Decoding the Ubiquitin Code: Mass Spectrometry Approaches for Mapping Chain Architectures in Disease and Therapy
This article provides a comprehensive overview of how mass spectrometry (MS) has revolutionized our understanding of the ubiquitin code.
Decoding the Ubiquitin Code: Mass Spectrometry Approaches for Mapping Chain Architectures in Disease and Therapy
Abstract
This article provides a comprehensive overview of how mass spectrometry (MS) has revolutionized our understanding of the ubiquitin code. Aimed at researchers and drug development professionals, it covers the foundational principles of diverse ubiquitin chain architectures—including homotypic, mixed, and branched chains—and their distinct cellular functions. The content details state-of-the-art MS methodologies, from shotgun proteomics and Ub-AQUA to innovative techniques like Ub-clipping and UbiChEM-MS, for identifying substrates, mapping modification sites, and elucidating chain topology. It further addresses key methodological challenges and optimization strategies, compares the capabilities of different MS and enrichment approaches, and explores the direct implications of these discoveries for understanding disease mechanisms and developing targeted therapeutics, such as PROTACs.
The Ubiquitin Code: Unveiling the Complexity of Chain Architectures and Cellular Signals
Ubiquitination, the post-translational attachment of the 76-amino-acid protein ubiquitin to substrate proteins, represents a sophisticated regulatory language that extends far beyond its initial characterization as a degradation signal. The diverse outcomes of ubiquitination are encrypted in what is known as the "ubiquitin code"—the specific topologies formed when ubiquitin molecules polymerize via one of their seven lysine residues (K6, K11, K27, K29, K33, K48, K63) or the N-terminal methionine (M1) [1]. While K48-linked chains predominantly target substrates for proteasomal degradation, and K63-linked chains regulate proteasome-independent signaling pathways such as DNA repair and inflammation, the functional repertoire of atypical ubiquitin linkages is rapidly expanding [1]. This whitepaper explores the sophisticated cellular functions of ubiquitin signaling beyond protein degradation, framed within the context of advanced mass spectrometry techniques that are deciphering these complex ubiquitin chain architectures with unprecedented precision.
Table 1: Ubiquitin Linkage Types and Their Known Cellular Functions
| Linkage Type | Structural Feature | Primary Cellular Functions |
|---|---|---|
| K48-linked | Closed conformation | Proteasomal degradation [1] |
| K63-linked | Open conformation | DNA repair, inflammatory signaling, endocytosis [1] |
| K29-linked | - | Proteotoxic stress response, cell cycle regulation [1] |
| K11-linked | - | Cell cycle regulation, protein degradation [1] |
| K27-linked | - | Autoimmunity, innate immune response, tumorigenesis [1] |
| K33-linked | - | Protein trafficking, signal transduction [1] |
| K6-linked | - | Mitophagy [1] |
| M1/Linear | - | Immune response [1] |
| Branched | Mixed linkages | Increased degradative potential [2] |
Non-Degradative Ubiquitin Signaling in Cellular Processes
DNA Damage Response and Repair
The cellular response to DNA damage represents a paradigm of non-degradative ubiquitin signaling. Recent research using the UBIMAX (UBiquitin target Identification by Mass spectrometry in Xenopus egg extracts) platform has identified the actin-organizing protein Dbn1 as a major target of DNA damage-induced ubiquitylation [3]. This modification is triggered by DNA double-strand breaks (DSBs) and occurs via a conserved mechanism driven by ATM-mediated phosphorylation of a previously uncharacterized β-Trcp1 degron containing an SQ motif. The E3 ligase SCFβ-Trcp1 targets Dbn1 for ubiquitylation in response to DNA damage, representing a specialized branch of the DNA damage response that directly connects apical DNA damage response kinases to ubiquitin signaling [3]. This pathway operates independently of proteosomal degradation for certain signaling functions, highlighting the diverse outcomes of damage-induced ubiquitination.
Cell Cycle Regulation and Proteotoxic Stress Response
K29-linked ubiquitin chains have emerged as significant players in cell cycle regulation and cellular stress pathways. Despite being one of the most abundant atypical ubiquitin linkages, comparable in quantity to K63-linked chains and second only to K48-linked ubiquitin [1], their functions remained poorly characterized until recently. Using a specially developed synthetic antigen-binding fragment (sAB-K29) that specifically recognizes K29-linked polyubiquitin, researchers discovered that K29-linked ubiquitination is enriched in the midbody during mitotic telophase [1]. Experimental downregulation of K29-linked ubiquitination through specific deubiquitinating enzymes (DUBs) resulted in cell cycle arrest at the G1/S phase transition, establishing a critical functional role for this linkage in cell cycle progression [1].
Furthermore, K29-linked ubiquitination displays pronounced involvement in cellular stress response pathways. Under proteotoxic stress conditions—including unfolded protein response, oxidative stress, and heat shock—K29-linked ubiquitin forms distinctive puncta within cells [1]. This spatial reorganization suggests specialized compartmentalization of stress response mechanisms mediated by this specific ubiquitin linkage, potentially facilitating the management of misfolded proteins or coordinating stress-activated signaling cascades.
Immune Signaling and Host Defense
Ubiquitin signaling plays multifaceted roles in immune regulation through both degradative and non-degradative mechanisms. Recent research has illuminated a sophisticated host-pathogen interaction centered on ubiquitin: the E3 ligase RNF213 mediates ubiquitylation of bacterial lipopolysaccharides as a host-protective mechanism, which the pathogen Shigella flexneri counteracts by deploying the effector protein IpaH1.4 to hijack the ubiquitylation machinery [2]. This effector specifically targets protective host factors like RNF213 and LUBAC for degradation, effectively removing them from the cellular environment [2].
Simultaneously, deubiquitinating enzymes (DUBs) have gained attention as regulators of immune responses. The DUB BRISC (BRCC36 isopeptidase complex) moderates immune signaling by deubiquitinating interferon receptors. Chemical inhibitors termed BLUEs selectively inactivate BRISC, thereby increasing degradative ubiquitylation of interferon receptors and attenuating immune responses [2]. This mechanism highlights how the balance between ubiquitination and deubiquitination precisely controls immune signaling intensity, offering therapeutic opportunities for immune modulation.
Analytical Frameworks: Deciphering Ubiquitin Signaling
Advanced Mass Spectrometry Approaches
The complexity of ubiquitin signaling necessitates sophisticated analytical methods capable of mapping modification sites and chain architectures simultaneously. Traditional mass spectrometry approaches have struggled to define both the site of ubiquitination and the topology of attached Ub chains on intact protein substrates concurrently [4]. To address this limitation, researchers have developed UbqTop, a custom computational platform that predicts Ub chain topology from tandem MS (MS2) fragmentation data using a Bayesian-like scoring algorithm [4].
This integrated strategy represents the first methodology enabling simultaneous determination of ubiquitin modification sites and chain architecture using top-down mass spectrometry (TD-MS). When dealing with complex substrates, the approach combines TD-MS with selective Asp-N proteolysis, which digests the substrate while preserving intact Ub chains [4]. This innovative workflow enables direct, site-resolved mapping of Ub chain topology on proteins, including the resolution of isomeric chains and branched architectures, establishing a powerful new framework for proteoform-level analysis of ubiquitin signaling with unprecedented structural resolution [4].
Diagram 1: Top-down MS workflow for ubiquitin analysis
The UBIMAX Platform for Global Ubiquitin Profiling
The UBIMAX platform represents a significant advancement for global detection of dynamic protein ubiquitylation under precise and adaptable conditions [3]. This method enables the enrichment of ubiquitin-conjugated proteins and quantification of regulation of protein ubiquitylation in response to specific cellular stimuli. The system has been rigorously benchmarked by investigating DNA double-strand break-responsive ubiquitylation events, successfully identifying previously known targets while also revealing novel substrates like Dbn1 [3].
The UBIMAX workflow involves supplementing Xenopus egg extracts with His₆-tagged ubiquitin, initiating reactions with specific DNA damage stimuli (undamaged plasmid, linearized plasmid for DSBs, or plasmid-protein crosslinks for DPCs), followed by enrichment of ubiquitin-conjugated proteins and quantitative liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis [3]. This approach has demonstrated exceptional efficiency in specifically detecting ubiquitin-conjugated proteins, with hierarchical clustering analysis of Z-scored ubiquitylated protein abundances robustly differentiating response patterns to various DNA treatments [3].
Table 2: Quantitative Ubiquitylation Changes Detected by UBIMAX in Response to DNA Damage
| Protein Target | DSB-Induced Fold Change | Statistical Significance | Functional Category |
|---|---|---|---|
| Dbn1 | >5x increase | FDR ≤ 0.01 | Actin organization |
| Ku80 | >3x increase | FDR ≤ 0.05 | DNA repair complex |
| Mre11 | >4x increase | FDR ≤ 0.01 | DNA damage sensing |
| Rpa1 | >3x increase | FDR ≤ 0.05 | Single-strand binding |
| Chfr | >2x increase | FDR ≤ 0.05 | Checkpoint control |
Experimental Insights: Methodologies and Reagents
Key Experimental Protocols
UBIMAX Protocol for DNA Damage-Induced Ubiquitylation
The UBIMAX protocol begins with Xenopus egg extracts supplemented with His₆-ubiquitin at an optimal concentration of 0.1 µg/µL [3]. Reactions are initiated by adding either buffer control (no DNA), undamaged plasmid DNA, linearized plasmid DNA (for DSBs), or plasmids carrying specific protein crosslinks (for DPCs). For inhibition controls, extracts can be pre-treated with ubiquitin E1 inhibitor (5-10 µM), ATM inhibitor (10-20 µM), neddylation E1 inhibitor (1-5 µM), or proteasome inhibitor (MG262, 10-50 µM) [3]. Following a 30-minute incubation at 21-23°C, reactions are terminated, and ubiquitin-conjugated proteins are enriched using His-pulldown under denaturing conditions (6M guanidine hydrochloride, pH 8.0). Samples are then subjected to tryptic digestion and analyzed by LC-MS/MS using a high-resolution mass spectrometer (Orbitrap series). Data processing includes intensity-based absolute quantification (iBAQ) normalization, with significance determined by two-tailed Student's t-test with permutation-based FDR control (s0 = 0.1, 2500 randomization rounds) to ensure FDR ≤ 0.05 [3].
K29-Linked Ubiquitin Detection Using sAB-K29
For specific detection of K29-linked ubiquitin chains, researchers developed a synthetic antigen-binding fragment (sAB-K29) selected from a phage display library (Library E) based on a humanized antibody Fab scaffold [1]. Biotinylated K29-linked diubiquitin required for selection is chemically synthesized using established methods incorporating a polyethylene glycol (PEG) linker between the diUb and biotin moieties, followed by refolding via standard dialysis and verification by circular dichroism spectroscopy [1]. For structural studies, K29-linked diUb can be enzymatically prepared using UBA1 (E1), UBE2L3 (E2), and UBE3C (E3) enzymes, with subsequent purification using vOTU treatment to remove K48-linked chains and anion exchange chromatography [1]. Crystallization of the sAB-K29/K29-diUb complex is achieved using reservoir solutions containing 0.1 M phosphate-citrate and 40% v/v PEG 300, with diffraction data collection at 2.9 Å resolution [1]. For cellular detection, sAB-K29 is used in pull-down assays, immunofluorescent imaging, and immunoblotting applications at nanomolar concentrations.
Diagram 2: Dbn1 ubiquitination pathway in DNA damage response
Essential Research Reagents and Tools
Table 3: Key Research Reagents for Studying Ubiquitin Signaling
| Reagent/Tool | Specificity/Function | Research Application |
|---|---|---|
| sAB-K29 | Synthetic antigen-binding fragment recognizing K29-linked diUb [1] | Detection and pull-down of endogenous K29-linked ubiquitin chains |
| UBIMAX Platform | Global ubiquitin target identification by mass spectrometry [3] | System-wide profiling of stimulus-regulated ubiquitylation events |
| UbqTop Software | Bayesian scoring of MS2 fragmentation data [4] | Computational determination of ubiquitin chain topology |
| His₆-Ubiquitin | Affinity-tagged ubiquitin for enrichment [3] | Purification of ubiquitin-conjugated proteins from complex mixtures |
| vOTU DUB | Viral OTU deubiquitinase that cleaves most linkages except K29 [1] | Selective removal of non-K29 ubiquitin chains for purification |
| BLUE Inhibitors | Selective inactivation of BRISC DUB complex [2] | Modulation of interferon signaling through increased receptor ubiquitination |
| E1 Inhibitor | Blocks ubiquitin activation [3] | Negative control for ubiquitin-dependent processes |
| SCFβ-Trcp1 siRNA | Knockdown of specific E3 ligase [3] | Functional validation of ubiquitin ligase-substrate relationships |
Implications for Therapeutic Development
The expanding understanding of non-degradative ubiquitin signaling has opened new avenues for therapeutic intervention, particularly in oncology and inflammatory diseases. The clinical importance of understanding the molecular rules of ubiquitin writers (E1-E2-E3 enzymes), erasers (DUBs), and readers is evident in the rapidly growing field of targeted protein degradation (TPD) [2]. Academic laboratories and pharmaceutical companies are actively generating novel molecular glues and proteolysis targeting chimeras (PROTACs), while expanding the arsenal of E3 ligases that can be harnessed to degrade otherwise difficult-to-target substrates [2].
Recent work has demonstrated how the E3 ligase GID4 can be leveraged via custom PROTACs to target clinically relevant substrates [2]. Simultaneously, the development of specific DUB inhibitors like the BLUE compounds that selectively inactivate BRISC highlights the therapeutic potential of modulating deubiquitination enzymes to fine-tune immune responses [2]. Additionally, the unique structural characteristics of E3 ligases such as Hakai—particularly its HYB domain that recognizes tyrosine-phosphorylated substrates through an antiparallel dimerization mechanism—present novel targeting opportunities for allosteric inhibitors in cancer treatment [5]. These advances underscore the transition from basic understanding of ubiquitin biology to targeted therapeutic applications that exploit the full complexity of the ubiquitin code.
The landscape of ubiquitin signaling extends far beyond its canonical role in protein degradation, encompassing sophisticated regulatory functions in DNA damage response, cell cycle control, immune signaling, and stress adaptation. The development of advanced mass spectrometry technologies—including top-down approaches with computational topology prediction and global profiling platforms like UBIMAX—has been instrumental in deciphering the complex ubiquitin code. As our understanding of linkage-specific functions and architectural diversity deepens, so too do opportunities for therapeutic intervention targeting specific nodes within the ubiquitin signaling network. The continued integration of structural biology, proteomics, and chemical biology will undoubtedly reveal further complexity and therapeutic potential within this essential regulatory system.
Ubiquitination is a critical post-translational modification (PTM) that controls a vast array of cellular processes, including protein degradation, DNA repair, immune signaling, and cell cycle progression [6] [7]. The remarkable functional diversity of ubiquitin signaling stems from its ability to form polymers of various architectures and topologies. A ubiquitin chain's topology—defined by the spatial arrangement of its subunits and the identities of the linkages between them—determines its specific cellular function [8] [9]. This architectural complexity allows the ubiquitin system to encode intricate biological information, forming a sophisticated "ubiquitin code" that is interpreted by cellular machinery [9]. While homotypic chains (linked through a single lysine type) have well-characterized functions, recent research has revealed an expanded complexity through heterotypic and branched chains, which greatly increase the signaling versatility of the ubiquitin system [7] [10]. This technical guide provides a comprehensive overview of ubiquitin chain types and topologies, with a specific focus on mass spectrometry-based methodologies for their analysis, framed within the context of ongoing research aimed at deciphering this complex signaling language.
Ubiquitin Chain Architectures: Structural and Functional Diversity
Ubiquitin chains are classified based on their linkage patterns and overall architecture. The 76-amino acid ubiquitin protein contains seven internal lysine residues (K6, K11, K27, K29, K33, K48, K63) and an N-terminal methionine (M1) that can serve as attachment points for subsequent ubiquitin molecules, enabling polymer formation [7] [9] [10].
Table 1: Classification of Ubiquitin Chain Architectures
| Chain Type | Structural Description | Key Functional Associations | Examples |
|---|---|---|---|
| Monoubiquitination | Single ubiquitin on substrate lysine | Endocytosis, histone regulation, DNA repair [8] [11] | Histone H2A K13/15 [11] |
| Multi-Monoubiquitination | Multiple single ubiquitins on different lysines of same substrate | Endocytic trafficking, protein activity regulation [7] | - |
| Homotypic Chains | Uniform linkage through same acceptor site | Specialized functions based on linkage type [7] | K48 (degradation), K63 (signaling) [7] |
| Mixed Heterotypic Chains | Multiple linkage types, each ubiquitin modified at single site | Increased signaling complexity [7] [10] | - |
| Branched Heterotypic Chains | One or more ubiquitins simultaneously modified at ≥2 different sites | Proteasomal targeting, signal regulation [7] [12] | K11/K48, K29/K48, K48/K63 [7] |
Table 2: Common Ubiquitin Chain Linkages and Their Functions
| Linkage Type | Primary Functions | Structural Features | Proteasomal Degradation |
|---|---|---|---|
| K48-linked | Canonical proteasomal degradation signal [7] | Compact conformations [9] | Yes [7] |
| K63-linked | DNA repair, NF-κB signaling, endocytosis, kinase activation [7] | Extended, open conformations [9] | No (typically) |
| M1-linked (Linear) | NF-κB activation, inflammatory signaling [10] | Extended rigid structure [9] | Context-dependent |
| K11-linked | Cell cycle regulation, ER-associated degradation (ERAD) [7] [12] | Compact conformations [9] | Yes (especially in branched with K48) [12] |
| K29-linked | Proteasomal degradation, ubiquitin fusion degradation pathway [7] | - | Yes |
| K33-linked | Kinase regulation, intracellular trafficking [10] | - | No |
| K6-linked | DNA damage response, mitophagy [7] | - | Context-dependent |
| K27-linked | Kinase activation, immune signaling [10] | - | - |
The structural biology of ubiquitin reveals remarkable molecular stability derived from its compact β-grasp fold, where a five-stranded β sheet cradles a central α helix, minimizing exposed surface area [9]. This stability allows ubiquitin to maintain structural integrity under various conditions while participating in diverse protein-protein interactions. The conformational flexibility of ubiquitin chains further contributes to their functional diversity, with different linkages adopting distinct geometries that are specifically recognized by ubiquitin-binding domains (UBDs) in effector proteins [9].
Branched Ubiquitin Chains: Emerging Complexity
Branched ubiquitin chains represent a sophisticated layer of signaling complexity where a single ubiquitin subunit is modified at two or more different sites, creating fork-like structures [7]. These chains account for approximately 10-20% of all ubiquitin polymers in cells and exhibit specialized functions [12]. The synthesis of branched chains often involves collaboration between E3 ligases with distinct linkage specificities or individual E3s that can recruit multiple E2 enzymes [7].
Table 3: Characterized Branched Ubiquitin Chain Types
| Branched Linkage | Synthetic Mechanism | Biological Context | Functional Outcome |
|---|---|---|---|
| K11/K48 | APC/C with UBE2C & UBE2S E2s [7] | Cell cycle progression, proteotoxic stress [12] | Enhanced proteasomal degradation [12] |
| K48/K63 | TRAF6 & HUWE1 or ITCH & UBR5 E3 pairs [7] | NF-κB signaling, apoptosis [7] | Signal conversion (non-degradative to degradative) [7] |
| K29/K48 | Ufd4 & Ufd2 collaboration in yeast [7] | Ubiquitin fusion degradation pathway [7] | Proteasomal targeting [7] |
| K6/K48 | Parkin (RBR E3) [7] | Mitochondrial quality control [7] | Proteasomal degradation [7] |
Recent structural studies have revealed that K11/K48-branched chains are recognized by the 26S proteasome through a multivalent mechanism involving RPN2, RPN10, and RPN1, explaining their priority as degradation signals [12]. This specialized recognition demonstrates how branched architectures can create unique interaction surfaces that are differentially interpreted by the cellular machinery.
Analytical Challenges in Ubiquitin Topology Characterization
Complete characterization of the ubiquitinome presents significant challenges due to the extraordinary structural diversity of ubiquitin modifications. Theoretically, a tetrameric ubiquitin chain can exist in 819 different isomeric structures when considering all possible linkage combinations and branching patterns [8]. This complexity is further compounded by several factors:
- Low Abundance: Many functionally important ubiquitin modifications, including branched chains, exist at low stoichiometry relative to their protein substrates [10].
- Dynamic Nature: Ubiquitination is a reversible modification, with deubiquitinases (DUBs) constantly processing chains and altering their topology [6].
- Linkage Cross-Talk: Multiple chain types can coexist on the same substrate, creating complex signaling outcomes that are difficult to deconvolute [10].
- Technical Limitations: Traditional antibody-based methods lack the resolution to distinguish between closely related topological isomers and often suffer from reproducibility issues [8].
Traditional analytical approaches, including immunoprecipitation with linkage-specific antibodies and reiterative use of selective deubiquitinases (DUBs), have provided valuable insights but offer limited resolution for complete topological characterization [8]. The tryptic diglycine (GG) remnant method, while excellent for identifying ubiquitination sites, provides no information about chain architecture [8]. These limitations have driven the development of advanced mass spectrometry-based approaches that can directly interrogate ubiquitin chain structure.
Mass Spectrometry Methodologies for Ubiquitin Chain Analysis
Mass spectrometry has emerged as the premier technology for comprehensive ubiquitin chain characterization due to its ability to identify linkage types, quantify abundance, and determine overall topology. Both bottom-up and top-down approaches offer complementary advantages for ubiquitin research.
Top-Down Mass Spectrometry for Intact Chain Analysis
Top-down mass spectrometry (TD-MS) analyzes intact proteins and protein complexes without proteolytic digestion, preserving valuable information about combinations of modifications and overall architecture [8] [4]. For ubiquitin chain analysis, TD-MS provides unparalleled insights into chain topology, branching patterns, and linkage combinations.
Protocol: Top-Down LC-MS/MS Analysis of Polyubiquitin Chains [8]
Sample Preparation:
- Reconstitute lyophilized polyubiquitin samples in water:acetonitrile (97.5:2.5) with 0.1% formic acid to a final concentration of at least 30 μg/mL.
- Ensure protein purity by SDS-PAGE evaluation prior to analysis.
Liquid Chromatography:
- System: Ultra-high-performance liquid chromatograph (e.g., Ultimate 3000).
- Columns: PepSwift RP-4H monolith trap column (100 μm × 5 mm) for desalting and concentration; ProSwift RP-4H monolith analytical column (200 μm × 25 cm) for separation.
- Mobile Phases: A) water-acetonitrile (97.5:2.5) with 0.1% formic acid; B) water-acetonitrile (25:75) with 0.1% formic acid.
- Loading: Load 3 μL sample onto trap column at 5 μL/min for 5 minutes for desalting and concentration.
- Separation: Linear gradient from 5% to 55% mobile phase B over 20 minutes at 1.5 μL/min flow rate with column temperature maintained at 35°C.
Tandem Mass Spectrometry:
- Instrument: High-resolution mass spectrometer (e.g., Orbitrap Fusion Lumos).
- Fragmentation Techniques: Combined electron transfer dissociation and collision-induced dissociation (ETciD) or electron transfer dissociation and higher-energy CID (EThcD).
- Key Parameters: Ion routing multipole pressure at 0.01-0.03 mTorr; in-source fragmentation energy at 10V; mass resolution of 120,000 at 200 m/z for both precursor and fragment ions.
This top-down approach is compatible with all ubiquitin linkage and chain types, can be extended to ubiquitin-like proteins, and benefits from continuing advances in LC-MS/MS instrumentation and interpretation software [8].
Integrated Strategies for Complex Substrates
Recent methodological advances have addressed the challenge of analyzing ubiquitin modifications on intact protein substrates. Shestoperova et al. (2025) developed an integrated strategy that combines selective Asp-N proteolysis with top-down mass spectrometry and a custom computational platform called UbqTop that predicts ubiquitin chain topology from MS2 fragmentation data using a Bayesian-like scoring algorithm [4]. This approach enables simultaneous determination of ubiquitination site and chain architecture, providing proteoform-level analysis of ubiquitin signaling with unprecedented structural resolution [4].
For branched chain identification, specialized workflows have been developed that include:
- Ubiquitin Absolute Quantification (Ub-AQUA) with stable isotope-labeled internal standards to quantify specific linkage types [12].
- Bispecific antibodies that recognize unique combinatorial epitopes [10].
- Chemical biology approaches using ubiquitin variants with point mutations (e.g., R54A) that facilitate mass spectrometry detection of branched chains [10].
Quantitative Proteomic Approaches
Understanding the dynamics of ubiquitin signaling requires quantitative assessment of modification stoichiometry and temporal changes. Several mass spectrometry-based quantitative approaches have been successfully applied to ubiquitin research:
- Stable Isotope Labeling with Amino acids in Cell culture (SILAC): Metabolic labeling that allows precise relative quantification across multiple conditions [6].
- Tandem Mass Tagging (TMT): Isobaric labeling that enables multiplexing of up to 10 samples, significantly improving throughput [6].
- Label-Free Quantification: Avoids chemical labeling but requires more instrument time and careful normalization [6].
Advanced implementations such as MultiNotch MS3 have been developed to address ratio compression problems in TMT experiments, significantly improving quantification accuracy for ubiquitin-modified peptides [6]. These quantitative approaches are particularly powerful when combined with enrichment strategies for ubiquitinated peptides or proteins, enabling system-wide analysis of ubiquitin signaling dynamics.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Essential Research Reagents for Ubiquitin Chain Analysis
| Reagent/Material | Specifications | Application & Function | Example Sources |
|---|---|---|---|
| Synthetic Ubiquitin Conjugates | 0.3-1 mg, 9-15% synthesis yield; purity verified by SDS-PAGE [8] | Method development and standardization; positive controls | In-house synthesis [8] |
| Linkage-Specific Antibodies | Validated for specific ubiquitin linkages (K11, K48, K63, etc.) [10] | Immunoprecipitation, Western blot validation | Commercial vendors |
| DUBs (Deubiquitinases) | Linkage-specific (e.g., UCHL5 for K11/K48 branches) [12] | Controlled disassembly for linkage validation; functional studies | Recombinant expression |
| Ubiquitin Variants | Point mutants (K63R, R54A) for specific applications [10] [12] | Block specific linkages; facilitate branched chain detection | Site-directed mutagenesis |
| Monolithic LC Columns | PepSwift RP-4H trap (100μm×5mm); ProSwift RP-4H analytical (200μm×25cm) [8] | High-resolution separation of ubiquitin conjugates | Thermo Fisher [8] |
| Stable Isotope-Labeled Ubiquitin | Heavy amino acids (13C6, 15N2 Lys/Arg) for SILAC/AQUA [6] | Absolute quantification of specific linkage types | Commercial vendors |
| Cross-Linking Reagents | MS-cleavable cross-linkers (e.g., DSSO) | Stabilize transient E2/E3/Ub complexes for structural MS | Commercial vendors |
| Computational Tools | UbqTop platform with Bayesian scoring algorithm [4] | Prediction of ubiquitin chain topology from MS2 data | Custom development [4] |
The field of ubiquitin research continues to evolve rapidly, with emerging evidence revealing additional layers of complexity in ubiquitin signaling. Recent studies have identified non-canonical ubiquitination events involving modification of serine, threonine, cysteine, and N-terminal α-amine groups, further expanding the ubiquitin code [9]. Additionally, ubiquitin itself is subject to other post-translational modifications including phosphorylation, acetylation, and ADP-ribosylation, creating complex combinatorial signaling scenarios [9].
Future methodological developments will likely focus on improving sensitivity and throughput for analyzing low-abundance branched chains, developing computational tools for automated topology prediction, and integrating ubiquitin chain analysis with other omics approaches to obtain system-wide understanding of ubiquitin signaling networks. The continued refinement of mass spectrometry-based approaches will be crucial for deciphering the complex language of ubiquitin signaling and understanding its implications in health and disease.
As these technologies mature, we anticipate increasingly sophisticated insights into how ubiquitin chain topology controls cellular decision-making processes, potentially opening new avenues for therapeutic intervention in cancer, neurodegenerative diseases, and immune disorders where ubiquitin signaling is disrupted.
Ubiquitylation is a critical post-translational modification that controls a wide variety of vital processes in eukaryotes, including cell division, differentiation, protein quality control, gene expression, DNA repair, and signal transduction [7]. The versatility of ubiquitin as a cellular signal stems from its capacity to form a diverse array of structures—from single monomers to complex polymers—that can be recognized by different effector proteins to dictate varied cellular outcomes [7] [13]. While early understanding centered on homotypic chains (uniform chains of the same linkage type), recent research has revealed an additional layer of complexity: branched ubiquitin chains, where a single ubiquitin subunit is modified simultaneously on at least two different acceptor sites [7]. The architecture of these chains—encompassing their linkage composition, length, and three-dimensional topology—has emerged as a critical functional determinant that directly controls the fate, activity, and interactions of modified substrates. This review examines the architectural challenge in ubiquitin biology, focusing on why chain architecture is a fundamental determinant of function and how cutting-edge mass spectrometry techniques are cracking this complex code.
The Structural Vocabulary of Ubiquitin Chains
Ubiquitin chains are classified based on the types of linkages connecting adjacent ubiquitin monomers. This structural vocabulary is fundamental to understanding their functional consequences.
- Homotypic Chains: These are linked uniformly through the same acceptor lysine (K6, K11, K27, K29, K33, K48, K63) or the N-terminal methionine (M1). They are often associated with specific cellular functions. For example, K48-linked chains primarily target proteins for degradation by the proteasome, while K63-linked chains and M1-linked linear chains regulate non-proteolytic processes like DNA repair, NF-κB signaling, and autophagy [7] [13].
- Heterotypic Chains: These contain more than one type of linkage and can be further divided into:
- Mixed Chains: Contain multiple linkage types, but each ubiquitin monomer is modified on only one acceptor site.
- Branched Chains: Comprise one or more ubiquitin subunits that are simultaneously modified on at least two different acceptor sites [7]. This branching greatly increases the complexity of ubiquitylation signals, analogous to branched oligosaccharides on the cell surface [7].
Table 1: Common Branched Ubiquitin Chain Architectures and Their Proposed Functions
| Branched Linkage | Biological Context | Proposed Function | Synthesizing Enzymes |
|---|---|---|---|
| K11/K48 | Mitosis | Target substrates for degradation during cell division [7] | APC/C with E2s UBE2C and UBE2S [7] |
| K48/K63 | NF-κB signaling; Apoptosis | Conversion of a non-degradative signal to a degradative mark [7] | TRAF6 & HUWE1; ITCH & UBR5 [7] |
| K29/K48 | Ubiquitin Fusion Degradation (UFD) pathway | Protein quality control, targeting for degradation [7] | Ufd4 & Ufd2 (in yeast) [7] |
| K6/K48 | - | Unidentified, but detected in vitro and in cells [7] | Parkin, NleL [7] |
Why Architecture Dictates Function
The architecture of a ubiquitin chain is not merely a structural curiosity; it is a primary determinant of its functional output. This relationship is governed by several key principles.
Specific Decoding by Ubiquitin-Binding Domains (UBDs)
The cellular interpretation of the ubiquitin code is carried out by effector proteins containing ubiquitin-binding domains (UBDs). There are over 20 different families of UBDs, including UIM, UBA, UBZ, and NZF, each with a unique structural fold [13]. These domains can exhibit remarkable specificity for particular chain architectures. The specificity originates from:
- Multimeric Interactions: Many UBDs synergistically bind multiple ubiquitin subunits within a chain. This allows them to sense both the linkage type and the overall topology of the chain [13].
- Linkage-Specific Contacts: UBDs can make unique contacts with the regions that link ubiquitin molecules into a polymer, enabling them to distinguish between, for example, a K48 linkage and a K63 linkage [13].
Therefore, a branched K48/K63 chain presents a unique "molecular barcode" that can be specifically recognized by a distinct set of UBDs, which would not bind effectively to either a homotypic K48 or K63 chain. This specific recognition then triggers the appropriate downstream outcome, such as proteasomal degradation or activation of a kinase complex.
Mechanism of Branched Chain Synthesis
The synthesis of branched chains is a highly regulated process that underscores their functional importance. Two primary mechanisms have been identified:
- Collaboration Between E3 Ligase Pairs: This is a common theme, involving two E3 ligases with distinct linkage specificities working in tandem. For instance, in the apoptotic response, the E3 ITCH first modifies the substrate TXNIP with non-proteolytic K63-linked chains. This initial mark is then recognized by the UBA domain of a second E3, UBR5, which attaches K48 linkages, resulting in a branched K48/K63 chain that targets TXNIP for proteasomal degradation [7]. This collaboration allows for the temporal regulation and conversion of a non-degradative signal into a degradative one.
- Action of a Single E3 with Multiple E2s or Innate Branching Capability: Some E3s can orchestrate branching alone. The Anaphase-Promoting Complex/Cyclosome (APC/C) recruits two different E2s—UBE2C (which builds initiating chains) and UBE2S (which extends K11 linkages)—to cooperatively assemble branched K11/K48 chains on mitotic substrates [7]. Other E3s, like certain HECT family members, possess an intrinsic ability to synthesize branched chains even with a single E2 [7].
The following diagram illustrates the collaborative synthesis of a branched ubiquitin chain.
Diagram 1: Collaborative synthesis of a branched chain by two E3 ligases.
The Analytical Challenge: Mapping Architecture with Mass Spectrometry
The low stoichiometry of ubiquitination, the multiplicity of modification sites, and the sheer structural diversity of chains make the characterization of ubiquitin chain architecture a significant technical challenge [14]. Traditional methods like immunoblotting are low-throughput and offer limited structural insight [14]. Mass spectrometry (MS) has therefore become the cornerstone technology for decoding the ubiquitin code.
Evolution of Methodologies for Ubiquitin Analysis
Several MS-based strategies have been developed to enrich and identify ubiquitinated proteins and their modification sites.
- Ubiquitin Tagging-Based Approaches: These involve expressing affinity-tagged ubiquitin (e.g., His- or Strep-tagged) in cells. Ubiquitinated substrates are then purified using the tag and identified by MS. A key advantage is the identification of the ubiquitination site via a characteristic 114.04 Da mass shift on the modified lysine after tryptic digestion [14]. While cost-effective, this method can co-purify non-ubiquitinated proteins and may not perfectly mimic endogenous ubiquitination [14].
- Antibody-Based Enrichment: This approach uses anti-ubiquitin antibodies (e.g., P4D1, FK1/FK2) or linkage-specific antibodies (e.g., for K48 or K63) to enrich endogenously ubiquitinated proteins from cell lysates or patient tissues without genetic manipulation [14]. The high cost of antibodies and potential for non-specific binding are notable limitations.
- UBD-Based Enrichment: Proteins containing ubiquitin-binding domains (UBDs), such as tandem-repeated UBD entities (TUBEs), can be used to affinity-purify ubiquitinated proteins. TUBEs exhibit high affinity for ubiquitin chains and can also protect chains from deubiquitinase (DUB) activity during purification [14].
A Revolutionary Approach: Top-Down Mass Spectrometry for Intact Ubiquitin Chains
A major limitation of traditional (bottom-up) MS is the need for proteolytic digestion, which destroys the intact chain architecture. An emerging solution is Top-Down Mass Spectrometry (TD-MS). A groundbreaking 2025 preprint by Shestoperova et al. describes an integrated strategy for the simultaneous determination of ubiquitination sites and chain architecture on intact protein substrates [4].
The core of this method is UbqTop, a custom computational platform that uses a Bayesian-like scoring algorithm to predict Ub chain topology directly from tandem MS (MS2) fragmentation data of the intact molecule. To analyze complex protein substrates, the method combines TD-MS with selective Asp-N proteolysis, which digests the substrate protein while leaving the ubiquitin chains intact, enabling direct mapping of the chain topology to a specific site [4]. This workflow is illustrated below.
Diagram 2: Top-down MS workflow for intact ubiquitin chain analysis.
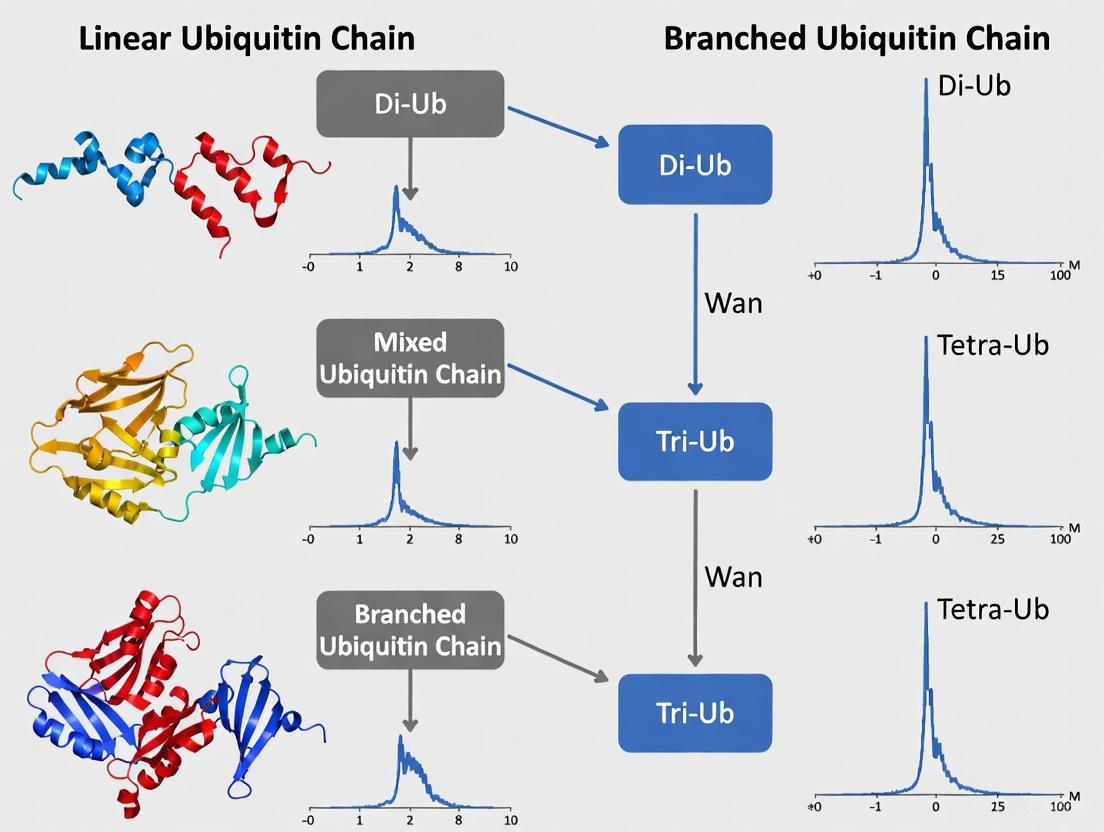
This TD-MS approach offers unparalleled structural resolution, as it can distinguish between isomeric chains and directly resolve branched architectures without inferring information from digested peptides, setting a new standard for proteoform-level analysis of ubiquitin signaling [4].
Table 2: Key Quantitative Proteomics Methods for Ubiquitin Signaling
| Method | Principle | Application in Ubiquitin Research | Advantages | Limitations |
|---|---|---|---|---|
| SILAC (Stable Isotope Labeling with Amino acids in Cell culture) [6] | Metabolic incorporation of "light" or "heavy" isotopic forms of amino acids into proteins for relative quantification. | Quantifying changes in ubiquitinated protein levels across different cellular states (e.g., stimulated vs. unstimulated). | Accurate in vivo labeling; allows multiplexing (e.g., triple SILAC). | Requires metabolically active cells; can't be used on clinical tissues. |
| TMT (Tandem Mass Tagging) [6] | Post-isolation, isobaric chemical labeling of tryptic peptides for relative quantification. | High-throughput screening of ubiquitination sites and dynamics across multiple conditions (up to 10-plex). | High multiplexing capacity; applicable to any protein sample. | Reporter ion signal compression requires MS3 for accurate quantification [6]. |
| Absolute Quantification [6] | Use of internal standard peptides of known concentration to determine absolute abundance. | Determining the stoichiometry of ubiquitination (i.e., what fraction of a substrate protein is ubiquitinated). | Provides crucial stoichiometric and kinetic data. | Requires synthetic isotope-labeled standards; more complex data analysis. |
The Scientist's Toolkit: Essential Reagents and Methodologies
To effectively study ubiquitin chain architecture, researchers rely on a suite of specialized reagents and tools.
Table 3: Essential Research Reagent Solutions for Ubiquitin Architecture Studies
| Tool / Reagent | Function | Key Application |
|---|---|---|
| Linkage-Specific Ub Antibodies [14] | Immuno-enrichment and detection of ubiquitin chains with a specific linkage (e.g., K48, K63). | Western blotting, immunofluorescence, and enrichment for MS analysis of specific chain types. |
| TUBEs (Tandem Ubiquitin Binding Entities) [14] | High-affinity capture of diverse ubiquitinated substrates; protects chains from DUBs. | Purification of endogenous ubiquitinated proteins for downstream assays; stabilization of polyUb chains. |
| Activity-Based DUB Probes | Chemical tools that covalently bind to active deubiquitinases. | Profiling DUB activity and specificity towards different chain architectures; identifying DUBs that reverse specific signals. |
| UbqTop Computational Platform [4] | Bayesian-like scoring algorithm for interpreting top-down MS2 spectra of intact ubiquitin chains. | Direct identification of ubiquitin chain topology, including branched architectures, from TD-MS data. |
| Stable Cell Lines Expressing Tagged Ubiquitin (e.g., His-, Strep-) [14] | Enables affinity-based purification of ubiquitinated proteins from cellular lysates. | System-wide profiling of ubiquitination sites (ubiquitinome) under different conditions. |
The architecture of ubiquitin chains—particularly the emerging complexity of branched polymers—is a fundamental functional determinant that expands the vocabulary of the ubiquitin code far beyond simple homotypic chains. The specific topology of a chain dictates its functional outcome by governing its recognition by a dedicated set of ubiquitin-binding effectors, thereby controlling critical processes like targeted degradation and cell signaling. Fully elucidating this architectural language is paramount for understanding cellular physiology and disease pathogenesis. The development of innovative technologies, especially top-down mass spectrometry coupled with advanced computational biology, is providing the necessary structural resolution to meet this analytical challenge. As these methodologies mature and become more accessible, they will unlock a deeper, proteoform-level understanding of ubiquitin signaling, paving the way for novel therapeutic strategies that target specific ubiquitin chain architectures in diseases such as cancer and neurodegeneration.
Ubiquitination is a fundamental post-translational modification that controls diverse cellular processes in eukaryotes. The versatility of ubiquitin signaling stems from its capacity to form polymers (polyubiquitin chains) of different topologies, which are specialized for distinct cellular functions [15]. While homotypic chains, linked uniformly through a single ubiquitin acceptor site, have been well-characterized, recent research has uncovered the profound functional significance of more complex architectures, particularly branched ubiquitin chains [15] [16]. In these structures, at least one ubiquitin moiety within the chain is simultaneously modified at two or more distinct acceptor sites, creating a bifurcated architecture that significantly expands the informational capacity of the ubiquitin code [15] [16].
This review focuses on the emerging roles of branched ubiquitin chains in two critical and interconnected cellular processes: cell cycle regulation and proteasomal degradation. We examine the latest structural insights into how these chains are recognized and processed, summarize quantitative data on their functional properties, detail the experimental methodologies enabling their study, and provide a toolkit for researchers investigating these complex signals. Understanding these architectures is paramount for a comprehensive view of ubiquitin signaling in health and disease, particularly within the context of mass spectrometry-based proteomic research.
Molecular Mechanisms of Branched Ubiquitin Chain Signaling
Architecture and Synthesis
Branched ubiquitin chains are remarkably diverse in terms of their chemical linkages, structures, and the biological information they transmit [15]. Theoretically, 28 different trimeric branched ubiquitin chain types containing two different linkages can be formed, though only a subset have been identified in cells and linked to biological functions [16].
- Common Branched Chain Types: Physiologically relevant branched chains include those consisting of K11/K48, K29/K48, and K48/K63 linkages [15]. Among these, K11/K48-branched chains are the best characterized, acting as a priority signal for proteasomal degradation during cell cycle progression and proteotoxic stress [12].
- Mechanisms of Synthesis: Branched chain formation often involves collaboration between pairs of E3 ligases with distinct linkage specificities.
- During NF-κB signaling, TRAF6 (synthesizing K63-linked chains) collaborates with HUWE1 (attaching K48 linkages) to produce branched K48/K63 chains [15].
- In the apoptotic response, the HECT E3s ITCH and UBR5 similarly collaborate to form branched K48/K63 chains on the pro-apoptotic regulator TXNIP, converting a non-degradative K63-linked signal into a degradative one [15].
- The APC/C (Anaphase-Promoting Complex/Cyclosome), a multisubunit RING E3, employs a different mechanism. It cooperates with two different E2s, UBE2C (Ubiquitin-conjugating enzyme E2 C) and UBE2S (Ubiquitin-conjugating enzyme E2 S), to form branched K11/K48 chains on substrates during mitosis [15]. UBE2C first attaches short chains containing mixed linkages, and UBE2S then extends them with K11 linkages, resulting in branched polymers [15].
The following diagram illustrates the collaborative synthesis of branched K48/K63 chains by two E3 ligases, a common mechanism for creating these complex signals.
Recognition and Processing by the Proteasome
Branched ubiquitin chains, particularly K11/K48 chains, are recognized as a priority signal for proteasomal degradation [12]. Recent cryo-EM structures of the human 26S proteasome in complex with a K11/K48-branched ubiquitin chain have revealed a multivalent substrate recognition mechanism.
- Multivalent Ubiquitin Binding: The proteasome recognizes the K11/K48-branched chain through a tripartite interface involving:
- The canonical K48-linkage binding site formed by RPN10 and RPT4/5.
- A previously unknown K11-linked Ub binding site at a groove formed by RPN2 and RPN10.
- RPN2 also recognizes an alternating K11-K48 linkage through a conserved motif, similar to the K48-specific T1 site of RPN1 [12].
- Role of Deubiquitinases (DUBs): The proteasome-associated DUB UCH37 (also known as UCHL5) plays a critical role in processing branched chains. UCH37 is recruited to the proteasome by RPN13 and exhibits a unique debranching activity, preferentially cleaving the K48 linkage in branched ubiquitin chains [17]. This activity facilitates the clearance of stress-induced inclusions and promotes the recycling of the proteasome for subsequent rounds of substrate processing [17]. RPN13 enhances UCH37's branched-chain specificity by restricting linear Ub chains from accessing its active site [17].
The molecular basis for the specific recognition of K11/K48-branched chains by the human 26S proteasome is illustrated below.
Quantitative Analysis of Branched Ubiquitin Chain Functions
The functional impact of branched ubiquitin chains can be quantified through their abundance, degradation efficiency, and enzymatic processing rates. The following tables summarize key quantitative data from recent studies.
Table 1: Cellular Abundance and Degradation Efficiency of Branched Ubiquitin Chains
| Branched Chain Type | Cellular Abundance | Proteasomal Degradation Efficiency | Key Biological Context |
|---|---|---|---|
| K11/K48 | Prevalent, ~10-20% of total Ub polymers [12] [17] | Highly efficient ("fast-tracking") [12] | Cell cycle progression (mitosis), proteotoxic stress [12] |
| K48/K63 | Detected in significant amounts [15] | Promotes degradation [16] | NF-κB signaling, apoptosis [15] [16] |
| K29/K48 | Identified in cells [15] | Promotes proteasomal degradation [16] | Ubiquitin Fusion Degradation (UFD) pathway [15] |
| K6/K48 | Detected in vitro or in cells [15] | Enhanced clearance (via UCH37) [17] | Substrate for UCH37 debranching [17] |
Table 2: Enzymatic Processing Rates of Branched Ubiquitin Chains by UCH37
| Ubiquitin Chain Architecture | Relative Hydrolysis Rate by UCH37 | Effect of RPN13 Binding |
|---|---|---|
| Branched K6/K48 Ub3 | 100 (Reference = High) | Further enhancement of activity [17] |
| Branched K11/K48 Ub3 | ~10-fold slower than K6/K48 | Enhancement of activity [17] |
| Branched K48/K63 Ub3 | ~100-fold slower than K6/K48 | Enhancement of activity [17] |
| Linear K48-linked Ub3 | Low (1) | Strong inhibition of activity [17] |
Note: Ub3 denotes a trimeric ubiquitin chain. Data derived from in vitro deubiquitination assays [17].
Experimental Protocols for Studying Branched Ubiquitin Chains
Investigating the architecture and function of branched ubiquitin chains requires specialized methodological approaches. Below are detailed protocols for key techniques in the field.
Enzymatic Assembly of Defined Branched Ubiquitin Trimers
This protocol describes a widely used method to generate branched ubiquitin trimers of defined linkage types for biochemical and structural studies [16].
- Preparation of Proximal Ubiquitin: Start with a C-terminally blocked proximal ubiquitin unit (e.g., ubiquitin 1-72, which lacks the C-terminal tail, or a ubiquitin mutant where the terminal glycine is mutated, Ub-D77).
- First Ligation Step:
- Incubate the blocked proximal ubiquitin with a distal ubiquitin mutant (e.g., Ub-K48R,K63R) and a linkage-specific E2/E3 enzyme pair.
- Example: To form a K63 linkage, use the E2 enzymes UBE2N (Ubc13) and UBE2V1 (Mms2).
- Purify the resulting dimer (e.g., Ub1-72- K63-Ub) using size-exclusion or ion-exchange chromatography.
- Second Ligation Step (Branching):
- Incubate the dimer from step 2 with another distal ubiquitin mutant (e.g., Ub-K48R,K63R) and an E2/E3 pair specific for the second desired linkage.
- Example: To add a K48 branch, use the E2 enzyme UBE2R1 (Cdc34) or UBE2K.
- Purify the final branched trimer product (e.g., K48/K63-branched ubiquitin).
- Validation: Confirm the identity, linkage, and architecture of the assembled chain using intact mass spectrometry and linkage-specific antibodies.
Chemical Synthesis of Branched Ubiquitin Chains
Chemical synthesis offers unparalleled control for incorporating specific modifications and building complex architectures [16].
- Ubiquitin Monomer Synthesis:
- Synthesize ubiquitin building blocks using Solid Phase Peptide Synthesis (SPPS) or via Native Chemical Ligation (NCL) of SPPS-generated fragments.
- Incorporate desired mutations, isotopic labels, or functional groups (e.g., fluorescent tags, warheads) during synthesis.
- Assembly of Branched Core ("isoUb" strategy):
- Create a core unit where a fragment of the distal ubiquitin (e.g., residues 46-76) is linked via a pre-formed isopeptide bond to a fragment of the proximal ubiquitin (e.g., residues 1-45).
- This core contains an N-terminal cysteine and a C-terminal hydrazide for further ligation.
- Chain Elongation:
- Use sequential NCL reactions to attach additional ubiquitin building blocks to the core, elongating the chain as needed.
- Folding and Purification:
- Refold the synthesized polypeptide into the native ubiquitin structure under appropriate buffer conditions.
- Purify the final branched chain to homogeneity using chromatography.
Mass Spectrometry-Based Analysis of Cellular Ubiquitin Chain Linkage
This protocol outlines the steps for identifying and quantifying branched ubiquitin linkages from cellular samples, a cornerstone of ubiquitin proteomics [14].
- Cell Lysis and Denaturation:
- Lyse cells in a denaturing buffer (e.g., containing SDS) to preserve ubiquitination states and inactivate DUBs.
- Enrichment of Ubiquitinated Proteins:
- Use an enrichment strategy to isolate ubiquitinated proteins from the complex lysate. Common methods include:
- Proteolytic Digestion:
- Digest the enriched proteins with trypsin. A key feature of ubiquitination is that trypsin cleavage leaves a di-glycine remnant (GG-signature, mass shift of +114.04 Da) on the modified lysine residue.
- Peptide Fractionation:
- Fractionate the resulting peptides by liquid chromatography to reduce sample complexity.
- Mass Spectrometry Analysis:
- Analyze the peptides using a high-resolution tandem mass spectrometer (LC-MS/MS).
- Use the detected GG-signature peptides to identify ubiquitination sites on substrates.
- Linkage Type Determination:
- For linkage analysis, map the GG-signature peptides to ubiquitin itself. The modified lysine (K6, K11, K27, K29, K33, K48, K63) or N-terminal methionine (M1) in ubiquitin indicates the chain linkage type.
- The presence of a ubiquitin peptide with two different GG-modified lysines provides strong evidence for a branched chain architecture [12] [14].
The workflow for this proteomic analysis is summarized in the following diagram.
The Scientist's Toolkit: Research Reagent Solutions
The study of branched ubiquitin chains relies on a suite of specialized reagents and tools. The following table catalogs essential solutions for researchers in this field.
Table 3: Key Research Reagents for Branched Ubiquitin Chain Studies
| Reagent / Tool | Function / Application | Key Characteristics |
|---|---|---|
| Linkage-Specific Ubiquitin Antibodies | Immunoblotting, immunofluorescence, and enrichment of ubiquitin chains with specific linkages (e.g., K48, K63, K11) [14]. | Critical for validating chain architecture; available for several homotypic linkages. Development of antibodies specific for branched architectures is an ongoing challenge. |
| Tandem Ubiquitin-Binding Entities (TUBEs) | High-affinity enrichment of polyubiquitinated proteins from cell lysates; protect chains from DUBs during purification [14]. | Based on tandem repeats of ubiquitin-associated domains (UBA); more effective than single UBDs for purification. |
| Defined Branched Ubiquitin Chains | Structural studies (e.g., Cryo-EM, X-ray crystallography), in vitro enzymatic assays (DUB/proteasome), binding studies. | Produced via enzymatic assembly or chemical synthesis; allow for precise control over linkage and architecture [16]. |
| Activity-Based Probes (ABPs) | Profiling DUB activity and specificity; identifying DUBs that process branched chains [16]. | Contain an electrophilic warhead that covalently traps DUBs in their active site; can be based on ubiquitin with defined linkages. |
| DUB Inhibitors (e.g., for UCHL5) | Functional studies to probe the role of specific DUBs in cellular processes involving branched chains [17]. | b-AP15 is a known inhibitor of UCHL5 and USP14; used to study consequences of branched chain accumulation. |
| Stable Tagged Ubiquitin Exchange (StUbEx) System | Global proteomic profiling of ubiquitination sites by replacing endogenous ubiquitin with affinity-tagged (e.g., His-, Strep-) ubiquitin in cells [14]. | Enables high-throughput mapping of ubiquitination sites via mass spectrometry; can be combined with linkage-specific ubiquitin mutants. |
| Genetic Code Expansion Systems | Site-specific incorporation of non-canonical amino acids into ubiquitin for click chemistry-based assembly of non-hydrolysable branched chains [16]. | Allows generation of chains resistant to DUB activity, useful for tracking and pull-down experiments. |
Branched ubiquitin chains represent a sophisticated layer of regulation within the ubiquitin-proteasome system. Their role as potent signals for proteasomal degradation, particularly during critical processes like cell cycle progression, is now firmly established. The recent structural elucidation of how the proteasome recognizes K11/K48-branched chains through a multivalent mechanism involving RPN2 provides a molecular framework for understanding this "fast-tracking" effect [12]. Furthermore, the discovery of specialized processing enzymes like the debranching DUB UCH37 highlights the cellular investment in regulating these complex signals [17].
From a methodological perspective, the field is advancing rapidly. The development of robust techniques for the enzymatic and chemical synthesis of defined branched chains, coupled with sophisticated mass spectrometry-based proteomics, is enabling the detailed characterization of their architecture and function [16] [14]. However, challenges remain, including the development of specific detection tools for branched chains and the need to fully elucidate the complete network of E3 ligases and DUBs that govern their dynamics in vivo.
Future research will undoubtedly focus on mapping the full spectrum of biological processes controlled by these complex signals and further unraveling the molecular mechanisms of their recognition. As these tools and understandings mature, they will provide novel insights into disease mechanisms and potentially reveal new therapeutic targets for conditions ranging from cancer to neurodegenerative disorders, where ubiquitin-mediated proteostasis is fundamentally disrupted.
Mass Spectrometry in Action: Techniques for Mapping Sites and Chain Architecture
The systematic identification of enzyme substrates represents a critical challenge in molecular biology, particularly for complex post-translational modifications like ubiquitination. This technical guide delineates how the integration of affinity enrichment methodologies with shotgun proteomics has revolutionized high-throughput substrate identification. By leveraging mass spectrometry-based proteomics, researchers can now systematically profile ubiquitinated substrates, identify modification sites, and decipher ubiquitin chain architectures. This whitepaper provides a comprehensive overview of current methodologies, detailed experimental protocols, and strategic frameworks for implementing these techniques in drug discovery and basic research, with particular emphasis on understanding the complexities of ubiquitin signaling networks.
Ubiquitination is a versatile post-translational modification (PTM) that regulates diverse fundamental features of protein substrates, including stability, activity, and localization [18]. This complexity arises from the ability of ubiquitin to form various polymer architectures—including monoubiquitination, multiple monoubiquitination, and polyubiquitin chains with different linkage types [18]. The versatility of ubiquitin signaling presents significant analytical challenges that conventional biochemical approaches cannot address in a high-throughput manner.
Traditional methods for identifying ubiquitinated substrates relied heavily on immunoblotting with anti-ubiquitin antibodies followed by mutagenesis of putative ubiquitinated lysine residues [18]. While useful for studying individual proteins, these approaches are time-consuming, low-throughput, and ill-suited for system-wide analyses. The emergence of affinity enrichment techniques coupled with shotgun proteomics has transformed the field by enabling researchers to profile hundreds to thousands of ubiquitination events in a single experiment, providing unprecedented insights into the ubiquitin code and its functional consequences in health and disease [18].
Core Principles of Shotgun Proteomics
Fundamental Workflow and Instrumentation
Shotgun proteomics represents the cornerstone of modern high-throughput protein analysis. This bottom-up approach characterizes complex protein mixtures through a series of well-orchestrated steps: enzymatic digestion of proteins into peptides, liquid chromatographic separation of these peptides, tandem mass spectrometry (MS/MS) analysis, and bioinformatic processing of the resulting data [19] [20]. The typical workflow begins with protein extraction and digestion, most commonly using trypsin, which cleaves proteins at the C-terminal side of lysine and arginine residues [21].
The digested peptides are then separated using high-performance liquid chromatography (LC), typically employing reverse-phase columns that separate peptides based on hydrophobicity [20]. This separation is crucial for reducing sample complexity before mass spectrometry analysis. The eluted peptides are ionized, most commonly via electrospray ionization (ESI), and introduced into the mass spectrometer where they are first analyzed as intact species (MS1 spectra) before the most abundant ions are selected for fragmentation (MS2 spectra) [21]. The resulting fragmentation patterns allow for determination of peptide sequences and identification of post-translational modifications through database searching against theoretical spectra generated from in silico digestion of protein databases [19].
Quantitative Approaches in Shotgun Proteomics
A significant advantage of shotgun proteomics is its compatibility with various quantitative strategies, which can be broadly categorized into label-based and label-free approaches [22]. Label-based methods incorporate stable isotopes into proteins or peptides, either metabolically (e.g., SILAC - Stable Isotope Labeling with Amino acids in Cell culture) or chemically (e.g., TMT - Tandem Mass Tag, iTRAQ - Isobaric Tag for Relative and Absolute Quantitation) [22] [19]. These tags allow for accurate relative quantification across multiple samples by creating mass signatures detectable in MS analysis.
Label-free quantification provides a cost-effective alternative, particularly advantageous when analyzing large sample sets such as clinical cohorts [22]. Two primary label-free strategies exist: intensity-based methods that compare peak areas of precursor ions, and spectral counting approaches that utilize the number of identified MS/MS spectra per protein as a quantitative measure [22]. More recent acquisition methods like Data-Independent Acquisition (DIA) or Sequential Window Acquisition of All Theoretical Mass Spectra (SWATH-MS) have further enhanced the reproducibility and depth of label-free quantification by systematically fragmenting all ions within predefined m/z windows [21].
Figure 1: Core shotgun proteomics workflow encompassing sample preparation through data analysis.
Affinity Enrichment Strategies for Ubiquitinated Substrates
Ubiquitin Tagging-Based Approaches
Ubiquitin tagging methodologies employ genetically engineered ubiquitin constructs containing affinity tags to purify ubiquitinated substrates from complex biological mixtures. The most commonly used tags include His, Flag, HA, and Strep tags, which allow for selective enrichment using corresponding affinity resins [18]. In this approach, cells are engineered to express tagged ubiquitin, which becomes incorporated into the cellular ubiquitination machinery. Following cell lysis, ubiquitinated proteins are captured using tag-specific resins—Ni-NTA for His tags or Strep-Tactin for Strep tags—and subsequently identified by mass spectrometry [18].
The pioneering work by Peng et al. demonstrated the power of this approach by identifying 110 ubiquitination sites on 72 proteins from Saccharomyces cerevisiae using 6× His-tagged ubiquitin [18]. Subsequent methodological refinements, such as the Stable Tagged Ubiquitin Exchange (StUbEx) system, have enhanced the efficiency of tagged ubiquitin incorporation, leading to the identification of hundreds to thousands of ubiquitination sites [18]. Despite its utility, this strategy has limitations, including potential artifacts from overexpression of tagged ubiquitin, incompatibility with human tissue samples, and co-purification of endogenous proteins that interact nonspecifically with the affinity resins [18].
Antibody-Based Enrichment Methods
Antibody-based approaches leverage immunorecognition to isolate endogenously ubiquitinated proteins without genetic manipulation of the ubiquitin system. Pan-specific anti-ubiquitin antibodies (e.g., P4D1, FK1, FK2) recognize ubiquitin regardless of linkage type and enable enrichment of the total ubiquitinated proteome [18]. For example, Denis et al. utilized FK2 affinity chromatography to identify 96 ubiquitination sites from MCF-7 breast cancer cells [18].
The development of linkage-specific ubiquitin antibodies has further expanded the utility of this approach by enabling characterization of ubiquitin chain architecture. Antibodies specific for M1-, K11-, K27-, K48-, and K63-linked chains allow researchers to investigate the biological functions associated with specific ubiquitin linkages [18]. Nakayama et al. employed a K48-linkage specific antibody to demonstrate abnormal accumulation of K48-linked polyubiquitination on tau proteins in Alzheimer's disease [18]. While powerful, antibody-based methods face challenges related to cost, availability, and potential off-target binding, which must be carefully controlled through appropriate experimental design.
Ubiquitin-Binding Domain (UBD) Based Approaches
Ubiquitin-binding domains (UBDs) present in various ubiquitin receptors, including some E3 ligases, deubiquitinases (DUBs), and ubiquitin receptors, can be harnessed to enrich ubiquitinated proteins [18]. These domains recognize ubiquitin chains in a general or linkage-selective manner, providing an alternative to antibody-based enrichment. Early implementations utilized single UBDs but suffered from low affinity, leading to the development of tandem-repeated UBD constructs with enhanced avidity [18].
This approach offers several advantages, including the ability to study endogenous ubiquitination under physiological conditions and compatibility with various sample types, including clinical specimens. Additionally, the selectivity of certain UBDs for specific ubiquitin chain types enables structural insights into ubiquitin chain architecture. However, careful validation is necessary to confirm binding specificity and minimize false positives from non-specific interactions [18].
Figure 2: Affinity enrichment strategies for ubiquitinated substrates.
Comparative Analysis of Enrichment Methodologies
Table 1: Comparison of Affinity Enrichment Methods for Ubiquitinated Substrates
| Method | Principle | Throughput | Advantages | Limitations |
|---|---|---|---|---|
| Ubiquitin Tagging | Expression of affinity-tagged ubiquitin (His, Strep, FLAG) in cells [18] | High | Relatively low cost; Easy implementation; Comprehensive substrate profiling | Requires genetic manipulation; Potential artifacts; Not suitable for human tissues |
| Antibody-Based Enrichment | Immunoaffinity purification using ubiquitin antibodies [18] | Medium | Works with endogenous ubiquitin; Compatible with tissues and clinical samples; Linkage-specific options available | High cost; Potential non-specific binding; Limited antibody availability |
| UBD-Based Approaches | Enrichment using ubiquitin-binding domains [18] | Medium | Studies physiological ubiquitination; Linkage-selective options; Compatible with clinical samples | Variable affinity; Requires validation of specificity; Limited commercial reagents |
Advanced Techniques for Ubiquitin Chain Architecture Analysis
Analyzing Branched Ubiquitin Chains
Recent research has highlighted the importance of branched ubiquitin chains in regulating diverse cellular processes, including protein degradation and signal transduction [7]. Unlike homotypic chains composed of a single linkage type, branched chains contain ubiquitin monomers modified at multiple sites, creating complex structures that can encode specialized biological information [7]. Mass spectrometry-based proteomics has been instrumental in deciphering these complex ubiquitin architectures.
Branched chains with defined physiological functions include K11/K48, K29/K48, and K48/K63 linkages, while other branched configurations (K6/K11, K6/K48, K27/K29, K29/K33) have been detected but their functions remain less characterized [7]. The synthesis of branched chains often involves collaboration between E3 ligases with distinct linkage specificities. For instance, TRAF6 and HUWE1 cooperate to produce branched K48/K63 chains during NF-κB signaling, while ITCH and UBR5 generate similar structures during apoptotic responses [7]. Advanced proteomic strategies employing linkage-specific reagents are essential for comprehensive analysis of these complex ubiquitin signals.
Proteomic Profiling of Ubiquitination Sites
The identification of specific ubiquitination sites represents a crucial step in understanding substrate regulation. Mass spectrometry enables precise mapping of ubiquitination sites through detection of a characteristic 114.04 Da mass shift on modified lysine residues, corresponding to the diglycine remnant left after tryptic digestion [18]. However, the low stoichiometry of ubiquitination at individual sites presents a significant challenge, necessitating effective enrichment strategies prior to MS analysis.
Several specialized methodologies have been developed to enhance the sensitivity of ubiquitination site identification. The biotin switch technique (BST) and its variations, such as SNO Site Identification (SNOSID) and SNO resin-assisted capture (SNO-RAC), incorporate biotin labeling for streptavidin-based enrichment of modified peptides [19]. These approaches significantly improve the depth of ubiquitin site mapping by reducing sample complexity and enriching low-abundance modified peptides, enabling identification of hundreds to thousands of ubiquitination sites in a single experiment.
Experimental Protocols for Substrate Identification
Ubiquitin Tagging and Enrichment Protocol
Materials Required:
- Plasmid encoding tagged ubiquitin (e.g., His- or Strep-tagged ubiquitin)
- Appropriate cell line for transfection
- Lysis buffer (e.g., 6 M guanidine hydrochloride, 0.1 M Na₂HPO₄/NaH₂PO₄, 10 mM imidazole, pH 8.0)
- Affinity resin (Ni-NTA agarose for His tags or Strep-Tactin resin for Strep tags)
- Wash buffer (e.g., 8 M urea, 0.1 M Na₂HPO₄/NaH₂PO₄, 10 mM imidazole, pH 8.0)
- Elution buffer (e.g., 200 mM imidazole or biotin-containing buffer for Strep tags)
Procedure:
- Transfect cells with plasmid encoding tagged ubiquitin and incubate for 24-48 hours to allow expression.
- Harvest cells and lyse in denaturing lysis buffer to preserve ubiquitination status and inhibit deubiquitinases.
- Incubate lysate with appropriate affinity resin for 2-4 hours at room temperature with gentle agitation.
- Pellet resin and wash extensively with wash buffer to remove non-specifically bound proteins.
- Elute bound proteins using elution buffer or by boiling in SDS-PAGE loading buffer.
- Process eluted proteins for MS analysis by reduction, alkylation, and tryptic digestion.
- Desalt peptides using C18 solid-phase extraction before LC-MS/MS analysis.
Critical Considerations:
- Include control samples expressing untagged ubiquitin to identify non-specifically bound proteins.
- Optimize transfection conditions to avoid proteotoxicity from ubiquitin overexpression.
- Use protease and deubiquitinase inhibitors throughout the procedure to preserve ubiquitination.
Immunoaffinity Enrichment Protocol for Endogenous Ubiquitome
Materials Required:
- Anti-ubiquitin antibody (e.g., FK1, FK2, or linkage-specific antibodies)
- Protein A/G agarose beads
- Cell lysis buffer (e.g., RIPA buffer with protease inhibitors)
- Wash buffer (e.g., PBS with 0.1% Triton X-100)
- Elution buffer (low pH glycine solution or SDS-containing buffer)
Procedure:
- Lyse cells or tissue in appropriate buffer using mechanical disruption if needed.
- Clarify lysate by centrifugation at high speed to remove insoluble material.
- Pre-clear lysate by incubation with protein A/G beads for 30 minutes to reduce non-specific binding.
- Incubate pre-cleared lysate with anti-ubiquitin antibody for 2-4 hours at 4°C.
- Add protein A/G beads and incubate for an additional 1-2 hours to capture antibody complexes.
- Pellet beads and wash extensively with wash buffer.
- Elute bound proteins using low pH buffer or SDS-PAGE loading buffer.
- Process eluted proteins for MS analysis through digestion and desalting.
Critical Considerations:
- Titrate antibody amount to maximize enrichment efficiency while minimizing non-specific binding.
- For linkage-specific antibodies, validate specificity using appropriate controls.
- Consider cross-linking antibodies to beads to reduce background from antibody leakage.
Table 2: Research Reagent Solutions for Ubiquitin Proteomics
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| Affinity Tags | 6× His-tag, Strep-tag, FLAG-tag [18] | Genetic fusion to ubiquitin for purification of ubiquitinated substrates |
| Enrichment Resins | Ni-NTA Agarose, Strep-Tactin Resin [18] | Selective capture of tagged ubiquitin conjugates |
| Ubiquitin Antibodies | P4D1, FK1, FK2, Linkage-specific antibodies [18] | Immunoaffinity enrichment of endogenous ubiquitinated proteins |
| Mass Spectrometry Standards | TMT, iTRAQ, SILAC reagents [22] [19] | Multiplexed quantification of ubiquitination changes across conditions |
| Ubiquitin-Binding Domains | Tandem UBD constructs [18] | Alternative enrichment tools for specific ubiquitin chain types |
Data Analysis and Bioinformatics Strategies
The analysis of mass spectrometry data from ubiquitin proteomics experiments requires specialized bioinformatic approaches to maximize identification confidence and biological insight. Initial protein identification typically involves searching MS/MS spectra against appropriate protein databases using search engines such as MaxQuant, Proteome Discoverer, or FragPipe. Special consideration should be given to the inclusion of ubiquitination as a variable modification, allowing identification of the characteristic diglycine remnant on lysine residues [18].
Following database searching, rigorous filtering should be applied to eliminate low-confidence identifications. Common criteria include requiring a minimum of one unique peptide per protein, setting false discovery rate thresholds (typically ≤1% at both peptide and protein levels), and applying statistical cutoffs for quantitative data [19]. For label-free quantification, normalization strategies must be implemented to account for technical variation across samples. Subsequent bioinformatic analysis often includes annotation of ubiquitination sites with structural and functional information, pathway enrichment analysis to identify biological processes regulated by ubiquitination, and network analysis to visualize relationships between ubiquitinated proteins [18].
The integration of affinity enrichment strategies with shotgun proteomics has fundamentally transformed our ability to identify ubiquitination substrates and decipher the complex language of ubiquitin signaling. These methodologies have enabled researchers to move from studying individual ubiquitination events to system-wide analyses that capture the dynamic nature of the ubiquitin-modified proteome. As mass spectrometry technology continues to advance with improvements in sensitivity, speed, and resolution, and as new enrichment tools with enhanced specificity are developed, we can anticipate even deeper insights into the regulatory functions of ubiquitination.
Future directions in the field will likely focus on increasing analytical sensitivity to study ubiquitination in limited clinical samples, developing more sophisticated methods for characterizing mixed and branched ubiquitin chains, and integrating ubiquitin proteomics with other omics technologies to obtain a more comprehensive understanding of cellular regulation. These advancements will continue to drive discoveries in basic ubiquitin biology while also facilitating the identification of novel therapeutic targets for diseases characterized by ubiquitination dysregulation, such as cancer and neurodegenerative disorders.
The ubiquitin-proteasome system (UPS) represents a crucial regulatory mechanism in eukaryotic cells, controlling protein stability, function, and localization. Central to understanding this complex post-translational modification is the precise mapping of ubiquitination sites, which has been revolutionized by the identification of the characteristic GlyGly remnant left on trypsinized peptides. This technical guide explores the fundamental principles, methodological approaches, and advanced applications of GlyGly remnant-based ubiquitinomics, framing this powerful methodology within the broader context of ubiquitin chain architecture research. We provide detailed experimental protocols, quantitative datasets, and visualization tools to equip researchers with practical resources for implementing these techniques in both basic research and drug discovery applications.
Protein ubiquitination is a versatile post-translational modification (PTM) that regulates diverse cellular functions, including protein degradation, activity, and localization [14]. This modification occurs through a sequential enzymatic cascade involving E1 activating, E2 conjugating, and E3 ligase enzymes, which ultimately conjugate the C-terminal glycine of ubiquitin to the ε-amino group of a lysine residue on substrate proteins [23] [14]. The complexity of ubiquitin signaling arises from the ability of ubiquitin itself to be modified on any of its seven lysine residues or N-terminal methionine, forming diverse polyubiquitin chains with distinct biological functions [14].
Trypsin digestion of ubiquitinated proteins yields a unique signature that has become the cornerstone of modern ubiquitinomics. Due to the Arg-Gly-Gly sequence at the C-terminus of ubiquitin, tryptic cleavage generates a diGly (GlyGly) remnant—a 114.04 Dalton modification—that remains attached to the modified lysine residue of substrate-derived peptides [24] [25]. This characteristic mass shift serves as a diagnostic signature for mass spectrometry-based identification of ubiquitination sites. The development of specific antibodies recognizing this diGly modification has enabled highly selective enrichment of these peptides from complex biological samples, facilitating comprehensive mapping of ubiquitination sites across the proteome [24] [26].
Table 1: Key Characteristics of the DiGly Remnant
| Characteristic | Description | Significance |
|---|---|---|
| Origin | C-terminal Gly-Gly sequence of ubiquitin after tryptic cleavage | Creates consistent signature across all ubiquitinated proteins |
| Mass Shift | +114.04 Da on modified lysine | Diagnostic mass spectrometry signature |
| Chemical Structure | ε-amino group of lysine conjugated to GlyGly moiety | Antibody generation for selective enrichment |
| Specificity | Distinguishes ubiquitination from other PTMs | Enables specific mapping of ubiquitination sites |
Methodological Foundations of DiGly Proteomics
Antibody-Based Enrichment of DiGly-Modified Peptides
The core technology enabling diGly proteomics is the development of monoclonal antibodies that specifically recognize the diGly remnant on trypsinized peptides. The foundational work describing this approach utilized a carefully designed antigen in which histone III-S was modified with Boc-Gly-Gly-NHS to form amide-linked adducts on lysine amines [24]. After removal of the Boc protecting group, the resulting GlyGly-modified histone was used to generate the GX41 monoclonal antibody, which demonstrated pronounced specificity for peptides containing diglycine-modified lysines while showing negligible reactivity with unmodified proteins or proteins containing internal Gly-Gly sequences [24].
Critical validation experiments demonstrated that this antibody could selectively enrich peptides containing Gly-Gly-modified lysines from complex mixtures while excluding peptides with N-terminal Gly-Gly sequences [24]. The enrichment process achieves remarkable efficiency, with nearly 100% yield of target peptides and enrichment factors exceeding 50-fold compared to non-modified peptides [24]. This high specificity and efficiency make antibody-based enrichment particularly suitable for detecting low-stoichiometry ubiquitination events in complex biological samples.
Workflow for Ubiquitinome Profiling
A standardized workflow for diGly proteomics has been established and refined across multiple studies [23] [27] [24]:
Cell Lysis with Deubiquitinase Inhibition: Lysis is performed in buffers containing deubiquitinase inhibitors such as chloroacetamide to preserve endogenous ubiquitination states [24].
Protein Digestion: Proteins are digested with trypsin, which cleaves after arginine residues and generates the characteristic diGly-modified peptides.
Immunoaffinity Enrichment: DiGly-modified peptides are enriched using anti-diGly remnant antibodies conjugated to solid supports.
Liquid Chromatography-Mass Spectrometry: Enriched peptides are separated by nanoLC and analyzed by high-resolution tandem MS.
Data Analysis: MS/MS spectra are searched against protein databases with specific variable modifications for diGly-linked lysines.
This workflow has enabled the identification of unprecedented numbers of ubiquitination sites, with one comprehensive study reporting approximately 19,000 diGly-modified lysine residues within about 5,000 proteins in human cells [26].
Diagram 1: Experimental workflow for diGly proteomics, highlighting key steps in ubiquitination site mapping.
Advanced Applications in Ubiquitin Chain Architecture
Quantitative DiGly Proteomics for E3 Ligase Substrate Identification
The combination of diGly proteomics with quantitative proteomic strategies has enabled the identification of substrates for specific E3 ubiquitin ligases. A prime example is the application of this approach to identify substrates of HUWE1, an E3 ligase implicated in cancer and intellectual disabilities [23]. Researchers implemented an inducible RNA interference system coupled with stable isotope labeling with amino acids in cell culture (SILAC) to quantitatively monitor changes in diGly peptide abundance following HUWE1 knockdown [23].
In this experimental design, cells expressing inducible HUWE1 shRNA were cultured in light (R0K0) or heavy (R10K6) SILAC media, with shRNA induction followed by proteomic analysis of diGly peptides [23]. This approach revealed DNA damage-inducible transcript 4 (DDIT4) as a novel HUWE1 substrate, which was subsequently validated through biochemical assays demonstrating that HUWE1 directly interacts with and regulates DDIT4 ubiquitination and stability [23]. This methodology provides a powerful general strategy for identifying novel E3 ligase substrates that may represent candidates for therapeutic modulation in the UPS.
Ub-Clipping for Elucidating Branched Ubiquitin Chains
While traditional diGly proteomics provides comprehensive site identification, it typically loses information about polyubiquitin chain architecture. The recently developed Ub-clipping methodology addresses this limitation by utilizing an engineered viral protease (Lbpro*) that cleaves ubiquitin after Arg74, leaving the signature GlyGly dipeptide attached to modified residues [28]. This approach collapses complex polyubiquitin chains into GlyGly-modified monoubiquitin species that retain information about branching patterns.
Strikingly, Ub-clipping analysis revealed that 10-20% of ubiquitin in polymers exists as branched chains in cellular environments [28]. Furthermore, application of this method to study Parkin-mediated mitophagy demonstrated that this process predominantly exploits mono- and short-chain polyubiquitin, with phosphorylated ubiquitin moieties not being further modified [28]. These findings highlight the power of Ub-clipping to provide unprecedented insights into the combinatorial complexity of the ubiquitin code.
Table 2: Quantitative Findings from DiGly Proteomics Studies
| Study Focus | System | Key Quantitative Findings | Reference |
|---|---|---|---|
| Global Ubiquitinome | HEK293 cells | 19,000 diGly sites on 5,000 proteins | [26] |
| HUWE1 Substrates | BT-549 cells | Identification of DDIT4 as novel substrate | [23] |
| Ubiquitin Branching | Multiple cell lines | 10-20% of ubiquitin in polymers is branched | [28] |
| Linkage Composition | Parkin-mediated mitophagy | K6 (38-43%), K11 (11-12%), K48 (27-33%), K63 (16%) | [28] |
| Early Ubiquitinome | HEK293 cells | 374 diGly sites on 236 proteins | [24] |
Experimental Protocols
Detailed Methodology for DiGly Proteomics
The following protocol outlines the key steps for conducting diGly proteomics experiments, adapted from multiple sources [23] [27] [24]:
Cell Lysis and Protein Extraction
- Culture cells in appropriate medium (e.g., RPMI-Flex for SILAC experiments)
- Lyse cells in buffer containing 50mM Tris-HCl (pH 8.0), 150mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate
- Supplement lysis buffer with 5mM chloroacetamide to inhibit deubiquitinases
- Include complete protease inhibitor cocktail
- Clarify lysates by centrifugation at 16,000 × g for 15 minutes
Protein Digestion and Peptide Preparation
- Reduce proteins with 5mM dithiothreitol at 56°C for 30 minutes
- Alkylate with 15mM iodoacetamide at room temperature for 30 minutes in the dark
- Quench alkylation with 10mM dithiothreitol
- Digest proteins with sequencing-grade trypsin (1:50 w/w) overnight at 37°C
- Acidify peptides with trifluoroacetic acid to pH < 3
- Desalt peptides using C18 solid-phase extraction cartridges
DiGly Peptide Enrichment
- Reconstitute peptides in immunoaffinity purification buffer (50mM MOPS-NaOH, pH 7.2, 10mM Na2HPO4, 50mM NaCl)
- Incubate with anti-diGly antibody conjugated to beads for 2 hours at 4°C with rotation
- Wash beads three times with IAP buffer followed by three washes with HPLC-grade water
- Elute peptides with 0.1% trifluoroacetic acid
Mass Spectrometry Analysis
- Separate peptides using nanoflow LC system with C18 column
- Perform MS analysis on high-resolution tandem mass spectrometer
- Use data-dependent acquisition with top-15 method for MS/MS
- Set mass tolerance of 10 ppm for precursor ions and 0.02 Da for fragment ions
- Search data against appropriate protein database with diGly (K-ε-GG; +114.042 Da) as variable modification
Protocol for Ub-Clipping Analysis
The Ub-clipping method provides complementary information about ubiquitin chain architecture [28]:
Sample Preparation and Lbpro* Digestion
- Incubate ubiquitinated samples or cell lysates with Lbpro* (2µg/µL) in reaction buffer (50mM HEPES, pH 7.5, 100mM NaCl, 5mM DTT)
- Include 1M urea in reaction to inhibit endogenous DUBs and E3 ligases
- Digest at 37°C for 2 hours
- Terminate reaction by acidification or heating at 95°C for 5 minutes
Analysis of GlyGly-Modified Ubiquitin
- Separate reaction products by SDS-PAGE or direct MS analysis
- For intact mass analysis, use high-resolution mass spectrometer in positive ion mode
- Quantify different GlyGly-modified ubiquitin species by peak integration
- Calculate branching percentage as (di-GlyGly ubiquitin + tri-GlyGly ubiquitin) / total ubiquitin × 100
Diagram 2: Ub-clipping workflow for analyzing branched ubiquitin chain architecture.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for DiGly Proteomics
| Reagent/Category | Specific Examples | Function and Application |
|---|---|---|
| Anti-diGly Antibodies | GX41 monoclonal antibody [24], Commercial K-ε-GG antibodies [29] | Immunoaffinity enrichment of diGly-modified peptides from tryptic digests |
| Ubiquitin Variants | His6-tagged ubiquitin [24], K63R ubiquitin mutant [12], Strep-tagged ubiquitin [14] | Affinity purification of ubiquitinated proteins; linkage-specific studies |
| Enrichment Tools | TUBEs (tandem ubiquitin-binding entities) [28], Linkage-specific antibodies [14] | Enrichment of polyubiquitinated proteins or specific chain types |
| Specialized Enzymes | Lbpro* [28], Trypsin [24] | Ub-clipping methodology; generation of diGly remnants |
| Mass Spec Standards | Boc-Gly-Gly-lysozyme [29], Synthetic diGly peptides [29] | Quality control and evaluation of enrichment efficiency |
| Inhibitors | Chloroacetamide [24], Proteasome inhibitors (MG132) [23] | Preservation of ubiquitination states by inhibiting DUBs and proteasomal degradation |
Discussion and Future Perspectives
The development of diGly remnant-based proteomics has fundamentally transformed our ability to study the ubiquitin-modified proteome. From initial studies identifying hundreds of ubiquitination sites [24], the field has advanced to comprehensive analyses mapping tens of thousands of sites across diverse biological systems [26]. This methodological progression has been paralleled by increasing sophistication in quantitative approaches, enabling not only site identification but also dynamic monitoring of ubiquitination changes in response to cellular perturbations [23] [26].
The emerging recognition that branched ubiquitin chains constitute a substantial fraction (10-20%) of cellular polyubiquitin [28] highlights the limitations of conventional diGly proteomics and the need for architectural insights provided by methods like Ub-clipping. These branched architectures significantly expand the signaling capacity of the ubiquitin system and appear to play particularly important roles in processes requiring precise degradation timing, such as cell cycle progression and proteotoxic stress response [30] [12]. The structural basis for recognition of K11/K48-branched ubiquitin chains by the 26S proteasome has recently been elucidated, explaining the molecular mechanism underlying their priority as degradation signals [12].
Future methodological developments will likely focus on integrating multiple dimensions of ubiquitin signaling—including site identification, chain architecture, and crosstalk with other post-translational modifications—within unified analytical frameworks. Additionally, applying these techniques to clinical samples and animal models will enhance our understanding of ubiquitination dysregulation in disease pathogenesis. As these methods become more accessible and comprehensive, they will continue to crack the molecular mechanisms of ubiquitin signaling in both physiological and pathological contexts, opening new avenues for therapeutic intervention in cancer, neurodegenerative diseases, and other ubiquitination-related disorders [14].
Ubiquitination is a crucial post-translational modification that regulates diverse cellular functions, including protein degradation, DNA repair, and signal transduction [14]. The versatility of ubiquitin signaling stems from its ability to form complex polyubiquitin chain architectures through different linkage types between the C-terminus of one ubiquitin molecule and specific lysine residues (K6, K11, K27, K29, K33, K48, K63) or the N-terminal methionine (M1) of another [14]. A key regulatory feature of ubiquitylation is that the identity of ubiquitin chain linkage types can control downstream processes [6]. Until recently, a significant knowledge gap existed in our understanding of ubiquitin chain-linkage composition in mammalian tissues and cells under physiological conditions [31]. Defining this in vivo ubiquitin chain-linkage landscape is essential for understanding the functional importance of different ubiquitin chain types in biological processes and disease states.
The emergence of sophisticated quantitative proteomic approaches has set a new standard for elucidating biochemical mechanisms of ubiquitin-driven signaling systems [6] [32]. Among these, Ubiquitin Absolute Quantification by Parallel Reaction Monitoring (Ub-AQUA-PRM) represents a cutting-edge methodology that enables precise, absolute quantification of ubiquitin chain linkages. This technical guide provides an in-depth examination of Ub-AQUA methodology, its applications in characterizing ubiquitin chain architectures, and its integration with complementary approaches for comprehensive ubiquitin signaling analysis.
Technical Foundations of Ub-AQUA Methodology
Core Principles of Absolute Quantification
Ub-AQUA combines synthetic heavy isotope-labeled ubiquitin peptides with high-resolution mass spectrometry to achieve absolute quantification of ubiquitin chain linkages [31] [12]. The method relies on parallel reaction monitoring (PRM), a targeted mass spectrometry technique that provides high specificity and sensitivity for detecting and quantifying predefined peptide targets. The fundamental principle involves spiking known quantities of synthetic, heavy isotope-labeled internal standard peptides corresponding to specific ubiquitin chain linkages into biological samples before mass spectrometry analysis [31].
These signature peptides are derived from tryptic digests of ubiquitin chains and contain the specific lysine-glycine-glycine (K-ε-GG) remnants that remain after trypsin digestion at modified lysine residues [33]. Each signature peptide represents a specific ubiquitin linkage type based on the lysine residue involved in the isopeptide bond. The heavy isotope-labeled synthetic peptides serve as internal standards for precise quantification, as they behave identically to their endogenous counterparts during sample preparation and mass spectrometry analysis but can be distinguished by their mass difference [31].
Experimental Workflow and Optimization
The optimized Ub-AQUA-PRM workflow involves several critical steps that have been refined to enable high-throughput analysis while maintaining analytical rigor [31]:
Sample Preparation Optimization: Critical parameters include urea concentration (8 M), Tris-HCl buffer (50 mM), and NaCl concentration (75 mM) for optimal protein extraction and denaturation while maintaining enzyme compatibility for subsequent digestion steps [33].
Dual Protease Digestion: Sequential digestion with Lys-C (1:100 protease:protein ratio) at room temperature for 4 hours, followed by trypsin digestion (1:25 protease:protein ratio) at 37°C overnight ensures complete digestion while minimizing missed cleavages that could compromise quantification accuracy [33].
Chromatographic Separation: Nano-liquid chromatography methods have been optimized to achieve separation of ubiquitin peptides in 10-minute LC-MS/MS runs, enabling high-throughput screening of multiple samples [31].
Mass Spectrometry Parameters: PRM analysis is typically performed on high-resolution mass spectrometers such as Q-Exactive Hybrid Quadrupole-Orbitrap instruments, with full MS scan resolution of 70,000 and MS2 resolution of 17,500 for precise quantification [33].
The following diagram illustrates the core experimental workflow for Ub-AQUA:
Quantitative Profiling of Ubiquitin Chain Landscapes
Tissue-Specific Ubiquitin Chain Composition
Application of Ub-AQUA-PRM to murine tissues has revealed striking tissue-specific differences in ubiquitin chain-linkage composition [31]. The table below summarizes key quantitative findings from comprehensive profiling across multiple tissue types:
Table 1: Ubiquitin Chain-Linkage Composition Across Murine Tissues
| Tissue Type | Total Ubiquitin Levels | Predominant Linkage Types | Notable Findings | Biological Significance |
|---|---|---|---|---|
| Heart Tissue | Variable between tissues | K63 > K48 > K11 > K6 > K29 > K33 > K27 | Significant enrichment of atypical K33-linked chains | Potential role in contractile function and cardiac physiology |
| Skeletal Muscle | Variable between tissues | K63 > K48 > K11 > K6 > K29 > K33 > K27 | Enrichment of K33-linked ubiquitin chains | Possible specialization for muscle-specific signaling pathways |
| Brain Tissue | Variable between tissues | K48-linked chains particularly abundant | - | Alignment with protein quality control in post-mitotic neurons |
| Bone Marrow-Derived Macrophages | Variable between tissues | K63-linked chains predominant | - | Consistent with inflammatory signaling and NF-κB pathway activation |
| Overall Tissue Patterns | Tissue-specific differences observed | Polyubiquitin chains: small proportion of total ubiquitin | Atypical chains enriched in specific tissues | Suggests functional specialization of ubiquitin linkages |
Specialized Functions of Atypical Ubiquitin Chains
The quantitative data obtained through Ub-AQUA has been particularly illuminating regarding the prevalence and potential functions of less-studied "atypical" ubiquitin chain linkages. K33-linked chains, traditionally among the least characterized ubiquitin linkages, show remarkable enrichment in contractile tissues such as heart and skeletal muscle [31]. This tissue-specific pattern suggests specialized roles for K33 linkages in muscle biology and contractile function that were previously unappreciated.
Similarly, K11/K48-branched ubiquitin chains have been identified as priority degradation signals recognized by the human 26S proteasome [12]. Ub-AQUA analysis revealed that these branched chains account for approximately 10-20% of ubiquitin polymers in cellular contexts and are particularly important for the timely degradation of mitotic regulators, misfolded nascent polypeptides, and pathological Huntingtin variants [12]. The ability to absolutely quantify these complex chain architectures has been instrumental in understanding their functional significance in proteostasis maintenance.
Integrated Ub-AQUA Experimental Protocol
Sample Preparation and Digestion
The following detailed protocol ensures optimal recovery and quantification of ubiquitin chain linkages from biological samples:
Protein Extraction and Denaturation:
- Homogenize tissue or cell samples in 8 M urea, 50 mM Tris-HCl, 75 mM NaCl buffer [33]
- Perform reduction with 10 mM DTT at 56°C for 30 minutes
- Alkylate with 55 mM chloroacetamide at room temperature for 30 minutes in the dark
- Quench excess alkylating agent with 40 mM DTT in 50 mM NH₄HCO₃
Internal Standard Addition:
- Spike known quantities of heavy isotope-labeled ubiquitin signature peptides
- Use peptide mixtures covering all linkage types (K6, K11, K27, K29, K33, K48, K63, M1)
- Maintain consistent spike-in ratios across all samples in an experiment
Proteolytic Digestion:
- First digestion: Lys-C (1:100 w/w protease:protein) at room temperature for 4 hours
- Second digestion: Trypsin (1:25 w/w protease:protein) at 37°C overnight
- Acidify with 1% trifluoroacetic acid to stop digestion
- Desalt using C18 solid-phase extraction cartridges or StageTips
LC-MS/MS Analysis and Quantification
Chromatographic Separation:
- Use nano-liquid chromatography system (e.g., UltiMate 3000 RSLCnano)
- Column: C18 reversed-phase analytical column (75 μm × 25 cm)
- Gradient: 2-30% acetonitrile in 0.1% formic acid over 60 minutes
- Flow rate: 300 nL/min
Mass Spectrometry Parameters:
- Instrument: High-resolution mass spectrometer (e.g., Q-Exactive Hybrid Quadrupole-Orbitrap)
- Full MS scan: Resolution 70,000, scan range 300-1500 m/z
- PRM scans: Resolution 17,500, HCD fragmentation with normalized collision energy 35%
- Isolation window: 2 m/z for targeted precursor ions
Data Processing and Absolute Quantification:
- Process raw data using software such as Skyline or MaxQuant
- Integrate peak areas for light (endogenous) and heavy (internal standard) peptide forms
- Calculate absolute quantities based on standard curves generated from heavy peptides
- Normalize values to total protein content or ubiquitin levels as appropriate
The Ubiquitin Signaling Network: Pathways and Analytical Integration
Ubiquitin signaling does not function in isolation but is integrated with other regulatory networks within the cell. Ub-AQUA provides a quantitative framework for understanding these complex relationships, particularly the crosstalk between ubiquitination and phosphorylation events [6]. The following diagram illustrates key ubiquitin signaling pathways and the integration points where Ub-AQUA provides critical quantitative data:
Advanced Applications and Complementary Techniques
Integration with Structural Biology and Functional Studies
Ub-AQUA methodology has proven particularly powerful when integrated with structural biology approaches. Recent cryo-EM studies of the human 26S proteasome in complex with K11/K48-branched ubiquitin chains utilized Ub-AQUA to characterize the chain-linkage composition of samples used for structural analysis [12]. This integration revealed a multivalent substrate recognition mechanism involving previously unknown ubiquitin binding sites, explaining the molecular basis for prioritized degradation of substrates marked with K11/K48-branched chains.
The methodology has also been applied to characterize ubiquitin chain architectures in extracellular vesicles, revealing that approximately 15% of proteins in human urinary exosomes are ubiquitinated with various topologies (Lys63 > Lys48 > Lys11 > Lys6 > Lys29 > Lys33 > Lys27) [33]. This application demonstrates how Ub-AQUA can provide insights into ubiquitination in physiological fluids and potential biomarker discovery.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagents for Ub-AQUA and Ubiquitin Chain Analysis
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| Linkage-Specific Antibodies | K48-specific, K63-specific, K11-specific antibodies | Immunoblotting validation and enrichment of specific chain types |
| Ubiquitin Mutants | K63R, K48R, K11R ubiquitin variants | Control for linkage specificity in enzymatic assays |
| Recombinant Enzymes | Rsp5-HECTGML engineered ligase, UCHL5 DUB | Generation and processing of specific ubiquitin chain types |
| Mass Spec Standards | Heavy isotope-labeled ubiquitin peptides (K-ε-GG) | Absolute quantification internal standards for Ub-AQUA |
| Affinity Reagents | TUBEs (Tandem Ubiquitin Binding Entities) | Enrichment of ubiquitinated proteins from complex mixtures |
| Computational Tools | UbqTop platform, X! Tandem, MaxQuant | Data analysis and ubiquitin chain topology prediction |
Future Perspectives and Methodological Advancements
The continued evolution of Ub-AQUA methodology promises to further enhance our understanding of ubiquitin signaling complexity. Emerging approaches include the integration of top-down mass spectrometry strategies that enable simultaneous determination of ubiquitination sites and chain architecture on intact protein substrates [4]. Computational platforms such as UbqTop, which utilizes Bayesian-like scoring algorithms to predict ubiquitin chain topology from tandem MS fragmentation data, represent powerful complements to Ub-AQUA quantification [4].
Additionally, methods combining selective proteolysis (e.g., Asp-N digestion) with intact ubiquitin chain preservation are being developed to address the challenge of analyzing complex substrates [4]. These advancements, coupled with improved sensitivity of mass spectrometry instrumentation, will enable more comprehensive profiling of ubiquitin chain architectures under physiological and pathological conditions.
As these methodologies mature, Ub-AQUA will continue to set the standard for quantitative analysis of ubiquitin chain linkages, providing critical insights into the biochemical mechanisms underlying ubiquitin-driven signaling systems in health and disease.
Ubiquitination is a crucial post-translational modification that regulates diverse cellular processes, with its functional versatility arising from architecturally complex polyubiquitin chains. While mass spectrometry (MS) has enabled mapping of individual ubiquitin modifications, the architectural complexity of polyubiquitin signals has remained largely inaccessible. We introduce Ub-clipping as a groundbreaking methodology that utilizes an engineered viral protease, Lbpro*, to directly analyze polyubiquitin chain architecture. This approach reveals that a significant proportion (10-20%) of ubiquitin in polymers exists as branched chains and provides unprecedented insight into combinatorial ubiquitin modifications. Applied to PINK1/Parkin-mediated mitophagy, Ub-clipping demonstrates predominant utilization of mono- and short-chain polyubiquitin with specific phosphorylation patterns, offering new perspectives on the ubiquitin code's complexity.
The ubiquitin code represents one of the most sophisticated post-translational modification systems in eukaryotic cells, governing virtually all cellular processes through targeted protein degradation, signaling transduction, and trafficking. Ubiquitin's versatility stems from its capacity to form architecturally diverse polymers, wherein individual ubiquitin moieties can be modified on one or multiple residues, and further complicated by secondary modifications such as phosphorylation and acetylation [28] [34]. While conventional MS approaches have successfully identified thousands of ubiquitination sites, they fundamentally lack the capacity to preserve and analyze the higher-order architecture of polyubiquitin chains [14]. This architectural information is critical, as different chain structures encode distinct cellular signals—linear chains versus branched structures can determine functional outcomes in processes ranging from proteasomal degradation to DNA repair and immune signaling.
The limitations of traditional trypsin-based MS methods become particularly evident when analyzing complex ubiquitin architectures. Tryptic digestion generates GlyGly-modified peptides that identify modification sites but simultaneously destroys the structural integrity of polyubiquitin chains, making it impossible to discern whether multiple modifications originate from branched structures or mixed populations of homotypic chains [28]. This gap in our analytical capabilities has hindered progress in understanding how ubiquitin chain architecture contributes to signal specificity in biological pathways. The development of Ub-clipping represents a paradigm shift by enabling direct interrogation of polyubiquitin chain architecture, thereby uncovering previously inaccessible dimensions of the ubiquitin code.
The Ub-Clipping Methodology: Principles and Advantages
Core Mechanism of Lbpro* Protease
Ub-clipping utilizes an engineered viral protease, Lbpro, derived from the leader protease of foot-and-mouth disease virus. This engineered enzyme features a point mutation (L102W) that enhances its capability to target all types of diubiquitin and improves catalytic efficiency, particularly in cleaving branched triubiquitin structures [28]. Unlike conventional proteases that completely liberate ubiquitin from modified substrates, Lbpro exhibits unique "clippase" activity by specifically hydrolyzing the peptide bond after Arg74 in ubiquitin, thereby generating two characteristic products: a truncated ubiquitin (residues 1-74) and a GlyGly-modified ubiquitin (1-74) that retains the signature dipeptide remnant on formerly modified lysine residues [28].
This partial cleavage mechanism preserves the GlyGly modification signature while collapsing complex polyubiquitin samples into monoubiquitin species that remain amenable to further analysis. The protease's ability to function in conditions containing 1 M urea further enhances its utility by inhibiting endogenous ubiquitin ligases and deubiquitinases during processing, thus preserving native ubiquitination states [28]. The preservation of GlyGly modifications on what effectively becomes "clipped" monoubiquitin enables subsequent quantification of multiply modified branch-point ubiquitin moieties, providing direct evidence of branched chain architectures that were previously undetectable.
Comparative Advantages Over Existing Methods
Traditional methodologies for ubiquitin analysis, including tryptic digestion-based MS, limited trypsinolysis, and ubiquitin chain restriction (UbiCRest), each present significant limitations for architectural studies. Trypsin-based approaches destroy chain integrity, while UbiCRest requires specialized expertise and optimization for different chain types [28]. Ub-clipping addresses these limitations through several distinct advantages:
- Architectural Preservation: By collapsing chains to modified monoubiquitin, Ub-clipping transforms the challenging analysis of complex polymer architecture into a simpler analysis of modified monoubiquitin species.
- Branch Point Identification: The intact mass analysis of generated monoubiquitin enables direct detection and quantification of ubiquitin molecules bearing multiple GlyGly modifications, unequivocally identifying branch points within chains.
- Compatibility with Complex Mixtures: Lbpro* remains active in cell lysate conditions, enabling global ubiquitin linkage composition analysis from complex biological samples.
- Background Reduction: The specific cleavage mechanism minimizes non-ubiquitin peptide background, enhancing sensitivity for ubiquitin-derived signals.
Table: Comparison of Ubiquitin Chain Analysis Methods
| Method | Architectural Insight | Branched Chain Detection | Throughput | Technical Complexity |
|---|---|---|---|---|
| Ub-clipping | Direct assessment of chain architecture | Direct quantification via multi-GlyGly ubiquitin | High | Moderate |
| Tryptic Digestion + MS | None - destroys chain integrity | Indirect inference only | High | Low |
| UbiCRest | Limited to linkage composition | Limited to specific combinations | Low | High |
| Middle-down MS | Moderate for homotypic chains | Possible but challenging | Moderate | High |
Experimental Framework and Workflows
Core Ub-Clipping Protocol
The standard Ub-clipping protocol involves several critical steps that ensure accurate analysis of ubiquitin chain architecture:
Sample Preparation: Cell lysates or purified ubiquitinated proteins are prepared in buffers compatible with Lbpro* activity. For cellular studies, lysis in conditions containing 1 M urea is recommended to inhibit endogenous deubiquitinases and ligases [28].
Lbpro* Treatment: Incubate samples with Lbpro* protease (preferably the L102W mutant) at appropriate concentrations. The enzyme-to-substrate ratio and incubation time should be optimized for different sample types, though the protease exhibits robust activity across various conditions [28].
Product Separation: Following proteolysis, the reaction products are separated by SDS-PAGE. The characteristic 8 kDa monoubiquitin band is excised for further analysis [28].
Mass Spectrometry Analysis: Process the gel-purified monoubiquitin for MS analysis. Two complementary approaches are recommended:
- Intact Mass Analysis: Direct LC-MS analysis of clipped monoubiquitin reveals different modification states based on mass shifts corresponding to single, double, or triple GlyGly modifications [28].
- AQUA Quantification: Use absolute quantitation with stable isotope-labeled standards to determine linkage composition from the collapsed monoubiquitin pool [28].
Data Interpretation: Interpret mass spectra to identify GlyGly-modified species. The key insight comes from detecting ubiquitin molecules with multiple GlyGly modifications, which directly indicate branch points in the original polyubiquitin chains.
Specialized Applications and Modifications
TUBE-based Enrichment: For samples with abundant free monoubiquitin that can skew branching calculations, tandem ubiquitin-binding entity (TUBE) pulldowns are recommended prior to Lbpro* treatment. This enrichment selectively isolates polyubiquitinated proteins, removes free monoubiquitin, and enables more accurate estimation of chain length and branching frequency [28].
Phospho-ubiquitin Analysis: Ub-clipping successfully analyzes phosphorylated ubiquitin chains, as demonstrated in PINK1/Parkin mitophagy studies. Lbpro* cleaves phospho-ubiquitin chains similarly to unmodified chains, enabling assessment of phosphorylation in specific chain contexts [28].
Substrate-focused Studies: For directed analysis of specific ubiquitinated substrates, immunopurification of the target protein followed by Ub-clipping enables characterization of its ubiquitination status and associated chain architectures [28].
Ub-Clipping Workflow: From Complex Samples to Architectural Insights
Key Research Applications and Findings
Discovery of Prevalent Branched Ubiquitin Chains
Ub-clipping has revealed the unexpected prevalence of branched ubiquitin chains in cellular systems. Quantitative analysis of Lbpro*-generated monoubiquitin from whole cell lysates identified low but consistent levels of di-GlyGly and tri-GlyGly modified ubiquitin, accounting for approximately 0.5% of total ubiquitin across multiple cell lines [28]. When focusing specifically on the polyubiquitin pool through TUBE enrichment, the branching frequency increases dramatically, with 4-7% of all ubiquitin in TUBE pulldowns modified with two GlyGly modifications [28]. This translates to an estimated 10-20% of ubiquitin molecules existing in branched chain contexts, suggesting branching represents a fundamental feature of ubiquitin chain architecture rather than a rare anomaly.
The discovery of abundant branching has profound implications for understanding ubiquitin signaling specificity. Branched chains may create unique structural motifs recognized by specific effector proteins, potentially explaining how the limited number of ubiquitin-binding domains in the genome can discriminate between the myriad ubiquitin signals in cells. Furthermore, different E2 enzymes exhibit distinct propensities for branch formation—UBE2D3, for instance, generates approximately 10% of ubiquitin modified with two, three, or even four GlyGly groups, highlighting how enzyme specificity contributes to architectural diversity [28].
Application to PINK1/Parkin-Mediated Mitophagy
The PINK1/Parkin pathway represents a paradigm for context-dependent ubiquitin signaling, where damaged mitochondria are marked for destruction through a carefully orchestrated ubiquitination cascade. Ub-clipping analysis of this pathway has provided unprecedented insights into the architectural specifics of mitophagic signaling. Contrary to expectations of extended polyubiquitin chains, Ub-clipping revealed that Parkin produces predominantly short chains with a ratio of GlyGly-modified to unmodified ubiquitin of approximately 1:2, indicating limited chain elongation [28].
Moreover, analysis of phosphorylated ubiquitin chains in this pathway demonstrated that phospho-ubiquitin moieties generated by PINK1 are not further modified, suggesting these specific modifications serve as terminal signals in the ubiquitin hierarchy [28]. The linkage composition analysis through Ub-clipping confirmed Parkin's production of mixed linkage chains, with Lys6 (38-43%), Lys11 (11-12%), Lys48 (27-33%), and Lys63-linkages (16%) all represented, consistent with previous reports [28]. These findings collectively paint a more nuanced picture of mitophagic signaling, where short, potentially branched chains with specific phosphorylation patterns create a complex ubiquitin landscape that directs selective autophagic clearance.
Table: Quantitative Findings from Ub-Clipping Applications
| Application | Parameter Measured | Finding | Biological Significance |
|---|---|---|---|
| Global Cellular Analysis | Branched chain frequency | 10-20% of ubiquitin in polymers | Branched chains are common architectural features |
| UBE2D3 Reactions | Multiple modification rate | ~10% with ≥2 GlyGly modifications | Specific E2 enzymes promote branching |
| PINK1/Parkin Mitophagy | Chain length | GlyGly:unmodified ~1:2 | Predominantly short chains in mitophagy |
| Parkin Specificity | Linkage composition | Mixed linkages (K6, K11, K48, K63) | Architectural and linkage complexity |
Essential Research Reagents and Tools
Successful implementation of Ub-clipping requires several critical reagents and tools that researchers should include in their experimental design:
Table: Essential Research Reagents for Ub-Clipping
| Reagent/Tool | Function | Application Notes |
|---|---|---|
| Lbpro* (L102W mutant) | Engineered viral protease that cleaves ubiquitin after Arg74 | Preferred over wild-type for improved efficiency with all diubiquitin types |
| TUBEs (Tandem Ubiquitin-Binding Entities) | Enrich polyubiquitinated proteins; remove free monoubiquitin | Critical for accurate branching quantification by eliminating unmodified ubiquitin |
| Anti-GlyGly Antibody | Detect GlyGly-modified proteins after Lbpro* treatment | Visualizes collapsed ubiquitinated proteins near their original molecular weights |
| AQUA Standards | Absolute quantification of ubiquitin linkage composition | Enables precise measurement of linkage frequencies from clipped monoubiquitin |
| Urea-containing Buffers | Inhibit endogenous DUBs and ligases during processing | Preserves native ubiquitination states during sample preparation |
Ub-clipping represents a transformative methodology that finally provides access to the architectural dimension of the ubiquitin code. By collapsing complex polyubiquitin chains into analytically accessible monoubiquitin units while preserving branch-point information, this technique bridges a critical technological gap in ubiquitin research. The discovery that 10-20% of ubiquitin in polymers exists in branched configurations fundamentally changes our understanding of ubiquitin chain architecture and suggests new mechanisms for signal diversification.
The application of Ub-clipping to biologically relevant systems like PINK1/Parkin-mediated mitophagy demonstrates its power to reveal previously inaccessible details of ubiquitin signaling pathways, from chain length characteristics to the positioning of secondary modifications within chain architectures. As this methodology gains broader adoption, it promises to illuminate architectural aspects of ubiquitin signaling in diverse physiological and pathological contexts, potentially uncovering new regulatory principles and therapeutic opportunities in ubiquitin-related diseases.
Future developments will likely focus on increasing sensitivity for low-abundance samples, combining Ub-clipping with other proteomic approaches for comprehensive ubiquitinome characterization, and adapting the methodology for single-cell analyses. The integration of Ub-clipping with emerging structural biology techniques may further enable correlation of architectural features with three-dimensional structures, ultimately providing a complete picture of how ubiquitin chain architecture dictates functional outcomes in cellular signaling.
Protein ubiquitination is a fundamental post-translational modification that regulates virtually every cellular process in eukaryotic cells, from protein degradation to DNA repair and immune signaling [35]. The versatility of ubiquitin signaling stems from the ability of ubiquitin itself to become modified, forming complex polymeric chains. While the study of homotypic chains (composed of a single linkage type) has dominated the field for decades, recent methodological advances have revealed an additional layer of complexity: branched ubiquitin chains. In these architectures, a single ubiquitin molecule within a polymer is modified at two or more distinct sites, creating a bifurcated or branched structure [36] [16].
The emergence of branched chains significantly expands the ubiquitin code's information capacity. However, their detection and characterization present unique challenges. Standard bottom-up proteomic approaches, which involve proteolytic digestion of proteins into peptides prior to mass spectrometric analysis, inherently collapse the structural information of the intact chain [37] [35]. This limitation has obscured the true complexity of the cellular ubiquitome. To address this gap, the UbiChEM-MS (Ubiquitin Chain Enrichment Middle-Down Mass Spectrometry) workflow was developed, providing a powerful strategy to detect and characterize branched ubiquitin conjugates directly from a cellular context [37] [38]. This technical guide details the principles, protocols, and applications of UbiChEM-MS, framing it within the broader thesis that a complete understanding of ubiquitin signaling requires analytical techniques capable of resolving complex chain architectures.
The UbiChEM-MS Workflow: Principles and Procedures
The UbiChEM-MS methodology integrates biochemical enrichment, controlled enzymatic digestion, and high-resolution mass spectrometry to preserve and identify branched chain topology.
Core Principle of Middle-Down Analysis
UbiChEM-MS is classified as a middle-down mass spectrometry approach. It occupies a strategic middle ground between bottom-up and top-down proteomics. Unlike bottom-up methods that digest proteins into short peptides, UbiChEM-MS uses minimal trypsinolysis to cleave ubiquitin specifically at its C-terminal tail, leaving the core ubiquitin structure (residues 1-74) intact [37] [39]. This generates a mixture of ubiquitin-derived species whose masses report directly on the chain's architecture:
- Ub1-74: Represents an unmodified, C-terminal ubiquitin unit.
- GG-Ub1-74: Represents a ubiquitin unit modified by a single glycine-glycine (GlyGly) remnant, indicating it was internally linked within a linear chain.
- 2xGG-Ub1-74: Represents a ubiquitin unit modified by two GlyGly remnants, providing definitive evidence that this specific ubiquitin molecule was a branch point within the chain [37] [39].
The detection of 2xGG-Ub1-74 is the hallmark signature for a branched ubiquitin chain, and its mass, combined with advanced fragmentation techniques like electron-transfer dissociation (ETD), can be used to identify which lysine residues are involved in the branching [37].
Detailed Experimental Protocol
The following section provides a step-by-step protocol for implementing the UbiChEM-MS workflow.
Step 1: Enrichment of Ubiquitin Conjugates
The first critical step is the isolation of polyubiquitin chains from complex cell lysates. This is typically achieved using ubiquitin-binding entities (UBEs).
- Tandem Ubiquitin Binding Entities (TUBEs): Recombinant proteins comprising multiple ubiquitin-associated (UBA) domains, which exhibit high affinity for polyubiquitin chains of various linkages and protect them from deubiquitinase (DUB) activity during purification [37].
- Linkage-Selective Binders: For targeted studies, domains with known linkage specificity can be employed. For example, the NZF1 domain from the deubiquitinase TRABID shows selectivity for K29-linked chains [37].
- Procedure:
- Prepare cell lysate from the tissue or cultured cells under denaturing conditions (e.g., using a buffer containing 1-2% SDS) to preserve ubiquitination states and inhibit DUBs.
- Dilute the lysate to reduce SDS concentration and incubate with bead-immobilized TUBEs or selective UBDs for 2-4 hours at 4°C.
- Wash the beads extensively with a non-denaturing buffer to remove non-specifically bound proteins.
- Elute the bound ubiquitin conjugates using a low-pH buffer (e.g., 0.1-0.5% formic acid) or by directly denaturing in a compatible MS buffer.
Step 2: Minimal Trypsinolysis and Sample Preparation
The enriched ubiquitin conjugates are then subjected to controlled digestion.
- Procedure:
- Reconstitute the eluted ubiquitin conjugates in an ammonium bicarbonate buffer (pH ~8.0).
- Add sequencing-grade trypsin at an enzyme-to-substrate ratio of approximately 1:50 (w/w).
- Incubate the reaction at 37°C for 1-2 hours—a significantly shorter duration than the overnight digestion typical of bottom-up proteomics [37] [39].
- Quench the reaction by acidification with trifluoroacetic acid or formic acid.
- Desalt the resulting Ub1-74, GG-Ub1-74, and 2xGG-Ub1-74 species using a C18 solid-phase extraction tip or column before MS analysis.
Step 3: High-Resolution Mass Spectrometry and Data Analysis
The digested samples are analyzed using high-resolution mass spectrometry.
- Instrumentation: A high-resolution mass spectrometer (e.g., Orbitrap-based instrument) coupled to nano-flow liquid chromatography is recommended.
- Data Acquisition:
- Perform MS1 scans at a high resolution (e.g., 120,000 at m/z 200) to accurately measure the masses of the various Ub1-74 species.
- For identification of branch point lysines, isolate 2xGG-Ub1-74 ions and fragment them using ETD or EThcD (ETD with supplemental activation). ETD is preferred as it preserves labile post-translational modifications like the GlyGly remnant and generates informative c- and z-ions for sequence determination [37].
- Data Interpretation:
- Identify Ub1-74 species by matching their observed monoisotopic masses to theoretical values.
- The presence of a mass corresponding to a ubiquitin unit with two GlyGly modifications (a mass shift of +114.0429 Da per GlyGly at each lysine) confirms a branch point.
- Use ETD fragmentation spectra to pinpoint the specific lysine residues modified within the 2xGG-Ub1-74 species.
The following diagram illustrates the core workflow and principle of UbiChEM-MS.
Essential Research Reagents and Tools
Successful implementation of UbiChEM-MS relies on a suite of specialized reagents. The table below catalogues the key solutions required for the workflow.
Table 1: Essential Research Reagent Solutions for UbiChEM-MS
| Reagent / Tool | Function / Description | Specific Examples / Notes |
|---|---|---|
| Tandem Ubiquitin Binding Entities (TUBEs) | High-affinity enrichment of diverse polyubiquitin chains from cell lysates; protects chains from deubiquitinases [37]. | Recombinant proteins with multiple UBA domains (e.g., from Prof. Komander's lab). Critical for isolating endogenous-level chains. |
| Linkage-Specific Binders | Enrichment of ubiquitin chains containing a specific linkage type, enabling targeted studies of branched chains [37]. | NZF1 domain from TRABID (binds K29 linkages); other linkage-specific UBDs (e.g., UBAN for M1-linkages). |
| Specific Ubiquitin Mutants | Used as controls or in follow-up experiments to validate linkage specificity and branch point identity [39]. | Single-lysine Ub mutants (e.g., Ub K48R, K63R); lysine-less Ub (Ub K0). |
| Linkage-Specific DUBs | Validation of chain linkage composition through enzymatic digestion (UbiCRest assay) [36] [39]. | OTUB1 (K48-specific), OTUD1 (K63-specific), Cezanne (K11-specific). Used to corroborate UbiChEM-MS findings. |
| High-Resolution Mass Spectrometer | Accurate mass measurement of intact Ub1-74 species and sequence analysis via fragmentation [37] [38]. | Orbitrap-based instruments (e.g., Thermo Scientific Orbitrap Exploris or Fusion series) are ideal for middle-down analysis. |
| Electron-Transfer Dissociation (ETD) | Fragmentation technique that preserves labile modifications (like GlyGly) and enables mapping of branch point lysines [37]. | Preferred over collision-based methods (CID/HCD) for locating modified lysines on Ub1-74 peptides. |
Key Findings and Biological Significance Enabled by UbiChEM-MS
The application of UbiChEM-MS has yielded quantitative insights into the prevalence and dynamics of branched ubiquitin chains, fundamentally advancing our understanding of ubiquitin signaling.
Quantitative Profiling of Branched Chains
UbiChEM-MS has transformed branched chains from biochemical curiosities into quantifiable signaling entities. Research employing this technique has provided critical data on their abundance and regulation.
Table 2: Quantitative Findings on Branched Ubiquitin Chains from UbiChEM-MS Studies
| Branched Chain Type | Enrichment Method | Cellular Condition | Abundance (Approx. % of Isolated Chains) | Biological Implication |
|---|---|---|---|---|
| Mixed Linkage Branched | TUBEs | Normal / Untreated | ~1% | Basal levels of branching exist endogenously [37]. |
| Mixed Linkage Branched | TUBEs | Proteasome Inhibition | ~4% | Branching is dynamic and responsive to proteotoxic stress [37]. |
| K29-linked Branched | NZF1 (K29-selective) | Normal / Untreated | ~4% | K29-branching may constitute a larger fraction of its specific chain type [37]. |
| K11/K48 Branched | K11-specific Antibody | Mitotic Arrest | ~3-4% of total Ub population | Specific branch type (K11/K48) is cell-cycle regulated [40] [39]. |
Functional Roles in Cellular Signaling and Disease
The quantitative data gathered via UbiChEM-MS underpins several major functional themes for branched ubiquitin chains:
- Regulation of Proteasomal Degradation: The discovery that K11/K48-branched chains are enriched during mitosis and upon proteasome inhibition suggests they act as a potent degradation signal [40] [39]. These hybrid chains are proposed to be more efficient than homotypic K48 chains in targeting substrates for proteasomal destruction, potentially by enhancing affinity for proteasomal receptors [36].
- Control of NF-κB Signaling: The K48/K63-branched chain has been identified as a critical regulator of NF-κB activity. This atypical chain type is edited by specific deubiquitinases to fine-tune the amplitude and duration of inflammatory signaling pathways [36].
- Integration of Signaling Inputs: Branched chains can function as coincidence detectors, integrating signals from different ubiquitin conjugation pathways. For example, the formation of a K48/K63 branch on a substrate might require the sequential action of two different E3 ligases, thereby ensuring degradation only occurs when multiple conditions are met [36].
The following diagram summarizes how different branched chain topologies drive distinct biological outcomes, as revealed by UbiChEM-MS and related methods.
Comparison with Other Methodologies and Future Perspectives
UbiChEM-MS occupies a unique niche in the ubiquitin researcher's toolkit. Unlike bottom-up proteomics, it preserves key topological information, and unlike full top-down MS (which analyzes intact proteins), it reduces complexity to a analytically tractable level, making it suitable for profiling cellular conjugates [4] [35]. The recent development of UbqTop, a computational platform for analyzing top-down MS data of ubiquitinated proteins, represents a complementary future direction for intact proteoform analysis [4].
Future advancements in UbiChEM-MS will likely focus on increasing throughput and sensitivity to enable system-wide profiling of branched chain dynamics. Coupling the workflow with more selective ubiquitin binders and improved bioinformatic pipelines will further deconvolute the staggering complexity of the ubiquitin code. As these tools mature, our understanding of how branched ubiquitin chains contribute to cellular homeostasis and disease pathogenesis will deepen, opening new avenues for therapeutic intervention in conditions like cancer and neurodegenerative disorders where ubiquitin signaling is disrupted.
Navigating Analytical Challenges: Optimization Strategies for Ubiquitin MS
Protein ubiquitination is a versatile post-translational modification (PTM) that regulates diverse fundamental features of protein substrates, including stability, activity, and localization [14]. This complexity arises from the ability of ubiquitin to form various conjugates, ranging from a single ubiquitin monomer (monoubiquitination) to polymers (polyubiquitin chains) with different lengths and linkage types [14]. The ubiquitin code's complexity is further enhanced by the possibility of mixed or branched (heterotypic) chain architectures [41]. Unsurprisingly, dysregulation of the intricate balance between ubiquitination and deubiquitination is implicated in numerous pathologies, including cancer and neurodegenerative diseases [14].
A critical challenge in characterizing protein ubiquitination is its very low stoichiometry under normal physiological conditions [14]. Furthermore, the dynamic and heterogeneous nature of ubiquitin signals means that modified proteins often appear as high-molecular weight 'smears' rather than defined species on gels, complicating analysis [41]. To decipher the biological functions encoded by ubiquitination, researchers must therefore employ sophisticated enrichment strategies to overcome the analytical sensitivity limits imposed by its low abundance and immense complexity. This guide details the core methodologies enabling the in-depth analysis of the ubiquitinome within the broader context of understanding ubiquitin chain architectures through mass spectrometry research.
Core Enrichment Methodologies for Ubiquitinome Analysis
Effective analysis of ubiquitination relies on strategies to isolate ubiquitinated proteins or peptides from complex biological lysates, thereby reducing sample complexity and enabling the identification of low-stoichiometry modification sites.
Antibody-Based Immunoenrichment
Immunoenrichment utilizes antibodies raised against ubiquitin or specific motifs associated with the modification to selectively capture ubiquitinated species.
- Anti-diGly Remnant Enrichment: After tryptic digestion of ubiquitinated proteins, a signature diglycine (diGly) remnant with a mass shift of 114.04 Da remains on the modified lysine residue. Antibodies specifically recognizing this K-ε-GG motif allow for the immunopurification of these modified peptides from complex peptide mixtures [42]. This highly efficient method enables the routine identification of over 23,000 diGly peptides from cell lysates and has been successfully applied to various biological sources, including cultured cells and brain tissue [42].
- Anti-Ubiquitin and Linkage-Specific Antibodies: Antibodies such as P4D1 and FK1/FK2, which recognize all ubiquitin linkages, can be used to enrich ubiquitinated proteins prior to digestion [14]. Additionally, linkage-specific antibodies (e.g., for K48, K63, M1) are available to isolate proteins modified with specific chain types, providing insight into the chain linkage involved [14]. For example, a K48-linkage specific antibody revealed the abnormal accumulation of K48-linked polyubiquitinated tau in Alzheimer's disease [14].
Affinity Tagging in Living Cells
This approach involves engineering cells to express ubiquitin tagged with an affinity handle, allowing for the purification of ubiquitinated substrates.
- Common Tags: His-tags and Strep-tags are the most commonly used affinity tags [14]. Cells are engineered to express tagged ubiquitin, which is incorporated into the cellular ubiquitination machinery. Subsequently, ubiquitinated proteins are purified using compatible resins, such as Ni-NTA for His-tags or Strep-Tactin for Strep-tags [14].
- Cellular Systems: The Stable Tagged Ubiquitin Exchange (StUbEx) system is an example where endogenous ubiquitin is replaced with a His-tagged variant, enabling the identification of hundreds of ubiquitination sites from human cell lines [14].
- Considerations: While cost-effective and relatively easy, this method requires genetic manipulation and may introduce artifacts, as the tagged ubiquitin may not perfectly mimic endogenous ubiquitin. It is also infeasible for use directly on animal or patient tissues [14].
Ubiquitin-Binding Domain (UBD)-Based Capture
Proteins containing Ubiquitin-Binding Domains (UBDs) can be exploited to bind and enrich endogenously ubiquitinated proteins under physiological conditions. To overcome the low affinity of single UBDs, Tandem-repeated Ub-binding entities (TUBEs) have been developed. These reagents exhibit high affinity for ubiquitin and can protect polyubiquitin chains from deubiquitinases (DUBs) and proteasomal degradation during cell lysis [14]. UBD-based approaches are valuable for studying endogenous ubiquitination without the need for tagging.
Table 1: Comparison of Key Enrichment Strategies for Ubiquitinated Proteins/Peptides
| Strategy | Principle | Key Advantage | Key Limitation |
|---|---|---|---|
| Anti-diGly Remnant [42] | Immunoaffinity purification of tryptic peptides containing the K-ε-GG motif. | High specificity and sensitivity; directly identifies modification sites. | Requires tryptic digestion; provides no direct information on chain architecture. |
| Full Protein Immunoprecipitation [14] | Uses anti-ubiquitin or linkage-specific antibodies to pull down ubiquitinated proteins. | Works on endogenous proteins; linkage-specific options available. | High cost of antibodies; potential for non-specific binding. |
| Affinity Tagging (e.g., His/Strep) [14] | Expression of epitope-tagged ubiquitin in cells; affinity purification of conjugates. | Relatively low-cost; good for screening in cell culture. | Not suitable for tissues; potential for artifacts from tag. |
| UBD/TUBE-Based [14] | Utilizes high-affinity ubiquitin-binding domains to capture ubiquitinated proteins. | Captures endogenous proteins; protects chains from degradation. | Can have linkage preferences; requires characterization of binder. |
Characterizing Ubiquitin Chain Architecture
Identifying the site of ubiquitination is only the first step; understanding the topology of the attached polyubiquitin chain is crucial for deciphering its functional consequences.
UbiCRest: Deubiquitinase-Based Chain Restriction
UbiCRest is a qualitative method that uses a panel of linkage-specific deubiquitinating enzymes (DUBs) to decipher chain types and architecture [41].
- Principle: A ubiquitinated substrate or purified polyubiquitin chains are treated with specific DUBs in parallel reactions. The resulting cleavage patterns are then analyzed by gel electrophoresis (e.g., immunoblotting) [41].
- Application: The method can demonstrate that a protein is ubiquitinated, identify the linkage type(s) present, and assess whether chains are homotypic or heterotypic (mixed/branched) [41]. For instance, the Lys48-specific DUB OTUB1 and the Lys63-specific DUB AMSH can be used to probe for the presence of these respective linkage types.
- Workflow: The substrate is incubated with a toolkit of DUBs (e.g., USP21 for a positive control as it cleaves all linkages, OTUB1 for Lys48, Cezanne for Lys11). The change in the ubiquitination smear or band pattern after treatment reveals the linkages present [41].
UbiCRest Workflow for Linkage Analysis
Ub-Clipping for Architectural Insights
Ub-clipping is an innovative methodology that provides deep insight into polyubiquitin chain architecture, including the identification of branched chains.
- Principle: The technique uses an engineered viral protease, Lbpro*, to incompletely remove ubiquitin from substrates. Instead of fully cleaving, it leaves the signature C-terminal Gly-Gly dipeptide attached to the modified lysine residue [43].
- Revealing Branch Points: A key advantage is that monoubiquitin generated by Lbpro* retains Gly-Gly-modified residues if they were sites of further ubiquitination. This enables the quantification of multiply Gly-Gly-modified branch-point ubiquitin molecules. Studies using Ub-clipping have revealed that a significant proportion (10–20%) of ubiquitin in polymers exists as branched chains [43].
- Application in Mitophagy: The analysis of depolarized mitochondria with Ub-clipping showed that PINK1/parkin-mediated mitophagy predominantly employs mono- and short-chain polyubiquitin, where phosphorylated ubiquitin moieties are not further elongated [43].
The Scientist's Toolkit: Essential Reagents and Materials
Successful ubiquitinome analysis requires a suite of specific reagents and tools. The following table details key research reagent solutions.
Table 2: Research Reagent Solutions for Ubiquitin Enrichment and Analysis
| Reagent / Tool | Function / Application | Example / Source |
|---|---|---|
| PTMScan Ubiquitin Remnant Motif Kit | Immunoaffinity enrichment of diGly-modified peptides from tryptic digests. | Cell Signaling Technology (Catalog #5562) [42]. |
| Linkage-Specific Anti-Ub Antibodies | Enrichment or detection of proteins modified with specific ubiquitin chain types (e.g., K48, K63, M1). | Commercially available from various suppliers (e.g., MilliporeSigma, Cell Signaling Technology) [14]. |
| Tandem Ubiquitin Binding Entities (TUBEs) | High-affinity capture of endogenous ubiquitinated proteins; protect chains from DUBs. | Recombinant proteins; details in Hjerpe et al. (EMBO Rep. 2009) [14]. |
| Recombinant Deubiquitinases (DUBs) | Enzymatic tools for UbiCRest analysis to determine ubiquitin chain linkage and architecture. | Can be purified in-house [41] or obtained commercially (e.g., OTUB1, AMSH, USP21). |
| Lbpro* Protease | Engineered viral protease for Ub-clipping to reveal chain branching and architecture. | Described in Swatek et al. (Nature 2019) [43]. |
| Stable Isotope-Labeled Amino Acids | For metabolic labeling (SILAC) in quantitative ubiquitinome profiling. | Lysine-8 (13C6;15N2), Arginine-10 (13C6;15N4) from Cambridge Isotope Laboratories [42]. |
Integrated Experimental Workflow: From Cell Culture to Ubiquitinome
A typical state-of-the-art workflow for deep ubiquitinome profiling integrates several of the strategies discussed above, as exemplified by the diGly remnant enrichment protocol.
Integrated Ubiquitinome Profiling Workflow
This workflow involves:
- Cell Culture and Treatment: Cells (e.g., HeLa, U2OS) are cultured, often treated with proteasome inhibitors like Bortezomib to stabilize ubiquitinated proteins [42].
- Protein Preparation and Digestion: Cells are lysed, proteins are extracted, and digestion is performed with trypsin (or LysC). This step generates peptides with the K-ε-GG motif on ubiquitinated lysines [42].
- Peptide Pre-Fractionation: To increase depth, peptides are often fractionated offline using high-pH reverse-phase chromatography prior to enrichment, reducing sample complexity [42].
- diGly Peptide Enrichment: The fractionated peptides are subjected to immunopurification using anti-diGly antibodies [42].
- Mass Spectrometry Analysis: Enriched peptides are analyzed by nanoflow liquid chromatography coupled to tandem mass spectrometry (nanoLC-MS/MS). The 114.04 Da mass shift on lysine is used for unambiguous site identification [42].
The field of ubiquitin research has been revolutionized by the development of highly specific enrichment strategies that overcome the inherent challenge of low stoichiometry. Methods like diGly immunoenrichment, UbiCRest, and Ub-clipping provide complementary layers of information, from site identification to the complex architecture of polyubiquitin chains. As these methodologies continue to evolve and are integrated with advanced mass spectrometry, they will undoubtedly unlock deeper insights into the molecular mechanisms governed by the ubiquitin code in health and disease, providing valuable targets for drug development.
The intricate post-translational modification of protein ubiquitination regulates diverse cellular functions, including protein stability, activity, and localization [18]. Unraveling the complexity of ubiquitin signaling—from identifying specific substrates and modification sites to determining chain linkage and architecture—is fundamental to understanding its role in both normal physiology and disease pathologies such as cancer and neurodegenerative disorders [18]. Central to this research is the initial critical step of enriching low-abundance ubiquitinated proteins from complex biological mixtures. This technical guide provides an in-depth comparison of the two predominant enrichment strategies: tagged ubiquitin systems and antibody-based approaches, framing their application within the broader context of mass spectrometry-driven research on ubiquitin chain architectures.
Technical Comparison of Enrichment Methodologies
The selection between tagged ubiquitin and antibody-based enrichment methods involves significant trade-offs in experimental design, specificity, and applicability. The table below summarizes the core characteristics, advantages, and limitations of each approach.
Table 1: Comparison of Ubiquitin Enrichment Methodologies for Mass Spectrometry
| Feature | Tagged Ubiquitin Approach | Antibody-Based Approach |
|---|---|---|
| Basic Principle | Ectopic expression of ubiquitin fused to an affinity tag (e.g., His, Strep) [18] | Immunoaffinity purification using antibodies against ubiquitin or specific linkages [18] |
| Key Advantages | - Relatively low-cost and easy protocol [18]- High-yield enrichment from engineered cells [18] | - Applicable to endogenous ubiquitin under physiological conditions [18]- Enables study of native tissues and clinical samples [18]- Compatible with linkage-specific analysis using specialized antibodies [18] |
| Key Limitations | - Potential for artifacts as tagged Ub may not fully mimic endogenous Ub [18]- Genetic manipulation required; not feasible for all systems (e.g., patient tissues) [18]- Co-purification of non-specific proteins (e.g., histidine-rich proteins) [18] | - High cost of high-quality antibodies [18]- Potential for non-specific binding [18]- General ubiquitin antibodies (e.g., FK2) do not distinguish linkage types [18] |
| Best Suited For | High-throughput screening of ubiquitinated substrates in tractable cell culture systems [18] | Profiling endogenous ubiquitination in physiological, pathological, or therapeutic contexts [18] |
Experimental Workflows for Ubiquitin Enrichment
The following diagrams illustrate the core experimental workflows for both tagged ubiquitin and antibody-based enrichment strategies, culminating in mass spectrometric analysis.
Advanced Methods for Decoding Ubiquitin Chain Architecture
While enrichment identifies ubiquitinated proteins, specialized techniques are required to decipher the architecture of polyubiquitin chains, which is critical for understanding functional outcomes.
Ub-AQUA/PRM Mass Spectrometry
The Ubiquitin-Absolute Quantification/Parallel Reaction Monitoring (Ub-AQUA/PRM) method provides direct and highly sensitive measurement of all eight ubiquitin linkage types simultaneously [44]. This targeted mass spectrometry approach uses isotopically labeled synthetic peptides corresponding to the signature tryptic peptides of each ubiquitin linkage as internal standards for absolute quantification [44]. PRM, performed on quadrupole-equipped Orbitrap instruments, offers high sensitivity and accuracy by monitoring fragment ions (MS2) with a high-resolution analyzer, making it ideal for quantifying linkage stoichiometry from complex samples [44].
Ub-clipping for Architectural Insight
Ub-clipping is an innovative methodology that utilizes an engineered viral protease, Lbpro*, to incompletely cleave ubiquitin after Arg74 [28]. This unique activity "clips" complex polyubiquitin chains into monoubiquitin units that retain the signature C-terminal Gly-Gly modification on the lysine residue(s) where another ubiquitin was attached [28]. By collapsing the complex architecture into simplified monoubiquitin, subsequent intact mass spectrometry analysis can reveal the number of Gly-Gly modifications on each ubiquitin molecule, directly identifying branch points and quantifying their abundance [28]. This method revealed that a significant fraction (10-20%) of ubiquitin in cellular polymers exists in branched chains [28].
Table 2: Key Research Reagents for Ubiquitin Enrichment and Analysis
| Reagent / Tool | Primary Function | Application Notes |
|---|---|---|
| Tandem Ubiquitin Binding Entities (TUBEs) | High-affinity enrichment of polyubiquitinated proteins; protects chains from deubiquitinases (DUBs) [28] | Used to isolate polyubiquitin prior to Ub-clipping to remove free monoubiquitin and enable chain length estimation [28] |
| Linkage-specific Ub Antibodies | Immunoaffinity enrichment of ubiquitin chains with a specific linkage (e.g., K48, K63) [18] | Enables study of linkage-specific biology; used in immunoblotting or enrichment for MS (e.g., K48-specific antibody revealed tau accumulation in Alzheimer's [18]) |
| AQUA Peptides | Isotopically labeled internal standards for absolute quantification of ubiquitin linkages by MS [44] | Spiked into samples after trypsin digestion; allow precise measurement of the stoichiometry of each ubiquitin chain linkage type [44] |
| Lbpro* Protease | Engineered viral protease for Ub-clipping; cleaves ubiquitin after Arg74 [28] | Core enzyme in Ub-clipping workflow; generates GlyGly-modified monoubiquitin for architectural analysis of chains and branches [28] |
The choice between tagged ubiquitin and antibody-based enrichment is not a matter of identifying a superior technique but of selecting the right tool for the specific biological question. Tagged ubiquitin systems offer a powerful, cost-effective solution for high-throughput discovery in genetically modifiable cell systems. In contrast, antibody-based methods provide the key to investigating endogenous ubiquitination under physiological and pathological conditions, including clinical samples. For a comprehensive understanding of the ubiquitin code, the initial enrichment step must often be coupled with advanced architectural analysis techniques like Ub-AQUA/PRM and Ub-clipping. Together, these methodologies provide researchers with a robust toolkit to dissect the complex roles of ubiquitin signaling in health and disease.
The ubiquitin code represents one of the most sophisticated post-translational regulatory systems in eukaryotic cells, controlling virtually all cellular processes through diverse ubiquitin chain architectures. These chains can be homotypic (uniform linkage), mixed (multiple linkage types in linear sequence), or branched (multiple linkages on a single ubiquitin moiety), with each topology encoding distinct functional consequences for the modified substrate [7]. The remarkable complexity of ubiquitin signaling—particularly the recently appreciated significance of branched chains—poses unique challenges for biochemical analysis. Branched ubiquitin chains, comprising subunits modified simultaneously on at least two different acceptor sites, dramatically expand the informational content of ubiquitylation signals and play crucial roles in cell signaling and protein degradation pathways [7]. Preserving these delicate architectural features during cell lysis is paramount for accurate experimental interpretation, as deubiquitinating enzymes (DUBs) remain active during extraction and can rapidly obliterate the very signals researchers seek to capture.
DUBs constitute a family of approximately 100 human enzymes that hydrolyze ubiquitin-protein bonds, functioning as key editors of the ubiquitin code [45]. During conventional cell lysis procedures, the sudden disruption of cellular compartments and dilution of endogenous inhibitors unleashes latent DUB activity, potentially leading to wholesale remodeling of ubiquitin chain architecture. This problem is particularly acute for the study of branched chains, whose complex structures may present multiple substrates for different DUB families. This technical guide provides a comprehensive framework for controlling DUB activity during cell extraction to preserve endogenous ubiquitin chain architecture, with specific emphasis on methodologies compatible with downstream mass spectrometry analysis.
Methodological Approaches for Controlling DUB Activity During Lysis
Chemical Inhibition Strategies
DUB-Specific Inhibitors: Strategic application of DUB inhibitors during lysis represents the most direct approach to preserving ubiquitin chain architecture. Both broad-spectrum and linkage-specific DUB inhibitors have been characterized and can be deployed depending on experimental goals. For general preservation of all ubiquitin chain types, pan-DUB inhibitors such as ubiquitin-vinyl sulfone (Ub-VS) and ubiquitin-propargylamide (Ub-PA) provide effective solutions [45]. These mechanism-based inhibitors covalently modify the active site cysteine of cysteine protease DUBs, irreversibly inactivating them during cell extraction. When specific chain types are of interest, linkage-selective inhibitors can be employed; for example, OTUB1 selectively inhibits K48-linkage cleavage while AMSH-family inhibitors selectively block K63-chain disassembly [46].
Cysteine-Directed Alkylating Agents: Since the majority of DUBs are cysteine proteases, inclusion of alkylating agents in lysis buffers provides additional protection against non-specific DUB activity. N-ethylmaleimide (NEM) and iodoacetamide effectively modify active site cysteines, rendering DUBs catalytically incompetent. These agents offer the advantage of acting rapidly at room temperature, making them ideal for the initial critical moments of cell disruption when DUB activity poses the greatest threat to ubiquitin chain integrity.
Table 1: Chemical Inhibitors for DUB Control During Lysis
| Inhibitor | Final Concentration | Mechanism of Action | Specificity | Compatibility with Downstream MS |
|---|---|---|---|---|
| Ub-Vinyl Sulfone (Ub-VS) | 1-5 µM | Covalent active site modification | Broad-spectrum (cysteine DUBs) | May interfere with trypsin digestion |
| N-Ethylmaleimide (NEM) | 5-10 mM | Alkylation of cysteine residues | All cysteine-dependent enzymes | Can be quenched with DTT before processing |
| PR-619 | 10-20 µM | Reversible DUB inhibitor | Broad-spectrum DUB inhibition | Compatible with label-free quantification |
| ZnCl₂ | 1-5 mM | Inhibition of JAMM metalloproteases | JAMM family DUBs | May interfere with phosphopeptide analysis |
Lysis Buffer Composition and Physical Conditions
The composition of lysis buffer and physical conditions during cell disruption critically influence DUB activity. While traditional lysis buffers often include reducing agents to maintain protein solubility, these compounds (DTT, β-mercaptoethanol) potently activate cysteine-dependent DUBs and should be omitted during initial extraction when preserving ubiquitin architecture is paramount [47]. Similarly, ATP is often included in lysis buffers to support ubiquitin conjugation, but may inadvertently stimulate certain DUB activities and should be carefully evaluated in pilot experiments.
Physical parameters including temperature and time require strict control. Performing lysis procedures at 4°C slows enzymatic activity but does not completely eliminate DUB function, necessitating combination with chemical inhibition strategies. Rapid processing is essential—the interval between cell disruption and complete inhibition of DUB activity should be minimized to under 30 minutes to prevent significant chain remodeling [48]. Mechanical lysis methods such as bead beating or high-pressure homogenization generate heat that can accelerate DUB activity, highlighting the need for efficient cooling systems during processing [48].
Table 2: Optimized Lysis Buffer Composition for Ubiquitin Chain Preservation
| Component | Final Concentration | Purpose | Considerations for Ubiquitin Studies |
|---|---|---|---|
| Tris-HCl pH 7.5 | 50 mM | Buffer capacity | Neutral pH minimizes non-specific hydrolysis |
| Sucrose | 250 mM | Osmotic stabilization | Maintains subcellular compartments initially |
| NaCl | 150 mM | Ionic strength | Reduces non-specific protein interactions |
| NEM | 10 mM | DUB inhibition | Must be fresh; light-sensitive |
| EDTA | 5 mM | Metalloprotease inhibition | Inhibits JAMM family DUBs |
| Protease Inhibitor Cocktail | 1X | General protease inhibition | Select formulations lacking DUB activators |
| Ub-VS | 5 µM | Specific DUB inhibition | Added immediately before use |
| Glycerol | 10% | Protein stabilization | aids in maintaining complex integrity |
Experimental Protocols for DUB Activity Control and Validation
Controlled Lysis Protocol for Ubiquitin Architecture Preservation
Materials and Reagents:
- HA-Ub-VS probe (Boston Biochem, #U-212)
- N-ethylmaleimide (Sigma-Aldrich, #04259)
- Sucrose (Sigma-Aldrich, #S0389)
- Tris-HCl pH 7.5 (Thermo Fisher, #15567027)
- EDTA (Thermo Fisher, #15575020)
- Glass beads, acid-washed (Sigma-Aldrich, #G4649)
- Pre-cold centrifugation equipment capable of maintaining 4°C
Procedure:
- Preparation of Inhibitor-Enhanced Lysis Buffer: Prepare fresh lysis buffer containing 50 mM Tris-HCl (pH 7.5), 250 mM sucrose, 5 mM EDTA, and 10 mM NEM. Keep on ice until use. Add HA-Ub-VS to a final concentration of 5 µM immediately before lysing cells.
Cell Harvesting: Culture cells to 70-80% confluence. Wash cells twice with ice-cold PBS containing 10 mM NEM to pre-inactivate surface DUBs. Scrape cells gently in PBS+NEM and pellet at 500 × g for 5 minutes at 4°C [47].
Rapid Mechanical Lysis: Resuspend cell pellet in 2 volumes of prepared lysis buffer. Add an equal volume of acid-washed glass beads. Vortex at maximum speed for 30-second bursts with 30-second cooling intervals on ice for a total of 5 minutes [47] [48].
Clarification: Centrifuge the lysate at 800 × g for 30 seconds to settle beads. Transfer supernatant to a fresh tube. Centrifuge at 5,000 × g for 5 minutes to remove nuclei and unbroken cells. Collect supernatant as whole cell lysate.
Validation of DUB Inhibition: Take a small aliquot (20 µg) of lysate and incubate with 2 µL of 1.35 µM HA-Ub-VS for 1 hour at 37°C. Process remaining lysate for downstream applications [47].
Western Blot Analysis: Separate proteins by SDS-PAGE using 4-20% gradient gels. Transfer to PVDF membrane and probe with anti-HA antibody (1:10,000) to confirm DUB inhibition by absence of labeling [47].
Activity-Based Profiling of DUB Inhibition Efficiency
The effectiveness of DUB control during lysis can be quantitatively assessed using activity-based protein profiling (ABPP). This approach utilizes ubiquitin-based probes that covalently modify active DUBs, enabling their detection and quantification.
Procedure:
- Sample Preparation: Divide optimized lysates into two aliquots. Treat one aliquot with HA-Ub-VS probe (50 nM final) for 1 hour at 37°C. Reserve the second aliquot as a negative control.
Affinity Enrichment: Incubate probe-labeled lysates with anti-HA agarose beads for 2 hours at 4°C. Wash beads extensively with lysis buffer containing 0.1% Triton X-100.
On-Bead Digestion: Reduce and alkylate proteins on beads. Digest with trypsin (1:50 w/w) overnight at 37°C.
Mass Spectrometric Analysis: Analyze resulting peptides by LC-MS/MS using a 2-hour gradient. Identify and quantify DUB peptides using MaxQuant software with label-free quantification enabled [45].
Data Interpretation: Compare DUB identification and intensity between inhibition-optimized lysates and controls. Effective DUB inhibition is demonstrated by reduced probe labeling while maintaining protein identification counts.
Diagram 1: Workflow for preservation of ubiquitin architecture during cell lysis. Critical inhibition steps ensure DUB activity is controlled before analysis.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for Controlled DUB Studies
| Reagent/Category | Specific Examples | Function/Application | Key Features |
|---|---|---|---|
| Activity-Based Probes | HA-Ub-Vinyl Sulfone, HA-Ub-Propargylamide | Covalent labeling of active DUBs; validation of inhibition | Hemagglutinin tag for detection; broad DUB reactivity [47] |
| DUB Inhibitors | PR-619, N-Ethylmaleimide (NEM), ZnCl₂ | Pan-DUB inhibition during lysis | PR-619: reversible; NEM: irreversible cysteine alkylation [45] |
| Linkage-Specific DUBs | OTUB1 (K48-specific), AMSH (K63-specific), OTULIN (M1-specific) | UbiCRest analysis of chain linkage type | Recombinant enzymes for controlled chain digestion [41] |
| Affinity Resins | Anti-HA Agarose, Streptavidin Beads | Enrichment of probe-labeled DUBs or ubiquitylated proteins | Compatible with MS sample preparation [45] |
| Mass Spec Standards | 15N-labeled ubiquitin, TMTpro 16-plex | Quantitative proteomics; absolute ubiquitin quantification | Internal standards for accurate quantification [46] |
Integration with Downstream Mass Spectrometry Analysis
Preserving ubiquitin chain architecture during lysis is particularly critical for mass spectrometry-based analyses, which offer the highest resolution view of ubiquitin signaling complexity. The methodological approaches described herein are specifically optimized for compatibility with downstream MS workflows. For global ubiquitin site identification, the preserved architectural integrity enables more accurate mapping of ubiquitylation sites and relative quantification of different chain types [45]. For specialized analyses focusing on branched chains, which have been implicated in crucial regulatory processes including NF-κB signaling and cell cycle control, maintaining structural integrity during lysis is absolutely essential for correct interpretation [7].
The UbiCRest approach, which utilizes linkage-specific DUBs to decipher chain architecture, particularly benefits from well-preserved samples [41]. In this method, purified ubiquitin chains or modified proteins are treated with a panel of DUBs with known specificities, followed by gel-based analysis of cleavage products. Samples prepared with proper DUB control during lysis yield clearer, more interpretable banding patterns that accurately reflect endogenous chain architecture rather than artifacts of sample preparation.
Diagram 2: UbiCRest workflow for ubiquitin chain architecture analysis. Starting with preserved lysates enables accurate identification of homotypic, mixed, and branched chains.
The integrity of ubiquitin chain architecture throughout experimental processing is foundational to accurate interpretation of ubiquitin signaling biology. By implementing the controlled lysis strategies outlined in this guide—combining targeted chemical inhibition with optimized physical parameters—researchers can significantly improve the fidelity of their ubiquitin analyses. These methodologies provide a robust framework for exploring the complex landscape of ubiquitin signaling, particularly as interest grows in the functions of branched and heterotypic chains in physiological and pathological processes. As mass spectrometry technologies continue to advance, enabling ever-more detailed interrogation of ubiquitin chain architecture, the importance of proper sample preparation at the initial lysis stage only becomes more pronounced.
The precise quantification of protein dynamics is a cornerstone of modern proteomics, enabling groundbreaking discoveries in biomarker identification and therapeutic development. This whitepaper explores the integration of stable isotope labeling with the targeted mass spectrometry approach Parallel Reaction Monitoring (PRM) to achieve unprecedented quantitative accuracy and dynamic range. Within the specific context of ubiquitin signaling—a complex post-translational regulatory system—we demonstrate how these methodologies facilitate the precise characterization of ubiquitin chain architectures on protein substrates. The combination of robust isotopic tagging and high-resolution targeted analysis provides researchers with a powerful framework for elucidating intricate cellular processes, offering significant potential for advancing drug discovery pipelines.
Mass spectrometry (MS) has become an indispensable tool in proteomics, providing deep insights into the complex protein landscapes of biological systems. The predominant "bottom-up" approach involves enzymatically digesting proteins into peptides, which are then separated by liquid chromatography and analyzed by MS-based techniques [49]. A critical limitation of conventional MS, however, is that signal intensity does not directly correlate with abundance due to variations in peptide ionization efficiency [49]. To overcome this, stable isotope labeling techniques were developed, allowing for the accurate relative quantification of proteins across different samples by introducing a predictable mass difference [49].
Ubiquitination is a crucial post-translational modification that regulates a vast array of cellular processes, including protein degradation, endocytosis, and DNA damage repair [4]. Its complexity arises from the ability of ubiquitin to form polymeric chains of varying linkage types, lengths, and branching topologies on substrate proteins. This diversity of "ubiquitin chain architectures" creates a sophisticated regulatory code that current mainstream proteomic methods struggle to decipher completely. Often, these methods cannot simultaneously define the exact site of ubiquitination and the topology of the attached ubiquitin chain on an intact protein substrate [4]. This gap in analytical capability hinders a full understanding of ubiquitin signaling dynamics. New, integrated strategies combining specific enzymatic digestion, advanced MS acquisition, and custom computational platforms like UbqTop are now emerging to enable proteoform-level analysis with unprecedented structural resolution [4].
Stable Isotope Labeling: Principles and Methods
Stable isotope labeling allows for the precise comparison of peptide abundance from different experimental conditions by incorporating heavy isotopes (e.g., ²H, ¹³C, ¹⁵N) into proteins or peptides. This incorporation creates a consistent mass shift detectable by a mass spectrometer, enabling relative quantification. The primary labeling strategies are metabolic, chemical, and enzymatic.
Metabolic Labeling: SILAC and Beyond
Metabolic labeling introduces stable isotopes at the earliest stage possible—during cell culture. The most common method is Stable Isotope Labeling by Amino acids in Cell culture (SILAC), where cells are cultured in media containing heavy forms of essential amino acids (e.g., lysine and arginine) [49]. After several cell divisions, these labeled amino acids are fully incorporated into the entire proteome. Upon mixing light and heavy samples, tryptic peptides (except for the C-terminal one) will exhibit a predictable mass shift, simplifying data interpretation [49]. SILAC is renowned for its high quantitative accuracy as it minimizes variability from sample preparation steps post-culture.
A novel advancement in this area is the Neutron Encoded (NeuCode) SILAC method. Instead of using large mass differences, NeuCode utilizes the subtle mass defects arising from the binding energies of different stable isotopes (e.g., ¹²C/¹³C, ¹H/²H) [49]. These tiny mass differences (e.g., 6-36 mDa) are indistinguishable on low-resolution instruments but can be resolved using high-resolution mass spectrometers like Orbitrap instruments, enabling highly multiplexed quantification without increasing spectral complexity [49].
Chemical Labeling: Isobaric and Isobaric Tags
Chemical labeling introduces isotopes via a chemical reaction with specific amino acid functional groups (e.g., amine groups) after protein digestion. Early reagents like the Isotope-Coded Affinity Tag (ICAT) targeted cysteine thiol groups, but this limited analysis to cysteine-containing peptides [49]. A more general approach is dimethylation, which labels the N-termini and ε-amino groups of lysine residues. This reaction is efficient, cost-effective, and can be multiplexed (e.g., 5-plex) [49]. Dimethylation also enhances peptide ionization efficiency, potentially improving sensitivity [49].
Isobaric tagging, used in methods like Tandem Mass Tags (TMT) and Isobaric Tags for Relative and Absolute Quantitation (iTRAQ), is another powerful chemical strategy. These tags are isobaric, meaning they have the same overall mass, and consist of a balance group and a reporter group. The key feature is that peptides from different samples, labeled with different isobaric tags, are indistinguishable in the MS1 spectrum. However, upon fragmentation in the MS2 stage, the linker between the balance and reporter group breaks, releasing low-mass reporter ions whose intensities provide quantitative information for each sample [49]. While powerful, these methods can suffer from ratio distortion due to co-isolated interfering peptides; however, advanced instrumentation and computational corrections are mitigating this issue.
Table 1: Comparison of Common Stable Isotope Labeling Methods
| Method | Type | Principle | Multiplexing Capacity | Key Advantage | Key Limitation |
|---|---|---|---|---|---|
| SILAC [49] | Metabolic | Incorporation of "heavy" amino acids during cell growth | 2-3 plex (standard), 6+ plex (NeuCode) | High accuracy; minimal post-harvest variability | Limited to cell culture or simple organisms |
| Dimethylation [49] | Chemical | Labeling of amine groups with formaldehyde/cyanoborohydride | 2-5 plex | Cost-effective, simple, improves ionization | Potential for side reactions |
| Isobaric Tags (e.g., TMT) [49] | Chemical | MS2-based quantification via reporter ions | 6-18 plex | High multiplexing, reduces MS1 complexity | Susceptible to ratio compression |
| ¹⁸O Labeling [49] | Enzymatic | Incorporation of ¹⁸O during enzymatic digestion | 2-plex | Applicable to diverse sample types | Limited multiplexing |
Parallel Reaction Monitoring (PRM): A Targeted Paradigm
Selected Reaction Monitoring (SRM) on triple quadrupole instruments has been the gold standard for targeted proteomics due to its high reproducibility and sensitivity. However, a new paradigm, Parallel Reaction Monitoring (PRM), leverages the capabilities of high-resolution and accurate mass (HR/AM) instruments, such as quadrupole-Orbitrap systems, to overcome several limitations of SRM [50].
In PRM, the first quadrupole isolates the precursor ion of a target peptide, much like in SRM. The key difference lies in the final stage: instead of a third quadrupole monitoring a few pre-selected fragments, the isolated precursor is fragmented, and all resulting product ions are detected in a parallel, high-resolution mass analysis [50]. This approach offers several distinct advantages:
- High Selectivity: The high mass accuracy of the fragment ions dramatically increases confidence in peptide identification and quantification by reducing chemical noise [50].
- Minimal Method Development: There is no need to pre-define and optimize transitions for each peptide, as all fragments are recorded. This simplifies experimental setup [50].
- Retrospective Analysis: The full MS2 spectrum is stored, allowing researchers to re-interrogate the data for different fragments or confirm the presence of unexpected modifications post-acquisition.
- Wider Dynamic Range: Studies have shown that PRM can yield quantitative data over a wider dynamic range than SRM in complex matrices because of its superior selectivity in the mass-to-charge domain [50].
Diagram 1: PRM targeted proteomics workflow.
Integrated Strategy for Ubiquitin Analysis
To address the specific challenge of analyzing ubiquitinated proteins, an integrated strategy combining selective proteolysis, top-down mass spectrometry (TD-MS), and custom computation has been developed [4]. This workflow is designed to preserve intact ubiquitin chains and simultaneously determine the site of ubiquitination and the chain architecture.
- Selective Asp-N Proteolysis: The protein substrate is selectively digested with the Asp-N enzyme, which cleaves the substrate while leaving the attached ubiquitin chains intact. This step simplifies the analysis by reducing the size and complexity of the substrate without destroying the topological information of the ubiquitin modification [4].
- Top-Down Mass Spectrometry: The resulting ubiquitinated peptides, with intact chains, are analyzed using TD-MS. This approach fragments the entire molecule without a prior digestion step, preserving information about the connectivity and modifications on the intact proteoform [4].
- Computational Analysis with UbqTop: The complex MS2 fragmentation data is processed by UbqTop, a custom computational platform. This software utilizes a Bayesian-like scoring algorithm to predict the ubiquitin chain topology directly from the fragmentation spectrum, enabling the resolution of isomeric chains and branched architectures [4].
Diagram 2: Integrated ubiquitin chain analysis workflow.
Quantitative Data Analysis and Dynamic Range
The quantitative analysis of isotope labeling data, especially from complex experiments involving fractional labeling, requires sophisticated computational tools. The Least-Squares Fourier Transform Convolution (LS-FTC) approach provides a general solution for this [51]. It calculates theoretical isotope distributions—including those from complex fractional atomic labeling (e.g., ¹⁵N) or fractional residue labeling (e.g., ¹³C-Isoleucine)—and uses least-squares fitting to determine both the relative abundance of different species and the extent of isotopic labeling from experimental spectra [51]. This method is exact and avoids the distortions that can arise from polynomial-based calculations that require truncation.
When comparing quantification methods, PRM demonstrates distinct performance advantages. A key study found that PRM and SRM showed statistically equal linearity over their quantifiable ranges. However, PRM achieved quantification over a wider dynamic range in the presence of a complex yeast background matrix. This expanded range is attributed to PRM's high selectivity in the mass-to-charge domain, which more effectively filters out chemical noise from co-eluting peptides [50].
Table 2: Quantitative Performance: PRM vs. SRM [50]
| Performance Metric | Parallel Reaction Monitoring (PRM) | Selected Reaction Monitoring (SRM) |
|---|---|---|
| Run-to-Run Reproducibility | High | High |
| Dynamic Range | Wider dynamic range in complex matrices | Narrower dynamic range compared to PRM |
| Measurement Accuracy | High (with high-resolution fragments) | High |
| Linearity | Statistically equal to SRM | Statistically equal to PRM |
| Method Development | Minimal; no need for optimized transitions | Extensive; requires pre-defined transitions |
| Data Completeness | Records all fragments; allows retrospective analysis | Limited to pre-selected transitions |
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for Isotope Labeling and Ubiquitin Studies
| Item / Reagent | Function / Application |
|---|---|
| SILAC Media Kits | Commercially available media formulations containing stable isotope-labeled amino acids (e.g., Lys-8, Arg-10) for metabolic labeling of cells in culture [49]. |
| Isobaric Tagging Kits (e.g., TMTpro) | Multiplexed chemical tagging reagents (e.g., 16-plex) for comparing multiple samples simultaneously in a single LC-MS run via MS2-based reporter ions [49]. |
| NeuCode Lysine/Arginine | Specialty amino acids with specific isotopic configurations (e.g., ⁶H⁸) for high-level multiplexing in SILAC experiments using high-resolution mass spectrometers [49]. |
| Asp-N Protease | A specific protease used in ubiquitin studies to digest protein substrates while leaving the ubiquitin chains intact, enabling topology analysis [4]. |
| UbqTop Software | A custom computational platform that utilizes a Bayesian-like scoring algorithm to predict ubiquitin chain topology from top-down MS2 fragmentation data [4]. |
| High-Resolution Mass Spectrometer | Instrumentation (e.g., quadrupole-Orbitrap) essential for PRM, NeuCode, and top-down MS analyses due to requirements for high mass accuracy and resolution [49] [4] [50]. |
The synergistic combination of stable isotope labeling and targeted PRM analysis represents a powerful frontier in quantitative proteomics. This approach provides researchers with a robust framework for achieving high quantitative accuracy, sensitivity, and a wide dynamic range. By applying this integrated methodology to the complex challenge of deciphering the ubiquitin code, scientists can now move beyond simple identification to a deeper, proteoform-level understanding of ubiquitin site and chain architecture. This technical advancement paves the way for significant discoveries in fundamental cell biology and offers a more precise toolkit for identifying and validating therapeutic targets in the drug development pipeline.
Validating the Signal: Correlating MS Data with Function and Disease
In the complex field of ubiquitin chain architecture analysis, the integration of mass spectrometry (MS) data with linkage-specific antibodies and biochemical assays represents a fundamental methodological challenge. Ubiquitination regulates numerous cellular processes through the attachment of polyubiquitin chains that vary dramatically in linkage type, length, and branching topology [4]. Beyond the canonical K48-linked homotypic polyubiquitination for proteasome-targeted proteolysis, emerging research has revealed the biological significance of complex chain architectures, particularly K11/K48-branched ubiquitin chains, which are now known to be involved in fast-tracking protein turnover during cell cycle progression and proteotoxic stress [12]. The structural basis for how the human 26S proteasome recognizes these K11/K48-branched chains was only recently elucidated through cryo-EM studies, demonstrating a multivalent substrate recognition mechanism that involves previously unknown ubiquitin binding sites [12].
The critical importance of cross-validation stems from the inherent limitations of any single analytical approach. Mass spectrometry, while powerful for identifying ubiquitination sites and chain topology, requires complementary validation through biochemical and immunological methods to confirm linkage specificity and biological relevance. Similarly, linkage-specific antibodies provide exceptional sensitivity but may exhibit cross-reactivity or fail to recognize novel or complex branched structures. This whitepaper establishes a comprehensive framework for cross-validation methodologies that integrate these disparate data sources to achieve unprecedented structural resolution in ubiquitin signaling research, with direct implications for drug development targeting the ubiquitin-proteasome system.
Experimental Methodologies for Ubiquitin Chain Analysis
Mass Spectrometry Approaches
Top-Down Mass Spectrometry for Intact Ubiquitin Chain Analysis Recent advancements in top-down mass spectrometry (TD-MS) have enabled simultaneous determination of ubiquitination sites and chain architecture without proteolytic digestion. The core innovation in this approach is the integration of TD-MS with custom computational platforms, such as UbqTop, which utilizes Bayesian-like scoring algorithms to predict Ub chain topology from tandem MS (MS2) fragmentation data [4]. This method preserves the intact ubiquitin chain structure, allowing direct mapping of Ub chain topology on proteins. For complex substrates, researchers combine this with selective Asp-N proteolysis, which digests the protein substrate while preserving intact Ub chains, enabling proteoform-level analysis of ubiquitin signaling with unprecedented structural resolution [4].
Bottom-Up Mass Spectrometry with Ubiquitin Absolute Quantification (Ub-AQUA) For targeted quantification of specific ubiquitin linkages, the bottom-up MS approach with Ub-AQUA provides exceptional specificity and sensitivity. This methodology involves:
- Sample Denaturation and Reduction: 8M urea, 5mM TCEP, 25-50mM ammonium bicarbonate
- Alkylation: 10mM iodoacetamide (30min, room temperature, dark)
- Proteolytic Digestion: Trypsin/Lys-C mixture (1:20-1:50 enzyme:substrate), 37°C overnight
- Peptide Desalting: C18 solid-phase extraction columns
- LC-MS/MS Analysis: Reverse-phase nanoflow chromatography coupled to high-resolution tandem mass spectrometer
- Data Analysis: Heavy isotope-labeled internal standard peptides for absolute quantification of specific ubiquitin linkages [12]
Linkage-Specific Antibody Applications
Linkage-specific antibodies remain indispensable tools for validating ubiquitin chain structures identified by MS approaches. These antibodies enable:
- Western Blot Validation: Confirming presence of specific ubiquitin linkages in complex samples
- Immunoprecipitation: Enriching for proteins modified with specific chain types
- Immunofluorescence: Spatial localization of specific ubiquitin signals in cells
Critical methodological considerations include antibody validation using defined ubiquitin chains, optimization of blocking conditions to minimize non-specific binding, and appropriate controls including knockout/knockdown samples where possible. For K11/K48-branched chain detection, recent studies have successfully employed K11-linkage specific antibodies in combination with K48-linkage reagents to confirm the presence of branched structures [12].
Biochemical and Functional Assays
Ubiquitin Chain Binding Assays Proteasomal ubiquitin receptor binding assays provide functional validation of ubiquitin chain architecture. The protocol involves:
- Immobilized Ubiquitin Chain Preparation: Biotinylated ubiquitin chains coupled to streptavidin beads
- Receptor Incubation: Recombinant ubiquitin receptors (RPN1, RPN10, RPN13) incubated with immobilized chains
- Binding Buffer: 25mM Tris-HCl (pH 7.5), 150mM NaCl, 0.1% Triton X-100, 1mM DTT, 0.5mg/mL BSA
- Wash Conditions: Three washes with binding buffer without BSA
- Elution and Detection: SDS-PAGE and Western blotting with receptor-specific antibodies [12]
Deubiquitinase (DUB) Specificity Profiling DUBs exhibit remarkable specificity for different ubiquitin chain types. UCHL5, for instance, preferentially recognizes and processes K11/K48-branched Ub chains from proteasomal substrates [12]. The DUB assay protocol includes:
- Enzyme Preparation: RPN13:UCHL5 complex (catalytically inactive C88A mutant for binding studies)
- Substrate: Fluorescently labeled ubiquitin chains (Sic1PY-Ubn with Alexa647 and fluorescein labeling)
- Reaction Conditions: 50mM Tris-HCl (pH 7.5), 100mM NaCl, 1mM DTT, 0.1mg/mL BSA, 37°C
- Reaction Monitoring: Fluorescence detection or Western blotting with linkage-specific antibodies
Integrated Cross-Validation Workflow
The following computational diagram illustrates the comprehensive cross-validation workflow that integrates these methodological approaches, with color-coding to distinguish between experimental techniques and validation nodes:
Figure 1: Integrated cross-validation workflow for ubiquitin chain architecture analysis. The diagram illustrates how mass spectrometry approaches (red), linkage-specific antibodies (yellow), and biochemical assays (blue) converge through data integration to generate validated structural models.
Quantitative Cross-Validation Data Analysis
Method Comparison Statistics
Robust statistical analysis is essential for demonstrating correlation between different analytical methods. The following table presents key validation parameters derived from cross-validation studies between multiplex LC-MS/MS and reference methods for monoclonal antibody quantification, which provides an excellent model for ubiquitin research:
Table 1: Cross-Validation Performance Metrics Between Multiplex LC-MS/MS and Reference Methods
| Validation Parameter | Bevacizumab | Cetuximab | Ipilimumab | Nivolumab | Pembrolizumab | Rituximab | Trastuzumab |
|---|---|---|---|---|---|---|---|
| Linear Range (µg/mL) | 2-100 | 2-100 | 2-100 | 2-100 | 2-100 | 2-100 | 2-100 |
| Inter-Assay Precision (% CV) | 1.5-3.3 | 1.7-4.1 | 3.2-8.9 | 4.1-14.6 | 2.1-6.3 | 2.5-7.4 | 2.1-8.9 |
| Inter-Assay Accuracy (%) | 100.5-105.4 | 97.8-104.2 | 93.1-107.1 | 91.3-104.5 | 95.2-103.8 | 96.4-104.7 | 95.1-104.9 |
| Mean Absolute Bias vs. Reference (%) | 6.2 | 8.7 | 11.3 | 19.9 | 9.5 | 7.1 | 3.0 |
| Signal-to-Noise at LLOQ | 33 | 41 | 8 | 11 | 25 | 10 | 30 |
Data adapted from cross-validation of multiplex LC-MS/MS method for simultaneous quantification of monoclonal antibodies [52]. LLOQ: Lower Limit of Quantification.
Ubiquitin Linkage Quantification Data
The integration of Ub-AQUA with linkage-specific antibodies enables comprehensive quantification of ubiquitin chain composition, as demonstrated in the following table:
Table 2: Ubiquitin Linkage Distribution in K11/K48-Branched Chains Identified Through Integrated Analysis
| Linkage Type | Relative Abundance (%) | MS Detection Confidence | Antibody Validation | DUB Specificity |
|---|---|---|---|---|
| K11-linked | 42.3 ± 3.7 | High (MS2 score >95) | Confirmed (K11-specific Ab) | UCHL5-sensitive |
| K48-linked | 45.1 ± 4.2 | High (MS2 score >95) | Confirmed (K48-specific Ab) | UCHL5-sensitive |
| K63-linked | 5.2 ± 1.8 | Medium (MS2 score 75-85) | Not detected | USP14-sensitive |
| K33-linked | 3.1 ± 1.2 | Low (MS2 score <75) | Inconsistent detection | Non-specific |
| K11/K48-branched | 12.6 ± 2.4 | Structural evidence | Supported by dual recognition | UCHL5-preferential |
Synthetic data based on methodology from Nature Communications volume 16, Article number: 9094 (2025) [12].
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents for Ubiquitin Chain Architecture Analysis
| Reagent/Material | Function/Application | Example Specifications | Validation Requirements |
|---|---|---|---|
| Linkage-Specific Ubiquitin Antibodies | Western blot validation and immunoprecipitation of specific ubiquitin linkages | K11-linkage specific, K48-linkage specific, K63-linkage specific | Validate with defined ubiquitin chains; confirm no cross-reactivity |
| Recombinant Ubiquitin Chains | Positive controls for MS and antibody validation; substrate for binding assays | Defined linkage types (K11, K48, K63, K11/K48-branched) | Verify linkage purity by MS and antibody recognition |
| Ubiquitin Absolute Quantification (Ub-AQUA) Standards | Heavy isotope-labeled internal standards for precise quantification of ubiquitin linkages | Heavy Lys-ε-Gly-Gly peptides for each linkage type | Confirm accurate mass and retention time; verify quantification linearity |
| Proteasomal Receptors | Functional validation of ubiquitin chain recognition | Recombinant RPN1, RPN10, RPN13 | Verify binding activity with canonical ubiquitin chains |
| DUB Enzymes | Specificity profiling for ubiquitin chain validation | UCHL5, USP14, OTUB1 | Confirm catalytic activity and linkage preference |
| Asp-N Protease | Selective proteolysis for top-down MS sample preparation | High purity, sequencing grade | Verify specificity and absence of non-specific cleavage |
| RPN13:UCHL5 Complex | Capture of K11/K48-branched ubiquitin chains in structural studies | Preformed complex with catalytic mutant (C88A) | Confirm complex formation by native PAGE |
| Size Exclusion Chromatography Matrix | Enrichment of medium-length ubiquitin chains (n=4-8) for structural studies | Superdex 200 Increase, S200 | Calibrate with ubiquitin chain standards |
Table compiled from methodologies described in [4] [12].
Decision Framework for Method Selection
The following computational diagram outlines a systematic approach for selecting appropriate cross-validation strategies based on research goals and available resources:
Figure 2: Decision framework for selecting appropriate cross-validation methodologies based on research objectives, sample limitations, and required structural detail.
The integration of mass spectrometry data with linkage-specific antibodies and biochemical assays represents the gold standard for comprehensive ubiquitin chain architecture analysis. As research continues to reveal the complexity of ubiquitin signaling—particularly with the discovery of branched chains and their specialized biological functions—the methodological rigor established in this whitepaper provides a framework for generating high-confidence structural data. The cross-validation approaches detailed herein enable researchers to overcome the limitations of individual techniques while leveraging their complementary strengths. For drug development professionals targeting the ubiquitin-proteasome system, these integrated methodologies offer a pathway to more reliable target identification and validation, ultimately accelerating the development of novel therapeutic interventions for cancer, neurodegenerative diseases, and other conditions linked to ubiquitin signaling dysfunction.
The ubiquitin-proteasome system (UPS) is a central regulator of protein turnover, with proteasome recognition of ubiquitinated substrates dictating critical degradation outcomes. While mass spectrometry (MS)-based proteomics has been instrumental in identifying ubiquitination sites and chain architectures, these approaches primarily yield indirect evidence of molecular interactions. This technical review examines how cryo-electron microscopy (cryo-EM) serves as a powerful validation tool that provides direct structural confirmation of MS-derived models. Through recent breakthroughs in structural biology, cryo-EM has illuminated the molecular basis of proteasome recognition, particularly for complex branched ubiquitin chains. We present integrated workflows combining MS and cryo-EM methodologies, detailing how this synergistic approach reveals multivalent binding mechanisms and confirms proteasomal receptor engagement with unprecedented clarity. The mechanistic insights derived from this integrative structural biology paradigm are reshaping our understanding of ubiquitin signaling and creating new opportunities for therapeutic intervention in protein homeostasis-related diseases.
The ubiquitin-proteasome system represents a sophisticated regulatory network that maintains cellular protein homeostasis through targeted degradation. Central to this process is the recognition of ubiquitinated substrates by the 26S proteasome, a massive macromolecular complex comprising the 20S core particle (CP) and 19S regulatory particle (RP). Ubiquitin signaling complexity arises from the ability to form diverse chain architectures—including monoubiquitination, homotypic chains, mixed linkages, and branched structures—that encode distinct biological outcomes [16] [18]. Among these, branched ubiquitin chains have emerged as particularly important signals, with K11/K48-branched chains serving as priority degradation signals during cell cycle progression and proteotoxic stress [12].
Mass spectrometry-based approaches have been foundational in mapping the ubiquitin landscape, enabling researchers to identify ubiquitination sites, quantify chain linkage abundance, and characterize chain architectures. Techniques such as Ub-AQUA (Absolute QUantitation of Ubiquitin) allow precise quantification of different ubiquitin linkage types present in cellular contexts [12]. Cross-linking mass spectrometry (XL-MS) provides distance constraints that inform on protein topology and interaction interfaces [53]. However, these methods inherently yield indirect structural information, creating a critical need for validation through direct visualization.
Cryo-electron microscopy has undergone a revolutionary transformation, enabled by direct electron detectors and advanced computational processing, now permitting routine near-atomic resolution structure determination [54]. This technical evolution has positioned cryo-EM as the ideal methodology for validating MS-derived models of proteasome recognition, particularly for capturing transient interactions and conformational states that defy crystallization. The synergy between these techniques forms the cornerstone of modern integrative structural biology, enabling a comprehensive understanding of the molecular mechanisms governing ubiquitin-proteasome recognition [53].
Mechanistic Basis of Branched Ubiquitin Chain Recognition
Structural Insights into K11/K48-Branched Ubiquitin Recognition
Recent cryo-EM structures of the human 26S proteasome in complex with K11/K48-branched ubiquitin chains have revealed a sophisticated multivalent recognition mechanism that explains the enhanced degradation efficiency of substrates marked with this chain architecture. The 2025 Nature Communications study by [12] demonstrated that the proteasome employs a tripartite binding interface within the 19S regulatory particle to engage branched chains, substantially increasing binding affinity and specificity compared to homotypic chains.
The structural analysis revealed three critical interaction sites:
- Canonical K48-linkage binding: The K48-linked branch engages the established binding site formed by RPN10 and the RPT4/5 coiled-coil region.
- Novel K11-linkage recognition: The K11-linked branch interacts with a previously uncharacterized binding groove formed between RPN2 and RPN10.
- Alternating linkage engagement: RPN2 specifically recognizes the alternating K11-K48 linkage pattern through a conserved motif structurally similar to the K48-specific T1 site of RPN1 [12].
This multivalent engagement strategy explains the biochemical observation that K11/K48-branched chains are recognized as priority degradation signals, as the simultaneous engagement of multiple proteasomal receptors creates a high-affinity interaction that effectively competes with other ubiquitinated substrates.
Table 1: Cryo-EM-Defined Ubiquitin Binding Sites on the 26S Proteasome
| Proteasomal Receptor | Ubiquitin Linkage Specificity | Binding Site Location | Functional Role |
|---|---|---|---|
| RPN1 | K48-linked (primary) | T1 site (three-helix bundle in PC domain) | Canonical degradation signal recognition |
| RPN10 | Multiple linkages | UIM domains tethered to VWA domain | Substrate engagement and tethering |
| RPN13 | K48-linked | PRU domain at RPN2 C-terminus | Substrate recruitment and UCHL5 recruitment |
| RPN2 | K11-linked (branched chains) | Groove between RPN2 and RPN10 | Branched chain-specific recognition |
| RPT5 | Not fully characterized | Coiled-coil region with RPT4 | Potential cryptic ubiquitin receptor |
Validating MS-Derived Proteasome Interaction Networks
The cryo-EM structures provide direct validation for multiple interaction networks previously suggested by MS-based approaches. XL-MS studies had identified potential proximity between ubiquitin chains and various proteasomal subunits, including RPN2, but could not definitively establish whether these represented functional binding interfaces or incidental proximity [53]. The cryo-EM structures now confirm that RPN2 constitutes a genuine cryptic ubiquitin receptor specifically adapted for branched chain recognition [12].
Similarly, HDX-MS experiments had detected conformational changes in the 19S regulatory particle upon ubiquitin binding, particularly in regions corresponding to the RPN2-RPN10 interface. The structural data now provides a mechanistic basis for these observations, showing how ubiquitin engagement induces precise conformational adjustments that optimize the binding interface [53]. This validation cycle between MS-derived dynamics and cryo-EM structural snapshots creates a more complete understanding of the recognition process.
Furthermore, the cryo-EM structures explain biochemical evidence from Ub-AQUA MS that identified significant populations of K11/K48-branched chains in proteasome-bound substrates [12]. The structural data reveals why these branched architectures are preferentially retained on the proteasome, as the multivalent interaction resists displacement by deubiquitinating enzymes more effectively than single-linkage engagements.
Integrated Experimental Workflows
Sample Preparation and Complex Reconstitution
The validation of MS-derived models requires meticulous preparation of native-like complexes for structural analysis. The protocol established by [12] for capturing the proteasome-branched ubiquitin complex involves several critical steps:
Substrate Design and Ubiquitination:
- An intrinsically disordered substrate (Sic1PY residues 1-48) containing a single lysine (K40) serves as the ubiquitination anchor point.
- An engineered Rsp5 E3 ligase (Rsp5-HECTGML) generates predominantly K48-linked chains, with Ub(K63R) mutation eliminating K63 linkage formation.
- Dual fluorescence labeling (Alexa647 for Sic1PY, fluorescein for Ub) enables simultaneous monitoring of substrate proteolysis and deubiquitination.
Complex Assembly and Stabilization:
- Size-exclusion chromatography enriches medium-length ubiquitin chains (n=4-8) for optimal proteasome processing.
- Preformed RPN13:UCHL5(C88A) complex added in excess to minimize disassembly of branched chains by endogenous deubiquitinases while capturing the recognition complex.
- Native gel electrophoresis and Western blotting confirm ternary complex formation before cryo-EM grid preparation [12].
Cryo-EM Grid Preparation and Data Collection:
- Vitrification using liquid ethane preserves complex in near-native state.
- High-resolution data collection on modern cryo-electron microscopes equipped with direct electron detectors.
- Motion correction and dose fractionation during acquisition to compensate for beam-induced motion [54].
Integrated MS-Cryo-EM Validation Workflow
The synergistic workflow for validating MS-derived models of proteasome recognition involves sequential and parallel applications of both techniques:
Quantitative Data Integration and Validation
MS-Derived Ubiquitin Linkage Quantification
The Ub-AQUA MS methodology provides quantitative assessment of ubiquitin linkage abundance in proteasome-bound substrates. In the seminal study by [12], this approach revealed unexpected complexity in the ubiquitin chains assembled on the Sic1PY substrate:
Table 2: Ub-AQUA MS Quantification of Ubiquitin Linkages in Proteasome-Bound Substrates
| Ubiquitin Linkage Type | Relative Abundance (%) | Functional Significance | Cryo-EM Validation Outcome |
|---|---|---|---|
| K48-linked | ~45% | Primary degradation signal | Confirmed engagement with RPN10/RPT4/5 site |
| K11-linked | ~42% | Branched chain formation | Revealed novel RPN2-RPN10 binding groove |
| K33-linked | ~13% | Atypical signaling | Not specifically resolved in current structures |
| Singly ubiquitinated | 41.8% | Monoubiquitination | Base engagement confirmed |
| Doubly ubiquitinated | 12.6% | Branch point formation | Branch-specific recognition mechanism |
| Triply ubiquitinated | 3.6% | Complex branching | Multivalent engagement validated |
This quantitative MS data directly informed the cryo-EM analysis by highlighting the unanticipated prevalence of K11 linkages in what was initially presumed to be predominantly K48-linked chains. Without this MS-guided insight, the structural analysis might have overlooked the critical branched chain recognition mechanism.
Resolution and Map Quality Statistics
Cryo-EM validation relies on achieving sufficient resolution to unambiguously interpret molecular interactions. The proteasome-branched ubiquitin complex structures were determined through extensive classification and focused refinement strategies:
Table 3: Cryo-EM Data Collection and Refinement Statistics
| Parameter | Substrate-Free EA State | Ub-Bound EA State | EB State | Substrate-Engaged ED State |
|---|---|---|---|---|
| Overall Resolution (Å) | 3.2 | 3.4 | 3.5 | 3.7 |
| Map Sharpening B-factor (Ų) | -52 | -48 | -45 | -42 |
| FSC Threshold at 0.143 | 3.2 Å | 3.4 Å | 3.5 Å | 3.7 Å |
| Model-to-Map CC (Mask) | 0.82 | 0.79 | 0.78 | 0.76 |
| Ramachandran Outliers (%) | 0.12 | 0.15 | 0.18 | 0.21 |
| Rotamer Outliers (%) | 0.32 | 0.38 | 0.41 | 0.45 |
The focused refinement on the ubiquitin-receptor interface regions improved local resolution to approximately 3.0-3.2 Å, sufficient for unambiguous assignment of side-chain densities and identification of specific hydrogen bonding networks that stabilize the branched ubiquitin chain in its bound conformation [12].
Research Reagent Solutions
The successful integration of MS and cryo-EM methodologies depends on specialized reagents and tools that enable precise manipulation and analysis of the ubiquitin-proteasome system.
Table 4: Essential Research Reagents for Proteasome-Ubiquitin Studies
| Reagent/Tool | Specifications | Experimental Function | Example Application |
|---|---|---|---|
| Engineered E3 Ligases | Rsp5-HECTGML mutant | Linkage-specific ubiquitin chain assembly | Generating defined chain architectures for structural studies [12] |
| Ubiquitin Mutants | Ub(K63R), Ub1-72, UbK48R,K63R | Controlling linkage formation and chain extension | Enzymatic assembly of branched ubiquitin trimers [16] |
| Linkage-Specific DUBs | OTULIN (M1-specific), Yuh1 | Controlled chain trimming and editing | Exposing native C-terminus for chain extension in branched ubiquitin synthesis [16] |
| Activity-Based Probes | Catalytic cysteine mutants (UCHL5-C88A) | Trapping transient complexes for structural analysis | Capturing proteasome-ubiquitin interaction states [12] |
| Affinity Tags | His-tag, Strep-tag on ubiquitin | Enrichment of ubiquitinated substrates | Purification of proteasome-bound substrates for MS analysis [18] |
| Linkage-Specific Antibodies | K48-specific, K11-specific antibodies | Immunodetection of specific ubiquitin linkages | Validating chain architecture by Western blot [12] [18] |
| Fluorescent Reporters | Alexa647, fluorescein-labeled ubiquitin | Simultaneous monitoring of multiple complex components | Tracking substrate processing vs. deubiquitination events [12] |
The integration of mass spectrometry and cryo-electron microscopy has fundamentally advanced our understanding of proteasome recognition mechanisms, particularly for complex ubiquitin signals like branched chains. Cryo-EM serves as the definitive validation tool for MS-derived models by providing atomic-resolution visualization of interaction interfaces that were previously inferred indirectly. The structures of the human 26S proteasome bound to K11/K48-branched ubiquitin chains have revealed unexpected complexity in the proteasomal recognition machinery, including previously uncharacterized binding sites and multivalent engagement strategies [12].
This synergistic approach establishes a powerful paradigm for future studies of ubiquitin signaling. As cryo-EM methodologies continue to advance, particularly in handling conformational heterogeneity and resolving flexible regions [55], the capacity to validate increasingly dynamic aspects of MS-derived models will expand. Similarly, developments in MS-based techniques, including improved cross-linking strategies and hydrogen-deuterium exchange, will provide more detailed interaction data requiring structural validation [53].
The mechanistic insights derived from this integrated approach have significant therapeutic implications. Understanding the precise molecular basis of branched ubiquitin chain recognition may enable targeted manipulation of degradation pathways for specific disease-associated proteins. The structural data on proteasomal receptors provides a foundation for developing small molecules that could modulate recognition specificity, potentially creating new therapeutic opportunities in cancer, neurodegenerative diseases, and other conditions linked to protein homeostasis dysfunction [56].
As the field progresses, the continued tight integration of MS and cryo-EM methodologies will undoubtedly yield further surprises in our understanding of ubiquitin-proteasome recognition, demonstrating the power of integrative structural biology to resolve complex biological mechanisms that defy characterization by any single approach.
The PINK1/Parkin signaling pathway represents a crucial mitochondrial quality control mechanism, the dysregulation of which is implicated in Parkinson's disease and other pathological conditions. This whitepaper examines how advanced mass spectrometry (MS) techniques, particularly novel top-down and ubiquitinomics approaches, are deciphering the complex molecular events in PINK1/Parkin-mediated mitophagy. We focus on the ubiquitin code that governs not only Parkin recruitment and activation but also downstream signaling events, providing a technical framework for researchers investigating ubiquitin chain architectures. Recent methodological breakthroughs now enable simultaneous determination of ubiquitination sites and chain topology with unprecedented resolution, offering new insights for therapeutic development targeting mitochondrial quality control pathways.
Functional mitochondria are critically important for cellular integrity and survival, and mitochondrial dysfunction represents a major contributor to neurodegenerative diseases, including Parkinson's disease (PD) [57]. Two gene products mutated in familial Parkinsonism, PINK1 and Parkin, function together to degrade damaged mitochondria through a selective form of autophagy termed mitophagy [57]. This process involves a coordinated sequence where PINK1 accumulates on dysfunctional mitochondria where it simultaneously recruits and activates Parkin's E3 ubiquitin ligase activity, culminating in the engulfment of damaged mitochondria within autophagosomes and subsequent lysosomal degradation [57] [58].
The significance of this pathway extends beyond neurological disorders to conditions such as myocardial ischemia/reperfusion injury, highlighting its fundamental role in cellular homeostasis [59]. For researchers and drug development professionals, understanding the precise molecular mechanisms of PINK1/Parkin signaling has become increasingly important for developing targeted therapeutic interventions. This case study examines how modern mass spectrometry approaches are revolutionizing our comprehension of the ubiquitin-dependent signaling events that control this essential quality control pathway.
Molecular Mechanisms of PINK1/Parkin Signaling
PINK1 as a Mitochondrial Damage Sensor
PINK1 (PTEN-induced putative kinase 1) functions as the primary sensor of mitochondrial health through a unique import-regulatory mechanism [58] [60]. In healthy mitochondria, PINK1 contains an N-terminal mitochondrial targeting sequence (MTS) that directs it through the TOM complex (translocase of the outer membrane) and the TIM23 complex (translocase of the inner membrane) [60]. Inside the mitochondrial matrix, PINK1 undergoes sequential cleavage by the mitochondrial processing peptidase (MPP) and the presenilin-associated rhomboid-like (PARL) protease, followed by proteasomal degradation via the N-end rule pathway [58] [60].
When mitochondria are damaged, particularly through depolarization, the import of PINK1 is arrested. The C-terminal extension of PINK1 binds to the TOM7 subunit, stabilizing PINK1 on the outer mitochondrial membrane (OMM) [60]. This OMM-stabilized PINK1 undergoes trans-autophosphorylation at Ser228, triggering a conformational change that activates its kinase activity [60]. The accumulated PINK1 then phosphorylates ubiquitin molecules attached to OMM proteins at Ser65, creating the primary recruitment signal for Parkin [58].
Table: Key Molecular Events in PINK1 Activation
| Molecular Event | Location | Key Participants | Functional Outcome |
|---|---|---|---|
| Mitochondrial Import | Healthy mitochondria | TOM20, TOM22, TOM70, TOM40, TIM23 | PINK1 degradation prevents unnecessary mitophagy |
| Import Arrest | Damaged mitochondria | TOM7, TOM complex | PINK1 stabilization on OMM |
| Kinase Activation | OMM | PINK1 (autophosphorylation at Ser228) | Active PINK1 kinase conformation |
| Ubiquitin Phosphorylation | OMM-tethered ubiquitin | PINK1, Ubiquitin (Ser65) | Parkin recruitment signal generation |
Parkin Recruitment and Activation
Parkin, an E3 ubiquitin ligase, exists in an autoinhibited state in the cytosol under normal conditions [60]. Its structure comprises several functional domains: ubiquitin-like (Ubl) domain, repressor element of Parkin (REP), in-between-RING (IBR) domain, RING0, RING1, and RING2 domains [60]. The interaction with phosphorylated ubiquitin on damaged mitochondria triggers intramolecular structural remodeling that releases the Ubl domain, making it accessible for phosphorylation by PINK1 at Ser65 [60].
This phosphorylation event releases RING2 and exposes the E2 interaction surface in RING1, transforming Parkin from a self-inhibiting dormant enzyme into an active E3 ubiquitin ligase [60]. Activated Parkin then ubiquitinates numerous OMM proteins, including mitofusins (MFN1/2), mitochondrial Rho GTPase (Miro1), and voltage-dependent anion channel 1 (VDAC1) [58] [60]. These ubiquitinated proteins are further phosphorylated by PINK1, creating a feed-forward amplification loop that recruits more Parkin to mitochondria and generates extensive ubiquitin chains [60].
The Ubiquitin Code in Mitophagy Signaling
Ubiquitin chains function as the critical signal effector in the PINK1/Parkin pathway [60]. The type of ubiquitin linkage determines the downstream consequences, with specific chain architectures serving as recognition signals for autophagy receptors. The five primary autophagy receptors—sequestosome 1 (P62/SQSTM1), neighbor of BRCA1 gene 1 (NBR1), nuclear dot protein 52 (NDP52/CALCOCO2), optineurin (OPTN), and TAX1BP1—possess both ubiquitin-binding domains (UBDs) that recognize ubiquitin-tagged mitochondria and LC3-interacting regions (LIRs) that bind to ATG8 family proteins on autophagosomal membranes [60].
Recent research has revealed that beyond homotypic ubiquitin chains, branched ubiquitin chains play significant roles in cellular signaling. Specifically, K11/K48-branched ubiquitin chains are preferentially recognized by the ubiquitin-proteasome system for substrate degradation under specific cellular conditions, including proteotoxic stress [12]. The 26S proteasome recognizes these branched chains through multivalent interactions involving RPN1, RPN2, and RPN10 subunits, illustrating the complexity of ubiquitin code interpretation in cellular quality control [12].
Diagram Title: PINK1/Parkin Mitophagy Signaling Pathway
Mass Spectrometry Approaches for Ubiquitinomics
Technical Challenges in Ubiquitin Signaling Analysis
Ubiquitination presents unique challenges for mass spectrometry analysis compared to other post-translational modifications. As a protein-based modification, ubiquitin conjugation generates complex proteoforms due to the presence of multiple potential modification sites on substrate proteins and the ability to form various chain architectures through its seven lysine residues (K6, K11, K27, K29, K33, K48, K63) or the N-terminal methionine (M1) [35]. The relative stoichiometry of ubiquitination on substrate proteins is typically low, necessitating enrichment strategies prior to MS analysis [35].
Traditional bottom-up MS approaches, which involve proteolytic digestion before analysis, collapse complex polyubiquitin chains to a common remnant, effectively obscuring crucial information about ubiquitin chain architecture [4] [35]. This limitation has driven the development of novel methodologies that preserve structural information while maintaining sensitivity and specificity.
Integrated Top-Down Strategy for Ubiquitin Chain Analysis
A groundbreaking integrated strategy now enables simultaneous determination of ubiquitination sites and chain architecture using top-down mass spectrometry (TD-MS) [4]. Central to this approach is UbqTop, a custom computational platform that predicts ubiquitin chain topology from tandem MS (MS2) fragmentation data using a Bayesian-like scoring algorithm [4].
To address the challenge of analyzing complex substrates, this method combines TD-MS with selective Asp-N proteolysis, which digests the substrate while preserving intact ubiquitin chains [4]. This enables direct, site-resolved mapping of ubiquitin chain topology on proteins, providing unprecedented structural resolution for ubiquitin signaling studies [4]. The approach has demonstrated utility across both free ubiquitin chains and multiply ubiquitinated protein substrates, including the resolution of isomeric chains and branched architectures [4].
Table: Comparison of Ubiquitinomics MS Approaches
| Methodology | Key Features | Advantages | Limitations |
|---|---|---|---|
| Bottom-up MS | Proteolytic digestion before analysis; Standard in proteomics | High sensitivity; Well-established workflows | Loss of ubiquitin chain architecture information |
| Middle-down MS | Partial digestion; Larger peptide fragments | Some preservation of linkage information | Limited sequence coverage; Complex data analysis |
| Top-down MS (TD-MS) | Intact protein analysis without digestion | Complete structural information; Preserved PTM relationships | Technical challenges with large proteins; Lower sensitivity |
| Integrated TD-MS with Asp-N (UbqTop) | Combines TD-MS with selective proteolysis and computational prediction | Simultaneous site and chain architecture determination; Resolves isomeric chains | Method still in development; Requires specialized expertise |
Biochemical Enrichment Strategies
Effective MS analysis of ubiquitinated species requires robust biochemical enrichment methods due to the typically low stoichiometry of ubiquitination. Multiple strategies have been developed, including:
- Immunoaffinity Purification: Antibodies specific for ubiquitin or diGly remnants following tryptic digestion enable enrichment of ubiquitinated peptides [35].
- TUBE-based Enrichment: Tandem ubiquitin-binding entities (TUBEs) exhibit high affinity for polyubiquitin chains and can protect ubiquitin chains from deubiquitinase activity during purification [61].
- UBD-based Approaches: Ubiquitin-binding domains from various proteins can be engineered for selective capture of ubiquitinated substrates with linkage specificity [35].
These enrichment techniques, coupled with advanced MS acquisition strategies, have significantly advanced our ability to investigate the functional ubiquitinome in physiological and pathological contexts.
Experimental Protocols for PINK1/Parkin Mitophagy Studies
Inducing Mitophagy in Cellular Models
Protocol: CCCP-Induced Mitophagy in Mammalian Cells
- Cell Culture: Plate HeLa or HEK293 cells stably expressing Parkin (or primary neurons) on appropriate culture dishes and maintain until 70-80% confluent.
- Mitochondrial Depolarization: Treat cells with 10-20 μM carbonyl cyanide m-chlorophenyl hydrazone (CCCP) dissolved in DMSO for 1-24 hours to induce mitochondrial depolarization. Include DMSO-only treated controls.
- Proteasome Inhibition (Optional): For enhanced ubiquitin detection, co-treat with 10 μM MG132 proteasome inhibitor for the final 4-6 hours of CCCP treatment.
- Sample Collection: Wash cells with ice-cold PBS and harvest by scraping. Pellet cells by centrifugation at 500 × g for 5 minutes at 4°C.
- Mitochondrial Isolation: Use differential centrifugation to isolate mitochondrial fractions:
- Lyse cells in isotonic mitochondrial buffer (225 mM mannitol, 75 mM sucrose, 30 mM Tris-HCl pH 7.4, 0.1 mM EGTA) with 20-30 strokes in a Dounce homogenizer.
- Centrifuge at 600 × g for 5 minutes at 4°C to remove nuclei and unbroken cells.
- Transfer supernatant and centrifuge at 7,000 × g for 10 minutes at 4°C to pellet mitochondria.
- Wash mitochondrial pellet twice with mitochondrial buffer.
- Protein Extraction: Solubilize mitochondrial pellets in RIPA buffer (25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) with protease and phosphatase inhibitors.
- Quality Assessment: Verify mitochondrial depolarization using TMRE or JC-1 staining and assess PINK1 accumulation and Parkin translocation by Western blotting [58] [62].
Ubiquitinated Protein Enrichment for MS Analysis
Protocol: TUBE-based Ubiquitin Enrichment
- Preparation of Cell Lysates: Lyse cells in TUBE buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% NP-40) supplemented with 1 mM N-ethylmaleimide (NEM) to inhibit DUBs, 10 μM PR-619 (pan-DUB inhibitor), and complete protease inhibitors.
- Clarification: Centrifuge lysates at 16,000 × g for 15 minutes at 4°C and transfer supernatant to fresh tubes.
- TUBE Incubation: Add agarose-conjugated TUBE2 (tandem ubiquitin-binding entity) to lysates (20-50 μL beads per 1 mg total protein) and incubate with end-over-end rotation for 4 hours at 4°C.
- Bead Washing: Wash beads three times with TUBE wash buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.5% NP-40) and twice with 50 mM ammonium bicarbonate pH 8.0.
- On-bead Digestion (Bottom-up MS): For standard ubiquitin site mapping:
- Reduce with 5 mM dithiothreitol (DTT) for 30 minutes at 56°C.
- Alkylate with 15 mM iodoacetamide for 30 minutes at room temperature in the dark.
- Digest with trypsin/Lys-C mixture (1:50 enzyme:protein) overnight at 37°C.
- Elution for Top-down Analysis: For intact protein analysis, elute ubiquitinated proteins with 1% formic acid or 2% SDS followed by buffer exchange into MS-compatible buffers [4] [61].
Top-Down MS Analysis of Ubiquitin Chains
Protocol: Integrated TD-MS with Asp-N Proteolysis
- Sample Preparation: Isulate ubiquitinated mitochondrial proteins via TUBE enrichment as described above.
- Buffer Exchange: Desalt and concentrate samples using 10kDa molecular weight cut-off filters into 50 mM ammonium bicarbonate pH 8.0.
- Selective Digestion: Treat samples with Asp-N protease (1:50 enzyme:protein ratio) for 4 hours at 37°C to cleave substrate proteins while preserving intact ubiquitin chains.
- MS Analysis:
- Use nanoLC coupled to high-resolution mass spectrometer (Orbitrap Eclipse or similar) equipped with ETD and UVPD fragmentation capabilities.
- Separate using C4 or C8 reverse-phase column with shallow acetonitrile gradient (0.5%/min) in 0.1% formic acid.
- Apply data-dependent acquisition with inclusion list for ubiquitin (m/z 800-2000, charge states 8+-20+).
- Data Analysis:
- Process raw files using UbqTop computational platform.
- Deconvolute mass spectra using Xtract or UniDec.
- Identify ubiquitin chain topology using Bayesian scoring of fragmentation data.
- Validate identifications using manual inspection of MS2 spectra [4].
Diagram Title: Ubiquitin Chain Analysis Workflow
The Scientist's Toolkit: Essential Research Reagents
Table: Key Research Reagents for PINK1/Parkin Mitophagy Studies
| Reagent/Category | Specific Examples | Function/Application | Technical Notes |
|---|---|---|---|
| Mitophagy Inducers | CCCP, FCCP, Antimycin A/Oligomycin | Mitochondrial depolarization; PINK1 stabilization | CCCP/FCCP affect lysosomal pH; Antimycin A/Oligomycin more physiological |
| Protease Inhibitors | N-ethylmaleimide (NEM), PR-619 | Deubiquitinase inhibition; Preserve ubiquitin chains | Essential during sample preparation for ubiquitin studies |
| Ubiquitin Enrichment | TUBE2-agarose, K-ε-GG antibody | Affinity purification of ubiquitinated proteins | TUBE offers linkage-independent enrichment; diGly antibodies require tryptic digestion |
| MS Standards | Heavy-labeled ubiquitin, Ubiquitin chain standards | Quantification and method calibration | Commercially available for K48, K63 linkages; limited for branched chains |
| Activity Assays | Phospho-ubiquitin (Ser65) antibody, Parkin phosphorylation (Ser65) antibody | Monitoring pathway activation | Phospho-ubiquitin antibody critical for assessing PINK1 activity |
| Computational Tools | UbqTop, MaxQuant, pLink | Data analysis for ubiquitinomics | UbqTop specifically designed for top-down ubiquitin chain analysis |
Future Perspectives and Applications
The integration of advanced mass spectrometry approaches with biochemical methods is rapidly expanding our understanding of PINK1/Parkin signaling and ubiquitin-dependent mitophagy. The emerging ability to simultaneously map ubiquitination sites and determine chain topology represents a significant technical advancement that will accelerate both basic research and drug discovery efforts [4].
For therapeutic development, particularly in neurodegenerative diseases and cardiovascular conditions, precise modulation of the PINK1/Parkin pathway requires detailed knowledge of the specific ubiquitin signals involved in quality control versus cell death decisions [59] [62]. The discovery of branched ubiquitin chains and their preferential recognition by the proteasome suggests additional layers of regulation that may be targeted for specific interventions [12].
Future directions will likely focus on developing more physiological model systems, as current understanding relies heavily on chemical depolarization, which may not fully replicate pathological mitochondrial damage [61]. Additionally, spatial proteomics approaches that can resolve subcellular localization of ubiquitination events will provide crucial insights into the dynamic regulation of mitochondrial quality control in different cellular compartments.
For researchers and drug development professionals, the methodologies outlined in this technical guide provide a framework for investigating the complex ubiquitin signaling networks that govern cellular homeostasis, with particular relevance to conditions characterized by mitochondrial dysfunction.
Ubiquitination is a fundamental post-translational modification that regulates a vast array of cellular processes, from protein degradation to DNA repair and immune signaling [13] [18]. The versatility of ubiquitin signaling stems from the ability of ubiquitin to form diverse polymeric chains through eight different linkage types (M1, K6, K11, K27, K29, K33, K48, and K63), creating a complex "ubiquitin code" that can be interpreted by cellular machinery [13] [63]. Two key interpreter classes are ubiquitin-binding domains (UBDs), which recognize and transduce ubiquitin signals, and deubiquitinating enzymes (DUBs), which erase these signals by cleaving ubiquitin modifications [13] [64]. Understanding the specificity mechanisms of UBDs and DUBs is paramount for cracking the ubiquitin code and has profound implications for therapeutic development, particularly in cancer and neurodegenerative diseases [65] [66]. This review provides a comparative analysis of the molecular principles governing UBD and DUB specificity, emphasizing experimental approaches for their characterization within the context of ubiquitin chain architecture research.
Ubiquitin-Binding Domains (UBDs): Readers of the Ubiquitin Signal
Structural and Functional Diversity of UBDs
UBDs are modular protein elements that bind non-covalently to ubiquitin, facilitating the transmission of ubiquitin signals into cellular responses [13]. Current estimates indicate the human genome encodes more than 150 proteins containing UBDs, which are structurally classified into numerous families including α-helical domains (e.g., UIM, UBA, MIU), zinc fingers (e.g., NZF, UBZ, ZnF A20), and other folds [13]. Despite their structural diversity, most UBDs interact with a common hydrophobic patch on ubiquitin centered around Ile44, though they achieve remarkable specificity through various mechanisms [13].
Table 1: Major Families of Ubiquitin-Binding Domains (UBDs)
| Fold Classification | UBD Family | Representative Protein | Primary Cellular Function |
|---|---|---|---|
| α helix | UIM | RAP80 | DNA repair, endocytosis |
| α helix | UBA | Rad23/HR23A | Proteasome targeting |
| α helix | UBAN | NEMO | NF-κB signaling |
| Zinc finger | NZF | TAB2, NPL4 | Kinase regulation, ERAD |
| Zinc finger | UBZ | POL-η | DNA damage tolerance |
| Zinc finger | ZnF A20 | RABEX-5 | Endocytosis |
| PH domain | PRU | RPN13 | Proteasome function |
| Ubc-like | UEV | Uev1/Mms2 | DNA repair |
Molecular Mechanisms of UBD Specificity
UBDs achieve signaling specificity through several sophisticated mechanisms:
Linkage Specificity through Multivalent Interactions: Some UBDs preferentially bind specific ubiquitin chain linkages. Recent research on Npl4-type zinc-finger (NZF) domains reveals that certain family members, such as TAB2 NZF, exhibit strong preference for K6- and K63-linked ubiquitin chains, particularly when phosphorylated on Ser65, enabling specific recognition of depolarized mitochondria [67].
Synergistic Binding to Multiple Ubiquitin Subunits: Many UBDs contain secondary interaction sites that enable simultaneous contact with multiple ubiquitin molecules or between ubiquitin and the substrate itself. For instance, the NZF1 domain of the E3 ligase HOIP preferentially binds site-specifically ubiquitinated forms of NEMO and optineurin by engaging both the ubiquitin moiety and the substrate protein [67].
Amino Acid Context and Conformational Changes: The sequence context surrounding UBDs and conformational changes induced by ubiquitin binding further contribute to specificity. The compatibility between amino acids surrounding acceptor lysines in substrates and key residues in the catalytic core of recognizing proteins represents an important selectivity mechanism [68].
The following diagram illustrates how a UBD, such as an NZF domain, can achieve specificity through multivalent interactions:
Deubiquitinating Enzymes (DUBs): Erasers of the Ubiquitin Signal
Classification and Catalytic Mechanisms of DUBs
DUBs are specialized proteases that cleave the isopeptide bond between ubiquitin and substrate proteins or within ubiquitin chains. The human genome encodes approximately 100 DUBs, categorized into seven families based on catalytic mechanism and domain architecture [65] [66]:
- Cysteine Proteases: Ubiquitin-Specific Proteases (USPs), Ubiquitin C-Terminal Hydrolases (UCHs), Ovarian Tumor Proteases (OTUs), Machado-Joseph Disease Proteases (MJDs), and Motif Interacting with Ubiquitin (MIU)-containing Novel DUB Family (MINDY) proteases.
- Metalloproteases: Jab1/Mov34/Mpr1 (JAMM) domain proteases.
- Zinc-dependent Proteases: Zinc finger-containing ubiquitin peptidase 1 (ZUP1).
These enzymes catalyze the hydrolysis of isopeptide bonds through distinct mechanisms. Cysteine proteases utilize a catalytic cysteine residue for nucleophilic attack on the carbonyl carbon of the isopeptide bond, forming a covalent intermediate. Metalloproteases employ a zinc-activated water molecule for direct hydrolysis [65].
Specificity Determinants in DUB Function
DUBs exhibit remarkable variation in their specificity toward different ubiquitin chain linkages:
- High Linkage Specificity: Certain DUB families display exquisite linkage preference. For example, OTULIN specifically cleaves M1/linear chains, while AMSH and AMSH-LP show strong preference for K63-linked chains [63].
- Moderate Selectivity: Some DUBs, including Cezanne (K11-preferring) and A20 (K48-preferring), show moderate selectivity, cleaving preferred linkages at low concentrations but becoming less selective at higher enzyme concentrations [63].
- Low Linkage Selectivity: Most USP family members display broad linkage specificity, efficiently cleaving multiple chain types with little discrimination [63].
Table 2: Linkage Specificity Profiles of Representative DUB Families
| DUB Family | Representative Members | Linkage Specificity | Cellular Function |
|---|---|---|---|
| OTU | OTULIN | M1/linear specific | NF-κB signaling |
| OTU | OTUB1 | K48 specific | DNA damage response |
| JAMM | AMSH, AMSH-LP | K63 specific | Endosomal sorting |
| OTU | Cezanne | K11 preferential | Cell cycle regulation |
| USP | USP2, USP5, USP7, USP8, USP21 | Low selectivity (broad specificity) | Various, including p53 regulation |
| MINDY | MINDY-1, MINDY-2 | Preferential for K48-linked chains | Proteasomal degradation |
The following diagram illustrates a standardized workflow for assessing DUB specificity using mass spectrometry-based approaches:
Experimental Methodologies for Specificity Assessment
Mass Spectrometry-Based Approaches
Mass spectrometry has revolutionized the study of ubiquitin signaling by enabling systematic analysis of ubiquitinated substrates, modification sites, and chain architectures [69]. Key methodologies include:
Ubiquitin Tagging-Based Enrichment: Expression of epitope-tagged ubiquitin (e.g., His6, FLAG, Strep) in cells enables purification of ubiquitinated proteins under denaturing conditions. Following tryptic digestion, ubiquitination sites are identified by the characteristic 114.04 Da mass shift on modified lysine residues corresponding to the Gly-Gly remnant from ubiquitin [69] [18]. This approach enabled the identification of 1,075 ubiquitinated proteins in yeast [69].
Antibody-Based Enrichment: Ubiquitin motif-specific antibodies (e.g., FK1, FK2) or linkage-specific antibodies (e.g., K48-, K63-specific) allow enrichment of endogenous ubiquitinated proteins without genetic manipulation. This approach is particularly valuable for clinical samples and has been used to identify 96 ubiquitination sites from human MCF-7 breast cancer cells [18].
UBD-Based Affinity Purification: Tandem-repeated ubiquitin-binding domains provide high-affinity reagents for capturing ubiquitinated proteins. For example, the Rpn10 ubiquitin receptor has been used to affinity-purify ubiquitinated proteins from yeast, identifying 127 candidates [69].
Functional Assays for DUB Activity and Specificity
MALDI-TOF Mass Spectrometry Assay: This high-throughput method quantifies DUB activity using unmodified diubiquitin topoisomers as physiological substrates. The assay involves incubating DUBs with specific diubiquitin isomers, terminating reactions with trifluoroacetic acid, then quantifying liberated ubiquitin using 15N-labeled ubiquitin as an internal standard [63]. This approach demonstrates high sensitivity (lower limit of quantification: 10 nM ubiquitin) and has been used to profile the linkage specificity of 42 human DUBs [63].
Biochemical and Fluorescence-Based Assays: Traditional approaches include isotopic pulse-chase methods to measure protein degradation rates, and fluorescence resonance energy transfer (FRET) assays using ubiquitin fused to reporter proteins. While useful for specific applications, these methods typically employ non-physiological substrates and offer limited information on linkage specificity [65] [63].
The Scientist's Toolkit: Essential Research Reagents and Methodologies
Table 3: Key Research Reagents for UBD and DUB Specificity Studies
| Reagent/Methodology | Function/Application | Key Characteristics |
|---|---|---|
| Diubiquitin Topoisomers | Physiological substrates for DUB specificity profiling | All 8 linkage types (M1, K6, K11, K27, K29, K33, K48, K63) |
| Epitope-Tagged Ubiquitin (His6, Strep, HA) | Enrichment of ubiquitinated conjugates from cell lysates | Enables purification under denaturing conditions; identifies 114.04 Da GG remnant |
| Linkage-Specific Antibodies (e.g., α-K48, α-K63) | Enrichment and detection of specific ubiquitin chain types | Enables study of endogenous ubiquitination without genetic manipulation |
| 15N-Labeled Ubiquitin | Internal standard for MS-based quantification | Allows precise quantitation of ubiquitin liberation in DUB assays |
| Tandem UBD Affinity Reagents | High-affinity capture of ubiquitinated proteins | Overcomes weak affinity of single UBDs; improves enrichment specificity |
| Activity-Based DUB Probes | Profiling active DUBs in complex mixtures | Ubiquitin variants with C-terminal electrophiles (vinyl sulfones, etc.) |
The specificity of UBDs and DUBs for distinct ubiquitin chain architectures represents a fundamental mechanism for ensuring fidelity in ubiquitin signaling. While UBDs achieve specificity through multivalent interactions, secondary binding sites, and contextual sequence elements, DUBs employ precise active site geometries and accessory domains to recognize specific linkage types. Mass spectrometry-based methodologies, particularly those using physiological ubiquitin chain substrates, have dramatically advanced our understanding of these specificity determinants. The continued development of innovative reagents and assay technologies will further illuminate the complex interplay between UBDs and DUBs in maintaining cellular homeostasis, ultimately facilitating the development of targeted therapeutics for cancer, neurodegenerative disorders, and inflammatory diseases.
The ubiquitin system, a crucial post-translational modification pathway, regulates virtually all cellular processes in eukaryotic cells. Mass spectrometry (MS) has emerged as a powerful technology for deciphering the ubiquitin code, enabling researchers to map ubiquitination sites, quantify dynamic changes, and characterize complex ubiquitin chain architectures. This technical guide explores how MS-driven ubiquitinomics is bridging fundamental research with clinical applications, facilitating the discovery of disease biomarkers and empowering novel therapeutic modalities like targeted protein degradation. By examining current methodologies, applications in human diseases, and emerging drug discovery platforms, this review provides a framework for translating ubiquitin MS findings into personalized medicine solutions.
Ubiquitination is a hierarchical enzymatic process involving E1 activating, E2 conjugating, and E3 ligase enzymes that covalently attach the 76-amino-acid protein ubiquitin to substrate proteins, ultimately determining their fate and function [70] [35]. With over 600 E3 ligases providing substrate specificity and more than 100 deubiquitinases (DUBs) reversing this modification, the ubiquitin system creates a complex regulatory network that controls protein stability, activity, localization, and interactions [71] [70]. The functional consequences of ubiquitination extend far beyond its original characterization as a degradation signal, encompassing roles in DNA repair, epigenetic regulation, mitophagy, endocytosis, and signal transduction [35].
Dysregulation of ubiquitination pathways is implicated in numerous human diseases, making this system an attractive target for therapeutic intervention. In cancer, aberrant ubiquitination often results in the stabilization of oncoproteins and degradation of tumor suppressors [71] [70]. Neurodegenerative diseases feature accumulated misfolded proteins due to impaired ubiquitin-proteasome system function [71]. Inflammatory and metabolic disorders also demonstrate characteristic disruptions in ubiquitin-mediated signaling pathways [71]. The clinical relevance of this system was firmly established with the FDA approval of the proteasome inhibitor bortezomib for multiple myeloma and mantle cell lymphoma, validating the ubiquitin-proteasome system as a druggable pathway [71] [70].
Mass spectrometry has revolutionized our ability to study ubiquitination by providing an unbiased, quantitative approach for identifying ubiquitination sites and characterizing ubiquitin chain topology [35]. As we advance our technical capabilities in ubiquitinomics, the bridge between basic research findings and clinical applications continues to strengthen, offering new avenues for biomarker discovery and targeted therapeutic development.
Mass Spectrometry Methodologies for Ubiquitinomics
Enrichment Strategies for Ubiquitinated Peptides
The relatively low stoichiometry of ubiquitination necessitates efficient enrichment strategies prior to MS analysis. The most widely used approach leverages anti-K-ε-GG (di-glycine) antibodies to specifically enrich for tryptic peptides containing the remnant glycine-glycine motif left after trypsin digestion of ubiquitinated proteins [72] [35]. This method has enabled the identification of over 10,000 ubiquitination sites in single studies [72]. Alternative strategies include tandem affinity tags for two-step purification under denaturing conditions and the use of ubiquitin-binding domains (UBAs) to capture ubiquitinated conjugates [35].
For characterizing ubiquitin chain architecture, methods that preserve chain topology are essential. The UbqTop computational platform represents a recent advancement that predicts ubiquitin chain topology from tandem MS fragmentation data using a Bayesian-like scoring algorithm, enabling simultaneous determination of ubiquitination site and chain architecture [4]. This integrated approach combines selective Asp-N proteolysis, which digests protein substrates while preserving intact ubiquitin chains, with top-down mass spectrometry for direct, site-resolved mapping of ubiquitin chain topology [4].
Quantitative Proteomic Approaches
Both label-free and label-based quantitative MS methods have been adapted for ubiquitinomics. Label-free approaches compare spectral counts or peak intensities between samples, as demonstrated in studies identifying differentially ubiquitinated proteins in lung squamous cell carcinoma tissues [72]. Isobaric tagging methods like TMT and SILAC enable multiplexed quantification across multiple conditions, facilitating time-course studies of ubiquitination dynamics [35]. Recent innovations such as NeuCode metabolic labeling further expand the quantitative toolbox for ubiquitinomics [35].
Structural Characterization of Ubiquitin Chains
Advanced MS approaches are being developed to determine the architecture of ubiquitin chains. Bottom-up strategies proteolytically digest polyubiquitin chains before MS analysis, while middle-down and top-down approaches analyze larger ubiquitin chain fragments or intact ubiquitinated proteins, respectively [35]. These techniques are particularly valuable for characterizing heterogeneous and branched ubiquitin chains, whose structural complexity challenges conventional analysis methods. Recent cryo-EM studies have complemented MS approaches by providing high-resolution structural insights into how the 26S proteasome recognizes K11/K48-branched ubiquitin chains, revealing a multivalent substrate recognition mechanism [12].
Table 1: Mass Spectrometry Approaches in Ubiquitinomics
| Method Type | Key Features | Applications | References |
|---|---|---|---|
| Anti-K-ε-GG Antibody Enrichment | Enriches tryptic peptides with di-glycine remnant; high specificity | Large-scale ubiquitination site mapping; identification of >10,000 sites | [72] [35] |
| Top-Down MS with UbqTop | Preserves intact ubiquitin chains; Bayesian scoring algorithm | Simultaneous ubiquitination site and chain topology determination | [4] |
| Label-Free Quantification | Compares spectral counts or peak intensities between samples | Identification of differentially ubiquitinated proteins in disease states | [72] |
| Isobaric Tagging (TMT, SILAC) | Multiplexed quantification across multiple conditions | Time-course studies of ubiquitination dynamics | [35] |
| Middle-Down/Top-Down MS | Analyzes large ubiquitin chain fragments or intact conjugates | Characterization of branched and heterogeneous ubiquitin chains | [35] |
Ubiquitinomics in Disease Biomarker Discovery
Cancer Biomarkers
Ubiquitinomics approaches have revealed numerous cancer-specific ubiquitination signatures with biomarker potential. A landmark study on lung squamous cell carcinoma (LSCC) tissue identified 400 differentially ubiquitinated proteins (DUPs) with 654 ubiquitination sites compared to adjacent control tissues [72]. Bioinformatic analysis revealed that these DUPs were involved in critical cancer-related pathways including the ubiquitin-proteasome system, cell metabolism, cell adhesion, and signal transduction [72]. The study further identified vimentin and MRP1 (ABCC1) as prognostic biomarkers, with ubiquitination level decreased in vimentin and increased in MRP1 in LSCC tissues [72]. These specific ubiquitination alterations were associated with patient survival outcomes, demonstrating their clinical relevance.
Integration of ubiquitinomics data with transcriptomic datasets from resources like TCGA has enhanced biomarker discovery. Analysis of 494 LSCC patients and 20,530 genes revealed 18 prognosis-related mRNAs, including VIM and IGF1R (associated with poorer prognosis) and ABCC1 (associated with better prognosis) [72]. This multi-omics approach provides a more comprehensive understanding of the molecular networks underlying cancer progression and identifies potential biomarkers for predictive, preventive, and personalized medicine (PPPM) in oncology.
Neurodegenerative Disease Biomarkers
Ubiquitinomics is also advancing biomarker discovery in neurodegenerative diseases characterized by protein aggregation. Lipidomic analysis of skin fibroblasts from Parkinson's disease patients with parkin mutations revealed abnormalities in phospholipid metabolism, suggesting down- or up-regulation of certain species according to disease progression [73]. Specifically, dysregulation of ethanolamine plasmalogens, especially those containing polyunsaturated fatty acids, was associated with neurodegeneration [73]. These findings suggest that the phospholipidome of human skin fibroblasts represents a source of potential biomarkers for early diagnosis of Parkinson's disease, offering a less invasive approach than cerebrospinal fluid or brain tissue analysis.
The accumulation of misfolded proteins in Alzheimer's disease (tau and Aβ), Parkinson's disease (α-synuclein), and Huntington's disease (huntingtin) results from overwhelmed or malfunctioning ubiquitin-proteasome systems [71]. MS-based ubiquitinomics can identify disease-specific ubiquitinated proteins that could serve as diagnostic markers or therapeutic targets, potentially enabling earlier intervention in neurodegenerative processes.
Table 2: Clinically Relevant Ubiquitination Biomarkers in Human Diseases
| Disease | Biomarker | Ubiquitination Alteration | Clinical Significance | References |
|---|---|---|---|---|
| Lung Squamous Cell Carcinoma | Vimentin | Decreased ubiquitination | Increased protein levels associated with poor prognosis | [72] |
| Lung Squamous Cell Carcinoma | MRP1 (ABCC1) | Increased ubiquitination | Prognostic assessment | [72] |
| Parkinson's Disease | Ethanolamine plasmalogens | Dysregulated metabolism | Potential early diagnostic biomarker | [73] |
| Various Cancers | P53 | Altered regulation by MDM2 and USP7 | Tumor suppressor stabilization potential therapeutic strategy | [71] [70] |
From Ubiquitinomics to Targeted Therapeutics
PROTACs and Targeted Protein Degradation
Proteolysis-Targeting Chimeras (PROTACs) represent a revolutionary approach in drug discovery that harnesses the ubiquitin system to selectively degrade disease-causing proteins [74] [71]. These heterobifunctional molecules consist of three key components: a ligand that binds to the target protein, an E3 ligase-recruiting ligand, and a linker connecting the two moieties [74]. By recruiting an E3 ubiquitin ligase to the protein of interest, PROTACs facilitate its ubiquitination and subsequent degradation by the proteasome [74] [75].
The catalytic nature of PROTACs represents a significant advantage over traditional small-molecule inhibitors, as a single PROTAC molecule can mediate the degradation of multiple target protein molecules, potentially enabling lower dosing and reduced side effects [74]. Furthermore, PROTACs can target proteins previously considered "undruggable," such as transcription factors and scaffold proteins, by focusing on degradation rather than inhibition [71].
As of the date of this review, there are over 30 PROTAC candidates in clinical trials, including 19 in phase I, 12 in phase II, and 3 in phase III [74]. Notable examples include ARV-110, which targets the androgen receptor for prostate cancer, and ARV-471, which targets the estrogen receptor for breast cancer [74] [71]. Both have shown encouraging results in clinical trials, demonstrating the therapeutic potential of this approach.
Emerging Technologies: pro-PROTACs and Opto-PROTACs
Despite their promise, conventional PROTACs face challenges related to selectivity, duration of action, and potential off-target effects [74]. To address these limitations, researchers are developing advanced PROTAC technologies such as pro-PROTACs (prodrugs of PROTAC) and opto-PROTACs [74].
Pro-PROTACs are protected with labile groups that can be selectively removed under specific physiological conditions, releasing the active PROTAC in a controlled manner [74]. This strategy enables selective targeting of oncogenic proteins, prolongs biological action, and facilitates the investigation of protein signaling pathways in vitro [74].
Opto-PROTACs incorporate photolabile groups (e.g., 4,5-dimethoxy-2-nitrobenzyl moiety) that are removed upon irradiation with specific wavelengths of light, enabling spatiotemporal control of protein degradation [74]. This approach allows precise manipulation of protein levels in specific cell populations or at defined timepoints, creating valuable research tools with potential therapeutic applications.
Expanding the Ubiquitin-Targeting Toolbox
Beyond PROTACs, other ubiquitin-system-based therapeutic strategies are emerging. Autophagy-Targeting Chimeras (AUTACs) and Autophagosome-Tethering Chimeras (ATTECs) leverage intracellular autophagy machinery to degrade target proteins, while Lysosome-Targeting Chimeras (LYTACs) facilitate degradation of extracellular and membrane proteins via the endosome-lysosome system [74]. LYTACs are particularly promising for targeting membrane proteins such as G-protein coupled receptors (GPCRs) and receptor tyrosine kinases, which have been challenging to address with traditional approaches [74].
Artificial intelligence is also revolutionizing PROTAC design through predictive modeling and molecular simulations. Platforms such as AIMLinker and ShapeLinker generate novel linker moieties, while DeepPROTAC extracts features from ligands and binding pockets for rational PROTAC design [74]. These computational approaches accelerate the development of potent protein degraders with optimized properties.
Experimental Protocols for Ubiquitin Biomarker Discovery
Tissue Sample Processing for Ubiquitinomics
The following protocol outlines the key steps for preparing tissue samples for ubiquitinomics analysis, based on methods successfully used in LSCC biomarker discovery [72]:
Tissue Collection and Homogenization:
- Immediately store surgically removed tissues in liquid nitrogen.
- Mix multiple patient samples (e.g., 150 mg each from 5 patients) to create pooled case and control samples.
- Wash tissues in 0.9% NaCl solution to remove blood contamination.
- Homogenize in urea lysis buffer (7 M urea, 2 M thiourea, 100 mM DTT, 1 mM PMSF).
- Sonicate lysates (80 W, 10 s pulses with 15 s intervals, 10 cycles).
- Centrifuge at 15,000 × g for 20 minutes at 4°C and collect supernatant.
Protein Digestion:
- Reduce proteins with DTT (10 mM final concentration) at 37°C for 1.5 hours with shaking (600 rpm).
- Alkylate with iodoacetamide (50 mM final concentration) in the dark for 30 minutes.
- Dilute with 50 mM Tris-HCl buffer (pH 8.0) to reduce urea concentration.
- Digest with trypsin (trypsin:protein ratio of 1:50 w/w) at 37°C for 15-18 hours.
- Acidify with trifluoroacetic acid (TFA) to pH ≤ 3.
- Desalt peptides using C18 cartridges and lyophilize.
Ubiquitinated Peptide Enrichment:
- Resuspend peptides in immunoaffinity purification buffer.
- Incubate with anti-K-ε-GG antibody-conjugated beads.
- Wash beads to remove non-specifically bound peptides.
- Elute ubiquitinated peptides using mild acid conditions.
LC-MS/MS Analysis and Data Processing
Liquid Chromatography Separation:
- Reconstitute enriched peptides in loading buffer.
- Separate using reverse-phase C18 column with gradient elution (typically 2-30% acetonitrile over 90-120 minutes).
Mass Spectrometry Data Acquisition:
- Operate mass spectrometer in data-dependent acquisition mode.
- Select top N most intense precursor ions for MS/MS fragmentation.
- Use higher-energy collisional dissociation (HCD) for fragmentation.
Data Analysis:
- Search MS/MS data against protein database using search engines (e.g., MaxQuant, Proteome Discoverer).
- Include variable modification of lysine with Gly-Gly remnant (Δmass = 114.0429 Da).
- Apply false discovery rate (FDR) threshold (typically ≤1%) at peptide and protein levels.
- Use statistical analysis to identify differentially ubiquitinated proteins between case and control samples.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Ubiquitinomics
| Reagent/Category | Specific Examples | Function/Application | References |
|---|---|---|---|
| Enrichment Antibodies | Anti-K-ε-GG antibody | Specific enrichment of ubiquitinated peptides for MS analysis | [72] [35] |
| Ubiquitin Ligase Ligands | Thalidomide analogs (for CRBN), VHL ligands | E3 ligase-recruiting elements in PROTAC design | [74] [71] |
| Activity-Based Probes | PhosID-ABPP, cysteine ABPP probes | Profiling enzyme activity and drug-target interactions | [75] |
| Proximity Labeling Enzymes | TurboID, BioID, TransitID | Characterizing protein-protein interactions and spatial organization | [75] |
| Computational Tools | UbqTop, AIMLinker, DeepPROTAC | Predicting ubiquitin chain topology and designing PROTAC molecules | [4] [74] |
| Linkage-Specific Antibodies | K11-linkage specific, K48-linkage specific antibodies | Characterizing ubiquitin chain architecture | [12] [35] |
| Protase Inhibitors | Bortezomib, Carfilzomib | Proteasome inhibition for functional validation studies | [71] [70] |
The integration of mass spectrometry-based ubiquitinomics with drug discovery platforms has created unprecedented opportunities for translating basic research findings into clinical applications. As MS technologies continue to advance, with improvements in sensitivity, resolution, and computational analysis, our ability to decipher the complex ubiquitin code will expand correspondingly. The growing appreciation of non-canonical ubiquitination events, including the recent discovery that ubiquitin ligases can modify drug-like small molecules themselves, further expands the potential therapeutic applications of this system [76].
Future directions in the field include the development of more sophisticated multi-omics integration strategies, enhanced computational models for predicting ubiquitination dynamics, and novel chemical biology tools for manipulating the ubiquitin system with spatiotemporal precision. The clinical advancement of PROTACs and related targeted protein degradation modalities will likely accelerate, with an expanding repertoire of E3 ligases being harnessed for therapeutic purposes. Additionally, the identification of ubiquitination-based biomarkers in accessible biofluids may enable non-invasive diagnosis and monitoring of various diseases.
As we continue to build bridges between ubiquitin MS findings and clinical applications, the promise of personalized medicine approaches based on an individual's ubiquitinome profile comes closer to realization. By leveraging the sophisticated tools and methodologies outlined in this technical guide, researchers and drug development professionals can contribute to this rapidly advancing field, ultimately improving patient outcomes through a deeper understanding of the ubiquitin system in health and disease.
Ubiquitin MS Data Translation Pathway: This diagram illustrates the translational pathway from mass spectrometry data generation to clinical applications, highlighting key biomarker and drug discovery applications that bridge basic research and clinical practice.
Conclusion
Mass spectrometry has fundamentally transformed our ability to decipher the complex language of ubiquitin chain architectures, moving from simple linkage identification to holistic analysis of branched and modified chains. Techniques like Ub-clipping and UbiChEM-MS have revealed that a significant proportion of cellular ubiquitin polymers are branched, with profound implications for signal specificity, particularly in proteasomal targeting. The integration of MS data with structural biology and functional assays is now cracking long-standing mechanistic codes, such as how the proteasome prioritizes K11/K48-branched chains for degradation. Future directions will involve further technological refinement to achieve single-cell and spatial ubiquitinomics, a deeper exploration of the crosstalk between ubiquitination and other PTMs, and the direct application of these insights to develop next-generation therapeutics, including expanded E3 ligase-targeting drugs and degraders for currently undruggable proteins. For researchers and drug developers, mastering these MS methodologies is no longer optional but essential for driving innovation in biomedicine.
References
- 1. K29-Linked Ubiquitin Regulates Proteotoxic Stress... Signaling [https://pmc.ncbi.nlm.nih.gov/articles/PMC8717942/]
- 2. Focus on ubiquitylation and protein degradation [https://www.nature.com/articles/s41594-025-01672-9]
- 3. Colding-Christensen CS et al. (2023), Profiling ubiquitin ... signalling [https://www.xenbase.org/xenbase/literature/article.do?method=display&articleId=60481]
- 4. Computationally Driven Top-Down Mass Spectrometry of ... [https://pubmed.ncbi.nlm.nih.gov/40766653/]
- 5. emerging roles of the E3 ubiquitin ligase Hakai [https://pmc.ncbi.nlm.nih.gov/articles/PMC11749156/]
- 6. Quantifying Ubiquitin Signaling - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC4441763/]
- 7. Emerging functions of branched ubiquitin chains [https://www.nature.com/articles/s41421-020-00237-y]
- 8. Analysis of the topology of ubiquitin chains - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6989142/]
- 9. Review Ubiquitin—A structural perspective [https://www.sciencedirect.com/science/article/abs/pii/S1097276524010104]
- 10. Analysis of ubiquitin signaling and chain topology cross-talk [https://www.sciencedirect.com/science/article/abs/pii/S1874391920300026]
- 11. Mechanism of nucleosomal H2A K13/15 monoubiquitination and... [https://www.nature.com/articles/s41589-024-01750-x?error=cookies_not_supported&code=b550523c-9362-4885-b662-a614ec2bedeb]
- 12. Structural basis of K11/K48-branched ubiquitin chain ... [https://www.nature.com/articles/s41467-025-64719-x]
- 13. Ubiquitin binding domains — from structures to functions - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7359374/]
- 14. Current methodologies in protein ubiquitination characterization [https://cellandbioscience.biomedcentral.com/articles/10.1186/s13578-022-00870-y]
- 15. functions of Emerging | branched Discovery ubiquitin chains Cell [https://www.nature.com/articles/s41421-020-00237-y?error=cookies_not_supported&code=76f4eb8c-f3a3-45e8-af62-eb25991a9ea2]
- 16. Emerging tools and methods to study cell signalling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12224918/]
- 17. binding and deubiquitination by UCH37... Branched ubiquitin chain [https://elifesciences.org/articles/72798]
- 18. Current methodologies in protein ubiquitination ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9373315/]
- 19. Shotgun Proteomics - an overview [https://www.sciencedirect.com/topics/medicine-and-dentistry/shotgun-proteomics]
- 20. Advanced Proteomics Techniques [https://www.technologynetworks.com/proteomics/articles/exploring-proteomics-techniques-from-mass-spectrometry-to-shotgun-405802]
- 21. Mass spectrometry-based proteomics as an emerging tool in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10464495/]
- 22. Comprehensive Workflow of Mass Spectrometry-based ... [https://pubmed.ncbi.nlm.nih.gov/34842228/]
- 23. Quantitative Lys-ϵ-Gly-Gly (diGly) Proteomics Coupled ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4200252/]
- 24. Global analysis of lysine ubiquitination by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2946519/]
- 25. Characterizing Ubiquitination Sites by Peptide-based ... [https://www.sciencedirect.com/science/article/pii/S1535947620334502]
- 26. Systematic and Quantitative Assessment of the Ubiquitin- ... [https://www.sciencedirect.com/science/article/pii/S1097276511006757]
- 27. Ubiquitin diGLY Proteomics as an Approach to Identify and ... [https://pubmed.ncbi.nlm.nih.gov/30242721/]
- 28. Insights into ubiquitin chain architecture using Ub-clipping [https://pmc.ncbi.nlm.nih.gov/articles/PMC6823057/]
- 29. Examining ubiquitinated peptide enrichment efficiency ... [https://www.sciencedirect.com/science/article/abs/pii/S000326971630255X]
- 30. Emerging tools and methods to study cell signalling ... [https://pubmed.ncbi.nlm.nih.gov/40380883/]
- 31. Targeted proteomic analysis reveals enrichment of atypical ... [https://www.sciencedirect.com/science/article/pii/S1874391920303316]
- 32. Review Quantifying Ubiquitin Signaling [https://www.sciencedirect.com/science/article/pii/S1097276515001343]
- 33. Deubiquitylation of Protein Cargo Is Not an Essential Step in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4858939/]
- 34. Insights into ubiquitin chain architecture using Ub - clipping [https://pubmed.ncbi.nlm.nih.gov/31413367/]
- 35. Proteomic approaches to study ubiquitinomics [https://www.sciencedirect.com/science/article/abs/pii/S1874939923000354]
- 36. Getting to the Root of Branched Chains... | SpringerLink Ubiquitin [https://link.springer.com/protocol/10.1007/978-1-0716-2859-1_2]
- 37. Chain Enrichment Ubiquitin - Middle Mass Spectrometry... Down [https://pubmed.ncbi.nlm.nih.gov/28291339/]
- 38. Towards Understanding the Role of Branched Chains in... Ubiquitin [https://search.library.wisc.edu/digital/AAYV3V4AN323CL8J]
- 39. : Detection Methods, Biological Functions... Branched Ubiquitination [https://www.mdpi.com/1420-3049/25/21/5200]
- 40. - Middle Mass Spectrometry Enables... | Semantic Scholar Down [https://www.semanticscholar.org/paper/Middle-Down-Mass-Spectrometry-Enables-of-Branched-Valkevich-Sanchez/bfabb311f5c62c6f14ceeebe2b9941292e1aa274]
- 41. Deubiquitinase–based analysis of ubiquitin chain ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5011418/]
- 42. Detection of Protein Ubiquitination Sites by Peptide Enrichment and... [https://app.jove.com/t/59079/detection-protein-ubiquitination-sites-peptide-enrichment-mass]
- 43. Insights into ubiquitin chain architecture using Ub-clipping [https://www.nature.com/articles/s41586-019-1482-y]
- 44. Methods to measure ubiquitin chain length and linkage [https://www.sciencedirect.com/science/article/abs/pii/S0076687918305159]
- 45. Comprehensive Landscape of Active Deubiquitinating ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6727631/]
- 46. Screening of DUB and specificity by MALDI-TOF activity ... mass [https://www.nature.com/articles/ncomms5763?error=cookies_not_supported&code=45d14c4c-7a35-4b0d-8e53-32fe2b57c626]
- 47. Method for Measuring the Activity of Deubiquitinating Enzymes in Cell ... [https://www.jove.com/t/52784/method-for-measuring-activity-deubiquitinating-enzymes-cell-lines]
- 48. A Review on Macroscale and Microscale Cell Methods - PMC Lysis [https://pmc.ncbi.nlm.nih.gov/articles/PMC6190294/]
- 49. A Tutorial Review of Labeling Methods in Mass ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11342459/]
- 50. Parallel Reaction Monitoring for High Resolution and ... [https://www.sciencedirect.com/science/article/pii/S1535947620341499]
- 51. Quantitative Analysis of Isotope Distributions In Proteomic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2502059/]
- 52. Cross-Validation of a Multiplex LC-MS/MS Method for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8401780/]
- 53. State-of-the-Art and Future Directions in Structural Proteomics [https://pmc.ncbi.nlm.nih.gov/articles/PMC12546877/]
- 54. High-resolution protein modeling through Cryo-EM and AI [https://pmc.ncbi.nlm.nih.gov/articles/PMC12590954/]
- 55. Exploring the Bottleneck in Cryo-EM Dynamic Disorder ... [https://www.mdpi.com/2673-4125/5/3/39]
- 56. E3 ubiquitin ligases in signaling, disease, and therapeutics [https://www.sciencedirect.com/science/article/pii/S0968000425001860]
- 57. Deciphering the Molecular Signals of PINK1/Parkin ... [https://pubmed.ncbi.nlm.nih.gov/27291334/]
- 58. Mechanisms of PINK1, ubiquitin and Parkin interactions in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11105328/]
- 59. The PINK1/Parkin signaling pathway-mediated mitophagy [https://www.sciencedirect.com/science/article/pii/S1043661824004110]
- 60. The mitophagy pathway and its implications in human ... [https://www.nature.com/articles/s41392-023-01503-7]
- 61. Regulatory Roles of PINK1-Parkin and AMPK in Ubiquitin ... [https://www.frontiersin.org/journals/physiology/articles/10.3389/fphys.2020.608474/full]
- 62. Mitophagy: Molecular Mechanisms, New Concepts on Parkin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8750607/]
- 63. Screening of DUB activity and specificity by MALDI-TOF ... [https://www.nature.com/articles/ncomms5763]
- 64. of Deubiquitinase Specificity and Regulation Mechanisms [https://pubmed.ncbi.nlm.nih.gov/28498721/]
- 65. Deubiquitinase dynamics: methodologies for understanding ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12123204/]
- 66. Identification of substrates for human deubiquitinating ... [https://www.sciencedirect.com/science/article/pii/S1084952122000039]
- 67. Secondary interactions in ubiquitin-binding domains ... [https://www.sciencedirect.com/science/article/pii/S221112472400874X]
- 68. Molecular Basis for Lysine Specificity in the Yeast Ubiquitin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2863694/]
- 69. Dissecting the ubiquitin pathway by mass spectrometry - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC1828906/]
- 70. The ubiquitin system, disease , and drug discovery [https://bmcbiochem.biomedcentral.com/articles/10.1186/1471-2091-9-S1-S7]
- 71. Pathways in Ubiquitination - Creative Proteomics Drug Discovery [https://www.creative-proteomics.com/resource/ubiquitination-pathways-in-drug-discovery.htm]
- 72. Label-free quantitative identification of abnormally ubiquitinated... [https://link.springer.com/article/10.1007/s13167-019-00197-8]
- 73. Searching for Potential Lipid Biomarkers of Parkinson's Disease in... [https://pubmed.ncbi.nlm.nih.gov/31284683/]
- 74. Recent Advances in the Development of Pro-PROTAC for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12473374/]
- 75. Recent advances in chemical proteomics for protein ... [https://www.sciencedirect.com/science/article/abs/pii/S1367593125000377]
- 76. Selective ubiquitination of drug-like small molecules by the ... [https://www.nature.com/articles/s41467-025-63442-x]