A Step-by-Step Guide to K48-Linked Ubiquitin Immunoblotting: From Sample Prep to Validation
This detailed protocol provides researchers and drug development professionals with a comprehensive framework for the reliable detection of K48-linked polyubiquitin chains via immunoblotting.
A Step-by-Step Guide to K48-Linked Ubiquitin Immunoblotting: From Sample Prep to Validation
Abstract
This detailed protocol provides researchers and drug development professionals with a comprehensive framework for the reliable detection of K48-linked polyubiquitin chains via immunoblotting. Covering foundational principles, a step-by-step methodological workflow, essential troubleshooting for common pitfalls, and rigorous validation techniques, this article synthesizes current best practices to ensure accurate interpretation of the proteasome-targeting ubiquitin code in diverse experimental contexts.
Decoding the K48 Ubiquitin Code: Significance and Antibody Specificity
The Biological Role of K48-Linked Ubiquitin in Proteasomal Degradation
The K48-linked polyubiquitin chain is a fundamental signal in the ubiquitin-proteasome system (UPS), primarily directing substrate proteins for proteasomal degradation [1]. Since the landmark discovery by Chau et al. that revealed K48-linked chains as the topology signaling protein degradation, our understanding of the ubiquitin code has expanded tremendously [2]. This specific linkage, formed when the carboxyl group of a ubiquitin's C-terminal glycine conjugates to the epsilon-amino group of lysine 48 on the preceding ubiquitin molecule, acts as a primary recognition signal for the 26S proteasome [1] [3].
The process of K48-linked ubiquitination involves a well-defined enzymatic cascade. First, a ubiquitin-activating enzyme (E1) activates ubiquitin in an ATP-dependent manner. The activated ubiquitin is then transferred to a ubiquitin-conjugating enzyme (E2). Finally, a ubiquitin ligase (E3) facilitates the transfer of ubiquitin to the target protein, with specific E2-E3 combinations determining linkage specificity [4] [5]. The 26S proteasome recognizes K48-linked ubiquitinated substrates through ubiquitin receptors such as RPN10 and RPN13 within its 19S regulatory particle, leading to substrate unfolding and degradation by the 20S core proteolytic chamber [3].
Table 1: Major Ubiquitin Linkage Types and Their Primary Functions
| Linkage Type | Primary Biological Function |
|---|---|
| K48-linked | Proteasomal degradation [1] |
| K63-linked | Immune signaling, DNA repair, protein trafficking [2] [6] |
| K11-linked | Cell cycle regulation, proteasomal degradation (often in branched chains) [3] |
| K6-linked | DNA damage response, antiviral signaling [5] |
| M1-linked (Linear) | NF-κB signaling, immune response [2] |
Biological Mechanisms and Current Research
Structural Recognition by the Proteasome
Recent structural biology advances, particularly cryo-EM studies, have revealed how the human 26S proteasome recognizes K48-linked ubiquitin chains. The proteasome employs a multivalent substrate recognition mechanism where K48-linked chains are primarily engaged at the canonical binding site formed by RPN10 and RPT4/5 coiled-coil domains [3]. This specific interaction ensures the selective degradation of proteins marked with K48 linkages over other chain types.
The recognition mechanism becomes more complex with branched ubiquitin chains. K11/K48-branched chains, which account for 10-20% of all ubiquitin polymers and are increasingly recognized as potent proteasomal targeting signals, engage additional proteasomal ubiquitin receptors [3]. These branched chains form a tripartite binding interface with the 19S regulatory particle, with RPN2 acting as a critical ubiquitin receptor that recognizes the K48-linkage extending from a K11-linked ubiquitin [3]. This elaborate recognition system allows the proteasome to prioritize substrates marked with specific ubiquitin architectures, particularly under cellular stress conditions.
K48-Ubiquitin in Immune Regulation and Cellular Homeostasis
K48-linked ubiquitination plays a critical role in immune cell function and signaling regulation. In dendritic cells (DCs), K48-linked ubiquitination determines antigen degradation and controls the endosomal recruitment of p97 and Sec61 complex proteins, which are essential components for cross-presentation [7]. This process enables DCs to present exogenous antigens on MHC class I molecules, initiating cytotoxic T lymphocyte (CTL) responses against viruses and tumors.
Research has demonstrated that nicotine treatment significantly increases K48-linked ubiquitination in bone marrow-derived dendritic cells (BM-DCs), enhancing their cross-presentation capacity and CTL priming efficiency [7]. Specifically, the mannose receptor (MR), an important antigen receptor in DCs, undergoes K48-linked ubiquitination following nicotine treatment, facilitating the endosomal recruitment of p97 and Sec61 necessary for antigen translocation and processing [7]. This mechanism contributes to superior adaptive immunity and highlights the therapeutic potential of modulating K48-linked ubiquitination in DC-mediated immune therapy.
The concept of ubiquitin chain editing represents another crucial regulatory mechanism in immune signaling. Studies using linkage-specific antibodies have revealed that signaling adaptors like RIP1 and IRAK1 initially acquire K63-linked polyubiquitin chains to activate signaling pathways, while at later time points, these are replaced by K48-linked chains that target them for proteasomal degradation [8]. This temporal switching of linkage types provides an elegant mechanism for attenuating innate immune responses and maintaining cellular homeostasis.
Experimental Analysis of K48-Linked Ubiquitination
Determining Ubiquitin Chain Linkage: A Step-by-Step Protocol
Understanding the experimental determination of ubiquitin chain linkage is essential for researchers investigating the ubiquitin-proteasome system. The following protocol utilizes ubiquitin mutants to specifically identify chain linkages [4].
Table 2: Required Reagents for Ubiquitin Linkage Determination
| Reagent | Stock Concentration | Working Concentration | Function |
|---|---|---|---|
| E1 Enzyme | 5 µM | 100 nM | Activates ubiquitin for conjugation |
| E2 Enzyme | 25 µM | 1 µM | Determines linkage specificity with E3 |
| E3 Ligase | 10 µM | 1 µM | Substrate-specific ubiquitin ligase |
| Wild-type Ubiquitin | 1.17 mM (10 mg/mL) | ~100 µM | Positive control for ubiquitination |
| Ubiquitin K-to-R Mutants | 1.17 mM (10 mg/mL) | ~100 µM | Identify essential lysine for linkage |
| Ubiquitin K-Only Mutants | 1.17 mM (10 mg/mL) | ~100 µM | Verify linkage specificity |
| MgATP Solution | 100 mM | 10 mM | Energy source for conjugation |
| 10X E3 Reaction Buffer | 10X | 1X (50 mM HEPES, pH 8.0) | Optimal reaction conditions |
Procedure:
Reaction Setup: Prepare two sets of nine in vitro ubiquitin conjugation reactions (25 µL each). The first set includes wild-type ubiquitin and seven ubiquitin K-to-R mutants (K6R, K11R, K27R, K29R, K33R, K48R, K63R). The second set includes wild-type ubiquitin and seven ubiquitin K-Only mutants (each containing only one lysine with the remaining six mutated to arginine). Include a negative control without ATP [4].
Reaction Composition:
- 2.5 µL 10X E3 Ligase Reaction Buffer
- 1 µL Ubiquitin or Ubiquitin mutant (~100 µM final)
- 2.5 µL MgATP Solution (10 mM final)
- Substrate (5-10 µM final)
- 0.5 µL E1 Enzyme (100 nM final)
- 1 µL E2 Enzyme (1 µM final)
- E3 Ligase (1 µM final)
- dH₂O to 25 µL [4]
Incubation: Incubate all reactions in a 37°C water bath for 30-60 minutes.
Reaction Termination:
- For direct analysis: Add 25 µL 2X SDS-PAGE sample buffer
- For downstream applications: Add 0.5 µL 500 mM EDTA (20 mM final) or 1 µL 1 M DTT (100 mM final) [4]
Analysis: Analyze by Western blotting using an anti-ubiquitin antibody. Interpretation:
- In K-to-R mutant reactions, the mutant that fails to form chains indicates the essential lysine for linkage.
- In K-Only mutant reactions, only the mutant retaining that specific lysine will form chains [4].
Diagram 1: Ubiquitin Linkage Determination Workflow
K48-Linkage Specific Immunoblotting Protocol
Materials:
- K48-linkage specific polyubiquitin antibody (e.g., Cell Signaling Technology #4289) [1]
- Standard Western blotting equipment
- Cell lysates prepared with proteasome inhibitor (e.g., MG-132) to preserve ubiquitination [5]
- Appropriate HRP-conjugated secondary antibodies
Procedure:
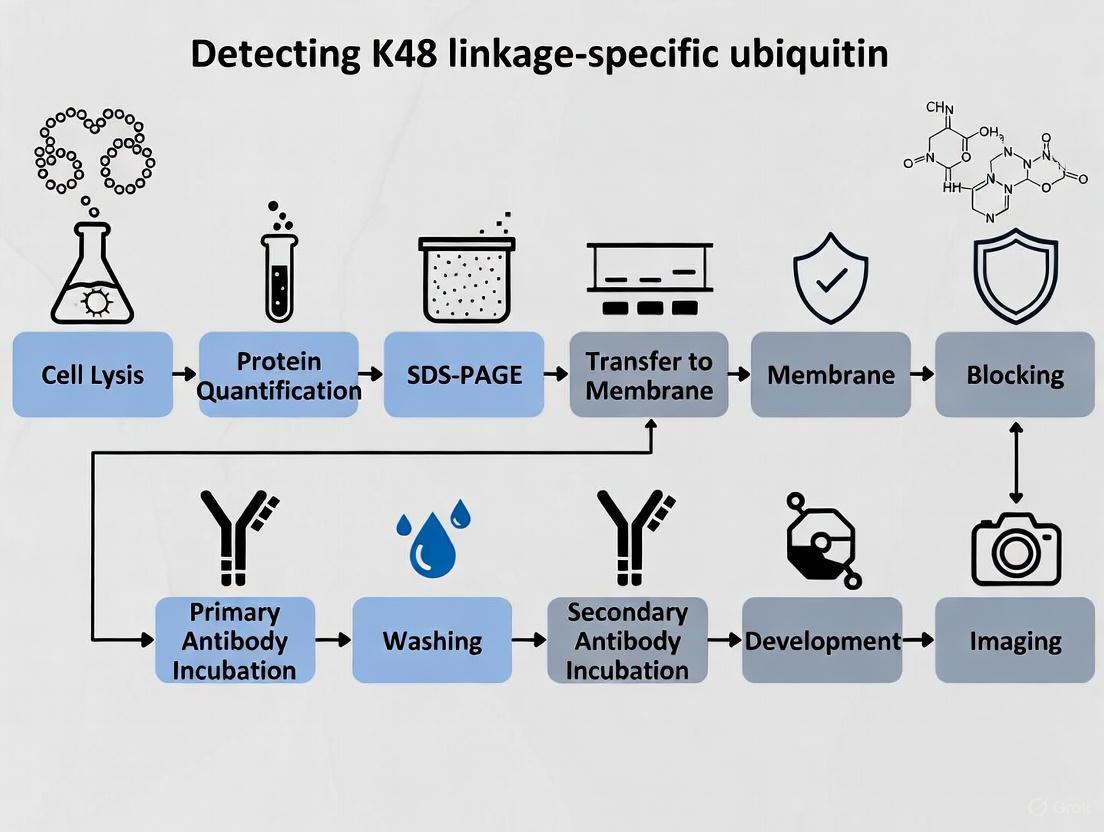
Sample Preparation: Treat cells with 5-25 µM MG-132 for 1-2 hours before harvesting to preserve ubiquitin signals. Avoid overexposure to prevent cytotoxicity [5].
Protein Separation: Separate proteins by SDS-PAGE using appropriate percentage gels based on target protein size.
Membrane Transfer: Transfer to PVDF or nitrocellulose membrane using standard protocols.
Blocking: Block membrane with 5% non-fat dry milk in TBST for 1 hour at room temperature.
Primary Antibody Incubation: Incubate with K48-linkage specific polyubiquitin antibody at 1:1000 dilution in blocking buffer overnight at 4°C [1].
Secondary Antibody Incubation: Incubate with appropriate HRP-conjugated secondary antibody for 1 hour at room temperature.
Detection: Develop using enhanced chemiluminescence substrate.
Troubleshooting: The K48-linkage specific antibody (#4289) demonstrates slight cross-reactivity with linear polyubiquitin chains but shows no cross-reactivity with monoubiquitin or polyubiquitin chains formed by linkages to different lysine residues [1]. Always include appropriate controls to distinguish specific signals.
Advanced Research Applications and Techniques
Quantitative Analysis of Ubiquitin Chain Degradation
Recent technological advances like UbiREAD (Ubiquitinated Reporter Evaluation After Intracellular Delivery) have enabled systematic comparison of intracellular degradation kinetics for different ubiquitin chains [9]. This approach has revealed that K48-Ub3 serves as a minimal cellular proteasomal targeting signal, with chains of three or more ubiquitins triggering degradation within minutes [9].
Table 3: Degradation Kinetics of Different Ubiquitin Chain Types
| Ubiquitin Chain Architecture | Degradation Rate | Deubiquitination Rate | Key Characteristics |
|---|---|---|---|
| K48-Ub3 | Fast (minutes) | Low | Minimal proteasomal signal [9] |
| K48-Ub4+ | Very Fast | Low | Conventional degradation signal [9] |
| K63-Ub3+ | Slow | High | Rapid deubiquitination [9] |
| K48/K63-Branched (K48-anchored) | Fast | Moderate | Substrate degradation [9] |
| K48/K63-Branched (K63-anchored) | Slow | High | Rapid deubiquitination [9] |
Research using UbiREAD has demonstrated that in K48/K63-branched chains, the substrate-anchored chain identity determines the degradation and deubiquitination behavior, establishing that branched chains are not simply the sum of their parts but exhibit a functional hierarchy [9]. This has profound implications for understanding how complex ubiquitin signals are decoded in cellular regulation.
Emerging Concepts: Branched Chains and Linkage Interplay
While K48-linked homotypic chains remain the classic degradation signal, recent research has illuminated the importance of branched ubiquitin chains in proteasomal targeting. The K11/K48-branched ubiquitin chains are preferentially recognized by the 26S proteasome and mediate fast-tracking of protein turnover during cell cycle progression and proteotoxic stress [3].
Ubiquitin interactome studies have identified branch-specific ubiquitin interactors, including histone ADP-ribosyltransferase PARP10/ARTD10, E3 ligase UBR4, and huntingtin-interacting protein HIP1 [6] [10]. These proteins specifically recognize the unique architecture of branched chains containing both K48 and K63 linkages, which make up approximately 20% of all K63 linkages in cells [6].
The development of sophisticated research tools has been crucial for these discoveries. Ubiquitin interactor pulldown coupled with mass spectrometry has enabled researchers to elucidate K48- and K63-linked interactomes, including novel heterotypic branch- and chain length-specific binders [6] [10]. These approaches typically utilize:
- Enzymatically synthesized ubiquitin chains of defined linkage and length
- Streptavidin resin for immobilization
- Deubiquitinase inhibitors (CAA or NEM) to preserve chain integrity
- Quantitative mass spectrometry for interactor identification [6]
Diagram 2: Ubiquitination Cascade and Functional Fate
Research Reagent Solutions
Table 4: Essential Research Reagents for K48-Ubiquitin Studies
| Reagent Category | Specific Examples | Research Application |
|---|---|---|
| K48-Linkage Specific Antibodies | Rabbit mAb D9D5 (CST #8081) [7]; CST #4289 [1] | Western blot detection of K48-linked chains |
| Ubiquitin Mutants | K48R-Ubiquitin [7]; Panel of K-to-R and K-Only mutants [4] | Linkage determination in conjugation assays |
| Proteasome Inhibitors | MG-132 [7] [5] | Preserve ubiquitinated proteins in cell lysates |
| Deubiquitinase Inhibitors | Chloroacetamide (CAA), N-Ethylmaleimide (NEM) [6] | Prevent chain disassembly during pulldowns |
| Ubiquitin Traps | ChromoTek Ubiquitin-Trap Agarose/Magnetic beads [5] | Immunoprecipitation of ubiquitinated proteins |
| Recombinant Enzymes | E1, E2 (CDC34 for K48), E3 ligases [4] | In vitro ubiquitination assays |
| Ubiquitin Linkage Kits | UbiCRest kit [6] | Linkage identification by DUB sensitivity |
These specialized research tools enable the comprehensive study of K48-linked ubiquitination, from basic detection to sophisticated mechanistic investigations. The K48-linkage specific antibodies are particularly valuable as they demonstrate minimal cross-reactivity with other linkage types, allowing specific detection of the degradation signal [1]. When planning experiments, researchers should consider that the Ubiquitin-Trap cannot differentiate between linkage types but serves as an excellent tool for initial enrichment, with linkage specificity determined through subsequent Western blotting using linkage-specific antibodies [5].
Ubiquitin is a small regulatory protein that can be covalently attached to substrate proteins through a process called ubiquitination. When multiple ubiquitin molecules are connected, they form polyubiquitin chains with distinct biological functions determined by the specific lysine residue used for linkage. Among the seven possible lysine linkages (K6, K11, K27, K29, K33, K48, and K63), K48-linked polyubiquitin chains represent the most well-characterized and abundant type, primarily serving as a potent signal for proteasomal degradation [11] [12]. This linkage is crucial for maintaining cellular homeostasis by directing damaged, misfolded, or short-lived regulatory proteins to the 26S proteasome for destruction [11].
The development of linkage-specific ubiquitin antibodies represents a breakthrough in ubiquitin research, enabling scientists to distinguish between different ubiquitin chain architectures in biological systems. Unlike pan-ubiquitin antibodies that recognize all ubiquitinated proteins regardless of linkage type, linkage-specific antibodies like those targeting K48 linkages provide exquisite specificity for decoding the "ubiquitin code" [13]. These reagents have revealed sophisticated regulatory mechanisms such as "ubiquitin chain editing," where proteins initially modified with K63-linked chains (typically involved in signaling) are later modified with K48-linked chains to direct them for degradation, effectively attenuating signaling pathways [13].
Mechanism of K48-Linkage Specific Antibodies
Molecular Basis of Specificity
K48-linkage specific antibodies achieve their remarkable specificity through precise molecular recognition of the unique structural epitope presented by K48-linked ubiquitin chains. The foundational mechanism was elucidated in a seminal 2008 study that described the development and characterization of the first linkage-specific ubiquitin antibodies [13]. Through cocrystal structure analysis of an anti-K63 linkage Fab bound to K63-linked diubiquitin, researchers demonstrated that these antibodies recognize the unique surface topology created when two ubiquitin molecules are connected through a specific lysine residue [13].
The K48-linkage specific antibody detects polyubiquitin chains formed specifically by Lys48 residue linkage while demonstrating only slight cross-reactivity with linear polyubiquitin chains and no detectable cross-reactivity with monoubiquitin or polyubiquitin chains formed through different lysine residues (K6, K11, K27, K29, K33, or K63) [11]. This specificity is achieved because the antibody's antigen-binding site accommodates the unique isopeptide bond and adjacent surface features that are exclusive to the K48 linkage interface between two ubiquitin molecules.
Generation and Validation
K48-linkage specific antibodies are typically produced using synthetic peptides corresponding to the Lys48 branch of the human diubiquitin chain as immunogens [11]. For example, the Cell Signaling Technology K48-linkage Specific Polyubiquitin Antibody (#4289) is generated by immunizing animals with such a peptide, followed by purification through protein A and peptide affinity chromatography [11]. Similarly, the Abcam anti-Ubiquitin (linkage-specific K48) antibody [EP8589] is a recombinant rabbit monoclonal antibody (RabMAb) produced using patented hybridoma technology, ensuring high batch-to-batch consistency [14] [15].
The specificity of these antibodies is rigorously validated using comprehensive approaches. Western blot analysis against panels of recombinant ubiquitin chains with different linkage types (K6, K11, K27, K29, K33, K48, K63) confirms minimal cross-reactivity with non-K48 linkages [14]. Additional validation includes immunofluorescence, immunohistochemistry on formalin-fixed paraffin-embedded tissues, intracellular flow cytometry, and functional assays in cell culture models [14] [15] [16].
Table 1: Commercially Available K48-Linkage Specific Antibodies
| Product Name | Clone | Host Species | Clonality | Applications | Specificity Validation |
|---|---|---|---|---|---|
| K48-linkage Specific Polyubiquitin Antibody #4289 | Not specified | Rabbit | Polyclonal | WB | Slight cross-reactivity with linear chains only [11] |
| Anti-Ubiquitin (linkage-specific K48) antibody [EP8589] | EP8589 | Rabbit | Monoclonal | WB, IHC-P, ICC/IF, Flow Cyt (Intra) | Specific for K48; tested against other linkage types [14] |
| Alexa Fluor 647 Anti-Ubiquitin (linkage-specific K48) antibody [EP8589] | EP8589 | Rabbit | Monoclonal | ICC/IF | Specific staining in HeLa cells [15] |
Application Notes and Protocols
Western Blotting for K48-Linked Polyubiquitin Detection
Western blotting remains the most widely used application for K48-linkage specific antibodies, enabling detection of endogenous K48-linked ubiquitin chains in cell and tissue lysates.
Sample Preparation
Cell Lysis: Harvest cells and lyse using RIPA buffer (25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) or similar lysis buffer supplemented with fresh protease inhibitors (e.g., 1 mM PMSF) and deubiquitinase (DUB) inhibitors such as 10-20 mM N-ethylmaleimide (NEM) or 5-10 mM chloroacetamide (CAA) to prevent chain disassembly during processing [6] [17]. The choice of DUB inhibitor is critical, as NEM provides more complete chain stabilization but may have more off-target effects, while CAA is more cysteine-specific but allows partial chain disassembly [6] [17].
Protein Quantification: Determine protein concentration using Bradford, BCA, or similar assay. Prepare samples in 1× Laemmli sample buffer, ensuring equal protein loading across gels (typically 20-40 μg per lane for whole cell lysates). Denature samples at 95°C for 5-10 minutes to disrupt non-covalent ubiquitin-binding interactions that might cause smearing or high molecular weight artifacts.
Electrophoresis and Transfer
Gel Electrophoresis: Separate proteins using 4-20% gradient SDS-PAGE gels to resolve polyubiquitin chains across a broad molecular weight range (approximately 26 kDa for diubiquitin up to >150 kDa for longer chains) [14]. Run gels at constant voltage (100-150V) until the dye front reaches the bottom.
Membrane Transfer: Transfer proteins to PVDF membrane using wet or semi-dry transfer systems. PVDF is preferred over nitrocellulose for better retention of small ubiquitin polymers. Confirm complete transfer with Ponceau S staining if necessary.
Immunoblotting
Blocking: Block membranes with 5% non-fat dry milk (NFDM) in TBST (Tris-buffered saline with 0.1% Tween-20) for 1 hour at room temperature with gentle agitation [14]. Alternative blocking buffers such as 3-5% BSA in TBST may also be used.
Primary Antibody Incubation: Incubate membrane with K48-linkage specific antibody diluted in blocking buffer. Optimal dilution varies by product:
- Cell Signaling #4289: 1:1000 dilution in 5% BSA/TBST [11]
- Abcam EP8589: 1:1000 to 1:2000 dilution in 5% NFDM/TBST [14] Incubate overnight at 4°C with gentle agitation.
Washing and Secondary Antibody: Wash membrane 3× for 5-10 minutes each with TBST. Incubate with appropriate HRP-conjugated secondary antibody (e.g., goat anti-rabbit IgG) diluted 1:2000 to 1:10000 in blocking buffer for 1 hour at room temperature [14]. Wash again 3× with TBST.
Detection: Develop blots using enhanced chemiluminescence (ECL) substrate according to manufacturer's instructions. Expose to X-ray film or capture using a digital imaging system. Multiple exposure times may be necessary to visualize both strong and weak signals.
Figure 1: Western Blot Workflow for K48-Linked Ubiquitin Detection
Immunofluorescence and Immunohistochemistry Protocols
K48-linkage specific antibodies can be used for spatial localization of K48-linked ubiquitin chains within cells and tissues, providing insights into subcellular compartmentalization of protein degradation signals.
Immunofluorescence (ICC/IF)
Cell Culture and Fixation: Plate cells on glass coverslips and culture until desired confluence. Fix cells with either:
- 4% formaldehyde for 10 minutes at room temperature [15], or
- 100% methanol for 5 minutes at -20°C [15]
Permeabilization and Blocking: Permeabilize fixed cells with 0.1% Triton X-100 in PBS for 5-10 minutes [15]. Block non-specific binding with 1% BSA/10% normal goat serum/0.3M glycine in 0.1% PBS-Tween for 1 hour at room temperature.
Antibody Staining: Incubate with K48-linkage specific antibody diluted in blocking buffer:
- Abcam EP8589 (unconjugated): 1:100 to 1:500 dilution [14] [15]
- Fluorescently conjugated variants: 1:100 dilution [15] Incubate overnight at 4°C or for 1-2 hours at room temperature.
Detection and Mounting: Wash 3× with PBS, then incubate with appropriate fluorescent secondary antibody (if using unconjugated primary) diluted 1:1000 in blocking buffer for 1 hour at room temperature protected from light [14]. Counterstain nuclei with DAPI (1.43 μM) for 5 minutes [14]. Mount coverslips using antifade mounting medium.
Imaging: Image using confocal or fluorescence microscope with appropriate filter sets. Include controls without primary antibody to assess non-specific secondary antibody binding.
Immunohistochemistry (IHC)
Tissue Preparation: Use formalin-fixed, paraffin-embedded tissue sections (4-5 μm thickness) mounted on charged slides. Bake slides at 60°C for 30 minutes to ensure adhesion.
Deparaffinization and Antigen Retrieval: Deparaffinize sections in xylene and rehydrate through graded ethanol series to water. Perform heat-mediated antigen retrieval using citrate buffer (pH 6.0) or EDTA buffer (pH 8.0) at 95-100°C for 20-30 minutes [14] [16]. Cool slides for 20-30 minutes before proceeding.
Immunostaining: Quench endogenous peroxidase activity with 3% H₂O₂ for 10 minutes. Block with 10% normal serum for 1 hour. Incubate with K48-linkage specific antibody (e.g., 1 μg/ml for EP8589 clone) for 16 minutes at 37°C or overnight at 4°C [14]. Detect using appropriate HRP-based detection system (e.g., OptiView DAB IHC Detection Kit) according to manufacturer's instructions [14]. Counterstain with hematoxylin, dehydrate, clear, and mount.
Table 2: Optimal Conditions for K48-Linked Ubiquitin Detection Across Applications
| Application | Recommended Fixation | Antigen Retrieval | Antibody Dilution | Incubation Conditions |
|---|---|---|---|---|
| Western Blot | Denaturing lysis buffer | Not applicable | 1:1000 - 1:2000 | Overnight, 4°C [11] [14] |
| Immunofluorescence | 4% formaldehyde or 100% methanol | 0.1% Triton X-100 permeabilization | 1:100 - 1:500 | Overnight, 4°C [14] [15] |
| Immunohistochemistry | Formalin-fixed, paraffin-embedded | Heat-mediated, EDTA buffer pH 8.5 | 1 μg/ml | 16 minutes, 37°C [14] |
| Flow Cytometry (Intracellular) | 80% methanol | 0.1% PBS-Tween | 1:100 | 30 minutes, 22°C [14] [16] |
Troubleshooting and Optimization
High Background Signal: Increase blocking time or try alternative blocking agents (BSA, serum, commercial blocking reagents). Optimize antibody concentration and increase wash stringency (increase salt concentration to 150-500 mM NaCl or add 0.1% Tween-20 to wash buffers).
Weak or No Signal: Confirm antigen preservation by testing different fixation methods. Optimize antigen retrieval conditions (pH, time, temperature). Increase primary antibody concentration or incubation time. Include positive control samples known to contain K48-linked ubiquitin chains.
Non-Specific Bands (Western Blot): Ensure complete denaturation of samples (verify by re-heating samples and adding fresh DTT). Test antibody specificity using recombinant ubiquitin chains of different linkages [14]. Include negative controls using lysates from cells treated with proteasome inhibitors (e.g., MG132) which should accumulate K48-linked chains.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Reagents for K48-Linked Ubiquitin Research
| Reagent Category | Specific Examples | Function/Application | Usage Notes |
|---|---|---|---|
| K48-Specific Antibodies | CST #4289; Abcam EP8589 clone | Detection of K48-linked ubiquitin chains | Validate specificity with linkage panels; clone EP8589 has extensive validation data [11] [14] |
| DUB Inhibitors | N-ethylmaleimide (NEM); Chloroacetamide (CAA) | Prevent ubiquitin chain disassembly during processing | NEM more potent but may have off-target effects; CAA more specific but allows partial digestion [6] [17] |
| Proteasome Inhibitors | MG132; Bortezomib; Lactacystin | Accumulate K48-linked ubiquitinated proteins | Use as positive control; treat cells 4-6 hours before harvesting [11] |
| Recombinant Ubiquitin Chains | K48-Ub2-7; Linkage-specific ubiquitin chains | Antibody validation controls | Essential for confirming specificity; available from various suppliers [14] |
| Detection Systems | HRP-conjugated secondaries; ECL substrates; Fluorescent secondaries | Signal detection and visualization | Choose based on application sensitivity requirements [14] |
K48-linkage specific antibodies represent powerful tools for deciphering the ubiquitin code, particularly for understanding processes related to targeted protein degradation. Their mechanism of action relies on exquisite molecular recognition of the unique structural epitope formed when ubiquitin molecules are linked through K48 residues. When applied using optimized protocols with appropriate controls and validation, these antibodies enable researchers to detect, quantify, and localize K48-linked ubiquitin chains across multiple experimental platforms from western blotting to immunohistochemistry. As research continues to reveal the complexity of ubiquitin chain architectures—including homotypic, mixed linkage, and branched chains—the precise specificity of these reagents becomes increasingly valuable for understanding cellular regulation and developing therapeutic interventions targeting the ubiquitin-proteasome system.
Ubiquitin is a small regulatory protein that can be covalently attached to target proteins through a process called ubiquitination. When multiple ubiquitin molecules form chains through specific lysine residues, they create polyubiquitin chains that determine the fate of the modified protein [18]. Among the eight possible ubiquitin chain linkages (K6, K11, K27, K29, K33, K48, K63, and Met1), K48-linked polyubiquitin chains primarily target proteins for degradation by the 26S proteasome, playing a fundamental role in maintaining cellular protein homeostasis [18] [19]. This proteasomal degradation pathway is essential for critical cellular processes including cell cycle regulation, stress response, and apoptosis [18]. The specific detection and analysis of K48-linked ubiquitin chains therefore provides crucial insights into protein turnover regulation and has significant implications for understanding disease mechanisms, particularly in cancer and neurodegenerative disorders [19].
The development of linkage-specific ubiquitin antibodies has revolutionized the study of ubiquitin signaling by enabling researchers to distinguish between different ubiquitin chain architectures without resorting to complex mass spectrometry techniques. This application note focuses on commercially available K48-linkage specific antibodies, their validation, and implementation in various experimental workflows, with particular emphasis on immunoblotting protocols relevant to pharmaceutical and basic research applications.
Commercial K48-Linkage Specific Antibodies
The market offers several well-characterized K48-linkage specific antibodies suitable for diverse research applications. The table below summarizes two prominent commercial options and their key specifications:
Table 1: Commercial K48-Linkage Specific Antibodies
| Product Name | Clone | Host Species | Clonality | Applications | Specificity Notes |
|---|---|---|---|---|---|
| K48-linkage Specific Polyubiquitin Antibody #4289 [18] | Not specified | Rabbit | Polyclonal | Western Blot | Detects polyubiquitin chains formed by Lys48 linkage; slight cross-reactivity with linear polyubiquitin chain; no cross-reactivity with monoubiquitin or other lysine-linked chains |
| Anti-Ubiquitin (linkage-specific K48) [EP8589] [14] | EP8589 | Rabbit | Monoclonal (Recombinant) | WB, IHC-P, ICC/IF, Flow Cytometry | Recognizes polyubiquitin chains formed by Lys-48 (K48) residue linkage; validated across multiple species |
Both antibodies demonstrate excellent specificity for K48-linked ubiquitin chains over other linkage types. The Cell Signaling Technology antibody (#4289) is a polyclonal preparation generated using a synthetic peptide corresponding to the Lys48 branch of the human diubiquitin chain, while the Abcam antibody (EP8589) represents a recombinant monoclonal platform offering high batch-to-batch consistency [18] [14] [19].
Recommended Working Dilutions
For Western blot applications, the recommended dilutions are:
- Cell Signaling Technology #4289: 1:1000 dilution [18]
- Abcam EP8589: 1:1000 to 1:2000 dilution (depending on sample type and preparation) [14]
Proper validation of linkage specificity is crucial for accurate data interpretation. The EP8589 antibody has been extensively validated against various linkage types, showing no cross-reactivity with K6-, K11-, K27-, K29-, K33-, K63-linked diubiquitin or monoubiquitin in Western blot assays [14].
The Ubiquitin-Proteasome Pathway: Mechanism of K48-Linked Protein Degradation
The ubiquitin-proteasome system represents the primary pathway for targeted protein degradation in eukaryotic cells. The process involves a cascade of enzymatic reactions that ultimately lead to the attachment of a K48-linked polyubiquitin chain to target proteins, marking them for destruction. Understanding this pathway is essential for contextualizing antibody-based detection methods.
Diagram 1: The K48 Ubiquitin-Proteasome Degradation Pathway. This diagram illustrates the sequential enzymatic cascade where E1, E2, and E3 enzymes mediate the attachment of a K48-linked polyubiquitin chain to a target protein, leading to its recognition and degradation by the 26S proteasome.
The 26S proteasome recognizes K48-linked ubiquitin chains through specialized ubiquitin receptors located within its 19S regulatory particle, including RPN1, RPN10, and RPN13 [3]. Recent structural studies using cryo-EM have revealed that the proteasome can also recognize more complex ubiquitin architectures, such as K11/K48-branched ubiquitin chains, through multivalent binding interfaces involving RPN2 in addition to the canonical receptors [3]. These branched chains appear to function as a priority degradation signal under specific cellular conditions, including cell cycle progression and proteotoxic stress [3].
Experimental Protocols for K48 Linkage Detection
Western Blot Protocol for K48-Linked Polyubiquitin Detection
The following protocol provides a standardized method for detecting K48-linked polyubiquitin chains using linkage-specific antibodies:
Table 2: Key Reagents for K48 Linkage Detection by Western Blot
| Reagent | Function | Specifications |
|---|---|---|
| K48-linkage Specific Antibody | Primary detection | Rabbit polyclonal (#4289) or monoclonal (EP8589) |
| HRP-conjugated Secondary Antibody | Signal generation | Anti-rabbit IgG, suitable for Western blot |
| Cell Lysis Buffer | Protein extraction | RIPA buffer with protease inhibitors and N-ethylmaleimide |
| Gel Electrophoresis System | Protein separation | Standard SDS-PAGE setup (8-16% gradient recommended) |
| Transfer System | Protein transfer | PVDF or nitrocellulose membrane |
| Blocking Solution | Reduce background | 5% non-fat dry milk or BSA in TBST |
| Chemiluminescent Substrate | Signal detection | HRP-compatible substrate for enhanced sensitivity |
Procedure:
Sample Preparation:
- Lyse cells or tissues in RIPA buffer supplemented with protease inhibitors and 10-20mM N-ethylmaleimide (to inhibit deubiquitinating enzymes).
- Quantify protein concentration and prepare samples with 2X Laemmli buffer.
- Denature samples at 95°C for 5 minutes.
Gel Electrophoresis:
- Load 20-50μg of total protein per lane on an 8-16% gradient SDS-PAGE gel.
- Run gel at constant voltage (100-120V) until the dye front reaches the bottom.
Protein Transfer:
- Transfer proteins to PVDF membrane using wet or semi-dry transfer systems.
- Confirm transfer with Ponceau S staining if necessary.
Immunoblotting:
- Block membrane with 5% non-fat dry milk in TBST for 1 hour at room temperature.
- Incubate with primary antibody (diluted 1:1000 in blocking buffer) overnight at 4°C with gentle agitation.
- Wash membrane 3×10 minutes with TBST.
- Incubate with HRP-conjugated secondary antibody (1:2000-1:10000 dilution) for 1 hour at room temperature.
- Wash membrane 3×10 minutes with TBST.
Detection:
- Develop blot with enhanced chemiluminescent substrate.
- Image using a digital imaging system capable of detecting linear signal range.
Troubleshooting Notes:
- For heavily ubiquitinated samples, high molecular weight smearing is expected rather than discrete bands.
- Always include a positive control (e.g., cells treated with proteasome inhibitor MG132) to enhance K48-ubiquitinated protein detection.
- To confirm specificity, pre-incubate antibody with K48-linked diubiquitin antigen (where available) for competition experiments.
Determining Ubiquitin Chain Linkage Using Biochemical Methods
While linkage-specific antibodies provide a convenient detection method, complementary biochemical approaches can confirm ubiquitin chain linkage. The following protocol utilizes ubiquitin mutants to determine chain linkage specificity:
Diagram 2: Experimental Workflow for Ubiquitin Linkage Determination. This workflow outlines the key steps in determining ubiquitin chain linkage using ubiquitin mutants in conjunction with Western blot analysis.
Materials and Reagents:
- E1 Enzyme (5μM stock)
- E2 Enzyme (25μM stock) - choice depends on E3 compatibility
- E3 Ligase (10μM stock) - specific to your system
- 10X E3 Ligase Reaction Buffer (500mM HEPES pH 8.0, 500mM NaCl, 10mM TCEP)
- Wild-type Ubiquitin (1.17mM, 10mg/mL)
- Ubiquitin K-to-R Mutants (each lysine mutated to arginine)
- Ubiquitin K-Only Mutants (only one lysine available)
- MgATP Solution (100mM)
Procedure:
Reaction Setup (25μL volume):
- Set up nine separate reactions containing:
- Reaction 1: Wild-type ubiquitin
- Reactions 2-8: Seven different ubiquitin K-to-R mutants (K6R, K11R, K27R, K29R, K33R, K48R, K63R)
- Negative control: Replace MgATP with dH₂O
- Add to each tube:
- 2.5μL 10X E3 Ligase Reaction Buffer
- 1μL ubiquitin or ubiquitin mutant (~100μM final)
- 2.5μL MgATP Solution (10mM final)
- Substrate protein (5-10μM final)
- 0.5μL E1 Enzyme (100nM final)
- 1μL E2 Enzyme (1μM final)
- XμL E3 Ligase (1μM final)
- dH₂O to 25μL total volume
- Set up nine separate reactions containing:
Incubation:
- Incubate reactions in a 37°C water bath for 30-60 minutes
Reaction Termination:
- For direct Western analysis: Add 25μL 2X SDS-PAGE sample buffer
- For downstream applications: Add 0.5μL 500mM EDTA (20mM final) or 1μL 1M DTT (100mM final)
Analysis:
- Analyze reactions by Western blot using anti-ubiquitin antibody
- Interpretation: A K-to-R mutant that fails to form polyubiquitin chains indicates the essential lysine for linkage
- Confirm with K-Only mutants where only the wild-type and the specific K-Only mutant should form chains
This biochemical approach provides orthogonal validation for antibody-based linkage detection and is particularly valuable when characterizing novel E3 ligases or detecting mixed linkage chains [4].
Advanced Research Applications and Methodologies
Novel Protein Ubiquitination Strategies
Recent methodological advances have enabled more precise study of K48-linked ubiquitination. The SpyTag/SpyCatcher system represents an innovative approach for generating homogenously ubiquitinated proteins that bypasses the promiscuity of enzymatic methods [20]. This system combines chemical synthesis and protein expression to create defined ubiquitin conjugates, allowing researchers to investigate how ubiquitin chain length affects proteasomal degradation [20].
Using this methodology, researchers have demonstrated that while the 26S proteasome primarily trims ubiquitin chains from conjugated substrates, the 20S proteasome can degrade both the substrate and the attached ubiquitin tag, revealing unexpected flexibility in proteasomal processing [20]. These findings challenge traditional models of ubiquitin-proteasome system function and highlight the importance of using well-defined ubiquitinated substrates.
Structural Insights into K48 Chain Recognition
Recent cryo-EM studies of human 26S proteasome in complex with K11/K48-branched ubiquitin chains have revealed novel aspects of substrate recognition [3]. These structures demonstrate a multivalent substrate recognition mechanism involving previously unknown ubiquitin binding sites on RPN2 in addition to the canonical receptors RPN10 and RPN13 [3]. This structural information provides molecular-level explanation for the preferential recognition of certain ubiquitin chain architectures and represents a significant advance in understanding the specificity of ubiquitin-mediated proteasomal degradation.
K48-linkage specific antibodies are indispensable tools for studying the ubiquitin-proteasome system, enabling researchers to specifically detect proteins targeted for proteasomal degradation. When used according to standardized protocols and in conjunction with biochemical validation methods, these reagents provide robust and reproducible results that advance our understanding of protein turnover regulation. The continuing development of novel ubiquitination methods and structural insights into proteasomal recognition mechanisms will further enhance the utility of these antibodies in both basic research and drug discovery contexts.
Immunoblotting Protocol for K48 Linkage-Specific Ubiquitin Antibody Research
Ubiquitination is a crucial post-translational modification wherein a small 8-kDa protein, ubiquitin, is covalently attached to target proteins. This process involves a sequential enzymatic cascade comprising ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3), which collectively confer substrate specificity [21]. Ubiquitin itself contains seven lysine residues (K6, K11, K27, K29, K33, K48, and K63) that can be utilized to form polyubiquitin chains, with the linkage type determining the functional consequence for the modified protein [22] [23]. Among these, K48-linked polyubiquitin chains primarily target proteins for proteasomal degradation, representing one of the most well-characterized ubiquitin signaling pathways [22] [23] [21]. In contrast, K63-linked chains typically regulate non-proteolytic functions including protein trafficking, DNA repair, and signal transduction [23].
The critical role of K48-linked ubiquitination extends to numerous cellular processes, with particular importance in DNA damage response pathways. Following DNA double-strand breaks, K48-linked polyubiquitin chains accumulate at damage sites where they facilitate the proteasomal degradation of barrier proteins such as JMJD2A, JMJD2B, and L3MBTL1, which otherwise compete with the DNA damage mediator 53BP1 for binding to methylated histone H4K20 [23]. This degradation is orchestrated by the E3 ubiquitin ligases RNF8 and RNF168, which promote K48-linked ubiquitination in a manner that enables 53BP1 focus formation and subsequent repair activation [23]. The ability to accurately detect and quantify these specific ubiquitin chains using linkage-specific antibodies is therefore fundamental to advancing our understanding of cellular stress response mechanisms, protein turnover regulation, and the development of targeted therapeutic interventions.
K48 Linkage-Specific Antibody Characterization
The specificity validation of K48 linkage-specific antibodies is paramount for generating reliable immunoblotting data. These antibodies are designed to distinguish K48-linked polyubiquitin chains from other linkage types, including K6, K11, K27, K29, K33, K63, and linear ubiquitin chains. The table below summarizes the key characteristics of two commercially available K48 linkage-specific antibodies:
Table 1: Characterization of K48 Linkage-Specific Antibodies
| Product Name | Clone/Code | Reactivity | Applications | Specificity Profile | Recommended Dilution |
|---|---|---|---|---|---|
| K48-linkage Specific Polyubiquitin Antibody [22] | #4289 | All Species Expected | Western Blot (1:1000) | • Detects K48-linked polyubiquitin chains.• Slight cross-reactivity with linear polyubiquitin.• No cross-reactivity with monoubiquitin or other lysine-linked chains. | 1:1000 (Western Blot) |
| Anti-Ubiquitin (linkage-specific K48) antibody [14] | EP8589 (ab140601) | Human, Mouse, Rat | WB, IHC-P, ICC/IF, Flow Cytometry (Intra) | • Specific for K48 linkage.• Verified with linkage-specific recombinant proteins. | 1:1000 (Western Blot, purified antibody)1/100 - 1/200 (Other applications) |
The Cell Signaling Technology antibody #4289 is a rabbit polyclonal antibody produced using a synthetic peptide corresponding to the Lys48 branch of human diubiquitin chain, with purification via protein A and peptide affinity chromatography [22]. The Abcam antibody EP8589 (ab140601) is a recombinant rabbit monoclonal antibody (RabMAb) that demonstrates specificity across multiple applications including Western Blot, immunohistochemistry, immunocytochemistry, and flow cytometry [14]. Specificity validation for this antibody includes Western blot analysis against a panel of linkage-specific ubiquitin dimers (K6, K11, K27, K29, K33, K48, K63), confirming selective detection of only the K48-linked form [14]. Both antibodies detect endogenous proteins without cross-reactivity to monoubiquitin, providing reliable tools for studying endogenous K48-linked ubiquitination.
Quantitative Western Blot Methodology
Transitioning from qualitative to quantitative Western blotting requires meticulous optimization to ensure data accuracy and reproducibility. The process demands careful attention to linear range detection, appropriate normalization strategies, and stringent protocol standardization.
Normalization Strategies for Accurate Quantification
Normalization is essential for correcting technical variations during sample preparation, electrophoresis, and transfer. The most common approaches include:
Housekeeping Protein (HKP) Normalization: This method utilizes constitutively expressed proteins such as β-actin, GAPDH, or α-tubulin as loading controls. However, HKPs can become saturated at common loading amounts (30-50 μg), resulting in non-linear responses and inaccurate normalization [24]. Each HKP must be validated to ensure consistent expression across experimental conditions.
Total Protein Normalization (TPN): This growingly popular approach normalizes target signal to the total protein loaded in each lane, effectively addressing limitations of HKP normalization. Fluorescent total protein stains (e.g., No-Stain Protein Labeling Reagent) provide superior linearity across a wide dynamic range of protein loads (R² = 0.9990 compared to R² = 0.8332-0.9438 for HKPs) [24]. TPN is particularly valuable when experimental treatments affect traditional housekeeping protein expression.
Table 2: Optimization Parameters for Quantitative Western Blotting
| Parameter | Common Pitfalls | Optimization Strategies | Impact on Quantification |
|---|---|---|---|
| Protein Loading | • Overloading high-abundance targets• Saturation of signal | • Load 1-10 μg for high-abundance proteins• 10-40 μg for low-abundance targets• Use precise protein assays (e.g., BCA) | Prevents signal saturation; maintains linear relationship between load and intensity [24] |
| Antibody Dilution | • Too concentrated: saturation, high background• Too dilute: poor sensitivity | • Titrate both primary and secondary antibodies• Test combinations (e.g., 1:500-1:5000 primary, 1:50,000-1:250,000 secondary) | Maximizes linear signal range; reduces background [24] |
| Detection Substrate | • Ultrasensitive substrates cause saturation• Standard ECL lacks sensitivity | • Use extended duration substrates (e.g., SuperSignal West Dura) for quantitative applications• Match substrate sensitivity to target abundance | Ensures wide dynamic range, linear response, and long signal half-life [24] |
Systematic Workflow for Quantitative Analysis
A systematic approach to Western blotting incorporates critical validation steps to minimize errors and variability [25]. The workflow begins with protein extraction using appropriate lysis buffers compatible with ubiquitination studies (typically containing protease and deubiquitinase inhibitors). Following protein quantification using sensitive assays (e.g., Qubit Protein BR Assay or Pierce Rapid Gold BCA Protein Assay), samples should be prepared in loading buffer with minimal heating to preserve ubiquitin chains.
During electrophoresis and transfer, optimization ensures complete transfer of high molecular weight polyubiquitinated species. For immunodetection, antibody concentrations must be titrated to establish the combined linear range where both the target and normalization signals respond linearly to protein load. This can be achieved by running a dilution series of lysates and probing with both the K48-linkage specific antibody and the normalization control. Finally, image acquisition should utilize calibrated imaging systems that avoid pixel saturation, with subsequent densitometric analysis employing validated software algorithms.
Experimental Protocols
Protocol: Determining Ubiquitin Chain Linkage Using Mutant Ubiquitin Panel
This protocol utilizes ubiquitin mutants to definitively determine the linkage type of polyubiquitin chains formed in vitro or detected in cellular systems [4].
Table 3: Research Reagent Solutions for Ubiquitin Linkage Determination
| Reagent | Function/Purpose | Stock Concentration | Working Concentration |
|---|---|---|---|
| E1 Enzyme | Ubiquitin-activating enzyme; initiates ubiquitination cascade | 5 µM | 100 nM |
| E2 Enzyme | Ubiquitin-conjugating enzyme; works with E3 to specify chain topology | 25 µM | 1 µM |
| E3 Ligase | Ubiquitin ligase; confers substrate specificity | 10 µM | 1 µM |
| Wild-type Ubiquitin | Positive control for ubiquitination reactions | 1.17 mM (10 mg/mL) | ~100 µM |
| Ubiquitin K to R Mutants | Identify required lysine; chain formation blocked in specific mutant | 1.17 mM (10 mg/mL) | ~100 µM |
| Ubiquitin K Only Mutants | Verify linkage specificity; chains form only with correct mutant | 1.17 mM (10 mg/mL) | ~100 µM |
| 10X E3 Ligase Reaction Buffer | Provides optimal reaction conditions | 10X (500 mM HEPES, pH 8.0, 500 mM NaCl, 10 mM TCEP) | 1X |
| MgATP Solution | Energy source for ubiquitination cascade | 100 mM | 10 mM |
Procedure:
Reaction Setup for K-to-R Mutants: Set up nine parallel 25 µL reactions containing:
- 2.5 µL 10X E3 Ligase Reaction Buffer
- 1 µL ubiquitin (wild-type or individual K-to-R mutants: K6R, K11R, K27R, K29R, K33R, K48R, K63R)
- 2.5 µL MgATP Solution
- Substrate (5-10 µM final)
- 0.5 µL E1 Enzyme (100 nM final)
- 1 µL E2 Enzyme (1 µM final)
- E3 Ligase (1 µM final)
- dH₂O to 25 µL Include a negative control replacing MgATP with dH₂O [4].
Incubation: Incubate reactions at 37°C for 30-60 minutes.
Reaction Termination:
- For direct analysis: Add 25 µL 2X SDS-PAGE sample buffer
- For downstream applications: Add 0.5 µL 500 mM EDTA (20 mM final) or 1 µL 1 M DTT (100 mM final) [4]
Analysis: Separate proteins by SDS-PAGE, transfer to membrane, and perform Western blot with anti-ubiquitin antibody. Lack of chain formation in a specific K-to-R mutant indicates requirement of that lysine for linkage.
Verification with K-Only Mutants: Repeat with Ubiquitin K-Only mutants (K6, K11, K27, K29, K33, K48, K63 Only). Chain formation should occur only with wild-type ubiquitin and the specific K-Only mutant corresponding to the linkage type [4].
Diagram 1: K48 ubiquitination cascade
Protocol: Quantitative Western Blot for K48-Linked Polyubiquitin Detection
Solutions and Reagents:
- RIPA lysis buffer supplemented with protease inhibitors and 20 mM N-ethylmaleimide (NEM) to inhibit deubiquitinases
- Precast gels (4-12% Bis-Tris) for optimal separation of polyubiquitinated proteins
- K48 linkage-specific primary antibody (e.g., #4289 or ab140601)
- HRP-conjugated secondary antibody
- Enhanced chemiluminescent substrate with extended dynamic range (e.g., SuperSignal West Dura)
- Total protein stain (e.g., No-Stain Protein Labeling Reagent) for normalization
Procedure:
Sample Preparation:
- Lyse cells in RIPA buffer with inhibitors
- Quantify protein concentration using compatible assay (e.g., BCA assay)
- Prepare samples in loading buffer with minimal heating (65°C for 10 minutes instead of 95°C to preserve ubiquitin chains)
- Load appropriate amount (1-20 μg based on target abundance) alongside pre-stained molecular weight markers
Electrophoresis and Transfer:
- Run samples at constant voltage until adequate separation
- Transfer to PVDF or nitrocellulose membrane using standardized transfer system
- For total protein normalization: stain membrane with fluorescent total protein label according to manufacturer's instructions and image prior to immunodetection
Immunodetection:
- Block membrane with 5% non-fat dry milk or BSA in TBST for 1 hour
- Incubate with K48 linkage-specific primary antibody at optimized dilution (typically 1:1000) overnight at 4°C
- Wash membrane 3×10 minutes with TBST
- Incubate with HRP-conjugated secondary antibody at appropriate dilution (typically 1:50,000-1:250,000) for 1 hour at room temperature
- Wash membrane 3×10 minutes with TBST
Detection and Analysis:
- Incubate with chemiluminescent substrate and image using digital imaging system with multiple exposure times
- Ensure no pixel saturation in bands of interest
- Quantify band intensity using densitometry software
- Normalize K48-linked ubiquitin signal to total protein or validated housekeeping protein
Diagram 2: Quantitative Western blot workflow
Troubleshooting and Technical Considerations
Common Challenges in K48-Linked Ubiquitin Detection
Smearing or High Background: This may indicate overloading of protein samples, insufficient washing, or non-specific antibody binding. Reduce protein load, optimize blocking conditions (consider different blocking agents), and titrate antibody concentrations.
Lack of Signal: Potential causes include inefficient transfer, antibody degradation, or insufficient ubiquitination. Verify transfer efficiency with Ponceau S or total protein stain, check antibody functionality with positive control lysates (e.g., MG132-treated cells), and ensure proper inhibition of deubiquitinases during sample preparation.
Unexpected Banding Patterns: K48-linked polyubiquitin typically appears as a high molecular weight smear with discrete bands corresponding to multi-ubiquitinated species. Discrete bands at lower molecular weights may indicate monoubiquitination or non-specific binding. Include linkage-specific controls when possible.
Validation of Specificity
For critical applications, confirm K48 linkage specificity through:
- siRNA knockdown of specific E3 ligases known to generate K48 linkages
- Proteasome inhibitor treatment (MG132, bortezomib) to accumulate K48-linked ubiquitinated proteins
- Linkage competition assays with recombinant K48-linked ubiquitin chains
- Validation with independent methods such as mass spectrometry or linkage-specific immunoprecipitation
The protocols and methodologies outlined herein provide a robust framework for investigating K48-linked ubiquitination using linkage-specific antibodies, enabling researchers to generate quantitative, reproducible data that advances our understanding of this critical regulatory pathway in cellular homeostasis and disease pathogenesis.
Optimized Immunoblotting Protocol for K48-Linked Ubiquitin Detection
In the analysis of protein ubiquitylation, particularly when employing K48-linkage-specific ubiquitin antibodies for immunoblotting, the preservation of the native ubiquitin landscape is paramount. K48-linked polyubiquitin chains primarily target proteins for degradation by the 26S proteasome, making their accurate detection crucial for understanding protein regulation, cell cycle control, and apoptosis [26]. However, the inherent activity of deubiquitinating enzymes (DUBs) during sample preparation can rapidly dismantle these chains, leading to significant underestimation of ubiquitylation levels and erroneous biological conclusions [27]. This application note details a robust and critical sample preparation protocol incorporating N-ethylmaleimide (NEM) and iodoacetamide (IAA), two cysteine-directed DUB inhibitors, to ensure the reliable capture and detection of K48-linked ubiquitin signals.
The Critical Role of DUB Inhibition in Ubiquitin Immunoblotting
Deubiquitinating enzymes represent a large family of proteases that catalyze the removal of ubiquitin from modified proteins. During cell lysis, the compartmentalization of DUBs is lost, and the changing chemical environment can trigger their activity, leading to the rapid degradation of labile ubiquitin chains before they can be analyzed [27]. This is especially problematic for signaling pathways regulated by dynamic ubiquitylation, such as those involving NF-κB, p53, and cell cycle regulators.
The use of linkage-specific antibodies, such as those targeting K48-linked chains, demands particularly high integrity of the ubiquitin chains, as these antibodies often recognize a specific conformational epitope present only in the assembled chain [26] [14]. Even partial cleavage of a K48 chain by a nonspecific DUB can destroy the antigenic site, rendering the modification undetectable by western blot. Therefore, the inclusion of DUB inhibitors like NEM and IAA in the lysis buffer is not an optional optimization but a fundamental requirement for generating quantitatively accurate and biologically relevant data on K48-linked ubiquitylation.
Mechanism of Action: NEM and IAA
Both NEM and IAA function as covalent cysteine protease inhibitors. They permanently inactivate the vast majority of DUBs, which belong to the cysteine protease family, by modifying the catalytic cysteine residue in their active site.
- N-Ethylmaleimide (NEM): This compound acts as an alkylating agent. Its maleimide group reacts rapidly with the thiol group (-SH) of the catalytic cysteine, forming a stable thioether bond that blocks the nucleophilic attack required for cleaving the isopeptide bond in ubiquitin chains [27].
- Iodoacetamide (IAA): Similarly, IAA is a haloacetamide-derived alkylating agent. It iodinates the cysteine thiol group, irreversibly inhibiting enzyme activity [27].
The sequential or combined use of these inhibitors ensures broad-spectrum and sustained inhibition of DUB activity throughout the sample preparation process, from the moment of cell disruption until the proteins are denatured by heating in SDS-PAGE sample buffer.
Comprehensive Reagent Preparation
Lysis Buffer with DUB Inhibitors
A well-formulated lysis buffer is the foundation for preserving ubiquitin modifications. The following table summarizes the components of a recommended lysis buffer, adapted from established protocols for studying the ubiquitin-proteasome system [28] [27].
Table 1: Composition of Lysis Buffer with DUB Inhibitors
| Component | Final Concentration | Function and Notes |
|---|---|---|
| Tris-HCl (pH 7.4-7.5) | 50 mM | Maintains physiological pH for protein stability. |
| Sucrose | 250 mM | Provides osmotic support to stabilize organelles. |
| Sodium Chloride (NaCl) | 100-200 mM | Controls ionic strength; can be adjusted to reduce non-specific binding. |
| MgCl₂ | 5 mM | Essential cofactor for some ATP-dependent processes. |
| ATP | 1 mM | Helps maintain the activity of ubiquitin-system enzymes during initial lysis. |
| Dithiothreitol (DTT) | Omit or add post-lysis | A reducing agent that MUST BE OMITTED from the initial lysis buffer as it will inactivate NEM and IAA. |
| N-Ethylmaleimide (NEM) | 5-25 mM | Broad-spectrum, irreversible cysteine protease/DUB inhibitor. Prepare fresh. |
| Iodoacetamide (IAA) | 5-20 mM | Broad-spectrum, irreversible cysteine protease/DUB inhibitor. Prepare fresh. |
| Protease Inhibitor Cocktail | 1X | Inhibits serine, aspartic, and metallo-proteases to prevent general protein degradation. |
Preparation Notes:
- A 500 mM stock solution of NEM should be prepared fresh in ethanol or isopropanol immediately before use, as it is susceptible to hydrolysis in aqueous solutions.
- A 500 mM stock solution of IAA should be prepared fresh in deionized water. IAA is light-sensitive, so the tube should be wrapped in foil.
- It is critical to omit DTT or β-mercaptoethanol from the lysis buffer until after the DUB inhibition step is complete, as these reducing agents will compete with and neutralize NEM and IAA.
The Scientist's Toolkit: Key Research Reagents
The following table lists essential reagents and materials required for the successful preparation and analysis of K48-linked ubiquitin conjugates.
Table 2: Essential Research Reagents for Ubiquitin Sample Preparation and Analysis
| Item | Function / Application | Example / Note |
|---|---|---|
| N-Ethylmaleimide (NEM) | Irreversible DUB inhibitor for sample preparation. | Use at 5-25 mM final concentration in lysis buffer [27]. |
| Iodoacetamide (IAA) | Irreversible DUB inhibitor for sample preparation. | Use at 5-20 mM final concentration in lysis buffer [27]. |
| K48-linkage Specific Antibody | Detection of K48-linked polyubiquitin chains by western blot. | e.g., Cell Signaling Technology #4289 or Abcam ab140601 [26] [14]. |
| HA-Ubiquitin Probes | Activity-based probes for monitoring functional DUB activity in lysates. | e.g., HA-Ub-VS (Vinyl Sulfone); useful for protocol validation [28]. |
| Ubiquitin Mutant Libraries | Determining ubiquitin chain linkage in in vitro assays. | K-to-R and K-Only mutants are critical for linkage mapping [4]. |
| DUB Inhibitors (e.g., VLX1570) | Selective chemical probes for specific DUBs in functional studies. | Used in proteomics-based substrate identification [29]. |
Step-by-Step Experimental Protocol
Cell Harvesting and Lysis
This protocol is designed for cultured cells and can be adapted for tissue samples with a homogenization step [28] [27].
- Pre-cool Equipment: Pre-cool a centrifuge to 4°C and place lysis buffer on ice.
- Harvest Cells: Remove media from cultured cells and wash with ice-cold Phosphate Buffered Saline (PBS). Gently scrape or trypsinize cells and collect them in a conical tube.
- Pellet Cells: Centrifuge the cell suspension at 290 × g for 5 minutes at 4°C to form a pellet. Carefully aspirate the supernatant.
- Wash Pellet: Resuspend the cell pellet in 5 mL of ice-cold PBS by gentle pipetting. Repeat the centrifugation and aspiration. This step removes residual serum containing proteins.
- Weigh/Lyse: For cell pellets, estimate the volume. Add twice the pellet volume of ice-cold lysis buffer containing NEM (10-25 mM) and IAA (10-20 mM). For tissues, use a mass-to-volume ratio of 1:9 (tissue mass:lysis buffer volume) [28].
- Vortex/Incubate: Immediately vortex the mixture vigorously for 15-30 seconds to ensure rapid and complete resuspension of the pellet. Incubate the lysate on ice for 15-30 minutes with occasional vortexing to allow for complete cell lysis and DUB inhibition.
Clarification and Post-Lysis Processing
- Clarify Lysate: Centrifuge the lysate at >15,000 × g for 10 minutes at 4°C to pellet nuclei, unbroken cells, and cellular debris.
- Transfer Supernatant: Carefully transfer the clarified supernatant (the total cell lysate) to a new, pre-chilled microcentrifuge tube.
- Add Reducing Agent (Optional): At this stage, after DUB inhibition is complete, DTT can be added to the lysate (e.g., to a final concentration of 1-5 mM) to reduce disulfide bonds in proteins, which may improve subsequent gel separation.
- Protein Quantification: Determine the protein concentration of the lysate using a compatible assay such as the bicinchoninic acid (BCA) assay.
Sample Derivatization for Western Blot
- Prepare Sample: Aliquot an amount of lysate corresponding to 20-50 µg of total protein into a new tube. Adjust the volume to a desired constant (e.g., 15-20 µL) using deionized water or lysis buffer.
- Add Laemmli Buffer: Add an equal volume of 2X Laemmli sample buffer to the protein aliquot.
- Denature Proteins: Heat the samples at 70-80°C for 5-10 minutes or at 95°C for 3-5 minutes. Avoid boiling for extended periods, as this can lead to protein aggregation, particularly of ubiquitylated proteins. The heating step serves to fully denature proteins and inactivate any residual enzymatic activity.
- Cool and Load: Briefly centrifuge the samples to bring down condensation and load onto a pre-cast gel (e.g., a 4-20% Tris-glycine gradient gel) for western blot analysis [28].
Workflow Visualization
The following diagram illustrates the critical decision points and steps in the sample preparation protocol, highlighting where DUB inhibitors are essential.
Diagram 1: Sample preparation workflow with DUB inhibition.
Troubleshooting and Validation
Common Pitfalls and Solutions
- Weak or No K48 Signal: This is often due to incomplete DUB inhibition. Ensure NEM and IAA stocks are fresh and that no reducing agents (DTT, β-mercaptoethanol) are present in the lysis buffer. Increase inhibitor concentrations within the recommended range.
- High Background Smearing: This is characteristic of ubiquitylated proteins. Ensure adequate clarification of the lysate and use of a gradient gel (e.g., 4-20%) can improve resolution. Avoid over-boiling samples.
- Inconsistent Results Between Preps: Always prepare fresh inhibitor stocks and use consistent lysis and incubation times. Normalize protein loading carefully across samples using a quantitative assay like BCA.
Validating DUB Inhibition
A powerful method to validate the efficacy of your DUB inhibition protocol is to use activity-based ubiquitin probes (ABPs), such as Hemagglutinin (HA)-tagged Ubiquitin Vinyl Sulfone (HA-Ub-VS) [28]. In this orthogonal assay:
- Split your cell lysate into two aliquots immediately after clarification.
- Incubate one aliquot with the HA-Ub-VS probe.
- Analyze both aliquots by western blot using an anti-HA antibody.
- Successful DUB Inhibition: In the NEM/IAA-treated sample, active DUBs are already blocked, so little to no HA-Ub-VS labeling will occur.
- Failed DUB Inhibition: If DUBs remain active, they will be covalently labeled by the HA-Ub-VS probe, appearing as multiple bands on the anti-HA blot, indicating that the inhibitor concentrations or conditions need optimization.
Integration with Downstream K48 Ubiquitin Analysis
The quality of sample preparation directly impacts the success of all downstream analyses. For K48-linkage-specific immunoblotting, a well-preserved lysate will show a characteristic laddering pattern above the protein of interest, representing polyubiquitin chains of increasing length. The use of validated, linkage-specific antibodies is critical, as they demonstrate minimal cross-reactivity with other linkage types (e.g., K63) or monoubiquitin [26] [14]. The protocol described here ensures that the observed signal is a true reflection of the cellular K48-linked ubiquitylation state, providing a reliable foundation for investigating its role in fundamental biological processes and drug discovery efforts targeting the ubiquitin-proteasome system [29].
Proteasome Inhibition Strategies to Preserve Ubiquitinated Targets
The ubiquitin-proteasome system (UPS) serves as the primary pathway for targeted intracellular protein degradation in eukaryotic cells [30]. This process is essential for maintaining cellular homeostasis by regulating the concentration of specific proteins and disposing of damaged or misfolded polypeptides. Ubiquitination, the covalent attachment of ubiquitin to target proteins, is a central mechanism within this system. Ubiquitin itself is a small, highly conserved regulatory protein that can be attached to substrate proteins via a cascade of enzymes: E1 (ubiquitin-activating), E2 (ubiquitin-conjugating), and E3 (ubiquitin-ligase) enzymes [31] [30]. The specificity of this system is largely determined by the E3 ubiquitin ligases, which recognize specific substrate proteins.
A critical aspect of ubiquitin signaling is the formation of polyubiquitin chains, where additional ubiquitin molecules are linked to one of the seven lysine residues (K6, K11, K27, K29, K33, K48, K63) or the N-terminal methionine (M1) of the preceding ubiquitin molecule [31] [30]. Among these, the K48-linked polyubiquitin chain is the principal signal for targeting proteins for degradation by the 26S proteasome [31] [30]. The 26S proteasome is a multi-subunit complex comprising a 20S core particle (CP) that carries out the proteolytic activity, and a 19S regulatory particle (RP) that recognizes, unfolds, and translocates ubiquitinated substrates into the core [30] [3]. The UPS has been implicated in a wide range of biological processes, including cell cycle progression, differentiation, stress response, and apoptosis [31]. Furthermore, dysregulation of the UPS is increasingly recognized as a crucial mechanism in pathological conditions such as cancer, where it can mediate tumor immune evasion by regulating the stability of immune checkpoint proteins like PD-L1 [30].
Diagram 1: The Ubiquitin-Proteasome Pathway and Inhibitor Action
For researchers studying cellular processes involving targeted protein degradation, the ability to experimentally preserve and detect K48-ubiquitinated proteins is paramount. This application note provides detailed methodologies for using proteasome inhibitors to stabilize K48-linked polyubiquitin conjugates and subsequently detect them using linkage-specific antibodies, thereby enabling accurate analysis of ubiquitination dynamics.
The Rationale for Proteasome Inhibition in Ubiquitination Studies
In a dynamic cellular environment, K48-ubiquitinated proteins are rapidly degraded by the proteasome, resulting in transient signals that are challenging to capture and measure accurately. Proteasome inhibitors are therefore indispensable tools that act as molecular traps, allowing for the accumulation of polyubiquitinated substrates that would otherwise be swiftly processed [30]. This stabilization is a critical prerequisite for the reliable detection and analysis of ubiquitination events using techniques such as western blotting.
The strategic importance of this approach is highlighted in cancer biology research, where the stability of key regulatory proteins is often controlled by the UPS. For instance, the E3 ubiquitin ligase SPOP normally promotes the K48-linked ubiquitination and degradation of PD-L1, an immune checkpoint protein [30]. In some cancers, competitive binding by other proteins like ALDH2 or SGLT2 disrupts SPOP's interaction with PD-L1, leading to PD-L1 stabilization and enhanced tumor immune evasion [30]. Similarly, other E3 ligases such as TRIM21 and ARIH1 have been documented to mediate K48-linked ubiquitination of PD-L1, with their activity being modulated by various cellular kinases and signaling pathways [30]. Pharmacological inhibition of the proteasome in such experimental settings allows researchers to "freeze" this dynamic process, making it possible to investigate the complex regulatory mechanisms governing protein stability and their implications for disease and therapy.
Experimental Protocols for Proteasome Inhibition and Detection
Proteasome Inhibition and Cell Lysis
Materials:
- Cell culture of interest
- Proteasome inhibitor (e.g., MG-132, Bortezomib, Lactacystin)
- Dimethyl sulfoxide (DMSO)
- Phosphate-buffered saline (PBS), ice-cold
- RIPA Lysis Buffer (or similar) supplemented with protease inhibitor cocktail
Procedure:
- Inhibitor Preparation: Prepare a stock solution of the chosen proteasome inhibitor in DMSO. MG-132 is commonly used at a final concentration of 10-20 µM.
- Cell Treatment:
- Aspirate the culture medium from cells grown to 70-90% confluence.
- Add fresh medium containing the pre-determined optimal concentration of the proteasome inhibitor. A vehicle control (DMSO only) must be included in parallel.
- Incubate cells for the desired duration (typically 4-16 hours). Note that prolonged inhibition can induce cellular stress.
- Cell Harvest and Lysis:
- Aspirate the medium and wash cells twice with ice-cold PBS.
- Lyse cells directly in the culture dish by adding an appropriate volume of ice-cold RIPA buffer supplemented with protease inhibitors.
- Scrape the cells and transfer the lysate to a microcentrifuge tube.
- Incubate on ice for 30 minutes, with brief vortexing every 10 minutes.
- Clarify the lysate by centrifugation at 14,000 x g for 15 minutes at 4°C.
- Transfer the supernatant (whole cell lysate) to a new pre-chilled tube.
- Protein Quantification: Determine the protein concentration of each lysate using a standard assay (e.g., BCA or Bradford assay). Lysates can be used immediately or stored at -80°C.
Detection of K48-Linked Polyubiquitin by Western Blot
Key Reagent: K48-linkage Specific Polyubiquitin Antibody (#4289, Cell Signaling Technology) [31]
- Reactivity: All species expected
- Application/Dilution: Western Blot (1:1000)
- Specificity: Specifically detects polyubiquitin chains formed via Lys48 linkage. It demonstrates slight cross-reactivity with linear polyubiquitin chains but no cross-reactivity with monoubiquitin or polyubiquitin chains formed by linkages to other lysine residues [31].
Materials:
- Clarified cell lysates (20-30 µg per lane is a standard starting point)
- K48-linkage Specific Polyubiquitin Antibody (#4289)
- Electrophoresis and western blot transfer systems
- PVDF or nitrocellulose membrane
- Blocking buffer (e.g., 5% BSA or non-fat dry milk in TBST)
- HRP-conjugated secondary antibody
- Chemiluminescent substrate
- Imaging system capable of detecting chemiluminescence or fluorescence
Procedure:
- Gel Electrophoresis and Transfer:
- Dilute protein lysates in Laemmli sample buffer.
- Denature samples by heating at 95-100°C for 5 minutes.
- Load equal amounts of protein (e.g., 20-30 µg) onto a SDS-PAGE gel.
- Run the gel at constant voltage until the dye front reaches the bottom.
- Transfer proteins from the gel to a PVDF or nitrocellulose membrane using a standard wet or semi-dry transfer system.
- Immunoblotting:
- Block the membrane with 5% BSA in TBST for 1 hour at room temperature to reduce nonspecific binding.
- Incubate the membrane with the anti-K48-linkage Specific Polyubiquitin Antibody (diluted 1:1000 in blocking buffer) overnight at 4°C with gentle agitation [31].
- Wash the membrane 3 times for 10 minutes each with TBST.
- Incubate with an appropriate HRP-conjugated secondary antibody (diluted as per manufacturer's instructions) for 1 hour at room temperature.
- Wash the membrane 3 times for 10 minutes each with TBST.
- Detection:
- Develop the blot using a enhanced chemiluminescence (ECL) substrate according to the manufacturer's protocol.
- Image the blot using a digital imaging system. Ensure that the image is not over-saturated, especially when performing quantitative analysis.
Diagram 2: Experimental Workflow for K48-Ubiquitin Detection
Quantitative Analysis and Normalization
For publication-quality quantitative western blot data, proper normalization is essential to account for variability in protein loading and transfer efficiency. The field is increasingly moving away from traditional housekeeping proteins (HKPs) like GAPDH or β-actin, as their expression can vary significantly with experimental conditions, cell type, and pathology [32]. Total Protein Normalization (TPN) is now considered the gold standard for quantitative western blotting [32] [33].
Total Protein Normalization Workflow:
- Total Protein Stain: After western blot transfer but before blocking, stain the membrane with a total protein stain such as No-Stain Protein Labeling Reagent or Coomassie-based stains. Alternatively, fluorescently-labeled total protein stains can be used in parallel with target detection in multiplexed fluorescent western blots [32].
- Image Acquisition: Image the total protein stain to visualize all proteins in each lane.
- Target Detection: Proceed with the immunodetection of K48-linked polyubiquitin as described above.
- Quantification:
- Use imaging software to quantify the signal intensity of each band in both the total protein stain image and the K48-ubiquitin image.
- For each lane, calculate the normalized K48-ubiquitin signal using the formula:
Normalized Signal = (K48-Ubiquitin Band Intensity) / (Total Protein Stain Intensity for the same lane). - Compare the normalized values across experimental conditions to determine relative changes in K48-linked ubiquitination.
Table 1: Common Proteasome Inhibitors and Their Properties
| Inhibitor | Typical Working Concentration | Mechanism of Action | Primary Use |
|---|---|---|---|
| MG-132 | 10 - 20 µM | Reversible peptide aldehyde; inhibits chymotrypsin-like activity of the 20S proteasome. | General laboratory research; short-term treatments. |
| Bortezomib | 10 - 100 nM | Reversible inhibitor targeting the chymotrypsin-like site. | Clinical (oncology); approved for multiple myeloma. |
| Lactacystin | 10 - 20 µM | Irreversibly binds to the β-subunit of the 20S proteasome. | Fundamental research; long-term inhibition studies. |
| Carfilzomib | 5 - 50 nM | Irreversible epoxyketone inhibitor; highly specific for the chymotrypsin-like site. | Clinical (oncology); relapsed/refractory multiple myeloma. |
Research Reagent Solutions
Table 2: Essential Reagents for K48-Ubiquitin Immunoblotting
| Reagent | Function / Role | Example Product / Catalog Number |
|---|---|---|
| K48-linkage Specific Antibody | Primary antibody for specific detection of K48-linked polyubiquitin chains in western blot. | Cell Signaling Technology (CST) #4289 [31] |
| Proteasome Inhibitor | Inhibits the 26S proteasome, preventing degradation of ubiquitinated proteins and enabling their accumulation. | MG-132 (CST #2194), Bortezomib (PS-341) |
| Protease Inhibitor Cocktail | Added to lysis buffer to prevent non-specific protein degradation by cellular proteases during sample preparation. | Various commercial tablets or solutions (e.g., EDTA-free) |
| Total Protein Stain | Used for accurate normalization of target protein signal to total protein loaded, superior to housekeeping proteins. | No-Stain Protein Labeling Reagent [32] |
| HRP-conjugated Secondary Antibody | Conjugated antibody for detection of the primary antibody, used with chemiluminescent substrates. | Anti-rabbit IgG, HRP-linked Antibody (CST #7074) |
| Enhanced Chemiluminescent (ECL) Substrate | Enzyme substrate that produces light upon reaction with HRP, enabling visualization of protein bands. | Various commercial kits |
Data Interpretation and Troubleshooting
Expected Results and Analysis
Successful proteasome inhibition will be evidenced by a marked increase in the smeared signal for K48-linked polyubiquitin in the inhibitor-treated lanes compared to the DMSO vehicle control lane on the western blot. This smear represents the heterogeneous population of polyubiquitinated proteins in the cell, which is a normal and expected result. A sharp, discrete band may indicate a specific, highly abundant ubiquitinated protein or potential non-specific antibody binding.
For quantitative comparisons, the normalized K48-ubiquitin signal (target signal / total protein stain) should be calculated for each sample. Statistical analysis (e.g., t-test, ANOVA) can then be performed to determine the significance of changes between experimental groups.
Troubleshooting Common Issues
- High Background Signal: Ensure sufficient washing after primary and secondary antibody incubations. Optimize the concentration of the primary and secondary antibodies. Try a different blocking agent (e.g., switch from milk to BSA).
- Weak or No Signal: Confirm the activity of the proteasome inhibitor with a positive control. Check antibody dilutions and ensure the antibody is validated for western blot. Verify that the ECL substrate is functional and the imaging exposure time is adequate.
- Saturation of Signal: If bands appear saturated or overexposed in the image, reduce the amount of protein loaded or decrease the exposure time during imaging. Quantitative analysis must be performed within the linear dynamic range of detection [32] [33].
- Lack of Change with Inhibition: The inhibitor may be inactive. Prepare a fresh stock solution and confirm its efficacy. The treatment time might be too short; consider a longer incubation (up to 16 hours, monitoring for cytotoxicity).
The strategic application of proteasome inhibitors is a fundamental technique for stabilizing and studying K48-linked polyubiquitinated proteins. When combined with a highly specific K48-linkage antibody and robust quantitative western blot practices—particularly total protein normalization—this protocol provides a reliable method for investigating the dynamics of the ubiquitin-proteasome pathway. This approach is vital for advancing research in areas ranging from basic cell biology to the development of novel therapeutic strategies aimed at modulating protein stability in disease.
Choosing the Right Gel and Buffer System for Optimal Ubiquitin Chain Separation
The separation of ubiquitin chains by SDS-PAGE is a foundational technique for studying the ubiquitin-proteasome system. However, the diverse molecular weights and structural complexities of polyubiquitin chains present significant separation challenges. For researchers focusing on K48-linked ubiquitination—the primary signal for proteasomal degradation—optimizing electrophoretic conditions is particularly critical for accurate interpretation of experimental results. This application note provides detailed protocols and data-driven recommendations for achieving superior resolution of ubiquitin chains, specifically tailored for K48 linkage-specific ubiquitin antibody research.
The Challenge of Ubiquitin Chain Separation
Ubiquitin chains present unique separation challenges in immunoblotting experiments. A single ubiquitin monomer is approximately 8.6 kDa, and proteins can be modified by chains containing 20 or more ubiquitins, adding over 170 kDa to their molecular mass. This results in a smear of polyubiquitin chains that typically stretches toward the top of the gel. Furthermore, the presence of different ubiquitin linkage types—including K48-linked, K63-linked, and complex branched chains—creates a heterogeneous mixture that requires optimal separation for accurate analysis [34].
Gel and Buffer System Optimization
Quantitative Comparison of Separation Systems
Table 1: Optimal Gel and Buffer Systems for Ubiquitin Chain Separation
| Separation System | Optimal Ubiquitin Chain Length | Key Advantages | Limitations |
|---|---|---|---|
| MES Buffer with Pre-Poured Gradient Gels | 2-5 ubiquitins | Superior resolution of small ubiquitin oligomers | Less effective for longer chains |
| MOPS Buffer with Pre-Poured Gradient Gels | 8+ ubiquitins | Excellent resolution of longer polyubiquitin chains | Reduced resolution of shorter chains |
| Tris-Acetate (TA) Buffer | 40-400 kDa protein range | Superior for high molecular weight proteins | Not ubiquitin chain-specific |
| Tris-Glycine (TG) Buffer with 8% Acrylamide | Up to 20 ubiquitins | Good separation across a wide range of chain lengths | Requires low percentage acrylamide |
| Tris-Glycine (TG) Buffer with 12% Acrylamide | Mono-ubiquitin and short oligomers | Enhanced detection of mono-ubiquitin and short chains | Compromised resolution of longer chains |
The selection of appropriate running buffers is crucial for achieving optimal ubiquitin chain separation. MES (2-(N-morpholino) ethane sulfonic acid) buffer provides superior resolution of relatively small ubiquitin oligomers comprising 2-5 ubiquitins, whereas MOPS (3-(N-morpholino) propane sulfonic acid) buffer yields better resolution for polyubiquitin chains containing eight or more ubiquitins. For overall separation of proteins in the molecular mass range of 40-400 kDa, Tris-acetate (TA) buffer generally provides superior performance [34].
When using single-concentration gels (approximately 8% acrylamide) with Tris-glycine (TG) buffer, researchers can successfully separate individual ubiquitin chains comprising up to 20 ubiquitins. However, to detect mono-ubiquitin and short ubiquitin oligomers, the acrylamide concentration must be increased to around 12%, though this comes at the expense of reducing resolution for longer polyubiquitin chains [34].
Workflow for Ubiquitin Chain Analysis
The following diagram illustrates the complete workflow for optimal ubiquitin chain analysis, from sample preparation to detection:
Essential Methodological Considerations
Preservation of Ubiquitination State
Maintaining the ubiquitination state of proteins during sample preparation is paramount. Protein ubiquitylation is reversible and can be lost through hydrolysis catalyzed by deubiquitylases (DUBs). It is therefore essential to include DUB inhibitors in cell lysis buffers, particularly during immunoprecipitation or pull-down experiments where extracts may be incubated for several hours under non-denaturing conditions [34].
Both iodoacetamide (IAA) and N-ethylmaleimide (NEM) effectively alkylate the active site cysteine residues of DUBs. While typical concentrations of 5-10 mM are used in many publications, research indicates that up to 10-fold higher concentrations may be necessary to preserve the ubiquitylation status of some proteins and ubiquitin chains. For K63-Ub chains and M1-Ub chains, high concentrations of NEM generally provide better preservation than IAA [34].
When planning to identify ubiquitylation sites by mass spectrometry, NEM is recommended over IAA because the covalent adduct formed by IAA with cysteine residues has a molecular mass identical to the Gly–Gly dipeptide that remains attached to lysine residues after trypsin digestion, potentially interfering with analysis [34].
Proteasome Inhibition
For studies focusing on K48-linked ubiquitination, proteasome inhibition is often necessary. Proteins modified by K48-linked polyubiquitin chains are rapidly targeted to the 26S proteasome for degradation. Treatment with proteasome inhibitors such as MG132 blocks protein degradation and preserves the ubiquitylated forms of proteins, thereby facilitating detection [34].
Research Reagent Solutions for K48 Ubiquitin Research
Table 2: Essential Research Reagents for K48-Linked Ubiquitin Studies
| Reagent / Tool | Specific Function | Application Notes |
|---|---|---|
| K48-linkage Specific Antibodies (e.g., #4289, ab140601, PA5-120616) | Specific detection of K48-linked polyubiquitin chains | Validate specificity using linkage-specific recombinant ubiquitins; slight cross-reactivity with linear chains reported for some antibodies [35] [14] [36] |
| Tandem Ubiquitin Binding Entities (TUBEs) | Affinity enrichment of polyubiquitinated proteins with nanomolar affinity | Enable capture of endogenous ubiquitination events; available in pan-specific and linkage-specific (K48, K63) formats [37] [17] |
| Deubiquitylase (DUB) Inhibitors (NEM, IAA, CAA) | Preserve ubiquitination state by inhibiting DUB activity | NEM generally preferred for MS applications; concentration optimization required for different sample types [34] [17] |
| Linkage-Specific Recombinant Ubiquitins | Positive controls for antibody validation and linkage assignment | Essential for validating antibody specificity and optimizing separation conditions [14] |
| Proteasome Inhibitors (e.g., MG132) | Stabilize K48-ubiquitinated proteins by blocking degradation | Critical for detecting endogenous K48-ubiquitinated substrates; note potential cytotoxic effects with prolonged incubation [34] |
Advanced Applications: Branching and Linkage Complexity
Recent research has revealed that ubiquitin chain topology is considerably more complex than previously recognized, with branched chains accounting for a significant proportion of ubiquitin polymers in cells. Among these, K11/K48-branched ubiquitin chains have emerged as particularly important for fast-tracking protein turnover during cell cycle progression and proteotoxic stress [3].
The recognition of K11/K48-branched ubiquitin chains involves a multivalent substrate recognition mechanism by the human 26S proteasome, including a previously unknown K11-linked ubiquitin binding site at the groove formed by RPN2 and RPN10, in addition to the canonical K48-linkage binding site formed by RPN10 and RPT4/5 [3]. This complexity underscores the importance of optimal separation techniques to resolve these different ubiquitin architectures.
Troubleshooting and Optimization Guidelines
Smearing or Poor Resolution: Optimize buffer system selection based on target chain length. For shorter chains (2-5 ubiquitins), switch to MES buffer systems. For longer chains (8+ ubiquitins), MOPS buffer typically provides better resolution.
Loss of Ubiquitination Signal: Ensure adequate DUB inhibition during cell lysis. Consider increasing concentrations of NEM (up to 50 mM) or using combination approaches with other DUB inhibitors.
Incomplete Transfer: For high molecular weight ubiquitinated proteins, ensure efficient transfer by using appropriate membrane materials and extended transfer times.
Non-Specific Antibody Binding: Include linkage-specific recombinant ubiquitin controls to verify antibody specificity and optimize blocking conditions.
Optimal separation of ubiquitin chains requires careful consideration of both gel composition and running buffer systems. The MES/MOPS buffer systems with gradient gels provide the most flexibility for resolving different chain lengths, while Tris-glycine systems with appropriate acrylamide concentrations offer a practical alternative for laboratories with standard equipment. For K48 linkage-specific research, combining these separation optimization strategies with robust sample preservation methods and appropriate detection reagents ensures accurate and reproducible analysis of this critical protein modification. As research continues to reveal the complexity of ubiquitin chain biology, these methodological foundations become increasingly important for advancing our understanding of ubiquitin-mediated processes in health and disease.
Standard Western Blot Procedure and Recommended Antibody Dilutions
Ubiquitination is a crucial post-translational modification that regulates numerous cellular processes, including protein degradation, signal transduction, and DNA repair. Specifically, K48-linked polyubiquitin chains primarily target proteins for proteasomal degradation, making their detection essential for understanding cellular protein homeostasis [38]. The Western blot, or immunoblot, has evolved from a simple qualitative technique to a powerful quantitative tool for measuring relative changes in protein expression [32]. For today's top journals, employing rigorous quantitative methodologies including total protein normalization and providing high-quality, transparent data are paramount for publication [32]. This application note provides a standardized protocol for the detection and quantification of K48-linked ubiquitin chains, framed within the broader context of reliable immunoblotting practices required for drug development research.
Key Biological Context: The Ubiquitin-Proteasome Pathway
The ubiquitin-proteasome pathway involves a cascade of E1 (activation), E2 (conjugation), and E3 (ligation) enzymes that ultimately attach ubiquitin to target proteins. When ubiquitin molecules form a chain through linkages at the lysine 48 (K48) residue, the modified protein is typically directed to the 26S proteasome for degradation [38]. This process is fundamental to regulating the concentration of key regulatory proteins, and its dysregulation is implicated in various diseases, including cancer and neurodegenerative disorders, making it a significant area for therapeutic intervention.
The following diagram illustrates this primary signaling pathway for K48-linked polyubiquitination:
Critical Experimental Design for Quantitative Data
Attaining reliable, quantitative data from Western blots requires careful experimental planning to minimize variability and ensure biological relevance. Key considerations include:
- Biological vs. Technical Replicates: Incorporate both biological replicates (different samples per condition) and technical replicates (same sample tested multiple times) to account for both biological and experimental variance [39].
- Appropriate Controls: Always include relevant control groups such as untreated versus treated, normal versus disease state, or time-zero time points [39].
- Sample Handling Consistency: Standardize procedures for sample collection, freezing, storage, and thawing to prevent artifactual protein degradation or modification [39].
The following workflow outlines the comprehensive quantitative Western blot procedure:
Detailed Standard Protocols
Sample Preparation Protocol
Proper sample preparation is critical for obtaining accurate and reproducible results. Inconsistent handling can lead to protein degradation, post-collection modifications, and ultimately, unreliable data.
- Cell Lysis: Use ice-cold RIPA buffer (1% NP-40 or Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 150 mM NaCl, 50 mM Tris-HCl, pH 7.8) supplemented with protease inhibitors. For adherent cells, add buffer directly to the plate, scrape, and pipette the lysate up and down. For cell pellets, resuspend in buffer [39].
- Tissue Homogenization: Snap-freeze tissue in liquid nitrogen and dice into 1 mm pieces on dry ice. Add to ice-cold RIPA buffer and homogenize using a Dounce homogenizer (25 strokes on ice). Sonicate on ice (5 × 20 seconds at 50% power) and clear extracts by centrifugation at 34,000 ×g at 4°C for 30 minutes [39].
- Protein Quantification: Measure total protein concentration using a detergent-compatible assay (e.g., RC DC protein assay from Bio-Rad). Dilute homogenates to at least 2 mg/mL to enable loading between 10 μg and 80 μg per lane of a 1 mm mini gel [39].
- Storage: Store supernatants at -80°C or in liquid nitrogen for long-term preservation. Avoid repeated freeze-thaw cycles [39].
Determining Linear Dynamic Range
A fundamental step in quantitative Western blotting is establishing the linear dynamic range for detection, which ensures that signal intensity is directly proportional to the amount of protein loaded.
- Dilution Series: Create a 1/2 dilution series of a pooled sample from all lysates in the study, starting from 100 μg total protein load over at least 12 dilutions [39].
- Electrophoresis and Transfer: Load dilutions on a TGX stain-free SDS-PAGE gel and transfer to a low-fluorescence PVDF membrane using a semi-dry or wet transfer system [39].
- Immunodetection and Imaging: Incubate with primary and secondary antibodies, develop with an imager-compatible chemiluminescent substrate (e.g., Clarity, Bio-Rad), and capture signals using a CCD-camera-based imager (e.g., ChemiDoc MP, Bio-Rad) [39].
- Analysis: Using densitometry software (e.g., Image Lab, Bio-Rad), plot relative density versus fold dilution for each antibody. Identify the linear range where consistent, 1/2 decreases in density are obtained, and select the protein load corresponding to the middle of this range for subsequent experiments [39].
Electrophoresis and Immunoblotting Protocol
- Gel Electrophoresis: Load predetermined optimal protein amounts (determined from linear range analysis) onto SDS-polyacrylamide gels. For K48-linked ubiquitin detection, a 4-20% gradient gel is recommended to resolve various polyubiquitinated species. Run at constant voltage until the dye front reaches the bottom [14] [38].
- Protein Transfer: Transfer proteins to a PVDF or nitrocellulose membrane using a wet or semi-dry transfer system. The Trans-Blot Turbo system (Bio-Rad) is recommended for efficient, consistent transfers. Verify transfer efficiency with total protein stains if performing TPN [39].
- Blocking: Block membranes with 5% non-fat dry milk (NFDM) in TBST or suitable blocking buffer for 1 hour at room temperature with gentle agitation [14].
- Antibody Incubation:
- Primary Antibody: Incubate with K48-linkage specific ubiquitin antibody at the recommended dilution (refer to Table 1 for specific products) in blocking buffer overnight at 4°C with agitation [14] [38].
- Washing: Wash membrane 4 times for 3-5 minutes each with TBST [39].
- Secondary Antibody: Incubate with species-appropriate HRP-conjugated secondary antibody (e.g., Goat Anti-Rabbit IgG H&L (HRP)) at 1:20000 dilution in blocking buffer for 1 hour at room temperature [14].
- Final Washing: Repeat washing step with TBST (4 × 3-5 minutes) [39].
- Detection: Develop blots with enhanced chemiluminescence (ECL) substrate suitable for your imaging system. For quantitative work, use substrates with a wide dynamic range (e.g., Clarity, Bio-Rad) [39].
K48 Linkage-Specific Ubiquitin Antibodies and Dilutions
The following table summarizes key commercially available antibodies for detecting K48-linked ubiquitin chains, with validated application conditions:
Table 1: K48 Linkage-Specific Ubiquitin Antibodies for Western Blotting
| Product Name | Supplier | Catalog Number | Recommended Dilution | Observed Band Sizes | Specificity Notes |
|---|---|---|---|---|---|
| Anti-Ubiquitin (linkage-specific K48) [EP8589] | Abcam | ab140601 | 1:1000 [14] | 26 kDa, 38 kDa, 39 kDa, 42 kDa, 78 kDa [14] | Rabbit monoclonal; detects polyubiquitin chains formed by Lys48 linkage |
| K48-linkage Specific Polyubiquitin Antibody | Cell Signaling Technology | 4289 | 1:1000 [38] | Varies based on protein target | Rabbit polyclonal; slight cross-reactivity with linear polyubiquitin chain |
Data Normalization and Analysis
The Critical Shift from Housekeeping Proteins to Total Protein Normalization
For quantitative Western blot analysis, appropriate normalization is essential to distinguish experimental variability from true biological changes. Traditional methods using housekeeping proteins (HKPs) like GAPDH, β-actin, or tubulin are falling out of favor with major journals due to significant limitations [32].
Key limitations of HKP normalization include:
- HKP expression varies with cell type, developmental stage, tissue pathology, and experimental conditions [32].
- HKPs are typically highly abundant, leading to early signal saturation and narrow linear dynamic range [32].
- HKPs can interact with experimental components, leading to co-migration with target proteins or antibody cross-reactivity [32].
Total Protein Normalization (TPN) is now considered the gold standard for quantitative Western blotting [32]. TPN normalizes the target protein signal to the total amount of protein in each lane, which:
- Is not affected by experimental manipulations [32]
- Provides a larger dynamic range for detection [32]
- Offers information about electrophoresis and transfer quality [32]
TPN can be achieved with total protein stains (e.g., Coomassie, Ponceau S) or fluorescent labeling technologies (e.g., No-Stain Protein Labeling Reagent, Thermo Fisher) followed by high-resolution imaging [32].
Densitometric Analysis and Calculation
For quantitative analysis, use background-subtracted densitometry values from imaging software:
- Measure Target Protein Signal: Obtain densitometry values for K48-linked ubiquitin bands.
- Normalization Factor: Calculate total protein signal for each lane using TPN.
- Normalized Target Signal: Divide target protein signal by normalization factor for each sample.
- Fold Change Calculation: Compare normalized values between experimental groups to determine fold changes in K48-linked ubiquitination.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagent Solutions for K48 Ubiquitin Western Blotting
| Reagent / Solution | Function | Example Products |
|---|---|---|
| RIPA Lysis Buffer | Protein extraction from cells and tissues; contains detergents and salts to solubilize proteins while maintaining integrity [39] | Various commercial formulations |
| Protease Inhibitor Cocktail | Prevents protein degradation during extraction and storage [39] | Complete Mini (Roche) |
| K48-linkage Specific Antibodies | Specific detection of K48-linked polyubiquitin chains [14] [38] | ab140601 (Abcam), #4289 (CST) |
| HRP-conjugated Secondary Antibodies | Signal generation for chemiluminescent detection [14] | Goat Anti-Rabbit IgG H&L (HRP) |
| Chemiluminescent Substrate | Enzyme substrate for signal detection and visualization [39] | Clarity (Bio-Rad) |
| Total Protein Normalization Reagents | Accurate normalization for quantitative analysis [32] | No-Stain Protein Labeling Reagent (Thermo Fisher) |
| PVDF Membranes | Protein immobilization after transfer [39] | Low-fluorescence PVDF |
Journal Publication Guidelines
Major scientific journals have implemented specific guidelines for Western blot publication to ensure data integrity:
- Nature: Strongly discourages quantitative comparisons between samples on different gels/blots and requires that loading controls be run on the same blot. High-contrast gels and blots are discouraged [32].
- Cell Press: Requires minimal image processing and demands transparency in explaining all processing steps in figure legends. All accepted papers are screened for image irregularities [32].
- Journal of Biological Chemistry (JBC): Has specific guidelines for quantitation and presentation, including requirements for describing antibody products, prohibiting excessive cropping, and requiring inclusion of molecular weight markers [32].
- General Image Integrity: Most journals prohibit the use of touch-up tools that deliberately obscure manipulations. Adjustments of brightness or contrast are acceptable only if they do not eliminate information present in the original [32].
Troubleshooting and Best Practices
- Antibody Validation: For each target protein and antibody, confirm specificity and determine the linear dynamic range of detection [25] [39].
- Loading Control Selection: Implement total protein normalization instead of traditional housekeeping proteins for more reliable quantification [32].
- Image Acquisition: Use CCD-camera-based imaging systems with wide dynamic range for accurate quantitation of both weak and strong signals [39].
- Experimental Replication: Perform a minimum of three independent biological replicates to ensure statistical significance of results [39].
- Documentation: Maintain detailed records of all experimental parameters, including antibody lot numbers, exposure times, and processing steps for publication transparency [32].
Special Considerations for Cell Lines and Tissue Lysates
Ubiquitination is a crucial post-translational modification that regulates virtually every cellular process, with linkage-specific polyubiquitin chains dictating diverse functional outcomes. Among the eight possible ubiquitin chain linkages, lysine 48 (K48)-linked polyubiquitin serves as the primary signal for proteasomal degradation, making it a critical focus in proteostasis research and drug development [40] [41]. The accurate detection of K48-linked ubiquitination in cell lines and tissue lysates presents significant technical challenges due to the presence of multiple ubiquitin chain types, the dynamic nature of the ubiquitin-proteasome system, and the susceptibility of ubiquitin chains to deubiquitinating enzymes (DUBs) [37]. This application note provides detailed methodologies and considerations for researchers investigating K48-linked ubiquitination using linkage-specific antibodies, with emphasis on maintaining chain integrity and assay specificity across different biological sample types.
Biological Context: K48 Ubiquitin Signaling Pathways
K48-linked polyubiquitin chains represent one of the most abundant linkage types in cells and function as the canonical signal for targeting proteins to the 26S proteasome for degradation [17] [41]. This modification is orchestrated through a sequential enzymatic cascade involving E1 (activating), E2 (conjugating), and E3 (ligating) enzymes, with the E3 ligases providing substrate specificity. The resulting isopeptide-linked chains, comprised of four or more ubiquitin molecules, are recognized by proteasomal subunits, leading to substrate degradation and ubiquitin recycling [40]. In contrast, other linkage types such as K63-linked chains primarily regulate non-proteolytic functions including signal transduction, protein trafficking, and DNA repair [37]. The diverse functions mediated by different ubiquitin linkages underscore the critical importance of linkage-specific detection methods for accurate biological interpretation.
Table 1: Functions of Major Ubiquitin Linkage Types
| Linkage Type | Primary Functions | Key Signaling Pathways |
|---|---|---|
| K48-linked | Proteasomal degradation, cell cycle regulation, apoptosis | p53 degradation, IκB degradation, Bcl-2 regulation |
| K63-linked | Signal transduction, protein trafficking, DNA repair, inflammation | NF-κB signaling, NLRP3 inflammasome activation, MAPK pathways |
| K11-linked | Proteasomal degradation, cell cycle regulation | Cell cycle progression, ER-associated degradation |
| K6-linked | DNA damage repair | DNA damage response pathways |
| M1-linked (linear) | NF-κB inflammatory signaling | NF-κB activation, immune response |
Key Technical Challenges in K48 Ubiquitin Detection
Preservation of Ubiquitin Chain Integrity
A primary challenge in detecting endogenous K48-linked ubiquitination is the rapid deubiquitination by cellular DUBs during sample preparation. The use of deubiquitinase inhibitors is essential immediately upon cell lysis to preserve polyubiquitin chains [37] [17]. Research indicates that N-ethylmaleimide (NEM) and chloroacetamide (CAA) are effective cysteine protease DUB inhibitors, though they exhibit different properties: NEM provides more complete DUB inhibition but has potential off-target effects, while CAA is more cysteine-specific but may allow partial chain disassembly [17] [6]. The selection of appropriate DUB inhibitors must balance preservation of ubiquitin chains with minimal perturbation of ubiquitin-binding surfaces.
Antibody Specificity Considerations
Linkage-specific antibodies for K48 ubiquitin, such as Cell Signaling Technology's #4289 Rabbit monoclonal antibody or abcam's ab140601 Rabbit Recombinant Monoclonal [EP8589], demonstrate high specificity for K48-linked polyubiquitin chains but may show slight cross-reactivity with linear polyubiquitin chains [40] [14]. These antibodies typically do not recognize monoubiquitin or polyubiquitin chains formed through other lysine residues (K6, K11, K27, K29, K33, K63) [40]. However, appropriate controls must be included to verify specificity, particularly when analyzing complex samples like tissue lysates that may contain multiple ubiquitin chain types.
Optimized Protocols for Sample Preparation
Cell Line Lysis and Processing
Materials and Reagents:
- Appropriate cell culture medium
- Phosphate-buffered saline (PBS), ice-cold
- Lysis buffer: 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% NP-40, 1 mM EDTA
- Freshly added DUB inhibitors: 10 mM NEM or 25 mM CAA
- Protease inhibitor cocktail (without EDTA)
- Phosphatase inhibitors (if studying phosphorylated proteins)
- BCA or Bradford protein assay kit
Procedure:
- Cell Treatment and Harvest: Grow cells to 70-90% confluence. Apply experimental treatments (e.g., proteasome inhibitors like MG132 for 4-6 hours to accumulate ubiquitinated species). Rapidly aspirate medium and wash cells with ice-cold PBS.
- Cell Collection: For adherent cells, scrape in ice-cold PBS; for suspension cells, pellet by centrifugation at 500 × g for 5 minutes at 4°C.
- Cell Lysis: Lyse cells in pre-chilled lysis buffer with freshly added DUB inhibitors (1:100 dilution). Use 100-200 μL lysis buffer per 1 × 10^6 cells. Incubate on ice for 15-30 minutes with occasional vortexing.
- Clarification: Centrifuge lysates at 16,000 × g for 15 minutes at 4°C. Transfer supernatant to a fresh pre-chilled tube.
- Protein Quantification: Determine protein concentration using BCA or Bradford assay. Adjust samples to equal concentrations with lysis buffer.
- Sample Preparation for Immunoblotting: Mix protein lysate with 4× Laemmli buffer, heat at 95°C for 5-10 minutes, and immediately place on ice.
Table 2: Troubleshooting Cell Line Preparation
| Problem | Potential Cause | Solution |
|---|---|---|
| Weak or no K48 signal | Incomplete DUB inhibition | Freshly prepare DUB inhibitors; increase inhibitor concentration; reduce lysis time |
| High background | Non-specific antibody binding | Include linkage-specific controls; optimize antibody dilution; increase blocking time |
| Smearing in Western blot | Protein aggregation | Ensure complete denaturation; include fresh DTT in sample buffer; brief sonication of lysates |
| Inconsistent results between replicates | Variable inhibitor activity | Prepare fresh inhibitor stocks; standardize lysis timing; use single-use aliquots |
Tissue Lysate Preparation
Materials and Reagents:
- Tissue homogenization buffer: 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS
- Freshly added DUB inhibitors: 10 mM NEM or 25 mM CAA
- Complete protease inhibitor cocktail
- Dounce homogenizer or mechanical homogenizer
- BCA or Bradford protein assay kit
Procedure:
- Tissue Collection and Preservation: Excise tissue rapidly and immediately freeze in liquid nitrogen. Store at -80°C until use. For optimal results, process tissues immediately without freezing when possible.
- Tissue Homogenization: Weigh tissue and add 5-10 volumes (w/v) of ice-cold homogenization buffer with freshly added DUB inhibitors. Homogenize with 15-20 strokes in a Dounce homogenizer or using a mechanical homogenizer on ice.
- Lysate Incubation: Rotate homogenate at 4°C for 30 minutes to ensure complete extraction.
- Clarification: Centrifuge at 16,000 × g for 20 minutes at 4°C. Collect supernatant carefully, avoiding lipid layer and pellet.
- Protein Quantification and Preparation: Determine protein concentration and prepare samples for Western blotting as described for cell lines.
Experimental Workflow and Quality Control
The following diagram illustrates the complete workflow for preparing and analyzing cell lines and tissue lysates for K48-linked ubiquitin detection:
Quality Control Measures
Specificity Verification:
- Include linkage-specific controls when possible, such as recombinant K48-linked and K63-linked diubiquitin
- Validate antibody specificity using ubiquitin mutants (K48R) that prevent K48 chain formation [4]
- Perform peptide competition assays to confirm signal specificity
Signal Optimization:
- For Western blotting, use 4-20% gradient gels to resolve high molecular weight polyubiquitin smears
- Transfer using low methanol content transfer buffer (≤10%) for efficient transfer of ubiquitinated proteins
- Optimize antibody dilution (typically 1:1000 for most commercial K48-linkage specific antibodies) [40] [14]
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for K48 Ubiquitin Research
| Reagent | Function | Example Products |
|---|---|---|
| K48-linkage Specific Antibodies | Detection of K48-linked polyubiquitin chains | CST #4289 [40], Abcam ab140601 [14], Thermo Fisher MA5-35382 [42] |
| Deubiquitinase (DUB) Inhibitors | Preserve ubiquitin chains during processing | N-ethylmaleimide (NEM), Chloroacetamide (CAA) [17] [6] |
| Proteasome Inhibitors | Accumulate ubiquitinated proteins | MG132, Bortezomib, Lactacystin |
| Ubiquitin Mutants | Specificity controls; linkage determination | K48-only Ubiquitin, K48R Ubiquitin Mutant [4] |
| Tandem Ubiquitin Binding Entities (TUBEs) | Affinity enrichment of polyubiquitinated proteins | K48-TUBEs, Pan-TUBEs [37] |
| Recombinant Ubiquitin Chains | Positive controls for linkage specificity | K48-linked Ub2-7, K63-linked Ub2-7 [14] |
Advanced Applications and Methodologies
TUBE-Based Ubiquitin Enrichment
Tandem Ubiquitin Binding Entities (TUBEs) provide a powerful alternative to antibody-based methods for studying linkage-specific ubiquitination. These engineered ubiquitin-binding domains with nanomolar affinities for polyubiquitin chains can be employed to capture endogenous ubiquitinated proteins from cell lysates [37]. K48-specific TUBEs effectively differentiate between ubiquitination events induced by different stimuli, such as distinguishing K48-linked ubiquitination mediated by PROTACs from K63-linked ubiquitination induced by inflammatory stimuli like L18-MDP in RIPK2 studies [37]. This approach is particularly valuable for studying ubiquitination dynamics in native cellular contexts.
Ubiquitin Linkage Determination Protocol
For researchers requiring definitive linkage characterization, the ubiquitin mutant-based protocol provides a reliable method for determining ubiquitin chain linkage [4]:
Materials:
- Wild-type ubiquitin
- Seven ubiquitin lysine-to-arginine (K-to-R) mutants (K6R, K11R, K27R, K29R, K33R, K48R, K63R)
- Seven ubiquitin "K-only" mutants (each containing only one lysine)
- E1 activating enzyme, E2 conjugating enzyme, E3 ligase
- 10X E3 ligase reaction buffer
- MgATP solution
Procedure:
- Set up separate conjugation reactions with wild-type ubiquitin and each K-to-R mutant
- Incubate at 37°C for 30-60 minutes with E1, E2, E3 enzymes
- Terminate reactions with SDS-PAGE sample buffer or DTT/EDTA
- Analyze by Western blotting with anti-ubiquitin antibody
- The reaction that fails to form polyubiquitin chains indicates the essential lysine for linkage
- Confirm results using "K-only" ubiquitin mutants [4]
This systematic approach enables definitive identification of ubiquitin chain linkages and can validate antibody specificity in complex biological samples.
Accurate detection of K48-linked ubiquitination in cell lines and tissue lysates requires meticulous attention to sample preparation, particularly the preservation of ubiquitin chains through appropriate DUB inhibition and validation of antibody specificity. The protocols and considerations outlined in this application note provide researchers with a robust framework for investigating this critical protein modification, enabling more reliable interpretation of K48-linked ubiquitination in both basic research and drug discovery contexts. As the field advances, methodologies such as TUBE-based enrichment and mass spectrometry-based interactome screens continue to enhance our understanding of the complex ubiquitin code and its physiological significance [17] [6].
Troubleshooting K48 Ubiquitin Blots: Solving Smears, Weak Signals, and High Background
In the study of the ubiquitin-proteasome pathway, K48-linked polyubiquitin chains are a primary signal targeting proteins for degradation by the 26S proteasome [43]. Researchers using K48-linkage specific antibodies in western blots frequently encounter a classic "smear" pattern—a continuous distribution of signal across a wide molecular weight range. Rather than being a technical artifact, this smear often represents a genuine biological outcome: the heterogeneous population of target proteins at various stages of polyubiquitination. This application note details the interpretation of this pattern and provides optimized protocols to ensure accurate data collection and analysis within the context of K48-linkage specific ubiquitin research.
Interpretation of the K48 Ubiquitin Smear
A smear in a western blot probed with a K48-linkage specific antibody indicates the presence of ubiquitinated proteins of varying molecular weights. This heterogeneity arises because substrate proteins of different sizes can be modified by K48-linked polyubiquitin chains of varying lengths.
- Biological Significance: The K48-linked ubiquitin smear is functionally significant. Proteins polyubiquitinated with K48-linked chains are primarily targeted for proteasomal degradation [43]. The smear thus represents a snapshot of the cellular pool of proteins marked for destruction, playing critical roles in regulating cell cycle progression, differentiation, stress response, and apoptosis [43].
- Specificity of the Signal: K48-linkage specific antibodies, such as Cell Signaling Technology's #4289, are highly specific. They detect polyubiquitin chains formed specifically through Lys48 linkages and demonstrate only slight cross-reactivity with linear polyubiquitin chains, with no observed cross-reactivity to monoubiquitin or polyubiquitin chains formed through linkages to other lysine residues (e.g., K63) [43]. This specificity confirms that the observed smear is likely a true K48-linked ubiquitination signal.
Optimization and Troubleshooting Guide
A diffuse smear can also result from suboptimal experimental conditions. The table below outlines common issues and verified solutions to achieve high-quality, interpretable data.
Table 1: Troubleshooting Guide for K48 Ubiquitin Western Blots
| Problem | Potential Cause | Recommended Optimization |
|---|---|---|
| High background noise | Incomplete blocking or non-specific antibody binding | Extend blocking time; optimize antibody dilution in a compatible buffer (e.g., 5% non-fat dry milk/TBST) [14]. |
| Non-specific bands | Antibody cross-reactivity or over-exposure | Include appropriate controls; titrate primary and secondary antibodies to determine the optimal dilution [43] [14]. |
| Weak or absent signal | Low ubiquitination levels or inefficient transfer | Use a positive control (e.g., recombinant K48-linked ubiquitin chains); validate protein transfer efficiency with a total protein stain [44] [32]. |
| Excessive smear intensity | Protein overload or signal saturation | Reduce the total protein load (e.g., 15-20 µg for neuronal isolates); use a fluorescent detection system with a wider linear dynamic range [44]. |
| Uneven or "smiley" bands | Excessive heat during electrophoresis | Run gels at a lower voltage (e.g., 180 V) to prevent overheating and ensure even protein migration [44]. |
Critical Experimental Considerations
- Sample Preparation: Use an appropriate extraction buffer like RIPA buffer (containing protease inhibitors) for homogenization. Centrifuge samples at 20,000 x g for 20 minutes at 4°C to isolate solubilized proteins [44].
- Protein Quantitation and Loading: Accurately determine protein concentration using an assay (e.g., BCA, Bradford) with a standard curve yielding an R-squared value ≥ 0.99. Load samples sequentially across gels to avoid bias from uneven transfer [44].
- Normalization Strategy: For quantitative analysis, Total Protein Normalization (TPN) is now considered the gold standard over housekeeping proteins (HKPs). TPN is less variable, provides a larger dynamic range, and is increasingly required by top scientific journals [32]. This can be achieved with a total protein stain or a fluorogenic labeling method used prior to immunoblotting [32].
Detailed Protocol for Quantitative K48 Ubiquitin Detection
This protocol is optimized for quantitative fluorescent western blotting (QFWB), which provides a linear detection profile superior to traditional chemiluminescence [44].
Sample Preparation and Gel Electrophoresis
- Homogenization: Manually macerate tissue and homogenize in a suitable cold extraction buffer (e.g., RIPA) at approximately 1:10 (w/v) [44].
- Clarification: Centrifuge homogenate at 20,000 x g for 20 min at 4°C. Collect the supernatant containing solubilized protein [44].
- Protein Assay: Determine protein concentration using a colorimetric assay. Ensure all samples are compared against the same standard curve [44].
- Sample Denaturation: Dilute protein samples to desired concentration (e.g., 15 µg in 10 µL dH₂O). Add 5 µL of loading buffer, vortex, and heat at 98°C for 2 minutes [44].
- Gel Electrophoresis:
- Use a 4-12% Bis-Tris gradient gel for broad molecular weight separation [44].
- Choose MES running buffer for better resolution of proteins between 3.5-160 kDa [44].
- Load molecular weight standards and samples. Run gel at 80 V for 4 min, then increase to 180 V for approximately 50 min, or until the dye front reaches the gel's foot [44].
Protein Transfer and Total Protein Normalization
- Membrane Transfer: Use a semi-dry or wet transfer system to transfer proteins from the gel to a PVDF or nitrocellulose membrane [45].
- Total Protein Stain (for Loading Control Gel): For the second of two identical gels, use a total protein stain to visualize all transferred proteins. This serves as the superior loading control for normalization [44] [32].
Immunoblotting with K48-linkage Specific Antibody
- Blocking: Incubate the membrane in a blocking buffer (e.g., 5% non-fat dry milk in TBST) for 1 hour at room temperature to prevent non-specific antibody binding [45].
- Primary Antibody Incubation: Incubate membrane with the anti-K48-linkage specific primary antibody (e.g., Cell Signaling Technology #4289 at 1:1000 dilution [43] or abcam ab140601 at 1/1000 dilution [14]) in blocking buffer overnight at 4°C with gentle agitation.
- Washing: Wash the membrane 3 times for 5 minutes each with TBST.
- Secondary Antibody Incubation: Incubate with a fluorescently-labeled secondary antibody (e.g., IRDye 800CW Goat anti-Rabbit) at a manufacturer-recommended dilution in blocking buffer for 1 hour at room temperature, protected from light.
- Final Washing: Wash the membrane 3 times for 5 minutes each with TBST, then briefly rinse with TBS or PBS to remove residual detergent.
Imaging and Quantitation
- Fluorescent Imaging: Image the membrane using a digital fluorescence imager (e.g., LI-COR Odyssey).
- Quantitative Analysis:
- Use the imaging system's software to quantify the signal intensity for both the K48 ubiquitin smear and the total protein stain in each lane.
- For the K48 signal, you may quantify the entire smear or defined regions of interest.
- Normalize the K48 ubiquitin signal to the total protein signal in each respective lane to obtain the relative abundance of K48-linked ubiquitinated proteins [44] [32].
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for K48-linkage Specific Ubiquitin Research
| Reagent | Function & Specificity | Example Product & Details |
|---|---|---|
| K48-linkage Specific Antibody | Detects polyubiquitin chains formed specifically via Lys48 linkages; minimal cross-reactivity [43]. | CST #4289: Rabbit polyclonal, validated for WB (1:1000). Specific for K48 chains, slight cross-reactivity with linear chains only [43]. |
| K48-linkage Specific mAb | Monoclonal antibody offering high lot-to-lot consistency for detecting K48 linkages [14]. | abcam ab140601 [EP8589]: Recombinant rabbit monoclonal, validated for WB, ICC/IF, IHC-P, Flow Cytometry. Works on Human, Mouse, Rat samples [14]. |
| Fluorescent Secondary Antibody | Enables quantitative fluorescent detection with a wide linear dynamic range, superior to ECL [44]. | IRDye 800CW Goat anti-Rabbit IgG: Used with LI-COR Odyssey imager for quantitative Western blotting [44]. |
| Total Protein Stain/Label | Provides accurate loading control by staining all proteins on the membrane, used for Total Protein Normalization (TPN) [32]. | Invitrogen No-Stain Protein Labeling Reagent: Fluorescent label for fast, sensitive TPN without destaining steps [32]. |
| Recombinant K48-linked Ubiquitin Chains | Essential positive control to verify antibody specificity and experimental workflow [6] [14]. | Recombinant K48-linked-Ub2-7: Used to confirm specific antibody recognition in Western blot assays [14]. |
Workflow and Pathway Visualization
K48 Ubiquitin Western Blot Workflow
The following diagram illustrates the optimized quantitative workflow for detecting K48-linked polyubiquitin chains, from sample preparation to analysis.
The K48-Linked Ubiquitin-Proteasome Pathway
This diagram outlines the core biological pathway of K48-linked ubiquitination, from substrate tagging to proteasomal degradation.
The classic "smear" observed in K48-linkage specific ubiquitin western blots is a feature, not a flaw. It provides valuable insight into the dynamic landscape of proteins marked for proteasomal degradation. By understanding its biological significance and implementing the optimized quantitative protocols and troubleshooting strategies outlined here—particularly the adoption of total protein normalization and fluorescent detection—researchers can transform this complex pattern into robust, reproducible, and publication-quality data. Adherence to these methods ensures accurate interpretation of the critical role K48-linked ubiquitination plays in cellular regulation and disease.
The detection of K48-linked polyubiquitin chains is fundamental to advancing our understanding of the ubiquitin-proteasome system, a critical pathway in cellular homeostasis and a major target in drug development. However, immunoblotting for this specific ubiquitin linkage is notoriously challenging, often resulting in weak or no signal. This failure frequently stems from two core areas: the loss of ubiquitin signal during sample preparation due to enzymatic activity, and suboptimal antibody application. This application note provides a detailed, evidence-based framework to troubleshoot and optimize these specific aspects of your immunoblotting protocol, ensuring the reliable detection of K48-linked ubiquitination required for high-quality research.
The Critical Role of Lysis Conditions in Preserving K48-Ubiquitin Signal
The inherent reversibility of ubiquitin modifications makes them exceptionally vulnerable to degradation during and after cell lysis. The preservation of K48-linked chains is therefore not merely a preliminary step but a decisive factor for successful detection.
Inhibition of Deubiquitylases (DUBs)
DUBs are cysteine proteases that rapidly cleave ubiquitin chains upon cell lysis. Their effective inhibition is non-negotiable. While 5-10 mM concentrations of alkylating agents like N-ethylmaleimide (NEM) or Iodoacetamide (IAA) are commonly used, evidence suggests these may be insufficient. Research indicates that up to 10-fold higher concentrations (50-100 mM) are necessary to fully preserve the ubiquitination status of certain proteins and specific ubiquitin chain types, such as K63 and M1 linkages [34]. For K48-linked chains, which are also DUB substrates, this robust inhibition is equally critical.
- NEM vs. IAA: NEM is generally more stable and is the preferred choice, especially if subsequent mass spectrometry is planned, as the IAA-cysteine adduct can interfere with tryptic peptide analysis [34]. IAA is light-sensitive and degrades within minutes.
- Chelating Agents: Include EDTA or EGTA (5-10 mM) in the lysis buffer to chelate heavy metal ions, thereby inactivating metalloproteinase-family DUBs [34].
A summary of recommended DUB inhibitors is provided in the table below:
Table 1: Deubiquitylase (DUB) Inhibitors for Lysis Buffer
| Inhibitor | Recommended Concentration | Key Considerations |
|---|---|---|
| N-Ethylmaleimide (NEM) | 20-100 mM | More stable; preferred for mass spectrometry. Essential for preserving K63/M1 chains [34]. |
| Iodoacetamide (IAA) | 20-100 mM | Light-sensitive; degrades rapidly. Adducts may interfere with MS [34]. |
| EDTA/EGTA | 5-10 mM | Inactivates metalloproteinase DUBs. A mandatory addition to the lysis buffer [34]. |
Proteasome Inhibition
As K48-linked polyubiquitin primarily targets proteins for degradation by the 26S proteasome, inhibiting the proteasome is often required to accumulate sufficient levels of modified proteins for detection. MG132 is a widely used proteasome inhibitor.
- Application: Treat cells with 10-20 µM MG132 for 4-6 hours prior to lysis. This blocks degradation and allows ubiquitylated proteins to accumulate.
- Caveat: Prolonged treatment (e.g., 12-24 hours) can induce cytotoxic stress responses that may indirectly alter ubiquitylation patterns [34].
For a complete overview, the following workflow diagram outlines the key steps in sample preparation to preserve ubiquitin signals.
Optimizing Immunoblotting with K48 Linkage-Specific Antibodies
Even with perfectly preserved samples, a weak signal can result from suboptimal antibody use. K48 linkage-specific antibodies, such as the rabbit monoclonal [EP8589] (ab140601) from Abcam or the polyclonal #4289 from Cell Signaling Technology, are powerful but require precise optimization [14] [46].
Antibody and Buffer Optimization
The key to a strong, specific signal lies in the fine-tuning of antibody concentrations and buffer composition.
Antibody Dilution: A critical parameter to balance signal and background.
- For ab140601, a starting dilution of 1:1000 in 5% non-fat dry milk (NFDM)/TBST is recommended for western blot, with validated use from 1:200 to 1:1000 [14].
- For #4289, a 1:1000 dilution is standard [46].
- Troubleshooting: High background necessitates increasing the dilution (e.g., to 1:2000 or 1:5000), while no signal may require a more concentrated antibody (e.g., 1:200-1:500) [47] [48].
Blocking Buffer: The choice of blocking agent can significantly impact the signal-to-noise ratio.
- 5% Non-Fat Dry Milk (NFDM) is a common and effective blocker used with ab140601 [14]. However, it can sometimes mask the signal of low-abundance targets.
- Alternative Blockers: For weaker signals or high background, try 3-5% BSA in TBST or commercial, purified protein blockers (e.g., Thermo Scientific StartingBlock Buffer), which may offer superior sensitivity [48].
Wash Stringency: Incorporate 0.05-0.1% Tween-20 in your TBST wash buffer. Increase wash volume, duration (e.g., 3 x 10 minutes), and agitation to remove non-specifically bound antibody effectively [47] [49]. For persistent background, a high-salt wash (e.g., with 0.5 M NaCl) can be effective.
Gel Electrophoresis and Transfer Considerations
The physical separation and transfer of polyubiquitylated proteins present unique challenges due to their high molecular weight and heterogeneous size.
- Gel Selection: Use 8-10% Tris-Glycine or Tris-Acetate gels for optimal resolution of high molecular weight smears typical of polyubiquitylated proteins. Tris-Acetate systems are particularly recommended for proteins above 150 kDa [34] [48].
- Transfer Conditions: Ensure complete transfer of large ubiquitin conjugates.
- Membrane: Use 0.45 µm PVDF membrane, pre-activated in methanol [50].
- Buffer: For tank transfer, consider adding 0.01-0.05% SDS to the transfer buffer to facilitate the elution of large proteins from the gel.
- Conditions: Use a high current (200 mA) for 60-90 minutes on ice to prevent overheating and ensure efficient transfer [50].
Table 2: Key Optimization Parameters for K48-Ubiquitin Immunoblotting
| Parameter | Recommended Starting Point | Optimization Tips for Weak Signal |
|---|---|---|
| Primary Antibody Dilution | 1:1000 | For weak signal: try 1:200 - 1:500. For high background: try 1:2000 - 1:5000 [14] [47]. |
| Blocking Buffer | 5% NFDM / TBST | If over-blocking is suspected, switch to 3-5% BSA or a commercial, sensitive blocker [48]. |
| Wash Stringency | 3 x 5 min with 0.05% TBST | Increase to 3 x 10 min with 0.1% TBST. For stubborn background, use a high-salt wash buffer [47]. |
| Gel Percentage | 8-10% | Use Tris-Acetate gels for superior resolution of high molecular weight smears (>150 kDa) [34] [48]. |
| Transfer Time/Current | 200 mA for 60 min | For proteins >100 kDa, extend time to 90 min. Always perform on ice or with a cooling unit [50]. |
The interrelationship between sample preparation and immunoblotting, and the iterative process of optimization, can be visualized as follows.
The Scientist's Toolkit: Key Research Reagents
Successful detection of K48-linked ubiquitin relies on a suite of specific reagents. The table below details essential materials and their functions.
Table 3: Essential Research Reagents for K48-Ubiquitin Immunoblotting
| Reagent | Function / Application | Example |
|---|---|---|
| K48 Linkage-Specific Antibody | Primary antibody for detecting K48-polyUb chains in WB, IHC, IF. | Anti-Ubiquitin (K48) [EP8589] (ab140601) [14]; K48-linkage Specific Polyubiquitin Antibody #4289 [46] |
| DUB Inhibitors | Preserve ubiquitin chains in cell lysates by alkylating DUB active sites. | N-Ethylmaleimide (NEM); Iodoacetamide (IAA) [34] |
| Proteasome Inhibitor | Allows accumulation of K48-ubiquitylated proteins destined for degradation. | MG132 [34] |
| HRP-Conjugated Secondary Antibody | For chemiluminescent detection of the primary antibody. | Goat Anti-Rabbit IgG (H&L) (HRP) [14] |
| Enhanced Chemiluminescent (ECL) Substrate | A sensitive substrate for HRP, crucial for detecting low-abundance ubiquitin signals. | SuperSignal West Atto (Thermo Fisher) [48] |
Mastering the detection of K48-linked polyubiquitin chains requires a dual-focused strategy that addresses both the lability of the modification and the technical nuances of immunoblotting. By implementing robust lysis protocols featuring high concentrations of DUB inhibitors and systematically optimizing antibody conditions, researchers can overcome the common pitfalls of weak or absent signals. The protocols and data-driven recommendations provided here serve as a comprehensive guide for obtaining reliable, high-quality data, thereby accelerating research in protein degradation, cell signaling, and targeted protein degradation therapeutics.
Minimizing High Background and Non-Specific Bands
In the specific context of K48-linked ubiquitin research, achieving clean Western blot results is paramount for accurate data interpretation. K48-linked polyubiquitin chains primarily target proteins for degradation by the 26S proteasome, making their specific detection crucial in studies of protein turnover, cell cycle regulation, and proteostasis [3]. However, the high sensitivity required to detect these often low-abundance modifications makes K48 immunoblotting particularly susceptible to high background and non-specific bands, which can obscure critical results and lead to erroneous conclusions. This application note provides a detailed, systematic framework to troubleshoot and resolve these issues, enabling researchers to generate reliable, publication-quality data specific to linkage-specific ubiquitin studies.
Troubleshooting High Background and Non-Specific Bands
High background and non-specific bands typically arise from a limited set of common issues within the Western blotting workflow. The table below summarizes the primary causes and their corresponding solutions, which are detailed in the subsequent protocol section.
Table 1: Summary of Common Issues and Solutions for High Background and Non-Specific Bands
| Issue | Primary Cause | Recommended Solution |
|---|---|---|
| Uniform High Background | Incomplete blocking of the membrane [51] [52] | Optimize blocking agent, concentration, and incubation time [53] [51]. |
| Excessive antibody concentration [54] [52] | Titrate both primary and secondary antibodies to find the optimal dilution [53] [24]. | |
| Inadequate washing [51] | Increase wash number, duration, and include detergent [53] [52]. | |
| Non-Specific Bands | Low antibody specificity or cross-reactivity [54] | Validate antibody specificity; incubate primary antibody at 4°C [54] [55]. |
| Incomplete blocking [54] | Use an engineered blocking buffer designed to reduce non-specific binding [54]. | |
| Membrane handling issues | Ensure membrane does not dry out; use fresh, valid membranes [51] [52]. |
The logical relationship between the common problems and the corrective actions in the Western blot workflow can be visualized as a troubleshooting flowchart.
Detailed Experimental Protocols
Optimization of Blocking Conditions
Objective: To prevent non-specific binding of antibodies to the membrane, thereby reducing high background.
Materials:
- Blocking agent (e.g., Bovine Serum Albumin (BSA) or non-fat dry milk)
- Tris-Buffered Saline (TBS) or Phosphate-Buffered Saline (PBS)
- Tween-20 detergent
- Orbital shaker
Method:
- Prepare Blocking Buffer: Freshly prepare a 1-5% (w/v) solution of your blocking agent in TBS or PBS. For K48-linkage specific ubiquitin detection, BSA is often preferred as it minimizes interference [51] [52]. Add 0.1% Tween-20 (TBST or PBST) to enhance blocking efficiency.
- Block Membrane: Following the transfer step, place the membrane in the blocking buffer. Ensure the membrane is fully submerged and agitate on an orbital shaker.
- Optimize Incubation: Block for a minimum of 1 hour at room temperature. If background persists, extend the blocking time to 2 hours or overnight at 4°C [51] [52].
- Probe Membrane: Proceed directly to the primary antibody incubation step without washing the membrane.
Notes: Always prepare blocking buffer fresh to avoid bacterial contamination, which can cause high background. If using a phospho-specific antibody, BSA is strongly recommended over milk, as casein in milk is a phosphoprotein and can increase background [52].
Antibody Titration and Incubation
Objective: To identify the lowest antibody concentration that provides a strong specific signal with minimal background.
Materials:
- Primary antibody (e.g., anti-Ubiquitin (linkage-specific K48) antibody [EP8589] (ab140601))
- HRP- or fluorophore-conjugated secondary antibody
- Blocking buffer (as optimized in Protocol 3.1)
- Wash buffer (e.g., TBST)
Method:
- Prepare Antibody Dilutions: Using the vendor's recommended dilution as a starting point, prepare a series of primary antibody dilutions in blocking buffer (e.g., 1:500, 1:1000, 1:2000, 1:5000).
- Incubate with Primary Antibody: Apply the diluted primary antibody to individual strips of your membrane. Incubate for 1 hour at room temperature with agitation. For best results, especially with problematic antibodies, incubate overnight at 4°C, as lower temperatures can significantly reduce non-specific binding [54] [52].
- Wash Membrane: Perform 3-5 washes for 5-10 minutes each with a generous volume of wash buffer (e.g., TBST) under agitation [53] [51].
- Incubate with Secondary Antibody: Dilute the HRP- or fluorophore-conjugated secondary antibody in blocking buffer. A starting dilution of 1:10,000 to 1:20,000 is often effective. Incubate for 1 hour at room temperature with agitation.
- Wash Membrane: Repeat the wash procedure from step 3.
- Detect and Analyze: Proceed with detection. The optimal dilution is the one that yields the strongest target signal with the cleanest background.
Notes: A control blot without the primary antibody can help determine if the secondary antibody is contributing to background [52].
Validation of Antibody Specificity for K48-Linked Ubiquitin
Objective: To confirm that the detected signal originates specifically from K48-linked ubiquitin chains.
Method:
- Genetic Controls: The most robust validation involves comparing signals from wild-type cell lines or tissues with those from knockout or knockdown models where the target protein is absent [55]. The absence of the band in the knockout sample confirms specificity.
- Peptide Competition: Pre-incubate the primary antibody with a molar excess of the antigenic peptide used to generate the antibody. The specific band should be significantly reduced or eliminated compared to the untreated antibody control.
- Linkage Specificity Check: For K48-linkage specific antibodies, validate using a panel of defined recombinant ubiquitin chains (e.g., K11-, K48-, K63-linked). The antibody should react strongly only with the K48-linked chains and show minimal to no cross-reactivity with other linkage types, as demonstrated in specificity tests for antibodies like ab140601 [14].
Quantitative Analysis and Normalization
For quantitative Western blotting, especially when assessing changes in ubiquitination levels, proper normalization is critical to account for experimental variability.
Table 2: Comparison of Normalization Methods for Quantitative Western Blotting
| Normalization Method | Principle | Advantages | Limitations | Recommendation for Ubiquitin Research |
|---|---|---|---|---|
| Housekeeping Proteins (HKPs) | Normalizes to a constitutively expressed protein (e.g., β-actin, GAPDH, α-tubulin) [24] [55]. | Well-established; easy to implement. | Expression can vary under experimental conditions [24] [55]; signal can saturate at common loading amounts [24]. | Must validate HKP stability for each experimental condition. Use with caution in treatments affecting cellular proteostasis. |
| Total Protein Normalization (TPN) | Normalizes the target signal to the total protein loaded in each lane, using stains like Ponceau S or fluorescent labels [24] [55]. | More stable and reliable as it doesn't rely on a single protein; wide dynamic range [24]. | Ponceau S is less sensitive; post-transfer staining may interfere with immunodetection if not removed [55]. | Highly recommended. Use fluorescent total protein stains (e.g., No-Stain Protein Labeling Reagent) for superior linearity and dynamic range [24]. |
Key Consideration for Quantitation: Ensure that both the target protein band and the normalization control band (HKP or total protein) are within the linear range of detection and are not saturated. Oversaturated bands invalidate quantitation [24] [55]. Always perform a dilution series of your sample to establish the linear range for your specific protein targets.
The Scientist's Toolkit: Essential Research Reagents
The following table lists key reagents and their optimized applications for achieving clean and specific detection in ubiquitin Western blotting.
Table 3: Key Research Reagent Solutions for Ubiquitin Immunoblotting
| Reagent | Function/Description | Application Note |
|---|---|---|
| K48 Linkage-Specific Antibody (e.g., Anti-Ubiquitin (linkage-specific K48) [EP8589] (ab140601)) [14] | Rabbit monoclonal antibody specific for K48-linked polyubiquitin chains. | Validated for WB, ICC/IF, IHC-P, Flow Cytometry; shows specificity against K48 linkages over K6, K11, K27, K29, K33, and K63 [14]. |
| BSA (Bovine Serum Albumin) | Protein-based blocking agent. | Preferred over milk for blocking when detecting post-translational modifications like ubiquitination; reduces risk of cross-reactivity [51] [52]. |
| Engineered Blocking Buffers (e.g., Azure Chemi/Fluorescent Blot Blocking Buffer) [54] | Specially formulated buffers designed to minimize non-specific antibody binding. | Can be superior to standard BSA or milk for difficult antibodies, as they are designed to enhance specific interactions [54]. |
| Tween-20 | Non-ionic detergent. | Added to wash buffers (e.g., at 0.1%) to help remove unbound and non-specifically bound antibodies, reducing background [53] [51]. |
| Nitroc ellulose Membrane | Low-fluorescence membrane with high protein binding capacity. | Can yield lower background than PVDF membranes and is recommended if high background persists with PVDF [51] [52]. |
| HRP Chemiluminescent Substrate (e.g., SuperSignal West Dura) [24] | Enhanced chemiluminescent (ECL) substrate for horseradish peroxidase. | Ideal for quantitative work due to its wide dynamic range, long signal half-life, and reduced tendency to saturate compared to ultra-sensitive substrates [24]. |
| Total Protein Stain (e.g., No-Stain Protein Labeling Reagent, Ponceau S) [24] [55] | Reagent for staining all proteins on a membrane for normalization. | Provides a more reliable loading control than housekeeping proteins. Fluorescent stains offer superior linearity and dynamic range [24]. |
Common Artifacts and How to Avoid Them
Immunoblotting with K48 linkage-specific ubiquitin antibodies is a powerful technique for studying the ubiquitin-proteasome pathway, a critical regulator of protein degradation in cellular processes. However, the accurate detection of K48-linked polyubiquitin chains is susceptible to specific experimental artifacts. This application note details common pitfalls encountered during this process and provides validated protocols to ensure data reliability, enabling researchers to draw confident conclusions about proteasomal targeting and protein turnover.
Common Artifacts and Mitigation Strategies
The table below summarizes the primary artifacts, their causes, and key solutions for K48 linkage-specific ubiquitin immunoblotting.
Table 1: Common Artifacts in K48 Linkage-Specific Ubiquitin Immunoblotting
| Artifact | Potential Cause | Impact on Data | Solution |
|---|---|---|---|
| Non-Specific Band Detection | Antibody cross-reactivity with other ubiquitin chain types (e.g., linear, K63) or non-ubiquitinated proteins [56]. | False-positive signals; misinterpretation of K48-ubiquitinated targets. | Validate antibody specificity using linkage-defined ubiquitin standards (see Protocol 1) [14] [4]. |
| Smearing or High Background | Incomplete protein transfer, over-saturation of signal, or poor membrane blocking. | Obscures specific bands; difficult to interpret molecular weights. | Optimize transfer conditions; titrate primary antibody; use high-quality blocking buffers. |
| Loss of Ubiquitin Signal | Inadequate deubiquitinase (DUB) inhibition during lysis, leading to chain disassembly [17]. | Underestimation of K48-ubiquitination; false negatives. | Include effective DUB inhibitors (e.g., NEM, CAA) in lysis buffer (see Protocol 2) [17]. |
| Multiple Non-Specific Bands | Non-specific antibody binding or protein aggregation. | Difficulty identifying true ubiquitin ladder. | Include a ubiquitin mutant (K48R) negative control; ensure proper sample preparation and denaturation. |
Critical Experimental Protocols
Protocol 1: Verification of Antibody Specificity Using Defined Ubiquitin Chains
This protocol is essential for confirming that an antibody specifically recognizes K48-linked chains and does not cross-react with other linkage types [14] [4].
Materials:
- K48 linkage-specific antibody (e.g., Cell Signaling Technology #4289 [56] or Abcam ab140601 [14])
- Wild-type ubiquitin
- Set of ubiquitin lysine-to-arginine (K-to-R) mutants (e.g., K6R, K11R, K27R, K29R, K33R, K48R, K63R)
- Set of "K-Only" ubiquitin mutants (only one lysine remains, others are arginine)
- E1 activating enzyme, E2 conjugating enzyme, E3 ligase (as required)
- 10X E3 Ligase Reaction Buffer (500 mM HEPES, pH 8.0, 500 mM NaCl, 10 mM TCEP)
- MgATP solution (100 mM)
- SDS-PAGE and Western Blot equipment
Procedure:
- Set Up Ubiquitination Reactions: Prepare two sets of nine 25 µL reactions in microcentrifuge tubes.
- Set 1 (K-to-R Mutants): Reactions containing wild-type ubiquitin and each of the seven K-to-R mutants.
- Set 2 (K-Only Mutants): Reactions containing wild-type ubiquitin and each of the seven K-Only mutants.
- A negative control for each set replaces MgATP with dH₂O.
Reaction Master Mix (for a single 25 µL reaction):
- dH₂O (to a final volume of 25 µL)
- 10X E3 Ligase Reaction Buffer: 2.5 µL
- Ubiquitin (or mutant): 1 µL (~100 µM final)
- MgATP Solution: 2.5 µL (10 mM final)
- Substrate protein: X µL (5-10 µM final)
- E1 Enzyme: 0.5 µL (100 nM final)
- E2 Enzyme: 1 µL (1 µM final)
- E3 Ligase: X µL (1 µM final)
Incubation: Incubate all reaction tubes in a 37°C water bath for 30-60 minutes.
Termination: Stop the reactions by adding 25 µL of 2X SDS-PAGE sample buffer.
Analysis:
- Separate the reaction products by SDS-PAGE and transfer to a membrane.
- Perform a Western blot using the K48 linkage-specific antibody.
- Interpretation:
- In Set 1, only the reaction with the K48R mutant should show an absence of polyubiquitin chains, confirming K48-linkage dependence.
- In Set 2, only the reaction with the K48-Only mutant should produce polyubiquitin chains, verifying linkage specificity [4].
Protocol 2: Preservation of Ubiquitin Chains in Cell Lysates
Rapid and effective inhibition of deubiquitinases (DUBs) during cell lysis is critical to prevent the degradation of ubiquitin chains before analysis [17].
Materials:
- Lysis buffer (e.g., RIPA) pre-chilled to 4°C
- DUB inhibitors: N-Ethylmaleimide (NEM) or Chloroacetamide (CAA). Prepare fresh stock solutions.
Procedure:
- Prepare Lysis Buffer: Add DUB inhibitors to the lysis buffer immediately before use. Recommended final concentrations are 10-25 mM for NEM or 5-10 mM for CAA [17].
- Cell Lysis: Place culture dishes on ice. Aspirate media and wash cells with ice-cold PBS.
- Immediately add the pre-chilled lysis buffer containing DUB inhibitors to the cells.
- Harvest Cells: Scrape adherent cells and transfer the lysate to a pre-chilled microcentrifuge tube.
- Incubate: Keep the lysates on ice for 10-30 minutes with occasional vortexing.
- Clarify: Centrifuge the lysates at >12,000 × g for 15 minutes at 4°C.
- Transfer: Carefully transfer the supernatant (cleared lysate) to a new tube. Keep samples on ice and proceed with protein quantification and immunoblotting promptly.
Visualizing the Experimental Workflow
The diagram below outlines the core workflow for a reliable K48 ubiquitin immunoblotting experiment, integrating key steps to prevent artifacts.
The Scientist's Toolkit: Key Research Reagents
The following table lists essential reagents for studying K48-linked ubiquitination, as featured in the protocols and literature.
Table 2: Essential Reagents for K48-Linked Ubiquitin Research
| Reagent | Function / Role in Experiment | Example |
|---|---|---|
| K48 Linkage-Specific Antibodies | Detect and validate K48-linked polyubiquitin chains in Western blot. | Rabbit mAb [EP8589] (ab140601) [14]; Polyclonal Antibody #4289 [56] |
| Defined Ubiquitin Mutants | Control for antibody specificity and determine chain linkage (K-to-R, K-Only) [4]. | Ubiquitin K48R, K63R; Ubiquitin K48-Only |
| Deubiquitinase (DUB) Inhibitors | Preserve endogenous ubiquitin chains during cell lysis by inhibiting DUB activity [17]. | N-Ethylmaleimide (NEM), Chloroacetamide (CAA) |
| Tandem Ubiquitin Binding Entities (TUBEs) | Affinity matrices to enrich and stabilize polyubiquitinated proteins from lysates, protecting them from DUBs [37]. | K48-linkage specific TUBEs |
| E1, E2, E3 Enzyme System | Reconstitute ubiquitination in vitro to generate defined chains for antibody validation [4]. | Commercial in vitro ubiquitination kits |
Validating Your Results: Confirming Specificity and Functional Relevance
Using Ubiquitin Mutants (K-to-R and K-Only) for Linkage Confirmation
Protein ubiquitination is a fundamental post-translational modification that regulates virtually all aspects of eukaryotic cell biology, with K48-linked polyubiquitin chains constituting approximately 40% of cellular ubiquitin linkages and serving as the primary signal for proteasomal degradation [6] [57]. The specific linkage type refers to the lysine residue (or N-terminal methionine) through which ubiquitin monomers connect, forming structurally and functionally distinct polyubiquitin chains. Accurate identification of K48-linked ubiquitination is therefore crucial for understanding proteasomal targeting and protein turnover mechanisms central to cellular homeostasis.
The complexity of the "ubiquitin code" presents a significant analytical challenge, as proteins can be modified with various chain types, including all seven lysine-based linkages (K6, K11, K27, K29, K33, K48, K63) and M1-linked linear chains [57]. To address this, the use of ubiquitin mutants in biochemical assays has emerged as a powerful method for definitive linkage confirmation, particularly when investigating specific linkages such as K48 with linkage-specific antibodies [4].
Theoretical Basis for Using Ubiquitin Mutants
The Ubiquitin Mutant Strategy
The strategic use of ubiquitin mutants leverages the enzyme specificity within the ubiquitination cascade to experimentally control chain formation. This approach utilizes two complementary sets of ubiquitin mutants:
- Lysine-to-Arginine (K-to-R) Mutants: These ubiquitin variants contain arginine substitutions at all but one lysine residue, preventing chain formation through the mutated positions. For example, the K48R mutant cannot form chains via K48 linkages [4].
- Lysine-Only (K-Only) Mutants: These variants retain only a single lysine residue with all other lysines mutated to arginine, restricting chain formation exclusively through that specific lysine [4].
When employed in well-controlled in vitro ubiquitination reactions, these mutants provide a genetic tool to establish or confirm the linkage specificity of ubiquitin-processing enzymes, including E2 conjugating enzymes, E3 ligases, and linkage-specific antibodies [4].
Molecular Mechanism of Linkage Determination
The experimental power of this approach stems from the steric and chemical constraints imposed by the mutations. Arginine substitution preserves the positive charge while eliminating the nucleophilic ε-amino group necessary for isopeptide bond formation with the C-terminal glycine of an incoming ubiquitin molecule. This effectively blocks chain elongation through the mutated residue without significantly altering ubiquitin's overall structure or function in most cases [4].
Recent structural biology studies have reinforced the validity of this approach. Cryo-EM analyses of HECT E3 ligases like Tom1 have revealed how specific ubiquitin-binding architectures contribute to linkage specificity, demonstrating that the molecular environment surrounding the growing ubiquitin chain directly influences which lysine residues are accessible for linkage formation [58].
Experimental Protocol for Linkage Confirmation
This protocol details the use of ubiquitin mutants to confirm K48 linkage specificity in conjunction with immunoblotting using K48 linkage-specific antibodies.
Materials and Reagents
Table 1: Essential Reagents for Ubiquitin Linkage Determination
| Reagent | Specifications | Function in Experiment |
|---|---|---|
| E1 Enzyme | 5 µM stock concentration [4] | Activates ubiquitin in an ATP-dependent manner, initiating the ubiquitination cascade |
| E2 Enzyme | 25 µM stock concentration [4] | Determines linkage specificity in partnership with E3 ligase |
| E3 Ligase | 10 µM stock concentration [4] | Provides substrate specificity and collaborates with E2 to determine linkage type |
| Wild-type Ubiquitin | 1.17 mM (10 mg/mL) [4] | Positive control for normal ubiquitination |
| Ubiquitin K-to-R Mutants | K6R, K11R, K27R, K29R, K33R, K48R, K63R; 1.17 mM [4] | Identify lysine residues essential for chain formation |
| Ubiquitin K-Only Mutants | K6-only, K11-only, K27-only, K29-only, K33-only, K48-only, K63-only; 1.17 mM [4] | Verify specific lysine residue sufficient for chain formation |
| 10X E3 Ligase Reaction Buffer | 500 mM HEPES (pH 8.0), 500 mM NaCl, 10 mM TCEP [4] | Provides optimal pH, ionic strength, and reducing conditions |
| MgATP Solution | 100 mM [4] | Essential energy cofactor for E1-mediated ubiquitin activation |
| K48-linkage Specific Antibody | e.g., Cell Signaling Technology #4289 or Abcam ab140601 [59] [14] | Selective detection of K48-linked polyubiquitin chains |
Step-by-Step Procedure
Experimental Setup
Reaction Preparation: Set up two parallel sets of nine ubiquitination reactions (25 µL each) in microcentrifuge tubes:
- Set A: Wild-type ubiquitin + seven K-to-R mutants + negative control (no ATP)
- Set B: Wild-type ubiquitin + seven K-Only mutants + negative control (no ATP)
Reaction Assembly: Combine components in the following order to minimize non-specific interactions:
- dH₂O (to reach 25 µL final volume)
- 2.5 µL 10X E3 Ligase Reaction Buffer (1X final concentration)
- 1 µL ubiquitin or ubiquitin mutant (approximately 100 µM final concentration)
- 2.5 µL MgATP Solution (10 mM final concentration)
- Substrate protein (volume dependent on stock concentration; 5-10 µM final)
- 0.5 µL E1 Enzyme (100 nM final)
- 1 µL E2 Enzyme (1 µM final)
- E3 Ligase (volume dependent on stock concentration; 1 µM final) [4]
Incubation: Transfer all reaction tubes to a 37°C water bath and incubate for 30-60 minutes to allow ubiquitination to proceed [4].
Reaction Termination and Sample Processing
Termination Method Selection:
Western Blot Analysis:
- Separate proteins by SDS-PAGE and transfer to PVDF or nitrocellulose membrane
- Perform immunoblotting using K48 linkage-specific antibody (e.g., 1:1000 dilution for Cell Signaling Technology #4289) [59]
- Include appropriate controls for antibody specificity
Figure 1: Experimental workflow for ubiquitin linkage confirmation using K-to-R and K-Only mutants. The pathway shows the parallel processing of mutant sets leading to definitive K48 linkage confirmation.
Expected Results and Interpretation
Table 2: Interpretation of Experimental Outcomes with Ubiquitin Mutants
| Ubiquitin Variant | Expected Result if K48-Specific | Interpretation |
|---|---|---|
| Wild-type Ubiquitin | Robust polyubiquitin chain formation | Positive control for ubiquitination capacity |
| K48R Mutant | Only monoubiquitination observed; no chains | K48 residue is essential for chain formation |
| Other K-to-R Mutants (K6R, K11R, K27R, K29R, K33R, K63R) | Polyubiquitin chain formation | These lysines are not required for chain formation |
| K48-Only Mutant | Polyubiquitin chain formation | K48 residue alone is sufficient for chain formation |
| Other K-Only Mutants (K6-only, K11-only, etc.) | No polyubiquitin chain formation | These individual lysines cannot support chain formation |
The power of this dual-mutant approach is its ability to provide both loss-of-function (K-to-R) and gain-of-function (K-Only) evidence for linkage specificity. A true K48-linked chain will only form when K48 is available, both in the wild-type context and in the K48-only mutant, while being abolished specifically in the K48R mutant [4].
Figure 2: Data interpretation workflow for K48 linkage confirmation. The pattern of chain formation across different mutant conditions provides definitive evidence for K48 linkage specificity.
Technical Considerations and Troubleshooting
Antibody Validation and Specificity
When using K48 linkage-specific antibodies for immunoblotting, it is crucial to validate their performance. Commercial antibodies such as Cell Signaling Technology #4289 and Abcam ab140601 have demonstrated specificity for K48-linked chains, though some may show slight cross-reactivity with linear polyubiquitin chains [59] [14]. The mutant strategy described herein serves as an excellent internal control for antibody specificity within the experiment.
Recent methodological advances highlight that proper antibody validation should include testing against a panel of defined ubiquitin chains, as some commercial reagents may exhibit unexpected cross-reactivities [57]. The inclusion of both K48R and K48-only mutants in your experimental design provides this validation internally.
Addressing Experimental Complexities
Several technical challenges may arise during linkage determination experiments:
- Mixed Linkage Chains: If all K-to-R mutants still produce chains, this may indicate mixed linkage or linear (M1-linked) chains [4]. In this case, supplementary approaches such as mass spectrometry or specialized DUB treatments may be necessary [6].
- Branched Chains: Recent research has revealed that heterotypic branched chains containing multiple linkage types are common in cells, with K48/K63-branched ubiquitin comprising up to 20% of all K63 linkages [6]. This complexity may yield ambiguous results with the basic mutant approach.
- Enzyme Specificity: Some E3 ligases exhibit preference for specific linkages. For example, the HECT ligase Tom1 contains a "structural ubiquitin" binding site that contributes to its K48 linkage specificity [58]. Understanding your E3 ligase's inherent preferences can help interpret results.
Advanced Applications and Recent Methodological Developments
The ubiquitin mutant approach has evolved beyond basic linkage confirmation to address increasingly complex questions in ubiquitin signaling:
Integration with Novel Tools
Recent studies have combined ubiquitin mutants with engineered deubiquitinases (enDUBs) to achieve substrate-specific linkage editing in live cells [60]. For example, enDUBs with K48 specificity can be used to validate findings from in vitro ubiquitination assays in cellular contexts.
Studying Branched Ubiquitin Chains
The ubiquitin mutant strategy has been adapted to investigate the complexity of branched ubiquitin chains. A 2024 study used ubiquitin interactor pulldowns coupled with mass spectrometry to identify novel K48/K63-branched ubiquitin interactors, revealing branched chain-specific readers including PARP10, UBR4, and HIP1 [6]. Such studies typically require more sophisticated mutant combinations but operate on the same fundamental principle.
Cell-Based Ubiquitin Replacement
Cutting-edge research now employs ubiquitin replacement cell lines in which endogenous ubiquitin is replaced with specific ubiquitin mutants. A 2025 study used this approach to profile system-wide impacts of ablating individual ubiquitin linkages, revealing essential roles for K48-, K63- and K27-linkages in cell proliferation [61]. This represents a powerful extension of the in vitro mutant approach to physiological contexts.
The use of ubiquitin K-to-R and K-Only mutants remains an indispensable methodological approach for confirming ubiquitin linkage specificity, particularly when validating K48-linked chain formation in conjunction with linkage-specific antibodies. This protocol provides a robust framework for researchers to definitively establish linkage specificity, with the complementary mutant sets providing both loss-of-function and gain-of-function evidence. As the complexity of ubiquitin signaling continues to expand, with recent discoveries of branched chains and non-canonical linkages, this fundamental approach maintains its relevance as a cornerstone of ubiquitin biochemistry.
Incorporating Linkage-Specific Deubiquitinases (DUBs) for Validation
Within the ubiquitin-proteasome system, the specific topology of polyubiquitin chains determines the functional fate of modified proteins. K48-linked polyubiquitin chains predominantly target substrates for proteasomal degradation, making them a critical focus in protein regulation studies [62] [63]. Validation of K48-linkage specificity in immunoblotting experiments requires rigorous methodological approaches, among which incorporation of linkage-specific deubiquitinases (DUBs) stands as a gold standard. This application note details the systematic use of linkage-specific DUBs as validation tools within immunoblotting protocols for K48-linked ubiquitin research, providing researchers with a framework to confirm antibody specificity and interpret experimental results with greater confidence.
The diversity of ubiquitin chain linkages—including seven lysine-based chains (K6, K11, K27, K29, K33, K48, K63) and linear (M1) chains—creates a complex "ubiquitin code" that demands highly specific detection methods [63] [64]. While commercial K48-linkage specific antibodies are available, their validation remains paramount. Linkage-specific DUBs, which cleave particular ubiquitin chain types with high fidelity, provide an exceptional tool for experimental validation when incorporated into immunoblotting workflows [27].
Theoretical Foundation: Ubiquitin Code and DUB Specificity
The Ubiquitin Signaling Landscape
Ubiquitination involves a sequential enzymatic cascade utilizing E1 (activating), E2 (conjugating), and E3 (ligating) enzymes to attach ubiquitin to substrate proteins. Polyubiquitin chains form when additional ubiquitin molecules attach to one of seven lysine residues or the N-terminal methionine of the previously conjugated ubiquitin [62] [63]. The resulting chain topology determines the molecular outcome for the modified protein:
- K48-linked chains: Primarily target proteins for proteasomal degradation [62] [63]
- K63-linked chains: Regulate non-proteolytic functions including signal transduction, DNA repair, and endosomal trafficking [63]
- K11-linked chains: Involved in cell division and endoplasmic-reticulum-associated degradation (ERAD) [63]
- M1-linked linear chains: Important in NF-κB signaling and inflammatory responses [64]
This linkage-specific functional specialization necessitates precise analytical tools for accurate interpretation of ubiquitination experiments.
Deubiquitinating Enzymes as Linkage-Specific Tools
Deubiquitinating enzymes (DUBs) counterbalance ubiquitination by removing ubiquitin chains from modified proteins. Approximately 100 DUBs encoded in the human genome display varying degrees of linkage specificity [64] [65]. While some DUB families (particularly USPs) show broad specificity, others (especially OTU family members) exhibit remarkable linkage selectivity [63] [64]. This inherent specificity provides the foundation for their use as validation tools in ubiquitin research.
Table 1: Linkage Specificities of Selected Deubiquitinases
| DUB | Family | Reported Linkage Specificity | Cellular Function |
|---|---|---|---|
| OTUB1 | OTU | Preferentially cleaves K48 linkages [65] | Regulation of proteasomal degradation |
| OTUD1 | OTU | Specific for K63 linkages [65] | DNA damage response |
| OTULIN | OTU | Highly specific for M1-linear linkages [64] | Negative regulator of NF-κB signaling |
| CYLD | USP | Preferentially cleaves K63 and M1 linkages [64] | Tumor suppressor, NF-κB regulation |
| Cezanne | OTU | Specific for K11 linkages [64] | Regulation of cell cycle and transcription |
| Ubp2 | USP | Preferentially cleaves K63 linkages [63] | Regulation of protein localization |
| Ubp3 | USP | Preferentially cleaves K48 linkages [63] | Proteasomal degradation regulation |
| USP21 | USP | Broad specificity across multiple linkages [65] | General housekeeping DUB functions |
Experimental Protocol: DUB Validation for K48-Linked Ubiquitin Detection
Reagent Preparation and Optimization
Critical Reagent Components
The following reagents are essential for implementing DUB validation protocols:
- Linkage-specific DUBs: Recombinant purified OTUB1 (K48-specific) and OTUD1 (K63-specific) serve as excellent controls for specificity validation [65]. Commercial sources provide these with varying purity levels.
- DUB reaction buffer: 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 5 mM DTT. DTT maintains reducing conditions essential for cysteine protease DUB activity [64].
- Protease inhibitors: Include N-ethylmaleimide (NEM) or iodoacetamide (IAA) in cell lysis buffers to inhibit endogenous DUB activity during sample preparation [27].
- K48-linkage specific antibody: Commercial antibodies such as Cell Signaling Technology #4289 or Abcam ab140601 have been validated for K48 specificity [62] [14].
Sample Preparation Considerations
Proper sample preparation is critical for preserving ubiquitin signals:
- Rapid lysis: Use hot SDS-lysis (95°C for 10 minutes) to instantly denature proteins and prevent artefactual deubiquitination [27].
- DUB inhibition: Include 10-20 mM NEM or 5-10 mM IAA in lysis buffers to inhibit endogenous DUB activity [27].
- Proteasome inhibition: When studying proteasomal targets, treat cells with 10-20 μM MG-132 for 4-6 hours before lysis to accumulate ubiquitinated species [66].
DUB Validation Workflow Implementation
The following diagram illustrates the complete experimental workflow for validating K48-linkage specificity using DUBs:
Step-by-Step Protocol
Prepare protein samples using appropriate lysis conditions with DUB inhibitors as described in section 3.1.2.
Divide lysates into four equal aliquots (minimum 20 μg per treatment) and pre-clear by brief centrifugation.
Set up DUB treatment reactions as follows:
- Tube 1: Protein lysate + DUB reaction buffer (no enzyme control)
- Tube 2: Protein lysate + K48-specific DUB (e.g., OTUB1, 100-500 nM)
- Tube 3: Protein lysate + K63-specific DUB (e.g., OTUD1, 100-500 nM)
- Tube 4: Protein lysate + broad-specificity DUB (e.g., USP21, 100-500 nM)
Incubate reactions at 37°C for 30-60 minutes. Optimize incubation time based on preliminary experiments to achieve partial rather than complete cleavage.
Terminate reactions by adding 4X SDS-PAGE sample buffer and heating at 95°C for 5 minutes.
Perform Western blotting using standard protocols with K48-linkage specific antibodies. Include loading controls to normalize for protein content.
Expected Results and Interpretation
The following diagram illustrates the expected Western blot results and their interpretation:
Valid K48-specific detection is confirmed when:
- K48-specific DUB treatment abolishes or dramatically reduces the K48-immunoreactive signal
- K63-specific DUB treatment causes minimal or no reduction in K48 signal
- Broad-specificity DUB treatment causes partial reduction of signal
- The no-DUB control shows strong smeared signal characteristic of polyubiquitinated proteins
Advanced Applications and Methodological Considerations
Integration with Other Ubiquitin Validation Methods
While DUB validation provides strong evidence for linkage specificity, combining this approach with complementary methods strengthens conclusions:
- Ubiquitin mutant panels: Utilize ubiquitin mutants (K-to-R and K-only) in transfection experiments to confirm chain linkage [4].
- Mass spectrometry: Employ quantitative proteomics to directly identify linkage types [63] [65].
- Tandem ubiquitin-binding entities (TUBEs): Use TUBEs with linkage specificity to enrich particular chain types before immunoblotting [27].
Troubleshooting Common Experimental Issues
Table 2: Troubleshooting DUB Validation Experiments
| Problem | Potential Cause | Solution |
|---|---|---|
| No signal change with any DUB | DUB enzyme inactive; Incorrect buffer conditions | Verify DUB activity with fluorogenic substrate; Check DTT concentration and pH |
| Complete signal loss with all DUBs | Excessive DUB concentration or incubation time | Titrate DUB concentration (50-500 nM); Reduce incubation time (15-45 min) |
| High background in no-DUB control | Incomplete DUB inhibition during lysis | Increase NEM concentration; Use rapid SDS-lysis method |
| Non-specific banding pattern | Antibody cross-reactivity | Include additional controls with linkage-specific DUBs for other linkages |
| Poor protein transfer | High molecular weight ubiquitin conjugates | Extend transfer time; Use Tris-acetate buffers for better high MW transfer |
Quantitative Considerations for DUB Validation
For rigorous quantification of DUB effects:
- Normalize band intensity to loading controls
- Analyze multiple exposure times to avoid signal saturation
- Use chemiluminescent substrates with wide dynamic range
- Consider digital imaging systems for quantitative analysis
The cleavage efficiency of linkage-specific DUBs can vary significantly. OTU family DUBs typically show higher linkage specificity but may have slower kinetics compared to more promiscuous USP family DUBs [64] [65].
Research Reagent Solutions
Table 3: Essential Reagents for DUB Validation Studies
| Reagent Category | Specific Examples | Function/Application | Key Considerations |
|---|---|---|---|
| K48-linkage Specific Antibodies | CST #4289 [62]; Abcam ab140601 [14] | Detection of K48-linked polyubiquitin chains | Validate specificity with DUB cleavage; Check species reactivity |
| Linkage-Specific DUBs | OTUB1 (K48-specific) [65]; OTUD1 (K63-specific) [65] | Validation of antibody specificity; Chain linkage determination | Verify activity with control substrates; Optimize concentration |
| DUB Inhibitors | N-ethylmaleimide (NEM); Iodoacetamide (IAA) [27] | Preservation of ubiquitin signals during sample preparation | Use fresh preparations; Include in all lysis buffers |
| Ubiquitin Mutants | K48R ubiquitin; K48-only ubiquitin [4] | Control for antibody specificity; Determination of chain linkage | Use with transfection systems; Confirm expression levels |
| Proteasome Inhibitors | MG-132; Bortezomib [66] | Accumulation of proteasomal substrates | Titrate for cell type; Limit exposure time to reduce toxicity |
| Ubiquitin Binding Domains | Tandem-repeated UBA domains [27] | Enrichment of polyubiquitinated proteins | Consider linkage specificity; Optimize binding conditions |
Incorporating linkage-specific deubiquitinases into immunoblotting protocols provides a robust method for validating K48-linked ubiquitin antibody specificity. This approach leverages the inherent biochemical specificity of DUBs to create a controlled experimental framework for verifying detection of authentic K48-linked polyubiquitin chains. When properly implemented with appropriate controls and optimized conditions, DUB validation significantly enhances the reliability and interpretation of ubiquitin immunoblotting data, ultimately strengthening conclusions about the role of K48-linked ubiquitination in cellular processes.
The expanding toolkit of well-characterized linkage-specific DUBs, combined with improved commercial antibodies and standardized protocols, continues to advance the field of ubiquitin research. By adopting these validation methodologies, researchers can address the critical challenge of antibody specificity that often complicates interpretation of ubiquitin immunoblots, particularly in the context of drug development where accurate target validation is essential.
Within the framework of a broader thesis on immunoblotting protocols for ubiquitin research, the precise detection of specific polyubiquitin linkages is paramount. Among the various chain types, K48-linked polyubiquitin chains are of particular interest due to their primary role in targeting proteins for proteasomal degradation [67] [68]. The reliability of such research hinges critically on the performance of linkage-specific antibodies. This application note provides a detailed, side-by-side analysis of a K48-linkage specific antibody, summarizing its quantitative performance data and presenting robust, validated protocols for its application in Western blotting.
K48-Linked Ubiquitin and Proteasomal Degradation
K48-linked ubiquitin chains constitute a major degradation signal in the ubiquitin-proteasome system (UPS) [68]. The proteasome recognizes these chains through intrinsic receptors such as Rpn10 and Rpn13 [3] [69]. Recent structural studies have revealed that the human proteasome can also recognize more complex signals like K11/K48-branched ubiquitin chains, but it does so by engaging multiple receptor sites, underscoring the fundamental importance of the K48 linkage as a core degradation signal [3]. The diagram below illustrates the pathway from ubiquitination to proteasomal degradation.
The following table summarizes the key characteristics and performance data of Cell Signaling Technology's K48-linkage Specific Polyubiquitin Antibody #4289 for use in Western blotting applications [67].
Table 1: Performance Specifications of K48-Linkage Specific Polyubiquitin Antibody #4289
| Parameter | Specification |
|---|---|
| Antibody Name | K48-linkage Specific Polyubiquitin Antibody #4289 |
| Reactivities | All Species Expected |
| Application | Western Blotting |
| Recommended Dilution | 1:1000 |
| Sensitivity | Endogenous |
| Specificity | Detects polyubiquitin chains formed by Lys48 linkage. Demonstrates slight cross-reactivity with linear polyubiquitin chains. No cross-reactivity with monoubiquitin or other lysine-linked chains (K6, K11, K27, K29, K33, K63). |
| Immunogen | Synthetic peptide corresponding to the Lys48 branch of the human diubiquitin chain. |
| Purification | Protein A and peptide affinity chromatography. |
Detailed Experimental Protocol
Western Blotting Procedure
This protocol is optimized for the detection of endogenous K48-linked polyubiquitin chains using Antibody #4289 [67].
Materials & Reagents
- Lysis Buffer: Use a RIPA or similar denaturing lysis buffer supplemented with 1x protease inhibitor cocktail and 5-10 mM N-ethylmaleimide (NEM) or chloroacetamide (CAA) to inhibit deubiquitinases (DUBs) and preserve ubiquitin chains [6].
- Primary Antibody: K48-linkage Specific Polyubiquitin Antibody #4289 [67].
- Secondary Antibody: HRP-conjugated anti-rabbit IgG.
- Blocking Buffer: 5% w/v Non-Fat Dry Milk in TBST.
Methodology
- Sample Preparation:
- Lyse cells or tissues directly in an appropriate volume of hot lysis buffer (e.g., 2X Laemmli buffer) and immediately boil for 10 minutes. This rapid denaturation is critical to prevent chain disassembly by DUBs.
- Cool samples and briefly sonicate or pass through a needle to shear genomic DNA.
- Centrifuge at >12,000 x g for 10 minutes to remove insoluble debris.
Gel Electrophoresis and Transfer:
- Load 20-50 µg of total protein per well on a 4-20% gradient SDS-PAGE gel.
- Separate proteins by electrophoresis and transfer to a nitrocellulose or PVDF membrane using standard wet or semi-dry transfer methods.
Immunoblotting:
- Blocking: Incubate the membrane in Blocking Buffer for 1 hour at room temperature with gentle agitation.
- Primary Antibody Incubation: Dilute the K48-linkage Specific Antibody #4289 to 1:1000 in Blocking Buffer or a recommended antibody diluent. Incubate with the membrane overnight at 4°C with gentle agitation.
- Washing: Wash the membrane 3 times for 5 minutes each with TBST.
- Secondary Antibody Incubation: Incubate with HRP-conjugated anti-rabbit IgG (diluted as per manufacturer's instructions) for 1 hour at room temperature.
- Washing: Repeat the TBST wash step 3 times for 5 minutes each.
- Detection: Develop the blot using enhanced chemiluminescence (ECL) reagents and visualize with a digital imager.
Troubleshooting Notes
- High Background: Ensure adequate blocking and washing. Titrate the antibody dilution if necessary.
- Weak or No Signal: Confirm that DUB inhibitors (NEM/CAA) were freshly added to the lysis buffer. Check antibody dilution and ECL reagent sensitivity.
- Specificity Concerns: The antibody may show slight cross-reactivity with linear polyubiquitin chains. For critical experiments, validation with linkage-specific DUBs (e.g., OTUB1 for K48 linkages) is recommended [6].
The Scientist's Toolkit: Essential Research Reagents
The table below lists key reagents and tools essential for research involving K48-linked ubiquitination.
Table 2: Key Research Reagent Solutions for K48-Linked Ubiquitin Research
| Reagent/Tool | Function/Description | Example Use |
|---|---|---|
| K48-linkage Specific Antibodies | Selective detection of K48-linked polyubiquitin chains in techniques like Western blotting. | Detecting endogenous K48-ubiquitinated proteins in cell lysates [67]. |
| Linkage-Specific TUBEs (Tandem Ubiquitin Binding Entities) | High-affinity ubiquitin-binding matrices that can be engineered for linkage specificity to capture polyubiquitinated proteins. | Enriching K48- or K63-ubiquitinated proteins from cell lysates for downstream analysis; used in high-throughput assays [68]. |
| DUB Inhibitors (NEM, CAA) | Cysteine alkylators that inhibit the activity of deubiquitinating enzymes, thereby preserving polyubiquitin chains during sample preparation. | Added to cell lysis buffers to prevent the degradation of ubiquitin chains before analysis [6]. |
| Linkage-Specific DUBs (e.g., OTUB1) | Deubiquitinases that selectively cleave specific ubiquitin linkages. Used as enzymatic tools for linkage validation. | Confirmatory cleavage of K48-linked chains in a specificity assay (UbiCRest) [6]. |
| PROTACs (Proteolysis Targeting Chimeras) | Heterobifunctional molecules that recruit a target protein to an E3 ubiquitin ligase, leading to its K48-linked ubiquitination and degradation. | Inducing targeted degradation of specific proteins of interest to study function [68] [70]. |
Experimental Workflow for Linkage-Specific Analysis
The following diagram outlines a recommended workflow for analyzing K48-linked ubiquitination, integrating the key reagents and the antibody discussed in this note.
Cross-Validation with Complementary Methods (e.g., Ubiquitin Traps, Mass Spectrometry)
The K48-linked polyubiquitin chain is a principal signal for targeting substrate proteins to the 26S proteasome for degradation. The advent of K48-linkage-specific antibodies has been instrumental in deciphering this pathway. However, the complexity of the ubiquitin code, characterized by multiple chain linkage types and architectures, necessitates that data obtained with these antibodies be rigorously cross-validated. Relying solely on immunoblotting can lead to misinterpretation due to potential antibody cross-reactivity or the presence of heterotypic chains. This application note details robust, complementary methodologies to validate K48-linked ubiquitination, thereby ensuring the reliability of conclusions drawn from immunoblotting experiments.
Key Research Reagents for Ubiquitin Analysis
A successful experimental workflow relies on a well-characterized toolkit. The table below summarizes essential reagents for studying K48-linked ubiquitination.
Table 1: Key Research Reagents for K48-Linked Ubiquitin Analysis
| Research Reagent | Function and Application | Examples / Specifics |
|---|---|---|
| K48-linkage Specific Antibodies | Detect K48-linked polyubiquitin chains in techniques like Western blotting, immunoprecipitation, and immunofluorescence. [71] [14] | Rabbit monoclonal [EP8589] (Abcam ab140601); Rabbit polyclonal (CST #4289). |
| Ubiquitin Mutants (Lys-to-Arg) | Determine ubiquitin chain linkage by in vitro reconstitution. A mutation in the specific lysine used for chain formation will abrogate polyubiquitination. [4] | Pan-lysine mutant (K0); Single K-to-R mutants (e.g., K48R). |
| Ubiquitin Mutants (Lys-Only) | Verify chain linkage. Only the mutant with the correct lysine residue will support chain formation in vitro. [4] | Single lysine mutants (e.g., K48-Only). |
| Recombinant Enzymatic System | Reconstitute ubiquitination for linkage determination assays in a controlled environment. [4] | E1 activating enzyme, E2 conjugating enzyme (e.g., Cdc34 for K48), E3 ligase, ATP, and reaction buffer. |
| Tandem Ubiquitin-Binding Entities (TUBEs) | Affinity-based enrichment of ubiquitinated proteins from complex mixtures while protecting chains from deubiquitinases. [72] | Multi-domain proteins with high affinity for ubiquitin chains. |
| AQUA (Absolute Quantification) Peptides | Synthetic, isotopically labeled peptides with a diglycine (diGly) remnant for precise, mass spectrometry-based quantification of ubiquitination sites and linkage abundance. [73] | Peptides corresponding to specific ubiquitin linkage signatures (e.g., K48-diGly). |
In Vitro Linkage Determination with Ubiquitin Mutants
This biochemical approach provides a direct and accessible method to confirm the linkage specificity of an ubiquitination event.
Experimental Protocol
This protocol involves two sequential sets of in vitro ubiquitination reactions [4].
Reaction Setup (Linkage Determination):
- Set up nine 25 µL reactions, each containing:
- 1X E3 Ligase Reaction Buffer (50 mM HEPES pH 8.0, 50 mM NaCl, 1 mM TCEP).
- 100 nM E1 enzyme.
- 1 µM E2 enzyme.
- 1 µM E3 ligase.
- 5-10 µM substrate protein.
- 10 mM MgATP.
- Approximately 100 µM of one of the following ubiquitin types:
- Reaction 1: Wild-type Ubiquitin
- Reactions 2-8: Individual Ubiquitin K-to-R Mutants (K6R, K11R, K27R, K29R, K33R, K48R, K63R)
- Negative Control: Wild-type Ubiquitin, but replace MgATP with dH₂O.
- Incubate at 37°C for 30-60 minutes.
- Terminate reactions by adding SDS-PAGE sample buffer.
- Set up nine 25 µL reactions, each containing:
Reaction Setup (Linkage Verification):
- Repeat step 1, but replace the K-to-R mutants with the set of Ubiquitin K-Only Mutants (K6-Only, K11-Only, etc.).
Analysis:
- Analyze all reaction products by SDS-PAGE and Western blotting using an anti-ubiquitin antibody.
- Interpretation:
- In the K-to-R set, the reaction that fails to form high molecular weight polyubiquitin chains (showing only mono-ubiquitination) identifies the critical lysine for linkage (e.g., K48R indicates K48-linkage) [4].
- In the K-Only set, only the reaction with the ubiquitin mutant containing the correct lysine (e.g., K48-Only) and the wild-type control will form polyubiquitin chains, thereby verifying the linkage [4].
The following diagram illustrates the logical workflow and expected outcomes for this method:
Mass Spectrometry-Based Validation
Mass spectrometry (MS) offers an unbiased, high-resolution strategy to characterize ubiquitination beyond immunoblotting.
Middle-Down/Top-Down with Ion Mobility (IM-MS)
This method separates intact ubiquitin chains based on their size and shape (collision cross-section, CCS), providing information on chain length and linkage-specific conformations.
- Workflow: A mixture of ubiquitin dimers is infused directly into the mass spectrometer via electrospray ionization (ESI). Ions are separated in a drift tube ion mobility (IM) cell filled with an inert gas before mass analysis [73].
- Quantitative Analysis: The IM spectra for each pure di-ubiquitin isomer serves as a reference standard. Using multiple linear regression analysis, the relative abundance of each isomer in a complex biological mixture can be quantified based on its unique CCS profile [73]. This approach has been shown to yield results consistent with the bottom-up AQUA method [73].
- Application: Ideal for analyzing the composition and heterogeneity of ubiquitin chains synthesized in vitro or purified from cellular environments.
Bottom-Up Proteomics and Cross-Linking MS (XL-MS)
These powerful techniques provide deep insights into ubiquitination sites and the architecture of the proteasome itself.
- Bottom-Up Proteomics with AQUA: Ubiquitinated proteins are enriched (e.g., via TUBEs or immunoprecipitation) and digested with trypsin. This generates a characteristic diGly remnant on modified lysines, which are identified by LC-MS/MS. Using synthetic AQUA peptides allows for absolute quantification of specific ubiquitin linkages [72] [73].
- In-Situ Cross-Linking Mass Spectrometry (XL-MS): This method captures protein-protein interactions and structural conformations within their native cellular environment. A cell-permeable cross-linker (e.g., BSP) fixes interacting proteins inside living cells before lysis and affinity purification. The cross-linked peptides are then analyzed by MS, providing distance restraints that can be used for structural modeling [74]. This technique has revealed compartment-specific proteasome interactomes and dynamic states, including interactions with ubiquitin-associated proteins and deubiquitinases like USP15 [74].
The workflow below integrates K48-linkage specific antibodies with downstream MS validation:
To ensure robust and publication-ready results, a multi-faceted approach is recommended. The following table provides a concise summary of the cross-validation methods.
Table 2: Summary of Complementary Methods for Cross-Validation
| Method | Key Application | Key Outcome | Throughput |
|---|---|---|---|
| Ubiquitin Mutants (in vitro) | Linkage determination of a specific substrate-E3 pair. | Identifies the specific lysine linkage used for chain formation. [4] | Low (Targeted) |
| Bottom-Up MS + AQUA | Site-specific mapping and absolute quantification of ubiquitin linkages. | Provides a quantitative profile of endogenous K48-linked ubiquitination sites. [72] [73] | High (Global) |
| Ion Mobility MS (IM-MS) | Conformational and linkage analysis of ubiquitin chains. | Quantifies relative abundance of different ubiquitin chain isomers in a mixture. [73] | Medium |
| In-Situ Cross-Linking MS | Mapping proteasome interactions and architecture in native cellular environments. | Reveals compartment-specific interactors and conformational states of the 26S proteasome. [74] | High (Global) |
In conclusion, while K48-linkage-specific antibodies are invaluable for initial discovery, their data must be fortified with orthogonal evidence. The integration of in vitro biochemical assays with advanced mass spectrometry techniques creates a powerful framework for the unequivocal characterization of K48-linked ubiquitination, ultimately leading to more reliable and impactful scientific findings.
Correlating K48 Signal with Functional Degradation Assays
Ubiquitination is a crucial post-translational modification that regulates diverse cellular processes, with K48-linked polyubiquitin chains serving as the primary signal for proteasomal degradation [75] [76]. This application note provides detailed methodologies for correlating K48 ubiquitination signals with functional degradation outcomes, enabling researchers to validate proteasomal targeting in physiological contexts. We present integrated experimental workflows that combine linkage-specific detection with quantitative degradation kinetics, addressing the critical need for orthogonal validation in ubiquitin-proteasome system (UPS) research.
The following conceptual workflow illustrates the core experimental approach for establishing correlation between K48 ubiquitination and protein degradation:
Core Principles: K48 Ubiquitination and Degradation
K48-linked polyubiquitin chains represent the canonical degradation signal recognized by the 26S proteasome [75] [76]. Unlike other linkage types that mediate non-proteolytic functions, K48 linkages specifically direct substrate proteins to proteasomal degradation through recognition by proteasomal ubiquitin receptors [3]. Recent research has revealed that chain length and topology significantly influence degradation efficiency, with K48-Ub3 identified as the minimal signal for efficient proteasomal targeting [77].
Quantitative Degradation Kinetics of K48-Linked Ubiquitin Chains
Table 1: Intracellular Degradation Kinetics of K48-Ubiquitinated Substrates
| Ubiquitin Chain Type | Minimal Degradation Signal | Degradation Half-Life | Cellular System | Key Findings |
|---|---|---|---|---|
| K48-Ub4-GFP | K48-Ub3 | ~1 minute | RPE-1 cells | Rapid degradation plateau at ~6 min with ~30% GFP remaining [77] |
| K48-Ub4-GFP | - | 1-2.2 minutes | Multiple cell lines (THP-1, U2OS, A549, HeLa, 293T) | Consistent rapid degradation across diverse mammalian cells [77] |
| K48 vs. K63 chains | K48-Ub3 | K48: Minutes; K63: Minimal degradation | RPE-1 cells | K63-ubiquitinated substrate rapidly deubiquitinated rather than degraded [77] |
| K48/K63-branched chains | Substrate-anchored chain dependent | Varies by branch configuration | RPE-1 cells | Branchpoint identity determines degradation vs. deubiquitination outcome [77] |
Experimental Protocols
UbiREAD: Ubiquitinated Reporter Evaluation After Intracellular Delivery
The UbiREAD platform enables systematic comparison of ubiquitin chain degradation capacities by delivering bespoke ubiquitinated proteins into living cells and monitoring their fate at high temporal resolution [77].
Protocol: UbiREAD Degradation Assay
Day 1: Preparation of Ubiquitinated GFP Reporters
- Synthesize Ubiquitinated GFP: Prepare Ub chains of defined length and composition using distal Ub mutants (e.g., K48R for K48 chains) to prevent further elongation [77]
- Conjugate to GFP substrate: Link pre-assembled Ub chains to mono-ubiquitinated GFP degradation substrate (engineered for efficient proteasomal degradation) [77]
- Validate chain integrity: Confirm chain composition and linkage specificity via UbiCRest analysis with linkage-specific deubiquitinases (e.g., OTUB1 for K48 chains) [77] [6]
- Quantify protein concentration: Adjust to 1-2 mg/mL in electroporation-compatible buffer
Day 2: Intracellular Delivery and Kinetic Analysis
- Harvest and wash cells: Use RPE-1, THP-1, or other mammalian cell lines at 70-80% confluence
- Electroporation: Deliver 5-10 µg of ubiquitinated GFP using optimized electroporation parameters (100-150V, 10-20 ms pulse) [77]
- Immediate processing: Aliquot cells for time-point analysis (20 seconds to 20 minutes post-delivery)
- Dual readout:
- Flow cytometry: Fix cells in 4% formaldehyde and analyze GFP fluorescence loss
- In-gel fluorescence: Lyse cells in ice-cold RIPA buffer with protease inhibitors, resolve by SDS-PAGE, and visualize GFP signal
Day 3: Data Analysis and Validation
- Calculate degradation kinetics: Plot remaining GFP fluorescence versus time and determine half-life using non-linear regression
- Confirm proteasome dependence: Include controls with 10 µM MG132 (proteasome inhibitor) and 1 µM TAK243 (E1 inhibitor) [77]
- Assess deubiquitination: Monitor appearance of free GFP band as indicator of competing deubiquitination activity
The experimental workflow for the UbiREAD methodology is visualized below:
Chain-Selective TUBE-Based Ubiquitination Capture
Tandem Ubiquitin Binding Entities (TUBEs) provide an alternative approach for capturing endogenous ubiquitination events with linkage specificity, enabling researchers to monitor ubiquitination of native proteins without requiring substrate purification or recombinant delivery [68].
Protocol: TUBE-Based Capture of Endogenous K48 Ubiquitination
Day 1: Cell Treatment and Lysis
- Treat cells with experimental conditions: Include appropriate controls (e.g., proteasome inhibitors, pathway activators)
- Prepare lysis buffer: 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% NP-40, 1 mM DTT, supplemented with:
- Protease inhibitor cocktail
- 10 mM N-ethylmaleimide (NEM) or 5 mM chloroacetamide (CAA) as deubiquitinase inhibitors [6]
- Lyse cells: Use ice-cold lysis buffer with brief sonication (3 × 5-second pulses) followed by centrifugation at 16,000 × g for 15 minutes at 4°C
Day 1: Ubiquitin Affinity Capture
- Prepare TUBE resin: Use 20 µL K48-TUBE magnetic agarose beads per sample
- Pre-clear lysate: Incubate with control beads for 30 minutes at 4°C
- Ubiquitin capture: Incubate 500 µg lysate with K48-TUBE beads for 2 hours at 4°C with rotation
- Wash beads: 3 × with wash buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.1% NP-40)
Day 2: Detection and Analysis
- Elute bound proteins: Use 2× Laemmli buffer with 5% β-mercaptoethanol at 95°C for 10 minutes
- Western blot analysis:
- Resolve proteins by SDS-PAGE (4-20% gradient gel)
- Transfer to PVDF membrane
- Probe with K48-linkage specific antibody (#4289, Cell Signaling Technology) at 1:1000 dilution [75]
- Detect with HRP-conjugated secondary antibodies and chemiluminescence
- Reprobe membrane: Validate target protein ubiquitination with target-specific antibodies
K48 Linkage-Specific Immunoblotting
Standard immunoblotting with K48-linkage specific antibodies provides a complementary approach for detecting K48 ubiquitination, though this method requires careful optimization to ensure specificity.
Protocol: K48 Linkage-Specific Western Blotting
Sample Preparation
- Lyse cells in RIPA buffer containing:
- 50 mM Tris-HCl (pH 8.0)
- 150 mM NaCl
- 1% NP-40
- 0.5% sodium deoxycholate
- 0.1% SDS
- 5 mM NEM or 10 mM CAA as deubiquitinase inhibitors [6]
- Denature samples: Heat at 95°C for 5 minutes in Laemmli buffer
- Resolve proteins: Use 4-20% gradient SDS-PAGE gels for optimal separation of ubiquitin chains
Immunoblotting
- Transfer to PVDF membrane: Use wet transfer system at 100V for 60 minutes
- Block membrane: 5% non-fat milk in TBST for 1 hour at room temperature
- Primary antibody incubation:
- K48-linkage specific polyubiquitin antibody (#4289, Cell Signaling Technology) [75]
- Dilution: 1:1000 in 5% BSA/TBST
- Incubate overnight at 4°C with gentle agitation
- Secondary antibody incubation:
- HRP-conjugated anti-rabbit IgG
- Dilution: 1:5000 in 5% non-fat milk/TBST
- Incubate 1 hour at room temperature
- Detection: Use enhanced chemiluminescence substrate and image with appropriate system
Table 2: Experimental Optimization Parameters for K48 Ubiquitination Detection
| Parameter | Recommended Conditions | Purpose | Considerations |
|---|---|---|---|
| Deubiquitinase Inhibition | 10 mM NEM or 5 mM CAA | Preserve endogenous ubiquitin chains during processing | NEM more potent but less specific; CAA more specific but less potent [6] |
| Proteasome Inhibition | 10 µM MG132 for 4-6 hours | Accumulate ubiquitinated substrates | Extended treatment (>8 hours) may cause cytotoxic effects [76] |
| Gel Percentage | 4-20% gradient SDS-PAGE | Optimal separation of polyubiquitin chains | Higher percentage gels better for short chains; lower for long chains |
| Antibody Specificity | Validation with linkage-defined standards | Ensure specific K48 linkage detection | K48 antibody #4289 shows slight cross-reactivity with linear chains [75] |
| Detection Method | Chemiluminescence with high-sensitivity substrates | Enhance detection of low-abundance species | Digital imaging systems recommended for quantitative analysis |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for K48 Ubiquitination and Degradation Studies
| Reagent/Solution | Specific Example | Function/Application | Supplier/Reference |
|---|---|---|---|
| K48-linkage Specific Antibody | Polyclonal Antibody #4289 | Detection of K48-linked polyubiquitin chains in Western blot | Cell Signaling Technology [75] |
| Proteasome Inhibitor | MG132 | Reversible proteasome inhibition to stabilize ubiquitinated proteins | Multiple suppliers [77] [76] |
| E1 Inhibitor | TAK243 | Blocks ubiquitin activation, confirming UPS dependence | Multiple suppliers [77] |
| Deubiquitinase Inhibitors | NEM, Chloroacetamide (CAA) | Prevent chain disassembly during processing | Sigma-Aldrich [6] |
| Tandem Ubiquitin Binding Entities (TUBEs) | K48-TUBE, Pan-TUBE | Affinity enrichment of linkage-specific ubiquitinated proteins | LifeSensors [68] |
| Ubiquitin Chain Standards | Defined K48-Ub2, Ub3, Ub4 | Method validation and quantification controls | Enzymatically synthesized [77] [6] |
| UbiCRest Kit | Linkage-specific DUB panel | Ubiquitin chain linkage validation | LifeSensors [77] [6] |
Data Interpretation and Troubleshooting
Correlating K48 Signal with Degradation Outcomes
Successful correlation requires orthogonal validation approaches:
- Quantitative comparison: Normalize K48 ubiquitination signals to degradation rates across multiple time points
- Threshold determination: Establish the minimal K48 ubiquitination level required to initiate degradation for your specific protein of interest
- Kinetic profiling: Compare the timing of K48 signal appearance with degradation onset
Common Technical Challenges and Solutions
- Weak K48 signal: Optimize deubiquitinase inhibition and increase protein loading; consider TUBE-based enrichment prior to immunoblotting [68]
- High background in Western blots: Include linkage-defined ubiquitin standards to validate antibody specificity [75]
- Discrepancy between K48 signal and degradation: Investigate potential competing modifications (e.g., branched chains, alternative linkages) that may alter degradation efficiency [77] [3]
- Cell-type specific differences: Validate assays in multiple cell lines, as degradation kinetics can vary between cell types [77]
The integrated methodologies presented herein enable robust correlation between K48 ubiquitination signals and functional degradation outcomes. By combining quantitative degradation kinetics with linkage-specific detection approaches, researchers can validate the functional consequences of K48 ubiquitination in physiologically relevant contexts. These protocols support critical applications in drug discovery, particularly for characterizing targeted protein degradation therapeutics such as PROTACs, and in fundamental research elucidating the complexities of the ubiquitin-proteasome system.
Conclusion
Mastering K48-linked ubiquitin immunoblotting requires a meticulous approach that integrates rigorous sample preservation, optimized electrophoretic separation, and robust validation. By understanding the foundational biology, adhering to a detailed methodological protocol, proactively troubleshooting common issues, and confirming specificity through complementary techniques, researchers can reliably decode this critical degradation signal. As research progresses to encompass complex ubiquitin architectures like branched chains, these refined immunoblotting skills will remain essential for advancing our understanding of protein homeostasis in health, disease, and therapeutic development.
References
- 1. K48-linkage Specific Polyubiquitin Antibody #4289 [https://www.cellsignal.com/products/primary-antibodies/k48-linkage-specific-polyubiquitin-antibody/4289?srsltid=AfmBOorS5JyGQFju-h4XlDI-jdc8OR22aY4__L_X4cIkscc0c6j4AjpJ]
- 2. Breaking the K48-Chain: Linking Ubiquitin Beyond Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11730971/]
- 3. Structural basis of K11/K48-branched ubiquitin chain ... [https://www.nature.com/articles/s41467-025-64719-x]
- 4. Protocol for Determining Ubiquitin Chain Linkage [https://www.rndsystems.com/resources/protocols/determine-ubiquitin-chain-linkage]
- 5. How to Better Study Ubiquitination [https://www.ptglab.com/news/blog/how-to-better-study-ubiquitination/?srsltid=AfmBOooBqNLlYlAlwphj0dvYZTYNcY9GLuC_JCe7DoGzDCDndDG_T6N5]
- 6. K48- and K63-linked ubiquitin chain interactome reveals ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11109483/]
- 7. K48-Linked Ubiquitination Contributes to Nicotine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8003133/]
- 8. Ubiquitin Chain Editing Revealed by Polyubiquitin Linkage ... [https://www.sciencedirect.com/science/article/pii/S0092867408009598]
- 9. UbiREAD deciphers proteasomal degradation code of ... [https://www.sciencedirect.com/science/article/pii/S1097276525001522]
- 10. K48- and K63-linked ubiquitin chain interactome reveals ... [https://www.life-science-alliance.org/content/7/8/e202402740/tab-figures-data]
- 11. K48-linkage Specific Polyubiquitin Antibody #4289 [https://www.cellsignal.com/products/primary-antibodies/k48-linkage-specific-polyubiquitin-antibody/4289?srsltid=AfmBOoo9jj3siHuBGg788Q9Aqc2Bn5qXE1dneQtV2umO0EayqLMq-Ejt]
- 12. Beyond K48 and K63: non-canonical protein ubiquitination [https://cmbl.biomedcentral.com/articles/10.1186/s11658-020-00245-6]
- 13. Ubiquitin chain editing revealed by polyubiquitin linkage- ... [https://pubmed.ncbi.nlm.nih.gov/18724939/]
- 14. Anti-Ubiquitin (linkage-specific K48) antibody [EP8589] [https://www.abcam.com/en-us/products/primary-antibodies/ubiquitin-linkage-specific-k48-antibody-ep8589-ab140601]
- 15. Alexa Fluor® 647 Anti-Ubiquitin (linkage-specific K48) ... [https://www.abcam.com/en-us/products/primary-antibodies/alexa-fluor-647-ubiquitin-linkage-specific-k48-antibody-ep8589-ab204476]
- 16. Anti-Ubiquitin (linkage-specific K48) antibody [EP8589] [https://www.abcam.com/en-us/products/primary-antibodies/ubiquitin-linkage-specific-k48-antibody-ep8589-bsa-and-azide-free-ab271911]
- 17. K48- and K63-linked ubiquitin chain interactome reveals ... [https://www.life-science-alliance.org/content/7/8/e202402740]
- 18. K48-linkage Specific Polyubiquitin Antibody #4289 [https://www.cellsignal.com/products/primary-antibodies/k48-linkage-specific-polyubiquitin-antibody/4289?srsltid=AfmBOoqJdfWO5b2N0IsPv2r9rdLJRJuIboaphHSbRjQy8sEgyYp8lk66]
- 19. PE Anti-Ubiquitin (linkage-specific K48) antibody [EP8589] [https://www.abcam.com/en-us/products/primary-antibodies/pe-ubiquitin-linkage-specific-k48-antibody-ep8589-ab303017]
- 20. SpyTag/SpyCatcher-mediated protein ubiquitination to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12434616/]
- 21. Ubiquitination - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/ubiquitination]
- 22. K48-linkage Specific Polyubiquitin Antibody #4289 [https://www.cellsignal.com/products/primary-antibodies/k48-linkage-specific-polyubiquitin-antibody/4289?srsltid=AfmBOoq1T1sJwCJl0A8J6MRyAAk5Z6-hNdUglb-wSsRxXqeOvS4NQGO0]
- 23. K48-linked ubiquitination and protein degradation regulate ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3411168/]
- 24. Quantitative Western Blot Analysis [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/quantitative-western-blot-analysis.html]
- 25. A systematic approach to quantitative Western blot analysis [https://www.sciencedirect.com/science/article/pii/S000326971930750X]
- 26. K48-linkage Specific Polyubiquitin Antibody #4289 [https://www.cellsignal.com/products/primary-antibodies/k48-linkage-specific-polyubiquitin-antibody/4289?srsltid=AfmBOoq2eU42XiUtzLkOvJFeg_okeOWSPglJbweP_tQUaclpbGymk-S3]
- 27. Optimising methods for the preservation, capture and ... [https://www.sciencedirect.com/science/article/pii/S0006291X15305015]
- 28. Method for Measuring the Activity of Deubiquitinating ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4542631/]
- 29. Proteomics-Based Identification of DUB Substrates Using ... [https://www.sciencedirect.com/science/article/pii/S2451945620303779]
- 30. Molecular mechanisms and potential targeting strategies of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12511930/]
- 31. K48-linkage Specific Polyubiquitin Antibody #4289 [https://www.cellsignal.com/products/primary-antibodies/k48-linkage-specific-polyubiquitin-antibody/4289?srsltid=AfmBOopiT4KEP8pRvTyY3vx4jB-zAfH2u88uYnbG5LXYH9mQoyqXvzxq]
- 32. 2024 Guide to Quantitative Western Blot Publication [https://www.thermofisher.com/blog/life-in-the-lab/2023-guide-to-quantitative-western-blot-publication/]
- 33. Quantitative Western Blot Overview [https://bio.licor.com/bio/help/empiria_studio/software/empiria_studio/quantitative-blots/quantitative-blot-overview.html]
- 34. Optimising methods for the preservation, capture and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4709362/]
- 35. K48-linkage Specific Polyubiquitin Antibody #4289 [https://www.cellsignal.com/products/primary-antibodies/k48-linkage-specific-polyubiquitin-antibody/4289?srsltid=AfmBOorIUDjygHJqQe6yA5Fyhf64SfYgCdqmJa6Cv7TG8Ox9_2ALTrQH]
- 36. Ub-K48 Polyclonal Antibody (PA5-120616) [https://www.thermofisher.com/antibody/product/Ub-K48-Antibody-Polyclonal/PA5-120616]
- 37. Unraveling chain specific ubiquitination in cells using tandem ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12217038/]
- 38. K48-linkage Specific Polyubiquitin Antibody #4289 [https://www.cellsignal.com/products/primary-antibodies/k48-linkage-specific-polyubiquitin-antibody/4289?srsltid=AfmBOorBZutiB2TW5YhFpaNXPt413Cg-pCl-lHIDRiFfLqeGfnEzRr7J]
- 39. The Design of a Quantitative Western Blot Experiment [https://pmc.ncbi.nlm.nih.gov/articles/PMC3971489/]
- 40. K48-linkage Specific Polyubiquitin Antibody #4289 [https://www.cellsignal.com/products/primary-antibodies/k48-linkage-specific-polyubiquitin-antibody/4289?srsltid=AfmBOop3x-ySt-m2hVuRF5rNqHq_-tT67YyR3oXPq_oP1sFce-y_TB8w]
- 41. Ubiquitination detection techniques - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC10625345/]
- 42. Anti-Ub-K48 Antibodies | Invitrogen [https://www.thermofisher.com/antibody/primary/target/ub-k48]
- 43. K48-linkage Specific Polyubiquitin Antibody #4289 [https://www.cellsignal.com/products/primary-antibodies/k48-linkage-specific-polyubiquitin-antibody/4289?srsltid=AfmBOoo9MKedHPBo2WdCVZRk7ZYYI0BVUWlnC7ZwUEE7OBGZ-JXwsBrk]
- 44. A Guide to Modern Quantitative Fluorescent Western ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4354265/]
- 45. How to Run a Western Blot Assay: An Illustrated Diagram [https://www.bio-techne.com/applications/western-blotting/western-blot-illustrated-assay]
- 46. K48-linkage Specific Polyubiquitin Antibody #4289 [https://www.cellsignal.com/products/primary-antibodies/k48-linkage-specific-polyubiquitin-antibody/4289?srsltid=AfmBOooIIh0u6FZa6ZJ3FgVF0__2Y6biYE-WROur_qMT62t6CkBZ4YWD]
- 47. 5 Tips for Reducing Non-specific Signal on Western Blots [https://www.biossusa.com/blogs/news/5-tips-for-reducing-non-specific-signal-on-western-blots?srsltid=AfmBOopQhYB9Ibu5EOhGZWVt_h4R0uq8Ktf5Jhg23IKXYkGk0vggA5wP]
- 48. Tips and Tricks | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-gel-electrophoresis-information/western-blot-tips-tricks.html]
- 49. 7 Tips to Improve Your Western Blot [https://www.technologynetworks.com/tn/how-to-guides/7-tips-to-improve-your-western-blot-htg-305298]
- 50. Ubiquitin (linkage-specific Lys 48) Antibody [D12J20] [https://www.selleckchem.com/antibodies/ubiquitin-linkage-specific-lys-48-antibody-d12j20.html]
- 51. A Scientist's Guide to Conquering High Background in ... - clyte [https://www.clyte.tech/post/guide-to-conquering-high-background-in-western-blotting?srsltid=AfmBOopAfULCzZWgVXupjUyoJQlXTFjTmnKgOQ9WxE1bBap00XM3MLLz]
- 52. 5 Tips for Reducing Non-specific Signal on Western Blots [https://www.biossusa.com/blogs/news/5-tips-for-reducing-non-specific-signal-on-western-blots?srsltid=AfmBOooWrno7qnZBfWrJB9QUYyMxQwXxij6cfy6ZgZPkwtdULXrHoW2N]
- 53. Help! Why do my Western blots look terrible? [https://azurebiosystems.com/blog/help-why-do-my-western-blots-look-terrible/]
- 54. Western Blot Troubleshooting: Non-specific bands [https://azurebiosystems.com/blog/western-blot-troubleshooting-what-to-do-about-non-specific-bands/]
- 55. A brief guide to good practices in pharmacological ... [https://www.nature.com/articles/s41401-020-00539-7]
- 56. K48-linkage Specific Polyubiquitin Antibody #4289 [https://www.cellsignal.com/products/primary-antibodies/k48-linkage-specific-polyubiquitin-antibody/4289?srsltid=AfmBOooGLWIvL7VNpexsjc8laRqLiYkbBZN2KJQ1HQYa1HeIgiPSno52]
- 57. The Molecular Toolbox for Linkage Type‐Specific Analysis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12118340/]
- 58. Structural ubiquitin contributes to K48 linkage specificity of ... [https://www.sciencedirect.com/science/article/pii/S2211124725004590]
- 59. K48-linkage Specific Polyubiquitin Antibody #4289 [https://www.cellsignal.com/products/primary-antibodies/k48-linkage-specific-polyubiquitin-antibody/4289?srsltid=AfmBOoofaHNiCwgbZg5sSoyTSIc4jhOGsmBXXxUmlub0JhEov08tPzk6]
- 60. Decoding polyubiquitin regulation of KV7. 1 (KCNQ1) ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12218124/]
- 61. Functional landscape of ubiquitin linkages couples K29- ... [https://link.springer.com/article/10.1038/s44318-025-00599-7]
- 62. K48-linkage Specific Polyubiquitin Antibody #4289 [https://www.cellsignal.com/products/primary-antibodies/k48-linkage-specific-polyubiquitin-antibody/4289?srsltid=AfmBOopTRkOKkeyo22dEXI7uCjWRLJfB9zuBzg9s65JM7OzBaOUmeKJg]
- 63. Ubiquitin Linkage Specificity of Deubiquitinases Determines ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7115106/]
- 64. A Human DUB Protein Array for Clarification of Linkage ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7344921/]
- 65. Neutron-encoded diubiquitins to profile linkage selectivity ... [https://www.nature.com/articles/s41467-023-37363-6]
- 66. An optimized protocol to detect protein ubiquitination ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10050764/]
- 67. K48-linkage Specific Polyubiquitin Antibody #4289 [https://www.cellsignal.com/products/primary-antibodies/k48-linkage-specific-polyubiquitin-antibody/4289?srsltid=AfmBOoo971xjSID6QUdHxOdnKpHJ_rVhtvXY-UsvWWyiONJYyXdve4Bq]
- 68. Unraveling chain specific ubiquitination in cells using ... [https://www.nature.com/articles/s41598-025-07242-9]
- 69. Structural basis for the recognition of K48-linked Ub chain ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6443662/]
- 70. Research Article Optimization of the PROTAC linker region ... [https://www.sciencedirect.com/science/article/pii/S0021925825003692]
- 71. K48-linkage Specific Polyubiquitin Antibody #4289 [https://www.cellsignal.com/products/primary-antibodies/k48-linkage-specific-polyubiquitin-antibody/4289?srsltid=AfmBOooOLA-O5eRZTs2TDBo-vmAMh8lsnjBVRHlNIORFDjGKZEta3_bU]
- 72. Current methodologies in protein ubiquitination characterization [https://cellandbioscience.biomedcentral.com/articles/10.1186/s13578-022-00870-y]
- 73. Quantitative Analysis of Diubiquitin Isomers Using Ion ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10712319/]
- 74. In-situ cross-linking mass spectrometry reveals ... [https://www.nature.com/articles/s41467-025-65752-6]
- 75. K48-linkage Specific Polyubiquitin Antibody #4289 [https://www.cellsignal.com/products/primary-antibodies/k48-linkage-specific-polyubiquitin-antibody/4289?srsltid=AfmBOooMMFw4JdslVcNxuoeyxiMXS-Q2_7aFfem2V4oY6aDX99Ef0Gcm]
- 76. How to Better Study Ubiquitination [https://www.ptglab.com/news/blog/how-to-better-study-ubiquitination/?srsltid=AfmBOorSGLJtCA8hFsRCJA8ayEnhp58E98o2evrAQPb_NSUDIMB0RSyZ]
- 77. UbiREAD deciphers proteasomal degradation code of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7617769/]