Advanced Enrichment Strategies for K11-Linked Polyubiquitin Chains: From Basic Principles to Cutting-Edge Applications
This comprehensive review explores the rapidly evolving field of K11-linked polyubiquitin chain enrichment, addressing critical needs for researchers studying cell cycle regulation, proteostasis, and targeted protein degradation.
Advanced Enrichment Strategies for K11-Linked Polyubiquitin Chains: From Basic Principles to Cutting-Edge Applications
Abstract
This comprehensive review explores the rapidly evolving field of K11-linked polyubiquitin chain enrichment, addressing critical needs for researchers studying cell cycle regulation, proteostasis, and targeted protein degradation. We systematically cover foundational knowledge of K11 chain biology and structural properties, detail current methodological approaches including linkage-specific antibodies, TUBEs, and engineered DUBs, provide troubleshooting guidance for common technical challenges, and present validation frameworks for assessing enrichment specificity and efficiency. With recent structural insights revealing unique K11/K48-branched chain recognition by the 26S proteasome and emerging applications in PROTAC development, this resource equips scientists with practical strategies to advance research in ubiquitin signaling and therapeutic targeting.
Understanding K11-Linked Ubiquitin Chains: Biological Significance and Structural Foundations
Cellular Functions and Biological Roles of K11 Linkages
Ubiquitination is a crucial post-translational modification that controls diverse cellular processes, with specificity determined by the architecture of polyubiquitin chains. Among the various chain topologies, K11-linked polyubiquitination has emerged as a critical regulatory signal, particularly in cell cycle control and protein degradation. Unlike canonical K48-linked chains that primarily target substrates for proteasomal degradation, K11-linked chains exhibit specialized functions and are notably upregulated during specific cellular events such as mitosis. This application note details the cellular functions, quantitative dynamics, and experimental methodologies for studying K11-linked ubiquitin chains, providing researchers with essential tools for advancing research in ubiquitin signaling and proteostasis. The content is framed within enrichment strategies for K11-linked polyubiquitin chain research, offering comprehensive guidance for investigators in this specialized field.
Biological Functions and Significance
Key Cellular Roles of K11-Linked Ubiquitin Chains
Cell Cycle Regulation: K11-linked chains are dramatically upregulated during mitosis and are essential for timely degradation of mitotic regulators [1] [2]. The anaphase-promoting complex/cyclosome (APC/C) utilizes K11 linkages to control the destruction of key cell cycle proteins including Aurora kinases, Polo-like kinase, and KIFC1, thereby ensuring proper mitotic progression [3]. Inhibition of K11-chain formation causes significant mitotic defects and stabilization of APC/C substrates.
Proteasomal Targeting: K11-linked ubiquitin chains function as potent degradation signals for the 26S proteasome [4] [2]. Structural studies reveal that K11/K48-branched ubiquitin chains are recognized as priority degradation signals by the human 26S proteasome through a specialized multivalent recognition mechanism involving RPN2 and RPN10 subunits [5]. This branched topology enables accelerated substrate turnover during proteotoxic stress and cell cycle progression.
Endoplasmic Reticulum-Associated Degradation (ERAD): K11 linkages play a specialized role in ERAD pathways, where they contribute to the degradation of misfolded proteins from the endoplasmic reticulum [4] [6]. The ubiquitin-conjugating enzyme Ubc6 has been identified as a key enzyme that primarily synthesizes K11-linked chains for ERAD substrate targeting.
Proteostasis Maintenance: Under conditions of proteotoxic stress, including proteasome inhibition and heat shock, K11-linked chains accumulate significantly and contribute to the clearance of misfolded proteins and pathological aggregates, such as Huntingtin variants [5] [1]. This highlights their importance in cellular stress response pathways.
Quantitative Analysis of K11 Linkage abundance
Table 1: Quantitative abundance and dynamics of K11-linked ubiquitin chains
| Measurement Parameter | Value | Context | Reference |
|---|---|---|---|
| Overall abundance in asynchronous human cells | ~2% of ubiquitin conjugate pool | Steady-state levels | [1] |
| Abundance in yeast | 28.0% ± 1.4% of polyUb linkages | Second most abundant linkage type | [4] |
| Mitotic upregulation | Dramatic increase | During mitotic exit in human cells | [2] [3] |
| Response to proteasome inhibition | 4-5 fold accumulation | After MG132 treatment in yeast | [4] |
| Branched chain occurrence | 10-20% of Ub polymers | K11/K48-branched topology | [5] |
Experimental Analysis of K11 Linkages
Structural Characterization Techniques
Cryo-EM Analysis of K11/K48-Branched Chain Recognition
Objective: Determine the structural basis for proteasomal recognition of K11/K48-branched ubiquitin chains.
Methodology:
- Complex Reconstitution:
- Reconstitute human 26S proteasome complex with polyubiquitinated substrate (Sic1PY with single lysine K40)
- Use engineered Rsp5-HECTGML E3 ligase with Ub(K63R) variant to prevent K63-linkage formation
- Include preformed RPN13:UCHL5(C88A) complex to minimize disassembly of branched chains [5]
Sample Preparation:
- Introduce dual fluorescence labeling (Alexa647 for Sic1PY, fluorescein for Ub)
- Fractionate by size-exclusion chromatography to enrich medium-length Ub chains (n=4-8)
- Verify complex formation by native gel electrophoresis with Western blotting and fluorescence imaging
Structural Analysis:
- Acquire cryo-EM data and perform extensive classification
- Conduct focused refinements to resolve Ub-binding regions
- Generate 3D reconstructions resembling substrate-bound states (EA, EB, and ED states) [5]
Key Findings: The cryo-EM structures revealed a multivalent recognition mechanism where K11/K48-branched chains simultaneously engage:
- A novel K11-linked Ub binding site at the RPN2/RPN10 groove
- The canonical K48-linkage binding site formed by RPN10 and RPT4/5
- An alternating K11-K48-linkage recognition site on RPN2 [5]
Table 2: Essential reagents for structural analysis of K11-linked ubiquitin chains
| Reagent | Specification | Function | Source/Reference |
|---|---|---|---|
| Rsp5-HECTGML E3 ligase | Engineered variant | Generates K48-linked chains | [5] |
| Ubiquitin (K63R) variant | Site-specific mutant | Prevents K63-linkage formation | [5] |
| Sic1PY substrate | Residues 1-48 of S. cerevisiae Sic1 | Ubiquitination substrate with single lysine | [5] |
| UCHL5(C88A) | Catalytic mutant | Branched chain stabilization without disassembly | [5] |
| K11-linkage specific antibody | Linkage-specific | Detection of K11 chains in Western blot | [2] |
Functional Analysis of K11 Linkages in Cell Cycle
Live-Cell Degradation Assay for APC/C Substrates
Objective: Quantify the contribution of K11 linkages to mitotic substrate degradation kinetics.
Methodology:
- Cell Synchronization and Treatment:
- Synchronize U2OS cells at G1/S boundary using double thymidine block
- Release into fresh medium and collect samples at time points through mitosis
- Monitor mitotic progression by Histone H3 phosphorylation [3]
UBE2S Depletion:
- Transfert with UBE2S-specific siRNA to abrogate K11-chain formation
- Include appropriate non-targeting siRNA controls
Substrate Ubiquitination Analysis:
- Express GFP-tagged substrates (Aurora A/Venus, Aurora B/Venus)
- Purify substrates from mitotic exit cells using immunoprecipitation
- Analyze ubiquitination profiles using K11-linkage specific antibody and GFP antibody [3]
Ubiquitin Chain Restriction (UbiCRest) Analysis:
- Treat purified ubiquitinated substrates with linkage-specific DUBs:
- USP21 (non-specific control)
- Cezanne (K11-specific)
- OTUB1 (K48-specific) [3]
- Monitor chain topology changes by Western blotting
- Treat purified ubiquitinated substrates with linkage-specific DUBs:
Live-Cell Degradation Kinetics:
- Track substrate degradation in single cells using live-cell imaging
- Compare degradation rates between control and UBE2S-depleted cells [3]
Key Findings: UBE2S depletion specifically impaired degradation of APC/C substrates during mitotic exit, despite the presence of significant K48-linked ubiquitination, establishing K11 linkages as critical determinants of degradation timing [3].
Linkage Quantification Methods
Mass Spectrometry-Based Absolute Quantification (Ub-AQUA)
Objective: Precisely quantify the abundance of K11 linkages in complex biological samples.
Methodology:
- Sample Preparation:
- Isolate ubiquitinated proteins by affinity purification
- Digest with trypsin to generate GG-tagged linkage-specific peptides
- Spike in known quantities of heavy isotope-labeled internal standard peptides [4]
LC-MS/MS Analysis:
- Separate peptides by reversed-phase liquid chromatography
- Analyze by tandem mass spectrometry with multiple reaction monitoring (MRM)
- Quantify native peptides by comparison to heavy standards [4]
Data Analysis:
- Calculate absolute amounts of each linkage type
- Normalize to total protein content or ubiquitin pool
- Express as percentage distribution of all ubiquitin linkages [4]
Key Findings: This approach revealed K11 linkages constitute approximately 28% of all polyubiquitin linkages in yeast, making them the second most abundant linkage type after K48 linkages [4].
Research Reagent Solutions
Table 3: Essential research reagents for K11-linked ubiquitin chain studies
| Reagent Category | Specific Examples | Application | Key Features |
|---|---|---|---|
| Linkage-Specific Antibodies | K11-linkage specific monoclonal antibody [2] | Immunoblotting, Immunofluorescence | Specifically recognizes K11 linkages without cross-reactivity |
| Enzymatic Tools | UBE2S (E2 enzyme) [3] | In vitro ubiquitination | K11-specific chain elongation |
| UBE2C (E2 enzyme) [1] [3] | In vitro ubiquitination | Chain initiation with K11 preference | |
| Cezanne (OTUD7B) deubiquitinase [3] | Chain restriction analysis | K11-linkage specific cleavage | |
| Ubiquitin Mutants | Ubiquitin(K11R) [6] | Genetic studies | Prevents K11-linked chain formation |
| Ubiquitin(K63R) [5] | Biochemical studies | Prevents K63 linkage interference | |
| Structural Biology Tools | RPN13:UCHL5(C88A) complex [5] | Cryo-EM studies | Stabilizes branched chains for structural analysis |
| Cell-Based Reporters | Aurora A/Venus fusions [3] | Live-cell degradation assays | Real-time tracking of substrate turnover |
K11-linked polyubiquitin chains represent a sophisticated regulatory mechanism that expands the functional complexity of the ubiquitin-proteasome system. Their specialized role in cell cycle control, particularly through the APC/C, and their function as priority signals for proteasomal degradation make them essential for cellular homeostasis. The experimental approaches detailed herein provide researchers with robust methodologies for investigating K11 linkage biology, from structural analysis to functional assessment in cellular contexts. As research in this field advances, the development of additional linkage-specific reagents and more sensitive detection methods will further enhance our understanding of how K11 linkages coordinate with other ubiquitin signals to control fundamental biological processes. The enrichment strategies presented form a foundation for continued exploration of this critical aspect of ubiquitin signaling.
Structural Characteristics Distinguishing K11 Chains from Other Linkages
K11-linked polyubiquitin chains represent a critical non-canonical ubiquitin signaling modality with specialized functions in cell cycle regulation and protein degradation. Unlike the well-characterized K48 and K63 linkages, K11 chains possess unique structural properties that enable distinct functional outcomes and recognition by specific cellular machinery. This application note details the structural characteristics that differentiate K11 linkages from other ubiquitin chain types, providing essential context for developing effective enrichment strategies in K11 chain research. We present comprehensive structural data, experimental protocols for chain synthesis and analysis, and key reagent solutions to support research in this evolving field.
Structural Characteristics of K11-Linked Ubiquitin Chains
K11-linked di-ubiquitin (K11-Ub2) adopts solution conformations that are distinct from both K48-linked and K63-linked chains, as determined by nuclear magnetic resonance (NMR) spectroscopy and small-angle neutron scattering (SANS) [7]. Importantly, these solution structures are inconsistent with previously published crystal structures of K11-Ub2, highlighting the importance of physiological conditions for structural analysis [7].
- Interdomain Flexibility: K11-Ub2 exhibits unique conformational dynamics in solution, with the relative orientation of ubiquitin units differing significantly from K48 and K63 linkages
- Salt-Dependent Compaction: Increasing salt concentration compacts K11-Ub2 and strengthens interactions between the two Ub units [7]
- Chemical Shift Perturbations: NMR analysis reveals distinct chemical shift perturbation patterns in K11-Ub2, with the proximal Ub showing significant perturbations clustered around K11, primarily resulting from isopeptide bond formation rather than novel Ub/Ub interfaces [7]
Comparative Structural Features
Table 1: Structural Comparison of Major Ubiquitin Linkage Types
| Linkage Type | Overall Conformation | Inter-Ubiquitin Interface | Structural Response to Ionic Conditions | Receptor Binding Characteristics |
|---|---|---|---|---|
| K11-linked | Compact, distinct from K48/K63 [7] | Unique interaction surfaces | Compacts with increasing salt concentration [7] | Intermediate affinity with different binding modes [7] |
| K48-linked | Closed conformation | Canonical hydrophobic patches | Less sensitive to ionic changes | High affinity for proteasomal receptors |
| K63-linked | Open, extended conformation | Minimal ubiquitin interface | Stable across ionic conditions | Non-proteolytic signaling complexes |
| M1-linked | Linear, rigid structure | Head-to-tail linkage | - | NF-κB signaling, cell death regulation [8] |
Structural Basis for Functional Specialization
The unique conformation of K11-linked chains creates specific interaction surfaces that are differentially recognized by ubiquitin receptors:
- Proteasomal Recognition: K11-linked chains interact with ubiquitin-receptor proteins from both proteasomal and non-proteasomal pathways, but with intermediate affinity and different binding modes compared to K48-linked or K63-linked di-ubiquitin [7]
- Branched Chain Formation: K11 linkages frequently form branched chains with K48 linkages, creating a specialized "priority degradation signal" recognized by the proteasome through multivalent interactions [5]
- Distinct Binding Surfaces: The hydrophobic patches (L8, I44, V70) on K11-linked chains are positioned differently than in other linkage types, creating unique epitopes for receptor recognition [7]
Experimental Protocols for K11 Chain Analysis
Enzymatic Synthesis of K11-Linked Ubiquitin Chains
Table 2: Key Reagents for K11-Linked Ubiquitin Chain Synthesis
| Reagent | Specifications | Function in Protocol | Alternative Options |
|---|---|---|---|
| UBE2S E2 Enzyme | K11-specific elongating E2 [1] [9] | Catalyzes specific K11 linkage formation | None (linkage-specific) |
| UBE2C E2 Enzyme | Chain-initiating E2 for APC/C substrates [1] | Initiates ubiquitin chain formation | UBE2D (less specific) |
| Recombinant Ubiquitin | With chain-terminating mutations (e.g., K48R, K63R) [7] | Prevents alternative linkage formation | Non-mutated ubiquitin (requires linkage validation) |
| E1 Activating Enzyme | Ubiquitin-activating enzyme | Primes ubiquitin for transfer | Standard commercial preparations |
| APC/C E3 Ligase | Anaphase-Promoting Complex/Cyclosome [1] | Physiological E3 for K11 chains | Other E3s with K11 specificity |
Step-by-Step Synthesis Protocol
Step 1: Reaction Mixture Preparation
- Prepare 50 μM recombinant ubiquitin in reaction buffer (50 mM Tris-HCl, pH 7.5, 50 mM NaCl, 10 mM MgCl₂, 2 mM ATP)
- Add E1 enzyme (100 nM), UBE2C (500 nM for initiation), and UBE2S (500 nM for elongation) [9]
- Include linkage-preserving mutations in ubiquitin (e.g., K48R, K63R) to prevent alternative linkage formation [7]
Step 2: Chain Assembly
- Initiate reaction by adding ATP to 2 mM final concentration
- Incubate at 30°C for 2-4 hours with gentle agitation
- Monitor reaction progress by SDS-PAGE and western blotting with linkage-specific antibodies
Step 3: Purification
- Terminate reaction by placing on ice
- Purify K11-linked chains by ion-exchange chromatography or affinity purification using K11-linkage specific binders
- Validate chain length and linkage specificity by mass spectrometry and immunoblotting
Structural Characterization Protocol
NMR Analysis of K11 Chain Conformation
Sample Preparation:
- Prepare isotopically labeled (¹⁵N, ¹³C) K11-Ub2 using recombinant expression
- Exchange into NMR buffer (25 mM phosphate, pH 6.8, 50 mM NaCl) with 10% D₂O
- Consider varying salt conditions (0-150 mM NaCl) to assess conformational changes [7]
Data Collection:
- Acquire ¹H-¹⁵N TROSY-HSQC spectra at 25°C
- Collect residual dipolar coupling (RDC) measurements using 5% C12E5/hexanol alignment medium [7]
- Perform chemical shift perturbation analysis comparing K11-Ub2 to monomeric ubiquitin
Data Analysis:
- Map chemical shift perturbations to ubiquitin structure to identify interaction surfaces
- Calculate alignment tensors from RDCs to determine relative ubiquitin orientations
- Compare experimental data with back-calculated RDCs from crystal structures to validate solution conformation [7]
Functional Interaction Assays
Proteasomal Binding Assays
Surface Plasmon Resonance Protocol:
- Immobilize proteasomal subunits (RPN1, RPN10, RPN13) on CMS sensor chips
- Flow K11-linked ubiquitin chains at varying concentrations (0.1-10 μM) over the surface
- Compare binding kinetics with K48-linked and K63-linked chains
- Analyze data to determine association/dissociation constants and binding stoichiometry
Deubiquitinase Specificity Profiling
- Incubate K11-linked chains with candidate deubiquitinases (DUBs)
- Monitor chain disassembly over time by SDS-PAGE and western blotting
- Include control reactions with other linkage types to establish specificity
- Identify DUBs with preference for K11 linkages (e.g., Cezanne) [9]
Research Reagent Solutions
Table 3: Essential Research Tools for K11-Linked Ubiquitin Studies
| Reagent Category | Specific Examples | Key Applications | Availability |
|---|---|---|---|
| Linkage-Specific Antibodies | K11-linkage specific antibody (clone 2A3/2E6) [10] [11] | Immunoblotting, immunofluorescence | Commercial/Research use |
| E2 Enzymes | UBE2S (K11-specific elongator) [1] [9] | In vitro chain synthesis | Recombinant expression |
| E3 Ligase Systems | APC/C complex with UBE2C and UBE2S [1] | Physiological chain assembly | Recombinant complex |
| Ubiquitin Mutants | K11-only (K6R, K27R, K29R, K33R, K48R, K63R) [7] | Specific chain synthesis | Recombinant expression |
| Structural Standards | K11-Ub2 NMR structure (PDB) [7] | Structural comparisons, modeling | PDB database |
| Reference Chains | Defined K11-linked ubiquitin chains (Ub2-Ub4) [9] | Assay standards, controls | In-house synthesis |
K11 Chain Signaling Pathways and Experimental Workflows
Figure 1: K11-Linked Ubiquitin Chain Synthesis and Function. This workflow illustrates the sequential process of K11-linked chain assembly and its functional consequences in targeted protein degradation.
K11-linked ubiquitin chains possess distinct structural characteristics that differentiate them from canonical ubiquitin linkages and enable their specialized functions in cellular regulation. Their unique conformation, salt-dependent compaction, and specific recognition by proteasomal components make them a critical signaling modality for targeted protein degradation, particularly during cell cycle progression. The experimental protocols and reagent solutions presented here provide a foundation for comprehensive analysis of K11 chains, enabling researchers to develop effective enrichment strategies and advance our understanding of this important ubiquitin signaling pathway. As research progresses, the structural insights into K11 linkages will continue to inform drug development efforts targeting the ubiquitin-proteasome system.
K11/K48-Branched Ubiquitin Chains as Priority Degradation Signals
K11/K48-branched ubiquitin chains represent a sophisticated topological arrangement within the ubiquitin code, where a single ubiquitin molecule is simultaneously modified at both lysine 11 (K11) and lysine 48 (K48) residues, creating a branched architecture. These chains function as priority degradation signals, efficiently targeting substrate proteins for proteasomal degradation during critical cellular processes such as cell cycle progression and the management of proteotoxic stress [5] [1]. Unlike homotypic chains composed of a single linkage type, branched chains expand the informational content of the ubiquitin code, enabling more specialized recognition by cellular machinery [12].
The biological significance of K11/K48-branched chains is profound. They facilitate the timely degradation of key regulatory proteins, including mitotic regulators, misfolded nascent polypeptides, and pathogenic variants like Huntingtin in Huntington's disease [5]. Recent cryo-EM structural studies have revealed that the 26S proteasome recognizes these branched chains through a multivalent mechanism, distinguishing them from homotypic K48 chains and explaining their enhanced efficiency in targeting substrates for degradation [5].
Structural Mechanisms of Proteasomal Recognition
The human 26S proteasome recognizes K11/K48-branched ubiquitin chains through a specialized multivalent binding mechanism that involves distinct ubiquitin receptors within the 19S regulatory particle. Cryo-EM structures have illuminated a tripartite binding interface that specifically accommodates the branched architecture [5].
Key Binding Sites and Receptor Interactions
The proteasome employs a coordinated system to recognize the K11/K48-branched chain, engaging multiple ubiquitin-binding sites simultaneously:
- K48-Linkage Binding Site: The canonical K48-linkage is recognized by a binding pocket formed by RPN10 and the RPT4/5 coiled-coil domain, which is also used for recognizing homotypic K48 chains [5].
- K11-Linkage Binding Site: A previously unidentified binding site for K11-linkages was discovered at a groove formed between RPN2 and RPN10, providing specificity for the K11 branch of the chain [5].
- Alternating Linkage Recognition: RPN2 contributes to chain recognition through a conserved motif that interacts with the alternating K11-K48-linkage pattern, similar to the K48-specific T1 binding site of RPN1 [5].
Table 1: Proteasomal Ubiquitin Receptors and Their Roles in K11/K48-Branched Chain Recognition
| Receptor | Binding Specificity | Functional Role |
|---|---|---|
| RPN2 | K11-linked Ub and alternating K11-K48 linkage | Cryptic ubiquitin receptor; forms binding groove with RPN10 |
| RPN10 | K48-linked Ub (via UIM domains) and K11-linked Ub | Canonical receptor; bridges multiple binding sites |
| RPN1 | K48-linkage (T1 site) | May contribute to K48 branch recognition |
| RPN13 | Ub binding via PRU domain | Shuttling factor; recruits UCHL5 DUB |
This multivalent recognition system allows the proteasome to preferentially engage substrates tagged with K11/K48-branched chains, facilitating their rapid degradation even in the presence of competing substrates modified with homotypic chains [5].
Analytical Methods and Detection Strategies
Enrichment Techniques for Ubiquitinated Proteins
The analysis of K11/K48-branched ubiquitin chains requires specialized enrichment strategies due to their low abundance and complex architecture. Several well-established methods can be employed:
- Tandem Ubiquitin Binding Entities (TUBE): GST-TUBE pulldowns effectively enrich polyubiquitinated proteins from cell lysates. The protocol involves incubating cell supernatant containing 1 mg total protein with 10 μg GST-TUBE and Glutathione sepharose 4B overnight at 4°C with constant rotation, followed by washing and elution [13].
- Linkage-Specific Antibodies: Antibodies that specifically recognize K11- or K48-linkages can be used for immunoprecipitation, though cross-reactivity can be a limitation [14].
- Ubiquitin-Binding Domains (UBDs): Tandem UBA domains exhibit high affinity for polyubiquitin chains and can be used as GST-fusion proteins for enrichment. A study using four sequential UBA domains (GST-qUBA) demonstrated a threefold increase in enrichment efficiency compared to direct methods [15].
Linkage Verification and Mapping
Confirming the presence and topology of K11/K48-branched chains requires multiple orthogonal approaches:
- UbiCRest Assay: This method uses linkage-specific deubiquitinases (DUBs) to selectively disassemble ubiquitin chains. For K11/K48-branched chains, K48-specific DUBs (like OTUB1) and K63-specific DUBs (like AMSH) can be employed to verify linkage composition [16].
- Mass Spectrometry-Based Absolute Quantification (Ub-AQUA): This quantitative proteomics approach uses heavy isotope-labeled ubiquitin peptides as internal standards to precisely quantify different linkage types present in a sample [5] [17].
- Lbpro* Ub Clipping: The Lbpro* protease selectively cleaves ubiquitin chains, allowing detection of branched species through intact mass spectrometry. This method revealed that 12.6% of ubiquitin chains were doubly ubiquitinated and 3.6% were triply ubiquitinated in studied samples, indicating branching [5].
Table 2: Quantitative Analysis of Linkage Types in Polyubiquitin Chains
| Linkage Type | Abundance | Detection Method | Biological Function |
|---|---|---|---|
| K11 | ~2% in async cells, increases during mitosis | Ub-AQUA, linkage-specific antibodies | Cell cycle regulation, proteotoxic stress response |
| K48 | Most abundant linkage | Ub-AQUA, immunoblotting | Canonical proteasomal degradation signal |
| K63 | Second most abundant | Ub-AQUA, linkage-specific antibodies | Signaling, DNA repair, endocytosis |
| K11/K48 Branched | 10-20% of Ub polymers | Lbpro* clipping, intact MS | Priority degradation signal |
Synthesis and Regulation of Branched Chains
Enzymatic Assembly Pathways
The synthesis of K11/K48-branched ubiquitin chains involves coordinated actions of specific E2 enzymes and E3 ligases that determine the chain topology:
- APC/C Pathway: The anaphase-promoting complex/cyclosome (APC/C) collaborates with two E2 enzymes—UBE2C (UbcH10) for chain initiation and UBE2S for K11-specific elongation—to build branched K11/K48 chains on substrates during mitosis [1] [12]. UBE2C first attaches short chains containing mixed linkages, then UBE2S extends these with K11 linkages, creating branch points [12].
- UBR5 Pathway: The E3 ligase UBR5 can generate K11/K48-branched chains through an alternative mechanism by attaching K48 linkages to preformed K11-linked chains, in contrast to the APC/C which assembles K11 linkages on preformed K48 chains [12].
The following diagram illustrates the collaborative synthesis of K11/K48-branched ubiquitin chains by the APC/C with its E2 enzymes:
Deubiquitination and Regulation
The stability and signaling duration of K11/K48-branched chains are regulated by deubiquitinating enzymes (DUBs), particularly UCHL5 (UCH37). UCHL5 is recruited to the proteasome through its interaction with RPN13 and exhibits preferential activity toward K11/K48-branched chains, providing a editing mechanism that controls the degradation fate of modified substrates [5]. The DUB activity of UCHL5 is activated upon binding to RPN13, creating a regulatory checkpoint at the proteasome [5].
Experimental Protocols
Protocol 1: TUBE-Based Enrichment of Ubiquitinated Proteins
This protocol details the enrichment of polyubiquitinated proteins using Tandem Ubiquitin Binding Entities (TUBE) for subsequent analysis of K11/K48-branched chains [13].
Materials:
- Lysis Buffer: 1% IGEPAL CA-630, 50 mM Tris-HCl (pH 7.5), 120 mM NaCl, 1 mM EDTA, protease inhibitors
- GST-TUBE protein
- Glutathione sepharose 4B
- BCA protein assay kit
- DUB inhibitors: Chloroacetamide (CAA) or N-ethylmaleimide (NEM)
Procedure:
- Cell Lysis: Lyse confluent cells in 10 cm dishes on ice with 1 mL lysis buffer.
- Clarification: Centrifuge lysates at 14,000 rpm for 10 min at 4°C to remove debris.
- Protein Quantification: Assay supernatant protein concentration using BCA assay.
- DUB Inhibition: Add CAA (preferred) or NEM to appropriate concentration (typically 5-10 mM) to prevent chain disassembly.
- Binding Reaction: Incubate 1 mg total protein with 10 μg GST-TUBE and Glutathione sepharose 4B in a total volume of 1 mL lysis buffer.
- Overnight Incubation: Rotate mixture at 4°C for 16 hours.
- Washing: Wash complexes twice with lysis buffer and once with PBS.
- Elution: Elute bound proteins at 95°C for 5 min in Laemmli buffer.
Notes: Comparison of DUB inhibitors reveals differential effects on ubiquitin interactors, with CAA generally preferred over NEM due to better specificity and fewer side reactions [16].
Protocol 2: Linkage Verification via UbiCRest Assay
This protocol verifies ubiquitin chain linkage composition using linkage-specific DUBs [16].
Materials:
- Enriched ubiquitin chains (from Protocol 1)
- Linkage-specific DUBs: OTUB1 (K48-specific), AMSH (K63-specific)
- Appropriate DUB reaction buffers
Procedure:
- Sample Preparation: Divide enriched ubiquitin chains into aliquots for different DUB treatments.
- DUB Digestion: Incubate each aliquot with the appropriate linkage-specific DUB:
- OTUB1 for K48-linkage verification
- AMSH for K63-linkage verification
- Incubation: Conduct reactions at 37°C for 2-4 hours.
- Analysis: Analyze digestion patterns by Western blotting using linkage-specific antibodies or mass spectrometry.
Interpretation: K11/K48-branched chains will show partial resistance to individual DUB treatments but complete disassembly with combination treatments.
Research Reagent Solutions
Table 3: Essential Research Reagents for K11/K48-Branched Ubiquitin Chain Studies
| Reagent Category | Specific Examples | Function/Application |
|---|---|---|
| Enrichment Tools | GST-TUBE, GST-qUBA | High-affinity enrichment of polyubiquitinated proteins |
| Linkage-Specific Antibodies | K11-linkage specific, K48-linkage specific | Detection and immunoprecipitation of specific linkages |
| DUB Inhibitors | Chloroacetamide (CAA), N-ethylmaleimide (NEM) | Prevention of chain disassembly during processing |
| Linkage-Specific DUBs | OTUB1 (K48-specific), AMSH (K63-specific) | Linkage verification via UbiCRest assay |
| Mass Spec Standards | Heavy isotope-labeled ubiquitin peptides (Ub-AQUA) | Absolute quantification of linkage types |
| Proteasomal Components | Recombinant RPN2, RPN10, RPN13 | Structural and binding studies of recognition mechanisms |
| E2 Enzymes | UBE2C (UbcH10), UBE2S | In vitro reconstitution of branched chain synthesis |
Functional Significance and Research Applications
The study of K11/K48-branched ubiquitin chains provides critical insights into fundamental biological processes and potential therapeutic interventions:
- Cell Cycle Regulation: During mitosis, K11/K48-branched chains on APC/C substrates ensure timely degradation of mitotic regulators, facilitating proper cell division [1]. Disruption of this process can lead to chromosomal instability and carcinogenesis [1].
- Proteostasis Maintenance: Under proteotoxic stress conditions, K11/K48-branched chains target misfolded proteins and pathological aggregates (e.g., Huntingtin) for efficient clearance, maintaining cellular protein homeostasis [5].
- Disease Connections: Aberrant formation or recognition of K11/K48-branched chains has been implicated in cancer and neurodegenerative diseases, making the enzymatic machinery involved a potential therapeutic target [5] [1].
The following workflow diagram outlines the key stages for experimental analysis of K11/K48-branched ubiquitin chains:
K11/K48-branched ubiquitin chains represent a sophisticated priority degradation signal in the ubiquitin-proteasome system, characterized by their unique branched topology and specialized recognition by the 26S proteasome. The experimental approaches outlined in this application note—including TUBE-based enrichment, linkage-specific verification, and advanced mass spectrometry techniques—provide researchers with robust methodologies to investigate these complex ubiquitin signals. As research in this field advances, further elucidation of the structural basis for branched chain recognition and the regulatory mechanisms governing their synthesis and removal will undoubtedly reveal new therapeutic opportunities for diseases characterized by proteostasis dysfunction.
K11-linked polyubiquitin chains are crucial regulators of cell division, serving as potent signals for the proteasomal degradation of key mitotic regulators [1]. The formation of these chains is catalyzed by a dedicated enzymatic machinery, primarily involving specific E2 conjugating enzymes and E3 ligases that act in a coordinated manner [18] [1]. This application note details the core enzymes, experimental protocols, and reagent solutions essential for researching K11-linked ubiquitination, providing a structured framework for scientists developing enrichment strategies for these chains. Understanding this machinery is fundamental for investigating cell cycle control and developing therapeutic interventions targeting the ubiquitin-proteasome system.
Core Enzymatic Machinery
The synthesis of K11-linked ubiquitin chains is a two-step process involving distinct E2 enzymes for chain initiation and elongation, working in concert with specific E3 ligases.
E2 Enzymes in K11 Linkage Formation
E2 enzymes are the central determinants of linkage specificity in K11-linked chain formation. The major E2s involved exhibit a clear division of labor.
- UBE2C (UbcH10): Chain Initiator - UBE2C is the primary E2 responsible for the initial attachment of ubiquitin to substrate proteins and the formation of short, K11-linked chains [1]. Its activity is regulated by an N-terminal APC/C-targeting motif and initiation motifs in substrate proteins [1]. Overexpression of UBE2C can destabilize the spindle checkpoint and has been linked to error-prone chromosome segregation and tumorigenesis [1].
- UBE2S (E2-EPF): Chain Elongator - UBE2S specializes in the processive elongation of K11-linked chains on substrates that have been pre-ubiquitinated by UBE2C or other initiating E2s like UbcH5 [18]. It copurifies with the APC/C and is essential for the timely degradation of APC/C substrates [18].
- UbcH5 Family: Promiscuous Initiators - Enzymes in the UbcH5 family (e.g., UbcH5A) are promiscuous E2s with an innate preference for synthesizing K11-, K48-, and K63-linked chains [19]. Structural studies of UbcH5A have revealed an interaction interface between ubiquitin and residues adjacent to the E2 catalytic cysteine that is critical for K11 linkage formation [19].
Table 1: Key E2 Enzymes in K11-Linked Ubiquitin Chain Formation
| E2 Enzyme | Role in K11 Synthesis | Key Features | Linkage Specificity |
|---|---|---|---|
| UBE2C (UbcH10) | Chain initiation | - APC/C-specific- Rate-limiting for substrate degradation- Levels peak during mitosis | Preferentially forms short K11-linked chains [1] |
| UBE2S (E2-EPF) | Chain elongation | - Processive chain extension |
Highly specific for K11 linkage [18] |
| UbcH5A | Promiscuous initiation | - Widely used with many E3s- Structural determinants near active site guide K11 specificity | Innate preference for K11, K48, and K63 [19] |
E3 Ligases in K11 Linkage Formation
E3 ligases provide substrate specificity and work with the aforementioned E2s to build K11-linked chains.
- Anaphase-Promoting Complex/Cyclosome (APC/C) - The APC/C is the primary E3 ligase known to assemble homogeneous K11-linked chains during mitosis [1]. When activated, it drives a dramatic increase in cellular K11-linked chains, which are essential for the degradation of mitotic regulators like securin and cyclin B [18] [2]. The APC/C employs a sequential mechanism: it uses UBE2C to initiate ubiquitination and then recruits UBE2S to elongate K11-linked chains [18] [1].
- RNF114 and RNF166: Reader-Writer E3s - Recent research has identified a new class of "reader-writer" E3 ligases. RNF114 and RNF166 can recognize mono-ADP-ribose ubiquitylation (MARUbylation) marks via a tandem Di19-UIM module (MARUbe-binding domain, M-UBD) and subsequently extend them with K11-linked polyubiquitin chains [20]. This exemplifies how K11-specific E3s can be recruited to pre-existing modification sites to add a degradation signal.
The following diagram illustrates the coordinated enzymatic cascade for the formation of K11-linked polyubiquitin chains by the APC/C.
Experimental Protocols for Studying K11 Linkage Formation
This section provides detailed methodologies for key experiments used to dissect the mechanisms of K11-linked chain formation.
In Vitro Ubiquitination Assay with Single-Lysine Substrates
This protocol, adapted from foundational studies, uses simplified substrates to precisely analyze ubiquitin chain topology [18].
Principle: By using a substrate with a single lysine residue, chain formation is restricted to a single site, eliminating the complexity of multi-ubiquitination and allowing clear analysis of chain linkage preference.
Procedure:
- Substrate Preparation: Generate a lysine-less version of a model APC/C substrate (e.g., securin or cyclin B N-terminal fragment) by mutating all lysines to arginine. Then, revert one key lysine residue (e.g., Lys48 in securin) to create a "single-lysine" substrate [18].
- Reaction Setup: Assemble a 20-50 µL reaction mixture containing:
- 50-100 nM purified APC/C (E3)
- 100 nM E1 activating enzyme
- Option A (Initiation): 1 µM UBE2C (E2)
- Option B (Elongation): 1 µM UBE2S (E2) with a pre-ubiquitinated substrate
- 2 µM single-lysine substrate
- 10-20 µM wild-type ubiquitin or linkage-specific ubiquitin mutants (e.g., Ub-K11-only, Ub-K48-only)
- Reaction buffer: 50 mM Tris-HCl (pH 7.5), 5 mM MgCl₂, 2 mM ATP
- Incubation: Incubate at 30°C for 60-90 minutes.
- Termination and Analysis: Stop the reaction with SDS-PAGE loading buffer. Analyze the products by immunoblotting using anti-substrate and anti-ubiquitin antibodies. Linkage specificity can be confirmed using K11-linkage specific antibodies [2].
Linkage Specificity Analysis Using Ubiquitin Mutants
This method quantitatively assesses an enzyme's inherent preference for forming K11 linkages versus other chain types.
Principle: Using a panel of ubiquitin mutants where only a single lysine (e.g., K11, K48, K63) is available for chain formation forces the synthesis of homotypic chains of a defined linkage, which can then be quantified.
Procedure:
- Reagent Preparation: Obtain or purify a panel of single-lysine ubiquitin mutants (K6-only, K11-only, K27-only, K29-only, K33-only, K48-only, K63-only).
- Parallel Reactions: Set up separate in vitro ubiquitination reactions for each ubiquitin mutant, using the E2-E3 pair of interest (e.g., UBE2S with APC/C).
- Product Quantification: Analyze the reactions by SDS-PAGE and stain with Coomassie blue or use immunoblotting. Quantify the formation of high molecular weight polyubiquitin chains for each linkage.
- Data Analysis: Calculate the efficiency of chain formation for each linkage type. UBE2S, for instance, will produce significantly longer chains with Ub-K11-only compared to other mutants [18].
Table 2: Quantitative Linkage Preference of E2 Enzymes in APC/C-Catalyzed Reactions
| Ubiquitin Mutant (Available Lysine) | Chain Length with UBE2C (Initiation) | Chain Length with UBE2S (Elongation) | Relative Efficiency for Degradation |
|---|---|---|---|
| K11-only | Short chains (di-/tri-Ub) [1] | Long chains (>6 Ub) [18] | Essential [18] |
| K48-only | Short to medium chains | Medium chains | Not essential [18] |
| K63-only | Short to medium chains | Medium chains | Not essential [18] |
| K6, K27, K29, K33-only | Very short chains (mono-/di-Ub) | Very short chains (mono-/di-Ub) | Inefficient |
The Scientist's Toolkit: Key Research Reagents
Critical reagents for experimental research on K11-linked polyubiquitination are summarized in the table below.
Table 3: Essential Research Reagents for K11-Linked Ubiquitination Studies
| Reagent Category | Specific Example | Function and Application in Research |
|---|---|---|
| E2 Enzymes | Recombinant UBE2C / UbcH10 | Used in initiation reactions to prime substrates with the first ubiquitin or short chains [18] [1]. |
| Recombinant UBE2S / E2-EPF | Used to study processive elongation of K11-linked chains on pre-ubiquitinated substrates [18]. | |
| E3 Ligases | Purified APC/C complex | The primary E3 for K11-chain formation; essential for reconstituting mitotic ubiquitination in vitro [18] [2]. |
| Recombinant RNF114 / RNF166 | Used to study K11-chain extension on non-canonical marks like MARUbylation [20]. | |
| Ubiquitin Mutants | Single-Lysine Ubiquitin (e.g., Ub-K11-only) | Determines linkage specificity of E2s/E3s by forcing formation of homotypic chains [18]. |
| Dominant-Negative Ubiquitin (e.g., Ub-K11R) | Inhibits specific linkage formation in degradation assays to test functional importance [18]. | |
| Specialized Antibodies | K11-linkage Specific Antibody | Validates the presence and abundance of K11 chains in cells (e.g., in mitosis) and in vitro assays [2]. |
| Chemical Tools | Proteasome Inhibitors (e.g., MG132) | Causes accumulation of ubiquitinated proteins, allowing easier detection of K11-linked chains in cells [2]. |
Visualization of an Advanced K11-Ubiquitination Workflow
The following diagram outlines a modern, multi-step experimental workflow for studying K11-linked chain formation, incorporating insights from recent research on complex E3 systems.
Natural Abundance and Detection Challenges of K11 Chains in Cellular Contexts
Ubiquitylation is an essential post-translational modification that controls diverse cellular processes, including cell division, protein quality control, and signal transduction [12]. Unlike other ubiquitin chain types, K11-linked chains exhibit unique dynamic regulation and functional specialization, particularly in controlling the timely degradation of cell cycle regulators [1]. These chains can exist in homogenous forms (uniform K11 linkages), mixed forms (alternating linkages in a linear chain), or branched forms (where a single ubiquitin molecule is modified at multiple sites, such as K11/K48 branches) [12] [1]. The structural complexity of K11-containing chains creates both opportunities for specialized signaling and significant challenges for detection and study, necessitating sophisticated enrichment and analytical strategies.
Quantitative Profiling of K11 Chain Abundance
The abundance of K11 linkages is highly dynamic and varies considerably depending on cellular context, cell cycle stage, and environmental conditions. The following table summarizes key quantitative findings from proteomic studies.
Table 1: Cellular Abundance and Dynamics of K11-Linked Ubiquitin Chains
| Cellular Context | Reported Abundance | Regulating Factors | Key References |
|---|---|---|---|
| Asynchronously dividing human cells | ~2% of total ubiquitin conjugate pool [1] | Baseline activity of E2/E3 enzymes | [1] |
| Activated mitosis | Dramatic increase (specific fold-change not quantified) [1] [21] | APC/C activation, Ube2S activity | [1] [21] |
| Proteotoxic stress (e.g., proteasome inhibition, heat shock) | Significant accumulation [1] | Cellular stress response pathways | [1] |
| Cell cycle exit (differentiation) | Decreased levels [1] | Downregulation of mitotic machinery | [1] |
| Overall branched ubiquitin chains | 10–20% of total ubiquitin polymers [5] | Collaboration of multiple E3 ligases | [5] |
The low basal abundance of K11 linkages in asynchronous cells presents a fundamental detection challenge, as signal from more prevalent chain types (e.g., K48 and K63) can easily obscure the K11 signal. Furthermore, the formation of branched chains containing K11 linkages, such as K11/K48, adds another layer of complexity, as standard detection methods often fail to distinguish these branched architectures from homotypic chains [12] [5].
Methodologies for Detection and Enrichment
Linkage-Specific Antibodies and TUBEs
Immunoblotting with linkage-specific antibodies remains a widely used method. However, the efficacy of K11-linkage-specific antibodies can be compromised in complex lysates due to low abundance and potential epitope masking in branched chains [21].
A more robust approach involves Tandem Ubiquitin Binding Entities (TUBEs). These are engineered, high-affinity reagents composed of multiple ubiquitin-associated (UBA) domains that bind polyubiquitin chains with nanomolar affinity [22] [23]. Linkage-specific TUBEs can be immobilized on solid surfaces, such as 96-well plates, to selectively enrich for particular chain types from cell lysates before detection with a target-specific antibody.
Table 2: Research Reagent Solutions for K11 Chain Research
| Research Reagent | Primary Function | Application in K11 Research | Considerations |
|---|---|---|---|
| K11-Linkage Specific Antibodies | Immunodetection of K11 linkages | Western blotting, immunofluorescence | Validate specificity with ubiquitin mutants (K11R); may not recognize branched chains effectively. |
| Linkage-Specific TUBEs | Affinity enrichment of specific Ub chains | High-throughput pull-down of K11 chains from lysates | Superior to antibodies for enriching low-abundance chains; requires optimization of lysis buffer. |
| Mutant Ubiquitin Plasmids (e.g., Ub-K11R, Ub-K11-only) | Define linkage requirement in cells | Transfect cells to test if a process depends on K11 linkage | Overexpression may cause artifacts; use stable, inducible systems for best results. |
| E2/E3 Enzyme Tools (e.g., Ube2S, APC/C) | In vitro reconstitution of K11 chains | Define minimal machinery and create defined chains for structural studies | Essential for proving direct synthesis of K11 or K11/K48-branched chains. |
Protocol 3.1: High-Throughput Capture of Endogenous K11-Ubiquitinated Proteins Using TUBE-Based Platform
Materials:
- K11-linkage specific TUBE-coated 96-well plates
- Cell lysis buffer (e.g., 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% NP-40, 1 mM EDTA) supplemented with 1 mM DTT and protease/deubiquitinase inhibitors (e.g., 10 mM N-Ethylmaleimide)
- Wash buffer (lysis buffer without detergents)
- Target protein-specific primary antibody
- HRP-conjugated secondary antibody
- Chemiluminescent detection reagent
Procedure:
- Cell Lysis: Harvest and lyse cells in the provided lysis buffer. Centrifuge at 16,000 × g for 15 minutes at 4°C to remove insoluble debris.
- Protein Quantification: Determine protein concentration of the supernatant. Use 50-100 µg of total protein per well of the TUBE plate.
- Capture: Apply the diluted lysate to the TUBE-coated plate. Incubate for 2 hours at 4°C with gentle shaking.
- Washing: Aspirate the lysate and wash each well 3-4 times with cold wash buffer to remove non-specifically bound proteins.
- Detection: Incubate with primary antibody against the protein of interest (e.g., anti-RIPK2) diluted in wash buffer for 1 hour. Wash again. Then, incubate with an HRP-conjugated secondary antibody for 1 hour. After final washes, develop with chemiluminescent reagent and read on a compatible plate reader [23].
Mass Spectrometry-Based Approaches
Mass spectrometry (MS) is a powerful tool for definitive identification of ubiquitin linkage types, including K11. The workflow typically involves digesting purified ubiquitin conjugates with trypsin, which generates a characteristic di-glycine remnant on modified lysines that can be detected by LC-MS/MS.
Protocol 3.2: Ub-AQUA (Absolute QUAntitation) Mass Spectrometry for Linkage Quantification
Materials:
- Heavy isotope-labeled internal standard peptides for each ubiquitin linkage type
- Denaturing lysis buffer (e.g., 6 M Guanidine-HCl)
- Anti-ubiquitin antibodies or Pan-TUBEs for ubiquitin enrichment
- Sequencing-grade trypsin
- Reverse-phase LC-MS/MS system
Procedure:
- Ubiquitin Conjugate Enrichment: Lyse cells in denaturing buffer to inactivate DUBs. Enrich for ubiquitinated proteins using pan-specific TUBEs or immunoprecipitation with an anti-ubiquitin antibody.
- Spike-in Standards: Add a known quantity of heavy isotope-labeled K11-linkage diagnostic peptide (and other linkage standards) to the enriched sample.
- Digestion: Reduce, alkylate, and digest the enriched proteins with trypsin.
- LC-MS/MS Analysis: Analyze the resulting peptide mixture by LC-MS/MS. Monitor the transition ions for the light (endogenous) and heavy (standard) forms of the K11-specific peptide.
- Data Analysis: Quantify the absolute amount of K11-linkages in the original sample by comparing the peak area of the endogenous peptide to that of the spiked internal standard [5]. This method confirmed nearly equal amounts of K11- and K48-linked Ub in branched chains assembled in vitro [5].
Enzymatic and Chemical Tools forIn VitroReconstitution
Defined K11-linked and K11/K48-branched chains can be synthesized in vitro for use as standards or in functional assays.
Protocol 3.3: Enzymatic Assembly of K11/K48-Branched Ubiquitin Trimers
Materials:
- E1 activating enzyme
- E2 enzymes: UBE2C (or UBE2D family) and UBE2S
- E3 enzyme: Anaphase-Promoting Complex/Cyclosome (APC/C)
- Ubiquitin mutants: Ub1-72 (C-terminally truncated), UbK48R, K63R
- Reaction buffer: 50 mM Tris-HCl (pH 7.5), 5 mM MgCl₂, 2 mM ATP
Procedure:
- Chain Initiation: Incubate APC/C, UBE2C (the initiating E2), and UbK48R, K63R with the proximal Ub1-72 to build the initial chain. UBE2C preferentially assembles short chains with mixed linkages, including K11.
- Chain Branching: Add the elongating E2, UBE2S, which is specific for K11 linkages. UBE2S recognizes the initial chain and attaches K11-linked ubiquitins, thereby creating a branched architecture on the proximal ubiquitin [21].
- Purification: Purify the assembled branched trimer using size-exclusion or ion-exchange chromatography. Analyze the linkage composition by MS and Ub-AQUA.
Diagram 1: Synthesis of K11/K48-branched ubiquitin chains by APC/C. The model shows sequential action of two distinct E2 enzymes, UBE2C and UBE2S, recruited by the APC/C to build a branched ubiquitin chain on a substrate protein.
K11-linked ubiquitin chains, while of low abundance under basal conditions, constitute a critical regulatory signal, especially during cell division and stress response. Their accurate detection is complicated by their dynamic nature and presence in complex branched polymers. A multi-faceted strategy combining high-affinity enrichment tools like TUBEs, definitive MS-based quantification, and in vitro reconstitution with defined enzyme systems provides the most robust framework for advancing research in this field. These protocols offer researchers a pathway to overcome the historical challenges in studying K11 chains and to fully elucidate their specific roles in health and disease.
Practical Enrichment Methodologies: From Antibodies to Affinity Tools
Ubiquitination is a crucial post-translational modification that regulates virtually all aspects of eukaryotic cell biology. The specificity of ubiquitin signaling is largely determined by the architecture of polyubiquitin chains, where ubiquitin molecules are connected through specific lysine residues. Among the various linkage types, K11-linked polyubiquitin chains have emerged as critical regulators of cell division, serving as priority degradation signals during mitosis and proteotoxic stress [1] [2] [5]. The development of linkage-specific antibodies has revolutionized our ability to decipher this complex ubiquitin code, enabling precise detection and investigation of specific ubiquitin chain types in biological systems.
The significance of K11-linked chains in cell cycle control underscores the importance of targeted enrichment strategies. These chains are highly upregulated during mitosis and are predominantly assembled by the anaphase-promoting complex/cyclosome (APC/C) in conjunction with the E2 enzymes Ube2C and Ube2S [1] [2]. Unlike the well-characterized K48-linked chains that primarily target proteins for proteasomal degradation, K11-linked chains exhibit unique structural properties and distinct receptor binding modes, allowing them to be specifically recognized by the proteasomal machinery [7] [5]. This review comprehensively addresses the development, specificity, and applications of linkage-specific antibodies, with particular emphasis on their role in advancing K11-linked polyubiquitin chain research.
The Ubiquitin Signaling Landscape
Ubiquitin can form at least twelve structurally and functionally distinct polyubiquitin chain linkages, including eight amide-linked types (M1, K6, K11, K27, K29, K33, K48, K63) and four recently discovered oxyester-linked types (Ser20, Thr12, Thr14, Thr22, Thr55) [24]. The linkage type determines the chain's conformation and consequently its biological function, creating an elaborate "ubiquitin code" that cells utilize to control key signaling pathways [1] [24].
Table 1: Characteristics of Major Ubiquitin Chain Linkages
| Linkage Type | Relative Abundance | Primary Functions | Key Structural Features |
|---|---|---|---|
| K48-linked | ~40% (most abundant) | Proteasomal degradation [24] | Compact conformation [7] |
| K63-linked | ~30% (second most abundant) | DNA repair, NF-κB signaling, endocytosis [24] | Extended conformation [7] |
| K11-linked | ~2% (upregulated in mitosis) | Cell cycle regulation, ERAD [1] [7] | Unique compact conformation distinct from K48/K63 [7] |
| M1-linked (linear) | Variable | NF-κB activation, inflammation [24] | Extended rigid structure [24] |
| K27/K29/K33-linked | Low abundance | Immune signaling, proteostasis [24] | Not well characterized |
K11-linked chains play particularly important roles in cell cycle progression and proteostasis maintenance. During mitosis, K11-linked chains dramatically increase in abundance and are essential for the targeted degradation of mitotic regulators [1] [2]. More recently, K11/K48-branched ubiquitin chains have been identified as particularly efficient degradation signals that are preferentially recognized by the 26S proteasome, highlighting the complexity and functional importance of these chain types [5].
Figure 1: K11-Linked Ubiquitin Signaling Pathway. K11-linked chains are assembled by APC/C and Ube2S on mitotic substrates, leading to proteasomal recognition and degradation, which enables proper cell cycle progression.
Development of Linkage-Specific Antibodies
Strategic Antigen Design
Generating antibodies that specifically recognize particular ubiquitin linkages presents unique challenges due to the structural similarity between different chain types and the lability of the isopeptide bond. Successful development requires carefully designed antigens that mimic the native ubiquitin linkage while resisting enzymatic cleavage [25].
Two primary strategies have emerged for antigen preparation:
- Thiolysine-mediated ligation produces antigens with native isopeptide bonds
- Click chemistry approaches create proteolytically stable analogs using amide triazole isosteres that closely mimic the native isopeptide linkage [25]
The incorporation of full-length ubiquitin in a proteolytically stable form significantly increases the likelihood of generating high-quality antibodies with strong specificity, as this approach presents more complete epitopes to the immune system [25].
K11 Linkage-Specific Antibody Development
The pioneering development of a K11 linkage-specific antibody demonstrated the feasibility of generating highly specific reagents for atypical ubiquitin chains [2]. This antibody was engineered using antigens that presented the unique structural epitopes of K11-linked diubiquitin, which crystallographic studies revealed adopts a distinct conformation from K48- or K63-linked diubiquitin [2].
The specificity of this antibody was rigorously validated through multiple approaches:
- Linkage specificity profiling against a panel of different ubiquitin chain types
- Immunoblot analysis demonstrating specific recognition of K11-linked chains in mitotic cell extracts
- Functional studies showing ablation of K11 signal upon APC/C inhibition [2]
This breakthrough reagent enabled the critical discovery that K11-linked chains are highly upregulated during mitosis and are predominantly generated by APC/C [2].
Research Reagent Solutions for K11-Linked Ubiquitin Research
Table 2: Essential Research Reagents for K11-Linked Ubiquitin Studies
| Reagent Type | Specific Examples | Applications | Key Features |
|---|---|---|---|
| Linkage-Specific Antibodies | K11-linkage specific mAb [2] | Western blot, immunofluorescence | Specifically detects K11 linkages without cross-reactivity |
| Ubiquitin Mutants | Ubiquitin K11R, K11-only mutants [26] | In vitro ubiquitination assays | Determines chain linkage specificity |
| E2 Enzymes | Ube2S (K11-specific) [1] | In vitro chain assembly | Specifically assembles K11-linked chains |
| E3 Ligases | APC/C [1] [2] | In vitro ubiquitination | Major cellular source of K11 linkages |
| DUBs | UCHL5 [5] | Branch analysis, proteasome studies | Preferentially processes K11/K48-branched chains |
| Proteasomal Components | RPN1, RPN10, RPN2 [5] | Binding studies, structural biology | Recognize K11/K48-branched chains |
The availability of these specialized reagents has created a comprehensive toolkit for investigating K11-linked ubiquitination. Particularly valuable are the ubiquitin mutants (K-to-R and K-only), which enable definitive determination of chain linkage through in vitro ubiquitination assays [26]. The recent identification of proteasomal receptors with specificity for K11/K48-branched chains has further enhanced our ability to study the functional consequences of this modification [5].
Experimental Protocols
Determining Ubiquitin Chain Linkage Using Ubiquitin Mutants
This established protocol utilizes ubiquitin mutants to definitively determine the linkage specificity of ubiquitin chains formed in in vitro ubiquitination assays [26].
Materials and Reagents:
- E1 activating enzyme (5 μM)
- E2 conjugating enzyme (25 μM)
- E3 ligase (10 μM)
- 10X E3 ligase reaction buffer (500 mM HEPES, pH 8.0, 500 mM NaCl, 10 mM TCEP)
- Wild-type ubiquitin (1.17 mM)
- Ubiquitin K-to-R mutants (K6R, K11R, K27R, K29R, K33R, K48R, K63R)
- Ubiquitin K-only mutants (K6-only, K11-only, K27-only, K29-only, K33-only, K48-only, K63-only)
- MgATP solution (100 mM)
- Substrate protein of interest
Procedure:
- Reaction Setup: Set up two sets of nine parallel ubiquitination reactions (25 μL each):
- Set 1: Wild-type ubiquitin + seven K-to-R mutants + negative control (no ATP)
- Set 2: Wild-type ubiquitin + seven K-only mutants + negative control (no ATP)
Reaction Composition:
Incubation: Incubate reactions at 37°C for 30-60 minutes
Termination:
- For SDS-PAGE analysis: Add 25 μL 2X SDS-PAGE sample buffer
- For downstream applications: Add 0.5 μL EDTA (20 mM final) or 1 μL DTT (100 mM final)
Analysis: Resolve reactions by SDS-PAGE and perform Western blotting with anti-ubiquitin antibody
Data Interpretation:
- In Set 1 (K-to-R mutants), the reaction containing the mutant lacking the specific lysine required for chain formation will show only monoubiquitination
- In Set 2 (K-only mutants), only the reaction containing the specific lysine required for chain formation will show polyubiquitination
- If all K-to-R mutants support chain formation, the chains may be M1-linked (linear) or contain mixed linkages [26]
Figure 2: Experimental Workflow for Ubiquitin Chain Linkage Determination. This diagram illustrates the parallel approach using K-to-R and K-only ubiquitin mutants to definitively identify ubiquitin chain linkage types.
Enrichment and Detection of K11-Linked Chains from Cellular Systems
Materials:
- K11 linkage-specific antibody [2]
- Cell lysates from synchronized mitotic cells
- Protein A/G agarose beads
- Lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% NP-40, protease inhibitors)
- Wash buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% NP-40)
- Elution buffer (0.1 M glycine, pH 2.5)
Procedure:
- Cell Synchronization: Synchronize cells in mitosis using nocodazole (100 ng/mL, 16-18 hours)
- Lysis: Prepare cell lysates in lysis buffer (1-2 mg/mL total protein)
- Pre-clearing: Incubate lysate with protein A/G beads for 30 minutes at 4°C
- Immunoprecipitation:
- Incubate pre-cleared lysate with K11 linkage-specific antibody (1-2 μg per mg lysate)
- Rotate overnight at 4°C
- Add protein A/G beads and incubate for 2-4 hours
- Washing: Wash beads 3-4 times with wash buffer
- Elution: Elute bound proteins with elution buffer and neutralize with 1 M Tris-HCl, pH 8.0
- Analysis: Analyze by Western blotting with relevant antibodies
Applications in K11-Linked Polyubiquitin Chain Research
Cell Cycle Studies
K11 linkage-specific antibodies have been instrumental in elucidating the role of ubiquitination in cell cycle control. These reagents enabled the discovery that K11-linked chains are highly upregulated during mitosis and that the APC/C is the primary E3 ligase responsible for their formation [2]. Immunofluorescence studies using these antibodies have revealed the spatial and temporal regulation of K11-linked chains throughout cell division.
Proteasomal Recognition Studies
The development of K11-specific antibodies facilitated critical studies on proteasomal recognition of ubiquitin chains. Recent cryo-EM structures have revealed that K11/K48-branched ubiquitin chains are recognized through a multivalent binding mechanism involving RPN2, RPN10, and RPT4/5 within the 26S proteasome [5]. This specialized recognition mechanism explains the preferential degradation of substrates modified with K11/K48-branched chains.
Disease Relevance
K11-linked chains have been implicated in various pathological conditions, including cancer and neurodegenerative diseases. Quantitative assessments using linkage-specific antibodies have revealed alterations in K11-linked chain homeostasis in cellular models of proteotoxic stress, suggesting potential therapeutic applications for modulating this pathway [5].
Linkage-specific antibodies represent indispensable tools for deciphering the complex language of ubiquitin signaling. The development of K11 linkage-specific antibodies in particular has transformed our understanding of cell cycle regulation and proteasomal targeting. As these reagents continue to improve in specificity and affinity, and as new methodologies for antigen design emerge, we can anticipate further insights into the multifaceted roles of K11-linked ubiquitination in health and disease. The continued refinement of enrichment strategies and detection methods will undoubtedly accelerate both basic research and drug discovery efforts targeting the ubiquitin-proteasome system.
Tandem Ubiquitin Binding Entities (TUBEs) for K11 Chain Enrichment
K11-linked polyubiquitin chains represent a critical regulatory signal within the ubiquitin-proteasome system, distinct from the canonical K48-linked degradation signals and K63-linked signaling chains. Recent research has illuminated the specialized functions of K11 linkages, particularly their role as a priority degradation signal that fast-tracks protein turnover during specific cellular processes [5]. The structural biology of K11 chain recognition reveals sophisticated mechanisms employed by cellular machinery, especially the 26S proteasome, to decode and execute the instructions embedded within these chains.
The significance of K11 linkages is particularly evident in cell cycle regulation and proteotoxic stress response, where their presence accelerates substrate degradation compared to K48 homotypic chains [5]. This accelerated degradation pathway is crucial for maintaining cellular homeostasis during periods of rapid protein production or external stress. Furthermore, K11 linkages frequently occur in branched ubiquitin chains, most notably in conjunction with K48 linkages (K11/K48-branched chains), creating complex topological structures that enhance proteasomal recognition and processing efficiency [5] [27].
Despite their biological importance, studying K11-linked ubiquitination presents significant technical challenges due to their typically low abundance relative to major chain types, their presence in complex heterotypic and branched architectures, and the historical lack of specific enrichment tools. This application note details how Tandem Ubiquitin Binding Entities (TUBEs) provide researchers with a powerful methodology to overcome these barriers, enabling precise capture and analysis of K11-linked ubiquitin chains in various experimental contexts.
K11 Ubiquitin Chain Biology and Signaling Pathways
Structural Recognition and Proteasomal Targeting
The human 26S proteasome recognizes K11/K48-branched ubiquitin chains through a multivalent substrate recognition mechanism that involves previously unidentified binding sites. Cryo-EM structures have revealed that the proteasome employs a tripartite binding interface within its 19S regulatory particle to engage with branched chains [5]. This interface includes:
- A novel K11-linked Ub binding site at the groove formed by RPN2 and RPN10 subunits
- The canonical K48-linkage binding site formed by RPN10 and RPT4/5 coiled-coil
- An RPN2 recognition site for alternating K11-K48-linkages through a conserved motif similar to the K48-specific T1 site of RPN1 [5]
This sophisticated recognition system explains the molecular mechanism underlying preferential recognition of K11/K48-branched ubiquitin chains as a priority signal for proteasomal degradation, significantly expanding our understanding of ubiquitin-mediated proteostasis.
Biological Functions of K11-Linked Chains
K11-linked ubiquitin chains serve essential functions in multiple critical cellular processes:
Table 1: Key Biological Functions of K11-Linked Ubiquitin Chains
| Biological Process | Specific Role | Functional Significance |
|---|---|---|
| Cell Cycle Progression | Timely degradation of mitotic regulators | Ensures proper cell division dynamics and fidelity |
| Proteotoxic Stress Response | Clearance of misfolded nascent polypeptides | Maintains protein homeostasis under stress conditions |
| Neurodegenerative Pathways | Degradation of pathological Huntingtin variants | Potential therapeutic target for Huntington's disease |
| Protein Quality Control | Accelerated degradation of specific substrates | Fast-tracking mechanism for urgent protein turnover |
The expanded functionality of K11 linkages underscores the necessity of specialized tools for their study, particularly as research continues to reveal new biological contexts where these chains exert regulatory influence.
TUBE Technology for K11 Chain Enrichment
Fundamental Principles of TUBEs
Tandem Ubiquitin Binding Entities (TUBEs) are engineered, high-affinity reagents composed of multiple ubiquitin-associated (UBA) domains arranged in tandem to achieve nanomolar affinity for polyubiquitin chains. This architectural design enables TUBEs to overcome the inherent limitations of single UBDs, which typically exhibit only millimolar affinity and are insufficient for efficient capture of ubiquitinated proteins from complex lysates [28].
The fundamental advantages of TUBE technology include:
- Protection from deubiquitinating enzymes (DUBs): TUBEs shield polyubiquitin chains from the activity of deubiquitinases during sample preparation, preserving the native ubiquitination state
- Prevention of proteasomal degradation: By occupying ubiquitin binding sites, TUBEs prevent recognition and destruction of ubiquitinated substrates by the proteasome during lysis
- Linkage-specific recognition: Engineered TUBE variants exhibit selective binding for specific ubiquitin chain linkages, including K11 linkages
While pan-selective TUBEs capture all ubiquitin linkage types (K6, K11, K27, K29, K33, K48, K63, and M1), the development of chain-specific TUBEs has revolutionized linkage-specific ubiquitination research by enabling precise isolation of particular chain types [29] [30].
K11 Chain Recognition by TUBEs
Although the search results do not explicitly detail commercially available K11-specific TUBEs, the demonstrated success of K48 and K63-specific TUBEs establishes the feasibility of this approach for K11 chains [29] [22]. The principle of linkage specificity in TUBEs is achieved through strategic engineering of UBA domain sequences to recognize the unique structural features presented by each ubiquitin linkage type.
For K11 linkages, specialized TUBEs would theoretically target the distinct conformation adopted by K11-linked chains, which differs from both the compact conformation of K48-linked chains and the extended structure of K63-linked chains. The development of such reagents would fill a critical gap in the ubiquitin research toolbox, enabling direct enrichment of K11-modified proteins without the need for genetic manipulation or antibody-based methods that may lack sufficient specificity or affinity.
Experimental Protocols for K11 Chain Enrichment Using TUBEs
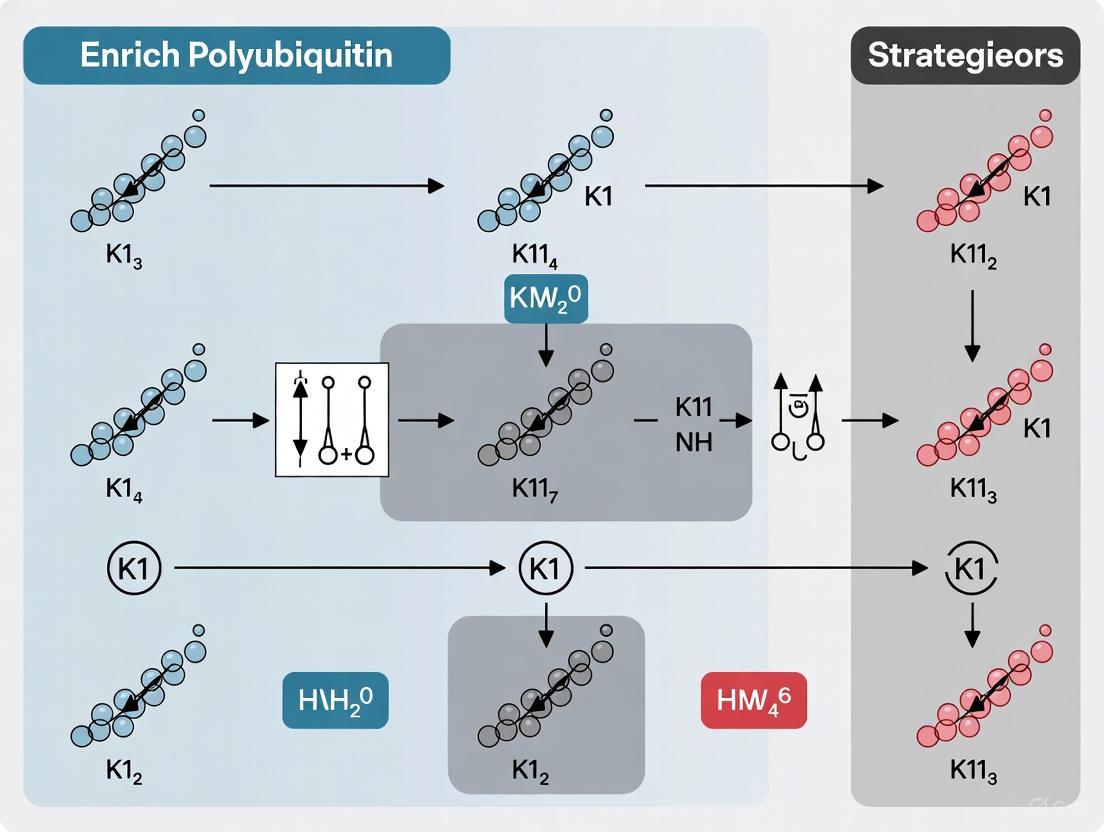
Protocol 1: Enrichment of K11-Ubiquitinated Proteins from Cell Lysates
This protocol describes a method for capturing K11-ubiquitinated proteins from mammalian cell cultures using K11 chain-specific TUBEs, adapted from established TUBE methodologies [29] [23].
Materials and Reagents:
- K11 chain-specific TUBEs (immobilized on magnetic beads or agarose resin)
- Cell lysis buffer: 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% NP-40, 1 mM EDTA, 10% glycerol, supplemented with DUB inhibitors (50 μM PR-619) and proteasome inhibitor (10 μM MG-132)
- Wash buffer: 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.1% NP-40, 1 mM EDTA
- Elution buffer: 100 mM Tris-HCl (pH 6.8), 4% SDS, 20% glycerol, 200 mM DTT
- Phosphate-buffered saline (PBS)
Procedure:
- Cell Lysis and Preparation: Harvest cells by centrifugation at 500 × g for 5 minutes. Wash twice with ice-cold PBS. Lyse cells in lysis buffer (1 mL per 10⁷ cells) by gentle vortexing. Incubate on ice for 30 minutes with occasional mixing.
- Clarification: Centrifuge lysates at 16,000 × g for 15 minutes at 4°C. Transfer supernatant to a fresh tube and determine protein concentration using a compatible assay (e.g., BCA assay).
- TUBE Incubation: Incubate 500 μg - 2 mg of clarified lysate with 20 μL of K11-TUBE beads for 2 hours at 4°C with end-over-end rotation.
- Washing: Collect beads using a magnetic separator or gentle centrifugation (500 × g for 1 minute). Wash three times with 1 mL wash buffer, incubating for 5 minutes with rotation during each wash.
- Elution: After final wash, completely remove wash buffer and resuspend beads in 40 μL elution buffer. Heat at 95°C for 10 minutes with shaking at 1,000 rpm. Separate supernatant containing eluted proteins from beads using magnetic separation or centrifugation.
- Downstream Analysis: Eluted proteins can be analyzed by Western blotting using linkage-specific antibodies or processed for mass spectrometry-based proteomics.
Critical Considerations:
- Maintain inhibitors throughout lysis and binding steps to preserve ubiquitination
- Avoid excessive vortexing or pipetting that may shear polyubiquitin chains
- Include controls with pan-TUBEs and bare beads to assess specificity
Protocol 2: DRUSP-TUBE Workflow for Enhanced K11 Chain Recovery
The Denatured-Refolded Ubiquitinated Sample Preparation (DRUSP) method significantly enhances ubiquitinated protein recovery by addressing limitations of native lysis conditions [31]. When combined with TUBE enrichment, this approach provides superior results for K11 chain analysis.
Materials and Reagents:
- Denaturing lysis buffer: 50 mM Tris-HCl (pH 7.5), 8 M urea, 1% SDS, 10 mM N-ethylmaleimide (DUB inhibitor)
- Refolding buffer: 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% NP-40, 1 mM EDTA
- K11 chain-specific TUBEs (immobilized)
- Amicon Ultra centrifugal filters (10 kDa MWCO, Millipore)
- Standard wash and elution buffers as in Protocol 1
Procedure:
- Denaturing Lysis: Lyse cells directly in denaturing lysis buffer (1 mL per 10⁷ cells) by brief sonication or vigorous pipetting. Incubate at room temperature for 15 minutes with shaking.
- Clarification: Centrifuge at 16,000 × g for 15 minutes at room temperature. Transfer supernatant to a fresh tube.
- Buffer Exchange and Refolding: Transfer denatured lysate to Amicon Ultra centrifugal filter devices. Add 4 mL refolding buffer and concentrate to approximately 500 μL by centrifugation according to manufacturer's instructions. Repeat this dilution and concentration step twice to ensure complete buffer exchange and protein refolding.
- TUBE Enrichment: Recover refolded lysate from filter device and proceed with TUBE enrichment as described in Protocol 1, steps 3-6.
Validation Data: The DRUSP method demonstrates remarkable improvement over conventional approaches:
- Approximately 3-fold stronger ubiquitin signal compared to control methods [31]
- Approximately 10-fold improvement in enriching ubiquitinated proteins overall [31]
- Enhanced quantitative accuracy and reproducibility for ubiquitinomics studies
Research Reagent Solutions for K11 Ubiquitination Studies
A comprehensive toolkit is essential for successful investigation of K11-linked ubiquitination. The following table details essential reagents and their specific applications in K11 chain research.
Table 2: Research Reagent Solutions for K11-Linked Ubiquitin Studies
| Reagent Category | Specific Examples | Applications and Functions |
|---|---|---|
| Chain-Specific TUBEs | K11-TUBEs, K48-TUBEs, K63-TUBEs, Pan-TUBEs | Linkage-specific enrichment of ubiquitinated proteins; protection from DUBs and proteasomal degradation [29] [30] |
| Linkage-Specific Antibodies | Anti-K11, Anti-K48, Anti-K63 ubiquitin antibodies | Detection and validation of specific ubiquitin linkages by Western blotting and immunofluorescence |
| Enzymatic Tools | UBE2S (K11-specific E2), Engineered E3 ligases | In vitro assembly of K11-linked chains; reconstitution of ubiquitination cascades |
| DUB Inhibitors | PR-619 (broad-spectrum), Linkage-specific DUB inhibitors | Preservation of ubiquitination signals during sample preparation |
| Branched Chain Assembly Systems | Ubiquitin mutants (e.g., Ub1-72, UbK48R,K63R), Specific E2/E3 combinations | Generation of defined K11/K48-branched ubiquitin chains for structural and functional studies [27] |
| Mass Spectrometry Standards | K11-linked diUb AQUA peptides, SILAC-labeled ubiquitin | Absolute quantification of K11 chain abundance in complex samples |
Data Analysis and Interpretation
Validation of K11-Specific Enrichment
Proper validation is crucial for confirming the specificity of K11 chain enrichment. Researchers should implement a multi-faceted validation strategy:
- Linkage Specificity Controls: Compare K11-TUBE enrichment to parallel experiments with K48-TUBEs, K63-TUBEs, and pan-TUBEs to demonstrate linkage selectivity
- Competition Assays: Pre-incubate K11-TUBEs with free K11-linked diUb or tetraUb chains to compete binding, demonstrating specificity through reduced substrate recovery
- Mass Spectrometry Verification: Utilize MS-based ubiquitin remnant motif profiling to confirm ubiquitination sites and linkage types through characteristic peptide signatures
Quantitative Assessment of K11 Ubiquitination
Recent advances in quantitative ubiquitinomics enable precise measurement of K11 chain dynamics:
Table 3: Quantitative Approaches for K11 Chain Analysis
| Method | Principle | Applications | Considerations |
|---|---|---|---|
| Ub-AQUA | Absolute quantification using heavy isotope-labeled ubiquitin peptides | Precise measurement of K11 chain abundance relative to other linkages | Requires specialized instrumentation and expertise in peptide quantification |
| SILAC/TMT with TUBE Enrichment | Metabolic or chemical labeling combined with affinity enrichment | Comparative analysis of K11 ubiquitination across multiple conditions | Potential introduction of bias during enrichment step |
| DUB Profiling | Treatment with linkage-specific deubiquitinases followed by Western blot | Semi-quantitative assessment of K11 chain contribution to total ubiquitination | Provides relative rather than absolute quantification |
Tandem Ubiquitin Binding Entities represent a transformative technology for studying K11-linked ubiquitin chains, enabling researchers to overcome historical limitations in specificity, affinity, and preservation of native ubiquitination states. The protocols and methodologies detailed in this application note provide a robust framework for investigating the biological functions of K11 linkages, particularly their emerging roles in prioritized protein degradation and cellular stress response pathways.
As research continues to elucidate the complex signaling capabilities of K11 chains, especially in the context of branched ubiquitin architectures, TUBE-based approaches will remain essential tools for deciphering the ubiquitin code. The integration of TUBE technology with advanced mass spectrometry techniques and structural biology approaches promises to unlock new dimensions of understanding regarding ubiquitin-mediated regulation of cellular processes.
Ubiquitin-Binding Domain (UBD) Based Capture Strategies
Ubiquitin-Binding Domain (UBD) based capture strategies represent a powerful methodology in proteomics for the enrichment and study of ubiquitinated proteins, particularly those modified with specific chain linkages such as K11-polyubiquitin. These strategies leverage the natural affinity of UBDs for ubiquitin moieties to isolate ubiquitinated conjugates from complex biological samples. Unlike antibody-based approaches, which can exhibit linkage bias and have limited affinity, UBD-based tools can be engineered for high affinity and pan-specificity, enabling unbiased enrichment of all ubiquitin linkage types [32] [30]. This is crucial for researching K11-linked chains, which often function in concert with other linkages (e.g., K11/K48-branched chains) to regulate critical processes like mitotic progression and proteasomal degradation [33] [5]. The application of these strategies provides researchers with a robust means to decipher the complex ubiquitin code within cellular systems.
UBD-Based Tools: Characteristics and Selection
Various UBD-based tools have been developed, each with distinct properties and applications. The selection of an appropriate tool is critical for experimental success, particularly when targeting specific chain architectures like K11-linked or K11/K48-branched chains.
Table 1: Comparison of Key UBD-Based Affinity Reagents
| Reagent Name | Composition / Type | Affinity & Specificity | Key Advantages | Primary Applications |
|---|---|---|---|---|
| TUBE(Tandem Ubiquitin-Binding Entity) | Multiple tandem UBA domains [30] | High affinity for polyUb; pan-selective for all linkages (K6, K11, K27, K29, K33, K48, K63, M1) [30] | Shields chains from DUBs; preserves native architecture during isolation [30] | Pulldown and proteomic identification of ubiquitinated proteins; studying dynamic ubiquitin remodeling [30] |
| ThUBD(Tandem Hybrid UBD) | Engineered fusion of different UBDs [32] | Unbiased, high-affinity capture of all ubiquitin chain types [32] | 16-fold wider linear range for capturing polyUb proteins compared to TUBE; superior sensitivity [32] | High-throughput, sensitive quantification of global and target-specific ubiquitination using coated plates; PROTAC development [32] |
| Linkage-Specific TUBEs | Tandem UBA domains with linkage preference | Specific for K48 or K63 linkages [30] | Enables deep, linkage-focused exploration of the ubiquitinome [30] | Targeted enrichment of substrates modified with specific chain types [30] |
Detailed Experimental Protocols
High-Throughput Capture Using ThUBD-Coated Plates
This protocol details a high-sensitivity method for capturing ubiquitinated proteins from cell lysates using 96-well plates coated with ThUBD [32].
Reagents and Equipment: Recombinant ThUBD protein [32], Corning 3603 96-well plates [32], cell lysis buffer (e.g., RIPA), proteasome inhibitor (e.g., MG132), PBS-T (PBS with 0.1% Tween-20), blocking buffer (5% BSA in PBS), detection primary antibody (e.g., anti-ubiquitin), HRP-conjugated secondary antibody, and chemiluminescent substrate.
Procedure:
- Plate Coating: Coat each well of a 96-well plate with 1.03 µg of ThUBD protein in PBS. Incubate overnight at 4°C.
- Blocking: Remove the coating solution and block plates with 200 µL of blocking buffer for 2 hours at room temperature.
- Sample Preparation and Incubation: Prepare whole-cell lysates from treated cells. Wash coated plates three times with PBS-T. Add up to 100 µg of protein lysate per well and incubate for 2 hours at room temperature with gentle shaking.
- Washing and Detection: Wash wells five times with PBS-T. Add primary antibody diluted in blocking buffer and incubate for 1 hour. After washing, add HRP-conjugated secondary antibody and incubate for 1 hour. Perform final washes and develop signal with chemiluminescent substrate. Quantify the chemiluminescence.
Affinity Enrichment with TUBEs for Mass Spectrometry
This protocol describes the use of pan-selective TUBEs for enriching ubiquitinated proteins from complex proteomes for subsequent mass spectrometric analysis [30].
Reagents and Equipment: Pan-selective TUBEs (immobilized on agarose beads), lysis buffer (e.g., with SDS and DUB inhibitors), benzonase, wash buffer (e.g., with 150-500 mM NaCl), urea buffer (8 M urea, 100 mM Tris-HCl, pH 8.0), digestion buffer (50 mM AMBC, 5% ACN, pH 8.0), Trypsin Gold [34].
Procedure:
- Cell Lysis and Pre-Clearance: Lyse cells in a buffer containing DUB inhibitors to preserve ubiquitin chains. Benzonase can be added to digest nucleic acids. Centrifuge the lysate to remove insolubles and pre-clear with control beads.
- TUBE Pulldown: Incubate the pre-cleared lysate with TUBE-coated beads for 2-4 hours at 4°C.
- Stringent Washing: Wash the beads sequentially with lysis buffer and high-salt wash buffer to reduce non-specific binding.
- On-Bead Digestion and Peptide Preparation: Denature proteins with urea buffer. Reduce, alkylate, and digest proteins on-bead with Trypsin Gold (e.g., 20 ng/µL, 37°C, 15 hours) [34]. Acidify the resulting peptide mixture, desalt, and concentrate for LC-MS/MS analysis.
Workflow for K11/K48-Branched Chain Analysis
The following diagram illustrates the core experimental workflow for analyzing K11/K48-branched ubiquitin chains using UBD-based strategies.
The Scientist's Toolkit: Key Research Reagents
Successful implementation of UBD-based capture strategies requires a set of essential reagents. The table below details key solutions for studying K11-linked polyubiquitination.
Table 2: Essential Research Reagents for UBD-Based Ubiquitin Capture
| Reagent / Tool | Function / Role in Experiment | Key Features & Considerations |
|---|---|---|
| Pan-Selective TUBEs | High-affinity enrichment of polyubiquitinated proteins with all linkage types from cell lysates [30]. | Protects ubiquitin chains from deubiquitinating enzymes (DUBs) and proteasomal degradation during processing [30]. |
| ThUBD | Unbiased, high-affinity capture of proteins modified with any ubiquitin chain type, used in plate-based or pulldown formats [32]. | Exhibits a 16-fold wider linear range and superior sensitivity compared to TUBEs, ideal for high-throughput applications [32]. |
| Linkage-Specific DUBs (for UbiCRest) | Enzymatic dissection of ubiquitin chain architecture following enrichment to confirm linkage composition (e.g., K11/K48-branched) [35]. | Used in parallel digestions; some DUBs have preferences for multiple linkages, which requires careful interpretation of results [35]. |
| DUB Inhibitors | Added to cell lysis and purification buffers to prevent the cleavage of ubiquitin chains by endogenous deubiquitinases, preserving the native ubiquitome [30]. | Critical for maintaining the integrity of labile chains like K11/K48-branched structures during sample preparation. |
| K11/K48-Bispecific Antibody | Immunoprecipitation and detection of heterotypic K11 and K48 ubiquitin chains, such as those in cell cycle regulation [35]. | Provides direct evidence for the coexistence of K11 and K48 linkages, though it may not distinguish branched from mixed chains without additional methods [35]. |
| UB-AQUA/PRM Peptides | Isotopically labeled internal standards for absolute quantification of all eight ubiquitin linkage types via targeted mass spectrometry [34]. | Enables direct comparison of linkage stoichiometry across samples; requires a targeted LC-MS/MS setup like a Q Exactive instrument [34]. |
Concluding Remarks
UBD-based capture strategies, utilizing reagents like TUBEs and ThUBDs, provide researchers with a versatile and powerful set of tools for the enrichment and study of the ubiquitinome. Their high affinity and ability to unbiasedly capture diverse chain linkages make them particularly valuable for investigating complex polyubiquitin signals, such as K11-linked and K11/K48-branched chains. The continued development and refinement of these protocols, especially in high-throughput formats, are accelerating our understanding of ubiquitin signaling in health and disease, and supporting critical efforts in targeted protein degradation drug discovery.
Engineered Deubiquitinases (enDUBs) for Linkage-Selective Analysis
Within the complex language of the ubiquitin code, K11-linked polyubiquitin chains have emerged as critical regulators in essential cellular processes, particularly in cell cycle progression and proteotoxic stress response [5]. During mitotic exit, cells exhibit a sharp increase in K11 linkages, which are synthesized by the coordinated action of the APC/C complex and the E2 enzyme UBE2S to ensure the timely degradation of mitotic regulators such as Aurora kinases and Polo-like kinase [3]. Beyond homotypic chains, K11/K48-branched ubiquitin chains constitute a distinct topological signal that functions as a priority degradation signal, efficiently recruiting substrates to the 26S proteasome through multivalent interactions with ubiquitin receptors RPN1, RPN2, and RPN10 [5].
Despite their established biological significance, research progress on K11-linked chains has been hampered by a lack of methods to specifically manipulate these linkages on individual proteins in live cells. This application note details how engineered deubiquitinases (enDUBs) bridge this critical methodological gap, providing researchers with a powerful tool to dissect the K11-linked polyubiquitin chain functions within their physiological contexts.
enDUB Technology: Principle and Linkage Specificity
Engineered deubiquitinases (enDUBs) represent a targeted protein stabilization method that enables the selective removal of polyubiquitin chains from specific proteins of interest in live cells. The core enDUB architecture consists of a catalytic domain from a linkage-selective deubiquitinase fused to a GFP-targeted nanobody (e.g., LaG16) [36]. This design facilitates the physical recruitment of the DUB activity to a target protein fused to GFP or YFP, enabling substrate-specific deubiquitination.
The linkage specificity of the enDUB system is conferred by the selective catalytic domains of naturally occurring DUBs. For K11-linked chain analysis, the catalytic domain of Cezanne is the primary tool due to its pronounced preference for hydrolyzing K11 linkages [37]. The table below summarizes the key enDUB constructs and their linkage specificities.
Table 1: Linkage-Selective enDUB Toolkit for Polyubiquitin Chain Analysis
| enDUB Construct | Catalytic Domain Source | Polyubiquitin Linkage Specificity | Primary Application in K11 Research |
|---|---|---|---|
| nanoCezanne | Cezanne | K11-specific | Probing ER retention & endosomal trafficking |
| nanoOTUD4 | OTUD4 | K48-specific | Control for studying canonical degradation |
| nanoOTUD1 | OTUD1 | K63-specific | Control for studying endocytosis & signaling |
| nanoTRABID | TRABID | K29/K33-specific | Probing ER retention & degradation |
| nanoUSP21 | USP21 | Non-specific | Pan-deubiquitination control |
Application Note: Decoding the K11-Linked Ubiquitin Code on KCNQ1
Quantitative Analysis of Polyubiquitin Linkages on KCNQ1
The application of the enDUB toolkit to the cardiac potassium channel KCNQ1 (KV7.1) provides a compelling case study. Mass spectrometry analysis of YFP-KCNQ1 expressed in HEK293 cells revealed a complex ubiquitin landscape, with K48 (72%) and K63 (24%) linkages being dominant, while K11 and other linkages constituted minor but functionally significant populations [37]. This quantitative profiling establishes a baseline for understanding the polyubiquitin composition on an ion channel substrate.
Table 2: Functional Outcomes of Linkage-Selective Deubiquitination on KCNQ1
| enDUB Treatment | Impact on KCNQ1 Surface Density | Impact on KCNQ1 Ionic Currents | Proposed Mechanism of Action |
|---|---|---|---|
| nanoCezanne (K11) | Significant Increase | Significant Increase | Reduces ER retention/degradation and enhances endocytosis/recycling |
| nanoTRABID (K29/K33) | Significant Increase | Significant Increase | Reduces ER retention/degradation |
| nanoOTUD1 (K63) | Moderate Increase | Moderate Increase | Reduces endocytosis and enhances recycling |
| nanoOTUD4 (K48) | Decrease | Decrease | Unexpectedly reduces forward trafficking |
| nanoUSP21 (Non-specific) | Moderate Increase | Moderate Increase | General stabilization across multiple pathways |
Mechanistic Insights into K11 Chain Function
The data from KCNQ1 studies demonstrate that K11 linkages exert a net retention effect on the channel, primarily achieved through two distinct mechanisms: promoting ER retention/degradation and enhancing endocytosis while reducing recycling [37] [38]. This dual regulatory role highlights the multifaceted nature of the K11 ubiquitin code in controlling protein localization. Furthermore, the regulatory code is mutable, as the effects of enDUBs on KCNQ1 observed in HEK293 cells differed in cardiomyocytes, emphasizing the context-dependent nature of ubiquitin signaling [37].
Experimental Protocols
Protocol 1: Constructing a Linkage-Selective enDUB
This protocol describes the molecular cloning strategy for generating a K11-specific enDUB [37] [36].
- Amplify DUB Catalytic Domain: Perform PCR amplification of the Cezanne catalytic domain (residues 1-340, or a similar defined fragment) from a cDNA source using primers that add appropriate restriction sites.
- Amplify Nanobody Sequence: Similarly, amplify the LaG16 anti-GFP nanobody sequence from a plasmid template (e.g., pCMV-LaG16).
- Plasmid Assembly: Digest both PCR products and the recipient mammalian expression vector (e.g., pcDNA3.1) with the chosen restriction enzymes. Purify the digested fragments.
- Ligation and Transformation: Ligate the DUB insert and nanobody insert into the prepared vector backbone in a single reaction. Transform the ligation product into competent E. coli cells.
- Screening and Validation: Screen colonies by colony PCR and analytical restriction digest. Confirm the final construct by Sanger sequencing to ensure in-frame fusion and error-free amplification.
- Protein Expression Validation: Transfert the purified plasmid into HEK293T cells and analyze lysates by western blotting 48 hours post-transfection using antibodies against the DUB or a tag to confirm expression.
Protocol 2: Validating enDUB Activity and Specificity on a Substrate
This protocol outlines steps to confirm that the engineered enDUB selectively deubiquitinates a target GFP-fused protein [37].
- Cellular Reconstitution: Co-transfect HEK293 cells with three plasmid constructs:
- pcDNA3.1-YFP-Protein of Interest (POI)
- pcDNA3.1-enDUB (e.g., nanoCezanne)
- pCMV-HA-Ubiquitin (or a linkage-restricted mutant, e.g., K11-only Ub)
- Immunoprecipitation: 48 hours post-transfection, lyse cells in RIPA buffer. Immunoprecipitate the YFP-POI complex using GFP-Trap beads. Incubate lysates with beads for 2 hours at 4°C, then wash 3x with lysis buffer.
- Western Blot Analysis: Elute immunoprecipitated proteins in SDS sample buffer and resolve by SDS-PAGE. Transfer to a PVDF membrane and probe with the following antibodies:
- Anti-HA (to detect conjugated ubiquitin)
- Anti-GFP (to assess total pulled-down YFP-POI)
- Linkage-specific antibodies (e.g., anti-K11-linkage specific antibody)
- Functional Assay (Ion Channels): For electrophysiological functional analysis, plate cells on glass coverslips. Co-transfect YFP-POI (e.g., KCNQ1) with the enDUB at a 1:3 ratio. 48 hours post-transfection, use patch-clamp electrophysiology to record ionic currents. A successful K11-specific enDUB (nanoCezanne) should significantly increase current density compared to controls.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for enDUB and K11 Ubiquitin Research
| Reagent / Tool | Function / Specificity | Key Application in Research |
|---|---|---|
| Cezanne Catalytic Domain | K11-linkage selective DUB activity | Core component of the K11-specific enDUB [37] |
| Anti-GFP Nanobody (LaG16) | Binds GFP/YFP with high affinity | Targets the enDUB to the substrate of interest [36] |
| K11-linkage Specific Antibody | Recognizes K11-linked polyUb chains | Validating enDUB specificity and profiling substrates [3] |
| K11-only Ubiquitin Mutant | Allows only K11 chain formation (all other lysines mutated) | Defining specific roles of K11 chains in substrate regulation [3] |
| UBE2S (E2 Enzyme) | APC/C-associated E2 that elongates K11 chains | Enzyme for in vitro reconstitution of K11 chains [3] |
| Activity-Based Probes (ABPs) | Ubiquitin-based probes with electrophilic warheads | Profiling endogenous DUB activity and specificity [39] |
Visualizing Experimental Workflows and Signaling Pathways
Tagged Ubiquitin Systems for Controlled Expression and Purification
The study of specific ubiquitin chain types, particularly K11-linked polyubiquitin, is crucial for advancing our understanding of regulated protein degradation in health and disease. K11-linked polyubiquitin chains have been identified as critical regulators of cell cycle progression, mitotic exit, and endoplasmic reticulum-associated degradation (ERAD) [2] [3] [4]. These chains constitute a significant proportion (approximately 28%) of the cellular ubiquitin conjugate pool and function as potent proteasomal targeting signals [4]. Research into K11-linked ubiquitination has revealed its role in the timely degradation of key mitotic regulators, including Aurora kinases and Polo-like kinase, during cell cycle progression [3].
A significant technical challenge in this field is the purification and characterization of proteins specifically modified with K11-linked chains from the complex cellular environment. To address this, tagged ubiquitin systems have been developed, enabling high-yield expression and affinity purification of ubiquitinated proteins. This application note details robust methodologies for utilizing these systems, with emphasis on their application for enriching and studying K11-linked polyubiquitin chains.
The Role of K11-Linked Polyubiquitin Chains in Cellular Regulation
K11-linked polyubiquitin chains are dynamically regulated signaling entities with distinct structural and functional characteristics. Structural analyses using NMR and SANS reveal that K11-linked di-ubiquitin (K11-Ub2) adopts unique conformations in solution that are distinct from K48-linked or K63-linked chains [7]. These unique conformational properties allow K11-linked chains to be differentially recognized by downstream receptor proteins.
Table 1: Key Functional Roles of K11-Linked Polyubiquitin Chains
| Biological Process | Function of K11 Linkages | Key Enzymes Involved | Reference |
|---|---|---|---|
| Cell Cycle Control | Target mitotic regulators (e.g., Aurora kinases) for degradation during mitotic exit | APC/C, UBE2C, UBE2S | [2] [3] |
| ERAD | Mediate degradation of misfolded proteins from the endoplasmic reticulum | Ubc6 | [4] |
| Proteotoxic Stress | Facilitate clearance of misfolded proteins and pathological Huntingtin variants | Not specified | [5] |
| General Proteasomal Targeting | Serve as efficient degradation signal, often in branched chains with K48 linkages | Various E2/E3 combinations | [5] [4] |
The anaphase-promoting complex/cyclosome (APC/C) functions with specific E2 enzymes (UBE2C for priming and UBE2S for K11-specific chain elongation) to generate K11 linkages on key mitotic substrates [3]. Quantitative studies demonstrate that depleting K11 linkages through UBE2S knockdown stabilizes anaphase substrates, confirming their critical role in degradation timing during mitotic exit [3]. Furthermore, recent cryo-EM structures reveal that the human 26S proteasome recognizes K11/K48-branched ubiquitin chains through a multivalent mechanism involving RPN2 and RPN10, explaining the priority degradation signal conferred by this chain architecture [5].
Figure 1: K11-Linked Ubiquitination Pathway. This diagram illustrates the enzymatic cascade for K11-linked chain assembly, highlighting the roles of UBE2C (priming) and UBE2S (K11-specific elongation) E2 enzymes working with the APC/C E3 ligase.
Tagged Ubiquitin Systems: Principles and Selection
Tagged ubiquitin systems involve the genetic fusion of an affinity tag to the ubiquitin molecule, enabling one-step purification of ubiquitin conjugates from complex cell lysates. The two primary design strategies are N-terminal tagging and C-terminal tagging, each with distinct advantages for specific research applications.
System Comparison and Selection Criteria
Table 2: Comparison of Tagged Ubiquitin Systems
| System Feature | His-Tagged Ubiquitin | Avi-Tagged Ubiquitin | Strep-Tagged Ubiquitin |
|---|---|---|---|
| Tag Example | 6xHis | AviTag (for biotinylation) | Strep-tag II |
| Affinity Resin | Ni-NTA or Co²⁺-NTA | Streptavidin | Strep-Tactin |
| Elution Method | Imidazole or low pH | Cleavage (e.g., 3C protease) | Desthiobiotin |
| Key Advantage | Cost-effective, high yield | High specificity, native elution | High specificity, gentle elution |
| Potential Drawback | Co-purification of histidine-rich proteins | Requires biotinylation step | Higher resin cost |
| Reported Yield Range | Up to 53 mg/L [40] | Not specified | Not specified |
| Compatibility with K11 Studies | Excellent for conjugate purification | Excellent for specific substrate studies | Excellent for interactome studies |
The pHUE vector (His-Ubiquitin-Epitope tag) represents an efficient E. coli-based system for expressing proteins as histidine-tagged ubiquitin fusions [40]. This system enables simple one-step purification of fusion proteins by immobilized metal affinity chromatography (IMAC), with reported yields up to 53 mg/L of culture for certain fusion proteins [40]. The histidine-tagged ubiquitin can be effectively used in conjugation assays to generate Ni²⁺-binding ubiquitin-protein conjugates, facilitating the purification of ubiquitinated species [41].
For researchers specifically interested in K11-linked ubiquitination, the choice of system depends on the experimental goals. His-tagged ubiquitin systems are ideal for initial conjugate purification and ubiquitination site mapping, while the Avi-tagged system described in Section 4.2 offers advantages for purifying natively mono-ubiquitinated or polyubiquitinated forms of specific proteins of interest.
Experimental Protocols
Protocol 1: Expression and Purification Using the pHUE System
This protocol describes the expression and purification of ubiquitinated proteins using the histidine-tagged ubiquitin pHUE vector system [40].
Materials
- pHUE vector (modified pET15b with His-tagged ubiquitin)
- BL21(DE3) E. coli competent cells
- Ni-NTA agarose resin
- Luria-Bertani (LB) medium with ampicillin (100 µg/mL)
- Isopropyl β-D-1-thiogalactopyranoside (IPTG)
- Lysis Buffer: 1x PBS, 20 mM imidazole, 10% (w/v) glycerol, 1 mM DTT
- Wash Buffer: Lysis Buffer with 40 mM imidazole
- Elution Buffer: Lysis Buffer with 250-500 mM imidazole
Method
- Cloning and Transformation: Clone the gene of interest into the pHUE vector multiple cloning site. Transform into BL21(DE3) competent cells and plate on LB-agar containing ampicillin.
- Protein Expression: Inoculate a single colony into 50 mL LB with ampicillin and grow overnight at 37°C. Dilute the culture 1:50 into fresh medium and grow at 37°C until OD600 reaches 0.6-0.8. Induce expression with 0.4-1.0 mM IPTG and incubate for 4-16 hours at appropriate temperature (20-37°C based on protein solubility).
- Cell Harvest and Lysis: Harvest cells by centrifugation at 6,000 × g for 15 minutes at 4°C. Resuspend pellet in Lysis Buffer supplemented with lysozyme (1 mg/mL) and protease inhibitors. Lyse by sonication on ice.
- Clarification: Centrifuge lysate at 16,000 × g for 40 minutes at 4°C. Collect supernatant containing soluble ubiquitin fusion proteins.
- Affinity Purification: Incubate supernatant with Ni-NTA resin (pre-equilibrated in Lysis Buffer) for 2 hours with gentle agitation at 4°C. Load resin into a column and wash with 10-20 column volumes of Wash Buffer.
- Elution: Elute the His-tagged ubiquitin fusion protein with Elution Buffer. Collect 1 mL fractions and analyze by SDS-PAGE.
- Cleavage (Optional): For tag removal, incubate purified fusion protein with histidine-tagged deubiquitinating enzyme (e.g., Usp2 catalytic domain) [40]. Remove the enzyme and cleaved ubiquitin using a second Ni-NTA step.
Figure 2: His-Tagged Ubiquitin Workflow. Experimental workflow for expression and purification using the pHUE histidine-tagged ubiquitin system.
Protocol 2: Avi-Tagged Ubiquitin for Native Ubiquitinated Protein Purification
This protocol describes a method for generating natively mono-ubiquitinated or polyubiquitinated proteins using an N-terminally biotinylated Avi-tagged ubiquitin, particularly useful for studying substrates of the Fanconi anemia-BRCA pathway and other DNA repair proteins [42].
Materials
- Avi-ubiquitin construct (N-terminal 10xHis tag, AviTag, 3C protease site, ubiquitin)
- BirA biotin ligase
- Streptavidin resin
- E1, E2, and E3 enzymes for in vitro ubiquitination
- Cleavage Buffer: 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM DTT
- HRV 3C protease
Method
- Biotinylated Avi-Ubiquitin Preparation: Co-express the Avi-ubiquitin construct with BirA biotin ligase in E. coli to achieve in vivo biotinylation. Purify using Ni-NTA chromatography as described in Protocol 1, steps 3-6.
- In Vitro Ubiquitination: Set up ubiquitination reactions containing E1 enzyme, specific E2 enzyme (select based on desired linkage), E3 ligase, Avi-ubiquitin, ATP-regenerating system, and substrate protein. For K11-linked chains, utilize UBE2S as the E2 enzyme [3]. Incubate at 30°C for 1-3 hours.
- Affinity Capture: Incubate the ubiquitination reaction with streptavidin resin for 1 hour at 4°C to capture biotinylated ubiquitin conjugates.
- Washing: Wash resin extensively with appropriate buffer to remove non-specifically bound proteins.
- Elution by Proteolytic Cleavage: Incubate resin with HRV 3C protease in Cleavage Buffer to release the natively ubiquitinated protein from the AviTag. Alternatively, use low pH elution (pH 4.5) if maintaining the tag is not an issue [42] [41].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for K11-Linked Ubiquitin Research
| Reagent / Tool | Function / Application | Example / Source | Utility in K11 Research |
|---|---|---|---|
| K11 Linkage-Specific Antibody | Detection and enrichment of K11-linked chains | Matsumoto et al., 2010 [2] | Validate K11 chain formation; monitor mitotic upregulation |
| UBE2S (E2 Enzyme) | K11-specific chain elongation | Recombinant expression [3] | Generate homotypic K11 chains in vitro |
| Cezanne (DUB) | K11-linkage specific deubiquitinase | Recombinant expression [3] | Confirm K11 linkage presence (UbiCRest analysis) |
| UbcH10 (E2 Enzyme) | K11-chain initiation with APC/C | Recombinant expression [7] | Recapitulate APC/C-mediated ubiquitination |
| His-Tagged Ubiquitin | Affinity purification of ubiquitin conjugates | pHUE vector system [40] | Purify K11-ubiquitinated substrates for proteomics |
| Avi-Tagged Ubiquitin | Purification of natively ubiquitinated proteins | His-Avi-3C-Ub construct [42] | Generate pure K11-ubiquitinated complexes for biochemistry |
| Lbpro* Ub Clipping Agent | Branch mapping of ubiquitin chains | Tandem ubiquitin binding entity (TUBE) [5] | Identify K11/K48-branched chains |
Applications in K11-Linked Polyubiquitin Chain Research
The tagged ubiquitin systems described herein are particularly valuable for researching K11-linked polyubiquitin chains, which exhibit unique conformational properties and function as critical degradation signals in specific cellular contexts [7]. These systems enable:
Identification of K11-Linked Substrates: His-tagged ubiquitin enables purification of ubiquitinated proteins for subsequent identification of K11-specific substrates using linkage-specific antibodies or mass spectrometry [4] [28]. This approach identified Ubc6 as a K11 linkage-specific substrate involved in ERAD [4].
Mechanistic Studies of K11 Chain Recognition: Purified K11-linked conjugates facilitate structural studies of chain recognition. Recent cryo-EM structures of K11/K48-branched chains bound to the human 26S proteasome revealed a specialized multivalent recognition mechanism involving RPN2 and RPN10, distinct from canonical K48-chain recognition [5].
Functional Analysis of K11 Chain Formation: The Avi-tagged system allows purification of natively ubiquitinated protein complexes to study the functional consequences of K11 ubiquitination without interference from other ubiquitin forms [42]. This is particularly useful for studying the role of K11 linkages in DNA repair complexes.
Quantification of K11 Chain Dynamics: Tagged ubiquitin systems combined with linkage-specific tools enable quantitative analysis of K11 chain accumulation during proteasomal inhibition or in specific cell cycle phases, revealing their regulation during mitosis [2] [3].
Troubleshooting and Technical Considerations
- Low Ubiquitination Efficiency: Optimize E2:E3 enzyme combinations and ratios. For K11 linkages, ensure UBE2S is present for chain elongation [3].
- Incomplete Cleavage: For Avi-tagged systems, verify 3C protease activity and optimize incubation time and temperature.
- Non-specific Binding in His-tag Purification: Include 40-60 mM imidazole in wash buffers and consider using a step-wise imidazole gradient [40].
- Low Yield of Ubiquitinated Proteins: Consider proteasome inhibition (e.g., MG132) to stabilize ubiquitin conjugates before purification [4].
- Linkage Specificity Verification: Always confirm K11 linkage formation using linkage-specific antibodies [2] or DUBs (e.g., Cezanne) [3] following purification.
Mass Spectrometry-Based Approaches for K11 Chain Verification
K11-linked polyubiquitin chains represent a critical, though less characterized, component of the ubiquitin code. Unlike the canonical K48-linked chains that predominantly target substrates for proteasomal degradation and K63-linked chains that function in non-proteolytic signaling, K11 linkages exhibit unique functional versatility. These chains are highly upregulated during mitosis and play essential roles in cell cycle regulation through the anaphase-promoting complex/cyclosome (APC/C), which ubiquitinates mitotic regulators with K11-linked chains for proteasomal degradation [1] [2]. Beyond their degradative function, K11 linkages also participate in non-proteolytic pathways, including cytokine signaling and NF-κB activation [7] [1]. Structural studies reveal that K11-linked di-ubiquitin adopts distinct conformations in solution that differ from both K48-linked and K63-linked chains, enabling unique interactions with ubiquitin-receptor proteins [7]. Furthermore, K11 linkages can form branched ubiquitin chains in combination with K48 linkages, creating a potent degradation signal that is preferentially recognized by the 26S proteasome [5]. The verification and quantification of these chains through mass spectrometry-based approaches are therefore essential for understanding their diverse cellular functions.
Mass Spectrometry Methods for Ubiquitin Chain Analysis
Ubiquitin-Absolute Quantification (Ub-AQUA) with Parallel Reaction Monitoring (PRM)
The Ub-AQUA/PRM method represents a targeted proteomics approach for direct and highly sensitive measurement of the stoichiometry of all eight ubiquitin linkage types simultaneously. This technique utilizes isotopically labeled signature peptides (AQUA peptides) for each linkage type as internal standards for absolute quantification [34]. The PRM methodology, performed on quadrupole-equipped Orbitrap instruments, provides quantitative data over a wide dynamic range from complex biological samples by measuring fragment ions (MS2) with a high-resolution Orbitrap analyzer, enabling both high sensitivity and accuracy [34].
Table: Key Steps in Ub-AQUA/PRM Sample Preparation and Analysis
| Step | Description | Key Considerations |
|---|---|---|
| Protein Digestion | Trypsin digestion of ubiquitinated proteins generates signature peptides specific to each linkage type. | Trypsin cleaves after lysine and arginine residues, producing characteristic peptides for each ubiquitin linkage. |
| AQUA Peptide Addition | Isotopically labeled signature peptides for all eight linkage types are added to samples as internal standards. | Added in known quantities before LC-MS/MS analysis to enable absolute quantification. |
| Liquid Chromatography | Peptide separation using nanoflow liquid chromatography. | Reduces sample complexity and improves ionization efficiency. |
| Mass Spectrometry Analysis | Parallel reaction monitoring on Q Exactive series instruments. | High-resolution, accurate-mass measurements of both precursor and fragment ions. |
| Data Analysis | Quantification based on fragment ion chromatograms of light (sample) and heavy (standard) peptides. | Enables precise determination of ubiquitin chain linkage abundance. |
Experimental Protocol: Ub-AQUA/PRM for K11 Linkage Quantification
Sample Preparation Protocol:
- Extract proteins from biological samples of interest (e.g., mitotic cells, tissue samples).
- Enrich ubiquitinated proteins using ubiquitin-binding entities or immunoprecipitation with ubiquitin-specific antibodies.
- Reduce and alkylate proteins using dithiothreitol and iodoacetamide standard protocols.
- Digest proteins with trypsin (20 ng/μL) in 50 mM AMBC, 5% ACN (pH 8.0) at 37°C for 15 hours.
- Add AQUA peptide mixture (25 fmol/injection) to the extracted peptides.
- Acidify samples with 0.1% TFA containing 0.05% H2O2 and incubate at 4°C overnight.
LC-MS/MS Analysis:
- Perform chromatographic separation using Easy nLC 1200 or similar nanoflow LC system.
- Set up PRM method on Q Exactive or Orbitrap Fusion Lumos mass spectrometer:
- Full MS scan: 70,000 resolution, 375-1500 m/z scan range
- PRM scans: 17,500 resolution, 1 m/z isolation window, normalized collision energy 27
- Target AGC value: 2e5, maximum injection time: 120 ms
- Acquire data for both endogenous peptides and heavy isotope-labeled AQUA standards.
Data Analysis:
- Process raw data using Skyline or similar software for targeted proteomics.
- Integrate peak areas for fragment ions of both light and heavy peptides.
- Calculate ratios of light to heavy peptides for each ubiquitin linkage type.
- Determine absolute amounts based on known quantities of AQUA peptides added.
This protocol enables highly sensitive and specific quantification of K11-linked ubiquitin chains in complex biological samples, with the capacity to detect as little as 25 fmol of ubiquitin linkage [34].
Complementary Methods for K11 Chain Verification
Linkage Determination Using Ubiquitin Mutants
A biochemical approach for determining ubiquitin chain linkage utilizes ubiquitin mutants in in vitro conjugation assays. This method employs two sets of ubiquitin variants: Ubiquitin Lysine to Arginine (K to R) Mutants and Ubiquitin K Only Mutants [26]. The protocol involves setting up multiple in vitro ubiquitination reactions with different ubiquitin mutants to identify the specific lysine residues required for chain formation.
Table: Ubiquitin Mutant Approach for Linkage Determination
| Reaction Type | Ubiquitin Variant | Interpretation of Results |
|---|---|---|
| K-to-R Series | Individual K-to-R mutants (K6R, K11R, K27R, K29R, K33R, K48R, K63R) | The mutant that fails to form chains indicates the essential linkage lysine. |
| K-Only Series | Single lysine mutants (all lysines except one mutated to arginine) | Only the mutant with the correct lysine will form chains, confirming linkage type. |
| Wild-type Control | Unmodified ubiquitin | Positive control for chain formation. |
| Negative Control | No ATP | Confirms reaction dependency on ubiquitination machinery. |
Experimental Protocol:
- Set up conjugation reactions (25 μL volume) containing:
- 2.5 μL 10X E3 Ligase Reaction Buffer (500 mM HEPES, pH 8.0, 500 mM NaCl, 10 mM TCEP)
- 1 μL Ubiquitin or Ubiquitin mutant (approximately 100 μM)
- 2.5 μL MgATP Solution (10 mM)
- Substrate (5-10 μM)
- E1 Enzyme (100 nM)
- E2 Enzyme (1 μM)
- E3 Ligase (1 μM)
- Incubate reactions at 37°C for 30-60 minutes.
- Terminate reactions with SDS-PAGE sample buffer or EDTA/DTT.
- Analyze by Western blot using anti-ubiquitin antibodies.
- Interpret results: If K11R mutant shows impaired chain formation while other mutants form chains normally, this indicates K11-linked chain formation [26].
Structural Characterization by NMR and SANS
Solution nuclear magnetic resonance (NMR) spectroscopy and small-angle neutron scattering (SANS) provide detailed structural information about K11-linked ubiquitin chains. These techniques have revealed that K11-linked di-ubiquitin adopts distinct conformations in solution that differ from both K48-linked and K63-linked chains, as well as from published crystal structures of K11-linked di-ubiquitin [7]. Residual dipolar coupling (RDC) measurements in NMR studies provide information on intermolecular orientation and positioning in protein-protein complexes, enabling the determination of solution structures under near-physiological conditions [7].
Quantitative Profiling of K11-Linked Ubiquitin Chains
Proteomic Profiling of K11-Dependent Pathways
Quantitative proteomics approaches have been instrumental in identifying biological pathways regulated by K11-linked ubiquitin chains. Whole proteome mass spectrometry analysis comparing wild-type yeast strains with ubiquitin K11R mutant strains (which cannot form K11-linked chains) revealed significant downregulation of methionine biosynthesis enzymes, indicating a previously unappreciated role for K11 linkages in regulating the SCFMet30-Met4 network [43]. This approach enabled the identification of K11 linkage-specific substrates, including Ubc6, a ubiquitin-conjugating enzyme involved in endoplasmic reticulum-associated degradation (ERAD) [44].
Table: Quantitative Proteomics Analysis of K11R Mutant Yeast Strains
| Proteomic Parameter | Wild-Type Strain | K11R Mutant Strain | Biological Significance |
|---|---|---|---|
| Methionine Pathway Enzymes | Normal expression | Significantly downregulated | K11 chains regulate sulfur amino acid metabolism |
| Ubc6 Protein Levels | Normal stability | Increased stability | Ubc6 is a K11 linkage-specific substrate |
| ERAD Efficiency | Normal degradation of ERAD substrates | Impaired degradation | K11 linkages function in ERAD pathway |
| Cell Cycle Regulators | Normal turnover | Altered degradation kinetics | K11 chains crucial for mitotic progression |
| Global Proteome Changes | 4,580 proteins identified | Similar coverage, specific changes | High coverage (68.2%) enables pathway analysis |
Analysis of Branched Ubiquitin Chains Containing K11 Linkages
Recent advances in mass spectrometry have enabled the quantification of branched ubiquitin chains containing K11 linkages. The K11/K48-branched ubiquitin chains represent a priority signal for proteasomal degradation, particularly during cell cycle progression and proteotoxic stress [5]. The Ub-AQUA method has been adapted to quantify these complex chain topologies by utilizing specific signature peptides that uniquely identify branched structures [34]. Cryo-EM structural studies have revealed that the human 26S proteasome recognizes K11/K48-branched ubiquitin chains through a multivalent mechanism involving a novel K11-linked ubiquitin binding site at the groove formed by RPN2 and RPN10, in addition to the canonical K48-linkage binding site [5].
The Scientist's Toolkit: Essential Research Reagents
Table: Key Research Reagents for K11-Linked Ubiquitin Chain Studies
| Reagent / Tool | Function / Application | Experimental Use |
|---|---|---|
| Ubiquitin K11R Mutant | Prevents K11-linked chain formation | Linkage determination in vitro conjugation assays [26] |
| K11 Linkage-Specific Antibodies | Immunodetection of K11 linkages | Western blot, immunoprecipitation [2] |
| AQUA Peptides for K11 | Absolute quantification internal standard | Ub-AQUA/PRM mass spectrometry [34] |
| Ube2S E2 Enzyme | K11-specific ubiquitin-conjugating enzyme | In vitro assembly of K11-linked chains [7] |
| APC/C E3 Ligase | Major cellular source of K11 linkages | Study of mitotic ubiquitination [1] [2] |
| RPN13:UCHL5 Complex | Branched chain recognition and processing | Structural studies of proteasomal recognition [5] |
| TUBE Reagents | Tandem ubiquitin-binding entities | Enrichment of ubiquitinated proteins [34] |
Mass spectrometry-based approaches, particularly the Ub-AQUA/PRM method, provide powerful tools for the verification and quantification of K11-linked ubiquitin chains in biological systems. These techniques enable researchers to not only identify the presence of K11 linkages but also to determine their absolute abundance and investigate their functional roles in critical cellular processes. The integration of biochemical methods using ubiquitin mutants with advanced mass spectrometry and structural techniques offers a comprehensive framework for deciphering the complex ubiquitin code and its implications for cell cycle regulation, protein quality control, and targeted drug development. As research in this field advances, these methodologies will continue to evolve, providing increasingly sophisticated insights into the multifaceted functions of K11-linked ubiquitin chains in health and disease.
Overcoming Technical Challenges: Optimization and Pitfall Avoidance
Addressing Low Abundance and Stoichiometry Challenges
K11-linked polyubiquitin chains play critical roles in cellular regulation, from cell cycle progression to proteotoxic stress response [5] [7]. Despite constituting a significant fraction of cellular polyubiquitin, their low relative abundance and complex stoichiometry present substantial challenges for detailed molecular characterization [27]. These chains exhibit unique conformational properties distinct from K48-linked or K63-linked chains, creating specific binding surfaces that affect interactions and downstream fate of modified proteins [7] [27]. This application note provides integrated methodologies to overcome these technical barriers, enabling robust isolation and analysis of K11-linked ubiquitin chains for therapeutic discovery and basic research.
Quantitative Assessment of K11-linked Ubiquitin Challenges
Table 1: Key Challenges in K11-linked Polyubiquitin Research
| Challenge | Quantitative Impact | Technical Consequence |
|---|---|---|
| Relative Abundance | K11 linkages nearly as abundant as K48 in yeast [7]; Branched chains account for 10-20% of Ub polymers [5] | Masking by more abundant chain types; Requirement for specific enrichment strategies |
| Structural Complexity | 28 theoretical trimeric branched chain types with two different linkages [27] | Difficulties in generating defined reference standards; Antibody cross-reactivity |
| Cellular Dynamics | Upregulated during anaphase of mitotic cycle [7] | Context-dependent stoichiometry requiring precise cellular timing for capture |
| Analytical Sensitivity | K11-K48 branched chains prioritize proteasomal degradation [5] | Rapid turnover necessitates stabilization methods for accurate quantification |
Strategic Experimental Framework
Integrated Workflow for K11-linked Chain Analysis
The following diagram outlines the core experimental strategy for addressing abundance and stoichiometry challenges in K11-linked ubiquitin research:
Research Reagent Solutions
Table 2: Essential Research Reagents for K11-linked Ubiquitin Studies
| Reagent Category | Specific Examples | Function & Application |
|---|---|---|
| Linkage-specific Antibodies | K11-linkage specific antibodies [5] | Immunoprecipitation and western blot detection of K11 chains |
| Ubiquitin Mutants | UbK48R,K63R; Ub1-72; UbD77 [27] | Controlled enzymatic assembly of defined chain architectures |
| Enzymatic Assembly Tools | UBE2S (K11-specific E2) [7]; UBE2N/UBE2V1 (K63-specific) [27] | In vitro synthesis of reference standards for quantification |
| Deubiquitinase Probes | UCHL5(C88A) catalytic mutant [5]; OTULIN [27] | Branch point stabilization and linkage verification |
| Mass Spec Standards | Ub-AQUA (Absolute Quantification) peptides [5] | Precise quantification of linkage abundance in complex samples |
Detailed Experimental Protocols
Protocol 1: K11/K48-branched Ubiquitin Chain Enrichment
Background: K11/K48-branched chains function as priority degradation signals but require stabilization for analysis [5].
Materials:
- Lysis Buffer: 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% NP-40, 1 mM EDTA, protease inhibitors
- Preformed RPN13:UCHL5(C88A) complex (catalytically inactive) [5]
- K11-linkage specific antibody
- Protein A/G magnetic beads
Procedure:
- Cell Lysis and Stabilization
- Harvest cells during mitotic phase (K11 abundance peak) [7]
- Lyse cells in pre-chilled lysis buffer with 10 mM N-ethylmaleimide (NEM) to inhibit DUBs
- Clarify lysate by centrifugation at 16,000 × g for 15 minutes at 4°C
Complex Reconstitution
- Incubate clarified lysate with 50 μg preformed RPN13:UCHL5(C88A) complex for 1 hour at 4°C
- Add K11-linkage specific antibody (1:100 dilution) and incubate overnight at 4°C with gentle rotation
Immunoprecipitation
- Add Protein A/G magnetic beads (50 μL bead slurry per sample)
- Incubate 2 hours at 4°C with rotation
- Wash beads 3× with ice-cold lysis buffer
- Elute with 2× Laemmli buffer at 95°C for 10 minutes
Validation: Analyze by western blot using K11 and K48 linkage-specific antibodies sequentially.
Protocol 2: Mass Spectrometry-based Linkage Quantification
Background: Ub-AQUA enables absolute quantification of linkage stoichiometry in complex samples [5].
Materials:
- Synthetic AQUA peptides for K11 and K48 linkages
- Lbpro* protease (linkage-specific clipping) [5]
- Trypsin/Lys-C mix
- Reverse-phase C18 LC-MS columns
Procedure:
- Sample Preparation
- Resuspend purified ubiquitin chains in 50 mM ammonium bicarbonate
- Reduce with 5 mM DTT (30 minutes, 60°C)
- Alkylate with 15 mM iodoacetamide (30 minutes, room temperature, dark)
Proteolytic Digestion
- Option A: Digest with Lbpro* (1:50 enzyme:substrate) for K11-specific clipping [5]
- Option B: Digest with Trypsin/Lys-C (1:50) overnight at 37°C for general ubiquitin mapping
Spike-in Quantification
- Add known quantities of synthetic AQUA peptides before LC-MS analysis
- Use heavy isotope-labeled internal standards for each linkage type
LC-MS Parameters
- Column: C18, 1.7 μm, 1.0 × 150 mm
- Gradient: 5-35% acetonitrile in 0.1% formic acid over 60 minutes
- MS: Data-independent acquisition (DIA) mode for comprehensive peptide capture
Data Analysis: Calculate linkage abundance by comparing peak areas of endogenous peptides to spiked AQUA standards.
Protocol 3: Enzymatic Assembly of Defined Branched Chains
Background: Controlled synthesis of reference standards bypasses natural abundance limitations [27].
Materials:
- Proximal ubiquitin mutant (Ub1-72 or UbK48R,K63R)
- Distal ubiquitin units (UbK48R,K63R for sequential ligation)
- E1 activating enzyme, UBE2S (K11-specific E2), UBE2R1 (K48-specific E2)
- Reaction buffer: 50 mM Tris-HCl (pH 8.0), 5 mM MgCl₂, 2 mM ATP
Procedure:
- First Ligation (K11 linkage)
- Combine proximal Ub mutant (100 μM) with distal UbK48R,K63R (120 μM)
- Add E1 (0.5 μM), UBE2S (5 μM) in reaction buffer with 2 mM ATP
- Incubate 2 hours at 37°C
- Verify conjugation by non-reducing SDS-PAGE
Second Ligation (K48 branch)
- To the K11-linked dimer, add fresh distal UbK48R,K63R (120 μM)
- Add E1 (0.5 μM), UBE2R1 (5 μM) with 2 mM ATP
- Incubate 2 hours at 37°C
Purification
- Resolve reaction products by anion-exchange chromatography
- Pool fractions containing branched trimer
- Concentrate and exchange into storage buffer (20 mM Tris pH 7.5, 50 mM NaCl)
Quality Control: Verify chain architecture by DUB cleavage profiling with linkage-specific deubiquitinases.
Structural Characterization Techniques
Cryo-EM Analysis of Proteasome-Branched Chain Complexes
The structural basis of K11/K48-branched chain recognition involves a multivalent mechanism as illustrated below:
Application Notes: For structural studies, reconstitute human 26S proteasome with K11/K48-branched tetra-ubiquitin and RPN13:UCHL5(C88A) complex as described in [5]. Use cryo-EM grid preparation with 2.5-3.5 μM complex applied to ultrAuFoil grids. This approach revealed the K11-linked Ub binding site at the RPN2-RPN10 groove and the canonical K48-linkage binding site formed by RPN10 and RPT4/5 coiled-coil [5].
Solution NMR for Dynamic Conformation Analysis
Key Findings: K11-linked di-ubiquitin (K11-Ub₂) adopts distinct conformations from K48-linked or K63-linked chains in solution [7]. Residual Dipolar Coupling (RDC) measurements demonstrate that individual Ub units in K11-Ub₂ maintain structural integrity similar to monoUb (PDB ID 1D3Z) despite isopeptide linkage [7].
Experimental Parameters:
- Sample: 0.5-1 mM K11-Ub₂ in 20 mM phosphate buffer (pH 6.8), 50 mM NaCl
- Alignment: 5% C12E5/hexanol for RDC measurements
- NMR: ¹H-¹⁵N TROSY-HSQC with non-uniform sampling
- Temperature: 298K for physiological relevance
Troubleshooting and Optimization
Common Technical Challenges
Table 3: Troubleshooting Guide for K11-linked Ubiquitin Studies
| Problem | Potential Cause | Solution |
|---|---|---|
| Low K11 signal in MS | Competition from abundant K48/K63 chains | Pre-enrichment with K11-specific antibodies prior to Ub-AQUA |
| Incomplete branched chain assembly | Non-optimal E2 enzyme ratios | Titrate UBE2S and UBE2R1 concentrations (2-10 μM range) |
| Proteasome complex instability | RPN13:UCHL5 dissociation | Use preformed complex with catalytic mutant (C88A) [5] |
| NMR spectral broadening | Conformational dynamics | Adjust salt concentration (50-150 mM NaCl) to modulate compactness [7] |
Method Validation Approaches
- Linkage Specificity: Validate K11 enrichment by parallel reaction monitoring (PRM) MS with linkage-specific signature peptides
- Stoichiometric Accuracy: Use Ub-AQUA with heavy isotope-labeled internal standards for absolute quantification [5]
- Functional Relevance: Verify biological activity through proteasome binding assays comparing degradation rates of substrates modified with K11/K48-branched vs homotypic chains
The integrated methodologies presented here provide a comprehensive framework for overcoming the low abundance and stoichiometry challenges inherent in K11-linked polyubiquitin research. By combining strategic enrichment approaches, controlled enzymatic synthesis of defined chain architectures, and multi-modal structural characterization, researchers can advance our understanding of these complex signaling molecules. The precise molecular mechanisms underlying K11-linked chain recognition, particularly in branched configurations, offer promising avenues for therapeutic intervention in cancer and neurodegenerative diseases where ubiquitin pathway dysregulation is increasingly implicated.
Minimizing Cross-Reactivity with Other Ubiquitin Linkages
The study of K11-linked polyubiquitin chains is fundamental to understanding essential cellular processes, particularly the precise regulation of cell division and mitosis [1]. However, a significant technical challenge complicates this research: the inherent cross-reactivity of many detection and enrichment reagents with other ubiquitin linkage types. This cross-reactivity stems from the high structural similarity between different ubiquitin chains, as ubiquitin molecules linked through different lysine residues (e.g., K11, K48, K63) share identical amino acid sequences and differ only in their three-dimensional topology [1] [28]. For researchers focusing on K11-linked chains, this problem is particularly acute due to the co-existence of K11 linkages with other types, especially K48, in complex branched chain architectures [5] [27]. The presence of these heterotypic K11/K48-branched chains, which account for a substantial fraction of cellular ubiquitin polymers and function as priority degradation signals, means that isolation of pure homotypic K11 chains is exceptionally difficult [5] [27]. This application note addresses this critical challenge by presenting validated strategies and detailed protocols to minimize cross-reactivity, thereby enabling more accurate characterization of K11-linked ubiquitination in complex biological systems.
Strategic Approaches for Specific K11-Linked Ubiquitin Analysis
The complexity of the ubiquitin code necessitates a clear understanding of where cross-reactivity arises in experimental workflows. Three primary areas present the greatest challenges:
- Antibody-based Enrichment: Traditional anti-ubiquitin antibodies (e.g., P4D1, FK1/FK2) recognize ubiquitin epitopes regardless of linkage type, while many linkage-specific antibodies still exhibit significant cross-reactivity with structurally similar linkages [28].
- Ubiquitin-Binding Domains (UBDs): Most natural UBDs show preference rather than absolute specificity for particular linkage types, with binding affinities often overlapping across multiple chain architectures [28].
- Mass Spectrometry Analysis: Without proper enrichment strategies, the low stoichiometry of K11-linkages compared to K48-linkages (approximately 2% vs. higher abundance in asynchronously dividing cells) makes specific detection challenging [1] [45].
Comparative Analysis of Enrichment and Detection Methods
Table 1: Methodologies for Specific K11-Linked Ubiquitin Analysis
| Method Category | Specific Technique | Cross-Reactivity Concerns | Suitable Applications |
|---|---|---|---|
| Antibody-based | Commercial K11-linkage specific antibodies | Variable between lots; may recognize K11/K48 branched chains [28] | Immunoblotting, immunofluorescence, enrichment for MS |
| UBD-based | Tandem-repeated Ub-binding entities (TUBEs) | Moderate specificity; requires validation with linkage-specific DUBs [28] | Stabilization and pull-down of ubiquitinated proteins |
| MS-based Proteomics | diGly antibody enrichment with DIA/SRM | Minimal when optimized; can distinguish linkages via signature peptides [45] [46] | Global ubiquitinome profiling, site identification, linkage quantification |
| Biochemical Tools | Linkage-specific DUBs | High specificity when properly characterized [27] | Validation of linkage type, controlled chain disassembly |
| Chemical Biology | Genetically encoded ubiquitin mutants | Minimal when properly designed [27] | In vitro chain assembly, mechanism studies |
Table 2: Quantitative Performance of Mass Spectrometry Methods for Ubiquitin Analysis
| MS Parameter | Data-Dependent Acquisition (DDA) | Data-Independent Acquisition (DIA) | Selected Reaction Monitoring (SRM) |
|---|---|---|---|
| Typical K11 Site IDs (Single Run) | ~20,000 diGly sites [45] | ~35,000 diGly sites [45] | Targeted quantification of specific linkages [46] |
| Quantitative Accuracy (CV) | 15% of sites with CV <20% [45] | 45% of sites with CV <20% [45] | <15% for optimized transitions [46] |
| Linkage Specificity | Moderate (depends on library) | High (with comprehensive library) | Excellent (targeted approach) |
| Best Application | Discovery-phase screening | Comprehensive ubiquitinome profiling | Validation and precise quantification |
Experimental Protocols for Cross-Reactivity Minimization
Protocol 1: K11-Enriched Ubiquitinome Profiling Using diGly Antibody with DIA-MS
Principle: This protocol leverages an anti-diGly antibody to enrich for ubiquitinated peptides followed by data-independent acquisition mass spectrometry (DIA-MS) for comprehensive identification of K11-linked ubiquitination sites with minimal cross-reactivity [45].
Materials:
- Anti-K-ε-GG antibody (Cell Signaling Technology, PTMScan Ubiquitin Remnant Motif Kit)
- Trypsin (sequencing grade)
- C18 StageTips for desalting
- Liquid chromatography system coupled to high-resolution mass spectrometer
- Spectral library containing K11 linkage signatures [45]
Procedure:
- Protein Extraction and Digestion:
- Lyse cells in 8 M urea buffer containing protease and phosphatase inhibitors
- Reduce proteins with 5 mM dithiothreitol (60°C, 30 min)
- Alkylate with 15 mM iodoacetamide (room temperature, 30 min in dark)
- Dilute urea concentration to 2 M and digest with trypsin (1:50 w/w, 37°C, 16 h)
diGly Peptide Enrichment:
- Acidify digested peptides to pH ~2 with trifluoroacetic acid
- Desalt peptides using C18 StageTips
- Resuspend peptides in immunoaffinity purification (IAP) buffer
- Incubate with anti-K-ε-GG antibody (1:8 antibody:peptide ratio) for 2 h at 4°C with rotation [45]
- Wash beads extensively with IAP buffer followed by water
- Elute diGly peptides with 0.15% trifluoroacetic acid
DIA-MS Analysis:
- Separate peptides using 25 cm C18 column with 90-min gradient
- Acquire data using optimized DIA method with 46 variable windows [45]
- Use MS2 resolution of 30,000 for improved identification
- Analyze data using spectral library generated from K11-enriched samples
Critical Steps for Cross-Reactivity Minimization:
- Pre-fractionate samples by basic reversed-phase chromatography to separate highly abundant K48-linked peptides that compete for antibody binding [45]
- Use a hybrid spectral library approach combining DDA and direct DIA searches to improve K11 linkage identification
- Implement cross-validation with linkage-specific antibodies to confirm K11-specific sites
Protocol 2: Structural Validation of K11 Linkages Using Cryo-EM
Principle: This protocol utilizes cryo-electron microscopy (cryo-EM) to visually confirm K11-linkages within branched ubiquitin chains, providing structural validation to complement biochemical approaches [5].
Materials:
- Human 26S proteasome purified from HEK293 cells
- Recombinant RPN13:UCHL5 complex
- Sic1PY substrate with single lysine residue (K40)
- Engineered Rsp5-HECTGML E3 ligase
- Quantifoil R1.2/1.3 300 mesh gold grids
Procedure:
- Sample Preparation:
- Reconstitute K11/K48-branched ubiquitin chains using Rsp5-HECTGML ligase and Ub(K63R) variant to eliminate K63 linkages [5]
- Form functional complex with 26S proteasome and catalytically inactive UCHL5(C88A) to stabilize branched chains
- Verify complex formation by native gel electrophoresis and Western blotting
Cryo-EM Grid Preparation:
- Apply 3.5 μL sample to glow-discharged grids
- Blot for 3-4 seconds at 100% humidity, 4°C
- Plunge-freeze in liquid ethane using Vitrobot
Data Collection and Processing:
- Collect movies on 300 keV cryo-EM microscope with K3 direct electron detector
- Process data using cryoSPARC with extensive classification
- Focus refinements on regions with additional density indicating K11/K48-branched ubiquitin chains
Validation of K11 Specificity:
- Identify multivalent recognition interfaces involving RPN2 groove for K11-linkage specificity [5]
- Confirm alternating K11-K48 linkage pattern through proximal ubiquitin positioning
- Distinguish from homotypic K48 chains by absence of RPN2-RPN10 groove binding
Figure 1: Integrated workflow combining mass spectrometry and structural approaches for specific K11-linked ubiquitin analysis.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for K11-Linked Ubiquitin Studies
| Reagent Category | Specific Examples | Function in K11 Research | Specificity Considerations |
|---|---|---|---|
| Linkage-Specific Antibodies | Anti-K11 ubiquitin linkage antibody | K11 chain detection and enrichment | Validate with K11-deficient samples; may cross-react with K11/K48 branched chains [28] |
| E2/E3 Enzyme Pairs | Ube2S-APC/C complex | K11-specific chain initiation and elongation [1] | Ube2C/UbcH10 initiates K11 chains; Ube2S extends K11 linkages specifically [1] |
| DUBs | UCHL5 (with RPN13) | Preferentially processes K11/K48-branched chains [5] | Use catalytically inactive mutants (C88A) for complex stabilization during structural studies [5] |
| Ubiquitin Mutants | Ub(K48R), Ub(K63R), Ub(1-72) | Assembly of defined linkage chains [27] | Eliminate competing linkages during in vitro reconstitution; enables clean K11 chain assembly |
| Mass Spectrometry Standards | Heavy isotope-labeled K11 signature peptides | Absolute quantification of K11 linkages [46] | Synthetic peptides with K11-GG signature enable precise SRM quantification without cross-reactivity |
| Proteasome Subunits | Recombinant RPN2, RPN10, RPN13 | Study K11 chain recognition mechanisms [5] | RPN2 contains unique K11-binding groove distinct from K48-binding sites |
Troubleshooting Guide for Cross-Reactivity Issues
Problem: Persistent K48 Signal in K11 Enrichments
- Potential Cause: Co-enrichment of K11/K48-branched chains, which are natural cellular species [5] [27]
- Solution: Implement sequential enrichment with K48-specific DUB pretreatment to isolate pure K11 chains
- Validation: Use orthogonal method (SRM) to quantify K11/K48 ratio in enriched samples [46]
Problem: Low Yield of K11 Sites in Global Ubiquitinome Analysis
- Potential Cause: Competition from abundant K48 peptides during diGly antibody enrichment [45]
- Solution: Pre-fractionate samples by basic reversed-phase chromatography to separate linkage types before enrichment
- Optimization: Increase peptide input to 1 mg with 31.25 μg antibody for optimal K11 recovery [45]
Problem: Inconsistent K11 Linkage-Specific Antibody Performance
- Potential Cause: Lot-to-lot variability in commercial antibodies
- Solution: Validate each antibody lot with defined ubiquitin chains of known linkage
- Alternative Approach: Develop in-house reagents using linkage-specific UBDs or DNA-encoded ubiquitin sensors
Minimizing cross-reactivity in K11-linked ubiquitin research requires a multifaceted approach that combines careful reagent selection, orthogonal validation methods, and an understanding of the natural complexity of ubiquitin chain architectures. The strategies outlined in this application note—particularly the optimized DIA-MS workflow and structural validation approaches—provide researchers with robust methods to specifically study K11 linkages despite the challenging cellular context of heterotypic and branched chains. As the field advances, emerging technologies including genetic code expansion for ubiquitin engineering, improved linkage-specific binders using phage display, and single-molecule ubiquitin chain sequencing platforms promise to further enhance our ability to dissect the specific functions of K11-linked ubiquitination in health and disease. By implementing these carefully validated protocols and maintaining rigorous standards for linkage specificity, researchers can overcome the persistent challenge of cross-reactivity to unlock the unique biological functions of K11-linked ubiquitin chains in cell cycle regulation and beyond.
Optimization of Lysis and Buffer Conditions to Preserve K11 Chains
Ubiquitination is a vital post-translational modification that regulates diverse cellular processes, with the functional outcome largely determined by the linkage type of the polyubiquitin chain. Among the eight possible homogenous chain types, K11-linked polyubiquitin chains have emerged as crucial regulators of cell division and protein degradation [1]. During mitosis, K11-linked chains are highly upregulated and facilitate the targeted degradation of cell cycle regulators by the 26S proteasome [2]. Beyond their role in the cell cycle, K11 linkages are also involved in non-proteolytic signaling pathways and can form complex heterotypic chains, including K11/K48-branched ubiquitin chains that are preferentially recognized by the proteasomal system for accelerated degradation [5].
The analysis of K11-linked ubiquitination presents unique challenges due to the structural and biochemical properties of these chains. K11-linked di-ubiquitin adopts distinct conformations in solution that differ from both K48- and K63-linked chains [7]. These structural differences, combined with the relatively low abundance of K11 linkages in asynchronous cells (approximately 2% of the ubiquitin conjugate pool) and their dynamic regulation during cell cycle progression, necessitate specialized preservation and capture strategies [1]. This application note provides optimized methods for preserving K11-linked ubiquitin chains during cell lysis and immunoblotting analysis, framed within the broader context of enrichment strategies for K11-linked polyubiquitin research.
Key Challenges in K11 Chain Preservation
The lability of K11-linked ubiquitin chains during sample preparation poses significant technical challenges. Several factors contribute to this instability:
- Deubiquitinase (DUB) Activity: Cellular DUBs remain active during cell lysis if not properly inhibited, leading to rapid chain disassembly [47]. Some DUBs exhibit linkage-specific preferences, with UCHL5 showing particular activity toward K11/K48-branched chains [5].
- Structural Sensitivity: The unique conformation and dynamics of K11-linked chains in solution may render them more susceptible to chemical or enzymatic degradation compared to canonical linkages [7].
- Competing Ubiquitin Interactions: The intermediate affinity of K11-linked chains for various ubiquitin-receptor proteins may result in displacement or rearrangement during processing [7].
Optimized Lysis and Buffer Formulations
Based on current literature and practical experience, the following buffer formulations have been optimized specifically for preserving K11-linked ubiquitin chains.
Table 1: Optimized Lysis Buffer Composition for K11 Chain Preservation
| Component | Concentration | Function | Rationale |
|---|---|---|---|
| Tris-HCl | 50 mM, pH 7.4-7.6 | Buffering | Maintains physiological pH for ubiquitin stability |
| Sodium Chloride | 150 mM | Ionic strength | Mimics physiological conditions; affects K11-Ub2 conformation [7] |
| NP-40 Alternative | 1% | Detergent | Membrane solubilization while preserving protein interactions |
| Glycerol | 10% (v/v) | Stabilizer | Prevents protein aggregation and stabilizes ubiquitin conformations |
| N-Ethylmaleimide (NEM) | 10-25 mM | DUB inhibitor | Irreversibly inhibits cysteine-dependent DUBs; critical for chain preservation [47] |
| Iodoacetamide (IAA) | 5-10 mM | Alkylating agent | Complements NEM for comprehensive DUB inhibition |
| EDTA | 5-10 mM | Chelating agent | Inhibits metalloprotease DUBs |
| Sodium Orthovanadate | 1-2 mM | Phosphatase inhibitor | Preserves phosphorylation status of ubiquitin and substrates |
| PMSF | 1 mM | Serine protease inhibitor | Broad-spectrum protease inhibition |
| Commercial Protease Inhibitor Cocktail | 1X | Multi-component inhibition | Provides additional protection against various protease classes |
| Ubiquitin Aldehyde | 0.5-1 µM | DUB inhibitor | Specific, potent inhibition of ubiquitin C-terminal hydrolases |
Table 2: Specialized Buffer Additives for Specific Applications
| Additive | Concentration | Application Context | Effect on K11 Chains |
|---|---|---|---|
| Dimethyl pimelidiate (DMP) | 5-10 mM | Crosslinking for weak interactions | Stabilizes transient ubiquitin-protein interactions |
| ATP | 1-2 mM | Energy-dependent processes | Maintains E1/E2/E3 enzyme complexes relevant to K11 synthesis |
| MgCl₂ | 5 mM | ATP-dependent processes | Cofactor for enzymatic activities in ubiquitin system |
Step-by-Step Protocol for K11 Chain Preservation
Cell Lysis and Sample Preparation
- Pre-chill all equipment and solutions to 4°C to minimize enzymatic activity during processing.
- Prepare fresh lysis buffer with all inhibitors added immediately before use, as NEM and IAA degrade in aqueous solution.
- Wash cells with ice-cold PBS containing 1 mM NEM to inhibit surface DUB activity before lysis.
- Lyse cells using 3-5 volumes of lysis buffer relative to cell pellet volume.
- Incubate on ice for 15-30 minutes with gentle vortexing every 5 minutes.
- Clarify lysates by centrifugation at 16,000 × g for 15 minutes at 4°C.
- Transfer supernatant to pre-chilled microcentrifuge tubes, avoiding lipid layers and pellets.
- Process immediately for immunoprecipitation or add Laemmli buffer for direct Western analysis.
Critical Control Experiments
To validate K11 chain preservation, include these essential controls:
- DUB Inhibition Control: Process samples with and without NEM/IAA to demonstrate DUB inhibition efficacy.
- Time Course Analysis: Process samples at different time points after lysis to assess chain stability.
- Linkage Specificity Control: Include known K11 chain standards when available [2].
- Proteasome Inhibition Pre-treatment: For cellular studies, pre-treat cells with 10 µM MG-132 for 4-6 hours to accumulate ubiquitinated substrates, particularly important for K11 chains due to their proteasomal targeting function [1] [2].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for K11-Linked Ubiquitin Chain Research
| Reagent/Solution | Specific Function | Application Notes |
|---|---|---|
| K11 Linkage-Specific Antibodies | Selective detection of K11 linkages in immunoblotting | Validate specificity using linkage-defined ubiquitin standards [2] |
| Chain-Selective TUBEs (Tandem Ubiquitin Binding Entities) | High-affinity capture of polyubiquitinated proteins with linkage preference | K63-TUBEs do not capture K48 linkages, demonstrating linkage specificity that can be exploited for K11 enrichment [23] |
| Ube2S Enzyme | K11-specific E2 conjugating enzyme | Useful for generating K11-linked chains in vitro [1] [7] |
| UCHL5/UCH37 Inhibitors | Selective inhibition of K11/K48-branched chain DUB | b-AP15 and related compounds help preserve K11/K48-branched chains [5] |
| K11-Ub2 Structural Standards | Reference for structural studies and method validation | Available through commercial suppliers or in-house expression [7] |
| RPN1/RPN10 Proteasomal Receptors | Study of K11/K48-branched chain recognition | Recombinant forms for binding assays [5] |
Experimental Workflow and Signaling Pathways
The following diagram illustrates the complete workflow for K11 chain preservation and analysis, integrating the key steps and critical control points:
K11 Chain Preservation and Analysis Workflow
The diagram above outlines the critical steps for successful preservation and analysis of K11-linked ubiquitin chains, emphasizing the importance of inhibitor pre-treatments and specialized lysis conditions.
Troubleshooting Guide
Table 4: Common Issues and Solutions in K11 Chain Preservation
| Problem | Potential Cause | Solution |
|---|---|---|
| Poor K11 signal despite strong total ubiquitin | Incomplete DUB inhibition or preferential K11 chain degradation | Increase NEM concentration to 25 mM; add ubiquitin aldehyde; reduce time between lysis and analysis |
| High background in K11 immunoblots | Antibody cross-reactivity with other linkages | Include linkage-specific competition with other ubiquitin chain types; optimize antibody dilution |
| Loss of K11 signal during IP | Weak interaction with capture reagents | Use crosslinking agents like DMP; try chain-selective TUBEs with high affinity for K11 chains [23] |
| Inconsistent results between experiments | Variation in inhibitor preparation or lysis time | Prepare fresh inhibitor stocks for each experiment; standardize lysis duration across samples |
The preservation of K11-linked ubiquitin chains requires careful attention to lysis conditions and inhibitor selection. The optimized protocols presented here leverage our current understanding of K11 chain biology, including their unique structural properties [7], regulation during cell division [1] [2], and recognition by the proteasomal system [5]. By implementing these methods, researchers can more accurately investigate the fascinating roles of K11-linked ubiquitination in cell cycle control, protein degradation, and cellular signaling pathways.
Strategies for Distinguishing Branched versus Homotypic K11 Chains
Ubiquitin chains exist in multiple topological forms, including homotypic chains (composed of a single linkage type) and more complex branched chains (where at least one ubiquitin moiety is modified at two or more distinct lysine residues). Among these, K11-linked polyubiquitin chains have emerged as critical regulators, particularly in cell cycle control and proteasomal degradation [2] [12]. Accurately distinguishing branched K11 chains from their homotypic counterparts is essential for deciphering their unique biological functions, which include fast-tracking protein degradation during mitosis and proteotoxic stress [5].
This application note provides a detailed framework of biochemical, analytical, and functional strategies to differentiate these complex architectures, supporting advanced research in ubiquitin signaling and targeted drug development.
Analytical and Biochemical Enrichment Strategies
Effective study of K11 chains requires methods to selectively enrich and analyze them from complex biological samples. The table below summarizes key reagent-based enrichment strategies.
Table 1: Reagent Solutions for Enriching K11-Linked Ubiquitin Chains
| Research Reagent | Type | Key Features & Function | Considerations |
|---|---|---|---|
| Linkage-Specific Antibodies [2] [28] | Antibody | Engineered to specifically recognize the unique conformation of K11-linked diubiquitin; used for immunoblotting, immunofluorescence, and immunoprecipitation. | Specificity must be rigorously validated; may not distinguish branched from homotypic K11 chains alone. |
| Tandem Ubiquitin-Binding Entities (TUBEs) [28] | Engineered Ub-Binding Domain | Tandem repeats of ubiquitin-associated domains (UBA) with high affinity for ubiquitin chains; can be linkage-specific or general; protects chains from DUBs during purification. | General TUBEs enrich all ubiquitinated proteins; linkage-specific TUBEs for K11 are desirable but not commercially widespread. |
| Affinity-Tagged Ubiquitin (e.g., His-, Strep-) [28] | Tagged Ubiquitin | Enables purification of ubiquitinated proteins from cell lysates under denaturing conditions via affinity resin (e.g., Ni-NTA for His-tag). | May introduce artifacts; does not provide linkage information without downstream analysis. |
The workflow for distinguishing chain topology typically begins with enrichment, followed by downstream linkage and architectural analysis.
Methods for Linkage and Architectural Analysis
Following enrichment, precise methodologies are required to decode the chain's architecture. The techniques below can be used sequentially or in parallel to confirm the presence of K11 linkages and identify branching.
Ubiquitin Chain Cleavage and Mass Spectrometry
Mass spectrometry (MS) is a powerful tool for defining ubiquitin chain architecture. Two primary approaches are detailed here.
Protocol: Linkage-Specific Deubiquitinase (DUB) Assay
- Reagent Preparation: Resuspend the enriched ubiquitinated proteins in an appropriate reaction buffer.
- Enzymatic Digestion: Split the sample into aliquots. Incubate each aliquot with a different linkage-specific DUB:
- OTULIN: Cleaves M1-linked (linear) chains [27].
- vOTU: Broad-spectrum DUB, useful as a control for complete chain disassembly.
- DUB of interest with known specificity for K11, K48, or K63.
- Reaction Control: Include a no-enzyme control and a sample with a pan-specific DUB.
- Termination and Analysis: Stop the reaction with SDS-PAGE loading buffer. Analyze the cleavage products by immunoblotting using K11-linkage-specific antibodies and total ubiquitin antibodies. The persistence of K11 signal after K48-specific DUB treatment suggests the K11 chain is part of a branched structure resistant to that DUB.
Protocol: Ubiquitin Absolute Quantification (Ub-AQUA) MS
This targeted proteomics method relies on synthetic, stable isotope-labeled internal standard peptides corresponding to tryptic ubiquitin remnants for each linkage type [5].
- Sample Digestion: Digest the enriched ubiquitinated proteins with trypsin. This generates a characteristic "di-glycine" (diGly) remnant (mass shift of 114.04 Da) on the modified lysine, which is isolated from the substrate protein or from an internal lysine within another ubiquitin.
- Spike-in Standards: Add a known quantity of synthetic diGly-linked peptides for all ubiquitin linkages (K6, K11, K27, K29, K33, K48, K63, M1).
- LC-MS/MS Analysis: Analyze the peptide mixture using liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) in selected reaction monitoring (SRM) or parallel reaction monitoring (PRM) mode.
- Data Quantification: Quantify the amount of each endogenous linkage by comparing its signal to the corresponding heavy isotope-labeled standard. The co-quantification of K11 and K48 (or other linkages) in a single sample strongly indicates the presence of a heterotypic chain. Distinguishing mixed from branched chains requires additional methods.
Biochemical and Functional Interaction Assays
The unique three-dimensional structure of branched chains creates distinct interaction surfaces recognized by specific proteins.
Protocol: In Vitro Proteasome Binding and Degradation Assay
This assay leverages the finding that K11/K48-branched ubiquitin chains are recognized by the 26S proteasome as a "priority degradation signal" [5] [12].
- Reconstitute Substrate: Prepare a model substrate (e.g., Sic1PY) modified with either homotypic K11 or branched K11/K48 ubiquitin chains. This can be achieved using engineered E3 ligases like Rsp5-HECTGML or combinations of E2/E3 enzymes in vitro [5].
- Form Complex: Incubate the ubiquitinated substrate with purified human 26S proteasome in the presence of an excess of catalytically inactive UCHL5(C88A) complexed with RPN13. This helps stabilize the branched chain for binding studies [5].
- Analyze Interaction:
- For Binding: Use native gel electrophoresis or size-exclusion chromatography to analyze complex formation. Alternatively, use cryo-EM to visualize the complex, which can reveal multivalent engagement of the branched chain with RPN2 and RPN10 receptors [5].
- For Degradation: Monitor substrate turnover over time by SDS-PAGE and immunoblotting, comparing the degradation rates of substrates tagged with homotypic K11 versus branched K11/K48 chains. Branched chains typically lead to accelerated degradation.
Table 2: Quantitative Comparison of Degradation Signals
| Ubiquitin Chain Type | Relative Degradation Rate by Proteasome | Key Proteasomal Receptors Involved | Cellular Context |
|---|---|---|---|
| Homotypic K48 | High (canonical signal) | RPN10, RPN13 [5] | General protein turnover |
| Homotypic K11 | Moderate | RPN10 [5] | Mitotic progression [2] |
| Branched K11/K48 | Very High (fast-track) | Multivalent: RPN2 (novel site) and RPN10 [5] | Cell cycle, proteotoxic stress [5] |
| Branched K48/K63 | Substrate-dependent | Not specified in results | NF-κB signaling, p97 processing [12] |
Synthesis of Defined Chains for Experimental Control
A critical step in validating any distinction strategy is the use of well-defined ubiquitin chains as controls. The following protocols enable the production of homotypic and branched chains for use as standards in the above assays.
Protocol: Enzymatic Assembly of Branched K11/K48 Trimers
This method uses sequential ligation with linkage-specific enzymes and ubiquitin mutants [27].
- Prepare Components:
- Proximal Ubiquitin: Use a C-terminally blocked ubiquitin (e.g., Ub(^{1-72})) or a ubiquitin where all lysines except the one for the second branch are mutated to arginine (e.g., Ub(^{K48R, K63R}) for a K11/K48 branch).
- Distal Ubiquitins: Use ubiquitin mutants that can only form the desired linkage (e.g., Ub(^{K48R, K63R}) for K11 linkage; Ub(^{K11R, K63R}) for K48 linkage).
- First Ligation: Attach the first distal ubiquitin (e.g., for the K11 branch) to the proximal ubiquitin using a K11-specific E2/E3 combination (e.g., UBE2S with APC/C) [2] [12].
- Second Ligation: Attach the second distal ubiquitin (e.g., for the K48 branch) to a different lysine on the same proximal ubiquitin using a K48-specific enzyme (e.g., UBE2R1/UBE2K).
- Purification: Purify the resulting branched trimer using size-exclusion or ion-exchange chromatography.
Protocol: Chemoenzymatic Assembly Using Photocaging
This advanced method allows for more flexible chain extension using wild-type ubiquitin [27].
- NVOC Protection: Chemically synthesize ubiquitin moieties where the target lysine residues are protected with a photolabile 6-nitroveratryloxycarbonyl (NVOC) group.
- First Elongation: Perform K63-specific elongation using protected ubiquitin.
- Deprotection: Expose the product to UV light to remove the NVOC group, revealing the native lysine.
- Second Elongation: Perform K48-specific elongation on the deprotected lysine.
- Purification: Purify the final branched tetramer product. The resulting chains are structurally native and can be recognized by all known ubiquitin receptors and DUBs.
Distinguishing branched from homotypic K11 ubiquitin chains requires a multi-faceted approach. No single method is sufficient; confidence is achieved by converging evidence from enrichment strategies, linkage-specific MS, DUB profiling, and functional proteasomal engagement assays. The ongoing development of new tools, including more specific antibodies, recombinant UBDs, and refined chemical biology techniques, will continue to enhance the resolution and accuracy of these strategies. By applying this integrated protocol, researchers can precisely decode the complex signals encoded in the ubiquitin code, advancing both basic science and drug discovery.
Quality Control Measures for Enrichment Reagents and Procedures
The study of K11-linked polyubiquitin chains represents a critical frontier in ubiquitin signaling research, particularly due to their established role in cell cycle regulation and proteasomal degradation [5]. Unlike the more well-characterized K48- and K63-linked chains, K11 linkages often form branched architectures with K48 linkages, creating a specialized degradation signal that fast-tracks substrates to the proteasome [5] [3]. This technical note outlines robust quality control measures for reagents and procedures used in the enrichment and study of K11-linked polyubiquitin chains, providing researchers with a framework for generating high-quality, reproducible data.
The complexity of the ubiquitin code demands exceptional specificity in analytical tools. K11 linkages constitute a significant proportion of ubiquitin chains during specific cellular processes such as mitotic exit, where they can comprise up to 30-40% of total ubiquitin conjugates in synchronized cell populations [3]. Proper enrichment and characterization are therefore essential for understanding their unique functions in proteostasis maintenance, mitotic progression, and cellular stress response [5] [24].
Research Reagent Solutions for K11-Linked Ubiquitin Research
A diverse molecular toolbox is required for specific detection and enrichment of K11-linked ubiquitin chains. The table below summarizes essential reagents validated for K11-chain research.
Table 1: Key Research Reagents for K11-Linked Polyubiquitin Chain Analysis
| Reagent Category | Specific Examples | Function and Application | Key Characteristics |
|---|---|---|---|
| Linkage-Specific Antibodies | Anti-K11-linkage antibody [3] | Immunoblotting, immunofluorescence | Validated via UBE2S knockdown; detects abrupt increase in K11 chains during mitotic exit [3] |
| Engineered Ubiquitin-Binding Domains (UBDs) | RPN1, RPN10, RPN2 constructs [5] | Affinity enrichment, structural studies | RPN2 recognizes alternating K11-K48 linkages; RPN10 binds K11 chains via a novel binding groove [5] |
| Catalytically Inactive Deubiquitinases (DUBs) | Cezanne (K11-specific) [3] | Linkage verification, UbiCRest analysis | Cleaves K11 linkages specifically; used in ubiquitin chain restriction (UbiCRest) assays [3] |
| Ubiquitin Mutants | Ub(K63R) variant [5] | Background reduction in chain assembly | Prevents formation of competing K63-linked chains during in vitro ubiquitination assays [5] |
| Linkage-Specific E2 Enzymes | UBE2S [3] | In vitro reconstitution of K11 chains | APC/C-associated E2 that specifically elongates K11-linked chains in mitotic exit [3] |
Quality Control for Enrichment Reagents
Validation of Linkage-Specific Antibodies
The cornerstone of specific K11-chain detection is rigorous antibody validation. Anti-K11 linkage antibodies must be validated using a multi-tiered approach:
- Genetic Validation: Demonstrate significant signal reduction upon siRNA-mediated knockdown of UBE2S, the E2 enzyme responsible for K11-chain assembly [3]. This approach confirmed the specificity of K11 antibodies in studies of Aurora kinase degradation during mitotic exit.
- Enzymatic Validation: Treat samples with the K11-specific deubiquitinase Cezanne, which should abolish detection signal [3]. Compare with other linkage-specific DUBs (e.g., OTUB1 for K48 linkages) which should have minimal effect on K11 signal.
- Cross-reactivity Profiling: Test antibody reactivity against all eight homotypic ubiquitin chain types (K6, K11, K27, K29, K33, K48, K63, M1) using purified chain assemblies. Signal should be exclusive to K11 linkages.
For branched chain studies, additional validation is required. Antibodies should be tested against K11/K48-branched ubiquitin chains, which are physiologically relevant substrates recognized by the 26S proteasome via multivalent interactions with RPN2 and RPN10 [5].
Quality Assessment of Affinity Reagents
Beyond antibodies, various affinity reagents including affimers, engineered UBDs, and macrocyclic peptides require stringent quality control [24]:
- Binding Specificity: Assess using surface plasmon resonance (SPR) or isothermal titration calorimetry (ITC) against a panel of ubiquitin chain types.
- Structural Confirmation: For crystallographically validated binders like RPN2, ensure proper folding via circular dichroism or nuclear magnetic resonance spectroscopy.
- Functional Validation: In enrichment applications, demonstrate that bound material can be specifically eluted with free K11-linked diubiquitin but not other linkage types.
Experimental Protocols for K11-Chain Analysis
Enrichment and Detection Workflow
The following diagram illustrates the core workflow for the enrichment and detection of K11-linked polyubiquitin chains:
Protocol: Immunoaffinity Enrichment of K11-Linked Chains
Materials:
- Lysis Buffer: 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% NP-40, 1 mM EDTA, supplemented with fresh 10 mM N-ethylmaleimide (NEM), 1× protease inhibitor cocktail, and 25 μM PR-619 (broad-spectrum DUB inhibitor)
- K11-linkage specific antibody (validated as in Section 3.1)
- Protein A/G magnetic beads
- Wash Buffer: 50 mM Tris-HCl (pH 7.5), 300 mM NaCl, 0.5% NP-40, 1 mM EDTA
- Elution Buffer: 0.1 M glycine-HCl (pH 2.5)
Procedure:
- Cell Preparation and Lysis:
- Synchronize cells if studying cell cycle-related processes (e.g., double thymidine block for mitotic studies) [3].
- Treat cells with 10 μM MG-132 for 4 hours prior to collection to enhance ubiquitinated protein recovery.
- Lyse 1×10⁷ cells in 1 mL ice-cold lysis buffer with gentle rotation for 30 minutes at 4°C.
- Clarify lysates by centrifugation at 16,000 × g for 15 minutes at 4°C.
Pre-clearing:
- Incubate lysate with control IgG-coupled beads for 1 hour at 4°C to reduce non-specific binding.
- Retain supernatant for immunoprecipitation.
Immunoaffinity Enrichment:
- Incubate pre-cleared lysate with K11-specific antibody (5 μg per 1 mg total protein) for 2 hours at 4°C.
- Add protein A/G magnetic beads and incubate for an additional 2 hours.
- Pellet beads and wash three times with 1 mL wash buffer.
Elution:
- Elubate captured proteins with 50 μL elution buffer for 10 minutes at room temperature.
- Neutralize with 5 μL 1 M Tris-HCl (pH 8.0).
Quality Control Steps:
- Include parallel control with isotype-matched IgG throughout the procedure.
- Confirm enrichment specificity by treating an aliquot of eluate with K11-specific DUB Cezanne [3].
- Analyze inputs, flow-through, and eluates by immunoblotting with K11-linkage specific antibodies.
Protocol: Ubiquitin Chain Restriction (UbiCRest) Analysis
UbiCRest analysis provides enzymatic validation of linkage identity using linkage-specific deubiquitinases.
Materials:
- Purified ubiquitinated substrates (from Section 4.2)
- K11-specific DUB Cezanne
- K48-specific DUB OTUB1
- General DUB USP21
- DUB Reaction Buffer: 50 mM Tris-HCl (pH 7.5), 50 mM NaCl, 1 mM DTT
Procedure:
- Sample Preparation:
- Divide purified ubiquitinated substrates into four 20 μL aliquots.
- Set up reactions containing:
- Tube 1: No enzyme control
- Tube 2: 100 nM Cezanne (K11-specific)
- Tube 3: 100 nM OTUB1 (K48-specific)
- Tube 4: 100 nM USP21 (general DUB)
DUB Digestion:
- Incubate reactions for 2 hours at 37°C.
- Terminate reactions by adding 5× SDS-PAGE loading buffer with 20 mM NEM.
Analysis:
- Resolve samples by SDS-PAGE and transfer to PVDF membrane.
- Probe with linkage-specific antibodies.
- Interpret results as follows:
Table 2: Expected UbiCRest Results for Different Chain Types
| Chain Architecture | No DUB Control | Cezanne (K11) | OTUB1 (K48) | USP21 (General) |
|---|---|---|---|---|
| Homotypic K11 | High MW smearing | Complete cleavage | No effect | Complete cleavage |
| Homotypic K48 | High MW smearing | No effect | Complete cleavage | Complete cleavage |
| K11/K48-Branched | High MW smearing | Partial cleavage | Partial cleavage | Complete cleavage |
This protocol was used to validate the presence of K11 linkages on Aurora A, where Cezanne treatment specifically removed K11 linkages while OTUB1 had minimal effect [3].
Quantitative Assessment and Troubleshooting
Performance Metrics for Enrichment Procedures
Establish quantitative metrics to evaluate enrichment procedure success:
- Enrichment Factor: Calculate as (RPMeMS / RPMsMS) where RPM is reads per million in enriched versus standard metagenomic sequencing [48]. Successful K11-enrichment should yield factors >30.
- Signal-to-Noise Ratio: Compare band intensity in specific antibody IP versus control IgG IP. Aim for >10:1 ratio.
- Linkage Specificity Index: Calculate as (1 - (signal after specific DUB treatment / untreated signal)) × 100%. Values should exceed 80% for high-specificity enrichments.
Troubleshooting Common Issues
Table 3: Troubleshooting Guide for K11-Chain Enrichment
| Problem | Potential Causes | Solutions |
|---|---|---|
| High Background | Non-specific antibody binding | Increase salt concentration in wash buffer to 300-500 mM NaCl; include pre-clearing step |
| Incomplete Enrichment | Insufficient antibody or incubation time | Optimize antibody:protein ratio; extend incubation time to 4 hours |
| Linkage Cross-reactivity | Antibody specificity issues | Validate with full panel of homotypic chains; use UbiCRest for confirmation |
| Low Yield | DUB activity during processing | Add multiple DUB inhibitors (NEM, PR-619) to lysis buffer; work quickly at 4°C |
| Branched Chain Loss | Method optimized for homotypic chains | Use receptors like RPN2 that specifically recognize branched configurations [5] |
Implementing rigorous quality control measures for enrichment reagents and procedures is essential for reliable research on K11-linked polyubiquitin chains. The protocols outlined here, emphasizing validation through genetic, enzymatic, and analytical methods, provide a framework for generating robust, reproducible data. As research continues to reveal the complex functions of K11 linkages in cell cycle regulation, proteotoxic stress response, and pathogen immunity [5] [49], these standardized approaches will facilitate deeper understanding of this critical ubiquitin signaling pathway.
Method Validation and Comparative Analysis of Enrichment Platforms
K11-linked polyubiquitin chains represent a significant non-canonical ubiquitin signaling modality, constituting approximately 28% of the conjugated ubiquitin pool in yeast cells, a abundance nearly equivalent to the canonical K48-linked chains [4]. These chains are now recognized as critical signals for targeted protein degradation, particularly during key cellular processes such as cell cycle progression and the management of proteotoxic stress [5] [3]. The anaphase-promoting complex/cyclosome (APC/C) utilizes the E2 enzyme UBE2S to assemble K11 linkages, which are essential for the timely degradation of mitotic regulators like Aurora kinases and Polo-like kinase, facilitating efficient mitotic exit [3]. Furthermore, K11 linkages frequently form branched architectures with K48-linked chains (K11/K48-branched), creating a potent proteasomal targeting signal that is preferentially recognized and processed by the 26S proteasome [5] [12].
Despite their established biological significance, the study of K11-linked ubiquitin chains presents considerable challenges. The transient nature of ubiquitination, the low stoichiometry of modified proteins within complex lysates, and the molecular complexity introduced by heterotypic and branched chains necessitate the use of highly specific and robust enrichment tools [27] [14] [50]. Consequently, rigorous benchmarking of these tools is paramount to ensure data accuracy and reliability. This application note provides a detailed framework for assessing the sensitivity and specificity of K11 enrichment methodologies, serving as a critical resource for researchers aiming to decipher the K11-linked ubiquitin code.
A diverse array of reagents has been developed to capture and analyze K11-linked ubiquitin chains. The following table summarizes the key tools and their applications.
Table 1: Key Research Reagent Solutions for K11-Linked Ubiquitin Research
| Reagent Type | Example Product/Name | Key Features and Function | Considerations for K11 Specificity |
|---|---|---|---|
| Linkage-Specific Antibodies | K11-linkage specific antibody [3] [14] | Immunoblotting, immunofluorescence; detects endogenous K11 chains. | Specificity must be validated via competing ubiquitin linkages and DUB digestion [47]. |
| Pan-Ubiquitin Binders | ChromoTek Ubiquitin-Trap [50] | Nanobody-based; pulldown of monoUb, all polyUb chains, and ubiquitylated proteins. | Not linkage-specific; requires downstream MS or immunoblotting with linkage-specific antibodies for K11 confirmation. |
| Tandem UBDs (TUBEs) | Tandem-repeated UBA domains [14] [47] | High-affinity capture; protects ubiquitin chains from DUBs during lysis. | General enrichment; K11 identity must be confirmed with linkage-specific tools. |
| Linkage-Specific DUBs | Cezanne (K11-specific) [3] | Enzymatic tool to confirm K11 identity by selectively cleaving this linkage. | Used as an analytical tool to validate K11 enrichment specificity in immunoblotting. |
| Activity-Based Probes | N/A | Chemically designed probes to profile DUB activity and specificity towards K11 chains. | Emerging technology; useful for characterizing DUBs that regulate K11 signaling [27]. |
| Reference Ubiquitin Chains | Recombinant K11-linked chains [27] [12] | Synthesized enzymatically or chemically; serve as essential positive controls. | Critical for benchmarking antibody specificity and optimizing enrichment protocols. |
Benchmarking Methodologies: A Practical Guide
Specificity Validation of K11-Binding Reagents
The cornerstone of reliable K11 research is confirming that tools specifically recognize K11 linkages over other ubiquitin chain types.
Protocol: Specificity Assessment by Immunoblotting
- Sample Preparation: Obtain a panel of recombinantly synthesized homotypic ubiquitin chains (K11, K48, K63, etc.) [27] [12]. Spot or load 200 ng of each chain type onto a nitrocellulose (NC) or polyvinylidene fluoride (PVDF) membrane, allowing it to air-dry completely [47].
- Membrane Processing: Block the membrane with a suitable blocking agent (e.g., 5% BSA in TBST) for 1 hour at room temperature.
- Antibody Probing: Incubate the membrane with the primary K11-linkage specific antibody, diluted in blocking buffer, for 2 hours at room temperature or overnight at 4°C.
- Signal Detection: Proceed with standard incubation with a horseradish peroxidase (HRP)-conjugated secondary antibody and chemiluminescent detection.
- Analysis: A specific antibody will produce a strong signal only for the K11-linked chain spot, with minimal to no cross-reactivity against other linkages.
Protocol: Specificity Validation using Linkage-Specific DUBs
- Enrichment: Use the K11-specific antibody or a pan-ubiquitin enrichment tool (e.g., Ubiquitin-Trap) to isolate ubiquitinated proteins from a cell lysate.
- On-Bead Digestion: Divide the captured material. Resuspend one aliquot in the appropriate reaction buffer containing the K11-specific DUB Cezanne (1-2 µg). The other aliquot serves as an untreated control and should be incubated with buffer alone [3].
- Incubation: Incubate both samples for 1-2 hours at 37°C with gentle agitation.
- Detection: Terminate the reaction, elute the proteins, and analyze by immunoblotting using the K11-specific antibody or a pan-ubiquitin antibody.
- Interpretation: Specific enrichment of K11 chains is demonstrated by a significant reduction (>70%) in K11 signal in the Cezanne-treated sample compared to the control, indicating that the enriched material was a bona fide substrate for the K11-specific enzyme [3].
Sensitivity Assessment of K11 Enrichment Workflows
Sensitivity benchmarking determines the lower detection limit of a given workflow, which is crucial for studying low-abundance ubiquitination events.
Protocol: Determining Limit of Detection (LOD)
- Spike-In Standard: Serially dilute recombinant K11-linked ubiquitin chains (e.g., from 1000 ng to 1 ng) into a constant volume of complex background, such as non-transfected HEK293T cell lysate.
- Enrichment and Detection: Subject each spiked sample to the full enrichment workflow (e.g., immunoprecipitation with a K11 antibody or Ubiquitin-Trap). Analyze the eluates by immunoblotting with a K11-specific antibody.
- Quantification: The LOD is defined as the lowest amount of spiked K11 chain that yields a reproducible signal distinguishable from the background of the negative control (lysate only).
Protocol: Quantitative Mass Spectrometry Benchmarking
- Stable Isotope Labeling: Employ Stable Isotope Labeling by Amino acids in Cell culture (SILAC) for quantitative comparisons [4]. Use "heavy"-labeled cells as a universal standard across all experiments.
- Comparative Enrichment: Process "light"-labeled test samples using different enrichment tools (e.g., K11 antibody vs. TUBEs vs. Ubiquitin-Trap).
- MS Analysis and Data Processing: Digest the enriched proteins, analyze by LC-MS/MS, and quantify the relative abundance of the K11-specific signature peptide (generating a GG-modified K11 peptide upon trypsin digestion) [14] [4].
- Metric Calculation: The tool that yields the highest heavy/light (H/L) ratio for the K11 GG-peptide, along with the greatest number of unique K11-modified substrate peptides, demonstrates superior sensitivity.
Table 2: Summary of Key Quantitative Metrics for Benchmarking
| Performance Metric | Experimental Approach | Interpretation and Benchmarking Goal |
|---|---|---|
| Signal-to-Noise Ratio | Immunoblotting with a panel of homotypic ubiquitin chains. | A high ratio indicates minimal cross-reactivity. Goal: Signal for K11 >> signal for any other linkage. |
| Limit of Detection (LOD) | Immunoblotting of serially diluted K11 chains spiked into cell lysate. | Defines the lowest detectable amount. Goal: A lower LOD indicates higher sensitivity. |
| Enrichment Fold-Change | Quantitative MS (e.g., SILAC) comparing K11 peptide abundance before and after enrichment. | Measures the degree of specific enrichment. Goal: A higher fold-enrichment for K11 peptides versus other linkages indicates superior specificity. |
| DUB Validation Score | Percentage reduction in signal after Cezanne treatment. | Confirms the identity of the enriched chains. Goal: >70% signal loss confirms high specificity of the enriched material [3]. |
Experimental Workflow for K11 Analysis
The following diagram illustrates a recommended integrated workflow for the specific and sensitive enrichment of K11-linked ubiquitin chains, incorporating the benchmarking controls described above.
Analysis of Branched K11/K48 Chains
It is critical to recognize that K11 linkages often exist not as homotypic chains but as heterotypic branched chains, most notably in conjunction with K48 linkages (K11/K48-branched) [5] [12]. Recent structural studies have revealed that the 26S proteasome possesses specialized receptors, including specific sites on RPN2, that directly recognize and bind these K11/K48-branched chains, leading to the accelerated degradation of the modified substrate [5]. This finding has profound implications for enrichment and interpretation.
When a K11-specific antibody is used, it may pull down a mixture of homotypic K11 chains and K11/K48-branched species. Therefore, the degradation phenotype observed for a substrate enriched with a K11-specific tool might be attributable to the presence of these highly efficient branched degradation signals. To deconvolute this, researchers should employ UbiCRest analysis, a methodology that uses a panel of linkage-specific DUBs [3] [47]. Sequential or parallel digestion with K11-specific (Cezanne) and K48-specific (OTUB1) DUBs, followed by immunoblotting, can help infer the architecture of the enriched chains. The recent development of methods to synthesize defined branched ubiquitin chains enzymatically or chemically provides essential standards for benchmarking tools against these complex architectures [27] [12].
Comparative Analysis of Antibody, TUBE, and enDUB Performance
K11-linked polyubiquitin chains represent a critical, though less characterized, form of ubiquitin signaling that plays essential regulatory roles in eukaryotic cells. Unlike the canonical K48-linked chains that primarily target proteins for proteasomal degradation, K11-linked chains exhibit diverse functional capabilities spanning both degradative and non-proteolytic pathways [1]. These chains are highly upregulated during mitosis and are assembled primarily by the anaphase-promoting complex/cyclosome (APC/C), an E3 ubiquitin ligase that controls cell cycle progression by targeting key regulatory proteins for destruction [2]. Quantitative mass spectrometry studies have revealed that K11 linkages can be as abundant as K48 linkages in yeast, while in human cells, they represent approximately 2% of the ubiquitin conjugate pool in asynchronous cells but increase dramatically during mitosis [1] [7]. The structural properties of K11-linked di-ubiquitin (K11-Ub2) reveal conformations distinct from K48-linked or K63-linked chains, enabling unique interactions with ubiquitin-receptor proteins [7]. Recent research has also highlighted the importance of K11/K48-branched ubiquitin chains, which serve as priority signals for proteasomal degradation during cell cycle progression and proteotoxic stress [5]. This application note provides a comprehensive comparison of three principal methodologies—specific antibodies, Tandem Ubiquitin Binding Entities (TUBEs), and engineered deubiquitinases (enDUBs)—for the detection and enrichment of K11-linked polyubiquitin chains, along with detailed protocols for their implementation in research settings.
Performance Metrics and Applications
The following table summarizes the key characteristics, advantages, and limitations of each technology for K11-linked ubiquitin chain research:
Table 1: Comparative analysis of methods for studying K11-linked polyubiquitin chains
| Method | Mechanism of Action | Key Advantages | Primary Limitations | Optimal Applications |
|---|---|---|---|---|
| K11-Linkage Specific Antibodies | Immunorecognition of unique K11-linked ubiquitin chain conformation | High specificity for K11 linkage; wide commercial availability; applicable to multiple techniques (Western blot, IF, IHC) | Cannot distinguish homotypic K11 from branched chains; may have variable affinity between lots; typically require denaturing conditions | Detection of endogenous K11 chains; cellular localization studies; quantitative assessment of chain levels in different conditions |
| TUBE (Tandem Ubiquitin Binding Entities) | Affinity purification using multiple ubiquitin-associated domains in tandem | Broad recognition of diverse ubiquitin linkages; preservation of labile ubiquitin modifications; protection from deubiquitinases | Lack of specificity for K11 linkage alone; requires secondary methods for linkage verification; may not recognize all chain conformations equally | Enrichment of ubiquitinated substrates from complex mixtures; study of ubiquitin dynamics in live cells; proteomic analysis of ubiquitome |
| enDUB (Engineered Deubiquitinases) | Targeted deubiquitination using engineered enzymes with specific substrate recognition | Unprecedented specificity through protein design; functional validation of chain dependency; applicable to live-cell studies | Requires genetic engineering; limited commercial availability; potential off-target effects if specificity is imperfect | Functional dissection of K11 signaling; rescue of trafficking-deficient mutants; therapeutic development for ubiquitination-related diseases |
Quantitative Performance Assessment
Table 2: Quantitative performance metrics for K11-linked ubiquitin detection methods
| Performance Parameter | K11-Specific Antibodies | TUBE-Based Approaches | enDUB Platforms |
|---|---|---|---|
| Detection Sensitivity | ~100 fmol (Western blot); highly dependent on antibody quality [51] | Not specifically quantified for K11 chains; generally high for ubiquitin enrichment | Demonstrated functional rescue at nanomolar concentrations in cellular models [52] |
| Linkage Specificity | High for designed epitopes; some cross-reactivity possible with similar linkages | Low; binds multiple linkage types simultaneously | Exceptionally high when properly engineered; dependent on targeting moiety |
| Temporal Resolution | Limited to endpoint measurements | Can monitor dynamics with appropriate experimental design | Suitable for real-time monitoring in live cells |
| Throughput Capability | High (compatible with automated platforms) | Moderate to high | Currently low to moderate |
| Quantitative Accuracy | Good with proper controls and standardization | Variable; requires careful normalization | High for functional readouts |
| Cellular Compatibility | Mostly fixed or lysed cells | Live cell applications possible with tagged TUBEs | Excellent for live-cell applications |
Research Reagent Solutions
Table 3: Essential research reagents for K11-linked polyubiquitin chain studies
| Reagent Category | Specific Examples | Function and Application | Considerations for Use |
|---|---|---|---|
| Linkage-Specific Detection Reagents | K11 linkage-specific antibody [2] | Direct immunodetection of K11 chains in Western blot, immunofluorescence, and immunohistochemistry | Validate specificity using linkage-deficient controls; optimize concentration for each application |
| Ubiquitin Affinity Reagents | Tandem Ubiquitin Binding Entities (TUBEs) | Affinity purification of polyubiquitinated proteins; protection from deubiquitinases | Choose appropriate tag (GST, His, etc.) for downstream applications; use protease inhibitors during purification |
| Engineered Enzymatic Tools | enDUBs (e.g., enDUB-O1) [52] | Targeted deubiquitination of specific substrates; functional validation of ubiquitination effects | Verify catalytic activity of enzyme; optimize expression levels to minimize off-target effects |
| Activity-Based Probes | Fluorophore-conjugated Connectase (N-Cnt) [51] | Direct in-gel fluorescence detection of tagged proteins; quantitative analysis without Western blot | Requires target proteins with N-terminal CnTags; offers exceptional sensitivity (0.1 fmol detection limit) |
| Cellular Model Systems | LQT1 cardiomyocyte model [52] | Physiological validation of K11 chain function in disease-relevant contexts | Ensure proper characterization of model system; include appropriate controls for disease mechanisms |
| Proteasome Recognition Assays | 26S proteasome complex reconstitution [5] | Study of K11/K48-branched chain recognition and degradation by proteasome | Requires sophisticated biochemical expertise; multiple validation methods recommended |
Detailed Experimental Protocols
Protocol 1: Detection of K11-Linked Ubiquitin Chains Using Linkage-Specific Antibodies
Background: K11 linkage-specific antibodies were engineered through structural characterization of K11-linked diubiquitin, which revealed a distinct conformation differing from K48- or K63-linked diubiquitin [2]. These antibodies recognize the unique structural epitope presented by K11-linked chains and have been instrumental in demonstrating the cell cycle-dependent regulation of these chains.
Procedure:
- Sample Preparation: Lyse cells in RIPA buffer (1% NP-40, 0.5% deoxycholate, 0.1% SDS) supplemented with 10 mM N-ethylmaleimide to inhibit deubiquitinases.
- Protein Separation: Separate 20-50 μg of total protein by SDS-PAGE using 4-12% Bis-Tris gels with MES or MOPS running buffer.
- Protein Transfer: Transfer to PVDF membrane using standard wet transfer protocols at 100V for 60 minutes.
- Blocking: Incubate membrane in 5% non-fat dry milk in TBST for 1 hour at room temperature.
- Primary Antibody Incubation: Incubate with K11 linkage-specific antibody at manufacturer's recommended dilution in blocking buffer overnight at 4°C.
- Washing: Wash membrane 3 times for 10 minutes each with TBST.
- Secondary Antibody Incubation: Incubate with appropriate HRP-conjugated or fluorescently-labeled secondary antibody for 1 hour at room temperature.
- Detection: Develop using enhanced chemiluminescence or fluorescence imaging systems.
Validation: Confirm antibody specificity using (1) cells treated with proteasome inhibitors (e.g., MG132) which should increase K11 chain levels, and (2) siRNA knockdown of APC/C components which should decrease mitotic K11 chains [2].
Figure 1: Workflow for detection of K11-linked ubiquitin chains using linkage-specific antibodies
Protocol 2: enDUB-Mediated Targeted Deubiquitination and Functional Rescue
Background: Engineered deubiquitinases (enDUBs) represent a groundbreaking approach for targeted ubiquitin chain removal. These tools fuse the catalytic domain of deubiquitinases (e.g., OTUD1) with specific substrate-targeting modules (e.g., nanobodies) to achieve precise deubiquitination of selected proteins [52]. This technology has demonstrated remarkable efficacy in rescuing trafficking-deficient mutant ion channels underlying diseases like Long QT syndrome type 1 (LQT1) and cystic fibrosis.
Procedure:
- enDUB Construct Design:
- Select appropriate targeting moiety (e.g., anti-GFP nanobody for GFP-tagged substrates)
- Fuse with catalytic domain of OTUD1 deubiquitinase using flexible linkers
- Incorporate fluorescent protein (e.g., CFP) via P2A self-cleaving peptide for expression tracking
Cell Transfection:
- Culture HEK293 cells in appropriate medium
- Transfect with enDUB plasmid using preferred transfection method
- Include controls: catalytically dead enDUB (enDUB-O1*), untargeted enDUB
Functional Validation:
- For electrophysiological studies: Perform whole-cell patch clamping 48-72 hours post-transfection
- For trafficking assays: Analyze surface expression by flow cytometry using extracellular epitope tags
- For biochemical studies: Assess ubiquitination status by immunoprecipitation and Western blot
Data Analysis:
- Quantify functional rescue relative to wild-type and mutant controls
- Determine statistical significance using appropriate tests (e.g., one-way ANOVA with post-hoc testing)
Application Note: enDUB treatment of LQT1 cardiomyocytes expressing the G589D KCNQ1 mutation normalized action potential duration (APD90 = 324±36 ms, n=11) compared to untreated mutant cells (APD90 = 753±93 ms, n=13), demonstrating functional rescue of pathological electrophysiological signatures [52].
Figure 2: enDUB workflow for targeted deubiquitination and functional rescue
Protocol 3: In-Gel Fluorescence Detection Using Connectase-Based Labeling
Background: Traditional Western blotting suffers from multiple limitations including variable transfer efficiency, antibody quality issues, and limited quantification capabilities. The Connectase-based in-gel fluorescence method provides an antibody-free alternative that offers superior sensitivity, quantification accuracy, and reproducibility [51]. This method uses a highly specific protein ligase (Connectase) to selectively fuse fluorophores to target proteins containing a recognition sequence (CnTag).
Procedure:
- CnTagged Protein Preparation:
- Engineer target proteins with N-terminal CnTags (PGAFDADPLVVEI) with 5-amino acid linkers (e.g., AAAGA)
- Express and purify proteins using standard protocols
Fluorophore-Connectase Conjugate Preparation:
- Incubate equimolar concentrations (5 μM) of Connectase and fluorescent peptide substrate (Cy5.5-RELASKDPGAFDADPLVVEI) for 1 minute
- Resulting conjugate (N-Cnt) contains approximately 25% fluorophore-loaded enzyme
Protein Labeling:
- Mix 6.67 nM of N-Cnt reagent with protein sample containing CnTagged POI
- Incubate ≥5 minutes for qualitative analysis or 30 minutes for quantitative analysis at room temperature
Gel Electrophoresis and Detection:
- Separate samples on polyacrylamide gel without protein transfer
- Analyze directly using fluorescence imager or scanner
- For storage, fix gels with 50% methanol/10% acetate
Performance Notes: This method demonstrates exceptional sensitivity with detection limits of ~0.1 fmol (3 pg of a 30 kDa protein), approximately three orders of magnitude more sensitive than typical Western blots [51]. The signal-to-substrate relationship is sigmoidal, with half-maximal signal at ~3 fmol and saturation above 25 fmol.
Technical Considerations and Troubleshooting
Method Selection Guidance
Choosing the appropriate method for K11-linked ubiquitin chain research depends on the specific research question and experimental context. Antibody-based approaches remain the most accessible for initial screening and localization studies, while TUBE-based methods offer advantages for proteomic analyses and enrichment of ubiquitinated substrates. enDUB technology provides unprecedented specificity for functional studies but requires more specialized expertise.
For investigators new to the field, we recommend beginning with validated K11 linkage-specific antibodies to establish baseline understanding of K11 chain behaviors in their experimental systems. Those with specific hypotheses about individual protein substrates may benefit from enDUB approaches, while researchers interested in global ubiquitination changes should consider TUBE-based enrichment strategies.
Troubleshooting Common Issues
- High Background in Antibody Detection: Optimize antibody concentration and include appropriate controls including competition with free K11-linked diubiquitin if available.
- Incomplete enDUB Activity: Verify catalytic activity of enDUB constructs and ensure proper subcellular targeting. Consider testing multiple expression levels.
- Poor Recovery with TUBE Enrichment: Include protease and deubiquitinase inhibitors in all buffers, and optimize binding/washing conditions for specific TUBE reagents.
- Specificity Validation: Always confirm K11 linkage specificity using multiple complementary methods, particularly when working with novel systems or conditions.
The expanding toolkit for studying K11-linked polyubiquitin chains, encompassing specific antibodies, TUBEs, and enDUBs, provides researchers with powerful options for investigating this important regulatory modification. Each method offers distinct advantages and limitations, making them complementary rather than competitive approaches. Antibodies provide accessibility and ease of use for detection, TUBEs enable broad ubiquitin enrichment, and enDUBs offer unprecedented specificity for functional studies. The continued refinement of these technologies, particularly in understanding K11/K48-branched chains and their recognition by the proteasome [5], will further enhance our ability to decipher the complex ubiquitin code and develop novel therapeutic strategies for diseases characterized by dysregulated ubiquitination.
Validation Through Genetic and Pharmacological Approaches
K11-linked polyubiquitin chains are essential regulators of critical cellular processes, with well-characterized roles in cell cycle control and proteasomal degradation. Their study requires robust validation strategies to confirm chain identity, functionality, and physiological relevance. This application note provides a comprehensive framework for validating K11-linked ubiquitin chains through integrated genetic and pharmacological approaches, enabling researchers to accurately decipher their specialized functions in cellular signaling and homeostasis. The methods outlined here are particularly crucial given that K11-linkages can represent approximately 2% of ubiquitin conjugates in asynchronous human cells but increase dramatically during mitosis [1]. Furthermore, the structural characterization of K11-linked chains reveals distinct conformations that differ from both K48- and K63-linked chains, necessitating specialized validation techniques [7].
Genetic Approaches for Validating K11-Linked Ubiquitin Chains
Genetic methods provide powerful tools for establishing the non-redundant functions of K11-linked ubiquitin chains in vivo. These approaches are particularly valuable for uncovering physiological roles that may be obscured by redundancy or low abundance in biochemical assays.
Ubiquitin Mutant Strain Engineering
The foundation of genetic analysis for ubiquitin linkage function lies in engineering yeast strains expressing ubiquitin mutants that perturb specific chain types.
Table 1: Ubiquitin Mutant Strains for K11-Linked Chain Analysis
| Strain Type | Genetic Composition | Functional Consequence | Key Applications |
|---|---|---|---|
| K11R Mutant | All ubiquitin loci modified to express K11R ubiquitin | Prevents formation of K11-linked chains | Identification of K11-specific functions; genetic interaction screening |
| K11-Only Mutant | All ubiquitin loci modified to express ubiquitin with only K11 available | Restricts polyubiquitination exclusively to K11 linkages | Functional validation of K11 linkages; substrate identification |
| Wild-type Ubiquitin | Unmodified ubiquitin loci | Normal ubiquitin function | Control for comparison with mutant strains |
A critical methodological consideration is that Saccharomyces cerevisiae encodes ubiquitin at four genomic loci (UBI1-4), requiring modification of all loci to ensure complete replacement with the desired ubiquitin variant [6] [53]. The UBI1, UBI2, and UBI3 loci express ubiquitin fused to ribosomal proteins RPL40A, RPL40B, and RPS31, respectively, while UBI4 encodes a polyubiquitin precursor. Engineering must preserve ribosomal protein expression while altering ubiquitin coding sequence [53]. The SK1 yeast strain background is recommended for these studies due to its high sporulation efficiency (~92%), which is essential when selecting for the multiple genetic modifications required [53].
Synthetic Genetic Array (SGA) Analysis
SGA methodology enables systematic identification of genetic interactions between K11-linked ubiquitin chains and other genes:
Diagram 1: SGA Workflow for K11 Linkage Analysis
This approach identified synthetic genetic interactions between K11R ubiquitin mutants and genes involved in threonine biosynthesis, leading to the discovery that K11 linkages are important for efficient threonine import [6]. Additionally, strong genetic interactions were observed with subunits of the anaphase-promoting complex (APC), confirming the role of K11 linkages in cell cycle regulation conserved from yeast to humans [6].
Conditional Expression of Single-Lysine Ubiquitin
For lethal ubiquitin mutations, conditional expression systems enable studying K11 linkage function:
- Clone single-lysine ubiquitin (K11-only) under a repressible promoter (e.g., GAL1)
- Integrate into a ubiquitin deletion strain alongside a wild-type ubiquitin expression plasmid with a counter-selectable marker (e.g., URA3)
- Repress mutant expression and maintain with wild-type plasmid
- Shift to permissive conditions and counter-select against wild-type ubiquitin plasmid
- Assess viability and phenotype under exclusive K11-linked chain formation [53]
This approach has demonstrated that the yeast APC modifies substrates with K11-linkages in vitro, and these chains contribute to normal APC-substrate turnover in vivo [6].
Pharmacological and Biochemical Validation Methods
Biochemical approaches provide direct evidence for K11-linked chain formation and function, allowing quantitative assessment under controlled conditions.
Linkage-Specific Antibody Detection
K11 linkage-specific antibodies enable direct detection and quantification of endogenous K11-linked chains:
Table 2: Experimental Detection of K11-Linked Chains During Cell Cycle
| Experimental Condition | K11-Linked Chain Abundance | Functional Interpretation |
|---|---|---|
| Asynchronous cells | Low (~2% of total ubiquitin conjugates) [1] | Baseline K11 linkage formation |
| Mitotic cells | Highly upregulated [2] | K11 linkages function in mitotic progression |
| APC/C inhibition | Strongly reduced [2] | APC/C is major source of mitotic K11 chains |
| Proteasome inhibition | Increased accumulation [2] | K11 chains act as proteasomal degradation signals |
Protocol for K11-linked chain detection using specific antibodies:
- Synchronize cells in mitosis using nocodazole (5 µg/mL, 16-18 hours)
- Prepare cell lysates in RIPA buffer with protease and deubiquitinase inhibitors
- Separate proteins by SDS-PAGE (4-12% gradient gels recommended)
- Transfer to PVDF membrane and block with 5% BSA
- Incubate with anti-K11 linkage-specific antibody (1:1000 dilution, overnight at 4°C)
- Detect with HRP-conjugated secondary antibody and chemiluminescence
- Normalize to total ubiquitin or loading controls
This methodology revealed that K11-linked chains are highly upregulated in mitotic human cells precisely when APC/C substrates are degraded [2].
In Vitro Ubiquitin Chain Linkage Determination
A definitive biochemical approach for determining ubiquitin chain linkage utilizes ubiquitin lysine mutants in reconstituted ubiquitination reactions:
Diagram 2: Linkage Determination Using Ubiquitin Mutants
Detailed protocol for determining K11 linkage specificity:
Materials:
- E1 activating enzyme (5 µM)
- E2 conjugating enzyme (e.g., Ube2S for K11 linkages, 25 µM)
- E3 ligase (e.g., APC/C, 10 µM)
- 10X E3 ligase reaction buffer (500 mM HEPES, pH 8.0, 500 mM NaCl, 10 mM TCEP)
- Wild-type ubiquitin, ubiquitin K-to-R mutants, K11-only ubiquitin mutant (1.17 mM)
- MgATP solution (100 mM)
Procedure:
- Set up nine 25 µL reactions containing:
- 2.5 µL 10X E3 reaction buffer
- 1 µL ubiquitin (wild-type or mutant, ~100 µM final)
- 2.5 µL MgATP solution (10 mM final)
- 5-10 µM substrate
- 0.5 µL E1 enzyme (100 nM final)
- 1 µL E2 enzyme (1 µM final)
- 1 µM E3 ligase
- dH₂O to 25 µL
Incubate at 37°C for 30-60 minutes
Terminate reactions with SDS-PAGE sample buffer or 20 mM EDTA
Analyze by western blotting with anti-ubiquitin antibody
Interpretation:
- If K11R mutant prevents chain formation → chains are K11-linked
- If K11-only mutant supports chain formation → confirmation of K11 linkage specificity [26]
This approach confirmed Ube2S as the primary elongating E2 for K11-linked chains with the APC/C [1].
Structural Validation of K11-Linked Chains
Structural analysis provides mechanistic insights into how K11-linked chains are assembled, recognized, and distinguished from other linkage types.
Solution NMR Characterization
NMR spectroscopy reveals the dynamic structural properties of K11-linked chains in near-physiological conditions:
- Prepare isotopically labeled (¹⁵N, ¹³C) K11-linked diubiquitin (K11-Ub₂) using enzymatic assembly with E1 and K11-specific E2 enzyme Ube2S
- Collect ¹H-¹⁵N TROSY-HSQC spectra of each ubiquitin unit in K11-Ub₂
- Measure chemical shift perturbations (CSPs) relative to monomeric ubiquitin
- Determine residual dipolar couplings (RDCs) in aligned media
- Validate with small-angle neutron scattering (SANS) for solution conformation
This methodology demonstrated that K11-linked diubiquitin adopts distinct conformations from K48- or K63-linked chains, with unique dynamical properties that enable specific recognition by downstream receptors [7]. The CSP pattern for the proximal ubiquitin unit shows significant perturbations clustered around K11, primarily resulting from isopeptide bond formation rather than novel Ub/Ub interfaces [7].
Recognition of K11/K48-Branched Ubiquitin Chains
Recent cryo-EM structures reveal how K11/K48-branched ubiquitin chains are recognized by the 26S proteasome:
- Reconstitute K11/K48-branched ubiquitin chains on substrate proteins using engineered E3 ligases
- Form complexes with human 26S proteasome and auxiliary proteins (RPN13, UCHL5)
- Solve cryo-EM structures to visualize binding interfaces
- Identify specific contacts between branched chains and proteasomal subunits
This approach identified a multivalent recognition mechanism where:
- RPN2 recognizes K48-linkages extending from K11-linked ubiquitin
- A unique groove formed by RPN2 and RPN10 binds K11-linked ubiquitin branches
- This specialized recognition explains the priority degradation signal of K11/K48-branched chains [5]
Branched K11/K48-triUb possesses a unique hydrophobic interface between distal ubiquitins and exhibits enhanced affinity for proteasomal subunit Rpn1, providing a structural basis for its efficient proteasomal targeting [54].
Research Reagent Solutions
Table 3: Essential Research Reagents for K11-Linked Ubiquitin Chain Studies
| Reagent Category | Specific Examples | Function in K11 Research |
|---|---|---|
| Ubiquitin Mutants | K11R, K11-only ubiquitin | Determine linkage specificity and function |
| E2 Enzymes | Ube2C (UbcH10), Ube2S | K11-linked chain initiation and elongation |
| E3 Ligases | APC/C (Anaphase-Promoting Complex) | Major cellular source of K11 linkages |
| Linkage-Specific Antibodies | Anti-K11 linkage antibody | Detect endogenous K11-linked chains |
| Proteasomal Subunits | Rpn1, Rpn10, Rpn13 | Study recognition of K11-linked chains |
| DUBs | UCHL5, USP14 | Investigate K11 chain disassembly |
| Structural Tools | Isotopically labeled ubiquitin, cross-linkers | Determine K11 chain architecture |
Integrated Validation Workflow
A comprehensive validation strategy for K11-linked ubiquitin chains should integrate multiple approaches:
- Initial Detection: Use K11 linkage-specific antibodies to detect endogenous chains
- Genetic Validation: Engineer ubiquitin mutant strains to establish physiological function
- Biochemical Confirmation: Employ in vitro reconstitution with ubiquitin mutants
- Structural Analysis: Determine solution and bound conformations of K11-linked chains
- Functional Assessment: Measure degradation efficiency and proteasomal recognition
This multifaceted approach has been instrumental in establishing the critical role of K11-linked chains in cell cycle regulation, where they function as priority degradation signals for mitotic regulators [1] [2] [5]. The conserved function from yeast to humans highlights the fundamental importance of these validation methodologies across model systems [6].
K11-linked polyubiquitination is a critical post-translational modification that regulates the timely degradation of cell cycle regulators, ensuring accurate progression through mitosis. Unlike the well-characterized K48-linked chains which serve as the canonical proteasomal degradation signal, K11-linked chains have emerged as specialized regulators of mitotic progression, particularly through their assembly by the Anaphase-Promoting Complex/Cyclosome (APC/C) [1]. In higher eukaryotes, these atypical chains are highly upregulated during mitosis and control the degradation of key mitotic substrates [1] [2]. This case study details the experimental approaches for analyzing K11-linked ubiquitin chains, with a focus on enrichment strategies and functional assessment in the context of cell cycle regulation.
Biological Background and Significance
The Ubiquitin Code in Cell Cycle Regulation
Protein ubiquitination involves a coordinated enzymatic cascade of E1 (activating), E2 (conjugating), and E3 (ligase) enzymes that covalently attach ubiquitin to substrate proteins [55]. The modification can be reversed by deubiquitinating enzymes (DUBs) [28]. Ubiquitin itself contains seven lysine residues (K6, K11, K27, K29, K33, K48, K63) and an N-terminal methionine (M1), each capable of forming structurally and functionally distinct polyubiquitin chains [1] [28]. The linkage specificity forms a "ubiquitin code" that determines the functional outcome for modified substrates [1].
Table 1: Major Ubiquitin Linkage Types and Their Primary Functions
| Linkage Type | Primary Known Functions | Key Regulatory Roles |
|---|---|---|
| K48-linked | Proteasomal degradation [4] | Major degradation signal |
| K63-linked | Non-proteolytic signaling [4] | DNA repair, kinase activation, inflammation |
| K11-linked | Cell cycle control, ERAD [1] [4] | Mitotic regulation via APC/C |
| K11/K48-branched | Accelerated proteasomal degradation [5] | Cell cycle progression, proteotoxic stress |
K11 Linkages in Mitotic Control
K11-linked chains demonstrate striking cell cycle-dependent regulation, with abundance peaking dramatically during mitosis [1] [2]. Quantitative proteomic studies reveal that K11 linkages can represent approximately 28% of the total ubiquitin conjugate pool in yeast, while comprising about 2% in asynchronously dividing human cells [1] [4]. During mitosis, this percentage increases substantially, coinciding with the degradation of APC/C substrates [2]. The APC/C is the primary E3 ligase responsible for assembling homogenous K11-linked chains during cell division, working in concert with the E2 enzymes Ube2C (initiator) and Ube2S (elongator) [1].
Experimental Strategies for K11 Chain Analysis
Enrichment Methodologies for K11-Linked Chains
Linkage-Specific Antibodies
The development of K11 linkage-specific antibodies has revolutionized the detection and enrichment of K11-linked ubiquitin chains from native cellular environments [2] [56]. These antibodies specifically recognize the unique conformational epitope presented by K11-linked diubiquitin, which adopts a distinct structure from K48- or K63-linked chains [2] [7].
Table 2: Key Reagent Solutions for K11-Linked Ubiquitin Research
| Research Reagent | Function/Application | Key Features |
|---|---|---|
| K11-linkage specific antibody [2] [56] | Immunodetection and immunoenrichment of K11 chains | High specificity; detects endogenous chains |
| Tandem Ubiquitin Binding Entities (TUBEs) [29] | Affinity enrichment of polyubiquitinated proteins | Pan-specific or linkage-selective variants available |
| Ubiquitin binding domains (UBDs) [28] | Enrichment of ubiquitinated proteins | Can be engineered for linkage specificity |
| Chain-terminating ubiquitin mutants (K11R) [4] | Genetic analysis of K11 chain function | Allows functional assessment of K11 linkages |
Experimental Protocol: Antibody-Based Detection of Endogenous K11-Linked Chains
- Cell Culture and Synchronization: Culture cells and synchronize at desired cell cycle stage (e.g., mitotic arrest using nocodazole).
- Cell Lysis: Lyse cells in modified RIPA buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 1 mM EDTA) supplemented with protease inhibitors and 10 mM N-ethylmaleimide to preserve ubiquitin chains.
- Immunoprecipitation: Incubate cell lysates with K11 linkage-specific antibody conjugated to Protein A/G beads for 4 hours at 4°C.
- Washing: Wash beads 3-5 times with lysis buffer containing 300-500 mM NaCl to reduce non-specific binding.
- Elution and Analysis: Elute bound proteins with Laemmli buffer for immunoblotting or specific elution buffers for mass spectrometry analysis.
Affinity-Based Enrichment Strategies
Tandem Ubiquitin Binding Entities (TUBEs) offer an alternative enrichment strategy with high affinity for polyubiquitin chains. While pan-specific TUBEs bind all chain types, the development of linkage-selective TUBEs enables more specific isolation of K11-linked chains [29]. These reagents protect polyubiquitin chains from proteasomal degradation and deubiquitination during extraction, preserving the native ubiquitome architecture.
Ubiquitin tagging approaches utilizing epitope-tagged ubiquitin (e.g., His-, HA-, or Strep-tags) allow purification of ubiquitinated proteins under denaturing conditions, minimizing co-purification of non-specifically bound proteins [28]. However, these require genetic manipulation of cells and may not fully replicate endogenous ubiquitination dynamics.
Analytical and Detection Methods
Mass Spectrometry-Based Quantification
Absolute quantification of ubiquitin linkages can be achieved through mass spectrometry with isotope-labeled internal standards [4]. This approach enables precise measurement of K11 chain abundance under different physiological conditions.
Experimental Protocol: Absolute Quantification of Ubiquitin Linkages by Mass Spectrometry
- Ubiquitin Conjugate Enrichment: Isulate ubiquitinated proteins from cell lysates by TUBE-based affinity purification or immunoprecipitation.
- Trypsin Digestion: Digest enriched proteins with trypsin, which cleaves ubiquitin after arginine 74, leaving a di-glycine (GG) remnant on modified lysines.
- Spike-in Standards: Add known quantities of heavy isotope-labeled, GG-modified linkage-specific peptides as internal standards.
- LC-MS/MS Analysis: Analyze peptides by liquid chromatography coupled to tandem mass spectrometry.
- Quantification: Calculate the abundance of native K11-linked peptides by comparing their signal intensity to the heavy isotope-labeled standards.
Table 3: Quantitative Analysis of Polyubiquitin Linkage Abundance in Yeast
| Linkage Type | Percent Abundance (%) | Fold-Increase After Proteasomal Inhibition |
|---|---|---|
| K6 | 10.9 ± 1.9 | 4-5 fold |
| K11 | 28.0 ± 1.4 | 4-5 fold |
| K27 | 9.0 ± 0.1 | ~2 fold |
| K29 | 3.2 ± 0.1 | 4-5 fold |
| K33 | 3.5 ± 0.1 | ~2 fold |
| K48 | 29.1 ± 1.9 | ~8 fold |
| K63 | 16.3 ± 0.2 | No significant change |
Data derived from quantitative mass spectrometry analyses [4].
Functional Validation Using Genetic Approaches
Genetic manipulation of ubiquitin pathways provides critical functional validation of K11 chain involvement in specific cellular processes:
K11-Specific Mutants: Expression of ubiquitin mutant K11R (lysine 11 mutated to arginine) disrupts formation of K11-linked chains, allowing assessment of the functional consequences [4].
APC/C Inhibition: Chemical or genetic inhibition of APC/C activity dramatically reduces K11-linked chain formation during mitosis, establishing the central role of this E3 ligase in mitotic K11 chain assembly [2].
Functional Rescue Experiments: Complementation with wild-type ubiquitin in cells expressing only K11R ubiquitin mutants tests whether K11 linkages are specifically required for mitotic progression.
K11 Linkages in APC/C-Mediated Proteolysis
The APC/C Enzyme System
The APC/C is a multi-subunit E3 ubiquitin ligase that orchestrates mitotic progression by targeting key regulatory proteins for degradation. The APC/C assembles K11-linked ubiquitin chains through a coordinated two-step mechanism:
- Chain Initiation: Ube2C (UbcH10) catalyzes the transfer of the first ubiquitin to the substrate and formation of short, preferentially K11-linked chains [1].
- Chain Elongation: Ube2S specifically extends K11-linked chains, processively building homogenous K11-linked polymers [1].
The recognition of substrates by APC/C is facilitated by degron sequences such as D-boxes and KEN-boxes, which are recognized by coactivators Cdc20 or Cdh1 [1].
Structural Insights into K11 Chain Recognition
Recent structural studies have revealed how the 26S proteasome recognizes K11-linked ubiquitin chains. Cryo-EM analyses demonstrate that K11/K48-branched ubiquitin chains engage in multivalent interactions with the proteasome, explaining their enhanced efficiency in targeting substrates for degradation [5]. Key findings include:
- RPN2 contains a previously unidentified ubiquitin-binding site that specifically recognizes K11 linkages
- K11/K48-branched chains establish tripartite binding interfaces with the 19S regulatory particle
- This multivalent recognition explains the priority degradation of substrates modified with K11/K48-branched chains during cell cycle progression
Advanced Research Applications
Therapeutic Targeting of Ubiquitin Pathways
Understanding K11-linked ubiquitination has significant implications for therapeutic development, particularly in targeted protein degradation technologies:
PROTACs (Proteolysis-Targeting Chimeras) are heterobifunctional molecules that recruit E3 ubiquitin ligases to target proteins, inducing their ubiquitination and degradation [55]. While current PROTACs primarily utilize a limited set of E3 ligases (CRBN, VHL), understanding linkage-specific ubiquitination could enable the development of next-generation degraders with enhanced specificity.
Molecular Glue Degraders are monovalent small molecules that induce proximity between E3 ligases and target proteins, leading to target ubiquitination and degradation [55]. Examples include immunomodulatory drugs (IMiDs) such as thalidomide derivatives that recruit novel substrates to the CRL4CRBN E3 ligase.
K11 Linkages in Disease Contexts
Beyond their role in normal cell cycle regulation, K11-linked chains have been implicated in pathological conditions:
Cancer: Overexpression of Ube2C, the initiating E2 for K11 chain formation, is observed in various cancers and linked to error-prone chromosome segregation and tumorigenesis [1].
Neurodegenerative Diseases: Aberrant K11-linked ubiquitination has been associated with the accumulation of pathological protein aggregates in neurodegenerative conditions [5].
The analysis of K11-linked polyubiquitin chains requires a multifaceted experimental approach combining specific enrichment strategies, sensitive detection methodologies, and functional validation. The development of K11 linkage-specific antibodies and chain-selective TUBEs has significantly advanced our ability to study these atypical chains in native biological contexts. The central role of K11 linkages in APC/C-mediated proteolysis during cell division highlights their fundamental importance in cell cycle regulation. Continued refinement of analytical techniques will further elucidate the diverse functions of K11-linked ubiquitination in both physiological and pathological processes, potentially opening new avenues for therapeutic intervention in cancer and other proliferation-associated disorders.
K11-linked polyubiquitin chains are crucial regulatory modifications that coordinate essential cellular processes, most notably the targeted degradation of specific substrates during cell cycle progression and proteotoxic stress. Unlike the canonical K48-linked chains, K11-linked chains constitute a distinct ubiquitin code that is rapidly decoded by the proteasome to ensure the timely destruction of key regulatory proteins. Research has revealed that K11 linkages are highly upregulated during mitosis and are preferentially assembled on cell cycle regulators by the anaphase-promoting complex/cyclosome (APC/C) in conjunction with the E2 enzyme UBE2S [3] [2]. The molecular basis for this efficient degradation signal lies in the unique structural properties of K11-linked chains and their recognition by specific receptors within the 26S proteasome, particularly through a specialized binding site involving RPN2 and RPN10 subunits [5]. This application note provides detailed methodologies for interrogating K11-linked ubiquitination through functional degradation assays and quantitative signaling readouts, enabling researchers to decipher this complex regulatory pathway with high specificity and precision.
Quantitative Analysis of K11-Linked Ubiquitination in Mitotic Exit
Experimental Protocol: Cell-Based Ubiquitination Assay for K11 Linkage Quantification
Purpose: To quantitatively measure K11-linked ubiquitination on specific substrates during mitotic exit.
Materials and Reagents:
- U2OS cells (or other appropriate cell line)
- Double thymidine block solution (2 mM thymidine in culture medium)
- Synchronization reagents (e.g., nocodazole for mitotic arrest)
- Plasmid DNA encoding GFP-tagged substrates (e.g., AurA-Venus, AurB-Venus)
- Transfection reagent
- UBE2S-specific siRNA
- Control siRNA
- Lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, supplemented with protease inhibitors and deubiquitinase inhibitors)
- GFP-Trap magnetic beads or equivalent GFP-nanobody conjugated beads
- K11-linkage specific antibody [2]
- GFP antibody
- Secondary antibodies compatible with detection system
- Immunoblotting equipment and reagents
Procedure:
- Cell Synchronization:
- Culture U2OS cells and synchronize at G1/S phase boundary using double thymidine block (2 mM thymidine for 18h, release for 9h, followed by a second thymidine block for 17h) [3].
- Release cells into fresh medium and collect samples at 2-hour intervals over a 14-hour time course to capture mitotic entry and exit.
- Confirm synchronization efficiency by monitoring histone H3 phosphorylation and cyclin B levels via immunoblotting.
UBE2S Depletion:
- Transfect cells with UBE2S-specific siRNA or control siRNA using standard transfection protocols 48 hours prior to synchronization.
- Validate knockdown efficiency by immunoblotting with UBE2S-specific antibody.
Substrate Expression and Purification:
- Transiently or stably express GFP-tagged substrates (e.g., Aurora A, Aurora B) in synchronized cells.
- At desired time points (typically 10-12 hours after release for mitotic exit), harvest cells and lyse in pre-chilled lysis buffer.
- Clarify lysates by centrifugation at 16,000 × g for 15 minutes at 4°C.
- Incubate supernatant with GFP-Trap magnetic beads for 2 hours at 4°C with gentle rotation.
- Wash beads three times with wash buffer (identical to lysis buffer but with 0.1% NP-40).
- Elute bound proteins using 2× Laemmli buffer with heating at 95°C for 5 minutes.
Ubiquitination Analysis:
- Resolve eluted proteins by SDS-PAGE (4-12% gradient gels recommended).
- Transfer to PVDF membranes and probe with K11-linkage specific antibody and GFP antibody.
- Quantify band intensities using appropriate imaging software.
- Normalize K11-ubiquitination signals to total substrate levels (GFP signal).
Data Interpretation and Quantitative Readouts
The following table summarizes typical quantitative data obtained from K11-ubiquitination assays during mitotic exit:
Table 1: Quantitative Profile of K11-Linked Ubiquitination on APC/C Substrates During Mitotic Exit
| Substrate | Total Ubiquitination (Relative Units) | K11-Specific Ubiquitination (Relative Units) | Reduction after UBE2Si (%) | Degradation Half-life (min) |
|---|---|---|---|---|
| Aurora A | 1.00 ± 0.15 | 0.45 ± 0.08 | 52.3 ± 6.1 | 28.4 ± 3.2 |
| Aurora B | 1.00 ± 0.12 | 0.38 ± 0.07 | 61.8 ± 5.7 | 31.7 ± 2.9 |
| Nek2A | 1.00 ± 0.18 | 0.51 ± 0.09 | 48.9 ± 7.2 | 25.3 ± 4.1 |
| Polo-like Kinase 1 | 1.00 ± 0.14 | 0.42 ± 0.06 | 57.4 ± 6.5 | 35.2 ± 3.8 |
Data adapted from [3]. Values represent mean ± SEM from at least three independent experiments.
The workflow for this quantitative assay can be visualized as follows:
Figure 1: Experimental workflow for quantitative analysis of K11-linked ubiquitination during mitotic exit.
Functional Degradation Assays for K11-Linked Substrates
Experimental Protocol: Live-Cell Degradation Tracking
Purpose: To monitor degradation kinetics of K11-ubiquitinated substrates at single-cell resolution in real-time.
Materials and Reagents:
- Stable cell lines expressing GFP-tagged substrates (e.g., Aurora A-GFP, Aurora B-GFP)
- Live-cell imaging chamber with environmental control (37°C, 5% CO2)
- Confocal or epifluorescence microscope with time-lapse capability
- Image analysis software (e.g., ImageJ, MetaMorph)
- Culture medium for live-cell imaging (without phenol red)
- SiRNA targeting UBE2S and control siRNA
- Proteasome inhibitor (e.g., MG132) as control
Procedure:
- Cell Preparation:
- Seed stable cell lines expressing GFP-tagged substrates in imaging chambers 24 hours prior to experimentation.
- Transfert with UBE2S-specific or control siRNA 48 hours before imaging.
Live-Cell Imaging:
- Synchronize cells in mitosis using 9 μM nocodazole for 4 hours.
- Wash out nocodazole thoroughly to initiate mitotic exit.
- Immediately place chambers in live-cell imaging system maintained at 37°C with 5% CO2.
- Acquire images every 3-5 minutes for 4-6 hours using a 20× or 40× objective.
- Maintain focus throughout the time course using hardware autofocus systems.
Data Analysis:
- Track individual cells from nuclear envelope breakdown through cytokinesis.
- Quantify GFP fluorescence intensity in the cytoplasm over time.
- Normalize fluorescence values to initial intensity at mitotic entry.
- Plot degradation curves and calculate half-lives using exponential decay models.
- Compare degradation kinetics between UBE2S-depleted and control cells.
Data Interpretation and Quantitative Readouts
Table 2: Degradation Kinetics of K11-Ubiquitinated Substrates in Live Cells
| Experimental Condition | Aurora A Half-life (min) | Aurora B Half-life (min) | Nek2A Half-life (min) | Degradation Completion (% at 60 min) |
|---|---|---|---|---|
| Control siRNA | 28.4 ± 3.2 | 31.7 ± 2.9 | 25.3 ± 4.1 | 78.3 ± 5.2 |
| UBE2S siRNA | 52.7 ± 4.8 | 58.9 ± 5.1 | 47.6 ± 6.3 | 41.6 ± 6.7 |
| MG132 Treatment | 215.6 ± 28.3 | 198.4 ± 22.7 | 189.2 ± 25.4 | 12.4 ± 3.8 |
Data adapted from [3]. Degradation half-lives represent mean ± SEM from at least 50 cells per condition across three independent experiments.
Structural Validation of K11/K48-Branched Ubiquitin Chain Recognition
Experimental Protocol: Cryo-EM Analysis of Proteasome-K11/K48 Ubiquitin Complexes
Purpose: To determine the structural basis of K11/K48-branched ubiquitin chain recognition by the 26S proteasome.
Materials and Reagents:
- Purified human 26S proteasome
- Engineered Rsp5 E3 ligase (Rsp5-HECTGML) for K48-linkage formation [5]
- Sic1PY substrate (residues 1-48 of S. cerevisiae Sic1 protein with single lysine K40)
- Ubiquitin mutants (K63R variant)
- RPN13:UCHL5 complex (with UCHL5 catalytic cysteine mutation C88A)
- Size-exclusion chromatography columns
- Cryo-EM grids (ultrauFoil or Quantifoil)
- Vitrification system (e.g., Vitrobot)
- High-end cryo-electron microscope
- Image processing software (e.g., RELION, cryoSPARC)
Procedure:
- Complex Reconstitution:
- Generate polyubiquitinated Sic1PY (Sic1PY-Ubn) using Rsp5-HECTGML and Ub(K63R).
- Fractionate reaction products by size-exclusion chromatography to enrich medium-length chains (n=4-8 ubiquitins).
- Confirm chain linkage composition by Lbpro* Ub clipping and intact mass spectrometry [5].
- Incubate enriched Sic1PY-Ubn with human 26S proteasome and RPN13:UCHL5(C88A) complex for 30 minutes at 30°C.
Cryo-EM Sample Preparation and Data Collection:
- Apply 3.5 μL of complex to freshly plasma-cleaned cryo-EM grids.
- Blot and vitrify grids in liquid ethane using standard vitrification procedures.
- Collect multi-frame movies on a 300 keV cryo-electron microscope with K3 direct electron detector.
- Target nominal magnification of 105,000× (0.826 Å/pixel) with total exposure of 50 e-/Å2.
Image Processing and 3D Reconstruction:
- Perform motion correction and CTF estimation on collected movies.
- Pick particles using template-based or AI-driven approaches.
- Conduct multiple rounds of 2D and 3D classification to isolate homogeneous complexes.
- Refine selected particles to obtain high-resolution reconstructions.
- Build atomic models through iterative refinement against the cryo-EM density.
Data Interpretation and Structural Insights
The structural analysis reveals a multivalent recognition mechanism for K11/K48-branched ubiquitin chains involving:
Novel K11-Linked Ub Binding Site: A previously unknown binding site for K11-linked Ub at the groove formed by RPN2 and RPN10 subunits [5].
Canonical K48-Linkage Binding: Simultaneous engagement of K48-linkage at the canonical binding site formed by RPN10 and RPT4/5 coiled-coil [5].
Alternating Linkage Recognition: RPN2 recognition of alternating K11-K48-linkage through a conserved motif similar to the K48-specific T1 binding site of RPN1 [5].
The molecular recognition process can be visualized as follows:
Figure 2: Molecular recognition pathway of K11/K48-branched ubiquitin chains by the 26S proteasome.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for K11-Linked Ubiquitin Studies
| Reagent Category | Specific Examples | Function/Application | Key Features |
|---|---|---|---|
| Linkage-Specific Antibodies | K11-linkage specific antibody [2] | Detection and quantification of K11-linked chains | Specifically recognizes K11 linkages without cross-reactivity to other chain types |
| E2 Enzymes | UBE2S (K11-specific elongator) [3] | In vitro assembly of K11-linked chains | Collaborates with APC/C to build K11 linkages on substrates |
| Deubiquitinases | Cezanne (K11-specific DUB) [3] | Validation of K11 linkage identity | Selectively cleaves K11 linkages without affecting other chain types |
| Ubiquitin Mutants | Ub(K63R) [5] | Specific chain assembly | Prevents formation of K63 linkages during in vitro ubiquitination |
| Proteasome Subunits | RPN13:UCHL5 complex [5] | Structural studies of chain recognition | Captures K11/K48-branched chains on proteasome for structural analysis |
| Substrate Reporters | GFP-tagged Aurora kinases [3] | Live-cell degradation tracking | Enables real-time monitoring of substrate degradation kinetics |
| Structural Tools | Engineered Rsp5 E3 ligase (Rsp5-HECTGML) [5] | In vitro generation of specific linkages | Produces K48-linked chains with minimal branching |
Integrated Workflow for Comprehensive K11-Linked Ubiquitin Analysis
The most powerful insights emerge from integrating multiple approaches to study K11-linked ubiquitination. The following comprehensive workflow connects biochemical, cellular, and structural methodologies:
Figure 3: Integrated workflow for comprehensive analysis of K11-linked ubiquitination, connecting biochemical, cellular, and structural approaches.
This application note provides a comprehensive framework for studying K11-linked ubiquitination through functional degradation assays and signaling outcomes. The integrated approach enables researchers to bridge molecular mechanisms with cellular phenotypes, offering powerful insights into how this specialized ubiquitin code coordinates essential biological processes.
Conclusion
The strategic enrichment of K11-linked polyubiquitin chains has evolved from a technical challenge to an achievable goal with diverse research applications. Current methodologies—including linkage-specific antibodies, TUBEs, and engineered DUBs—provide researchers with multiple avenues for reliable K11 chain isolation and analysis. The recent structural elucidation of K11/K48-branched chain recognition by the 26S proteasome underscores the biological importance of these chains in priority degradation signaling. As the field advances, future developments will likely focus on improving enrichment specificity, adapting methods for single-cell analysis, and expanding applications in drug discovery, particularly for PROTAC development and targeted protein degradation therapeutics. The continued refinement of K11 chain enrichment strategies will undoubtedly yield new insights into ubiquitin-coding principles and open novel therapeutic avenues for manipulating protein degradation pathways.
References
- 1. K11-linked ubiquitin chains as novel regulators of cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3205209/]
- 2. K11-Linked Polyubiquitination in Cell Cycle Control ... [https://www.sciencedirect.com/science/article/pii/S109727651000523X]
- 3. Efficient APC/C substrate degradation in cells undergoing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4666129/]
- 4. Quantitative Proteomics Reveals the Function of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2668214/]
- 5. Structural basis of K11/K48-branched ubiquitin chain ... [https://www.nature.com/articles/s41467-025-64719-x]
- 6. Genetic analysis reveals functions of atypical polyubiquitin ... [https://elifesciences.org/articles/42955]
- 7. Unique structural, dynamical, and functional properties of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3802530/]
- 8. Linear Ubiquitin Chains: Cellular Functions and Strategies ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2019.00915/full]
- 9. Synthesis and Analysis of K -Linked Ubiquitin 11 | SpringerLink Chains [https://link.springer.com/protocol/10.1007/978-1-61779-474-2_15]
- 10. RCSB PDB - 3NOB: Structure of K -linked di-ubiquitin 11 [https://www.rcsb.org/structure/3NOB]
- 11. K11- and K48-Linked Ubiquitin Chains Interact with p97 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4160081/]
- 12. Emerging functions of branched ubiquitin chains | Cell Discovery [https://www.nature.com/articles/s41421-020-00237-y]
- 13. Protein enrichment from cell lysates [https://bio-protocol.org/exchange/minidetail?id=16954785&type=30]
- 14. Current methodologies in protein ubiquitination ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9373315/]
- 15. Enrichment and analysis of rice seedling ubiquitin-related ... [https://www.sciencedirect.com/science/article/abs/pii/S0168945214002192]
- 16. K48- and K63-linked ubiquitin chain interactome reveals ... [https://www.life-science-alliance.org/content/7/8/e202402740]
- 17. The K48-K63 Branched Ubiquitin Chain Regulates NF-κB ... [https://pubmed.ncbi.nlm.nih.gov/27746020/]
- 18. UBE2S drives elongation of K11-linked ubiquitin chains by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2802591/]
- 19. Modulation of K11-Linkage Formation by Variable Loop ... [https://www.sciencedirect.com/science/article/abs/pii/S0022283611002646]
- 20. RNF114 and RNF166 exemplify reader-writer E3 ligases ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12583694/]
- 21. Enhanced protein degradation by branched ubiquitin chains - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4028144/]
- 22. Unraveling Chain specific Ubiquitination in Cells Using ... [https://lifesensors.com/unraveling-chain-specific-ubiquitination-in-cells-using-tandem-ubiquitin-binding-entities-in-a-high-throughput-assay/]
- 23. Unraveling chain specific ubiquitination in cells using tandem ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12217038/]
- 24. The Molecular Toolbox for Linkage Type‐Specific Analysis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12118340/]
- 25. Strategy for Development of Site-Specific Ubiquitin ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2020.00111/full]
- 26. Protocol for Determining Ubiquitin Chain Linkage [https://www.rndsystems.com/resources/protocols/determine-ubiquitin-chain-linkage]
- 27. Emerging tools and methods to study cell signalling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12224918/]
- 28. Current methodologies in protein ubiquitination characterization [https://cellandbioscience.biomedcentral.com/articles/10.1186/s13578-022-00870-y]
- 29. Unraveling chain specific ubiquitination in cells using ... [https://www.nature.com/articles/s41598-025-07242-9]
- 30. Mass Spectrometry Proteomics using TUBEs [https://lifesensors.com/mass-spectromtry-proteomics-using-tubes/]
- 31. Super Enhanced Purification of Denatured-Refolded ... [https://www.sciencedirect.com/science/article/pii/S1535947624001427]
- 32. A High-Throughput Method for Specific, Rapid, Precise and ... [https://www.sciencedirect.com/science/article/abs/pii/S0039914025015681]
- 33. Branching via K11 and K48 Bestows Ubiquitin Chains with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6996796/]
- 34. Methods to measure ubiquitin chain length and linkage [https://www.sciencedirect.com/science/article/abs/pii/S0076687918305159]
- 35. Branched Ubiquitination: Detection Methods, Biological ... [https://www.mdpi.com/1420-3049/25/21/5200]
- 36. Targeted deubiquitination rescues distinct... | Nature Methods [https://www.nature.com/articles/s41592-020-00992-6?error=cookies_not_supported]
- 37. Decoding polyubiquitin regulation of KV7. 1 functional expression with... [https://pubmed.ncbi.nlm.nih.gov/39345403/]
- 38. Abstract Mo063: Hacking the ubiquitin code to distinctively modulate... [https://www.bohrium.com/paper-details/abstract-mo063-hacking-the-ubiquitin-code-to-distinctively-modulate-ion-channel-functional-expression/1063150405006917657-8999]
- 39. Evaluating enzyme activities and structures of DUBs - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7126802/]
- 40. An efficient system for high-level expression and easy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2286746/]
- 41. Utility of polyhistidine-tagged ubiquitin in the purification ... [https://www.sciencedirect.com/science/article/pii/S0021925820805900]
- 42. Preparation and purification of mono-ubiquitinated proteins ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7039436/]
- 43. Proteomics links ubiquitin chain topology change to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7751889/]
- 44. Article Quantitative Proteomics Reveals the Function of ... [https://www.sciencedirect.com/science/article/pii/S0092867409000890]
- 45. Data-independent acquisition method for ubiquitinome ... [https://www.nature.com/articles/s41467-020-20509-1]
- 46. Characterizing the connectivity of poly-ubiquitin chains by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3057100/]
- 47. Optimising methods for the preservation, capture and ... [https://www.sciencedirect.com/science/article/pii/S0006291X15305015]
- 48. Empirical assessment of the enrichment-based ... [https://www.nature.com/articles/s41598-024-75120-x]
- 49. The ubiquitin system: a critical regulator of innate immunity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5037286/]
- 50. How to Better Study Ubiquitination [https://www.ptglab.com/news/blog/how-to-better-study-ubiquitination/?srsltid=AfmBOoqu96R61e2dEL64L-28fWld2RARY4LtIku07vTaZbNHa-LPApA3]
- 51. Specific, sensitive and quantitative protein detection by in- ... [https://www.nature.com/articles/s41467-023-38147-8]
- 52. Targeted deubiquitination rescues distinct trafficking- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9335257/]
- 53. A genetic approach to study polyubiquitination in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6499080/]
- 54. Branching via K11 and K48 Bestows Ubiquitin Chains with ... [https://www.sciencedirect.com/science/article/pii/S0969212619303491]
- 55. Applications of protein ubiquitylation and deubiquitylation in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11087986/]
- 56. K11-linked polyubiquitination in cell cycle control revealed ... [https://pubmed.ncbi.nlm.nih.gov/20655260/]