Evaluating K-ε-GG Remnant Antibody Specificity: A Comprehensive Guide for Proteomics and Drug Development
This article provides a systematic evaluation of K-ε-GG remnant antibody specificity, a cornerstone technology for ubiquitinomics.
Evaluating K-ε-GG Remnant Antibody Specificity: A Comprehensive Guide for Proteomics and Drug Development
Abstract
This article provides a systematic evaluation of K-ε-GG remnant antibody specificity, a cornerstone technology for ubiquitinomics. It covers the foundational principles of the K-ε-GG signature and antibody development, details methodological workflows from sample preparation to mass spectrometry, and offers troubleshooting strategies for common pitfalls. A critical comparison of commercial antibodies and validation techniques equips researchers and drug development professionals to select optimal reagents, maximize data quality, and advance discoveries in disease mechanisms and therapeutic targeting.
The K-ε-GG Signature: Unraveling the Molecular Basis of Ubiquitin Detection
The evolution of ubiquitinomics from its serendipitous discovery in a chromosomal protein to a sophisticated proteomics discipline represents a transformative journey in molecular biology. This review chronicles the key historical milestones that enabled the transition from initial biochemical characterization to the development of modern mass spectrometry-based technologies for system-wide ubiquitination site mapping. We focus specifically on evaluating the technical performance and specificity of anti-K-ε-GG remnant antibodies, the cornerstone reagents in contemporary ubiquitinomics research. By comparing experimental workflows, enrichment efficiencies, and quantitative capabilities across different methodologies, this analysis provides researchers with a comprehensive framework for selecting appropriate protocols and reagents for ubiquitination studies. The refined techniques discussed herein have dramatically expanded our understanding of the ubiquitin code's complexity, enabling identification of over 20,000 endogenous ubiquitination sites in single experiments and opening new avenues for therapeutic intervention in ubiquitin-related pathologies.
The ubiquitin field represents one of the most compelling narratives in modern biology, beginning with an accidental discovery and culminating in a Nobel Prize-winning revelation of a fundamental cellular regulatory mechanism. Ubiquitin was first isolated in 1974 from cattle thymus as a lymphocyte differentiation-promoting factor, initially mistaken for a thymic hormone [1]. The subsequent realization that this protein appeared universally across eukaryotic cells earned it the name "ubiquitin" [1]. The true breakthrough came in 1977 when Goldknopf and Busch characterized the A24 protein in chromatin, discovering it contained ubiquitin linked via an isopeptide bond to histone H2A [1] [2]. This seminal work established that ubiquitin could be conjugated to other proteins and set the stage for understanding its regulatory significance.
The late 1970s and early 1980s brought the crucial recognition that non-lysosomal intracellular proteolysis depended on both ATP and ubiquitin [1]. The elaborate enzymatic cascade governing ubiquitin conjugation—involving E1 activating, E2 conjugating, and E3 ligase enzymes—was gradually elucidated, revealing an intricate post-translational regulatory system of remarkable complexity [3]. The importance of this pathway was cemented in 2004 when Aaron Ciechanover, Avram Hershko, and Irwin Rose received the Nobel Prize in Chemistry for their discovery of ubiquitin-mediated protein degradation [3].
The emergence of ubiquitinomics as a distinct proteomics discipline represents the convergence of this biochemical knowledge with advanced mass spectrometry technologies. As a transformative methodology, ubiquitinomics enables system-wide identification and quantification of ubiquitination sites, revealing the astonishing complexity of ubiquitin signaling in cellular regulation [4]. This review traces this technological evolution, with particular emphasis on evaluating the specificity and performance of K-ε-GG remnant antibodies that form the foundation of modern ubiquitination site mapping.
Historical Progression of Key Methodological Advancements
Table 1: Historical Timeline of Key Developments in Ubiquitin Research
| Year | Development | Significance |
|---|---|---|
| 1974 | Initial isolation from cattle thymus | First identification of ubiquitin (misidentified as thymic hormone) [1] |
| 1977 | Characterization of A24 chromosomal protein | Discovery of ubiquitin-protein conjugation via isopeptide bond with histone H2A [2] |
| Late 1970s-1980s | ATP and ubiquitin-dependent proteolysis | Established ubiquitin's central role in non-lysosomal protein degradation [1] |
| 2000s | Development of anti-K-ε-GG antibodies | Created specific reagents for ubiquitination site enrichment [2] |
| 2012 | Refined enrichment protocols | Enabled identification of >20,000 ubiquitination sites in single experiments [5] |
The journey from initial protein characterization to modern proteomic analysis has been marked by several transformative technological breakthroughs. The foundational insight emerged from understanding that tryptic digestion of ubiquitinated proteins yields a characteristic signature—a diglycine (K-ε-GG) remnant attached via isopeptide bond to the modified lysine residue of the substrate protein [2]. This discovery, first made during analysis of the A24 protein, established the conceptual framework for all subsequent ubiquitination site mapping methodologies.
Early approaches to ubiquitination site identification relied on Edman sequencing and required substantial protein quantities, limiting throughput and sensitivity [2]. The first mass spectrometry-based identifications of K-GG peptides focused on specific targets like Lys-48-linked polyubiquitin and the yeast G-protein coupled receptor Gpa1 [2]. These proof-of-concept studies demonstrated that liquid chromatography-tandem mass spectrometry (LC-MS/MS) could effectively localize ubiquitination sites, but methodological constraints initially restricted applications to individual proteins or simple mixtures.
The critical turning point came with the development of immunoaffinity reagents capable of specifically enriching K-ε-GG-containing peptides from complex proteomic digests [2]. This methodology, adapted from similar approaches for phosphorylated tyrosine peptides, leveraged the unique N-terminus created by the attached diglycine remnant following tryptic digestion [2]. The commercial availability of highly specific anti-K-ε-GG antibodies dramatically transformed the ubiquitinomics landscape, enabling researchers to transition from identifying hundreds of ubiquitination sites to mapping thousands to tens of thousands of sites in individual experiments [5] [4].
Figure 1: The methodological evolution of ubiquitinomics from initial protein discovery to modern high-throughput technologies, highlighting key transitional developments that enabled progressively higher-resolution analysis.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagents for K-ε-GG Enrichment Studies
| Reagent / Resource | Function / Application | Specifications | Research Context |
|---|---|---|---|
| Anti-K-ε-GG Antibodies | Immunoaffinity enrichment of ubiquitinated peptides | Rabbit polyclonal; recognizes K-ε-GG motif independent of flanking sequence [6] [7] | Core enrichment reagent; critical for specificity and depth of ubiquitinome coverage [5] |
| Cross-linking Reagents | Immobilize antibodies to solid support | Dimethyl pimelimidate (DMP) in sodium borate buffer [5] | Redces antibody leaching; improves sample-to-sample reproducibility [5] |
| Fractionation Columns | Off-line peptide separation | Zorbax 300 Extend-C18; basic reversed-phase chromatography [5] | Reduces sample complexity; improves identification of low-abundance peptides [5] |
| StageTips | Micro-scale peptide desalting | C18 membrane; small sample volumes [5] | Sample cleanup and concentration before MS analysis [5] |
| SILAC Reagents | Metabolic labeling for quantification | Arg-0/6/10 and Lys-0/4/8 variants [5] | Enables precise quantification of ubiquitination dynamics [5] |
The modern ubiquitinomics toolkit centers around anti-K-ε-GG remnant antibodies, which specifically recognize the diglycine signature left at ubiquitination sites after tryptic digestion [6] [7]. These antibodies form the foundation of enrichment protocols, with their specificity and affinity directly determining experimental success. Commercial versions include rabbit polyclonal antibodies that detect the ubiquitin remnant motif across human, mouse, and rat samples, with applications in Western blot, ELISA, and most importantly, immunoaffinity enrichment for mass spectrometry [6] [7].
Protocol refinements have introduced antibody cross-linking as a crucial step, typically using dimethyl pimelimidate (DMP) to covalently link antibodies to solid supports [5]. This innovation significantly reduces antibody leaching during enrichment procedures, improving sample-to-sample reproducibility and minimizing contamination of eluted peptides with antibody fragments [5]. For comprehensive ubiquitinome analysis, off-line fractionation using basic reversed-phase chromatography represents another essential tool, with non-contiguous pooling strategies effectively reducing sample complexity while maintaining comprehensive coverage [5].
Quantitative ubiquitinomics heavily relies on stable isotope labeling methods, particularly Stable Isotope Labeling by Amino acids in Cell culture (SILAC), which enables precise measurement of ubiquitination dynamics in response to cellular perturbations [5]. The combination of these reagents in optimized workflows has progressively improved ubiquitination site identification from mere hundreds to approximately 20,000 distinct sites in single experiments, representing a dramatic advancement in analytical capability [5].
Comparative Analysis of K-ε-GG Antibody Performance
The performance of anti-K-ε-GG antibodies for ubiquitin remnant enrichment must be evaluated across multiple parameters, including specificity, sensitivity, reproducibility, and quantitative accuracy. Systematic optimization studies have revealed that antibody input requirements, cross-linking efficiency, and peptide-to-antibody ratios critically influence experimental outcomes [5].
Table 3: Performance Comparison of Ubiquitin Enrichment Methodologies
| Methodology | Enrichment Specificity | Typical Sites Identified | Sample Input Requirements | Quantitative Capabilities | Key Limitations |
|---|---|---|---|---|---|
| Protein-level Enrichment | Moderate | Hundreds to low thousands | High (10+ mg) | Limited by protein-level labeling | Co-enrichment of interacting proteins; lower specificity [2] |
| Early K-ε-GG Peptide Enrichment | High | 1,000-5,000 sites | Moderate to high (5-35 mg) | Compatible with SILAC and TMT | Required multiple replicates for depth; antibody inconsistency [5] |
| Optimized K-ε-GG Cross-linked Workflow | Very high | ~20,000 sites | Moderate (5 mg protein input) | Excellent quantitative precision | Technical complexity; requires protocol optimization [5] |
| Ubiquitin Pan Nanobody | High for proteins | Dozens of ubiquitylated proteins | Moderate | Compatible with label-free quantification | Protein-level only; no site-specific information [8] |
Comparative studies demonstrate that optimized K-ε-GG antibody-based enrichment substantially outperforms alternative methodologies. For instance, when analyzing RNF111/Arkadia E3 ubiquitin ligase substrates, the diGly remnant peptide immunoprecipitation method successfully identified SKIL ubiquitylation among 108 potential RNF111 substrates, while a ubiquitin pan nanobody approach detected 52 potential substrates including SKI and SKIL but lacked site-specific resolution [8]. This highlights the critical advantage of peptide-level enrichment for precise ubiquitination site mapping.
The refinement of K-ε-GG antibody workflows has yielded remarkable improvements in experimental efficiency. Where earlier approaches required 35mg of protein input and multiple replicates to identify >5,000 ubiquitination sites, current optimized protocols routinely achieve ∼20,000 nonredundant K-ε-GG site quantifications from just 5mg of protein input per SILAC channel in triple-encoded experiments [5]. This represents a 10-fold improvement in protein input efficiency while simultaneously increasing site identification.
The specificity of K-ε-GG antibodies has proven exceptional, with minimal cross-reactivity reported against similar modifications. This specificity is particularly evident when compared to antibodies developed for other ubiquitin-like modifications, such as the anti-VG-ε-K antibodies used for UFMylation studies, which showed 6- to 17-fold enhanced specificity for VG-ε-K-containing peptides over GG-ε-GG peptides [9]. This discrimination between highly similar modification signatures underscores the precision of well-validated remnant antibodies.
Experimental Protocols for Ubiquitin Remnant Enrichment
Cell Culture and Protein Preparation
For comprehensive ubiquitinome analysis, Jurkat E6-1 cells are cultured in SILAC RPMI 1640 media deficient in l-arginine and l-lysine and supplemented with 10% dialyzed fetal bovine serum [5]. Cells undergo approximately six doublings with heavy isotope-labeled amino acids (Arg-0/6/10 and Lys-0/4/8) to ensure complete metabolic labeling. Prior to harvest, cells are typically treated for 4 hours with proteasome inhibitors (e.g., 2-5μM MG-132) or DMSO vehicle control to stabilize ubiquitinated substrates [5]. Cell pellets are lysed in denaturing conditions using 8M urea buffer containing 50mM Tris-HCl (pH 7.5), 150mM NaCl, protease inhibitors, and deubiquitinase inhibitors (50μM PR-619) to preserve ubiquitination signatures [5]. Protein concentrations are determined by bicinchoninic acid (BCA) assay, followed by reduction with dithiothreitol (DTT), carbamidomethylation with iodoacetamide, and overnight digestion with sequencing-grade trypsin at an enzyme-to-substrate ratio of 1:50 [5].
Peptide Fractionation and Antibody Cross-linking
To reduce sample complexity, digested peptides are fractionated using offline basic reversed-phase chromatography on a Zorbax 300 Extend-C18 column (9.4 × 250mm, 300Å, 5μm) with a 64-minute gradient from 2% to 60% solvent B (90% MeCN, 5mM ammonium formate, pH 10) at 3ml/min flow rate [5]. Eighty fractions are collected and pooled in a non-contiguous manner into eight final fractions to maximize separation of co-eluting peptides [5]. For antibody preparation, anti-K-ε-GG antibody beads are washed with 100mM sodium borate (pH 9.0) and cross-linked with 20mM dimethyl pimelimidate (DMP) for 30 minutes at room temperature [5]. After blocking with 200mM ethanolamine (pH 8.0), beads are washed and stored in immunoprecipitation buffer (IAP: 50mM MOPS, pH 7.2, 10mM sodium phosphate, 50mM NaCl) at 4°C until use [5].
Immunoaffinity Enrichment and Mass Spectrometry Analysis
Dried peptide fractions are resuspended in 1.5ml IAP buffer and incubated with cross-linked anti-K-ε-GG antibody beads (typically 31μg antibody per fraction) for 1 hour at 4°C with rotation [5]. Beads are washed four times with ice-cold PBS, and K-ε-GG peptides are eluted with two 50μl aliquots of 0.15% trifluoroacetic acid (TFA) [5]. Eluted peptides are desalted using C18 StageTips and analyzed by LC-MS/MS on high-resolution tandem mass spectrometers. For quantitative experiments, SILAC-based quantification or isobaric labeling approaches like TMT can be employed, with data processed using specialized computational pipelines for ubiquitination site identification and quantification [5] [9].
Figure 2: Comprehensive workflow for K-ε-GG remnant ubiquitinomics, highlighting critical steps in sample preparation, antibody-based enrichment, and mass spectrometric analysis that enable high-confidence ubiquitination site identification and quantification.
Applications in Basic Research and Drug Discovery
The refined methodologies for ubiquitination site mapping have dramatically expanded our understanding of ubiquitin signaling in both physiological and pathological contexts. In basic research, quantitative ubiquitinomics has revealed the astonishing scope of ubiquitin regulation upon proteasome inhibition and identified specific protein classes, including newly synthesized proteins and chromatin-related proteins, that undergo dramatic changes in ubiquitination status following drug treatment [5]. The application of these techniques has illuminated previously unappreciated complexity in diverse biological processes, from TGF-β signaling regulation through RNF111-mediated ubiquitination of SKI and SKIL repressors [8] to the intricate regulation of neuronal function at glutamatergic synapses [3].
In drug discovery contexts, ubiquitinomics approaches provide powerful tools for characterizing the specificity and mechanisms of action for therapeutic compounds targeting the ubiquitin-proteasome system [4]. As the ubiquitin system becomes increasingly recognized as a therapeutic target in cancers, neurodegenerative diseases, and inflammatory disorders, the ability to comprehensively profile ubiquitination changes in response to candidate compounds represents a critical advancement [4]. For instance, ubiquitinomics can identify novel substrates of E3 ligases targeted by molecular glues or validate the specificity of PROTAC compounds, accelerating the development of more precise therapeutic interventions [4].
The application of these methodologies to human disease samples has yielded particularly insightful findings. Recent work analyzing ubiquitin-fold modifier 1 (UFM1) modifications in skeletal muscle biopsies from people living with amyotrophic lateral sclerosis (ALS) revealed extensive changes in myosin UFMylation, demonstrating how remnant-based enrichment approaches can illuminate pathological mechanisms in human disorders [9]. Similarly, integrated proteomic and SUMOylome analyses in glioma tissues have identified novel regulatory axes in tumor progression, highlighting the potential for post-translational modification mapping to reveal new therapeutic targets [10].
The journey from the initial characterization of the A24 chromosomal protein to contemporary ubiquitinomics exemplifies how technological innovation drives biological discovery. The development and refinement of anti-K-ε-GG remnant antibodies represent a cornerstone achievement in this narrative, enabling researchers to transition from studying individual ubiquitination events to system-wide analyses of the ubiquitin code. The current state-of-the-art methodologies, incorporating antibody cross-linking, advanced fractionation, and high-resolution mass spectrometry, now support the quantification of approximately 20,000 distinct endogenous ubiquitination sites in single experiments—a 10-fold improvement over earlier approaches [5].
Despite these remarkable advances, challenges remain in comprehensively capturing the entire ubiquitinome, particularly low-abundance substrates and tissue-specific modifications. Future methodological developments will likely focus on improving sensitivity for limited clinical samples, enhancing quantification accuracy for dynamic ubiquitination changes, and integrating ubiquitinomics with other 'omics datasets to provide more holistic views of cellular regulation. The continued refinement of pan-specific antibodies for ubiquitin-like modifications, building on successes like the anti-VG-ε-K antibodies for UFMylation studies [9], will further expand our ability to simultaneously monitor multiple post-translational modification networks.
As ubiquitinomics methodologies mature and become more accessible, their application across diverse biological and clinical contexts will undoubtedly yield new insights into the intricate regulatory functions of the ubiquitin system. The historical progression from initial biochemical characterization to modern proteomic profiling stands as a testament to the power of technological innovation in illuminating fundamental biological processes, with the A24 protein serving as the foundational discovery that launched an entire field of inquiry.
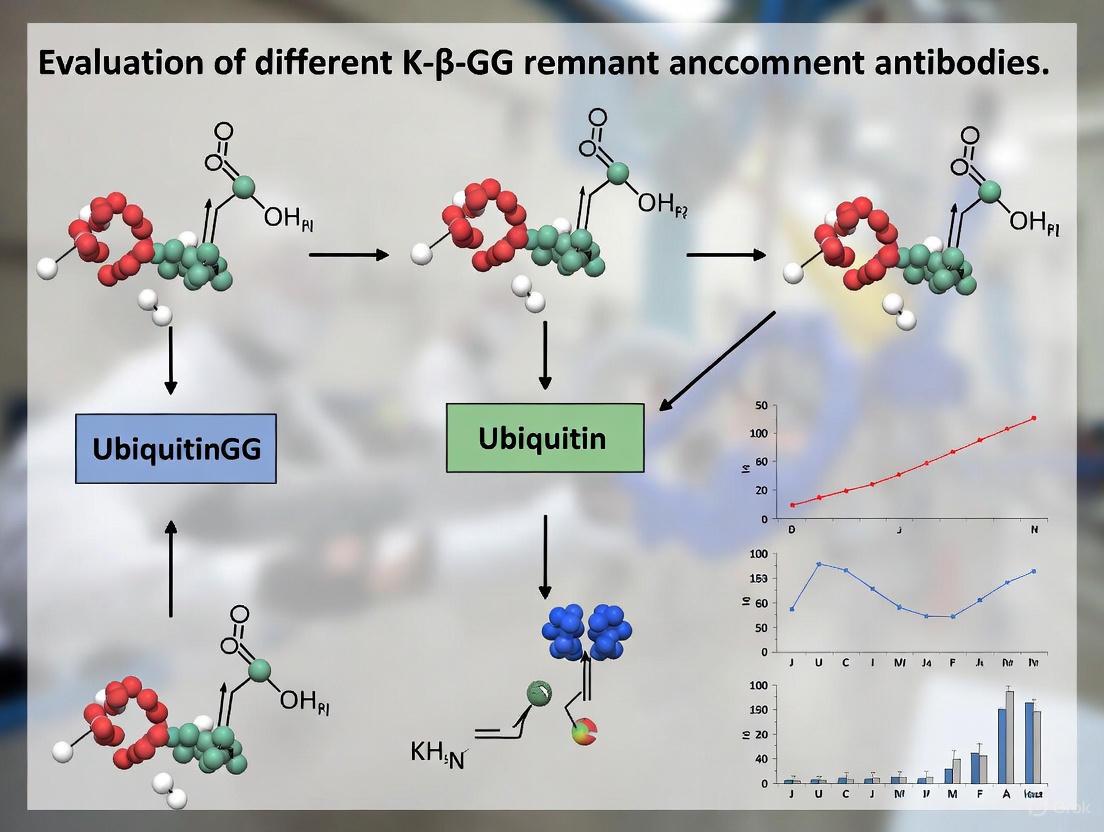
The identification of protein ubiquitination sites by mass spectrometry has been revolutionized by a specific trypsin-dependent mechanism that generates a recognizable signature on modified lysines. This proteolytic process creates a di-glycine (K-ε-GG) remnant, which serves as a diagnostic handle for immunoaffinity enrichment and subsequent mass spectrometric analysis. This guide objectively compares the performance of various antibody-based enrichment approaches for this ubiquitin remnant, detailing their specific applications, limitations, and experimental considerations to inform researcher selection for specific ubiquitinome profiling goals.
Protein ubiquitination is an essential post-translational modification regulating diverse cellular processes including proteasomal degradation, signal transduction, and DNA repair [11]. The covalent attachment of ubiquitin to substrate proteins occurs via an isopeptide bond between the C-terminal carboxyl group of ubiquitin and the ε-amino group of a lysine residue in the target protein [12]. For decades, the identification of specific ubiquitination sites remained analytically challenging due to the low stoichiometry of modified proteins and the complexity of ubiquitin chain architectures.
A critical breakthrough emerged from understanding trypsin digestion behavior toward ubiquitinated proteins. Trypsin cleaves after arginine and lysine residues, and the C-terminal sequence of ubiquitin is Arg-Gly-Gly [13] [11]. When trypsin encounters ubiquitin conjugated to a substrate protein, it cleaves after the arginine residue, leaving a diglycine remnant (approximately 114.04 Da) attached to the modified lysine's ε-amino group on the substrate-derived peptide [12] [13]. This K-ε-GG modification serves as a stable, mass-detectable signature of the original ubiquitination event, enabling development of targeted enrichment strategies.
The Diagnostic DiGlycine Remnant: Generation and Recognition
The Trypsin-Mediated Mechanism
The trypsin digestion process converts heterogeneous ubiquitinated proteins into peptides bearing a consistent, recognizable modification. The molecular transformation involves:
- Ubiquitin C-terminal sequence: Ubiquitin terminates with the sequence ...Arg-Gly-Gly at its C-terminus
- Trypsin cleavage specificity: Trypsin recognizes and cleaves after the arginine residue within ubiquitin
- Remnant formation: The two glycine residues remain conjugated via an isopeptide bond to the ε-amino group of the modified lysine on the substrate peptide
- Mass signature: The resulting K-ε-GG modification produces a characteristic 114.04 Da mass shift detectable by mass spectrometry [12]
This conserved tryptic signature enabled the development of immunoaffinity reagents that specifically recognize the K-ε-GG remnant, dramatically improving the capacity for large-scale ubiquitination site mapping from complex biological samples [13] [11].
Specificity Considerations for K-ε-GG Antibodies
A critical consideration in ubiquitin remnant profiling is that the K-ε-GG signature is not absolutely specific to ubiquitination. The ubiquitin-like modifiers NEDD8 and ISG15 also generate a diglycine remnant upon trypsin digestion due to their similar C-terminal sequences [13]. However, experimental evidence indicates that in most biological contexts, the vast majority (>94%) of K-ε-GG identifications represent genuine ubiquitination events rather than modification by NEDD8 or ISG15 [13]. This high prevalence makes the K-ε-GG enrichment approach particularly valuable for comprehensive ubiquitinome profiling, though researchers investigating crosstalk with specific ubiquitin-like modifiers should consider complementary experimental approaches.
Comparative Analysis of DiGlycine Remnant Enrichment Approaches
Antibody Performance and Technological Evolution
The development and refinement of anti-K-ε-GG antibodies has dramatically improved the scale and precision of ubiquitination site identification. Current platforms demonstrate significant differences in their capabilities and applications.
Table 1: Comparison of DiGlycine Remnant Enrichment Technologies
| Technology/Product | Key Features | Typical Identifications | Specificity | Primary Applications |
|---|---|---|---|---|
| Standard K-ε-GG Antibody [13] [14] | Immunoaffinity enrichment of tryptic K-ε-GG peptides | ~11,000-20,000 sites in single experiments [14] [15] | Recognizes canonical lysine ubiquitination sites | Global ubiquitinome profiling, quantitative ubiquitination studies |
| PTMScan Ubiquitin Remnant Motif Kit [16] | Commercial kit format with bead-conjugated antibody | Hundreds to >1,000 ubiquitinated sequences | Specific for di-glycine tag from trypsin-digested ubiquitin | Standardized ubiquitination site mapping, clinical samples |
| HS Ubiquitin/SUMO Remnant Motif Kit [16] | Magnetic bead version with higher sensitivity/specificity | Improved recovery of low-abundance sites | Enhanced specificity for K-ε-GG remnant | Challenging samples, low-input applications, high-sensitivity needs |
| Anti-GGX Antibodies [17] | Selective for N-terminal diglycine motifs | 73 putative UBE2W substrates identified [17] | Does not recognize K-ε-GG peptides; specific for linear N-terminal GGX | N-terminal ubiquitination studies, UBE2W substrate identification |
Performance Considerations and Limitations
Each enrichment approach presents distinct advantages and limitations that researchers must consider during experimental design:
- Standard K-ε-GG antibodies provide the most comprehensive coverage of conventional lysine ubiquitination sites but cannot distinguish ubiquitination from NEDDylation or ISG15ylation without additional controls [13]
- Commercial PTMScan kits offer standardized protocols and consistency across experiments but provide less flexibility for protocol modification compared to researcher-configured approaches [16]
- High-sensitivity versions improve detection of low-abundance ubiquitination events but typically at higher cost per sample [16]
- Anti-GGX antibodies enable specialized investigation of N-terminal ubiquitination but offer limited utility for studying conventional lysine ubiquitination [17]
Experimental Protocols for Ubiquitin Remnant Enrichment
Standard K-ε-GG Enrichment Workflow
The following optimized protocol enables routine identification of >10,000 ubiquitination sites from cell lines or tissue samples [13] [18]:
Sample Preparation (Days 1-2)
- Cell Lysis: Lyse cells or tissue in fresh urea lysis buffer (8 M urea, 50 mM Tris HCl pH 8.0, 150 mM NaCl) supplemented with protease and deubiquitinase inhibitors (e.g., 50 μM PR-619, 1 mM PMSF) [13]
- Protein Reduction and Alkylation: Reduce disulfide bonds with 1-5 mM dithiothreitol (37°C, 30 min) followed by alkylation with 10 mM iodoacetamide or chloroacetamide (room temperature, 20 min in darkness)
- Protein Digestion: First, digest with LysC (1:100 enzyme:protein) for 2-3 hours at room temperature. Then dilute urea concentration to 2 M and add trypsin (1:100 enzyme:protein) for overnight digestion at room temperature [13]
- Peptide Cleanup: Desalt peptides using C18 solid-phase extraction cartridges. Wash with 0.1% TFA and elute with 50% acetonitrile/0.1% formic acid [13]
Peptide Fractionation (Day 2)
- Basic pH Reversed-Phase Fractionation: Fractionate peptides using high-pH reversed-phase chromatography (pH 10) with increasing acetonitrile gradients (5-90%). Concatenate fractions to reduce analysis time while maintaining depth [13] [18]
Immunoaffinity Enrichment (Days 3-4)
- Antibody Cross-Linking: Cross-link anti-K-ε-GG antibody to protein A agarose beads using dimethyl pimelimidate to prevent antibody leakage and improve signal-to-noise ratio [13] [14]
- Peptide Enrichment: Incubate fractionated peptides with cross-linked antibody beads for 1.5-2 hours at 4°C with gentle rotation
- Wash and Elution: Wash beads extensively with ice-cold PBS and IAP buffer, then elute enriched peptides with 0.15% trifluoroacetic acid [13] [16]
Mass Spectrometric Analysis (Day 5)
- LC-MS/MS Analysis: Analyze enriched peptides by nanoflow liquid chromatography coupled to tandem mass spectrometry using high-resolution instruments (e.g., Orbitrap Fusion Lumos) [12]
- Data Analysis: Process raw data using search engines (e.g., MaxQuant) with specific settings for the 114.04 Da diglycine modification on lysine residues [18]
Critical Protocol Modifications for Enhanced Performance
Several methodological refinements significantly improve ubiquitination site identifications:
- Antibody cross-linking: Chemical cross-linking of antibodies to beads substantially reduces contamination from antibody fragments and non-specific peptides, improving signal-to-noise ratio in mass spectrometry analyses [13] [14]
- Offline high-pH fractionation: Pre-enrichment fractionation by basic pH reversed-phase chromatography significantly increases ubiquitination site identifications by reducing sample complexity prior to immunoaffinity purification [12] [13]
- Fresh urea preparation: Always prepare urea lysis buffer fresh to prevent protein carbamylation, which creates artificial modifications and compromises protein identification [13]
- Comprehensive protease inhibition: Include deubiquitinase inhibitors (e.g., PR-619) in lysis buffers to preserve endogenous ubiquitination states during sample preparation [13]
Essential Research Reagent Solutions
Table 2: Key Research Reagents for DiGlycine Remnant Studies
| Reagent/Category | Specific Examples | Function/Purpose | Considerations |
|---|---|---|---|
| Anti-K-ε-GG Antibodies | PTMScan Ubiquitin Remnant Motif Kit [16] | Immunoaffinity enrichment of ubiquitinated peptides | Commercial standard; enables consistent results across laboratories |
| Cell Culture Reagents | SILAC amino acids (Lys-8, Arg-10) [13] | Metabolic labeling for quantitative ubiquitinome studies | Enables precise quantification of ubiquitination dynamics |
| Protease Inhibitors | PR-619, PMSF, Aprotinin, Leupeptin [13] | Preserve endogenous ubiquitination states during processing | PR-619 specifically inhibits deubiquitinases |
| Digestion Enzymes | Trypsin, LysC [12] [13] | Protein digestion to generate K-ε-GG peptides | Sequential LysC/trypsin digestion improves efficiency |
| Chromatography Materials | C18 solid-phase extraction cartridges, Basic pH RP columns [12] [13] | Peptide cleanup and fractionation | High-pH fractionation significantly enhances coverage |
| Mass Spectrometry Systems | Orbitrap Fusion Lumos, EASY-nanoLC 1200 [12] | High-sensitivity detection of enriched peptides | High-resolution instrumentation essential for confident identifications |
The trypsin digestion process that generates the diagnostic diGlycine remnant has fundamentally transformed ubiquitin research, enabling systematic mapping of ubiquitination sites at an unprecedented scale. The continued refinement of anti-K-ε-GG antibodies and associated methodologies now supports the routine identification of >20,000 distinct ubiquitination sites in single experiments. When selecting enrichment approaches, researchers must consider the specific biological questions being addressed—whether comprehensive ubiquitinome profiling requiring standard K-ε-GG antibodies, or specialized investigation of N-terminal ubiquitination demanding the emerging class of anti-GGX reagents. As these technologies continue to evolve alongside advances in mass spectrometry sensitivity and computational analysis, our capacity to decipher the complex regulatory networks controlled by protein ubiquitination will undoubtedly expand, offering new insights into fundamental biology and therapeutic opportunities.
Ubiquitination is a fundamental post-translational modification (PTM) that regulates diverse cellular functions, including protein degradation, activity modulation, and signal transduction [19]. This versatility stems from the complexity of ubiquitin conjugates, which can range from single ubiquitin monomers to polymers of different lengths and linkage types [19]. The critical breakthrough in studying this modification came with the development of antibodies specifically targeting the di-glycine remnant (K-ε-GG) that remains attached to lysine residues after tryptic digestion of ubiquitinated proteins [5] [20] [21]. These antibodies have dramatically transformed the proteomic landscape by enabling researchers to enrich and identify thousands of endogenous ubiquitination sites, moving the field from identifying only several hundred sites to routinely quantifying over 20,000 distinct sites in single experiments [5] [21]. This article examines the core technology behind anti-K-ε-GG antibodies, their mechanism of recognition, and their performance compared to alternative methodologies for ubiquitin characterization.
The Molecular Basis of K-ε-GG Recognition
The Trypsin-Mediated Generation of the Di-Glycine Signature
The fundamental mechanism that enables specific recognition of ubiquitination sites begins with proteolytic processing of ubiquitinated proteins. When trypsin digests ubiquitinated proteins, it cleaves after arginine and lysine residues in both the substrate protein and the attached ubiquitin molecule [21]. However, a unique signature emerges at the site of modification: the C-terminal glycine (G76) of ubiquitin forms an isopeptide bond with the epsilon amino group of a lysine residue in the substrate protein [19] [21]. Trypsin cleavage leaves a di-glycine remnant (Gly-Gly) covalently attached to the modified lysine side chain, creating the K-ε-GG motif that serves as a definitive signature of ubiquitination [20] [21]. This signature has a mass shift of 114.04 Da on the modified lysine residue, which can be detected by mass spectrometry [19].
Antibody Recognition of the K-ε-GG Motif
Anti-K-ε-GG antibodies are highly specific reagents that recognize this di-glycine remnant attached to lysine residues [5] [6]. The commercialization of these antibodies represented a watershed moment in ubiquitin research, as they provide the specificity needed to isolate low-abundance ubiquitinated peptides from complex protein digests [5] [20]. The recognition is so specific that these antibodies can distinguish the K-ε-GG motif from other similar modifications, enabling researchers to profile ubiquitination sites on a proteome-wide scale with unprecedented sensitivity and specificity [5] [21].
Table 1: Key Characteristics of Anti-K-ε-GG Antibodies
| Feature | Description | Significance |
|---|---|---|
| Target Epitope | Di-glycine remnant (K-ε-GG) on lysine residues | Specific signature of ubiquitination after trypsin digestion |
| Recognition Specificity | High specificity for K-ε-GG motif | Minimal cross-reactivity with other modifications |
| Commercial Availability | Widely available from multiple vendors (e.g., Thermo Fisher, Cell Signaling Technology) | Accessible to research community [5] [6] |
| Applications | Western blot, ELISA, immunoaffinity enrichment for mass spectrometry | Versatile use across multiple experimental platforms [6] |
| Species Reactivity | Human, Mouse, and others | Broad applicability across model systems [6] |
Performance Comparison: Anti-K-ε-GG Antibodies Versus Alternative Methodologies
Comparison of Ubiquitin Enrichment Techniques
While anti-K-ε-GG antibodies have revolutionized ubiquitin site identification, several alternative methods exist for enriching ubiquitinated proteins or peptides. Each approach has distinct advantages and limitations that researchers must consider when designing experiments.
Table 2: Performance Comparison of Ubiquitin Enrichment Methodologies
| Methodology | Mechanism | Sensitivity | Throughput | Key Limitations |
|---|---|---|---|---|
| Anti-K-ε-GG Antibodies | Immunoaffinity enrichment of tryptic peptides with di-glycine remnant [5] | High (∼20,000 sites from 5mg protein) [5] | High with optimized workflows | Requires tryptic digestion; may not preserve linkage information |
| Ubiquitin Tagging (His/Strep) | Affinity purification of ubiquitinated proteins using tagged ubiquitin [19] | Moderate (110-753 sites in early studies) [19] | Moderate | Potential artifacts from tagged ubiquitin expression; co-purification of non-ubiquitinated proteins [19] |
| Pan-Ubiquitin Antibodies (e.g., FK1/FK2) | Immunoaffinity enrichment of ubiquitinated proteins [19] | Moderate (e.g., 96 sites in MCF-7 cells) [19] | Moderate | Lower specificity; may enrich all ubiquitinated proteins regardless of linkage |
| UBD-Based Approaches (TUBEs) | Tandem-repeated ubiquitin-binding entities enrich ubiquitinated proteins [19] | Varies with affinity | Moderate | Low affinity of single UBDs limited early applications [19] |
| Linkage-Specific Antibodies | Immunoaffinity enrichment of specific ubiquitin linkage types [19] | High for specific linkages | Moderate to high | Limited to specific linkage types; higher cost |
Quantitative Performance of Anti-K-ε-GG Workflows
Substantial improvements in anti-K-ε-GG workflows have dramatically enhanced their performance. Systematic optimization of key pre-analytical variables, including antibody cross-linking, peptide input requirements, and off-line fractionation protocols, has enabled identification of approximately 20,000 distinct endogenous ubiquitination sites from moderate protein input (5 mg) in SILAC experiments [5]. This represents a 10-fold improvement over earlier methods [5]. The development of innovative protocols like the UbiFast method has further increased sensitivity, allowing quantification of approximately 10,000 ubiquitylation sites from as little as 500 μg of peptide per sample [21]. This method utilizes on-antibody TMT labeling while peptides are bound to anti-K-ε-GG antibodies, preventing derivatization of the di-glycyl remnant primary amine and significantly improving relative yield of K-ε-GG peptides to 85.7% compared to 44.2% with in-solution labeling [21].
Experimental Protocols for K-ε-GG Immunoaffinity Enrichment
Standard Enrichment Workflow
A refined and practical workflow for K-ε-GG enrichment enables comprehensive ubiquitination site mapping [5]:
Cell Lysis and Digestion: Cells are lysed in denaturing conditions (8 M urea, 50 mM Tris-HCl, pH 7.5, 150 mM NaCl) with protease and deubiquitinase inhibitors (e.g., PR-619). Protein concentrations are determined by BCA assay, and 5 mg of protein per condition is typically used for SILAC experiments [5].
Reduction and Alkylation: Proteins are reduced with 5 mM dithiothreitol (DTT) for 45 minutes at room temperature, then alkylated using 10 mM iodoacetamide for 30 minutes in the dark [5].
Trypsin Digestion: Lysates are diluted to 2 M urea and digested overnight at 25°C with sequencing-grade trypsin at an enzyme-to-substrate ratio of 1:50 [5].
Desalting: Digested samples are acidified with formic acid and desalted using C18 Sep-Pak SPE cartridges [5].
Basic Reversed-Phase Fractionation: Peptides are fractionated using basic pH reversed-phase chromatography (pH 10) with a 64-minute linear gradient. Fractions are pooled in a noncontiguous manner into 8 fractions to reduce complexity [5].
Antibody Cross-Linking: Anti-K-ε-GG antibody beads are cross-linked using 20 mM dimethyl pimelimidate (DMP) in 100 mM sodium borate (pH 9.0) for 30 minutes at room temperature to stabilize the antibody for repeated use [5].
Immunoaffinity Enrichment: Peptide fractions are resuspended in immunoprecipitation buffer (50 mM MOPS, pH 7.2, 10 mM sodium phosphate, 50 mM NaCl) and incubated with cross-linked anti-K-ε-GG antibody beads for 1 hour at 4°C [5].
Wash and Elution: Beads are washed four times with ice-cold PBS, and K-ε-GG peptides are eluted with 0.15% trifluoroacetic acid (TFA) [5].
Mass Spectrometry Analysis: Eluted peptides are desalted using C18 StageTips and analyzed by LC-MS/MS [5].
Advanced UbiFast Protocol for Multiplexed Analysis
The UbiFast protocol addresses the challenge of multiplexed quantification with TMT reagents [21]:
Peptide Enrichment: K-ε-GG peptides are enriched from 0.5-1 mg of Jurkat cell peptides using anti-K-ε-GG antibodies [21].
On-Antibody TMT Labeling: While still bound to antibodies, peptides are labeled with TMT reagents (0.4 mg) for 10 minutes, protecting the di-glycyl remnant from derivatization [21].
Reaction Quenching: The labeling reaction is quenched with 5% hydroxylamine [21].
Peptide Elution and Analysis: TMT-labeled K-ε-GG peptides from multiple samples are combined, eluted from antibodies, and analyzed by LC-MS/MS with FAIMS to improve quantitative accuracy [21].
Visualization of Key Workflows and Biological Mechanisms
Diagram 1: K-ε-GG antibody workflow for ubiquitin site identification. This diagram illustrates the complete process from ubiquitinated proteins to site identification, highlighting the critical role of anti-K-ε-GG antibodies in peptide enrichment.
Diagram 2: Molecular mechanism of K-ε-GG remnant formation. This diagram shows how ubiquitin attaches to substrate proteins and how trypsin cleavage generates the K-ε-GG signature recognized by specific antibodies.
Essential Research Reagent Solutions
Table 3: Key Research Reagents for K-ε-GG-Based Ubiquitination Studies
| Reagent Category | Specific Examples | Function in Workflow |
|---|---|---|
| Anti-K-ε-GG Antibodies | PTMScan Ubiquitin Remnant Motif Kit (Cell Signaling Technology); Thermo Fisher PA5-120707 [5] [6] | Specific recognition and enrichment of K-ε-GG modified peptides |
| Protease Inhibitors | Aprotinin, Leupeptin, PMSF [5] | Prevent protein degradation during cell lysis |
| Deubiquitinase Inhibitors | PR-619 [5] [22] | Preserve endogenous ubiquitination states by blocking deubiquitinating enzymes |
| Proteasome Inhibitors | MG-132 [5] [22] | Accumulate ubiquitinated proteins by blocking proteasomal degradation |
| Cross-linking Reagents | Dimethyl pimelimidate (DMP) [5] | Stabilize antibodies on beads for repeated use |
| Digestion Enzymes | Sequencing-grade trypsin [5] | Generate K-ε-GG peptides from ubiquitinated proteins |
| Chromatography Media | C18 Sep-Pak cartridges; Zorbax 300 Extend-C18 column [5] | Desalting and fractionation of peptides before enrichment |
| Mass Spectrometry Tags | SILAC reagents; Tandem Mass Tags (TMT) [5] [21] | Enable quantitative comparisons across experimental conditions |
Anti-K-ε-GG antibodies represent a cornerstone technology in ubiquitin research, providing unparalleled specificity for proteome-wide mapping of ubiquitination sites. Their mechanism of action—targeting the trypsin-generated di-glycine remnant—offers a robust and specific approach to identify this biologically critical modification. When compared to alternative methodologies such as ubiquitin tagging approaches, pan-ubiquitin antibodies, and UBD-based enrichment, anti-K-ε-GG antibodies consistently demonstrate superior performance in both sensitivity and specificity for site identification [5] [19] [21]. Continued refinement of experimental workflows, including antibody cross-linking, optimized fractionation, and innovative labeling strategies like the UbiFast protocol, has further enhanced their utility, making comprehensive ubiquitin profiling accessible to researchers across biological and translational disciplines [5] [21]. As the field advances, these antibodies will undoubtedly continue to play a vital role in deciphering the complex ubiquitin code and its implications in health and disease.
The specificity of an antibody is its defining characteristic, determining its ability to uniquely recognize a target epitope amidst a complex biological milieu. For researchers investigating post-translational modifications, particularly ubiquitination, specificity is not merely a performance metric but a fundamental prerequisite for data validity. The development of anti-di-glycine remnant (K-ε-GG) antibodies revolutionized ubiquitination research by enabling the systematic enrichment and mass spectrometry-based identification of thousands of endogenous ubiquitination sites from cell lines and tissues [23] [19]. This guide provides a comprehensive evaluation of performance metrics for K-ε-GG remnant antibodies and related reagents, presenting structured experimental data and protocols to empower scientists in making informed reagent selections for their ubiquitination research.
Quantitative Performance Comparison of Ubiquitination Site Enrichment Antibodies
The performance of antibodies targeting ubiquitination remnants can be evaluated through their enrichment efficiency, specificity, and quantitative precision. The table below summarizes key characteristics of major antibody types used in ubiquitination research.
Table 1: Performance Comparison of Antibodies for Ubiquitination Site Enrichment
| Antibody Type / Clone | Specificity | Enrichment Efficiency | Key Applications | Limitations |
|---|---|---|---|---|
| K-ε-GG (Commercial) | Isopeptide-linked diglycine on lysine | ~20,000 ubiquitination sites per SILAC experiment [23] | Global ubiquitin profiling [23] [24] | Cannot distinguish ubiquitination from other Ub-like modifications [19] |
| GGX Clones (1C7, 2B12, 2E9, 2H2) | Linear N-terminal diglycine motifs [17] | Identified 73 putative UBE2W substrates [17] | Specific detection of N-terminal ubiquitination [17] | Minimal cross-reactivity with K-ε-GG peptides [17] |
| Linkage-Specific Ub Antibodies | Specific polyUb linkages (M1, K48, K63) [19] | Varies by linkage type | Studying chain architecture-specific functions [19] | High cost; potential non-specific binding [19] |
| VG-ε-K (Anti-UFM1) | UFMylation remnant sites [25] | >200 UFMylation sites from mouse tissues [25] | UFMylome characterization [25] | Specialized for UFM1 modification only |
Experimental Protocols for Specificity Validation
Rigorous experimental validation is crucial for confirming antibody specificity. Below are detailed methodologies for key validation approaches cited in the literature.
Phage Display for Antibody Discovery and Specificity Screening
The anti-GGX monoclonal antibodies were discovered using a comprehensive phage display workflow:
- Library Construction: Single-chain Fv (scFv) libraries were constructed from rabbits immunized with a Gly-Gly-Met (GGM) peptide [17].
- Biopanning: Three rounds of plate-based biopanning were performed against the GGM peptide with counter-selection against the K-ε-GG peptide to eliminate cross-reactive clones [17].
- Specificity Profiling: Reformatted IgGs were screened via ELISA against a panel of 19 GGX peptides (where X represents different amino acids) to determine recognition patterns [17].
- Structural Validation: X-ray crystallography of Fab-peptide complexes at 2.85 Å resolution revealed the structural basis for selective linear diglycine recognition [17].
Immunoaffinity Enrichment and Mass Spectrometry Workflow
The refined K-ε-GG enrichment protocol for global ubiquitination site mapping involves:
- Protein Digestion: Cells or tissues are lysed and proteins digested with trypsin, which cleaves ubiquitinated proteins to leave a di-glycine remnant (~114 Da) on modified lysines [23] [19].
- Peptide Enrichment: Digested peptides are incubated with anti-K-ε-GG antibody cross-linked to protein A/G beads [23].
- Offline Fractionation: Basic pH reverse-phase fractionation (typically 12-24 fractions) reduces sample complexity prior to enrichment [23].
- LC-MS/MS Analysis: Enriched peptides are separated by liquid chromatography and analyzed by tandem mass spectrometry [23] [24].
- Data Analysis: Ubiquitination sites are identified by searching for the di-glycine remnant mass shift on lysine residues [19].
In Vivo Validation Using Genetic Models
For UFMylation studies, the anti-VG-ε-K antibody was validated through:
- Knockdown Approaches: siRNA-mediated knockdown of the E3 ligase UFC1 in mouse models demonstrated concomitant down-regulation of identified UFMylation sites [25].
- Disease Relevance Assessment: Analysis of human amyotrophic lateral sclerosis (ALS) muscle biopsies revealed prominent increases in myosin UFMylation, establishing pathological relevance [25].
Figure 1: Mass spectrometry-based workflow for ubiquitination site identification using anti-K-ε-GG antibodies.
The Scientist's Toolkit: Essential Research Reagents
Successful ubiquitination profiling requires a carefully selected set of reagents and methodologies. The table below outlines essential solutions for comprehensive ubiquitination research.
Table 2: Essential Research Reagents for Ubiquitination Studies
| Reagent / Method | Function | Key Features |
|---|---|---|
| Anti-K-ε-GG Antibodies | Enrichment of canonical ubiquitination sites | Enables identification of >10,000 sites; compatible with SILAC/TMT quantification [23] [24] |
| Anti-GGX Antibodies (1C7, 2B12, 2E9, 2H2) | Specific detection of N-terminal ubiquitination | Minimal cross-reactivity with K-ε-GG; broad specificity at third position [17] |
| Linkage-Specific Ub Antibodies | Enrichment of specific polyUb chain types | Studies of chain-specific signaling; available for M1, K48, K63 linkages [19] |
| Tandem Ub-Binding Entities (TUBEs) | Protection of Ub chains from DUBs; affinity enrichment | Tandem UBDs increase affinity; can preserve labile ubiquitination [19] |
| Strep/His-Tagged Ub | Affinity purification of ubiquitinated proteins | Alternative to antibody-based enrichment; enables substrate identification [19] |
| DUB Inhibitors | Preservation of ubiquitination during preparation | Prevents loss of signal during sample processing [19] |
Advanced Applications and Emerging Trends
Investigating Ubiquitination in Aging and Disease
Recent applications of K-ε-GG antibodies have revealed significant insights into age-related changes:
- Brain Aging: Quantitative ubiquitylome analysis of mouse brains revealed 29% of altered ubiquitination sites in aged mice were independent of protein abundance changes, indicating genuine alterations in modification stoichiometry [24].
- Dietary Interventions: Dietary restriction modified the brain ubiquitylome, partially rescuing age-related ubiquitination changes [24].
- Organ Specificity: Age-related ubiquitination signatures showed minimal correlation between brain and liver, indicating tissue-specific regulation of the ubiquitylome [24].
Addressing Specificity Challenges with Computational Approaches
Emerging computational methods are complementing experimental approaches for antibody characterization:
- Machine Learning: Graph neural networks are being developed to predict antibody-antigen binding affinity, though current models face challenges with generalizability due to limited experimental training data [26].
- Structure-Based Prediction: Integrated AI approaches combining AlphaFold2 with inverse folding models show promise for improving antibody specificity prediction [27].
Figure 2: Specificity challenges in ubiquitination remnant antibodies and validation strategies.
The evaluation of antibody specificity extends beyond simple validation experiments to encompass a comprehensive assessment of performance across multiple metrics. For K-ε-GG remnant antibodies and related reagents, researchers must consider enrichment efficiency, specificity for intended targets, quantitative precision, and applicability to specific biological questions. The experimental protocols and performance data presented here provide a framework for critical assessment of these essential research tools. As the field advances, integration of rigorous experimental validation with emerging computational approaches will further enhance our ability to precisely characterize antibody specificity, ultimately strengthening the foundation of ubiquitination research and therapeutic development.
Optimized Workflows: Integrating K-ε-GG Antibodies into Ubiquitin Profiling Protocols
In mass spectrometry-based ubiquitinomics, sample preparation is the crucial foundation for obtaining high-quality, reproducible data. The initial step of cell lysis can significantly influence protein recovery, the preservation of post-translational modifications, and ultimately, the depth of ubiquitinome coverage. Within this context, the choice between sodium deoxycholate (SDC) and urea-based lysis buffers represents a critical methodological decision that researchers must make. This comparison guide objectively evaluates the performance of these two common lysis approaches within the broader framework of research aimed at evaluating the specificity of K-ε-GG remnant antibodies. For researchers, scientists, and drug development professionals, selecting the optimal lysis protocol directly impacts the ability to comprehensively profile ubiquitin signaling in cellular processes, disease mechanisms, and drug response pathways.
The following diagram outlines the key decision points and considerations in the SDC versus urea lysis workflow for ubiquitinome studies:
Quantitative Performance Comparison: SDC Demonstrates Clear Advantages
Direct comparative studies reveal significant performance differences between SDC and urea lysis buffers in ubiquitinome profiling. When measuring key metrics of identification numbers, reproducibility, and specificity, SDC consistently outperforms traditional urea-based methods.
Table 1: Performance Comparison of SDC vs. Urea Lysis for Ubiquitinome Analysis
| Performance Metric | SDC-Based Lysis | Urea-Based Lysis | Improvement | Experimental Context |
|---|---|---|---|---|
| K-GG Peptide Identifications | 26,756 peptides | 19,403 peptides | 38% increase with SDC [28] | HCT116 cells treated with MG-132; 4 workflow replicates [28] |
| Quantification Precision | Higher percentage of peptides with CV < 20% [28] | Lower percentage of precisely quantified peptides [28] | Significant improvement in reproducibility [28] | Benchmarking using proteasome inhibitor-treated cells [28] |
| Enrichment Specificity | Maintained or improved specificity [28] | Standard specificity | Comparable or better for SDC [28] | Immunoaffinity purification of K-GG remnant peptides [28] |
| Sample Input Requirements | 20-times less protein input needed [28] | Higher input requirements | Substantial reduction with SDC workflow [28] | Comparison against UbiSite method [28] |
| Method-Induced Artifacts | No di-carbamidomethylation of lysine residues [28] | Potential for di-carbamidomethylation [28] | Reduced artifacts with SDC [28] | Incubation at high temperatures with CAA alkylation [28] |
Beyond the specific comparison with urea, the optimized SDC-based lysis protocol has been benchmarked against other advanced methodologies. When compared to the UbiSite approach—which relies on urea lysis and immunoaffinity purification of longer ubiquitin remnant peptides (K-GGRLRLVLHLTSE) generated by Lys-C digestion—the SDC workflow achieved a much better enrichment specificity while requiring only 1/10th of the MS acquisition time per sample [28]. This makes the SDC approach particularly advantageous for applications where sample material is limited or when processing large sample series.
Detailed Experimental Protocols for Implementation
SDC-Based Lysis and Digestion Protocol
The superior performance of SDC-based lysis stems from its optimized composition and processing steps. The following protocol has been specifically validated for ubiquitinome studies:
Lysis Buffer Composition: 1% SDC in 100 mM Tris-HCl (pH 8.5), supplemented with chloroacetamide (CAA) for immediate cysteine protease inactivation [28] [29]. The use of CAA rather than iodoacetamide is crucial as it does not induce unspecific di-carbamidomethylation of lysine residues, even when incubated at high temperatures [28].
Cell Lysis Procedure: Resuspend cell pellets in SDC buffer and incubate at room temperature. For efficient homogenization, either sonication (10 cycles of 5-second pulses at 25% power with 10-second intervals on ice) or mechanical disruption (BeatBox system at high speed for 2×10 minutes) can be employed [29]. Immediate boiling of samples after lysis together with high concentrations of CAA increases ubiquitin site coverage by rapidly inactivating cysteine ubiquitin proteases through alkylation [28].
Protein Digestion: Determine protein concentration using BCA assay. For digestion, use 100 μg protein aliquots. Reduce proteins with TCEP (final concentration ~5 mM) for 20 minutes at 37°C with shaking at 750 rpm. Alkylate with CAA (final concentration ~15 mM) in the dark for 15 minutes. Dilute the SDC concentration to approximately 0.5% to prevent inhibition of trypsin. Add trypsin/Lys-C protease mix at 1:30 (w/w) enzyme-to-protein ratio and digest overnight at 37°C with shaking at 750 rpm [29].
Peptide Cleanup: Acidify samples with TFA to a final concentration of 0.5-1% to precipitate SDC. Centrifuge at 13,000g for 10 minutes and collect supernatant. Desalt using C18 columns (e.g., GL Sciences MonoSpin C18) and elute with 70% acetonitrile, 0.2% formic acid [29]. Alternative desalting with amide columns (e.g., GL Sciences MonoSpin amide) has also been successfully employed with SDC-digested samples [29].
Traditional Urea-Based Lysis Protocol
The conventional urea-based method provides a reference point for comparison:
Lysis Buffer Composition: 8 M urea in 100 mM Tris-HCl (pH 8.5) [28] [29]. Some protocols use 8 M urea with 50 mM NEM (N-ethylmaleimide) in PBS for embryo lysis in tissue-specific ubiquitination studies [30].
Cell Lysis Procedure: Resuspend cell pellets in urea buffer. For sonication approach, use 10 cycles of 5-second pulses at 25% power with 10-second intervals on ice. Alternatively, employ mechanical disruption (BeatBox system at high speed for 2×10 minutes) [29]. In tissue-specific applications, dounce homogenization in urea buffer is commonly used [30].
Protein Digestion: Dilute urea samples to 2 M final concentration using 50 mM HEPES or similar buffer. Reduce with TCEP (final concentration ~5 mM) for 20 minutes at 37°C. Alkylate with CAA (final concentration ~15 mM) in the dark for 15 minutes. Digest with trypsin/Lys-C mix at 1:30 (w/w) enzyme-to-protein ratio overnight at 37°C [29].
Peptide Cleanup: Acidify with TFA to stop digestion. Desalt using C18 columns and elute with 70% acetonitrile, 0.2% formic acid [29].
Integration with Downstream Ubiquitinomics Workflows
The compatibility of SDC lysis with advanced mass spectrometry techniques positions it as the optimal choice for modern ubiquitinome profiling. When combined with data-independent acquisition (DIA) mass spectrometry and neural network-based data processing, SDC-based sample preparation has enabled the identification of over 70,000 ubiquitinated peptides in single MS runs—more than tripling the identification numbers achievable with data-dependent acquisition (DDA) approaches [28]. This dramatic improvement in coverage is coupled with significant gains in quantitative precision, with median coefficients of variation (CVs) for quantified K-GG peptides of approximately 10% [28].
The streamlined SDC workflow integrates seamlessly with K-ε-GG antibody-based enrichment, which is central to ubiquitin remnant profiling. This antibody specifically recognizes the di-glycine–lysine residue left on modified peptides after trypsin digestion of ubiquitinated proteins [31]. The higher enrichment specificity achieved with SDC lysis [28] ensures more efficient utilization of these valuable immunoenrichment reagents. Furthermore, SDC's compatibility with low sample input requirements (20-times less protein input compared to some urea-based methods) [28] makes it particularly valuable for precious clinical samples or applications where material is limited.
For research focusing on K-ε-GG remnant antibody specificity, the reduced method-induced artifacts with SDC lysis provide cleaner input material for enrichment. The absence of di-carbamidomethylation of lysine residues—which can mimic ubiquitin remnant K-GG peptides in terms of mass tag added (both 114.0249 Da) [28]—minimizes false positives and ensures that antibody enrichment truly targets ubiquitin-derived modifications.
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of optimized ubiquitinome profiling requires specific reagents and materials. The following table details key research solutions for SDC-based ubiquitinomics:
Table 2: Essential Research Reagents for SDC-Based Ubiquitinome Analysis
| Reagent/Material | Function in Workflow | Specific Application Notes |
|---|---|---|
| Sodium Deoxycholate (SDC) | Denaturing detergent for efficient protein extraction | Use at 1% concentration in Tris-HCl buffer (pH 8.5); precipitates at low pH for easy removal [28] [29] |
| Chloroacetamide (CAA) | Alkylating agent for cysteine modification | Preferred over iodoacetamide to prevent di-carbamidomethylation of lysine residues [28] |
| K-ε-GG Antibody | Immunoaffinity enrichment of ubiquitinated peptides | Recognizes di-glycine-lysine remnant after trypsin digestion; critical for ubiquitinome specificity [31] |
| Trypsin/Lys-C Mix | Proteolytic digestion of proteins | Generates K-ε-GG remnant peptides; 1:30 enzyme-to-protein ratio recommended [29] |
| C18 Desalting Columns | Peptide cleanup and buffer exchange | Remove SDC and other contaminants prior to MS analysis [29] |
| Tris(2-carboxyethyl)phosphine (TCEP) | Reducing agent for disulfide bonds | More stable than DTT; use at 5 mM concentration for 20 minutes at 37°C [29] |
| Trifluoroacetic Acid (TFA) | Acidification for SDC precipitation and digestion termination | 0.5-1% final concentration effectively precipitates SDC for easy removal [29] |
The comprehensive comparison between SDC and urea lysis methods demonstrates a clear advantage for SDC-based protocols in ubiquitinome studies. With 38% higher identification rates of K-GG peptides, improved reproducibility, reduced sample input requirements, and minimal method-induced artifacts [28], SDC emerges as the superior choice for researchers seeking maximum ubiquitinome coverage. These performance advantages hold significant implications for drug development professionals investigating ubiquitin signaling in disease mechanisms or evaluating compounds targeting deubiquitinases and ubiquitin ligases.
The integration of optimized SDC lysis with advanced DIA mass spectrometry and neural network-based data processing represents the current state-of-the-art in ubiquitinomics [28]. This powerful combination enables unprecedented depth and quantitative precision in profiling ubiquitination dynamics, providing researchers with a robust platform for exploring the multifaceted roles of ubiquitin signaling in cellular regulation. As research continues to refine K-ε-GG remnant antibody specificity and applications, the foundation of optimized sample preparation with SDC ensures that resulting data reflects biological reality rather than methodological artifacts.
For the scientific community focused on ubiquitin biology, adopting SDC-based lysis protocols can substantially enhance research outcomes, providing more comprehensive and reliable ubiquitinome datasets. This advancement directly supports the broader thesis of improving antibody specificity and methodological rigor in ubiquitin remnant profiling, ultimately accelerating our understanding of this crucial regulatory system in health and disease.
In the field of proteomics, particularly in the study of post-translational modifications such as ubiquitination, achieving sufficient depth of coverage remains a significant analytical challenge. The low stoichiometry of endogenous ubiquitination presents a major hurdle for mass spectrometry-based detection, necessitating highly effective fractionation and enrichment strategies [32]. Basic reversed-phase chromatography has emerged as a powerful technique to address this challenge, enabling researchers to significantly boost the number of identifications in ubiquitylome studies. This technique serves as a critical sample preparation step prior to immunoaffinity enrichment using K-ε-GG remnant antibodies, which specifically recognize the diglycine remnant left on ubiquitinated peptides after tryptic digestion [5] [32].
The evaluation of K-ε-GG antibody specificity is paramount for obtaining reliable ubiquitylome data, and the effectiveness of this evaluation is heavily dependent on the prefractionation strategy employed. As researchers strive to identify thousands of ubiquitination sites from complex biological samples, basic reversed-phase chromatography has proven instrumental in reducing sample complexity and mitigating the effects of dynamic range that often limit proteomic analyses [5]. This guide provides a comprehensive comparison of basic reversed-phase chromatography with alternative fractionation techniques, supported by experimental data and detailed methodologies relevant to ubiquitination research.
Theoretical Foundation of Basic Reversed-Phase Chromatography
Principles of Reversed-Phase Chromatography
Reversed-phase chromatography (RPC) operates on the principle of hydrophobic interactions, where a non-polar stationary phase and a polar mobile phase are used to separate compounds based on their hydrophobicity [33] [34]. This configuration reverses the traditional normal-phase chromatography approach, which uses a polar stationary phase and non-polar mobile phase [33]. In RPC, hydrophobic molecules in the polar mobile phase tend to adsorb to the hydrophobic stationary phase, while hydrophilic molecules pass through the column and are eluted first [33]. The more hydrophobic the molecule, the more strongly it will bind to the stationary phase, requiring a higher concentration of organic solvent for elution [33] [35].
The separation mechanism involves the partitioning of analytes between the mobile phase and the hydrophobic stationary phase, with retention governed by the hydrophobic effect [34]. This effect drives the association of non-polar regions of analytes with the stationary phase to minimize their exposure to the aqueous mobile phase. The extent of retention depends on the surface chemistry of the stationary phase, the hydrophobicity of the analyte, and the composition of the mobile phase [33].
Basic pH Specificity and Mechanism
Basic reversed-phase chromatography utilizes the same fundamental principles as conventional RPC but operates at elevated pH (typically pH 10) using mobile phases modified with ammonium formate or similar basic buffers [5]. This basic environment alters the ionization state of acidic residues on peptides, suppressing their negative charges and increasing their hydrophobicity [5]. The result is a different selectivity profile compared to acidic RPC, providing an orthogonal separation mechanism that complements traditional acidic pH separations.
At basic pH, the ionization of silanol groups on silica-based stationary phases is also enhanced, potentially introducing secondary interactions that can improve separation selectivity for certain compound classes [33]. This altered selectivity is particularly beneficial for complex peptide mixtures, as it distributes analytes differently across the separation window, thereby increasing peak capacity and resolution when combined with acidic RPC in a two-dimensional separation scheme [5].
Figure 1: Basic Reversed-Phase Workflow for Ubiquitylome Analysis. This diagram illustrates the key steps in using basic reversed-phase chromatography as a front-end fractionation technique prior to K-ε-GG immunoaffinity enrichment and LC-MS/MS analysis.
Comparative Analysis of Fractionation Techniques
Performance Comparison of Fractionation Methods
Various fractionation techniques have been employed in proteomic workflows to reduce sample complexity and enhance proteome coverage. The table below provides a systematic comparison of basic reversed-phase chromatography with alternative fractionation methods, with particular emphasis on their utility in ubiquitylome analyses.
Table 1: Comparison of Fractionation Techniques for Ubiquitylome Analysis
| Fractionation Technique | Mechanism of Separation | Compatibility with K-ε-GG Workflow | Typical Number of Ubiquitination Sites Identified | Key Advantages | Major Limitations |
|---|---|---|---|---|---|
| Basic Reversed-Phase Chromatography | Hydrophobicity at high pH | Excellent | ~20,000 sites (with optimized workflow) [5] | High peak capacity, orthogonal to acidic RPC, compatible with MS | Requires high pH stable columns, additional sample handling |
| Acidic Reversed-Phase Chromatography | Hydrophobicity at low pH | Good | ~5,000-10,000 sites (varies with sample input) [32] | MS-compatible, high resolution | Limited orthogonality to MS separation |
| Strong Cation Exchange (SCX) | Electrostatic interactions | Moderate | ~5,000 sites (with 2-3 mg protein input) [32] | Orthogonal to RPC, high capacity | Incompatible with direct MS analysis, requires desalting |
| Hydrophilic Interaction Liquid Chromatography (HILIC) | Polarity | Good | Limited data available | Retains hydrophilic compounds, orthogonal to RPC | Suffers from decreased chromatographic resolution, long equilibration times [36] |
Orthogonality in Separation Mechanisms
The power of basic reversed-phase chromatography lies in its orthogonality to both the subsequent immunoaffinity enrichment and the final LC-MS/MS separation. While traditional acidic RPC separates peptides based on hydrophobicity at low pH, basic RPC provides a different selectivity profile by suppressing the negative charges on acidic amino acids [5]. This orthogonality is crucial for comprehensive ubiquitylome analysis, as it distributes the ubiquitinated peptides across multiple fractions based on different physicochemical properties, thereby reducing the complexity of each fraction subjected to K-ε-GG enrichment.
Studies have demonstrated that the combination of basic reversed-phase fractionation with K-ε-GG antibody enrichment enables the identification of approximately 20,000 distinct endogenous ubiquitination sites from moderate protein input amounts (5 mg per SILAC channel) [5]. This represents a significant improvement over earlier approaches that required substantially more protein input or multiple experimental replicates to achieve comparable coverage.
Experimental Protocols for Basic Reversed-Phase Chromatography
Detailed Methodology for Basic Reversed-Phase Fractionation
The following protocol has been optimized for ubiquitylome analysis and is adapted from established methodologies in the field [5]:
Column Preparation:
- Utilize a Zorbax 300 Extend-C18 column (9.4 × 250 mm, 300 Å, 5 μm) or equivalent that is stable at high pH.
- Condition the column with initial mobile phase (2% acetonitrile, 5 mM ammonium formate, pH 10) at a flow rate of 3 mL/min for at least 30 minutes to establish a stable baseline [5].
Sample Preparation:
- Desalt digested peptide samples using C18 solid-phase extraction cartridges.
- Resuspend the dried peptide sample in 1.8 mL of basic reversed-phase solvent A (2% acetonitrile, 5 mM ammonium formate, pH 10) [5].
- Centrifuge at 20,000 × g for 10 minutes to remove any insoluble material that could clog the column.
Chromatographic Separation:
- Inject the sample and run the following gradient at a flow rate of 3 mL/min:
- Initial increase to 8% solvent B (90% acetonitrile, 5 mM ammonium formate, pH 10) at 1.1% B/min
- 38-minute linear gradient from 8% B to 27% B (0.5% B/min)
- Successive ramps to 31% B (1% B/min), 39% B (0.5% B/min), and 60% B (3% B/min) [5]
- Collect 80 fractions across the entire separation window.
Fraction Pooling:
- Pool fractions in a non-contiguous manner into 8-12 final fractions for subsequent K-ε-GG enrichment.
- For example, combine fractions 1, 9, 17, 25, 33, 41, 49, 57, 65, and 73 to create the first pooled fraction, and continue this pattern for the remaining fractions [5].
- Dry the pooled fractions completely using a SpeedVac concentrator before proceeding to immunoaffinity enrichment.
K-ε-GG Immunoaffinity Enrichment Protocol
Following basic reversed-phase fractionation, perform K-ε-GG enrichment using the following optimized protocol:
Antibody Cross-Linking (Optional but Recommended):
- Wash anti-K-ε-GG antibody beads three times with 1 mL of 100 mM sodium borate, pH 9.0.
- Resuspend beads in 1 mL of 20 mM dimethyl pimelimidate (DMP) and incubate at room temperature for 30 minutes with rotation.
- Wash beads twice with 1 mL of 200 mM ethanolamine, pH 8.0, then incubate in 1 mL of 200 mM ethanolamine for 2 hours at 4°C with rotation [5].
- Wash cross-linked beads three times with ice-cold IAP buffer (50 mM MOPS, pH 7.2, 10 mM sodium phosphate, 50 mM NaCl) and store at 4°C until use.
Peptide Enrichment:
- Resuspend dried basic RP fractions in 1.5 mL of IAP buffer.
- Incubate with cross-linked anti-K-ε-GG antibody beads (31 μg antibody per fraction) for 1 hour at 4°C on a rotating platform [5].
- Wash beads four times with 1.5 mL of ice-cold PBS.
- Elute K-ε-GG peptides with two applications of 50 μL of 0.15% trifluoroacetic acid.
- Desalt eluted peptides using C18 StageTips prior to LC-MS/MS analysis [5].
Research Reagent Solutions
Successful implementation of basic reversed-phase chromatography for ubiquitylome analysis requires specific reagents and materials. The following table details essential research reagent solutions and their functions in the experimental workflow.
Table 2: Essential Research Reagents for Basic Reversed-Phase Ubiquitylome Analysis
| Reagent/Material | Function | Specifications | Alternative Options |
|---|---|---|---|
| Anti-K-ε-GG Antibody | Immunoaffinity enrichment of ubiquitinated peptides | Rabbit polyclonal, recognizes diglycine remnant on Lys residues [6] | Commercial kits (PTMScan Ubiquitin Remnant Motif Kit) [5] |
| High pH Stable C18 Column | Basic reversed-phase separation | Zorbax 300 Extend-C18 (9.4 × 250 mm, 300 Å, 5 μm) [5] | Other high pH stable C18 columns with similar dimensions |
| Ammonium Formate, pH 10 | Mobile phase buffer for basic RPC | 5 mM in water, pH adjusted with ammonium hydroxide [5] | Ammonium bicarbonate, ammonium acetate (less ideal) |
| Dimethyl Pimelimidate (DMP) | Antibody cross-linking reagent | 20 mM in sodium borate buffer, pH 9.0 [5] | Other homobifunctional cross-linkers (e.g., DSS, BS3) |
| IAP Buffer | Immunoaffinity enrichment buffer | 50 mM MOPS, pH 7.2, 10 mM sodium phosphate, 50 mM NaCl [5] | Commercial IAP buffers or similar physiological buffers |
| C18 StageTips | Micro-solid phase extraction for sample clean-up | Empore C18 disks or commercial StageTips [5] | C18 ZipTips, in-house prepared C18 columns |
Quantitative Assessment of Technique Performance
Enhancement of Ubiquitination Site Identifications
The implementation of basic reversed-phase chromatography as a front-end fractionation technique has demonstrated remarkable improvements in ubiquitylome coverage. Research shows that this approach enables the identification of approximately 20,000 distinct endogenous ubiquitination sites in a single SILAC experiment using moderate amounts of protein input (5 mg per SILAC channel) [5]. This represents a substantial advancement compared to earlier methodologies that identified only several hundred ubiquitination sites without sophisticated fractionation [32].
The quantitative improvement can be attributed to several factors: (1) reduced sample complexity in each fraction, minimizing ion suppression effects during MS analysis; (2) improved enrichment efficiency due to lower competition for antibody binding sites; and (3) enhanced detection of low-abundance ubiquitinated peptides that would otherwise be masked by more abundant species in unfractionated samples.
Comparison with Alternative Enrichment Strategies
Recent methodological advances have introduced alternative approaches for ubiquitinated protein enrichment, including the use of ubiquitin pan nanobodies that recognize all ubiquitin chains and monoubiquitination [8]. While these methods show promise, the diGly immunoaffinity approach preceded by basic reversed-phase fractionation remains the gold standard for comprehensive ubiquitylome analysis.
A comparative study evaluating both methods demonstrated that the diGly approach enabled the detection of 108 potential RNF111 substrates, while the ubiquitin pan nanobody method identified 52 potential substrates, including key targets in the TGF-β signaling pathway such as SKI and SKIL [8]. This highlights the continued importance of basic reversed-phase fractionation combined with K-ε-GG antibodies for maximal coverage in ubiquitylome studies.
Figure 2: Evolution of Ubiquitination Site Identifications with Advanced Fractionation. This diagram illustrates the progressive improvement in ubiquitination site identifications achieved with advancing fractionation methodologies, culminating in basic reversed-phase chromatography combined with K-ε-GG enrichment.
Technical Considerations and Optimization Strategies
Critical Parameters for Method Optimization
Successful implementation of basic reversed-phase chromatography for ubiquitylome analysis requires careful attention to several technical parameters:
pH Control and Buffer Selection:
- Maintain precise pH control at 10.0 ± 0.1 using freshly prepared ammonium formate buffer.
- Use high-purity ammonia solution for pH adjustment to minimize MS contamination.
- Verify pH using a properly calibrated pH meter, as small variations can significantly alter retention times and selectivity.
Gradient Optimization:
- Adjust gradient slopes to distribute peptides evenly across the separation window.
- Implement shallower gradients in regions with high peptide density to maintain resolution.
- Balance separation time with practical considerations for sample throughput.
Fractionation Scheme:
- Determine the optimal number of fractions based on sample complexity and analytical goals.
- For discovery-level analyses aiming for maximum coverage, 8-12 fractions typically provide an optimal balance between depth of coverage and instrument time.
- For targeted analyses, fewer fractions may be sufficient, focusing on regions where peptides of interest elute.
Troubleshooting Common Issues
Poor Chromatographic Resolution:
- Cause: Column degradation or contamination from previous samples.
- Solution: Implement rigorous column cleaning protocols with high organic washes (e.g., 80% acetonitrile) between runs. Replace column if performance does not improve.
Low Ubiquitinated Peptide Recovery:
- Cause: Inefficient elution from antibody beads or peptide loss during clean-up steps.
- Solution: Optimize elution conditions (e.g., increased TFA concentration, alternative acids) and ensure proper conditioning of StageTips or other clean-up devices.
High Non-Specific Binding:
- Cause: Inadequate blocking of antibody beads or insufficient washing.
- Solution: Extend blocking time with ethanolamine and increase number of wash steps with IAP buffer or PBS.
Basic reversed-phase chromatography represents a powerful fractionation technique that significantly enhances the depth and coverage of ubiquitylome analyses when combined with K-ε-GG immunoaffinity enrichment. The method's strength lies in its orthogonality to both the subsequent enrichment step and the final LC-MS/MS separation, effectively reducing sample complexity and mitigating dynamic range limitations that often plague ubiquitination studies.
The experimental data presented in this guide demonstrates that basic reversed-phase fractionation enables the identification of approximately 20,000 ubiquitination sites from moderate protein input, representing a substantial improvement over alternative fractionation methods. While the technique requires careful method optimization and specific reagents, its implementation provides researchers with a robust tool for comprehensive ubiquitylome characterization.
As the field continues to evolve, basic reversed-phase chromatography will likely remain a cornerstone technique for ubiquitination research, particularly when integrated with emerging enrichment strategies and advancing mass spectrometry technologies. The continued refinement of this approach promises to further our understanding of the critical regulatory roles played by protein ubiquitination in health and disease.
Post-translational modification by ubiquitin represents a crucial regulatory mechanism in eukaryotic cells, controlling processes ranging from protein degradation to signal transduction [2]. When proteins are modified by ubiquitin and subsequently digested with the protease trypsin, a unique signature remnant is left behind: a diglycine (Gly-Gly) motif attached via an isopeptide bond to the epsilon amino group of a lysine residue (K-ε-GG) [2] [37]. This K-ε-GG remnant serves as a specific molecular tag for previously ubiquitinated proteins, enabling researchers to identify ubiquitination sites on a proteome-wide scale [2].
The development of high-specificity antibodies targeting the K-ε-GG motif has revolutionized the study of ubiquitination, transforming our ability to detect endogenous ubiquitination sites by mass spectrometry [5]. Before the commercial availability of these antibodies, proteomics experiments were limited to identifying only several hundred ubiquitination sites, severely constraining global ubiquitination studies [5]. Modern immunoaffinity enrichment techniques using these antibodies now enable researchers to routinely identify and quantify thousands of distinct endogenous ubiquitination sites in a single experiment, providing unprecedented insights into the ubiquitin regulatory network [5] [2].
Key Reagents and Materials for K-ε-GG Enrichment
Table 1: Essential research reagents for K-ε-GG immunoaffinity enrichment
| Reagent Category | Specific Examples | Function/Purpose |
|---|---|---|
| Primary Antibodies | PTMScan Ubiquitin Remnant Motif (K-ε-GG) Antibody (Cell Signaling Technology); Rabbit Polyclonal Anti-K-ε-GG (Thermo Fisher, PA5-120707) | Specifically binds to the diglycine remnant attached to lysine residues for immunoaffinity capture |
| Chromatography Supports | Protein A Agarose Beads; Magnetic Bead Versions (PTMScan HS) | Solid support for antibody immobilization |
| Buffers & Solutions | IAP Buffer (50 mM MOPS, pH 7.2, 10 mM sodium phosphate, 50 mM NaCl); Urea Lysis Buffer (8 M urea); Cross-linking Reagents (Dimethyl Pimelimidate - DMP) | Maintain optimal binding conditions and sample integrity |
| Sample Preparation | Sequencing Grade Trypsin; C18 Solid-Phase Extraction Cartridges; Basic Reversed-Phase Chromatography Columns | Protein digestion and peptide fractionation prior to enrichment |
| Elution & Cleaning | 0.15% Trifluoroacetic Acid (TFA); C18 StageTips | Elute captured peptides from antibodies and desalt for MS analysis |
Commercially Available K-ε-GG Enrichment Solutions
The commercial landscape for K-ε-GG enrichment reagents includes several well-established options, primarily based on immunoaffinity principles. The PTMScan Technology platform from Cell Signaling Technology represents one of the most comprehensive systems, employing a proprietary methodology for peptide enrichment by immunoprecipitation using specific bead-conjugated antibodies in conjunction with LC-MS/MS for quantitative profiling of ubiquitination sites [37]. This platform includes both standard and high-sensitivity magnetic bead formats, with the PTMScan HS Ubiquitin/SUMO Remnant Motif Kit offering enhanced performance characteristics [37].
Alternative antibody sources include Thermo Fisher Scientific's rabbit polyclonal antibody targeting the Ubiquitin Remnant Motif (K-epsilon-GG), validated for use in applications such as ELISA and Western Blot, though its application in enrichment protocols may require additional optimization [6]. When selecting commercial options, researchers must consider whether the product includes the antibody alone or as part of a complete kit with optimized buffers and protocols, as this significantly impacts experimental reproducibility and ease of implementation.
Step-by-Step K-ε-GG Enrichment Protocol
Sample Preparation and Digestion
Begin with cell lysis in a denaturing buffer containing 8 M urea, 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, and appropriate protease inhibitors to preserve ubiquitination signatures while ensuring complete disruption of cellular structures [5]. Following protein quantification using a BCA assay, reduce proteins with 5 mM dithiothreitol (DTT) for 45 minutes at room temperature, then alkylate with 10 mM iodoacetamide for 30 minutes in the dark [5]. Dilute the urea concentration to 2 M with 50 mM Tris-HCl (pH 7.5) before digesting overnight at 25°C with sequencing-grade trypsin at an enzyme-to-substrate ratio of 1:50 [5]. After digestion, acidify samples with formic acid and desalt using C18 solid-phase extraction cartridges [5].
Peptide Pre-Fractionation
For deep ubiquitinome coverage, offline basic reversed-phase fractionation is recommended prior to immunoaffinity enrichment. Resuspend desalted peptides in basic RP solvent A (2% MeCN, 5 mM ammonium formate, pH 10) and separate using a C18 column with a 64-minute gradient from 8% to 60% solvent B (90% MeCN, 5 mM ammonium formate, pH 10) [5]. Collect 80 fractions which can be pooled non-contiguously into 8-12 fractions to reduce sample complexity while maintaining resolution [5]. This fractionation strategy significantly enhances the number of unique ubiquitination sites identified by reducing sample complexity during subsequent LC-MS/MS analysis.
Antibody Cross-Linking
Antibody cross-linking is a critical step that significantly improves enrichment performance by minimizing antibody leaching during immunoprecipitation. Wash anti-K-ε-GG antibody beads three times with 100 mM sodium borate (pH 9.0), then resuspend in 1 mL of 20 mM dimethyl pimelimidate (DMP) and incubate at room temperature for 30 minutes with rotation [5] [38]. Wash cross-linked beads twice with 200 mM ethanolamine (pH 8.0), then block in 1 mL of 200 mM ethanolamine for 2 hours at 4°C with rotation [5] [38]. Finally, wash beads three times with ice-cold IAP buffer before resuspending in the same buffer for immediate use or storage at 4°C [5].
Immunoaffinity Enrichment
Resuspend dried peptide fractions in 1.5 mL of IAP buffer and incubate with cross-linked anti-K-ε-GG antibody beads for 1-2 hours on a rotating unit at 4°C [5] [38]. The antibody-to-peptide ratio should be optimized for specific experimental needs, with typical amounts ranging from 31 μg to 250 μg of antibody per enrichment [5]. Following incubation, wash beads four times with 1.5 mL of ice-cold PBS to remove non-specifically bound peptides [5]. Elute captured K-ε-GG peptides with two applications of 50 μL of 0.15% trifluoroacetic acid (TFA), then desalt using C18 StageTips before LC-MS/MS analysis [5] [38].
Performance Optimization and Critical Parameters
Antibody and Peptide Input Optimization
Systematic optimization of antibody-to-peptide ratios is essential for achieving maximum enrichment efficiency. Research indicates that using 31 μg of antibody with peptide inputs derived from 5 mg of protein per SILAC channel enables routine quantification of approximately 20,000 nonredundant K-ε-GG sites in a single experiment, representing a significant improvement over earlier methodologies [5]. Titration experiments comparing antibody amounts of 31, 62, 125, and 250 μg have demonstrated that lower antibody amounts can be sufficient when combined with optimized sample preparation and fractionation protocols, potentially reducing reagent costs without compromising results [5].
Cross-Linking for Enhanced Performance
The implementation of antibody cross-linking protocols using dimethyl pimelimidate (DMP) represents a crucial technical advancement that substantially improves enrichment performance. Cross-linking minimizes antibody leaching during immunoprecipitation and subsequent washing steps, thereby reducing background signal and improving the specificity of K-ε-GG peptide capture [5] [38]. This modification is particularly important for experiments requiring multiple washes or extended processing times, as it maintains the integrity of the antibody-bead conjugation throughout the enrichment procedure.
Performance Comparison and Experimental Data
Table 2: Performance metrics for K-ε-GG enrichment methods
| Performance Parameter | Traditional Methods (Pre-antibody) | Modern K-ε-GG Immunoaffinity | Optimized with Cross-linking |
|---|---|---|---|
| Ubiquitination Sites Identified | Few hundred sites per experiment [5] | ~5,000 sites with standard protocols [5] | ~20,000 sites with optimized workflow [5] |
| Protein Input Requirements | High (up to ~35 mg) [5] | Moderate (5-10 mg) [5] | Low (5 mg) per SILAC channel [5] |
| Antibody Usage | Not applicable | Higher amounts typically required | 31 μg per enrichment [5] |
| Technical Replicates Needed | Multiple replicates for comprehensive coverage [5] | Reduced replicates | Single experiment sufficient for deep coverage [5] |
| Key Technical Features | Epitope-tagged ubiquitin and IMAC resins [2] | Direct immunoaffinity capture | Antibody cross-linking and advanced fractionation [5] |
The performance advantages of modern K-ε-GG immunoaffinity enrichment are demonstrated through quantitative proteomic experiments. In one optimized workflow, researchers achieved identification of approximately 20,000 distinct endogenous ubiquitination sites from a triple-encoded SILAC experiment starting with just 5 mg of protein input per channel [5]. This represents a ten-fold improvement over previously published methods and highlights how protocol refinements dramatically enhance experimental outcomes [5].
The specificity and sensitivity of K-ε-GG antibodies have been rigorously validated through multiple experimental approaches. These antibodies recognize the diglycine remnant of ubiquitin left on protein substrates after trypsin digestion, enabling highly selective enrichment of ubiquitinated peptides from complex trypsin-digested cell samples [37]. When combined with liquid chromatography tandem mass spectrometry (LC-MS/MS), this approach provides quantitative profiles of hundreds to over a thousand non-redundant ubiquitinated sequences, offering a comprehensive view of the ubiquitin-modified proteome without preconceived biases about where these modifications occur [37].
Troubleshooting and Technical Considerations
Common Challenges and Solutions
- Low Recovery of Ubiquitinated Peptides: Ensure proper antibody cross-linking and optimize incubation times. Cross-linking with DMP significantly improves performance by preventing antibody leaching [5] [38].
- High Background Signal: Implement more stringent washing conditions with ice-cold PBS and consider adjusting antibody-to-peptide ratios. Non-contiguous fraction pooling can also reduce complexity [5].
- Incomplete Digestion: Verify trypsin activity and use sequencing-grade enzyme at proper ratios. Ensure complete reduction and alkylation before digestion [5].
- Sample Loss During Processing: Use C18 StageTips for desalting and concentration, as these minimize sample loss compared to traditional columns [5] [38].
Quality Control Measures
- Buffer Compatibility: Always use recommended IAP buffer (50 mM MOPS, pH 7.2, 10 mM sodium phosphate, 50 mM NaCl) for immunoprecipitation to maintain optimal binding conditions [5] [37].
- Proper Storage: Antibody beads supplied in IAP buffer with 50% glycerol should be stored at -20°C without aliquoting to maintain stability [37].
- Process Validation: Include positive controls when possible and validate method performance using standardized samples to ensure consistency between experiments.
The refinement of K-ε-GG immunoaffinity enrichment protocols represents a significant advancement in ubiquitin research, enabling comprehensive analysis of ubiquitination sites at an unprecedented scale. Through optimization of antibody cross-linking, peptide input ratios, and pre-fractionation strategies, researchers can now routinely identify and quantify tens of thousands of ubiquitination sites from modest protein inputs [5]. These technical improvements have transformed our ability to study the ubiquitin proteome, providing insights into regulatory mechanisms underlying numerous cellular processes and disease states.
As the field continues to evolve, further enhancements in antibody specificity, enrichment efficiency, and integration with complementary methods will undoubtedly expand the applications and capabilities of K-ε-GG enrichment. The standardized protocols and performance comparisons presented here provide researchers with a solid foundation for implementing these powerful techniques in their own investigations of the ubiquitin system.
In modern proteomics and metabolomics, the choice of mass spectrometry (MS) acquisition mode is pivotal, fundamentally determining the depth, sensitivity, and reproducibility of analytical results. The evolution from traditional Data-Dependent Acquisition (DDA) to the increasingly prominent Data-Independent Acquisition (DIA) represents a paradigm shift, particularly for applications requiring high quantitative accuracy across large sample cohorts. Within the specific context of evaluating the specificity of different K-ε-GG remnant antibodies—a critical focus in ubiquitinomics and post-translational modification (PTM) research—the quantitative performance of the MS method is paramount. DIA-MS emerges as a powerful solution, strategically designed to overcome the limitations of its predecessors by offering an unparalleled combination of broad proteome coverage and exceptional reproducibility. This guide provides a objective comparison of DIA-MS against other common acquisition modes, supported by experimental data, to inform method selection for advanced research and drug development.
The core principle differentiating MS acquisition modes lies in how precursor ions are selected for fragmentation and analysis. Data-Dependent Acquisition (DDA), a long-standing cornerstone of discovery proteomics, operates by selecting the most abundant precursor ions from an MS1 survey scan for subsequent MS/MS fragmentation. This intensity-based selection is inherently stochastic, leading to inconsistencies in peptide identification across replicate runs, especially for low-abundance species. In contrast, Data-Independent Acquisition (DIA) eliminates this stochasticity by systematically fragmenting all ions within predefined, sequential mass-to-charge (m/z) windows across the full scanning range. This deterministic acquisition strategy ensures that every detectable ion is fragmented and recorded, producing comprehensive and highly reproducible datasets [39] [40]. Targeted modes like Multiple Reaction Monitoring (MRM) and Parallel Reaction Monitoring (PRM) offer the highest sensitivity and precision for quantifying predefined targets but lack the discovery potential of untargeted methods due to their limited scale [39].
Comparative Analysis of MS Acquisition Modes
Principles and Workflows
A deep understanding of the operational workflows for each MS acquisition mode is essential for appreciating their comparative strengths and limitations. The following diagram illustrates the core decision logic and steps involved in DDA, DIA, and MRM/PRM.
Direct Performance Comparison: DDA vs. DIA
Robust experimental studies have systematically benchmarked the performance of DIA against traditional DDA, consistently highlighting DIA's superiority in quantitative applications. One such study employed a gold-standard spike-in sample set with known concentrations of proteins in a complex background, allowing for precise assessment of quantification accuracy, reproducibility, and specificity. The key findings demonstrated that DIA is superior to DDA in quantification reproducibility, specificity, and accuracy. Furthermore, DIA notably outperformed DDA in the quantification of low-abundance protein amounts, a critical factor when analyzing rare PTMs or signaling proteins [41].
A separate multi-laboratory assessment involving 11 sites worldwide provided further compelling evidence for DIA's robustness. In this extensive inter-laboratory study, SWATH-MS (a specific implementation of DIA) consistently detected and quantified over 4,000 proteins from HEK293 cells with high reproducibility across all sites. The study concluded that the acquisition of reproducible quantitative proteomics data by multiple labs is achievable with SWATH-MS, establishing it as a reproducible method for large-scale protein quantification [42]. This level of inter-lab consistency is exceptionally difficult to achieve with the stochastic precursor selection of DDA.
The table below summarizes the key characteristics of all four major acquisition modes based on experimental findings:
Table 1: Comprehensive Comparison of Mass Spectrometry Acquisition Modes
| Feature | DDA (Data-Dependent Acquisition) | DIA (Data-Independent Acquisition) | MRM (Multiple Reaction Monitoring) | PRM (Parallel Reaction Monitoring) |
|---|---|---|---|---|
| Acquisition Type | Untargeted | Untargeted | Targeted | Targeted |
| Principle | Selects top N intense precursors | Fragments all ions in sequential m/z windows | Monitors predefined precursor-product ion transitions | Monitors predefined precursors; records all fragments |
| Quantitative Reproducibility | Moderate (stochastic sampling) | High (deterministic sampling) [41] [42] | Very High (gold standard) | Very High |
| Proteome Coverage | Broad, but incomplete | Very Broad and Deep [40] | Limited to ~100 targets/injection | Limited to ~100 targets/injection |
| Sensitivity | Good for high-abundance ions | Excellent, especially for low-abundance analytes [41] | Excellent for targeted ions | Excellent for targeted ions |
| Best Applications | Discovery proteomics, spectral library generation | Large-scale quantitative studies, biomarker discovery [39] [40] | Validated clinical assays, pharmacokinetics | Targeted protein verification, PTM analysis |
The advantages of DIA extend beyond proteomics into metabolomics. A comparative study of untargeted metabolomics methods found that DIA demonstrated superior reproducibility, with a coefficient of variance (CV) of 10% across detected compounds over three measurements, compared to 17% for DDA. DIA also detected and identified the highest number of metabolic features and showed better consistency in compound identification [43].
Experimental Protocols for DIA-MS
Detailed DIA-MS Workflow
Implementing a robust DIA-MS workflow involves several critical steps, from sample preparation to computational data extraction. The protocol below is adapted from methodologies used in the cited comparative studies [41] [42].
Sample Preparation:
- Extract proteins from cells or tissue of interest (e.g., C2C12 cell lysate as used in benchmark studies [41]).
- Perform reduction, alkylation, and enzymatic digestion (typically with trypsin) to create a peptide mixture.
- Desalt the resulting peptides using solid-phase extraction (e.g., C18 cartridges) to remove interfering salts.
Spectral Library Generation (Library-Based Analysis):
- Fractionation: To build a comprehensive spectral library, a portion of the sample can be pre-fractionated using high-pH reversed-phase chromatography or SDS-PAGE to maximize peptide coverage [41].
- DDA Analysis: Analyze the fractionated and non-fractionated samples using a DDA method on a high-resolution mass spectrometer. This generates a collection of MS/MS spectra that map peptide sequences to their fragmentation patterns.
- Database Search: Process the DDA files with search engines (e.g., Proteome Discoverer, Spectronaut Pulsar) against a relevant protein sequence database to identify peptides and proteins, creating a project-specific spectral library.
DIA Data Acquisition:
- Inject the experimental samples for DIA analysis.
- The mass spectrometer is programmed to cycle through a series of consecutive, isolated m/z windows (e.g., 64 variable windows covering 400-1000 m/z) throughout the entire chromatographic elution [42].
- In each cycle, the instrument performs one full MS1 scan, followed by MS2 scans for all isolation windows. All product ions within each window are recorded by a high-resolution mass analyzer (Orbitrap or time-of-flight).
DIA Data Analysis:
- The complex DIA data files are processed using peptide-centric software such as Spectronaut, OpenSWATH, or DIA-NN [42].
- The software uses the spectral library to query for specific peptides in the DIA data, extracting and integrating the chromatographic peaks for their corresponding fragment ions.
- Peptide and protein abundances are quantified based on the extracted ion chromatograms of the fragments, and statistical analysis is performed to identify differentially abundant proteins, controlling for false discovery rate (FDR).
Key Research Reagent Solutions
The following table details essential materials and reagents used in a typical DIA-MS workflow, as inferred from the experimental methodologies in the search results.
Table 2: Research Reagent Solutions for DIA-MS Workflows
| Item | Function in the Workflow | Examples / Specifications |
|---|---|---|
| Cell Lysate | Complex biological background for spike-in studies or direct analysis. | C2C12 (mouse myoblast) cells [41]. |
| Stable Isotope-Labeled Standard (SIS) Peptides | Internal standards for absolute quantification and assessment of quantification accuracy. | Synthetically produced peptides with heavy isotopes [42]. |
| Spectral Library | Reference database for peptide identification from multiplexed DIA MS2 spectra. | Generated from project-specific DDA runs with or without pre-fractionation [41] [40]. |
| Trypsin | Proteolytic enzyme for digesting proteins into peptides for MS analysis. | Sequencing-grade modified trypsin. |
| LC-MS Grade Solvents | High-purity solvents for liquid chromatography to minimize background noise. | Water, acetonitrile, formic acid. |
| Chromatographic Column | Separates peptides prior to mass spectrometry analysis to reduce complexity. | C18 reversed-phase column (e.g., 30 cm x 75 µm) [42]. |
| Data Analysis Software | Extracts and quantifies peptide signals from complex DIA data. | Spectronaut, OpenSWATH, DIA-NN [41] [42]. |
Visualization of DIA-MS Data Analysis Logic
The process of analyzing DIA data is distinct from that of DDA. It relies on a targeted data extraction strategy to deconvolute the highly complex, multiplexed MS2 spectra. The following diagram outlines the core logical workflow for this process.
The compelling body of evidence from controlled spike-in studies and large-scale multi-laboratory assessments firmly establishes Data-Independent Acquisition (DIA) as a next-generation MS methodology that successfully bridges the gap between discovery-oriented and targeted proteomics. When evaluating critical reagents such as K-ε-GG remnant antibodies, where specificity and quantitative accuracy are non-negotiable, DIA-MS provides a compelling solution. Its deterministic acquisition strategy delivers unprecedented reproducibility across technical replicates and laboratories, while its deep, systematic coverage of the proteome ensures that low-abundance modified peptides are not missed. By leveraging DIA-MS, researchers and drug development professionals can generate highly robust, data-rich datasets, thereby enhancing the reliability of findings in PTM research and accelerating the path to biomarker discovery and therapeutic innovation.
Maximizing Specificity and Yield: Critical Troubleshooting for K-ε-GG Enrichment
Antibody cross-linking represents a foundational technique in molecular biology for creating robust, reusable affinity reagents. This process involves covalently attaching antibodies to solid supports such as magnetic beads or chromatography resins, preventing antibody co-elution with the target antigen during immunoaffinity purification. For research focused on post-translational modifications, particularly ubiquitination studies using K-ε-GG remnant antibodies, effective cross-linking is paramount for obtaining high-quality data with minimal background interference. The strategic selection of cross-linking protocols directly influences key performance metrics including signal-to-noise ratio, antibody longevity, and reagent consumption, making it a critical consideration for researchers and drug development professionals seeking to optimize experimental outcomes while conserving valuable antibodies.
The fundamental challenge in immunoprecipitation workflows lies in balancing efficient target antigen recovery against reduced non-specific binding. Traditional methods that rely on non-covalently attached antibodies often suffer from antibody leakage, leading to substantial background interference in downstream analyses like mass spectrometry and 2D-PAGE [44]. Cross-linking addresses this issue but introduces its own complexities, as the choice of cross-linker chemistry and conjugation strategy significantly impacts antibody orientation, binding capacity, and ultimately, the specificity of the enrichment process—particularly crucial when working with low-abundance targets such as ubiquitinated peptides [44] [5].
Comparative Analysis of Cross-Linking Reagents and Methods
Conventional Cross-Linking Reagents
The most commonly used cross-linkers in laboratory practice are amine-reactive compounds that target primary amines in lysine residues and protein N-termini. Among these, dimethyl pimelimidate (DMP) and bis(sulfosuccinimidyl) suberate (BS3) have been extensively characterized for immunoprecipitation applications, each offering distinct advantages and limitations for K-ε-GG antibody immobilization.
Table 1: Performance Comparison of Common Cross-Linking Reagents
| Cross-Linker | Reaction Specificity | Non-Specific Binding | Target Yield | Ig Leakage | Cost Considerations |
|---|---|---|---|---|---|
| DMP | Primary ε-amines of lysines at pH 9-10 [44] | Higher non-specific binding [44] | Higher overall target yield [44] | Minimal leakage after cross-linking [44] | Lower cost (~30× less than BS3) [44] |
| BS3 | Primary amines with additional reactivity toward tyrosines, serines, and threonines [44] | Significantly lower non-specific binding [44] | Reduced target signal compared to DMP [44] | Complete elimination of leakage [44] | Higher cost, but can be used at reduced concentrations [44] |
| Preactivation Method | Oriented immobilization via Fc region [45] | Not reported | Maximum binding capacity [45] | Not applicable | No antibody modification required [45] |
As evidenced in Table 1, BS3 generally produces superior results in terms of reduced non-specific binding and complete elimination of immunoglobulin leakage, though potentially at the cost of reduced target yield. DMP, while generating higher background, may be preferable when maximizing recovery of low-abundance targets is the primary objective. Cost-conscious laboratories can consider using BS3 at reduced concentrations (50% of manufacturer recommendations) while maintaining excellent western blot signal-to-noise ratios [44].
Advanced Orientation-Control Strategies
Recent methodological advances have focused on controlling antibody orientation during immobilization to maximize antigen-binding capacity. Conventional cross-linking approaches often result in random antibody orientation, potentially obscuring antigen-binding domains and reducing overall reagent efficiency [45].
The preactivation cross-linking method represents a significant innovation in this area. This two-step approach first activates Protein A or Protein G with "slow" cross-linkers, removes excess reagent, and then adds antibodies for oriented conjugation via their Fc regions [45]. This strategy ensures optimal presentation of antigen-binding domains while still achieving covalent stabilization, resulting in signals "much higher than a traditional method" according to comparative studies [45].
For K-ε-GG antibody applications specifically, research demonstrates that cross-linking with 20 mM dimethyl pimelimidate in 100 mM sodium borate (pH 9.0) for 30 minutes at room temperature effectively stabilizes the antibody-bead complex while maintaining specificity for ubiquitin remnant motifs [5]. This protocol has enabled the identification of approximately 20,000 distinct endogenous ubiquitination sites from moderate protein inputs, highlighting the efficacy of optimized cross-linking for high-sensitivity applications [5].
Experimental Protocols for Cross-Linking Optimization
Standard BS3 and DMP Cross-Linking Procedures
BS3 Cross-Linking Protocol (for Protein A/G Beads):
- Wash antibody-bound Protein A/G beads with PBS (pH 7.4) to remove unbound antibody.
- Resuspend beads in freshly prepared BS3 solution (1-5 mM in PBS, pH 7.4).
- Incubate with rotation for 30 minutes at room temperature.
- Quench the reaction by adding Tris-HCl (pH 7.5) to a final concentration of 20-50 mM and incubate for 15 minutes.
- Wash beads thoroughly with appropriate buffer before use or storage.
- Note: Cost-saving can be achieved by reducing BS3 concentration to 50% of manufacturer's recommendation without significant performance loss [44].
DMP Cross-Linking Protocol (for Anti-K-ε-GG Antibodies):
- Wash antibody-coated beads three times with 1 mL of 100 mM sodium borate (pH 9.0).
- Resuspend beads in 1 mL of 20 mM dimethyl pimelimidate (DMP) in borate buffer.
- Incubate at room temperature for 30 minutes with rotation.
- Wash twice with 1 mL of 200 mM ethanolamine (pH 8.0).
- Resuspend in 1 mL of 200 mM ethanolamine and incubate for 2 hours at 4°C to block residual reactive groups.
- Wash three times with ice-cold IAP buffer (50 mM MOPS, pH 7.2, 10 mM sodium phosphate, 50 mM NaCl) [5].
- Store cross-linked antibodies at 4°C in IAP buffer until use.
Preactivation Cross-Linking for Oriented Immobilization
Protein A Preactivation Method:
- Immobilize Protein A on the desired solid support using standard methods.
- Wash with coupling buffer (e.g., phosphate buffer, pH 7-8).
- Activate with a "slow" cross-linker such as succinimidyl iodoacetate (SIA) or sulfo-SIAB (1-5 mM in DMSO, then diluted in buffer).
- Incubate for 30-60 minutes at room temperature.
- Remove excess cross-linker by thorough washing.
- Add antibody without purification and incubate for 1-2 hours for conjugation.
- Block any remaining active sites with ethanolamine or Tris buffer [45].
This oriented immobilization approach is particularly valuable for antibodies where maintaining binding capacity is crucial, and can be applied even to antibodies in crude cell culture supernatants or raw sera without purification [45].
Impact on K-ε-GG Ubiquitin Remnant Research
The application of optimized cross-linking protocols has proven particularly transformative in ubiquitin proteomics, where detection of endogenous ubiquitination sites requires exceptional specificity. The commercial availability of anti-di-glycine remnant (K-ε-GG) antibodies has revolutionized ubiquitination site detection by mass spectrometry, but achieving comprehensive coverage of the ubiquitinome demands meticulous attention to reagent preparation [5] [6].
Table 2: Performance Metrics in K-ε-GG Enrichment with Cross-Linking Optimization
| Parameter | Without Cross-Linking | DMP Cross-Linking | BS3 Cross-Linking | Preactivation Method |
|---|---|---|---|---|
| Non-specific binding | High [44] | Increased [44] | Significantly reduced [44] | Not reported |
| Antibody leakage | Significant [44] | Minimal [44] | None detected [44] | Not applicable |
| K-ε-GG site identification | Limited by background | Improved | Optimal for high-specificity applications | Not reported |
| Reusability | Limited | Good | Excellent | Not reported |
Implementation of cross-linked anti-K-ε-GG antibodies in conjunction with off-line basic reversed-phase fractionation has enabled researchers to routinely identify and quantify approximately 20,000 distinct endogenous ubiquitination sites from just 5 mg of protein input—representing a 10-fold improvement over earlier methods [5]. This dramatic enhancement in performance underscores the critical importance of cross-linking strategies in maximizing the value of precious research samples and reagents.
For K-ε-GG antibodies specifically, the recommended workflow incorporates cross-linked antibodies at approximately 31 μg per enrichment reaction, with peptide inputs of 1-5 mg, followed by desalting using C18 StageTips prior to LC-MS/MS analysis [5]. This optimized approach minimizes background while conserving antibody reagents across multiple uses.
Research Reagent Solutions for Cross-Linking Workflows
Table 3: Essential Reagents for Antibody Cross-Linking Protocols
| Reagent | Function | Application Notes |
|---|---|---|
| Protein A/G Magnetic Beads | Antibody capture platform | Paramagnetic beads show lower non-specific binding than Sepharose/agarose [44] |
| Dimethyl Pimelimidate (DMP) | Amine-reactive cross-linker | Preferred for maximum target yield; cost-effective [44] [5] |
| BS3 | Homobifunctional NHS ester cross-linker | Superior for reducing background; used at reduced concentrations to conserve reagents [44] |
| Sodium Borate Buffer (pH 8-9) | Alkaline cross-linking buffer | Optimal for DMP reactions [5] |
| Anti-K-ε-GG Antibodies | Ubiquitin remnant enrichment | Specificity verified for ubiquitination site mapping [6] [46] |
| IAP Buffer | Immunoaffinity purification buffer | Maintains antibody stability during K-ε-GG enrichment [5] |
| DSPE-PEG2000-DBCO | Lipophilic conjugate | For nanobody-assisted antibody capture on LNPs [47] |
Visualization of Cross-Linking Workflows
Comparative Cross-Linking Methods Diagram
K-ε-GG Antibody Cross-Linking Workflow
Antibody cross-linking protocols offer powerful strategies for reducing background interference and conserving valuable reagents in ubiquitin proteomics and other affinity-based applications. The selection of appropriate cross-linking methodology should be guided by specific research objectives: DMP for maximum target recovery, BS3 for superior signal-to-noise ratio, and preactivation methods for optimal antibody orientation when binding capacity is paramount.
For K-ε-GG ubiquitin remnant research specifically, the implementation of cross-linked antibodies has demonstrated remarkable improvements in experimental outcomes, enabling identification of thousands of ubiquitination sites from modest protein inputs. The strategic integration of these protocols—coupled with appropriate fractionation and detection methods—provides researchers with a robust framework for advancing our understanding of ubiquitin-mediated cellular processes while maximizing the utility of precious antibody reagents across multiple experimental cycles.
In the field of ubiquitin proteomics, the anti-K-ε-GG antibody has revolutionized the detection of endogenous protein ubiquitination sites by mass spectrometry, enabling researchers to identify thousands of ubiquitination sites and understand their regulatory roles in cellular processes [5] [48]. However, the unpredictable nature of usable input amounts from samples and undefined antibody titer presents significant challenges in experimental variability and reproducibility [49]. Input titration—the systematic optimization of protein input and antibody quantity—has emerged as a crucial methodology for balancing experimental cost, depth of coverage, and quantitative accuracy.
The commercialization of antibodies recognizing lysine residues modified with a di-glycine remnant (K-ε-GG) has dramatically transformed ubiquitination studies, enabling identification of >50,000 ubiquitylation sites in human cells [48]. Despite these successes, achieving comprehensive ubiquitome coverage often requires substantial protein input (up to ~35 mg) and multiple experimental replicates, creating cost and practical barriers for many laboratories [5]. Through systematic titration approaches, researchers can now achieve routine quantification of approximately 20,000 distinct endogenous ubiquitination sites from moderate protein inputs (5 mg), representing a 10-fold improvement over earlier methods [5] [50].
Experimental Protocols for Titration Optimization
Antibody Amount Titration Methodology
The foundation of effective input titration lies in systematically varying antibody amounts while holding peptide input constant. The refined protocol involves several critical steps:
Cell Culture and Lysis: Jurkat E6-1 cells are grown in RPMI 1640 media with appropriate supplements. For SILAC experiments, cells are cultured in custom RPMI media deficient in l-arginine and l-lysine with stable isotope-labeled amino acids [5]. Cells are treated with proteasome inhibitors (e.g., MG-132) prior to harvest to enhance ubiquitinated peptide recovery. Lysis is performed in denaturing conditions using buffer containing 8 M urea, 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, and protease inhibitors [5].
Protein Digestion and Preparation: Protein concentrations are estimated using bicinchoninic acid (BCA) assay. Proteins are reduced with dithiothreitol (DTT), alkylated with iodoacetamide, and digested overnight with sequencing-grade trypsin at an enzyme-to-substrate ratio of 1:50 [5]. Following digestion, samples are acidified with formic acid and desalted using C18 solid-phase extraction cartridges [5].
Antibody Titration Protocol: The key titration step involves incubating constant peptide amounts with varying antibody quantities (e.g., 31, 62, 125, or 250 μg) [5]. Cross-linked anti-K-ε-GG antibody beads are prepared by washing with sodium borate buffer and incubating with dimethyl pimelimidate (DMP) for cross-linking [5]. Peptide fractions are resuspended in immunoprecipitation buffer (IAP: 50 mM MOPS, pH 7.2, 10 mM sodium phosphate, 50 mM NaCl) and incubated with cross-linked antibody beads for 1 hour at 4°C [5].
Enrichment and Analysis: Following incubation, antibody beads are washed extensively with ice-cold PBS, and K-ε-GG peptides are eluted using 0.15% trifluoroacetic acid (TFA) [5]. Eluted peptides are desalted using C18 StageTips prior to LC-MS/MS analysis [5].
Protein Input Normalization Strategy
Building on principles from chromatin immunoprecipitation, researchers have adapted a quick, DNA-based measurement method to quantify chromatin input, enabling accurate normalization of antibody amounts to the optimal titer in individual immunoprecipitation reactions [49]. This approach involves:
Rapid Chromatin Quantification: DNA content of chromatin input (DNAchrom) is directly measured from 0.2% of total input using a Qubit assay, a high-sensitivity method specific to double-stranded DNA [49]. This measurement typically takes less than 5 minutes and shows strong linear correlation (R² = 0.99) with purified DNA amounts [49].
Titer Determination: Chromatin input is prepared from fixed cells and DNAchrom is measured. Optimal antibody titer is determined by performing immunoprecipitations with different antibody amounts ranging from 0.05 to 10.0 μg per 10 μg of DNAchrom [49]. The optimal range is identified by measuring both yield (DNA amount after IP divided by input DNA amount) and fold enrichment (% enrichment of positive genomic loci versus negative loci) [49].
Titer Normalization: The optimal antibody-to-DNAchrom ratio is defined as "titer 1" (T1), which represents the ideal balance between signal and background [49]. This normalized approach ensures consistent immunoprecipitation conditions across samples with varying input amounts.
Quantitative Comparison of Titration Strategies
Table 1: Comparison of Antibody Titration Approaches for K-ε-GG Enrichment
| Titration Parameter | Traditional Approach | Optimized Titration | Automated Workflow |
|---|---|---|---|
| Antibody Amount | Not systematically optimized | 31 μg antibody per enrichment | Magnetic bead-conjugated K-ε-GG |
| Protein Input | Up to 35 mg per sample | 5 mg protein per SILAC channel | 500 μg input per TMT sample |
| Identified Sites | Several hundred | ~20,000 ubiquitination sites | ~20,000 sites (TMT10-plex) |
| Processing Time | Manual, time-consuming | ~2 hours manual processing | ~2 hours automated processing |
| Cost Efficiency | High reagent consumption | 10-fold improvement | High throughput (96 samples/day) |
| Reproducibility | Variable between experiments | Improved consistency | Significantly reduced variability |
Table 2: Impact of Antibody Titer on Enrichment Specificity and Yield
| Antibody Amount (μg) | CHIP Yield (%) | Fold Enrichment | Specificity Assessment | Recommended Applications |
|---|---|---|---|---|
| 0.05 | 0.1% | 202-fold | High specificity, low yield | Limited material applications |
| 0.25 | 0.5% | 150-fold | Optimal balance | Standard quantitative studies |
| 1.0 | 2.1% | 80-fold | Moderate specificity | High-throughput screening |
| 5.0 | 4.3% | 35-fold | Reduced specificity | Preliminary screening only |
| 10.0 | 5.4% | 18-fold | High background, low specificity | Not recommended |
Research Reagent Solutions for Ubiquitin Proteomics
Table 3: Essential Research Reagents for K-ε-GG Ubiquitin Proteomics
| Reagent/Category | Specific Examples | Function in Workflow | Optimization Tips |
|---|---|---|---|
| K-ε-GG Antibodies | PTMScan Ubiquitin Remnant Motif Kit (Cell Signaling #5562); Ubiquitin Remnant Motif Antibody (Thermo Fisher PA5-120707) [51] [52] | Enrichment of di-glycine modified peptides from trypsin-digested ubiquitylated proteins | Cross-linking to beads prevents antibody leakage; titration essential for optimal performance |
| Cell Lysis Reagents | 8M Urea, 50mM Tris-HCl, 150mM NaCl, Complete Protease Inhibitor, 5mM N-Ethylmaleimide (NEM) [48] | Effective protein extraction while preserving ubiquitination status | NEM inhibits deubiquitinases; urea denatures proteins effectively |
| Digestion Enzymes | LysC (Wako), Sequencing-grade Trypsin (Sigma) [5] [48] | Generation of appropriate peptide fragments with C-terminal K-ε-GG remnants | Two-step digestion (LysC followed by trypsin) improves efficiency |
| Chromatography Media | Sep-Pak tC18 reverse phase column (Waters) [5]; Basic RP fractionation with Zorbax 300 Extend-C18 [5] | Peptide desalting and pre-fractionation to reduce complexity | Basic pH fractionation improves separation of hydrophobic peptides |
| Automation Platforms | Magnetic bead-conjugated K-ε-GG antibody (HS mag anti-K-ε-GG) [50] | High-throughput processing with improved reproducibility | Enables processing of 96 samples in a single day with reduced variability |
Workflow Visualization: Titration-Optimized Ubiquitin Proteomics
Titration Optimized K-ε-GG Workflow - This diagram illustrates the integrated titration approach for ubiquitin proteomics, highlighting the critical antibody optimization steps in red that enable balanced cost-effectiveness and experimental depth.
Advanced Applications and Methodological Comparisons
The UbiFast method represents a significant advancement in ubiquitin proteomics by combining TMT multiplexing with K-ε-GG enrichment. This approach employs on-antibody TMT labeling, where K-ε-GG peptides are labeled with TMT reagents while still bound to the anti-K-ε-GG antibody, allowing the NHS-ester group of the TMT reagent to react with peptide N-terminal amine groups and ε-amine groups of lysine residues, but not the primary amine of the di-glycyl remnant [50]. Automation of this method using magnetic bead-conjugated K-ε-GG antibody (HS mag anti-K-ε-GG) and a magnetic particle processor has further enhanced reproducibility and throughput, enabling processing of up to 96 samples in a single day with significantly reduced variability across experimental replicates [50].
Comparative studies have evaluated alternative enrichment strategies, including ubiquitin pan nanobody approaches that recognize all ubiquitin chains and monoubiquitylation [8]. While the diGlycine remnant immunoprecipitation with K-ε-GG antibody successfully identified SKIL ubiquitylation among 108 potential RNF111 substrates, the ubiquitin pan nanobody method detected 52 potential substrates including SKI, SKIL, and RNF111, demonstrating complementary strengths for comprehensive ubiquitylome analysis [8].
The principles of titration-based normalization extend beyond ubiquitin proteomics to other applications, including Western blotting. The recently developed titration-based Western blotting (t-WB) method uses serial dilutions of protein samples to generate regression curves for precise protein quantification, eliminating the need for housekeeping protein normalization and resolving biases inherent to classical protocols [53]. This approach demonstrates how titration methodologies continue to evolve and transform experimental design across multiple domains of biological research.
Input titration represents a fundamental methodology for balancing experimental cost, depth of coverage, and quantitative accuracy in ubiquitin proteomics. The systematic optimization of antibody quantity and protein input enables researchers to achieve comprehensive ubiquitome coverage from moderate protein inputs, with identified sites increasing from several hundred to approximately 20,000 in a single experiment [5] [50]. The development of automated workflows using magnetic bead-conjugated antibodies further enhances reproducibility and throughput while maintaining cost-effectiveness [50].
As ubiquitin proteomics continues to evolve toward more complex sample types including primary tissues and clinical specimens [50], the implementation of robust titration strategies becomes increasingly critical. By adopting these optimized approaches, researchers can maximize the return on investment for precious research samples while generating high-quality, reproducible data that advances our understanding of ubiquitin signaling in health and disease.
In the rigorous field of ubiquitin proteomics, the specificity of an antibody is paramount. For researchers employing anti-K-ε-GG remnant antibodies to map ubiquitination sites, two of the most persistent adversaries are contamination by keratin and nonspecific binding. These factors introduce significant noise, obscuring true biological signals and compromising data quality. A systematic evaluation of experimental protocols and reagents is essential to combat these issues. This guide objectively compares the performance of different methodological approaches, supported by experimental data, to provide scientists and drug development professionals with strategies for achieving cleaner, more reliable results in their ubiquitin profiling studies.
Experimental Protocols for Specific Enrichment
The following detailed methodologies are adapted from refined workflows designed for large-scale ubiquitination site identification.
Optimized Sample Preparation and Digestion
This protocol is designed to minimize sample degradation and contamination during the initial processing stages [5].
- Cell Lysis: Use a denaturing lysis buffer (8 M Urea, 50 mM Tris-HCl, pH 7.5, 150 mM NaCl) supplemented with protease inhibitors (e.g., 2 μg/mL aprotinin, 10 μg/mL leupeptin, 1 mM PMSF) and deubiquitinase inhibitors (e.g., 50 μM PR-619). Keep samples at 4°C during lysis.
- Protein Handling: Estimate protein concentration using a BCA assay. Use a high input (e.g., 5-15 mg of protein) to ensure sufficient target material for subsequent enrichment.
- Reduction and Alkylation: Reduce proteins with 5 mM dithiothreitol (DTT) for 45 minutes at room temperature. Subsequently, alkylate with 10 mM iodoacetamide for 30 minutes in the dark.
- Trypsin Digestion: Dilute the lysate to 2 M urea with 50 mM Tris-HCl, pH 7.5. Digest with sequencing-grade trypsin at an enzyme-to-substrate ratio of 1:50, overnight at 25°C.
- Peptide Desalting: Acidify peptides with formic acid (FA) and desalt using a C18 solid-phase extraction cartridge (e.g., a 500-mg tC18 Sep-Pak). Condition the cartridge with methanol, acetonitrile, and trifluoroacetic acid (TFA) before loading the sample. Wash with 0.1% TFA and elute with 50% acetonitrile, 0.1% FA. Dry the eluate completely.
Off-line Basic Reversed-Phase Fractionation
Prior to immunoprecipitation, pre-fractionation significantly reduces sample complexity and interference [5].
- Column: Use a Zorbax 300 Extend-C18 column (9.4 x 250 mm, 300 Å, 5 μm) on an HPLC system.
- Solvents: Use Solvent A (2% Acetonitrile, 5 mM Ammonium Formate, pH 10) and Solvent B (90% Acetonitrile, 5 mM Ammonium Formate, pH 10).
- Gradient: Employ a 64-minute linear gradient from 8% B to 60% B at a flow rate of 3 mL/min.
- Fraction Pooling: Collect 80 fractions and pool them in a non-contiguous manner into 8 super-fractions (e.g., combine fractions 1, 9, 17, ... 73). This pooling strategy reduces variance and improves LC-MS/MS analysis.
Antibody Cross-linking for Reduced Nonspecific Binding
Cross-linking the antibody to the solid support prevents antibody leaching and co-elution, a major source of nonspecific MS signals [5].
- Wash: Wash anti-K-ε-GG antibody beads three times with 100 mM sodium borate, pH 9.0.
- Cross-link: Resuspend the beads in 20 mM dimethyl pimelimidate (DMP) in borate buffer and incubate for 30 minutes at room temperature with rotation.
- Quench: Wash beads twice with 200 mM ethanolamine, pH 8.0, and then incubate in the same buffer for 2 hours at 4°C to block unreacted sites.
- Storage: Wash the cross-linked beads three times with ice-cold Immunoaffinity Purification (IAP) Buffer (50 mM MOPS, pH 7.2, 10 mM Sodium Phosphate, 50 mM NaCl). Resuspend in IAP buffer and store at 4°C.
Immunoaffinity Enrichment and Elution
This is the core step for specifically isolating K-ε-GG-containing peptides [5] [54].
- Resuspension and Incubation: Resuspend each dried peptide fraction in 1.5 mL of IAP buffer. Incubate with the cross-linked anti-K-ε-GG antibody beads (e.g., 31 μg antibody per fraction) for 1 hour at 4°C with rotation.
- Washing: After incubation, wash the beads four times with 1.5 mL of ice-cold PBS to remove nonspecifically bound peptides thoroughly.
- Elution: Elute the captured K-ε-GG peptides with two applications of 50 μL of 0.15% TFA.
- Desalting: Desalt the eluted peptides using C18 StageTips or similar micro-scale solid-phase extraction before MS analysis.
Quantitative Comparison of Methodological Performance
The following tables summarize key experimental data that highlight the impact of different strategies on enrichment specificity and yield.
Table 1: Impact of Antibody Input on K-ε-GG Peptide Identification
This data, derived from systematic titration experiments, helps optimize reagent use and minimize nonspecific binding by avoiding antibody over-saturation [5].
| Antibody Input (μg) | Peptide Input (mg) | Number of Unique K-ε-GG Sites Identified | Key Observations |
|---|---|---|---|
| 31 μg | 5 mg | ~20,000 | Optimal efficiency; cost-effective for high yields. |
| 62 μg | 5 mg | Data not specified | Diminishing returns; minimal gain in sites identified. |
| 125 μg | 5 mg | Data not specified | Increased reagent cost with no significant benefit. |
| 250 μg | 5 mg | Data not specified | Potential for increased nonspecific binding; not cost-effective. |
Table 2: Comparison of K-ε-GG Enrichment Workflow Components
This table compares the performance of different protocol components, illustrating how a refined workflow combats contamination and improves data quality [5].
| Workflow Component | Standard Protocol | Refined / Optimized Protocol | Impact on Specificity and Yield |
|---|---|---|---|
| Protein Input | Lower input (e.g., 1-2 mg) | High input (5-15 mg) | Increases depth of coverage, enabling identification of low-abundance sites. |
| Antibody Use | Direct use from vial | Cross-linked to beads | Reduces antibody-derived contaminants in MS; improves signal-to-noise. |
| Pre-Enrichment Fractionation | None or simple pooling | High-pH reverse-phase with non-contiguous pooling | Reduces sample complexity, minimizing competitive binding and interference. |
| Reported Site Identification | Several hundred to ~5,000 sites | ~20,000 distinct sites in a single experiment | 10-fold improvement demonstrates superior specificity and comprehensiveness. |
Visualizing the Optimized Workflow
The following diagram illustrates the refined experimental workflow, highlighting key steps that are critical for minimizing contamination and nonspecific binding.
The Scientist's Toolkit: Essential Research Reagent Solutions
The following reagents are critical for implementing the high-specificity workflows described above.
Table 3: Key Reagents for Optimized K-ε-GG Enrichment
| Research Reagent | Function / Role in Specificity | Examples / Specifications |
|---|---|---|
| Anti-K-ε-GG Antibody | Core reagent for immunoaffinity purification; specificity dictates enrichment quality. | Rabbit monoclonal from CST PTMScan Kit [54]; also available as polyclonal from Thermo Fisher [6]. |
| Protease & DUB Inhibitors | Preserve native ubiquitination state by preventing protein degradation and ubiquitin removal. | Aprotinin, Leupeptin, PMSF, PR-619 [5]. |
| Cross-linking Reagent (DMP) | Covalently immobilizes antibody to beads, preventing leaching and contaminating IgG peptides. | Dimethyl Pimelimidate [5]. |
| IAP Buffer | Provides optimal pH and ionic strength for antibody-antigen interaction during enrichment. | 50 mM MOPS, pH 7.2, 10 mM Sodium Phosphate, 50 mM NaCl [5] [54]. |
| Fractionation Column | Reduces sample complexity pre-enrichment, minimizing competitive binding. | Zorbax 300 Extend-C18, 9.4 x 250 mm [5]. |
| C18 Solid Phase | For desalting and cleaning peptide samples before and after enrichment. | tC18 Sep-Pak cartridges, C18 StageTips [5]. |
Discussion and Comparative Analysis
The data clearly demonstrate that a holistic approach to workflow refinement is more effective than focusing on a single variable. While the choice of antibody is fundamental, its performance is heavily influenced by sample preparation, cross-linking, and pre-fractionation. The cross-linking of antibodies directly addresses the problem of nonspecific binding and contamination by preventing the co-elution of antibody fragments, which are a major source of keratin and other interfering signals in mass spectrometry. Furthermore, the use of high-pH reversed-phase fractionation prior to enrichment reduces the complexity of the peptide mixture presented to the antibody, thereby minimizing competitive binding and improving the isolation of true K-ε-GG peptides.
Commercial offerings, such as the PTMScan Ubiquitin Remnant Motif Kit from Cell Signaling Technology, provide a standardized system that incorporates these principles, offering researchers a vetted path to high-quality data [54]. The market also includes polyclonal alternatives from suppliers like Thermo Fisher Scientific, which can be suitable for various applications [6] [55]. The critical takeaway is that the strategies of cross-linking, high-input protein, and sophisticated fractionation, when combined, enable the routine identification of over 20,000 ubiquitination sites—a benchmark that is unattainable with older, less refined methods [5] [23]. By systematically implementing these protocols, researchers can significantly mitigate the challenges of keratin and nonspecific binding, ensuring that their conclusions about ubiquitin biology are built upon a foundation of robust and specific proteomic data.
The study of protein ubiquitination has been revolutionized by antibodies targeting the di-glycine (K-ε-GG) remnant left on trypsinized peptides. However, a significant challenge persists: the same K-ε-GG motif is also generated from modifications by ubiquitin-like modifiers (UBLs), including SUMO (Small Ubiquitin-Related Modifier). This cross-reactivity can compromise data interpretation, as detected changes may reflect alterations in SUMOylation rather than genuine ubiquitination events. The specificity of K-ε-GG antibodies is therefore paramount for accurate ubiquitinome profiling, particularly in studies investigating complex cellular signaling and disease mechanisms where both modifications play distinct roles. This guide objectively compares the performance of different K-ε-GG antibody platforms and enrichment strategies in addressing this challenge, providing researchers with experimental data and methodologies to ensure specificity in their ubiquitination studies.
Fundamental Differences Between Ubiquitin and SUMO Remnants
Although tryptic digestion of both ubiquitinated and SUMOylated proteins produces peptides with a K-ε-GG motif, key biochemical differences allow for discrimination.
- Ubiquitin Remnant: Trypsin cleaves after the C-terminal glycine-76 of ubiquitin, leaving a Gly-Gly moiety attached via an isopeptide bond to the ε-amino group of a lysine residue in the substrate protein. The resulting peptide is characterized by a K-ε-GG remnant where the ubiquitin-derived sequence ends with two glycine residues.
- SUMO Remnant: SUMO proteins (SUMO-1, -2, -3) possess a short C-terminal sequence following the Gly-Gly motif. Trypsin cleavage after the Gly-Gly motif is inefficient. Instead, wild-type alpha-lytic protease (WaLP), a serine endopeptidase, is required to cleave at the carboxyl terminal side of alanine, serine, threonine, or valine to properly expose the C-terminal Gly-Gly motif of SUMO [56]. Therefore, the predicted SUMO sequence of the studied organism must contain AGG, SGG, TGG, or VGG at the C-terminus to be reactive with K-ε-GG antibodies after WaLP digestion [56].
This enzymatic distinction forms the basis for experimental protocols designed to ensure specificity for ubiquitin-derived peptides.
Experimental Protocols for Validating Specificity
Protease-Based Specificity Validation
A critical method to confirm antibody specificity for ubiquitin over SUMO involves differential protease digestion.
Protocol: WaLP Digestion for SUMO-Specific Cleavage [56]
- Divide Sample: Split the trypsin-digested peptide sample into two aliquots.
- Treatment Aliquot: Incubate one aliquot with wild-type alpha-lytic protease (WaLP). WaLP specifically cleaves at the C-terminal side of A, S, T, or V, generating the K-ε-GG remnant from SUMO-modified peptides that were inaccessible to trypsin.
- Control Aliquot: Incubate the other aliquot with a buffer-only control.
- Parallel Enrichment: Enrich both aliquots using the same K-ε-GG antibody protocol.
- Data Analysis: Identify peptides that are significantly enriched in the WaLP-treated sample compared to the control. These peptides originate from SUMOylated proteins. True ubiquitin-derived peptides will be present in both samples, as their K-ε-GG motif is already exposed by trypsin.
Automated Immunoprecipitation with Magnetic Beads
Automation improves reproducibility and reduces variability in enrichment, which is crucial for consistent specificity.
Protocol: Automated UbiFast with Magnetic Beads [50]
- Lysis and Digestion: Lyse cells or tissue in 8 M urea buffer supplemented with protease and deubiquitinase inhibitors (e.g., PR-619). Reduce disulfide bonds with dithiothreitol (DTT), alkylate with iodoacetamide (IAA), and digest proteins sequentially with Lys-C and trypsin.
- Peptide Cleanup: Desalt peptides using C18 solid-phase extraction cartridges.
- Automated Immunoprecipitation: Use a magnetic particle processor for all subsequent steps. Resuspend dried peptide aliquots (e.g., 500 μg) in Immunoaffinity Purification (IAP) buffer. Incubate with magnetic bead-conjugated K-ε-GG antibody (e.g., HS mag anti-K-ε-GG). This bead-conjugated format eliminates the need for a cross-linking step and prevents antibody co-elution.
- Washing: Perform multiple automated wash steps with IAP buffer to remove non-specifically bound peptides.
- On-Bead TMT Labeling: While peptides are bound to the antibody, label them with Tandem Mass Tag (TMT) reagents. This "on-antibody" labeling strategy is crucial because it prevents the TMT label from modifying the N-terminus of the di-glycine remnant, which would block antibody recognition [50].
- Peptide Elution and Analysis: Combine labeled samples, elute peptides from the beads, and analyze by LC-MS/MS.
Table 1: Key Reagents for Specific Ubiquitin Remnant Enrichment
| Reagent / Kit | Type | Key Feature | Role in Ensuring Specificity |
|---|---|---|---|
| PTMScan HS Ubiquitin/SUMO Kit [56] | Antibody Kit | Includes specificity information for SUMO | Acknowledges cross-reactivity and provides guidance for SUMO motif verification (AGG, SGG, TGG, VGG). |
| HS mag anti-K-ε-GG [50] | Magnetic Bead-Conjugated Antibody | Irreversibly coupled to magnetic beads | Enables automation, reduces manual variability, and improves reproducibility of enrichment, indirectly supporting more consistent specificity. |
| Wild-type Alpha-Lytic Protease (WaLP) [56] | Serine Endopeptidase | Cleaves C-terminal to A, S, T, V | Critical tool for experimental validation and discrimination of SUMO-derived K-ε-GG peptides. |
| Tandem Mass Tag (TMT) [50] | Isobaric Label Reagent | Enables multiplexing | On-antibody labeling preserves the K-ε-GG motif for antibody binding during enrichment. |
Performance and Specificity Data Comparison
Direct comparative studies provide the most objective data for evaluating antibody performance and specificity.
Table 2: Quantitative Performance Comparison of Enrichment Methods
| Parameter | Traditional Agarose Beads (Manual) [50] | Magnetic Beads (Automated UbiFast) [50] | Implication for Specificity |
|---|---|---|---|
| Total Ubiquitylation Sites Identified | ~10,000-15,000 (from TMT10-plex) | ~20,000 (from TMT10-plex, 500 μg input) | Greater depth suggests more comprehensive coverage, potentially including lower-abundance true ubiquitin sites. |
| Processing Time | ~6-8 hours (manual) | ~2 hours (automated for 10-plex) | Reduced processing time minimizes peptide degradation and non-specific interactions. |
| Reproducibility (Inter-replicate Variability) | Higher variability | Significantly reduced variability | Improved reproducibility increases confidence in the quantitative data for true ubiquitination events. |
| Throughput | Limited by manual steps | Up to 96 samples per day | Enables larger experimental designs with more biological replicates, strengthening statistical conclusions. |
| SUMO Cross-Reactivity | Not explicitly quantified; requires WaLP validation. | Not explicitly quantified; requires WaLP validation. | Both platforms require enzymatic validation (WaLP digest) to empirically determine and account for SUMO cross-reactivity. |
The data demonstrates that the magnetic bead-based automated method offers substantial improvements in depth, speed, and reproducibility over traditional agarose-based manual methods [50]. While neither study explicitly quantifies the percentage of captured peptides that are SUMO-derived, the requirement for WaLP digestion in specificity protocols confirms that cross-reactivity is a real concern for both platforms [56].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Ubiquitinome Analysis
| Item | Function | Specificity Consideration |
|---|---|---|
| K-ε-GG Motif Antibody | Immunoaffinity enrichment of ubiquitin and UBL remnant peptides. | The primary tool; choice between agarose-coupled and magnetic bead-conjugated formats affects reproducibility and throughput. |
| Deubiquitinase Inhibitors (e.g., PR-619) | Preserves ubiquitin conjugates during cell lysis by inhibiting deubiquitinating enzymes (DUBs) [50] [22]. | Prevents the loss of the K-ε-GG motif during sample preparation, ensuring a accurate representation of the ubiquitinome. |
| Wild-type Alpha-Lytic Protease (WaLP) | Specifically cleaves SUMO proteins to expose their C-terminal Gly-Gly motif [56]. | The essential reagent for experimentally discriminating SUMO-derived K-ε-GG peptides from ubiquitin-derived ones. |
| Magnetic Bead Processor | Automates wash and elution steps in immunoprecipitation. | Dramatically reduces variability and hands-on time, leading to more robust and reproducible enrichments [50]. |
| Tandem Mass Tags (TMT) | Enables multiplexed quantitative analysis of up to 18 samples simultaneously. | The "on-antibody" labeling protocol is critical to prevent tags from blocking antibody binding to the K-ε-GG motif [50]. |
Achieving specificity for ubiquitin over SUMO using K-ε-GG antibodies is a critical, addressable challenge. The move towards automated platforms with magnetic bead-conjugated antibodies represents a significant advancement, offering superior reproducibility and depth of coverage. However, the core issue of SUMO cross-reactivity remains and must be actively managed.
For researchers designing ubiquitinome studies, the following evidence-based recommendations are provided:
- Validate Empirically: Do not assume absolute specificity. Incorporate WaLP digestion protocols to experimentally determine the proportion of SUMO-derived peptides in your specific system.
- Embrace Automation: Where possible, adopt magnetic bead-based automated enrichment to minimize variability and enhance the robustness of your quantitative data.
- Verify Motifs: Check the predicted C-terminal sequences of SUMO proteins in your model organism (for AGG, SGG, TGG, VGG) to confirm potential for cross-reactivity [56].
- Use Appropriate Controls: Include controlled digestions with and without WaLP as part of method development and validation to build a system-specific understanding of antibody cross-reactivity.
By applying these rigorous experimental practices, researchers can confidently interpret their ubiquitinome data, ensuring that reported changes genuinely reflect regulatory ubiquitination events.
Visualized Workflows
Diagram 1: Experimental workflow for validating SUMO cross-reactivity using WaLP protease.
Diagram 2: Automated UbiFast workflow for specific and multiplexed ubiquitinome analysis.
Benchmarking K-ε-GG Reagents: A Comparative Analysis of Commercial Antibodies and Validation Methods
The development of antibodies specific to the di-glycine remnant (K-ε-GG) has revolutionized the study of protein ubiquitylation by mass spectrometry. This modification remains attached to substrate lysine residues after tryptic digestion of ubiquitylated proteins, serving as a specific marker for ubiquitin modification sites [21]. The commercialization of these highly specific reagents transformed a previously challenging field, enabling researchers to move from identifying only several hundred ubiquitination sites to routinely profiling tens of thousands of distinct sites in single experiments [5] [15]. This breakthrough has opened new avenues for understanding the extensive role of ubiquitin in cellular regulation, protein degradation, and disease pathways, particularly in cancer progression, immune disorders, and neurological diseases [21]. Within this landscape, several commercial platforms have emerged, with Cell Signaling Technology's PTMScan portfolio representing a leading solution that continues to evolve with technological advancements such as magnetic bead-based workflows and high-sensitivity formulations.
Platform Comparison: Technical Specifications and Performance Metrics
The following table provides a detailed comparison of key commercial K-ε-GG antibody platforms, highlighting their core technologies and performance characteristics.
| Platform / Product Name | Core Technology | Bead Format | Key Workflow Features | Reported Performance |
|---|---|---|---|---|
| PTMScan HS Ubiquitin/SUMO Remnant Motif Kit (#59322) [57] | Magnetic bead conjugation with near-covalent antibody linkage | Magnetic beads | Simplified washing via magnetism; reduced antibody contamination; optimized buffers | Improved yield and specificity; compatible with automation [58] |
| Traditional PTMScan Ubiquitin Remnant Motif Kit (#5562) [59] | Antibody conjugated to protein A agarose beads | Agarose beads | Standard immunoaffinity purification; requires centrifugation | Established methodology; higher sensitivity magnetic bead version available [59] |
| UbiFast Method [21] | Anti-K-ε-GG antibody with on-bead TMT labeling | Not specified | TMT labeling while peptides are bound to antibody; enables multiplexing | >10,000 ubiquitylation sites from 500 μg peptide per sample [21] |
| ThermoFisher Antibody (PA5-120707) [6] | Rabbit polyclonal antibody | Not specified | Western Blot, ELISA applications | Limited application data provided |
Experimental Protocols and Workflow Methodologies
Standard PTMScan Ubiquitin Remnant Workflow
The foundational protocol for K-ε-GG enrichment using PTMScan technology involves several critical steps. Cells or tissues are first lysed in a urea-containing buffer to denature proteins and preserve modifications. Cellular proteins are then digested with trypsin, which cleaves after arginine and lysine residues, generating peptides with the K-ε-GG remnant at previously ubiquitylated sites. The resulting peptides are purified by reversed-phase, solid-phase extraction before undergoing immunoaffinity purification using the bead-conjugated PTMScan Motif antibody. After extensive washing to remove unbound peptides, the captured PTM-containing peptides are eluted with dilute acid and prepared for LC-MS/MS analysis [59].
Advanced PTMScan HS Workflow Innovations
The PTMScan HS (High Sensitivity) platform incorporates significant improvements over traditional methods. The key innovation involves a near-covalent strength antibody-bead linkage that prevents antibody contamination during the elution step, thereby preventing liquid chromatography column clogging and increasing PTM peptide yield. Furthermore, the HS kit utilizes magnetic beads rather than agarose, which dramatically simplifies the workflow by eliminating multiple centrifugation steps during bead washing and elution. The buffers have also been optimized with changes to bind and wash buffers that help reduce non-specific peptide binding, improving overall specificity [57].
The UbiFast Method for Multiplexed Analysis
A significant methodological innovation is the UbiFast protocol, which enables highly multiplexed ubiquitylation profiling from limited sample amounts. This approach addresses a fundamental limitation of traditional methods: the inability of anti-K-ε-GG antibodies to enrich peptides when the N-terminus of the di-glycyl remnant is derivatized with isobaric tags like TMT. UbiFast circumvents this by performing TMT labeling while K-ε-GG peptides are still bound to the anti-K-ε-GG antibody. This protects the di-glycyl remnant amine from derivatization while allowing labeling of peptide N-termini and lysine side chains. Optimized conditions (0.4 mg TMT reagent, 10 min labeling) achieve >92% labeling efficiency for antibody-bound K-ε-GG peptides. This advancement enables quantification of approximately 10,000 ubiquitylation sites from as little as 500 μg of peptide per sample in a TMT 10-plex experiment [21].
Automation-Compatible Workflows
Recent advancements have focused on adapting K-ε-GG enrichment protocols for automation platforms to increase throughput and reproducibility. PTMScan HS kits with magnetic beads are compatible with bead-handler robots like the ThermoFisher KingFisher Apex, which move magnetic beads across wells containing samples and wash buffers. Testing has demonstrated that automated enrichment performs as well as manual preparation but with greater handling ease and scalability to larger sample sizes [58]. For non-magnetic bead conjugates, hybrid platforms like the Agilent AssayMAP Bravo system can be used, where specialized tips are pre-loaded with antibody. This approach has demonstrated 30-135% higher PTM peptide identifications compared to manual preparation when using a bidirectional aspirate program [58].
Research Reagent Solutions Toolkit
The table below outlines essential materials and reagents required for implementing K-ε-GG enrichment workflows, based on protocols described in the literature.
| Reagent / Kit | Function / Application | Key Features |
|---|---|---|
| PTMScan IAP Buffer (#9993) [59] | Immunoaffinity purification buffer | Optimized binding and wash conditions; supplied with PTMScan kits |
| Anti-K-ε-GG Antibody Bead Conjugate [59] | Enrichment of ubiquitin remnant peptides | Specific for di-glycine tag left after trypsin digestion; agarose or magnetic formats |
| Dimethyl Pimelimidate (DMP) [5] | Antibody cross-linking | Stabilizes antibody-bead linkage; reduces antibody contamination |
| Tandem Mass Tag (TMT) Reagents [21] | Multiplexed quantitative proteomics | Enables comparison of multiple samples; requires on-antibody labeling for ubiquitylome |
| C18 Solid-Phase Extraction Cartridges [5] | Peptide cleanup and desalting | Pre-enrichment peptide purification; available in various sizes (e.g., 500-mg tC18 Sep-Pak) |
| LysC/Trypsin Protease [60] | Protein digestion | Generates K-ε-GG-containing peptides; sequencing grade recommended |
Workflow Diagram of K-ε-GG Enrichment and Analysis
The following diagram illustrates the core experimental workflow for K-ε-GG-based ubiquitylation site profiling, integrating both standard and advanced methodological approaches.
Performance Optimization and Methodological Refinements
Significant improvements to the K-ε-GG enrichment workflow have been developed through systematic optimization of key parameters. Antibody cross-linking using dimethyl pimelimidate (DMP) in sodium borate buffer stabilizes the antibody-bead linkage, significantly reducing antibody leakage during elution and improving peptide yield [5]. Off-line basic reversed-phase fractionation prior to immunoenrichment, using a high-pH solvent system and non-contiguous pooling of fractions, dramatically improves depth of coverage [5]. Input amount optimization has enabled the identification of approximately 20,000 distinct ubiquitination sites from moderate protein inputs (5 mg per SILAC channel), representing a 10-fold improvement over earlier methods [5] [15]. The integration of FAIMS (High-Field Asymmetric Waveform Ion Mobility Spectrometry) technology further improves quantitative accuracy for PTM analysis by reducing sample complexity prior to MS detection [21].
These refined workflows have been successfully incorporated into sophisticated multi-omic platforms such as the MONTE (Multi-Omic Native Tissue Enrichment) workflow, which enables serial analysis of HLA immunopeptidome, ubiquitylome, proteome, phosphoproteome, and acetylome from a single tissue sample. This approach demonstrates that ubiquitylome analysis can be effectively integrated with other omic modalities without compromising data quality [60].
The evolution of commercial K-ε-GG antibody platforms has fundamentally transformed ubiquitin research, enabling comprehensive, site-specific ubiquitinome profiling at unprecedented scale and sensitivity. The PTMScan HS system represents a significant advancement over traditional agarose bead-based methods through its magnetic bead format, near-covalent antibody linkage, and optimized buffers that collectively improve yield, specificity, and workflow simplicity. Parallel innovations like the UbiFast method with on-antibody TMT labeling have addressed critical limitations in multiplexed analysis of limited samples. These technological advancements, combined with optimization strategies such as antibody cross-linking and pre-enrichment fractionation, provide researchers with powerful tools to investigate the complex landscape of protein ubiquitylation in biological systems and disease models. As these platforms continue to evolve with increasing automation compatibility and integration into multi-omic workflows, they will further accelerate discoveries in ubiquitin biology and therapeutic development.
In the field of ubiquitination research, the specificity of anti-diglycine remnant (K-ε-GG) antibodies is paramount for generating reliable proteomic data. These antibodies are essential tools for enriching and identifying endogenous ubiquitination sites through mass spectrometry (MS), enabling researchers to decipher the complex regulatory roles of protein ubiquitination in cellular processes and disease pathogenesis. As the ubiquitin field advances, rigorous antibody validation has become increasingly critical to address what some researchers have termed a "replication crisis" in the life sciences, where insufficiently validated reagents contribute to irreproducible results. This guide objectively compares two cornerstone validation methodologies—synthetic peptide controls and knockout/knockdown controls—within the broader context of evaluating K-ε-GG remnant antibody specificity, providing researchers with experimental frameworks and data to inform their reagent selection and validation practices.
Comparative Analysis of Validation Techniques
Table 1: Key Performance Indicators for Specificity Validation Techniques
| Validation Technique | Primary Validation Metric | Typical Experimental Workflow | Key Advantages | Inherent Limitations |
|---|---|---|---|---|
| Synthetic Peptide Controls | Signal-to-Noise Ratio; Absence of off-target peptide enrichment | Spiked-in peptide recovery; Peptide microarray; Competitive ELISA | Defines exact epitope; High-throughput compatible; Provides quantitative binding data | May not mimic native protein context; Requires knowledge of exact epitope |
| Knockout/Knockdown Controls | Signal Disappearance in Target-Depleted Samples | CRISPR-Cas9; RNAi; Western Blotting/MS analysis of KO cells | Confirms specificity in a complex biological milieu; Tests antibody in its final application | Can be time-consuming and costly; Potential for compensatory biological changes |
Table 2: Quantitative Performance Data from Published Studies
| Study Focus | Antibody Type / Target | Validation Method(s) Employed | Key Quantitative Outcome | Reference |
|---|---|---|---|---|
| Ubiquitin Remnant Enrichment | Anti-K-ε-GG Antibody | Refined protocol with optimized peptide input & cross-linking | Identification of ~20,000 distinct ubiquitination sites in a single experiment | [23] [5] |
| N-terminal Ubiquitination | Anti-GGX Monoclonal Antibodies | Specificity profiling against 19 GGX peptides | No cross-reactivity with K-ε-GG peptides; Broad recognition of 14/19 GGX motifs | [17] |
| Antibody Specificity Pipeline | Various | Capture WB with antibody arrays and MS | High-throughput implementation of IWGAV validation concepts | [61] |
Detailed Experimental Protocols
Protocol 1: Specificity Validation Using Synthetic Peptide Controls
Synthetic peptides serve as high-specificity reference materials for confirming antibody binding to the intended epitope.
- Peptide Design and Synthesis: Design peptides corresponding to the tryptic K-ε-GG remnant motif. The sequence should typically be 7-25 amino acids long, incorporating the diglycine-modified lysine centrally. Include control peptides with sequences found in non-target proteins or with near-neighbor modifications to test cross-reactivity. Peptides should be synthesized to a high chemical purity (>95%), with identity verified by LC-MS and amino acid analysis [62].
- Experimental Setup (ELISA or Microarray): Immobilize the synthetic peptides on a solid surface. For a high-throughput assessment of specificity, peptide microarrays can be used to probe hundreds to thousands of sequences simultaneously. Incubate the anti-K-ε-GG antibody with the peptide-coated surface across a range of concentrations. Include a negative control peptide with an unmodified lysine and a positive control with the canonical K-ε-GG motif [17].
- Data Analysis and Interpretation: Quantify the binding signal for each peptide. The antibody is considered specific if it shows strong binding only to the K-ε-GG peptide and significantly lower signal (e.g., >10-fold difference) for negative controls and non-target peptides. This data provides a quantitative measure of affinity and cross-reactivity [63].
Protocol 2: Specificity Validation Using Knockout Controls
Genetic knockout controls provide biological confirmation of antibody specificity by removing the target epitope.
- Generation of Knockout Cell Lines: Use CRISPR-Cas9 to disrupt the gene(s) encoding target proteins in a relevant cell line (e.g., HEK293 or HeLa). Alternatively, use RNA interference (RNAi) for transient knockdown. Target multiple proteins simultaneously if the antibody is expected to recognize a common motif across numerous substrates. Validate the knockout efficiency by qPCR and confirm the absence of target proteins by MS [64].
- Sample Preparation and Analysis: Culture both wild-type and knockout cells under identical conditions. Prepare protein lysates from both cell lines using a denaturing lysis buffer (e.g., containing 8 M urea) to preserve post-translational modifications. Perform a standard immunoaffinity enrichment protocol with the anti-K-ε-GG antibody, followed by LC-MS/MS analysis [5].
- Data Analysis and Interpretation: Process the MS data to identify and quantify K-ε-GG peptides. A specific antibody will show a dramatic reduction or complete absence of K-ε-GG peptide signals derived from the knocked-out proteins in the knockout sample compared to the wild-type. The persistence of strong signals in the knockout sample indicates non-specific binding [64] [63].
Conceptual Workflows and Relationships
Research Reagent Solutions
Table 3: Essential Research Reagents for Antibody Validation
| Reagent / Material | Primary Function in Validation | Application Example | Key Considerations |
|---|---|---|---|
| PTMScan Ubiquitin Remnant Motif Kit | Immunoaffinity enrichment of K-ε-GG peptides from complex digests | Profiling ubiquitination sites in cell lysates; Ideal for researchers establishing the workflow [65]. | Contains bead-conjugated antibody; Optimized IAP buffer included. |
| Synthetic K-ε-GG Peptide Controls | Define antibody specificity and epitope recognition | Positive controls in ELISA; Spike-in standards for MS recovery assays [62]. | Require high purity (>95%); Should mimic tryptic remnant sequence. |
| Stable Isotope-Labeled Peptide Standards | Internal standards for quantitative MS | Absolute quantification of ubiquitination sites; Normalization for technical variation [62]. | Typically ^13C/^15N-labeled; Sequence identical to endogenous target. |
| CRISPR-Cas9 Knockout Cell Lines | Provide biological negative controls | Confirm absence of off-target signal in a complex proteome [64]. | Requires validation of knockout efficiency via MS. |
| Linkage-Specific Ub Antibodies | Characterize polyubiquitin chain architecture | Enrich proteins with specific Ub linkages (e.g., K48, K63) [19]. | Complements K-ε-GG site mapping. |
The rigorous validation of anti-K-ε-GG antibodies is a non-negotiable prerequisite for generating trustworthy data in ubiquitination research. As detailed in this guide, both synthetic peptide controls and knockout/knockdown controls offer complementary and powerful means to confirm antibody specificity. Synthetic peptides provide a reductionist, high-throughput approach to precisely define the epitope, while knockout controls offer ultimate biological verification within the complex native proteomic environment. The most robust validation strategy, as endorsed by international working groups, leverages multiple pillars of evidence. Researchers should therefore integrate these techniques to build a compelling case for antibody specificity, thereby ensuring the reproducibility and reliability of their findings in the rapidly advancing field of ubiquitin proteomics.
The development of antibodies specific for the di-glycine remnant (K-ε-GG) left on ubiquitinated peptides after trypsin digestion has revolutionized the study of ubiquitination, enabling system-wide profiling of this crucial post-translational modification [5] [21]. These antibodies form the cornerstone of ubiquitin proteomics, allowing researchers to enrich low-abundance ubiquitinated peptides from complex biological samples for identification and quantification by mass spectrometry [66]. As the field has matured, multiple methodological approaches have emerged, each with distinct performance characteristics regarding enrichment specificity, reproducibility, and dynamic range. This guide objectively compares the performance of these key methodologies, providing researchers with critical experimental data to inform their proteomic study design.
Comparative Performance Analysis of K-ε-GG Enrichment Methodologies
The evolution of K-ε-GG enrichment protocols has focused on improving sensitivity, specificity, and throughput. The table below summarizes the quantitative performance metrics of three established methodologies.
Table 1: Performance Comparison of K-ε-GG Enrichment Methodologies
| Methodology | Protein Input | Sites Identified | Quantitative Precision | Throughput | Key Applications |
|---|---|---|---|---|---|
| Standard Immunoaffinity Enrichment [5] | 5 mg per SILAC channel | ~20,000 sites | High (SILAC-based) | Moderate | In-depth site discovery from cell lines |
| UbiFast (On-Antibody TMT) [21] | 0.5 mg per TMT channel | ~10,000 sites | High (TMT-based, 85.7% relative yield) | High (∼5 hours) | Multiplexed studies from tissue/primary cells |
| Targeted PRM-MS [67] | Variable | 78 ribosomal sites (targeted) | Very High (Absolute quantification) | Mid-throughput | Validation and precise quantification of specific sites |
Detailed Experimental Protocols
Refined Standard Immunoaffinity Enrichment
The optimized workflow for large-scale ubiquitination site identification involves several critical steps that maximize recovery and specificity [5].
Cell Lysis and Digestion: Cells are lysed in 8 M urea buffer containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, and protease inhibitors. Proteins are reduced with 5 mM dithiothreitol, alkylated with 10 mM iodoacetamide, and diluted to 2 M urea before overnight digestion with trypsin at a 1:50 enzyme-to-substrate ratio at 25°C [5].
Peptide Pre-Fractionation: Digested peptides are desalted and subjected to offline basic reversed-phase chromatography using a Zorbax 300 Extend-C18 column with a 64-minute gradient from 2% to 60% solvent B (90% MeCN, 5 mM ammonium formate, pH 10). Fractions are pooled non-contiguously into eight fractions to reduce complexity while maintaining resolution [5].
Antibody Cross-Linking and Enrichment: Anti-K-ε-GG antibody beads are cross-linked using 20 mM dimethyl pimelimidate in 100 mM sodium borate (pH 9.0) for 30 minutes, followed by blocking with 200 mM ethanolamine (pH 8.0). Peptide fractions are incubated with cross-linked beads for 1 hour at 4°C, washed extensively with ice-cold PBS, and K-ε-GG peptides are eluted with 0.15% trifluoroacetic acid [5].
UbiFast On-Antibody TMT Labeling
The UbiFast method addresses the limitation that anti-K-ε-GG antibodies do not efficiently bind TMT-labeled di-glycine remnants, enabling highly multiplexed quantification [21].
Peptide Enrichment and On-Bead Labeling: K-ε-GG peptides are enriched from 0.5-1 mg of digested peptide material using anti-K-ε-GG antibody beads. While peptides are bound to the antibody, TMT labeling is performed directly on the beads using 0.4 mg of TMT reagent for 10 minutes, effectively protecting the di-glycine remnant from derivatization. The reaction is quenched with 5% hydroxylamine [21].
Peptide Combining and Analysis: TMT-labeled K-ε-GG peptides from multiple samples are combined and eluted from the antibody beads. The pooled sample can be analyzed by single-shot LC-MS/MS without additional fractionation, significantly reducing instrument time. The incorporation of High-field Asymmetric Waveform Ion Mobility Spectrometry (FAIMS) further improves quantitative accuracy for post-translational modification analysis [21].
Targeted PRM-MS for Ubiquitination Stoichiometry
For precise quantification of specific ubiquitination sites, a targeted Parallel Reaction Monitoring Mass Spectrometry (PRM-MS) approach offers superior sensitivity and quantitative accuracy [67].
Synthetic Standard Design: Isotopically labeled reference peptides corresponding to known ubiquitination sites are synthesized with a C-terminal heavy lysine (13C6,15N2) or arginine (13C6,15N4) and include the di-Gly remnant (K-GG) at the modified lysine residue [67].
Sample Processing and Analysis: Samples are digested and enriched using standard K-ε-GG protocols. Enriched peptides are spiked with heavy labeled synthetic standards and analyzed by LC-PRM-MS. The retention times and mass-to-charge ratios of the heavy standards guide scheduled acquisition, enabling precise quantification of endogenous ubiquitinated peptides based on the heavy-to-light ratio [67].
Visualizing Experimental Workflows
The following diagrams illustrate the key methodological differences between the main enrichment and quantification strategies.
Standard K-ε-GG Enrichment Workflow
UbiFast On-Antibody TMT Labeling
The Scientist's Toolkit: Essential Research Reagents
Successful ubiquitin proteomics requires specific reagents and materials optimized for K-ε-GG remnant enrichment. The following table details essential components.
Table 2: Essential Research Reagents for K-ε-GG Proteomics
| Reagent/Material | Function | Specifications | Commercial Examples |
|---|---|---|---|
| K-ε-GG Antibody | Immunoaffinity enrichment of ubiquitinated peptides | High specificity for tryptic di-glycine remnant | PTMScan Ubiquitin Remnant Motif Kit [66] |
| Cross-linking Reagent | Immobilizes antibody to beads to prevent leakage | Dimethyl pimelimidate (DMP) in borate buffer, pH 9.0 [5] | Sigma-Aldrich DMP |
| Isobaric Tags | Multiplexed quantification of ubiquitination sites | Tandem Mass Tag (TMT) 10/11-plex reagents [21] | Thermo Scientific TMT |
| Fractionation Column | Reduces sample complexity pre-enrichment | Zorbax 300 Extend-C18, 9.4 × 250 mm, 300 Å [5] | Agilent Basic RP Columns |
| Protease | Generates K-ε-GG remnant from ubiquitinated proteins | Sequencing-grade trypsin, 1:50 enzyme:substrate ratio [5] | Promega Trypsin |
The quantitative comparison presented herein demonstrates that methodological selection for K-ε-GG enrichment should be guided by specific research objectives. The refined standard immunoaffinity protocol provides exceptional depth for discovery-phase studies, capable of identifying approximately 20,000 ubiquitination sites with moderate throughput requirements [5]. The UbiFast on-antibody TMT labeling method offers superior multiplexing capacity and reduced sample input requirements, making it ideal for larger-scale comparative studies, particularly when working with precious tissue samples or primary cell models [21]. For validation studies requiring absolute quantification of specific ubiquitination events, targeted PRM-MS provides unmatched sensitivity and quantitative precision, enabling measurement of modification stoichiometry across a dynamic range exceeding four orders of magnitude [67]. Understanding these performance characteristics allows researchers to strategically implement K-ε-GG enrichment methodologies that best address their specific biological questions while optimizing resource allocation.
The di-glycine remnant (K-ε-GG) represents a crucial molecular signature in proteomics research, serving as a specific marker for protein ubiquitination. When ubiquitin is conjugated to target proteins and the resulting complexes are digested with trypsin, a characteristic di-glycine moiety remains attached to the modified lysine residue, creating the K-ε-GG motif. The development of highly specific antibodies targeting this motif has revolutionized the study of ubiquitination by enabling researchers to enrich, identify, and quantify endogenous ubiquitination sites on a proteome-wide scale using mass spectrometry [5] [68].
The specificity and performance of these antibodies vary significantly across different commercial sources and product formats, making application-based selection essential for research success. For discovery-based proteomics aimed at identifying thousands of ubiquitination sites, requirements for antibody performance differ substantially from targeted studies focusing on specific proteins or pathways. This guide provides an objective comparison of available K-ε-GG remnant antibodies, supported by experimental data and detailed methodologies, to help researchers select the optimal reagents for their specific research contexts [5] [69].
Comparative Analysis of K-ε-GG Antibody Products and Performance
Table 1: Commercial K-ε-GG Remnant Antibodies and Key Characteristics
| Product Name | Host/Source | Clonality | Reactivity | Recommended Applications | Key Features |
|---|---|---|---|---|---|
| PTMScan Ubiquitin Remnant Motif Kit [68] | Rabbit | Not Specified | Universal | Peptide Enrichment for MS | High sensitivity and specificity for ubiquitin remnant motif |
| PTMScan HS K-ε-GG Remnant Magnetic Beads [69] | Rabbit IgG | Recombinant | Universal | Peptide Enrichment for MS | Superior lot-to-lot consistency, animal-free manufacturing |
| Ubiquitin Remnant Motif Antibody [6] | Rabbit | Polyclonal | Human, Mouse | WB, ELISA | Detects K-ε-GG in human and mouse samples |
| pan-Ubiquitin Remnant Motif Antibody [70] | Rabbit | Polyclonal | Human, Mouse, Rat | WB, ELISA | Recognizes di-glycine remnant independent of flanking sequence |
The performance of K-ε-GG antibodies in discovery proteomics has been demonstrated in optimized workflows. A landmark study utilizing the PTMScan Ubiquitin Remnant Motif Kit achieved the identification of approximately 20,000 distinct endogenous ubiquitination sites in a single SILAC experiment using only 5 mg of protein input per channel. This represented a 10-fold improvement over previously published methods and established a new benchmark for comprehensive ubiquitinome analysis [5]. The critical factors enabling this depth of coverage included optimized antibody cross-linking procedures, off-line basic reversed-phase fractionation with noncontiguous pooling, and precise control of antibody-to-peptide ratios [5].
For researchers requiring higher consistency, the PTMScan HS magnetic beads offer a recombinant antibody solution with superior lot-to-lot consistency due to animal-free manufacturing processes. This product is specifically designed for high-sensitivity applications where reproducibility across multiple experiments is essential [69].
Experimental Designs and Workflows for Ubiquitinome Analysis
Optimized Sample Preparation Protocol for Discovery Proteomics
The following methodology, adapted from Udeshi et al. (2012), outlines an optimized workflow for large-scale ubiquitination site identification [5]:
Cell Culture and Lysis: Grow Jurkat E6-1 cells in SILAC media for metabolic labeling. Treat cells with proteasome inhibitors (e.g., 2-5 μM MG-132) for 4 hours prior to harvest to stabilize ubiquitinated proteins. Lyse cells in denaturing buffer (8 M urea, 50 mM Tris-HCl pH 7.5, 150 mM NaCl) supplemented with protease inhibitors (aprotinin, leupeptin, PMSF) and deubiquitinase inhibitors (PR-619) [5].
Protein Digestion: Determine protein concentration using BCA assay. Reduce proteins with 5 mM dithiothreitol (45 min, room temperature) and alkylate with 10 mM iodoacetamide (30 min, room temperature in dark). Dilute lysates to 2 M urea with 50 mM Tris-HCl (pH 7.5) and digest overnight at 25°C with sequencing-grade trypsin at 1:50 enzyme-to-substrate ratio [5].
Peptide Cleanup and Fractionation: Acidify digested peptides with formic acid and desalt using C18 solid-phase extraction cartridges. Fractionate peptides using basic reversed-phase chromatography on a Zorbax 300 Extend-C18 column with a 64-minute gradient from 2% to 60% solvent B (90% acetonitrile, 5 mM ammonium formate, pH 10). Pool fractions in a noncontiguous manner into 8 final fractions to reduce complexity while maintaining resolution [5].
Antibody Cross-Linking and K-ε-GG Peptide Enrichment: Wash anti-K-ε-GG antibody beads with 100 mM sodium borate (pH 9.0). Cross-link antibodies to beads using 20 mM dimethyl pimelimidate (30 min, room temperature). Block with 200 mM ethanolamine (pH 8.0, 2h, 4°C). Incubate fractionated peptides with cross-linked antibody beads (31 μg antibody per fraction) for 1 hour at 4°C. Wash beads extensively with ice-cold PBS, and elute K-ε-GG peptides with 0.15% trifluoroacetic acid [5].
Mass Spectrometry Analysis: Desalt eluted peptides using C18 StageTips and analyze by LC-MS/MS using high-resolution instruments. Identify and quantify ubiquitination sites using appropriate software platforms, with SILAC labeling enabling precise quantification of changes in ubiquitination following cellular perturbations [5].
Key Research Reagent Solutions for Ubiquitin Studies
Table 2: Essential Research Reagents for K-ε-GG Studies
| Reagent / Solution | Function | Example Formulation / Notes |
|---|---|---|
| IAP Buffer [68] | Immunoaffinity purification buffer | 50 mM MOPS, pH 7.2, 10 mM sodium phosphate, 50 mM NaCl; optimal for antibody-antigen binding |
| Urea Lysis Buffer [5] | Protein denaturation and extraction | 8 M urea, 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, plus protease and deubiquitinase inhibitors |
| Basic Reversed-Phase Solvents [5] | High-pH peptide separation | Solvent A: 2% acetonitrile, 5 mM ammonium formate, pH 10; Solvent B: 90% acetonitrile, 5 mM ammonium formate, pH 10 |
| Cross-Linking Reagents [5] | Stabilize antibody-bead conjugation | Dimethyl pimelimidate (DMP) in sodium borate buffer, pH 9.0 |
| Proteasome Inhibitors [5] | Stabilize ubiquitinated proteins | MG-132 (2-5 μM), PR-619 (5 μM); added 4 hours pre-harvest |
Theoretical Framework: Understanding Antibody Specificity in Molecular Recognition
The fundamental challenge in selecting research antibodies lies in understanding the physical principles governing molecular recognition. Traditional "lock-and-key" and "induced-fit" models provide simplified explanations but fail to adequately represent the complexity of antibody-antigen interactions, particularly in explaining how the same antibody can exhibit both high specificity and appropriate cross-reactivity [71].
The energy landscape theory offers a more comprehensive framework for understanding antibody performance. In this model, antibody-antigen binding is conceptualized as a dynamic process where molecular conformations follow successive pathways toward thermodynamically favorable states. High-affinity interactions correspond to deep, sharply defined energy wells characterized by substantial negative free energy changes (ΔG approximately -7 to -14 kcal/mol). These interactions result from precise geometric and chemical complementarity at the antibody-antigen interface, enabling extensive non-covalent interactions including hydrogen bonding, van der Waals forces, and hydrophobic packing [71].
In contrast, lower-affinity or "cross-reactive" binding corresponds to broad, shallow energy basins on the molecular energy landscape. These interactions arise from less structurally refined molecular interfaces and are characterized by fewer stabilizing interactions and more rapid dissociation rates (k_off typically 10⁻¹ to 10¹ s⁻¹). Rather than representing mere background noise, these transient interactions reflect a functional mode of recognition that enables antibodies to engage with multiple structurally related antigens [71].
This theoretical framework has practical implications for antibody selection. For discovery proteomics where the goal is identification of thousands of ubiquitination sites, antibodies with a balanced specificity that allows recognition of diverse K-ε-GG motifs in varying sequence contexts are essential. For targeted studies, antibodies with deep energy wells for specific epitopes may be preferable, even if this comes at the expense of broader reactivity [71].
Ubiquitination Pathway and Proteomics Workflow
The ubiquitin-proteasome pathway involves a cascade of enzymatic reactions. Ubiquitin is first activated by E1 enzymes, transferred to E2 carrier proteins, and finally delivered to target proteins via E3 ligases, forming a covalent bond with lysine residues. After trypsin digestion, the C-terminal -RGG sequence of ubiquitin is cleaved, leaving a di-glycine remnant (K-ε-GG) on the modified lysine. This remnant serves as the recognition site for K-ε-GG specific antibodies, enabling enrichment and mass spectrometry analysis [68] [69].
The optimized workflow for comprehensive ubiquitinome analysis begins with sample preparation including protein extraction and tryptic digestion. Peptides are then fractionated using basic reversed-phase chromatography to reduce sample complexity. K-ε-GG-containing peptides are specifically enriched using anti-K-ε-GG antibodies before analysis by high-resolution mass spectrometry. This workflow enables identification of thousands of ubiquitination sites from moderate protein input amounts [5].
Application-Based Selection Guidelines
Discovery Proteomics Applications
For large-scale discovery proteomics aimed at identifying thousands of ubiquitination sites across the proteome, the PTMScan Ubiquitin Remnant Motif Kit and PTMScan HS Magnetic Beads demonstrate superior performance. The critical factors for success in these applications include:
High Specificity and Sensitivity: The PTMScan technology has been proven to identify approximately 20,000 distinct ubiquitination sites in a single experiment, representing the current gold standard for comprehensive ubiquitinome analysis [5].
Optimized Workflow Integration: These antibodies perform best when integrated with the complete optimized workflow including basic reversed-phase fractionation, antibody cross-linking, and appropriate peptide-to-antibody ratios [5].
Reproducibility: The recombinant HS magnetic beads offer superior lot-to-lot consistency, which is essential for long-term discovery projects requiring multiple experiments over time [69].
Targeted Studies and Validation Applications
For targeted studies focusing on specific proteins, pathways, or validation of candidate ubiquitination sites, alternative antibody formats may be more appropriate:
Western Blot and ELISA Applications: The pan-ubiquitin remnant motif antibodies from Thermo Fisher and Assay Genie provide cost-effective solutions for techniques like Western blotting and ELISA, where the priority is detecting specific ubiquitinated proteins rather than comprehensive proteome coverage [6] [70].
Species-Specific Considerations: For studies in common model organisms (human, mouse, rat), the broad reactivity of polyclonal antibodies offers sufficient coverage, while more specialized models may require additional validation [6] [70].
Resource Allocation: Targeted studies with limited sample amounts or budget constraints can benefit from the more accessible polyclonal antibodies, provided appropriate validation controls are included [6].
The selection of K-ε-GG remnant antibodies should be driven by specific research objectives and experimental designs. For discovery proteomics requiring comprehensive ubiquitinome coverage, the PTMScan platform provides unmatched performance when implemented with optimized workflows. For targeted applications and validation studies, more accessible polyclonal antibodies offer practical alternatives. As mass spectrometry technologies continue to advance and the theoretical understanding of antibody specificity evolves, application-based reagent selection becomes increasingly crucial for generating reliable, reproducible data in ubiquitin research.
Conclusion
The specificity of K-ε-GG remnant antibodies is the critical determinant for successful and reliable ubiquitinomics. This synthesis underscores that rigorous evaluation, from foundational recognition principles to optimized application workflows, is essential. The ongoing refinement of these reagents, coupled with advanced mass spectrometry techniques like DIA-MS, continues to propel the field forward. Future directions will involve developing antibodies with even greater specificity to distinguish ubiquitin from UBLs, creating standardized validation benchmarks, and applying these powerful tools to map dynamic ubiquitination networks in disease models, thereby accelerating the discovery of novel drug targets in oncology and neurodegeneration.
References
- 1. History... [https://www.chm.bris.ac.uk/motm/ubiquitin/ubiquitin_pliki/page0003.htm]
- 2. Characterizing Ubiquitination Sites by Peptide-based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3518114/]
- 3. Historical perspective and progress on protein ubiquitination ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8831078/]
- 4. Ubiquitinomics: History, methods, and applications in basic ... [https://pubmed.ncbi.nlm.nih.gov/35353442/]
- 5. Refined Preparation and Use of Anti-diglycine Remnant (K- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3591673/]
- 6. Ubiquitin Remnant Motif (K-Epsilon-GG) Antibodies [https://www.thermofisher.com/antibody/primary/target/ubiquitin%20remnant%20motif%20(k-epsilon-gg)]
- 7. pan-Ubiquitin Remnant Motif (K-eps... Antibody [https://www.assaygenie.com/anti-pan-ubiquitin-remnant-motif-k-gg-antibody-cab20303/?srsltid=AfmBOory6ppEWKkzTTuDaU1up1xhuL8WuCsDPB1Ulc7kQdSwSQ3SFw_O]
- 8. Quantitative Ubiquitylome Analysis Reveals the Specificity ... [https://www.sciencedirect.com/science/article/pii/S1535947621001456]
- 9. Site-specific quantification of the in vivo UFMylome reveals ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12146631/]
- 10. Integrated 4D label-free proteome and SUMOylated ... [https://www.sciencedirect.com/science/article/pii/S2772892725000276]
- 11. The new landscape of protein ubiquitination - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3242807/]
- 12. Detection of Protein Ubiquitination Sites by Peptide Enrichment and... [https://app.jove.com/t/59079/detection-protein-ubiquitination-sites-peptide-enrichment-mass]
- 13. Large-Scale Identification of Ubiquitination Sites by Mass ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4725055/]
- 14. Refined preparation and use of anti-diglycine remnant (K-ε ... [https://www.broadinstitute.org/publications/broad4541]
- 15. Refined Preparation and Use of Anti-diglycine Remnant (K- ... [https://www.sciencedirect.com/science/article/pii/S1535947620330565]
- 16. PTMScan® Ubiquitin Remnant Motif (K-epsilon-GG) Kit [https://www.cellsignal.com/products/proteomic-analysis-products/ubiquitin-remnant-motif-k-epsilon-gg-kit/5562?srsltid=AfmBOooRV4_tvjGUy-KbfxNcw6Kw-cJ_IVcoatzqkePdMFhWeareHgXW]
- 17. Antibody toolkit reveals N-terminally ubiquitinated ... [https://www.nature.com/articles/s41467-021-24669-6]
- 18. Large-scale identification of ubiquitination sites by... | Nature Protocols [https://www.nature.com/articles/nprot.2013.120?error=cookies_not_supported&code=d4e91ab1-4b58-46a9-b52a-5e5fd1eb1fbc]
- 19. Current methodologies in protein ubiquitination characterization [https://cellandbioscience.biomedcentral.com/articles/10.1186/s13578-022-00870-y]
- 20. Characterizing Ubiquitination Sites by Peptide-based ... [https://www.sciencedirect.com/science/article/pii/S1535947620334502]
- 21. Rapid and deep-scale ubiquitylation profiling for biology ... [https://www.nature.com/articles/s41467-019-14175-1]
- 22. Methods for Quantification of in vivo Changes in Protein ... [https://www.sciencedirect.com/science/article/pii/S1535947620304576]
- 23. Refined preparation and use of anti-diglycine remnant (K-ε ... [https://pubmed.ncbi.nlm.nih.gov/23266961/]
- 24. Aging and diet alter the protein ubiquitylation landscape in ... [https://www.nature.com/articles/s41467-025-60542-6]
- 25. Site-specific quantification of the in vivo UFMylome reveals ... [https://www.sciencedirect.com/science/article/pii/S2667237525000840]
- 26. Investigating the volume and diversity of data needed for ... [https://www.nature.com/articles/s43588-025-00823-8]
- 27. How AI Is Finally Cracking the Code of Antibody Specificity [https://www.ailurus.bio/post/beyond-structure-how-ai-is-finally-cracking-the-code-of-antibody-specificity]
- 28. Time-resolved in vivo ubiquitinome profiling by DIA-MS ... [https://www.nature.com/articles/s41467-021-25454-1]
- 29. In-Depth Comparison of Reagent-Based Digestion ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11923677/]
- 30. Deciphering Tissue-specific Ubiquitination by Mass ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3475722/]
- 31. Comprehensive Ubiquitome Analysis of Nicotiana ... [https://www.mdpi.com/2079-7737/14/6/656]
- 32. Application of quantitative proteomics to the integrated ... [https://clinicalproteomicsjournal.biomedcentral.com/articles/10.1186/s12014-015-9086-5]
- 33. Reversed-phase chromatography [https://en.wikipedia.org/wiki/Reversed-phase_chromatography]
- 34. Reversed-Phase Chromatography Overview [https://www.creative-proteomics.com/proteinseq/resource/reversed-phase-chromatography-overview.htm]
- 35. Reverse Phase Chromatography Techniques [https://chromtech.com/blog/reverse-phase-chromatography-techniques/]
- 36. Systematic Comparison of Reverse Phase and Hydrophilic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3689152/]
- 37. PTMScan® Pilot Ubiquitin Remnant Motif (K-epsilon-GG) Kit [https://www.cellsignal.com/products/proteomic-analysis-products/pilot-ubiquitin-remnant-motif-k-e-gg-kit/14482?srsltid=AfmBOopa_2mHs2N0GC79jd80O1dfj1FpqJshGza8-fs_6EibAM_-cr0w]
- 38. Enrichment of ubiquitinated (K-ε-GG) peptides [https://bio-protocol.org/exchange/minidetail?id=2242819&type=30]
- 39. DDA vs. DIA vs. MRM vs. PRM [https://www.metwarebio.com/mass-spectrometry-acquisition-modes-dda-vs-dia-vs-mrm-vs-prm/]
- 40. Data-independent acquisition (DIA) [https://pmc.ncbi.nlm.nih.gov/articles/PMC8674493/]
- 41. Reproducibility, Specificity and Accuracy of Relative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6944235/]
- 42. Multi-laboratory assessment of reproducibility, qualitative ... [https://www.nature.com/articles/s41467-017-00249-5]
- 43. mass spectrometry based- untargeted metabolomics ... [https://www.sciencedirect.com/science/article/pii/S2001037025002090]
- 44. Antibody cross-linking and target elution protocols used for immunoprecipitation significantly modulate signal-to noise ratio in downstream 2D-PAGE analysis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3162493/]
- 45. Preactivation Crosslinking—An Efficient Method for the Oriented Immobilization of Antibodies - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6632165/]
- 46. pan-Ubiquitin Remnant Motif (K-eps... Antibody [https://www.assaygenie.com/anti-pan-ubiquitin-remnant-motif-k-gg-antibody-cab20303/?srsltid=AfmBOooFcvYwW7JlquTZeNhOXSGwGBcyKpO3En_OXdV4VE34SKhBVl9y]
- 47. A versatile antibody capture system drives specific in vivo ... [https://www.nature.com/articles/s41565-025-01954-9]
- 48. Ubiquitin diGLY proteomics as an approach to identify and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6791129/]
- 49. Titration-based normalization of antibody amount improves ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-023-09253-0]
- 50. Automating UbiFast for High-throughput and Multiplexed ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9357436/]
- 51. PTMScan Motif Antibody Kits [https://www.cellsignal.com/applications/proteomics/ptmscan-motif-antibody-kits?srsltid=AfmBOorf9UvVnNe6N1VakcEzrPJElp3OEQ-cgmgAyk7r9yMjTO3JgvCc]
- 52. Ubiquitin Remnant Motif (K-Epsilon-GG) Polyclonal Antibody [https://www.thermofisher.com/antibody/product/Ubiquitin-Remnant-Motif-K-Epsilon-GG-Antibody-Polyclonal/PA5-120707]
- 53. Titration-WB: A methodology for accurate quantitative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12161570/]
- 54. PTMScan® Pilot Ubiquitin Remnant Motif (K-epsilon-GG) Kit [https://www.cellsignal.com/products/proteomic-analysis-products/pilot-ubiquitin-remnant-motif-k-e-gg-kit/14482?srsltid=AfmBOoplbSCkRrFpM73xkIyM30U6inRpDFRRTrx2aEzzgTN2uD5I5jFB]
- 55. 多克隆 抗 市场规模,2032 年增长报告 体 [https://www.marketresearchfuture.com/zh-cn/reports/polyclonal-antibodies-market-30221]
- 56. PTMScan ® HS Ubiquitin/SUMO Remnant Motif (K-epsilon- ... [https://www.cellsignal.com/products/proteomic-analysis-products/hs-ubiquitin-sumo-remnant-motif-k-epsilon-gg-kit/59322?srsltid=AfmBOooqBly92rvkbCcndO7xIiCYkmpG3K73JGT94F7rlfdtEDQZFqug]
- 57. How is the PTMScan® HS Ubiquitin/SUMO Remnant Motif ... [https://www.cellsignal.com/learn-and-support/technical-support/how-is-the-ptmscan-hs-ubiquitin-sumo-remnant-motif-k-gg-kit-59322-different-from-traditional-agarose-bead-ptmscan/000001576?srsltid=AfmBOoqwzmE8gt0HsSTFe3PpjWTgWq97xUtSDyjuZ8xXyPLhHryuln4R]
- 58. Automating PTM Profiling for Mass Spec: Scalable Proteomics ... [https://blog.cellsignal.com/high-throughput-proteomics-automating-ptm-enrichment-for-mass-spec]
- 59. PTMScan® Ubiquitin Remnant Motif (K-epsilon-GG) Kit [https://www.cellsignal.com/products/proteomic-analysis-products/ubiquitin-remnant-motif-k-epsilon-gg-kit/5562?srsltid=AfmBOoqI9TPMV8VigHzx6udOSSiYWVL4G9V5E4DdjbR4u8mn9367MamY]
- 60. Workflow enabling deepscale immunopeptidome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10070353/]
- 61. A high-throughput pipeline for validation of antibodies [https://pubmed.ncbi.nlm.nih.gov/30377371/]
- 62. : High- Peptide Reference Material for Assay... Control Specificity [http://biomonitoring2.org/peptide-control-high-specificity-reference-material-for-assay-validation-protein-quantification-and-analytical-calibration/]
- 63. Ten Basic Rules of Antibody Validation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5813849/]
- 64. Guide to Antibody Validation Techniques [https://www.neobiotechnologies.com/resources/antibody-validation-methods/?srsltid=AfmBOoqMmicmdrAyk6GJhaPzup42SYU3J2o0F0nkrSB7poRP6At4prwj]
- 65. PTMScan® Ubiquitin Remnant Motif ( K - ε - GG ) Kit [https://www.cellsignal.com/products/proteomic-analysis-products/ubiquitin-remnant-motif-k-e-gg-kit/5562]
- 66. PTMScan® Ubiquitin Remnant Motif (K-epsilon-GG) Kit [https://www.cellsignal.com/products/proteomic-analysis-products/ubiquitin-remnant-motif-k-epsilon-gg-kit/5562?srsltid=AfmBOorwCVN9bDvKdPliJ-fENtTh579YFojNsq3zkM1X7i0MbrGAND7N]
- 67. Context specific ubiquitin modification of ribosomes regulates ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11838502/]
- 68. PTMScan® Ubiquitin Remnant Motif (K-epsilon-GG) Kit [https://www.cellsignal.com/products/proteomic-analysis-products/ubiquitin-remnant-motif-k-epsilon-gg-kit/5562?srsltid=AfmBOopkLfd62LcfQ9_EjEgmtgp3g1-CYWj4qmDkiEnLrmMI1Rumx2C0]
- 69. PTMScan ® HS K-epsilon-GG Remnant Magnetic ... [https://www.cellsignal.com/products/proteomic-analysis-products/hs-k-epsilon-gg-remnant-magnetic-immunoaffinity-beads/34608?srsltid=AfmBOooJNHsIVrCMeoy1QvRYZl6BVw4ktcTiychiXd-2RisDaXbd-xeV]
- 70. pan-Ubiquitin Remnant Motif (K-eps... Antibody [https://www.assaygenie.com/anti-pan-ubiquitin-remnant-motif-k-gg-antibody-cab20303/?srsltid=AfmBOoqWzP6O2yolIsd9z86vGNwnHxSFzVV5R1RuSvSnIpM_SLxlbdo1]
- 71. Reconciling specificity and non-specificity in antibody binding [https://pmc.ncbi.nlm.nih.gov/articles/PMC12443689/]