses of PTMs across a wide range of mass shifts without being limited to predetermined modification types [76]. This approach has revealed novel temporal patterns in N-terminal N α acetylation, C-terminal glutamylation, and asparagine deamidation in prokaryotic systems.
Figure 1: Proximity Labeling Workflow for Detecting Transient Interactions
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 4: Key Research Reagent Solutions for Prokaryotic Ubl Studies
| Reagent Category | Specific Examples | Function in Research | Considerations for Prokaryotic Systems |
|---|---|---|---|
| PL Enzymes | BirA∗ (BioID), BioID2, TurboID, miniTurbo, APEX2 [77] | Catalyze proximity-dependent biotinylation of interacting proteins | Size may interfere with bait function; optimization required for different bacterial systems |
| Biotin Derivatives | Biotin-phenol (for peroxidase-based PL), free biotin (for ligase-based PL) [77] | Serve as substrates for PL enzymes | Membrane permeability and effective concentration critical |
| Enrichment Matrices | Streptavidin beads/magnetic beads [77] | Capture biotinylated proteins for purification | Non-specific binding can be issue; rigorous washing conditions needed |
| Proteomic Tools | LC-MS/MS systems, unbiased PTM search algorithms (MODa) [76] | Identify and quantify modified peptides | Must accommodate small sample amounts and complex backgrounds |
| Chemical Crosslinkers | Membrane-permeable and impermeable crosslinkers | Stabilize transient interactions before lysis | Optimization needed to balance preservation vs. introduction of artifacts |
| Ubiquitin-like Probes | Active-site directed probes for JAB domains and other deubiquitinases [26] | Identify and characterize deconjugating enzymes | Limited availability for prokaryotic-specific enzymes |
Detailed Experimental Protocols
Proximity Labeling for Transient Interaction Capture
The following protocol outlines the implementation of TurboID for capturing transient interactions in prokaryotic systems, based on optimized methodologies from recent studies [77]:
Construct Preparation: Fuse the TurboID enzyme to your protein of interest (bait) using appropriate expression vectors. Consider including a cleavable linker (e.g., TEV protease site) between the bait and TurboID to enable competitive elution after affinity capture.
Expression Optimization: Transform the construct into your target prokaryotic system and optimize expression conditions to minimize toxicity while maintaining sufficient labeling efficiency. For TurboID, this typically involves testing induction conditions and temperature.
Biotin Supplementation: Add biotin to the growth medium at optimized concentrations (typically 50-500 μM) for the required labeling duration. For capturing transient interactions, shorter labeling times (10-30 minutes) are preferable.
In Vivo Labeling: Allow the labeling reaction to proceed under appropriate growth conditions. Control temperature carefully as it affects enzyme activity.
Cell Lysis: Harvest cells and lyse using gentle conditions to preserve protein complexes while ensuring complete disruption. Include protease inhibitors and, if studying redox-sensitive modifications, antioxidants.
Affinity Capture: Incubate lysates with streptavidin-coated beads for 1-2 hours at 4°C with gentle agitation. Use sufficient bead capacity to capture all biotinylated proteins.
Stringent Washing: Wash beads extensively with modified RIPA buffer (50 mM HEPES, pH 7.5, 500 mM NaCl, 0.1% SDS, 1% Triton X-100, 1 mM EDTA, 1 mM EGTA) followed by urea wash (2 M urea in 50 mM Tris, pH 7.5) to remove nonspecific binders.
On-Bead Digestion: Digest captured proteins directly on beads using sequencing-grade trypsin (typically 1:50 enzyme-to-substrate ratio) in 50 mM ammonium bicarbonate buffer overnight at 37°C.
Peptide Cleanup: Desalt peptides using C18 StageTips or similar solid-phase extraction methods prior to LC-MS/MS analysis.
LC-MS/MS Analysis: Analyze peptides using high-resolution mass spectrometry with data-dependent acquisition or data-independent acquisition methods for improved quantification.
Enrichment Strategies for Low-Abundance Conjugates
For comprehensive identification of low-abundance Ubl conjugates, a multi-dimensional enrichment approach is recommended:
Affinity-Based Enrichment: Utilize affinity tags (e.g., His-tags, FLAG-tags) on Ubl proteins to enrich for conjugated species. Perform under denaturing conditions to preserve transient modifications.
Immunoaffinity Purification: Employ antibodies specific for Ubl proteins or their modifications. This approach benefits from the commercial availability of high-quality ubiquitin antibodies, though cross-reactivity with prokaryotic Ubls must be verified.
Tandem Ubiquitin Binding Entities (TUBEs): Although developed for eukaryotic ubiquitin, the principle can be adapted for prokaryotic Ubls by identifying natural binding domains or engineering specific binders.
Chemical Enrichment Strategies: Implement chemistries that target specific features of Ubl modifications, such as di-glycine remnant enrichment following tryptic digestion.
Figure 2: Prokaryotic Ubiquitin-like Signaling Pathway with Recycling Mechanism
Data Analysis and Interpretation Frameworks
The analysis of proteomic data from prokaryotic Ubl studies requires specialized computational approaches to address the challenges of modification heterogeneity and low abundance. Several key considerations should guide data analysis:
Database Searching Strategies: Implement open search algorithms that can identify unexpected modifications rather than being limited to predetermined mass shifts. The MODa algorithm has proven particularly valuable for this purpose, as it uses de novo sequence-tag matching and spectral alignment to identify modifications across a wide mass range (+/- 200 Da) without prior assumptions about modification types [76].
False Discovery Rate Control: Apply stringent statistical thresholds to account for the increased search space in modification-aware analyses. Use target-decoy approaches at both the peptide-spectrum match level and the modification site localization level.
Temporal Pattern Analysis: When time-course data is available, employ clustering algorithms to identify modification patterns that correlate with growth phases or specific environmental conditions. This approach has revealed condition-specific modifications in E. coli that would be missed in single-time-point analyses [76].
Integration with Genomic Context: Leverage the conserved gene neighborhoods observed in prokaryotic Ubl systems to prioritize putative interactions [26] [20]. Proteins encoded together in operons or conserved gene clusters are more likely to represent biologically relevant interaction partners.
The field of prokaryotic ubiquitin-like signaling is rapidly evolving, with new methodologies continuously emerging to address the persistent challenges of studying transient modifications and low-abundance conjugates. The experimental hurdles outlined in this guide—particularly the transient nature of interactions and the low abundance of conjugates—require sophisticated methodological approaches that are still being optimized for prokaryotic systems.
Future advancements will likely come from several directions: First, the continued development of more efficient proximity labeling enzymes with faster kinetics and smaller sizes will improve our ability to capture fleeting interactions. Second, advances in mass spectrometry sensitivity and scanning speeds will enhance detection of low-abundance modified species. Third, the integration of structural biology with proteomics will provide mechanistic insights into the recognition specificities of prokaryotic Ubl systems.
For researchers and drug development professionals, understanding these experimental challenges and the methodologies to address them is crucial for advancing our knowledge of prokaryotic cell biology and developing novel antibacterial strategies. The evolutionary conservation between prokaryotic and eukaryotic ubiquitin-like systems suggests that insights gained from bacterial studies may also inform our understanding of human biology and disease mechanisms.
As methodologies continue to improve, we anticipate that the coming years will bring significant breakthroughs in our understanding of the functional roles and regulatory mechanisms of ubiquitin-like signaling in prokaryotic systems, with potential applications in antibiotic development, biotechnology, and fundamental microbiology.
The evolutionary arms race between bacterial pathogens and their hosts has led to the development of sophisticated bacterial effector proteins that manipulate host cellular processes, particularly the ubiquitin system. This comparison guide classifies these effector mechanisms into two primary categories: "rule followers" that structurally and mechanistically mimic host ubiquitin machinery, and "rule breakers" that employ novel folds and distinct biochemical strategies. We objectively compare the performance of diverse effector classes through structured experimental data, detailed methodologies, and pathway visualizations, providing researchers and drug development professionals with a framework for understanding these virulence mechanisms within the broader context of prokaryotic-eukaryotic ubiquitin system evolution.
Ubiquitination is a crucial post-translational modification pathway in eukaryotic cells, governing protein degradation, signal transduction, and immune responses [78]. This pathway typically follows a conserved three-enzyme cascade involving E1 (activating), E2 (conjugating), and E3 (ligating) enzymes that coordinate to attach ubiquitin to substrate proteins [79]. The profound importance of the ubiquitin system in host immunity has made it a prime target for bacterial pathogens. Despite lacking conventional ubiquitin systems of their own, bacteria have evolved secreted effector proteins that extensively manipulate host ubiquitination pathways [78]. Recent research reveals that bacteria not only manipulate host ubiquitin systems but also possess ancient, functional ubiquitination-like pathways themselves, suggesting these systems may have first arisen in bacteria [4] [5]. This guide systematically compares the two dominant strategies employed by bacterial effectors: those that follow established rules of eukaryotic ubiquitination and those that break these conventions to achieve pathogenesis.
Comparative Analysis of Bacterial Effector Mechanisms
Bacterial effectors targeting host ubiquitination pathways can be classified into two primary categories based on their structural and mechanistic relationship to eukaryotic ubiquitin machinery. The table below provides a comprehensive comparison of these effector classes, their representative examples, and their functional characteristics.
Table 1: Classification and Characteristics of Bacterial Effector Mechanisms
| Effector Class | Representative Examples | Pathogen Source | Structural & Mechanistic Features | Cellular Targets & Functions |
|---|---|---|---|---|
| Rule Followers | AvrPtoB | Pseudomonas syringae | U-box domain with structural homology to RING-type E3 ligases; conserved E2-binding interface and linchpin residue [78] | Targets Fen, CERK1, FLS2, BAK1 kinases for degradation; suppresses plant immunity [79] |
| NleG | Enterohemorrhagic E. coli (EHEC) | RING-finger/U-box mimic; interacts with human E2 enzymes similar to eukaryotic E3 counterparts [78] [80] | Specific intracellular targets unknown; associated with virulence [80] | |
| SopA, NleL | Salmonella, EHEC | HECT-like ligases with conserved catalytic cysteine; interacts with E2 enzyme UbcH7 on same surface as eukaryotic ligases [79] [80] | SopA regulates inflammation; NleL involved in pedestal formation [79] | |
| Rule Breakers | IpaH Family (IpaH9.8, IpaH4.5) | Shigella, Salmonella | Novel E3 ligase (NEL) domain distinct from HECT/RING; LRR domain for substrate recognition; dramatic reorientation upon substrate binding [79] [80] | IpaH9.8 ubiquitinates NEMO/IKKγ and Ste7; IpaH4.5 targets p65; suppresses inflammation and splicing [79] |
| OspI | Shigella flexneri | Deamidase activity; does not mimic DUBs but modifies E2 enzyme [80] | Deamidates UBC13 (Q100 to E); prevents TRAF6 polyubiquitylation and NF-κB signaling [79] [80] | |
| Cif | EHEC, Burkholderia pseudomallei | Deamidase/cycloamidease activity targeting NEDD8 and ubiquitin [79] | Inhibits Cullin-RING ligase (CRL) activation; blocks host protein degradation [79] |
Experimental Approaches for Studying Effector Mechanisms
Structural Characterization Protocols
X-ray Crystallography of Effector Complexes:
- Purpose: Determine atomic-scale molecular information about effector-host interactions, conformational changes, and substrate specificity [79].
- Methodology:
- Clone and express effector proteins (e.g., E1BilD, E2BilB, UblBilA) in E. coli, often requiring co-expression for soluble complex formation [5].
- Purify complexes using affinity chromatography (Ni-NTA for tagged proteins) followed by size exclusion chromatography.
- Crystallize using vapor diffusion methods with optimization of pH, precipitant, and temperature.
- Collect X-ray diffraction data at synchrotron facilities and solve structures using molecular replacement or MAD/SAD phasing [5].
- Key Applications: Revealed architectural parallels between bacterial E1:E2:Ubl complexes and eukaryotic ubiquitination machinery [5].
Yeast Two-Hybrid Screening:
- Purpose: Identify novel interactions between effectors and host ubiquitin system components [78].
- Methodology:
- Fuse effector protein to DNA-binding domain (bait).
- Fuse host cell cDNA library to activation domain (prey).
- Co-transform into yeast reporter strain and plate on selective media.
- Sequence positive clones from colonies growing on selective media to identify interacting partners.
- Key Applications: Initial identification of AvrPtoB interactions with ubiquitin system components [78].
Functional Ubiquitination Assays
In Vitro Ubiquitination Assays:
- Purpose: Demonstrate direct E3 ligase activity of bacterial effectors and identify required components [78] [79].
- Methodology:
- Purify recombinant effector protein, mammalian E1 enzyme, specific E2 enzyme (e.g., UbcH5), ubiquitin, and ATP.
- Combine components in reaction buffer (e.g., 50mM Tris-HCl pH 7.5, 5mM MgCl₂, 2mM ATP).
- Incubate at 30°C for 1-2 hours.
- Terminate reaction with SDS loading buffer and analyze by western blotting using anti-ubiquitin antibodies or detect auto-ubiquitination [78].
- Key Applications: Confirmed E3 ligase activity for AvrPtoB and NleG family effectors [78].
Effector Screening in Insect Cells:
- Purpose: Identify effectors that rescue arbovirus replication in non-permissive lepidopteran cells [81].
- Methodology:
- Generate library of bacterial effectors (210 effectors from 7 pathogens) in expression vectors.
- Transfect restrictive insect cells (e.g., LD652) with effector plasmids.
- Infect cells with arboviruses (VSV, SINV, RRV, ONNV) that normally undergo abortive replication.
- Measure viral replication via plaque assays, qRT-PCR, or fluorescent reporter expression [81].
- Key Applications: Identified IpaH4 as E3 ubiquitin ligase that targets SHOC2 and PSMC1 to promote arbovirus replication [81].
Bacterial Ubiquitin Pathway Manipulation
The diagram below illustrates the five primary mechanisms by which bacterial effectors manipulate the host ubiquitin system, highlighting both rule-following and rule-breaking strategies.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagents for Studying Bacterial Effector Mechanisms
| Reagent / Method | Specific Examples | Research Application | Key Experimental Function |
|---|---|---|---|
| Eukaryotic E1/E2 Enzymes | Human UBA1, UBA6, UbcH5, UbcH7 | In vitro ubiquitination assays | Provide basal ubiquitination machinery for testing effector E3 activity [78] [79] |
| Antibody Reagents | Anti-ubiquitin, anti-NEMO, anti-p65, anti-IκBα | Western blot, immunoprecipitation | Detect ubiquitination status and substrate degradation [79] |
| Cell-Based Systems | Lepidopteran LD652 cells, Drosophila models, human cell lines | Effector screening, functional characterization | Provide cellular context for studying effector function in innate immunity [82] [81] |
| Expression Vectors | Tightly-regulated metallothionein promoter, UAS/Gal4 system | Controlled effector expression in hosts | Enable expression of toxic effectors in tissue-specific or inducible manner [82] |
| Structural Biology | X-ray crystallography, AlphaFold predictions | Determining effector-host complexes | Reveal atomic-scale interaction mechanisms and conformational changes [79] [5] |
The classification of bacterial effectors as rule followers or rule breakers provides a valuable framework for understanding host-pathogen interactions and the evolution of ubiquitin signaling systems. Rule-following effectors demonstrate remarkable convergent evolution with eukaryotic enzymes, while rule-breaking effectors reveal novel structural and mechanistic approaches to ubiquitin manipulation [78]. From a therapeutic perspective, rule-breaking effectors represent particularly attractive drug targets due to their absence from human biochemistry, potentially offering greater specificity and reduced off-target effects. Recent discoveries of functional ubiquitination-like pathways in bacteria themselves [4] [5] further highlight the ancient evolutionary origins of these systems and open new avenues for antimicrobial development. Future research should focus on characterizing unstudied effector families, developing high-throughput screening methods for effector inhibitors, and exploiting bacterial effector proteins as molecular tools to identify novel antiviral host factors [81], ultimately advancing our ability to combat infectious diseases through targeted therapeutic interventions.
The ubiquitin system, a cornerstone of eukaryotic cell biology, has long been characterized as a reversible post-translational modification. This reversibility is maintained by deubiquitinases (DUBs) that cleave the isopeptide bond between ubiquitin and substrate lysine residues, providing dynamic control over protein fate. However, recent discoveries have revealed that bacterial pathogens have evolved a remarkable mechanism to overcome this fundamental reversibility through a novel class of effector proteins termed ubiquitin C-terminal clippases (UCCs). Unlike conventional DUBs, UCCs permanently inactivate ubiquitin by cleaving within its C-terminal sequence, thereby creating an irreversible modification that disrupts host cell defenses [83].
This review examines bacterial UCCs as unique virulence factors, comparing their mechanisms, specificities, and functional consequences with canonical eukaryotic DUBs and bacterial effectors that follow traditional ubiquitin manipulation rules. We present comprehensive experimental data and methodologies that highlight the distinctive biochemical properties of UCCs and their potential applications in both basic research and therapeutic development.
Ubiquitin Signaling: Canonical versus Bacterial Pathogen Strategies
The Eukaryotic Ubiquitin System
In eukaryotic cells, ubiquitination involves a coordinated enzymatic cascade comprising E1 activating, E2 conjugating, and E3 ligase enzymes that attach ubiquitin to substrate proteins. The canonical process typically creates an isopeptide bond between the C-terminal glycine (Gly76) of ubiquitin and the ε-amino group of a lysine residue on target proteins [84] [21]. This modification is inherently reversible through the action of deubiquitinating enzymes (DUBs), which hydrolyze the isopeptide linkage to release ubiquitin for recycling [85].
The ubiquitin code encompasses remarkable diversity, with different chain linkages directing distinct cellular outcomes:
- K48-linked chains: Primarily target substrates for proteasomal degradation [21] [85]
- K63-linked chains: Regulate non-degradative processes including DNA repair and kinase activation [21] [85]
- M1-linked linear chains: Critical for NF-κB signaling and inflammatory responses [21] [85]
Bacterial Manipulation of Host Ubiquitination
Intracellular bacterial pathogens secrete effector proteins that extensively manipulate host ubiquitin signaling to overcome immune defenses. These effectors employ diverse strategies:
- Rule-following effectors: Proteins like Pseudomonas syringae AvrPtoB mimic eukaryotic E3 ligases, utilizing structurally similar RING or U-box domains to hijack the host ubiquitination cascade [78].
- Rule-breaking effectors: Enzymes such as the Legionella pneumophila SidE family bypass the canonical E1-E2-E3 cascade entirely, creating phosphoribosyl-linked ubiquitination on serine residues through a single enzymatic step [84].
Among these strategies, UCCs represent a particularly ingenious mechanism that permanently alters the ubiquitin molecule itself, creating an irreversible modification that subverts host cell signaling.
Bacterial Ubiquitin C-terminal Clippases: Mechanism and Specificity
The Clippase Discovery
Hermanns et al. (2025) identified a novel class of bacterial deubiquitinases within the Josephin family of cysteine proteases that cleave ubiquitin at an unprecedented location [83]. Unlike conventional DUBs that hydrolyze the isopeptide bond after Gly76, these UCCs cleave between Arg74 and Gly75 within the ubiquitin C-terminal tail (Figure 1). This cleavage leaves a characteristic Gly-Gly dipeptide attached to the substrate lysine while releasing a truncated ubiquitin missing the last two C-terminal residues [83].
Structural Basis for Clippase Activity
Structural studies of bacterial Josephins complexed with ubiquitin substrates reveal the molecular basis for this atypical cleavage specificity:
- Parachlamydia PcJOS: In complex with linear (M1-linked) diubiquitin, this clippase positions the catalytic site to hydrolyze the peptide bond before the diglycine motif through extensive contacts with both distal and proximal ubiquitin molecules [83].
- Burkholderia pyrrocinia BpJOS: This promiscuous clippase exhibits stable interaction primarily at the S1 ubiquitin-binding site, positioning the Arg74-Gly75 bond at the catalytic center through unique alpha helices following the active-site cysteine [83].
- Comparison with conventional DUBs: The related Pigmentiphaga aceris PaJOS enzyme, a conventional DUB, contains structural elements (primarily α2-η1-α3) that displace the S1-bound ubiquitin to position the catalytic cysteine at the conventional isopeptide cleavage site [83].
Table 1: Comparison of Bacterial Josephin Enzymes with Clippase versus DUB Activity
| Enzyme | Organism | Cleavage Site | Linkage Specificity | Activity Type |
|---|---|---|---|---|
| PcJOS | Parachlamydia sp. | Arg74-Gly75 | M1-linear specific | Clippase |
| BpJOS | Burkholderia pyrrocinia | Arg74-Gly75 | Promiscuous | Clippase |
| PaJOS | Pigmentiphaga aceris | After Gly76 | Conventional DUB | DUB |
Experimental Characterization of UCC Activity
Methodologies for Detecting Clippase Activity
Diubiquitin Cleavage Assays
Protocol: Purified Josephin enzymes are incubated with a panel of diubiquitin substrates containing different linkage types (K6, K11, K27, K29, K33, K48, K63, M1) in appropriate reaction buffer (e.g., 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM DTT). Reactions are terminated at timepoints by adding SDS-PAGE loading buffer, and products analyzed by immunoblotting with ubiquitin-specific antibodies or mass spectrometry [83].
Key finding: Several bacterial Josephins cleaved between Arg74-Gly75 instead of after Gly76, with some enzymes showing broad linkage specificity while others were highly specific for linear (M1) ubiquitin linkages [83].
Structural Analysis via X-ray Crystallography
Protocol: Josephin enzymes are co-crystallized with diubiquitin substrates or ubiquitin suicide probes. Crystals are flash-frozen, and diffraction data collected at synchrotron sources. Structures are solved by molecular replacement using known Josephin and ubiquitin coordinates [83].
Key finding: Co-crystal structures revealed distinct ubiquitin-binding modes between clippases and conventional DUBs that determine cleavage site specificity [83].
Predictive Modeling with AlphaFold
Protocol: AlphaFold2 is used to predict structures of uncharacterized bacterial Josephin proteins in complex with ubiquitin. Predicted structures are compared to solved clippase and DUB complexes to classify potential cleavage specificity [83].
Key finding: Predictions from AlphaFold modeling perfectly matched experimental results for seven tested Josephin proteins (six predicted and confirmed as clippases, one as conventional DUB) [83].
Quantitative Analysis of UCC Enzymatic Properties
Table 2: Enzymatic Properties of Characterized Bacterial Ubiquitin Clippases
| Enzyme | Cleavage Site | Specific Activity* (min⁻¹) | Linkage Preference | Ubiquitin-Like Protein Specificity |
|---|---|---|---|---|
| PcJOS | Arg74-Gly75 | 2.8 ± 0.3 | M1-linear | Ubiquitin, NEDD8 |
| BpJOS | Arg74-Gly75 | 4.2 ± 0.5 | Promiscuous | Ubiquitin, NEDD8 |
| Lbpro (viral) | Varied | 0.15 ± 0.02 | ISG15 preference | ISG15 > Ubiquitin |
| Data represent approximate values based on experimental characterization described in [83] |
Functional Consequences of Ubiquitin Clipping
Irreversible Inactivation of Ubiquitin Signaling
The clippase mechanism produces two molecular outcomes with distinct functional consequences:
Truncated ubiquitin: Lacking the C-terminal Gly75-Gly76 residues, the cleaved ubiquitin cannot be activated by E1 enzymes or conjugated to substrates, permanently removing it from the ubiquitin pool [83].
Blocked substrate lysine: The residual Gly-Gly dipeptide attached to the substrate lysine prevents further ubiquitination at this site, creating a permanently modified residue that cannot participate in subsequent ubiquitin signaling [83].
This dual action represents a sophisticated bacterial strategy to disrupt host ubiquitin-dependent defenses more permanently than conventional DUBs, which leave both ubiquitin and substrate lysine available for future modification cycles.
Predicted Roles in Bacterial Pathogenesis
While direct evidence for the role of UCCs in bacterial infection is still emerging, their activities suggest several plausible functions in pathogenesis:
- Disruption of xenophagy: By cleaving ubiquitin coats on intracellular bacteria or surrounding vacuoles, UCCs may prevent recognition by autophagic machinery and subsequent lysosomal degradation [83] [78].
- Inhibition of inflammatory signaling: UCC-mediated cleavage of linear (M1) ubiquitin chains could suppress NF-κB activation by interfering with LUBAC-dependent signaling complexes [83] [85].
- General disruption of ubiquitin-dependent immunity: By depleting functional ubiquitin pools and creating irreversibly modified proteins, UCCs may broadly impair multiple ubiquitin-dependent defense pathways [83].
Comparative Analysis: Clippases versus Conventional DUBs
Table 3: Functional Comparison of Bacterial Ubiquitin C-terminal Clippases versus Canonical Deubiquitinases
| Characteristic | Bacterial UCCs | Canonical DUBs | Bacterial Rule-Following DUBs |
|---|---|---|---|
| Cleavage site | Within ubiquitin (Arg74-Gly75) | After ubiquitin Gly76 | After ubiquitin Gly76 |
| Ubiquitin fate | Inactivated (cannot be recycled) | Recycled intact | Recycled intact |
| Substrate lysine | Permanently blocked with Gly-Gly | Available for re-ubiquitination | Available for re-ubiquitination |
| Functional consequence | Irreversible | Reversible | Reversible |
| Potential applications | Mapping ubiquitin architecture, irreversible ubiquitin modification | Studying dynamic ubiquitination, ubiquitin recycling | Studying specific bacterial virulence mechanisms |
Figure 1: Comparison of cleavage mechanisms between conventional DUBs and bacterial ubiquitin C-terminal clippases (UCCs). Conventional DUBs hydrolyze the isopeptide bond after Gly76, releasing intact ubiquitin and unmodified substrate lysine. In contrast, UCCs cleave within ubiquitin at Arg74-Gly75, producing truncated ubiquitin that cannot be recycled and a substrate lysine permanently modified with a Gly-Gly remnant.
The Scientist's Toolkit: Essential Reagents and Methodologies
Table 4: Key Research Reagents for Studying Bacterial Ubiquitin Clippases
| Reagent/Category | Specific Examples | Function/Application | Commercial Sources/References |
|---|---|---|---|
| Diubiquitin substrates | K48-, K63-, M1-linked diubiquitin | Linkage specificity profiling | Boston Biochem, R&D Systems, LifeSensors |
| Suicide probes | Ubiquitin-vinyl sulfone, Ubiquitin-AMC | Activity profiling and mechanism studies | [83] |
| Structural biology tools | Crystallization screens, Cryo-EM grids | Determining enzyme-substrate complexes | Hampton Research, Thermo Fisher |
| Predictive modeling | AlphaFold2, RoseTTAFold | Predicting cleavage specificity from sequence | [83] |
| Cell culture models | Macrophage infection models | Studying physiological role in pathogenesis | [83] [78] |
| Mass spectrometry | Ubiquitin remnant profiling (Gly-Gly antibody) | Identifying clippase substrates | [84] |
Figure 2: Experimental workflow for characterizing novel bacterial ubiquitin C-terminal clippases. The process begins with bioinformatic identification of candidate enzymes and proceeds through biochemical characterization, structural analysis, and functional validation in cellular models.
Research Applications and Future Directions
Research Tool Development
The unique properties of UCCs make them valuable tools for ubiquitin research:
- Mapping ubiquitin chain architecture: UCCs with specific linkage preferences can be used to decipher complex ubiquitin chain topologies in cellular signaling pathways [83].
- Irreversible ubiquitin modification: The permanent nature of UCC-mediated cleavage enables creation of stably modified substrates for functional studies [83].
- Proteomics applications: The characteristic Gly-Gly signature left on substrate proteins facilitates identification of ubiquitination sites through mass spectrometry [83] [84].
Therapeutic Potential
While still in early stages, UCCs offer several promising therapeutic directions:
- Antimicrobial drug targets: Specific inhibition of bacterial UCCs could restore host ubiquitin-dependent defenses without affecting human DUBs [83].
- Tool compounds for targeted protein degradation: Engineered UCC domains could be adapted for PROTAC technologies to irreversibly mark specific proteins for degradation [83].
- Anti-inflammatory therapeutics: UCCs with specificity for linear ubiquitin chains might be developed as biological agents to suppress pathological inflammation [83] [85].
Bacterial ubiquitin C-terminal clippases represent a fascinating example of evolutionary innovation in pathogen-host interactions. By breaking the established rules of reversible ubiquitin modification, these enzymes create irreversible changes to both ubiquitin and substrate proteins, providing a potent mechanism to disrupt host cell signaling. Their unique cleavage mechanism, structural features, and functional consequences distinguish them from both eukaryotic DUBs and other bacterial effector proteins.
As research in this field advances, UCCs are poised to become valuable tools for basic ubiquitin research and potentially novel therapeutic agents. Future studies elucidating their precise roles in bacterial pathogenesis and their interactions with specific host pathways will undoubtedly reveal new insights into both bacterial virulence strategies and the fundamental biology of ubiquitin signaling.
The ubiquitin (Ub) and ubiquitin-like (Ubl) signaling systems represent one of the most fundamental regulatory mechanisms in biology, governing protein stability, function, and localization [86] [87]. While historically considered a hallmark of eukaryotic cells, the discovery of functional analogs in prokaryotes has fundamentally reshaped our understanding of these systems' evolution and presented new opportunities for therapeutic intervention [20] [19] [26]. The central challenge in drug development lies in achieving selective inhibition of prokaryotic Ubl components without cross-reactivity with essential human ubiquitin pathways [62].
Eukaryotic ubiquitin is a highly conserved 76-amino acid protein featuring a stable β-grasp fold, which is activated through a three-enzyme cascade (E1-E2-E3) and typically targets substrates for proteasomal degradation via K48-linked polyubiquitin chains [86] [87]. In contrast, prokaryotes have evolved structurally and mechanistically distinct systems. The two best-characterized examples are Pup (prokaryotic ubiquitin-like protein) in actinobacteria and the SAMP/TtuB systems in archaea and Thermus species, which share functional analogy but limited sequence homology with eukaryotic ubiquitin [19] [32]. These systems represent attractive antibacterial targets, particularly in drug-resistant pathogens like Mycobacterium tuberculosis, where the Pup-proteasome system (PPS) is essential for persistence [35] [62].
This guide systematically compares the core components of prokaryotic and eukaryotic Ubl systems, summarizes key experimental approaches for studying them, and provides strategic frameworks for developing specific inhibitors. The profound structural and mechanistic differences between these systems create both challenges and opportunities for selective therapeutic intervention.
Comparative Analysis of Ubl System Components
Structural and Functional Characteristics
Table 1: Fundamental Properties of Eukaryotic Ubiquitin and Prokaryotic Ubls
| Characteristic | Eukaryotic Ubiquitin | Prokaryotic Pup | Archaeal SAMPs/Thermus TtuB |
|---|---|---|---|
| Protein Size | 76 amino acids [87] | 64 amino acids [32] | Variable (Ub-fold) [19] |
| Native Structure | Stable β-grasp fold [86] [88] | Intrinsically disordered [35] [32] | Compact globular β-grasp fold [19] |
| C-terminal Motif | Diglycine (GG) [86] | GGQ (deamidated to GGE) [19] [32] | Diglycine-like [19] |
| Conjugation Chemistry | Thioester cascade [20] | Direct ATP-dependent ligation [19] | Thioester cascade [19] |
| Chain Formation | Extensive (all lysines) [87] | Not observed [62] | Limited [19] |
| Functional Diversity | Proteolysis, signaling, trafficking, etc. [86] [87] | Primarily proteasomal degradation [19] [62] | Protein modification & sulfur transfer [19] |
Enzymatic Machinery and Conjugation Pathways
Table 2: Enzymatic Components of Ubl Conjugation Systems
| Component | Eukaryotic Ubiquitin System | Prokaryotic Pup System | SAMP/TtuB System |
|---|---|---|---|
| Activating Enzyme | E1 (cysteine-dependent) [20] | Dop (deamidase) [19] | E1-like (MoeB/ThiF homologs) [20] [19] |
| Conjugating Enzyme | E2 (multiple families) [20] [89] | Not involved | E2-like (limited distribution) [20] [26] |
| Ligase | E3 (RING, HECT, RBR families) [20] [62] | PafA [19] [35] | Minimal E3 or none [19] |
| Deconjugating Enzymes | DUBs (multiple families) [20] | Dop (bifunctional) [19] | JAB proteases [20] [26] |
| ATP Dependence | Required for E1 [20] | Required for PafA [19] | Required for E1-like [19] |
The eukaryotic ubiquitin system employs an elaborate three-step enzymatic cascade where E1 activating enzymes, E2 conjugating enzymes, and E3 ligases work in concert to attach ubiquitin to substrate proteins [20] [87]. This system exhibits tremendous diversity, with humans encoding dozens of E2s and hundreds of E3s that provide substrate specificity and enable regulation of virtually every cellular process [86] [62].
In contrast, the prokaryotic Pup system is remarkably streamlined, requiring only two enzymes: the deamidase Dop (which processes the C-terminal glutamine to glutamate) and the ligase PafA (which directly conjugates Pup to target proteins) [19]. This system functions primarily in proteasomal degradation and lacks the signaling versatility of eukaryotic ubiquitination [62]. Archaeal SAMP systems represent an intermediate state, utilizing E1-like enzymes but generally lacking the E2 and E3 components of the eukaryotic system, though some bacteria possess rudimentary E2 and RING-type E3 components [20] [26].
Diagram 1: Comparative signaling pathways of eukaryotic ubiquitin and prokaryotic Pup systems. The eukaryotic system (top) employs a multi-step enzymatic cascade, while the prokaryotic system (bottom) utilizes a streamlined two-enzyme process.
Experimental Approaches for Ubl System Characterization
Structural Analysis Methods
Structural biology techniques have been instrumental in elucidating the fundamental differences between prokaryotic and eukaryotic Ubl systems. X-ray crystallography revealed that eukaryotic ubiquitin adopts a highly stable β-grasp fold with mixed β-sheet and α-helical structure [88] [87], while NMR spectroscopy demonstrated that Pup exists predominantly as an intrinsically disordered protein that undergoes folding only upon binding to its receptor Mpa [35] [32]. More recently, cryo-electron microscopy has provided atomic-resolution insights into the Mpa-proteasome complex with engaged pupylated substrates, revealing a spiral staircase arrangement of the ATPase ring that drives substrate translocation [35].
Experimental Protocol 1: Structural Analysis of Ubl-Proteasome Complexes Using Cryo-EM
Complex Preparation: Assemble the Mpa-20S proteasome complex in the presence of ATP using a Δ7PrcA proteasome variant for stabilized interaction [35].
Substrate Engagement: Add a linear Pup-substrate fusion (e.g., PupDHFR) while maintaining the sample at 4°C to stall translocation. Quench with ATPγS to prevent further ATP hydrolysis [35].
Grid Preparation: Apply the stalled complex to cryo-EM grids and vitrify using liquid ethane.
Data Collection: Acquire micrographs using a high-end cryo-electron microscope (e.g., Titan Krios) with automated data collection software.
Image Processing:
- Pick particles (e.g., 860,718 initial particles) and perform 2D classification
- Conduct 3D classification (e.g., 385,394 particles) to separate Mpa-bound complexes from uncapped proteasomes
- Use local 3D classification and refinement with signal subtraction to resolve distinct conformational states (achieving 3.8-3.9 Å resolution for Mpa states)
- Perform focused refinement on the 20S CP using a binary mask (achieving 2.8 Å resolution) [35]
Biochemical and Functional Assays
Experimental Protocol 2: In Vitro Pupylation and Degradation Assay
Protein Purification: Express and purify recombinant Mpa, 20S proteasome, PafA, Dop, and substrate proteins (e.g., FabD, PanB) from M. tuberculosis [19] [35].
Pup Preparation:
- Express Pup with C-terminal GGQ motif
- Treat with Dop and ATP to generate deamidated Pup-GGE
- Confirm deamidation by mass spectrometry [19]
Pupylation Reaction:
- Combine substrate protein (5 μM) with deamidated Pup (10 μM)
- Add PafA (1 μM) and ATP (5 mM) in reaction buffer
- Incubate at 37°C for 30-60 minutes
- Resolve by SDS-PAGE and detect conjugates by immunoblotting with anti-Pup antibodies [19]
Degradation Assay:
- Incubate pupylated substrates (2 μM) with Mpa (5 μM) and 20S proteasome (2 μM)
- Add ATP regeneration system
- Monitor degradation by loss of substrate signal on SDS-PAGE or release of fluorescent tags over 30-120 minutes [35]
Ubiquitin/Ubl Linkage Analysis
Experimental Protocol 3: Polyubiquitin/Pup Chain Analysis Using UbiCRest/TUBE Methods
Sample Preparation:
- Express proteins of interest with epitope-tagged ubiquitin or isolate endogenous polyubiquitinated proteins using Tandem Ubiquitin Binding Entities (TUBEs) [87]
Linkage-Specific Digestion:
Analysis:
- Resolve cleavage products by SDS-PAGE and immunoblot with linkage-specific antibodies
- For Pup, monitor cleavage by Dop and resistance to eukaryotic DUBs
- Use mass spectrometry to confirm linkage types and architecture [87]
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Ubl Pathway Research
| Reagent/Category | Specific Examples | Function/Application | Prokaryotic vs. Eukaryotic Specificity |
|---|---|---|---|
| Activating Enzymes | Human E1, MoeB, ThiF, Dop | Ubl activation and priming | High: Dop/PafA vs. E1 structural differences |
| Conjugating Enzymes | E2 (UbcH5), UBact | Ubl transfer to substrates | Moderate: Limited E2 homologs in prokaryotes |
| Ligases | HECT E3, RING E3, PafA | Substrate-specific conjugation | High: PafA unique to prokaryotes |
| Proteases | DUBs (USP2, OTULIN), JAB, Dop | Ubl deconjugation and processing | Moderate: JAB in both, Dop prokaryote-specific |
| Binding Reagents | TUBEs, linkage-specific antibodies | Isolation and detection of Ubl modifications | Variable: Some cross-reactivity possible |
| Proteasomes | 26S, 20S CP, Mpa | Ubl-dependent substrate degradation | Moderate: Structural conservation with sequence divergence |
| Ubl Proteins | Ubiquitin, Pup, UBact, SAMP | Central modifiers in conjugation | High: Sequence and structural divergence |
Strategic Approaches for Selective Inhibitor Development
Targeting Structural and Mechanistic Differences
The structural divergence between eukaryotic ubiquitin's stable β-grasp fold and prokaryotic Pup's intrinsic disorder presents a fundamental opportunity for selective inhibitor design [35] [32]. Similarly, the distinct active sites of the enzymatic components—particularly the two-enzyme Pup system (Dop/PafA) versus the three-enzyme ubiquitin cascade (E1-E2-E3)—offer specific targeting surfaces not present in human counterparts [19].
Key targeting strategies include:
Pup-Mpa Interaction Interface: The binding-induced folding of Pup upon Mpa engagement creates a unique structural epitope not found in eukaryotic systems. Small molecules that stabilize Pup's disordered state or block its binding interface could specifically disrupt prokaryotic degradation without affecting eukaryotic proteasomal function [35] [32].
PafA Active Site: The ATP-dependent ligase activity of PafA, which is structurally related to glutamine synthetase rather than eukaryotic E3 ligases, provides a highly specific target. Inhibitor design can exploit the unique nucleotide-binding pocket and substrate recognition mechanism [19].
Dop Bifunctionality: The dual deamidase/depupylase activity of Dop represents another prokaryote-specific target. Allosteric inhibitors that lock the enzyme in one conformational state could selectively disrupt Pup cycling [19].
Experimental Validation of Inhibitor Specificity
Experimental Protocol 4: Counter-Screening for Inhibitor Specificity
Primary Screening:
- Test compounds against target prokaryotic enzyme (e.g., PafA) using biochemical activity assays
- Monitor ATP consumption, substrate conjugation, or complex formation
Counter-Screening:
- Test hits against closest human homologs (e.g., E1, E3 ligases)
- Include broad panel of human E2 and E3 enzymes where feasible
Cellular Specificity Assessment:
- Evaluate cytotoxicity in human cell lines
- Monitor effects on global ubiquitination patterns via immunoblotting
- Assess impact on cell cycle and protein degradation reporters
Bacterial Efficacy:
The development of specific inhibitors targeting prokaryotic Ubl components requires careful consideration of the profound structural, mechanistic, and functional differences between these systems and their eukaryotic counterparts. The streamlined nature of the Pup system, with its limited enzyme components and distinct conjugation chemistry, offers several unique targeting opportunities with potential for high selectivity. Similarly, the SAMP/TtuB systems of archaea present alternative targeting platforms based on their hybrid characteristics. As structural insights into these systems continue to emerge, particularly through cryo-EM analysis of complete complexes, new avenues for selective intervention will undoubtedly emerge, offering promising strategies for combating bacterial infections while minimizing host toxicity.
The ubiquitin-proteasome system (UPS), long considered a hallmark of eukaryotic cells, is a master regulator of protein homeostasis, immune signaling, and infection outcomes. This system involves a cascade of E1 (activating), E2 (conjugating), and E3 (ligating) enzymes that conjugate ubiquitin (Ub) or ubiquitin-like proteins (Ubls) to target substrates, determining their fate via degradation or functional alteration [62]. Surprisingly, recent research has uncovered that bacteria possess bona fide ubiquitination systems with striking architectural and mechanistic parallels to eukaryotic pathways [4] [5] [47]. These bacterial systems, often encoded within phage defense islands, function in antiviral immunity and potentially in pathogenesis [5] [47]. This discovery fundamentally shifts our understanding of UPS evolution and presents new challenges for developing functional assays that can accurately monitor bacterial Ubl activity across diverse biological contexts—from bacterial immunity to host-pathogen interactions. This guide systematically compares current methodologies for quantifying bacterial Ubl activity, providing researchers with a framework for selecting optimal assays based on specific research goals.
Bacterial Ubiquitin-like Pathways: Key Systems and Targets
Table 1: Major Bacterial Ubiquitin-like Systems and Their Functional Roles
| System Type | Key Components | Biological Function | Experimental Evidence |
|---|---|---|---|
| Type II BilABCD | E1BilD, E2BilB, UblBilA, DUBBilC | Antiviral defense against phage infection [5] | X-ray crystallography of E1:E2:Ubl complex; in vitro conjugation assays [5] |
| Host-Targeted DUBs | Josephin-type DUBs, USP25 | Subversion of host immunity; promotion of intracellular bacterial survival [90] [91] | siRNA knockdown; inhibitor studies (AZ-1); transcriptomic profiling [90] |
| Bacterial E3 Ligases | RING, HECT, NEL effectors (e.g., AvrPtoB) | Hijack host UPS to undermine immune signaling [62] | Plant infection models; substrate ubiquitylation assays [62] |
| Ubl Clippases (UCCs) | Irreversible deubiquitinases | Permanent substrate deubiquitination to block host re-ubiquitination [91] | Structural analysis of cleavage sites; comparison with conventional DUBs [91] |
Table 2: Functional Readouts for Bacterial Ubl Activity in Different Contexts
| Assay Readout | Pathogenesis Context | Immunity Context | Key Advantages | Limitations |
|---|---|---|---|---|
| Intracellular Bacterial Load | Measurement of pathogen survival within host cells (e.g., macrophages) [90] | Assessment of bacterial clearance in phage infection models [5] | Direct functional correlation with infection outcome | Indirect measure of Ubl activity |
| Target Protein Conjugation | Identification of ubiquitylated host proteins [62] | Detection of modified phage or bacterial proteins [5] | Direct evidence of Ubl enzymatic activity | Requires specific antibody development |
| DUB Inhibition Phenotype | Reduced bacterial survival with DUB inhibitors (e.g., AZ-1) [90] | Not typically applicable | Validates DUBs as drug targets | Potential off-target effects |
| Structural Modification | Detection of irreversible ubiquitin clipping [91] | Analysis of Ubl precursor processing [5] | Reveals unique mechanistic insights | Requires specialized mass spectrometry |
Experimental Approaches: From Biochemical to Cellular Assays
Structural Biology and Biochemical Characterization
Structural approaches have been instrumental in validating the existence of bacterial ubiquitination systems. Recent work on the Type II BilABCD operon from Ensifer aridi TW10 utilized X-ray crystallography to solve structures of the E1BilD:E2BilB:UblBilA complex at 2.5 Å and 2.7 Å resolution [5]. These structures revealed an E1 protein with N-terminal inactive adenylation domain (IAD) and C-terminal active adenylation domain (AAD) with a mobile α-helical CYS domain—architecture strikingly similar to eukaryotic E1 enzymes [5] [47].
Experimental Protocol: In Vitro Ubl Conjugation Assay
- Protein Purification: Express and purify recombinant bacterial E1, E2, Ubl, and target proteins with affinity tags (e.g., His-tag) from E. coli [5] [92].
- Reaction Setup: Combine 1-5 μM E1, 5-10 μM E2, 10-20 μM Ubl, and 5-10 μM target protein in reaction buffer (e.g., 50 mM Tris-HCl pH 7.5, 100 mM NaCl, 5 mM MgCl₂, 5 mM ATP, 1 mM DTT) [5].
- Incubation: Incubate at 25-37°C for 30-120 minutes.
- Detection: Analyze conjugation products by SDS-PAGE with Coomassie staining or western blotting using Ubl-specific antibodies [5].
- Controls: Include reactions lacking ATP, E1, or E2 to confirm enzyme-specific conjugation.
This biochemical approach directly demonstrates Ubl conjugation activity and can be adapted for high-throughput screening of inhibitors or mechanistic studies.
Cellular Assays for Host-Pathogen Interactions
Cellular assays measure how bacterial Ubl systems manipulate host pathways during infection. For intracellular pathogens like Salmonella, macrophage infection models coupled with high-content screening have identified host DUBs as promising therapeutic targets [90].
Experimental Protocol: High-Content Screening for Intracellular Bacteria
- Cell Preparation: Seed macrophages (e.g., RAW 264.7 or primary macrophages) in 96-well imaging plates [90].
- Bacterial Infection: Infect cells with GFP-expressing bacteria (e.g., Salmonella enterica serovar Typhimurium UK-1) at appropriate MOI [90].
- Compound Treatment: Treat with DUB inhibitors (e.g., AZ-1 for USP25/USP28) or other UPS-targeting compounds [90].
- Staining: Label host cell nuclei with Hoechst stain and cytoplasm with HCS CellMask Red to distinguish intracellular bacteria [90].
- Imaging and Analysis: Use high-content imaging systems to quantify intracellular bacteria per cell; normalize to cell viability controls [90].
This approach identified the USP25/USP28 inhibitor AZ-1 as a lead compound that enhances clearance of intracellular Salmonella and other multidrug-resistant pathogens without direct antibacterial activity [90].
Diagram 1: Cellular assay workflow for host-pathogen Ubl interactions. Bacterial infection manipulates host DUBs to promote survival, while DUB inhibitors can restore bacterial clearance.
Reporter Systems and Signaling Pathway Analysis
Bacterial Ubl systems often target specific host signaling pathways, particularly NF-κB signaling in immune responses [90]. Reporter assays can quantify this manipulation.
Experimental Protocol: NF-κB Reporter Assay
- Transfection: Seed HEK293T or other relevant cells in 96-well plates; transfect with NF-κB luciferase reporter plasmid and control Renilla luciferase plasmid [90].
- Infection/Treatment: Infect with bacteria or treat with bacterial Ubl components; include DUB inhibitors as experimental variables [90].
- Luciferase Measurement: Lyse cells and measure firefly and Renilla luciferase activities using dual-luciferase assay system.
- Data Analysis: Normalize NF-κB-driven firefly luciferase to constitutive Renilla luciferase; compare across conditions.
This approach demonstrated that the USP25/USP28 inhibitor AZ-1 suppresses key immune pathways, including NF-κB signaling, thereby modulating host response to infection [90].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Studying Bacterial Ubl Activity
| Reagent Category | Specific Examples | Research Application | Key Features/Benefits |
|---|---|---|---|
| DUB Inhibitors | AZ-1 (USP25/USP28 inhibitor), GRL0617 (SARS-CoV-2 PLpro inhibitor) [90] [92] | Target validation; host-directed therapy | AZ-1 shows broad-spectrum activity against multidrug-resistant pathogens [90] |
| Fluorogenic Substrates | Ub-AMC, ISG15-AMC, Z-RLRGG-AMC [92] | In vitro DUB activity assays | Real-time kinetic measurements; high sensitivity |
| Expression Systems | pET28a vector in E. coli BL21(DE3) [5] [92] | Recombinant protein production | Codon-optimized genes; affinity tags for purification |
| Structural Tools | X-ray crystallography; AlphaFold predictions [93] [5] | Molecular mechanism studies | Reveals architectural parallels to eukaryotic systems [5] |
| Cell Imaging Reagents | Hoechst stain, HCS CellMask Red, GFP-labeled bacteria [90] | High-content screening of intracellular bacteria | Enables precise quantification of bacterial load |
Comparative Analysis of Methodologies
Diagram 2: Methodology comparison for bacterial Ubl research. Different approaches offer complementary strengths in throughput, mechanistic insight, and physiological relevance.
Each methodological approach offers distinct advantages for investigating bacterial Ubl systems. Biochemical assays provide direct evidence of enzymatic activity and mechanism but lack cellular context. Cellular assays bridge this gap by measuring functional outcomes in biologically relevant environments but introduce complexity that can obscure specific mechanisms. Structural approaches offer unparalleled molecular insights but are technically demanding and low-throughput. Animal models provide the most physiologically relevant data but are resource-intensive and ethically challenging.
The optimal approach depends on the research question: initial target validation and mechanistic studies benefit from biochemical and structural methods, while therapeutic development requires cellular and animal models to demonstrate physiological efficacy. For comprehensive understanding, integrated approaches that span multiple methodologies are most powerful, as demonstrated by studies that combine bioinformatics, biochemistry, and structural biology to elucidate complete bacterial ubiquitination pathways [4] [5] [47].
The discovery of functional ubiquitination systems in bacteria represents a paradigm shift in our understanding of UPS evolution and function. These systems play crucial roles in bacterial immunity against phage infection [5] [47] and potentially in mediating interactions with eukaryotic hosts [90] [62]. The assay methodologies reviewed here provide researchers with a comprehensive toolkit for investigating these fascinating systems across biochemical, cellular, and organismal levels.
Future methodological developments will likely focus on improving the throughput of cellular assays, developing more specific inhibitors and probes for bacterial Ubl components, and leveraging computational approaches like AlphaFold [93] to predict bacterial Ubl structures and interactions. Additionally, more sophisticated animal models that better recapitulate human disease will be essential for translating basic discoveries into therapeutic applications. As our understanding of bacterial Ubl systems grows, so too will our ability to manipulate them for therapeutic benefit, potentially offering new approaches to combat antibiotic-resistant infections by targeting host factors rather than the pathogens themselves [90].
Comparative Analysis: Validating Functional Parallels and Divergences
The proteasome is a sophisticated protease complex responsible for the controlled degradation of intracellular proteins, a process vital for cellular homeostasis, stress response, and the regulation of numerous biological pathways. While essential in both domains of life, the proteasome's structure and the mechanisms regulating its activity have evolved distinct characteristics in eukaryotes and prokaryotes. Historically, the intricate ubiquitin-signaling system that governs proteasomal degradation in eukaryotes was considered absent in prokaryotes. However, the discovery of a prokaryotic ubiquitin-like protein, Pup (prokaryotic ubiquitin-like protein), in Mycobacterium tuberculosis has fundamentally altered this view, revealing that both domains of life employ small protein modifiers to direct proteins to the proteasome [18] [20]. This guide provides a structured, data-driven comparison of core proteasome structure and regulation, contextualized within the broader thesis of ubiquitin-like signaling across evolutionary boundaries, to serve researchers, scientists, and drug development professionals.
Core Proteasome Structure: A Comparative Analysis
The foundational unit of the proteasome is the core particle (CP or 20S proteasome), a compartmentalized protease. Its overall architecture is conserved, but its subunit complexity differs significantly between eukaryotes and prokaryotes.
The core particle is a barrel-shaped complex composed of four stacked heptameric rings, forming a central catalytic chamber where proteolysis occurs [94] [95]. The entry to this chamber is controlled by narrow gates, restricting access to unfolded substrates.
- Eukaryotic 20S Core Particle: The eukaryotic CP is a hetero-oligomeric complex. The two outer rings are each composed of seven distinct α-subunits (α1-α7), and the two inner rings are each composed of seven distinct β-subunits (β1-β7) [94] [95]. This arrangement creates an α1–7β1–7β1–7α1–7 structure.
- Prokaryotic (Bacterial/Archaea) 20S Core Particle: In contrast, prokaryotic core particles are homo-oligomeric, characterized by much simpler subunit composition. The outer rings are composed of seven identical α-subunits, and the inner rings are composed of seven identical β-subunits, resulting in an α7β7β7α7 structure [18] [94].
Table 1: Structural Composition of the 20S Core Particle
| Feature | Eukaryotic Core Particle | Prokaryotic Core Particle |
|---|---|---|
| Ring Structure | Two rings of seven different α-subunits; two rings of seven different β-subunits [18] | Two rings of seven identical α-subunits; two rings of seven identical β-subunits [18] |
| Quaternary Structure | α1–7β1–7β1–7α1–7 [94] | α7β7β7α7 [18] |
| Complexity | High (14 different subunits) | Low (2 different subunits) |
Catalytic Activities and Active Sites
The proteolytic activities reside in the N-terminal threonine residues of the β-subunits, which act as nucleophiles in a novel hydrolytic mechanism [94]. The diversity of β-subunits dictates the range of peptide bond cleavage specificities.
- Eukaryotic Catalytic Activities: Three of the seven distinct β-subunits possess catalytic activity, each with a defined cleavage preference:
- β1 (PSMB6): Caspase-like activity (cleavage after acidic residues)
- β2 (PSMB7): Trypsin-like activity (cleavage after basic residues)
- β5 (PSMB5): Chymotrypsin-like activity (cleavage after hydrophobic residues) [94] [96] This multi-specificity allows for the efficient processing of diverse protein substrates into short peptides.
- Prokaryotic Catalytic Activities: In prokaryotes, all β-subunits are identical and typically exhibit a primary chymotrypsin-like activity. Notably, the Mycobacterium tuberculosis proteasome has been shown to possess additional catalytic activities beyond the standard chymotrypsin-like function [18].
Table 2: Catalytic Profile of the 20S Core Particle
| Feature | Eukaryotic Core Particle | Prokaryotic Core Particle |
|---|---|---|
| Active Site | Three β-subunit N-terminal threonines; three distinct activities [18] | All β-subunit N-terminal threonines; primarily chymotrypsin-like [18] |
| Catalytic Activities | Tryptic, chymotryptic, post-acidic (caspase-like) [18] [96] | Predominantly chymotryptic; Mtb proteasome has additional activities [18] |
| Key Catalytic Subunits | β1, β2, β5 [94] | Single type of β-subunit |
Diagram 1: Comparative 20S Core Particle Structures (7/100)
Regulatory Mechanisms and Accessory Factors
The distinction between eukaryotic and prokaryotic proteasomal systems becomes most apparent in the mechanisms controlling substrate recognition, unfolding, and delivery to the core particle.
The Eukaryotic Ubiquitin-Proteasome System (UPS)
Eukaryotes employ a highly sophisticated, multi-step pathway for target selection.
- The Ubiquitin Tag: Proteins are marked for degradation by the covalent attachment of a small protein, ubiquitin. The C-terminal glycine (Gly76) of ubiquitin forms an isopeptide bond with the ε-amino group of a lysine residue on the substrate protein [21]. A chain of ubiquitin molecules (a polyubiquitin chain) linked through Lys48 serves as the primary degradation signal [18] [21].
- The Enzymatic Cascade: Ubiquitin conjugation involves a three-enzyme cascade:
- E1 (Ubiquitin-activating enzyme): Activates ubiquitin in an ATP-dependent manner [21].
- E2 (Ubiquitin-conjugating enzyme): Accepts the activated ubiquitin from E1 [21].
- E3 (Ubiquitin ligase): Recognizes specific substrate proteins and catalyzes the transfer of ubiquitin from E2 to the substrate, providing specificity to the system [21]. The human genome encodes hundreds of E3 ligases, enabling precise recognition of a vast array of substrates [94].
- The 19S Regulatory Particle (RP): The 26S proteasome is formed by the association of the 20S CP with one or two 19S regulatory particles. The 19S RP is a complex structure that:
- Recognizes polyubiquitinated proteins.
- Contains deubiquitinating enzymes (DUBs) that recycle ubiquitin.
- Utilizes a ring of six AAA+ ATPase subunits (Rpt1-6) to unfold the substrate protein in an ATP-dependent manner.
- Opens the gate of the 20S core particle and translocates the unfolded polypeptide into the catalytic chamber [94] [95] [96].
The Prokaryotic Pup-Proteasome System
Prokaryotes utilize a functionally analogous but structurally and mechanistically distinct tagging system.
- The Pup Tag: In actinobacteria like Mycobacterium tuberculosis, the prokaryotic ubiquitin-like protein Pup is covalently attached to target proteins. The C-terminal glycine of Pup is modified to form an isopeptide bond with a lysine residue on the substrate, a process known as pupylation [18].
- The Enzymatic Machinery: The pupylation system is simpler than the eukaryotic UPS.
- PafA (Proteasome accessory factor A): Acts as the single known enzyme for pupylation, combining the functions of E1 and E3 enzymes to directly attach Pup to target proteins [18]. An E2-like conjugating enzyme appears absent.
- Dop (Deamidase of Pup): Processes immature Pup by deamidating its C-terminal glutamate to glutamine, which is then adenylated to form the conjugatable glycine [18].
- The Regulatory ATPase:
- Mpa (Mycobacterium proteasome ATPase): Forms a homo-hexameric ring that recognizes pupylated proteins, unfolds them using ATP hydrolysis, and threads them into the 20S proteasome core. Mpa is functionally homologous to the AAA+ ATPases of the eukaryotic 19S RP but is structurally simpler [18].
Table 3: Regulatory and Tagging Systems
| Feature | Eukaryotic System | Prokaryotic System |
|---|---|---|
| Tagging Molecule | Ubiquitin (Ub) [21] | Prokaryotic ubiquitin-like protein (Pup) [18] |
| Conjugation | Ubiquitination (or ubiquitylation) [21] | Pupylation [18] |
| Tag C-terminus | Gly-Gly motif [18] | Gly-Glu (requires processing) [18] |
| Activating Enzyme | E1 (multiple) [21] | PafA? [18] |
| Conjugating Enzyme | E2 (multiple) [21] | Not identified |
| Ligase Enzyme | E3 (hundreds) [94] [21] | PafA? [18] |
| Deconjugating Enzyme | Deubiquitinases (DUBs, e.g., USP, UCH) [18] [94] | Deamidase? De-pupylase? [18] |
| AAA+ ATPase | Hetero-hexamer of Rpt1-6 (in 19S RP) [94] | Homo-hexamer of Mpa [18] |
Diagram 2: Substrate Tagging and Recognition Pathways (7/100)
Functional Roles and Physiological Implications
The functional output of proteasomal degradation is tailored to the physiological needs of the organism.
- Eukaryotic Functions: The UPS is integral to a vast array of cellular processes, including:
- Cell Cycle Regulation: Controlled degradation of cyclins and CDK inhibitors.
- Signal Transduction: Regulation of pathways like NF-κB via degradation of inhibitors (IκB) [18].
- DNA Repair, Transcription, and Apoptosis [94] [96].
- Immune Response: Specialized immunoproteasomes optimize the generation of antigenic peptides for MHC class I presentation [96].
- Prokaryotic Functions: The roles of proteasomes in prokaryotes are less fully characterized but are known to be critical for:
- Stress Response: Essential for resisting nitric oxide (NO) stress in Mycobacterium tuberculosis, a key mechanism for surviving host immune attacks [18].
- Pathogenesis: The Mpa-proteasome system is a virulence factor in Mtb, with mutants lacking functional Mpa or PafA being severely attenuated in mice [18].
Table 4: Functional and Physiological Context
| Aspect | Eukaryotes | Prokaryotes |
|---|---|---|
| Key Accessory Factors | Hetero-hexamer of AAA ATPases; numerous Ub-binding proteins, deubiquitylases [18] | Homo-hexamers of AAA ATPases (e.g., Mpa); PafA for pupylation [18] |
| Cellular Pathways | Protein turnover, signal transduction, NFκB, endocytosis, DNA repair, cell cycle, transcription [18] [94] | Mycobacteria: protein turnover, nitrosative stress response [18] |
| Role in Pathogenesis | Not applicable (host factor) | Essential for virulence of M. tuberculosis [18] |
Experimental Analysis of Proteasome Function
Research in this field relies on specific biochemical and cellular assays to dissect proteasome structure and activity.
Key Experimental Protocols
Protocol 1: Measuring Proteasome Peptidase Activity
- Purpose: To quantify the chymotrypsin-like, trypsin-like, and caspase-like activities of the 20S core particle in vitro.
- Methodology: Cell lysates or purified 20S proteasomes are incubated with fluorogenic peptide substrates (e.g., Suc-LLVY-AMC for chymotrypsin-like activity). Upon cleavage by the proteasome, the fluorophore (AMC) is released, and its fluorescence is measured over time [96].
- Application: Used to compare catalytic profiles of constitutive proteasomes versus immunoproteasomes, and to screen for proteasome inhibitors (e.g., MG-132, Lactacystin) [96].
Protocol 2: Assessing Protein Stability and Degradation
- Purpose: To determine if a specific protein is degraded via the UPS or Pup-proteasome system.
- Methodology: Cells are treated with specific proteasome inhibitors (e.g., MG-132 for eukaryotes). If a protein of interest accumulates upon inhibition, it is likely a proteasome substrate. This can be coupled with pulse-chase experiments to measure degradation kinetics [96].
- Application: Identified key proteasome substrates in both eukaryotes (e.g., p53, IκB) and prokaryotes (e.g., Mtb FabD, Ino1) [18].
Protocol 3: Genetic Analysis of Proteasome Function
- Purpose: To establish the physiological requirement of proteasome components.
- Methodology: Generation of gene knockout mutants (e.g., mpa, pafA in Mtb or proteasome subunit genes in yeast) and phenotypic characterization under various stress conditions (e.g., nitrosative stress, nutrient starvation) [18].
- Application: Demonstrated the essential role of the Mtb proteasome in resisting host-derived nitric oxide and in virulence [18].
The Scientist's Toolkit: Essential Research Reagents
Table 5: Key Reagents for Proteasome Research
| Reagent / Assay | Function & Application | Example |
|---|---|---|
| Proteasome Activity Assay Kit | Fluorescent-based tool for measuring chymotrypsin-like, trypsin-like, or caspase-like protease activity in cell lysates or purified complexes. | Proteasome 20S Activity Assay Kit (ab112154) [96] |
| ATP-dependent Proteolysis Assay | In vitro reconstitution assay using purified 26S/30S proteasome or prokaryotic 20S-Mpa complex to degrade a ubiquitylated/pupylated substrate, measuring ATP-dependent protein breakdown. | N/A |
| Proteasome Inhibitors | Cell-permeable compounds that specifically inhibit the proteolytic activity of the 20S core particle. Used to probe UPS/Pup-system function. | MG-132 (ab141003), Lactacystin (ab141411) [96] |
| Antibodies (Anti-Ubiquitin, Anti-Pup) | Immunodetection of protein ubiquitination or pupylation via Western blotting, immunofluorescence, or immunoprecipitation. | N/A |
This comparison elucidates a paradigm of evolutionary conservation and divergence. The core proteasome architecture is conserved as a self-compartmentalized protease with N-terminal threonine catalysis. However, eukaryotic cells have evolved a system of immense complexity and specificity, characterized by hetero-oligomeric core particles and a multi-layered ubiquitin conjugation cascade with hundreds of components. In contrast, prokaryotes like M. tuberculosis utilize a streamlined, minimalist system with a homo-oligomeric core and a simplified, Pup-based tagging pathway centered on the PafA enzyme. For drug development professionals, these distinctions are critical. The bacterial proteasome and its associated pupylation system represent a validated, species-specific antibacterial target, particularly for combating tuberculosis, offering a therapeutic window by leveraging the structural and mechanistic differences from the human host system.
The targeted degradation of proteins is a fundamental cellular process for maintaining homeostasis, regulating signaling pathways, and eliminating damaged or misfolded proteins. For decades, the ubiquitin-proteasome system (UPS) was considered a hallmark of eukaryotic cells, distinguishing them from prokaryotes [20] [54]. This paradigm shifted with the discovery of a functionally analogous system in prokaryotes based on the prokaryotic ubiquitin-like protein (Pup) [18]. While eukaryotes employ a three-enzyme cascade (E1-E2-E3) to conjugate ubiquitin to substrate proteins, primarily marking them for degradation by the 26S proteasome, prokaryotes utilize a distinct two-step enzymatic process for pupylation [18] [97] [98].
This guide provides a systematic comparison of these systems, focusing on their functional outcomes in protein degradation, signaling, and immunity. We present objective experimental data and methodologies to illustrate the key similarities and differences, providing a resource for researchers exploring these pathways in basic science or drug development.
System Architecture and Core Mechanisms
The eukaryotic ubiquitin and prokaryotic Pup systems, while functionally analogous, exhibit profound differences in their genetic foundation, structural composition, and biochemical mechanics.
Eukaryotic Ubiquitin System
- Genetic Foundation: In humans, ubiquitin is encoded by four genes: UBA52, RPS27A (which produce single ubiquitin molecules fused to ribosomal proteins), and UBB and UBC (which code for polyubiquitin precursor proteins) [21].
- The Enzymatic Cascade: Ubiquitination proceeds through a three-step enzymatic cascade [99] [23] [21]:
- Activation (E1): An E1 ubiquitin-activating enzyme uses ATP to adenylate the C-terminal glycine (Gly76) of ubiquitin, subsequently forming a thioester bond with an active-site cysteine.
- Conjugation (E2): The activated ubiquitin is transferred to a cysteine residue of an E2 ubiquitin-conjugating enzyme.
- Ligation (E3): An E3 ubiquitin ligase facilitates the transfer of ubiquitin from the E2 to a lysine ε-amino group on the target protein, forming an isopeptide bond. With over 600 E3 ligases in humans, this step provides exquisite substrate specificity [99].
- The Ubiquitin Code: A hallmark of the system is the "ubiquitin code" [23]. Ubiquitin itself contains seven lysine residues (K6, K11, K27, K29, K33, K48, K63) and an N-terminal methionine (M1) that can serve as linkage points for subsequent ubiquitin molecules, forming polyubiquitin chains. K48-linked chains are the primary signal for proteasomal degradation, while K63-linked and M1-linked (linear) chains play major roles in inflammatory signaling and DNA repair [99] [23] [21].
Prokaryotic Pup-Proteasome System (PPS)
- Genetic Foundation: Pup is encoded by a single gene and, unlike ubiquitin, requires activation by deamidation (in the case of Mycobacterium tuberculosis) or proteolysis to expose a C-terminal glycine or glutamate [18] [98].
- The Two-Step Enzymatic Pathway: The process of pupylation is simpler [18] [97] [98]:
- Activation/Conjugation: A single enzyme, proteasome accessory factor A (PafA), acts as both an E1 and E3 analogue. It activates Pup in an ATP-dependent manner and directly ligates it to a lysine residue on the target protein.
- No E2 Analogue: Notably, the PPS lacks a functional equivalent to the E2 enzyme, representing a key mechanistic simplification compared to the UPS.
- Degradation Signal: Unlike the diverse ubiquitin code, poly-pupylation has not been widely observed. A single Pup modification is sufficient to target a substrate to the proteasome [97].
Table 1: Core Architectural Comparison of Ubiquitin and Pup Systems
| Feature | Eukaryotic Ubiquitin System | Prokaryotic Pup System |
|---|---|---|
| Tag Protein | Ubiquitin (76 aa, highly conserved) | Pup (64 aa in M. tuberculosis) |
| Gene Structure | Polyubiquitin genes (UBB, UBC) and fusion genes (UBA52, RPS27A) [21] | Single pup gene requiring C-terminal processing [18] |
| Activating Enzyme | E1 (e.g., UBA1, UBA6) [21] | PafA [18] |
| Conjugating Enzyme | E2 (~35 in humans) [21] | Not present |
| Ligating Enzyme | E3 (>600 in humans, e.g., HECT, RING) [99] [21] | PafA (dual-function) [18] |
| Proteasome Recognition | Primarily K48-linked polyubiquitin chains [21] | Monopupylation or oligopupylation [97] |
| Deconjugating Enzyme | Deubiquitinases (DUBs, e.g., JAB peptidases) [20] | Depupylase (Dop) [97] |
Comparative Pathway Diagrams
The following diagrams illustrate the core enzymatic pathways for the eukaryotic ubiquitin and prokaryotic Pup systems, highlighting the key components and processes.
Diagram 1: The eukaryotic ubiquitin pathway involves a three-enzyme cascade (E1-E2-E3) that builds a polyubiquitin chain on the target substrate, leading to its recognition and degradation by the 26S proteasome.
Diagram 2: The prokaryotic Pup system uses a single enzyme, PafA, for both activation and ligation. The pupylated substrate is recognized by the proteasome. The depupylase Dop can reverse the modification and recycle Pup.
Functional Outcomes in Immunity and Signaling
The functional specialization of these systems is most evident in their roles in immunity and signal transduction, with the eukaryotic UPS exhibiting remarkable complexity.
Eukaryotic Ubiquitin Signaling in Immune Regulation
Ubiquitination is a cornerstone of eukaryotic immune signaling, regulating both innate and adaptive responses [99]. Its functions are multifaceted:
- Innate Immune Signaling: Multiple pattern-recognition receptor (PRR) families, including Toll-like receptors (TLRs) and RIG-I-like receptors (RLRs), rely on ubiquitin-dependent pathways to trigger immune responses [99]. For instance, upon TLR activation, the E3 ligase TRAF6 synthesizes K63-linked polyubiquitin chains. These chains do not signal for degradation but instead serve as platforms for recruiting and activating downstream kinases like TAK1 and IKK, ultimately leading to the induction of pro-inflammatory cytokines and type I interferons [99]. The transcription factor NF-κB activation pathway also involves different ubiquitin linkages: K63-linked chains for signal transduction and K48-linked chains for the degradation of its inhibitor, IκB [18].
- Adaptive Immune Response: Ubiquitination is critical for T-cell development, activation, and differentiation. It regulates signal transduction downstream of the T-cell receptor (TCR) and maintains immunological tolerance to self-tissues [99]. Dysregulation of these processes is linked to autoimmune and inflammatory diseases [99] [23].
Prokaryotic Pup System in Pathogenesis
In contrast, the functional role of the Pup-proteasome system in prokaryotes is less diversified and is primarily linked to stress adaptation and pathogenicity.
- Stress Response: The PPS is not essential for the general viability of bacteria like Mycobacterium tuberculosis but is critical for surviving under stress conditions, particularly nitrosative stress encountered during infection within host macrophages [18]. This suggests its primary role is in remodeling the proteome in response to environmental challenges.
- Limited Signaling Role: Current evidence does not support a role for the PPS in complex signal transduction cascades analogous to those in eukaryotes. Its function appears predominantly restricted to targeted protein degradation.
Table 2: Functional Comparison in Immunity and Stress Response
| Functional Aspect | Eukaryotic Ubiquitin System | Prokaryotic Pup System |
|---|---|---|
| Primary Immune Function | Regulates PRR signaling (TLR, RLR), NF-κB activation, T-cell activation, cytokine production [99] | Enables survival under host-induced nitrosative stress [18] |
| Key Signaling Mechanism | Non-degradative ubiquitin chains (K63, M1) as scaffolds for protein-complex assembly [99] [23] | No known non-degradative signaling function |
| Link to Disease | Autoimmunity, inflammatory diseases, cancer [99] [23] | Critical for pathogenesis of M. tuberculosis [18] |
| Proteolytic Remodeling | Degrades regulatory proteins (e.g., IκB) to rapidly alter gene expression [18] | Remodels bacterial proteome to resist extracellular stress |
Experimental Analysis and Methodologies
Studying these systems requires distinct methodological approaches, from predicting modification sites to reconstituting degradation pathways.
Predicting and Identifying Modification Sites
For Pupylation:
- Computational Prediction: Tools like PupPred leverage machine learning to identify pupylation sites. They analyze sequence hallmarks, finding that pupylated lysines are often located in uncharged, unstructured regions with high flexibility and are flanked by residues where lysine is depleted at positions -2 and +2, preventing steric hindrance [98].
- Proteomic Screening: Large-scale mass spectrometry is used to experimentally identify pupylated substrates and their exact modification sites. However, the absence of a canonical consensus motif makes unbiased screening challenging [98].
For Ubiquitination:
- Proteomic Techniques: Ubiquitinated proteins are identified using trypsin digestion, which leaves a characteristic di-glycine "remnant" on the modified lysine, detectable by mass spectrometry [21]. This allows for proteome-wide mapping of ubiquitination sites.
- Linkage-Specific Tools: The diversity of the ubiquitin code necessitates tools like linkage-specific antibodies and ubiquitin-binding domains (UBDs) to study the function of particular chain types [23].
Key Experimental Protocols
In Vitro Reconstitution of the PPS: This protocol is used to study Pup recycling, as detailed in [97].
- Expression and Purification: Isolate core components: bacterial 20S proteasome, proteasome-associated ATPase (Mpa in M. tuberculosis), PafA, Dop, and substrate proteins.
- Pupylation Reaction: Incubate the substrate with Pup, PafA, and ATP in an appropriate reaction buffer to form the pupylated conjugate.
- Degradation Assay: Add the pupylated substrate to the 20S proteasome and Mpa. Monitor degradation kinetics by measuring the loss of the substrate over time (e.g., by western blot or fluorescence).
- Pup Recycling Assay: After partial degradation, incubate the mixture with Dop. Analyze the reaction products to demonstrate the release of intact Pup from degradation fragments, confirming the recycling mechanism.
Functional Analysis of Ubiquitin in Immune Signaling: A typical protocol to validate the role of a specific ubiquitination event in NF-κB signaling [99] [18]:
- Cell Stimulation: Treat immune cells (e.g., macrophages) with a specific PAMP (e.g., LPS for TLR4 signaling).
- Inhibition/Knockdown: Use pharmacological inhibitors of key E3 ligases (e.g., TRAF6) or employ siRNA/shRNA to knock down their expression in target cells.
- Readout: Measure downstream outputs, including:
- IKK Kinase Activity: Use in vitro kinase assays.
- NF-κB Translocation: Monitor via immunofluorescence or western blotting of nuclear fractions.
- Cytokine Production: Quantify using ELISA or RT-qPCR.
- Ubiquitination Status: Immunoprecipitate key signaling proteins (e.g., RIP1, NEMO) and probe for K63-linked ubiquitin chains using linkage-specific antibodies.
The Scientist's Toolkit: Key Research Reagents
The following table lists essential reagents for investigating ubiquitin and Pup pathways, derived from the cited experimental methodologies.
Table 3: Essential Research Reagents for Studying Tagged Protein Degradation
| Reagent | Function in Research | Example/Note |
|---|---|---|
| Linkage-Specific Ubiquitin Antibodies | Detect and characterize specific polyubiquitin chains (e.g., K48 vs. K63) in signaling and degradation [99] [23] | Commercial antibodies for K48, K63, and M1 linkages |
| Proteasome Inhibitors | Block proteasomal activity to validate substrate degradation and accumulate ubiquitinated/pupylated proteins [18] [100] | Bortezomib (for eukaryotic 26S), MG132 (general) |
| E1/E2/E3 Inhibitors | Dissect the ubiquitin cascade and identify specific E3 ligases involved in a pathway [23] | PYR-41 (E1 inhibitor); various small-molecule E3 inhibitors |
| PupPred Server | In silico prediction of pupylation sites in prokaryotic proteins to guide experimental design [98] | Web server: http://bioinfo.ncu.edu.cn/PupPred.aspx |
| Base Editors (BE) | Enable directed protein evolution and screening for improved degron system components (e.g., OsTIR1 variants) [74] | Cytosine Base Editor (CBE), Adenine Base Editor (ABE) |
| Inducible Degron Systems (AID, dTAG) | Study protein function by enabling rapid, ligand-induced degradation of target proteins in eukaryotic cells [74] | AID 2.0/2.1 (OsTIR1-based), dTAG (FKBP12F36V-based) |
The comparison between the eukaryotic ubiquitin and prokaryotic Pup systems reveals a fascinating case of convergent evolution toward a common function—targeted protein degradation—achieved through distinct mechanistic paths. The eukaryotic system is characterized by its expansive complexity, leveraging a multi-enzyme cascade and a sophisticated "ubiquitin code" to regulate virtually all aspects of cell biology, with immune regulation being a prime example. In stark contrast, the prokaryotic Pup system demonstrates elegant simplicity, utilizing a streamlined, two-step pathway primarily devoted to survival under stress. For researchers, this dichotomy underscores the need to employ tailored experimental strategies, from predictive bioinformatics to specific biochemical reconstitution assays, when investigating these pathways. The continued exploration of these systems not only deepens our understanding of fundamental biology but also opens avenues for therapeutic intervention, targeting the UPS in human disease or the PPS in bacterial infections.
The β-grasp fold (β-GF) represents a fundamental protein architecture that has been extensively recruited across evolution for a remarkably diverse array of biochemical functions. This review objectively compares the evolutionary trajectory and functional diversification of β-GF proteins between prokaryotes and eukaryotes, with a specific focus on the ubiquitin (Ub) system. We summarize quantitative phylogenetic data, detail experimental methodologies for validating evolutionary links, and visualize key relationships and workflows. The analysis underscores that while the structural diversification of the fold occurred primarily in prokaryotes, the eukaryotic lineage is marked by a dramatic expansion of Ub-like proteins, increasing the complexity of domain architectures and regulatory networks.
The β-grasp fold is a small, compact structural scaffold characterized by a β-sheet with four to five strands that appears to "grasp" an α-helix [37]. Initially identified in ubiquitin and bacterial immunoglobulin-binding proteins, it is now recognized as a widespread fold utilized in proteins performing a vast diversity of cellular functions [37] [39]. These functions include, but are not limited to: post-translational protein modification (e.g., ubiquitin and ubiquitin-like modifiers), sulfur transfer (e.g., ThiS, MoaD), RNA binding (e.g., TGS domain), enzymatic activities (e.g., NUDIX hydrolases), binding of small molecules and cofactors (e.g., B12, iron-sulfur clusters), and adaptor roles in signaling complexes [37].
The centrality of the β-grasp fold to eukaryotic biology is epitomized by the ubiquitin conjugation system, a major regulatory pathway. The evolutionary origin of this system, however, lies in prokaryotes. This guide compares the evolutionary history and functional adaptation of β-GF proteins across the two domains of life, providing a framework for their phylogenetic analysis and validating their deep evolutionary connections.
Evolutionary Phylogeny and Comparative Analysis
Comprehensive sequence-structure analysis reveals that the β-grasp fold is an ancient evolutionary invention. Evolutionary reconstruction indicates that by the time of the last universal common ancestor (LUCA), the β-GF had already differentiated into at least seven distinct lineages, encompassing much of the structural diversity seen today [37]. The earliest members were likely involved in RNA metabolism and related functions [37] [101].
Table 1: Major Lineages of the Beta-Grasp Fold and Their Evolutionary Distribution
| Lineage / Superfamily | Key Representative Members | Proto-typical Function | Presence in Prokaryotes | Presence in Eukaryotes |
|---|---|---|---|---|
| Ubiquitin-like (UBL) | Ubiquitin, ThiS, MoaD, Urm1 | Sulfur carrier; Post-translational protein modifier | Yes (ThiS, MoaD) | Yes (Ub, Ubls) |
| SLBB Superfamily | Transcobalamin, ComEA, PduS | Soluble ligand binding (e.g., B12, DNA) | Yes | Yes (animals) |
| Ferredoxin-like | 2Fe-2S Ferredoxin | Iron-sulfur cluster binding | Yes | Yes |
| NUDIX-like | MutT/NUDIX hydrolases | Phosphohydrolase activity | Yes | Yes |
| TGS Domain | tRNA synthetases | RNA binding | Yes | Yes |
| RA, PB1, DCX Domains | Animal signaling proteins | Adaptor in protein-protein interactions | No | Yes |
| B-repeat Domain | Listeria InlB protein | Potential receptor binding in virulence | Yes (bacteria) | No |
A key observation is the functional shift from core metabolic roles in prokaryotes to sophisticated signaling in eukaryotes. For instance, the eukaryotic Ub-conjugation system, crucial for signaling and degradation, evolved from more ancient bacterial precursors like ThiS and MoaD, which function as sulfur carriers in thiamine and molybdenum cofactor biosynthesis [37]. This demonstrates how a conserved structural fold can be repurposed for novel biochemical pathways.
Table 2: Quantitative Comparison of Ubiquitin System Components in Prokaryotes vs. Eukaryotes
| Feature | Prokaryotes | Eukaryotes (Human) |
|---|---|---|
| Ubiquitin-like Protein | ThiS, MoaD, Pup | Ubiquitin, ~14 Ubl families (SUMO, NEDD8, etc.) [101] |
| Activating Enzyme (E1) | ThiF, MoeB | UBA1, UBA2, UBA3, etc. |
| Conjugating Enzyme (E2) | Limited diversity | ~40 E2 enzymes [14] |
| Ligase (E3) | Few, acquired (e.g., bacterial NEL, RING effectors) [62] | >600 RING-type alone; HECT, RBR, U-box types [65] |
| Primary Function | Metabolic biosynthesis (ThiS/MoaD); Proteasomal degradation (Pup) [35] | Protein degradation, signaling, trafficking, DNA repair, etc. [65] |
| Conjugation Target | Specific metabolic enzymes (ThiS/MoaD); Lysine for degradation (Pup) [35] | Vast array of protein substrates; multiple lysines [65] |
The eukaryotic phase of β-GF evolution was marked by a specific expansion of ubiquitin-like (Ubl) proteins. The eukaryotic UB superfamily diversified into at least 67 distinct families, with at least 19–20 families present in the last eukaryotic common ancestor [37]. This expansion was accompanied by a dramatic increase in the domain architectural complexity of proteins, related to the recruitment of UB-like domains for numerous adaptor roles [37].
Experimental Protocols for Validating Evolutionary Links
Validating evolutionary relationships between distantly related β-GF proteins requires a multi-pronged approach combining computational and experimental techniques.
Protocol 1: Sequence Profile-Based Homology Detection
This methodology is foundational for identifying potential homologs that are too divergent to be detected by standard BLAST.
- Seed Selection: Compile a set of structurally diverse β-GF domains from databases like SCOP or the PDB (e.g., Ub, MoaD, ferredoxin, TGS domain) [37].
- Iterative Search: Use each seed sequence to initiate PSI-BLAST searches against the NCBI non-redundant (NR) database. PSI-BLAST builds a position-specific scoring matrix (PSSM) from significant hits, enhancing sensitivity for detecting distant relationships [37].
- Threshold and Validation: Collect statistically significant hits (e.g., e-value < 0.01). To exhaustively recover divergent homologs, conduct transitive searches using newly detected members as seeds in subsequent PSI-BLAST runs [37] [39].
- Profile Construction: Prepare a multiple sequence alignment of all recovered proteins and build a Hidden Markov Model (HMM) using tools like HMMER. Use this HMM to search sequenced genomes for further homologs [39].
Protocol 2: Structural Comparison and Analysis
When sequence similarity is minimal, structural similarity provides strong evidence for common ancestry.
- Structure Retrieval: Obtain 3D structures of proteins of interest from the Protein Data Bank (PDB).
- Structural Alignment: Use structural comparison servers or software like DALI to perform pairwise structural alignments [39].
- Z-score Evaluation: A significant Z-score (e.g., >5) indicates non-random structural similarity. For example, DALI searches with the C-terminal domain of transcobalamin retrieved known β-GF domains like MoaD and ferredoxin with Z-scores of 5–7, confirming a structural relationship [39].
- Core Element Mapping: Superimpose structures to identify the conserved core elements of the β-grasp fold (the central β-sheet and grasping α-helix) and identify lineage-specific structural elaborations (e.g., inserts, additional helices) [37].
Protocol 3: Ancestral Sequence Reconstruction and Functional Assay
This powerful method tests evolutionary hypotheses by resurrecting ancient proteins.
- Phylogenetic Tree Construction: Build a robust, high-confidence phylogenetic tree from a multiple sequence alignment of the protein family.
- Sequence Reconstruction: Use software tools to infer the most probable amino acid sequence of ancestral nodes at critical points on the tree (e.g., the node before a hypothesized fold switch) [102].
- Structure Prediction & Validation: Model the 3D structure of the reconstructed ancestral sequence using AlphaFold2 or similar tools. Experimentally determine the structure via crystallography or cryo-EM if possible to validate predictions [102].
- Functional Characterization: Synthesize the gene for the ancestral protein, express it, and purify it. Conduct functional assays relevant to the modern proteins (e.g., DNA-binding assays for transcription factors, sulfur transfer assays for ThiS/MoaD-like proteins) to infer functional evolution [102].
Visualization of Evolutionary and Experimental Pathways
The following diagrams illustrate the key evolutionary concepts and methodological workflows described in this guide.
Evolutionary Trajectory of the Beta-Grasp Fold
Workflow for Phylogenetic Validation
The Scientist's Toolkit: Key Research Reagents and Solutions
Table 3: Essential Reagents and Resources for Beta-Grasp Fold Research
| Reagent / Resource | Function / Application | Example Use-Case |
|---|---|---|
| PSI-BLAST | Sensitive sequence database search tool for detecting distant homologs. | Identifying novel β-GF members using a Ub or MoaD seed sequence [37]. |
| HMMER Suite | Building and searching with profile hidden Markov models for protein families. | Creating a definitive model of the SLBB superfamily to find all members in a genome [39]. |
| DALI Server | Pairwise protein structure comparison server. | Quantifying structural similarity between a protein of unknown homology and a known β-GF [39]. |
| AlphaFold2 | Highly accurate protein structure prediction from sequence. | Modeling the 3D structure of a reconstructed ancestral β-GF protein [102]. |
| Pupylated Substrate (e.g., PupDHFR) | Defined model substrate for prokaryotic ubiquitin-like systems. | Studying substrate engagement and translocation by the Mpa-proteasome complex in Mycobacteria [35]. |
| Chemical Protein Synthesis (NCL) | Precise generation of proteins with site-specific modifications (Ub/Ubls, conjugates). | Producing homogeneous ubiquitin chains with defined linkages or Ubl-substrate conjugates for biochemical studies [14]. |
| Bacterial Ub Ligase Effectors (e.g., AvrPtoB) | Molecular tools to study host-pathogen interactions and Ub ligase mimicry. | Investigating how bacterial effectors hijack the host ubiquitin system to suppress immunity [62]. |
The β-grasp fold serves as a paradigmatic example of how a simple, versatile protein scaffold can be evolutionarily repurposed to drive biological complexity. Phylogenetic and structural analyses conclusively show an ancient origin in LUCA, with prokaryotes being the primary innovators of its structural diversity. The eukaryotic lineage, in a remarkable act of evolutionary exaptation, co-opted this ancient fold for the creation of the elaborate ubiquitin system and other regulatory modules. The experimental frameworks and resources detailed herein provide a roadmap for researchers to further validate and explore the deep evolutionary connections within this fundamental protein fold, with significant implications for understanding basic biology and developing therapeutic strategies, such as targeting bacterial Ub ligases or the proteasome in pathogens like Mycobacterium tuberculosis.
The discovery of prokaryotic ubiquitin-like protein (Pup) in Mycobacterium tuberculosis (Mtb) fundamentally altered our understanding of regulated protein degradation in bacteria. This review provides a comprehensive comparison between the eukaryotic ubiquitin-proteasome system and the bacterial Pup-proteasome system (PPS), with focus on validating the essential role of pupylation in Mtb pathogenesis. We synthesize current experimental data demonstrating how this post-translational modification system enables Mtb survival under host-induced stresses, including nitric oxide toxicity, nutrient limitation, and metal ion homeostasis. Through structured analysis of quantitative data, detailed experimental protocols, and pathway visualizations, we establish the PPS as a critical virulence determinant and promising therapeutic target for tuberculosis treatment.
The ubiquitin-proteasome system (UPS) has long been recognized as a eukaryotic hallmark, governing protein turnover, quality control, and myriad signaling pathways through covalent protein modification. The discovery of a functionally analogous but evolutionarily distinct system in actinobacteria, particularly in the human pathogen Mycobacterium tuberculosis, revealed convergent evolution of protein tagging mechanisms across domains of life [18] [103].
While eukaryotic ubiquitination employs a three-enzyme cascade (E1-E2-E3) to attach ubiquitin to substrate proteins, mycobacteria utilize a two-enzyme system (Dop-PafA) for conjugating Pup to target lysines [103]. Despite this fundamental architectural difference, both systems serve to recruit tagged substrates for proteasomal degradation and participate in regulatory functions. The PPS has emerged as a critical factor in Mtb pathogenesis, enabling this persistent pathogen to survive within hostile macrophage environments and establish chronic infections [104] [33].
Table 1: Comparative Features of Eukaryotic Ubiquitin and Bacterial Pup Systems
| Feature | Eukaryotic Ubiquitin System | Mycobacterial Pup System |
|---|---|---|
| Tag Protein | Ubiquitin (76 aa, structured β-grasp fold) | Pup (64 aa, intrinsically disordered) |
| Conjugation Machinery | E1 (activating), E2 (conjugating), E3 (ligating) enzymes | Dop (deamidase/depupylase), PafA (ligase) |
| Energy Source | ATP | ATP |
| Proteasome Interaction | Through ubiquitin receptors in 19S regulatory particle | Through Mpa/ARC ATPase |
| Reversibility | Deubiquitinases (DUBs) | Dop (depupylase activity) |
| Chain Formation | Polyubiquitin chains (various linkages) | Monopupylation (single Pup per lysine) |
| C-terminal Motif | Gly-Gly | Gly-Gly-Gln/Glu |
| Bond to Substrate | Isopeptide bond with C-terminal glycine carboxylate | Isopeptide bond with C-terminal glutamate side chain |
The Pup-Proteasome System: Core Components and Mechanism
Architectural Organization
The Pup-proteasome system (PPS) in Mtb is encoded within a dedicated gene locus containing dop (deamidase of Pup), pup, prcBA (proteasome core subunits), and pafA (proteasome accessory factor A), with mpa (mycobacterial proteasome ATPase) typically located in a separate operon [104] [103]. This genetic organization is conserved across actinobacteria, though some species have lost proteasomal subunits while retaining pupylation capability, suggesting non-degradative functions for Pup modification [103].
The 20S core particle (CP) forms the proteolytic heart of the system, composed of four stacked heptameric rings (α7β7β7α7) with the proteolytic active sites residing in the β-subunits [33]. This structure creates a gated chamber where substrate degradation occurs. Unlike eukaryotic proteasomes that exhibit multiple proteolytic activities, the bacterial CP primarily demonstrates chymotrypsin-like activity [18] [33].
Enzymatic Cascade
Pupylation proceeds through a carefully orchestrated two-step process:
Activation: In mycobacteria, Pup is synthesized with a C-terminal glutamine (PupQ) that must be deamidated to glutamate by Dop before conjugation can occur [103] [105]. This activation step is chemically equivalent to depupylation and ensures temporal control over the pupylation process.
Conjugation: PafA, the single Pup ligase, catalyzes the formation of an isopeptide bond between the γ-carboxylate of Pup's C-terminal glutamate and the ε-amino group of a lysine residue on the target protein [42]. Structural analyses indicate that PafA likely evolved from ancestral glutamine synthetase-like enzymes [104].
The reverse reaction, depupylation, is also mediated by Dop, which cleaves the isopeptide bond between Pup and substrate proteins, providing reversibility to this post-translational modification [103].
Substrate Recognition and Targeting
Contrary to initial assumptions of promiscuity, PafA exhibits remarkable selectivity toward genuine substrates. Recent research has revealed that surface lysine accessibility alone is insufficient for pupylation [42]. Instead, PafA recognizes specific structural motifs centered around exposed lysines, with electrostatic interactions playing a guiding role in substrate selection.
The proteasomal ATPase Mpa (also known as ARC in other actinobacteria) forms a hexameric ring that recognizes pupylated proteins through interaction with the N-terminal coiled-coil domains [104]. Upon binding, Pup undergoes a disorder-to-order transition, forming an extended helix that integrates with the Mpa coiled-coil structure [104]. This interaction initiates ATP-dependent unfolding and translocation of the target protein into the 20S CP for degradation.
Diagram 1: Pupylation Pathway and Proteasomal Degradation in M. tuberculosis
Experimental Validation: Key Methodologies and Data
In Vitro Pupylation Assays
Protocol: Comprehensive in vitro pupylation assays require purified components: target substrate proteins (e.g., FabD, PanB), Mtb PafA, Dop, Pup, and ATP in reaction buffer (typically 50 mM Tris-HCl pH 7.5, 10 mM MgCl₂, 1 mM ATP) [42]. Reactions are incubated at 37°C, and timepoints are analyzed by SDS-PAGE and immunoblotting using anti-Pup antibodies.
Key Findings: Experimental data reveal significant variation in pupylation efficiency among putative substrates identified in pupylome studies [42]. For instance, PanB undergoes complete modification within 2 hours under standard conditions, while adenosine kinase (Adk) requires approximately 20 hours for full pupylation. Some reported pupylome members, including malate dehydrogenase (MDH) and ClpP2, show minimal or no pupylation in vitro, suggesting they may represent false positives or extremely minor substrates [42].
Table 2: Pupylation Efficiency of Selected M. tuberculosis Substrates
| Substrate | Function | In Vitro Pupylation Efficiency | Documentated Physiological Impact |
|---|---|---|---|
| FabD | Malonyl CoA-ACP transacylase | Complete within 2 hours | Accumulates in PPS mutants; essential for fatty acid synthesis |
| PanB | Ketopantoate hydroxymethyltransferase | Complete within 2 hours | Accumulates in PPS mutants; involved in pantothenate biosynthesis |
| Ino1 | Inositol-1-phosphate synthase | Efficient pupylation | Implicated in redox stress response |
| SodA | Superoxide dismutase | Efficient pupylation | Critical for oxidative stress defense |
| Log | LoaP operon glycosylase | Efficient pupylation | Regulates NO sensitivity; degradation essential for virulence |
| Adk | Adenosine kinase | Requires 20 hours for completion | Minimal accumulation in PPS mutants |
| MDH | Malate dehydrogenase | Minimal pupylation | No documented functional impact |
Genetic Approaches to PPS Function
Protocol: Construction of targeted gene knockouts (Δmpa, ΔpafA, ΔprcBA, Δpup) in Mtb strains, complemented with single-copy chromosomal integration of wild-type or mutant alleles [106]. Mutant strains are subjected to various stress conditions in vitro, including nitric oxide exposure, nutrient limitation, and metal toxicity. Virulence is assessed in murine infection models by measuring bacterial loads in organs over time.
Key Findings:
Nitric Oxide Resistance: PPS mutants (Δmpa, ΔpafA) exhibit extreme sensitivity to nitric oxide and related reactive nitrogen intermediates in vitro [33] [106]. The substrate Log has been identified as a key mediator of this phenotype, with Δlog mutants suppressing NO sensitivity in PPS-deficient backgrounds [106].
Mouse Virulence Studies: PPS mutants are severely attenuated in mouse models of tuberculosis, failing to establish lethal infections [33] [105]. While deletion of log restores NO resistance in vitro, it only partially rescues virulence defects, indicating additional PPS substrates contribute to pathogenesis [106].
Nitrate Utilization: PPS mutants cannot use nitrate as a sole nitrogen source due to a specific defect in the nitrite reductase step of nitrate assimilation [106]. This metabolic limitation may impact bacterial survival in specific host niches.
Proteomic Identification of Pupylation Targets
Protocol: Affinity enrichment of pupylated proteins using tagged Pup variants (e.g., His-Pup, FLAG-Pup) from mycobacterial lysates, followed by mass spectrometric identification [42]. Quantitative proteomic approaches compare protein abundance in wild-type versus PPS mutant strains to identify substrates whose stability is regulated by pupylation.
Key Findings: Pupylome studies have identified hundreds of putative pupylation targets in Mtb and related mycobacteria [42]. However, these datasets show significant bias toward abundant proteins, with pupylome members being on average 4-fold more abundant than the general proteome [42]. This suggests current pupylomes may overrepresent highly expressed proteins rather than biologically relevant substrates.
Functional Roles in Pathogenesis: Experimental Evidence
Stress Adaptation and Survival in Macrophages
The PPS is essential for Mtb to resist host antimicrobial defenses and persist within macrophages. Multiple lines of evidence establish this critical role:
Oxidative and Nitrosative Stress: PPS mutants show heightened sensitivity to reactive nitrogen and oxygen intermediates [104] [33]. The regulated degradation of stress-response proteins like SodA and Log through pupylation enables Mtb to maintain redox homeostasis under challenging conditions.
Metal Homeostasis: The PPS participates in regulating intracellular levels of essential metals, including iron and copper [104]. Recent research demonstrates that Mtb hijacks the host ubiquitin system through UBE2O-mediated ferritin degradation to increase iron availability, while simultaneously utilizing its own PPS for metal adaptation [107].
Nutrient Limitation: During periods of nutrient scarcity, the PPS facilitates metabolic adaptation by remodeling the proteome. Proteasomal degradation generates amino acids that can be reused for synthesis of essential proteins or serve as carbon sources [104] [106].
Metabolic Regulation
Beyond stress response, the PPS plays specific roles in regulating metabolic pathways:
Nitrogen Metabolism: While not required for survival during complete nitrogen starvation, the PPS is essential for Mtb growth on nitrate as a sole nitrogen source [106]. PPS mutants accumulate nitrite due to impaired NirBD nitrite reductase activity, linking pupylation to nitrogen assimilation.
Chaperonin Regulation: The PPS regulates expression of groEL/groES chaperonin genes through degradation of the repressor HrcA [106]. This function is particularly important during growth in nitrate, where robust chaperonin expression is required for NirBD activity.
Interaction with Host Systems
Mtb has evolved mechanisms to exploit host ubiquitin systems while maintaining its own proteostatic regulation:
Ferritin Manipulation: Mtb infection triggers UBE2O-mediated ubiquitination and degradation of host ferritin, increasing intracellular iron availability for bacterial acquisition [107]. This hijacking of host ubiquitination depends on phosphorylation of UBE2O at serine 82.
Xenophagy Evasion: The Mtb surface protein Rv1468c contains a eukaryotic-like ubiquitin-associated (UBA) domain that directly binds host ubiquitin chains, recruiting autophagy receptor p62 to target bacteria to autophagosomes [108]. This mechanism paradoxically promotes bacterial clearance, suggesting Mtb may fine-tune this interaction to maintain optimal intracellular loads without triggering excessive inflammation.
Diagram 2: Parallel Host Ubiquitin and Bacterial Pup Pathways During Infection
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Investigating the Pup-Proteasome System
| Reagent/Condition | Application | Key Features/Experimental Use |
|---|---|---|
| PPS Mutant Strains (Δmpa, ΔpafA, ΔprcBA) | Genetic studies | Assess PPS function in stress resistance and virulence; essential for in vivo studies |
| Anti-Pup Antibodies | Detection | Immunoblotting to monitor pupylation status; validate substrate identification |
| Recombinant PafA | In vitro assays | Biochemical characterization of pupylation; substrate specificity studies |
| Recombinant Dop | In vitro assays | Deamidation of PupQ to PupE; depupylation activity studies |
| Tagged-Pup Variants (His-Pup, FLAG-Pup) | Affinity enrichment | Pupylome studies; identification of novel substrates |
| Proteasome Inhibitors (MLN-273, epoxomicin) | Functional inhibition | Chemical validation of proteasome-dependent processes |
| Nitric Oxide Donors (DETA/NO) | Stress challenge | Assess NO resistance in PPS mutants |
| Nitrate Minimal Media | Metabolic studies | Evaluate nitrogen assimilation function of PPS |
| UBE2O Inhibitors (Arsenic trioxide) | Host-pathogen interaction | Study iron acquisition mechanisms |
Comparative Analysis: Eukaryotic UPS vs. Bacterial PPS
The parallel evolution of ubiquitin-like tagging systems in eukaryotes and bacteria represents a fascinating case of convergent evolution. While both systems fulfill the fundamental need for targeted protein degradation, they have distinct characteristics:
Evolutionary Origins: The bacterial PPS appears to have originated through horizontal gene transfer and subsequent adaptation, with PafA and Dop evolving from glutamine synthetase-like ancestors rather than E1-E2-E3 enzymes [104] [103]. This independent origin explains the profound structural and mechanistic differences between the systems.
Architectural Simplicity: The PPS operates with remarkable economy, employing a single ligase (PafA) compared to hundreds of E3 ubiquitin ligases in eukaryotes [42]. This simplicity suggests different strategies for achieving substrate specificity, potentially relying more on structural motifs than dedicated recognition components.
Regulatory Dynamics: Both systems incorporate reversal mechanisms (deubiquitinases in eukaryotes, Dop depupylation activity in bacteria), allowing dynamic regulation of protein modification [103]. However, the integration of activation (deamidation) and reversal in a single enzyme in the PPS may enable more coordinated control.
Physiological Roles: While both systems contribute to protein quality control and stress adaptation, the PPS has particularly specialized in supporting bacterial survival under specific host-induced stresses, making it a key virulence determinant in pathogenic species like Mtb [104] [33].
The Pup-proteasome system represents a sophisticated adaptation that enables Mycobacterium tuberculosis to navigate the challenges of host infection. Through regulated protein pupylation and degradation, Mtb maintains proteostasis under stress, modulates metabolic pathways, and resists antimicrobial immune mechanisms. The experimental evidence summarized in this review validates the PPS as an essential virulence system and promising target for therapeutic intervention. The comparative analysis between bacterial and eukaryotic ubiquitin-like systems highlights both convergent functional solutions and distinct mechanistic implementations across domains of life. Future research elucidating the structural basis of substrate recognition and the regulatory networks controlling pupylation will further advance our understanding of this fascinating bacterial adaptation to pathogenic existence.
The Ubiquitin-Proteasome System (UPS) represents a cornerstone of eukaryotic innate immunity, regulating protein stability, signaling cascades, and antimicrobial responses. For decades, the UPS was considered exclusively eukaryotic, while prokaryotes were thought to possess only simplified, related systems for metabolic purposes. However, recent research has fundamentally upended this paradigm, revealing that bacteria not only possess ancestral ubiquitination machinery but have also evolved sophisticated effector proteins that directly hijack and manipulate the eukaryotic UPS. This biological arms race has driven the evolution of remarkable molecular countermeasures, with bacterial pathogens deploying ubiquitin ligases and other UPS-targeting effectors to neutralize host defenses. This comparison guide examines the intricate interplay between eukaryotic UPS defenses and prokaryotic effector strategies, providing structural, functional, and experimental data to illuminate this dynamic host-pathogen battlefield.
Evolutionary and Structural Foundations of Ubiquitin Systems
Eukaryotic UPS: A Complex Defense Apparatus
The canonical eukaryotic ubiquitin system comprises a sophisticated enzymatic cascade that tags proteins for degradation or functional modification. This system begins with E1 ubiquitin-activating enzymes that ATP-dependently activate ubiquitin, followed by E2 ubiquitin-conjugating enzymes that carry the activated ubiquitin, and culminates with E3 ubiquitin ligases that specifically transfer ubiquitin to target proteins [79]. The system generates diverse ubiquitin chain topologies, with K48-linked chains typically marking proteins for proteasomal degradation and K63-linked chains acting as regulatory signals in inflammation and immunity [79] [109]. Deubiquitinases (DUBs) provide counter-regulation by removing ubiquitin modifications, creating a dynamic system that eukaryotes employ to mount targeted immune responses against invading pathogens.
Prokaryotic Ubiquitin System Antecedents
Contrary to long-held assumptions, prokaryotes possess ubiquitination-related machinery with striking architectural parallels to eukaryotic systems. Bacterial operons such as the Type II BilABCD system encode complete ubiquitination pathways featuring E1, E2, Ubl (ubiquitin-like protein), and JAB-family deubiquitinase proteins [5]. Structural analyses reveal that bacterial E1 enzymes (E1BilD) contain N-terminal inactive adenylation domains (IAD) and C-terminal active adenylation domains (AAD) with mobile cysteine-containing domains, mirroring the architecture of eukaryotic E1s like human UBA6 and NAE1-UBA3 [5]. These bacterial systems function in antiviral defense, modifying virion structural proteins to protect against phage infection [5]. Additionally, bacteria possess simplified ubiquitin-like systems such as the Pup (prokaryotic ubiquitin-like protein) system in Mycobacteria, though this functions primarily in degradation without the regulatory complexity of eukaryotic ubiquitination [5] [62].
Table 1: Comparative Features of Eukaryotic and Prokaryotic Ubiquitin-Related Systems
| Feature | Eukaryotic UPS | Prokaryotic Ancestral Systems | Bacterial Effector Mimicry |
|---|---|---|---|
| Core Enzymes | E1, E2, E3 cascade | ThiF/MoeB (E1-like), ThiS/MoaD (Ubl) | E3 ligases (NEL, HECT, RING) |
| Structural Architecture | Multi-domain E1 with IAD, AAD, CYS domains | Bacterial E1 with IAD, AAD, CYS domains [5] | E3 domains structurally mimicking eukaryotic counterparts |
| Primary Functions | Protein degradation, signaling, immune regulation | Sulfur metabolism (ancestral) [20] | Host UPS manipulation, immune evasion |
| Ubiquitin Conjugation | Lysine isopeptide linkage | Acyl-persulfide (ThiS-ThiF) [20] | Lysine isopeptide linkage to host proteins |
| Deconjugation Enzymes | Multiple DUB families | JAB peptidases [20] | Effector-associated DUB activity |
| Phage/Antiviral Role | Indirect through immune signaling | Direct anti-phage activity [5] | Subversion of host antiviral defenses |
Bacterial Effector Arsenal: Molecular Weapons Against Host UPS
Pathogenic bacteria have evolved three primary strategic approaches to counteract eukaryotic UPS defenses, employing effectors delivered via specialized secretion systems (e.g., Type III, IV) that manipulate host ubiquitination processes to suppress immunity and promote infection.
E3 Ubiquitin Ligase Mimics
The most sophisticated bacterial countermeasures involve direct molecular mimicry of eukaryotic E3 ubiquitin ligases, which can be categorized into three major structural classes:
NEL (Novel E3 Ligase) Effectors: The IpaH family effectors from Shigella, Salmonella, and other pathogens feature a C-terminal NEL domain that structurally diverges from canonical HECT and RING domains while maintaining ubiquitin ligase functionality [79] [62]. These effectors use N-terminal leucine-rich repeats (LRR) for substrate recognition and their NEL domains to ubiquitinate specific host targets. For instance, Shigella IpaH9.8 ubiquitinates NEMO/IKKγ, an essential component of the NF-κB signaling pathway, leading to its degradation and suppression of inflammatory responses [79] [62]. IpaH4 similarly targets SHOC2 and PSMC1 for degradation to promote arbovirus replication in insect and human cells [110].
HECT-like Effectors: Bacterial effectors such as Salmonella SopA and EHEC NleL structurally and functionally resemble eukaryotic HECT-type E3 ligases, containing catalytic cysteine residues that form thioester intermediates with ubiquitin before transfer to substrates [79] [109]. SopA plays roles in Salmonella-induced inflammation and transepithelial migration of polymorphonuclear neutrophils, though its complete substrate profile remains under investigation [79] [109].
RING/U-box Effectors: The Pseudomonas syringae effector AvrPtoB exemplifies RING-type mimicry, containing a C-terminal domain structurally similar to eukaryotic RING domains that binds E2 enzymes [79] [62]. AvrPtoB ubiquitinates host kinases critical for plant immunity (e.g., Fen, CERK1, FLS2, BAK1), targeting them for proteasomal degradation to suppress effector-triggered immunity [79] [62].
UPS Pathway Manipulators
Beyond direct E3 mimicry, bacterial effectors employ alternative strategies to manipulate UPS function:
Deamidating Effectors: Shigella OspI deamidates Ubc13, altering its function in the NF-κB signaling pathway to suppress inflammatory responses [79]. Similarly, EPEC/EHEC Cif and related effectors deamidate NEDD8 and ubiquitin, inhibiting cullin-RING ligase (CRL) activation and thereby disrupting multiple host signaling pathways [79].
Kinase-like Effectors: Shigella, EHEC, and Yersinia OspG functions as a kinase that binds to E2 enzymes (e.g., UbcH5), potentially sequestering them or altering their activity to modulate ubiquitination dynamics during infection [79].
DUB-like Effectors: Salmonella SseL exhibits deubiquitinase activity that is required for successful infection in mice, presumably by reversing host ubiquitination events that would otherwise restrict bacterial replication [79].
Table 2: Major Classes of Bacterial Effectors Targeting Eukaryotic UPS
| Effector Class | Representative Examples | Bacterial Source | Mechanism of Action | Host Targets/Consequences |
|---|---|---|---|---|
| NEL E3 Ligases | IpaH family (IpaH9.8, IpaH4) | Shigella, Salmonella | Ubiquitinates host proteins for degradation | NEMO/IKKγ (NF-κB suppression) [79]; SHOC2, PSMC1 (viral restriction evasion) [110] |
| HECT-like E3 Ligases | SopA, NleL | Salmonella, EHEC | Forms thioester intermediate with Ub | Unknown substrates; inflammation regulation [79] [109] |
| RING/U-box E3 Ligases | AvrPtoB, LubX, NleG | P. syringae, L. pneumophila, EHEC | Scaffolds E2-substrate interactions | Plant immunity kinases (degradation) [79] [62] |
| Deamidases | OspI, Cif | Shigella, EPEC/EHEC | Deamidates Ub/Ubl-related proteins | Ubc13 (NF-κB suppression) [79]; NEDD8 (CRL inhibition) [79] |
| Kinases | OspG | Shigella, EHEC, Yersinia | Binds E2 enzymes | UbcH5 (ubiquitination modulation) [79] |
| DUBs | SseL | Salmonella | Removes ubiquitin from substrates | Unknown targets; promotes infection [79] |
Experimental Approaches and Methodologies
Structural Characterization Techniques
X-ray Crystallography: Structural insights into bacterial UPS manipulation have been achieved through X-ray crystallography of effector complexes. For example, structures of the bacterial E1BilD:E2BilB:UblBilA complex were determined at 2.5-2.7 Å resolution, revealing architectural parallels with eukaryotic E1:E2:Ub complexes [5]. Similarly, crystallography of AvrPtoB and IpaH effectors revealed their structural mimicry of eukaryotic RING and novel E3 ligase folds, respectively [79] [62].
Protocol Overview:
- Clone and co-express bacterial E1, E2, and Ubl genes in E. coli
- Purify complexes using affinity and size-exclusion chromatography
- Crystallize using vapor diffusion methods with optimized conditions
- Collect diffraction data at synchrotron facilities
- Solve structures via molecular replacement or experimental phasing
- Analyze interfaces and catalytic sites for mechanistic insights
Functional Ubiquitination Assays
In Vitro Reconstitution: Biochemical validation of bacterial E3 ligase activity involves reconstituting ubiquitination cascades with purified components [79] [62].
Standard Protocol:
- Purify bacterial effector proteins and host UPS components (E1, E2, ubiquitin)
- Set up reaction mixtures containing ATP, E1, E2, ubiquitin, and effector
- Include potential substrate proteins when testing specific ubiquitination targets
- Incubate at 30-37°C for timed intervals
- Terminate reactions with SDS loading buffer
- Analyze by western blotting with ubiquitin-specific and substrate-specific antibodies
- Confirm thioester intermediates under non-reducing conditions
Cellular Infection Models
Host-Pathogen Interaction Studies: Understanding effector function in physiological contexts employs cellular infection models with wild-type and effector-mutant bacteria [79] [110].
Methodological Approach:
- Infect cultured mammalian cells (e.g., HeLa, HEK293) or plant cells with pathogenic bacteria
- Compare infection outcomes between wild-type and isogenic effector-deletion strains
- Monitor substrate degradation via immunoblotting of cell lysates
- Assess immune signaling outputs (NF-κB activation, cytokine production)
- Use proteasome inhibitors (MG132) to confirm proteasome-dependent degradation
- Employ RNAi or CRISPR to knock down potential host targets and validate specificity
Table 3: Key Research Reagents for Studying UPS-Pathogen Interactions
| Reagent Category | Specific Examples | Research Applications | Key Features/Considerations |
|---|---|---|---|
| Recombinant Bacterial Effectors | IpaH family proteins (IpaH9.8, IpaH4), AvrPtoB, SopA, Cif | In vitro ubiquitination assays, structural studies, cellular transduction | Catalytic mutants (C/S) essential as negative controls; validate proper folding |
| Ubiquitination System Components | Human E1 (UBA1, UBA6), E2 enzymes (UbcH5, UbcH7), Ubiquitin (WT, mutant forms) | Reconstitution assays, chain topology studies | K48-only, K63-only ubiquitin mutants for chain specificity; fluorescent/HA-tagged versions for detection |
| Cell Culture Models | HeLa, HEK293, Caco-2, plant protoplasts | Cellular infection studies, transfection/transduction, immune signaling readouts | Select cell lines based on pathogen tropism; consider knockout lines for specific host factors |
| Infection-Relevant Bacterial Strains | Wild-type and isogenic effector mutants (ΔipaH, ΔsopA, etc.) | Physiological infection models, comparative virulence studies | Essential to confirm effector secretion and delivery; complement with plasmid expression |
| Proteasome Inhibitors | MG132, Bortezomib, Lactacystin | Validate proteasome-dependent degradation, stabilize ubiquitinated substrates | Titrate concentration to balance efficacy with cellular toxicity; include DMSO controls |
| Antibodies for Detection | Anti-ubiquitin (linkage-specific), anti-NEMO, anti-IκBα, anti-SHOC2, anti-PSMC1 | Immunoblotting, immunofluorescence, immunoprecipitation | Linkage-specific Ub antibodies valuable but require validation; species compatibility critical |
| Secretion System Inducers | Type III secretion inducers (low calcium, temperature) | Activate bacterial effector delivery in culture | Optimize induction timing relative to infection; monitor bacterial viability |
Data Presentation and Comparative Analysis
Quantitative Assessment of Effector Impacts
Table 4: Experimental Data from Key Bacterial Effector Studies
| Effector (Source) | Experimental System | Target Substrate | Quantitative Impact | Functional Consequence |
|---|---|---|---|---|
| IpaH9.8 (Shigella) | HEK293T transfection + infection | NEMO/IKKγ | >80% reduction in NEMO protein levels at 4h post-infection [79] | Suppression of NF-κB signaling and inflammatory cytokine production |
| AvrPtoB (P. syringae) | Plant protoplast transfection | Fen kinase | ~70% reduction in Fen levels; 10-fold increase in bacterial survival [62] | Suppression of effector-triggered immunity and programmed cell death |
| IpaH4 (Shigella) | LD652 and HEK293T cells | SHOC2, PSMC1 | ~60-70% reduction in target levels; 100-1000x increase in viral titers [110] | Rescue of restricted arbovirus replication in insect and human cells |
| SopA (Salmonella) | Polarized epithelial cell infection | Unknown | 5-fold increase in PMN transepithelial migration [109] | Enhanced inflammation and bacterial dissemination |
| Cif (EPEC/EHEC) | HeLa cell infection | NEDD8 | ~50% reduction in CRL activity; cell cycle arrest [79] | Inhibition of host cell division and potential modulation of immune signaling |
The evolutionary arms race between eukaryotic UPS defenses and prokaryotic effectors represents a remarkable example of molecular adaptation and counter-adaptation. Eukaryotes developed the UPS as a sophisticated regulatory system that was subsequently co-opted for antimicrobial defense, while bacteria responded by evolving diverse effector proteins that mimic, manipulate, or inhibit components of this system. The recent discovery of complete bacterial ubiquitination pathways with striking architectural similarities to eukaryotic systems suggests deep evolutionary roots for ubiquitin signaling and blurs the traditional boundaries between prokaryotic and eukaryotic biology.
Future research directions should focus on identifying the complete repertoire of host substrates for bacterial E3 ligases, understanding the structural determinants of effector specificity, and exploring the potential of these systems as targets for novel antimicrobial strategies. The emerging paradigm of trans-kingdom conservation in antiviral defense mechanisms further suggests that insights from bacterial effector studies may illuminate fundamental aspects of host-pathogen interactions across the tree of life. As research continues to unravel the complexity of these molecular interactions, we gain not only a deeper understanding of pathogenesis but also new perspectives on the evolution of cellular signaling systems themselves.
Conclusion
The discovery of functional ubiquitin-like protein conjugation systems in prokaryotes has fundamentally reshaped our understanding of this crucial regulatory pathway's evolution. While core structural and mechanistic principles are conserved, as seen in the recent identification of bona fide bacterial E1-E2-Ubl cascades, profound differences exist in enzymatic components and biological roles—from sulfur metabolism in ancient systems to phage defense and virulence in modern bacteria. For biomedical research, these insights are transformative. The intricate interplay between host ubiquitin signaling and bacterial effector proteins represents a rich landscape for therapeutic intervention. Future directions should focus on exploiting the unique aspects of prokaryotic Ubl systems, such as the Pup-proteasome pathway in mycobacteria or the newly described bacterial E1 enzymes, to develop novel antibiotics that selectively disarm pathogens. Furthermore, studying how bacterial effectors achieve specificity provides a blueprint for designing new classes of highly targeted molecular probes and drugs for manipulating the human ubiquitin system in diseases like cancer and neurodegeneration.
References
- 1. Molecular Architecture and Assembly of the Eukaryotic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3827779/]
- 2. The ubiquitin system: from cell signalling to disease ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7862391/]
- 3. Sent to Destroy: The Ubiquitin Proteasome System Regulates ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2826711/]
- 4. Spotlight Ubiquitous ubiquitin: From bacteria to eukaryotes [https://www.sciencedirect.com/science/article/pii/S096921262400371X]
- 5. A eukaryotic-like ubiquitination system in bacterial antiviral ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11476048/]
- 6. exploring the ubiquitin system for drug development [https://www.nature.com/articles/cr201631]
- 7. Ubiquitin modifications | Cell Research [https://www.nature.com/articles/cr201639]
- 8. Protein Engineering in the Ubiquitin System [https://www.sciencedirect.com/science/article/pii/S0031699724009578]
- 9. Ubiquitin-like protein [https://en.wikipedia.org/wiki/Ubiquitin-like_protein]
- 10. Origin and Function of Ubiquitin-like Protein Conjugation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2819001/]
- 11. Structural and Functional Insights to Ubiquitin-Like Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4118471/]
- 12. More than Meets the ISG : Emerging 15 in the DNA Damage... Roles [https://pmc.ncbi.nlm.nih.gov/articles/PMC7698331/]
- 13. Role of ubiquitin- and Ubl-binding proteins in cell signaling [https://www.sciencedirect.com/science/article/abs/pii/S0955067407000166]
- 14. Chemical approaches to explore ubiquitin-like proteins [https://pubs.rsc.org/en/content/articlehtml/2025/cb/d4cb00220b]
- 15. A comprehensive platform for the analysis of ubiquitin-like ... [https://www.nature.com/articles/srep40756]
- 16. and ISGylation modulates cancer stem cell-like characteristics in... ISG 15 [https://jeccr.biomedcentral.com/articles/10.1186/s13046-023-02751-9]
- 17. Structural and functional insights into Ubl domain-mediated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12495856/]
- 18. PROKARYOTIC UBIQUITIN-LIKE PROTEIN, ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3662484/]
- 19. Prokaryotic Ubiquitin-Like Protein Modification - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4757901/]
- 20. The prokaryotic antecedents of the ubiquitin-signaling system ... [https://genomebiology.biomedcentral.com/articles/10.1186/gb-2006-7-7-r60]
- 21. Ubiquitin [https://en.wikipedia.org/wiki/Ubiquitin]
- 22. Biochemistry, Ubiquitination - StatPearls - NCBI Bookshelf - NIH [https://www.ncbi.nlm.nih.gov/books/NBK556052/]
- 23. The ubiquitin system: from cell signalling to disease ... [https://www.nature.com/articles/s41418-020-00703-w]
- 24. Emerging drug development technologies targeting ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7112620/]
- 25. Targeting the ubiquitin system by fragment-based drug ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2022.1019636/full]
- 26. The prokaryotic antecedents of the ubiquitin-signaling system ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1779556/]
- 27. Mechanism of ubiquitin activation revealed by the structure of a bacterial MoeB-MoaD complex - PubMed [https://pubmed.ncbi.nlm.nih.gov/11713534/]
- 28. The dual role of ubiquitin-like protein Urm1 as a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4875326/]
- 29. RCSB PDB - 1JW9: Structure of the Native MoeB-MoaD Protein Complex [https://www.rcsb.org/structure/1JW9]
- 30. Ubiquitin-like prokaryotic MoaD as a fusion tag for expression ... [https://bmcbiotechnol.biomedcentral.com/articles/10.1186/1472-6750-14-5]
- 31. Recent progress in ubiquitin and ubiquitin-like protein (Ubl ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4822135/]
- 32. Prokaryotic ubiquitin-like protein [https://en.wikipedia.org/wiki/Prokaryotic_ubiquitin-like_protein]
- 33. The Pup-Proteasome System of Mycobacterium tuberculosis [https://pmc.ncbi.nlm.nih.gov/articles/PMC4212895/]
- 34. Structures of prokaryotic ubiquitin-like protein Pup in ... [https://www.nature.com/articles/s41467-021-26848-x]
- 35. Structural basis of prokaryotic ubiquitin-like protein ... [https://www.nature.com/articles/s41467-021-27787-3]
- 36. Global Analysis of Protein and Small-Molecule Substrates ... [https://www.sciencedirect.com/science/article/pii/S1535947625000738]
- 37. Small but versatile: the extraordinary functional and structural ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1949818/]
- 38. the extraordinary functional and structural diversity of the beta ... [https://pubmed.ncbi.nlm.nih.gov/17605815/]
- 39. A novel superfamily containing the β-grasp fold involved in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1796856/]
- 40. Prokaryotic Ubiquitin-Like Protein and Its Ligase/Deligase ... [https://www.sciencedirect.com/science/article/abs/pii/S0022283617301961]
- 41. The Mechanism of Mycobacterium smegmatis PafA Self ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4783102/]
- 42. Electrostatic interactions guide substrate recognition of the ... [https://www.nature.com/articles/s41467-023-40807-8]
- 43. E1, E2, E3 Enzyme and Their Regulators [https://www.bocsci.com/resources/e1-e2-e3-enzyme-and-their-regulators.html?srsltid=AfmBOoq1HBX3tVrcC4vr9lMK6Oeq_0yK2mDeo_ch6FV5YOftbpRL8_AL]
- 44. Ubiquitin/Proteasome [https://www.cellsignal.com/pathways/ubiquitin-proteasome-pathway?srsltid=AfmBOop7rTh35PU5g67pFJsagmd010d1l02IIkFRdRZmB2fFrQnB4YRF]
- 45. Allosteric Transitions Direct Protein Tagging by PafA, the ... [https://www.sciencedirect.com/science/article/pii/S0021925820672052]
- 46. Functional reconstruction of a eukaryotic-like E1/E2/(RING) ... [https://www.nature.com/articles/s41467-017-01162-7]
- 47. A eukaryotic-like ubiquitination system in bacterial ... - PubMed [https://pubmed.ncbi.nlm.nih.gov/39020180/]
- 48. Ubiquitin-like conjugation by bacterial cGAS enhances anti ... [https://www.nature.com/articles/s41586-023-05862-7]
- 49. Bacterial antiviral defense pathways encode eukaryotic-like ... [https://pubmed.ncbi.nlm.nih.gov/37808811/]
- 50. Article Structural diversity and oligomerization of bacterial ... [https://www.sciencedirect.com/science/article/abs/pii/S096921262500108X]
- 51. 8TYZ: Structure of a bacterial E1-E2-Ubl complex (form 2) [https://www.rcsb.org/structure/8TYZ]
- 52. 2MWS: Structure of the complex of ubiquitin ... [https://www.rcsb.org/structure/2MWS]
- 53. The Ubiquitin Conjugating Enzyme - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC7215765/]
- 54. Prokaryotes vs Eukaryotes: What Are the Key Differences? [https://www.technologynetworks.com/cell-science/articles/prokaryotes-vs-eukaryotes-what-are-the-key-differences-336095]
- 55. COMPARISON BETWEEN PROKARYOTIC AND ... [https://inlibrary.uz/index.php/imjrd/article/view/101348]
- 56. Characterization and regulation mechanism analysis of ... [https://bmcplantbiol.biomedcentral.com/articles/10.1186/s12870-021-03421-8]
- 57. Genome-wide identification of wheat Ubiquitin-conjugating ... [https://bmcplantbiol.biomedcentral.com/articles/10.1186/s12870-025-06931-x]
- 58. Chemical Tools for Probing the Ub/Ubl Conjugation Cascades [https://pubmed.ncbi.nlm.nih.gov/39313481/]
- 59. Functional Validation - 2025 UBC iGEM Wiki [https://2025.igem.wiki/ubc-vancouver/biocementing-bacteria/functional-validation]
- 60. Recent Advances in ADCs [https://njbio.com/antibody-drug-conjugates/]
- 61. The NEL Family of Bacterial E3 Ubiquitin Ligases - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9320238/]
- 62. Bacteria-host relationship: ubiquitin ligases as weapons of ... [https://www.nature.com/articles/cr201630]
- 63. Bacterial E3 Ligases That Exploit the Eukaryotic Ubiquitin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4133400/]
- 64. The emerging role and therapeutic implications of bacterial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10703165/]
- 65. E3 ubiquitin ligases: styles, structures and functions - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8607428/]
- 66. Bacterial Interference of Ubiquitination and Deubiquitination [https://pmc.ncbi.nlm.nih.gov/articles/PMC7173291/]
- 67. Targeting the Ubiquitin–Proteasome System for Cancer - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12495026/]
- 68. The Evolution and Diversification of Proteasome Inhibitors ... [https://chemrxiv.org/engage/chemrxiv/article-details/6853081ac1cb1ecda0b1e33f]
- 69. Emerging therapies targeting the ubiquitin proteasome ... [https://www.jci.org/articles/view/71602]
- 70. A new era in cancer therapy: targeting the Proteasome-Bcl-2 ... [https://jeccr.biomedcentral.com/articles/10.1186/s13046-025-03505-5]
- 71. Targeting the ubiquitin-proteasome system and drug ... [https://pubmed.ncbi.nlm.nih.gov/40844830/]
- 72. Unlocking new degradation mechanisms [https://www.news-medical.net/health/Unlocking-new-degradation-mechanisms-with-alternative-E3-ligases.aspx]
- 73. The ubiquitin-proteasome system in the tumor immune ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1522427/full]
- 74. Systematic comparison and base-editing-mediated ... [https://www.nature.com/articles/s41467-025-61848-1]
- 75. Characterizing host-microbe interactions with bacterial ... [https://www.nature.com/articles/s42003-025-07950-y]
- 76. Large-scale analysis of post-translational modifications in E ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5392934/]
- 77. Proximity labeling techniques for protein– ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12359232/]
- 78. Ubiquitin‐targeted bacterial effectors: rule breakers of the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10505922/]
- 79. Bacterial Effectors and Their Functions in the Ubiquitin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4197628/]
- 80. Five Mechanisms of Manipulation by Bacterial Effectors [https://journals.plos.org/plospathogens/article?id=10.1371/journal.ppat.1002823]
- 81. Exploiting bacterial effector proteins to uncover evolutionarily ... [https://journals.plos.org/plospathogens/article?id=10.1371/journal.ppat.1012010]
- 82. Bacterial effectors: learning on the fly - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC3427734/]
- 83. Bacterial Deubiquitinases with Ubiquitin Clippase Activity [https://pmc.ncbi.nlm.nih.gov/articles/PMC12255157/]
- 84. Deciphering non-canonical ubiquitin signaling: biology and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10897730/]
- 85. The Ubiquitination System within Bacterial Host–Pathogen ... [https://www.mdpi.com/2076-2607/9/3/638]
- 86. The Ubiquitin Tale: Current Strategies and Future Challenges [https://pmc.ncbi.nlm.nih.gov/articles/PMC11406696/]
- 87. Ubiquitin - an overview | ScienceDirect Topics [https://www.sciencedirect.com/topics/pharmacology-toxicology-and-pharmaceutical-science/ubiquitin]
- 88. and Evolution of Structure and... | SpringerLink Ubiquitin [https://link.springer.com/protocol/10.1007/978-1-61779-474-2_2]
- 89. A Giant Ubiquitin-conjugating Enzyme Related to IAP ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2132795/]
- 90. Targeting deubiquitinating enzymes and ubiquitin pathway ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12506015/]
- 91. Bacterial clippases shift ubiquitin [https://www.nature.com/articles/s41594-025-01545-1]
- 92. Structural and functional insights into Ubl domain-mediated ... [https://biologydirect.biomedcentral.com/articles/10.1186/s13062-025-00690-3]
- 93. PLIP 2025: introducing protein-protein interactions to the ... [https://pubmed.ncbi.nlm.nih.gov/40347107/]
- 94. The proteasome: Overview of structure and functions - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3524306/]
- 95. Proteasome [https://en.wikipedia.org/wiki/Proteasome]
- 96. Unlocking the proteasome: Structure and function [https://www.abcam.com/en-us/knowledge-center/cell-biology/proteasome]
- 97. Article The prokaryotic ubiquitin-like protein presents poor ... [https://www.sciencedirect.com/science/article/pii/S2211124721008457]
- 98. Systematic Analysis and Prediction of Pupylation Sites in Prokaryotic ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0074002]
- 99. Ubiquitin signaling in immune responses - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4822134/]
- 100. Modulation of the Ubiquitin-Proteasome System by ... [https://www.sciencedirect.com/science/article/pii/S095528632500289X]
- 101. Tracking of Ubiquitin Signaling through 3.5 Billion Years of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11354881/]
- 102. Identification of a covert evolutionary pathway between two ... [https://www.nature.com/articles/s41467-023-38519-0]
- 103. The pupylation pathway and its role in mycobacteria - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3511204/]
- 104. Survival in Hostile Conditions: Pupylation and the Proteasome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8223512/]
- 105. Molecular analysis of the prokaryotic ubiquitin-like protein ... [https://vivo.weill.cornell.edu/display/pubid20636328]
- 106. The Mycobacterium tuberculosis Pup-proteasome system ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6386731/]
- 107. Mycobacterium tuberculosis hijacks the UBE2O pathway to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12043076/]
- 108. A Mycobacterium tuberculosis surface protein recruits ... [https://www.nature.com/articles/s41467-019-09955-8]
- 109. Exploitation of Eukaryotic Ubiquitin Signaling Pathways by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1781473/]
- 110. Exploiting bacterial effector proteins to uncover evolutionarily ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11098378/]