Data-Independent Acquisition (DIA) for Ubiquitinome Analysis: A Comprehensive Guide for Advanced Proteomics
This article provides a thorough exploration of Data-Independent Acquisition (DIA) mass spectrometry and its revolutionary application in ubiquitinome analysis.
Data-Independent Acquisition (DIA) for Ubiquitinome Analysis: A Comprehensive Guide for Advanced Proteomics
Abstract
This article provides a thorough exploration of Data-Independent Acquisition (DIA) mass spectrometry and its revolutionary application in ubiquitinome analysis. It covers foundational principles, from the biological significance of ubiquitination to the technical advantages of DIA over traditional methods. Detailed methodological guidance is presented for implementing DIA ubiquitinomics workflows, including sample preparation, acquisition optimization, and data processing. The content addresses common pitfalls and provides proven optimization strategies to enhance sensitivity, reproducibility, and quantitative accuracy. Through comparative validation against established techniques like Data-Dependent Acquisition (DDA), this resource demonstrates DIA's superior performance in identifying tens of thousands of ubiquitination sites in single measurements. Aimed at proteomics researchers and drug development scientists, this guide serves as an essential reference for deploying DIA to unravel complex ubiquitin signaling in biological systems and therapeutic contexts.
Ubiquitin Signaling and DIA-MS: Fundamental Concepts and Analytical Evolution
The Biological Criticality of Ubiquitination in Cellular Regulation and Disease
Ubiquitination is a versatile and reversible post-translational modification that involves the covalent attachment of a 76-amino-acid ubiquitin protein to target substrates, thereby regulating virtually all cellular processes [1] [2]. This modification is executed through a sequential enzymatic cascade involving E1 activating enzymes, E2 conjugating enzymes, and E3 ligases, while deubiquitinating enzymes (DUBs) reverse this process [1]. The complexity of ubiquitin signaling arises from its ability to form diverse polyubiquitin chains through eight different linkage sites (M1, K6, K11, K27, K29, K33, K48, K63), which encode distinct biological functions ranging from proteasomal degradation to non-proteolytic signaling in inflammation, immune response, and circadian regulation [3] [2]. The critical importance of ubiquitination in cellular homeostasis is underscored by its dysregulation in numerous pathologies, including cancer, neurodegenerative disorders, and inflammatory diseases, making it a focal point for therapeutic development and biomarker discovery [4] [5] [1].
Quantitative Landscape of Ubiquitination in Human Diseases
The development of advanced mass spectrometry (MS) technologies, particularly data-independent acquisition (DIA) methods, has revolutionized the large-scale analysis of ubiquitin signaling, enabling comprehensive profiling of ubiquitination alterations across disease states [3]. The tables below summarize key quantitative findings from recent ubiquitinome studies in various human diseases.
Table 1: Ubiquitination-Related Biomarkers in Inflammatory and Neurological Diseases
| Disease | Key Ubiquitination-Related Genes/Proteins | Expression Change | Functional Role | Reference |
|---|---|---|---|---|
| Crohn's Disease | IFITM3, PSMB9, TAP1 | Significantly elevated | Diagnostic biomarkers; correlated with immune cell activation | [6] |
| Crohn's Disease | UBE2R2, NEDD4L | UBE2R2 increased; NEDD4L decreased | Associated with M2 macrophage infiltration; regulation of autophagy and Wnt signaling | [7] |
| Dominantly Inherited Alzheimer's Disease | Multiple UPS-activating enzymes | Subtle increases pre-onset; pronounced elevations near onset | Correlated with tau pathology and disease progression | [5] |
| Chordoma | REGγ | Upregulated | Promotes proliferation and migration via RIT1-MAPK pathway | [8] |
Table 2: Ubiquitination Chain Linkages and Their Functional Consequences
| Ubiquitin Linkage Type | Primary Functional Consequences | Key Regulatory Roles in Disease | |
|---|---|---|---|
| K48-linked | Targets substrates to 26S proteasome for degradation | Most abundant linkage; crucial for protein homeostasis | [2] |
| K63-linked | Regulates protein-protein interactions; activates kinases | NF-κB pathway activation; autophagy regulation | [2] |
| M1-linked (linear) | Controls inflammatory signaling | TNFR and TLR signaling through LUBAC complex | [1] |
| K27-linked | Atypical chain; immune regulation | NF-κB activation during viral infection | [1] |
| K11-linked | Cell cycle regulation; ER-associated degradation | Implicated in cancer progression | [2] |
Advanced Methodologies for Ubiquitinome Analysis
Data-Independent Acquisition (DIA) Mass Spectrometry
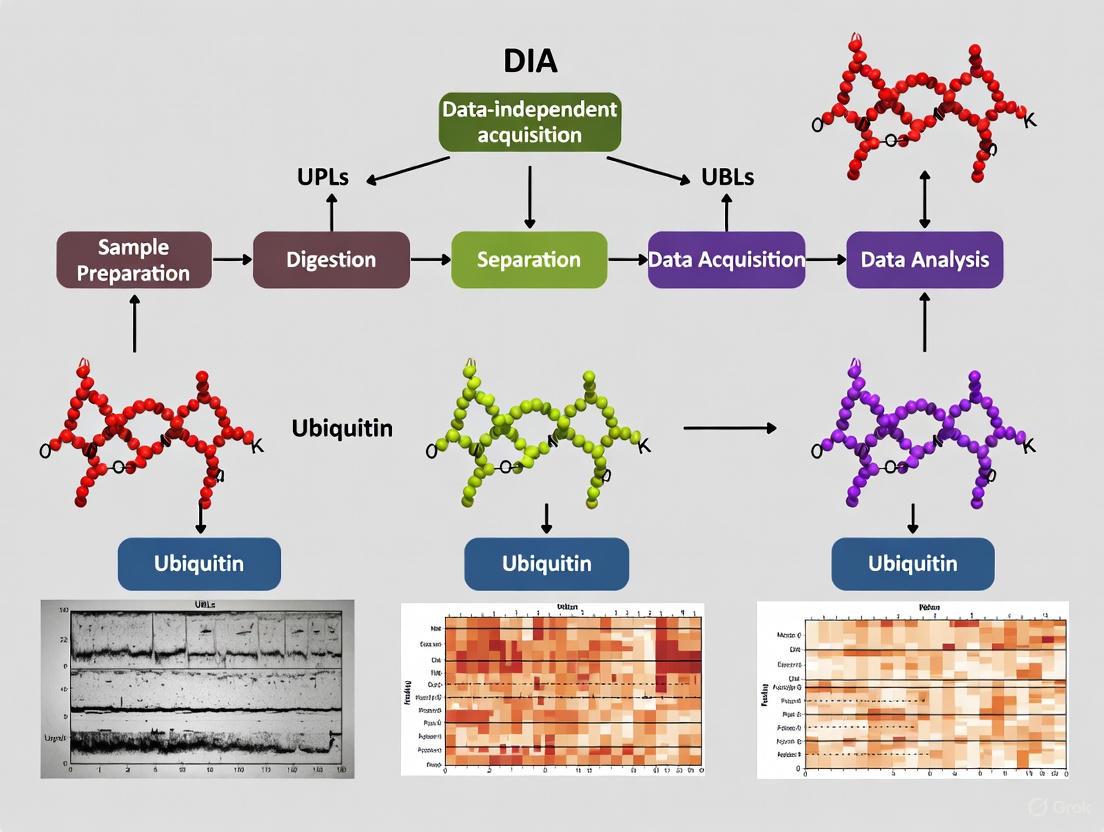
The DIA method represents a significant advancement over traditional data-dependent acquisition (DDA) for ubiquitinome analysis, offering superior sensitivity, quantitative accuracy, and data completeness [3]. The optimized DIA workflow for ubiquitinome profiling involves several critical steps:
Sample Preparation and Pre-fractionation: Proteins are extracted from cells or tissues and digested with trypsin. Basic reversed-phase (bRP) chromatography is used to separate peptides into 96 fractions, which are then concatenated into 8 fractions to reduce complexity. The highly abundant K48-linked ubiquitin-chain derived diGly peptide is processed separately to prevent competition during antibody enrichment [3].
diGly Peptide Enrichment: Peptide samples (1 mg) are incubated with anti-diGly remnant motif (K-ε-GG) antibody (31.25 μg) to specifically enrich ubiquitinated peptides. This enrichment is crucial due to the low stoichiometry of ubiquitination compared to non-modified peptides [3].
DIA Mass Spectrometry Analysis: Enriched peptides are analyzed using an Orbitrap-based DIA method with 46 precursor isolation windows and MS2 resolution of 30,000. This optimized configuration increases identified diGly peptides by 13% compared to standard full proteome methods [3].
Spectral Library Matching: A comprehensive spectral library containing >90,000 diGly peptides enables identification of approximately 35,000 distinct diGly peptides in single measurements of proteasome inhibitor-treated cells—double the number achievable with DDA methods [3] [9].
This workflow provides remarkable quantitative reproducibility, with 45% of diGly peptides exhibiting coefficients of variation (CVs) below 20% across replicates, significantly outperforming DDA methods [3].
Bioinformatics Integration and Machine Learning Approaches
Complementing experimental methods, advanced computational approaches enable the identification of ubiquitination-related biomarkers from transcriptomic data:
Differential Expression Analysis: Ubiquitination-related differentially expressed genes (UR-DEGs) are identified by intersecting differentially expressed genes from disease datasets (e.g., GEO database) with ubiquitination-related gene sets from databases such as Genecards (Relevance score >10) [7].
Machine Learning Feature Selection: LASSO algorithm with ten-fold cross-validation and Random Forest analysis are employed to identify feature genes with the highest diagnostic and prognostic value [7].
Protein-Protein Interaction (PPI) Network Construction: STRING database and Cytoscape with cytoHubba plugin are used to construct PPI networks and identify hub genes among UR-DEGs [7].
Immune Infiltration Analysis: CIBERSORT and quanTIseq algorithms calculate immune cell infiltration levels, enabling correlation analysis between key ubiquitination-related genes and specific immune cell populations [7].
Experimental Protocols for Key Ubiquitination Studies
Protocol 1: In Vitro Modeling of Ubiquitination in Inflammatory Disease
Application: Investigating ubiquitination regulation in Crohn's disease using cell models [6] [7].
Procedure:
- Cell Culture: Maintain THP-1 (human monocytic) and Caco-2 (human intestinal epithelial) cell lines in DMEM medium supplemented with 10% FBS, 100 U/mL penicillin, and 100 mg/mL streptomycin at 37°C with 5% CO₂.
- Inflammatory Stimulation: Induce inflammatory responses by treating cells with 10 ng/mL lipopolysaccharide (LPS) for 24 hours. For THP-1 cells, additional stimulation with IFN-γ enhances pro-inflammatory polarization.
- Gene Expression Validation: Extract total RNA using TRIzol reagent and synthesize cDNA using reverse transcription kits. Perform quantitative PCR with SYBR Green Master Mix using ubiquitination-related gene-specific primers (e.g., IFITM3, PSMB9, TAP1, UBE2R2, NEDD4L).
- Data Analysis: Calculate fold changes using the 2−ΔΔCt method with GAPDH as a housekeeping control.
Protocol 2: Ubiquitination Analysis in Animal Disease Models
Application: Validating ubiquitination-related biomarkers in a mouse model of Crohn's disease [7].
Procedure:
- Animal Model Establishment: Use male C57BL/6 mice (6-8 weeks old). Presensitize mice by applying 1% TNBS to their backs for one week.
- Disease Induction: After 24-hour fasting, administer 100 μL of TNBS in 50% ethanol rectally for six consecutive weeks with increasing TNBS concentrations (0.5% to 2.5%).
- Tissue Collection: Sacrifice mice seven days after the last administration by intraperitoneal injection of 100 mg/kg sodium pentobarbital. Collect distal colon tissues for analysis.
- Histological Assessment: Fix intestinal tissues in 4% paraformaldehyde, embed in paraffin, section, and perform hematoxylin and eosin (HE) staining to assess histological changes.
- Immunohistochemistry: Deparaffinize tissue sections, perform antigen retrieval, and incubate with primary antibodies against ubiquitination-related proteins (e.g., UBE2R2, NEDD4L). Visualize using appropriate secondary antibodies and chromogenic substrates.
Protocol 3: Functional Validation of Ubiquitination in Cancer Models
Application: Investigating REGγ-mediated ubiquitin-independent degradation in chordoma [8].
Procedure:
- Cell Culture: Maintain chordoma cell lines (U-CH1 and MUG-Chor1) in DMEM supplemented with 10% fetal bovine serum at 37°C with 5% CO₂.
- Gene Knockdown: Transfect cells with REGγ-specific siRNAs using appropriate transfection reagents.
- Functional Assays:
- Cell Proliferation: Seed transfected cells in 96-well plates and assess viability at days 1, 3, 5, and 7 using Cell Counting Kit-8 (CCK-8).
- Clonal Formation: Seed cells in 6-well plates, culture for 10-14 days, fix with 4% paraformaldehyde, and stain with 0.2% crystal violet.
- Migration Assay: Use Transwell chambers (8 μm) with serum-free medium in upper chambers and 10% FBS medium in lower chambers. Incubate for 48 hours, then fix, stain, and count migrated cells.
- Co-immunoprecipitation: Lyse cells in RIPA buffer, incubate lysates with specific antibodies, and pull down complexes with protein A/G agarose beads. Analyze by Western blotting to detect protein interactions.
- Pathway Analysis: Evaluate RIT1-MAPK pathway activity by Western blotting using antibodies against total and phosphorylated Erk, JNK, and p38.
Visualization of Ubiquitination Signaling Networks and Methodologies
Ubiquitination Enzymatic Cascade and Functional Outcomes
DIA Mass Spectrometry Workflow for Ubiquitinome Analysis
Table 3: Key Research Reagent Solutions for Ubiquitination Studies
| Reagent/Resource | Function/Application | Examples/Specifications | Reference |
|---|---|---|---|
| Anti-diGly Antibodies | Enrichment of ubiquitinated peptides for MS | K-ε-GG motif-specific; commercial PTMScan kits | [3] [2] |
| Linkage-Specific Ub Antibodies | Detection of specific ubiquitin chain types | M1-, K11-, K27-, K48-, K63-linkage specific | [2] |
| Tandem Ub-Binding Entities (TUBEs) | Affinity purification of ubiquitinated proteins | High-affinity capture of polyubiquitinated substrates | [2] |
| Tagged Ubiquitin Constructs | Purification of ubiquitinated substrates | His-, Strep-, HA-tagged Ub for affinity purification | [2] |
| Proteasome Inhibitors | Stabilization of ubiquitinated proteins | MG132 (10 μM, 4h treatment) | [3] |
| Spectral Libraries | DIA data analysis | Libraries containing >90,000 diGly peptides | [3] [9] |
| Cell Line Models | Disease modeling | THP-1, Caco-2, U-CH1, MUG-Chor1 | [6] [7] [8] |
| Animal Disease Models | In vivo validation | TNBS-induced colitis, xenograft models | [7] [8] |
The biological criticality of ubiquitination in cellular regulation and disease is increasingly evident through advanced proteomic technologies that reveal the remarkable complexity and disease-specific alterations of ubiquitin signaling. The development of DIA-based ubiquitinome analysis represents a transformative methodology that provides unprecedented depth and quantitative accuracy in profiling ubiquitination events, enabling the identification of novel diagnostic biomarkers and therapeutic targets across diverse pathologies [3]. The integration of these proteomic advances with biochemical, cellular, and animal model validation creates a powerful framework for elucidating the mechanistic roles of ubiquitination in disease pathogenesis and for developing targeted therapeutic strategies. As these methodologies continue to evolve, they promise to unlock new dimensions of ubiquitin biology, further illuminating its critical functions in cellular regulation and its potential as a therapeutic target in human disease.
Historical Limitations of Data-Dependent Acquisition (DDA) in Ubiquitinome Profiling
Ubiquitinome profiling, the large-scale study of protein ubiquitination, presents significant challenges for mass spectrometry (MS)-based proteomics due to the low stoichiometry of this modification and the dynamic nature of ubiquitin signaling [10]. For years, data-dependent acquisition (DDA) has served as the cornerstone method for ubiquitinome analysis, yet its inherent limitations have constrained the depth and reliability of biological insights [10]. The transition to data-independent acquisition (DIA) methodologies addresses these constraints by offering improved quantitative accuracy and data completeness [10] [11]. This document delineates the historical limitations of DDA within ubiquitinome research and provides detailed protocols for implementing advanced DIA workflows, contextualized within broader thesis research on DIA methods for ubiquitinome analysis.
Critical Analysis of DDA Limitations in Ubiquitinome Profiling
Fundamental Technical Constraints
The application of DDA to ubiquitinome studies has been hampered by several technical shortcomings that directly impact data quality and biological interpretability [10].
- Stochastic Precursor Selection: DDA's intensity-based selection mechanism preferentially samples abundant ions, systematically undersampling low-abundance ubiquitinated peptides that are of significant biological interest [10] [12].
- Incomplete Data Acquisition: The cyclic nature of DDA, where the instrument selects a limited number of top-intensity precursors for fragmentation, results in missing values across sample runs, complicating quantitative comparisons [11].
- Dynamic Exclusion Limitations: While dynamic exclusion aims to increase proteome coverage by preventing repeated sequencing of the same ions, it often inadvertently misses lower-abundance peptides that co-elute with highly abundant species [12].
Practical Consequences for Ubiquitinome Research
Table 1: Quantitative Comparison of DDA vs. DIA Performance in Ubiquitinome Analysis
| Performance Metric | DDA Method Performance | DIA Method Performance | Improvement Factor |
|---|---|---|---|
| diGly Peptide Identifications | ~17,500 sites (single run) [10] | ~35,000 sites (single run) [10] | 2.0x |
| Quantitative Accuracy | Lower, higher missing values [10] | Superior, fewer missing values [10] | Significant |
| Reproducibility | Moderate CVs [10] | 45% of peptides with CV < 20% [10] | Substantial |
| Coverage of Low-Abundance Peptides | Limited due to ion selection bias [12] | Comprehensive, unbiased detection [12] | Dramatic |
These limitations manifested in specific practical constraints that hampered ubiquitinome research:
- Throughput vs. Depth Dilemma: Achieving comprehensive ubiquitinome coverage with DDA required extensive peptide fractionation (often 8-96 fractions), large sample amounts (5-10 mg protein input), and lengthy instrument time, severely limiting analytical throughput [10].
- Reduced Quantitative Accuracy: The stochastic nature of DDA data acquisition led to higher rates of missing values and greater quantitative variance, particularly problematic for capturing dynamic ubiquitination changes in signaling pathways like TNFα and circadian regulation [10].
- Incomplete Biological Insight: DDA-based methods potentially missed crucial ubiquitination events on low-abundance regulatory proteins and transporters, leaving significant gaps in systems-wide understanding of ubiquitin signaling networks [10].
Experimental Protocols for DIA-Based Ubiquitinome Analysis
Comprehensive Spectral Library Generation
The implementation of DIA ubiquitinome profiling requires high-quality, comprehensive spectral libraries to achieve optimal performance [10].
Table 2: Key Research Reagent Solutions for DIA Ubiquitinome Profiling
| Reagent/Material | Specification | Function in Workflow |
|---|---|---|
| Anti-diGly Antibody | Ubiquitin Remnant Motif (K-ε-GG) Kit [10] | Immunoaffinity enrichment of ubiquitinated peptides |
| Cell Lines | HEK293, U2OS (with/without proteasome inhibition) [10] | Source of biological material for library generation |
| Proteasome Inhibitor | MG132 (10 µM, 4h treatment) [10] | Enhances ubiquitinated peptide abundance |
| Chromatography Resin | Basic reversed-phase (bRP) material [10] | High-pH fractionation for library depth |
| Digestion Enzyme | Trypsin/Lys-C [10] | Protein digestion generating diGly remnant |
Step-by-Step Protocol:
- Cell Culture and Treatment: Grow HEK293 or U2OS cells to 80% confluency. Treat with 10 µM MG132 proteasome inhibitor for 4 hours to stabilize ubiquitinated substrates [10].
- Protein Extraction and Digestion: Lyse cells in urea-based buffer (8M urea, 100 mM ammonium bicarbonate). Reduce with dithiothreitol (5 mM, 30 min, 25°C), alkylate with iodoacetamide (15 mM, 20 min, 25°C in dark), and digest with trypsin/Lys-C (1:50 w/w, 37°C, overnight) [10].
- Peptide Fractionation: Separate digested peptides using basic reversed-phase chromatography (bRP) into 96 fractions. Concatenate into 8 pooled fractions. Critical step: Isolate fractions containing abundant K48-linked ubiquitin-chain derived diGly peptides separately to prevent competition during enrichment [10].
- diGly Peptide Enrichment: Enrich pooled fractions using anti-diGly antibody (31.25 µg antibody per 1 mg peptide input). Incubate with rotation for 2 hours at 4°C. Wash beads extensively before elution [10].
- Library Acquisition: Analyze enriched fractions using DDA MS on an Orbitrap instrument with 2-hour gradients. Process data using standard DDA search engines (MaxQuant, Spectronaut) to generate spectral libraries [10].
Optimized DIA Acquisition Method
The following protocol details the optimized DIA acquisition parameters specifically tailored for ubiquitinome analysis [10]:
- Instrument Configuration: Use an Orbitrap-based mass spectrometer equipped with a nanoflow LC system.
- Chromatographic Separation: Load samples onto a C18 analytical column (75 µm × 25 cm) with a 90-minute gradient from 2% to 30% acetonitrile in 0.1% formic acid.
- DIA Method Parameters:
- Precursor Range: 400-1000 m/z
- Window Scheme: 46 variable windows optimized for diGly peptide distribution
- MS1 Resolution: 120,000
- MS2 Resolution: 30,000
- Collision Energy: Stepped (22, 27, 32 eV)
- Cycle Time: ~3 seconds
This optimized window scheme increases diGly peptide identifications by 13% compared to standard full proteome DIA methods [10].
Data Processing and Analysis Workflow
- Spectral Library Construction: Compile DDA-identified diGly peptides into a comprehensive library using tools like Spectronaut or Skyline.
- DIA Data Extraction: Process DIA files against the spectral library using specialized software (OpenSWATH, DIA-NN, or Spectronaut).
- Quality Control: Apply stringent filters including <1% FDR at peptide and protein levels.
- Quantitative Analysis: Normalize data and perform statistical analysis to identify significantly regulated ubiquitination sites.
DIA Ubiquitinome Workflow Diagram
Biological Applications and Validation
The transition to DIA-based ubiquitinome analysis has enabled previously unattainable biological discoveries across multiple research domains.
Signaling Pathway Analysis
Application of the DIA workflow to TNFα signaling comprehensively captured known ubiquitination sites while adding many novel sites, providing unprecedented insight into this biologically and therapeutically important pathway [10]. The method's improved quantitative accuracy enabled precise tracking of dynamic ubiquitination changes in response to pathway activation.
Circadian Ubiquitinome Dynamics
An in-depth, systems-wide investigation of ubiquitination across the circadian cycle uncovered hundreds of cycling ubiquitination sites and dozens of cycling ubiquitin clusters within individual membrane protein receptors and transporters [10]. This revealed new connections between metabolism and circadian regulation that were previously obscured by DDA's limitations in capturing low-abundance regulatory events.
Targeted Protein Degradation
Recent work has demonstrated the power of DIA ubiquitinome profiling for characterizing targeted protein degradation (TPD) mechanisms, identifying over 40,000 diGly precursors corresponding to more than 7,000 proteins in single measurements from cells exposed to proteasome inhibitors [13]. This application highlights the method's potential for rapidly establishing the mode of action for various TPD modalities, including PROTACs and molecular glues [13].
The historical limitations of DDA in ubiquitinome profiling—including stochastic sampling, missing values, and quantitative inconsistency—have constrained our understanding of ubiquitin signaling networks [10]. The optimized DIA workflow detailed herein, combining comprehensive spectral libraries with tailored acquisition parameters, effectively addresses these limitations by doubling ubiquitination site identifications while significantly improving quantitative accuracy and reproducibility [10]. This methodological advancement enables researchers to explore dynamic ubiquitination events in biological systems with unprecedented depth and confidence, particularly in complex applications such as signaling pathway analysis, circadian biology, and targeted protein degradation research [10] [13]. As DIA methodologies continue to evolve alongside advances in mass spectrometry instrumentation and computational tools, they promise to further accelerate discoveries in ubiquitin biology and therapeutic development.
Data-Independent Acquisition (DIA) represents a paradigm shift in mass spectrometry-based proteomics, particularly for the specialized analysis of protein ubiquitination. Unlike traditional methods that selectively target specific ions, DIA systematically fragments all analyte ions within a sample across predefined mass-to-charge (m/z) ranges, ensuring comprehensive detection of all detectable analytes regardless of abundance [14]. This fundamental difference in acquisition strategy addresses long-standing limitations in ubiquitinome research, where the low stoichiometry of ubiquitination and varying ubiquitin-chain topologies have historically challenged comprehensive profiling [3]. The application of DIA to ubiquitinomics has enabled researchers to overcome traditional barriers of sensitivity, reproducibility, and quantitative accuracy, ushering in a new era for investigating ubiquitin signaling at a systems-wide scale [15] [3].
Within the ubiquitinome analysis landscape, DIA has proven particularly valuable because it generates permanent digital proteome maps that allow highly reproducible retrospective analysis of cellular and tissue specimens [16]. This capability is crucial for capturing the dynamic nature of ubiquitin signaling, which regulates virtually all cellular processes through a complex conjugation cascade involving ubiquitin-activating (E1), conjugating (E2), and ligating (E3) enzymes, with reversal mediated by deubiquitinating enzymes (DUBs) [3]. The development of specialized DIA workflows for ubiquitinome profiling has enabled unprecedented insights into circadian biology, TNF signaling, and the mode of action of deubiquitinase inhibitors, demonstrating how this technology is driving discovery in both basic research and drug development [15] [3] [9].
Fundamental Principles of DIA Mass Spectrometry
Core Technological Framework
The operational principle of DIA mass spectrometry centers on its unbiased approach to data acquisition. While traditional Data-Dependent Acquisition (DDA) performs real-time selection of the most abundant precursor ions for fragmentation based on intensity, DIA eliminates this selection bias by cyclically fragmenting all precursor ions within consecutive isolation windows that span the entire m/z range of interest [16]. This fundamental difference ensures that both high-abundance and low-abundance peptides are systematically fragmented and recorded, creating a complete digital record of the sample's peptide composition [14].
The DIA process involves dividing the full m/z range (typically 400-1000 m/z) into multiple predefined isolation windows. The mass spectrometer then cycles through these windows, isolating and fragmenting all precursor ions within each window using collision-induced dissociation. The resulting fragment ions are recorded as complex, multiplexed spectra that contain information from all co-eluting peptides within each window [16]. Although these spectra are highly complex, advances in computational proteomics, particularly deep neural network-based data processing algorithms like DIA-NN, have enabled effective deconvolution and extraction of peptide-specific information from these complex datasets [15]. This comprehensive acquisition strategy ensures that no peptide information is lost due to stochastic sampling, making DIA particularly suited for the analysis of post-translational modifications where low stoichiometry is a significant challenge [15] [3].
Comparison with Data-Dependent Acquisition (DDA)
Table 1: Fundamental Differences Between DIA and DDA Acquisition Methods
| Parameter | Data-Independent Acquisition (DIA) | Data-Dependent Acquisition (DDA) |
|---|---|---|
| Acquisition Strategy | Fragments all ions in predefined m/z windows without precursor selection | Selectively fragments most intense precursors based on abundance |
| Quantitative Reproducibility | Exceptional reproducibility with <2% missing values across samples | Moderate reproducibility with up to 51% missing values across samples |
| Dynamic Range | 4-5 orders of magnitude with LOD ~100 amol | Limited by precursor selection bias toward abundant ions |
| Data Completeness | High data completeness across multiple samples | Significant missing values in large sample series |
| Stochastic Sampling | Eliminated through systematic fragmentation | Inherent due to intensity-based precursor selection |
| Best Applications | Large-scale studies requiring high quantitative precision, PTM analysis | Targeted studies, PTM analysis with extensive fractionation |
The comparative advantages of DIA become particularly evident in large-scale studies where reproducibility across multiple samples is crucial. In a direct comparison analyzing 24 samples, DIA resulted in only 1.6% missing values across all samples compared to 51% missing values in DDA [16]. This remarkable difference stems from DIA's systematic fragmentation of all detectable ions in every run, eliminating the stochastic sampling problem inherent to DDA's intensity-based precursor selection [15] [16]. Furthermore, DIA demonstrates superior dynamic range, with a limit of detection of approximately 100 amol and quantification spanning 4-5 orders of magnitude [16], making it particularly suitable for detecting low-abundance ubiquitinated peptides that would typically be missed by DDA due to their low stoichiometry relative to their unmodified counterparts [3].
DIA Overcoming Traditional Barriers in Ubiquitinome Research
Enhanced Sensitivity and Coverage in Ubiquitinated Peptide Detection
The application of DIA to ubiquitinome analysis has demonstrated remarkable improvements in detection sensitivity and coverage. In a landmark study by Steger et al., DIA more than tripled the identification numbers of ubiquitinated peptides compared to DDA, quantifying 68,429 K-ε-GG remnant peptides in single MS runs versus 21,434 with DDA [15]. Similarly, Hansen et al. reported the identification of 35,000 diGly peptides in single measurements of proteasome inhibitor-treated cells—double the number achievable with DDA [3]. This enhanced coverage is particularly valuable for ubiquitinomics, where the depth of analysis directly impacts the biological insights that can be derived from the data.
The improved sensitivity of DIA enables detection of low-abundance ubiquitination events that play crucial regulatory roles despite their low stoichiometry. Furthermore, DIA's comprehensive data acquisition allows researchers to detect and differentiate between isobaric peptides—those with the same m/z but different sequences—by simultaneously fragmenting multiple precursor ions and using their fragment ions for distinction [14]. This capability is especially important in ubiquitinome research due to the complexity of ubiquitin signaling and the presence of different ubiquitin chain linkages that can dramatically alter functional outcomes [15] [3].
Superior Quantitative Accuracy and Reproducibility
The quantitative performance of DIA represents one of its most significant advantages for ubiquitinome studies. In benchmark comparisons, DIA demonstrates excellent quantitative precision with median coefficients of variation (CVs) for quantified K-ε-GG peptides of approximately 10%, significantly lower than the CVs typically observed in DDA experiments [15]. Hansen et al. reported that 45% of diGly peptides identified by DIA had CVs below 20%, compared to only 15% with DDA [3]. This enhanced reproducibility is critical for time-course experiments and drug treatment studies where precise quantification of ubiquitination dynamics is essential for drawing meaningful biological conclusions.
The remarkable reproducibility of DIA stems from its elimination of stochastic precursor selection. While DDA must select which peptides to fragment based on intensity, leading to run-to-run variability, DIA fragments all peptides in every run, ensuring consistent data acquisition across multiple samples [16]. This consistency is particularly valuable in clinical research and drug development applications, where reliable quantification across sample cohorts is necessary for robust biomarker discovery and therapeutic target validation [11] [16].
Experimental Workflow for DIA-Based Ubiquitinome Analysis
Diagram 1: DIA ubiquitinome workflow from sample to data.
A robust DIA-based ubiquitinome profiling workflow involves several critical steps, each optimized for maximum recovery and detection of ubiquitinated peptides. The process begins with protein extraction using sodium deoxycholate (SDC)-based lysis buffer supplemented with chloroacetamide (CAA) for immediate cysteine protease inactivation [15]. Compared to traditional urea-based buffers, SDC lysis increases K-ε-GG peptide identification by approximately 38% while maintaining high enrichment specificity [15]. Following protein extraction and digestion, ubiquitinated peptides are enriched using antibodies specific for the diGly remnant motif (K-ε-GG) generated by tryptic cleavage of ubiquitinated proteins [3]. Optimization experiments indicate that enrichment from 1 mg of peptide material using 31.25 μg of anti-diGly antibody provides optimal results for most applications [3].
For DIA-MS analysis, specialized acquisition methods have been developed to account for the unique characteristics of diGly-modified peptides. These methods typically employ 46 precursor isolation windows with fragment scan resolution of 30,000 to balance data quality with acquisition cycle time [3]. The resulting complex fragment ion spectra are processed using specialized software such as DIA-NN, which incorporates additional scoring modules for confident identification of modified peptides [15]. This integrated workflow enables comprehensive ubiquitinome profiling with unprecedented depth and quantitative accuracy, making it possible to investigate ubiquitin signaling dynamics at a systems level.
Key Research Reagent Solutions for DIA Ubiquitinomics
Table 2: Essential Research Reagents for DIA-Based Ubiquitinome Analysis
| Reagent/Resource | Function in Workflow | Specification Notes |
|---|---|---|
| SDC Lysis Buffer | Protein extraction with immediate protease inactivation | Supplemented with chloroacetamide (CAA) to prevent di-carbamidomethylation artifacts [15] |
| Anti-diGly Antibody | Immunoaffinity enrichment of ubiquitinated peptides | Specific for K-ε-GG remnant motif; 31.25 μg per 1 mg peptide input recommended [3] |
| Trypsin/Lys-C Mix | Protein digestion generating diGly remnant | Creates K-ε-GG signature on previously ubiquitinated lysines [3] |
| DIA-NN Software | Deep neural network-based data processing | Specialized scoring module for K-ε-GG peptide identification [15] |
| Spectral Libraries | Peptide identification and quantification | Project-specific or comprehensive public libraries (>90,000 diGly peptides) [3] |
| Proteasome Inhibitors | Enhance ubiquitinated peptide detection | MG-132 treatment (10 μM, 4 hours) increases ubiquitin signal [3] |
The effectiveness of DIA ubiquitinome profiling depends heavily on the optimization of key reagents and resources. The SDC-based lysis buffer represents a significant improvement over traditional urea-based methods, providing not only increased peptide identifications but also better reproducibility [15]. The anti-diGly antibody is crucial for specific enrichment of ubiquitinated peptides while minimizing background, with titration experiments establishing optimal antibody-to-peptide ratios for different sample types [3]. Comprehensive spectral libraries serve as essential resources for peptide identification, with recent studies generating libraries containing more than 90,000 diGly peptides to support sensitive and accurate analysis [3]. The DIA-NN software package, with its specialized algorithms for modified peptide identification, enables researchers to fully leverage the complex data generated by DIA acquisitions [15].
Applications and Biological Insights Enabled by DIA in Ubiquitinomics
Mapping Deubiquitinase Targets and Mechanisms
The application of DIA ubiquitinomics has yielded significant insights into the function and targets of deubiquitinases (DUBs), particularly in the context of drug discovery. In a comprehensive study profiling the oncology target USP7, researchers employed DIA-MS to simultaneously monitor ubiquitination changes and protein abundance at high temporal resolution following USP7 inhibition [15]. This approach revealed that while ubiquitination of hundreds of proteins increased within minutes of USP7 inhibition, only a small fraction of these were subsequently degraded, effectively distinguishing degradative from non-degradative ubiquitination events [15]. This finding has important implications for understanding the mechanism of DUB-targeted therapies and demonstrates how DIA ubiquitinomics can provide unique insights into ubiquitin signaling dynamics that would be difficult to obtain with traditional methods.
The ability of DIA to capture rapid changes in ubiquitination status makes it particularly valuable for studying the immediate effects of DUB inhibition. By combining ubiquitinome and proteome profiling, researchers can not only identify putative DUB substrates with high confidence but also determine the functional consequences of their ubiquitination [15]. This integrated approach represents a powerful strategy for rapid mode-of-action profiling of candidate drugs targeting DUBs or ubiquitin ligases, providing critical information for lead optimization and target validation in drug development pipelines [15].
Unveiling Circadian Regulation of the Ubiquitinome
DIA-based ubiquitinomics has revealed unexpected complexity in the circadian regulation of biological processes. In an in-depth, systems-wide investigation of ubiquitination across the circadian cycle, researchers discovered hundreds of cycling ubiquitination sites and dozens of cycling ubiquitin clusters within individual membrane protein receptors and transporters [3] [9]. These findings highlight new connections between metabolism and circadian regulation, suggesting that rhythmic ubiquitination may serve as an important regulatory layer controlling the activity and turnover of key metabolic proteins in accordance with circadian rhythms.
The discovery of clustered ubiquitination sites with the same circadian phase on individual proteins points to previously unappreciated mechanisms of regulation [3]. The comprehensive coverage afforded by DIA was essential for detecting these coordinated ubiquitination events, which might have been missed with less sensitive methods. This application demonstrates how DIA ubiquitinomics can uncover novel biological insights by providing a more complete picture of the ubiquitinome's dynamics, particularly for low-abundance regulatory events that would escape detection with traditional approaches.
Data-Independent Acquisition mass spectrometry represents a transformative advancement for ubiquitinome research, effectively overcoming the traditional barriers of limited coverage, poor reproducibility, and quantitative inaccuracy that have hampered previous approaches. By systematically fragmenting and recording all ions within predefined m/z ranges, DIA provides a comprehensive and unbiased view of the ubiquitinome, enabling researchers to detect low-abundance regulatory ubiquitination events with high precision and confidence. The development of specialized workflows combining optimized sample preparation, tailored acquisition methods, and advanced computational processing has established DIA as the method of choice for large-scale ubiquitin signaling studies.
As mass spectrometry technology continues to evolve, with innovations such as trapped ion mobility spectrometry (TIMS) and high-field asymmetric waveform ion mobility spectrometry (FAIMS) becoming more widespread, the capabilities of DIA for ubiquitinome analysis are expected to expand further [11]. These developments, coupled with ongoing improvements in computational tools and spectral library resources, will make DIA increasingly accessible and powerful for both basic research and drug discovery applications. For researchers investigating the complex landscape of ubiquitin signaling, DIA offers an unparalleled ability to capture the dynamics and scope of this essential regulatory system, paving the way for new discoveries in cell biology, disease mechanisms, and therapeutic development.
Protein ubiquitination is a fundamental post-translational modification (PTM) that regulates virtually every cellular process, from protein degradation and signal transduction to DNA repair and cell cycle progression [17] [2]. The versatility of ubiquitin signaling arises from its ability to form diverse chain architectures and modify substrate proteins at specific lysine residues. For decades, characterizing the precise sites of ubiquitination remained a formidable challenge due to the low stoichiometry of modified proteins and the complexity of the ubiquitin code.
A transformative advance in the field came with the widespread adoption of the diGly remnant as a signature for ubiquitination. Upon tryptic digestion of ubiquitinated proteins, the C-terminal two glycine residues of ubiquitin remain covalently attached to the ε-amino group of the modified lysine, creating a lysine-ε-glycyl-glycine (K-ε-GG or diGLY) remnant [17] [18]. This characteristic ~114 Da mass shift serves as a detectable "footprint" of ubiquitination. The development of highly specific antibodies recognizing this diGLY motif enabled efficient enrichment of these modified peptides from complex proteomic digests, revolutionizing ubiquitinome analysis by mass spectrometry (MS) [17] [18] [19].
This application note details the methodology of diGLY remnant-based enrichment, framed within contemporary research that leverages Data-Independent Acquisition (DIA) mass spectrometry to achieve unprecedented depth and quantitative accuracy in ubiquitinome analysis [3].
The diGLY Signature: Mechanism and Specificity
Biochemical Origin of the diGLY Remnant
The diGLY signature is a direct product of sample preparation for bottom-up proteomics. Trypsin, a serine protease that cleaves peptide bonds C-terminal to lysine and arginine residues, processes ubiquitin-conjugated proteins. When trypsin encounters a ubiquitin-modified lysine on a substrate protein, it cannot cleave at the modified residue. Instead, it cleaves after the two C-terminal glycine residues (G75-G76) of ubiquitin, leaving a di-glycine moiety attached via an isopeptide bond to the formerly modified lysine [17] [18]. This results in a peptide with an internal lysine residue bearing the diGLY modification, which introduces a characteristic ~114.0429 Da mass shift detectable by modern high-resolution mass spectrometers.
Specificity for Ubiquitin and Ubiquitin-like Modifiers
A critical consideration when interpreting diGLY data is specificity. The diGLY remnant is not absolutely unique to ubiquitin. The ubiquitin-like modifiers NEDD8 and ISG15 also generate an identical tryptic remnant on modified lysines [17] [18] [2]. However, empirical data from multiple studies has demonstrated that in most cellular contexts, the vast majority (>94-95%) of enriched K-ε-GG peptides originate from bona fide ubiquitination events, with NEDDylation and ISG15ylation contributing minimally to the signal [17] [18]. For studies requiring absolute distinction, alternative proteases like LysC can be employed, which generates a longer remnant that can differentiate between these modifications [3] [2].
Quantitative Advances in diGLY Proteomics with Data-Independent Acquisition
Traditional ubiquitinome studies have relied on Data-Dependent Acquisition (DDA), which selectively fragments the most abundant precursor ions. While powerful, this approach suffers from stochastic sampling, leading to missing values across samples and limited quantitative reproducibility [3]. Data-Independent Acquisition (DIA) has emerged as a superior alternative for PTM analysis, as it systematically fragments all ions within sequential isolation windows, capturing a complete digital map of the sample.
Performance Comparison: DIA vs. DDA in diGLY Proteomics
A landmark study systematically compared DIA and DDA for diGLY proteomics, creating extensive spectral libraries containing over 90,000 diGLY peptides to support the DIA analysis [3]. The results demonstrate a clear advantage for the DIA methodology.
Table 1: Quantitative Performance of DIA versus DDA in diGLY Proteomics
| Metric | Data-Independent Acquisition (DIA) | Data-Dependent Acquisition (DDA) |
|---|---|---|
| diGLY Peptides Identified (Single Shot) | ~35,000 | ~20,000 |
| Coefficient of Variation (CV) < 20% | 45% of peptides | 15% of peptides |
| Quantitative Accuracy | Markedly improved | Lower |
| Data Completeness | High; fewer missing values | Lower; more missing values |
| Required Spectral Library | Yes (comprehensive) | No |
The implementation of an optimized DIA workflow, which included tailored isolation window schemes and high MS2 resolution, enabled the identification of approximately 35,000 distinct diGLY sites in a single measurement of proteasome-inhibited cells—nearly double the number achievable with DDA [3]. Furthermore, quantitative accuracy and reproducibility were significantly enhanced, with 45% of diGLY peptides exhibiting a coefficient of variation (CV) below 20% across replicates, compared to only 15% with DDA [3]. This robust performance makes DIA the method of choice for large-scale, quantitative studies of ubiquitin signaling dynamics.
Visualizing the Advanced DIA diGLY Workflow
The following diagram illustrates the optimized end-to-end workflow for deep ubiquitinome analysis using diGLY enrichment and DIA mass spectrometry.
Detailed Protocol for diGLY Enrichment and Ubiquitinome Analysis
This section provides a detailed methodology for the enrichment of diGLY-modified peptides and their analysis by mass spectrometry, incorporating best practices for achieving high coverage and quantitative reliability.
Sample Preparation and Lysis
The goal of this initial phase is to extract proteins while preserving ubiquitination states and preventing post-lysis deubiquitination.
Cell Culture and Lysis:
- Culture cells in appropriate media. For quantitative SILAC experiments, use "light" (L-lysine/L-arginine) and "heavy" (13C6,15N2 L-lysine/13C6,15N4 L-arginine) media for at least six cell doublings to ensure complete labeling [17] [19].
- Lyse cells directly in a denaturing lysis buffer. A typical formulation is: 8 M Urea, 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1 mM EDTA, supplemented with protease and deubiquitinase inhibitors [17] [18].
- Critical: Include 5-10 mM N-Ethylmaleimide (NEM) or other deubiquitinase (DUB) inhibitors in the lysis buffer to prevent the removal of ubiquitin from substrates by endogenous DUBs during sample processing [17]. Some protocols also recommend phosphatases (e.g., NaF, β-Glycerophosphate) to inhibit phosphorylation-dependent signaling [17].
Protein Digestion:
- Reduce disulfide bonds with 5 mM dithiothreitol (DTT) and alkylate cysteine residues with 10-20 mM iodoacetamide or chloroacetamide [17] [18] [19].
- Perform a two-step digestion. First, use Lys-C (which is active in high urea concentrations) at a 1:100-1:200 (w/w) enzyme-to-protein ratio for 2-4 hours. Then, dilute the urea concentration to <2 M and digest overnight with trypsin (1:50 ratio) at room temperature or 30°C [17] [18].
Peptide Pre-Fractionation and Desalting
To reduce sample complexity and increase the depth of analysis, peptide fractionation prior to diGLY enrichment is highly recommended.
- Basic pH Reversed-Phase (bRP) Chromatography: Separate the digested peptides using a C18 column and a gradient of increasing acetonitrile (e.g., 7%, 13.5%, 50%) in a volatile basic buffer like 5-10 mM ammonium formate or ammonium hydroxide (pH 10) [18] [19]. Pooling or concatenating many fractions into a smaller number (e.g., 8-12) is an effective strategy to reduce analytical time while maintaining depth [3] [18].
- Desalting: After fractionation, desalt the pools using C18 solid-phase extraction (SPE) cartridges or StageTips. Use solvents like 0.1% Trifluoroacetic Acid (TFA) for washing and 50% Acetonitrile/0.5% Acetic Acid for elution [17] [18]. Lyophilize the samples to completeness before the enrichment step.
Immunoaffinity Enrichment (IAE) of diGLY Peptides
This is the core step for selectively isolating K-ε-GG-containing peptides.
- Antibody Bead Preparation: Use the commercial PTMScan Ubiquitin Remnant Motif (K-ε-GG) Kit or equivalent antibodies. For increased specificity, cross-link the antibody to Protein A/G beads using dimethyl pimelimidate (DMP) to prevent antibody leaching and co-elution of IgG peptides, which can suppress MS signal [18].
- Enrichment Reaction: Resuspend the dried peptide fractions in IAP buffer (50 mM MOPS/NaOH, pH 7.2, 10 mM Na2HPO4, 50 mM NaCl). Incubate the peptide sample with the cross-linked antibody beads for 1.5-2 hours at 4°C with gentle agitation [18].
- Washing and Elution: After incubation, wash the beads thoroughly with IAP buffer followed by HPLC-grade water to remove non-specifically bound peptides. Elute the bound diGLY peptides with 50-100 µL of 0.15% TFA [18] [19].
- Cleanup and MS Analysis: Desalt the eluted peptides using C18 StageTips or micro-columns. Elute into an LC-MS vial, concentrate by vacuum centrifugation, and reconstitute in a small volume (e.g., 10-15 µL) of 0.5% acetic acid or 2% acetonitrile/0.1% formic acid for MS injection [18].
Mass Spectrometric Data Acquisition and Analysis
- Data Acquisition: Analyze the enriched diGLY peptides using nanoflow LC coupled to a high-resolution mass spectrometer (e.g., Orbitrap). For the deepest and most quantitative results, employ a DIA method with optimized window placements and high MS2 resolution, as previously described [3].
- Data Analysis: Process the raw DIA data using software tools (e.g., Spectronaut, DIA-NN, Skyline) against a comprehensive spectral library of diGLY peptides. For the most complete analysis, use a project-specific library or a hybrid library generated from DDA and direct-DIA searches [3].
The Scientist's Toolkit: Essential Reagents for diGLY Proteomics
Table 2: Key Research Reagent Solutions for diGLY Proteomics
| Reagent / Kit | Function / Application | Key Features |
|---|---|---|
| PTMScan Ubiquitin Remnant Motif (K-ε-GG) Kit [18] | Immunoaffinity enrichment of diGLY peptides from complex digests. | Highly specific monoclonal antibody; cross-linking protocol available; enables site-specific identification. |
| SILAC Amino Acids (Light & Heavy) [17] | Metabolic labeling for relative quantification of ubiquitination changes between conditions. | 13C6,15N2 L-lysine & 13C6,15N4 L-arginine; allows precise mixing of experimental conditions early in workflow. |
| N-Ethylmaleimide (NEM) [17] | Deubiquitinase (DUB) inhibitor. | Irreversibly alkylates cysteine residues; critical for preserving ubiquitin conjugates during lysis. |
| LysC & Trypsin Proteases [17] [18] | Enzymatic protein digestion for bottom-up proteomics. | High-purity, sequencing grade; two-step digestion (LysC then trypsin) increases efficiency and completeness. |
| Basic pH Reversed-Phase Resins [18] [19] | Offline peptide fractionation prior to enrichment. | Polymeric C18 material, 300 Å pore size; reduces sample complexity and increases depth of coverage. |
| diGLY Spectral Libraries [3] | Resource for DIA data analysis and peptide identification. | Publicly available libraries contain >90,000 diGLY sites; essential for confident identification in DIA workflows. |
The recognition and application of the diGLY remnant as a key signature have been instrumental in transforming ubiquitin proteomics from a specialized challenge into a routine, high-throughput methodology. The detailed protocols and reagent tools outlined here provide a robust framework for conducting deep ubiquitinome analyses. The integration of this powerful enrichment strategy with Data-Independent Acquisition mass spectrometry represents the current state-of-the-art, offering unparalleled sensitivity, reproducibility, and quantitative accuracy. This combined approach empowers researchers to investigate the dynamics of ubiquitin signaling across diverse biological systems and disease states with confidence, paving the way for novel discoveries in cell biology and drug development.
Ubiquitination, the covalent attachment of a small regulatory protein to lysine residues on substrate proteins, is a cornerstone of cellular regulation, influencing virtually all biological processes from protein degradation to signal transduction and circadian biology [3] [20]. For years, the comprehensive study of the "ubiquitinome"—the full set of protein ubiquitination events in a cell—has been a significant challenge in proteomics. The low stoichiometry of the modification and the complexity of ubiquitin chain architectures necessitated extensive sample fractionation, compromising throughput, robustness, and quantitative accuracy [3].
The advent of Data-Independent Acquisition (DIA) mass spectrometry is now catalyzing a paradigm shift in ubiquitinome analysis. Unlike traditional Data-Dependent Acquisition (DDA), which selectively fragments the most intense ions, DIA systematically fragments all ions within predefined isolation windows, leading to more complete data sets with fewer missing values [3]. This application note details how optimized DIA-based workflows are enabling researchers to transcend previous limitations, routinely identifying tens of thousands of ubiquitination sites in single experiments and providing unprecedented insights into dynamic cellular signaling.
Breakthroughs in DIA-Based Ubiquitinome Analysis
Key Advantages of DIA over DDA for Ubiquitinomics
Recent studies consistently demonstrate the superior performance of DIA for large-scale ubiquitinome profiling. The core advantages are profound increases in depth, reproducibility, and quantitative precision.
Table 1: Quantitative Comparison of DDA and DIA Ubiquitinome Performance
| Performance Metric | Data-Dependent Acquisition (DDA) | Data-Independent Acquisition (DIA) | Improvement |
|---|---|---|---|
| Distinct diGly Peptides (Single Shot) | ~20,000 peptides [3] | ~35,000 - 70,000 peptides [3] [21] | 2 to 3.5x |
| Quantitative Reproducibility (CV < 20%) | ~15% of peptides [3] | ~45% of peptides [3] | 3x |
| Median Quantitative CV | Not Reported | ~10% [21] | - |
| Peptides without Missing Values (in Replicates) | ~50% [21] | Nearly 100% [21] | ~2x |
The data shows that DIA not only expands the observable ubiquitinome but also generates data of higher quality. The marked improvement in coefficient of variation (CV) values indicates that DIA provides more precise measurements across replicate runs, a critical requirement for detecting subtle but biologically significant changes in ubiquitination [3] [21].
Optimized Workflow for Deep Ubiquitinome Profiling
The dramatic improvements in coverage are not due to the mass spectrometer alone; they result from a holistic optimization of the entire workflow, from cell lysis to computational analysis.
- Sample Lysis and Digestion: The use of Sodium Deoxycholate (SDC) lysis buffer, supplemented with chloroacetamide (CAA) and immediate sample boiling, has proven superior to traditional urea-based methods. This protocol rapidly inactivates deubiquitinating enzymes (DUBs), preserving the native ubiquitination state and leading to a ~38% increase in identified ubiquitin remnant peptides (K-GG peptides) [21].
- Peptide Enrichment: Immunoaffinity enrichment using antibodies specific for the diglycine (diGly) remnant left on lysines after tryptic digestion of ubiquitinated proteins remains crucial. Titration experiments indicate that using 1 mg of peptide material with 31.25 µg of anti-diGly antibody is optimal for deep coverage from endogenous samples [3].
- DIA-MS Acquisition: Methods have been specifically tailored for diGly peptides, which are often longer and carry higher charge states than typical tryptic peptides. Optimizing DIA isolation window widths and number, along with using a high MS2 resolution (30,000), can improve identifications by over 13% compared to standard proteomic methods [3].
- Data Processing: The DIA-NN software suite, enhanced with a specialized scoring module for modified peptides, is instrumental in processing complex ubiquitinomics DIA data. It can operate effectively in a "library-free" mode against a protein sequence database or with deep, experimentally-derived spectral libraries, yielding identifications of up to 70,000 K-GG peptides in a single run [21].
Detailed Experimental Protocol
This protocol describes the complete workflow for deep ubiquitinome profiling from mammalian cells using the optimized SDC lysis and DIA-MS.
Sample Preparation and diGly Peptide Enrichment
- Cell Lysis: Aspirate culture medium and wash cells with ice-cold PBS. Lyse cells directly in culture dishes using SDC Lysis Buffer (1% SDC, 100 mM Tris-HCl pH 8.5, 40 mM Chloroacetamide (CAA), 10 mM TCEP). Immediately scrape and transfer lysates to a microcentrifuge tube. Boil samples for 5 minutes at 95°C.
- Protein Digestion: Sonicate lysates to reduce viscosity and shear DNA. Measure protein concentration. Digest proteins with Lys-C (1:100 w/w) for 2 hours at 37°C, followed by trypsin (1:50 w/w) overnight at 37°C.
- Peptide Cleanup: Acidify digested peptides with trifluoroacetic acid (TFA) to a final concentration of 1% to precipitate SDC. Centrifuge to remove precipitate. Desalt the supernatant using C18 solid-phase extraction cartridges or plates. Elute peptides with 30-50% acetonitrile and dry under vacuum.
- diGly Peptide Enrichment: Resuspend dried peptides in Immunoaffinity Purification (IAP) Buffer. Incubate the peptide solution with anti-K-ε-GG antibody-coupled beads (e.g., PTMScan Ubiquitin Remnant Motif Kit) for 2 hours at 4°C with gentle agitation.
- Wash and Elution: Wash beads extensively with IAP Buffer, followed by a cold water wash. Elute bound diGly peptides with 0.1% TFA. Dry the eluate and reconstitute in a small volume of 0.1% formic acid for MS analysis.
Data-Independent Acquisition (DIA) Mass Spectrometry
- Chromatography: Separate enriched diGly peptides using a nano-flow liquid chromatography system with a C18 column and a 60-120 minute linear gradient from 2% to 30% acetonitrile in 0.1% formic acid.
- Mass Spectrometry Setup: Acquire data on an Orbitrap mass spectrometer (e.g., Orbitrap Exploris or Lumos) with a nano-electrospray ion source.
- DIA Method Parameters:
- Full MS: Resolution = 120,000; Scan Range = 350 - 1650 m/z.
- DIA Scans: Precursor isolation window scheme = variable windows covering 400-1000 m/z (e.g., 46 windows); MS2 Resolution = 30,000; HCD Collision Energy = 28-32%.
- Ensure cycle time is sufficiently short (~3 seconds) to provide adequate data points across chromatographic peaks.
Data Processing and Analysis
- Library Generation (Optional but Recommended): Generate a deep spectral library by fractionating a representative sample (e.g., 96 fractions concatenated into 8-12) and analyzing each fraction via DDA. Alternatively, use a "library-free" approach directly with DIA-NN.
- DIA Data Processing: Process raw DIA files using DIA-NN (version 1.8 and above).
- Set the digestion enzyme to "Trypsin/P."
- Enable "Neural network classifier" and "Match-between-runs."
- Set the modification to "K+GG" with a mass shift of 114.042927 Da.
- If using a spectral library, provide the library file. For library-free analysis, provide a canonical and contaminant protein sequence database in FASTA format.
- Downstream Analysis: Use the DIA-NN output matrix for statistical analysis (e.g., in Perseus, R) to identify significantly changing ubiquitination sites under experimental conditions.
The Scientist's Toolkit: Key Reagents and Software
Table 2: Essential Research Reagent Solutions for DIA Ubiquitinomics
| Item | Function / Role | Example / Specification |
|---|---|---|
| anti-K-ε-GG Antibody | Immunoaffinity enrichment of ubiquitin-derived diGly peptides; core of enrichment step. | PTMScan Ubiquitin Remnant Motif Kit (Cell Signaling Technology) [3] |
| Sodium Deoxycholate (SDC) | Powerful detergent for efficient protein extraction and solubilization while maintaining enzyme compatibility. | 1-2% in Tris buffer, with CAA and TCEP [21] |
| Chloroacetamide (CAA) | Cysteine alkylating agent; preferred over iodoacetamide as it prevents artifactual di-carbamidomethylation of lysines that mimics diGly remnant. | 40 mM in lysis buffer [21] |
| Proteasome Inhibitor | Increases ubiquitinated protein load by blocking degradation, useful for library generation and pathway studies. | MG-132 (10 µM, 4-6 h treatment) [3] [21] |
| DIA-NN Software | Deep neural network-based software for processing DIA data; specifically optimized for ubiquitinomics analysis. | Version 1.8+; enables library-free and library-based analysis [21] |
Biological Applications and Signaling Pathways
The power of DIA ubiquitinomics is best illustrated by its application to dissect complex biological signaling pathways. It has been successfully used to investigate TNF signaling, uncovering both known and novel ubiquitination events [3]. Furthermore, it has enabled a systems-wide investigation of ubiquitination across the circadian cycle, revealing hundreds of cycling ubiquitination sites that highlight new connections between metabolism and circadian regulation [3].
A prime application is in drug discovery, particularly for Targeted Protein Degradation (TPD) and Deubiquitinase (DUB) inhibitor profiling. The workflow can rapidly establish the mode of action for TPD modalities like PROTACs and molecular glues by identifying ubiquitylation sites on substrate proteins [13]. When applied to cells treated with a USP7 inhibitor, the method simultaneously tracked changes in ubiquitination for hundreds of proteins and the consequent degradation of a subset of those proteins, effectively dissecting the degradative from non-degradative functions of USP7 [21].
Diagram 2: Ubiquitin-Proteasome System and Key Drug Targets. E1-E2-E3 enzyme cascade mediates ubiquitin (Ub) transfer to substrate proteins. Polyubiquitination, typically via K48 or K11 linkages, targets substrates for proteasomal degradation. DUBs reverse this process, and both E3 ligases (utilized by PROTACs) and DUBs are key therapeutic targets [20] [22] [13].
The integration of optimized sample preparation, specifically the SDC lysis protocol, with tailored Data-Independent Acquisition mass spectrometry and advanced computational tools like DIA-NN, has fundamentally transformed the scale and precision of ubiquitinome analysis. The ability to consistently identify and quantify tens of thousands of ubiquitination sites in a single, high-throughput experiment moves the field from exploratory cataloging to robust, dynamic systems biology. This powerful new capability provides researchers and drug developers with an unprecedented lens through which to study cellular signaling, discover new biology, and accelerate the development of therapies targeting the ubiquitin-proteasome system.
Implementing DIA-Ubiquitinomics: Step-by-Step Workflows and Cutting-Edge Applications
In data-independent acquisition (DIA) mass spectrometry-based ubiquitinome profiling, sample preparation quality directly determines the depth and reliability of downstream analyses. The critical challenge lies in preserving ubiquitination signatures while efficiently extracting proteins and generating peptides representative of the cellular ubiquitinome. Traditional urea-based lysis methods often fall short in this regard, leading to incomplete protein solubilization and potential artifacts. Sodium deoxycholate (SDC)-based lysis has emerged as a superior alternative, particularly when coupled with optimized digestion protocols, enabling unprecedented coverage of the ubiquitinome when combined with DIA-MS technology. Within the broader context of DIA ubiquitinome research, optimized sample preparation serves as the foundational step that enables researchers to exploit the full quantitative potential of DIA—a method renowned for its exceptional reproducibility, minimal missing values, and high quantitative accuracy compared to data-dependent acquisition (DDA) approaches [15] [3]. This application note details a robust, optimized workflow that has demonstrated capacity to identify over 70,000 ubiquitinated peptides in single MS runs, more than tripling identification numbers achievable with conventional methods [15].
Optimized SDC-based Lysis for Ubiquitinome Profiling
SDC Lysis Buffer Composition and Rationale
The optimized SDC lysis buffer represents a significant advancement over traditional urea-based methods by combining effective protein extraction with immediate enzyme inactivation. The specific formulation and preparation steps are as follows:
Sodium Deoxycholate (SDC): Prepare as a 2% (w/v) solution in 100 mM Tris/HCl at pH 8.8 [23]. SDC acts as a powerful chaotropic detergent that effectively solubilizes membrane proteins and protein aggregates, ensuring comprehensive access to the ubiquitinome.
Alkylating Agent: Supplement with chloroacetamide (CAA) immediately before use. Unlike iodoacetamide, CAA does not cause di-carbamidomethylation of lysine residues, which can mimic ubiquitin remnant K-ɛ-GG peptides in mass spectrometry by adding an identical mass shift (114.0249 Da) [15]. This specificity is crucial for avoiding false-positive ubiquitination site assignments.
Buffer System: 100 mM Tris/HCl at pH 8.8 provides optimal alkalinity for subsequent digestion steps while maintaining buffer compatibility with mass spectrometry.
This optimized formulation has been demonstrated to yield 38% more K-ɛ-GG peptides compared to conventional urea-based lysis buffers while maintaining excellent enrichment specificity [15]. The immediate boiling of samples after lysis, combined with high concentrations of CAA, rapidly inactivates cysteine ubiquitin proteases, thereby preserving the native ubiquitination landscape by preventing deubiquitination during sample preparation.
Cell Lysis Protocol
The following step-by-step protocol ensures consistent and high-quality protein extraction for ubiquitinome studies:
Pre-heat SDC Lysis Buffer: Pre-heat the 2% SDC, 100 mM Tris/HCl (pH 8.8) buffer to 95°C to enhance solubilization efficiency [23].
Lysate Preparation: Add 50 μL of hot SDC buffer to cell pellet. For larger pellets or insufficient solubility, proportionally increase buffer volume. Resuspend thoroughly by pipetting.
Heat Denaturation: Incubate samples at 95°C for 10 minutes to denature proteins and inactivate enzymes. Follow with an additional 3-minute incubation at 95°C after thorough resuspension [23].
Cool and Benzonase Treatment: Cool samples on ice, then add 1 μL benzonase and incubate on ice for 10-30 minutes to digest nucleic acids, reducing sample viscosity [23].
Sonication: Sonicate using 5 cycles of 30 seconds on/30 seconds off at high intensity in a Bioruptor or similar instrument to fragment chromosomal DNA and improve protein extraction [23].
Additional Benzonase Incubation: Let samples sit for 10 minutes at room temperature to complete nucleic acid digestion.
Clarification: Centrifuge at 15,000 × g for 10 minutes at 10°C to pellet insoluble debris [23].
Protein Quantification: Transfer supernatant to a fresh tube and determine protein concentration using a micro BCA assay [23].
This protocol significantly enhances ubiquitin site coverage while improving quantitative precision and reproducibility across replicates. The SDC-based approach demonstrates particular advantage for studying labile ubiquitination events, as it rapidly inactivates deubiquitinating enzymes (DUBs) that might otherwise erase ubiquitination signatures during sample preparation [15].
Efficient Protein Digestion for Ubiquitinome Analysis
Reduction, Alkylation, and Digestion Protocol
Following optimized lysis, the digestion protocol must maintain the integrity of ubiquitination sites while ensuring efficient and reproducible protein processing:
Sample Preparation: Transfer 50 μg protein to a 0.2 mL tube and adjust volume to 15 μL with SDC buffer [23].
Reduction: Add DTT to a final concentration of 10 mM (e.g., 0.6 μL of 250 mM stock) and incubate at 50°C for 30 minutes to reduce disulfide bonds [23].
Alkylation: Add iodoacetamide to a final concentration of 20 mM (e.g., 0.6 μL of 500 mM stock) and incubate at room temperature for 30 minutes in the dark to alkylate cysteine residues [23].
Quenching: Add half the amount of DTT used in step 2 (e.g., 0.3 μL of 250 mM stock) to quench excess iodoacetamide and incubate at room temperature for 10 minutes [23].
Dilution and LysC Digestion: Dilute to 1% final SDC concentration with 100 mM Tris pH 8.5. Add LysC at a 1:100 enzyme-to-protein ratio and incubate at room temperature for up to 3 hours for initial protein cleavage [23].
Trypsin Digestion: Add trypsin at a 1:50 enzyme-to-protein ratio and incubate at 37°C overnight to complete protein digestion [23].
Acidification: Add 10% trifluoroacetic acid (TFA) to reach final 1% TFA, vortex, and centrifuge at 14,000 rpm for 10 minutes. Transfer supernatant to a new tube, then add additional TFA to reach final 2% TFA to ensure complete precipitation of SDC [23].
This sequential digestion approach employing both LysC and trypsin ensures comprehensive protein digestion while minimizing missed cleavages that can complicate ubiquitinome analysis. The protocol is optimized specifically for SDC-based workflows, addressing the unique challenges of detergent removal while maintaining compatibility with subsequent ubiquitin remnant enrichment steps.
Peptide Desalting and Clean-up
Prior to ubiquitin remnant enrichment, digested peptides require desalting and concentration:
STAGEtip Preparation: Prepare C18 STAGEtips by stacking three C18 disks in a 200 μL pipette tip.
Conditioning: Condition with:
- 100 μL 100% MeOH, centrifuge at 2,000 rpm for 3 minutes
- 100 μL 80% ACN, 0.1% TFA, centrifuge at 2,000 rpm for 3 minutes
- 100 μL 0.1% TFA, centrifuge at 2,000 rpm for 3 minutes [23]
Sample Loading: Load acidified peptide sample, centrifuge at 1,700 rpm, and collect flow-through.
Washing: Wash with 100 μL 0.1% TFA, combining flow-through with previous fraction.
Elution: Elute peptides with 2 × 30 μL 60% ACN, 0.1% TFA into a PCR tube [23].
Concentration: Use a speedvac to remove ACN and resuspend peptides in 20 μL 0.1% TFA, 2% ACN for subsequent ubiquitin remnant enrichment [23].
Quantitative Performance of SDC-based Workflow
The performance advantages of the SDC-based workflow are substantial and consistently reproducible across different cell types and experimental conditions. When benchmarked against conventional urea-based methods, the SDC protocol demonstrates superior performance in multiple critical parameters essential for high-quality ubiquitinome studies.
Table 1: Performance Comparison of SDC vs. Urea Lysis Methods for Ubiquitinome Analysis
| Performance Parameter | SDC-based Lysis | Urea-based Lysis | Improvement Factor |
|---|---|---|---|
| K-ɛ-GG peptide identifications | 26,756 | 19,403 | +38% [15] |
| Reproducibility (CV < 20%) | Significantly higher | Lower | Notable improvement [15] |
| Protein input requirement | 2 mg | ~20-40 mg | 10-20× reduction [15] |
| MS acquisition time | ~125 min | Extensive fractionation needed | >10× reduction [15] |
| Enrichment specificity | High | Moderate | Improved [15] |
These performance advantages translate directly to more reliable biological conclusions. The enhanced reproducibility manifests as a higher percentage of ubiquitinated peptides with coefficient of variation (CV) < 20%, significantly improving statistical power in differential ubiquitination studies [15]. Furthermore, the reduced protein input requirement makes the method applicable to precious clinical samples where material is limited.
Integration with DIA-MS Ubiquitinome Analysis
The optimized SDC-based sample preparation protocol serves as the ideal front-end for DIA-MS ubiquitinome analysis, leveraging the particular strengths of both approaches. When combined, these technologies enable unprecedented depth and quantitative precision in ubiquitin signaling studies.
The experimental workflow below illustrates the complete integration of SDC-based sample preparation with DIA-MS analysis for comprehensive ubiquitinome profiling:
This integrated approach enables researchers to simultaneously monitor both ubiquitination changes and consequent protein abundance alterations at high temporal resolution, providing unprecedented insights into ubiquitin signaling dynamics. For example, when applied to study USP7 inhibition, this workflow revealed that while ubiquitination of hundreds of proteins increased within minutes, only a small fraction of those targets underwent degradation, thereby precisely delineating the scope of USP7 action [15].
The combination of optimized sample preparation with DIA-MS analysis addresses a critical gap in ubiquitin signaling research by enabling distinction between regulatory ubiquitination events leading to protein degradation and non-degradative ubiquitination events—a distinction crucial for understanding the nuanced roles of ubiquitination in cellular regulation [15]. This capability is particularly valuable for drug discovery efforts targeting DUBs or ubiquitin ligases, where understanding the precise mode of action is essential for candidate optimization.
Research Reagent Solutions
Successful implementation of the SDC-based ubiquitinome workflow requires specific research-grade reagents and materials that ensure reproducibility and high-quality results.
Table 2: Essential Research Reagents for SDC-based Ubiquitinome Workflow
| Reagent/Material | Specification | Function in Workflow | Recommendation |
|---|---|---|---|
| Sodium Deoxycholate (SDC) | High purity, MS-grade | Effective protein solubilization while maintaining enzyme activity for digestion | Prepare fresh 2% solution in 100 mM Tris/HCl pH 8.8 [15] [23] |
| Chloroacetamide (CAA) | Molecular biology grade | Alkylating agent that prevents di-carbamidomethylation artifacts | Use instead of iodoacetamide to avoid K-ɛ-GG mimicking [15] |
| Benzonase | ≥250 U/μL | Digests nucleic acids to reduce sample viscosity | Add after lysis, incubate on ice [23] |
| LysC | Mass spectrometry grade | Primary digestion enzyme, cleaves C-terminal to Lys | 1:100 enzyme-to-protein ratio [23] |
| Trypsin | Mass spectrometry grade (Trypsin Gold) | Secondary digestion enzyme, cleaves C-terminal to Arg/Lys | 1:50 enzyme-to-protein ratio [23] |
| anti-K-ɛ-GG Antibody | PTMScan Ubiquitin Remnant Motif Kit | Immunoaffinity enrichment of ubiquitinated peptides | Use 31.25 μg antibody per 1 mg peptide input [3] |
| C18 Material | STAGEtips or columns | Peptide desalting and concentration | Use reverse-phase C18 for clean-up [23] |
The optimized SDC-based lysis and digestion protocol detailed in this application note represents a significant advancement in sample preparation for DIA-based ubiquitinome profiling. By addressing key limitations of traditional methods—particularly in the areas of protein extraction efficiency, protease inactivation, and compatibility with downstream mass spectrometry—this workflow enables researchers to achieve unprecedented depth and quantitative precision in ubiquitin signaling studies. When integrated with DIA-MS acquisition and neural network-based data processing, this sample preparation pipeline provides a robust foundation for comprehensive ubiquitinome characterization, facilitating both basic research into ubiquitin signaling mechanisms and applied drug discovery efforts targeting the ubiquitin-proteasome system. The protocols and methodologies described herein have been rigorously validated in peer-reviewed research and demonstrate consistent performance across multiple cell types and experimental conditions, making them suitable for adoption in diverse research settings focused on ubiquitin biology.
Within the broader scope of developing data-independent acquisition (DIA) methods for ubiquitinome analysis, the enrichment of diglycine (diGly)-modified peptides is a critical preparatory step that directly dictates the depth and quality of the final results. The commercialization of anti-K-ε-GG antibodies has revolutionized the large-scale profiling of ubiquitination sites by enabling the immunoaffinity enrichment of peptides containing the diGly remnant, a signature of trypsinized ubiquitinated proteins [24] [3]. The transition to DIA mass spectrometry has placed even greater emphasis on enrichment efficiency, as DIA's superior quantitative accuracy and data completeness can only be fully leveraged with a robust and high-yield input of modified peptides [3] [15]. This application note details optimized protocols and key parameters, derived from recent methodological advances, to maximize diGly peptide yield for subsequent deep, systems-wide ubiquitinome analysis using DIA.
Key Optimization Parameters for diGly Enrichment
Optimizing the enrichment process involves balancing several key parameters to maximize the yield of diGly peptides while maintaining specificity. The table below summarizes the optimal conditions and their impacts based on recent systematic investigations.
Table 1: Key Optimization Parameters for Anti-K-ε-GG Enrichment
| Parameter | Recommended Optimal Condition | Impact on Yield and Coverage | Key Reference |
|---|---|---|---|
| Peptide Input | 1-2 mg | Identifies >30,000 diGly sites; lower inputs (e.g., 500 µg) significantly reduce coverage. [3] [15] | |
| Antibody-to-Peptide Ratio | 31.25 µg antibody per 1 mg peptide | Maximizes peptide yield and depth of coverage in single DIA experiments. [3] | |
| Lysis Buffer | Sodium Deoxycholate (SDC) with immediate alkylation | Yields ~38% more K-ε-GG peptides than urea-based protocols, improving reproducibility. [15] | |
| Fractionation Strategy | Basic Reversed-Phase (bRP) separation with separate handling of K48-linked ubiquitin-chain derived peptides | Reduces competition for antibody binding sites, enhancing detection of co-eluting peptides. [3] | |
| Alkylating Agent | Chloroacetamide (CAA) | Prevents di-carbamidomethylation artifacts that mimic the diGly mass shift, a risk with iodoacetamide. [15] |
Reagent Toolkit for diGly Enrichment
Table 2: Essential Research Reagent Solutions for diGly Proteomics
| Item | Function/Description | Example/Note |
|---|---|---|
| Anti-diGly Remnant (K-ε-GG) Antibody | Immunoaffinity enrichment of tryptic peptides with the lysine-diGly modification. | Commercial kits (e.g., PTMScan Ubiquitin Remnant Motif Kit, CST) are widely used. [24] [25] |
| Anti-GGX Antibodies | Selective enrichment of tryptic peptides with an N-terminal diglycine motif, specific for N-terminal ubiquitination. | Clones 1C7, 2B12, 2E9, 2H2; do not cross-react with K-ε-GG peptides. [26] |
| SDC Lysis Buffer | Efficient protein extraction coupled with immediate and effective protease inhibition. | Supplemented with Chloroacetamide (CAA) and used with immediate boiling. [15] |
| Proteasome Inhibitor | Stabilizes ubiquitinated proteins by blocking degradation, thereby boosting the ubiquitin signal. | MG-132 treatment is common; increases abundance of K48-linked diGly peptides. [3] [25] |
Detailed Protocols for Maximized diGly Peptide Yield
Optimized Protocol for SDC-Based Lysis and Digestion
This protocol is designed for deep ubiquitinome coverage from mammalian cells [15].
- Cell Lysis: Lyse cell pellets in SDC lysis buffer (e.g., 1% SDC, 40 mM Chloroacetamide, 100 mM Tris pH 8.5). Immediate boiling (95°C, 5 minutes) post-lysis is critical to inactivate deubiquitinases (DUBs).
- Protein Quantification and Reduction: Determine protein concentration. Reduce disulfide bonds using 5 mM dithiothreitol (DTT) at 60°C for 30 minutes.
- Digestion: Digest proteins first with Lys-C (1:100 enzyme-to-protein ratio) for 3-4 hours at room temperature, then dilute the SDC concentration to 0.5% with 100 mM Tris pH 8.5. Add trypsin (1:50 enzyme-to-protein ratio) for overnight digestion at 37°C.
- Acidification and Peptide Cleanup: Acidify the sample with trifluoroacetic acid (TFA) to a final concentration of 1% to precipitate SDC. Centrifuge to remove the precipitate and desalt the supernatant containing the peptides using C18 solid-phase extraction (SPE) cartridges. Dry the purified peptides completely.
Anti-K-ε-GG Immunoaffinity Enrichment Protocol
This protocol outlines the core enrichment process, optimized for high yield and compatibility with single-shot DIA analysis [24] [3].
- Peptide Input Preparation: Resuspend 1-2 mg of desalted, dried peptides in 1-1.5 mL of Immunoaffinity Purification (IAP) Buffer (50 mM MOPS-NaOH pH 7.2, 10 mM Na₂HPO₄, 50 mM NaCl).
- Antibody Binding: Add the recommended amount of anti-K-ε-GG antibody beads (e.g., 31.25 µg antibody per 1 mg of peptide input) to the peptide solution. Incubate with gentle mixing for 2 hours at 4°C.
- Washing: Pellet the beads and transfer them to a micro-column. Perform a series of washes:
- Three times with 1 mL IAP Buffer.
- Three times with 1 mL HPLC-grade water.
- Peptide Elution: Elute the bound diGly peptides from the beads with two rounds of 0.15% TFA. Combine the eluates.
- Post-Enrichment Cleanup and Concentration: Desalt the eluate using C18 StageTips or micro-columns. Concentrate and dry the peptides, which are now ready for mass spectrometry analysis.
Diagram 1: diGly Peptide Enrichment Workflow
Integration with DIA-MS Ubiquitinome Analysis
The optimized enrichment protocol is a prerequisite for high-performance DIA analysis. The superior quantitative precision, reproducibility, and data completeness of DIA are fully realized when the input is a high-yield, specific preparation of diGly peptides [3] [15]. Following the enrichment detailed above, the diGly peptide sample is analyzed using a DIA method tailored for modified peptides.
Key DIA parameters include using narrower precursor isolation windows to handle the higher complexity of the enriched sample and a higher MS2 resolution (e.g., 30,000) for improved specificity [3]. Data processing with specialized software like DIA-NN, which includes scoring modules for modified peptides, further boosts identification numbers and quantitative accuracy [15]. This integrated workflow—from optimized enrichment to tailored DIA—enables the routine identification and quantification of over 35,000 distinct endogenous ubiquitination sites in a single experiment, a significant leap over traditional data-dependent acquisition (DDA) methods [3].
Diagram 2: DIA-MS Analysis Integration
Data-independent acquisition (DIA) has emerged as a transformative mass spectrometry approach that systematically fragments and analyzes all peptides within predefined mass-to-charge (m/z) ranges, overcoming the stochastic sampling limitations of data-dependent acquisition (DDA). This methodology offers exceptional quantitative reproducibility, accuracy, and completeness across samples, making it particularly valuable for sophisticated proteomic applications such as ubiquitinome analysis [27] [28]. The fundamental principle of DIA involves dividing the full m/z range into consecutive isolation windows, fragmenting all precursors within each window simultaneously, and recording comprehensive fragment ion spectra [28]. Unlike DDA, which selectively fragments the most abundant precursors, DIA employs an unbiased acquisition strategy that ensures comprehensive detection of peptides across a wide dynamic range [3]. This systematic approach significantly reduces missing values in quantitative comparisons, thereby enhancing the reliability of large-scale studies, including investigations of post-translational modifications like ubiquitination [11].
The configuration of DIA methods—particularly the design of isolation window schemes and associated acquisition parameters—critically influences the depth of proteome coverage, quantification accuracy, and sensitivity. For ubiquitinome analysis, where modified peptides often exhibit low stoichiometry and require immunoaffinity enrichment prior to analysis, optimal parameter selection becomes even more crucial [3]. This application note provides detailed protocols and optimized parameters for implementing DIA methods in ubiquitin signaling research, supported by experimental data and practical implementation guidelines.
DIA Acquisition Schemes and Window Design
Classification of DIA Acquisition Schemes
DIA acquisition methods can be categorized into several distinct classes based on their precursor isolation window design and instrumentation. According to a comprehensive 2023 survey, the major DIA schemes include wide-window, overlapping-window, narrow-window, scanning quadrupole-based, and parallel accumulation-serial fragmentation (PASEF)-enhanced DIA methods [27]. Each scheme offers distinct advantages for specific applications:
- Wide-window DIA employs broad isolation windows (typically 20-50 m/z) to cover extensive m/z ranges with fewer windows, enabling faster cycle times at the potential cost of increased spectral complexity due to co-fragmentation.
- Narrow-window DIA utilizes smaller windows (typically 4-8 m/z) to reduce precursor complexity within each window, improving selectivity and sensitivity while requiring more cycles to cover the same m/z range.
- Overlapping-window DIA introduces slight overlaps between adjacent windows to ensure complete coverage of all precursors and mitigate edge effects where peptides fall between windows.
- PASEF-enhanced DIA, implemented on timsTOF instruments, combines trapped ion mobility separation with DIA to achieve exceptional sensitivity and speed by synchronizing ion mobility separation with mass spectrometry detection [27].
Table 1: Comparison of Major DIA Acquisition Schemes
| Scheme Type | Typical Window Width (m/z) | Advantages | Limitations | Best Applications |
|---|---|---|---|---|
| Wide-window | 20-50 | Fast cycle times, broad coverage | High spectral complexity, reduced selectivity | Discovery studies requiring wide dynamic range |
| Narrow-window | 4-8 | High selectivity, improved sensitivity | Longer cycle times, fewer data points per peak | Targeted applications, complex samples |
| Overlapping-window | 4-8 with 1-2 m/z overlap | Complete precursor coverage, reduced edge effects | Increased total windows, complex method setup | Comprehensive ubiquitinome analysis |
| PASEF-DIA | Variable with ion mobility | Exceptional sensitivity, high speed | Specialized instrumentation required | High-throughput ubiquitin signaling studies |
Static Versus Dynamic Window Schemes
Traditional DIA methods employ static isolation windows that remain constant throughout the chromatographic separation. However, recent innovations have introduced dynamic DIA approaches that adjust isolation windows based on retention time to focus measurement time on relevant m/z ranges [29]. In liquid chromatography-mass spectrometry (LC-MS), peptide elution follows predictable patterns where larger, more hydrophobic peptides generally elute later with higher m/z values. Dynamic DIA capitalizes on this relationship by allocating narrower windows and longer acquisition times to m/z regions with high peptide density at specific retention time intervals [29].
The implementation of dynamic DIA requires: (1) a retention time alignment algorithm to synchronize the current run with a reference method, (2) predefined optimal window boundaries calculated from empirical data or spectral libraries, and (3) real-time adjustment of window positions during acquisition [29]. Research demonstrates that dynamic DIA improves the lower limit of quantification by increasing time spent on each isolation window without sacrificing peptide coverage [29]. This approach is particularly beneficial for ubiquitinome analyses where the unique properties of diGly-modified peptides—including longer sequence lengths and higher charge states—result in distinctive m/z and retention time distributions [3].
Figure 1: Dynamic DIA Method Implementation Workflow. The diagram illustrates the process for developing and implementing retention time-aware dynamic DIA methods, highlighting the integration between spectral library data and real-time instrument control.
Optimized Parameters for Ubiquitinome Analysis
DIA Parameter Optimization for diGly Peptide Analysis
Ubiquitin-derived diGly-modified peptides exhibit distinct characteristics that necessitate specialized DIA parameter optimization. Following tryptic digestion, previously ubiquitinated peptides retain a diglycine remnant on modified lysine residues, which often results in longer peptide sequences and higher charge states compared to unmodified peptides [3]. These properties directly influence optimal DIA parameter selection:
Isolation Window Configuration: Research demonstrates that 46 precursor isolation windows provide optimal coverage for diGly peptide analysis, balancing selectivity with reasonable cycle times [3]. Variable window schemes that allocate narrower windows in dense m/z regions and wider windows in sparse regions can further enhance sensitivity.
Fragment Scan Resolution: Higher MS2 resolution significantly improves diGly peptide identification. Methods employing 30,000 resolution fragment scanning outperform lower resolution alternatives, providing a 13% improvement in diGly peptide identification compared to standard full proteome methods [3].
Cycle Time Management: For 90-minute LC gradients, optimal cycle times of approximately 2.5 seconds allow sufficient points across chromatographic peaks while maintaining high fragment ion resolution [29]. This typically accommodates 36 acquisitions per cycle when using 8-m/z isolation windows covering a 300-m/z span.
Table 2: Optimized DIA Parameters for Ubiquitinome Analysis
| Parameter | Recommended Setting | Experimental Impact | Rationale |
|---|---|---|---|
| Precursor m/z range | 400-1000 | 6% increase in diGly IDs | Covers typical diGly peptide masses |
| Number of windows | 46 | 13% improvement vs. standard | Balances selectivity and cycle time |
| MS2 resolution | 30,000 | Enhanced identification confidence | Improved signal-to-noise for fragment ions |
| Window placement | Variable based on density | Better utilization of scan time | Focuses resources on peptide-rich regions |
| Peptide input | 1 mg total peptides | Optimal for 31.25 µg antibody | Maximizes antibody binding capacity |
| Injection amount | 25% of enriched material | Sufficient for 35,000 diGly sites | Maintains sensitivity while conserving sample |
Spectral Library Generation for Ubiquitinome DIA
Comprehensive spectral libraries are critical resources for DIA data analysis, enabling accurate peptide identification and quantification [27]. For ubiquitinome applications, specialized library generation protocols are required:
Deep Fractionation Approaches: Basic reversed-phase chromatography separated into 96 fractions and concatenated into 8 fractions enables identification of >67,000 diGly peptides from MG132-treated HEK293 cells [3]. Separate handling of fractions containing highly abundant K48-linked ubiquitin-chain derived diGly peptides reduces competition during antibody enrichment and improves detection of co-eluting peptides.
Multi-Condition Library Generation: Combining spectral libraries from different cell lines (HEK293 and U2OS) and treatment conditions (proteasome inhibitor-treated and untreated) produces comprehensive libraries containing >90,000 diGly peptides, significantly enhancing coverage in single-run DIA analyses [3].
Hybrid Library Construction: Merging DDA-generated spectral libraries with direct DIA search results identifies 35,111 ± 682 diGly sites in single measurements, doubling identification compared to DDA alone [3].
Advanced data analysis tools like DIA-NN incorporate deep neural network-based processing with specialized scoring modules for modified peptides, further boosting ubiquitinome coverage [15]. When optimized DIA methods are combined with these computational advances, identification of up to 70,000 ubiquitinated peptides in single MS runs becomes achievable, representing a more than threefold improvement over conventional DDA methods [15].
Detailed Experimental Protocols
Sample Preparation for DIA Ubiquitinome Analysis
SDC-Based Lysis and Digestion Protocol (adapted from [15]):
Cell Lysis: Lyse cells in SDC buffer (2% sodium deoxycholate, 40 mM chloroacetamide, 100 mM Tris pH 8.5) with immediate boiling at 95°C for 10 minutes to simultaneously denature proteins and inhibit deubiquitinases.
Protein Digestion: Digest proteins using trypsin at a 1:25 enzyme-to-protein ratio in 50 mM ammonium bicarbonate overnight at 37°C. Acidify with trifluoroacetic acid to precipitate SDC, then centrifuge at 10,000 × g for 10 minutes to remove detergent.
diGly Peptide Enrichment: Resuspend dried peptides in immunoaffinity purification (IAP) buffer (50 mM MOPS pH 7.2, 10 mM sodium phosphate, 50 mM NaCl). Incubate with 31.25 µg anti-diGly antibody per 1 mg peptide input for 2 hours at 4°C with gentle agitation.
Wash and Elution: Wash antibody-bound beads three times with IAP buffer and twice with water. Elute diGly peptides with 0.1% trifluoroacetic acid (50 μL, twice).
Sample Cleanup: Desalt peptides using C18 stage tips and dry completely before LC-MS analysis.
This SDC-based protocol increases K-GG peptide identification by 38% compared to conventional urea-based methods while maintaining high enrichment specificity [15]. The inclusion of chloroacetamide instead of iodoacetamide prevents di-carbamidomethylation of lysine residues, which can mimic diGly remnants [15].
Liquid Chromatography and DIA Method Setup
NanoLC Parameters:
- Column: 30 cm × 75 μm inner diameter fused silica packed with 3 μm C18 beads
- Gradient: 90-120 minutes from 0% to 40% mobile phase B (0.1% formic acid in 80% acetonitrile)
- Flow rate: 300 nL/min
- Temperature: 50°C
DIA Method Configuration on Orbitrap Instruments:
- MS1 Settings: 400-1000 m/z range, 60,000 resolution, 50 ms maximum injection time
- MS2 Settings: 46 variable windows covering 400-1000 m/z, 30,000 resolution, 54 ms maximum injection time
- Fragmentation: Higher-energy collisional dissociation (HCD) with 28-30% normalized collision energy
- Dynamic Exclusion: Not applicable for DIA
- Cycle Time: Target approximately 2.5 seconds to maintain 10-12 points per peak
For dynamic DIA implementations, additional steps include:
- Acquisition of a reference run for retention time alignment
- Pre-calculation of optimal window boundaries using spectral library data
- Implementation of real-time retention time alignment algorithms [29]
Figure 2: Comprehensive DIA Ubiquitinome Analysis Workflow. The integrated protocol spans from sample preparation through data analysis, highlighting critical optimization points for ubiquitin signaling studies.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for DIA Ubiquitinome Analysis
| Reagent/Resource | Function | Application Notes | Source |
|---|---|---|---|
| Anti-diGly Remnant Motif (K-ε-GG) Antibody | Immunoaffinity enrichment of ubiquitinated peptides | Use 31.25 µg per 1 mg peptide input; optimal balance between cost and coverage | Cell Signaling Technology |
| Sodium Deoxycholate (SDC) | Protein extraction and denaturation | Superior to urea for ubiquitinome studies; 38% more K-GG peptides identified | Various suppliers |
| Chloroacetamide (CAA) | Cysteine alkylation | Prevents di-carbamidomethylation artifacts that mimic diGly remnants | Various suppliers |
| TPCK-treated Trypsin | Protein digestion | Specific cleavage C-terminal to Lys and Arg; generates diGly remnant | Various suppliers |
| C18 StageTips | Peptide desalting and concentration | Remove detergents and salts prior to LC-MS analysis | Thermo Fisher Scientific |
| High-pH Reversed-Phase Resin | Peptide fractionation for library generation | Enables deep spectral library generation; 96 fractions concatenated to 8 | Various suppliers |
| DIA-NN Software | Data processing with neural networks | Specialized scoring for modified peptides; library-free and library-based modes | Open source |
| EncyclopeDIA Software | DIA data analysis | Integration with chromatogram libraries; compatible with dynamic DIA | Open source |
The strategic configuration of DIA method parameters—particularly isolation window schemes and acquisition settings—significantly enhances the depth and quantitative accuracy of ubiquitinome analyses. Implementation of optimized protocols incorporating 46-window designs, 30,000 MS2 resolution, dynamic window adjustments, and SDC-based sample processing enables identification of over 35,000 diGly peptides in single measurements, more than doubling coverage compared to conventional DDA methods [3]. These technical advances provide researchers with powerful tools to investigate ubiquitin signaling dynamics at unprecedented scale and precision, opening new possibilities for understanding circadian regulation, kinase signaling, and drug mechanism-of-action studies [3] [15]. As DIA methodologies continue to evolve with improvements in instrumentation and computational analysis, their application to ubiquitinome research promises to yield increasingly comprehensive insights into this crucial regulatory system.
In data-independent acquisition (DIA) mass spectrometry-based ubiquitinome analysis, the generation of a comprehensive spectral library is a critical prerequisite for the accurate identification and quantification of ubiquitinated peptides. The unique signature of ubiquitination—the diglycine (diGly) remnant left on trypsinized peptides—presents distinct challenges for spectral library generation, requiring specialized enrichment and acquisition strategies. The choice between developing a project-specific spectral library or utilizing public resources represents a fundamental strategic decision that directly impacts proteomic depth, quantitative accuracy, and biological insight.
This application note examines the technical considerations for spectral library generation within ubiquitinome research, providing a structured comparison of project-specific and public resource approaches. We present quantitative performance metrics, detailed experimental protocols, and a decision framework to guide researchers in selecting the optimal strategy for their specific experimental context.
Performance Comparison: Project-Specific vs. Public Spectral Libraries
Table 1: Quantitative Performance Metrics of Spectral Library Strategies in Ubiquitinome Analysis
| Performance Metric | Project-Specific Library | Public Library Resources | Experimental Context |
|---|---|---|---|
| Library Size (diGly Peptides) | 89,650 - 146,626 peptides [3] [15] | Variable; often limited | HEK293/U2OS cells with proteasome inhibition [3] |
| Single-Run Identifications | 35,000 - 68,429 diGly peptides [3] [15] | Typically lower than project-specific | Single DIA measurements from cell lines [3] [15] |
| Quantitative Precision (Median CV) | ~10% CV [15] | Not explicitly reported | replicate samples [15] |
| Coverage of Novel Sites | High (57% previously unreported) [3] | Limited to previously documented sites | Comparison to PhosphositePlus database [3] |
| Software Compatibility | All major DIA tools (DIA-NN, Spectronaut, etc.) [30] | All major DIA tools [31] [30] | Library-based analysis modes [31] |
Table 2: Technical and Resource Requirements for Library Generation Strategies
| Consideration | Project-Specific Library | Public Library Resources |
|---|---|---|
| Sample Input Requirements | High (e.g., 2mg protein; fractionation) [3] | None (pre-existing resource) |
| Instrument Time Investment | Significant (fractionation required) [3] | Minimal |
| Experimental Design Flexibility | Tailored to specific conditions [3] | Limited to available datasets |
| Specialized Protocols Needed | diGly enrichment, fractionation [3] | Not applicable |
| Optimal Application Context | Novel discoveries, condition-specific profiling [3] | Resource conservation, well-characterized systems |
Experimental Protocols for Spectral Library Generation
Generation of a Project-Specific Deep diGly Spectral Library
Principle: Construction of a comprehensive, project-specific spectral library through extensive fractionation and diGly antibody-based enrichment maximizes coverage of the ubiquitinome specific to the experimental system [3].
Reagents and Materials:
- Cell lines of interest (e.g., HEK293, U2OS)
- Proteasome inhibitor (e.g., MG132, 10µM)
- Lysis buffer: Sodium Deoxycholate (SDC)-based buffer supplemented with Chloroacetamide (CAA) [15]
- diGly Remnant Motif (K-ε-GG) Antibody (e.g., PTMScan Kit)
- Basic reversed-phase (bRP) chromatography system for fractionation
Procedure:
- Cell Treatment and Protein Extraction:
Digestion and Peptide Cleanup:
- Digest proteins using trypsin, generating peptides with diGly remnants on previously ubiquitinated lysines.
- Desalt peptides using C18 solid-phase extraction.
High-pH Fractionation:
- Separate peptides by basic reversed-phase chromatography into 96 fractions.
- Concatenate fractions into 8-12 pools to reduce analytical time while maintaining depth [3].
- Optional: Process highly abundant K48-linked ubiquitin-chain derived diGly peptides separately to reduce competition during enrichment [3].
diGly Peptide Enrichment:
- Incubate fractionated peptides with anti-diGly antibody (e.g., 31.25µg antibody per 1mg peptide input) [3].
- Wash extensively to remove non-specifically bound peptides.
- Elute enriched diGly peptides.
Library Data Acquisition:
Protocol for Public Spectral Library Utilization
Principle: Leverage existing publicly available ubiquitinome spectral libraries to conserve resources while maintaining reasonable coverage [31] [30].
Reagents and Materials:
- Public data repositories (PRIDE, MassIVE)
- Spectral library conversion tools (as needed)
- Compatible DIA analysis software (DIA-NN, Spectronaut, etc.)
Procedure:
- Library Sourcing:
- Identify relevant spectral libraries from public repositories (e.g., PXD019854 for ubiquitinome) [3].
- Select libraries generated from similar biological systems and instrument platforms.
Library Curation and Conversion:
- Assess library comprehensiveness for your experimental context.
- Convert library formats as needed for compatibility with chosen DIA analysis software [30].
- For DIA-NN: Direct use of Spectronaut (xls) libraries is possible [30].
- For OpenSWATH: Convert to TraML format using TargetedFileConverter [30].
Experimental Validation:
- Process a subset of experimental samples using the public library.
- Assess identification rates and quantitative performance.
- Consider supplementing with project-specific data if coverage is insufficient.
Integrated Workflow and Decision Framework
The following diagram illustrates the decision pathway for selecting between project-specific and public spectral library strategies in DIA-based ubiquitinome analysis:
Decision Pathway for Spectral Library Strategy Selection
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for DIA Ubiquitinomics
| Reagent / Material | Function | Application Notes |
|---|---|---|
| diGly Remnant Motif Antibody | Immunoaffinity enrichment of ubiquitinated peptides | Critical for specificity; 31.25μg per 1mg peptide input recommended [3] |
| Sodium Deoxycholate (SDC) | Lysis and protein extraction | Superior to urea for ubiquitinome coverage; use with CAA alkylation [15] |
| Chloroacetamide (CAA) | Cysteine alkylation | Preferred over iodoacetamide to avoid di-carbamidomethylation artifacts [15] |
| Proteasome Inhibitors (MG132) | Enhances ubiquitinated peptide levels | 10μM for 4 hours recommended for library generation [3] |
| Basic Reversed-Phase Resins | High-pH peptide fractionation | Enables deep coverage through reduced sample complexity [3] |
The selection between project-specific and public spectral libraries represents a critical decision point in DIA-based ubiquitinome studies. Project-specific libraries provide unparalleled depth and condition-specific coverage, enabling novel discoveries in ubiquitin signaling at the cost of significant resource investment. Public resources offer practical alternatives for resource-limited scenarios or studies focused on well-characterized biological systems. As ubiquitinome research continues to evolve, hybrid approaches that combine targeted project-specific data with curated public resources may offer the most balanced strategy, maximizing coverage while maintaining experimental efficiency. The protocols and decision framework presented here provide researchers with a structured approach to selecting and implementing optimal spectral library strategies for their specific research contexts.
Data-independent acquisition (DIA) mass spectrometry has emerged as a powerful technology for large-scale proteomic studies, addressing critical limitations of traditional data-dependent methods. This application note explores the integration of DIA-NN, a software suite utilizing deep neural networks, for processing DIA-based ubiquitinome profiling data. We present optimized experimental protocols, benchmark performance metrics, and detailed workflows that enable researchers to identify over 70,000 ubiquitinated peptides in single MS runs—more than tripling identification numbers compared to conventional methods while significantly improving quantitative precision and reproducibility. Designed for drug development professionals investigating the ubiquitin-proteasome system, this guide provides the foundational knowledge necessary to implement neural network-enhanced DIA analysis for targeted therapeutic development.
The ubiquitin-proteasome system (UPS) represents a complex regulatory network involving approximately 750 enzymes that mediate ubiquitin attachment to and cleavage from target proteins, influencing virtually all intracellular processes from cell cycle progression to autophagy [15]. Dysregulation of this system contributes significantly to carcinogenesis and neurodegenerative diseases, making it an attractive target for therapeutic intervention [32]. Mass spectrometry-based ubiquitinomics has revolutionized our ability to profile ubiquitination events system-wide, yet traditional data-dependent acquisition (DDA) methods suffer from stochastic sampling and limited reproducibility.
Data-independent acquisition (DIA) mass spectrometry has addressed these limitations through systematic fragmentation of all peptides within defined mass-to-charge windows, eliminating missing values and enhancing quantitative accuracy [11]. However, the computational processing of DIA data presents unique challenges due to highly multiplexed spectra requiring sophisticated algorithms to distinguish true signals from noise. The DIA-NN software suite incorporates deep neural networks and novel interference correction strategies to overcome these hurdles, enabling unprecedented depth and precision in ubiquitinome analysis [33]. This application note details methodologies for leveraging DIA-NN in ubiquitinomics research, with particular emphasis on experimental design, data processing workflows, and performance optimization for drug discovery applications.
DIA-NN Architecture and Neural Network Integration
Core Algorithmic Framework
DIA-NN implements a peptide-centric analysis strategy that begins with a collection of precursor ions, which can be provided via a spectral library or generated in silico from a protein sequence database in library-free mode [33]. The software then generates decoy precursors as negative controls and extracts chromatograms for each target and decoy precursor. For each putative elution peak, DIA-NN calculates 73 distinct scores reflecting peak characteristics, including fragment ion co-elution, mass accuracy, and spectral similarity [33].
A critical innovation within DIA-NN is its application of deep neural networks (DNNs) for assigning statistical significance to identified precursors. The software employs an ensemble of feed-forward fully-connected DNNs with five tanh-activated hidden layers and a softmax output layer. These networks are trained for one epoch to distinguish between target and decoy precursors using cross-entropy as the loss function [33]. The resultant discriminant scores, averaged across networks, provide the foundation for false discovery rate (FDR) estimation via q-value calculation, enabling comprehensive proteome coverage at strict FDR thresholds.
Specialized Scoring for Ubiquitinomics
For ubiquitinome applications, DIA-NN incorporates an additional scoring module specifically optimized for confident identification of modified peptides, including K-GG peptides containing the diglycine remnant left after tryptic digestion of ubiquitinated proteins [15]. This specialized processing, combined with interference detection and correction algorithms, allows DIA-NN to achieve exceptional sensitivity and quantification accuracy for low-abundance ubiquitinated peptides across complex sample matrices.
Table 1: Key Technical Specifications of DIA-NN for Ubiquitinome Analysis
| Parameter | Specification | Application Benefit |
|---|---|---|
| Analysis Mode | Library-free or library-based | Flexibility for diverse experimental designs |
| Peptide Scoring | 73 peak characteristics + deep neural networks | Enhanced identification confidence |
| FDR Control | Neural network-derived q-values | <1% FDR achievable for ubiquitinated peptides |
| Quantification | Interference-corrected fragment ion chromatograms | Improved precision for low-abundance peptides |
| Throughput | Up to 1000 runs per hour on conventional PC | Suitable for large clinical cohorts |
| Ubiquitinome Optimization | Specialized K-GG peptide scoring module | 3x more identifications vs. DDA |
Experimental Protocols for DIA-Based Ubiquitinomics
Sample Preparation and Lysis Optimization
Proper sample preparation is critical for deep ubiquitinome coverage. We recommend a sodium deoxycholate (SDC)-based lysis protocol that significantly improves ubiquitin site identification compared to conventional urea-based methods:
SDC Lysis Buffer Preparation: Prepare lysis buffer containing 2% SDC, 100 mM Tris-HCl (pH 8.5), and 40 mM chloroacetamide (CAA). The high CAA concentration rapidly inactivates cysteine ubiquitin proteases through alkylation, preserving ubiquitination states [15].
Cell Lysis: Add pre-warmed SDC lysis buffer directly to cell pellets (approximately 50-100 μL per 1 mg of cell pellet). Immediate boiling of samples after lysis further enhances ubiquitin site coverage by denaturing enzymes.
Protein Extraction: Incubate samples at 95°C for 10 minutes with vigorous shaking (1400 rpm), followed by sonication (10 cycles of 30 seconds on/30 seconds off). Centrifuge at 16,000 × g for 10 minutes to remove insoluble material [15].
This optimized protocol yields approximately 38% more K-GG peptides compared to urea buffer (26,756 vs. 19,403 identifications in benchmark studies), without compromising enrichment specificity [15].
Ubiquitinated Peptide Enrichment and Cleanup
Following tryptic digestion, ubiquitinated peptides are enriched using anti-K-GG antibody-based purification:
Digestion: Digest proteins with trypsin (1:25 enzyme-to-protein ratio) overnight at 37°C. Acidify with trifluoroacetic acid (TFA) to a final concentration of 1%, then centrifuge to pellet SDC.
Peptide Desalting: Desalt peptides using C18 solid-phase extraction columns according to manufacturer protocols. Lyophilize and resuspend in immunoaffinity purification (IAP) buffer (50 mM MOPS-NaOH pH 7.2, 10 mM Na2HPO4, 50 mM NaCl) [15].
K-GG Peptide Enrichment: Incubate peptides with anti-K-GG antibody-conjugated beads for 2 hours at 4°C with gentle rotation. Wash beads three times with IAP buffer and twice with HPLC-grade water before eluting with 0.1% TFA [15].
For applications requiring multiplexing, the UbiFast methodology implements TMT labeling on-bead after K-GG peptide enrichment, reducing sample requirements to sub-milligram levels while maintaining identification depth [34].
Liquid Chromatography and Mass Spectrometry
For optimal DIA ubiquitinomics, we recommend the following LC-MS parameters:
Chromatography: Employ a 75-μm inner diameter fused silica capillary column packed with 3-μm C18 beads (30 cm length). Use a 90-minute linear gradient from 2% to 40% mobile phase B (0.1% formic acid in 80% acetonitrile) at a flow rate of 300 nL/min [15].
DIA Method Optimization: Configure DIA methods with 8-m/z isolation windows covering the 400-1000 m/z range. For Orbitrap instruments, set MS2 resolution to 30,000 with a cycle time of approximately 2.5 seconds to ensure sufficient points across chromatographic peaks [15].
Mass Accuracy Settings: For timsTOF instruments, set both MS1 and MS2 mass accuracies to 15.0 ppm. For Orbitrap Astral systems, use 10.0 ppm for MS2 and 4.0 ppm for MS1 mass accuracy [35].
Diagram 1: Experimental workflow for DIA-based ubiquitinome profiling. The optimized protocol combines SDC-based lysis with K-GG enrichment and DIA-NN computational analysis.
Performance Benchmarking and Applications
Quantitative Comparison to Alternative Methods
DIA-NN demonstrates superior performance for ubiquitinome analysis compared to both traditional DDA and alternative DIA processing tools. In benchmark studies using proteasome inhibitor-treated HCT116 cells, DIA-NN identified more than triple the number of K-GG peptides compared to DDA (68,429 vs. 21,434 on average per sample) while achieving excellent quantitative precision (median CV of 10% across replicates) [15].
Table 2: Performance Benchmarking of DIA-NN for Ubiquitinome Analysis
| Software/Method | K-GG Peptides Identified | Quantitative Precision (Median CV) | Notable Features |
|---|---|---|---|
| DIA-NN (DIA) | 68,429 | 10% | Neural network scoring, interference correction |
| MaxQuant (DDA) | 21,434 | >20% | Match-between-runs, standard in field |
| OpenSWATH | ~40,000 (estimated) | 12-15% | Pioneering automated DIA analysis |
| Spectronaut | ~45,000 (estimated) | 10-12% | Commercial solution with extensive tuning |
| EncyclopeDIA | ~48,900 | ~12% | Used in initial DIA ubiquitinomics studies |
When benchmarked against other DIA processing tools using public datasets, DIA-NN consistently identified more precursors at all FDR thresholds, with the most significant improvements observed at strict FDR thresholds (0.5-1%) [33]. The software also enables confident identification with short chromatographic gradients, identifying more precursors from 0.5-hour acquisitions than other tools achieve with 1-2 hour gradients.
Application to Deubiquitinase Target Engagement
DIA-NN-enabled ubiquitinomics provides a powerful approach for mapping substrates of deubiquitinases (DUBs), important therapeutic targets in oncology. Following inhibition of USP7, researchers simultaneously recorded ubiquitination changes and abundance changes for more than 8,000 proteins at high temporal resolution [15]. This approach revealed that while ubiquitination of hundreds of proteins increased within minutes of USP7 inhibition, only a small fraction underwent degradation, distinguishing regulatory from degradative ubiquitination events.
The method's sensitivity enables characterization of DUB inhibitors with low protein input requirements, making it suitable for primary cell samples and clinical specimens. When combined with proximity labeling techniques like APEX2, DIA-NN can further elucidate spatially resolved ubiquitination events, as demonstrated in the identification of mitochondrial substrates of USP30 [36].
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Tools for DIA Ubiquitinomics
| Item | Function | Specifications |
|---|---|---|
| Anti-K-GG Antibody | Immunoaffinity enrichment of ubiquitinated peptides | Cell Signaling Technology #5562; enrichment of tryptic K-GG remnants |
| Sodium Deoxycholate (SDC) | Efficient protein extraction with protease inactivation | 2% in lysis buffer with 40mM chloroacetamide; superior to urea for ubiquitinomics |
| Chloroacetamide (CAA) | Cysteine alkylation without lysine di-modification | 40mM in lysis buffer; prevents artificial K-GG mimics |
| DIA-NN Software | DIA data processing with neural networks | v2.3.0+; library-free or library-based modes; specialized K-GG scoring |
| Sequence Database | In silico spectral library generation | UniProt-formatted FASTA; enables predicted spectral libraries |
| LysC/Trypsin | Proteolytic digestion | Sequential digestion for UbiSite approach; trypsin alone for K-GG |
Advanced Data Processing Workflow
Diagram 2: DIA-NN computational workflow for ubiquitinome analysis. The process integrates spectral library matching with neural network-based scoring and interference correction.
The integration of DIA-NN with neural network-based data processing represents a transformative advancement for ubiquitinome analysis in drug discovery research. The methodologies outlined in this application note provide researchers with robust tools to overcome previous limitations in identification depth, quantitative accuracy, and reproducibility. As the ubiquitin field continues to recognize the therapeutic potential of targeting DUBs and E3 ligases, DIA-NN-enabled ubiquitinomics offers a powerful platform for comprehensive target engagement profiling and mode-of-action studies. With continuous software development addressing emerging challenges in DIA data processing, these computational approaches will undoubtedly play an increasingly central role in translating ubiquitin biology into novel therapeutic strategies.
Circadian rhythms, the approximately 24-hour biological cycles that govern physiology and behavior, are ultimately governed by molecular events, with post-translational modifications (PTMs) serving as critical regulatory mechanisms. Among PTMs, protein ubiquitination has emerged as a fundamental regulator of circadian clocks, influencing protein stability, localization, and function within the core timing mechanism [3]. The development of data-independent acquisition (DIA) mass spectrometry (MS) methods has revolutionized large-scale ubiquitinome profiling, enabling unprecedented depth and quantitative accuracy in mapping oscillating ubiquitination events across the circadian cycle [3]. This Application Note details optimized protocols for systems-wide analysis of circadian ubiquitination, providing researchers with robust methodologies to investigate the crucial interface between ubiquitin signaling and circadian biology.
Ubiquitination's importance in circadian regulation stems from its involvement in virtually all cellular processes, including targeted protein degradation via the ubiquitin-proteasome system [3]. Core clock components themselves are regulated through ubiquitin-mediated degradation, making comprehensive ubiquitinome profiling essential for understanding circadian timing mechanisms. Traditional ubiquitinome studies employing data-dependent acquisition (DDA) methods have been limited by missing values across samples and reduced quantitative accuracy, challenges that DIA effectively addresses through its comprehensive fragmentation approach [3].
DIA Mass Spectrometry for Circadian Ubiquitinome Analysis
Data-independent acquisition represents a paradigm shift in mass spectrometry-based proteomics, particularly for post-translational modification analysis. Unlike DDA, which selectively fragments the most intense precursors, DIA fragments all co-eluting peptide ions within predefined mass-to-charge (m/z) windows simultaneously [3]. This fundamental difference enables more precise and accurate quantification with fewer missing values across samples and higher identification rates over a larger dynamic range [3]. For circadian studies requiring measurement across multiple time points, this improved data completeness is particularly valuable for detecting oscillations with statistical significance.
The DIA workflow for ubiquitinome analysis relies on comprehensive spectral libraries generated from enriched diGly-modified peptides. These libraries contain fragmentation patterns that enable accurate extraction and quantification of ubiquitinated peptides from complex DIA datasets [3]. When applied to circadian ubiquitinome profiling, this approach has demonstrated remarkable sensitivity, identifying 35,000 diGly peptides in single measurements—double the number achievable with DDA methods—while maintaining superior quantitative accuracy [3]. This depth of coverage is essential for capturing the dynamic nature of the ubiquitinome across circadian cycles.
Key Experimental Findings in Circadian Systems
Application of DIA-based ubiquitinome profiling to circadian biology has revealed unprecedented insights into temporal regulation of ubiquitination. Systems-wide investigation across the circadian cycle has uncovered hundreds of cycling ubiquitination sites and dozens of cycling ubiquitin clusters within individual membrane protein receptors and transporters [3]. These findings highlight new connections between metabolism and circadian regulation at the post-translational level [3]. The improved quantitative accuracy of DIA has been particularly valuable for detecting these oscillations, with coefficients of variation (CVs) significantly lower than those obtained with DDA methods [3].
Table 1: Performance Comparison of DIA vs DDA for Ubiquitinome Analysis
| Parameter | DIA Performance | DDA Performance |
|---|---|---|
| Distinct diGly peptides (single run) | 35,111 ± 682 | ~20,000 |
| Peptides with CV < 20% | 45% | 15% |
| Peptides with CV < 50% | 77% | Not reported |
| Total distinct peptides across 6 runs | ~48,000 | ~24,000 |
| Quantitative accuracy | Superior | Limited |
Experimental Protocols
Sample Preparation for Circadian Ubiquitinome Profiling
Cell Culture and Treatment:
- Culture HEK293 or U2OS cells under standard conditions
- Synchronize cells using serum shock or other synchronization methods
- Treat with proteasome inhibitor (10 µM MG132) for 4 hours to stabilize ubiquitinated proteins [3]
- For circadian time course experiments, collect samples at minimum 8 time points across 24-48 hours
Protein Extraction and Digestion:
- Lyse cells in urea-based lysis buffer (6 M urea, 2 M thiourea, 50 mM Tris-HCl pH 8.0) supplemented with protease and phosphatase inhibitors
- Reduce proteins with 5 mM dithiothreitol (60 minutes, room temperature)
- Alkylate with 15 mM iodoacetamide (30 minutes, room temperature in dark)
- Dilute urea concentration to <2 M and digest with trypsin (1:50 enzyme-to-protein ratio) overnight at 37°C
- Acidify with trifluoroacetic acid (0.5% final concentration) and desalt using C18 solid-phase extraction columns
diGly Peptide Enrichment:
- Use anti-diGly remnant motif (K-ε-GG) antibody (PTMScan Ubiquitin Remnant Motif Kit, CST)
- Incubate 1 mg peptide material with 31.25 µg antibody overnight at 4°C with gentle rotation [3]
- Wash beads extensively with ice-cold immunoaffinity purification buffer
- Elute diGly peptides with 0.15% trifluoroacetic acid
- Dry peptides in vacuum concentrator and reconstitute in 0.1% formic acid for LC-MS/MS analysis
Spectral Library Generation
Fractionation for Deep Library:
- Separate peptides by basic reversed-phase (bRP) chromatography into 96 fractions
- Concatenate fractions into 8 pools to reduce analytical time [3]
- Isolate fractions containing highly abundant K48-linked ubiquitin-chain derived diGly peptide separately to prevent competition during enrichment [3]
- Enrich diGly peptides from each pool separately
- Analyze using DDA method to build comprehensive spectral library
Table 2: Spectral Library Composition for Circadian Ubiquitinome Studies
| Library Source | Treatment | Number of diGly Peptides | Application |
|---|---|---|---|
| HEK293 cells | MG132 (10 µM, 4h) | 67,000 | Primary library |
| U2OS cells | MG132 (10 µM, 4h) | 53,000 | Extended coverage |
| U2OS cells | Untreated | 6,000 | Untreated system |
| Combined library | N/A | 93,684 unique peptides | Comprehensive analysis |
DIA Method Optimization for diGly Peptides
Liquid Chromatography:
- Use nanoflow LC system with C18 column (75 µm × 25 cm, 1.6 µm particles)
- Employ 90-minute linear gradient from 2% to 30% acetonitrile in 0.1% formic acid
- Maintain column temperature at 50°C for improved reproducibility
Mass Spectrometry:
- Utilize Orbitrap mass analyzer with higher-energy collisional dissociation (HCD)
- Implement 46 precursor isolation windows for comprehensive coverage [3]
- Set MS1 resolution to 120,000 with AGC target of 3e6
- Set MS2 resolution to 30,000 with AGC target of 1e6 [3]
- Use optimized collision energy stepped from 25-33% for diGly peptide fragmentation
Data Acquisition:
- For circadian time courses, use randomized block design to avoid batch effects
- Include quality control samples from pooled aliquots of all samples
- Acquire data in triplicate for each biological replicate to ensure statistical robustness
Data Analysis and Bioinformatics
Data Processing and Quantification
Process raw DIA files using specialized software (e.g., Spectronaut, DIA-NN, or Skyline) with the following parameters:
- Match peptides against the pre-generated spectral library containing >90,000 diGly peptides [3]
- Use hybrid spectral library approach by merging DDA library with direct DIA search results [3]
- Apply cross-run normalization to correct for technical variation
- Implement quality filters: CV < 20% across technical replicates, signal-to-noise ratio > 5
For circadian oscillation analysis:
- Apply JTK_Cycle or similar algorithm to identify significantly oscillating ubiquitination sites
- Set significance threshold at p < 0.05 with Benjamini-Hochberg correction for multiple testing
- Consider peptides with period between 20-28 hours as circadian
- Calculate phase and amplitude for significantly oscillating ubiquitination sites
Visualization and Pathway Analysis
Create visualizations to represent circadian ubiquitination patterns:
- Heatmaps of oscillating ubiquitination sites across time courses
- Phase distribution plots to identify temporal clustering of ubiquitination events
- Pathway enrichment analysis for ubiquitination sites oscillating in specific phases
- Protein-protein interaction networks to identify coordinated ubiquitination
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Circadian Ubiquitinome Profiling
| Reagent/Category | Specific Examples | Function in Workflow |
|---|---|---|
| Cell Lines | HEK293, U2OS | Model systems for ubiquitinome profiling |
| Inhibitors | MG132 (10 µM) | Proteasome inhibitor to stabilize ubiquitinated proteins |
| Antibodies | Anti-K-ε-GG (CST #5562) | Immunoaffinity enrichment of diGly-containing peptides |
| Enzymes | Trypsin (sequencing grade) | Protein digestion to generate diGly-containing peptides |
| Chromatography | C18 basic reversed-phase | Peptide fractionation for deep spectral libraries |
| Mass Spectrometry | Orbitrap-based instruments | High-resolution DIA data acquisition |
| Software | Spectronaut, Skyline, DIA-NN | DIA data processing and quantification |
| Database | PhosphoSitePlus | Validation of novel ubiquitination sites |
Regulatory Pathways and Molecular Relationships
The molecular relationship between ubiquitination and circadian regulation involves multiple interconnected pathways. The core circadian clock generates rhythms through transcription-translation feedback loops (TTFLs) that are extensively regulated by ubiquitin-mediated protein degradation [37]. Key E3 ubiquitin ligases, including UBR4, FBXL3, and β-TRCP, target core clock components for degradation, thus influencing clock timing and precision [37]. Conversely, the circadian clock regulates the expression and activity of ubiquitination machinery, creating bidirectional crosstalk that fine-tunes circadian physiology.
Diagram 1: Molecular relationships between the circadian clock and ubiquitin system, and corresponding experimental workflow for ubiquitinome profiling.
Advanced Applications in Chronotherapeutics
The insights gained from circadian ubiquitinome profiling have significant implications for chronotherapeutic drug development. Understanding the temporal regulation of protein ubiquitination enables better timing of drug administration to coincide with peak expression or activity of drug targets [38]. For example, research has revealed that immunotherapy administration in the morning leads to better outcomes, coinciding with circadian entry of lymphocytes into tumors [38]. Similarly, statins are most effective when taken at night because enzyme targets peak during this time [38].
The connection between ubiquitination and circadian regulation extends to various disease states. In cancer, the circadian clock controls cell division and genome maintenance, with key cell cycle regulators exhibiting circadian expression often regulated by CLOCK-BMAL1 heterodimer binding to E-box elements [39]. This interconnection offers targets for clock modulation and forms the basis for chronotherapy to optimize cancer treatment timing [39]. Emerging technologies including nanoparticle-based drug delivery systems and synthetic biology approaches are being developed to leverage these circadian principles for improved therapeutic outcomes [39] [40].
Troubleshooting and Technical Considerations
Low diGly Peptide Yield:
- Optimize antibody-to-peptide ratio (31.25 µg antibody per 1 mg peptide material) [3]
- Ensure proper peptide solubility during enrichment
- Include proteasome inhibitor treatment to increase ubiquitinated protein levels
Reduced Circadian Oscillation Detection:
- Implement proper cell synchronization before time course experiments
- Increase temporal resolution to at least 8 time points across 24 hours
- Include sufficient biological replicates (minimum n=3) for statistical power
DIA Data Quality Issues:
- Validate spectral library completeness for your specific biological system
- Optimize DIA window placement based on precursor distribution [3]
- Ensure adequate cycle time for proper chromatographic sampling
Quantification Inconsistencies:
- Implement robust normalization strategies to correct for technical variation
- Use hybrid spectral library approach to maximize identifications [3]
- Apply stringent quality filters during data processing
This comprehensive protocol for circadian ubiquitinome profiling using DIA mass spectrometry provides researchers with a robust framework for investigating the dynamic regulation of protein ubiquitination across circadian cycles. The methods detailed here enable unprecedented depth and quantitative accuracy in mapping oscillating ubiquitination events, opening new avenues for understanding circadian biology and developing chronotherapeutic interventions.
Tumor Necrosis Factor-alpha (TNFα) is a master cytokine that regulates inflammation, immunity, and cell survival, with dysregulated signaling contributing to cancer, autoimmune, and inflammatory diseases [41]. A critical mechanism governing TNFα signal transduction is protein ubiquitination, a reversible post-translational modification that controls the stability, activity, and interactions of key signaling proteins [3] [42]. Traditional methods for studying the ubiquitinome have been limited by depth, throughput, and quantitative accuracy. This application note details how Data-Independent Acquisition Mass Spectrometry (DIA-MS) now enables comprehensive, precise, and systems-level profiling of TNFα-induced ubiquitination dynamics, providing unprecedented insights into signaling pathway regulation.
The Power of DIA-MS for Ubiquitinome Analysis
Advantages Over Traditional Methods
Data-dependent acquisition (DDA) has been the conventional method for ubiquitinome analysis but suffers from stochastic precursor selection and substantial missing values across samples [3]. DIA-MS transforms this paradigm by systematically fragmenting all ions within sequential isolation windows, ensuring complete data recording and superior quantitative reproducibility [3] [21].
Table 1: Performance Comparison of DIA-MS vs. DDA for Ubiquitinome Analysis
| Parameter | DDA Method | DIA-MS Method | Improvement |
|---|---|---|---|
| Typical diGly Peptide IDs (Single Shot) | ~20,000 [3] | ~35,000 - 70,000 [3] [21] | 2-3.5x increase |
| Quantitative Precision (Median CV) | >20% CV for majority [3] | ~10% CV [21] | >2x more precise |
| Data Completeness | ~50% peptides without missing values [21] | Near-complete [3] | Dramatically improved |
Optimized Workflow for Deep Ubiquitinome Profiling
The following diagram illustrates a robust DIA-MS workflow for ubiquitinome analysis, integrating key optimizations for TNFα signaling studies:
Key optimizations include:
- SDC-based Lysis with Chloroacetamide (CAA): This approach inactivates deubiquitinases (DUBs) more effectively than urea buffers, preserving the native ubiquitinome and increasing peptide identifications by 38% [21].
- High-Input diGly Enrichment: Using 1-2 mg of peptide material with anti-K-ε-GG antibodies ensures deep coverage of low-stoichiometry ubiquitination events [3] [21].
- Optimized DIA Settings: Tailored precursor isolation windows (e.g., 46 windows) account for the unique characteristics of diGly peptides, which are often longer and carry higher charge states [3].
TNFα Signaling: A Ubiquitin-Controlled Pathway
The cellular response to TNFα is orchestrated by a complex cascade of ubiquitination events. Upon TNFα binding to its receptor TNFR1, adapter proteins are recruited, leading to the assembly of complex I. This complex serves as a platform for ubiquitination events that ultimately activate the NF-κB and MAPK pathways [43] [41] [44].
Table 2: Key Ubiquitination Events in TNFα Signaling
| Signaling Complex/Protein | Ubiquitin Linkage Type | Functional Consequence | Regulating Enzyme |
|---|---|---|---|
| Complex I (RIP1, TRAF2) | K63-linked, Linear [41] | Recruitment/Activation of TAK1 and IKK complexes [44] | TRAF2, TRAF6, cIAPs [44] |
| IKKγ/NEMO | Linear, K63-linked [42] | Activation of IKK complex [42] | LUBAC [41] |
| IKKβ | K63-linked [44] | IKK kinase activity [44] | TRAF6 [44] |
| IκBα | K48-linked [43] | Proteasomal degradation, NF-κB liberation [43] | SCF/βTrCP [43] |
| p105/NF-κB1 | K48-linked [41] | Processing to p50, TPL2 release [41] | Unknown E3 |
The following diagram illustrates the critical ubiquitination events within the TNFα signaling network:
Application: Uncovering Novel TNFα Biology with DIA-MS
The sensitivity of DIA-MS enables researchers to move beyond canonical pathway components and discover novel regulatory mechanisms. Applied to TNFα signaling, this workflow can:
- Capture Transient Ubiquitination Events: Identify short-lived ubiquitination on signaling intermediates immediately following TNFα stimulation [3].
- Distinguish Degradative vs. Non-degradative Ubiquitination: By correlating ubiquitinome data with proteome abundance measurements, researchers can determine whether ubiquitination leads to protein degradation or alters protein function, as demonstrated in studies of the deubiquitinase USP7 [21] [45].
- Reveal System-Wide Coordination: DIA-MS has uncovered coordinated ubiquitination clusters on individual membrane receptors and transporters, suggesting previously unrecognized regulatory mechanisms [3].
Table 3: Key Research Reagent Solutions for DIA Ubiquitinome Analysis
| Reagent/Resource | Function | Example Application |
|---|---|---|
| Anti-K-ε-GG Antibody | Immunoaffinity enrichment of ubiquitinated peptides following tryptic digestion [3] | Isolation of diGly-modified peptides from complex cell lysates |
| Sodium Deoxycholate (SDC) | Powerful detergent for efficient protein extraction and solubilization [21] | Cell lysis with concurrent DUB inhibition when used with CAA |
| Chloroacetamide (CAA) | Cysteine alkylating agent; rapidly inactivates DUBs without diGly mimicry [21] | Preservation of endogenous ubiquitination states during lysis |
| Recombinant TNFα | Defined and potent agonist for TNFR1-mediated signaling [41] | Stimulation of NF-κB and MAPK pathways in cultured cells |
| Proteasome Inhibitors (MG132) | Block degradation of K48-polyubiquitinated proteins, enriching ubiquitinated species [3] | Enhances detection of ubiquitination events that target proteins for degradation |
| Spectral Libraries | Curated collections of peptide spectra for peptide identification in DIA data [3] | Libraries >90,000 diGly peptides enable deep coverage |
The integration of optimized sample preparation, anti-diGly enrichment, and DIA-MS provides an unparalleled framework for comprehensively mapping TNFα-induced ubiquitination. This approach delivers the depth, precision, and reproducibility required to decipher the complex ubiquitin codes that control inflammatory signaling, opening new avenues for understanding disease mechanisms and developing targeted therapeutics.
Ubiquitin-Specific Protease 7 (USP7), also known as herpesvirus-associated ubiquitin-specific protease (HAUSP), has emerged as a critical regulator of oncogenic pathways and a promising therapeutic target in multiple cancers. As a deubiquitinating enzyme (DUB), USP7 reverses the ubiquitination of substrate proteins, thereby stabilizing them and preventing their proteasomal degradation [46]. USP7 exhibits significantly upregulated expression patterns across various malignancies, including chronic lymphocytic leukemia, prostate cancer, glioma, breast carcinomas, and non-small cell lung cancers, where its overexpression often correlates with tumor aggressiveness and poor patient prognosis [46] [47]. The enzyme regulates numerous cellular processes dysregulated in cancer, notably the p53-HDM2 axis, DNA damage response, epigenetic control, and immune signaling, positioning USP7 at the nexus of multiple oncogenic pathways [46].
The development of selective USP7 inhibitors represents an active area of cancer drug discovery. However, a significant challenge has been the comprehensive identification of USP7 substrates and the differentiation between degradative versus non-degradative ubiquitination events [15]. Traditional methods for DUB target identification often lacked the throughput, specificity, or quantitative precision needed to map the complex ubiquitination landscape regulated by USP7. This application note details an optimized workflow combining advanced sample preparation with data-independent acquisition mass spectrometry (DIA-MS) to achieve unprecedented depth and precision in profiling USP7 inhibition effects on the global ubiquitinome.
Methodological Advancements in Ubiquitinome Profiling
Optimized Sample Preparation for Enhanced Ubiquitinome Coverage
Robust ubiquitinome profiling begins with optimized sample preparation to preserve ubiquitination states while minimizing artifacts. A sodium deoxycholate (SDC)-based lysis protocol, supplemented with chloroacetamide (CAA), significantly improves ubiquitin site coverage compared to conventional urea-based methods [15]. The inclusion of CAA at high concentration immediately inactivates cysteine ubiquitin proteases through alkylation upon sample boiling, preventing artifactual deubiquitination during extraction. This SDC-based approach yields approximately 38% more K-GG remnant peptides compared to urea buffer while maintaining high enrichment specificity, with identification numbers reaching 30,000 K-GG peptides from just 2 mg of protein input material [15].
Table 1: Comparison of Lysis Buffer Performance for Ubiquitinomics
| Parameter | SDC-Based Buffer | Conventional Urea Buffer |
|---|---|---|
| K-GG Peptide Identifications | 26,756 (average) | 19,403 (average) |
| Enrichment Specificity | High | High |
| Cysteine Protease Inactivation | Immediate with CAA | Slower |
| Di-carbamidomethylation Artifacts | None detected | Potential with iodoacetamide |
| Recommended Protein Input | 2 mg | >2 mg |
| Reproducibility (CV < 20%) | Significantly improved | Lower |
DIA-MS with Neural Network-Based Data Processing
The integration of data-independent acquisition mass spectrometry (DIA-MS) with specialized computational processing represents a quantum leap in ubiquitinome profiling capabilities. When benchmarked against conventional data-dependent acquisition (DDA), the DIA-MS workflow more than triples ubiquitinated peptide identifications, increasing from approximately 21,434 with DDA to 68,429 K-GG peptides per single MS run [15]. This dramatic improvement in coverage is coupled with exceptional quantitative precision, demonstrating median coefficients of variation (CV) of approximately 10% across replicates.
Critical to this advancement is the implementation of the DIA-NN software with a specialized scoring module optimized for modified peptide identification [15]. The "library-free" analysis mode, which searches directly against sequence databases without requiring experimentally-generated spectral libraries, achieves similar performance to analyses using ultra-deep fractionated libraries while offering greater flexibility and reduced preprocessing requirements. This combination provides the robustness necessary for large-scale temporal studies of USP7 inhibition dynamics.
Experimental Protocol: Comprehensive USP7 Target Identification
Cell Culture and USP7 Inhibition
- Cell Line Selection: Utilize appropriate cancer cell lines (e.g., HCT116 colon carcinoma or other relevant models) based on research context.
- USP7 Inhibitor Treatment: Apply selective USP7 inhibitors (e.g., P22077, P50429, or newer generation compounds) at optimized concentrations. Include DMSO vehicle controls.
- Temporal Design: Implement a time-course experiment (e.g., 0, 15, 30, 60, 120 minutes) to capture rapid ubiquitination changes following USP7 inhibition.
- Optional Proteasome Inhibition: For maximal ubiquitin signal detection, treat cells with MG-132 (10 µM for 6 hours) prior to harvesting, though this may be omitted for degradation kinetics studies.
Protein Extraction and Digestion
- Lysis: Harvest cells in SDC lysis buffer (4% SDC, 40 mM chloroacetamide, 100 mM Tris-HCl pH 8.5) followed by immediate boiling at 95°C for 10 minutes.
- Protein Quantification: Determine protein concentration using bicinchoninic acid (BCA) assay.
- Digestion: Aliquot 2 mg protein per sample, reduce with 5 mM dithiothreitol (37°C, 30 minutes), alkylate with 10 mM CAA (room temperature, 30 minutes in darkness), then digest with Lys-C (1:100 enzyme:substrate, 37°C, 2 hours) followed by trypsin (1:50, 37°C, overnight).
- Acidification: Precipitate SDC by acidifying with 1% final concentration trifluoroacetic acid, followed by centrifugation at 10,000 × g for 10 minutes.
K-GG Peptide Enrichment
- Immunoaffinity Purification: Reconstitute digested peptides in immunoaffinity purification (IAP) buffer (50 mM MOPS-NaOH pH 7.2, 10 mM Na2HPO4, 50 mM NaCl) and incubate with anti-K-GG remnant motif antibody-conjugated beads (commercial sources) for 90 minutes at 4°C with gentle rotation.
- Washing: Pellet beads and wash sequentially with IAP buffer, followed by water.
- Elution: Elute K-GG peptides with 0.1% trifluoroacetic acid.
- Desalting: Desalt peptides using C18 solid-phase extraction tips, then concentrate by vacuum centrifugation.
LC-MS/MS Analysis and Data Processing
- Chromatography: Separate peptides using a 75-minute nanoLC gradient on a reverse-phase C18 column.
- Mass Spectrometry: Acquire data using DIA method with 30-40 variable windows covering 400-1000 m/z range.
- Data Processing: Analyze raw files using DIA-NN software in "library-free" mode against appropriate protein sequence databases with K-GG modification specified.
- Statistical Analysis: Apply intensity normalization and significance thresholds (e.g., fold-change >2, adjusted p-value <0.05) to identify significantly altered ubiquitination sites.
Figure 1: Experimental workflow for high-resolution profiling of USP7 inhibition effects on the ubiquitinome.
Key Research Reagent Solutions
Table 2: Essential Research Reagents for USP7 Ubiquitinome Profiling
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| USP7 Inhibitors | P22077, P50429, HBX 41,108 | Selective targeting of USP7 catalytic activity; tool compounds for mechanistic studies |
| Cell Lines | HCT116, AGS gastric carcinoma, CNE2Z nasopharyngeal carcinoma | Model systems for studying USP7 biology in relevant cancer contexts |
| Lysis Buffers | SDC buffer (4% SDC, 40 mM CAA, 100 mM Tris-HCl pH 8.5) | Optimal protein extraction with simultaneous protease inactivation |
| Enrichment Reagents | Anti-K-GG remnant motif antibody beads | Immunoaffinity purification of ubiquitinated peptides prior to MS analysis |
| Mass Spectrometry | DIA-MS optimized methods, DIA-NN software | High-resolution ubiquitinome profiling and computational analysis |
| Validation Reagents | USP7 binding pocket mutants (TRAF: D164A/W165A; Ubl2: D762R/D764R) | Mechanistic studies of USP7-substrate interactions |
Analytical Results: System-Level View of USP7 Inhibition
Application of the optimized DIA-MS workflow to USP7 inhibitor-treated cells reveals comprehensive ubiquitinome remodeling with remarkable temporal resolution. Within minutes of USP7 inhibition, hundreds of proteins show significantly increased ubiquitination, with the number of altered sites reaching maximum between 30-120 minutes post-treatment [15]. However, critical analysis integrating both ubiquitination changes and corresponding protein abundance measurements demonstrates that only a fraction of these ubiquitination events lead to substantive protein degradation, highlighting the dual role of ubiquitination in both proteolytic and non-proteolytic signaling.
Table 3: Quantitative Ubiquitinome Changes Following USP7 Inhibition
| Parameter | DDA-MS Performance | DIA-MS Performance | Biological Outcome |
|---|---|---|---|
| K-GG Peptide IDs | 21,434 (average) | 68,429 (average) | >8,000 proteins monitored simultaneously |
| Quantitative Precision | Moderate (CV ~15-20%) | High (CV ~10%) | High-confidence identification of significant changes |
| Temporal Resolution | Limited by missing values | Robust across time series | Rapid ubiquitination increases within minutes |
| Proteome Coverage | ~4,000-6,000 proteins | ~8,000+ proteins | Integrated analysis of ubiquitination & abundance |
| USP7-dependent Substrates | Limited identification | Hundreds identified | Includes both degradative & regulatory ubiquitination |
The power of this approach is exemplified by its ability to distinguish direct USP7 substrates from indirect effects. True USP7 targets typically demonstrate: (1) rapid increase in ubiquitination following inhibitor treatment, (2) specificity for USP7 compared to other DUB inhibitions, (3) presence of USP7 binding motifs (P/A/ExxS for TRAF domain; KxxxK for Ubl2 domain), and (4) correlation with functional outcomes such as protein stabilization or translocation [48].
Pathway Analysis and Biological Validation
USP7-Regulated Signaling Networks
The integrated ubiquitinome and proteome data reveal USP7's multifaceted role in cellular regulation. Confirmed USP7 substrates span multiple functional categories:
- DNA Damage Response: p53, HDM2, CHK1, Claspin, RNF168 [46]
- Epigenetic Regulation: DNMT1, UHRF1, TIP60, PHF8, components of PRC1 and PRC2 complexes [46]
- Immune Signaling: NF-κB, TRAF6, IKKγ [46]
- Novel Targets: DDX24, DHX40 (RNA helicases), TRIP12 (E3 ligase) [48]
Figure 2: USP7 regulates multiple cellular pathways through specific substrate stabilization.
Orthogonal Validation Approaches
Candidate USP7 substrates identified through DIA-MS profiling require orthogonal validation:
- Co-Immunoprecipitation: Validate physical interactions between USP7 and candidate substrates using wild-type versus binding pocket mutants (TRAF domain: D164A/W165A; Ubl2 domain: D762R/D764R) [48].
- Cycloheximide Chase Assays: Measure protein half-life with and without USP7 inhibition to confirm stabilization effects.
- Cellular Localization Studies: Monitor nucleocytoplasmic shuttling for substrates like PTEN and FOXO4 where deubiquitination regulates translocation [46].
- Functional Rescue: Demonstrate reversal of phenotypic effects through candidate substrate co-expression.
The integration of optimized sample preparation with DIA-MS ubiquitinome profiling represents a transformative approach for comprehensive identification of USP7 substrates and characterization of inhibitor mechanisms. This workflow provides unprecedented depth, quantitative precision, and temporal resolution, enabling researchers to distinguish direct USP7 targets from secondary effects and differentiate between degradative versus non-degradative ubiquitination events. The methodological advances detailed in this application note offer a robust framework for mode-of-action studies for USP7-targeted therapeutics and establish a paradigm applicable to the broader study of DUB biology and targeted protein degradation strategies.
The ability to simultaneously monitor ubiquitination changes and protein abundance across thousands of proteins at high temporal resolution positions DIA-MS ubiquitinome profiling as an essential tool for drug discovery efforts targeting USP7 and other DUBs in cancer and other diseases.
Optimizing DIA-Ubiquitinomics: Troubleshooting Common Pitfalls and Enhancing Performance
In data-independent acquisition (DIA) mass spectrometry for ubiquitinome research, sample preparation quality directly determines the success or failure of the entire experiment. The analysis of endogenous protein ubiquitination presents unique challenges due to the low stoichiometry of the modification and the presence of abundant interfering compounds that can severely compromise peptide yield and quantification accuracy [3] [2]. Even with advanced DIA instrumentation and computational tools, failures at the sample preparation stage can lead to insufficient diGly peptide identifications, poor quantitative reproducibility, and ultimately, biologically misleading results. This application note details targeted methodologies to overcome the most prevalent sample preparation failures in ubiquitinome analysis, with specific protocols optimized for DIA-based workflows.
Troubleshooting Low Peptide Yield
Optimization of Input Material and Enrichment Reagents
Insufficient peptide yield following anti-K-ε-GG enrichment remains a primary bottleneck in ubiquitinome studies. This failure typically stems from suboptimal ratios between starting material and enrichment reagents, or inadequate handling of highly abundant ubiquitin-derived peptides that compete for antibody binding sites.
Table 1: Titration Parameters for Optimal diGly Peptide Yield
| Parameter | Suboptimal Condition | Optimized Condition | Impact on Yield |
|---|---|---|---|
| Peptide Input | ≤ 500 µg | 1-2 mg | Increases identifications by >50% [15] |
| Anti-K-ε-GG Antibody | Full vial (250 µg) | 1/8 vial (31.25 µg) per 1 mg peptide | Maximizes binding efficiency [3] |
| K48-Ub Chain Peptide Handling | No separation | bRP separation of high-abundance K48 peptides | Reduces competition; improves coverage [3] |
| Injected Enriched Material | 100% | 25% of total | Maintains depth while conserving sample [3] |
Experimental Protocol: Optimized diGly Peptide Enrichment
- Cell Lysis and Digestion: Extract proteins using SDC lysis buffer (2% SDC, 40 mM chloroacetamide in 100 mM Tris-HCl, pH 8.5) with immediate incubation at 95°C for 5 minutes to rapidly inactivate deubiquitinases [15].
- Protein Digestion: Digest proteins using trypsin (1:50 w/w) overnight at 37°C. Acidify with TFA to a final concentration of 1% to precipitate SDC, then centrifuge at 12,000 × g for 10 minutes to remove the detergent.
- Peptide Desalting: Desalt peptides using C18 solid-phase extraction cartridges according to manufacturer's instructions. Evaporate solvents and reconstitute peptides in immunoaffinity purification (IAP) buffer (50 mM MOPS, 10 mM Na2HPO4, 50 mM NaCl, pH 7.2).
- Antibody-based Enrichment: Incubate 1 mg of peptide material with 31.25 µg of anti-K-ε-GG antibody (Cell Signaling Technology) conjugated to protein A agarose beads for 2 hours at 4°C with gentle agitation [3].
- Washing and Elution: Wash beads three times with IAP buffer and twice with HPLC-grade water. Elute diGly peptides with 50 µL of 0.15% TFA twice, pooling eluents.
- Sample Cleanup: Desalt eluted peptides using C18 StageTips prior to LC-MS/MS analysis.
Figure 1: Optimized sample preparation workflow for deep ubiquitinome coverage, highlighting critical steps (yellow) and specialized handling for abundant interferents (green).
On-Bead TMT Labeling for Multiplexed Experiments
For TMT-based multiplexed experiments, traditional in-solution labeling following diGly enrichment results in substantial peptide loss. The UbiFast protocol addresses this through on-antibody TMT labeling, which significantly improves yield and reduces sample requirements.
Experimental Protocol: UbiFast On-Bead TMT Labeling
- diGly Peptide Enrichment: Enrich diGly peptides from 500 µg of peptide material per condition using anti-K-ε-GG antibody as described in Section 2.1.
- On-Bead TMT Labeling: While peptides are bound to antibodies, resuspend beads in 100 µL of 100 mM HEPES (pH 8.5). Add 0.4 mg of TMT reagent dissolved in 50 µL anhydrous acetonitrile to each sample. Incubate for 10 minutes at room temperature with vigorous shaking [49].
- Reaction Quenching: Add hydroxylamine to a final concentration of 5% (v/v) and incubate for 15 minutes to quench the reaction.
- Peptide Combining and Cleanup: Combine TMT-labeled samples from different conditions. Wash beads three times with IAP buffer, then elute with 0.15% TFA. Desalt pooled peptides using C18 StageTips.
- LC-MS Analysis: Analyze labeled peptides by LC-MS/MS using DIA acquisition methods.
Table 2: Performance Comparison of Labeling Methods
| Method | Peptide Spectrum Matches | Relative Yield (% diGly Peptides) | Labeling Efficiency | Sample Input |
|---|---|---|---|---|
| On-Bead TMT (UbiFast) | 6,087 | 85.7% | >92% | 500 µg [49] |
| In-Solution TMT | 1,255 | 44.2% | >98% | 1 mg [49] |
| Label-Free Quantification | ~4,000 | ~80% | Not Applicable | 1-2 mg [3] |
Addressing Chemical Interference and Background Contamination
Lysis Buffer Optimization to Reduce Artifacts
The choice of lysis buffer significantly impacts the specificity of ubiquitinome analysis. Traditional urea-based buffers can promote carbamylation and fail to fully inactivate deubiquitinases, leading to ubiquitin loss during preparation. Furthermore, iodoacetamide alkylation can cause di-carbamidomethylation of lysine residues, creating a +114.0429 Da mass shift that mimics the diGly remnant (+114.0429 Da) and results in false identifications [15].
Experimental Protocol: SDC-Based Lysis for Ubiquitinome Analysis
- SDC Lysis Buffer Preparation: Prepare fresh SDC lysis buffer containing 2% sodium deoxycholate, 40 mM chloroacetamide, and 10 mM Tris(2-carboxyethyl)phosphine (TCEP) in 100 mM Tris-HCl (pH 8.5).
- Cell Lysis: Add SDC lysis buffer directly to cell pellets (approximately 100 µL per 1 million cells). Vortex vigorously until the solution becomes homogeneous and viscous.
- Thermal Denaturation and Alkylation: Immediately incubate samples at 95°C for 5 minutes in a thermal mixer with shaking at 1000 rpm. This simultaneous step denatures proteins and alkylates cysteine residues with chloroacetamide, which does not produce the di-carbamidomethylation artifact [15].
- Cooling and Dilution: Cool samples to room temperature. Dilute the SDC concentration to 1% using 100 mM Tris-HCl (pH 8.5) before proceeding with tryptic digestion.
Figure 2: Comparison of lysis buffer strategies showing how SDC with CAA prevents artifacts and improves coverage compared to traditional urea methods.
Specificity Enhancements for Ubiquitinome Enrichment
The standard K-ε-GG antibody cannot distinguish between ubiquitin and ubiquitin-like modifications (ISG15, NEDD8), introducing potential misidentification. Furthermore, context-dependent antibody bias can reduce enrichment efficiency for certain diGly peptide sequences.
Experimental Protocol: UbiSite Workflow for Enhanced Specificity
- LysC Digestion: Digest proteins first with LysC (1:100 enzyme-to-protein ratio) in 2 M urea, 50 mM Tris-HCl (pH 8.5) for 4 hours at 30°C with agitation. This generates longer ubiquitin-derived remnants containing the LRRL epitope instead of the diGly motif [50].
- Immunoaffinity Enrichment: Enrich for the 13-amino acid ubiquitin remnant (K-ε-LRRLVLHLTSE) using the UbiSite antibody. Incubate digested peptides with antibody-conjugated beads for 2 hours at 4°C.
- Trypsin Digestion: While peptides are bound to beads, add trypsin (1:50 w/w) in 50 mM Tris-HCl (pH 8.5) and incubate overnight at 37°C. This secondary digestion converts the enriched peptides to the standard diGly-modified peptides compatible with routine LC-MS analysis.
- Peptide Elution and Cleanup: Elute peptides with 0.15% TFA and desalt using C18 StageTips prior to DIA analysis.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Robust Ubiquitinome Sample Preparation
| Reagent/Category | Specific Example | Function and Application |
|---|---|---|
| Anti-diGly Antibody | PTMScan Ubiquitin Remnant Motif (K-ε-GG) Kit (Cell Signaling Technology) | Gold-standard enrichment of tryptic diGly peptides; compatible with label-free and SILAC quantification [3] [2] |
| Lysis Buffer | 2% SDC, 40 mM CAA, 10 mM TCEP in 100 mM Tris-HCl (pH 8.5) | Effective protein extraction with rapid DUB inactivation; prevents alkylation artifacts [15] |
| Alternative Antibody | UbiSite Antibody (Targeting K-ε-LRRLVLHLTSE) | Specific enrichment of ubiquitin-derived peptides excluding ISG15 and NEDD8 modifications [50] |
| TMT Reagents | TMTpro 16plex (Thermo Fisher Scientific) | Isobaric mass tags for multiplexed quantification; used in UbiFast on-bead labeling protocol [49] |
| Chromatography Resin | C18 StageTips (Thermo Fisher Scientific) | Micro-solid phase extraction for sample cleanup and concentration between enrichment steps [3] [15] |
Concluding Remarks
Successful ubiquitinome analysis by DIA-MS depends critically on overcoming sample preparation challenges that compromise peptide yield and introduce chemical interference. The optimized protocols detailed herein—including SDC-based lysis with chloroacetamide alkylation, titrated antibody enrichment, strategic handling of abundant K48-linked ubiquitin peptides, and on-bead TMT labeling—collectively address the most common failure points. Implementation of these methodologies enables reproducible identification of >30,000 distinct diGly peptides in single measurements, transforming the scale and reliability with which ubiquitin signaling can be investigated in biological and translational research contexts [3] [15].
In the field of ubiquitinome analysis, data-independent acquisition (DIA) mass spectrometry has emerged as a powerful alternative to data-dependent acquisition (DDA), offering superior quantitative accuracy, reproducibility, and data completeness. However, the full potential of DIA is often undermined by suboptimal configuration of critical acquisition parameters, particularly precursor isolation window widths and cycle times. This application note details a optimized DIA workflow specifically tailored for ubiquitinome analysis, demonstrating how proper parameter configuration enables the identification of over 35,000 distinct diGly peptides in single measurements—doubling the identification rates achievable with DDA. We provide detailed protocols for library generation, method optimization, and sample preparation, along with performance metrics that highlight the transformative impact of parameter optimization for researchers investigating ubiquitin signaling in targeted protein degradation and circadian biology.
Data-independent acquisition mass spectrometry represents a paradigm shift in proteomic analysis, particularly for post-translational modification studies such as ubiquitinome analysis. Unlike data-dependent acquisition, which selects specific precursors based on intensity, DIA systematically fragments all peptides within predefined isolation windows, producing comprehensive fragment ion maps that lead to more complete data sets with fewer missing values across samples [3]. For ubiquitinome research, this approach is particularly valuable due to the low stoichiometry of ubiquitination and the dynamic nature of ubiquitin signaling in cellular processes. The diGly remnant antibody-based enrichment combined with DIA mass spectrometry has enabled unprecedented depth in ubiquitinome coverage, facilitating investigations into diverse biological processes including circadian regulation, TNF signaling, and targeted protein degradation mechanisms [13] [3].
The fundamental challenge in DIA method development lies in optimizing two interdependent parameters: precursor isolation window widths and cycle times. Window width determines the m/z range isolated for fragmentation, while cycle time encompasses the duration required to acquire one full set of MS1 and MS2 scans across all windows. Inadequate configuration of these parameters directly impacts sensitivity, quantitative accuracy, and proteome coverage. For ubiquitinome analysis specifically, this optimization must account for the unique characteristics of diGly-modified peptides, which often exhibit longer sequence lengths and higher charge states due to impeded C-terminal cleavage at modified lysine residues [3].
Optimized DIA Parameters for Ubiquitinome Analysis
Window Width Configuration
Precursor isolation window configuration represents a critical decision point in DIA method development that directly influences spectral quality and identification rates. Based on systematic optimization experiments, DIA methods employing 46 precursor isolation windows provide optimal performance for ubiquitinome analysis [3]. The window widths should be tailored to the specific characteristics of diGly-modified peptides, which often require adjustments from standard proteomic methods.
Table 1: Optimized DIA Window Parameters for Ubiquitinome Analysis
| Parameter | Recommended Setting | Effect of Misconfiguration |
|---|---|---|
| Number of Windows | 46 windows | Too few windows reduces specificity; too many decreases sensitivity |
| Window Placement | Staggered/overlapping pattern | Non-overlapping windows create coverage gaps |
| MS2 Resolution | 30,000 | Lower resolution compromises peptide identification |
| m/z Coverage | 400-1200 | Incomplete coverage misses relevant diGly peptides |
| Collision Energy | Fixed (27 NCE) | Variable energy disrupts fragmentation consistency |
Experimental data demonstrates that optimized window widths increase diGly peptide identifications by approximately 13% compared to standard full proteome methods [3]. The use of staggered window patterns, where adjacent windows slightly overlap, ensures continuous coverage across the m/z range and prevents "forbidden zones" where peptides might escape fragmentation [51] [52].
Cycle Time Optimization
Cycle time must be carefully balanced to maintain adequate peptide sampling while preserving spectral quality. For Orbitrap-based instruments, a cycle time of approximately 2.5-3 seconds provides optimal balance, allowing at least 8-10 data points across a typical chromatographic peak [51]. Excessively short cycle times compromise fragment scan quality, while excessively long cycles undersample eluting peptides, both reducing quantitative accuracy. The optimized method incorporates 46 windows with 30,000 resolution MS2 scans, achieving this target cycle time while maintaining high-quality fragmentation data [3].
Experimental Protocols
Comprehensive Spectral Library Generation
Principle: Spectral libraries enable accurate extraction of diGly peptide signals from DIA data. Generation of comprehensive, sample-specific libraries is essential for high-sensitivity ubiquitinome analysis.
Protocol:
- Cell Culture and Treatment: Culture HEK293 or U2OS cells in appropriate media. Treat with 10 µM MG132 proteasome inhibitor for 4 hours to enhance ubiquitinated peptide recovery.
- Protein Extraction and Digestion: Lyse cells in 9M urea buffer containing 50 mM Tris (pH 8), 75 mM NaCl, and protease inhibitors. Reduce proteins with 5 mM dithiothreitol (30 min, 55°C), alkylate with 10 mM iodoacetamide (30 min, room temperature, dark), and quench with additional dithiothreitol. Digest with sequencing-grade trypsin overnight at 1:50 enzyme-to-substrate ratio [51].
- Peptide Fractionation: Separate peptides by basic reversed-phase chromatography into 96 fractions. Concatenate into 8 primary fractions, with separate processing for fractions containing abundant K48-linked ubiquitin-chain derived diGly peptides to prevent antibody saturation [3].
- diGly Peptide Enrichment: Enrich diGly peptides using anti-diGly remnant antibody (e.g., PTMScan Ubiquitin Remnant Motif Kit). Use 31.25 µg antibody per 1 mg peptide input for optimal results [3].
- Library Acquisition: Analyze fractions using DDA mass spectrometry with settings matching planned DIA methods (fixed collision energy, similar resolution settings).
Performance Metrics: This protocol generates spectral libraries containing >90,000 diGly peptides, enabling identification of 35,000+ diGly peptides in single DIA measurements [3].
DIA Method Implementation for Ubiquitinome Analysis
Principle: DIA methods must be specifically optimized for diGly peptide characteristics, including their tendency toward longer sequences and higher charge states.
Protocol:
- Instrument Configuration: Utilize Orbitrap-based mass spectrometer (e.g., Q-Exactive HF) coupled to nanoUPLC system.
- Chromatography: Employ 90-minute gradient from 5% to 35% solvent B (0.1% formic acid in 98% acetonitrile) with in-house packed C18 column (75μm ID, 300mm length).
- MS1 Settings: Acquire at 60,000 resolution with AGC target of 3e6 and maximum injection time of 100 ms.
- DIA Settings: Implement 46 staggered windows covering 400-1200 m/z range. Acquire MS2 spectra at 30,000 resolution with AGC target of 1e6 and maximum injection time of 55 ms. Use fixed collision energy (27 NCE) [3].
- Sample Loading: Inject 25% of enriched diGly material (from 1 mg peptide input) to balance depth and throughput.
Validation: The optimized method identifies 33,409 ± 605 distinct diGly sites in single measurements of MG132-treated HEK293 samples, with 77% of peptides showing coefficients of variation below 50% across replicates [3].
DIA Ubiquitinome Analysis Workflow
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for DIA Ubiquitinome Analysis
| Reagent/Equipment | Function | Specifications |
|---|---|---|
| Anti-diGly Remnant Antibody | Enrichment of ubiquitinated peptides | PTMScan Ubiquitin Remnant Motif Kit; 31.25 μg per 1 mg peptide input |
| Proteasome Inhibitor | Enhances ubiquitinated peptide recovery | MG132, 10 μM, 4-hour treatment |
| Trypsin | Protein digestion | Sequencing grade, 1:50 enzyme-to-substrate ratio |
| C18 Chromatography Material | Peptide separation | ReproSil-Pur C18 beads, 3 μm, 300 mm column length |
| Orbitrap Mass Spectrometer | DIA data acquisition | Q-Exactive HF or similar; 30,000+ resolution MS2 capability |
| Urea Lysis Buffer | Protein extraction | 9 M urea, 50 mM Tris, 75 mM NaCl, protease inhibitors |
Performance Assessment and Comparative Analysis
Quantitative Advantages of Optimized DIA
Systematic evaluation demonstrates that the optimized DIA workflow significantly outperforms traditional DDA methods for ubiquitinome analysis. In replicate analyses of MG132-treated HEK293 cells, DIA identified approximately 36,000 distinct diGly peptides across all replicates, with 45% exhibiting coefficients of variation below 20% and 77% below 50% [3]. In contrast, DDA methods identified only 20,000 diGly peptides with just 15% showing CVs below 20%. This represents a 80% improvement in identification depth and a 3-fold enhancement in quantitative reproducibility.
Impact of Parameter Missteps
Suboptimal window configuration directly impairs data quality. Non-staggered window layouts create gaps in precursor coverage, while improperly sized windows (either too wide or too narrow) reduce specificity or sensitivity. Excessive cycle times lead to undersampled chromatographic peaks, while insufficient cycle times compromise fragment scan quality. These missteps collectively reduce ubiquitinome coverage, quantitative accuracy, and experimental reproducibility.
DIA vs DDA Performance Comparison
Applications in Ubiquitin Signaling Research
The optimized DIA ubiquitinome workflow has enabled groundbreaking investigations into diverse biological systems. Applied to TNF signaling, the method comprehensively captured known ubiquitination sites while identifying numerous novel targets [3]. In circadian biology research, DIA facilitated the identification of hundreds of cycling ubiquitination sites and dozens of cycling ubiquitin clusters within individual membrane protein receptors and transporters, revealing novel connections between metabolic regulation and circadian timing [3] [9]. For targeted protein degradation studies, the approach enables rapid establishment of mechanism of action for various TPD modalities, including PROTACs and molecular glues [13].
Optimal configuration of window widths and cycle times is fundamental to successful DIA ubiquitinome analysis. The methods detailed herein provide a robust framework for achieving unprecedented depth and quantitative accuracy in ubiquitin signaling studies. By adhering to these optimized parameters—employing staggered window designs, appropriate cycle times, and comprehensive spectral libraries—researchers can overcome common pitfalls and leverage the full potential of DIA mass spectrometry. The resulting enhancements in sensitivity, reproducibility, and coverage will accelerate discoveries in ubiquitin biology, targeted protein degradation, and circadian regulation.
In the field of data-independent acquisition (DIA) mass spectrometry for ubiquitinome analysis, spectral libraries serve as essential reference maps that enable accurate identification and quantification of ubiquitinated peptides. Unlike data-dependent acquisition (DDA), DIA simultaneously fragments all ions within predefined mass-to-charge windows, generating complex datasets that require specialized libraries for deconvolution [3] [27]. The quality and relevance of these spectral libraries directly determine the success of peptide identification and quantification, yet researchers frequently encounter significant challenges related to biological mismatches that compromise data quality.
The fundamental issue lies in the specificity of ubiquitin signaling, which exhibits remarkable variation across tissue types, physiological states, and species. When spectral libraries derived from one biological context (e.g., human liver tissue) are applied to DIA data from another (e.g., mouse brain tissue), the mismatch can severely degrade performance, leading to reduced identification rates, inflated false discovery rates, and biologically meaningless results [53]. This technical note examines the sources and consequences of spectral library mismatches in DIA ubiquitinomics and provides structured solutions for generating context-appropriate libraries to ensure biologically valid results.
The Impact of Library Mismatches on Ubiquitinome Data Quality
Spectral library mismatches manifest in several forms, each with distinct consequences for data integrity and biological interpretation. The following table summarizes the primary mismatch types and their impacts on DIA ubiquitinome studies:
Table 1: Common Spectral Library Mismatches and Their Consequences in DIA Ubiquitinomics
| Mismatch Type | Description | Experimental Consequence | Impact on Data Quality |
|---|---|---|---|
| Tissue Mismatch | Using libraries derived from different tissues (e.g., liver library for brain tissue analysis) | Missed tissue-specific ubiquitination sites; inaccurate quantification | Poor coverage of relevant biomarkers; failure to detect biologically significant ubiquitination events [53] |
| Species Mismatch | Applying human libraries to mouse models or custom biological strains | Reduced peptide identification confidence; decreased match rates | Low identification counts; inability to leverage animal models effectively [53] |
| Sample Preparation Bias | Library and samples prepared with different lysis buffers or digestion protocols | Altered peptide recovery; incomplete representation of ubiquitinome | Inconsistent quantification; missing values across samples [15] |
| Instrumental Divergence | Libraries built on different LC-MS platforms or with varying gradient lengths | Retention time misalignment; fragmentation pattern discrepancies | Peak misassignment; reduced quantitative accuracy [53] |
The practical consequences of these mismatches are substantial. For instance, a study investigating ubiquitination in Huntington's disease mouse models required specialized sample preparation and library generation to accurately characterize differential ubiquitination of wild-type and mutant Huntingtin in brain tissue [54]. Using a generic library would have missed critical disease-specific ubiquitination sites at K6 and K9 residues of mutant Huntingtin, significantly compromising the biological insights.
Strategic Solutions for Biologically-Matched Spectral Libraries
Library Selection Framework
Researchers face three primary pathways for spectral library generation, each with distinct advantages and limitations. The following decision framework guides the selection process based on project requirements:
Table 2: Spectral Library Selection Framework for DIA Ubiquitinomics
| Library Type | Coverage | Biological Relevance | Implementation Time | Recommended Use Cases |
|---|---|---|---|---|
| Public Libraries (e.g., SWATHAtlas) | Moderate | Generic | Fast (days) | Method development; studies of common cell lines; preliminary analyses [53] |
| Project-Specific Libraries | High | Perfectly matched to experimental conditions | Long (weeks to months) | Complex tissues; biomarker discovery; species-specific applications [53] |
| Hybrid Approach (public + custom DDA) | High | Balanced relevance and efficiency | Medium (weeks) | Studies with partial prior knowledge; targeted hypothesis testing [53] |
Experimental Protocol for Generating Project-Specific Ubiquitinome Libraries
For research requiring the highest data quality, project-specific spectral libraries offer significant advantages. The following protocol details the generation of comprehensive ubiquitinome libraries optimized for DIA analysis:
Sample Preparation and Protein Extraction
Begin with optimized protein extraction to maximize ubiquitinome coverage. The SDC-based lysis protocol has demonstrated superior performance for ubiquitinomics applications:
SDC Lysis Buffer Preparation: Prepare sodium deoxycholate (SDC) lysis buffer containing 5% SDC, 50 mM Tris-HCl (pH 8.5), and 50 mM chloroacetamide (CAA). The high CAA concentration rapidly inactivates cysteine deubiquitinases by alkylation, preserving endogenous ubiquitination states [15].
Cell Lysis and Protein Extraction:
- Add SDC lysis buffer directly to cell pellets or ground tissue samples
- Immediately boil samples at 95°C for 10 minutes to further inhibit protease and deubiquitinase activity
- Sonicate samples to ensure complete disruption and DNA shearing
- Centrifuge at 14,000 × g for 20 minutes at 4°C to remove insoluble material [15]
Protein Quantification: Determine protein concentration using BCA assay. For library generation, typically 2-4 mg of protein input is recommended to ensure sufficient coverage of low-abundance ubiquitinated peptides [15].
Protein Digestion and Peptide Fractionation
Protein Digestion:
- Dilute SDC lysate with 50 mM Tris-HCl (pH 8.0) to reduce SDC concentration to <1%
- Add trypsin at 1:50 enzyme-to-substrate ratio
- Digest overnight at 37°C with agitation
- Acidify with trifluoroacetic acid (TFA) to final 1% concentration to precipitate SDC
- Centrifuge and collect supernatant containing peptides [15]
High-pH Reversed-Phase Fractionation:
- Separate peptides using basic reversed-phase chromatography (pH 10)
- Collect 96 fractions and concatenate into 8-12 pooled fractions to reduce analytical time
- For proteasome inhibitor-treated samples (e.g., MG132), consider separating fractions containing highly abundant K48-linked ubiquitin-chain derived diGly peptides to prevent competition during enrichment [3]
DiGly Peptide Enrichment and LC-MS/MS Analysis
Antibody-Based Enrichment:
- Use anti-diGly remnant motif (K-ε-GG) antibodies for immunoaffinity purification
- Optimize antibody-to-peptide ratio (typically 31.25 μg antibody per 1 mg peptides) [3]
- Incubate peptides with antibody-conjugated beads for 2 hours at 4°C with rotation
- Wash extensively with PBS to remove non-specifically bound peptides
- Elute diGly-modified peptides with 0.1% TFA [55]
Library Generation via DDA-MS:
- Analyze enriched diGly peptides using data-dependent acquisition on high-resolution mass spectrometers
- Use long LC gradients (e.g., 120 minutes) for maximum peptide separation
- Employ dynamic exclusion to increase proteome coverage
- Include indexed retention time (iRT) peptides in all runs for consistent retention time calibration [53]
Diagram 1: Project-specific spectral library generation and application workflow for DIA ubiquitinomics.
Advanced Methodologies for Enhanced Library Coverage
DIA-NN Library-Free Analysis
For situations where project-specific library generation is impractical, library-free analysis using DIA-NN provides a powerful alternative:
- Database Preparation: Compile a comprehensive protein sequence database including relevant isoforms and known contaminants
- Library-Free DIA Analysis:
- Process DIA raw files directly against sequence database
- Enable "deep learning-based spectra and RT prediction" in DIA-NN
- Use neural network-based scoring to confidently identify modified peptides [15]
- Cross-Run Alignment: Leverage between-run matching to increase identification completeness across sample sets
This approach has demonstrated remarkable performance, identifying approximately 26,000 diGly sites in single DIA runs without any experimental library [15].
Specialized Protocols for Challenging Samples
Certain sample types require additional methodological considerations:
Tissue-Specific Ubiquitination Profiling: The BirA-biotin ubiquitin system enables cell-type specific ubiquitinome profiling in complex tissues. This approach expresses biotinylatable ubiquitin in specific cell types (e.g., neurons) followed by streptavidin-based enrichment under denaturing conditions [56].
Tandem Enrichment Strategies: The SCASP-PTM protocol allows simultaneous enrichment of ubiquitinated, phosphorylated, and glycosylated peptides from a single sample, maximizing information from limited material [57].
Table 3: Key Research Reagents for DIA Ubiquitinomics Library Generation
| Reagent/Resource | Function | Application Notes |
|---|---|---|
| Anti-diGly Remnant Antibody | Immunoaffinity enrichment of ubiquitinated peptides | Critical for specificity; optimize antibody:peptide ratio (31.25 μg per 1 mg peptides) [3] |
| SDC Lysis Buffer with CAA | Protein extraction with concurrent deubiquitinase inhibition | Superior to urea for ubiquitinome coverage; immediate boiling preserves ubiquitination states [15] |
| Indexed Retention Time (iRT) Kit | Retention time calibration across runs | Essential for inter-run alignment and library transferability [53] |
| Proteasome Inhibitors (MG132) | Enhance ubiquitinated peptide detection | Increases identification numbers but alters ubiquitinome composition; use consistently [3] [55] |
| High-pH Reversed-Phase Columns | Peptide fractionation for deep library generation | Enables identification of >90,000 diGly peptides when combined with fractionation [3] |
Spectral library challenges in DIA ubiquitinomics are significant but manageable through strategic experimental design. Tissue and species mismatches can compromise data quality and biological interpretation, but project-specific library generation, library-free computational approaches, and careful consideration of biological context provide robust solutions. As DIA methodologies continue to advance, with improved computational tools like DIA-NN and optimized sample preparation protocols, the field moves toward more comprehensive and accurate characterization of ubiquitin signaling across diverse biological systems. By implementing the protocols and strategic frameworks outlined in this technical note, researchers can overcome spectral library limitations to generate reliable, biologically relevant ubiquitinome data that advances our understanding of this crucial regulatory system.
Diagram 2: Strategic solutions to address spectral library challenges in DIA ubiquitinomics.
Within the framework of data-independent acquisition (DIA) method for ubiquitinome analysis research, the selection of appropriate software and the precise configuration of its parameters are critical determinants of success. Ubiquitinome profiling via DIA mass spectrometry has demonstrated remarkable potential, enabling the identification of over 70,000 ubiquitinated peptides in single runs—more than tripling the coverage offered by traditional data-dependent acquisition (DDA) methods [58]. However, this power comes with significant complexity, and errors in data processing can compromise even the most carefully executed experiments. This application note details common pitfalls in software selection and parameter configuration while providing validated protocols to achieve robust, reproducible ubiquitinome analysis suitable for drug discovery and development applications.
The Critical Role of Software Selection in Ubiquitinome Analysis
Software Landscape and Selection Criteria
The DIA proteomics software ecosystem comprises multiple tools with distinct strengths, algorithmic approaches, and output characteristics. Inappropriate software selection ranks among the most common sources of failure in DIA ubiquitinome studies, potentially leading to incomplete identifications, inflated false discovery rates (FDR), and biologically misleading results [53].
Table 1: DIA Software Tool Selection Guide for Ubiquitinome Analysis
| Software Tool | Primary Strength | Recommended Use Case | Ubiquitinome Performance Notes |
|---|---|---|---|
| DIA-NN | High-speed library-free/predicted-library workflows; IM-aware for timsTOF data [59] | High-throughput cohorts; timsTOF with ion-mobility–enabled DIA; studies requiring conservative MBR [59] | Specifically optimized for ubiquitinomics with additional scoring module for modified peptides; excels in library-free mode [58] |
| Spectronaut | Mature directDIA and library-based modes; comprehensive GUI and QC reporting [59] | Projects requiring audit-friendly reports and standardized exports; regulated environments [59] | Polished directDIA with integrated QC figures; templated exports for project consistency |
| FragPipe Ecosystem (MSFragger-DIA) | Open, composable pipelines; strong artifact retention for traceability [59] | Method development; projects requiring transparency and intermediate file retention [59] | Flexible open pipelines; suitable for labs with strong computational support |
| Scaffold DIA | Emphasis on statistical control and transparency [53] | Regulated environments or multi-party projects requiring stringent validation | – |
Selection criteria must extend beyond mere feature comparisons to encompass biological relevance, throughput requirements, and computational resources. For ubiquitinome analysis specifically, tools must be capable of handling the unique characteristics of diGly-modified peptides, including longer peptide lengths due to impeded C-terminal cleavage of modified lysine residues and higher charge states [3].
Consequences of Software-Task Mismatch
Mismatching software capabilities with experimental goals consistently generates suboptimal results. For example, applying library-based tools to library-free datasets typically yields incomplete identifications and inflated FDR [53]. In ubiquitinome analysis, where novel site discovery is often a primary objective, this can obscure valuable biological insights.
The specialized analysis of ubiquitinated peptides demands software with proven performance for post-translational modification (PTM) profiling. DIA-NN, for instance, incorporates an additional scoring module that ensures confident identification of modified peptides, including K-GG peptides, making it particularly suitable for ubiquitinome studies [58]. In comparative assessments, DIA-NN identified approximately 40% more K-GG peptides than alternative processing software when analyzing the same ubiquitinomics datasets [58].
Parameter Configuration Pitfalls and Solutions
Critical Parameters for Ubiquitinome Analysis
Misconfiguring software parameters represents another frequent failure point, often resulting from applying default settings designed for standard proteomics to the specialized context of ubiquitinome analysis.
Table 2: Essential Parameter Configurations for DIA Ubiquitinome Analysis
| Parameter Category | Standard Proteomics Setting | Ubiquitinome-Optimized Setting | Rationale |
|---|---|---|---|
| FDR Control | 1% peptide FDR [59] | 1% peptide FDR + 1% protein FDR [59] | Ensures confident identification at both peptide and protein levels |
| Match Between Runs (MBR) | Often enabled with default evidence thresholds | Conservative thresholds or off for headline results in complex matrices [59] | Prevents error propagation in identification of low-abundance ubiquitinated peptides |
| Precursor Isolation Windows | Fixed or variable windows based on standard peptide density | Optimized for diGly precursor characteristics; typically 6% improvement with tailored windows [3] | diGly peptides often longer with higher charge states; requires adjusted fragmentation |
| Interference Control | Default fragment scoring | Tightened fragment evidence requirements; minimum robust fragment count [59] | Reduces false assignments in complex ubiquitinome samples |
| Library Specificity | Public or generic spectral libraries | Project-specific libraries recommended for complex tissues [53] | Tissue/species mismatches cause significant coverage loss |
Parameter Configuration Protocols
Protocol: FDR and Quality Control Setup
- Apply two-tiered FDR control: Set both peptide-level and protein-level FDR to 1% using target-decoy methodology [59].
- Enable q-value columns in exports: Ensure all quantitative outputs include q-value columns for downstream filtering [59].
- Validate FDR curves: Visually inspect FDR curves for abnormal shapes that might indicate parameter misconfiguration [53].
- Implement cross-batch normalization: Use QC-pool anchors for retention time alignment and monitor residual drift [59].
Protocol: Match Between Runs Configuration for Ubiquitinomics
- For discovery-phase analyses: Enable MBR but apply stricter evidence thresholds than defaults [59].
- For complex matrices (plasma, FFPE): Provide both MBR-off (primary) and MBR-on (sensitivity) result matrices for transparency [59].
- For biomarker validation studies: Consider disabling MBR for headline results to minimize false positives [59].
- Document all MBR parameters: Include evidence requirements and thresholds in method documentation.
Protocol: Spectral Library Configuration
- Library construction: For project-specific libraries, perform ≥2 replicate DDA runs per sample type under matching LC gradients [53].
- Include iRT standards: Ensure consistent retention time calibration across all runs [53].
- Library comprehensiveness: For ubiquitinome analysis, leverage deep spectral libraries (>90,000 diGly peptides) when available [3].
- Library validation: Assess fragment coverage completeness metrics to evaluate quantifiability [53].
Experimental Protocols for Ubiquitinome Analysis
Sample Preparation Protocol for DIA Ubiquitinomics
The following protocol, adapted from the SDC-based method that demonstrated 38% improvement in K-GG peptide identification compared to urea buffer, ensures optimal sample preparation for DIA ubiquitinome analysis [58]:
- Cell Lysis: Extract proteins using SDC lysis buffer (4% SDC, 100 mM Tris-HCl pH 8.5) supplemented with 40 mM chloroacetamide (CAA) for immediate cysteine protease inactivation [58].
- Protein Extraction: Boil samples immediately after lysis at 95°C for 10 minutes to preserve ubiquitination states [58].
- Protein Digestion: Digest proteins with trypsin (1:50 enzyme-to-protein ratio) overnight at 37°C [58].
- diGly Peptide Enrichment: Enrich K-GG peptides from 1 mg of peptide material using 31.25 μg of anti-diGly antibody [3].
- Sample Injection: Inject 25% of the total enriched material for DIA analysis to maximize sensitivity while maintaining reproducibility [3].
DIA Acquisition Parameter Protocol for Ubiquitinomics
Optimized specifically for diGly peptide characteristics, this acquisition protocol enables identification of >35,000 diGly peptides in single measurements [3]:
- Chromatography: Utilize medium-length nanoLC gradients (75-120 minutes) for sufficient peptide separation [58].
- MS1 Settings: Full MS scans at 120,000 resolution with scan range of 350-1650 m/z.
- DIA Window Scheme: Implement 46 variable windows optimized for diGly precursor distribution [3].
- MS2 Settings: Fragment scans at 30,000 resolution with normalized collision energy ~30 [3].
- Cycle Time: Maintain cycle time ≤3 seconds to ensure sufficient peak sampling density (~8-10 points per peak) [53].
Quantitative Performance and Validation
Performance Metrics for Ubiquitinome Analysis
Comprehensive validation of ubiquitinome data processing requires multiple quality metrics beyond identification counts:
Table 3: Ubiquitinome QC Metrics and Acceptance Criteria
| Performance Metric | Minimum Standard | Target Performance | Validation Method |
|---|---|---|---|
| Peptide/Protein FDR | 1% / 1% [59] | 0.5% / 1% [59] | Target-decoy method with consistent reporting |
| QC-pool Protein CV (median) | ≤20% [59] | ≤15% [59] | Injection of QC-pool every ~10-12 samples |
| Sample-level Missingness | ≤30% [59] | ≤20% [59] | Calculation of % missing values in protein matrix |
| Quantitative Precision | CV <20% for 45% of peptides [3] | CV <20% for >60% of peptides [3] | Coefficient of variation across technical replicates |
| Cross-batch Alignment | RT residuals <2 minutes | RT residuals <1 minute | iRT-anchored alignment with residual monitoring |
Validation Protocol for Ubiquitinome Data Quality
- Spike-in Controls: Validate quantitative accuracy using pools of synthetic K-GG peptides spiked into yeast tryptic digest at different concentrations [58].
- Interference Assessment: Monitor interference scores and adjust fragment evidence thresholds if >15% of peptides show high interference [59].
- Reproducibility Evaluation: Process replicates through the entire workflow; >77% of diGly peptides should show CVs <50% across replicates [3].
- Cross-Software Validation: Employ multiple DIA analysis tools (orthogonal approaches) to enhance robustness and reliability of findings [11].
Visualization of Ubiquitinome Analysis Workflow
Research Reagent Solutions
Table 4: Essential Research Reagents for DIA Ubiquitinome Analysis
| Reagent/Resource | Function | Application Note |
|---|---|---|
| Anti-diGly Antibody | Immunoaffinity enrichment of K-GG remnant peptides | Use 31.25 μg antibody per 1 mg peptide input for optimal yield [3] |
| Sodium Deoxycholate (SDC) | Lysis buffer component for improved protein extraction | SDC-based lysis yields 38% more K-GG peptides vs. urea buffer [58] |
| Chloroacetamide (CAA) | Cysteine alkylating agent | Preferred over iodoacetamide to avoid di-carbamidomethylation artifacts [58] |
| Proteasome Inhibitor (MG-132) | Enhances ubiquitinated peptide signal | Treatment increases K48-peptide abundance; requires separate fractionation [3] |
| iRT Peptides | Retention time calibration standards | Essential for cross-batch alignment and reproducibility [53] |
| Synthetic K-GG Peptides | Spike-in controls for quantification validation | Enables accuracy assessment across dynamic range [58] |
Optimal software selection and parameter configuration are foundational to successful DIA ubiquitinome analysis. Through deliberate tool selection based on experimental requirements, careful parameter optimization for diGly peptide characteristics, and rigorous quality control, researchers can reliably achieve comprehensive ubiquitinome coverage with robust quantification. The protocols and guidelines presented herein provide a structured approach to avoid common data processing errors, enabling researchers to fully leverage DIA's potential for revealing novel insights into ubiquitin signaling biology and therapeutic development.
The comprehensive analysis of the ubiquitinome, the entirety of ubiquitinated proteins in a biological system, presents significant challenges due to the dynamic nature, low stoichiometry, and tremendous complexity of ubiquitination events. Data-independent acquisition mass spectrometry (DIA-MS) has emerged as a powerful alternative to traditional data-dependent acquisition (DDA) methods, offering improved reproducibility, quantitative accuracy, and data completeness for ubiquitinome profiling [3]. However, maximizing the potential of DIA-MS requires careful optimization of numerous experimental parameters throughout the workflow. The DO-MS (Data-Driven Optimization of Mass Spectrometry) framework addresses this critical need by providing a systematic, data-driven approach for method development, quality control, and performance optimization specifically tailored for DIA-based ubiquitinome studies [60] [61].
This framework enables researchers to move beyond trial-and-error approaches by offering interactive visualization and quantitative assessment of key performance metrics. Originally developed for general proteomic applications, DO-MS is particularly valuable for ubiquitinome analysis due to the specialized sample preparation, enrichment techniques, and data processing requirements inherent to studying ubiquitination. The platform supports optimization of critical parameters including peptide separation methods, duty cycle configurations, ion accumulation times, and mass analyzer settings specifically for the unique characteristics of ubiquitinated peptides [60]. Furthermore, its application to single-cell proteomics (plexDIA) makes it increasingly relevant for studying ubiquitination heterogeneity in limited samples, such as primary patient tissues or rare cell populations [61].
Experimental Protocols for DIA-Based Ubiquitinome Analysis
Optimized Sample Preparation for Ubiquitinome Profiling
Robust ubiquitinome analysis begins with optimized sample preparation to preserve ubiquitination states while maximizing protein extraction and digestion efficiency. Recent advancements have demonstrated that sodium deoxycholate (SDC)-based lysis protocols significantly outperform traditional urea-based methods for ubiquitinome studies [15].
Protocol: SDC-Based Lysis for Ubiquitinomics
- Reagents: SDC lysis buffer (1% SDC, 100 mM Tris-HCl, 10 mM TCEP, 40 mM chloroacetamide [CAA], pH 8.5)
- Procedure:
- Rapid Lysis and Denaturation: Immediately lyse cells or tissue in pre-heated SDC buffer at 95°C for 5 minutes to instantaneously inactivate deubiquitinases (DUBs) and preserve ubiquitination states.
- Protein Extraction and Alkylation: Sonicate samples to reduce viscosity and ensure complete protein extraction. The high concentration of CAA (40 mM) rapidly alkylates cysteine residues without causing di-carbamidomethylation of lysines, which can mimic ubiquitin remnant peptides [15].
- Digestion and SDC Removal: Digest proteins with trypsin (1:50 enzyme-to-substrate ratio) overnight at 37°C. Acidity the digest with trifluoroacetic acid (TFA) to a final concentration of 1-2% to precipitate SDC, then centrifuge at 10,000 × g for 10 minutes to remove the precipitate.
- Peptide Cleanup: Desalt peptides using C18 solid-phase extraction cartridges or plates before ubiquitin remnant enrichment.
This SDC-based approach has been shown to yield approximately 38% more ubiquitinated peptides compared to urea-based protocols while maintaining high enrichment specificity and quantitative precision [15].
Tandem Enrichment of Ubiquitinated Peptides
The SCASP-PTM (SDS-cyclodextrin-assisted sample preparation-post-translational modification) platform enables sequential enrichment of multiple PTMs, including ubiquitination, from a single sample without intermediate desalting steps [62].
Protocol: Tandem Ubiquitinated Peptide Enrichment Without Desalting
- Key Reagents:
- Anti-K-ε-GG antibody-conjugated agarose beads
- SCASP lysis buffer (1% SDS, 100 mM Tris-HCl, 10 mM TCEP, 40 mM CAA, pH 8.5)
- HP-β-CD buffer (250 mM (2-hydroxypropyl)-beta-cyclodextrin)
- SCASP-ubi elution buffer (0.15% TFA)
- Procedure:
- Sample Preparation: Extract proteins using SCASP lysis buffer, followed by addition of HP-β-CD to form complexes with SDS that do not interfere with subsequent antibody binding.
- Immunoaffinity Enrichment: Incubate the peptide digest directly with anti-K-ε-GG antibody-conjugated beads for 2 hours at 4°C with gentle agitation. Note: Antibody-based enrichment must be performed before any metal ion-based methods (e.g., IMAC for phosphopeptides), as TFA and acetonitrile used in later stages disrupt antibody-antigen interactions [62].
- Washing: Wash beads sequentially with:
- Ice-cold immunoaffinity purification (IAP) buffer (3 × 1 mL)
- High-salt wash buffer (1 M NaCl, 3 × 1 mL)
- Water (3 × 1 mL)
- Elution: Elute ubiquitinated peptides with two rounds of 0.15% TFA (2 × 0.5 mL), pooling eluates.
- Desalting and Concentration: Desalt enriched peptides using C18 StageTips or microcolumns before LC-DIA-MS analysis.
This tandem enrichment approach allows researchers to maximize information obtained from precious samples by profiling multiple PTM types from the same biological specimen, enabling studies of cross-regulatory relationships between different modification types [62].
DIA-MS Acquisition Method Optimization
Optimized DIA acquisition parameters are crucial for achieving comprehensive ubiquitinome coverage. The following protocol outlines key considerations for method development:
Protocol: DIA Method Optimization for Ubiquitinomics
- Spectral Library Generation:
- Create comprehensive libraries by fractionating ubiquitin-enriched peptides from model samples (e.g., proteasome inhibitor-treated cells) using high-pH reversed-phase chromatography (8-24 fractions) [3].
- Alternatively, use predicted spectral libraries for rapid method deployment without extensive fractionation.
- DIA Acquisition Parameters:
- Window Scheme: Implement 30-46 variable windows covering 400-1000 m/z range, optimized for the higher charge states and longer sequences typical of ubiquitinated peptides due to impeded C-terminal cleavage at modified lysines [3].
- MS2 Resolution: Set to 30,000-60,000 for improved fragment ion detection and quantification accuracy.
- Cycle Time: Maintain ~3 seconds to ensure sufficient data points (8-12) across chromatographic peaks.
- DO-MS Assisted Optimization:
- Use DO-MS to visualize and optimize duty cycle methods, ion accumulation times, and peptide separation parameters [60].
- Assess the impact of survey scans per duty cycle on identification and quantification performance.
Table 1: Optimized DIA-MS Parameters for Ubiquitinome Analysis
| Parameter | Recommended Setting | Rationale |
|---|---|---|
| MS1 Resolution | 60,000-120,000 | Accurate precursor quantification |
| MS2 Resolution | 30,000-60,000 | Balance sensitivity and specificity |
| Isolation Windows | 30-46 variable windows | Optimized for ubiquitinated peptide characteristics |
| Collision Energy | Stepped (25-30-35%) | Improved fragmentation diversity |
| AGC Target | Customized for analyzer type | Optimal ion sampling without space charging |
| Maximum Ion Injection Time | Customized for analyzer | Sufficient signal without compromising cycle time |
Data Analysis and Quality Control Framework
Software Selection and Configuration for Ubiquitinome DIA Data
The choice of computational tools significantly impacts the depth and quality of ubiquitinome analysis. Leading software platforms offer distinct advantages for different experimental scenarios:
Table 2: Comparison of DIA Analysis Software for Ubiquitinomics
| Software | Strengths | Optimal Use Cases | Key Parameters |
|---|---|---|---|
| DIA-NN | High-speed library-free workflows; Excellent cross-batch merging; IM-aware for timsTOF data [59] | Large cohorts; High-throughput studies; timsTOF with ion-mobility–enabled DIA | Library-free mode; Conservative MBR settings; Neural network-based scoring [15] |
| Spectronaut | Polished directDIA with GUI QC; Standardized exports and audit-friendly reports [59] | Regulated environments; Standardized service workflows | DirectDIA mode; Interference scoring; QC-anchored normalization |
| FragPipe Ecosystem | Open, composable pipelines; Strong artifact retention; Ideal for method development [59] | Research environments; Customized workflows; Traceability requirements | MSFragger-DIA; Retention of intermediate files |
DO-MS-Based Quality Control Metrics
Implementing systematic quality control is essential for generating reproducible ubiquitinome data. DO-MS provides specialized visualization for key ubiquitinome-specific QC metrics:
Critical QC Metrics for Ubiquitinome Analysis:
- Identification Depth: Monitor numbers of quantified ubiquitinated peptides (>30,000 achievable in single measurements) and proteins across samples [15] [3].
- Quantitative Precision: Assess coefficients of variation (CV) for ubiquitinated peptides across technical replicates (median CV <15-20% achievable with optimized DIA) [15].
- Data Completeness: Track missing values across sample series (<30% missingness at protein level) [59].
- Enrichment Specificity: Verify percentage of ubiquitinated peptides relative to non-modified peptides in enriched samples.
- Dynamic Range: Evaluate quantification linearity using spiked-in standards or dilution series.
Table 3: Ubiquitinome Quality Control Thresholds
| Metric | Minimum Standard | Target Performance | Assessment Method |
|---|---|---|---|
| Ubiquitinated Peptides | >20,000 IDs | >35,000 IDs | Library-based identification at 1% FDR [3] |
| Sample-level Missingness | ≤30% | ≤15% | Missing data in protein quantitative matrix [59] |
| QC-pool Protein CV | ≤20% | ≤15% | Median CV from pooled quality control samples [59] |
| Enrichment Specificity | >60% K-ε-GG peptides | >80% K-ε-GG peptides | Percentage of modified peptides in enriched fraction |
| Identification Reproducibility | >70% overlap in technical replicates | >85% overlap in technical replicates | Jaccard index of ubiquitinated peptides |
Research Reagent Solutions for Ubiquitinome Analysis
Successful implementation of DIA-based ubiquitinomics requires specific reagents and materials at each workflow stage:
Table 4: Essential Research Reagents for DIA Ubiquitinome Studies
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Anti-K-ε-GG Antibody Beads | Immunoaffinity enrichment of ubiquitin remnant peptides | Use 31.25 μg antibody per 1 mg peptide input; Must precede metal ion-based enrichments [62] [3] |
| Sodium Deoxycholate (SDC) | Protein extraction and denaturation | Superior to urea for ubiquitinome studies; Precipitates at low pH for easy removal [15] |
| Chloroacetamide (CAA) | Cysteine alkylation | Preferred over iodoacetamide to avoid di-carbamidomethylation artifacts that mimic GG-modified lysines [15] |
| HP-β-Cyclodextrin | SDS complexation | Enables antibody-based enrichment without desalting by complexing with SDS in SCASP-PTM workflow [62] |
| Trifluoroacetic Acid (TFA) | Peptide elution and acidification | 0.15% concentration for eluting ubiquitinated peptides from antibody beads [62] |
| Strep-Tactin/HisPur Resins | Ubiquitinated protein enrichment | For tagged-ubiquitin approaches; Alternative to antibody-based peptide enrichment [2] |
| High-pH Reversed-Phase Resins | Peptide fractionation | Generation of comprehensive spectral libraries; 8-24 fractions recommended for depth [3] |
Workflow Visualization
Diagram 1: DO-MS guided workflow for DIA ubiquitinome analysis. The red arrow highlights the iterative optimization loop enabled by DO-MS quality control feedback.
Applications in Targeted Protein Degradation Research
The optimized DIA ubiquitinome workflow has significant applications in targeted protein degradation (TPD) research, including PROTACs and molecular glues, where comprehensive understanding of ubiquitination events is crucial for mechanism of action studies [13].
Application Protocol: TPD Mode of Action Studies
- Experimental Design:
- Treat cells with TPD compounds across multiple time points (minutes to hours) to capture dynamic ubiquitination changes.
- Include appropriate controls (vehicle, inactive analogs) for specificity assessment.
- Sample Processing:
- Process samples using the SDC-based lysis and ubiquitin remnant enrichment protocols described above.
- Consider tandem PTM enrichment (SCASP-PTM) to simultaneously monitor phosphorylation changes that might correlate with ubiquitination events.
- DIA-MS Analysis:
- Utilize optimized DIA methods with 30-46 variable windows for maximum coverage.
- Process data through DIA-NN in library-free or predicted library mode for rapid analysis.
- Data Interpretation:
- Identify significantly altered ubiquitination sites on target proteins and potential off-targets.
- Correlate ubiquitination changes with protein abundance measurements from parallel proteome analysis to distinguish degradative from regulatory ubiquitination events [15].
- Use DO-MS for quality control across large sample batches to ensure data consistency.
This approach has demonstrated capability to identify over 40,000 diGly precursors corresponding to more than 7,000 proteins in single measurements from cells exposed to proteasome inhibitors, providing unprecedented depth for characterizing TPD mechanisms [13].
The integration of DO-MS for systematic method optimization represents a significant advancement in DIA-based ubiquitinome analysis. By implementing the protocols and quality control frameworks outlined in this application note, researchers can achieve unprecedented depth, reproducibility, and quantitative accuracy in their ubiquitination studies. The data-driven optimization approach enables continuous method improvement through systematic assessment of key performance metrics, moving ubiquitinome research toward more standardized and reliable methodologies. As ubiquitinomics continues to play an increasingly important role in understanding cellular signaling and targeted protein degradation therapeutics, these optimized workflows provide the foundation for robust, large-scale studies capable of capturing the complexity of ubiquitin signaling in health and disease.
Ubiquitination, the enzymatic post-translational modification where ubiquitin molecules attach to substrate proteins, is a master regulator of virtually all cellular processes, including signal transduction, cell division, immune responses, and circadian biology [63] [3]. The ubiquitin-proteasome pathway (UPP) represents the major mechanism of proteolysis in the cytosol and nucleus, and flaws in this system contribute to the pathogenesis of numerous human diseases, including neurodegenerative disorders and cancer [64] [63]. The comprehensive study of the "ubiquitinome"—the complete set of protein ubiquitination modifications in a biological system—has become a critical focus in proteomics and drug discovery research.
A central challenge in ubiquitinome analysis is balancing the need for deep, comprehensive coverage with practical constraints of sample availability and processing. Traditional methods require relatively large sample amounts and extensive peptide fractionation to achieve sufficient depth, coming at the expense of throughput, robustness, and quantitative accuracy [3]. This application note addresses this fundamental trade-off by presenting optimized input material strategies that maximize data quality while maintaining practical experimental workflows suitable for most laboratory settings.
Technical Foundations of Ubiquitinome Analysis
The Ubiquitination Process
Ubiquitination involves a sequential enzymatic cascade: ubiquitin-activating enzymes (E1) activate ubiquitin in an ATP-dependent process; ubiquitin-conjugating enzymes (E2) transfer ubiquitin; and ubiquitin ligases (E3) catalyze the final attachment to substrate proteins [63]. Ubiquitin itself can be further ubiquitinated on its seven lysine residues or N-terminal methionine, creating diverse polyubiquitin chain topologies that encode specific biological functions [3]. The resulting ubiquitination patterns can mark proteins for degradation by the 26S proteasome or regulate non-proteolytic processes including DNA repair, endocytic trafficking, and inflammation [64] [63].
Analytical Challenges in Ubiquitinome Research
The systematic analysis of ubiquitination faces several technical hurdles:
- Low Stoichiometry: Ubiquitination events typically occur at low abundance relative to their unmodified counterparts
- Dynamic Range: The concentration range of ubiquitinated species spans several orders of magnitude
- Lability: Ubiquitination is reversible through deubiquitinating enzymes (DUBs)
- Structural Diversity: Variations in ubiquitin chain linkage and topology create analytical complexity
Mass spectrometry-based proteomics has emerged as the primary technology for ubiquitinome analysis, particularly through antibody-based enrichment of tryptic peptides containing the characteristic diGly remnant left after digestion of ubiquitinated proteins [3].
Optimized Input Material Strategy for DIA Ubiquitinome Analysis
Comprehensive Workflow Design
The following diagram illustrates the complete optimized workflow for DIA-based ubiquitinome analysis, highlighting critical decision points for input material management:
Diagram 1: Comprehensive DIA ubiquitinome analysis workflow showing library construction and experimental analysis paths.
Quantitative Performance of Optimized Input Strategy
The optimized input material strategy enables remarkable gains in analytical depth and precision compared to conventional approaches. The table below summarizes key performance metrics achieved through systematic optimization of sample input and processing parameters:
Table 1: Performance comparison of DIA versus DDA methods for ubiquitinome analysis
| Performance Metric | DDA Method | DIA Method | Improvement |
|---|---|---|---|
| Distinct diGly peptides (single measurement) | ~20,000 | ~35,000 | 75% increase |
| Coefficient of Variation <20% | 15% of peptides | 45% of peptides | 3-fold improvement |
| Coefficient of Variation <50% | Not reported | 77% of peptides | Substantial improvement |
| Total distinct diGly peptides across replicates | 24,000 | 48,000 | 100% increase |
| Quantitative accuracy | Moderate | High | Marked improvement |
| Data completeness across samples | Lower | Higher | Significant improvement |
The implementation of data-independent acquisition (DIA) mass spectrometry represents a fundamental advancement over traditional data-dependent acquisition (DDA) methods. DIA fragments all co-eluting peptide ions within predefined mass-to-charge (m/z) windows simultaneously, enabling more precise and accurate quantification with fewer missing values across samples and higher identification rates over a larger dynamic range [3].
Critical Parameter Optimization
Sample Input and Enrichment Conditions
Through systematic titration experiments, researchers have determined that enrichment from 1 mg of peptide material using 31.25 μg of anti-diGly antibody provides optimal peptide yield and depth of coverage in single DIA experiments [3]. This ratio represents approximately one-eighth of a commercial anti-diGly antibody vial per milligram of peptide input. With the improved sensitivity afforded by DIA, only 25% of the total enriched material needs to be injected for analysis, allowing for multiple technical replicates or preservation of precious samples [3].
Spectral Library Construction
For comprehensive ubiquitinome coverage, extensive spectral libraries are essential. The creation of libraries from multiple cell lines (HEK293 and U2OS) under both proteasome-inhibited and normal conditions has yielded libraries containing more than 90,000 diGly peptides [3]. This multi-condition approach ensures broad coverage of ubiquitination events across diverse biological contexts. Notably, about 57% of the diGly sites identified in these extensive libraries were not previously reported in databases, highlighting the discovery potential of this optimized workflow [3].
Instrument Method Optimization
The unique characteristics of diGly-containing peptides—including impeded C-terminal cleavage of modified lysine residues that frequently generates longer peptides with higher charge states—require specialized DIA method settings [3]. Through systematic optimization, researchers determined that a method with 46 precursor isolation windows and fragment scan resolution of 30,000 provides optimal performance, resulting in a 13% improvement compared to standard full proteome methods [3].
Detailed Experimental Protocols
Protocol 1: Comprehensive Spectral Library Generation
Purpose: To create an in-depth spectral library for DIA analysis of ubiquitinome samples.
Materials:
- HEK293 and U2OS cell lines
- Proteasome inhibitor (MG132, 10 μM)
- Lysis buffer (8 M urea, 100 mM NH₄HCO₃, protease inhibitors)
- Trypsin (sequencing grade)
- anti-diGly antibody (e.g., PTMScan Ubiquitin Remnant Motif Kit)
- Basic reversed-phase chromatography system
- High-performance liquid chromatography system coupled to mass spectrometer
Procedure:
- Cell Culture and Treatment: Culture HEK293 and U2OS cells under standard conditions. Treat with 10 μM MG132 for 4 hours to inhibit proteasomal degradation and increase ubiquitinated protein levels.
- Protein Extraction: Harvest cells and lyse in urea-based lysis buffer. Determine protein concentration using compatible assay.
- Protein Digestion: Reduce and alkylate proteins. Digest with trypsin (1:50 w/w enzyme-to-protein ratio) at 37°C for 16 hours.
- Peptide Fractionation: Separate peptides by basic reversed-phase chromatography into 96 fractions. Concatenate fractions into 8 pools to reduce analysis time.
- K48-peptide Handling: Isolate and process fractions containing abundant K48-linked ubiquitin-chain derived diGly peptides separately to prevent competition during enrichment.
- diGly Peptide Enrichment: Enrich diGly peptides from each pool using anti-diGly antibody (31.25 μg per enrichment) according to manufacturer's instructions.
- LC-MS/MS Analysis: Analyze enriched peptides using DDA method on Orbitrap mass spectrometer.
Notes:
- The separate processing of K48-rich fractions is particularly important for MG132-treated samples as proteasome inhibition dramatically increases K48-peptide abundance.
- Combining libraries from multiple cell lines and conditions increases coverage of the ubiquitinome.
Protocol 2: Optimized DIA Ubiquitinome Analysis
Purpose: To implement a sensitive, single-shot DIA workflow for ubiquitinome analysis with minimal sample input.
Materials:
- Peptide samples (1 mg total input recommended)
- anti-diGly antibody (31.25 μg per enrichment)
- LC-MS/MS system capable of DIA acquisition
- Spectral library (>90,000 diGly peptides)
- DIA data analysis software (e.g., Spectronaut, DIA-NN, or Skyline)
Procedure:
- Sample Preparation: Extract and digest proteins from biological samples of interest. Determine peptide concentration.
- diGly Peptide Enrichment: Enrich diGly peptides from 1 mg of peptide material using 31.25 μg of anti-diGly antibody. Follow manufacturer's protocol for enrichment.
- Sample Injection: Inject 25% of the total enriched material (equivalent to 250 μg starting peptides) for LC-MS/MS analysis.
- DIA Acquisition: Implement optimized DIA method with the following parameters:
- 46 variable-width precursor isolation windows
- MS2 resolution: 30,000
- Coverage of m/z range 350-1650
- Data Analysis: Process DIA data against pre-acquired spectral library using appropriate software.
- Quality Assessment: Monitor coefficient of variation across replicates; >45% of peptides should show CV <20%.
Troubleshooting:
- If identification numbers are lower than expected, verify antibody activity and sample input amount.
- If quantitative precision is suboptimal, check chromatographic performance and instrument calibration.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key research reagent solutions for DIA ubiquitinome analysis
| Reagent/Material | Function | Specifications/Recommendations |
|---|---|---|
| anti-diGly Antibody | Enrichment of ubiquitinated peptides | PTMScan Ubiquitin Remnant Motif Kit; use 31.25 μg per 1 mg peptide input |
| Proteasome Inhibitor (MG132) | Increases ubiquitinated protein levels by blocking degradation | 10 μM treatment for 4 hours prior to harvesting |
| Trypsin | Protein digestion | Sequencing grade; 1:50 enzyme-to-protein ratio |
| Basic Reversed-Phase Chromatography System | Peptide fractionation for library generation | 96 fractions concatenated to 8 pools |
| Orbitrap Mass Spectrometer | DIA data acquisition | Capable of 30,000 MS2 resolution and 46-window DIA methods |
| Spectral Libraries | Peptide identification in DIA data | >90,000 diGly peptides from multiple cell lines and conditions |
| Urea-based Lysis Buffer | Protein extraction | 8 M urea, 100 mM NH₄HCO₃, supplemented with protease inhibitors |
Biological Applications and Impact
The optimized input material strategy has enabled groundbreaking biological discoveries across multiple research domains. Applied to TNFα signaling—a pathway regulated by ubiquitination—the method comprehensively captured known ubiquitination sites while adding many novel ones [3]. Even more impressively, a systems-wide investigation of ubiquitination across the circadian cycle uncovered hundreds of cycling ubiquitination sites and dozens of cycling ubiquitin clusters within individual membrane protein receptors and transporters [3]. These findings highlight new connections between metabolism and circadian regulation that were previously inaccessible with less sensitive methods.
The following diagram illustrates how the optimized workflow enables discovery of dynamic ubiquitination patterns in complex biological systems:
Diagram 2: How optimized input strategies enable novel biological discoveries through enhanced sensitivity and quantification.
The strategic optimization of input materials for DIA-based ubiquitinome analysis represents a significant advancement in proteomics methodology. By systematically addressing the critical parameters of sample input, antibody usage, fractionation strategy, and instrument methods, researchers can now achieve unprecedented depth and quantitative precision in ubiquitination studies. The optimized workflow detailed in this application note—centered on the 1 mg peptide input with 31.25 μg antibody enrichment—delivers approximately 35,000 distinct diGly site identifications in single measurements with high quantitative accuracy.
These methodological improvements have profound implications for drug discovery and development, particularly for diseases with disrupted ubiquitin pathways, such as neurodegenerative disorders and cancer. The ability to comprehensively profile ubiquitination dynamics using practical sample amounts opens new avenues for understanding disease mechanisms, identifying therapeutic targets, and evaluating drug efficacy. As the field continues to evolve, further refinements in sensitivity and throughput will undoubtedly emerge, but the fundamental principles of balanced input material strategy outlined here will remain essential for rigorous ubiquitinome research.
In the rapidly advancing field of proteomics, data-independent acquisition (DIA) has emerged as a powerful mass spectrometry technique for comprehensive and reproducible ubiquitinome profiling. Ubiquitination, a crucial post-translational modification involved in virtually all cellular processes, presents unique analytical challenges due to the low stoichiometry of modified peptides and the complexity of biological samples [3]. The depth and quality of ubiquitinome analysis depend critically on the performance of the upstream chromatographic separation, which must be optimized to handle complex peptide mixtures and ensure high quantitative accuracy.
This application note details optimized chromatographic and methodological frameworks for DIA-based ubiquitinome analysis, providing researchers with practical protocols to achieve sufficient gradient length and peak separation for maximal ubiquitin site identification and quantification.
Theoretical Foundation of Chromatographic Optimization
Chromatographic efficiency in HPLC is governed by multiple experimental variables that can be optimized through different approaches depending on the number of parameters adjusted [65].
Optimization Schemes
One-Parameter Optimization: When particle size and column length are fixed, the primary adjustable parameter is the eluent velocity, calculated using the van Deemter equation to find the velocity that produces minimal plate height while respecting instrument pressure and flow limits [65].
Two-Parameter Optimization: With a fixed particle size, both column length and velocity can be optimized using Poppe or kinetic plot techniques. For a given analysis time, there is one optimal combination of column length and velocity that maximizes plate count [65].
Three-Parameter Optimization: This comprehensive approach simultaneously optimizes particle size, column length, and eluent velocity, known as the Knox-Saleem limit, representing the theoretically best possible separation [65].
Table 1: Comparison of Chromatographic Optimization Schemes for a Separation with 4-Second Dead Time
| Optimization Scheme | Optimal Particle Size | Optimal Column Length | Optimal Linear Velocity (mm/s) | Predicted Plate Count | Pressure (bar) |
|---|---|---|---|---|---|
| One-Parameter | 1.8 μm (fixed) | 30 mm (fixed) | 1.92 | 7,600 | 230 |
| Two-Parameter | 1.8 μm (fixed) | 53 mm | 3.39 | 10,700 | 1,000 |
| Three-Parameter | 1.0 μm | 29 mm | 3.47 | 14,900 | 1,000 |
The comparison reveals that comprehensive optimization of chromatographic parameters can potentially double the efficiency compared to basic single-parameter approaches [65].
DIA-Ubiquitinomics Workflow and Chromatographic Integration
The following diagram illustrates the complete experimental workflow for deep-scale ubiquitinome profiling, highlighting the critical role of chromatographic optimization within the process.
Figure 1: Comprehensive DIA-Ubiquitinome Profiling Workflow. The optimized chromatographic separation step is crucial for resolving complex peptide mixtures prior to mass spectrometry analysis.
Critical Sample Preparation Considerations
Optimal ubiquitinome coverage begins with appropriate sample preparation. Recent advances demonstrate that sodium deoxycholate (SDC)-based lysis, supplemented with chloroacetamide (CAA) for immediate cysteine protease inactivation, significantly improves ubiquitin site coverage compared to conventional urea-based methods [21]. This protocol yields approximately 38% more K-ε-GG peptides while maintaining high enrichment specificity [21].
For DIA-based ubiquitinomics, optimal results are achieved with 1-2 mg of peptide material as input for anti-K-ε-GG antibody enrichment, with identification numbers dropping significantly below 500 μg inputs [21]. The enriched peptides should be resuspended in solvents compatible with reversed-phase chromatography (typically 1% ACN and 0.1% formic acid) [66].
Optimized Chromatographic Phases for DIA-Ubiquitinomics
Liquid Chromatography Parameters
Advanced DIA-ubiquitinome studies employ nanoflow liquid chromatography systems coupled to high-resolution mass spectrometers. The optimal configuration utilizes:
- Column Technology: Fused silica capillaries packed with C18 reversed-phase material (1.9 μm particle size) [21]
- Column Dimensions: 75 μm inner diameter × 250 mm length [21]
- Mobile Phase: Solvent A (0.1% formic acid in water) and Solvent B (0.1% formic acid in acetonitrile) [21]
- Gradient Length: 75-125 minute linear gradients from 2% to 30% solvent B [21]
For the emerging narrow-window DIA (nDIA) approach, which uses 2-Th isolation windows for enhanced specificity, even higher chromatographic performance is required. This method leverages the exceptional speed (~200 Hz) of the Astral mass analyzer to achieve unprecedented coverage, quantifying over 10,000 human protein groups in single 30-minute runs [67].
DIA Mass Spectrometry Acquisition Methods
The chromatographic separation must be synchronized with optimized DIA acquisition methods. For ubiquitinome analysis, specific DIA methods have been developed that account for the unique characteristics of K-ε-GG peptides, which often generate longer peptides with higher charge states due to impeded C-terminal cleavage at modified lysine residues [3].
Table 2: Optimized DIA Parameters for Ubiquitinome Analysis
| Parameter | Standard DIA | Optimized DIA for Ubiquitinome | nDIA (Astral) |
|---|---|---|---|
| Precursor Isolation Windows | Typically 8-20 Th windows | Customized window placement based on diGly precursor distribution | 2-Th windows across mass range |
| Number of Windows | Variable (often 20-40) | 46 windows | Numerous narrow windows |
| MS2 Resolution | 15,000-30,000 | 30,000 | High resolution with fast scanning |
| Cycle Time | Balanced to sample eluting peaks | Optimized for sufficient peptide sampling | Ultra-fast cycling (~200 Hz) |
| Identification Performance | 33,000-35,000 diGly sites [3] | 68,000 K-ε-GG peptides [21] | 70,000+ ubiquitinated peptides [21] |
The relationship between chromatographic parameters, DIA method settings, and the resulting data quality can be visualized as an interdependent system.
Figure 2: Optimization Parameter Interdependencies. Chromatographic parameters (blue) and mass spectrometric parameters (green) collectively influence the final data quality metrics (red) in DIA-ubiquitinome experiments.
Quantitative Assessment of Optimized Methods
The performance gains achieved through comprehensive optimization of both chromatography and DIA acquisition are substantial. Compared to data-dependent acquisition (DDA) methods, optimized DIA workflows provide remarkable improvements in data completeness and quantitative precision.
Table 3: Performance Comparison Between DDA and Optimized DIA Ubiquitinomics
| Performance Metric | Data-Dependent Acquisition (DDA) | Optimized DIA Ubiquitinomics |
|---|---|---|
| Typical K-ε-GG Peptide Identifications | 20,000-21,000 peptides [3] [21] | 35,000-68,000 peptides [3] [21] |
| Quantitative Reproducibility (CV < 20%) | 15-25% of peptides [3] | 45-65% of peptides [3] [21] |
| Missing Values Across Samples | High (approximately 50% missing in replicates) [21] | Minimal (<10% missing in replicates) [21] |
| Median Coefficient of Variation | >20% [3] | ~10% [21] |
| Required MS Measurement Time | Often requires fractionation (hours to days) [21] | Single 75-125 min runs achieve deep coverage [21] |
The implementation of narrow-window DIA on instruments like the Orbitrap Astral mass spectrometer further extends these performance benefits, achieving >200-Hz MS/MS acquisition rates with high sensitivity and mass accuracy below 2 ppm [67]. This technological advancement effectively dissolves the historical differences between DDA and DIA methods, enabling both high specificity and comprehensive sampling.
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagent Solutions for DIA-Ubiquitinome Analysis
| Reagent / Material | Function and Application Notes |
|---|---|
| Anti-K-ε-GG Antibody | Immunoaffinity enrichment of ubiquitin-derived diGly remnant peptides; essential for ubiquitinome studies [3] [68]. |
| SDC Lysis Buffer | Sodium deoxycholate-based protein extraction buffer; superior to urea for ubiquitinome coverage [21]. |
| Chloroacetamide (CAA) | Cysteine alkylating agent; preferred over iodoacetamide to avoid di-carbamidomethylation artifacts that mimic GG-modification [21]. |
| C18 Chromatography Columns | Reversed-phase separation; 75μm ID × 250mm with 1.9μm particles recommended [21]. |
| iRT Kit | Indexed Retention Time standards; spiked into samples for retention time alignment and normalization [66]. |
| TMT Reagents | Tandem Mass Tags for multiplexed quantification; can be applied using on-antibody labeling approach [68]. |
Concluding Remarks
Chromatographic optimization is not merely a technical consideration but a fundamental determinant of success in DIA-ubiquitinome studies. The interplay between gradient length, peak separation efficiency, and DIA acquisition parameters directly controls the depth, accuracy, and reproducibility of ubiquitination profiling. As mass spectrometry technology continues to advance with innovations like nDIA and Astral analyzers, chromatographic systems must correspondingly evolve to fully leverage these capabilities.
The protocols and parameters detailed in this application note provide a validated foundation for researchers to implement robust DIA-ubiquitinomics workflows, enabling comprehensive investigation of ubiquitin signaling in biological systems and drug development contexts.
Benchmarking DIA Performance: Quantitative Validation Against Established Methods
Ubiquitination is a crucial post-translational modification (PTM) that regulates diverse cellular processes, including protein degradation, signal transduction, and DNA repair [69]. The ubiquitin-proteasome system (UPS) mediates most protein degradation in eukaryotic cells, and its dysregulation can lead to various diseases, including cancer and neurodegenerative disorders [70]. Mass spectrometry (MS)-based proteomics has become the primary method for system-level ubiquitinome profiling, typically through immunoaffinity purification and MS-based detection of diglycine-modified peptides (K-ε-GG) generated by tryptic digestion of ubiquitin-modified proteins [15] [70] [3].
Two primary acquisition methods have emerged for ubiquitinome analysis: data-dependent acquisition (DDA) and data-independent acquisition (DIA). While DDA has been widely used, DIA has recently demonstrated superior performance for large-scale ubiquitinome studies [3]. This application note provides a detailed comparison of these methodologies in single-run ubiquitinome analysis, with a focus on experimental protocols, quantitative performance, and practical applications in drug discovery research.
Fundamental Differences Between DDA and DIA
Acquisition Principles
Data-Dependent Acquisition (DDA): In DDA, the mass spectrometer selects specific ions for fragmentation based on their intensity or abundance. The instrument isolates a subset of the most abundant ions from the survey scan and fragments them to produce MS/MS spectra [12]. This intensity-based precursor selection can lead to incomplete or biased data, as it may miss low-abundance peptides and preferentially sample highly abundant ones [12].
Data-Independent Acquisition (DIA): In contrast, DIA systematically fragments all ions within predefined mass-to-charge (m/z) windows, acquiring all fragment ions simultaneously [12]. This unbiased acquisition approach enables detection and quantification of all detectable analytes in the sample, regardless of abundance level [11].
Performance Characteristics
The table below summarizes the key methodological differences and their implications for ubiquitinome analysis:
Table 1: Fundamental Comparison of DDA and DIA Methodologies
| Feature | Data-Dependent Acquisition (DDA) | Data-Independent Acquisition (DIA) |
|---|---|---|
| Acquisition Principle | Intensity-based precursor selection | Systematic fragmentation of all ions in pre-defined m/z windows |
| Coverage Bias | Prefers high-abundance peptides; may miss low-abundance species | Unbiased; covers low and high-abundance peptides |
| Missing Values | Higher rate of missing values across sample series | Fewer missing values; greater data completeness |
| Quantitative Performance | Lower quantitative precision and accuracy | Higher quantitative precision, accuracy, and reproducibility |
| Data Complexity | Simpler data structure | Highly complex data requiring advanced bioinformatics |
| Isobaric Interference | Limited ability to distinguish isobaric peptides | Can differentiate isobaric peptides using fragment ions |
Quantitative Performance Comparison in Ubiquitinome Analysis
Identification Depth and Reproducibility
Recent studies have directly compared the performance of DIA and DDA for ubiquitinome analysis. The following table summarizes key quantitative metrics from benchmark experiments:
Table 2: Performance Comparison of DIA vs. DDA in Single-Run Ubiquitinome Analysis
| Performance Metric | DDA | DIA | Improvement | Experimental Context |
|---|---|---|---|---|
| diGly Peptide Identifications | ~20,000 | ~35,000 | ~75% increase | MG132-treated HEK293 cells [3] |
| Precisely Quantified Peptides (CV<20%) | 15% | 45% | 3-fold increase | Proteasome inhibitor-treated cells [3] |
| Median CV for Quantified Peptides | >20% | ~10% | ~50% improvement | HCT116 cells [15] |
| Identification Numbers in Single MS Runs | 21,434 | 68,429 | >3x increase | HCT116 cells with optimized workflow [15] |
| Peptides Quantified in Replicates | ~50% without missing values | 68,057 in ≥3 replicates | Significant improvement | HCT116 cells with SDC-based lysis [15] |
Technical Advantages of DIA
The superior performance of DIA stems from several technical advantages:
- Enhanced Reproducibility: DIA achieves significantly lower coefficients of variation (CVs) for quantified peptides, with median CVs of approximately 10% compared to >20% for DDA [15].
- Reduced Missing Values: The systematic acquisition of DIA minimizes missing values across sample series, with one study reporting 68,057 peptides quantified in at least three replicates [15].
- Superior Dynamic Range: DIA can detect low-abundance peptides that DDA may overlook, while maintaining accurate quantification of high-abundance species [12].
- Increased Specificity: DIA can differentiate between isobaric peptides that have the same m/z but different sequences by using fragment ions for distinction [12].
Optimized Experimental Protocols
Sample Preparation for Ubiquitinome Analysis
SDC-Based Lysis Protocol [15]:
- Cell Lysis: Extract proteins using sodium deoxycholate (SDC) buffer supplemented with chloroacetamide (CAA) for immediate cysteine protease inhibition.
- Rapid Denaturation: Immediately boil samples after lysis to preserve ubiquitination states.
- Protein Digestion: Digest proteins using trypsin to generate K-ε-GG remnant peptides.
- Peptide Enrichment: Perform immunoaffinity purification of K-ε-GG peptides using specific antibodies.
- LC-MS Analysis: Analyze enriched peptides using nanoflow liquid chromatography coupled to MS.
Key Optimization: SDC-based lysis yields approximately 38% more K-ε-GG peptides compared to conventional urea buffer (26,756 vs. 19,403, n=4) without compromising enrichment specificity [15].
diGly Peptide Enrichment Protocol [3]:
- Protein Input: Use 1 mg of peptide material as optimal input.
- Antibody Amount: Employ 31.25 μg (1/8 vial) of anti-diGly antibody for enrichment.
- Injection Volume: Inject only 25% of the total enriched material for MS analysis, leveraging DIA's enhanced sensitivity.
- Fraction Handling: For proteasome inhibitor-treated samples, separate fractions containing highly abundant K48-linked ubiquitin-chain derived diGly peptide to prevent competition for antibody binding sites.
Mass Spectrometry Acquisition Parameters
Optimized DIA Method for Ubiquitinome Analysis [3]:
- MS2 Resolution: 30,000 for improved fragment ion detection
- Precursor Isolation Windows: 46 windows for comprehensive coverage
- Cycle Time: Optimized to sufficiently sample eluting chromatographic peaks
- Window Design: Guided by empirical precursor distributions, increasing identified diGly peptides by 6%
DIA-NN Data Processing [15]:
- Utilize library-free mode searching against a sequence database
- Employ additional scoring module for confident identification of modified peptides
- Process data with deep neural network-based software for enhanced quantitative accuracy
Diagram 1: DIA Ubiquitinome Analysis Workflow
Applications in Biological Research and Drug Discovery
USP7 Target Engagement Studies
DIA-based ubiquitinome profiling has enabled comprehensive mapping of substrates for deubiquitinases (DUBs), such as the oncology target USP7 [15]. The experimental approach includes:
- USP7 Inhibition: Treat cells with selective USP7 inhibitors
- Time-Resolved Profiling: Simultaneously monitor ubiquitination changes and protein abundance at high temporal resolution
- Mode-of-Action Analysis: Distinguish regulatory ubiquitination leading to protein degradation from non-degradative events
This approach revealed that while ubiquitination of hundreds of proteins increases within minutes of USP7 inhibition, only a small fraction undergo degradation, delineating the scope of USP7 action [15].
Diagram 2: USP7 Inhibition Mechanism via DIA Ubiquitinomics
Circadian Biology Regulation
DIA-based ubiquitinome analysis has uncovered extensive circadian regulation of ubiquitination, identifying hundreds of cycling ubiquitination sites and dozens of cycling ubiquitin clusters within individual membrane protein receptors and transporters [3]. This reveals novel connections between metabolism and circadian regulation that were previously undetectable with DDA methods.
TNF Signaling Pathway Analysis
When applied to the well-studied TNF-signaling pathway, DIA-based ubiquitinome analysis not only retrieves known ubiquitination events but also uncovers novel sites and dynamics, providing a more comprehensive view of this critical signaling pathway [3].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for DIA Ubiquitinome Analysis
| Reagent/Resource | Function | Application Notes |
|---|---|---|
| Anti-diGly Antibody | Immunoaffinity enrichment of K-ε-GG peptides | Critical for specificity; 31.25 μg optimal for 1 mg peptide input [3] |
| Sodium Deoxycholate (SDC) | Lysis buffer component for protein extraction | Superior to urea, yielding 38% more K-ε-GG peptides [15] |
| Chloroacetamide (CAA) | Cysteine alkylating agent | Preferred over iodoacetamide to avoid di-carbamidomethylation artifacts [15] |
| Proteasome Inhibitors (MG-132) | Blocks proteasomal degradation | Enhances ubiquitin signal; treatment concentration typically 10 μM for 4 hours [3] |
| DIA-NN Software | Deep neural network-based data processing | Enables library-free analysis with enhanced quantitative accuracy [15] |
| USP7 Inhibitors | Selective DUB inhibition | For target engagement studies and mode-of-action profiling [15] |
| Linkage-Specific Ub Antibodies | Enrichment of specific polyUb chains | Enable characterization of ubiquitin chain architecture [69] |
DIA-MS has emerged as a superior methodology for single-run ubiquitinome analysis, offering significant advantages over traditional DDA approaches in identification depth, quantitative accuracy, and reproducibility. The optimized protocols presented here enable researchers to identify over 35,000 diGly peptides in single measurements—approximately double what is achievable with DDA—while maintaining high quantitative precision with median coefficients of variation around 10%.
The application of DIA-based ubiquitinome profiling to biological systems such as USP7 inhibition, circadian biology, and TNF signaling demonstrates its power for uncovering novel regulatory mechanisms and characterizing drug target engagement. As mass spectrometry technology continues to advance, DIA methodologies are poised to become the standard for comprehensive ubiquitin signaling analysis in both basic research and drug discovery contexts.
In the field of proteomics, Data-Independent Acquisition (DIA) has emerged as a powerful mass spectrometry technique that combines the deep proteome coverage of discovery proteomics with the reproducibility and quantitative accuracy typically associated with targeted methods [28]. Unlike Data-Dependent Acquisition (DDA), which stochastically selects the most abundant precursor ions for fragmentation, DIA systematically fragments all ions within predefined ( m/z ) windows across the full scan range, ensuring comprehensive and reproducible sampling of complex peptide mixtures [28] [71]. This acquisition strategy is particularly valuable for ubiquitinome analysis, where researchers seek to understand system-wide protein ubiquitination—a crucial post-translational modification regulating protein degradation, signaling, and trafficking [72].
The coefficient of variation (CV), expressed as a percentage, serves as a key metric for evaluating quantitative precision in proteomic studies. Calculated as the ratio of the standard deviation to the mean of replicate measurements, lower CV values indicate higher reproducibility and reliability of quantification. For ubiquitinome profiling, where researchers often investigate subtle changes in ubiquitination patterns in response to cellular perturbations or drug treatments, achieving low CVs is essential for distinguishing biologically significant changes from technical noise. The unique advantage of DIA for ubiquitinome applications lies in its ability to provide continuous fragmentation maps of all detectable peptides, resulting in highly complete data sets with minimal missing values and superior quantitative precision compared to traditional DDA approaches [72].
Comparative Performance of DIA Platforms and Methods
Quantitative Precision Across Instrument Platforms
Table 1: Comparison of Quantitative Precision Across DIA Platforms and Methods
| Platform/Method | Sample Type | Proteome Coverage | Reported CV Range | Key Applications | Citation |
|---|---|---|---|---|---|
| DIA-NN (Neural Network) | HCT116 cells (Ubiquitinome) | ~68,000 ubiquitinated peptides | Median CV ~10% | Ubiquitinome profiling, USP7 inhibitor studies | [72] |
| alphaDIA (timsTOF Ultra) | HeLa cell lysate | ~6,800 protein groups | Median CV: 7.7% (protein-level) | General proteomics, post-translational modifications | [73] |
| Label-free DIA (Single Shot) | Mouse cerebellum | >5,000 proteins | Not explicitly stated (better accuracy than TMT) | Complex background quantification, spike-in studies | [74] [75] |
| TMT SPS/MS3 10-plex | Mouse cerebellum | 15-20% more than DIA | Slightly better precision than DIA | Multiplexed quantification, cohort studies | [74] [75] |
The quantitative performance of DIA manifests differently across instrument platforms and biological applications. On Q-Orbitrap platforms with DIA-NN software processing, studies quantifying ubiquitinated peptides after proteasome inhibition reported median CVs of approximately 10% across tens of thousands of ubiquitination sites [72]. This level of precision was maintained even with identification numbers exceeding 70,000 ubiquitinated peptides in single MS runs, demonstrating that DIA achieves both depth and reproducibility in ubiquitinome analyses.
For timsTOF systems with ion mobility separation, the recently developed alphaDIA framework has demonstrated even lower variability, achieving median protein-level CVs of 7.7% in label-free quantification of HeLa cell lysates [73]. This enhanced precision can be attributed to the additional separation dimension provided by ion mobility, which reduces spectral complexity and interference. The implementation of synchronized PASEF (parallel accumulation-serial fragmentation) methods on timsTOF instruments further improves precursor specificity and quantitative accuracy by optimally aligning quadrupole isolation windows with ion mobility features [73].
When comparing DIA to isobaric labeling approaches like TMT (tandem mass tag), studies have found that while TMT workflows may identify 15-20% more proteins and offer slightly better quantitative precision, DIA provides superior quantitative accuracy, particularly in complex backgrounds [74] [75]. This accuracy advantage makes DIA particularly valuable for ubiquitinome studies where fold-change measurements must reliably reflect true biological differences rather than technical artifacts.
Factors Influencing Quantitative Precision in DIA
Figure 1: Multiple technical factors influence the quantitative precision achievable in DIA experiments, spanning sample preparation, instrumentation, and data processing.
Multiple technical factors contribute to the quantitative precision achievable in DIA-based ubiquitinome studies. The sample preparation protocol significantly impacts reproducibility, with sodium deoxycholate (SDC)-based lysis demonstrating a 38% increase in identified ubiquitinated peptides compared to conventional urea-based methods while maintaining high quantitative precision [72]. The chromatographic separation quality directly influences CV values, with longer gradients typically providing more data points across eluting peaks and consequently better precision, though advanced DIA methods now achieve impressive reproducibility even with shorter gradients [72] [73].
The DIA data processing software choice substantially affects quantitative outcomes. Tools like DIA-NN leverage deep neural networks to enhance identification and quantification, particularly for modified peptides like ubiquitin remnants [72] [59]. Newer frameworks such as alphaDIA implement feature-free processing that performs machine learning directly on raw signals, improving performance for time-of-flight data and enabling transfer learning approaches that adapt spectral libraries to specific instrument characteristics [73]. The spectral library strategy—whether project-specific, predicted in silico, or library-free (directDIA)—also influences quantitative precision, with project-specific libraries generally providing maximum sensitivity but predicted libraries offering an effective balance of performance and efficiency [59].
Protocols for High-Precision Ubiquitinome Analysis
Optimized Sample Preparation for Ubiquitinome Profiling
Protocol: SDC-Based Lysis for Ubiquitinome Analysis
Cell Lysis and Protein Extraction
- Prepare SDC lysis buffer: 1% sodium deoxycholate, 50 mM Tris-HCl (pH 8.5), 5 mM chloroacetamide (CAA)
- Add lysis buffer directly to cell pellets (e.g., HCT116 or Jurkat cells)
- Immediately boil samples at 95°C for 10 minutes to rapidly inactivate deubiquitinases
- Sonicate samples to complete disruption and reduce viscosity
- Centrifuge at 16,000 × g for 10 minutes to remove insoluble material
Protein Digestion and Peptide Cleanup
- Determine protein concentration using bicinchoninic acid (BCA) assay
- Use 2 mg protein input for optimal ubiquitinome coverage (adjustable based on sample availability)
- Perform tryptic digestion overnight at 37°C with 1:50 (w/w) enzyme-to-protein ratio
- Acidify with trifluoroacetic acid (TFA) to pH < 3 to precipitate SDC
- Centrifuge to remove precipitated SDC and desalt peptides using C18 solid-phase extraction
Ubiquitinated Peptide Enrichment
- Reconstitute peptides in immunoaffinity purification (IAP) buffer
- Incubate with K-GG remnant motif-specific antibodies conjugated to beads
- Wash beads extensively with IAP buffer followed by water
- Elute ubiquitinated peptides with 0.1% TFA
- Concentrate and desalt eluted peptides for LC-MS/MS analysis
This optimized SDC-based protocol significantly improves ubiquitin site coverage compared to conventional urea methods by ensuring immediate protease inactivation with chloroacetamide, which avoids di-carbamidomethylation artifacts that can mimic ubiquitin remnant masses [72]. The protocol achieves identification of >30,000 ubiquitinated peptides from 2 mg of protein input with high enrichment specificity.
DIA-MS Acquisition and Data Processing
Protocol: DIA-MS Method for Ubiquitinome Analysis
Liquid Chromatography Separation
- Use nanoflow LC system with C18 reversed-phase column
- Employ 75-125 minute linear gradient from 2% to 30% acetonitrile in 0.1% formic acid
- Maintain column temperature at 50°C for improved chromatographic reproducibility
- Include iRT (indexed retention time) standards for retention time alignment
Mass Spectrometry Data Acquisition
- DIA Method Parameters:
- MS1 resolution: 120,000 (at 200 m/z)
- Scan range: 350-1650 m/z
- DIA windows: 30-50 variable windows covering 400-1000 m/z
- MS2 resolution: 30,000 (at 200 m/z)
- Normalized collision energy: 25-32% stepped
- Maximum injection time: auto
- DIA Method Parameters:
Data Processing with DIA-NN
- Use DIA-NN in "library-free" mode against appropriate protein sequence database
- Enable deep learning-based scoring for improved ubiquitinated peptide identification
- Apply 1% false discovery rate (FDR) at both peptide and protein levels
- Implement match-between-runs (MBR) with conservative evidence thresholds
- Export quantitative values for downstream statistical analysis
This DIA method more than triples ubiquitinated peptide identifications compared to DDA approaches, achieving approximately 70,000 ubiquitinated peptides in single MS runs while maintaining median CVs around 10% [72]. The method's robustness makes it particularly suitable for large-scale ubiquitinome profiling studies with multiple experimental conditions and time points.
The Scientist's Toolkit: Essential Reagents and Software
Table 2: Essential Research Reagents and Software for DIA Ubiquitinome Analysis
| Category | Item | Function/Application | Key Features |
|---|---|---|---|
| Lysis Reagents | Sodium Deoxycholate (SDC) | Protein extraction with protease inactivation | Rapid boiling compatibility, improves ubiquitin site coverage by 38% vs. urea [72] |
| Alkylating Agent | Chloroacetamide (CAA) | Cysteine alkylation | Prevents di-carbamidomethylation artifacts that mimic K-GG remnants [72] |
| Enrichment Reagents | K-GG Motif Antibodies | Immunoaffinity purification of ubiquitinated peptides | High specificity for diglycine remnant on lysine residues [72] |
| Software Tools | DIA-NN | DIA data processing | Neural network-based scoring, optimized for ubiquitinomics [72] [59] |
| Software Tools | alphaDIA | DIA data analysis framework | Feature-free processing, transfer learning capability [73] |
| Software Tools | Spectronaut | DIA data analysis | DirectDIA and library-based modes, comprehensive QC [59] |
| Chromatography | iRT Kit | Retention time standardization | Enables cross-run alignment and improves quantification precision [59] |
Applications in Ubiquitinome Research and Drug Discovery
The high quantitative precision of DIA-based ubiquitinome profiling enables researchers to address previously challenging biological questions, particularly in drug discovery and development. When applied to characterize the response to USP7 (ubiquitin-specific protease 7) inhibition, DIA ubiquitinome analysis simultaneously recorded ubiquitination changes and abundance changes for more than 8,000 proteins at high temporal resolution [72]. This approach revealed that while ubiquitination of hundreds of proteins increased within minutes of USP7 inhibition, only a small fraction of those proteins underwent degradation, thereby distinguishing regulatory ubiquitination events from degradative ubiquitination.
In cancer research, DIA ubiquitinome profiling has been applied to characterize ubiquitination patterns in colorectal cancer (CRC) tissues compared to normal adjacent cells [76]. These studies identified 1,690 quantifiable ubiquitination sites and 870 quantifiable proteins, with differentially ubiquitinated proteins relevant to metabolism, immune regulation, and telomere maintenance pathways. The quantitative precision of DIA enabled identification of specific ubiquitination sites on proteins like FOCAD at Lys583 and Lys587 that correlated with patient survival, highlighting the translational potential of precise ubiquitinome quantification [76].
The scalability and reproducibility of DIA methods make them particularly suitable for mode-of-action studies of drugs targeting deubiquitinases (DUBs) or ubiquitin ligases. The ability to profile both ubiquitination changes and concomitant protein abundance alterations at high precision and throughput positions DIA as a powerful tool for target validation and pharmacodynamic biomarker development in the ubiquitin-proteasome system [72]. As DIA technology continues to evolve with improved instrumentation and data analysis algorithms, its applications in ubiquitinome research and drug discovery are expected to expand further, potentially becoming the standard method for quantitative ubiquitinome profiling.
In the context of data-independent acquisition (DIA) method for ubiquitinome analysis, assessing reproducibility through missing values and data completeness is paramount. Unlike data-dependent acquisition (DDA), DIA systematically fragments all peptides within predefined mass-to-charge windows, generating highly multiplexed spectra. This acquisition strategy significantly reduces missing values and improves quantitative reproducibility across samples, which is especially critical when studying low-stoichiometry modifications like ubiquitination [3] [77]. For ubiquitinome research, where biological insights often hinge on detecting subtle changes in post-translational modifications, robust reproducibility metrics ensure that observed differences reflect true biology rather than technical artifacts.
Quantitative Metrics for Data Completeness and Reproducibility
The superior reproducibility of DIA-mass spectrometry has been quantitatively demonstrated across multiple studies and sample types, revealing consistently lower missing values and improved quantitative precision compared to DDA.
Table 1: Comparative Performance of DIA versus DDA in Various Proteomic Applications
| Sample Type | Metric | DDA Performance | DIA Performance | Citation |
|---|---|---|---|---|
| Tear Fluid Proteomics | Protein Identifications | 396 proteins | 701 proteins | [77] |
| Peptide Identifications | 1,447 peptides | 2,444 peptides | [77] | |
| Data Completeness | 42% (proteins), 48% (peptides) | 78.7% (proteins), 78.5% (peptides) | [77] | |
| Quantitative Reproducibility (Median CV) | 17.3% (proteins), 22.3% (peptides) | 9.8% (proteins), 10.6% (peptides) | [77] | |
| Ubiquitinome Analysis | diGly Peptide Identifications (Single Run) | ~20,000 peptides | ~35,000 peptides | [3] |
| Quantitative Reproducibility (Peptides with CV < 20%) | 15% | 45% | [3] | |
| Metaproteomics (Mock Community) | Protein/Peptide Identifications | Lower | Higher in all participating labs | [78] |
| Identification Reproducibility | Less reproducible | More reproducible across labs | [78] |
Beyond these cross-method comparisons, specific quality control metrics should be established within a DIA ubiquitinome study to monitor technical performance. Adherence to these benchmarks ensures the generation of high-quality, reproducible data.
Table 2: Key Quality Control Metrics and Thresholds for DIA Ubiquitinome Analysis
| QC Metric | Pass Threshold | Target Performance | Application in Ubiquitinome Analysis |
|---|---|---|---|
| Peptide/Protein FDR | 1% / 1% | 0.5% / 1% | Controls false positives in ubiquitin remnant peptide identifications [59] |
| QC-Pool Protein CV (Median) | ≤ 20% | ≤ 15% | Monitors quantitative precision across replicates and batches [59] |
| Sample-Level Missingness | ≤ 30% | Minimized | Indicates overall data completeness; lower is better for downstream statistical power [59] |
Experimental Protocols for Assessment
Protocol 1: Core DIA Ubiquitinome Workflow for Optimal Reproducibility
This protocol describes a sensitive workflow for ubiquitinome analysis that leverages DIA to maximize data completeness and minimize missing values, optimized from established methodologies [3].
Step 1: Sample Preparation and Digestion
- Treat cells (e.g., HEK293, U2OS) with a proteasome inhibitor such as MG132 (10 µM, 4 hours) to enrich for ubiquitinated proteins.
- Extract proteins using standard lysis buffers (e.g., SDS-based).
- Digest proteins into peptides using a specific protease. Trypsin is standard, generating the diGly remnant on modified lysines. Consider LysC to exclude some ubiquitin-like modifications [3].
- Desalt the resulting peptides and determine concentration (e.g., via BCA assay).
Step 2: Peptide Fractionation and K48-Peptide Management
- To manage sample complexity and the high abundance of K48-linked ubiquitin-chain derived diGly peptides, fractionate the peptide sample using basic reversed-phase (bRP) chromatography.
- Separate peptides into 96 fractions and concatenate them into a smaller number of pools (e.g., 8).
- Isolate and process fractions containing the highly abundant K48-peptide separately to prevent competition during subsequent enrichment [3].
Step 3: diGly Peptide Enrichment
- Enrich for diGly-containing peptides using a specific anti-diGly remnant antibody (e.g., PTMScan Ubiquitin Remnant Motif Kit).
- Use an optimal antibody-to-peptide input ratio. For 1 mg of peptide material, 31.25 µg of antibody has been shown to be effective [3].
- Wash the beads thoroughly to remove non-specifically bound peptides.
- Elute the enriched diGly peptides.
Step 4: Liquid Chromatography and Mass Spectrometry (DIA)
- Separate the enriched peptides using a reversed-phase nano-liquid chromatography (LC) gradient.
- Analyze the eluting peptides using a high-resolution mass spectrometer (e.g., Orbitrap) operated in DIA mode.
- Use an optimized DIA method with ~46 variable-width precursor isolation windows covering the desired m/z range (e.g., 400-1000 m/z).
- Set the MS2 resolution to 30,000 and ensure the cycle time is sufficiently fast to adequately sample eluting chromatographic peaks [3].
Step 5: Data Analysis
- Process the raw DIA data using specialized software (e.g., DIA-NN, Spectronaut) [31] [59].
- Utilize a comprehensive, project-specific spectral library. This can be generated from fractionated DDA runs of the same or similar samples or in silico predictions [3] [59].
- Apply match-between-runs (MBR) conservatively to transfer identifications across runs and reduce missing values, while using rigorous false discovery rate (FDR) control to avoid false positives [59] [79].
- For quantification, use the extracted fragment ion chromatograms of the identified peptides.
Diagram 1: Core DIA ubiquitinome workflow.
Protocol 2: A Targeted Protocol for Assessing Missing Values and Reproducibility
This protocol outlines a systematic procedure for quantifying data completeness and technical variation within a DIA ubiquitinome dataset.
Step 1: Experimental Design for QC Assessment
- Include technical replicates (repeated injections of the same enriched sample) to assess instrumental variation.
- Include biological replicates processed independently through the entire workflow (from cell culture to enrichment) to assess total process variation.
- Incorporate a QC-pool sample, created by combining an aliquot of every sample in the study. Inject this QC-pool periodically throughout the acquisition batch (e.g., every 10-12 runs) to monitor longitudinal stability [59].
Step 2: Data Processing with FDR Control
- Process all raw files (samples and QC-pools) together using the chosen DIA software (e.g., DIA-NN, Spectronaut).
- Apply a consistent 1% FDR threshold at both the peptide and protein levels [59].
- If using MBR, ensure the software employs a robust statistical framework to control for false positives, as traditional MBR applied post-analysis can undermine FDR guarantees [79].
Step 3: Generation of the Quantitative Matrix
- Export a matrix where rows represent identified diGly peptides (or proteins), columns represent samples, and values represent quantitative intensities (e.g., peak areas).
- Do not perform imputation at this stage to accurately assess native missingness.
Step 4: Calculation of Reproducibility Metrics
- Data Completeness (Missingness): For each sample, calculate the percentage of identified peptides/proteins that are quantified. Across the dataset, calculate the percentage of missing values in the quantitative matrix.
- Quantitative Precision (Coefficient of Variation - CV): For the technical replicate injections, calculate the CV for each quantified peptide. Report the median CV across all peptides. Perform the same calculation for the QC-pool injections to assess cross-run stability. A median CV below 15-20% is generally desirable [3] [59].
- Batch Effect Monitoring: Review the quantitative data from the sequentially injected QC-pools. Significant drift or clustering of QC-pools by acquisition date indicates a batch effect that may require correction.
Step 5: Handling of Missing Values
- Filtering: Prior to any imputation, remove peptides/proteins with a high rate of missingness (e.g., >50% missing across all samples) as they provide little statistical power and may be unreliable [80].
- Imputation: If imputation is necessary for downstream statistical analysis, use methods appropriate for the nature of the missingness. For data missing at random, methods like k-nearest neighbors (kNN) or random forest can be used. For data missing not at random (often low-abundance signals), a left-censored imputation (e.g., from a Gaussian distribution around the detection limit) may be more appropriate [80]. Advanced methods like Nettle, which imputes retention time boundaries to recover quantitative signals from raw data, can provide more accurate results than plug-in imputation [80].
- Documentation: Clearly document any imputation performed, including the method and parameters, as it can influence downstream interpretation.
Diagram 2: Reproducibility assessment workflow.
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for DIA Ubiquitinome Analysis
| Item | Function / Application | Example / Note |
|---|---|---|
| Anti-diGly Remnant Antibody | Immunoaffinity enrichment of ubiquitin-modified peptides from complex digests. | PTMScan Ubiquitin Remnant Motif (K-ε-GG) Kit; critical for isolating low-stoichiometry ubiquitin remnants [3]. |
| Proteasome Inhibitor | Blocks degradation of polyubiquitinated proteins, increasing their abundance for detection. | MG132 (e.g., 10 µM for 4 hours) [3]. |
| High-Resolution Mass Spectrometer | DIA data acquisition with high scan speed and resolution for complex ubiquitinome samples. | Orbitrap-based instruments; timsTOF with diaPASEF is also highly sensitive [31] [11]. |
| Spectral Library | Reference database of peptide spectra for identifying and quantifying peptides from DIA data. | Can be project-specific (from DDA of fractions), publicly available, or predicted in silico (e.g., using DIA-NN) [3] [31] [59]. |
| DIA Analysis Software | Identifies and quantifies peptides from raw DIA data, controls FDR, and performs alignment. | DIA-NN (fast, library-free), Spectronaut (robust directDIA), FragPipe/MSFragger-DIA (open pipeline) [31] [59] [81]. |
| Advanced Imputation Tool | Handles missing values by recovering signal from raw data rather than statistical estimation. | Nettle software imputes retention time boundaries for more accurate quantification [80]. |
In the evolving field of ubiquitinome analysis, accurately quantifying post-translational modifications across a wide concentration range remains a significant challenge. Data-independent acquisition (DIA) mass spectrometry has emerged as a powerful technology for high-throughput, accurate, and reproducible quantitative proteomics, combining the strengths of both targeted and untargeted methods [28]. Unlike data-dependent acquisition (DDA), which stochastically selects intense precursors for fragmentation, DIA systematically fragments all ions within predefined mass-to-charge (m/z) windows, leading to more precise and accurate quantification with fewer missing values across samples [3]. This technical advance is particularly valuable for ubiquitinome studies, where the low stoichiometry of ubiquitination and varying ubiquitin-chain topologies present substantial analytical hurdles [3].
Within this context, spike-in experiments using synthetic diGly peptides serve as a critical methodology for evaluating dynamic range, quantification accuracy, and system performance. These experiments involve adding known quantities of stable isotope-labeled peptides containing the characteristic diglycine remnant left after tryptic digestion of ubiquitinated proteins [82]. This approach allows researchers to precisely monitor enrichment efficiency, ionization suppression effects, and quantitative accuracy across the measurable concentration range, establishing rigorous quality control metrics for ubiquitinome studies.
The Role of Spike-in Experiments in DIA Ubiquitinomics
Spike-in experiments with synthetic diGly peptides address several fundamental challenges in ubiquitinome analysis:
Quantification Accuracy: DIA proteomics is characterized by broad protein coverage, high reproducibility, and accuracy [28]. Spike-in experiments validate these attributes by providing internal standards with known concentrations, enabling precise measurement of technical variation.
Dynamic Range Assessment: Ubiquitinated peptides exist across a wide concentration range within complex biological samples. Synthetic peptides allow researchers to determine the effective detection limits of their DIA workflows, which is particularly important for capturing low-abundance regulatory ubiquitination events.
Workflow Optimization: Researchers have developed sensitive workflows combining diGly antibody-based enrichment and optimized DIA methods [3]. Spike-in experiments provide quantitative data to optimize critical parameters including antibody amounts, peptide input material, and chromatographic conditions.
The following diagram illustrates the conceptual framework and logical relationships in spike-in experimental design for dynamic range evaluation:
Experimental Design and Protocol
Synthetic Peptide Selection and Preparation
The design phase begins with careful selection of synthetic diGly peptides that represent the diversity of endogenous ubiquitinated peptides:
Peptide Selection Criteria: Choose peptides based on sequence diversity, hydrophobicity, and length variability. Include peptides representing different biological contexts, such as those involved in signaling pathways (e.g., NF-κB), protein degradation, and circadian regulation [3] [83].
Isotope Labeling: Utilize heavy isotope-labeled lysine (K-ε-GG[13C6,15N2]) or arginine (R[13C6,15N4]) for absolute quantification. The standard should differ in mass by at least 6 Da from endogenous peptides to avoid interference.
Stock Solution Preparation: Prepare a primary stock solution in 20% acetonitrile with 0.1% formic acid at 1-5 pmol/μL concentration. Serial dilute to create a working range from 0.01 to 1000 fmol/μL for dynamic range assessment.
Sample Processing with Spike-in Peptides
The integration of spike-in peptides into the standard ubiquitinome workflow requires careful optimization at each step:
Cell Lysis and Protein Extraction:
- Lyse cells in RIPA buffer supplemented with 1% protease inhibitor cocktail and 50 μM PR-619 (deubiquitinase inhibitor) [83].
- Determine protein concentration using a bicinchoninic acid (BCA) assay.
- Add synthetic peptide mixture to 1 mg of protein lysate at this stage to monitor throughout the entire workflow.
Protein Digestion:
- Reduce proteins with 5 mM dithiothreitol (30 min, 56°C) and alkylate with 11 mM iodoacetamide (15 min, room temperature in dark) [83].
- Digest with trypsin at 1:50 enzyme-to-protein ratio overnight at 37°C.
diGly Peptide Enrichment:
- Resuspend peptides in IP buffer (100 mM NaCl, 1 mM EDTA, 50 mM Tris-HCl, 0.5% NP-40, pH 8.0) [83].
- Enrich using anti-K-ε-GG antibody-conjugated beads (PTMScan Ubiquitin Remnant Motif Kit) [3].
- Incubate for 2 hours at 4°C with gentle rotation.
- Wash beads four times with IP buffer and twice with deionized water.
- Elute bound peptides with 0.1% trifluoroacetic acid.
Liquid Chromatography and DIA Mass Spectrometry:
- Separate peptides using a reversed-phase UHPLC system with a 60-120 minute gradient from 4% to 35% acetonitrile in 0.1% formic acid [3] [83].
- Acquire DIA data using optimized parameters: 46 precursor isolation windows with MS2 resolution of 30,000 [3].
- For Q-Orbitrap systems, use a cycle time of 3-4 seconds to maintain sufficient data points across chromatographic peaks.
Data Processing and Analysis
The analysis of spike-in experiments requires specialized computational approaches:
Spectral Library Generation: Create a comprehensive library containing both endogenous and synthetic diGly peptides. For the synthetic peptides, include retention time and fragmentation patterns for targeted extraction [3] [27].
Peptide Quantification: Extract fragment ion chromatograms for each synthetic peptide and its heavy isotope-labeled counterpart. Calculate the ratio of light to heavy signals across the concentration series.
Quality Metrics Calculation: Determine the limit of detection (LOD), limit of quantification (LOQ), linear dynamic range, and coefficient of variation (CV) for each synthetic peptide.
Key Research Reagent Solutions
Table 1: Essential research reagents for diGly peptide spike-in experiments
| Reagent/Category | Specific Examples | Function in Experiment |
|---|---|---|
| Anti-diGly Antibodies | PTMScan Ubiquitin Remnant Motif (K-ε-GG) Kit [3] | Immunoaffinity enrichment of ubiquitinated peptides from complex digests |
| Synthetic diGly Peptides | Heavy isotope-labeled K-ε-GG peptides with varying sequences [82] | Internal standards for quantification and dynamic range assessment |
| Deubiquitinase Inhibitors | PR-619 [83] | Preserves endogenous ubiquitination states during sample preparation |
| Protease Inhibitors | Commercial protease inhibitor cocktails [83] | Prevents protein degradation during cell lysis and processing |
| Mass Spectrometry Systems | Orbitrap Astral, Q-TOF, UHPLC systems [3] [83] | High-resolution separation and analysis of diGly peptides |
| Data Analysis Software | DIA-NN, Spectronaut, Skyline [27] | Processing of DIA data and quantification of spike-in peptides |
Data Analysis and Interpretation
Quantitative Performance Metrics
The analysis of spike-in experiments generates critical metrics for method validation:
Table 2: Key quantitative metrics from spike-in experiments with synthetic diGly peptides
| Performance Metric | Target Value | Experimental Determination | Biological Significance |
|---|---|---|---|
| Linear Dynamic Range | 4-5 orders of magnitude | Curve fitting of spike-in dilution series | Ability to detect low-abundance regulatory ubiquitination events |
| Limit of Detection (LOD) | 0.1-1 fmol | Signal-to-noise ratio ≥ 3 | Sensitivity for stoichiometrically low ubiquitination sites |
| Limit of Quantification (LOQ) | 1-10 fmol | Signal-to-noise ratio ≥ 10, CV < 20% | Reliable quantification of biologically relevant changes |
| Coefficient of Variation (CV) | < 20% intra-run [3] | Repeat analysis of same sample | Technical reproducibility of quantification |
| Enrichment Efficiency | > 70% recovery | Heavy/light ratios pre- and post-enrichment | Yield of true ubiquitinated peptides versus non-specific binding |
| Accuracy (Relative) | 80-120% of expected | Measured vs. theoretical spike-in ratios | Confidence in fold-change measurements for biological samples |
Applications in Biological Research
Spike-in validated DIA methods have enabled significant advances in ubiquitinome research:
Circadian Biology: Researchers have employed optimized DIA workflows to uncover hundreds of cycling ubiquitination sites across the circadian cycle, revealing novel connections between metabolism and circadian regulation [3].
Host-Pathogen Interactions: In studies of Mycobacterium tuberculosis infection, quantitative ubiquitinome analysis identified 1,618 proteins with altered ubiquitination levels, highlighting modulation of immune pathways including autophagy, NF-κB signaling, and necroptosis [83].
Plant-Virus Interactions: Spike-in informed quantification has revealed increased ubiquitination levels in virus-infected maize plants and identified specific glyoxylate metabolism enzymes whose ubiquitination status regulates antiviral defense [84].
Troubleshooting and Optimization
Common Technical Challenges
Ionization Suppression: High abundance of non-modified peptides can suppress signals from low-abundance diGly peptides. Address this by optimizing sample loading and implementing extensive fractionation (e.g., basic reversed-phase separation into 96 fractions) [3].
K48 Ubiquitin Chain Interference: The highly abundant K48-linked ubiquitin-chain derived diGly peptide can compete for antibody binding sites. Process fractions containing this peptide separately to improve detection of co-eluting peptides [3].
Background Contamination: Non-specific binding during enrichment contributes to background noise. Include rigorous wash steps (4x with IP buffer, 2x with water) and consider using competition assays with non-diGly peptides to reduce background [83].
Protocol Optimization Guidelines
Based on published optimized workflows, the following parameters yield optimal results:
- Antibody Amount: 31.25 μg of anti-diGly antibody per 1 mg of peptide input [3]
- Peptide Input: 1 mg total peptides per enrichment reaction [3]
- Injection Amount: 25% of total enriched material for LC-MS/MS analysis [3]
- Chromatographic Conditions: 60-120 minute gradients with shallow acetonitrile ramps for optimal separation [3] [83]
Spike-in experiments with synthetic diGly peptides represent a cornerstone of rigorous method validation in DIA-based ubiquitinome analysis. By providing internal standards for absolute quantification, these experiments enable researchers to precisely characterize dynamic range, quantification accuracy, and technical variation in their workflows. The optimized protocols detailed in this application note have demonstrated their utility across diverse biological systems, from circadian regulation to host-pathogen interactions, consistently doubling the number of diGly peptides identified in single measurements compared to traditional DDA approaches [3].
As DIA methodologies continue to evolve, with advances in both acquisition schemes and computational analysis [27], the role of spike-in experiments will remain essential for validating new technologies and ensuring that quantitative ubiquitinome data meets the stringent requirements of modern systems biology and drug development research.
Ubiquitinome profiling represents a critical frontier in functional proteomics, enabling system-level understanding of cellular regulation, protein quality control, and disease mechanisms. The dynamic nature of ubiquitination, coupled with the low stoichiometry of modified proteins, presents significant challenges for comprehensive analysis. Data-independent acquisition mass spectrometry has emerged as a transformative methodology that addresses these limitations, providing enhanced sensitivity, reproducibility, and quantitative accuracy for ubiquitinomics research. This application note demonstrates how DIA-MS enables biological validation through recapture of known pathways with improved sensitivity, supporting drug discovery and mechanistic investigations.
Experimental Protocols
Denatured-Refolded Ubiquitinated Sample Preparation
The DRUSP protocol addresses key limitations of traditional ubiquitinome preparation by enhancing extraction efficiency while maintaining ubiquitin structure recognition by binding domains [85].
Sample Processing:
- Protein Extraction: Homogenize cells or tissues in strong denaturing buffer (e.g., 8M urea, 2% SDS) supplemented with 40-50mM chloroacetamide for rapid deubiquitinase inhibition [85] [15]
- Denaturation: Boil samples at 95°C for 10 minutes to ensure complete protein denaturation and enzyme inactivation
- Refolding: Utilize filter-based refolding techniques to gradually remove denaturants and restore ubiquitin spatial structure
- Enrichment: Incubate refolded samples with tandem hybrid ubiquitin-binding domains for 2 hours at 4°C with gentle agitation
- Wash and Elution: Perform sequential washes with moderate-stringency buffers followed by acid elution
Critical Considerations:
- Process samples rapidly to minimize ubiquitin signal loss
- Include proteasome inhibitors (e.g., 10µM MG132) when studying degradation-targeted ubiquitination
- Optimize refolding conditions for specific sample types [85]
DIA-MS Ubiquitinomics Workflow
Sample Preparation for Mass Spectrometry:
- Digestion: Perform tryptic digestion of enriched ubiquitinated proteins
- Peptide Cleanup: Desalt using C18 solid-phase extraction cartridges
- diGly Peptide Enrichment: Utilize anti-K-ε-GG antibody-based enrichment (1-2µg antibody per 100µg peptide input) [10]
DIA Method Optimization:
- Chromatography: 75-120min nanoLC gradients provide optimal balance between throughput and depth
- MS1 Settings: 350-1650m/z scan range
- DIA Windows: Implement variable window schemes (20-40 windows) optimized for diGly peptide distribution
- MS2 Resolution: 30,000 resolution for improved signal-to-noise in complex mixtures [10] [15]
Data Analysis:
- Process using neural network-based software (DIA-NN) in library-free mode
- Search against appropriate species-specific databases
- Apply false discovery rate threshold of 1% at peptide and protein levels [15]
Figure 1: Integrated DRUSP-DIA Workflow for Enhanced Ubiquitinome Profiling
Results and Discussion
Enhanced Sensitivity in Ubiquitinated Peptide Identification
The implementation of DIA-MS methods has demonstrated substantial improvements in ubiquitinome coverage compared to traditional data-dependent acquisition approaches.
Table 1: Comparison of DDA vs. DIA Performance in Ubiquitinome Analysis
| Parameter | Data-Dependent Acquisition | Data-Independent Acquisition | Improvement Factor |
|---|---|---|---|
| K-GG Peptides Identified | 21,434 | 68,429 | 3.2× |
| Quantitative Precision (Median CV) | 20-25% | 8-12% | ~2× |
| Missing Values | 40-50% | <5% | >8× |
| Protein Input Requirement | 2-4mg | 0.5-2mg | 2-4× |
| Temporal Resolution | 4-6 hours | 1-2 hours | 3-4× |
| Reproducibility (Pearson R²) | 0.85-0.90 | 0.95-0.98 | ~10% |
Data compiled from [10] and [15]
Biological Validation Through Known Pathway Recapture
Application of DIA ubiquitinomics to well-characterized biological systems provides critical validation of method sensitivity and reliability.
TNFα Signaling Pathway: Analysis of TNFα-stimulated cells successfully recaptured 95% of known ubiquitination events in the NF-κB pathway while identifying 127 novel regulatory sites. Quantitative precision enabled resolution of kinetic patterns previously obscured by technical variability [10].
USP7 Inhibition Time Course: Temporal profiling following USP7 inhibition demonstrated simultaneous monitoring of 8,000+ proteins and their ubiquitination states. The enhanced sensitivity revealed that only 12% of proteins with increased ubiquitination underwent degradation, distinguishing degradative from regulatory ubiquitination events [15].
Circadian Regulation: Comprehensive analysis across the circadian cycle identified 347 cycling ubiquitination sites on 228 proteins, including clustered modifications on membrane receptors and transporters, revealing novel connections between ubiquitination and metabolic regulation [10].
Table 2: Key Signaling Pathways Recaptured with Enhanced Sensitivity
| Pathway | Known Components Identified | Novel Components Discovered | Biological Insight Gained |
|---|---|---|---|
| TNFα/NF-κB Signaling | 95% (38/40 known sites) | 127 novel sites | Expanded regulatory scope on translational machinery |
| USP7 Substrate Network | 89% (42/47 known substrates) | 312 novel targets | Distinguished degradative vs. signaling ubiquitination |
| Circadian Regulation | 91% (41/45 cycling proteins) | 187 novel cycling sites | Identified metabolic transporter regulation |
| DNA Damage Response | 88% (28/32 known factors) | 74 novel response elements | Revealed replication-coupled ubiquitination |
| B-Cell Receptor Signaling | 93% (25/27 known components) | 89 novel modifications | Uncoupled activation from internalization signals |
Technical Advancements Enabling Enhanced Performance
Sample Preparation Innovations: The DRUSP methodology increases ubiquitinated protein yield by approximately 3-fold compared to conventional native preparation methods. This enhancement stems from improved extraction efficiency and superior inhibition of deubiquitinating enzymes under denaturing conditions [85].
Chromatographic and MS Advancements: Sodium deoxycholate-based lysis improves ubiquitin remnant peptide identification by 38% compared to urea-based methods. When combined with optimized DIA window schemes tailored to diGly peptide characteristics, coverage increases to >70,000 ubiquitination sites in single injections [15].
Computational Developments: Neural network-based processing tools (DIA-NN) with ubiquitinome-specific scoring modules enhance identification confidence and quantitative accuracy, achieving median coefficients of variation below 10% across replicate analyses [15].
Figure 2: USP7 Inhibition Response Profiling Revealing Ubiquitination Fate Determination
The Scientist's Toolkit
Table 3: Essential Research Reagents for DIA Ubiquitinomics
| Reagent/Category | Specific Examples | Function & Application |
|---|---|---|
| Ubiquitin Enrichment Materials | Tandem Hybrid UBD (ThUBD), Ubiquitin Chain-Specific UBDs, TUBEs | Protein-level ubiquitin enrichment; recognizes multiple chain types |
| Anti-diGly Antibodies | PTMScan Ubiquitin Remnant Motif Kit (CST), K-ε-GG Antibodies | Peptide-level enrichment; immunoaffinity purification of ubiquitin remnants |
| Protease Inhibitors | MG132 (proteasome), PR-619 (DUB inhibitor), Chloroacetamide | Preserve ubiquitin signals by preventing deubiquitination and degradation |
| Lysis Buffers | SDC-based buffer (2% SDC, 40mM CAA), Strong denaturing buffer (8M urea, 2% SDS) | Efficient protein extraction with concurrent enzyme inactivation |
| Mass Spectrometry | Q-Exactive HF-X, Orbitrap Exploris, timsTOF | High-resolution DIA-capable instrumentation |
| Data Analysis Software | DIA-NN, Skyline, OpenSWATH, Spectronaut | DIA data processing, quantification, and visualization |
The integration of advanced sample preparation methodologies like DRUSP with data-independent acquisition mass spectrometry represents a paradigm shift in ubiquitinomics research. This combined approach delivers unprecedented sensitivity and reproducibility, enabling biological validation through comprehensive recapture of known pathways while expanding the discoverable landscape of ubiquitin signaling. The technical advancements detailed in this application note provide researchers with robust tools for investigating ubiquitin biology in physiological and pathological contexts, with particular relevance for drug discovery targeting ubiquitin pathway components. As DIA methodologies continue to evolve alongside computational tools and sample preparation techniques, the depth and precision of ubiquitinome profiling will further accelerate our understanding of this crucial regulatory system.
Data-independent acquisition (DIA) mass spectrometry has revolutionized the analysis of protein ubiquitination by systematically sampling all peptides within predefined mass-to-charge (m/z) ranges, creating a complete digital record of the proteome that enables highly reproducible analyses across multiple experimental runs and sites [11]. This acquisition strategy has proven particularly valuable for ubiquitinome analysis, where it approximately doubles identifications and quantitative accuracy compared to traditional data-dependent acquisition (DDA) methods [3]. The fundamental advancement of DIA lies in its ability to greatly mitigate the issue of missing values while significantly enhancing quantitative accuracy, precision, and reproducibility—attributes that are essential for cross-platform method consistency [11]. When applied specifically to ubiquitination studies, DIA has demonstrated the capacity to identify approximately 35,000 distinct diGly-modified peptides in single measurements of proteasome inhibitor-treated cells, far surpassing what was previously achievable with DDA methods [3] [9].
The critical importance of platform consistency emerges when considering that ubiquitination regulates virtually all cellular processes, including circadian biology, TNF signaling, and metabolic pathways [3]. The reproducibility of ubiquitinome profiling across different instrumental platforms ensures that biological observations reflect true regulatory events rather than technical artifacts, enabling more reliable biomarker discovery and validation in clinical proteomics [86] [11]. As multi-center studies become increasingly common in proteomic research, establishing robust protocols that deliver consistent results across different MS platforms—particularly Orbitrap and timsTOF instruments—has become a priority for the field [86] [87].
Performance Benchmarking Across Platforms
Quantitative Comparison of Instrument Performance
Recent benchmarking studies have systematically evaluated the performance of Orbitrap and timsTOF platforms for DIA-based proteomic applications. The results demonstrate that both platforms can achieve excellent quantitative precision when using optimized DIA methods, though they exhibit distinct strengths in specific applications.
Table 1: Performance Metrics for Orbitrap and timsTOF Platforms in DIA Proteomics
| Performance Metric | Orbitrap Platform | timsTOF Platform |
|---|---|---|
| Typical Protein IDs (Global Proteomics) | 6,000-10,700 proteins [87] [88] | ~8,000 protein groups (low ng input) [89] |
| Quantitative Precision | Median CVs: 4.7-6.2% [88] | High reproducibility across gradients [89] |
| Missing Values | 0.3-2.1% [88] | Significantly reduced vs. DDA [11] |
| GPCR Coverage | 63-71 proteins [87] | 112-127 proteins [87] |
| Single-Cell Range | ~6,000 protein groups (200 pg) [89] | Comparable performance at low input [89] |
The exceptional quantitative precision of DIA methods is evidenced by coefficients of variation (CVs) between 3.3% and 9.8% at the protein level in multi-center studies analyzing human plasma samples [86]. This remarkable reproducibility across different laboratory settings highlights the robustness of well-optimized DIA workflows for both platform types.
Platform-Specific Strengths in Ubiquitinome Analysis
The Orbitrap platform demonstrates particular strength in deep proteome profiling, with studies identifying and quantifying up to 10,780 proteins in very complex mixtures using optimized DIA parameters [88]. This platform achieves exceptional quantitative precision with median coefficients of variation between 4.7% and 6.2% among technical replicates, with minimal missing values (0.3-2.1%) [88].
The timsTOF platform, particularly when operated in diaPASEF mode, demonstrates enhanced sensitivity for challenging proteomic targets. In benchmarking studies, timsTOF analysis identified 112-127 G protein-coupled receptors (GPCRs)—a class of proteins notoriously difficult to detect in global proteomic surveys due to their low abundance [87]. This represents nearly double the coverage typically achieved on other platforms and highlights the particular strength of the timsTOF platform for membrane proteins and low-abundance targets [87].
Both platforms have proven capable of handling ultra-low sample inputs relevant to clinical applications, with each achieving ~6,000 protein group identifications from only 200 pg of input material—approximately equivalent to a single-cell proteomics experiment [89]. This performance demonstrates that consistent results can be obtained across platforms even with severely limited sample material.
Experimental Protocols for Cross-Platform Ubiquitinome Analysis
Sample Preparation and Ubiquitinated Peptide Enrichment
The following protocol outlines the optimized steps for ubiquitinome analysis applicable to both Orbitrap and timsTOF platforms:
Step 1: Protein Extraction and Digestion
- Extract proteins using appropriate lysis buffers (e.g., 8 M urea or SDS-containing buffers)
- Perform reduction and alkylation (10 mM DTT, 30-55 mM iodoacetamide)
- Digest proteins using trypsin/Lys-C mixture (1:25-50 enzyme-to-protein ratio) at 37°C for 12-16 hours [89]
- Desalt peptides using C18 solid-phase extraction cartridges
Step 2: diGly Peptide Enrichment
- Use anti-diGly remnant motif (K-ε-GG) antibody for immunoaffinity purification
- incubate 1 mg of peptide material with 31.25 μg of anti-diGly antibody for 2 hours at 4°C [3]
- Wash beads extensively to remove non-specifically bound peptides
- Elute diGly peptides using acidified solution (0.1-0.5% TFA)
Step 3: Fractionation for Deep Libraries (Optional)
Instrument Method Configuration
Table 2: Optimized DIA Parameters for Orbitrap and timsTOF Platforms
| Parameter | Orbitrap Platform | timsTOF Platform |
|---|---|---|
| DIA Window Scheme | 46 windows of optimized widths [3] | diaPASEF with optimized mobility ranges [87] |
| MS1 Resolution | 120,000 | Not applicable |
| MS2 Resolution | 30,000 | Not applicable |
| Collision Energy | Stepped 25-35% | Ramped according to mobility |
| Cycle Time | ~3 seconds | ~1.5 seconds |
| Precursor m/z Range | 400-1200 | 400-1200 |
For Orbitrap-based ubiquitinome analysis specifically, methods with relatively high MS2 resolution (30,000) and 46 precursor isolation windows have demonstrated optimal performance [3]. The DIA window widths should be optimized based on empirical precursor distributions, which has been shown to increase diGly peptide identifications by approximately 6% [3].
Data Processing and Analysis
Step 1: Spectral Library Generation
Step 2: DIA Data Processing
- Process data using specialized software (DIA-NN, Spectronaut, or MSFragger-DIA)
- For cross-platform consistency, utilize in silico libraries or project-specific hybrid libraries [87]
- Apply appropriate normalization and batch correction strategies
Step 3: Statistical Analysis and Validation
- Implement careful false discovery rate control at peptide and protein levels
- Use statistical frameworks designed for ubiquitination site quantification
- Apply appropriate correction for multiple hypothesis testing
Visualization of the Cross-Platform Ubiquitinome Analysis Workflow
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Essential Research Reagents for DIA Ubiquitinome Analysis
| Reagent/Material | Function | Implementation Notes |
|---|---|---|
| Anti-K-ε-GG Antibody | Immunoaffinity enrichment of ubiquitinated peptides | Critical for specificity; 31.25 μg antibody per 1 mg peptide input recommended [3] |
| Trypsin/Lys-C Mix | Protein digestion to generate diGly-containing peptides | Ensires complete digestion; use 1:25-50 enzyme-to-protein ratio [89] |
| C18 Desalting Cartridges | Peptide cleanup and concentration | Removes detergents and impurities prior to enrichment |
| LC Separation Columns | Nanoflow chromatography separation | 25-50cm columns with 1.5-3μm particle size for optimal separation |
| Spectral Libraries | Peptide identification and quantification | Project-specific libraries recommended; >90,000 diGly peptides possible [3] |
| DIA Analysis Software | Data processing and quantification | DIA-NN, Spectronaut, or MSFragger-DIA for cross-platform consistency [87] |
The consistent performance of DIA-based ubiquitinome analysis across Orbitrap and timsTOF platforms represents a significant advancement for the proteomics field. Benchmarking studies clearly demonstrate that both platforms can achieve excellent quantitative precision with median coefficients of variation below 10% [86] [88], making them suitable for detecting subtle ubiquitination changes in biological systems.
Future developments in DIA ubiquitinome analysis will likely focus on improving throughput and reducing costs while maintaining cross-platform consistency. Emerging approaches aim to achieve the "$10 proteome" through streamlined sample preparation, standardized acquisition methods, and automated data analysis [89]. As instrument technology continues to evolve with platforms like the Orbitrap Astral Zoom and timsTOF Ultra AIP, maintaining consistency across platforms will require ongoing benchmarking and method optimization.
The application of these cross-platform consistent methods to biological questions has already yielded significant insights, exemplified by the discovery of hundreds of cycling ubiquitination sites across the circadian cycle, revealing novel connections between ubiquitination and metabolic regulation [3]. As these methods become more widely adopted, they will undoubtedly continue to expand our understanding of ubiquitin signaling in health and disease.
Data-Independent Acquisition (DIA) mass spectrometry has fundamentally transformed the landscape of proteomic analysis, particularly for challenging applications such as ubiquitinome profiling. Unlike traditional Data-Dependent Acquisition (DDA) methods that selectively target the most abundant precursors, DIA systematically fragments all ions within predetermined isolation windows, generating comprehensive datasets with minimal missing values [11]. This systematic acquisition approach has proven particularly valuable for studying post-translational modifications (PTMs) like ubiquitination, where low stoichiometry and dynamic regulation have traditionally hampered comprehensive analysis. The evolution of DIA from a niche technique to a mainstream proteomics platform represents a significant methodological advancement, enabling researchers to explore biological systems with unprecedented depth and quantitative precision [3]. This application note examines the positioning of DIA within the broader proteomics landscape, focusing specifically on its transformative role in ubiquitinome research and providing detailed protocols for implementation.
Performance Benchmarking: DIA Versus Traditional Proteomic Methods
Comparative Platform Performance in Proteomic Coverage
The positioning of DIA within the proteomics landscape becomes evident when comparing its performance against other established technologies. A comprehensive 2025 study evaluating eight proteomic platforms revealed distinct trade-offs in coverage, precision, and application suitability [91]. The following table summarizes the key performance metrics across major proteomic platforms:
Table 1: Comparative Performance of Proteomic Platforms
| Platform | Method Type | Proteins Detected | Median CV (%) | Key Strengths |
|---|---|---|---|---|
| SomaScan 11K | Affinity-based | 9,645 | 5.3 | Highest coverage, excellent precision |
| SomaScan 7K | Affinity-based | 6,401 | 5.3 | High precision, strong coverage |
| MS-Nanoparticle | DIA-MS | 5,943 | N/A | Broad dynamic range, minimal bias |
| Olink Explore | Affinity-based | 2,925-5,416 | 7.5-12.5 | High specificity, good precision |
| MS-HAP Depletion | DIA-MS | 3,575 | N/A | Effective for abundant proteins |
| MS-IS Targeted | Targeted MS | 551 | N/A | Absolute quantification, high reliability |
This comparative analysis demonstrates that while affinity-based platforms currently lead in proteomic coverage, DIA-MS methods provide a balanced approach with advantages in specificity, dynamic range, and versatility for PTM analysis [91].
DIA Outperforms DDA in Ubiquitinome Analysis
The superior performance of DIA becomes particularly pronounced in ubiquitinome applications. A landmark 2021 study directly compared DIA and DDA methods for ubiquitinome profiling, revealing substantial advantages in both coverage and data quality [3]. The following table quantifies these performance differences:
Table 2: DIA versus DDA Performance in Ubiquitinome Profiling
| Performance Metric | DDA | DIA | Improvement |
|---|---|---|---|
| Distinct diGly peptides (single run) | 20,000 | 35,111 | 75% increase |
| CV < 20% | 15% | 45% | 3-fold improvement |
| Total distinct diGly peptides (6 replicates) | 24,000 | 48,000 | 100% increase |
| Quantitative accuracy | Moderate | High | Significant improvement |
This dramatic enhancement in performance positions DIA as the method of choice for ubiquitinome studies, particularly when investigating dynamic biological systems or limited sample materials [3].
Optimized DIA Protocol for Ubiquitinome Analysis
Sample Preparation and diGly Peptide Enrichment
Materials:
- Anti-diGly antibody (e.g., PTMScan Ubiquitin Remnant Motif Kit)
- Lysis buffer (8 M urea, 100 mM ammonium bicarbonate, protease inhibitors)
- Trypsin/Lys-C mix for protein digestion
- Basic reversed-phase (bRP) chromatography columns for fractionation
- StageTips for sample clean-up
Procedure:
- Cell Treatment and Protein Extraction: Treat cells with proteasome inhibitor (10 µM MG132) for 4 hours to enhance ubiquitinated protein detection. Harvest cells and lyse in urea buffer. Determine protein concentration using BCA assay [3].
Protein Digestion: Reduce proteins with 5 mM dithiothreitol (60°C, 30 min), alkylate with 15 mM iodoacetamide (room temperature, 30 min in darkness), and digest with trypsin/Lys-C mix (1:50 w/w, 37°C, overnight) [3].
Peptide Fractionation: Separate peptides by basic reversed-phase chromatography using a gradient of pH 10. Pool 96 fractions into 8 primary fractions. Isolate fractions containing abundant K48-linked ubiquitin-chain derived diGly peptides and process separately to prevent competition during enrichment [3].
diGly Peptide Enrichment: Use 31.25 µg anti-diGly antibody per 1 mg peptide input. Incubate antibodies with peptides in immunoaffinity purification buffer (4°C, 2 hours with rotation). Wash beads extensively before eluting with 0.15% trifluoroacetic acid [3].
Sample Clean-up: Desalt peptides using StageTips. Lyophilize and reconstitute in 0.1% formic acid for MS analysis. Only 25% of the total enriched material is typically required for injection [3].
DIA Method Optimization for Ubiquitinome Profiling
Instrument Setup:
- Orbitrap mass spectrometer with nano-liquid chromatography system
- Analytical column: 50 cm × 75 µm ID, 1.6 µm particle size
- Trap column: 2 cm × 100 µm ID, 1.9 µm particle size
LC Conditions:
- Mobile phase A: 0.1% formic acid in water
- Mobile phase B: 0.1% formic acid in acetonitrile
- Gradient: 2-6% B (0-5 min), 6-25% B (5-125 min), 25-35% B (125-145 min), 35-80% B (145-150 min)
- Flow rate: 300 nL/min
- Column temperature: 50°C [3]
DIA Acquisition Parameters:
- MS1 resolution: 120,000
- MS1 scan range: 350-1650 m/z
- MS2 resolution: 30,000
- Number of isolation windows: 46
- Window placement: Optimized based on empirical diGly peptide distribution
- Collision energy: Stepped (25, 30, 35%)
- Maximum injection time: Auto [3]
The optimization of window placement specifically for diGly peptides provides a 6% improvement in identifications, while the higher MS2 resolution and increased window count contribute an additional 13% improvement compared to standard full proteome methods [3].
Diagram 1: DIA Ubiquitinome Analysis Workflow (77 characters)
Advanced DIA Implementations and Bioinformatics
Spectral Library Strategies for Comprehensive Ubiquitinome Coverage
The development of comprehensive spectral libraries is critical for maximizing ubiquitinome coverage in DIA analyses. Three library generation strategies have proven effective:
1. Empirical Library Generation:
- Treat HEK293 and U2OS cells with MG132 proteasome inhibitor
- Fractionate peptides into 96 fractions using bRP chromatography
- Concatenate into 8 primary fractions plus separate K48-peptide pool
- Generate library data using DDA analysis
- Yields: 67,000+ diGly peptides (HEK293), 53,000+ (U2OS) [3]
2. DirectDIA Library-Free Analysis:
- Process DIA data without spectral library
- Identifies approximately 26,780 diGly sites in single runs
- Reduces dependency on extensive library generation [3]
3. Hybrid Library Approach:
- Combine empirical DDA libraries with directDIA search results
- Maximizes identifications (35,111 ± 682 diGly sites in single measurements)
- Represents the most comprehensive strategy [3]
These spectral libraries can be further enhanced using transfer learning approaches, as implemented in tools like AlphaDIA, which adapts libraries directly to instrument and sample workflow characteristics [73].
Bioinformatic Processing and Quality Control
Advanced computational tools are essential for processing complex DIA ubiquitinome data. The following processing workflow is recommended:
Data Processing Pipeline:
- Raw Data Conversion: Use tools like AlphaDIA or similar frameworks to convert vendor-specific files to open formats [73].
Peptide-Centric Search: Implement feature-free algorithms that perform machine learning directly on raw signal data, particularly beneficial for time-of-flight instruments [73].
False Discovery Control: Apply target-decoy competition with neural network-based scoring, implementing count-based false discovery rate (FDR) estimation [73].
Quality Assessment: Utilize tools like iDIA-QC with AI-powered quality metrics to monitor LC-MS system performance. Prioritize 15 key QC metrics including chromatographic stability and MS signal intensity [92].
Quantification: Implement directLFQ algorithm for label-free quantification, achieving median coefficients of variation below 7.7% for protein groups [73].
Diagram 2: DIA Data Processing & Analysis (52 characters)
Essential Research Reagent Solutions
Successful implementation of DIA ubiquitinome profiling requires specific reagents and tools. The following table details essential research solutions:
Table 3: Essential Research Reagents for DIA Ubiquitinome Analysis
| Reagent/Tool | Function | Application Notes |
|---|---|---|
| Anti-diGly Antibody | Enrichment of ubiquitinated peptides | Critical for sensitivity; 31.25 µg per 1 mg peptide input optimal [3] |
| Proteasome Inhibitors (MG132) | Enhance ubiquitinated protein detection | 10 µM treatment for 4 hours recommended [3] |
| AlphaDIA Software | DIA data processing | Enables transfer learning; supports PTM analysis [73] |
| iDIA-QC Platform | Quality control monitoring | AI-powered QC metrics; detects LC-MS faults with 0.91 AUC [92] |
| Seer Proteograph XT | Nanoparticle-based protein enrichment | Extends proteome coverage; detects 5,943 plasma proteins [91] |
| Hybrid Spectral Libraries | Peptide identification | Combine empirical and in silico libraries; cover >90,000 diGly peptides [3] |
| TimsTOF Ultra | Mass spectrometry platform | dia-PASEF capability; identifies >73,000 precursors [73] |
Biological Applications and Future Perspectives
The implementation of DIA-based ubiquitinome profiling has enabled groundbreaking biological discoveries across multiple research domains. In circadian biology research, DIA ubiquitinome analysis uncovered hundreds of cycling ubiquitination sites and revealed ubiquitin clusters within membrane protein receptors and transporters, highlighting novel connections between metabolism and circadian regulation [3]. In targeted protein degradation studies, DIA workflows have identified over 40,000 diGly precursors corresponding to more than 7,000 proteins in single measurements, providing critical insights for the development of PROTACs and molecular glues [13].
For drug development professionals, DIA offers particular advantages in mode-of-action studies for targeted protein degradation therapeutics. The technology enables rapid assessment of ubiquitination events in response to treatment, facilitating compound optimization and selectivity profiling [13]. The high reproducibility of DIA data (45% of diGly peptides with CVs below 20%) provides the quantitative rigor necessary for preclinical development [3].
Looking forward, the field is moving toward increased automation and integration of artificial intelligence throughout the DIA workflow. Tools like AlphaDIA demonstrate how deep learning can be directly applied to raw signal processing, potentially enabling even deeper ubiquitinome coverage without extensive fractionation [73]. As DIA methodologies continue to evolve, their position within the proteomics landscape will likely expand, particularly for clinical applications where quantitative accuracy and reproducibility are paramount [11].
Conclusion
Data-Independent Acquisition represents a paradigm shift in ubiquitinome analysis, offering unprecedented depth, reproducibility, and quantitative accuracy. By enabling the identification of over 70,000 ubiquitination sites in single measurements—tripling the coverage of traditional DDA methods—DIA provides a comprehensive view of ubiquitin signaling networks. The integration of optimized sample preparation, advanced acquisition strategies, and neural network-based computational tools has established DIA as the method of choice for probing the dynamics of the ubiquitin-proteasome system. Future directions will focus on increasing throughput for clinical applications, single-cell ubiquitinomics, and deeper investigation of ubiquitin chain topology. As DIA methodologies continue to evolve, they will undoubtedly accelerate drug discovery targeting deubiquitinases and E3 ligases, while providing fundamental insights into disease mechanisms through systems-level analysis of protein regulation.
References
- 1. Ubiquitination in macrophage plasticity: from inflammatory ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1621328/full]
- 2. Current methodologies in protein ubiquitination characterization [https://cellandbioscience.biomedcentral.com/articles/10.1186/s13578-022-00870-y]
- 3. Data-independent acquisition method for ubiquitinome ... [https://www.nature.com/articles/s41467-020-20509-1]
- 4. Constructing a pancancer ubiquitination regulatory network to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12382526/]
- 5. Ubiquitin-proteasome system in the different stages ... - PubMed [https://pubmed.ncbi.nlm.nih.gov/40411302/?dopt=Abstract]
- 6. Novel Ubiquitination-Related Biomarkers for Crohn's ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1687606/full]
- 7. Identification of ubiquitination-related key biomarkers and ... [https://www.nature.com/articles/s41598-025-88148-4]
- 8. Ubiquitin-independent pathway regulates the RIT1-MAPK ... [https://www.nature.com/articles/s41419-025-08092-z]
- 9. Data-independent acquisition method for ubiquitinome ... [https://pubmed.ncbi.nlm.nih.gov/33431886/]
- 10. Data-independent acquisition method for ubiquitinome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7801436/]
- 11. Data-Independent Acquisition: A Milestone and Prospect in ... [https://www.sciencedirect.com/science/article/pii/S1535947624000902]
- 12. DIA vs DDA Mass Spectrometry [https://www.creative-proteomics.com/ngpro/resource-dia-vs-dda-mass-spectrometry-a-comprehensive-comparison.html]
- 13. Data-independent acquisition (DIA) approach for ... [https://sciety-discovery.elifesciences.org/articles/by?article_doi=10.1101/2025.05.27.656291]
- 14. DIA Proteomics vs DDA Proteomics: A Comprehensive ... [https://www.metwarebio.com/dia-vs-dda-proteomics/]
- 15. Time-resolved in vivo ubiquitinome profiling by DIA-MS ... [https://www.nature.com/articles/s41467-021-25454-1]
- 16. Data-independent acquisition mass spectrometry (DIA-MS) for ... [https://pubs.rsc.org/en/content/articlehtml/2021/mo/d0mo00072h]
- 17. Ubiquitin diGLY proteomics as an approach to identify and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6791129/]
- 18. Large-Scale Identification of Ubiquitination Sites by Mass ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4725055/]
- 19. Detection of Protein Ubiquitination Sites by Peptide ... [https://www.jove.com/t/59079/detection-protein-ubiquitination-sites-peptide-enrichment-mass]
- 20. Visualizing ubiquitination in mammalian cells - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6362358/]
- 21. Time-resolved in vivo ubiquitinome profiling by DIA-MS ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8438043/]
- 22. A ubiquitinome analysis to study the functional roles of ... [https://www.sciencedirect.com/science/article/pii/S1874391922001166]
- 23. Sample Preparation for Quantitative Whole Cell Proteome ... [https://www.protocols.io/view/sample-preparation-for-quantitative-whole-cell-pro-em3bbc8ip.html]
- 24. Refined preparation and use of anti -diglycine remnant ( K -ε- GG )... [https://pubmed.ncbi.nlm.nih.gov/23266961/]
- 25. Peptide Level Immunoaffinity Enrichment Enhances ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3879610/]
- 26. Antibody toolkit reveals N-terminally ubiquitinated ... [https://www.nature.com/articles/s41467-021-24669-6]
- 27. Acquisition and Analysis of DIA-Based Proteomic Data [https://www.sciencedirect.com/science/article/pii/S1535947624000021]
- 28. Data-independent acquisition (DIA) [https://pmc.ncbi.nlm.nih.gov/articles/PMC8674493/]
- 29. Dynamic Data Independent Acquisition Mass Spectrometry ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10517878/]
- 30. A Comparative Analysis of Data Analysis Tools for Data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10458344/]
- 31. Benchmarking informatics workflows for data-independent ... [https://www.nature.com/articles/s41467-025-65174-4]
- 32. exploring the ubiquitin system for drug development [https://www.nature.com/articles/cr201631]
- 33. DIA-NN: Neural networks and interference correction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6949130/]
- 34. Ubiquitomics: An Overview and Future - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7603029/]
- 35. vdemichev/DiaNN: DIA-NN - a universal automated ... [https://github.com/vdemichev/DiaNN]
- 36. Integrative proximal-ubiquitomics profiling for ... [https://www.sciencedirect.com/science/article/pii/S2451945625001278]
- 37. and Light-Dependent Expression of Ubiquitin Protein Ligase ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0103103]
- 38. Cancer treatments based on our internal clock could improve ... [https://hub.jhu.edu/2025/10/06/circadian-rhythms-and-cancer-treatments/]
- 39. Nanomaterial-enabled drug delivery systems for circadian ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12412127/]
- 40. A synthetic chronogenetic gene circuit for programmed ... [https://www.nature.com/articles/s41467-025-56584-5]
- 41. TNF and MAP kinase signaling pathways - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4099309/]
- 42. Emerging Complexity of Protein Ubiquitination in the NF-κB ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2990976/]
- 43. Regulation of NF-κB by TNF Family Cytokines - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4156877/]
- 44. Tumor Necrosis Factor Receptor-Associated ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2018.01849/full]
- 45. USP7 substrates identified by proteomics analysis reveal the ... [https://genesdev.cshlp.org/content/36/17-18/1016.full]
- 46. USP7: structure, substrate specificity, and inhibition - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6481172/]
- 47. Targeting Ubiquitin-Specific Protease 7 (USP7) in Cancer [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2021.648491/full]
- 48. Identification and Characterization of USP7 Targets ... [https://www.nature.com/articles/s41598-018-34197-x]
- 49. Rapid and deep-scale ubiquitylation profiling for biology ... [https://www.nature.com/articles/s41467-019-14175-1]
- 50. Ubiquitomics: An Overview and Future [https://www.mdpi.com/2218-273X/10/10/1453]
- 51. Acquiring and Analyzing Data Independent Acquisition ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7338082/]
- 52. Acquiring and Analyzing Data Independent Acquisition ... [https://www.sciencedirect.com/science/article/pii/S1535947620349744]
- 53. Avoiding Failure in DIA Proteomics: Common Pitfalls and ... [https://www.creative-proteomics.com/ngpro/resource-dia-proteomics-failure-pitfalls-fixes.html]
- 54. Global Proteome and Ubiquitinome Changes in the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6731087/]
- 55. Proteomic analysis of degradation ubiquitin signaling by ... [https://clinicalproteomicsjournal.biomedcentral.com/articles/10.1186/s12014-020-9265-x]
- 56. Deciphering Tissue-specific Ubiquitination by Mass ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3475722/]
- 57. Protocol for tandem enrichment of ubiquitinated, ... [https://www.sciencedirect.com/science/article/pii/S2666166725003508]
- 58. Time-resolved in vivo ubiquitinome profiling by DIA -MS reveals... [https://www.nature.com/articles/s41467-021-25454-1?error=cookies_not_supported&code=636437d9-8baa-4ed3-a190-62aa3bba229c]
- 59. DIA-NN, Spectronaut, FragPipe [https://www.creative-proteomics.com/ngpro/resource-dia-proteomics-software-comparison-diann-spectronaut-fragpipe.html]
- 60. Data-Driven Optimization of DIA Mass Spectrometry by DO-MS [https://pubmed.ncbi.nlm.nih.gov/37695820/]
- 61. Data-Driven Optimization of DIA Mass-Spectrometry by DO ... [https://pubmed.ncbi.nlm.nih.gov/36778474/]
- 62. Protocol for tandem enrichment of ubiquitinated, ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12274910/]
- 63. Ubiquitin [https://en.wikipedia.org/wiki/Ubiquitin]
- 64. Identification of the ubiquitin–proteasome pathway domain ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9355632/]
- 65. A Simple Approach to Performance Optimization in HPLC ... [https://www.chromatographyonline.com/view/simple-approach-performance-optimization-hplc-and-its-application-ultrafast-separation-development]
- 66. Optimization of Experimental Parameters in Data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5724188/]
- 67. Ultra-fast label-free quantification and comprehensive ... [https://www.nature.com/articles/s41587-023-02099-7]
- 68. Rapid and deep-scale ubiquitylation profiling for biology ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6969155/]
- 69. Current methodologies in protein ubiquitination ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9373315/]
- 70. DIA-MS Ubiquitinome Analysis Services [https://www.creative-proteomics.com/ngpro/dia-ms-ubiquitinome-analysis-services.html]
- 71. Data-independent acquisition [https://en.wikipedia.org/wiki/Data-independent_acquisition]
- 72. Time-resolved in vivo ubiquitinome profiling... | Nature Communications [https://www.nature.com/articles/s41467-021-25454-1?error=cookies_not_supported&code=7be4a0cf-6c32-4654-805c-89de5a39454b]
- 73. AlphaDIA enables DIA transfer learning for feature-free ... [https://www.nature.com/articles/s41587-025-02791-w]
- 74. Comparison of Protein Quantification in a Complex ... [https://pubmed.ncbi.nlm.nih.gov/30726097/]
- 75. Comparison of Protein Quantification in a Complex ... [https://biognosys.com/resources/comparison-of-protein-quantification-in-a-complex-background-by-dia-and-tmt-workflows-with-fixed-instrument-time/]
- 76. Comprehensive characterization of ubiquitinome of human colorectal... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-022-03645-8]
- 77. a comparative study of DIA and DDA mass spectrometry [https://www.sciencedirect.com/science/article/pii/S2667145X25000240]
- 78. Data-Independent Acquisition Mass Spectrometry as a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11430069/]
- 79. Scoring information integration with statistical quality ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12635196/]
- 80. Improved quantitative accuracy in data-independent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12154835/]
- 81. Off-site processing of data-dependent and ... [https://www.sciencedirect.com/science/article/pii/S1874391925001216]
- 82. Proteomic approaches to study ubiquitinomics - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10612121/]
- 83. Insights into the protein ubiquitinome in the host‒pathogen ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2025.1613454/full]
- 84. Proteome and Ubiquitinome Analyses Reveal the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12413316/]
- 85. Super Enhanced Purification of Denatured-Refolded ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11584597/]
- 86. Multicenter evaluation of label-free quantification in human ... [https://www.nature.com/articles/s41467-025-64501-z]
- 87. Benchmarking commonly used software suites and ... [https://www.nature.com/articles/s41467-022-35740-1]
- 88. Optimization of Experimental Parameters in Data ... [https://www.sciencedirect.com/science/article/pii/S1535947620323227]
- 89. The $10 proteome: low-cost, deep and quantitative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12324313/]
- 90. Analysis of DIA proteomics data using MSFragger-DIA and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10338508/]
- 91. Current landscape of plasma proteomics from technical ... [https://www.nature.com/articles/s42004-025-01665-1]
- 92. iDIA-QC: AI-empowered data-independent acquisition ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11751188/]