Decoding Cancer's Ubiquitination Blueprint: From Molecular Mechanisms to Clinical Applications
This comprehensive review synthesizes current knowledge on the systematic alterations in ubiquitination patterns between cancerous and normal tissues.
Decoding Cancer's Ubiquitination Blueprint: From Molecular Mechanisms to Clinical Applications
Abstract
This comprehensive review synthesizes current knowledge on the systematic alterations in ubiquitination patterns between cancerous and normal tissues. Ubiquitination, a crucial post-translational modification, is extensively dysregulated across cancer types, affecting tumor metabolism, immune evasion, cancer stem cell maintenance, and therapeutic responses. We explore foundational concepts of the ubiquitin-proteasome system, methodological approaches for profiling ubiquitination signatures, troubleshooting strategies for technical challenges, and validation frameworks for translating these findings into clinical applications. For researchers and drug development professionals, this article provides critical insights into ubiquitination-based biomarkers and emerging therapeutic strategies, including proteasome inhibitors, molecular glues, and PROTACs that are revolutionizing cancer treatment.
The Ubiquitin-Proteasome System: Core Mechanisms and Cancer-Specific Alterations
The ubiquitin-proteasome system (UPS) represents a crucial regulatory mechanism for maintaining cellular homeostasis, orchestrating the controlled degradation and modulation of vast numbers of intracellular proteins [1]. This system employs a sequential enzymatic cascade involving E1 (ubiquitin-activating), E2 (ubiquitin-conjugating), and E3 (ubiquitin-ligating) enzymes to tag substrate proteins with ubiquitin, a small 76-amino-acid protein [2] [3]. The human genome encodes a limited number of E1 enzymes (2), approximately 40 E2 enzymes, and an estimated 600-1000 E3 enzymes, which confer substrate specificity [1] [3]. The reverse reaction, deubiquitination, is performed by deubiquitinases (DUBs), with nearly 100 human DUB genes identified that counterbalance ubiquitination to maintain precise protein stability [2] [4]. This dynamic equilibrium ensures proper regulation of vital cellular processes including cell cycle progression, signal transduction, DNA repair, and apoptosis [2] [1]. The integrity of this system is fundamental to cellular health, and its dysregulation is a hallmark of numerous pathological states, most notably cancer.
Comparative Analysis of Ubiquitination Patterns in Cancerous vs. Normal Tissues
Comprehensive multi-omics studies have revealed distinct and pervasive dysregulation of ubiquitination machinery across various cancer types, impacting mRNA expression, protein stability, and enzymatic activity.
Pan-Cancer Genetic and Expression Alterations
Integrated genomic analyses demonstrate widespread perturbations of ubiquitination regulators (UBRs) in pan-cancer cohorts. A systematic investigation of UBRs revealed widespread genetic alterations and expression perturbations across multiple cancer types [5]. The expression patterns of UBRs show significant heterogeneity across different tissues, with the testis exhibiting the most distinct profile; however, in carcinogenesis, these expression programs become profoundly disrupted [5]. Furthermore, the relationship between UBR expression and cancer hallmark pathways is remarkably extensive, with 79 UBRs showing close correlation with the activity of 32 cancer hallmark-related pathways, indicating a global reprogramming of ubiquitin-mediated regulatory networks in malignancy [5].
Table 1: Ubiquitination Regulator Alterations in Pan-Cancer Analyses
| Analysis Type | Key Finding | Cancer Implications |
|---|---|---|
| Genetic Alterations | Somatic mutations and copy number variations in UBRs | Disrupted substrate specificity and protein turnover |
| mRNA Expression | Widespread expression perturbations of UBRs | Altered abundance of ubiquitination machinery components |
| Pathway Correlation | 79 UBRs correlated with 32 cancer hallmark pathways | Reprogrammed cellular signaling and metabolic networks |
| Clinical Relevance | >90% of UBRs affect cancer patient survival | Potential prognostic and therapeutic significance |
Tissue-Specific Ubiquitinome Reprogramming
Ubiquitinomics technologies, which utilize anti-K-ε-GG antibody-based enrichment of ubiquitinated peptides coupled with mass spectrometry, have enabled precise mapping of ubiquitination alterations in specific cancer types.
In sigmoid colorectal cancer, the first ubiquitinome analysis identified 1,249 ubiquitinated sites within 608 differentially ubiquitinated proteins (DUPs) compared to para-carcinoma control tissues [6]. Bioinformatics analysis revealed these DUPs participate in 35 significantly altered signaling pathways, including Salmonella infection, glycolysis/gluconeogenesis, and ferroptosis pathways [6]. The relationship between ubiquitination and corresponding gene expression revealed four distinct regulatory models (DUP-up/DEG-up; DUP-up/DEG-down; DUP-down/DEG-up; DUP-down/DEG-down), indicating complex layers of ubiquitination control in cancer pathogenesis [6].
In hepatocellular carcinoma (HCC), integrated multi-omics analysis demonstrated that ubiquitination-related genes are significantly upregulated in tumor tissues, with high expression levels correlating with poor patient prognosis [7]. These ubiquitination alterations were enriched in critical pathways including cell cycle regulation, DNA repair, metabolic reprogramming, and p53 signaling, collectively contributing to a tumor-permissive microenvironment through enhanced proliferation, matrix remodeling, and angiogenesis [7].
Table 2: Tissue-Specific Ubiquitinome Alterations in Cancer
| Cancer Type | Ubiquitination Alterations | Affected Pathways/Biological Processes | Clinical Correlation |
|---|---|---|---|
| Sigmoid Colorectal Cancer | 1,249 altered ubiquitination sites on 608 proteins | Glycolysis/gluconeogenesis, ferroptosis, Salmonella infection | 46 overall survival-related DUPs identified |
| Hepatocellular Carcinoma | Significant upregulation of ubiquitination-related genes | Cell cycle, DNA repair, metabolic reprogramming, p53 signaling | Poor prognosis associated with high UBR expression |
| Various Cancers | Dysregulation of Met1-linked ubiquitin chains | NF-κB activation, inflammatory signaling, infection response | Immune disorders and cancer progression |
E3 Ligase and DUB Dysregulation in Oncogenesis
Specific components of the ubiquitination machinery are frequently altered in cancer. The anaphase-promoting complex/cyclosome (APC/C) and Skp1-Cul1-F-box (SCF) E3 ligase complexes, crucial regulators of cell cycle progression, are frequently dysregulated, leading to uncontrolled proliferation and tumorigenesis [2]. The E3 ligase MDM2, which targets tumor suppressor p53 for degradation, is overactive in many cancers, effectively shutting down p53's protective functions [1] [3]. Conversely, tumor suppressor DUBs like BAP1 are frequently inactivated in cancer, disrupting their roles in regulating cell proliferation, differentiation, and metabolism [2].
The USP28 deubiquitinase is overexpressed in colon and lung cancers, where it stabilizes potent oncogenes including c-Myc, Notch1, and c-jun, driving tumor progression and therapeutic resistance [4]. These specific alterations represent potential therapeutic vulnerabilities in the ubiquitination system that can be exploited for cancer treatment.
Experimental Protocols for Ubiquitination Analysis
Ubiquitinomics Workflow for Profiling Ubiquitination Sites
The comprehensive identification and quantification of ubiquitination patterns in tissues requires specialized ubiquitinomics approaches.
Protocol: Label-Free Quantitative Ubiquitinomics
- Tissue Sample Preparation: Flash-freeze tissue samples in liquid nitrogen and homogenize in lysis buffer containing protease and phosphatase inhibitors [6].
- Protein Digestion: Digest proteins with trypsin following standard proteomics protocols [6].
- Ubiquitinated Peptide Enrichment: Incubate digested peptides with anti-K-ε-GG antibody beads (PTMScan Ubiquitin Remnant Motif Kit) to specifically enrich for ubiquitinated peptides [6].
- Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS): Separate enriched peptides using nano-liquid chromatography and analyze by high-resolution tandem mass spectrometry [6].
- Data Analysis: Process raw MS data using specialized software (MaxQuant, Skyline) to identify and quantify ubiquitination sites. Use bioinformatics tools for pathway enrichment analysis and network construction [6].
This protocol enables large-scale qualitative and quantitative analysis of ubiquitinated proteins without expensive isotope labels, making it accessible for multiple sample comparisons [6].
Functional Validation of Ubiquitination Regulators
Protocol: In Vitro Functional Assays for UBR Validation
- Gene Knockdown: Transfect HCC cells (Huh7, Hep3B) with shRNA plasmids specifically targeting UBRs of interest (e.g., UBE2C) using Lipofectamine 3000. Culture cells in DMEM with 10% FBS and select with puromycin for stable knockdown [7].
- qPCR Validation: Extract total RNA 48 hours post-transfection using RNA extraction kits. Synthesize cDNA and perform quantitative PCR with gene-specific primers to confirm knockdown efficiency [7].
- Phenotypic Assays:
- Transwell Assay: Seed 5×10^4 cells in serum-free medium into Transwell chambers (Matrigel-coated for invasion, uncoated for migration). Count migrated/invaded cells after 24 hours [7].
- Wound Healing Assay: Create uniform scratch in confluent cell monolayer and measure migration distance at 0 and 24 hours [7].
- CCK-8 Assay: Seed 2500 cells/well in 96-well plates, add CCK-8 reagent, and measure OD450 after 4 hours to assess viability [7].
- Clonogenic Assay: Seed 600 cells/well in 6-well plates and count colonies after 14 days [7].
These functional assays provide mechanistic insights into how specific UBRs influence cancer phenotypes including proliferation, invasion, and metastasis.
Diagram 1: Ubiquitin-Proteasome System in Normal Homeostasis and Cancer Dysregulation. The enzymatic cascade (E1-E2-E3) mediates ubiquitin transfer to substrates, targeting them for proteasomal degradation, while DUBs reverse this process. In cancer, specific components are dysregulated, disrupting protein homeostasis.
Therapeutic Targeting of Ubiquitination Machinery in Cancer
The profound dysregulation of ubiquitination pathways in cancer has motivated drug development targeting various components of the UPS, with several agents achieving clinical success.
Clinically Approved UPS-Targeting Therapies
Proteasome inhibitors represent the most successful class of UPS-targeting drugs, with bortezomib being the first therapeutic proteasome inhibitor approved by FDA for multiple myeloma and mantle cell lymphoma [1]. Bortezomib's boron atom forms high-affinity bonds with the catalytic site of the 26S proteasome, disrupting protein degradation and causing accumulation of pro-apoptotic proteins in malignant cells [1]. Carfilzomib, a second-generation proteasome inhibitor derived from the natural product epoxomicin, is approved for relapsed and refractory multiple myeloma [1]. Additional proteasome inhibitors in clinical development include marizomib, ixazomib, and CEP-18770, expanding therapeutic options for hematologic malignancies [1].
Emerging Investigational Agents
E1 enzyme inhibitors represent another strategic approach, with TAK-243 (MLN7243) being a potent, mechanism-based small-molecule inhibitor of the primary mammalian E1 enzyme, UAE [8]. TAK-243 treatment depletes cellular ubiquitin conjugates, disrupts signaling events, induces proteotoxic stress, and impairs cell cycle progression and DNA damage repair pathways, resulting in cancer cell death [8]. The NEDD8-activating enzyme (NAE) inhibitor MLN4924 (pevonedistat) exploits the neddylation pathway crucial for Cullin-RING ligase activity, inducing DNA damage and apoptosis in proliferating tumor cells [9]. MLN4924 forms a covalent adduct that mimics NEDD8-AMP, blocking NAE function and disrupting CRL-mediated protein turnover [9].
E2 enzyme inhibitors include CC0651, an allosteric inhibitor of CDC34 that causes conformational rearrangement interfering with ubiquitin discharge [9], and NSC697923 and BAY 11-7082, which inhibit UBE2N-UBE2V1 heterodimer formation and K63-specific polyubiquitin chain synthesis [9].
E3-targeting strategies have focused on disrupting specific E3-substrate interactions. Nutlin-3a and RG7112 target MDM2-p53 interaction, stabilizing p53 in cancers with wild-type TP53 [1]. Immunomodulatory drugs thalidomide, lenalidomide, and pomalidomide recruit novel substrates to the CRL4CRBN E3 ubiquitin ligase, leading to targeted degradation of disease-causing proteins [1].
Table 3: Targeted Therapies Against Ubiquitination System Components
| Target Class | Therapeutic Agent | Molecular Target | Development Status | Primary Cancer Indications |
|---|---|---|---|---|
| Proteasome | Bortezomib | 20S proteasome | FDA approved | Multiple myeloma, mantle cell lymphoma |
| Proteasome | Carfilzomib | 20S proteasome | FDA approved | Relapsed/refractory multiple myeloma |
| E1 Enzyme | TAK-243 (MLN7243) | UAE/UBA1 | Phase I/II trials | Advanced solid tumors, hematologic malignancies |
| E1-like Enzyme | MLN4924 (Pevonedistat) | NEDD8-activating enzyme | Phase II/III trials | Acute myeloid leukemia, myelodysplastic syndromes |
| E2 Enzyme | CC0651 | CDC34 | Preclinical | Preclinical models |
| E2 Enzyme | NSC697923 | UBE2N-UBE2V1 heterodimer | Preclinical | Lymphoma, multiple myeloma models |
| E3 Interaction | Nutlin-3a | MDM2-p53 interaction | Phase I trials | Cancers with wild-type p53 |
| E3 Recruitment | Lenalidomide | CRL4CRBN E3 ligase | FDA approved | Multiple myeloma, myelodysplastic syndromes |
The Scientist's Toolkit: Essential Research Reagents
Diagram 2: Ubiquitinomics Experimental Workflow for Cancer Research. The integrated approach combines ubiquitinated peptide enrichment, mass spectrometry quantification, bioinformatics analysis, and functional validation to decipher ubiquitination alterations in cancer.
Table 4: Essential Research Reagents for Ubiquitination Studies
| Reagent/Category | Specific Examples | Research Application | Key Features |
|---|---|---|---|
| Ubiquitination Enrichment Kits | PTMScan Ubiquitin Remnant Motif Kit (anti-K-ε-GG antibody) | Isolation of ubiquitinated peptides for mass spectrometry | High-affinity motif antibody; enables system-wide ubiquitinome profiling |
| Cell Culture Systems | Huh7, Hep3B hepatocellular carcinoma cells | Functional validation of UBRs in relevant cancer models | Well-characterized HCC models; transfertable with shRNA/siRNA |
| Gene Knockdown Tools | shRNA plasmids targeting UBE2C | Loss-of-function studies to determine UBR roles | Enables stable knockdown; puromycin selection |
| Phenotypic Assay Kits | Transwell chambers, CCK-8 assay, wound healing tools | Functional analysis of proliferation, invasion, migration | Quantifiable metrics for cancer hallmarks |
| Proteasome Inhibitors | Bortezomib, Carfilzomib | Control compounds for UPS disruption studies | FDA-approved; establish expected phenotypic outcomes |
| Bioinformatics Databases | TCGA (The Cancer Genome Atlas), STRING, UbiBrowser | Integrated analysis of ubiquitination networks | Clinical correlation data; protein-protein interaction networks |
| Pathway Analysis Tools | KEGG, Gene Ontology, GSVA | Interpretation of ubiquitination alterations in biological context | Identifies enriched pathways in cancer ubiquitinomes |
The comprehensive comparison of ubiquitination machinery in cancerous versus normal tissues reveals profound and systematic reprogramming of this critical regulatory system in malignancy. From widespread genetic and expression alterations of UBRs in pan-cancer analyses to tissue-specific ubiquitinome modifications in colorectal and hepatocellular carcinomas, the evidence consistently demonstrates that ubiquitination dysregulation is a fundamental hallmark of cancer. The integrated experimental approaches combining ubiquitinomics, functional validation, and therapeutic targeting provide powerful frameworks for deciphering this complexity. As drug development technologies advance, targeting specific components of the ubiquitination system—from E1 enzymes and E2 conjugating enzymes to the highly specific E3 ligases and DUBs—holds exceptional promise for precision cancer therapeutics. The continued refinement of research tools and methodologies will further accelerate our understanding of ubiquitination patterns in cancer, ultimately enabling more effective patient stratification and targeted interventions in the era of predictive, preventive, and personalized medicine.
Ubiquitination, a critical post-translational modification, regulates protein stability, localization, and function through a sequential enzymatic cascade involving E1 activating enzymes, E2 conjugating enzymes, and E3 ligases [10]. The ubiquitin-proteasome system (UPS) mediates approximately 80-90% of cellular protein degradation, playing fundamental roles in maintaining cellular homeostasis [11] [12]. Dysregulation of this system contributes significantly to tumorigenesis by affecting crucial cancer-associated processes including cell cycle progression, DNA repair, immune evasion, and metabolic reprogramming [7] [10].
Pan-cancer analyses have emerged as powerful approaches for identifying consistent molecular patterns across diverse cancer types. Systematic investigation of ubiquitination enzymes across malignancies provides valuable insights into shared oncogenic mechanisms and potential therapeutic vulnerabilities. This review synthesizes current evidence regarding dysregulated expression patterns of key ubiquitination enzymes across multiple cancer types, their prognostic significance, and implications for therapeutic development.
Expression Landscape of Ubiquitination Enzymes in Pan-Cancer
Systematic Pan-Cancer Overexpression Patterns
Comprehensive bioinformatics analyses integrating data from TCGA, GTEx, and other large-scale databases reveal consistent overexpression of specific ubiquitination enzymes across diverse cancer types. These patterns suggest fundamental roles in oncogenic processes that transcend tissue-specific differences.
Table 1: Pan-Cancer Expression Patterns of Key Ubiquitination Enzymes
| Enzyme | Enzyme Class | Overexpressed Cancers | Associated Clinical Outcomes |
|---|---|---|---|
| UBE2T | E2 conjugating enzyme | Multiple myeloma, breast cancer, renal cell carcinoma, ovarian cancer, cervical cancer, retinoblastoma [11] | Reduced OS and PFS [11] |
| UBA1 | E1 activating enzyme | Most cancer types [13] | Poor prognosis [13] |
| UBA6 | E1 activating enzyme | Most cancer types [13] | Poor prognosis [13] |
| TRIM56 | E3 ligase | CHOL, COAD, ESCA, GBM, HNSC, KIRC, KIRP, LIHC, PCPG, READ, SARC, STAD, THYM [14] | Varies by cancer type [14] |
UBE2T constitutes a critical component of the ubiquitin-proteasome system and demonstrates elevated expression across multiple tumor types, where its upregulation associates with poor clinical outcomes and prognosis [11]. Gene variation analysis identifies "amplification" as the predominant alteration in the UBE2T gene, followed by mutations, with data from the GSCALite database demonstrating high frequencies of UBE2T copy number variations across pan-cancer cohorts [11].
The UBA family enzymes UBA1 and UBA6, both classic ubiquitin-activating E1 enzymes, show heightened expression in most cancer types, which may associate with poor patient prognosis [13]. UBA1 participates in the ubiquitination of most proteins in the body, and its abnormal expression relates to malignant phenotypes in lung cancer, liver cancer, and colorectal cancer [13].
E3 ubiquitin ligase TRIM56 demonstrates differential expression across various tumors, with high expression in tumor tissues of cholangiocarcinoma, colon adenocarcinoma, esophageal cancer, glioblastoma multiforme, head and neck squamous cell carcinoma, clear cell renal carcinoma, and multiple other cancer types [14].
Tissue-Specific Expression Variations
While pan-cancer patterns reveal consistent dysregulation, significant tissue-specific variations exist in ubiquitination enzyme expression, reflecting the complex interplay between ubiquitination pathways and tissue context.
The UBE2T gene shows elevated mRNA expression in multiple myeloma, breast cancer, renal cell carcinoma, ovarian cancer, cervical cancer, and retinoblastoma compared with adjacent normal tissues [11]. Experimental validation in pancreatic cancer cell lines (PANC1, ASPC, BXPC3, MIA2, SW1990, and CAPAN1) confirms elevated UBE2T expression at both mRNA and protein levels compared to normal pancreatic epithelial cells (HPDE) [11].
TRIM56 exhibits particularly complex expression patterns across cancers, with high expression in normal tissues of BLCA (bladder urothelial carcinoma), BRCA (breast invasive carcinoma), CESC (cervical squamous carcinoma and adenocarcinoma), LUAD (lung adenocarcinoma), LUSC (lung squamous cell carcinoma), PRAD (prostate carcinoma), THCA (thyroid carcinoma), and UCEC (endometrial carcinoma), while showing high expression in tumor tissues of numerous other cancer types [14]. This complex expression profile suggests TRIM56 may play context-dependent roles in cancer development.
Prognostic Implications and Clinical Correlations
Survival Analysis Across Cancer Types
Ubiquitination enzyme expression patterns show significant correlations with patient survival outcomes, although these relationships vary by specific enzyme and cancer type, highlighting the context-dependent functions of the UPS in cancer biology.
Table 2: Prognostic Significance of Ubiquitination Enzymes in Various Cancers
| Enzyme | Cancer Type | Survival Correlation | Additional Clinical Associations |
|---|---|---|---|
| UBE2T | Pan-cancer | Poor OS and PFS [11] | Associated with advanced stage [11] |
| TRIM56 | BLCA, KIRC, MESO, SKCM | Longer OS [14] | Correlated with B cells, macrophages, CD4+, CD8+ T cell infiltration [14] |
| TRIM56 | COAD, GBM, LGG | Shorter OS [14] | Associated with age in LGG, BLCA, COAD [14] |
| UBA1/UBA6 | Pan-cancer | Poor prognosis [13] | Correlation with clinical stages [13] |
UBE2T upregulation associates with reduced overall survival (OS) and progression-free survival (PFS), indicating its potential role in tumor progression [11]. Functional analyses link elevated UBE2T expression to changes in key cellular processes including proliferation, invasion, and epithelial-mesenchymal transition [11].
TRIM56 demonstrates cancer-type-specific prognostic associations, with high expression correlating with shorter overall survival in COAD, GBM, and LGG, but longer OS in BLCA, KIRC, MESO, and SKCM [14]. In BLCA and KIRC, high TRIM56 expression closely associates with B cells, macrophages, and CD4+ and CD8+ T cell infiltration, potentially contributing to a favorable prognosis in these specific contexts [14].
UBA1 and UBA6 expression shows correlation with clinical stages in specific tumors, with their differential expression significantly correlated with patient survival rates, tumor grade, and cancer stage [13].
Immune Infiltration and Microenvironment Associations
The tumor immune microenvironment represents a critical interface between cancer cells and host defense mechanisms, with ubiquitination enzymes playing multifaceted roles in modulating immune responses across cancer types.
UBE2T expression demonstrates significant associations with tumor immune markers, checkpoint genes, and immune cell infiltration [11]. Similarly, UBA1 and UBA6 show close relationships with immune score, immune subtypes, and tumor infiltrating immune cells across various cancer types [13].
TRIM56 expression associates with various immune cell populations, and enrichment analysis suggests it may contribute to tumor development through immune-related pathways [14]. These findings position TRIM56 as a potential influencer of tumor immunity and prognostic outcomes, holding potential as both an immunotherapy target and prognostic marker [14].
Methodological Approaches in Ubiquitination Research
Experimental Techniques for Ubiquitination Analysis
Advanced mass spectrometry-based approaches have revolutionized the characterization of ubiquitination patterns in cancer tissues, providing comprehensive insights into the ubiquitinome.
Ubiquitinome Analysis Workflow
Liquid chromatography-mass spectrometry (LC-MS/MS) with anti-K-ε-GG-based ubiquitination peptide enrichment enables comprehensive ubiquitinome characterization [15]. This approach has identified 1,690 quantifiable sites and 870 quantifiable proteins in colorectal cancer samples, revealing that highly-ubiquitinated proteins specifically involve in biological processes such as G-protein coupling, glycoprotein coupling, and antigen presentation [16].
In lung adenocarcinoma research, ion mobility separation coupled with liquid chromatography-mass spectrometry provides accurate and reliable ubiquitylome and proteomic analysis of clinical samples [15]. This methodology enables identification of characteristic protein ubiquitination motifs distinct from other cancer types like lung squamous cell carcinoma [15].
Bioinformatics and Multi-Omics Integration
Computational approaches complement experimental methods in ubiquitination research, enabling large-scale pan-cancer analyses and integration with clinical outcomes.
The 'Gene DE' module of TIMER 2.0 database compares UBE2T expression levels between tumor tissues and adjacent normal tissues across various cancer types [11]. Similarly, UALCAN database provides protein expression data for ubiquitination enzymes in pan-cancer contexts, supporting TCGA-based cancer data analysis [11] [13].
Multi-omics analyses integrate genomic, transcriptomic, and proteomic data to comprehensively characterize the roles of ubiquitination enzymes. Gene Set Cancer Analysis (GSCA) database provides an integrated platform for analyzing genomic mutational landscape, including CNV/SNV modules that visualize the proportion of heterozygous/homozygous and amplification/deletion patterns [13].
Therapeutic Implications and Future Directions
Targeting Ubiquitination Enzymes in Cancer Therapy
The consistent dysregulation of ubiquitination enzymes across cancers positions them as attractive therapeutic targets, with several emerging strategies showing promise.
UBE2T expression demonstrates positive correlation with trametinib and selumetinib sensitivity, and negative correlation with CD-437 and mitomycin, suggesting potential therapeutic applications [11]. These associations indicate that UBE2T expression status may inform treatment selection and predict response to specific therapeutic agents.
Emerging anticancer strategies leveraging the UPS include proteolysis targeting chimeras (PROTACs) and molecular glues [12]. PROTAC technology represents a valuable platform for driving targeted protein degradation, with ARV-110 and ARV-471 progressing to phase II clinical trials [12]. Molecular glues such as CC-90009 facilitate ubiquitination-mediated degradation of specific targets by recruiting E3 ligase complexes and are in phase II trials for leukemia therapy [12].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Ubiquitination Studies
| Reagent/Resource | Application | Function |
|---|---|---|
| Anti-K-ε-GG antibody [15] | Ubiquitinated peptide enrichment | Specific recognition and isolation of ubiquitinated peptides for mass spectrometry |
| K-ε-GG binding resin (PTM-1104) [15] | Ubiquitinated peptide enrichment | Immunoaffinity purification of ubiquitinated peptides |
| Radio Immunoprecipitation Assay buffer [11] | Protein extraction | Cell lysis and protein extraction for western blotting |
| Trichloroacetic acid [15] | Protein precipitation | Concentration and purification of protein samples |
| Tetraethyl ammonium bromide [11] | Protein digestion | Buffer for tryptic digestion of proteins |
| TIMER 2.0 [11] | Bioinformatics analysis | Comparing gene expression levels between tumor and normal tissues |
| UALCAN [11] [13] | Cancer data analysis | TCGA-based cancer data analysis and protein expression retrieval |
| GSCA [13] | Genomic analysis | Analyzing genomic mutational landscape including CNV/SNV patterns |
Therapeutic Targeting Strategies
Comprehensive pan-cancer analyses reveal consistent dysregulation of ubiquitination enzymes across diverse cancer types, with significant implications for tumor progression, prognosis, and therapeutic responses. The ubiquitin-proteasome system represents a complex regulatory network with both universal and context-specific functions in cancer biology. Systematic characterization of ubiquitination patterns provides valuable insights into cancer mechanisms and identifies potential biomarkers for diagnosis, prognosis, and treatment selection. Emerging therapeutic strategies that target ubiquitination enzymes or exploit the UPS for targeted protein degradation hold significant promise for advancing cancer treatment. Future research should focus on elucidating the specific substrates targeted by dysregulated ubiquitination enzymes in different cancer contexts and developing more selective approaches to modulate their activity for therapeutic benefit.
Ubiquitination, one of the most abundant post-translational modifications in eukaryotic cells, serves as a critical regulatory mechanism controlling virtually all cellular processes. This complex process involves the coordinated action of E1 (activating), E2 (conjugating), and E3 (ligating) enzymes that attach ubiquitin molecules to target proteins, determining their stability, activity, localization, and interactions. In cancer research, understanding the dysregulation of ubiquitination patterns between cancerous and normal tissues has emerged as a pivotal area of investigation, revealing profound implications for tumor initiation, progression, and therapeutic resistance. This guide provides a comprehensive comparison of ubiquitination alterations in cancerous versus normal tissues, focusing on three fundamental biological processes: cell cycle control, DNA repair, and apoptosis, while equipping researchers with essential methodologies and tools for continued investigation in this field.
Ubiquitination Alterations in Cancer vs. Normal Tissues
Table 1: Comparative Ubiquitination Patterns in Cancer vs. Normal Tissues
| Biological Process | Key Ubiquitination Components | Normal Tissue Function | Cancer Tissue Alterations | Experimental Evidence |
|---|---|---|---|---|
| Cell Cycle Control | APC/C, SCF complex, SKP2, FBW7, CDC20 | Regulates timely degradation of cyclins and CDK inhibitors for proper cell cycle progression [17] [18] | Upregulated SKP2 degrades p27, enabling uncontrolled proliferation; FBW7 mutations stabilize oncoproteins like MYC [17] | SKP2-deficient mice show p27 accumulation and polyploidy; FBW7 mutations common in human cancers [17] |
| DNA Repair | BRCA1-BARD1, RNF4, USP7, Pellino1 | Maintains genomic integrity via controlled repair protein turnover [19] | Dysregulated E3 ligases impair repair mechanisms, increasing mutation rates [19] [20] | RNF4 and USP7 coordinate SLX4 stability in DNA damage response [20] |
| Apoptosis | USP1, USP22, Bcl-2 ubiquitination | Regulates programmed cell death through controlled degradation of pro- and anti-apoptotic factors [21] | Dysregulated USPs (USP1, USP22) lead to uncontrolled cell cycle progression and evasion of cell death [21] | USP22 dysregulation promotes abnormal DNA repair and tumorigenesis [21] |
| Metabolic Reprogramming | NEDD4, APC/CCDH1, SKP2 | Maintains balanced metabolic pathways appropriate for normal cellular demands [19] | E3 ligases reprogram metabolism toward glycolysis; SKP2 regulates Akt ubiquitination and glycolysis [19] | SKP2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, and Herceptin sensitivity [19] |
| Immune Evasion | PD-1/PD-L1 ubiquitination | Controls immune checkpoint protein turnover for proper immune homeostasis [21] | Altered ubiquitination of PD-1/PD-L1 enables immune evasion; UBE2C inhibits anti-tumor immunity [7] [21] | Ubiquitination regulates PD-1/PD-L1 degradation and antigen presentation [21] |
The ubiquitin-proteasome system (UPS) demonstrates widespread dysregulation across cancer types, with ubiquitination-related genes (UBRs) showing distinct expression patterns in cancerous versus normal tissues. Comprehensive pan-cancer analyses reveal that more than 90% of UBRs can affect cancer patient survival, with specific UBRs serving as valuable markers for prognostic classification [5]. Notably, UBA1 and UBA6, the two primary E1 activating enzymes, show significantly elevated expression in most cancer types compared to normal tissues, which correlates strongly with poor patient prognosis and advanced disease stages [22]. These alterations create a permissive environment for tumor development by disrupting the precise control of critical cellular processes.
Key Biological Processes Affected by Ubiquitination Dysregulation
Cell Cycle Control
The orderly progression through the cell cycle is predominantly regulated by the ubiquitin-proteasome system, which controls the periodic degradation of key regulatory proteins. In normal tissues, the anaphase-promoting complex/cyclosome (APC/C) and SCF (Skp1-Cul1-F-box protein) complex function as the primary E3 ubiquitin ligases responsible for the timed destruction of cyclins, CDK inhibitors, and other cell cycle regulators [17] [18]. These complexes ensure unidirectional cell cycle progression through specific ubiquitination signals: K11-linked ubiquitin chains primarily target APC/C substrates for degradation during mitosis, while K48-linked polyubiquitination by SCF complexes facilitates proteasomal degradation of various cell cycle regulators [18].
In cancer tissues, this precise regulatory system becomes fundamentally disrupted. SKP2, an F-box protein component of the SCF complex, is frequently overexpressed in human cancers, where it drives excessive degradation of the CDK inhibitor p27, enabling uncontrolled cell cycle progression [17]. Similarly, FBW7, another F-box protein that normally targets positive cell cycle regulators like MYC, JUN, and cyclin E for degradation, is often mutated in human cancers, leading to stabilization of oncoproteins [17]. The critical role of these regulators is evidenced by studies showing that Skp2-deficient mice exhibit accumulation of p27 and cyclin E, resulting in polyploidy and centrosome overduplication [17].
Diagram: Ubiquitination-Mediated Cell Cycle Control in Normal vs. Cancerous Tissues
DNA Repair
Ubiquitination plays an essential role in maintaining genomic integrity through regulated DNA damage response (DDR) pathways. In normal cellular conditions, ubiquitination controls the stability, activity, and recruitment of key DNA repair proteins, creating a coordinated response to DNA lesions. The BRCA1-BARD1 complex, an E3 ubiquitin ligase, monoubiquitinates histone proteins in response to DNA damage, facilitating repair processes [23]. Additionally, E3 ligases such as NEDD4, APC/CCDH1, FBXW7, and Pellino1 function as critical bridges connecting cellular metabolism with DNA damage response, ensuring appropriate repair resource allocation [19].
Cancer tissues exhibit substantial rewiring of these ubiquitination-mediated DNA repair mechanisms. The RNF4 and USP7 deubiquitinating enzymes coordinate spatial regulation of SLX4 stability within PML nuclear bodies, with dysregulation contributing to defective DNA repair in cancer [20]. Recent research has also revealed how ubiquitination controls post-replicative gap formation and repair in response to DNA replication stress, with specific E3 ligases and deubiquitinating enzymes regulating the choice between error-free and error-prone repair pathways [20]. These alterations create a permissive environment for genomic instability while simultaneously presenting therapeutic opportunities, as evidenced by the finding that ubiquitination of ABC transporters and DNA repair enzymes can facilitate chemotherapy resistance [21].
Apoptosis
The controlled process of programmed cell death is extensively regulated by ubiquitination, which determines the stability and activity of key pro-apoptotic and anti-apoptotic factors. In normal tissues, ubiquitination maintains appropriate apoptotic thresholds through K48 and K63 ubiquitin linkages that regulate proteins including Bcl-2, ACSL4, and p62, thereby controlling cellular survival decisions [21]. Specific deubiquitinating enzymes, particularly ubiquitin-specific proteases (USPs) such as USP1 and USP22, provide additional regulatory layers that ensure proper control of cell death pathways in response to cellular damage and stress signals.
Cancer tissues develop numerous mechanisms to evade apoptosis through ubiquitination pathway alterations. Dysregulation of USPs leads to uncontrolled cell cycle progression and abnormal DNA repair capacity, effectively disabling critical apoptotic triggers [21]. Additionally, cancer cells exploit ubiquitination mechanisms to inhibit ferroptosis and autophagy-related cell death pathways, further enhancing their survival potential. Therapeutic targeting of these pathways shows significant promise, as demonstrated by the ability of proteasome inhibitors, E3 ligase inhibitors, and PROTACs to restore apoptotic sensitivity in cancer cells [21].
Experimental Approaches for Ubiquitination Research
Key Methodologies for Ubiquitination Studies
Table 2: Experimental Protocols for Ubiquitination Analysis
| Method | Key Procedures | Applications in Cancer Research | Key Reagents |
|---|---|---|---|
| shRNA Knockdown | Plasmid construction targeting specific UBRs; viral transduction; puromycin selection; qRT-PCR validation [7] | Functional validation of ubiquitination enzymes; study of proliferation, invasion, metastasis | shRNA plasmids, polybrene, puromycin, qRT-PCR reagents |
| Transwell Assay | Seed 5×10⁴ cells in Matrigel-coated (invasion) or uncoated (migration) chambers; 24h culture; fix with 4% PFA; crystal violet staining; microscopic quantification [7] | Assess cancer cell migration and invasion capabilities post-UBR manipulation | Transwell chambers, Matrigel, paraformaldehyde, crystal violet |
| CCK-8 Assay | Seed 2500 cells/well in 96-well plates; add 10μL CCK8 reagent; incubate 4h; measure OD450 [7] | Quantify cell viability and proliferation following UBR modulation | Cell Counting Kit-8, 96-well plates, microplate reader |
| Wound Healing Assay | Create linear scratch with pipette tip at 80% confluence; wash with PBS; serum-free medium; document migration at 0h and 24h [7] | Evaluate two-dimensional cell migration capacity | 6-well plates, sterile pipette tips, serum-free medium |
| Single-cell RNA Sequencing | Quality control excluding cells with <200 or >7000 genes; normalize data; PCA/tSNE/UMAP dimensionality reduction; cluster with KNN [24] | Identify ubiquitination heterogeneity across tumor cell subtypes | 10X Genomics platform, Seurat package, clustering algorithms |
| Spatial Transcriptomics | Data normalization with LogNormalize; PCA dimensionality reduction; RCTD deconvolution for cell type annotation [24] | Map ubiquitination patterns within tumor architecture | Spacexr package, spatial transcriptomics platforms |
Multi-Omics Integration Approaches
Advanced multi-omics strategies have significantly enhanced our understanding of ubiquitination patterns in cancer. Integrated bioinformatics analysis combining multiple HCC datasets enables comprehensive assessment of ubiquitination status across various cell types in the tumor microenvironment, including plasma cells, fibroblasts, endothelial cells, and epithelial-mesenchymal transition (EMT) cells [7]. These approaches utilize ubiquitination scores to categorize cell types and integrate survival data with spatial transcriptomics to evaluate how different ubiquitination levels influence cancer progression.
Single-cell and spatial transcriptomic methodologies provide unprecedented resolution for mapping ubiquitination patterns within complex tumor architectures. As demonstrated in pancreatic cancer research, these techniques can identify distinct cell populations with high ubiquitination activity, such as endothelial cells exhibiting elevated ubiquitination scores (High_ubiquitin-Endo) that enriched interactions with fibroblasts and macrophages through WNT, NOTCH, and integrin pathways [24]. Spatial validation further confirms the localization patterns of these ubiquitination-active cell populations within tumor tissues.
Research Reagent Solutions
Table 3: Essential Research Reagents for Ubiquitination Studies
| Reagent Category | Specific Examples | Research Applications | Key Functions |
|---|---|---|---|
| E1 Enzyme Inhibitors | UBA1 inhibitors, UBA6 inhibitors [22] | Target validation, therapeutic efficacy studies | Block ubiquitin activation, global ubiquitination inhibition |
| E2 Enzyme Tools | UBE2C shRNA, UBE2C overexpression constructs [7] | Functional studies of specific E2 enzymes | Modulate specific ubiquitin-conjugating enzyme activity |
| E3 Ligase Modulators | TRIM9 constructs, SCF complex components [24] | Pathway analysis, substrate identification | Alter substrate-specific ubiquitination events |
| Deubiquitinase Reagents | USP inhibitors, DUB activity assays [21] | Study ubiquitination reversal, protein stabilization | Investigate deubiquitination processes and consequences |
| Ubiquitin Linkage Tools | Linkage-specific antibodies, ubiquitin mutants [23] | Ubiquitin chain topology studies | Detect specific ubiquitin linkage types (K48, K63, K11, etc.) |
| Proteasome Inhibitors | Bortezomib, MG132 [21] | Study protein degradation, therapeutic applications | Block proteasomal degradation, stabilize ubiquitinated proteins |
| Activity Assays | CCK-8, clonogenic formation, wound healing [7] | Functional consequence assessment | Measure proliferation, viability, migration phenotypes |
Signaling Pathways and Therapeutic Implications
The ubiquitin-proteasome system intersects with numerous oncogenic signaling pathways, creating complex regulatory networks that differ substantially between normal and cancerous tissues. Integrated analyses of ubiquitination regulators across pan-cancer datasets reveal that the expression of 79 UBRs closely correlates with the activity of 32 cancer hallmark-related pathways [5]. These correlations highlight the central positioning of ubiquitination networks in controlling fundamental cancer behaviors, with specific hub genes demonstrating excellent prognostic classification power for particular cancer types.
Diagram: Therapeutic Targeting of Ubiquitination Pathways in Cancer
Emerging therapeutic strategies increasingly focus on exploiting the differential ubiquitination patterns between normal and cancerous tissues. Proteasome inhibitors represent the first clinically successful category of UPS-targeting drugs, with additional approaches including E3 ligase inhibitors, PROTACs (Proteolysis Targeting Chimeras), and DUB inhibitors showing significant promise in preclinical and clinical development [21]. These approaches capitalize on the specific dependencies that cancer cells develop on altered ubiquitination pathways, creating therapeutic windows that can be exploited while sparing normal tissues.
The connection between ubiquitination and cancer immune evasion presents particularly compelling therapeutic opportunities. Ubiquitination regulates critical immune checkpoint proteins including PD-1 and PD-L1, with specific E3 ligases controlling their degradation and consequently shaping the anti-tumor immune response [21]. Additionally, ubiquitination mechanisms influence antigen presentation through major histocompatibility complex (MHC) class I molecules, further modulating immune recognition of cancer cells [23]. These insights have stimulated development of combination strategies that simultaneously target ubiquitination pathways and immune checkpoints, potentially enabling enhanced and durable anti-tumor immunity.
The comprehensive comparison of ubiquitination patterns between cancerous and normal tissues reveals profound alterations affecting core biological processes including cell cycle control, DNA repair, and apoptosis. These differences are not merely incidental but represent fundamental mechanisms driving tumor development and progression. The continuing development of sophisticated research tools—from single-cell omics technologies to targeted ubiquitination modulators—provides an expanding toolkit for deciphering this complex regulatory landscape. As our understanding of the ubiquitin code in cancer deepens, so too does the potential for innovative therapeutic strategies that specifically target the ubiquitination dependencies of cancer cells while sparing normal tissues, ultimately promising more effective and selective cancer treatments.
The ubiquitin-proteasome system (UPS) serves as a critical regulatory mechanism for protein homeostasis, dynamically controlling the stability, localization, and activity of numerous cellular proteins. This intricate process involves a sequential enzymatic cascade where E1 activating enzymes, E2 conjugating enzymes, and E3 ligases work in concert to attach ubiquitin chains to target proteins [25]. The specificity of this system is largely governed by E3 ubiquitin ligases, with the human genome encoding approximately 800 such enzymes [26]. Conversely, deubiquitinating enzymes (DUBs) counteract this process by removing ubiquitin chains, providing an additional layer of regulation [25]. In cancer biology, the precise balance of ubiquitination is frequently disrupted, leading to two fundamental pathogenic mechanisms: the excessive degradation of tumor suppressor proteins and the aberrant stabilization of oncoproteins. This imbalance creates a cellular environment conducive to uncontrolled proliferation, evasion of apoptosis, and ultimately, tumorigenesis. Understanding these opposing mechanisms provides critical insights into cancer pathogenesis and reveals potential therapeutic vulnerabilities for targeted intervention.
Molecular Mechanisms of Tumor Suppressor Degradation
The ubiquitin-mediated degradation of tumor suppressors represents a common pathway hijacked in numerous cancers. This process often involves the overexpression or hyperactivation of specific E3 ubiquitin ligases that target tumor suppressor proteins for proteasomal destruction.
Key Pathways and E3 Ligases
MDM2 and p53 Degradation: The MDM2 E3 ligase serves as a primary negative regulator of the p53 tumor suppressor. By ubiquitinating p53, MDM2 targets it for proteasomal degradation, thus controlling its cellular levels. In many cancers, MDM2 is amplified or overexpressed, leading to excessive p53 degradation and diminished tumor suppressor activity [27]. This pathway is particularly significant given p53's crucial role in cell cycle arrest, apoptosis, and DNA repair.
HPV E6-AP and p53 Inactivation: In cancers associated with high-risk human papillomavirus (HPV), the viral E6 protein recruits the cellular E6-AP E3 ligase to ubiquitinate p53, resulting in its degradation [27]. This mechanism effectively neutralizes p53's protective function and contributes to viral oncogenesis.
SCF Complexes in Cell Cycle Regulation: SCF (Skp1-Cul1-F-box protein) complexes constitute a major family of E3 ubiquitin ligases that regulate cell cycle progression. The F-box protein Skp2, in particular, targets the cell cycle inhibitors p27 and p21 for degradation, promoting uncontrolled proliferation in cancers such as lung, breast, and colon carcinomas [27].
Table 1: E3 Ubiquitin Ligases Implicated in Tumor Suppressor Degradation
| E3 Ligase | Tumor Suppressor Substrate | Cancer Associations | Biological Outcome |
|---|---|---|---|
| MDM2 | p53 | Sarcomas, Breast Cancer | Uncontrolled proliferation, genomic instability |
| E6-AP (with HPV E6) | p53 | Cervical, Head & Neck Cancers | Evasion of apoptosis, impaired DNA repair |
| SCFSkp2 | p27, p21 | Lung, Glioma, Gastric, Prostate | Enhanced cell cycle progression, proliferation |
| VHL | HIF-1α | Renal Cell Carcinoma | Tumor angiogenesis, metabolic adaptation |
Ubiquitin-Independent Degradation Pathways
Not all proteasomal degradation requires ubiquitination. An alternative pathway involves direct degradation of specific tumor suppressors by 20S proteasomes. The tumor suppressors p53 and p73 can be degraded through this ubiquitin-independent mechanism, which is regulated by NQO1 (NAD(P)H quinone oxidoreductase 1) in an NADH-dependent manner [28]. NQO1 physically interacts with both p53 and p73, protecting them from 20S proteasomal degradation, and interestingly, most cellular NQO1 is found associated with 20S proteasomes, suggesting it functions as a gatekeeper for this degradation pathway [28].
Mechanisms of Oncoprotein Stabilization in Cancer
While excessive degradation removes protective tumor suppressors, the inappropriate stabilization of oncoproteins provides equally powerful driving forces for tumor development. Multiple distinct molecular mechanisms can lead to oncoprotein stabilization in cancer cells.
Genetic Alterations in Degrons and E3 Ligases
Oncoproteins frequently acquire stabilizing mutations through tumor evolution:
Degron Mutations: Degrons are short amino acid sequences required for E3 ubiquitin ligase recognition. Mutations within these motifs allow oncoproteins to evade recognition and subsequent degradation. In colon cancer and melanoma, mutations substituting Ser33 and Ser37 with Ala within the β-catenin degron prevent its phosphorylation-dependent recognition by the E3 ligase β-TRCP, resulting in a stable and transcriptionally active protein [26]. Similarly, in Burkitt's lymphoma, the Thr58Ala mutation in c-Myc's degron abolishes binding to the Fbw7 E3 ligase, leading to c-Myc stabilization [26] [29].
E3 Ligase Inactivation: Genetic or epigenetic inactivation of E3 ligases that normally target oncoproteins for degradation represents another common mechanism. The Fbw7 ligase, which targets multiple oncoproteins including c-Myc, c-Jun, Notch, and cyclin E, is frequently mutated in cholangiocarcinomas, T-cell acute lymphocytic leukemia, and various solid tumors [26] [27]. Similarly, inactivation of the Cbl ubiquitin ligase stabilizes growth factor receptors like EGFR, leading to sustained oncogenic signaling [26].
Table 2: Mechanisms of Oncoprotein Stabilization in Human Cancers
| Mechanism | Representative Example | Affected Oncoprotein | Cancer Type |
|---|---|---|---|
| Degron Mutation | Ser33/37Ala in β-catenin degron | β-catenin | Colon Cancer, Melanoma |
| Degron Mutation | Thr58Ala in c-Myc degron | c-Myc | Burkitt's Lymphoma |
| E3 Ligase Inactivation | Fbw7 mutations | c-Myc, Cyclin E, Notch | T-ALL, Various Solid Tumors |
| E3 Ligase Inactivation | Cbl mutations | EGFR | Various Cancers |
| Ubiquitin-Independent Stabilization | NQO1 regulation | p53, p73 | Multiple Cancers |
Active Ubiquitin-Dependent Stabilization Pathways
Beyond passive evasion of degradation, cancer cells also activate specific pathways that actively stabilize oncoproteins. A central enzyme in one such pathway is the RNF4 ubiquitin ligase, which mediates a ubiquitin-dependent protein stabilization pathway that translates transient mitogenic signals into long-lasting protein stabilization, enhancing the activity of key oncoproteins [26]. This active stabilization pathway operates independently of the canonical degradation machinery and represents a promising therapeutic target.
Comparative Analysis of Ubiquitination Patterns: Cancerous vs. Normal Tissues
Advanced proteomic and ubiquitinomic technologies have enabled comprehensive characterization of ubiquitination patterns across cancer types, revealing fundamental differences between cancerous and normal tissues.
Global Ubiquitinome Alterations in Colorectal Cancer
A comprehensive ubiquitinomic analysis of colorectal cancer (CRC) tissues versus normal adjacent mucosa revealed significant alterations in the ubiquitination landscape [30]. This study identified 1,690 quantifiable ubiquitination sites and 870 quantifiable proteins, with 1,172 proteins showing up-regulated ubiquitination and 1,700 proteins demonstrating down-regulated ubiquitination in CRC cells [30]. Highly ubiquitinated proteins (those with ≥10 modification sites) were specifically involved in critical biological processes including G-protein coupling, glycoprotein coupling, and antigen presentation [30]. This widespread reprogramming of the ubiquitinome highlights the profound impact of ubiquitination dysregulation in cancer pathogenesis.
Hepatocellular Carcinoma and UBE2C Overexpression
In hepatocellular carcinoma (HCC), ubiquitination-related genes are significantly upregulated, with high expression levels correlating with poor patient prognosis [31]. Pathway analysis demonstrates that these genes are enriched in key processes including cell cycle regulation, DNA repair, metabolic reprogramming, and p53 signaling [31]. Notably, UBE2C, a critical E2 ubiquitin-conjugating enzyme, emerges as a key player in HCC progression. Experimental data confirms that UBE2C overexpression promotes HCC cell proliferation, invasion, and metastasis, while also contributing to immune evasion by potentially inhibiting anti-tumor immune responses [31].
Table 3: Ubiquitinome Alterations in Human Cancers: Quantitative Comparisons
| Cancer Type | Analytical Method | Key Findings | Prognostic Correlation |
|---|---|---|---|
| Colorectal Cancer | Ubiquitin-proteomic LC-MS/MS (n=6 patients) | 1,172 proteins with up-regulated ubiquitination; 1,700 with down-regulated ubiquitination | FOCAD ubiquitination at Lys583/Lys587 associated with survival |
| Hepatocellular Carcinoma | Bioinformatics analysis of multiple HCC datasets | Ubiquitination-related genes significantly upregulated | High ubiquitination score correlates with poor prognosis |
| Multiple Cancers | Machine learning (deepDegron) | Mutations affecting degrons drive tumorigenesis | Predicts impact of mutations on protein stability |
Experimental Methodologies for Ubiquitination Research
Ubiquitin-Proteomic Workflow
The comprehensive characterization of ubiquitination events relies on sophisticated proteomic approaches. A standard workflow begins with tissue collection and protein extraction, followed by tryptic digestion. For ubiquitinomic analyses, this typically involves enrichment of ubiquitinated peptides using specific antibodies [30]. Liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) enables the identification and quantification of ubiquitination sites. Data processing through search engines like MaxQuant against human protein databases allows for the identification of ubiquitination sites, with false discovery rate (FDR) correction to ensure data reliability [30]. Subsequent bioinformatic analyses, including Gene Ontology (GO) annotation and KEGG pathway enrichment, help interpret the functional significance of observed ubiquitination patterns.
Functional Validation Approaches
Gene Knockdown Studies: Plasmid-based shRNA or siRNA-mediated knockdown of target genes, such as UBE2C, followed by phenotypic assays, provides functional validation. Transfection efficiency is typically monitored using qRT-PCR to confirm knockdown efficacy [31].
Cell Proliferation and Viability Assays: The Cell Counting Kit-8 (CCK-8) assay provides a reliable method for assessing cell viability following experimental manipulations. This colorimetric assay measures the metabolic activity of cells and can detect changes in proliferation rates resulting from ubiquitination pathway perturbations [31].
Migration and Invasion Assays: Transwell chambers, either uncoated for migration studies or Matrigel-coated for invasion assays, enable quantitative assessment of cell migratory and invasive capabilities. These assays typically involve staining with crystal violet and microscopic quantification of cells that have traversed the membrane [31].
Wound Healing Assay: This simple yet effective method evaluates collective cell migration by creating a "wound" in a confluent cell monolayer and monitoring closure over time, typically 24 hours, using microscopy to measure migration distance [31].
Visualization of Key Pathways and Concepts
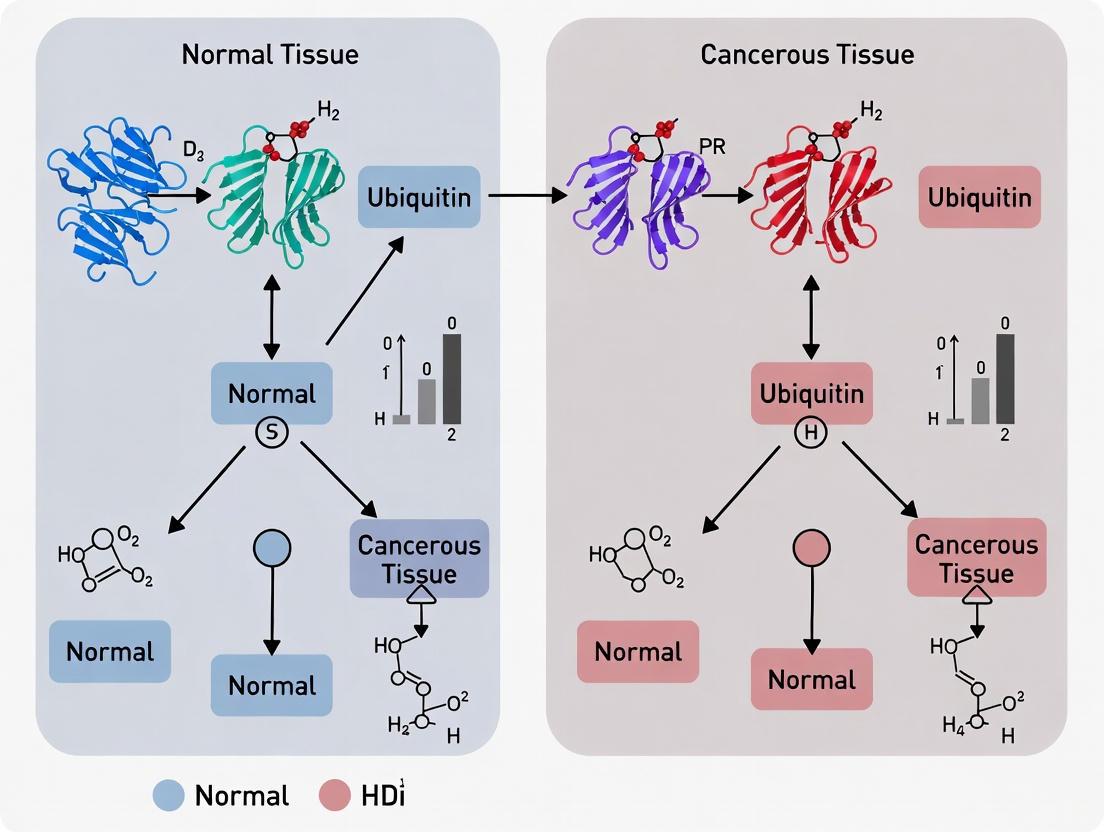
Diagram 1: The Ubiquitination Balance in Normal vs. Cancerous Tissues. This schematic illustrates the shift from balanced protein homeostasis in normal tissues to dysregulated ubiquitination in cancer, characterized by tumor suppressor degradation and oncoprotein stabilization.
Diagram 2: Experimental Workflow for Ubiquitinome Analysis. This flowchart outlines the key steps in comprehensive ubiquitination profiling, from sample preparation through LC-MS/MS analysis to functional validation of findings.
Table 4: Essential Research Tools for Ubiquitination Studies
| Reagent/Resource | Primary Function | Example Applications | Key Considerations |
|---|---|---|---|
| LC-MS/MS Systems | Identification and quantification of ubiquitination sites | Ubiquitinome characterization, site-specific quantification | High resolution and sensitivity required for modified peptide detection |
| Ubiquitin Motif Antibodies | Immunoenrichment of ubiquitinated peptides | Pull-down assays, Western blot validation | Specificity for ubiquitin remnants (e.g., diGly signature) after trypsin digestion |
| shRNA/siRNA Libraries | Targeted gene knockdown | Functional validation of E3 ligases, DUBs, and ubiquitination targets | Requires confirmation of knockdown efficiency (qRT-PCR) |
| Proteasome Inhibitors (e.g., MG132, Bortezomib) | Block protein degradation via proteasome | Stabilization of ubiquitinated proteins, half-life determination | Cytotoxicity at prolonged exposures requires careful dose optimization |
| CCK-8 Assay Kits | Cell viability and proliferation assessment | Measuring phenotypic effects of ubiquitination manipulations | More sensitive and stable alternative to MTT assays |
| Transwell Chambers | Cell migration and invasion quantification | Evaluating metastatic potential in cancer models | Matrigel coating required for invasion (vs. migration) assays |
| Public Databases (CPTAC, GEPIA, HPA) | Bioinformatics analysis of expression and ubiquitination | Correlation with clinical outcomes, tissue expression patterns | Essential for translational relevance and validation |
The intricate balance between tumor suppressor degradation and oncoprotein stabilization represents a fundamental aspect of cancer biology with profound therapeutic implications. The comprehensive characterization of ubiquitination patterns across different cancer types, coupled with mechanistic insights into the molecular pathways involved, provides a robust foundation for developing targeted interventions. Future research directions should focus on expanding the catalog of cancer-relevant ubiquitination events, particularly through pan-cancer ubiquitinome analyses, and developing more specific small-molecule inhibitors targeting oncogenic E3 ligases or stabilizing tumor suppressor proteins. The continuing evolution of proteomic technologies, combined with sophisticated bioinformatic tools and well-validated functional assays, promises to unravel the full complexity of the ubiquitination network in cancer and reveal novel opportunities for therapeutic exploitation.
Ubiquitination is a critical post-translational modification regulating protein degradation, signaling, and localization. This comparison guide analyzes tissue-specific ubiquitination signatures across major cancer types, contrasting them with normal tissue patterns to highlight heterogeneity and its implications for targeted therapy.
Comparative Analysis of Ubiquitinome Alterations
The following table summarizes key quantitative findings from recent ubiquitinome profiling studies across different cancer types, demonstrating the tissue-specific nature of these alterations.
Table 1: Tissue-Specific Ubiquitination Signature Comparison in Cancer vs. Normal Tissues
| Cancer Type | Tissue of Origin | Key Upregulated Ubiquitination Events | Key Downregulated Ubiquitination Events | Primary E3 Ligase/Deubiquitinase (DUB) Alterations | Functional Consequence |
|---|---|---|---|---|---|
| Colorectal Cancer (CRC) | Colon Epithelium | K48-linked polyUb on APC, p53 | K63-linked polyUb on Wnt/β-catenin pathway components | RNF43 loss (E3); USP7 overexpression (DUB) | Enhanced Wnt signaling, genomic instability |
| Lung Adenocarcinoma (LUAD) | Lung Alveolar Cells | K48-linked polyUb on PTEN | K63-linked polyUb on EGFR | SMURF1 overexpression (E3); USP9X downregulation (DUB) | Hyperactive PI3K/AKT and EGFR signaling |
| Triple-Negative Breast Cancer (TNBC) | Mammary Ductal Cells | K11-linked polyUb on BRCA1 | K48-linked polyUb on Cyclin E | TRIM25 overexpression (E3); BAP1 mutation (DUB) | Defective DNA repair, cell cycle progression |
| Glioblastoma (GBM) | Brain Glial Cells | K48-linked polyUb on PDCD4 | K63-linked polyUb on NF-κB pathway | MDM2 amplification (E3); OTUB1 overexpression (DUB) | Suppressed apoptosis, enhanced invasiveness |
Experimental Protocols for Ubiquitinome Profiling
The data in Table 1 is derived from studies utilizing the following core methodology.
Protocol: TUBE-based Ubiquitinated Peptide Enrichment and LC-MS/MS
- Tissue Lysis: Snap-frozen cancerous and adjacent normal tissues are homogenized in a denaturing lysis buffer (e.g., 8M Urea, 100mM NH₂HCO₃, pH 8.0) containing 10mM N-Ethylmaleimide (NEM) to inhibit deubiquitinating enzymes (DUBs) and 1x protease/phosphatase inhibitors.
- Protein Digestion: Proteins are reduced with Dithiothreitol (DTT), alkylated with Iodoacetamide (IAA), and digested with sequencing-grade Trypsin/Lys-C mix.
- Ubiquitin Remnant Enrichment: Digested peptides are incubated with Tandem Ubiquitin Binding Entities (TUBEs) conjugated to agarose beads. TUBEs specifically bind to ubiquitin chains, enriching for ubiquitinated peptides.
- Washing and Elution: Beads are washed extensively with IP buffer to remove non-specifically bound peptides. Ubiquitinated peptides are eluted with a low-pH buffer.
- LC-MS/MS Analysis: Enriched peptides are separated by nano-liquid chromatography (nano-LC) and analyzed by tandem mass spectrometry (MS/MS) on a high-resolution instrument (e.g., Q-Exactive HF-X).
- Data Analysis: MS/MS spectra are searched against a protein database using software (e.g., MaxQuant) with diGly (K-ε-GG) as a variable modification to identify ubiquitination sites. Quantitative differences are calculated using label-free or TMT-based methods.
Visualization of Core Signaling Pathways
The diagrams below illustrate key pathways where tissue-specific ubiquitination alterations drive oncogenesis.
Diagram 1: Wnt/β-catenin Ubiquitination in CRC
Diagram 2: EGFR-PTEN Axis Ubiquitination in LUAD
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Ubiquitination Signature Analysis
| Reagent / Material | Function in Research | Example Application |
|---|---|---|
| Tandem Ubiquitin Binding Entities (TUBEs) | High-affinity affinity matrices for enrichment of polyubiquitinated proteins and peptides from complex lysates. | Immunoprecipitation of ubiquitinated conjugates prior to MS/MS analysis. |
| diGly (K-ε-GG) Antibody | Antibody specific for the glycine-glycine remnant left on lysine after tryptic digestion of ubiquitinated proteins. | Enrichment of ubiquitinated peptides for mass spectrometry-based ubiquitinome profiling. |
| Proteasome Inhibitors (e.g., MG132, Bortezomib) | Block the 26S proteasome, preventing degradation of ubiquitinated proteins and leading to their accumulation. | Used in cell culture to stabilize ubiquitinated proteins for easier detection. |
| Deubiquitinase (DUB) Inhibitors (e.g., PR-619, P22077) | Broad-spectrum inhibitors of DUBs, preventing the cleavage of ubiquitin chains. | Preserve the endogenous ubiquitinome state during cell lysis and protein extraction. |
| Activity-Based Probes (UB-AMC, Ub-Rho110) | Fluorescently tagged ubiquitin substrates that report on DUB enzymatic activity in cell lysates. | Quantify functional DUB activity changes between cancer and normal tissues. |
| E3 Ligase-Specific Inhibitors (e.g., Nutlin-3 for MDM2) | Small molecules that block the interaction between a specific E3 ligase and its substrate. | Functional validation of E3 ligase roles in specific ubiquitination events. |
Advanced Techniques for Profiling Ubiquitination Landscapes in Malignancies
The comprehensive characterization of complex diseases like cancer requires interrogation across multiple molecular layers. While single-level omics analyses provide valuable insights, they often lack the resolving power to establish causal relationships between molecular alterations and phenotypic manifestations [32]. Multi-omics data integration strategies across different cellular function levels—including genomes, epigenomes, transcriptomes, proteomes, metabolomes, and ubiquitinomes—offer unparalleled opportunities to understand the underlying biology of cancer [32]. This integrated approach is particularly crucial for investigating ubiquitination patterns, a dynamic and reversible post-translational modification that regulates protein stability, cell differentiation, and immunity [33]. Dysregulation of ubiquitination processes can lead to destabilization of biological processes and contribute significantly to oncogenesis [33].
The integration of transcriptomic, proteomic, and ubiquitinomic data enables researchers to capture distinct dimensions of biological regulation: the transcriptome reflects dynamic gene activity, the proteome encodes functional effectors, and the ubiquitinome represents crucial post-translational modifications that determine protein fate and function [34]. When combined, these layers offer synergistic insights into disease mechanisms and treatment response that cannot be inferred from any single modality alone [34]. This approach has transformed our understanding of tumor heterogeneity, therapeutic resistance, and the intricate regulatory networks within the tumor microenvironment [35].
Methodological Frameworks for Multi-Omics Data Integration
Computational Integration Strategies
The integration of multi-omics data presents significant computational challenges due to the high-dimensional nature of each dataset and their inherent technical variations. Several sophisticated computational frameworks have been developed to address these challenges, each with distinct strengths and applications in cancer research.
Table 1: Computational Methods for Multi-Omics Data Integration
| Method Category | Specific Tools | Underlying Algorithm | Key Applications in Cancer Research |
|---|---|---|---|
| Matrix Factorization | Joint NMF | Non-negative Matrix Factorization | Identified novel signaling pathway perturbations and patient subgroups in ovarian cancer [32] |
| Statistical Integration | iCluster+/iClusterBayes | Gaussian Latent Variable Model | Revealed novel breast cancer subgroups with distinct clinical outcomes beyond classic expression subtypes [32] |
| Network-Based Approaches | PARADIGM | Probabilistic Graphical Models | Identified defects in homologous recombination in ovarian cancer, predicting PARP inhibitor response [32] |
| Bayesian Methods | BCC | Dirichlet Mixture Model | Integrated gene expression, miRNA, methylation, and proteomics without requiring pre-specified cluster numbers [32] |
| Dimensionality Reduction | JIVE | Principal Component Analysis Extension | Improved characterization of glioblastoma types by integrating miRNA and gene expression [32] |
The choice of integration strategy depends heavily on the research question and data characteristics. Unsupervised methods like iCluster and Joint NMF are valuable for discovering novel disease subtypes without prior knowledge of sample classifications [32]. In contrast, supervised and semi-supervised approaches incorporate known sample labels or partial labels to guide the integration process, which can be particularly useful for predictive modeling of treatment response [32]. Network-based methods such as PARADIGM leverage prior knowledge from pathway databases like KEGG to interpret multi-omics data within established biological contexts, enabling researchers to identify pathway-level perturbations that might be missed when analyzing individual molecular layers [32].
Experimental Workflows for Multi-Omics Data Generation
The generation of high-quality multi-omics data requires carefully optimized experimental workflows that preserve molecular integrity while enabling comprehensive profiling. Recent advances have enabled truly integrated data generation from the same tissue specimens, enhancing data comparability and biological relevance.
Figure 1: Integrated Spatial Multi-Omics Workflow. This framework enables transcriptomic, proteomic, and histologic data generation from the same tissue section, ensuring spatial consistency [36].
A key innovation in multi-omics experimental design is the ability to perform spatial transcriptomics and spatial proteomics on the same tissue section, as demonstrated in recent lung cancer studies [36]. This approach ensures consistency in tissue morphology and spatial context, overcoming limitations of analyzing adjacent sections. The workflow typically involves sequential processing of tissue sections through platforms such as 10x Genomics Xenium for transcriptomics, COMET hyperplex immunohistochemistry for proteomics, and standard H&E staining for histopathological annotation [36]. Computational registration using software like Weave then enables accurate alignment and annotation transfer across modalities, facilitating single-cell level comparisons of RNA and protein expression [36].
For ubiquitinomic analyses, researchers typically employ ubiquitination-related gene sets obtained from databases such as GeneCard (using a relevance score >10, resulting in ~405 genes) or curated lists of ubiquitination regulators (UBRs) categorized as writers (E1-E3 enzymes), readers (proteins with ubiquitin-binding domains), and erasers (deubiquitinases) [33] [24]. These gene sets enable the calculation of ubiquitination activity scores and facilitate the identification of hub genes through protein-protein interaction network analysis using topological algorithms like EPC, Degree, MNC, and Closeness [33].
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table 2: Essential Research Reagents and Platforms for Multi-Omics Studies
| Category | Specific Product/Platform | Key Function | Application Example |
|---|---|---|---|
| Spatial Transcriptomics | 10x Genomics Xenium | Targeted gene expression with spatial context | Human lung cancer panel with 289 genes [36] |
| Spatial Proteomics | COMET (Lunaphore) | Hyperplex immunohistochemistry for 40+ protein markers | Sequential staining of immune and tumor markers [36] |
| Cell Segmentation | CellSAM | Deep learning-based cell segmentation using nuclear/membrane markers | Integrates DAPI and pan-cytokeratin staining [36] |
| Data Integration Software | Weave (Aspect Analytics) | Registration and visualization of multi-omics data | Co-registration of ST, SP, and H&E data [36] |
| Ubiquitination Assays | Ubiquitin-activated E1 enzyme inhibitors (e.g., MLN4924) | Inhibition of ubiquitination initiation | Investigation of ubiquitination-dependent processes [33] |
| Single-Cell Analysis | Seurat R package | Processing and analysis of single-cell RNA-seq data | Identification of 12 cell types in pancreatic cancer [24] |
| Spatial Data Deconvolution | spacexr (RCTD method) | Cell type annotation in spatial transcriptomics | Mapping single-cell clusters to spatial contexts [24] |
The selection of appropriate research tools is critical for generating high-quality multi-omics data. For spatial transcriptomics, the 10x Genomics Xenium platform offers targeted gene expression profiling with subcellular resolution, typically using custom panels focused on cancer-relevant genes [36]. For spatial proteomics, the COMET system enables hyperplex immunohistochemistry through cyclical staining, imaging, and elution of off-the-shelf primary antibodies against 40 or more protein markers, providing comprehensive protein expression mapping [36]. The integration of these datasets requires sophisticated computational tools like Weave software, which employs non-rigid registration algorithms to align different spatial omics readouts, enabling accurate cross-modality analysis at single-cell resolution [36].
For ubiquitination-focused studies, researchers can leverage public databases such as Gene Set Cancer Analysis (GSCA) for genomic, pharmacogenomic, and immunogenomic analysis [37], and The Cancer Genome Atlas (TCGA) for multi-omics data across 33 cancer types [37]. Additionally, specialized platforms like CancerSEA enable single-cell functional state analysis by correlating gene expression with 14 predefined cancer-related functional states [37].
Comparative Analysis of Ubiquitination Patterns in Cancer vs. Normal Tissues
Genetic and Transcriptional Alterations in Ubiquitination Machinery
Multi-omics analyses have revealed extensive dysregulation of ubiquitination-related genes across cancer types, providing insights into their potential roles as oncogenic drivers or tumor suppressors.
Table 3: Ubiquitination Regulator Alterations in Lung Adenocarcinoma (LUAD)
| Ubiquitination Regulator | Mutation Frequency | CNV Pattern | Expression in Tumor vs Normal | Prognostic Impact |
|---|---|---|---|---|
| UBE2T | Low | High amplification | Overexpressed | Negative [33] |
| AURKA | No mutations detected | Not reported | Overexpressed | Negative [33] |
| CDC20 | Not specified | Not specified | Overexpressed | Negative [38] |
| BRCA1 | 3% (highest frequency) | Amplification leads to higher expression | Overexpressed | Negative [33] |
| BARD1 | 2% | Not specified | Overexpressed | Negative [33] |
| UBA1 | Not specified | Not specified | Overexpressed in most cancers | Negative in BRCA, COAD, KIRC, LUAD [13] |
| UBA6 | Not specified | Not specified | Variable expression | Context-dependent [13] |
In lung adenocarcinoma, comprehensive analysis of 17 hub ubiquitination regulators revealed widespread genetic alterations and expression perturbations [33]. Somatic mutations in these hub UBRs occurred in 68 of 616 LUAD patients (11.04% frequency), with BRCA1 showing the highest mutation frequency (3%), followed by BARD1 (2%) [33]. Copy number variation analysis demonstrated that specific UBRs like DTL and UBE2T exhibit higher frequencies of CNV amplification, while others like CDC34 and UBA7 show higher frequencies of CNV loss [33]. At the transcriptional level, most hub UBRs were dysregulated in cancer compared to normal tissues, with predominant overexpression patterns [33].
Similar patterns have been observed in pancreatic cancer, where single-cell RNA sequencing identified 12 distinct cell types, with endothelial cells exhibiting particularly high ubiquitination scores [24]. These High_ubiquitin-Endo cells showed enriched interactions with fibroblasts and macrophages through WNT, NOTCH, and integrin pathways, suggesting a role for ubiquitination in modulating the tumor microenvironment [24]. Through Summary-data-based Mendelian Randomization analysis, TRIM9 was prioritized as a pancreatic cancer-protective gene, showing downregulation in tumors and correlation with better survival [24].
Functional Consequences of Ubiquitination Dysregulation
The dysregulation of ubiquitination machinery in cancer has profound functional consequences, affecting key oncogenic pathways and therapeutic responses. In colorectal cancer, integrated multi-omics analysis revealed that approximately 80% of PTM pathways are dysregulated, with ubiquitination sustaining Wnt/β-catenin signaling and GALNT6-mediated glycosylation driving immune evasion through PD-L1 stabilization and CD8+ T cell exclusion [39]. Single-cell analysis further revealed GALNT6-specific enrichment in immune-excluded goblet cells, highlighting the cell-type-specific functions of ubiquitination-related processes [39].
In lung adenocarcinoma, E3 ubiquitin ligases such as CDC20 show significant overexpression and association with poor prognosis [38]. Gene set enrichment analysis demonstrated that high CDC20 expression enriches several key oncogenic pathways, including G2MCHECKPOINT, MTORC1SIGNALING, OXIDATIVE_PHOSPHORYLATION, and GLYCOLYSIS [38]. Further correlation analysis indicated that CDC20 is positively correlated with the expression of key genes in the mTORC1 signaling pathway (mTOR, S6K1, and 4E-BP1) and the autophagy-related gene ULK1 [38], suggesting a multifaceted role in regulating cancer cell metabolism and proliferation.
Figure 2: Functional Consequences of Ubiquitination Dysregulation in Cancer. Ubiquitination alterations impact multiple hallmarks of cancer through various molecular mechanisms [33] [39] [38].
Implications for Cancer Diagnostics, Prognostics, and Therapeutics
Clinical Translation and Biomarker Development
The integration of transcriptomic, proteomic, and ubiquitinomic data has significant implications for cancer diagnostics, prognostics, and therapeutics. In colorectal cancer, machine learning approaches applied to multi-omics data identified a 5-gene PTM Activity Signature (CCNB1IP1, GALNT6, NEDD4L, PSMD14, UBE2C) that perfectly distinguished patients with diseases from those without (AUC = 1.00) [39]. Mendelian randomization analysis further validated GALNT6 as a causal risk factor (OR = 1.10, 95%CI:1.01-1.18), with its inhibition synergizing with anti-PD-1 treatment to enhance CD8+ T cell infiltration [39].
In lung adenocarcinoma, ubiquitination-based stratification has proven valuable for patient classification. Based on the expression profiles of hub UBRs, patients can be classified into two ubiquitination subtypes with significantly different characteristics across multiple dimensions, including survival, expression level, mutation burden, female predominance, infiltration level, immune profile, and drug response [33]. Researchers have established scoring systems for evaluating individual patients' ubiquitination status, such as the UB_risk score, which correlates with immunotherapy response, with low-score patients more likely to benefit from immune checkpoint inhibitors [33].
Similar approaches in pancreatic cancer have identified TRIM9 as a key ubiquitination regulator with tumor suppressor activity [24]. Mechanistic studies revealed that TRIM9 promotes K11-linked ubiquitination and proteasomal degradation of HNRNPU, dependent on its RING domain [24]. In vivo experiments demonstrated that TRIM9 overexpression reduced tumor growth, an effect that could be rescued by HNRNPU co-expression [24], highlighting the potential for targeting this axis therapeutically.
Therapeutic Implications and Drug Development
The multi-omics characterization of ubiquitination patterns in cancer has uncovered promising therapeutic targets and biomarkers for treatment response. For E3 ubiquitin ligases in lung adenocarcinoma, drug sensitivity analysis revealed that sensitivity to multiple antitumor drugs increased when CCNF was highly expressed [38]. All five identified E3 ubiquitin ligase genes showed consistent immune correlations—negatively correlated with B cells and dendritic cells but positively related to neutrophil immune infiltration [38]—suggesting potential for combination immunotherapies.
Ubiquitination regulators represent particularly attractive therapeutic targets because of their enzymatic activity and central role in controlling protein stability. Many UBRs, such as E3 ubiquitin ligases and deubiquitinases, have been identified as potential drug targets for cancer therapy, and several small-molecule inhibitors targeting UBRs have been developed [33]. These include MLN4924 (targeting the E1 enzyme), Leucettamol A (targeting the E2 enzyme), nutlin (targeting the E3 enzyme), and compound G5 (targeting DUB activity) [33]. The multi-omics characterization of ubiquitination patterns provides a rational basis for selecting patients most likely to respond to these targeted agents.
For immune-based therapies, ubiquitination status assessment may help identify patients most likely to benefit from checkpoint inhibitors. Patients with low UB_risk scores in lung adenocarcinoma show better response to immunotherapy, likely due to differences in the tumor immune microenvironment [33]. Similarly, in colorectal cancer, GALNT6 inhibition synergizes with anti-PD-1 treatment to enhance CD8+ T cell infiltration [39], suggesting combination approaches that simultaneously target ubiquitination-related processes and immune checkpoints.
The integration of transcriptomic, proteomic, and ubiquitinomic data provides unprecedented insights into cancer biology and therapeutic opportunities. Through sophisticated computational integration methods and advanced experimental workflows, researchers can now reconstruct comprehensive molecular networks that capture the dynamic interplay between different regulatory layers in cancer cells. The systematic characterization of ubiquitination patterns across cancer types has revealed extensive dysregulation of ubiquitination machinery, with profound consequences for oncogenic pathway activation, immune evasion, metabolic reprogramming, and therapeutic resistance.
These findings have significant translational implications, enabling the development of novel diagnostic classifiers, prognostic biomarkers, and targeted therapeutic strategies. As multi-omics technologies continue to evolve—particularly single-cell and spatial approaches—our understanding of ubiquitination in cancer will become increasingly refined, potentially unlocking new opportunities for personalized cancer therapy based on comprehensive molecular profiling.
The analysis of ubiquitination patterns in cancerous versus normal tissues relies heavily on large-scale public genomic databases processed through standardized, yet varied, bioinformatics pipelines. The Cancer Genome Atlas (TCGA), Genotype-Tissue Expression (GTEx) project, and the Cancer Cell Line Encyclopedia (CCLE) represent three cornerstone resources for cancer research, providing transcriptomic, genomic, and proteomic data across thousands of samples. These resources enable researchers to investigate differential ubiquitination pathways by comparing normal tissue expression from GTEx against cancerous samples from TCGA, while CCLE facilitates experimental validation through in vitro models. However, each resource employs distinct bioinformatics processing methodologies that significantly impact downstream analyses, including ubiquitination pathway characterization. Understanding these pipelines' technical differences is crucial for accurately interpreting data on ubiquitin-related genes, E3 ligases, deubiquitinases, and their substrate patterns in malignancy.
The complexity of ubiquitination signaling—involving multi-step enzymatic cascades and diverse polyubiquitin chain topologies—demands particularly careful consideration of bioinformatic processing methods. As ubiquitination regulates key cancer pathways including p53, NF-κB, and HIF1α, variations in RNA-seq quantification between pipelines can substantially alter conclusions about ubiquitin pathway dysregulation in tumorigenesis. This guide provides a comprehensive comparison of TCGA, GTEx, and CCLE processing methodologies, experimental protocols for cross-database analyses, and visualization of ubiquitination pathways relevant to cancer research, offering drug development professionals a framework for robust comparative analyses of ubiquitination patterns in normal and cancerous tissues.
Database-Specific Bioinformatics Pipelines
TCGA (The Cancer Genome Atlas) Processing Pipeline
The TCGA database, managed by the NCI Genomic Data Commons (GDC), provides uniformly processed multi-omics data from over 11,000 patients across 33 cancer types. The standardized GDC RNA-seq processing pipeline serves as a primary resource for analyzing ubiquitination-related gene expression in tumor tissues. The pipeline begins with read alignment using STAR (Spliced Transcripts Alignment to the Reference) against the GRCh38 reference genome, followed by gene-level quantification via HTSeq to generate raw count data. Expression values are normalized using FPKM (Fragments Per Kilobase of Million mapped reads) and TPM (Transcripts Per Million) to facilitate cross-sample comparisons. For ubiquitination studies, this pipeline provides comprehensive expression profiles for E1 activating enzymes, E2 conjugating enzymes, E3 ligases, and deubiquitinating enzymes (DUBs) across cancer types, enabling systematic identification of dysregulated ubiquitination pathway components in tumorigenesis [40].
The GDC pipeline also offers harmonized somatic mutation calls, copy number variations, and DNA methylation data, allowing integrative analysis of genetic alterations affecting ubiquitination pathways. For instance, researchers can correlate mutations in E3 ligase genes like MDM2 with p53 expression levels across cancer types. The availability of paired clinical data further enables survival analyses based on ubiquitination-related gene expression patterns, supporting the identification of prognostic ubiquitination biomarkers in oncology [40].
GTEx (Genotype-Tissue Expression) Processing Pipeline
The GTEx project provides a critical reference of normal tissue gene expression, using a standardized pipeline optimized for cross-tissue comparability. The GTEx consortium employs the TOPMed pipeline for RNA-seq processing, which includes alignment using STAR to the GRCh38 reference genome and gene-level quantification with RNA-SeQC. Expression values are normalized using TPM, with careful attention to batch effect mitigation across multiple tissue collection sites. This normalization approach is particularly important for ubiquitination studies, as it enables accurate comparison of ubiquitin pathway component expression across diverse normal tissues, establishing baseline expression patterns for E3 ligases and DUBs in non-diseased states [40] [41].
A key feature of the GTEx pipeline for ubiquitination researchers is the availability of eQTL (expression Quantitative Trait Loci) mappings, which identify genetic variants that influence gene expression levels. The PredictDB pipeline builds upon GTEx data to create gene expression prediction models for 48 different human tissues, using a nested cross-validated elastic-net approach trained on European-ancestry samples only. These models help researchers connect genetic variations to expression changes in ubiquitination-related genes, potentially revealing regulatory mechanisms affecting ubiquitination pathways in normal physiology [41].
CCLE (Cancer Cell Line Encyclopedia) Processing Pipeline
The CCLE provides extensive molecular characterization of over 1,000 human cancer cell lines across 36 tumor types, serving as a bridge between TCGA tumor data and experimental models. The CCLE RNA-seq processing pipeline includes both RPKM (Reads Per Kilobase of Million mapped reads) and RSEM (RNA-Seq by Expectation-Maximization) normalized expression values, along with raw read counts for differential expression analysis. This multi-format availability allows ubiquitination researchers to apply their preferred normalization methods when analyzing E2 enzymes, E3 ligases, or DUB expression across cell lines [42] [43].
Beyond transcriptomics, CCLE offers comprehensive genetic data including merged mutation calls, ABSOLUTE copy number analysis, and structural variant calls. These datasets enable researchers to connect genetic alterations in ubiquitination pathway components with functional consequences across cell lines. For instance, researchers can identify cell lines with amplifications in the MDM2 E3 ligase gene and correlate this with p53 protein levels measured in companion proteomic datasets. The availability of miRNA expression, DNA methylation, histone modification, and metabolomics data further supports multi-layered investigations of ubiquitination regulation in cancer models [42] [43].
Comparative Analysis of Pipeline Methodologies
Quantitative Pipeline Comparison Metrics
Different RNA-seq processing pipelines can yield substantially different gene expression estimates, with important implications for ubiquitination research. A comprehensive study comparing five best-in-class RNA-seq processing pipelines applied to 6,690 human tumor and normal tissues found that while nearly 88% of protein-coding genes show similar expression profiles across pipelines, over 12% of protein-coding genes exhibit greater than four-fold expression differences when processed through different pipelines applied to the exact same RNA-seq reads. These discordantly quantified genes (DQ genes) include many disease-associated genes, with three-fold and two-fold expression differences affecting 22.04% and 57.6% of genes in TCGA data respectively [40].
Table 1: Impact of Pipeline Differences on Gene Expression Estimates
| Metric | TCGA Data | GTEx Data | Implications for Ubiquitination Research |
|---|---|---|---|
| Genes with >4-fold expression differences | 1,637 genes (~10%) | 1,214 genes (~7%) | Major quantitative differences for affected ubiquitination genes |
| Genes with >3-fold expression differences | 3,551 genes (~22.04%) | 2,621 genes (~15.87%) | Moderate differences affecting many ubiquitination pathway components |
| Genes with >2-fold expression differences | 9,279 genes (~57.6%) | 8,061 genes (~48.8%) | Widespread minor differences across most ubiquitination genes |
| Genes with divergent fold-change estimates | 1,958 genes (~95% of DQ genes) | Similar proportion | Impacts differential expression analysis for ubiquitination targets |
For ubiquitination research, these pipeline-induced variations are particularly concerning as they can significantly alter conclusions about ubiquitin pathway dysregulation in cancer. The correlation between mRNA and protein levels for approximately half of TCGA genes with available protein abundance data shows remarkably low correlation, complicating the prediction of actual ubiquitination activity from transcriptomic data alone. This is especially relevant for ubiquitination studies since E3 ligase activity often depends on post-translational regulation rather than simply mRNA abundance [40].
Cross-Platform Normalization Challenges
The comparison of ubiquitination patterns between TCGA tumors and GTEx normal tissues presents significant technical challenges due to fundamental differences in pipeline methodologies and normalization approaches. Principal Component Analysis (PCA) of FPKM-based expression values from different data sources shows clustering primarily by data source rather than biological similarity, with data source explaining over 50% of the total variability. While conversion to TPM units improves comparability, inter-batch differences between TCGA and GTEx samples may still confound comparisons despite unit normalization [40].
When analyzing cell lines as models of primary tumors, additional considerations emerge. A comprehensive pan-cancer analysis comparing CCLE cell lines to TCGA tumors across 22 tumor types found that tumor purity significantly impacts correlation results. In 75% of solid tumor types, cell lines were significantly more correlated with primary tumor samples in the top quartile of tumor purity than with those in the bottom quartile. This has implications for ubiquitination studies, as tumor samples with lower purity may show artificially reduced expression of cancer-cell-specific ubiquitination components due to dilution with non-malignant cells [44].
Table 2: Pipeline Methodologies Across Major Genomics Resources
| Processing Component | TCGA (GDC) | GTEx (TOPMed) | CCLE | Impact on Ubiquitination Analysis |
|---|---|---|---|---|
| Alignment Tool | STAR | STAR | Not specified in results | Consistent alignment for ubiquitination transcripts |
| Reference Genome | GRCh38 | GRCh38 | hg19 | Coordinate mapping challenges for ubiquitin genes |
| Expression Units | FPKM, TPM, counts | TPM, RPKM | RPKM, RSEM, counts | Normalization differences affect ubiquitin gene quantification |
| Gene Annotation | Gencode | Gencode | Gencode v19 | Version differences impact ubiquitin gene models |
| Batch Effect Correction | Combat | Not specified | Not specified | Affects cross-study ubiquitination comparisons |
| Tissue Specificity | Tumor tissues only | Normal tissues only | Cell lines only | Fundamental biological differences in ubiquitination |
Experimental Protocols for Ubiquitination Pattern Analysis
Cross-Database Ubiquitination Signature Identification
To identify differentially regulated ubiquitination components between cancerous and normal tissues, researchers can implement a standardized protocol integrating data from all three resources. Begin by downloading RNA-seq data for your cancer type of interest from TCGA, matched normal tissue data from GTEx, and relevant cancer cell line data from CCLE. For ubiquitination-focused analyses, curate a target gene list encompassing E1 activating enzymes (UBA1, UBA6), representative E2 conjugating enzymes (UBE2N, UBE2C, UBE2S), key E3 ligases (MDM2, SKP2, FBW7), and deubiquitinating enzymes (USP7, USP9X), plus major ubiquitination substrates (p53, c-MYC, PTEN, RBI) [45] [46].
Process all datasets through a uniform normalization pipeline to mitigate technical artifacts, converting all expression values to TPM units and applying cross-platform batch effect correction using established methods like ComBat. For TCGA data, account for tumor purity using available estimation algorithms, as purity differences can significantly affect ubiquitination pathway expression patterns. Perform differential expression analysis using established statistical methods (e.g., DESeq2, limma-voom) comparing TCGA tumor samples against GTEx normal tissues, applying multiple testing correction and setting significance thresholds appropriate for ubiquitination pathway analysis [40] [44].
Validate findings in CCLE cell lines by comparing expression patterns of significantly dysregulated ubiquitination components across relevant cancer types. For ubiquitination components showing consistent dysregulation, perform correlation analysis with corresponding protein abundance data where available (e.g., RPPA data in TCGA, mass spectrometry data in CCLE) to assess transcript-protein concordance. This integrated approach controls for platform-specific artifacts while leveraging the complementary strengths of each resource for comprehensive ubiquitination pathway analysis [43] [44].
Ubiquitination Pathway Activity Inference
Beyond simple expression profiling, researchers can infer alterations in ubiquitination pathway activity using multi-omics integration. Start by identifying ubiquitination substrates whose protein stability may be affected by dysregulated ubiquitination machinery, focusing on established relationships such as MDM2-p53, SKP2-p27, and FBW7-c-MYC. For these substrate-regulator pairs, analyze the relationship between E3 ligase or DUB expression and substrate protein levels using available proteomic data, noting that inverse correlations may indicate functional regulatory relationships [45] [46].
Infer ubiquitination pathway activation by examining expression patterns of ubiquitination-dependent signaling components. For NF-κB signaling, analyze expression of UBE2N and UBE2V1, which form K63-linked ubiquitin chains essential for IKK activation, alongside expression of NF-κB target genes. Similarly, for hypoxia response pathways, examine correlations between UBE2S expression (which promotes VHL degradation) and HIF target gene expression across cancer types. These analyses can reveal cancer-type-specific ubiquitination pathway alterations that may represent therapeutic vulnerabilities [46].
For experimental validation, select cell lines from CCLE that represent the ubiquitination dysregulation patterns observed in primary tumors. For instance, choose cell lines with high UBE2N/UBE2V1 expression for NF-κB pathway inhibition studies, or cell lines with elevated UBE2S expression for HIF pathway targeting. This systematic approach bridges bioinformatic discovery with experimental validation, facilitating the identification of targetable ubiquitination dependencies in cancer [44] [46].
Ubiquitination Signaling Pathways in Cancer
Key Ubiquitination Pathways in Tumorigenesis and Metastasis
Ubiquitination regulates numerous oncogenic and tumor suppressive pathways through diverse mechanisms. The ubiquitin-proteasome system (UPS) controls the stability of key regulatory proteins, with E3 ligases recognizing specific substrates for polyubiquitination and subsequent degradation. Beyond proteasomal degradation, ubiquitination regulates protein activity, localization, and interactions through monoubiquitination and non-degradative polyubiquitin chain types. Cancer cells exploit ubiquitination pathways to destabilize tumor suppressors, stabilize oncoproteins, and activate pro-survival signaling, making ubiquitination components promising therapeutic targets [45] [47].
The p53-MDM2 axis represents one of the most extensively studied ubiquitination pathways in cancer. Under normal conditions, the E3 ligase MDM2 maintains low p53 levels through polyubiquitination and proteasomal degradation. In many cancers, MDM2 amplification or overexpression leads to excessive p53 degradation, allowing uncontrolled cell proliferation. Additionally, the deubiquitinase USP7 stabilizes both MDM2 and p53 through deubiquitination, creating a complex regulatory network. Therapeutic strategies targeting MDM2-p53 interaction aim to reactivate p53 in tumors with wild-type p53, demonstrating the clinical relevance of understanding ubiquitination pathways [45].
Non-proteolytic ubiquitination signaling through K63-linked chains plays crucial roles in cancer progression and metastasis. The UBE2N-UBE2V1 complex specifically builds K63-linked ubiquitin chains that activate NF-κB and TGF-β signaling pathways, promoting expression of metastasis-associated genes including CNN2, IL13RA2, CD44, and VCAM-1. UBE2N is overexpressed in breast, pancreas, colon, prostate, lymphoma, and ovarian carcinomas, and is required for breast cancer metastasis to the lung in vivo, though interestingly not for primary tumor formation. This highlights the stage-specific roles of certain ubiquitination components in cancer progression [46].
Diagram 1: Key Ubiquitination Pathways in Cancer. This diagram illustrates major ubiquitination-mediated signaling pathways dysregulated in cancer, including protein degradation pathways, p53 regulation, NF-κB signaling, and hypoxia response mechanisms.
Cell Cycle and DNA Damage Response Regulation
Ubiquitination plays essential roles in regulating cell cycle progression and DNA damage response, with frequent dysregulation in cancer. The anaphase-promoting complex/cyclosome (APC/C) functions as a multi-subunit E3 ligase that controls metaphase-to-anaphase transition and mitotic exit by targeting cyclins and other cell cycle regulators for degradation. UBE2C (UBCH10) partners with APC/C to ensure proper chromosome alignment and segregation, with UBE2C overexpression causing chromosomal missegregation, aneuploidy, and mitotic slippage—hallmarks of cancer cells. Mice engineered to overexpress UBE2C develop elevated lung tumor burden and various other tumors, demonstrating its oncogenic potential [46].
Cyclin regulation by ubiquitination represents another key cancer-relevant pathway. Cyclin E, which controls G1 to S phase transition, is targeted for degradation by the SCF (Skp1-Cullin-F-box protein) complex to prevent premature S phase entry. In cancers, dysregulation of this ubiquitin-mediated degradation results in cyclin E stabilization and continuous cell cycle progression. Conversely, CDK inhibitors like p21 and p27 are targeted for degradation by SCF complexes, with downregulation of these inhibitors through overactive ubiquitination promoting unchecked cell cycle entry in many tumors [45].
In the DNA damage response, ubiquitination coordinates repair pathway activation and cell fate decisions. Key DNA damage response proteins including ATM, ATR, and BRCA1 undergo activating ubiquitination modifications following DNA damage. The RNF8/RNF168 E3 ligases promote K63-linked ubiquitination at DNA damage sites, facilitating recruitment of repair factors. BRCA1, a critical tumor suppressor in hereditary breast and ovarian cancers, is itself regulated by ubiquitination, with mutations affecting its ubiquitination pathway impairing DNA repair and increasing genomic instability. The interconnected roles of ubiquitination in both tumor suppressor regulation and genome integrity maintenance highlight its central importance in cancer prevention and progression [45].
The Scientist's Toolkit: Research Reagent Solutions
Essential Databases and Analysis Tools
Table 3: Essential Research Resources for Ubiquitination and Cancer Genomics
| Resource Category | Specific Resource | Function in Ubiquitination Research | Data Access |
|---|---|---|---|
| Primary Databases | TCGA (GDC Portal) | Provides multi-omics data for tumor tissues across 33 cancer types | https://portal.gdc.cancer.gov/ |
| GTEx Portal | Reference normal tissue expression for comparison with tumor data | https://gtexportal.org/ | |
| CCLE Database | Molecular characterization of 1000+ cancer cell lines for validation | https://sites.broadinstitute.org/ccle/ | |
| Processing Pipelines | GDC RNA-seq Pipeline | Standardized processing for TCGA data | https://docs.gdc.cancer.gov/ |
| GTEx TOPMed Pipeline | Normal tissue-specific processing optimized for cross-tissue comparison | https://github.com/broadinstitute/gtex-pipeline | |
| Recount2 | Uniformly processed RNA-seq data from TCGA, GTEx, and SRA | https://jhubiostatistics.shinyapps.io/recount/ | |
| Specialized Tools | PredictDB | Gene expression prediction models for 48 tissues trained on GTEx | https://github.com/hakyimlab/PredictDBPipelineGTEx_v7 |
| Ubiquitination Sites Database | Curated resource of experimentally verified ubiquitination sites | Various resources available | |
| Experimental Reagents | Ubiquitination Pathway Antibodies | Detect ubiquitin, E1/E2/E3 enzymes, DUBs in validation experiments | Commercial suppliers |
| Proteasome Inhibitors (MG132, Bortezomib) | Block protein degradation to study ubiquitination substrates | Commercial suppliers | |
| E1 Inhibitors (PYR-41, TAK-243) | Specific targeting of ubiquitination activation | Commercial suppliers |
Quality Control Considerations for Ubiquitination Studies
When designing studies of ubiquitination patterns in cancer, researchers should implement specific quality control measures to address common technical challenges. For transcriptomic analyses, carefully consider the impact of different normalization methods on ubiquitination pathway components, as E2 enzymes, E3 ligases, and DUBs may be differentially affected by standard normalization approaches. When comparing TCGA tumor data with GTEx normal tissues, apply cross-platform normalization and explicitly account for batch effects using established statistical methods [40].
For analyses incorporating cell line models, critically evaluate the relevance of specific cell lines to the primary tumors being studied. Reference pan-cancer analyses comparing CCLE cell lines to TCGA tumors have established that not all cell lines equally model their corresponding primary tumors, with correlation coefficients ranging from 0.66 in head and neck squamous cell carcinoma to 0.49 in liver hepatocellular carcinoma. Prioritize cell lines with higher correlation to primary tumors for validation studies, and consult resources that rank cell lines by representativeness for specific cancer types [44].
When interpreting expression data for ubiquitination components, consider the potential discordance between mRNA and protein levels, which is particularly relevant for ubiquitination pathway elements that may be regulated post-translationally. Where possible, integrate proteomic data from RPPA or mass spectrometry to confirm that transcript-level changes reflect protein-level alterations. For key ubiquitination substrates, consider implementing functional assays to directly measure protein stability and turnover rather than relying solely on expression data [40] [43].
Diagram 2: Experimental Workflow for Ubiquitination Pattern Analysis. This diagram outlines a systematic approach for comparing ubiquitination patterns between cancerous and normal tissues, incorporating key quality control checkpoints to ensure robust findings.
The comparative analysis of bioinformatics pipelines for processing TCGA, GTEx, and CCLE data reveals both opportunities and challenges for ubiquitination research in cancer. While each resource provides valuable molecular data, significant technical differences in processing methodologies can substantially impact conclusions about ubiquitination pathway dysregulation in malignancy. The finding that over 12% of protein-coding genes show greater than four-fold expression differences between pipelines underscores the importance of standardized processing when comparing ubiquitination components across databases. For drug development professionals, these pipeline differences represent a critical methodological consideration when prioritizing ubiquitination pathway targets for therapeutic intervention.
The integration of TCGA tumor data, GTEx normal tissue reference, and CCLE experimental models offers a powerful framework for identifying clinically relevant ubiquitination alterations in cancer. By implementing the experimental protocols and quality control measures outlined in this guide, researchers can more reliably identify dysregulated E3 ligases, DUBs, and ubiquitination substrates that drive oncogenesis. The expanding characterization of ubiquitination pathways in cancer, particularly through proteomic and metabolomic profiling in resources like CCLE, continues to enhance our understanding of how cancer cells exploit ubiquitination for survival and progression. As ubiquitination-targeted therapies advance toward clinical application, rigorous bioinformatic analysis using these major databases will remain essential for translating mechanistic insights into effective cancer treatments.
The tumor microenvironment (TME) represents a complex ecosystem where cancer cells coexist with immune cells, fibroblasts, endothelial cells, and various signaling molecules. Within this dynamic milieu, ubiquitination—a crucial post-translational modification—has emerged as a master regulator of protein stability, immune responses, and cellular homeostasis. The integration of single-cell RNA sequencing (scRNA-seq) technologies has revolutionized our ability to dissect the intricate ubiquitination patterns across individual cell types within tumors, moving beyond bulk tissue analysis to reveal unprecedented cellular heterogeneity. This technological synergy is illuminating how ubiquitination differentially influences cancer cells versus normal tissues, providing critical insights for targeted therapeutic development.
The ubiquitin-proteasome system encompasses a sophisticated enzymatic cascade involving E1 (activating), E2 (conjugating), and E3 (ligase) enzymes that collectively coordinate the attachment of ubiquitin chains to target proteins, thereby determining their degradation, localization, or activity [7] [48]. When dysregulated, this system contributes fundamentally to oncogenesis, metabolic reprogramming, and immune evasion across various cancer types [7] [49]. Single-cell technologies now enable researchers to map the expression patterns of ubiquitination-related genes (UbRGs) with cellular precision, creating high-resolution catalogs of how these regulatory mechanisms shape tumor progression and treatment resistance.
Technological Framework: Single-Cell RNA Sequencing Methodologies
Core Experimental Workflow
Single-cell RNA sequencing technologies have evolved significantly since their inception in 2009, with current platforms enabling the analysis of hundreds of thousands of individual cells simultaneously [50]. The standard workflow encompasses several critical stages:
Single-cell isolation and capture: Individual cells are isolated from fresh tumor tissues using methods such as fluorescence-activated cell sorting (FACS), magnetic-activated cell sorting, or microfluidic systems. This step requires careful tissue dissociation while minimizing artificial transcriptional stress responses that can occur at higher temperatures [50] [51]. Recent approaches often employ dissociation at 4°C or utilize single-nucleus RNA sequencing (snRNA-seq) for tissues difficult to dissociate, such as brain and muscle tissues [50].
Cell lysis and reverse transcription: Captured cells are lysed, and their mRNA transcripts are converted to complementary DNA (cDNA) using reverse transcriptase. This step incorporates unique molecular identifiers (UMIs) and cell barcodes to precisely track individual transcripts and their cell of origin, effectively controlling for amplification biases [50].
cDNA amplification and library preparation: The cDNA is amplified either through polymerase chain reaction (PCR) or in vitro transcription (IVT). PCR-based amplification is utilized in platforms such as Smart-seq2 and 10x Genomics, while IVT is employed in CEL-seq and MARS-Seq protocols [50].
Sequencing and data analysis: Amplified libraries are sequenced using high-throughput platforms such as Illumina NovaSeq, followed by computational analysis using tools like Seurat and Scanpy for cell clustering, differential expression analysis, and trajectory inference [50] [52].
Key scRNA-seq Platforms and Their Applications in Ubiquitination Research
Table 1: Comparison of Single-Cell RNA Sequencing Technologies
| Technology | Throughput | Amplification Method | UMI Utilization | Key Applications in Ubiquitination Research |
|---|---|---|---|---|
| 10x Genomics | High (thousands to millions of cells) | PCR-based | Yes | Characterizing ubiquitination heterogeneity across cell populations in TME [50] |
| Smart-seq2 | Low to medium (hundreds of cells) | PCR-based | No | Full-length transcript analysis of specific ubiquitination enzymes [50] |
| CEL-seq2 | Medium | IVT-based | Yes | Quantifying expression levels of ubiquitination-related genes [50] |
| MARS-seq | High | IVT-based | Yes | Large-scale profiling of UbRGs across cell types [50] |
| Drop-seq | High | PCR-based | Yes | Identifying rare cell populations with distinct ubiquitination signatures [50] |
Ubiquitination Heterogeneity Across Tumor Microenvironments
Hepatocellular Carcinoma (HCC)
In hepatocellular carcinoma, scRNA-seq analyses have revealed significant upregulation of ubiquitination-related genes in malignant cells compared to normal hepatocytes. These ubiquitination patterns distinctly shape the immunosuppressive landscape of the TME. UBE2C, a critical ubiquitin-conjugating enzyme, emerges as a central regulator—its overexpression correlates with poor prognosis and drives HCC cell proliferation, invasion, and metastasis through multiple pathways [7].
Experimental validation using transwell assays, CCK-8 assays, and wound healing assays confirmed that UBE2C knockdown significantly impairs HCC cell migration and viability [7]. Pathway enrichment analyses further connected ubiquitination signatures to critical cancer-related processes including cell cycle regulation, DNA repair mechanisms, metabolic reprogramming, and p53 signaling cascades [7]. These findings position UBE2C as both a prognostic biomarker and a promising therapeutic target in HCC precision medicine.
Breast Cancer
Comprehensive bioinformatics analyses of breast cancer samples have identified distinct molecular subtypes based on ubiquitination-related gene expression patterns. Researchers developed a prognostic model incorporating eight key UbRGs that effectively stratifies patients into high-risk and low-risk categories with significantly different overall survival outcomes [49] [53].
Functional experiments validated FBXL6 and PDZRN3 as crucial regulators of breast cancer development. FBXL6 knockdown suppressed tumor growth both in vitro and in vivo, while PDZRN3 overexpression demonstrated tumor-suppressive characteristics [49] [53]. Drug sensitivity analysis further revealed that ubiquitination signatures predict response to conventional therapies including tamoxifen, fulvestrant, cyclophosphamide, and targeted agents like lapatinib, highlighting their potential for guiding treatment selection [49].
Colorectal Cancer (CRC)
Ubiquitinome profiling of colorectal cancer tissues has identified dramatic alterations in ubiquitination patterns compared to normal adjacent mucosa. Mass spectrometry-based analyses detected 1,690 quantifiable ubiquitination sites across 870 proteins, with 1,172 proteins showing increased ubiquitination and 1,700 proteins demonstrating decreased ubiquitination in CRC tissues [30].
Notably, highly ubiquitinated proteins (containing ≥10 modification sites) were enriched in critical biological processes including G-protein coupling, glycoprotein metabolism, and antigen presentation pathways [30]. FOCAD emerged as a protein of particular interest, with increased ubiquitination at Lys583 and Lys587 residues correlating with patient survival outcomes, suggesting its potential role in RNA localization and translation processes within CRC cells [30].
Acral Melanoma
Single-cell RNA sequencing of acral melanoma subtypes has revealed striking tumor heterogeneity between subungual melanoma (SM) and plantar melanoma (PM). Malignant melanocytes from these distinct anatomical locations exhibited significant differences in gene expression profiles, copy number variations (CNVs), and pathway activation [51].
SM melanocytes demonstrated higher CNV signals and predominant activation of NF-kB signaling, while PM melanocytes preferentially utilized Wnt pathway signaling [51]. Cell-cell communication analysis further identified the EPHA3-EFNA1 axis as specifically active between cancer-associated fibroblasts (CAFs) and melanocytes in PM, while the TIGIT-NECTIN2 axis was expressed across both AM subtypes in T/NK cells and melanocytes, suggesting potential immunotherapeutic targets [51].
Experimental Methodologies for Validating Ubiquitination Functions
In Vitro Functional Assays
Table 2: Key Experimental Protocols for Ubiquitination Research
| Method | Experimental Procedure | Key Applications in Ubiquitination Research |
|---|---|---|
| Transwell Assay | Seed cells in serum-free medium in upper chamber; complete medium in lower chamber; incubate 24h; fix migrated cells with 4% PFA; stain with crystal violet; count cells [7] | Evaluate effect of UbRGs (e.g., UBE2C) on cancer cell migration and invasion [7] |
| CCK-8 Assay | Seed transfected cells in 96-well plates (2,500 cells/well); add CCK-8 reagent; incubate 4h; measure OD450 with microplate reader [7] | Assess cell viability and proliferation after modulation of ubiquitination enzymes [7] |
| Wound Healing Assay | Create scratch in confluent cell monolayer with pipette tip; wash with PBS; add serum-free medium; capture images at 0h and 24h; measure migration distance [7] | Quantify collective cell migration capabilities influenced by ubiquitination [7] |
| Colony Formation Assay | Seed 600 cells in 6-well plates; incubate for 14 days; stain and count visible colonies [7] | Determine long-term proliferative capacity after UbRG manipulation [7] |
| Western Blot Analysis | Lyse cells with RIPA buffer; separate proteins via SDS-PAGE; transfer to PVDF membrane; block with milk; incubate with primary and secondary antibodies; detect with ECL [53] | Verify protein expression levels and knockdown efficiency of ubiquitination enzymes [53] |
In Vivo and Ex Vivo Techniques
Beyond in vitro approaches, researchers employ sophisticated in vivo models and spatial techniques to validate ubiquitination mechanisms. Orthotopic xenograft models in immunocompromised mice have been instrumental in demonstrating the tumor-promoting effects of genes like FBXL6 in breast cancer [49]. Additionally, multiplex immunofluorescence staining enables simultaneous visualization of multiple ubiquitination-related proteins and immune markers in intact tissue sections, preserving spatial context that is lost in single-cell suspensions [51] [52].
Spatial transcriptomics techniques are increasingly combined with scRNA-seq to map ubiquitination signatures within specific tissue architectures, correlating gene expression patterns with histological features. This integrated approach has proven particularly valuable in characterizing the immune microenvironment of rare cancers such as small cell carcinoma of the ovary, hypercalcemic type (SCCOHT), where it revealed exhausted T cells and distinct tumor-associated macrophage subsets infiltrating the tumor mass [52].
Signaling Pathways and Molecular Mechanisms
Key Ubiquitination-Regulated Pathways in Cancer
Ubiquitination modulates several critical signaling cascades that drive tumor progression and immune evasion. The diagrams below illustrate two key ubiquitination-regulated pathways in cancer biology:
Figure 1: Ubiquitination-Regulated Signaling Pathways in Cancer. This diagram illustrates how key ubiquitination enzymes such as UBE2C and differentially expressed genes like CDK4 and GAB2 activate critical cancer-promoting pathways including NF-κB, Wnt signaling, p53 degradation, and immune evasion mechanisms [7] [51].
Ubiquitination-Mediated Immune Regulation
The tumor microenvironment exhibits complex ubiquitination-dependent immunoregulatory mechanisms that scRNA-seq has helped illuminate:
Figure 2: Ubiquitination-Mediated Immune Regulation in TME. This flowchart depicts how tumor-specific ubiquitination signatures influence immune cell infiltration, polarization of tumor-associated macrophages (TAMs), T-cell exhaustion states, and development of therapy resistance across cancer types [49] [52].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Key Research Reagent Solutions for scRNA-seq and Ubiquitination Studies
| Reagent/Category | Specific Examples | Function and Application |
|---|---|---|
| scRNA-seq Library Kits | GEXSCOPE Single Cell RNA Library Kits, 10x Genomics Chromium | High-throughput library construction for single-cell transcriptomics [51] [52] |
| Cell Dissociation Solutions | sCelLive Tissue Dissociation Solution, collagenase-based enzymes | Tissue-specific digestion into single-cell suspensions while preserving RNA integrity [51] [52] |
| Cell Preservation Media | GEXSCOPE Tissue Preservation Solution, RNAlater | Maintain cell viability and transcriptome stability during sample transport and processing [51] |
| Ubiquitination Assay Reagents | Ubiquitin-activating enzyme (E1) inhibitors, Proteasome inhibitors (MG132) | Modulate ubiquitination processes in functional validation experiments [7] |
| Cell Culture Reagents | DMEM/1640 media with 10% FBS, penicillin/streptomycin | Maintain cell lines for in vitro ubiquitination manipulation studies [7] [53] |
| Transfection Reagents | Lipofectamine 2000/3000, lentiviral transduction systems | Introduce UbRG overexpression constructs or shRNAs for functional studies [7] [53] |
| Antibodies | Anti-EFNA1, anti-TIGIT, anti-NECTIN2, anti-PD-1 | Validate protein expression and localization in ubiquitination studies [51] |
The integration of single-cell RNA sequencing with ubiquitination research has fundamentally transformed our understanding of tumor heterogeneity, revealing complex regulatory networks that operate in a cell-type-specific manner within the tumor microenvironment. These advanced technologies have enabled researchers to move beyond bulk tissue analyses to identify rare cell populations with distinct ubiquitination signatures, track the spatial organization of ubiquitination enzymes and their substrates, and uncover novel therapeutic vulnerabilities across various cancer types.
Future research directions will likely focus on developing multi-omics approaches that simultaneously capture transcriptomic, proteomic, and ubiquitinomic information from the same single cells. Additionally, the integration of spatial transcriptomics with ubiquitination mapping will provide unprecedented insights into how these modifications shape cellular crosstalk within intact tumor architectures. As these technologies continue to evolve, they promise to accelerate the development of novel ubiquitination-targeted therapies and biomarkers that can be deployed for personalized cancer treatment, ultimately improving outcomes for patients across diverse cancer types.
The contrasting ubiquitination patterns between cancerous and normal tissues—particularly the upregulation of specific E2 and E3 enzymes in malignant cells—highlight the therapeutic potential of selectively targeting these dysregulated pathways while sparing normal cellular functions. As single-cell technologies continue to advance, they will undoubtedly uncover additional layers of complexity in ubiquitination-mediated regulation, further expanding our opportunities for therapeutic intervention in cancer and other diseases.
The ubiquitin-proteasome system (UPS) represents a crucial post-translational modification mechanism that governs virtually all cellular processes, from protein degradation to signal transduction [5]. Recent advances in multi-omics technologies have revealed the extensive involvement of ubiquitination in shaping the tumor immune microenvironment (TIME), influencing immune cell infiltration, function, and ultimately, patient response to therapy [48] [54]. Ubiquitination involves a sequential enzymatic cascade comprising E1 activating enzymes, E2 conjugating enzymes, and E3 ligases, which collectively determine the specificity and outcome of substrate modification [55]. The dysregulation of this system in cancer cells not only drives tumor proliferation and survival but also creates an immunosuppressive milieu that facilitates immune evasion [7] [13]. This review synthesizes current evidence from multi-omics studies to compare ubiquitination patterns between cancerous and normal tissues, with a specific focus on how these modifications regulate immune infiltration across various cancer types, providing a foundation for developing novel immunotherapeutic strategies.
Comparative Ubiquitination Landscapes: Cancerous versus Normal Tissues
Pan-Cancer Alterations in Ubiquitination Machinery
Comprehensive genomic analyses across multiple cancer types have consistently demonstrated widespread dysregulation of ubiquitination-related genes (URGs) in tumor tissues compared to their normal counterparts. These alterations encompass expression changes, genetic mutations, and copy number variations that collectively contribute to tumor pathogenesis and immune modulation.
Table 1: Ubiquitination-Related Gene Alterations Across Cancer Types
| Gene Category | Example Genes | Alteration in Cancer | Associated Cancers | Immune Correlation |
|---|---|---|---|---|
| E1 Enzymes | UBA1, UBA6 | Upregulated | BRCA, COAD, KIRC, LUAD [13] [22] | Immune score, T-cell infiltration [13] |
| E2 Enzymes | UBE2C, UBE2N | Upregulated | HCC, LUAD [7] [56] | Negative correlation with B cells, DCs [38] |
| E3 Ligases | CDC20, CCNF, AURKA | Upregulated | LUAD [38] [57] | Neutrophil infiltration [38] |
| E3 Ligases (Regulatory) | CBLB, STUB1 | Variable | Multiple [54] | FOXP3+ Treg stability [54] |
The UBA family of E1 enzymes, particularly UBA1 and UBA6, demonstrates significant overexpression in numerous cancer types including breast cancer (BRCA), colorectal cancer (COAD), renal cancer (KIRC), and lung adenocarcinoma (LUAD) [13] [22]. This elevated expression correlates with advanced tumor stage and poorer patient survival, suggesting their critical role in tumor progression. At the E2 level, enzymes such as UBE2C show marked upregulation in hepatocellular carcinoma (HCC), where they promote cancer cell proliferation, invasion, and metastasis through regulation of cell cycle progression and immune evasion mechanisms [7]. Similarly, in lung adenocarcinoma, UBE2N overexpression associates with diminished immune cell infiltration and compromised antigen presentation, indicating its role in shaping an immunosuppressive TIME [56].
Tissue-Specific Ubiquitination Patterns
The expression patterns of URGs exhibit significant heterogeneity across different tissue types, with the most distinct pattern observed in testicular tissue [5]. This tissue-specific variation suggests specialized roles for ubiquitination in different physiological contexts, which may be co-opted during tumorigenesis. In non-cancerous tissues, URGs maintain cellular homeostasis through regulated protein degradation, DNA repair mechanisms, and proper immune cell function. However, in transformed tissues, these processes become subverted to support tumor growth and evade immune surveillance. For instance, in lung adenocarcinoma, multiple E3 ubiquitin ligases including CDC20, AURKA, CCNF, POC1A, and UHRF1 are significantly overexpressed compared to normal lung tissue and correlate with inferior patient outcomes [38] [57].
Methodologies for Ubiquitination-Mediated Immune Infiltration Analysis
Multi-Omics Integration Approaches
Advanced multi-omics strategies have enabled comprehensive profiling of the ubiquitinome and its relationship to immune infiltration:
Transcriptomic Analysis: Bulk RNA sequencing from databases like TCGA and GTEx allows comparison of URG expression between tumor and normal samples. Single-cell RNA sequencing (scRNA-seq) further resolves cell-type-specific expression patterns within the TIME [7] [56]. Analytical pipelines typically involve differential expression analysis, correlation with immune signatures, and survival association studies.
Proteomic Validation: Immunohistochemistry (IHC) on tissue microarrays validates protein-level expression of ubiquitination enzymes [57] [56]. For example, IHC confirmation of CDC20 overexpression in non-small cell lung cancer tissues provides translational relevance to transcriptomic findings [57].
Genetic Alteration Mapping: Analysis of somatic mutations and copy number variations (CNV) in URGs across pan-cancer cohorts identifies genetically disrupted pathways [5]. The GSCA database is commonly employed to characterize the mutational landscape of URGs and their clinical relevance [13] [22].
Immune Infiltration Quantification
Multiple computational approaches have been developed to quantify immune cell abundances from bulk transcriptomic data and correlate them with URG expression:
Table 2: Experimental Methods for Ubiquitination-Immune Infiltration Analysis
| Method Category | Specific Techniques | Application in Ubiquitination-Immune Analysis | Key Output Metrics |
|---|---|---|---|
| Computational Deconvolution | CIBERSORT, EPIC, TIMER | Estimating immune cell abundances from bulk tumor RNA-seq [13] [56] | Immune cell fractions, correlation with URG expression |
| Pathway Activity Assessment | GSVA, ssGSEA | Quantifying activity of immune-related pathways [5] [56] | Pathway enrichment scores, immune function activity |
| Spatial Analysis | Spatial transcriptomics, Immunofluorescence | Mapping URG expression in tissue architecture [7] [57] | Spatial localization of URGs in tumor vs. stromal regions |
| Functional Validation | CRISPR screens, In vitro co-culture | Testing causal relationships between URGs and immune phenotypes [56] | Immune cell-mediated killing, cytokine production |
The CIBERSORT algorithm is widely applied to estimate the relative proportions of 22 immune cell types based on gene expression signatures [13] [22]. This approach has revealed, for instance, that specific E3 ligases in LUAD show negative correlations with B cells and dendritic cells while demonstrating positive associations with neutrophil infiltration [38]. Single-sample gene set enrichment analysis (ssGSEA) further quantifies the activity of immune-related pathways and functions, allowing researchers to connect URG expression with specific immune processes [56].
Functional Validation Experiments
In vitro and in vivo models provide mechanistic insights into how URGs modulate immune cell function:
Immune Cell Culture Models: Co-culture systems incorporating tumor cells with peripheral blood mononuclear cells (PBMCs) or specific immune cell subsets enable assessment of how URG manipulation affects immune-mediated killing, cytokine production, and immune checkpoint expression [54]. For example, CRISPR-based screening in co-culture models has identified UBE2N as a promoter of immune evasion in LUAD [56].
Animal Models: Syngeneic mouse models allow evaluation of URG manipulation on immune infiltration in an intact tumor microenvironment. In vivo CRISPR screens in these models have proven valuable for identifying URGs that functionally regulate T-cell-mediated tumor control [56].
Molecular Interaction Studies: Immunofluorescence, co-immunoprecipitation, and molecular docking simulations characterize interactions between ubiquitination enzymes and immune signaling components [54] [57]. For instance, molecular docking has been employed to verify the binding capacity of potential therapeutic compounds to E3 ligases like CCNF [57].
Ubiquitination-Mediated Regulation of Immune Cell Function
T Cell Regulation
Ubiquitination exerts profound effects on T cell biology, particularly in regulating the stability and function of key transcription factors and surface receptors:
Diagram 1: E3 Ligase Regulation of FOXP3 in Treg Cells. Multiple E3 ubiquitin ligases differentially regulate FOXP3 stability and function through distinct ubiquitination mechanisms [54].
The stability of FOXP3, the master transcription factor for regulatory T cells (Tregs), is critically regulated by ubiquitination. Different E3 ligases employ distinct mechanisms to control FOXP3 function: STUB1 and CBLB promote FOXP3 degradation, thereby weakening Treg-mediated immunosuppression [54]. Conversely, RNF31 stabilizes FOXP3 through M1-linked linear ubiquitination, while Itch enhances FOXP3 nuclear localization via K63-linked ubiquitination [54]. The balance between these opposing forces determines Treg abundance and function within the TIME, significantly influencing anti-tumor immunity.
Innate Immune Cell Modulation
Ubiquitination pathways extensively regulate innate immune cells including dendritic cells, macrophages, and neutrophils:
Myeloid Cell Function: E3 ligases such as CUL4B regulate epigenetic modifications in myeloid cells, leading to altered expression of genes controlling cell growth and migration [54]. The ubiquitination status of cGAS and STING components critically determines innate immune sensing capabilities and subsequent dendritic cell activation [48].
Neutrophil Infiltration: In lung adenocarcinoma, E3 ligases including CDC20 demonstrate positive correlations with neutrophil infiltration, suggesting their role in recruiting or modulating these innate immune cells within tumors [38].
Therapeutic Implications and Research Reagents
Experimental Toolkit for Ubiquitination-Immune Studies
Table 3: Essential Research Reagents for Ubiquitination-Immune Infiltration Studies
| Reagent Category | Specific Examples | Research Application | Key Function |
|---|---|---|---|
| Databases | TCGA, GTEx, GEO, TISCH, UALCAN | Bioinformatics analysis of URG expression and immune correlation [13] [56] | Provide omics datasets, clinical annotations, and analysis tools |
| Cell Lines | Huh7, Hep3B (HCC); A549, H1299 (LUAD) | In vitro functional validation of URG manipulation [7] [56] | Model tumor cell-immune interactions in relevant cancer types |
| Antibodies | Anti-UBE2C, Anti-CDC20, Anti-FOXP3, Anti-CD markers | IHC, IF, Western blot, flow cytometry [7] [57] | Detect protein expression, localization, and immune cell identification |
| Genetic Tools | shRNA plasmids, CRISPR/Cas9 systems | URG knockdown/knockout studies [7] [56] | Establish causal relationships between URGs and immune phenotypes |
| Chemical Inhibitors | P5091 (USP7 inhibitor), HOIPIN-8 (RNF31 inhibitor) | Functional perturbation of ubiquitination pathways [54] | Test therapeutic targeting of specific ubiquitination enzymes |
Therapeutic Targeting Strategies
The growing understanding of ubiquitination in immune regulation has spurred development of several therapeutic approaches:
PROTACs Technology: Proteolysis Targeting Chimeras (PROTACs) represent an innovative strategy that harnesses E3 ubiquitin ligases to selectively degrade target proteins of interest [54]. These bifunctional molecules simultaneously bind to target proteins and E3 ligases, facilitating ubiquitination and degradation of specific oncoproteins or immune regulators.
Combination Immunotherapies: Targeting ubiquitination pathways alongside immune checkpoint blockade shows synergistic potential. For instance, USP5 inhibition enhances response to PD-(L)1 blockade by stabilizing YTHDF1 and modulating mTORC1 signaling [54]. Similarly, targeting E3 ligases that regulate PD-L1 stability, such as SPOP and FBXO22, may improve efficacy of existing immunotherapies [54].
Small Molecule Inhibitors: Developing specific inhibitors against oncogenic URGs represents another promising avenue. For example, three potential UBE2N-inhibiting compounds have been identified that suppress tumor cell viability and induce apoptosis in lung adenocarcinoma models [56].
Integrative analysis of ubiquitination patterns and immune infiltration provides critical insights into tumor immunology, revealing complex regulatory networks that connect protein modification systems with anti-tumor immunity. The consistent overexpression of specific URGs across cancer types, coupled with their associations with immune cell abundances and patient outcomes, highlights their potential as both prognostic biomarkers and therapeutic targets. Future research should focus on developing more selective modulators of ubiquitination pathways and validating their efficacy in combination with existing immunotherapies. The continuing refinement of multi-omics technologies and single-cell resolution approaches will further illuminate the precise mechanisms through which ubiquitination shapes the tumor immune landscape, ultimately guiding more effective cancer treatments.
Ubiquitination, a critical post-translational modification, has emerged as a pivotal regulator of oncogenesis and tumor suppression through its control of protein stability, activity, and localization. This process involves a coordinated enzymatic cascade comprising E1 activating, E2 conjugating, and E3 ligase enzymes, which conjugate ubiquitin molecules to target proteins, thereby influencing their degradation, signaling functions, and interactions [7]. The integration of pathway enrichment analysis with multi-omics datasets has revolutionized our understanding of how ubiquitination pathways are systematically altered in cancer tissues compared to normal counterparts. These computational approaches enable researchers to identify key biological processes, signaling networks, and cancer hallmarks that are controlled by ubiquitin-mediated regulation.
Mounting evidence demonstrates that ubiquitination significantly impacts multiple cancer hallmarks, including sustained proliferation, evasion of growth suppression, resistance to cell death, and activation of invasion and metastasis [7]. For instance, in hepatocellular carcinoma (HCC), ubiquitination-related genes are significantly upregulated in tumor tissues and correlate strongly with poor patient prognosis [7]. Similarly, in esophageal squamous cell carcinoma (ESCC), the mitochondrial E3 ubiquitin ligase RNF185 promotes cancer progression by regulating BAK1 ubiquitination, thereby maintaining mitochondrial integrity and preventing apoptosis [58]. The strategic application of pathway enrichment methods to ubiquitination datasets provides powerful insights into these cancer-specific regulatory mechanisms, offering potential avenues for therapeutic intervention.
Comparative Analysis of Pathway Enrichment Methods
Pathway enrichment analysis represents a cornerstone of bioinformatics approaches for interpreting large-scale omics data in cancer research. These methods statistically identify biological pathways that are significantly enriched with genes or proteins showing differential expression or modification in cancer states. When applied to ubiquitination datasets, these tools help unravel the complex regulatory networks that drive oncogenesis.
g:Profiler provides a comprehensive web server for functional enrichment analysis and gene list conversions, offering access to diverse annotation sources including Gene Ontology (GO), Molecular Signature Database (MsigDB), Reactome, Panther, KEGG, PathwayCommons, and WikiPathways [59]. Its user-friendly interface makes it particularly accessible for experimental biologists without programming expertise. The tool supports statistical correction for multiple testing and provides interactive visualizations of enrichment results, facilitating the identification of ubiquitination-related pathways altered in cancer tissues.
Gene Set Enrichment Analysis (GSEA) employs a fundamentally different approach that considers the entire expression dataset rather than just a pre-filtered list of significant genes [59]. This method ranks all genes based on their correlation with a phenotypic difference (e.g., cancerous vs. normal tissue) and then tests whether genes from predefined sets (e.g., ubiquitination pathways) are non-randomly distributed toward the top or bottom of this ranked list. GSEA is especially valuable for detecting subtle but coordinated changes in ubiquitination-related pathways that might be missed by threshold-based approaches.
ActiveDriver represents a specialized method for identifying post-translational modification sites, including ubiquitination and acetylation sites, that are significantly mutated in cancer [60]. This method integrates protein sequence features with mutation data to discover regulatory elements and protein interaction networks disrupted in tumors. Unlike traditional frequency-based approaches, ActiveDriver can identify significant co-occurrences of mutations in PTM sites even when mutation frequency is low, providing mechanistic hypotheses for how these mutations drive cancer progression [60].
Table 1: Comparison of Pathway Enrichment Methods for Ubiquitination Research
| Method | Primary Approach | Key Ubiquitination Applications | Statistical Strengths | Visualization Capabilities |
|---|---|---|---|---|
| g:Profiler | Overrepresentation analysis of gene lists | Functional interpretation of ubiquitinated protein lists | Multiple testing correction, integration of multiple databases | Interactive graphs, static diagrams |
| GSEA | Genome-wide rank-based enrichment | Identifying coordinated changes in ubiquitination pathways | No arbitrary significance thresholds, sensitive to subtle effects | Enrichment plots, network visualizations |
| ActiveDriver | PTM site-specific mutation analysis | Discovering mutated ubiquitination sites in cancer | Accounts for protein sequence features, mutation clustering | Protein diagrams with mutation hotspots |
Performance Metrics and Experimental Validation
The analytical performance of pathway enrichment methods must be rigorously evaluated through both computational metrics and experimental validation. In practical applications, these tools have demonstrated significant utility in identifying ubiquitination-related pathways dysregulated across multiple cancer types.
Comparative studies have shown that integrated approaches combining multiple enrichment methods yield the most biologically insightful results. For example, a multi-omics analysis of hepatocellular carcinoma integrated datasets from public repositories including TCGA and ICGC, applying both GSEA and g:Profiler to ubiquitination-related gene sets [7]. This approach revealed significant enrichment in critical cancer pathways including cell cycle regulation, DNA repair, metabolic reprogramming, and p53 signaling – all processes known to be regulated by ubiquitination. The findings were subsequently validated through experimental techniques including shRNA knockdown, transwell assays, and CCK-8 proliferation assays, which confirmed the functional role of identified ubiquitination genes in promoting HCC progression [7].
In pan-cancer analyses examining acetylation and ubiquitination site mutations across 3,200 tumor samples from The Cancer Genome Atlas, ActiveDriver identified significant mutation hotspots in known oncoproteins (TP53, AKT1, IDH1) and revealed candidate cancer driver genes with PTM-related mechanisms [60]. Pathway analysis further demonstrated that PTM mutations in ubiquitination sites accumulate in cancer-related processes including cell cycle, apoptosis, chromatin regulation, and metabolism [60]. These computational predictions were strengthened by clinical correlation analyses showing that many PTM-specific mutations associate with decreased patient survival across multiple cancer types.
Table 2: Experimental Validation Approaches for Ubiquitination Pathway Findings
| Validation Method | Application in Ubiquitination Studies | Key Readouts | Typical Workflow |
|---|---|---|---|
| shRNA/siRNA Knockdown | Functional validation of ubiquitination-related genes | Changes in proliferation, invasion, apoptosis | Design targeting sequences → Viral transduction → Selection → Phenotypic assays |
| CCK-8 Assay | Measuring cell viability after ubiquitination perturbation | Optical density at 450nm reflecting metabolic activity | Seed cells in 96-well plates → Add CCK-8 reagent → Incubate 4h → Measure absorbance |
| Transwell Assay | Assessing migration and invasion capabilities | Cell counts through membrane | Seed cells in serum-free medium in upper chamber → Culture 24h → Fix, stain, count |
| Wound Healing Assay | Measuring two-dimensional cell migration | Migration distance over time | Create scratch with pipette tip → Image at 0h and 24h → Quantify closure |
| Western Blot Analysis | Confirming protein expression and ubiquitination status | Protein levels, ubiquitin conjugation | Cell lysis → SDS-PAGE → Transfer to PVDF → Antibody incubation → Detection |
Experimental Design for Ubiquitination Pathway Analysis
Integrated Multi-Omics Workflow
A robust experimental framework for analyzing ubiquitination patterns in cancer versus normal tissues requires the integration of multiple complementary approaches. The following workflow diagram illustrates a comprehensive strategy that combines bioinformatics analysis with experimental validation to identify and verify cancer hallmarks regulated by ubiquitination:
Detailed Methodological Protocols
Ubiquitinome Profiling and Enrichment Analysis
The experimental workflow begins with comprehensive ubiquitinome profiling using affinity purification methods coupled with mass spectrometry to identify ubiquitination sites differentially regulated between cancerous and normal tissues. Following data acquisition, bioinformatic analysis proceeds through several methodical stages:
Differential Expression Analysis: Initial processing identifies differentially expressed genes (DEGs) and ubiquitinated proteins using tools like BioJupies for RNA-seq data or GEO2R for microarray datasets [59]. Statistical thresholds typically include adjusted p-values <0.05 and absolute log fold change >1-2, depending on the experimental design and sample size. For ubiquitination-specific analysis, focus is placed on genes involved in the ubiquitin-proteasome system, including E1, E2, and E3 enzymes, deubiquitinases (DUBs), and ubiquitin receptors.
Pathway Enrichment Execution: The resulting gene lists serve as input for pathway enrichment tools. For g:Profiler analysis, parameters include statistical scope set to "annotated genes," significance threshold of FDR <0.05, and inclusion of multiple data sources (GO, KEGG, Reactome, etc.) [59]. For GSEA, analysis employs 1,000 permutations, weighted enrichment statistic, and signal-to-noise ratio as the ranking metric. The Molecular Signatures Database (MsigDB) provides ubiquitination-relevant gene sets, including those for proteasome degradation, ubiquitin ligase complexes, and protein polyubiquitination.
Multi-omics Integration: Advanced analyses integrate ubiquitination data with other molecular layers, such as genomic mutations from whole-exome sequencing and transcriptomic profiles from RNA-seq. This integrated approach can reveal connections between mutation events in ubiquitination sites and pathway alterations, as demonstrated in pan-cancer studies that found significant accumulation of mutations in acetylation and ubiquitination sites affecting cancer-related processes [60].
Functional Validation Experiments
Candidate pathways and ubiquitination targets identified through enrichment analysis require rigorous experimental validation to confirm their biological relevance and functional roles in cancer progression:
Genetic Perturbation Experiments: shRNA or CRISPR/Cas9-mediated knockout of key ubiquitination-related genes is performed to assess functional consequences. As exemplified in ESCC research, RNF185 knockout cell lines generated using CRISPR/Cas9 demonstrated suppressed cell proliferation, induced apoptosis, and mitochondrial damage, validating its role in cancer progression [58]. Transfection protocols typically use lipofectamine-based methods with puromycin selection for stable cell lines, followed by confirmation of knockdown efficiency via qPCR and Western blot.
Phenotypic Assays: Multiple functional assays evaluate the impact of ubiquitination perturbations on cancer hallmarks:
- CCK-8 assays measure cell viability, with cells seeded in 96-well plates at 2,500 cells/well and absorbance measured at 450nm after 4 hours of CCK-8 reagent incubation [7].
- Transwell assays assess migration and invasion capabilities using chambers coated with (invasion) or without (migration) Matrigel, with cells fixed after 24 hours and stained with crystal violet for quantification [7].
- Wound healing assays evaluate two-dimensional migration by creating scratches with pipette tips and monitoring closure over 24 hours [7].
- Colony formation assays determine long-term proliferative potential by seeding 600 cells in 6-well plates and allowing 14 days for colony formation before staining and counting [7].
Molecular Mechanism Elucidation: Co-immunoprecipitation assays investigate protein-protein interactions and ubiquitination status, as demonstrated in ESCC studies showing RNF185-mediated BAK1 ubiquitination [58]. Western blot analysis examines activation of relevant signaling pathways using phospho-specific antibodies. For mitochondrial-related mechanisms, transmission electron microscopy (TEM) assesses ultrastructural changes, while ELISA measures mtDNA release into the cytosol [58].
Key Signaling Pathways in Ubiquitination-Driven Carcinogenesis
Pathway enrichment analyses applied to ubiquitination datasets have consistently identified several crucial cancer-related pathways that are frequently regulated by ubiquitin-mediated mechanisms. The following diagram illustrates the core signaling circuitry through which ubiquitination governs cancer hallmarks:
Experimentally Validated Ubiquitination Pathways
The integration of pathway enrichment analysis with experimental validation has elucidated several key mechanisms through which ubiquitination drives carcinogenesis:
p53 Pathway Regulation: Multiple enrichment analyses have identified p53 signaling as a central pathway regulated by ubiquitination in cancer [7] [60]. The E3 ligase MDM2 mediates ubiquitin-dependent degradation of p53, effectively suppressing its tumor suppressor functions. Additionally, deubiquitinating enzymes such as USP7 and USP10 can stabilize p53 by removing ubiquitin chains [7]. In hepatocellular carcinoma, ubiquitination-related genes show significant enrichment in p53 signaling pathways, and their high expression correlates with poor prognosis [7].
Mitochondrial Apoptosis Regulation: Research in esophageal squamous cell carcinoma has revealed a novel mechanism whereby the E3 ligase RNF185 promotes cancer progression by regulating BAK1 ubiquitination [58]. RNF185 maintains mitochondrial integrity by targeting BAK1, a pro-apoptotic protein, for ubiquitination. Loss of RNF185 leads to BAK1 accumulation, mitochondrial outer membrane permeabilization, and release of mitochondrial DNA, which activates the cGAS-STING-IRF3 pathway and promotes apoptosis [58]. This pathway represents a critical link between ubiquitination, mitochondrial homeostasis, and immune signaling in cancer.
Cell Cycle Control: Ubiquitination mechanisms exert precise control over cell cycle progression through regulation of cyclins, cyclin-dependent kinases (CDKs), and CDK inhibitors. Pathway enrichment analyses consistently identify cell cycle as a top pathway regulated by ubiquitination-related genes [7] [60]. The anaphase-promoting complex/cyclosome (APC/C), a multi-subunit E3 ubiquitin ligase, targets key cell cycle regulators for degradation, ensuring proper progression through mitosis. Dysregulation of these ubiquitination mechanisms contributes to uncontrolled proliferation in cancer.
Metabolic Reprogramming: Cancer cells undergo metabolic adaptations to support rapid growth and proliferation, and ubiquitination plays a crucial role in regulating these processes. Enrichment analyses have revealed significant involvement of ubiquitination-related genes in metabolic pathways, including glucose metabolism, lipid metabolism, and oxidative phosphorylation [7]. Ubiquitination regulates the stability and activity of metabolic enzymes and transporters, enabling cancer cells to adapt to nutrient availability and metabolic demands.
Research Reagent Solutions for Ubiquitination Studies
The experimental approaches discussed require specialized reagents and tools designed specifically for ubiquitination research. The following table details essential research reagent solutions for investigating ubiquitination pathways in cancer:
Table 3: Essential Research Reagents for Ubiquitination Pathway Analysis
| Reagent/Tool | Specific Application | Key Features | Example Use Cases |
|---|---|---|---|
| CRISPR/Cas9 Knockout Systems | Genetic perturbation of ubiquitination-related genes | Specific sgRNAs targeting E3 ligases, DUBs; lentiCRISPR v2 vector; puromycin selection | Generation of RNF185 knockout ESCC cell lines to study BAK1 ubiquitination [58] |
| Ubiquitination Site Enrichment Kits | Affinity purification of ubiquitinated proteins | Anti-ubiquitin antibodies; ubiquitin remnant motifs; compatibility with mass spectrometry | Ubiquitinome profiling to identify differentially ubiquitinated sites in cancer vs normal tissues |
| Pathway Enrichment Software | Bioinformatics analysis of ubiquitination pathways | g:Profiler, GSEA, ActiveDriver; multiple database integration; statistical significance testing | Identifying enriched pathways in ubiquitination-related gene lists from HCC datasets [59] [7] |
| Protein Interaction Assays | Studying ubiquitin-mediated protein complexes | Co-immunoprecipitation kits; crosslinkers; ubiquitin binding domains | Validating RNF185-BAK1 interaction in ESCC [58] |
| PTM-Specific Antibodies | Detection of ubiquitination events | Anti-ubiquitin; linkage-specific antibodies (K48, K63); phospho-ubiquitin antibodies | Western blot analysis of BAK1 ubiquitination status in mitochondrial fractions [58] |
| Activity-Based Probes | Monitoring deubiquitinase activity | Ubiquitin-based probes with warhead groups; fluorescent or biotin tags | Screening for DUB activity changes in cancer cells |
| Ubiquitination Assay Kits | In vitro ubiquitination reactions | Purified E1, E2, E3 enzymes; ubiquitin; ATP regeneration system | Reconstituting ubiquitination cascades for mechanistic studies |
The integration of pathway enrichment methods with multi-omics datasets and experimental validation provides a powerful framework for deciphering the complex roles of ubiquitination in cancer biology. Approaches such as g:Profiler, GSEA, and ActiveDriver each offer unique strengths for identifying cancer hallmarks regulated by ubiquitination, from overall pathway alterations to specific mutation hotspots in ubiquitination sites. The consistent identification of key pathways – including p53 signaling, cell cycle regulation, mitochondrial apoptosis, and metabolic reprogramming – across multiple cancer types highlights the fundamental importance of ubiquitination in oncogenesis.
As research in this field advances, the application of these methods to increasingly comprehensive datasets will undoubtedly reveal additional ubiquitination-dependent mechanisms in cancer. The continuing development of specialized reagents and tools for ubiquitination research will further enhance our ability to experimentally validate computational predictions. Ultimately, this integrated approach promises to identify novel therapeutic targets within the ubiquitin-proteasome system, potentially leading to more effective strategies for cancer treatment based on precision targeting of ubiquitination pathways.
Addressing Technical Challenges in Ubiquitination Research and Data Interpretation
Overcoming Artifacts in Ubiquitin Enrichment and Mass Spectrometry Detection
The precise characterization of the ubiquitinome is fundamental to advancing our understanding of cellular regulation in health and disease. Within cancer research, delineating ubiquitination patterns that distinguish cancerous from normal tissues offers immense potential for discovering novel therapeutic targets and biomarkers. However, the path to obtaining high-fidelity ubiquitinome data is fraught with technical challenges. Artifacts introduced during sample preparation, enrichment, and mass spectrometry analysis can severely compromise data accuracy, leading to false discoveries and erroneous biological conclusions. This guide objectively compares the performance of current mainstream methodologies for ubiquitin enrichment and detection, providing a structured framework for selecting optimal protocols to overcome these artifacts in the context of cancer ubiquitinome profiling.
Methodological Comparison: Performance and Pitfalls
The core strategies for enriching ubiquitinated peptides each present a unique balance of advantages and inherent limitations, which directly impact their suitability for sensitive comparative studies. The following table summarizes the key characteristics of these primary approaches.
Table 1: Comparison of Primary Ubiquitin Enrichment Methodologies
| Enrichment Method | Principle | Key Artifacts & Limitations | Best-Suited Applications |
|---|---|---|---|
| Ubiquitin Tagging [61] | Expression of affinity-tagged (e.g., His, Strep) ubiquitin in cells. Enrichment via tag-specific resins. | Co-purification of endogenous His-rich/biotinylated proteins; inability to mimic endogenous Ub perfectly; infeasible for patient tissues [61]. | Discovery-based profiling in engineered cell lines; relatively low-cost initial screens [61]. |
| Antibody-Based (K-ε-GG) [62] | Immunoaffinity purification of tryptic peptides containing the diGly remnant motif. | Antibody cross-reactivity; potential co-enrichment of non-specific peptides; high cost of high-quality antibodies [61] [63]. | Deep ubiquitinome profiling from any biological source, including clinical and tissue samples; linkage-specific analysis with dedicated antibodies [61] [62]. |
| UBD-Based (TUBEs) [61] | Use of Tandem Ubiquitin-Binding Entities for high-affinity enrichment of endogenous ubiquitinated proteins. | Potential linkage preference of certain UBDs; may enrich for non-covalently bound ubiquitin complexes [61]. | Studying endogenous ubiquitination under physiological conditions; stabilizing the ubiquitinome by protecting against DUBs [61]. |
Quantitative Performance Data in Ubiquitinomics
The transition from traditional Data-Dependent Acquisition (DDA) to modern Data-Independent Acquisition (DIA) mass spectrometry represents a significant leap in overcoming analytical artifacts. The quantitative data below highlight the performance disparities between these approaches and the impact of sample preparation.
Table 2: Quantitative Comparison of MS Acquisition and Lysis Methods
| Parameter | Data-Dependent Acquisition (DDA) | Data-Independent Acquisition (DIA) | Notes & Experimental Context |
|---|---|---|---|
| Typical K-GG Peptide ID (Single Run) | ~21,434 peptides [64] | ~68,429 peptides [64] | From proteasome inhibitor-treated HCT116 cells; 75-min gradient. |
| Quantitative Reproducibility (Median CV) | >20% [64] | ~10% [64] | Coefficient of Variation (CV) across replicates. |
| Missing Values in Replicates | ~50% of IDs [64] | Strongly reduced [64] | DIA's comprehensive sampling minimizes missing data. |
| Lysis Buffer: K-GG Peptide Yield | Urea: ~19,403 peptides [64] | SDC-based: ~26,756 peptides [64] | Compared from same HCT116 input; SDC boosts yield without sacrificing specificity [64]. |
Detailed Experimental Protocols
Adherence to optimized and meticulously detailed protocols is critical for minimizing variability and artifacts. Below are the core methodologies for the most robust current workflow.
Optimized SDC-Based Lysis and Protein Digestion
This protocol is designed for maximum protein extraction and efficient digestion while minimizing deubiquitinase activity and alkylation artifacts [64].
- Cell Lysis: Lyse cell pellets or ground tissue in ice-cold lysis buffer containing 50 mM Tris-HCl (pH 8.2), 0.5% Sodium Deoxycholate (SDC), and 10-40 mM Chloroacetamide (CAA). Immediate boiling for 5 minutes is crucial to rapidly inactivate DUBs [64].
- Protein Quantitation: Determine protein concentration using a colorimetric assay (e.g., BCA assay).
- Reduction and Alkylation: Reduce disulfide bonds with 5 mM dithiothreitol (30 min, 50°C). Alkylate free cysteines with the provided 10 mM CAA (15 min, room temperature in the dark). The use of CAA, instead of iodoacetamide, prevents di-carbamidomethylation of lysines, a modification that can mimic the K-ε-GG mass shift and create artifacts [64].
- Protein Digestion: First, digest with Lys-C (1:200 enzyme-to-substrate ratio) for 4 hours. Follow with an overnight digestion with trypsin (1:50 enzyme-to-substrate ratio) at 30°C [62].
- Peptide Cleanup: Acidify the digest to a final concentration of 0.5-1% Trifluoroacetic Acid (TFA) to precipitate SDC. Centrifuge at 10,000 x g for 10 minutes and collect the supernatant containing the peptides [62].
High-pH Fractionation and diGly Peptide Immunoenrichment
Prefractionation prior to enrichment dramatically reduces sample complexity, leading to deeper ubiquitinome coverage [62].
- Offline High-pH Reversed-Phase Fractionation:
- Use a C18 column with a polymeric stationary phase (300 Å pore size). A bed size of 0.5 g of resin per 10 mg of peptide digest is recommended [62].
- Load the acidified peptide supernatant onto the column.
- Wash with 10 column volumes of 0.1% TFA, followed by 10 volumes of H₂O.
- Elute peptides step-wise with 10 mM ammonium formate (pH 10) containing increasing concentrations of acetonitrile (e.g., 7%, 13.5%, and 50%). Pool and lyophilize fractions as needed [62].
- diGly Peptide Immunoprecipitation:
- Use commercial K-ε-GG motif-specific antibodies conjugated to protein A agarose beads.
- Resuspend lyophilized peptides in immunoaffinity purification buffer.
- Incubate the peptide mixture with the antibody beads for several hours at 4°C.
- Wash beads stringently to remove non-specifically bound peptides.
- Elute diGly peptides with a low-pH buffer (e.g., 0.1-0.5% TFA). Desalt the eluate using C18 StageTips before MS analysis [62].
DIA-MS Analysis and Data Processing
This protocol leverages DIA to achieve high coverage, reproducibility, and quantitative accuracy [64].
- Mass Spectrometry Acquisition:
- Use a nanoLC system coupled to a high-resolution tandem mass spectrometer capable of DIA.
- Employ a 75-125 min reversed-phase acetonitrile gradient for peptide separation.
- For DIA, set up a sequential window acquisition method that covers the entire precursor mass range (e.g., 400-1000 m/z) with window widths of 10-25 m/z [64].
- Data Processing with DIA-NN:
- Process the raw DIA data using the DIA-NN software suite in "library-free" mode.
- Search the data against the appropriate protein sequence database.
- Utilize the built-in neural network-based scoring to confidently identify and quantify K-GG modified peptides, ensuring a controlled false discovery rate (e.g., <1%) [64].
Visualizing the Optimized Ubiquitinomics Workflow
The following diagram synthesizes the core steps of the optimized protocol, highlighting key decision points for artifact mitigation.
The Scientist's Toolkit: Essential Research Reagents
Successful execution of the ubiquitinomics workflow depends on a set of key reagents, each with a specific function to ensure data quality.
Table 3: Essential Reagents for Ubiquitin Enrichment and Detection
| Research Reagent | Critical Function | Considerations for Artifact Prevention |
|---|---|---|
| K-ε-GG Motif Antibodies | Immunoaffinity purification of ubiquitinated peptides from tryptic digests [62]. | Quality and specificity vary by vendor; linkage-specific antibodies (e.g., K48, K63) enable targeted studies [61]. |
| Sodium Deoxycholate (SDC) | Powerful anionic detergent for efficient protein extraction and solubilization [64]. | Compatible with MS; precipitates in acid for easy removal, reducing MS ion suppression [64]. |
| Chloroacetamide (CAA) | Alkylating agent that covalently modifies cysteine residues [64]. | Preferred over iodoacetamide to avoid di-carbamidomethylation of lysines, a reaction that mimics the K-ε-GG mass shift [64]. |
| TUBEs (Tandem Ubiquitin-Binding Entities) | High-affinity capture of endogenous polyubiquitinated proteins, protecting them from DUBs [61]. | Can be used to stabilize the native ubiquitinome before moving to denaturing conditions and digestion [61]. |
| DIA-NN Software | Deep neural network-based data processing for DIA-MS data [64]. | Library-free mode allows deep analysis without prior fractionation, streamlining the workflow and improving reproducibility [64]. |
The choice of methodology in ubiquitinomics directly dictates the reliability and biological relevance of the findings, especially in the complex endeavor of comparing cancerous and normal tissues. While antibody-based diGly enrichment coupled with DIA-MS currently stands as the most robust and sensitive platform for large-scale studies, the integration of SDC-based lysis, CAA alkylation, and strategic prefractionation is non-negotiable for mitigating pervasive artifacts. By adopting this optimized, integrated workflow, researchers can generate ubiquitinome data of the highest quantitative quality, paving the way for the confident discovery of cancer-specific ubiquitination signatures with true diagnostic and therapeutic potential.
Ubiquitination is a critical post-translational modification that regulates virtually all cellular processes in eukaryotes. The covalent attachment of ubiquitin to substrate proteins can signal for proteasomal degradation or alter a protein's activity, localization, and interactions. Ubiquitin itself contains seven lysine residues (K6, K11, K27, K29, K33, K48, and K63) and an N-terminal methionine residue (M1) that can form polyubiquitin chains with distinct structures and functions [12] [65]. Among these, K48-linked ubiquitination primarily targets substrates for proteasomal degradation, representing the best-characterized ubiquitin signal. In contrast, K63-linked ubiquitination typically serves non-proteolytic functions regulating protein-protein interactions, kinase activation, intracellular trafficking, and DNA repair [66] [67]. The diversity of ubiquitin linkages creates a complex ubiquitin code that dictates specific cellular outcomes, with K48 and K63 linkages representing the most extensively studied and functionally distinct chain types.
In cancer biology, the balance between different ubiquitin linkages is frequently disrupted, contributing to tumor initiation, progression, and therapeutic resistance. Dysregulation of K48 and K63 linkages affects key oncogenic signaling pathways, including p53 stabilization, PI3K/Akt activation, and NF-κB signaling [68] [66]. The validation of specific ubiquitin linkage types has therefore become essential for understanding molecular mechanisms in cancer and developing targeted therapies. This guide provides a comprehensive comparison of experimental approaches for distinguishing between K48 and K63 ubiquitination patterns, with particular emphasis on applications in cancer research.
Methodologies for Validating K48 and K63 Ubiquitin Linkages
Linkage-Specific Antibodies and Immunoaffinity Enrichment
The most accessible method for detecting specific ubiquitin linkages employs linkage-specific antibodies that recognize the unique structural features of K48- or K63-linked ubiquitin chains. These antibodies enable various experimental techniques including western blotting, immunohistochemistry, and immunofluorescence. For enhanced detection sensitivity, researchers typically combine linkage-specific antibodies with immunoaffinity purification prior to analysis.
Table 1: Linkage-Specific Antibody Applications in Ubiquitin Research
| Application | Key Advantage | Limitation | Common Use in Cancer Research |
|---|---|---|---|
| Western Blotting | Semi-quantitative analysis of chain abundance | Cannot identify substrate proteins | Monitoring global ubiquitination changes in tumor vs. normal tissue |
| Immunohistochemistry | Spatial context within tissue architecture | Semi-quantitative | Correlation of ubiquitination patterns with clinical pathology |
| Immunofluorescence | Subcellular localization | Resolution limits for precise compartmentalization | Studying nuclear vs. cytoplasmic ubiquitination in cancer cells |
| Immunoprecipitation | Enrichment of specific linkage types | May not capture transient modifications | Pre-enrichment for proteomic analysis of ubiquitinated substrates |
The experimental workflow typically involves: (1) Cell lysis under denaturing conditions to preserve ubiquitination states and prevent deubiquitinase activity; (2) Immunoaffinity enrichment using linkage-specific antibodies conjugated to beads; (3) Stringent washing to remove non-specifically bound proteins; and (4) Downstream analysis including western blotting or mass spectrometry [68]. For tissue samples, proper fixation and antigen retrieval are critical for maintaining epitope integrity. Validation experiments should include controls with recombinant ubiquitin chains of known linkage to confirm antibody specificity.
Mass Spectrometry-Based Ubiquitinomics
Mass spectrometry represents the gold standard for comprehensive mapping of ubiquitination sites and linkage types. Advanced liquid chromatography-tandem mass spectrometry (LC-MS/MS) enables system-wide identification of ubiquitination events with precise linkage determination. The typical workflow involves: (1) Trypsin digestion of protein samples, which cleaves ubiquitin after arginine 74, leaving a di-glycine remnant on modified lysines; (2) Peptide enrichment using di-glycine-lysine-specific antibodies; (3) LC-MS/MS analysis with high-resolution mass accuracy; and (4) Bioinformatic processing to identify modified sites and quantify changes between samples [30].
In a comprehensive characterization of the colorectal cancer ubiquitinome, researchers identified 1,690 quantifiable ubiquitination sites and 870 quantifiable proteins using LC-MS/MS [30]. This approach revealed that highly ubiquitinated proteins (with ≥10 modification sites) were specifically involved in G-protein coupling, glycoprotein coupling, and antigen presentation. The study also identified five motif sequences frequently recognized by ubiquitin machinery in colorectal cancer cells, providing insights into substrate recognition patterns. For linkage-specific analysis, parallel reaction monitoring (PRM) methods can quantify predefined ubiquitin linkages with high sensitivity and reproducibility, while data-independent acquisition (DIA) approaches provide untargeted profiling of ubiquitin chain types.
Genetic and Chemical Biology Approaches
Genetic manipulation of ubiquitin system components provides functional validation of specific linkage types. Key approaches include:
- Expression of ubiquitin mutants where all lysine residues except one are mutated to arginine, forcing chain formation through a specific linkage [67]
- Knockdown or knockout of specific E2 enzymes or E3 ligases that preferentially catalyze certain linkage types
- Modulation of deubiquitinases (DUBs) with specificity for particular ubiquitin linkages
Chemical biology tools have greatly expanded the ubiquitin research arsenal. Activity-based probes can profile DUB activities with linkage specificity, while ubiquitin chain-specific inhibitors enable functional dissection of particular chain types. For example, the DUB CYLD specifically processes K63 linkages and linear ubiquitin chains, while USP14 primarily removes K48-linked chains [67]. The development of linkage-specific engineered DUBs as research reagents has provided powerful tools for validating ubiquitin chain types in cellular contexts.
Diagram Title: Experimental Framework for Ubiquitin Linkage Validation
Comparative Analysis of K48 and K63 Linkages in Cancer Signaling
Structural and Functional Properties
K48 and K63-linked ubiquitin chains exhibit distinct structural configurations that determine their functional specificity. K48-linked chains adopt compact conformations that are recognized by proteasomal receptors, facilitating substrate degradation. In contrast, K63-linked chains form more open, extended structures that serve as scaffolds for protein complex assembly and signaling activation [67]. These structural differences underlie their divergent roles in cellular regulation and cancer pathogenesis.
Table 2: Functional Comparison of K48 and K63 Ubiquitin Linkages
| Characteristic | K48-Linked Ubiquitination | K63-Linked Ubiquitination |
|---|---|---|
| Primary Function | Proteasomal degradation | Signaling regulation, endocytosis, DNA repair |
| Chain Structure | Compact, closed conformation | Extended, open conformation |
| Cellular Outcomes | Protein turnover, homeostasis | Kinase activation, complex assembly, trafficking |
| Key E2 Enzymes | UBE2D, UBE2R1, UBE2G1 | UBE2N/UEV1A complex, UBE2V1/UEV2 |
| Key E3 Ligases | MDM2, β-TrCP, HUWE1 | TRAF6, cIAP1/2, TRIP12, Pellino-1 |
| Specific DUBs | USP14, OTUB1, OTUD5 | CYLD, TRABID, OTUD1 |
| Role in Cancer | Tumor suppressor inactivation, oncogene stabilization | Oncogenic signaling activation, metastasis promotion |
Pathway-Specific Roles in Oncogenesis
The functional dichotomy between K48 and K63 linkages is exemplified in key cancer-relevant pathways. In the p53 tumor suppressor pathway, TRIM31 directly interacts with p53 to induce K63-linked ubiquitination while simultaneously inhibiting MDM2-mediated K48-linked ubiquitination, leading to p53 stabilization and activation in breast cancer [68]. This dual mechanism illustrates how coordinated regulation of different ubiquitin linkages controls the activity of critical tumor suppressors. Loss of TRIM31 promotes breast cancer progression by disrupting this balance, highlighting the pathological consequences of linkage-specific ubiquitination defects.
In the PI3K/Akt signaling pathway, K63-linked ubiquitination plays a crucial activating role. Multiple E3 ligases including TRAF6, Skp2, and others mediate K63-linked ubiquitination of Akt, enhancing its membrane localization and phosphorylation [66]. The E3 ligase TRAF2 and deubiquitinating enzyme OTUD7B control a K63-linked polyubiquitination switch of GβL that modulates mTORC2/AKT signaling homeostasis and activation [66]. SETDB1-mediated methylation facilitates K63-linked ubiquitination and activation of Akt, promoting tumor initiation. These regulatory mechanisms demonstrate how K63 linkages directly coordinate pro-survival signaling in cancer cells.
The Wnt/β-catenin pathway similarly employs linkage-specific ubiquitination for precise regulation. While K48-linked ubiquitination typically targets β-catenin for degradation, K63-linked ubiquitination stabilizes and activates β-catenin in cancer contexts. Rad6B mediates K63-linked ubiquitination of β-catenin at K394, regulating its stability and activity in breast cancer [66]. The deubiquitinating enzyme Trabid binds to and deubiquitinates APC, regulating the β-catenin destruction complex and promoting cancers through Wnt signaling enhancement [66].
Cancer-Relevant Case Studies of Linkage-Specific Ubiquitination
TRIM31-p53 Regulation in Breast Cancer
The TRIM31-p53 regulatory axis provides a compelling example of balanced K48 and K63 ubiquitination controlling tumor suppressor activity. In normal breast tissue, TRIM31 is expressed at relatively high levels and maintains p53 activity through a dual mechanism: (1) inducing K63-linked ubiquitination of p53 via its RING domain, and (2) suppressing MDM2-mediated K48-linked ubiquitination through competitive inhibition of MDM2-p53 interaction [68]. This results in p53 stabilization and activation of its tumor suppressor functions.
In breast cancer tissues, TRIM31 expression is significantly downregulated, leading to unbalanced p53 ubiquitination. The loss of TRIM31 shifts the equilibrium toward MDM2-mediated K48-linked ubiquitination, resulting in enhanced p53 degradation [68]. This molecular alteration correlates with larger tumor size, higher Ki67 expression, advanced TNM stage, and lymph node invasion. Experimentally, TRIM31 overexpression suppresses breast cancer cell proliferation, colony formation, migration, and invasion, while its knockdown has opposite effects. These phenotypic consequences are p53-dependent, as p53 knockdown reverses TRIM31-mediated growth and metastasis inhibition.
TRIP12-Mediated EZH2 Regulation in Lymphoma
In extranodal natural killer/T-cell lymphoma (ENKTL), the E3 ligase TRIP12 catalyzes K63-linked ubiquitination of EZH2 at K634, leading to EZH2 stabilization rather than degradation [69]. This non-proteolytic ubiquitination enhances the interaction between EZH2 and its binding partners SUZ12 or CDK1 and increases EZH2 T487 phosphorylation. The stabilized EZH2 promotes lymphoma cell migration through accelerated epithelial-mesenchymal transition (EMT).
This case illustrates several important concepts: (1) K63 ubiquitination can stabilize oncoproteins in specific cancer contexts; (2) the functional outcome of ubiquitination depends on both the linkage type and the modified residue; and (3) ubiquitination can influence protein function through mechanisms beyond degradation, including altered protein interactions and secondary modifications. The TRIP12-EZH2 axis could be targeted therapeutically, as dexamethasone treatment destabilizes both TRIP12 and EZH2 and obstructs ENKTL cell migration [69].
UBE2C in Hepatocellular Carcinoma
UBE2C, an E2 ubiquitin-conjugating enzyme, is significantly upregulated in hepatocellular carcinoma (HCC) tissues, with high expression correlating with poor patient prognosis [7]. Functional studies demonstrate that UBE2C overexpression promotes HCC cell proliferation, invasion, and metastasis. Pathway analysis reveals that UBE2C and other ubiquitination-related genes are enriched in key processes including cell cycle regulation, DNA repair, metabolic reprogramming, and p53 signaling.
UBE2C appears to play an additional role in immune evasion, potentially by inhibiting anti-tumor immune responses and reducing immune system recognition of tumor cells [7]. This example highlights how ubiquitinating enzymes can influence multiple cancer hallmarks simultaneously, and how comprehensive ubiquitinome analyses can identify clinically relevant biomarkers and therapeutic targets.
Diagram Title: K48 and K63 Ubiquitination in Cancer Pathways
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Ubiquitin Linkage Validation
| Reagent Category | Specific Examples | Primary Research Application | Considerations for Use |
|---|---|---|---|
| Linkage-Specific Antibodies | Anti-K48 ubiquitin, Anti-K63 ubiquitin, Anti-linear ubiquitin | Detection and enrichment of specific chain types | Verify specificity with recombinant chains; optimize for different applications (WB, IHC, IP) |
| Ubiquitin Mutants | K48-only (all K except K48 mutated to R), K63-only (all K except K63 mutated to R) | Functional studies of specific linkage types | Confirm expression levels; monitor potential dominant-negative effects |
| Recombinant Ubiquitin Chains | Defined K48-linked chains, Defined K63-linked chains, Mixed linkage chains | Antibody validation, in vitro assays, standards for mass spectrometry | Source from reputable suppliers; verify chain length and purity |
| Activity-Based Probes | Ubiquitin-based probes with warhead groups | DUB activity profiling, linkage specificity assessment | Optimize concentration and incubation time; include appropriate controls |
| E2/E3 Enzyme Modulators | E2 enzyme inhibitors, E3 ligase agonists/antagonists | Functional dissection of ubiquitination pathways | Consider potential off-target effects; use combinatorial approaches for specificity |
| DUB Inhibitors | Linkage-specific DUB inhibitors (e.g., CYLD inhibitors) | Functional validation of specific ubiquitin linkages | Assess selectivity across DUB family members; monitor cellular toxicity |
The validation of specific ubiquitin linkage types, particularly K48 and K63 chains, provides critical insights into molecular mechanisms driving cancer pathogenesis. The distinct structural and functional properties of these linkages enable precise control over protein fate and function, with K48 primarily directing proteasomal degradation and K63 regulating signaling activation and complex assembly. In cancer tissues, the balance between these linkages is frequently disrupted, contributing to tumor suppressor inactivation, oncogene stabilization, and metastatic progression.
Advanced methodologies including linkage-specific antibodies, mass spectrometry-based ubiquitinomics, and genetic and chemical biology approaches enable comprehensive mapping and functional validation of ubiquitin linkages in biological systems. The continuing development of more specific research reagents and analytical methods will further enhance our understanding of the ubiquitin code in cancer biology. As research progresses, targeting specific ubiquitin linkages or their regulatory components holds promise for developing novel cancer therapeutics that restore normal ubiquitination patterns in malignant cells.
Distinguishing Driver vs. Passenger Ubiquitination Events in Tumorigenesis
Ubiquitination, a pivotal post-translational modification, governs the stability, activity, and localization of thousands of proteins within eukaryotic cells [3]. This enzymatic process, mediated by a cascade of E1 activating, E2 conjugating, and E3 ligase enzymes, attaches the small protein ubiquitin to substrate proteins [70] [3]. The resulting ubiquitin code—comprising monoubiquitination or various polyubiquitin chain linkages—directs diverse cellular outcomes, from proteasomal degradation to signal transduction and DNA repair [46] [3]. The reverse process, deubiquitination, is executed by deubiquitinating enzymes (DUBs), adding a dynamic regulatory layer [25] [70].
In tumorigenesis, the precise machinery of ubiquitination is frequently disrupted. Cancer genomes harbor numerous mutations in genes encoding ubiquitination system components, but a central challenge lies in distinguishing driver events that confer a selective growth advantage from functionally inert passenger events [71] [72]. Driver mutations in ubiquitination pathways are causally involved in oncogenesis, promoting uncontrolled proliferation, metabolic reprogramming, evasion of cell death, and metastasis [46] [3]. Accurately identifying these driver events is paramount for understanding cancer biology and developing targeted therapies [71].
Molecular Signatures of Driver and Passenger Ubiquitination Events
Driver and passenger ubiquitination events exhibit distinct molecular, genetic, and functional characteristics. The table below summarizes the key differentiating features.
Table 1: Characteristic Features of Driver versus Passenger Ubiquitination Events
| Feature | Driver Ubiquitination Event | Passenger Ubiquitination Event |
|---|---|---|
| Functional Impact | Alters oncogenic or tumor suppressive pathways, conferring growth advantage [71] [72]. | No consequential contribution to tumor development or progression [72]. |
| Mutation Pattern | Recurrent, hotspot mutations in functional domains (e.g., E3 ligase substrate-binding regions) [60]. | Random, non-recurrent mutations scattered throughout the genome [72]. |
| Conservation & Selection | Sites under positive selection and evolutionarily conserved [60]. | Evolutionarily neutral, no selective pressure [72]. |
| Effect on Ubiquitination | Directly alters substrate ubiquitination (e.g., disrupting E3-SPOP binding in prostate cancer) or substrate recognition [60] [25]. | No functional disruption of ubiquitination signaling. |
| Pathway Consequence | Activates oncogenic signaling (e.g., NF-κB, mTOR) or inactivates tumor suppressors (e.g., PTEN) [73] [70] [46]. | No discernible impact on cancer-related cellular processes. |
| Frequency in Tumors | Low to moderate frequency, but statistically significant recurrence across samples [60] [71]. | Can be very high in number (~40-100 per tumor) but are biologically inert "hitchhikers" [72]. |
Experimental Approaches for Discriminating Ubiquitination Events
Genomic and Computational Methods
Identifying driver ubiquitination events requires sophisticated bioinformatic analyses of large-scale cancer genomic data.
- Site-Specific Mutation Enrichment Analysis (ActiveDriver): This computational method identifies genes with significant co-occurrence of missense mutations and post-translational modification (PTM) sites, including ubiquitination sites. It uses a Poisson regression model that accounts for protein disorder, direct and flanking PTM residues, and site density to predict cancer driver genes based on low-frequency mutations that cluster in functionally active sites [60].
- Mutation Clustering Analysis (OncoDriveClust): This method assesses the spatial clustering of mutations within protein sequences. Driver mutations often cluster in specific functional domains or active sites, whereas passenger mutations are randomly distributed. Applying this to ubiquitination sites helps pinpoint mutations that likely disrupt functional regions of E3 ligases or DUBs [60].
- Signature Analysis of Genomic Alterations: This approach differentiates between deletions targeting tumor suppressor genes (usually bi-allelic) and those occurring at fragile sites (often mono-allelic). This signature helps distinguish driver deletions in ubiquitination system components from passenger deletions [71].
Table 2: Key Experimental Methodologies for Characterizing Ubiquitination Events
| Methodology | Key Objective | Technical Output | Interpretation in Driver/Passenger Context |
|---|---|---|---|
| Mass Spectrometry-based Ubiquitinomics | System-wide profiling of ubiquitinated peptides [74]. | Identification and quantification of ubiquitination sites and chain topology. | Driver events show altered ubiquitination in cancer vs. normal tissues or correlate with pathway activity [74]. |
| RNAi/CRISPR Screens | Functional interrogation of E3 ligases and DUBs. | Fitness scores (e.g., essentiality for cell proliferation/survival). | Genes essential in specific cancer contexts are candidate drivers [74]. |
| Correlation with Pathway Activity | Linking UBR expression to oncogenic pathways. | Pearson correlation coefficients between UBR mRNA and pathway activity scores [74]. | UBRs strongly correlated with hallmark cancer pathways (e.g., mTOR, HIF-1) are likely drivers [74] [70]. |
| Analysis of Somatic Mutations & CNV | Identifying genetic alterations in UBRs in pan-cancer cohorts. | Mutation frequency, type, and copy number variation (CNV) data [74]. | Recurrent, non-silent mutations or amplifications/deletions suggest driver role [74]. |
Functional Validation in Cellular Models
Bioinformatic predictions require validation in biological systems to confirm driver functionality.
- In Vivo Metastasis Models: To demonstrate a driver role in metastasis, studies use techniques such as shRNA-mediated inhibition of specific E2 enzymes (e.g., UBE2N) in breast cancer cells, followed by tail-vein injection into mice. A significant suppression of lung metastasis formation confirms the protein's driver function in this process [46].
- Metabolic Assays: For ubiquitination events implicated in metabolic reprogramming, functional validation includes measuring extracellular acidification rate (ECAR) for glycolysis and oxygen consumption rate (OCR) for mitochondrial respiration after modulating the candidate E3 or DUB. Driver events will significantly alter these metabolic parameters [70].
- Protein Stability and Degradation Assays: Classic cycloheximide chase experiments can determine if a candidate mutation in an E3 ligase or substrate affects the half-life of the substrate protein. Altered degradation kinetics support a driver role [73] [70].
Key Signaling Pathways with Driver Ubiquitination Events
mTORC1 Signaling and Metabolic Reprogramming
The mTORC1 pathway is a critical hub for regulating cell growth and metabolism, and its ubiquitination represents a clear driver event in cancer.
Diagram 1: Ubiquitination in mTORC1 activation.
The E3 ligase TRAF6 catalyzes K63-linked polyubiquitination of mTOR in an amino acid-dependent manner. This specific ubiquitination event, a driver modification, facilitates the translocation of mTORC1 to the lysosomal surface, a prerequisite for its activation [70]. Once active, mTORC1 drives anabolic processes, enhancing glycolysis and biosynthesis to fuel tumor growth. Conversely, other E3 ligases like FBXW7 and FBX8 mediate K48-linked ubiquitination and degradation of mTOR, acting as tumor suppressors. Loss-of-function mutations in these ligases stabilize mTOR, contributing to oncogenesis [70].
PTEN Nuclear Import and Tumor Suppression
The tumor suppressor PTEN is regulated by a sophisticated ubiquitination switch that differentiates between its nuclear and cytoplasmic fates, a classic example of a driver mechanism.
Diagram 2: Ubiquitination switch regulating PTEN.
Monoubiquitination of PTEN at specific lysine residues (e.g., K289) is essential for its nuclear import, where it exerts critical tumor-suppressive functions [73]. This monoubiquitination is a driver event for tumor suppression. Notably, a K289E mutation in PTEN, identified in patients with Cowden syndrome, abrogates this monoubiquitination, sequestering PTEN in the cytoplasm and abolishing its nuclear tumor-suppressive role without affecting its catalytic activity [73]. In contrast, polyubiquitination of cytoplasmic PTEN targets it for proteasomal degradation, and dysregulation of this process can also drive tumorigenesis.
NF-κB Activation and Inflammation-Driven Cancer
The UBE2N/UBE2V1 (UBC13/UEV1A) E2 complex drives oncogenic signaling by catalyzing K63-linked polyubiquitin chains, which activate signaling pathways rather than target proteins for degradation.
Diagram 3: UBE2N/UBE2V1 in NF-κB activation.
This E2 complex is overexpressed in multiple cancers, including breast, pancreas, and colon carcinomas [46]. In breast cancer, UBE2N/UBE2V1 is required for TGFβ-mediated activation of TAK1 and p38, leading to the expression of metastasis-associated genes like MMP1, CD44, and VCAM-1 [46]. Crucially, while UBE2N is not required for primary tumor formation, it is essential for metastasis in in vivo models, categorizing its activity as a distinct driver of the metastatic cascade [46].
The Scientist's Toolkit: Research Reagent Solutions
Studying driver ubiquitination events requires a specialized set of research tools and reagents. The table below details key resources for functional characterization.
Table 3: Essential Research Reagents for Investigating Ubiquitination in Cancer
| Research Reagent / Tool | Primary Function | Key Application in Driver/Passenger Studies |
|---|---|---|
| ActiveDriver Software [60] | Computational prediction of significant PTM site mutations. | Initial bioinformatic screening of cancer genomics data (e.g., from TCGA) to identify genes with enriched mutations in ubiquitination sites. |
| Tandem Ubiquitin Binding Entities (TUBEs) | High-affinity capture of polyubiquitinated proteins from cell lysates. | Isolation of ubiquitinated proteins to assess global ubiquitination changes or specific substrate ubiquitination driven by candidate mutations. |
| Linkage-Specific Ubiquitin Antibodies (e.g., K48, K63) | Immunodetection of specific polyubiquitin chain linkages. | Determining the type of ubiquitin chain formed in a driver event (e.g., K63 for signaling vs. K48 for degradation) via Western blot or immunohistochemistry. |
| Proteasome Inhibitors (e.g., Bortezomib, Carfilzomib) [3] | Block degradation of polyubiquitinated proteins by the 26S proteasome. | Stabilizing ubiquitinated substrates to aid in their detection; used clinically to treat hematological malignancies. |
| shRNA/siRNA Libraries | Knockdown of specific E2s, E3s, or DUBs. | Functional validation of candidate driver genes through in vitro (proliferation, migration) and in vivo (xenograft) assays. |
| Ubiquitin Mutants (K-to-R, K-only) | Express ubiquitin where specific lysines are mutated to arginine (prevents chain formation) or are the only lysine available. | Defining the specific lysine residue used for polyubiquitination in a driver event and its functional consequence. |
Distinguishing driver from passenger ubiquitination events is a cornerstone of precision oncology. Driver events are characterized by their recurrence, functional impact on oncogenic or tumor-suppressive pathways, and clinical relevance, often correlating with patient survival and therapeutic response [60] [46]. The integration of multi-omics data, sophisticated computational tools like ActiveDriver, and robust functional validation in disease-relevant models is essential for accurate discrimination.
This distinction is not merely academic; it directly informs therapeutic discovery. Driver ubiquitination events represent promising drug targets [12] [3]. The successful development of proteasome inhibitors for blood cancers paved the way for a new generation of targeted agents, including molecular glues and PROTACs (Proteolysis-Targeting Chimeras), which hijack the ubiquitin system to degrade previously "undruggable" oncoproteins [12]. As our understanding of the ubiquitin code in cancer deepens, so too will our ability to develop innovative therapies that specifically target the drivers of tumorigenesis.
Batch Effect Correction in Multi-Cohort and Cross-Platform Analyses
Batch effects represent a fundamental challenge in modern biomedical research, particularly in large-scale omics studies that integrate data from multiple cohorts or technological platforms. These non-biological technical variations are introduced through differences in experimental conditions, reagent lots, handling personnel, instrumentation, or sequencing technologies, and can profoundly obscure genuine biological signals [75]. In the context of cancer research, specifically when comparing ubiquitination patterns between cancerous and normal tissues, batch effects can lead to misleading conclusions, reduced statistical power, and irreproducible findings if not adequately addressed [75].
The proliferation of multi-center consortium studies and the practice of merging publicly available datasets have magnified the batch effect problem, making effective correction methodologies essential for valid biological interpretation. Batch effects manifest differently across omics technologies—genomics, transcriptomics, proteomics, and metabolomics—each requiring specialized correction approaches tailored to their specific technical characteristics and data structures [75]. This guide provides a comprehensive comparison of batch-effect correction methods, their performance characteristics, and practical implementation protocols to support researchers in producing robust, reproducible analyses in ubiquitination-focused cancer research.
Batch effects arise throughout the experimental workflow, with different omics technologies sharing common sources while also exhibiting platform-specific challenges. During study design, flawed or confounded arrangements represent a critical source of cross-study irreproducibility, particularly when samples are not randomized or when batch groups correlate with biological factors of interest [75]. In the sample preparation and storage phase, variations in protocol procedures, reagent lots, and storage conditions introduce technical variations that propagate through subsequent analyses [75].
The data generation phase introduces platform-specific batch effects: in mass spectrometry-based proteomics, differences in liquid chromatography systems, mass spectrometer calibration, and instrument drift across runs create significant technical variations [76]; in single-cell RNA sequencing, the technology suffers from higher technical variations due to lower RNA input, higher dropout rates, and greater cell-to-cell heterogeneity compared to bulk RNA-seq [75]; for microarray and bulk RNA-seq, differences in sample processing dates, laboratory personnel, and RNA extraction kits introduce measurable batch effects [77] [78].
Impact on Ubiquitination Research and Cancer Studies
In the context of ubiquitination pattern analysis, batch effects can profoundly impact research outcomes. When investigating differences in ubiquitination between cancerous and normal tissues, technical variations can:
- Obscure genuine biological signals: Batch effects introduce noise that dilutes true ubiquitination differences between normal and cancerous tissues, reducing statistical power to detect clinically relevant biomarkers [75].
- Generate false discoveries: When batch effects correlate with disease status, they can lead to erroneous identification of ubiquitination markers that actually reflect technical artifacts rather than biological truth [75].
- Hinder reproducibility: The irreproducibility caused by batch effects has resulted in retracted papers and discredited research findings across omics disciplines, representing a significant economic and scientific burden [75].
Table 1: Common Sources of Batch Effects in Multi-Omics Studies
| Experimental Stage | Source of Batch Effects | Impact on Ubiquitination Research |
|---|---|---|
| Study Design | Confounded batch and biological groups | Inability to distinguish technical from biological variation in ubiquitination patterns |
| Sample Collection | Differences in collection protocols, stabilization methods | Variation in protein/ubiquitin preservation affecting quantification |
| Sample Processing | Differences in lysis methods, enrichment protocols | Inconsistent ubiquitinated peptide recovery |
| Data Generation | Instrument variability, reagent lots, sequencing platforms | Technical variation in ubiquitination site identification and quantification |
| Data Analysis | Different bioinformatics pipelines, normalization methods | Inconsistent ubiquitination pattern calling and quantification |
Comprehensive Comparison of Batch Correction Methods
Method Categories and Underlying Algorithms
Batch-effect correction algorithms (BECAs) can be categorized by their mathematical approaches and the omics data types they target. Empirical Bayes methods like ComBat adjust for mean shifts across batches using an empirical Bayesian framework, making them suitable for both microarray and bulk RNA-seq data [77] [79]. Nearest-neighbor approaches such as Mutual Nearest Neighbors (MNN) and Harmony identify correspondences between batches—either in high-dimensional space or reduced dimensions—to align datasets while preserving biological variation [80] [79]. Matrix factorization techniques including LIGER use integrative non-negative matrix factorization to decompose data into shared and batch-specific factors, particularly effective when biological differences exist between batches [79]. Deep learning approaches like NormAE employ neural networks to learn and remove non-linear batch effects while preserving biological signals [76].
Performance Comparison Across Omics Platforms
Different BECAs exhibit varying performance characteristics across omics platforms and experimental scenarios. For single-cell RNA-seq data, a comprehensive benchmark of 14 methods across ten datasets revealed that Harmony, LIGER, and Seurat 3 consistently outperformed other approaches in terms of batch mixing while preserving cell type purity [79]. Harmony demonstrated significantly shorter runtime, making it particularly suitable for large-scale datasets becoming common in single-cell studies [79].
In proteomics applications, recent benchmarking studies using the Quartet protein reference materials have demonstrated that protein-level correction generally outperforms peptide- or precursor-level correction across multiple quantification methods [76]. The Ratio method, which scales intensities relative to concurrently profiled reference samples, showed particularly robust performance, especially when batch effects were confounded with biological groups of interest [76].
For cross-platform transcriptomics integrating data from different technologies (e.g., polyA-enriched vs. ribo-depleted libraries), ComBat-Seq has proven effective at removing technical variations while preserving biological signals, as demonstrated in studies comparing Universal Human Reference (UHR) and Human Brain Reference (HBR) samples across different library preparation methods [77].
Table 2: Performance Comparison of Batch Correction Methods by Data Type
| Method | Primary Algorithm | Recommended Data Types | Strengths | Limitations |
|---|---|---|---|---|
| ComBat/ComBat-Seq | Empirical Bayes | Bulk RNA-seq, Microarrays | Effective mean adjustment, handles small batches | May over-correct with biological differences |
| Harmony | Mixture models | scRNA-seq, Proteomics | Fast runtime, good batch mixing | May oversmooth subtle biological differences |
| MNN/fastMNN | Mutual nearest neighbors | scRNA-seq, Cross-platform | Preserves biological structure | Computationally intensive for very large datasets |
| LIGER | Non-negative matrix factorization | scRNA-seq, Multi-batch | Separates technical and biological variation | Complex parameter tuning |
| Seurat 3 | CCA + anchor weighting | scRNA-seq, Multi-modal | Integrates diverse data types | Memory-intensive for large datasets |
| Ratio | Reference-based scaling | Proteomics, Metabolomics | Robust to confounded designs | Requires reference materials |
| RUV-III-C | Linear regression | Proteomics, Bulk RNA-seq | Handles unwanted variation in raw intensities | Requires negative control genes |
Experimental Protocols for Batch Correction
Protocol 1: Batch Correction for Multi-Cohort Transcriptomics Data
This protocol outlines the steps for correcting batch effects in RNA-seq data combining multiple cohorts or studies, such as when analyzing ubiquitination-related genes across different datasets.
Step 1: Data Preprocessing and Quality Control Begin with raw count data, ensuring proper normalization for library size differences. For bulk RNA-seq, use established normalization methods such as TMM (trimmed mean of M-values) or DESeq2's median of ratios. For cross-study integrations, apply variance stabilizing transformation (VST) to mitigate mean-variance relationships. Perform principal component analysis (PCA) to visualize initial batch separation and identify potential outliers.
Step 2: Batch Effect Assessment Quantitatively evaluate batch effects using metrics such as Principal Variance Component Analysis (PVCA) to determine the proportion of variance explained by batch versus biological factors. Visually inspect PCA plots colored by batch and biological conditions. In the context of ubiquitination studies, examine whether samples cluster more strongly by study origin than by cancer versus normal tissue classification.
Step 3: Method Selection and Implementation Based on data characteristics, select an appropriate correction method. For balanced designs with minimal biological differences between batches, ComBat-Seq applied directly to counts is effective [77]. For datasets with complex batch structures or when integrating across different platforms (e.g., polyA-selected vs. ribo-depleted libraries), consider Harmony or mutual nearest neighbor approaches. Implement the chosen method following package-specific requirements, ensuring proper parameter specification.
Step 4: Post-Correction Validation Re-run PCA on corrected data to visualize improvement in batch mixing. Use quantitative metrics such as average silhouette width (ASW) for biological group separation and kBET (k-nearest neighbor batch-effect test) for batch mixing. Validate that known biological signals (e.g., established ubiquitination markers) are preserved while technical artifacts are diminished.
Step 5: Downstream Analysis Proceed with differential expression analysis using batch-corrected data, including appropriate statistical models that account for any residual technical variation. For cross-study comparisons, consider meta-analysis approaches like RobustRankAggreg as complementary validation [78].
Protocol 2: Batch Correction for MS-Based Proteomics Data in Ubiquitination Studies
This protocol specifically addresses batch-effect correction in mass spectrometry-based proteomics data, with emphasis on ubiquitination pattern analysis.
Step 1: Level-Specific Preprocessing Process raw MS data through standard proteomics pipelines. Determine the optimal level for batch correction: precursor, peptide, or protein. Recent evidence suggests protein-level correction provides the most robust results for downstream analysis [76]. For ubiquitination-specific studies, ensure proper enrichment and identification of ubiquitinated peptides before aggregation to protein level.
Step 2: Imputation Strategy Selection Address missing values common in proteomics data. For missing-not-at-random (MNAR) values (typical of low-abundance peptides below detection limit), use half-LOD (limit of detection) replacement. For missing-at-random (MAR) values, consider K-nearest neighbor (KNN) imputation. Note that imputation choice can interact with batch correction efficacy [80].
Step 3: Batch Correction Implementation Apply correction methods appropriate for proteomics data. The Ratio method, which uses reference samples for intensity scaling, performs well particularly with confounded designs [76]. ComBat with inclusion of biological covariates (e.g., disease status) preserves true biological signal while removing technical variation [80]. For large-scale studies with injection order effects, consider methods like WaveICA2.0 that account for time-dependent signal drifts.
Step 4: Quality Assessment and Validation Evaluate correction efficacy using metrics such as coefficient of variation (CV) within technical replicates across batches. For ubiquitination studies, spiked-in standards or internal references provide quality controls. Use principal component analysis to confirm reduced batch separation and preserved biological group differences (cancer vs. normal tissue).
Step 5: Differential Ubiquitination Analysis Perform statistical analysis on corrected protein-level data to identify differentially ubiquitinated proteins between conditions. Confirm findings with orthogonal methods when possible, especially for candidate biomarkers.
Diagram 1: Proteomics Batch Correction Workflow. Protein-level correction generally provides the most robust results for downstream analysis.
Reference Materials and Quality Controls
Well-characterized reference materials are indispensable for batch monitoring and correction, particularly in large-scale studies. Quartet reference materials (D5, D6, F7, M8) provide multi-level quality control for proteomics studies, enabling accurate batch-effect assessment across multiple laboratories [76]. Universal Human Reference (UHR) RNA and Human Brain Reference (HBR) RNA serve as standards for transcriptomics applications, allowing performance evaluation across different sequencing platforms and library preparation methods [77]. Spiked-in standards (e.g., SIS peptides in proteomics, ERCC RNA controls in transcriptomics) enable quantitative assessment of technical variation and correction efficacy.
Computational Tools and Software Packages
Table 3: Essential Computational Tools for Batch-Effect Correction
| Tool/Package | Primary Function | Applicable Omics | Key Features |
|---|---|---|---|
| sva (R/Bioconductor) | Surrogate variable analysis, ComBat | Transcriptomics, Proteomics | Empirical Bayes framework, handles complex designs |
| Harmony (R) | Iterative clustering integration | scRNA-seq, Proteomics | Fast runtime, good scalability to large datasets |
| Seurat (R) | Multi-modal single-cell analysis | scRNA-seq, Spatial transcriptomics | Anchor-based integration, handles diverse datatypes |
| LIGER (R) | Integrative non-negative matrix factorization | scRNA-seq, Multi-omics | Separates shared and dataset-specific factors |
| batchelor (R/Bioconductor) | MNN correction in reduced dimensions | scRNA-seq, Bulk genomics | Fast mutual nearest neighbors implementation |
| Normalyzer (R) | Normalization method evaluation | Proteomics, Metabolomics | Comprehensive normalization assessment |
| PRONE (R) | Proteomics normalization evaluation | Proteomics | Specialized assessment for proteomics data |
Integration with Ubiquitination Pattern Analysis in Cancer Research
Special Considerations for Ubiquitination Studies
The analysis of ubiquitination patterns in cancer versus normal tissues presents unique challenges for batch-effect correction. Ubiquitinated peptides typically exhibit low stoichiometric abundance, making their quantification particularly vulnerable to technical variations and missing data issues. The dynamic nature of ubiquitination necessitates careful experimental design to distinguish true biological regulation from technical artifacts. Furthermore, the diversity of ubiquitin chain linkages (K48, K63, M1, etc.) with distinct functional consequences requires preservation of subtle pattern differences during correction procedures.
When designing multi-cohort ubiquitination studies, incorporate balanced reference samples across batches to monitor technical variation. Consider linkage-specific ubiquitin antibodies during enrichment to ensure consistent recovery across batches. For data analysis, employ correction methods that preserve quantitative relationships between different ubiquitination forms, as methods that over-aggregate or oversmooth may obscure important biological patterns.
Pathway Integration and Biological Validation
Batch-corrected ubiquitination data should be integrated with complementary omics layers and pathway information to enhance biological interpretation. The ubiquitin-proteasome system regulates numerous cancer-relevant processes including cell cycle progression, DNA repair, and immune recognition, with specific E3 ligases and deubiquitinases acting as oncogenes or tumor suppressors [12].
Diagram 2: Ubiquitination Pathway and Cancer Relevance. E3 ligases and deubiquitinases (DUBs) represent key regulatory nodes with therapeutic potential.
Following batch correction, ubiquitination patterns should be contextualized within cancer hallmark processes. For example, increased ubiquitination of tumor suppressors like p53 would be expected in certain cancers, while altered immune checkpoint regulator ubiquitination (e.g., PD-L1) might indicate immune evasion mechanisms [12]. Correlate ubiquitination changes with transcriptomic and proteomic data from the same samples to identify consistent regulatory patterns. Validate key findings using orthogonal methods such as immunoblotting or targeted mass spectrometry to ensure that batch correction has preserved true biological signals.
Effective batch-effect correction is not merely a technical preprocessing step but a fundamental component of rigorous omics research, particularly in multi-cohort studies of ubiquitination patterns in cancer. The optimal correction strategy depends on multiple factors: the specific omics platform, study design (balanced vs. confounded), sample size, and the specific biological questions being addressed. Based on current benchmarking studies, Harmony offers an excellent balance of performance and computational efficiency for single-cell data, while protein-level correction combined with Ratio or ComBat with covariates represents a robust approach for proteomics applications [79] [76].
Future directions in batch-effect correction will likely involve more sophisticated integration of multiple omics layers, leveraging cross-platform normalization strategies. The development of correction methods specifically designed for ubiquitination proteomics data, accounting for its unique stoichiometry and dynamic range, represents an important unmet need. As large-scale consortium studies continue to generate multi-cohort ubiquitination data, reference-based correction approaches using well-characterized standards will become increasingly essential for producing biologically meaningful and clinically relevant findings in cancer research.
The ubiquitin-proteasome system (UPS) represents a crucial regulatory network in cellular homeostasis, and its dysregulation is a hallmark of numerous cancers. In tumorigenesis, ubiquitination patterns undergo significant alterations compared to normal tissues, affecting critical processes from cell cycle regulation to immune modulation [7] [3]. The ability to accurately map and quantify these changes is fundamental to advancing both basic cancer biology and targeted drug discovery. Functional validation of ubiquitination dynamics requires sophisticated methodological approaches that can span from high-throughput screening to detailed mechanistic studies. This guide provides a comprehensive comparison of current technologies enabling researchers to dissect ubiquitination patterns in cancerous versus normal tissues, with particular emphasis on practical implementation, experimental design, and data interpretation.
The ubiquitination process involves a sequential enzymatic cascade comprising E1 activating enzymes, E2 conjugating enzymes, and E3 ligases, which together confer substrate specificity [3]. Deubiquitinating enzymes (DUBs) provide the counterbalance, removing ubiquitin modifications and completing the cycle of dynamic regulation [70]. In cancer research, comprehensive analysis requires tools that can capture this complexity across different spatial and temporal scales, from initial high-throughput discovery to targeted validation of specific ubiquitination events. The integration of these approaches provides a powerful framework for identifying novel therapeutic targets and understanding the mechanistic basis of oncogenesis and tumor progression.
Comparative Analysis of Ubiquitination Screening Platforms
Table 1: Comparison of High-Throughput Ubiquitination Screening Platforms
| Platform Name | Core Technology | Throughput | Key Applications | Sensitivity | Real-Time Monitoring | Complexity |
|---|---|---|---|---|---|---|
| UbiReal [81] | Fluorescence Polarization | High | E1/E2/E3 kinetics, DUB activity, inhibitor screening | High (nM range) | Yes | Moderate |
| URT-Dual-Luciferase [82] | Dual-Luciferase reporter with ubiquitin-reference technique | High | E3 ligase modulator screening, substrate degradation | Moderate | No (endpoint) | High |
| Cell-Based Ubiquitin-Reference Technique [82] | Ubiquitin fusion protein with internal reference | Medium | E3-substrate interactions, proteasomal degradation | Moderate | No | High |
Each platform offers distinct advantages depending on the research objectives. The UbiReal system utilizes fluorescence polarization to monitor all stages of ubiquitin conjugation and deconjugation in real time, providing kinetic data on E1 activation, E2~Ub discharge, E3-dependent ubiquitin chain formation, and DUB activity [81]. This platform excels in comprehensive pathway analysis and direct measurement of enzyme kinetics, making it ideal for mechanistic studies and compound screening.
The URT-Dual-Luciferase method integrates the ubiquitin-reference technique with a Dual-Luciferase system to create a cell-based assay that normalizes for experimental variability [82]. By using Renilla luciferase as an internal reference and firefly luciferase fused to a substrate of interest, this system accurately quantifies E3 ligase-dependent substrate degradation while compensating for differences in cell seeding density and transfection efficiency. This approach is particularly valuable for studying specific E3-substrate relationships in a more physiologically relevant cellular context.
Experimental Protocols for Key Methodologies
UbiReal Fluorescence Polarization Assay Protocol
The UbiReal platform provides a versatile approach for monitoring ubiquitination dynamics in real time using fluorescently-labeled ubiquitin. The protocol can be adapted for studying various components of the ubiquitination cascade:
Step 1: Reaction Setup
- Prepare reaction buffer (25 mM Tris, 100 mM NaCl, 5 mM MgCl2, 0.5 mM TCEP, pH 7.6)
- Add 50 nM fluorescently-labeled ubiquitin (F-Ub or T-Ub)
- Include ATP regeneration system (2 mM ATP, 10 mM creatine phosphate, 10 μg/mL creatine kinase)
Step 2: Enzyme Addition and Monitoring
- Initiate reaction by adding E1 enzyme (100 nM final concentration)
- Monitor fluorescence polarization every 30 seconds using a plate reader
- Sequentially add E2 (500 nM) and E3 (200 nM) enzymes as required
- For DUB assays, add DUB enzyme after ubiquitin chain formation
Step 3: Data Analysis
- Calculate polarization values in millipolarization units (mP)
- Plot mP versus time to visualize ubiquitination progression
- Determine kinetic parameters from the initial linear phase of the reaction
This assay has been successfully used to characterize E1 inhibitor PYR-41, demonstrating its utility for small molecule screening and mechanistic studies [81].
URT-Dual-Luciferase Cell-Based Screening Protocol
This method enables identification of E3 ligase modulators in a cellular context using SMURF1 and its substrate RHOB as a model system:
Step 1: Plasmid Construction
- Construct pRUF(RL-UbR48-FL)-RHOB fusion vector expressing:
- 3×FLAG-Renilla Luciferase-UbR48-3×FLAG-Firefly Luciferase-RHOB
- Use UbK48R mutant to prevent unintended ubiquitin conjugation
Step 2: Cell Transfection and Treatment
- Co-transfect HEK293T cells with pRUF-RHOB and SMURF1 expression vectors
- Seed transfected cells in 96-well plates (10,000 cells/well)
- Treat with compound library or controls (DMSO, MG-132) for 16-24 hours
Step 3: Luciferase Measurement and Analysis
- Lyse cells and measure Firefly and Renilla luciferase activities
- Calculate FL/RL ratio for each well
- Normalize data to DMSO controls
- Determine Z-factor to validate assay quality (target >0.5)
The internal reference (Renilla luciferase) corrects for variations in cell number, transfection efficiency, and compound toxicity, ensuring robust screening performance [82].
Workflow Visualization of Screening Platforms
Diagram 1: Comparative workflows for UbiReal and URT-Dual-Luciferase screening platforms.
Ubiquitination Signaling in Cancer Context
Table 2: Key Ubiquitination Regulators Altered in Cancer Tissues Versus Normal Tissues
| Ubiquitination Regulator | Expression in Cancer | Functional Role in Cancer | Associated Pathways |
|---|---|---|---|
| UBE2C [7] | Upregulated in HCC | Promotes cell proliferation, invasion, metastasis; correlates with poor prognosis | Cell cycle regulation, p53 signaling |
| UBA1 [22] | Upregulated in multiple cancers | Essential for ubiquitination initiation; supports tumor growth | Protein degradation, DNA repair |
| UBA6 [22] | Upregulated in specific cancers | Activates ubiquitin and FAT10; linked to immune infiltration | Immune modulation, p53 regulation |
| SMURF1 [82] | Context-dependent | Regulates TGFβ signaling, EMT, RHOB degradation | TGFβ pathway, epithelial-mesenchymal transition |
| TRAF6 [70] | Upregulated in cancer cells | Mediates K63-linked ubiquitination of mTOR | mTORC1 activation, metabolic reprogramming |
Understanding the molecular alterations in ubiquitination machinery between cancerous and normal tissues provides critical insights for assay selection and validation. Comprehensive pan-cancer analyses reveal that ubiquitination regulators (UBRs) demonstrate widespread expression perturbations across cancer types, with more than 90% associated with patient survival outcomes [5]. These alterations impact hallmark cancer pathways including cell cycle regulation, metabolic reprogramming, and immune evasion.
In hepatocellular carcinoma (HCC), ubiquitination-related genes are significantly upregulated, with high expression levels correlating with poor prognosis [7]. Pathway analysis shows enrichment in critical processes including DNA repair, metabolic reprogramming, and p53 signaling. Notably, the E2 conjugating enzyme UBE2C emerges as a key driver of HCC progression, promoting tumor cell proliferation, matrix remodeling, and angiogenesis through its effects on the tumor microenvironment [7].
The E1 activating enzymes UBA1 and UBA6 show particularly interesting patterns across cancer types. These essential initiation enzymes are frequently overexpressed in malignancies and correlate with advanced disease stage and poor survival [22]. Their expression patterns also associate with immune infiltration characteristics, suggesting roles in modulating the tumor immune microenvironment. This comprehensive understanding of ubiquitination alterations in cancer informs the selection of appropriate screening platforms and validation strategies.
Essential Research Reagent Solutions
Table 3: Essential Research Reagents for Ubiquitination Studies
| Reagent Category | Specific Examples | Function in Ubiquitination Research | Application Notes |
|---|---|---|---|
| E1 Enzymes | UBA1, UBA6 [22] | Initiate ubiquitination cascade | Essential for in vitro reconstitution assays |
| E2 Enzymes | UBE2C [7], UBE2D3 [81] | Carry activated ubiquitin | Determine chain-type specificity |
| E3 Ligases | SMURF1 [82], MDM2 [3] | Provide substrate specificity | Key therapeutic targets |
| DUBs | USP7, USP21 [81] [3] | Remove ubiquitin modifications | Counter-regulatory enzymes |
| Ubiquitin Mutants | K48R, K63R [82] [3] | Study chain-type specific functions | Define ubiquitin linkage roles |
| Proteasome Inhibitors | MG-132, Bortezomib [82] [3] | Block degradation of ubiquitinated proteins | Validate proteasome dependence |
| Fluorescent Ubiquitin | Fluorescein-Ub, TAMRA-Ub [81] | Enable real-time monitoring | Essential for UbiReal platform |
| Luciferase Reporters | Firefly, Renilla [82] | Quantify substrate degradation | Used in cell-based URT system |
The selection of appropriate research reagents is critical for successful ubiquitination studies. For in vitro assays, recombinant enzymes must exhibit high activity and purity, with careful attention to preservation of enzymatic function throughout purification and storage. E3 ligases and DUBs represent particularly attractive therapeutic targets due to their substrate specificity and central roles in cancer pathways [3].
Ubiquitin mutants serve as indispensable tools for dissecting the functional consequences of specific ubiquitin linkages. For example, the K48R mutant prevents formation of canonical degradation signals, while K63R disrupts signaling assemblies without affecting proteasomal targeting [82] [3]. These tools have been instrumental in defining the non-proteolytic functions of ubiquitination in processes such as DNA repair, kinase activation, and protein trafficking.
Emerging evidence suggests that ubiquitination regulators demonstrate tissue-specific expression patterns, with testicular tissue showing the most distinct profile [5]. This heterogeneity underscores the importance of selecting appropriate model systems and validation approaches when studying ubiquitination in specific cancer types or normal tissue counterparts.
Strategic Integration for Comprehensive Validation
Optimizing functional validation in ubiquitination research requires strategic integration of complementary approaches. The selection of appropriate screening platforms should be guided by research objectives, with high-throughput discovery followed by targeted mechanistic studies. Initial screens using cell-based systems like the URT-Dual-Luciferase assay can identify potential modulators of specific E3-substrate relationships, while mechanistic follow-up with biophysical approaches like UbiReal provides detailed kinetic characterization.
The integration of multi-omics analyses provides powerful validation of screening results, with ubiquitination regulator expression patterns, genetic alterations, and pathway activities offering orthogonal confirmation of functional significance [7] [5]. This comprehensive approach enables researchers to distinguish driver alterations from passenger events in carcinogenesis.
For research focused on the tumor microenvironment and immune interactions, consideration of ubiquitination effects on immune cell infiltration and function adds another dimension of complexity [7] [22]. The expanding toolkit for ubiquitination research, including PROTAC technology and targeted protein degradation approaches, offers exciting opportunities for therapeutic translation of basic research findings. By strategically combining high-throughput screening platforms with mechanistic follow-up studies, researchers can accelerate the development of novel cancer therapeutics targeting the ubiquitin-proteasome system.
Clinical Translation: Biomarker Discovery and Therapeutic Targeting Validation
Ubiquitination, a fundamental post-translational modification, plays a crucial regulatory role in protein stability, function, and localization within eukaryotic cells. The ubiquitin-proteasome system (UPS) mediates approximately 80-90% of cellular proteolysis and has emerged as a pivotal player in cancer pathogenesis [12]. Recent research has illuminated how dysregulation of ubiquitinating and deubiquitinating enzymes contributes to oncogenic processes across various cancer types, driving significant interest in ubiquitination-related genes (UbRGs) as potential prognostic biomarkers and therapeutic targets [12] [83].
The investigation of ubiquitination-based prognostic signatures represents a cutting-edge approach in oncology, leveraging multi-omics data to decipher the complex molecular underpinnings of tumor behavior. These signatures have demonstrated remarkable potential in prognostic prediction, immune landscape characterization, and therapeutic guidance across diverse malignancies including laryngeal cancer, breast cancer, cervical cancer, and diffuse large B-cell lymphoma [84] [49] [85]. This review systematically compares recently identified ubiquitination signatures, their correlation with patient survival, their relationship with treatment responses, and the experimental methodologies underpinning these discoveries.
Ubiquitination Machinery in Cellular Homeostasis and Cancer
The ubiquitination process involves a well-orchestrated enzymatic cascade comprising ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3). This system culminates in the covalent attachment of ubiquitin molecules to target proteins, marking them for proteasomal degradation or functional modification [12] [25]. E3 ubiquitin ligases, numbering approximately 1,000 in the human genome, confer substrate specificity and thus represent particularly attractive targets for therapeutic intervention [25]. Counterbalancing this process are deubiquitinating enzymes (DUBs) that remove ubiquitin modifications, creating a dynamic equilibrium essential for maintaining cellular protein homeostasis [25].
In cancer pathogenesis, disruption of the ubiquitin-proteasome system leads to aberrant stabilization of oncoproteins or accelerated degradation of tumor suppressors. As highlighted by recent research, "Ubiquitination, a pivotal posttranslational modification of proteins, plays a fundamental role in regulating protein stability. The dysregulation of ubiquitinating and deubiquitinating enzymes is a common feature in various cancers" [12]. This dysregulation affects all recognized cancer hallmarks, including evading growth suppressors, reprogramming energy metabolism, unlocking phenotypic plasticity, and polymorphic microbiomes [12].
Figure 1: The Ubiquitination-Debuquitination Cascade. This diagram illustrates the sequential enzymatic actions of E1 (activation), E2 (conjugation), and E3 (ligation) enzymes in attaching ubiquitin to substrate proteins, targeting them for degradation or signaling modification. Deubiquitinating enzymes (DUBs) reverse this process, creating a dynamic regulatory equilibrium. Created with DOT language.
Methodological Framework for Ubiquitination Signature Development
Bioinformatics Pipelines and Computational Approaches
The development of ubiquitination-based prognostic signatures typically follows a standardized bioinformatics workflow that integrates multi-omics data from public repositories such as The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) [84] [49] [85]. The general methodological framework involves several key steps:
Data Acquisition and Preprocessing: RNA-sequencing data and clinical information are obtained from TCGA and GEO databases. Samples with incomplete pathological or survival information are typically excluded to ensure analytical rigor. Data normalization is performed using standardized approaches such as transcripts per kilobase of exon model per million mapped reads (TPM) [84].
Identification of Ubiquitination-Related Genes: UbRGs are compiled from specialized databases including iUUCD 2.0 and UbiBrowser 2.0 [84] [49]. Differential expression analysis between tumor and normal tissues is conducted using R packages like "limma" with thresholds typically set at false discovery rate (FDR) < 0.05 and |log2 fold change| > 1 [84].
Molecular Subtyping: Unsupervised consensus clustering utilizing algorithms such as "ConsensusClusterPlus" is employed to categorize patients into distinct molecular subtypes based on UbRG expression patterns. This clustering is typically repeated 1,000 times to ensure stability [49] [85].
Prognostic Model Construction: Least absolute shrinkage and selection operator (LASSO) Cox regression analysis is applied to identify the most prognostically significant UbRGs while preventing overfitting. Ten-fold cross-validation determines the optimal penalty parameter [84] [49] [86]. Multivariate Cox regression then establishes the final prognostic signature, with risk scores calculated using the formula: Risk score = Σ(βi × Expi), where β represents the coefficient from Cox regression and Exp denotes gene expression level [84].
Validation and Performance Assessment: Signatures are validated internally and externally using independent datasets. Kaplan-Meier analysis with log-rank tests evaluates survival differences between risk groups. Receiver operating characteristic (ROC) curves assess predictive accuracy for 1-, 3-, and 5-year overall survival [84] [49].
Functional Characterization and Immune Landscape Analysis
Additional analytical components include functional enrichment analysis using Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways to elucidate biological processes associated with ubiquitination signatures [84] [85]. Immune microenvironment characterization is performed using algorithms such as CIBERSORT and ESTIMATE to evaluate immune cell infiltration patterns [84] [49]. Drug sensitivity prediction utilizes computational tools like "oncoPredict" to calculate half-maximal inhibitory concentration (IC50) values for various chemotherapeutic and targeted agents [49] [86].
Figure 2: Bioinformatics Workflow for Ubiquitination Signature Development. This diagram outlines the standardized computational pipeline for developing prognostic ubiquitination signatures, from initial data acquisition through final validation and functional characterization. Created with DOT language.
Comparative Analysis of Ubiquitination Signatures Across Cancers
Signature Genes and Prognostic Value
Table 1: Comparative Analysis of Ubiquitination-Based Prognostic Signatures Across Cancer Types
| Cancer Type | Key Signature Genes | Sample Size | Prognostic Power | Clinical Applications | Reference |
|---|---|---|---|---|---|
| Laryngeal Cancer | PPARG, LCK, LHX1 | 116 TCGA + 46 GEO | Strong discrimination of OS in TCGA and validation set | Immune landscape indication, chemotherapy vs. immunotherapy guidance | [84] |
| Breast Cancer | 8-gene signature (including FBXL6, PDZRN3) | 700 TCGA + 460 GEO | Reliable prognostic stratification | Immune infiltration assessment, drug sensitivity prediction | [49] |
| Cervical Cancer | 13-gene signature (KLHL22, UBXN11, FBXO25, etc.) | 283 TCGA | High predictive accuracy | Immune microenvironment characterization, mutation distribution | [85] |
| Diffuse Large B-Cell Lymphoma | CDC34, FZR1, OTULIN | 1,800 samples across 3 datasets | Effective risk stratification | Immune score correlation, drug sensitivity (Osimertinib) | [86] |
| Bladder Cancer | 7-gene signature (EMP1, CLSTN2, SERPINB2, etc.) | 404 TCGA + GEO validation | Robust prognosis prediction | Immunotherapy response prediction, synergistic drug effects | [87] |
Immune Microenvironment and Therapeutic Implications
The relationship between ubiquitination signatures and tumor immune microenvironment represents a particularly significant finding across multiple cancer types. In laryngeal cancer, the study demonstrated that "the low-risk group had a more activated immune function, higher infiltration of anti-cancer immune cells and stronger expression of immune-promoting cytokines than the high-risk group" [84]. Additionally, researchers observed distinct correlations between individual signature genes and immune profiles: "PPARG and LHX1 were negatively correlated, whereas LCK positively correlated, with the immuno-promoting microenvironment" [84].
These immune microenvironment differences translate directly to therapeutic implications. For laryngeal cancer patients, the ubiquitination signature could guide treatment selection: "chemotherapy would be more effective in high-risk patients, while immune checkpoint inhibitors would be more effective in low-risk patients" [84]. Similarly, in breast cancer, ubiquitination-based subtyping revealed significant differences in "immune cell infiltration and immune function" that correlated with treatment responses [49].
Table 2: Therapeutic Implications of Ubiquitination Signatures in Cancer
| Cancer Type | Chemotherapy Implications | Immunotherapy Implications | Targeted Therapy Implications | Reference |
|---|---|---|---|---|
| Laryngeal Cancer | More effective in high-risk patients | Immune checkpoint inhibitors more effective in low-risk patients | Potential for ubiquitination-targeted therapies | [84] |
| Breast Cancer | Differential sensitivity to tamoxifen, fulvestrant, cyclophosphamide | Correlated with immune infiltration patterns | Response variations to gefitinib, lapatinib | [49] |
| Diffuse Large B-Cell Lymphoma | Sensitivity to Osimertinib identified | Associated with immune scores and microenvironment | Specific vulnerability to Boehringer Ingelheim compound 2536 | [86] |
| Bladder Cancer | EMP1 inhibition synergized with oxaliplatin | Predictive of immune checkpoint inhibitor response | SLC26A8 identified as protective factor | [87] |
| Non-Small Cell Lung Cancer | USP5 regulates radiosensitivity | USP family members control PD-L1 stability | PROTACs and molecular glues show promise | [88] [83] |
Experimental Validation and Functional Studies
In Vitro and In Vivo Validation Approaches
The computational identification of ubiquitination signatures is consistently followed by experimental validation to confirm biological relevance and clinical applicability. Standard validation approaches include:
Western Blot and qRT-PCR: These techniques verify differential expression of signature genes at protein and mRNA levels in cancer cell lines compared to normal controls [84] [49]. For example, in laryngeal cancer, "dysregulation of the signature genes was confirmed in LC cell lines by Western blot" [84].
Gene Manipulation Studies: Knockdown or overexpression experiments elucidate functional roles of signature genes. In breast cancer research, "the effects of FBXL6 and PDZRN3 on breast carcinogenesis were experimentally verified" using siRNA and lentiviral transduction approaches [49]. Similarly, bladder cancer investigations demonstrated that "knockdown of SLC26A8 significantly promoted tumor progression in BLCA" [87].
Drug Sensitivity Assays: Cell viability assays measure IC50 values to confirm predicted chemosensitivity patterns. Research in bladder cancer validated that "EMP1 inhibition synergized with the antitumor effects of oxaliplatin in T24 and 5637 BLCA cell lines" [87].
Mechanistic Investigations: Co-immunoprecipitation and ubiquitination assays delineate molecular mechanisms. In NSCLC, studies revealed that "USP5 stabilizes the autophagy regulator Beclin1 to promote p53 degradation" [83], while separate research established that "USP5 regulates cancer progression and radiosensitivity by stabilizing HOXA10" [88].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for Ubiquitination Signature Investigation
| Reagent/Resource | Function/Application | Examples/Specifications | Reference |
|---|---|---|---|
| TCGA and GEO Datasets | Source of RNA-seq and clinical data | Preprocessed data from cancer patients and normal controls | [84] [49] |
| iUUCD 2.0 and UbiBrowser 2.0 | Ubiquitin-related gene databases | Comprehensive collections of UbRGs with functional annotations | [84] [49] |
| ConsensusClusterPlus | Molecular subtyping and classification | R package for unsupervised clustering | [49] [85] |
| glmnet R Package | LASSO Cox regression analysis | Variable selection and prognostic model construction | [84] [86] |
| CIBERSORT Algorithm | Immune cell infiltration analysis | Deconvolution of tumor immune microenvironment | [84] [49] |
| oncoPredict R Package | Drug sensitivity prediction | Calculation of IC50 values for chemotherapeutic agents | [49] [86] |
| TIDE Algorithm | Immunotherapy response prediction | Evaluation of tumor immune dysfunction and exclusion | [85] [87] |
Discussion and Clinical Translation
Concordance and Divergence Across Cancer Types
The comprehensive analysis of ubiquitination signatures across multiple malignancies reveals both conserved themes and cancer-specific peculiarities. A consistent observation across studies is the intimate connection between ubiquitination signatures and immune microenvironment composition. In laryngeal cancer, breast cancer, cervical cancer, and bladder cancer alike, ubiquitination-based risk stratification consistently correlates with distinct immune infiltration patterns and immune function activation [84] [49] [85]. This suggests a fundamental role for ubiquitination processes in shaping tumor-immune interactions regardless of tissue of origin.
Nevertheless, cancer-specific differences emerge in the particular biological processes most strongly associated with ubiquitination signatures. In cervical cancer, ubiquitination-related subtypes were particularly enriched in "covalent chromatin modification, mitochondrial protein complexes, and histone binding" pathways [85], while in DLBCL, ubiquitination signatures correlated strongly with "endocytosis-related mechanisms" [86]. These variations highlight the tissue-specific contexts through which ubiquitination processes influence cancer progression.
Therapeutic Implications and Clinical Applications
The potential clinical applications of ubiquitination signatures extend beyond mere prognostic prediction to active treatment guidance. As summarized in the laryngeal cancer study, these signatures are "valuable in clinical application, indicative of the immune microenvironment and beneficial for individualized treatment guidance" [84]. Several particularly promising clinical applications emerge across studies:
Treatment Selection Guidance: Ubiquitination signatures show consistent ability to predict differential responses to chemotherapy versus immunotherapy. The finding that "chemotherapy would be more effective in high-risk patients, while immune checkpoint inhibitors would be more effective in low-risk patients" [84] provides a clinically actionable decision-making framework.
Novel Therapeutic Target Identification: Signature genes frequently represent promising therapeutic targets themselves. For instance, in NSCLC, "USP5 stabilizes the autophagy regulator Beclin1 to promote p53 degradation" [83], suggesting USP5 inhibition as a potential therapeutic strategy. Similarly, in breast cancer, experimental validation of "FBXL6 and PDZRN3 on breast carcinogenesis" [49] nominates these genes as candidate drug targets.
Drug Repurposing Opportunities: Ubiquitination signatures can identify novel sensitivity patterns to existing drugs. The DLBCL study found "significant differences in immune scores and concentration for Boehringer Ingelheim compound 2536 and Osimertinib" between risk groups [86], suggesting these agents might be effectively repurposed in ubiquitination-defined DLBCL subtypes.
Limitations and Future Directions
Despite their promise, ubiquitination signature studies face several limitations that must be addressed in future research. Most signatures described to date derive from retrospective analyses of public databases, requiring prospective validation in clinically representative cohorts. The transition from transcriptomic signatures to functional protein ubiquitination status presents another challenge, as mRNA expression may not fully capture post-translational regulatory dynamics.
Technical considerations around implementation also merit attention. As noted in the bladder cancer study, the field requires "standardization of analytical pipelines and consensus on optimal UbRG panels" [87] to facilitate cross-study comparisons and clinical adoption. The integration of ubiquitination signatures with established clinical biomarkers represents another critical step for clinical translation.
Future research directions should prioritize several key areas. First, expanding ubiquitination signature validation across diverse racial and ethnic populations will ensure broader applicability. Second, integrating ubiquitination signatures with other molecular data types (genomic, proteomic, metabolomic) may yield more comprehensive predictive models. Finally, developing targeted therapeutic strategies based on signature findings—such as PROTACs or molecular glues targeting specific E3 ligases or DUBs—represents the ultimate translational objective.
Ubiquitination-based prognostic signatures represent a powerful emerging approach for dissecting cancer heterogeneity and guiding personalized treatment strategies. Across multiple cancer types, these signatures demonstrate robust prognostic value, consistent associations with immune microenvironment composition, and actionable implications for treatment selection. The convergence of findings from laryngeal cancer, breast cancer, cervical cancer, DLBCL, bladder cancer, and NSCLC strongly supports the fundamental importance of ubiquitination processes in cancer pathogenesis and treatment response.
While methodological standardization and prospective validation remain necessary before widespread clinical implementation, the consistent demonstration of ubiquitination signatures' predictive power across diverse malignancies suggests substantial clinical potential. As ubiquitination-targeted therapeutics continue to develop—including PROTACs, molecular glues, and specific E3 ligase or DUB inhibitors—these signatures may eventually guide not only treatment selection but also targeted ubiquitination modulation itself. The integration of ubiquitination signatures into multidimensional cancer diagnostic frameworks promises to advance personalized oncology by incorporating this crucial layer of post-translational regulatory information into clinical decision-making.
Ubiquitination, a crucial post-translational modification, has emerged as a fundamental regulatory mechanism in cancer pathogenesis across multiple tumor types. This reversible process involves the covalent attachment of ubiquitin molecules to target proteins, thereby regulating their stability, activity, and cellular localization through a coordinated enzymatic cascade involving E1 activating, E2 conjugating, and E3 ligase enzymes [7] [83]. The ubiquitin-proteasome system (UPS) mediates more than 80% of protein degradation in eukaryotic organisms, and its dysregulation affects critical cellular processes including cell proliferation, differentiation, DNA repair, immune inflammation, and signal transduction [22]. In recent years, comprehensive multi-omics analyses have revealed that ubiquitination patterns demonstrate remarkable heterogeneity across cancer stages and subtypes, offering new insights into tumor classification, prognostic assessment, and therapeutic targeting [89] [33]. This comparative analysis synthesizes current understanding of ubiquitination-related molecular subtypes, their clinical implications, and experimental approaches for investigating ubiquitination patterns in human malignancies.
Molecular Landscapes of Ubiquitination Across Cancer Types
Hepatocellular Carcinoma (HCC)
In hepatocellular carcinoma, ubiquitination plays a multifaceted role in shaping tumor progression and the tumor microenvironment. Integrated multi-omics analysis of HCC tissues has demonstrated significant upregulation of ubiquitination-related genes in tumor tissues compared to normal adjacent tissues (NATs), with high expression levels correlating with poor patient prognosis [7]. A comprehensive 4D-label-free proteomics study that profiled nine PTM types in HCC revealed 21,453 ubiquitinated sites, with 2,765 sites quantified across samples [89]. This extensive ubiquitinome analysis demonstrated that ubiquitination abundance, along with most other PTMs except lactylation and ubiquitination itself, significantly distinguished tumor lesions from matched NATs.
Ubiquitination in HCC predominantly affects pathways involved in cell cycle regulation, DNA repair, metabolic reprogramming, and p53 signaling [7]. Notably, the ubiquitin-conjugating enzyme UBE2C has been identified as a key regulator in HCC, where its overexpression promotes tumor cell proliferation, invasion, and metastasis through in vitro experimental validation including transwell, CCK8, and wound healing assays [7]. Furthermore, UBE2C appears to facilitate immune evasion by inhibiting anti-tumor immune responses and reducing immune recognition of tumor cells, highlighting its potential as both a prognostic biomarker and therapeutic target in HCC [7].
Lung Adenocarcinoma (LUAD)
Lung adenocarcinoma exhibits distinct ubiquitination patterns that have been systematically characterized through multi-omics approaches. Research has identified 17 hub ubiquitination regulators (UBRs) from protein-protein interaction networks correlated with cancer hallmark-related pathways, with widespread genetic and transcriptional alterations observed in LUAD samples [33]. These hub UBRs demonstrate significant dysregulation, with most showing overexpression in cancer patients compared to normal tissues, and their high expression typically correlating with detrimental effects on patient survival [33].
Single-cell RNA sequencing analysis of the LUAD microenvironment has revealed that malignant cells exhibit elevated scores for ubiquitination enzymes and ubiquitin-binding domains compared to normal epithelial cells [90]. This approach identified 53 ubiquitination-related molecules with prognostic significance, among which FGR, PSMD14, and ZBTB16 emerged as genes with particular prognostic value [90]. PSMD14, a critical deubiquitination enzyme, shows higher expression in epithelial and malignant cells and has been functionally validated to promote LUAD progression by stabilizing the AGR2 protein through reduced ubiquitination, leading to enhanced AGR2 stability and subsequent increases in cell viability, invasion, and migration [90].
Based on expression profiles of hub UBRs, LUAD patients can be classified into two ubiquitination subtypes with significant differences in survival rates, gender distribution, mutation burden, immune profiles, and therapeutic responses [33]. These molecular subtypes enable the development of ubiquitination-related risk (UBrisk) scoring systems that show predictive value for immunotherapy responses, with low-UBrisk patients more likely to benefit from immunotherapeutic interventions [33].
Breast Cancer (BC)
Breast cancer demonstrates extensive ubiquitination heterogeneity that has been leveraged for prognostic model development. Through analysis of 763 ubiquitination-related genes (UbRGs), researchers have established a reliable prognostic signature for breast cancer patients, identifying eight overall survival-associated UbRGs that effectively stratify patients into distinct risk categories [49]. This ubiquitination-based classification system demonstrates significant associations with tumor microenvironment characteristics and drug sensitivity patterns, particularly in response to endocrine therapy, chemotherapy, and targeted agents commonly used in breast cancer treatment [49].
Experimental validation in breast cancer models has confirmed the functional roles of specific ubiquitination-related genes in tumor progression. For instance, FBXL6 and PDZRN3 have been experimentally demonstrated to significantly influence breast carcinogenesis through in vitro and in vivo experiments [49] [53]. The ubiquitination patterns in breast cancer not only provide prognostic information but also offer insights into therapeutic resistance mechanisms, enabling more personalized treatment approaches based on ubiquitination profiles.
Bladder Cancer (BCa)
Bladder cancer exhibits distinct ubiquitination-related molecular subtypes with clinical implications. Research has identified four consensus molecular subtypes based on ubiquitination-related gene expression patterns, each demonstrating significantly different clinical characteristics, prognosis, PD-L1 expression levels, and tumor microenvironment composition [91]. These subtypes enable the development of a prognostic index utilizing six UbRGs (HLA-A, TMEM129, UBE2D1, UBE2N, UBE2T, and USP5) that effectively stratifies patients according to survival outcomes across multiple validation cohorts [91].
The ubiquitination-based classification system in bladder cancer demonstrates particular clinical utility for specific patient subgroups, showing enhanced prognostic performance in subtype 1 and 3 patients, older individuals, males, those with high-grade tumors, and patients with AJCC stage III-IV disease [91]. This molecular subtyping approach provides a framework for understanding the biological heterogeneity of bladder cancer and potentially guiding therapeutic decision-making.
Laryngeal Squamous Cell Carcinoma (LSCC)
In laryngeal squamous cell carcinoma, ubiquitination-related biomarkers show promising diagnostic and prognostic potential. Transcriptomics-based analysis has identified four key biomarkers (WDR54, KAT2B, NBEAL2, and LNX1) through integrative bioinformatics approaches combining differential expression analysis, weighted gene co-expression network analysis (WGCNA), and machine learning algorithms [92]. These biomarkers demonstrate significant associations with cancer pathways and show accurate performance in predicting clinical aspects of LSCC, as validated through receiver operating characteristic (ROC) curve analysis [92].
The ubiquitination landscape in LSCC appears to be regulated by specific transcription factors (BRD4, MYC, AR, and CTCF) that control the expression of these ubiquitination-related biomarkers [92]. Experimental validation using reverse transcription-quantitative PCR (RT-qPCR) has confirmed the expression patterns of KAT2B, LNX1, and NBEAL2 in clinical samples, supporting their potential utility as clinical biomarkers for LSCC diagnosis and treatment [92].
Table 1: Ubiquitination Patterns Across Cancer Types
| Cancer Type | Key Ubiquitination Regulators | Molecular Subtypes | Clinical Implications |
|---|---|---|---|
| Hepatocellular Carcinoma | UBE2C, MDM2, USP7, USP10 | Not specified | Poor prognosis, promotes proliferation and immune evasion [7] |
| Lung Adenocarcinoma | PSMD14, UBE2T, AURKA, CDC20, BRCA1 | Two ubiquitination subtypes | Distinct survival, gender ratio, mutation burden, and immunotherapy response [33] [90] |
| Breast Cancer | FBXL6, PDZRN3, RNF126 | Clusters based on 763 UbRGs | Prognostic stratification, drug sensitivity prediction [49] |
| Bladder Cancer | HLA-A, TMEM129, UBE2D1, UBE2N, UBE2T, USP5 | Four molecular subtypes | Prognostic stratification, associated with PD-L1 expression and TME [91] |
| Laryngeal Squamous Cell Carcinoma | WDR54, KAT2B, NBEAL2, LNX1 | Not specified | Diagnostic and prognostic biomarkers [92] |
Experimental Approaches for Ubiquitination Analysis
Multi-Omics Profiling Techniques
Comprehensive ubiquitination analysis requires integrated multi-omics approaches that simultaneously capture multiple dimensions of molecular information. The 4D-label-free proteomics technique combined with PTM-specific antibody enrichment represents a cutting-edge methodology for system-level ubiquitinome characterization [89]. This approach enables robust identification and quantification of thousands of PTM sites across multiple modification types, facilitating the detection of differentially modified sites between tumor and normal tissues. In typical experimental workflows, protein extracts from tumor tissues and paired normal adjacent tissues are digested, followed by peptide enrichment using PTM-specific antibodies, and ultimately analyzed through liquid chromatography-tandem mass spectrometry (LC-MS/MS) [89].
Bioinformatics processing of multi-omics data involves several critical steps: Gaussian kernel density estimation for assessing intensity distributions, principal component analysis for distinguishing modification patterns between sample types, and Spearman's correlation analysis for investigating relationships between modification abundance and protein expression [89]. Differential modification analysis typically employs thresholds such as log₂ (tumor vs. NAT) > 0.58 (fold change > 1.50) for upregulated sites and log₂ (tumor vs. NAT) < -0.58 (fold change < 0.67) for downregulated sites to identify significant alterations in ubiquitination patterns [89].
Single-Cell RNA Sequencing Applications
Single-cell RNA sequencing has emerged as a powerful tool for delineating ubiquitination heterogeneity within the tumor microenvironment. Analytical workflows for scRNA-seq data in ubiquitination research typically involve multiple stages: data normalization and quality control, cell clustering and annotation, malignant cell identification using tools like InferCNV, and assessment of ubiquitination enzyme activity through AUCell scoring [90]. This approach enables the characterization of cell-type-specific ubiquitination patterns and identifies subpopulations with distinct ubiquitination signatures that may drive tumor progression or therapeutic resistance.
The application of single-cell sequencing to ubiquitination analysis in LUAD has demonstrated that malignant cells exhibit elevated scores for ubiquitination enzymes and ubiquitin-binding domains compared to normal epithelial cells, highlighting the potential of this approach for identifying tumor-specific ubiquitination vulnerabilities [90]. Furthermore, integration of scRNA-seq data with bulk transcriptome analyses enables the validation of key ubiquitination-related molecules and their association with clinical outcomes.
Functional Validation Methodologies
Experimental validation of ubiquitination-related targets employs a range of molecular and cellular techniques to establish functional mechanisms:
Gene Knockdown Approaches: Plasmid-based shRNA systems or siRNA oligonucleotides are used to target specific ubiquitination enzymes, with knockdown efficiency typically validated through qRT-PCR and Western blot analysis [7] [53]. Stable knockdown cell lines are often established through lentiviral infection and puromycin selection [7].
In Vitro Functional Assays: Transwell assays evaluate cell migration and invasion capabilities; CCK-8 assays measure cell viability and proliferation; wound healing assays assess collective cell migration; and clonogenic formation assays determine colony-forming efficiency [7].
Protein Interaction and Stability Analysis: Co-immunoprecipitation assays investigate physical interactions between ubiquitination enzymes and substrate proteins; cycloheximide chase experiments monitor protein half-life; and ubiquitination assays detect specific protein ubiquitination status [90].
In Vivo Validation: Xenograft mouse models assess tumor formation and growth, providing physiological context for in vitro findings [90].
Diagram 1: Experimental workflow for ubiquitination pattern analysis
Comparative Ubiquitination Patterns: Normal vs. Cancerous Tissues
Quantitative Ubiquitination Alterations
Systematic comparisons between cancerous and normal tissues reveal consistent patterns of ubiquitination dysregulation across multiple cancer types. Large-scale proteomic studies in HCC have demonstrated that most PTMs, except N-glycosylation and malonylation, show increased abundance in tumor lesions compared to normal adjacent tissues [89]. This pattern of widespread ubiquitination upregulation in tumors suggests a fundamental role for ubiquitination in oncogenic processes.
The magnitude of ubiquitination alterations varies considerably across cancer types and specific ubiquitination enzymes. For instance, in the pan-cancer analysis of UBA family enzymes, UBA1 and UBA6 demonstrate significantly elevated expression in most cancer types compared to normal tissues, with expression levels correlating with poor prognosis, advanced clinical stages, and specific immune infiltration patterns [22]. These conserved alterations highlight the potential of ubiquitination enzymes as broad biomarkers for cancer detection and monitoring.
Cancer-Specific Ubiquitination Signatures
While general patterns of ubiquitination dysregulation exist across cancers, each cancer type also exhibits distinct ubiquitination signatures that reflect tissue-specific oncogenic mechanisms:
HCC: Shows enrichment of ubiquitination in proteins containing RRM1 domains, with phosphorylation and acylation of splicing factors like NCL at multiple residues (S67, K398, K646) cooperatively regulating RNA processing [89].
LUAD: Demonstrates predominant ubiquitination alterations in EGFR and KRAS signaling pathways, with specific E3 ligases and deubiquitinases contributing to targeted therapy resistance [83] [33].
Breast Cancer: Exhibits ubiquitination patterns associated with estrogen receptor status, with different ubiquitination signatures in triple-negative versus hormone receptor-positive subtypes [49].
Bladder Cancer: Shows four distinct ubiquitination molecular subtypes with differential responses to immunotherapy and varying PD-L1 expression levels [91].
These cancer-specific ubiquitination patterns not only provide insights into tissue-specific oncogenic mechanisms but also offer opportunities for developing targeted therapeutic interventions tailored to specific cancer types.
Table 2: Key Ubiquitination-Related Enzymes as Therapeutic Targets
| Enzyme | Cancer Type | Function | Therapeutic Approach |
|---|---|---|---|
| UBE2C | HCC | Promotes proliferation, invasion, immune evasion | Potential target for precision therapy [7] |
| PSMD14 | LUAD | Stabilizes AGR2 protein, promotes malignancy | Deubiquitinase inhibitor [90] |
| UBA1 | Pan-cancer | Ubiquitin-activating enzyme, elevated in multiple cancers | MLN4924 (E1 inhibitor) [83] [22] |
| FBXL6 | Breast Cancer | Promotes carcinogenesis | Targeted degradation [49] |
| USP21 | LSCC | Stabilizes AURKA through deubiquitination | Deubiquitinase inhibitor [92] |
Comprehensive ubiquitination research requires access to specialized databases and bioinformatics tools:
iUUCD 2.0 Database: Provides integrated annotations for ubiquitin and ubiquitin-like conjugation systems, containing 1,393 ubiquitination-related genes essential for UbRG identification [49] [92].
TCGA (The Cancer Genome Atlas): Offers multi-omics data across multiple cancer types, enabling comparative analysis of ubiquitination patterns [91] [49] [33].
GEO (Gene Expression Omnibus): Repository of transcriptome datasets for independent validation of ubiquitination signatures [49] [90] [92].
STRING Database: Facilitates protein-protein interaction network analysis for identifying hub ubiquitination regulators [33].
UALCAN Platform: Enables analysis of gene expression differences between cancer and normal tissues across multiple tumor types [22].
Experimental Reagents and Assay Systems
Functional validation of ubiquitination mechanisms requires specific research tools:
PTM-Specific Antibodies: Essential for enrichment of ubiquitinated peptides in proteomic studies; quality critically impacts detection sensitivity [89].
shRNA/siRNA Libraries: Enable targeted knockdown of ubiquitination enzymes; lentiviral systems facilitate stable gene suppression [7] [53].
Proteasome Inhibitors: Compounds like bortezomib used to investigate ubiquitination-dependent protein degradation [83].
PROTACs (Proteolysis Targeting Chimeras): Heterobifunctional molecules that recruit E3 ligases for targeted protein degradation [83].
Deubiquitinase Inhibitors: Small molecules targeting specific DUBs for functional studies and therapeutic exploration [83] [90].
Diagram 2: Ubiquitination enzymatic cascade and regulatory nodes
Comparative analysis of ubiquitination patterns across cancer stages and subtypes reveals both conserved and tissue-specific mechanisms of oncogenic transformation. The consistent upregulation of ubiquitination activity across multiple cancer types suggests a fundamental role in tumorigenesis, while the distinct molecular subtypes within each cancer indicate context-specific functions. The development of ubiquitination-based classification systems and risk scores demonstrates clinical potential for prognostic stratification and treatment selection, particularly in the context of immunotherapy.
Future research directions should focus on elucidating the spatial organization of ubiquitination patterns within the tumor microenvironment, understanding dynamic ubiquitination changes during disease progression and therapeutic intervention, and developing selective modulators of ubiquitination enzymes for targeted therapy. The integration of ubiquitination profiling into clinical decision-making represents a promising avenue for advancing precision oncology and improving patient outcomes across multiple cancer types.
The ubiquitin-proteasome system (UPS) is a crucial regulatory mechanism responsible for the controlled degradation of intracellular proteins in eukaryotic organisms, mediating more than 80% of protein degradation [13] [93]. This system utilizes a cascade of enzymatic reactions involving ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3) to tag proteins for proteasomal degradation or alter their function and localization [94] [93]. The UPS maintains cellular homeostasis by regulating essential processes including cell cycle progression, DNA damage repair, apoptosis, and immune response [13] [93]. Dysregulation of UPS components can lead to aberrant stabilization of oncoproteins or accelerated degradation of tumor suppressors, making various E1, E2, and E3 enzymes potential biomarkers and therapeutic targets in cancer [94]. This review comprehensively compares the biomarker potential of four key UPS enzymes—UBA1 (E1), UBA6 (E1), UBE2C (E2), and MDM2 (E3)—across different cancer types, focusing on their expression patterns, prognostic significance, and roles within the tumor immune microenvironment.
Enzyme-Specific biomarker Profiles and Case Studies
UBA1: The Canonical E1 Enzyme in Cancer
Biological Function: UBA1 is the primary and most abundant ubiquitin-activating enzyme in humans, initiating the ubiquitination cascade for most cellular proteins by activating ubiquitin in an ATP-dependent manner and transferring it to E2 enzymes [13] [95]. It is highly conserved across eukaryotes and represents a fundamental component of the protein degradation machinery [95].
Expression Patterns and Prognostic Significance: UBA1 demonstrates significantly elevated expression across multiple cancer types compared to normal tissues. A comprehensive pan-cancer analysis revealed UBA1 overexpression in 16 different tumor types, including breast cancer (BRCA), colorectal cancer (COAD), renal cancer (KIRC), and lung adenocarcinoma (LUAD) [13] [95]. This elevated expression correlates strongly with poor patient outcomes across several metrics. In breast cancer, high UBA1 expression is associated with worse overall survival (OS), disease-specific survival (DSS), and progression-free survival (PFS) [95]. The protein is also linked to advanced disease stages in specific tumor types, suggesting its potential role in cancer progression [13].
Table 1: UBA1 as a Cancer Biomarker Across Multiple Malignancies
| Cancer Type | Expression Pattern | Prognostic Significance | Clinical Correlations |
|---|---|---|---|
| Breast Cancer (BRCA) | Significantly elevated in tumor tissues [95] | Worse OS, DSS, and PFS [95] | Associated with T stage and histological type [95] |
| Colorectal Cancer (COAD) | Highly expressed in tumor tissues [13] | Poor prognosis [13] | Correlated with advanced stages [13] |
| Lung Adenocarcinoma (LUAD) | Elevated in cancer tissues [13] | Reduced survival [13] | Associated with tumor stage [13] |
| Renal Cancer (KIRC) | Overexpressed compared to normal tissue [13] | Poor prognosis [13] | Linked to advanced disease [13] |
| Glioblastoma (GBM) | Critical for cell survival [13] | NA (preclinical data) | Inhibition suppresses survival and proliferation [13] |
Functional Mechanisms in Tumorigenesis: UBA1 contributes to cancer progression through multiple mechanisms. In leukemia and myeloma models, UBA1 knockdown resulted in decreased ubiquitinated proteins and increased cancer cell death, while in vivo inhibition significantly reduced tumor weight and volume [13]. In glioblastoma, UBA1 inhibition reduced ubiquitination, induced endoplasmic reticulum stress and unfolded protein response, and suppressed survival, proliferation, and colony formation of GBM cell lines [13]. Beyond its direct roles in tumor cell survival, UBA1 significantly influences the tumor immune microenvironment. In breast cancer, elevated UBA1 expression demonstrates a negative correlation with infiltrating anti-tumor immune cells including mast cells, Th1 cells, dendritic cells, B cells, CD8+ T cells, and cytotoxic cells [95]. Functional enrichment analyses indicate UBA1's involvement in key inflammatory pathways such as TNF-α signaling, IL-6/JAK/STAT3 signaling, and KRAS signaling pathways [95].
Therapeutic Targeting Potential: The essential role of UBA1 in cancer cells positions it as a promising therapeutic target. Preclinical studies demonstrate that UBA1 silencing significantly impedes the growth and development of breast cancer cell lines [95]. Similarly, in colon cancer models, UBA1 knockdown inhibits tumor growth in xenograft models [96]. These findings collectively highlight UBA1's dual utility as both a prognostic biomarker and potential therapeutic target across diverse malignancies.
UBA6: The Non-Canonical E1 Enzyme with Tumor-Suppressive Properties
Biological Function: UBA6 is a non-canonical ubiquitin-activating enzyme found only in vertebrates and sea urchins, sharing approximately 40% sequence homology with UBA1 [13] [96]. This E1 enzyme exhibits dual specificity, capable of activating both ubiquitin and the ubiquitin-like protein FAT10 [13] [96]. UBA6 functions with its specific E2 enzyme, UBE2Z/Use1, while also sharing some E2 enzymes with UBA1 [96].
Expression Patterns and Functional Roles: Unlike UBA1 which generally demonstrates oncogenic properties, UBA6 exhibits context-dependent roles in cancer development, often functioning as a tumor suppressor. In breast cancer, UBA6 expression is low or undetectable in approximately 38% of invasive carcinomas, suggesting that its downregulation may contribute to cancer development [96]. Experimental models demonstrate that UBA6-deficient mammary epithelial cells fail to establish proper cell cycle arrest in response to detachment from extracellular matrix, fully engaged cell-cell contact, or growth factor deprivation [96]. Furthermore, UBA6-deficient cells undergo spontaneous epithelial-mesenchymal transition (EMT) under growth factor deprivation and exhibit accelerated kinetics of TGF-β-induced EMT [96].
Table 2: Comparative Analysis of E1 Enzymes UBA1 and UBA6 in Cancer
| Characteristic | UBA1 | UBA6 |
|---|---|---|
| Enzyme Type | Canonical E1 [13] | Non-canonical E1 [96] |
| Specificity | Ubiquitin only [13] | Ubiquitin and FAT10 [13] [96] |
| Primary Cancer Role | Oncogenic [13] [95] | Tumor suppressive [96] |
| Expression Pattern | Overexpressed in multiple cancers [13] [95] | Downregulated in breast and other cancers [96] |
| Immune Modulation | Suppresses anti-tumor immunity [95] | Regulates T cell function and inflammation [13] |
| Therapeutic Implications | Inhibition suppresses tumor growth [13] [95] | Loss promotes EMT and tumor progression [96] |
| Key Pathways | TNF-α, IL-6/JAK/STAT3, KRAS signaling [95] | EMT, Rho-GTPase signaling, cell polarity [96] |
Mechanisms of Tumor Suppression: UBA6 establishes a suppressive barrier against critical steps of mammary carcinogenesis including loss of polarity, anoikis resistance, and epithelial-mesenchymal transition [96]. The Rho-GTPase CDC42 has been identified as a specific target of UBA6-initiated ubiquitination and plays a key role in UBA6's function in controlling epithelial homeostasis [96]. UBA6 is required for proper mammary epithelial morphogenesis, with UBA6-deficient cells failing to form hollow lumen structures in 3D culture and instead developing tumor-like gigantic aggregates [96]. These deficiency also leads to loss of contact inhibition, with cells continuing to proliferate without proper cell cycle arrest and exhibiting mesenchymal characteristics including upregulated N-cadherin and vimentin with concurrent downregulation of E-cadherin and γ-catenin [96].
Immune Regulatory Functions: UBA6 plays distinct roles in immune regulation compared to UBA1. UBA6 expression increases in T cells but shows lower or absent expression in dendritic cells, macrophages, B cells and natural killer cells [13]. Deficiency of UBA6 in T cells leads to increased intracellular IFN-γ expression and subsequent multi-organ inflammation in mice [13]. This immunomodulatory function, combined with its role in regulating FAT10-mediated degradation of the tumor suppressor p53, positions UBA6 as a significant regulator of tumor immunogenicity and a potential target for enhancing anti-tumor immune responses [13].
UBE2C: The Cell Cycle-Regulating E2 Enzyme
Biological Function: UBE2C is a ubiquitin-conjugating enzyme (E2) that plays a critical role in cell cycle progression and checkpoint control by directing the polyubiquitination of key regulatory proteins [97]. It is particularly essential for the destruction of mitotic cyclins and the regulation of the spindle assembly checkpoint, ensuring proper chromosome segregation during cell division [97].
Expression Patterns and Prognostic Significance: UBE2C is frequently overexpressed in a wide range of human malignancies. In hepatocellular carcinoma (HCC), UBE2C expression is significantly elevated in tumor tissues compared to normal samples, with higher expression closely associated with advanced tumor grade and stage [98]. Patients with high UBE2C expression demonstrate significantly shorter survival compared to those with low expression, with a hazard ratio (HR) of 1.870 [98]. Similar patterns of UBE2C overexpression are observed in numerous other cancers including breast cancer, where it is highly expressed in microcalcification lesions, esophageal squamous cell carcinoma, and various other solid tumors and hematological malignancies [97] [98]. The expression level typically correlates with tumor aggressiveness, making UBE2C a valuable prognostic indicator across multiple cancer types [97].
Functional Mechanisms in Tumorigenesis: UBE2C promotes tumorigenesis primarily through its role in cell cycle regulation. Cells overexpressing UBE2C ignore mitotic spindle checkpoint signals and exhibit chromosomal instability, a hallmark of cancer [97]. UBE2C amplification occurs at the chromosome level in cancers in a manner similar to known oncogenes like c-Myc, which is directly upstream of UBE2C [97]. Protein-protein interaction analyses reveal high correlation between UBE2C and other cell cycle-related proteins, consistent with its fundamental role in mitotic regulation [98]. Beyond its cell cycle functions, UBE2C also influences the tumor immune microenvironment. In hepatocellular carcinoma, highly expressed UBE2C associates with an increased number of immunosuppressive molecules, suggesting potential mechanisms through which it may contribute to immune evasion [98].
Diagnostic and Therapeutic Implications: The consistent overexpression pattern across diverse malignancies positions UBE2C as a promising diagnostic biomarker. Its immunohistochemical detection has been proposed for integration into the diagnosis of thyroid malignancies and gliomas [97]. Furthermore, UBE2C demonstrates potential as an independent predictive marker for HCC patients, with its prognostic value for survival improved when combined with clinical stage information [98]. These findings collectively highlight UBE2C as both a robust prognostic biomarker and a potential therapeutic target deserving of further investigation.
MDM2: The Master Regulator of p53 and E3 Ligase
Biological Function: MDM2 (mouse double minute 2; HDM2 in humans) is a RING domain-containing E3 ubiquitin ligase that serves as the primary negative regulator of the tumor suppressor p53 [94]. It contains a p53-binding domain at its N-terminus and a RING domain with E3 ligase activity at its C-terminus, enabling it to both bind p53 and promote its ubiquitin-mediated degradation [94].
Oncogenic Mechanisms: MDM2 drives tumor development through both p53-dependent and independent mechanisms. Its best-characterized function involves binding p53 and promoting its proteasomal degradation, effectively neutralizing this critical tumor suppressor pathway [94]. In normal cellular conditions, MDM2 maintains p53 at low levels, but overexpression of MDM2 results in excessive p53 degradation, allowing uncontrolled cell proliferation [94]. MDM2 itself is frequently overexpressed in various cancers, and this overexpression correlates with poor prognosis and increased chemoresistance [94]. Beyond its regulation of p53, MDM2 also exhibits p53-independent oncogenic activities through interactions with other regulatory proteins including the retinoblastoma protein (pRb) and modulation of E2F1 activity [94].
Therapeutic Targeting Strategies: MDM2 represents a promising therapeutic target, particularly in cancers retaining wild-type p53. Several targeting approaches have been developed:
Compounds disrupting MDM2-p53 interaction: Several small molecule classes including Nutlin, RITA, and MI-17 have been developed to disrupt MDM2-p53 binding, leading to p53 stabilization and activation in cancer cells [94].
MDM2 E3 ligase inhibitors: The HLI98 series of small molecules was identified through high-throughput screening using an in vitro MDM2 autoubiquitination assay [94]. These compounds inhibit MDM3 activity and stabilize both p53 and MDM2, activating p53-dependent transcription and apoptosis [94].
PROTAC applications: MDM2 has emerged as a therapeutically pivotal candidate in proteolysis-targeting chimeras (PROTACs), serving both as an intrinsic E3 ligase for degrading other proteins of interest and as a direct target for degradation itself [99]. MDM2-harnessing PROTACs utilize MDM2's E3 ligase activity to degrade specific target proteins, while MDM2-targeted PROTACs directly degrade MDM2 itself, offering innovative approaches for therapeutic intervention [99].
Table 3: Comparative Biomarker Potential of UPS Enzymes Across Cancer Types
| Enzyme | Enzyme Class | Key Cancer Types | Expression in Cancer | Prognostic Value | Immune Microenvironment Role |
|---|---|---|---|---|---|
| UBA1 | E1 | Breast, colorectal, renal, lung adenocarcinoma [13] [95] | Overexpressed [13] [95] | Poor survival [13] [95] | Reduces cytotoxic cells, CD8+ T cells, DCs [95] |
| UBA6 | E1 | Breast cancer [96] | Downregulated [96] | Tumor suppressive role [96] | Regulates T cell function and inflammation [13] |
| UBE2C | E2 | Hepatocellular, breast, esophageal, thyroid [97] [98] | Overexpressed [97] [98] | Poor survival [97] [98] | Increases immunosuppressive molecules [98] |
| MDM2 | E3 | Various cancers with wild-type p53 [94] | Overexpressed [94] | Poor prognosis, chemoresistance [94] | Regulates p53-mediated immune responses [94] |
Comparative Analysis of Ubiquitination Patterns: Cancerous vs. Normal Tissues
The ubiquitination enzymes UBA1, UBA6, UBE2C, and MDM2 demonstrate distinctive yet interconnected roles in carcinogenesis, each contributing to the altered ubiquitination patterns observed in malignant tissues compared to their normal counterparts.
Divergent Expression Patterns: Malignant transformation is characterized by consistent overexpression of UBA1, UBE2C, and MDM2 across multiple cancer types, while UBA6 demonstrates frequent downregulation, particularly in invasive breast carcinomas [13] [96] [95]. These expression patterns reflect the distinct functional roles of these enzymes—UBA1, UBE2C, and MDM2 primarily drive oncogenic processes, while UBA6 appears to function as a tumor suppressor in specific contexts. The overexpression of UBE2C is particularly notable for its correlation with tumor grade and stage, suggesting its potential utility as a marker for disease progression [97] [98].
Prognostic Implications: Elevated expression of UBA1, UBE2C, and MDM2 consistently correlates with poor patient outcomes across various malignancies [13] [97] [95]. For UBA1, this association extends across overall survival, disease-specific survival, and progression-free survival in breast cancer patients [95]. Similarly, UBE2C overexpression predicts reduced survival in hepatocellular carcinoma patients with a hazard ratio of 1.870 [98]. MDM2 overexpression is linked to poor prognosis and increased resistance to chemotherapy [94]. In contrast, UBA6 loss is associated with features of aggressive disease including spontaneous EMT and loss of growth control, positioning it as a potential marker of disease progression [96].
Impact on Tumor Immune Microenvironment: These ubiquitination enzymes significantly influence the composition and function of the tumor immune microenvironment through distinct mechanisms. UBA1 expression demonstrates a negative correlation with multiple anti-tumor immune cell populations including cytotoxic cells, CD8+ T cells, and dendritic cells [95]. UBA6 plays a role in regulating T cell function and inflammation, with deficiency leading to increased IFN-γ expression and multi-organ inflammation in murine models [13]. UBE2C expression associates with increased immunosuppressive molecules in the tumor microenvironment [98], while MDM2 primarily modulates immune responses through its regulation of p53, which in turn controls various immunomodulatory pathways [94].
Methodologies for Studying Ubiquitination Enzymes in Cancer Research
Experimental Protocols for Biomarker Validation
Multi-Omics Database Mining: Comprehensive analysis of ubiquitination enzymes as cancer biomarkers typically begins with mining multi-omics data from public repositories. The Cancer Genome Atlas (TCGA) database serves as a primary resource for pan-cancer data including survival statistics, clinical information, stemness scores, and immunological subtypes [13] [22]. Researchers utilize platforms like UCSC Xena for data access and extraction, followed by differential expression analysis using statistical methods such as the Wilcox test to compare normal and malignant tissues [13] [22]. Additional databases including UALCAN provide clinical data across various cancer types for examining gene expression differences between specific cancers and normal tissues [13] [22]. The Human Protein Atlas (HPA) offers immunohistochemical expression data in both normal and malignant tissues for protein-level validation [13] [22].
Survival and Clinical Correlation Analysis: To establish prognostic significance, researchers typically perform survival analysis using R packages such as "survival" and "survminer" with patient data from TCGA [13] [22]. This involves categorizing patients into high and low expression groups based on median expression values and comparing survival outcomes using Kaplan-Meier curves and Cox proportional hazards models [13] [95]. Clinical correlation analysis examines relationships between enzyme expression and clinicopathological parameters such as tumor stage, grade, and histological type through univariate logistic regression [95]. Validation using independent datasets from Gene Expression Omnibus (GEO) through platforms like Km-Plotter further strengthens prognostic assessments [13] [22].
Immune Infiltration Assessment: Evaluating the relationship between ubiquitination enzymes and the tumor immune microenvironment employs several computational approaches. The TISIDB database is frequently utilized to examine associations between gene expression and immune subtypes [13] [22]. CIBERSORT algorithm enables assessment of correlations between target gene expression and the relative proportions of 22 types of tumor-infiltrating immune cells [13]. ESTIMATE and similar algorithms calculate immune scores, stromal scores, and estimate scores to characterize the tumor microenvironment [13]. Additional analyses often include examination of relationships with immune checkpoints, tumor mutational burden (TMB), and microsatellite instability (MSI) using R packages with statistical validation [13].
Table 4: Essential Research Reagents and Databases for Studying Ubiquitination Biomarkers
| Resource/Reagent | Type | Primary Application | Key Features |
|---|---|---|---|
| TCGA Database | Genomic database | Pan-cancer data analysis [13] [22] | Multi-omics data for 33+ cancer types with clinical annotations |
| UCSC Xena | Platform | TCGA data access and integration [13] [22] | User-friendly interface for genomic and clinical data |
| UALCAN | Web resource | Gene expression analysis [13] [22] | Query gene expression across cancer subtypes and normal tissues |
| Human Protein Atlas | Database | Protein expression validation [13] [22] | Immunohistochemistry data for normal and cancer tissues |
| TISIDB | Web portal | Immunogenomics analysis [13] [22] | Integrates tumor-immune system interactions |
| GSCA | Platform | Genomic mutational landscape [13] [22] | Analysis of SNV, CNV, and their survival impact |
| CIBERSORT | Algorithm | Immune cell infiltration estimation [13] | Deconvolutes immune cell fractions from bulk RNA-seq |
| ESTIMATE | Algorithm | Tumor microenvironment scoring [13] | Calculates immune and stromal scores |
| GeneMANIA | Web tool | Protein-protein interaction networks [13] [22] | Functional association data and interaction networks |
| CellMiner | Database | Drug sensitivity analysis [22] | NCI-60 drug screening data for correlation studies |
Visualizing Ubiquitination Enzyme Networks in Cancer
The following pathway diagrams illustrate the functional relationships and experimental workflows relevant to studying ubiquitination enzymes as cancer biomarkers.
Ubiquitination Cascade and Cancer Signaling Pathways
Ubiquitination Enzymes in Cancer Pathways
Multi-Omics Analysis Workflow for Biomarker Validation
Biomarker Validation Workflow
The comprehensive analysis of UBA1, UBA6, UBE2C, and MDM2 across multiple cancer types reveals both shared and distinctive roles in tumorigenesis, positioning them as valuable biomarkers with clinical utility. UBA1 demonstrates consistent overexpression and correlation with poor prognosis across diverse malignancies, coupled with significant effects on the tumor immune microenvironment. UBA6 exhibits tumor-suppressive properties in breast cancer models, with loss of expression promoting EMT and disease progression. UBE2C emerges as a robust prognostic indicator linked to cell cycle dysregulation and immune suppression. MDM2 remains a critical regulator of p53 function with well-established oncogenic roles and emerging applications in targeted protein degradation strategies.
Future research directions should focus on validating these biomarkers in prospective clinical trials, developing standardized detection assays for clinical implementation, and exploring therapeutic interventions targeting these enzymes either directly or through novel approaches like PROTAC technology. The integration of these ubiquitination enzymes into multi-marker panels combining traditional clinicopathological factors may enhance prognostic accuracy and guide personalized treatment decisions. Additionally, further investigation into the interplay between these enzymes and the tumor immune microenvironment may uncover novel immunotherapeutic strategies. As our understanding of the ubiquitin code in cancer biology continues to expand, so too will the clinical applications of these critical regulatory enzymes as biomarkers and therapeutic targets.
Ubiquitination, a crucial post-translational modification, regulates protein stability, activity, and localization, influencing fundamental cellular processes. Dysregulation of ubiquitination signaling is intimately linked to cancer pathogenesis, affecting tumor cell proliferation, invasion, immune evasion, and metabolic reprogrammation [7] [5]. Preclinical models capable of recapitulating these complex mechanisms are therefore indispensable for translating basic research into clinical applications. This guide systematically compares contemporary in vitro and in vivo models used for functional assessment of ubiquitination in cancer, providing researchers with a framework for model selection and methodological implementation within the context of comparing ubiquitination patterns in cancerous versus normal tissues.
In Vitro Models for Ubiquitination Research
In vitro models provide controlled environments for mechanistic studies and high-throughput screening of ubiquitination-targeting therapeutics.
Established Cancer Cell Lines
Experimental Protocols from Cited Studies:
- Cell Culture: Human hepatocellular carcinoma (HCC) cell lines (Huh7, Hep3B) are maintained in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/L penicillin, and 100 mg/L streptomycin at 37°C in a 5% CO₂ environment [7].
- Gene Manipulation: Plasmid-based shRNA or siRNA transfection using Lipofectamine 3000. For ubiquitination enzyme studies, UBE2C shRNA constructs are delivered via viral supernatants with polybrene, followed by puromycin selection (2.0 μg/mL) [7].
- Functional Assays:
- Transwell Invasion: Seed 5 × 10⁴ cells into Matrigel-coated chambers with serum-free medium upper chamber and complete DMEM lower chamber. Fix migrated cells after 24h with 4% paraformaldehyde, stain with 0.1% crystal violet, and quantify [7].
- CCK-8 Viability: Plate 2500 cells/well in 96-well plates. Add 10 μL CCK-8 reagent per well, incubate 4h at 37°C, and measure OD450 [7].
- Wound Healing: Create uniform scratch in confluent cell monolayer with sterile pipette tip. Monitor wound closure at 0h and 24h in serum-free medium using inverted microscopy [7].
Table 1: Quantitative Functional Data from Cancer Cell Line Studies
| Cancer Type | Ubiquitination Factor | Proliferation Change | Invasion/Migration Change | Key Pathways Affected |
|---|---|---|---|---|
| Hepatocellular Carcinoma | UBE2C overexpression | Significant increase (CCK-8) | Significant promotion (Transwell) | Cell cycle regulation, p53 signaling [7] |
| Prostate Cancer | OTUB1 knockdown | No significant effect | ~50% reduction (Matrigel invasion) | RhoA activation, p53 phosphorylation [100] |
| Prostate Cancer | OTUB1 catalytically inactive mutant | Not reported | Diminished invasion | Androgen signaling modulation [100] |
| Ovarian Cancer | Proteasome inhibition (MG132) | Not reported | Not reported | Altered ubiquitin occupancy at distinct sites [101] |
Primary Cell and Organoid Models
Patient-derived organoids represent advanced in vitro models that better preserve tumor heterogeneity and tissue architecture.
Experimental Protocol:
- Organoid Establishment: Culture tumor cells in specialized matrices (e.g., Matrigel) with tissue-specific growth factor cocktails. Generate from fresh tumor specimens obtained during surgical resection with appropriate ethical approval [102].
- Drug Screening: Utilize high-throughput platforms to test therapeutic agents on patient-derived organoids. Integration with whole-exome sequencing enables correlation of ubiquitination-related genetic alterations with drug response [102].
Applications: This model enables investigation of ubiquitination patterns in native tissue context and assessment of personalized therapeutic approaches targeting ubiquitination enzymes [102].
In Vivo Models for Ubiquitination Studies
In vivo models provide essential insights into tumor microenvironment interactions, metastatic progression, and therapeutic efficacy in whole-organism contexts.
Patient-Derived Xenograft (PDX) Models
Experimental Protocol:
- Implementation: Implant patient-derived tumor fragments subcutaneously or orthotopically into immunocompromised mice. Establish from clinical samples with preservation of tumor histopathology and molecular characteristics [102].
- Therapeutic Validation: Treat established PDX tumors with ubiquitination-targeting agents. Monitor tumor growth and metastatic dissemination, with OTUB1 knockdown xenografts showing reduced tumor growth and metastasis in prostate cancer models [100].
Advantages: PDX models maintain tumor heterogeneity and ubiquitination signatures similar to original patient tumors, enabling clinically relevant assessment of ubiquitination-modulating therapies [102].
Genetically Engineered Mouse Models (GEMMs)
GEMMs enable investigation of specific ubiquitination enzymes in tumor initiation and progression within intact immune microenvironments.
Applications: While not explicitly detailed in the search results, these models are referenced in the context of validating ubiquitination-related findings from in vitro studies in physiologically relevant systems [100].
Comparative Analysis of Model Systems
Table 2: Comprehensive Comparison of Preclinical Models for Ubiquitination Research
| Model Characteristic | Cancer Cell Lines | Patient-Derived Organoids | Patient-Derived Xenografts (PDX) | Genetically Engineered Models |
|---|---|---|---|---|
| Throughput Capacity | High | Medium | Low | Very Low |
| Experimental Duration | Days-Weeks | Weeks | Months | Months |
| Cost Considerations | Low | Medium | High | High |
| Tumor Heterogeneity | Low | High | High | Variable |
| Microenvironment Complexity | Limited | Moderate | High (human tumor mouse stroma) | High (murine) |
| Immune System Components | Absent | Limited | Absent (immunocompromised hosts) | Intact |
| Clinical Predictive Value | Moderate | High | High | Context-dependent |
| Ubiquitination Pattern Fidelity | Altered in culture | Better preserved | Well-preserved | Dependent on genetic design |
| Typical Applications | Mechanism screening, target validation | Personalized medicine, drug screening | Therapeutic efficacy, metastasis studies | Tumor initiation, immune interactions |
Technical Approaches for Ubiquitination Characterization
Comprehensive ubiquitination assessment requires specialized methodologies for detecting and quantifying this modification.
Ubiquitinated Protein Enrichment Strategies
- Ub Antibody-Based Enrichment: Utilize anti-K-ε-GG antibodies for ubiquitinated peptide enrichment from digested samples. Critical for ubiquitinome analyses of clinical tissues without genetic manipulation [15] [61].
- Ub Tagging-Based Approaches: Express His- or Strep-tagged ubiquitin in cells, enabling purification of ubiquitinated substrates under denaturing conditions. Identifies hundreds of ubiquitination sites but may not perfectly mimic endogenous ubiquitin [61].
- Ubiquitin-Binding Domain (UBD) Tools: Employ tandem-repeated Ub-binding entities (TUBEs) with nanomolar affinity to protect ubiquitinated proteins from deubiquitination and proteasomal degradation during analysis [61].
Proteomic Analysis of Ubiquitination
Liquid Chromatography-Mass Spectrometry (LC-MS/MS) Protocol:
- Sample Preparation: Digest enriched ubiquitinated peptides, desalt using C18 columns, and fractionate via basic reversed-phase liquid chromatography [101] [15].
- MS Analysis: Utilize tims-TOF Pro mass spectrometry with nanoElute UHPLC separation. Data processing with MaxQuant against reference proteome databases [15].
- Functional Interpretation: Computational assessment of relative ubiquitin occupancy changes in response to proteasome inhibition (e.g., MG132) to distinguish degradation versus non-degradation ubiquitin signaling [101].
Integrated Experimental Workflows
The following diagrams illustrate standardized workflows for ubiquitination analysis in preclinical models.
In Vitro to In Vivo Validation Pipeline
Ubiquitinome Characterization Workflow
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Ubiquitination Studies
| Reagent/Category | Specific Examples | Research Application | Technical Considerations |
|---|---|---|---|
| Cell Culture Media | DMEM, RPMI-1640 with 10% FBS | Maintenance of cancer cell lines | Vary composition based on cell type; use dialyzed FBS for SILAC [7] [101] |
| Gene Manipulation Tools | shRNA plasmids, siRNA pools, viral delivery systems | Ubiquitination enzyme knockdown/overexpression | Validate efficiency via qPCR/Western blot; use catalytically inactive mutants as controls [7] [100] |
| Ubiquitination Enrichment Reagents | Anti-K-ε-GG antibody beads, linkage-specific Ub antibodies | Proteomic ubiquitinome analysis | Linkage-specific antibodies enable chain architecture studies [15] [61] |
| Proteasome Inhibitors | MG132, PR-619 | Stabilization of ubiquitinated proteins | Confirm inhibition efficiency via immunoblotting for ubiquitin [101] |
| Mass Spectrometry Reagents | Stable isotopes (SILAC), trypsin, C18 desalting columns | Quantitative ubiquitinome profiling | Offline fractionation improves identification depth [101] [15] |
| Antibodies for Detection | Ubiquitin (P4D1, FK1/FK2), linkage-specific antibodies | Immunoblotting, immunohistochemistry | Linkage-specific antibodies available for K48, K63, M1 chains [61] |
The choice of preclinical models significantly influences the characterization of ubiquitination patterns in cancer research. Integrated model systems that combine the throughput of cell lines, the biological relevance of patient-derived organoids, and the physiological complexity of in vivo models provide the most comprehensive approach for validating ubiquitination-related therapeutic targets. The consistent observation that ubiquitination-related genes are upregulated in multiple cancer types and correlate with poor prognosis underscores the importance of these models in developing novel treatment strategies [7] [5]. As ubiquitination profiling technologies advance, particularly in mass spectrometry and computational analysis, preclinical validation approaches will continue to improve in their ability to predict clinical success of ubiquitination-targeting therapies, ultimately enabling more precise targeting of this critical regulatory system in cancer.
The Ubiquitin-Proteasome System (UPS) represents a sophisticated regulatory network that controls virtually all cellular processes through targeted protein degradation and modification. Comprising a cascade of E1 (activating), E2 (conjugating), and E3 (ligating) enzymes, along with deubiquitinases (DUBs) and the proteasome complex, the UPS maintains protein homeostasis by tagging proteins with ubiquitin molecules for proteasomal degradation or functional modification [9] [103]. In cancerous tissues, this system undergoes significant dysregulation, leading to altered ubiquitination patterns that drive tumorigenesis through enhanced proliferation, evasion of apoptosis, metabolic reprogramming, and immune modulation [7] [5] [103]. The clinical success of proteasome inhibitors in hematological malignancies has validated the UPS as a therapeutic target, spurring drug development efforts targeting specific UPS components with greater precision [104] [103]. This guide systematically compares experimental approaches for validating ubiquitination targets and designing clinical trials, providing researchers with methodologies to bridge the gap between basic discovery and clinical application.
Comparative Ubiquitination Landscapes: Malignant vs. Normal Tissues
Comprehensive multi-omics analyses have revealed distinct ubiquitination patterns between cancerous and normal tissues, highlighting potential therapeutic vulnerabilities. Pan-cancer studies demonstrate that ubiquitination regulators (UBRs) exhibit widespread genetic alterations and expression perturbations across cancer types [5]. Malignant cells show elevated expression of specific UBRs compared to normal counterparts, creating dependency on UPS components for survival—a phenomenon termed "ubiquitin addiction" [103].
Table 1: Key Ubiquitination-Related Genes Dysregulated in Cancer
| Gene | UPS Role | Cancer Type | Expression Change | Functional Impact |
|---|---|---|---|---|
| UBE2C | E2 conjugating enzyme | Hepatocellular Carcinoma | Upregulated [7] | Promotes proliferation, invasion, immune evasion [7] |
| UBA1 | E1 activating enzyme | Pan-cancer (including LC, CRC) [22] | Upregulated [22] | Correlates with poor prognosis, immune infiltration [22] |
| UBA6 | E1 activating enzyme | Pan-cancer (including BRCA, COAD) [22] | Upregulated [22] | Associated with advanced stage, worse survival [22] |
| PSMD14 | Proteasome subunit | Lung Adenocarcinoma [90] | Upregulated [90] | Stabilizes oncoproteins like AGR2, enhances malignancy [90] |
| SKP2 | E3 ligase | Multiple cancers [9] | Upregulated [9] | Degrades tumor suppressors (p27, p21) [9] |
The ubiquitination landscape exhibits tissue-specific patterns, with testicular tissue showing the most distinct UBR expression profile [5]. In cancer, these patterns become further distorted, with specific UBRs demonstrating strong associations with cancer hallmark pathways including cell cycle regulation, DNA repair, metabolic reprogramming, and p53 signaling [7] [5]. Single-cell RNA sequencing of lung adenocarcinoma tissues has revealed that malignant cells display elevated scores for ubiquitination-related enzymes and ubiquitin-binding domains compared to normal epithelial cells, highlighting the cell-type specificity of ubiquitination dysregulation within the tumor microenvironment [90].
Figure 1: Ubiquitination Cascade and Functional Outcomes. The sequential E1-E2-E3 enzyme cascade attaches ubiquitin to substrate proteins, determining their fate through different chain linkages.
Validating UPS Targets: From Multi-Omics to Functional Assays
Bioinformatics and Multi-Omics Profiling
Initial target validation begins with comprehensive molecular profiling to identify dysregulated UPS components. Recommended approaches include:
- Pan-cancer analysis: Integrate data from TCGA, GTEx, and GEO datasets to identify UBRs consistently altered across multiple cancer types [5] [22]. The UALCAN portal and Human Protein Atlas provide expression validation at mRNA and protein levels.
- Genetic alteration assessment: Analyze somatic mutations, copy number variations (CNVs), and single nucleotide variants (SNVs) in UBRs using resources like GSCA and cBioPortal [5] [22].
- Pathway correlation mapping: Employ Gene Set Variation Analysis (GSVA) to correlate UBR expression with cancer hallmark pathway activities [5].
- Single-cell RNA sequencing: Resolve UBR expression at cellular resolution within tumor ecosystems to identify cell-type specific dysregulation [90].
Experimental Validation of Candidate Targets
Following bioinformatics identification, functional validation employs standardized experimental protocols:
Table 2: Core Experimental Protocols for Functional Validation of UPS Targets
| Method | Key Procedure | Application in UPS Validation | Example from Literature |
|---|---|---|---|
| Gene Knockdown | shRNA plasmids with puromycin selection; viral transduction; qRT-PCR confirmation | Assess requirement of target for cancer cell viability | UBE2C knockdown in HCC cells [7] |
| CCK-8 Assay | Seed 2,500 cells/well in 96-well plates; add 10μL CCK8 reagent; measure OD450 after 4h | Quantify cell proliferation and viability | Viability assessment post-UPS target inhibition [7] |
| Transwell Assay | Seed 5×10^4 cells in Matrigel-coated (invasion) or uncoated (migration) chambers; count cells after 24h | Evaluate migratory and invasive capacity | UBE2C-dependent HCC cell invasion [7] |
| Wound Healing Assay | Create scratch with 200μL pipette tip; document closure at 0h and 24h with inverted microscope | Measure two-dimensional cell migration | Migration of Huh7 and Hep3B HCC cells [7] |
| Clonogenic Assay | Seed 600 cells in 6-well plates; incubate for 14 days; count formed colonies | Determine long-term proliferative capacity | Colony formation after UPS perturbation [7] |
| Protein Stability Assay | Co-immunoprecipitation; cycloheximide chase; ubiquitination pulldown | Identify substrates and measure half-life | PSMD14-AGR2 interaction in LUAD [90] |
Figure 2: UPS Target Validation Workflow. Comprehensive pathway from target identification through clinical translation.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for UPS Target Validation
| Reagent/Category | Specific Examples | Function/Application | Considerations |
|---|---|---|---|
| Gene Knockdown | shRNA plasmids (e.g., GenePharma); viral packaging systems (lentiviral/retroviral); polybrene | Stable gene silencing; functional validation | Confirm knockdown efficiency via qRT-PCR; include scrambled controls |
| Small Molecule Inhibitors | MLN4924 (NAE inhibitor); PYR-41 (UBA1 inhibitor); CC0651 (CDC34 inhibitor); NSC697923 (UBE2N inhibitor) [9] [104] | Pharmacological target validation; therapeutic potential | Assess specificity and off-target effects; determine IC50 values |
| Cell Culture Models | HCC lines (Huh7, Hep3B); LUAD lines; primary cancer cells; normal counterparts | In vitro functional studies | Use low passages; authenticate regularly; include multiple lines |
| Antibodies | Anti-ubiquitin; anti-K48/K63 linkage-specific; target-specific antibodies | Western blot, IP, IHC validation | Verify specificity; optimize concentrations |
| Proteasome Assays | Fluorogenic substrates (Suc-LLVY-AMC, Z-ARR-AMC); proteasome activity probes | Measure chymotrypsin-like, trypsin-like, caspase-like activity | Distinguish constitutive vs immunoproteasome |
| Ubiquitination Assays | HA-ubiquitin plasmids; TUBE reagents; DUB substrates | Monitor global/substrate-specific ubiquitination | Use linkage-specific reagents for precision |
| In Vivo Models | Patient-derived xenografts; genetically engineered mouse models; cell line-derived xenografts | Preclinical therapeutic efficacy | Choose immunocompromised strain based on application |
Clinical Trial Designs for Predictive Marker Validation
Translating UPS targets from bench to bedside requires thoughtfully designed clinical trials that can establish predictive value of biomarkers and demonstrate therapeutic efficacy. Two predominant trial designs have emerged for validating predictive biomarkers in oncology:
Marker by Treatment Interaction Design
This approach functions as a classical randomized clinical trial with upfront stratification for the marker. Patients are randomized to treatment arms regardless of marker status, but the design includes prospective stratification by the biomarker to evaluate:
- Differential treatment efficacy between marker-defined subgroups
- Statistical significance of the interaction between marker status and treatment effect
- Requires predefined biomarker cutoff values and analytical validation [105]
This design is optimal when preliminary evidence suggests the biomarker predicts response to a specific therapy, and when the biological rationale supports different effect sizes across marker subgroups.
Marker-Based Strategy Design
In this model, patients are first tested for the biomarker, then randomized to one of two strategies:
- Marker-based arm: Treatment selection guided by biomarker status
- Non-marker-based arm: Standard treatment regardless of biomarker status
The predictive value of the marker is assessed by comparing outcomes between all patients in the marker-based arm versus all patients in the non-marker-based arm [105]. This design more closely mirrors clinical practice and evaluates the overall utility of biomarker-guided treatment, though it requires larger sample sizes and may be confounded by effectiveness of assigned treatments.
Considerations for UPS-Targeted Trial Design
When designing clinical trials for UPS-targeted therapies, several unique factors warrant consideration:
- Combination strategies: Given pathway redundancies, UPS inhibitors may demonstrate maximal efficacy in combination with other targeted therapies or conventional cytotoxics
- Biomarker selection: For ubiquitination targets, potential biomarkers include substrate accumulation, ubiquitination signatures, or transcriptional signatures of pathway activation
- Resistance monitoring: Plan for sequential biomarker assessment to monitor emerging resistance mechanisms
- Leveraging real-world data: Tempus-style approaches using multimodal real-world data can inform trial design and biomarker selection [106]
The systematic validation of ubiquitination targets represents a frontier in precision oncology, offering potential for innovative therapeutic strategies against historically undruggable targets. The journey from bench to bedside requires rigorous multidisciplinary approaches integrating multi-omics discovery, functional validation, and thoughtfully designed clinical trials. As our understanding of ubiquitination patterns in cancer deepens through single-cell technologies and advanced proteomics, new vulnerabilities continue to emerge. The frameworks and methodologies presented in this guide provide a roadmap for researchers navigating this complex landscape, from initial target identification through clinical trial design. Continued innovation in targeting technologies—including PROTACs, molecular glues, and DUB inhibitors—promises to expand the therapeutic arsenal against cancer by leveraging the exquisite specificity of the ubiquitin-proteasome system for precision medicine.
Conclusion
The comprehensive analysis of ubiquitination patterns reveals their fundamental role in cancer pathogenesis, with distinct signatures differentiating malignant from normal tissues across diverse cancer types. Key takeaways include the consistent upregulation of specific E1/E2/E3 enzymes in tumors, the functional consequences of ubiquitination dysregulation on critical cancer hallmarks, and the validated utility of ubiquitination-related factors as prognostic biomarkers and therapeutic targets. Future directions should focus on developing more precise ubiquitination-profiling technologies, expanding pan-cancer ubiquitinome maps, advancing novel targeting approaches like PROTACs and molecular glues, and validating ubiquitination-based biomarkers in clinical trials. These efforts will ultimately enable personalized cancer therapies that exploit the ubiquitination system, offering new hope for patients with resistant or advanced malignancies.
References
- 1. Targeting ubiquitination for cancer therapies - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4976843/]
- 2. The ubiquitin system: from cell signalling to disease biology and new... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7862391/]
- 3. The role of ubiquitination in tumorigenesis and targeted drug discovery | Signal Transduction and Targeted Therapy [https://www.nature.com/articles/s41392-020-0107-0]
- 4. Deubiquitinating enzyme - Wikipedia [https://en.wikipedia.org/wiki/Deubiquitinating_enzyme]
- 5. Integrated analysis of the ubiquitination mechanism reveals the... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-023-09583-z]
- 6. Ubiquitinomics revealed disease- and stage-specific patterns relevant... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10439878/]
- 7. Frontiers | Integrating multi-omics and experimental techniques to... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1545472/full]
- 8. A small-molecule inhibitor of the ubiquitin activating enzyme for cancer treatment | Nature Medicine [https://www.nature.com/articles/nm.4474]
- 9. Drugging the undruggables: exploring the ubiquitin system for drug development | Cell Research [https://www.nature.com/articles/cr201631]
- 10. Frontiers | The role of ubiquitination and deubiquitination in cancer lipid metabolism [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2025.1464914/full]
- 11. Comprehensive pan ‑ cancer analysis reveals ubiquitin ‑conjugating... [https://www.spandidos-publications.com/10.3892/ol.2025.15237?text=fulltext]
- 12. Ubiquitination and deubiquitination in cancer: from mechanisms to novel therapeutic approaches | Molecular Cancer | Full Text [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-024-02046-3]
- 13. Frontiers | Multi-omics analysis identifies UBA family as potential... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1510503/full]
- 14. Comprehensive analysis of TRIM56’s prognostic value and immune... [https://www.nature.com/articles/s41598-025-97856-w?error=cookies_not_supported&code=5359da08-43af-478f-8766-21e0b7aef4c2]
- 15. Identification of ubiquitinated substrate proteins and their gene... [https://atm.amegroups.org/article/view/84238/html]
- 16. Comprehensive characterization of ubiquitinome of human colorectal... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9528151/]
- 17. ligases: Ubiquitin - cell and... | Nature Reviews cycle control Cancer [https://www.nature.com/articles/nrc1881?error=cookies_not_supported&code=be3a9082-97a9-477f-b3c8-50bc030e9a88]
- 18. signaling in Ubiquitin and tumorigenesis - PMC cell cycle control [https://pmc.ncbi.nlm.nih.gov/articles/PMC7862229/]
- 19. Ubiquitination Links DNA Damage and Repair Signaling to Cancer Metabolism - PubMed [https://pubmed.ncbi.nlm.nih.gov/37176148/]
- 20. DNA Replication Gaps, Cancer and Disease | Keystone Symposia [https://www.keystonesymposia.org/conferences/conference-listing/meeting/onpage-program/d52025]
- 21. The advancement of ubiquitination regulation in apoptosis, ferroptosis, autophagy, drug resistance and treatment of cancer - PubMed [https://pubmed.ncbi.nlm.nih.gov/40499632/]
- 22. Multi-omics analysis identifies UBA family as potential pan- cancer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11880792/]
- 23. Ubiquitination Enzymes in Cancer, Cancer Immune Evasion, and Potential Therapeutic Opportunities [https://www.mdpi.com/2073-4409/14/2/69]
- 24. Frontiers | Single- cell and multi-omics analysis identifies TRIM9 as... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1631708/full]
- 25. Frontiers | The role of ubiquitination and deubiquitination in urological tumours [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1532878/full]
- 26. From the Evasion of Degradation to Ubiquitin -Dependent Protein... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8469536/]
- 27. Ubiquitin becomes ubiquitous in cancer: Emerging roles of ubiquitin ligases and deubiquitinases in tumorigenesis and as therapeutic targets [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3023568/]
- 28. A mechanism of ubiquitin -independent proteasomal degradation of... [https://pubmed.ncbi.nlm.nih.gov/15687255/]
- 29. c-Myc proteolysis by the ubiquitin-proteasome pathway: stabilization of c-Myc in Burkitt's lymphoma cells - PubMed [https://pubmed.ncbi.nlm.nih.gov/10713166/?dopt=Abstract]
- 30. Comprehensive characterization of ubiquitinome of human colorectal... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-022-03645-8]
- 31. Integrating multi-omics and experimental techniques to decode... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12022440/]
- 32. - Multi approaches in omics with applications in tumor... cancer research [https://pmc.ncbi.nlm.nih.gov/articles/PMC7868685/]
- 33. Integrative Multi-Omics Analysis Reveals the Molecular Characteristics, Tumor Microenvironment, and Clinical Significance of Ubiquitination Mechanisms in Lung Adenocarcinoma - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12250011/]
- 34. - Integrative in Precision Medicine: From Molecular... Multi Omics [https://www.ijsrtjournal.com/article/Integrative-MultiOmics-in-Precision-Medicine-From-Molecular-Interconnectivity-to-SingleCell-Resolution]
- 35. Single-cell multi - omics in cancer immunotherapy: from tumor... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-025-02426-3]
- 36. Frontiers | An integrated approach for analyzing spatially resolved... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2025.1614288/full]
- 37. An in-silico pan-cancer bulk and single-cell profiling of transcription factors in protein autoubiquitination | Discover Oncology [https://link.springer.com/article/10.1007/s12672-025-03067-0]
- 38. Multi-omics analysis of core E3 ubiquitin ligase identifies prognostic biomarkers associated with immune infiltration and drug sensitivity in lung adenocarcinoma - PubMed [https://pubmed.ncbi.nlm.nih.gov/39895786/]
- 39. Integrated multi-omics analysis reveals PTM networks as key regulators of colorectal cancer progression and immune evasion | Discover Oncology [https://link.springer.com/article/10.1007/s12672-025-03445-8]
- 40. Variability in estimated gene expression among commonly used... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7026138/]
- 41. GitHub - hakyimlab/PredictDB_ Pipeline _ GTEx _v7: PredictDB Pipeline ... [https://github.com/hakyimlab/PredictDB_Pipeline_GTEx_v7]
- 42. Cancer Cell Line Encyclopedia (CCLE) [https://sites.broadinstitute.org/ccle]
- 43. Datasets | Cancer Cell Line Encyclopedia (CCLE) [https://sites.broadinstitute.org/ccle/datasets]
- 44. Comprehensive transcriptomic analysis of cell lines as models of primary tumors across 22 tumor types | Nature Communications [https://www.nature.com/articles/s41467-019-11415-2]
- 45. Ubiquitination and Protein Degradation in Cancer - Creative Proteomics [https://www.creative-proteomics.com/resource/ubiquitination-protein-degradation-in-cancer.htm]
- 46. The importance of regulatory ubiquitination in cancer and metastasis - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5397262/]
- 47. Ubiquitin - Wikipedia [https://en.wikipedia.org/wiki/Ubiquitin]
- 48. Frontiers | Editorial: The role of ubiquitination in disease development, progression, and prognosis [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1595755/full]
- 49. The Clinical Prediction Value of the Ubiquitination Model Reflecting the Microenvironment Infiltration and Drug Sensitivity in Breast Cancer [https://www.jcancer.org/v16p0784.htm]
- 50. Single‐cell RNA sequencing technologies and applications: A brief overview - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8964935/]
- 51. - Single unveils cell and... RNA sequencing tumor heterogeneity [https://www.nature.com/articles/s41598-024-57640-8?error=cookies_not_supported&code=b75bf10c-4ee2-4a23-b0bc-8403e75611cd]
- 52. - Single reveals the intra-tumoral cell ... RNA sequencing heterogeneity [https://ovarianresearch.biomedcentral.com/articles/10.1186/s13048-025-01649-8]
- 53. The Clinical Prediction Value of the Ubiquitination Model Reflecting the Microenvironment Infiltration and Drug Sensitivity in Breast Cancer - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11705054/]
- 54. Frontiers | Commentary: The ubiquitin-proteasome system in the tumor immune microenvironment: a key force in combination therapy [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1522427/full]
- 55. Frontiers | Ubiquitination in lipid metabolism reprogramming: implications for pediatric solid tumors [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1554311/full]
- 56. Frontiers | UBE2N as a novel prognostic and therapeutic biomarker of lung adenocarcinoma [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1636503/full]
- 57. Multi-omics analysis of core E3 ubiquitin ligase identifies prognostic biomarkers associated with immune infiltration and drug sensitivity in lung adenocarcinoma [https://www.jcancer.org/v16p1363.htm]
- 58. RNF185 promotes esophageal squamous cell carcinoma progression... [https://eurjmedres.biomedcentral.com/articles/10.1186/s40001-025-03357-x]
- 59. Pathway Enrichment Analysis: Open Access Tools - HSLS MolBio Workshops - LibGuides at Health Sciences Library System [https://hsls.libguides.com/MBISworkshops/PathwaysOpen]
- 60. Frequent mutations in acetylation and ubiquitination sites suggest... [https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-016-0311-2]
- 61. Current methodologies in protein ubiquitination characterization: from... [https://cellandbioscience.biomedcentral.com/articles/10.1186/s13578-022-00870-y]
- 62. Detection of Protein Ubiquitination Sites by Peptide Enrichment and... [https://www.jove.com/t/59079/detection-protein-ubiquitination-sites-peptide-enrichment-mass]
- 63. Dissecting the ubiquitin pathway by mass spectrometry - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC1828906/]
- 64. Time-resolved in vivo ubiquitinome profiling by DIA-MS reveals USP7 targets on a proteome-wide scale | Nature Communications [https://www.nature.com/articles/s41467-021-25454-1]
- 65. Ubiquitin chain diversity at a glance - PubMed [https://pubmed.ncbi.nlm.nih.gov/26906419/?dopt=Abstract]
- 66. Role of K63-linked ubiquitination in cancer | Cell Death Discovery [https://www.nature.com/articles/s41420-022-01204-0]
- 67. K63-Linked Ubiquitination in Kinase Activation and Cancer - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3355940/?q=instability]
- 68. Loss of TRIM31 promotes breast cancer progression through... [https://www.nature.com/articles/s41419-021-04208-3?error=cookies_not_supported]
- 69. EZH2 K63-polyubiquitination affecting migration in extranodal natural killer/T-cell lymphoma | Clinical Epigenetics | Full Text [https://clinicalepigeneticsjournal.biomedcentral.com/articles/10.1186/s13148-023-01606-6]
- 70. The role of ubiquitination and deubiquitination in cancer metabolism | Molecular Cancer | Full Text [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-020-01262-x]
- 71. genomes: Distinguishing between ' Cancer ' and ' driver ... passenger [https://www.news-medical.net/news/20100219/Cancer-genomes-Distinguishing-between-driver-and-passenger-mutations.aspx]
- 72. mutations in Passenger evolution cancer [https://www.oatext.com/passenger-mutations-in-cancer-evolution.php]
- 73. regulates PTEN nuclear import and Ubiquitination suppression tumor [https://pubmed.ncbi.nlm.nih.gov/17218261/]
- 74. Integrated analysis of the ubiquitination mechanism reveals the specific signatures of tissue and cancer - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10478310/]
- 75. Assessing and mitigating batch effects in large-scale omics studies | Genome Biology | Full Text [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03401-9]
- 76. Protein-level batch - effect ... | Nature Communications correction [https://www.nature.com/articles/s41467-025-64718-y?error=cookies_not_supported&code=65014dd1-7d5a-4ea8-bf0c-a7f02a997002]
- 77. Batch Correction Analysis | Griffith Lab [https://rnabio.org/module-03-expression/0003/06/02/Batch-Correction/]
- 78. to Batch differentially expressed genes... effect correction compare [https://www.biostars.org/p/9519800/]
- 79. A benchmark of batch - effect methods for single-cell RNA... correction [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1850-9]
- 80. Comparative evaluation of imputation and batch-effect correction for proteomics/peptidomics differential-expression analysis | medRxiv [https://www.medrxiv.org/content/10.1101/2025.08.14.25333694v1.full]
- 81. Frontiers | A High - Throughput Assay for Monitoring Ubiquitination in... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2019.00816/full]
- 82. A cell-based high - throughput method based on... screening [https://pmc.ncbi.nlm.nih.gov/articles/PMC6393603/]
- 83. Mechanistic Insights and Therapeutic Potentials of Ubiquitin‐Proteasome System in Non‐Small Cell Lung Cancer - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12240642/]
- 84. Frontiers | A novel ubiquitin -related genes-based signature ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1513948/full]
- 85. Identification and validation of a thirteen-gene signature based on... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12137841/]
- 86. A novel ubiquitination -based molecular signature predicts prognosis ... [https://link.springer.com/article/10.1007/s00277-025-06668-5]
- 87. Identification of E3 ubiquitin ligase-based molecular subtypes and... [https://cancerci.biomedcentral.com/articles/10.1186/s12935-025-03703-3]
- 88. Deubiquitinase USP5 regulates cancer progression and radiosensitivity by stabilizing HOXA10 in non-small cell lung cancer - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S0344033825002985?via=ihub]
- 89. Deciphering a profiling based on multiple post-translational... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-024-02199-1]
- 90. Bioinformatics analysis and single-cell RNA sequencing: elucidating the... [https://jtd.amegroups.org/article/view/77282/html]
- 91. Ubiquitination-Related Molecular Subtypes and a Novel Prognostic Index for Bladder Cancer Patients - PubMed [https://pubmed.ncbi.nlm.nih.gov/34776794/]
- 92. Transcriptomics-based exploration of ubiquitination-related biomarkers and potential molecular mechanisms in laryngeal squamous cell carcinoma | BMC Medical Genomics | Full Text [https://bmcmedgenomics.biomedcentral.com/articles/10.1186/s12920-025-02148-x]
- 93. Ubiquitination Enzymes in Cancer, Cancer Immune Evasion, and Potential Therapeutic Opportunities - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11763706/]
- 94. E3 Ubiquitin Ligases as Cancer Targets and Biomarkers - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC1601942/]
- 95. -Activating Ubiquitin ( Enzyme ) as a Prognostic E ... 1 UBA 1 Biomarker [https://www.mdpi.com/1422-0067/25/23/12696/xml]
- 96. The non-canonical ubiquitin activating enzyme UBA6 suppresses epithelial-mesenchymal transition of mammary epithelial cells - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5675648/]
- 97. -conjugating Ubiquitin : molecular biology, enzyme in... UBE 2 C role [https://pubmed.ncbi.nlm.nih.gov/22170434/]
- 98. Comprehensive analysis of the prognosis and tumor immune microenvironment of ubiquitin-conjugating enzyme transport-related gene UBE2C in hepatocellular carcinoma - PubMed [https://pubmed.ncbi.nlm.nih.gov/40410642/]
- 99. Dual functionality of MDM2 in PROTACs expands the horizons of targeted protein degradation | Biomarker Research | Full Text [https://biomarkerres.biomedcentral.com/articles/10.1186/s40364-025-00826-7]
- 100. OTUB1 de-ubiquitinating enzyme promotes prostate cancer cell... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-014-0280-2]
- 101. Proteomic analysis of degradation ubiquitin signaling by ubiquitin occupancy changes responding to 26S proteasome inhibition | Clinical Proteomics | Full Text [https://clinicalproteomicsjournal.biomedcentral.com/articles/10.1186/s12014-020-9265-x]
- 102. Personalized In and Vitro to Guide Precision... In Vivo Cancer Models [https://pubmed.ncbi.nlm.nih.gov/28331002/]
- 103. Drug Development Targeting the Ubiquitin–Proteasome System (UPS) for the Treatment of Human Cancers - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7226376/]
- 104. Emerging drug development technologies targeting ubiquitination for cancer therapeutics - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7112620/]
- 105. for predictive marker Clinical in cancer... trial designs validation [https://pubmed.ncbi.nlm.nih.gov/15774793/]
- 106. a novel Validating and informing target for... - Tempus trial design [https://www.tempus.com/resources/content/case-studies/validating-a-novel-target-and-informing-trial-design-for-a-first-in-class-therapeutic/]