HECT vs. RING E3 Ligases: Specificity for Atypical Ubiquitin Chains and Therapeutic Implications
This article provides a comprehensive comparison of HECT and RING-family E3 ubiquitin ligases, focusing on their distinct catalytic mechanisms and specificities for generating atypical ubiquitin chain linkages (K6, K11, K27,...
HECT vs. RING E3 Ligases: Specificity for Atypical Ubiquitin Chains and Therapeutic Implications
Abstract
This article provides a comprehensive comparison of HECT and RING-family E3 ubiquitin ligases, focusing on their distinct catalytic mechanisms and specificities for generating atypical ubiquitin chain linkages (K6, K11, K27, K29, K33). Aimed at researchers and drug development professionals, it explores the structural basis for linkage specificity, current methodologies for studying these enzymes, common experimental challenges, and validation strategies. The review synthesizes how understanding these differences opens avenues for developing targeted therapeutics, such as PROTACs, that exploit specific E3 ligase functions in diseases like cancer and immune disorders.
Decoding the Catalytic Engines: Foundational Mechanisms of HECT and RING E3 Ligases
The ubiquitin-proteasome system (UPS) is a highly complex, temporally controlled, and conserved pathway that serves as the primary mechanism for regulated intracellular protein degradation in eukaryotes [1] [2] [3]. By controlling the degradation of short-lived, misfolded, or damaged proteins, the UPS maintains cellular protein homeostasis and regulates a vast array of cellular processes, including immune response, apoptosis, cell cycle progression, cell differentiation, and signaling cascades [1]. The system functions through a hierarchical enzymatic cascade that culminates in the covalent attachment of ubiquitin to target proteins, marking them for destruction by the proteasome [1]. Understanding the precise mechanisms of this system is crucial, as dysregulation of UPS components is implicated in numerous diseases, including cancers, neurological disorders, and immune-related conditions [1] [4].
The fundamental process involves two discrete, successive steps: (1) covalent attachment of multiple ubiquitin molecules to the protein substrate through a three-enzyme cascade (E1-E2-E3), and (2) degradation of the tagged protein by the 26S proteasome complex with concomitant release of reusable ubiquitin [3]. This review will provide a comprehensive primer on the E1, E2, and E3 enzyme cascades, with a specific focus on comparing the mechanisms and specificities of two major E3 ligase families—RING and HECT—in generating atypical ubiquitin chains.
The Ubiquitin Conjugation Cascade: E1, E2, and E3 Enzymes
E1: Ubiquitin-Activating Enzyme
The ubiquitination pathway initiates with the E1 ubiquitin-activating enzyme, which serves as the molecular "alarm clock" that activates ubiquitin and begins the enzymatic cascade [2]. This activation occurs in an ATP-dependent two-step reaction where the E1 enzyme forms a high-energy thioester bond between its active site cysteine residue and the C-terminal glycine of ubiquitin [5] [4]. This reaction ultimately yields an E1-ubiquitin intermediate that represents the first committed step in the protein degradation pathway [5]. The structure of the UPS is hierarchical, with a single E1 enzyme or a very small number of E1s carrying out the activation of ubiquitin required for all subsequent modifications in the cell [3].
E2: Ubiquitin-Conjugating Enzyme
Following activation, ubiquitin is transferred to an E2 ubiquitin-conjugating enzyme (also known as ubiquitin-carrier proteins or ubiquitin-conjugating enzymes, UBCs) [3]. This transfer results in the formation of a thioester-linked E2-ubiquitin intermediate [5] [4]. The human genome encodes approximately 40 E2 enzymes, each capable of interacting with specific E3 ligases [6]. E2 enzymes function as the "baton passer" of the UPS, preparing to transfer the activated ubiquitin to the final enzymatic component of the cascade [2]. The E2 enzyme family provides an initial layer of specificity in the ubiquitination process, as different E2s can influence the type of ubiquitin chain formed on the substrate [7].
E3: Ubiquitin Ligase
The final and most crucial step in the cascade is mediated by E3 ubiquitin ligases, which are responsible for substrate recognition and the ultimate transfer of ubiquitin to the target protein [1] [5]. E3s achieve this by catalyzing the formation of an isopeptide bond between the C-terminal glycine of ubiquitin and the ε-amino group of a lysine residue on the substrate protein [5]. With approximately 600-700 E3 ligases encoded in the human genome, this enzyme family provides the remarkable specificity of the UPS, as each E3 recognizes a distinct set of substrates [1] [6]. The E3 ligase family is divided into three major categories based on their structural features and mechanisms of action: RING-type E3s, HECT-type E3s, and RBR-type E3s [8] [4].
Table 1: Key Characteristics of E1, E2, and E3 Enzymes in the Ubiquitin Cascade
| Enzyme | Number in Humans | Primary Function | Mechanism | Key Features |
|---|---|---|---|---|
| E1 (Activating) | 1-2 [8] | Ubiquitin activation | ATP-dependent formation of E1~Ub thioester | Initiates cascade; hierarchical role |
| E2 (Conjugating) | ~40 [6] | Ubiquitin transfer from E1 to E3 | Forms E2~Ub thioester intermediate | Influences chain topology; ~40 variants |
| E3 (Ligase) | ~600-700 [1] [6] | Substrate recognition & ubiquitin ligation | RING: Scaffold for direct transferHECT: Two-step with E3~Ub intermediateRBR: Hybrid mechanism | Provides substrate specificity; largest family |
E3 Ligase Families: Architectural and Mechanistic Divergence
RING E3 Ligases: Scaffold-Mediated Direct Transfer
RING (Really Interesting New Gene) E3 ligases represent the largest family of ubiquitin ligases, with over 600 members in humans [7] [4]. These E3s are characterized by a RING domain that coordinates two structural zinc ions through a cross-brace arrangement of cysteine and histidine residues [7] [4]. Rather than participating directly in catalysis, RING E3s function as scaffolds that simultaneously bind both the E2~Ub intermediate and the substrate protein, facilitating the direct transfer of ubiquitin from the E2 to the substrate without forming a covalent E3-ubiquitin intermediate [7] [8].
A critical feature of RING E3 mechanism is the "linchpin" residue—typically a cationic arginine—that stabilizes the closed conformation of the E2~Ub intermediate, optimizing it for nucleophilic attack by the substrate lysine [7]. Recent research has demonstrated that altering this single residue can dramatically modulate ubiquitin transfer ability, ranging from minor reduction to complete abolition of activity [7]. This linchpin residue forms a network of hydrogen bonds with both the E2 and ubiquitin, positioning the thioester bond for efficient transfer [7].
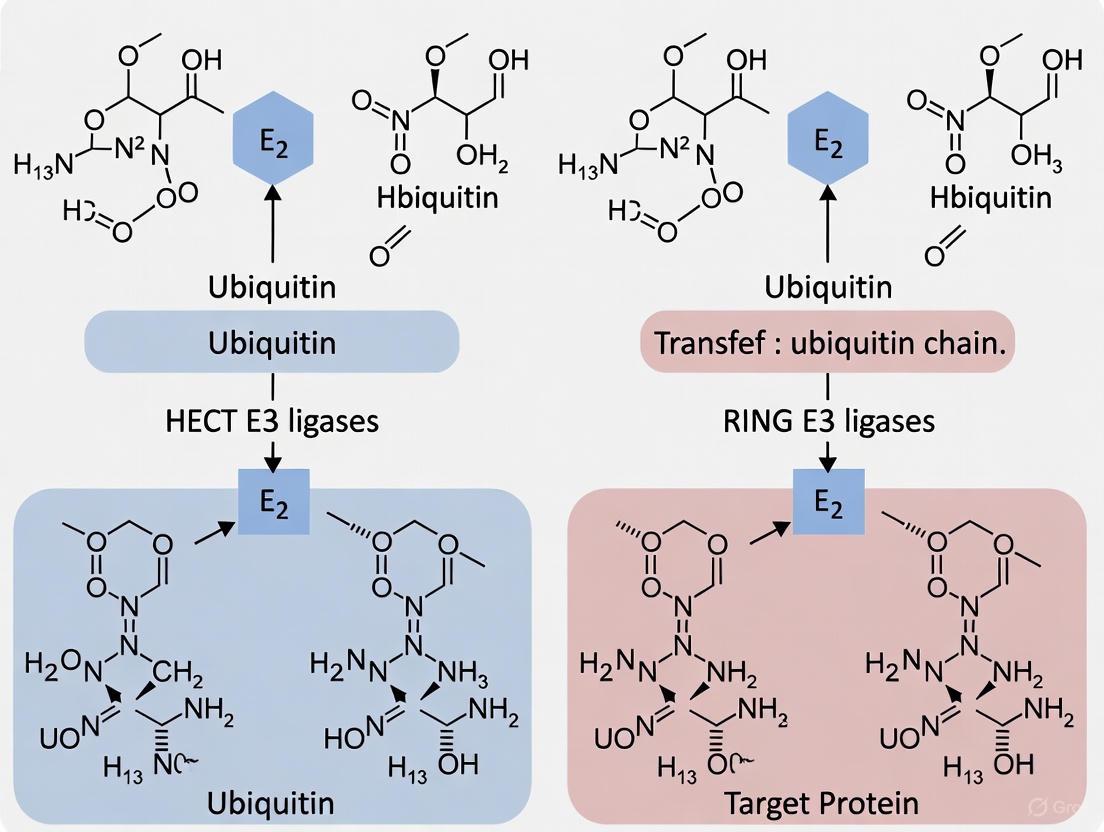
HECT E3 Ligases: Two-Step Catalytic Mechanism
HECT (Homologous to E6AP C-terminus) E3 ligases employ a distinct two-step catalytic mechanism that involves the direct participation of the E3 in the transfer reaction [9] [8]. The 28 human HECT E3s are characterized by a conserved C-terminal HECT domain of approximately 350 amino acids that contains an active site cysteine residue [4]. Unlike RING E3s, HECT E3s first accept the activated ubiquitin from the E2~Ub intermediate, forming a labile, thioester-linked E3~Ub intermediate, before subsequently transferring the ubiquitin to the substrate lysine [9] [8].
The HECT domain consists of two structural lobes: an N-lobe that binds the E2~Ub intermediate, and a C-lobe that contains the catalytic cysteine [9] [10]. These lobes are connected by a flexible hinge region that enables large-scale conformational changes during the ubiquitin transfer process [9]. Structural studies have revealed that HECT E3s adopt an "inverted-T conformation" during ubiquitin transfer from E2 to E3, followed by a reorientation into an "L conformation" for the final transfer to the substrate [10]. The N-terminal regions of HECT E3s, which are highly variable in length and sequence, are responsible for substrate recognition and regulatory interactions [4].
Table 2: Comparative Analysis of RING vs. HECT E3 Ligase Families
| Feature | RING E3 Ligases | HECT E3 Ligases |
|---|---|---|
| Catalytic Mechanism | Direct transfer from E2 to substrate [8] | Two-step mechanism with E3~Ub intermediate [9] [8] |
| Intermediate Formation | No covalent E3~Ub intermediate [8] | Covalent thioester-linked E3~Ub intermediate [9] |
| Catalytic Domain | RING domain (zinc-coordinating) [7] | HECT domain (~350 aa) with catalytic Cys [4] |
| Representative Members | RNF4, MIB1, RNF38, XIAP, RNF214 [7] [6] | NEDD4 family, HERC family, TRIP12, UBR5 [4] [10] |
| Structural Feature | Linchpin residue stabilizes E2~Ub closed state [7] | Bilobal HECT domain with flexible hinge [9] |
| Human Family Size | >600 members [7] [6] | 28 members [4] [6] |
Atypical Ubiquitin Chain Specificity: HECT vs. RING E3 Ligases
Ubiquitin Chain Diversity and Functional Consequences
Ubiquitin contains seven lysine residues (K6, K11, K27, K29, K33, K48, K63) and an N-terminal methionine (M1) that can serve as linkage sites for polyubiquitin chain formation [1] [4]. The topology of the ubiquitin chain determines the functional consequence for the modified substrate. While K48-linked chains primarily target substrates for proteasomal degradation, and K63-linked chains are involved in non-proteolytic signaling, the so-called "atypical" chains (including K6, K11, K27, K29, K33) have more specialized and less characterized functions [1] [4].
For instance, K29-linked chains have been associated with proteotoxic stress responses, and when formed as branched chains with K48 linkages, they play roles in regulating diverse substrates in response to oxidative, lipid, and pH stresses [10]. K27-linked chains participate in the DNA damage response, while K33-linked chains have been shown to negatively regulate T-cell receptor signaling [4]. The specificity for generating these atypical linkages varies considerably between different E3 ligase families.
HECT E3 Specificity for Atypical Chains
HECT E3 ligases demonstrate remarkable specificity for generating atypical ubiquitin chains. A prime example is TRIP12, a HECT E3 that specifically forges K29-linked ubiquitin chains and K29/K48-branched chains [10]. Recent structural studies utilizing cryo-EM have revealed that TRIP12 resembles a molecular pincer, with one side comprising tandem ubiquitin-binding domains that engage the proximal ubiquitin to direct its K29 residue toward the ubiquitylation active site [10]. The opposite side of the pincer—the HECT domain—precisely juxtaposes the donor and acceptor ubiquitins to ensure K29 linkage specificity [10].
The mechanism underlying TRIP12's specificity involves tight geometric constraints that position the epsilon amino group of the acceptor lysine precisely relative to the E3~Ub active site [10]. Biochemical assays have demonstrated that TRIP12 preferentially targets K48-linked di-ubiquitin chains over mono-ubiquitin or di-ubs with other linkages, and shows a strong preference for modifying K29 in the proximal ubiquitin of these chains [10]. This specificity is exquisitely sensitive to the acceptor lysine geometry, as side chains shorter or longer than the native lysine tetramethylene linker significantly impair or abolish branched chain formation [10].
RING E3 Versatility in Chain Formation
RING E3 ligases generally exhibit different mechanisms for determining chain specificity. Rather than directly catalyzing specific linkages through precise geometric constraints like HECT E3s, many RING E3s rely more heavily on their cognate E2 enzymes to determine chain topology [7]. The RING domain primarily functions to stabilize the closed conformation of the E2~Ub intermediate, with the identity of the "linchpin" residue playing a crucial role in modulating this stabilization [7].
Different RING E3s show varying degrees of linkage specificity. Some, like the RING-between-RING (RBR) family members, can exhibit strong preferences for specific chain types, while many canonical RING E3s display more versatility in the linkages they can generate [7]. The recent discovery that single substitutions in the RING domain linchpin residue can dramatically alter ubiquitin transfer efficiency suggests that RING E3 activity is highly tunable, potentially allowing for regulation of linkage specificity under different cellular conditions [7].
Diagram Title: Ubiquitin-Proteasome Pathway Overview
Experimental Approaches for Studying E3 Ligase Specificity
The BioE3 System for Substrate Identification
A significant challenge in E3 ligase research has been discriminating between genuine ubiquitination targets and mere interactors. The BioE3 system represents an innovative strategy designed to identify specific substrates of both RING and HECT E3 ligases by combining site-specific biotinylation of ubiquitin-modified substrates with BirA-E3 ligase fusion proteins [6].
The methodology involves several key steps:
- Generation of stable cell lines expressing a bioGEFUb construct under doxycycline control
- Introduction of BirA-E3 fusion proteins into these cells
- Growth in biotin-depleted media to control labeling timing
- Limited biotin labeling during active ubiquitination by the BirA-E3 fusion
- Streptavidin capture and LC-MS/MS identification of biotinylated substrates [6]
This system has been successfully applied to both RING-type E3s (RNF4, MIB1, MARCH5, RNF214) and HECT-type E3s (NEDD4), identifying both known and novel targets and providing insights into the biological roles of these enzymes [6]. The use of a modified AviTag with lower affinity for BirA (bioGEF) was crucial for reducing non-specific background labeling and enabling detection of transient ubiquitylation events [6].
Structural Biology Techniques
Advanced structural biology techniques have been instrumental in elucidating the mechanisms of linkage specificity in E3 ligases. Cryo-electron microscopy (cryo-EM) has enabled visualization of TRIP12 in complex with donor and acceptor ubiquitins, revealing how this HECT E3 positions the acceptor ubiquitin to direct K29-specific linkage [10]. X-ray crystallography has provided high-resolution insights into RING E3/E2~Ub complexes, demonstrating how the linchpin residue stabilizes the closed conformation of the E2~Ub intermediate [7].
Chemical biology approaches have been particularly valuable for trapping transient intermediates in the ubiquitination cascade. For example, the use of chemical warheads to create stable mimics of the transition state during ubiquitylation has allowed structural characterization of the E3~Ub-substrate complex that would otherwise be too transient to capture [10].
Biochemical Assays for Linkage Specificity
Biochemical pulse-chase assays using defined ubiquitin substrates have been essential for quantifying E3 specificity for different chain types. These assays typically employ fluorescently labeled donor ubiquitin that cannot serve as an acceptor (e.g., lysine-less ubiquitin) to track the formation of specific ubiquitin linkages [10]. By testing various acceptor ubiquitins with specific lysine mutations or different linkage types, researchers can determine an E3's preference for particular chain topologies.
Additionally, the use of semi-synthetic ubiquitin substrates containing lysine analogs with varying side chain lengths has provided insights into the geometric constraints of the E3 active site [10]. This approach demonstrated that TRIP12 activity is exquisitely sensitive to the number of methylene groups between the α-carbon and amino group of the acceptor lysine, with the native tetramethylene linker of lysine being optimal for K29/K48-branched chain formation [10].
Table 3: Key Research Reagents and Methods for Studying E3 Specificity
| Tool/Reagent | Function/Application | Key Features |
|---|---|---|
| BioE3 System [6] | Identification of E3-specific substrates | Proximity-dependent biotinylation; works with RING and HECT E3s |
| bioGEFUb [6] | Biotinylatable ubiquitin for BioE3 | Modified AviTag with reduced BirA affinity minimizes background |
| Tandem Ubiquitin-Binding Entities (TUBEs) [2] | Enrichment of polyubiquitinated proteins | High-affinity reagents composed of multiple UBA domains |
| Proteasome Inhibitors (e.g., MG132) [5] | Accumulation of ubiquitinated proteins | Allows detection of otherwise transient ubiquitination events |
| Semi-synthetic Ubiquitin Variants [10] | Probing active site geometry | Lysine analogs with modified side chain lengths |
| Pulse-Chase Biochemical Assays [10] | Quantifying linkage specificity | Uses defined ubiquitin substrates to track chain formation |
Research Reagent Solutions for the UPS Investigator
The study of ubiquitin-proteasome system mechanisms requires specialized reagents and tools. For investigators exploring E3 ligase specificity, several key resources have proven invaluable:
Ubiquitin Enrichment Tools: Tandem Ubiquitin-Binding Entities (TUBEs) are engineered, high-affinity reagents composed of multiple ubiquitin-associated (UBA) domains that bind polyubiquitin chains with enhanced affinity compared to single UBA domains, enabling more efficient enrichment of polyubiquitinated proteins from cell lysates [2]. These can be particularly useful when studying the endogenous ubiquitination status of specific substrates.
Proteasome Inhibitors: Compounds such as MG132 are essential tools for accumulating ubiquitinated proteins that would otherwise be rapidly degraded, facilitating their detection and analysis [5]. When treating cells with proteasome inhibitors, researchers typically observe increased global ubiquitination levels, as demonstrated by western blot analysis with anti-ubiquitin antibodies [5].
Activity-Based Probes: Recent advances in chemical biology have yielded activity-based probes that can trap E3~Ub intermediates or visualize specific ubiquitin linkages, providing insights into the dynamics of ubiquitination in live cells [10]. These probes often employ mechanism-based crosslinkers or warheads that covalently modify the active site of E3s during the ubiquitin transfer process.
Linkage-Specific Antibodies: The development of antibodies that specifically recognize particular ubiquitin linkage types has greatly facilitated the study of atypical chain formation. These reagents allow researchers to monitor changes in specific chain types under different physiological conditions or in response to E3 manipulation.
Diagram Title: RING vs HECT E3 Catalytic Mechanisms
The ubiquitin-proteasome system represents a sophisticated regulatory network that controls protein stability and function through the coordinated action of E1, E2, and E3 enzymes. The divergence in catalytic mechanisms between RING and HECT E3 ligases illustrates the evolution of distinct strategies for achieving substrate specificity and linkage diversity in ubiquitin signaling.
While RING E3s function primarily as scaffolds that facilitate direct ubiquitin transfer from E2 to substrate, HECT E3s employ a two-step mechanism involving a covalent E3~Ub intermediate that provides greater opportunity for regulating the timing and specificity of ubiquitination. This mechanistic difference likely underlies the observed specialization of certain HECT E3s, like TRIP12, for generating specific atypical ubiquitin linkages such as K29-linked and K29/K48-branched chains.
Recent methodological advances, including the BioE3 system for substrate identification and cryo-EM for structural characterization of transient intermediates, have dramatically accelerated our understanding of E3 specificity. These tools, combined with traditional biochemical approaches, continue to reveal the intricate mechanisms by which E3 ligases control cellular processes through targeted protein ubiquitination.
As the field progresses, key challenges remain: understanding how E3 activity is regulated in space and time, elucidating the code governing branched ubiquitin chain specificity, and developing targeted strategies for modulating specific E3-substrate interactions for therapeutic purposes. The continuing refinement of tools for studying ubiquitination, particularly those capable of capturing transient intermediates and mapping ubiquitin linkages in complex cellular environments, will be essential for addressing these questions and advancing both basic science and drug discovery efforts focused on the ubiquitin-proteasome system.
Ubiquitination is a crucial post-translational modification that governs virtually all eukaryotic cellular processes, with E3 ubiquitin ligases serving as the key specificity determinants in this system. Among the >600 human E3s, the Homologous to E6AP C-terminus (HECT) family represents a distinct class of 28 enzymes that employ a unique two-step catalytic mechanism involving a covalent E3~Ub thioester intermediate [11] [12]. Unlike RING E3s that function primarily as scaffolds, HECT E3s directly participate in catalysis by forming a transient thioester bond with ubiquitin before transferring it to substrate proteins [11] [9]. This fundamental mechanistic difference enables HECT E3s to override E2-specific linkage preferences and assemble atypical ubiquitin chains—including K29-linked and branched chains—that function in specialized biological pathways from protein quality control to DNA damage response [10] [12]. This review provides a comprehensive comparison of HECT versus RING E3 mechanisms, with particular focus on the structural basis for HECT E3 specificity toward atypical ubiquitin chain formation, supported by recent experimental findings and methodological advances.
Catalytic Mechanisms: Fundamental Differences Between HECT and RING E3 Ligases
The Two-Step Thioester Mechanism of HECT E3s
HECT E3s employ a conserved catalytic mechanism that fundamentally differs from RING-type E3s. The process begins with the HECT domain's N-lobe interacting with an E2~Ub thioester intermediate. ubiquitin is then transferred from the E2 to the catalytic cysteine residue within the HECT C-lobe, forming a labile HECT~Ub thioester intermediate [11] [9]. In the decisive second step, the HECT domain repositions into an L-shaped conformation to facilitate ubiquitin transfer from the E3 to specific lysine residues on substrate proteins [10]. This two-step mechanism allows HECT E3s to exert greater control over the specificity of ubiquitin chain linkages compared to RING E3s [9] [12].
The following diagram illustrates this unique catalytic cycle:
The Single-Step Scaffold Mechanism of RING E3s
In contrast to HECT E3s, RING-type E3s function as molecular scaffolds that facilitate direct ubiquitin transfer from E2~Ub to substrates without forming a covalent E3~Ub intermediate [11] [13]. The RING domain binds both the E2~Ub complex and substrate, positioning them in close proximity to enable ubiquitin transfer [7]. A critical feature of this mechanism is the "linchpin" residue—typically an arginine—within the RING domain that stabilizes the closed conformation of E2~Ub, optimizing the thioester bond for nucleophilic attack by the substrate lysine [7]. This single-step mechanism means RING E3s largely depend on their cognate E2 enzymes to determine linkage specificity.
Table 1: Fundamental Mechanistic Differences Between HECT and RING E3 Ligases
| Feature | HECT E3s | RING E3s |
|---|---|---|
| Catalytic Mechanism | Two-step with covalent E3~Ub intermediate | Single-step without covalent intermediate |
| Ubiquitin Transfer | E2 → HECT Cys → Substrate | E2 → Substrate |
| Key Structural Domains | Bilobal HECT domain (N-lobe, C-lobe) | RING domain (Zn²⁺ coordination) |
| Role of E3 | Direct catalytic participant | Allosteric activator/scaffold |
| Linkage Determination | Primarily by E3 | Primarily by E2 |
| Representative Members | NEDD4, HACE1, TRIP12, HUWE1 | CBL, MDM2, APC11, BRCA1/BARD1 |
Structural Basis for Atypical Ubiquitin Chain Formation by HECT E3s
Specialization for Atypical Linkages: K29 and Beyond
Recent structural studies have revealed how specific HECT E3s achieve remarkable specificity for atypical ubiquitin linkages. TRIP12, a human HECT E3 associated with neurodegenerative disorders, specializes in forming K29-linked ubiquitin chains and K29/K48-branched chains [10]. Cryo-EM structures of TRIP12 during active ubiquitination reveal a pincer-like architecture: one side consists of tandem ubiquitin-binding domains that engage the proximal ubiquitin and position its K29 residue toward the active site, while the HECT domain forms the opposite side, precisely juxtaposing donor and acceptor ubiquitins [10]. This specialized arrangement ensures K29 linkage specificity, with biochemical assays demonstrating that TRIP12 preferentially modifies K48-linked di-ubiquitin chains over other acceptors, targeting K29 on the proximal ubiquitin [10].
Similarly, the yeast HECT E3 Tom1 employs a "structural ubiquitin" mechanism to ensure fidelity in K48-linked chain assembly. Cryo-EM snapshots of Tom1 during active ubiquitination reveal a non-canonical ubiquitin-binding site within the solenoid-shaped region that coordinates a structural ubiquitin molecule, contributing to linkage specificity [14]. This mechanism illustrates how HECT E3s can use ubiquitin itself as a structural component to guide proper chain formation.
Structural Transitions and Linkage Switching
Some HECT E3s exhibit remarkable flexibility in their linkage specificity through structural transitions. Studies on WWP1, a NEDD4 family HECT E3, demonstrate that ubiquitin chain formation occurs in two distinct phases: an initial phase where chains are synthesized unidirectionally through Lys-63, followed by a second phase characterized by multidirectional elongation with mixed ubiquitin linkages and branched structures [15]. This linkage switching depends on a low-affinity, noncovalent ubiquitin-binding site within the HECT domain, revealing how conformational dynamics can enable a single E3 to generate diverse ubiquitin signals [15].
Table 2: Experimentally Determined Linkage Specificities of Characterized HECT E3s
| HECT E3 | Organism | Primary Linkages | Structural Features | Biological Functions |
|---|---|---|---|---|
| TRIP12 | Human | K29-linked, K29/K48-branched | Pincer-like architecture with tandem Ub-binding domains | DNA damage response, proteotoxic stress |
| Tom1 | Yeast | K48-linked | Non-canonical Ub-binding site in solenoid region | Ubiquitin chain amplification, quality control |
| WWP1 | Human | K63 > K48 > K11 | Low-affinity Ub-binding site in HECT domain | Sequential addition with linkage switching |
| HUWE1 | Human | Multiple atypical linkages | Extended region adjacent to HECT domain | Regulation of apoptosis, DNA repair |
| HACE1 | Human | K48-linked (on RAC1) | Ankyrin repeats, middle domain | Redox homeostasis, tumor suppression |
Experimental Approaches for Studying HECT E3 Mechanisms
Key Methodologies and Technical Advances
Understanding HECT E3 mechanisms has required sophisticated biochemical and structural approaches. Pulse-chase biochemical assays using fluorescently-labeled ubiquitin variants have been instrumental in characterizing linkage specificity and kinetics [10]. These assays typically employ lysine-free donor ubiquitin (*Ub(K0)) that can be tracked via SDS-PAGE migration, allowing researchers to monitor transfer through HECT E3s to specific acceptor ubiquitins with defined linkages [10].
Structural biology breakthroughs, particularly cryo-electron microscopy (cryo-EM), have provided unprecedented views of full-length HECT E3s during catalysis. For TRIP12, researchers employed mechanism-based crosslinking strategies to capture stable mimics representing transition states during ubiquitylation [10]. Similarly, studies on HACE1 combined cryo-EM with solution-based methods including small-angle X-ray scattering (SAXS) and hydrogen-deuterium exchange mass spectrometry (HDX-MS) to elucidate autoinhibition and substrate recognition mechanisms [16].
Chemical biology tools have proven invaluable for probing HECT E3 mechanisms. Activity-based probes (ABPs) featuring C-terminal reactive groups (e.g., Ub-MES) mimic ubiquitin's chemistry and enable covalent capture of HECT~Ub intermediates [13]. The UbFluor-SH assay conjugates fluorescein-thiol to Ub-MES, generating a probe that reacts with the HECT catalytic cysteine to form HECT~Ub while releasing fluorescent Fluor-SH, detectable by fluorescence polarization [13]. This approach enables high-throughput screening for HECT E3 inhibitors without confounding effects from E1, E2, or ATP.
The following workflow illustrates a typical experimental pipeline for structural mechanism determination:
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Studying HECT E3 Mechanisms
| Reagent/Tool | Function/Application | Key Features | Example Usage |
|---|---|---|---|
| Ub-MES | Activity-based probe | Mercaptoethanesulfonate-activated ubiquitin | Forms HECT~Ub intermediates without E1/E2 [13] |
| UbFluor-SH | HTS-compatible probe | Fluorescein-thiol conjugate of Ub-MES | Fluorescence polarization detection of HECT activity [13] |
| K0 Ubiquitin | Donor ubiquitin in assays | Lysine-free mutant (no acceptor sites) | Tracks single ubiquitin transfer events [10] |
| Linkage-specific Di-Ub | Defined acceptor substrates | Recombinant di-ubiquitin with specific linkages | Determines linkage preference in pulse-chase assays [10] |
| Chemical Crosslinkers | Trapping transient complexes | Covalent stabilization of E3-substrate complexes | Cryo-EM sample preparation (e.g., TRIP12, HACE1) [10] [16] |
| HECT Cys Mutants | Catalytic mechanism studies | Cysteine-to-serine/alanine mutations | Traps HECT~Ub intermediate; studies transfer steps [9] |
Discussion: Biological Implications and Therapeutic Opportunities
The specialized ability of HECT E3s to generate atypical ubiquitin linkages has profound biological implications. K29-linked chains have been associated with proteotoxic stress responses and quality control pathways, while K29/K48-branched chains serve as potent degradation signals [10] [12]. The finding that TRIP12 preferentially modifies K48-linked di-ubiquitin chains to create K29/K48-branched structures suggests a hierarchical organization of ubiquitin signaling, where HECT E3s can amplify or modify signals initiated by other E3s [10].
From a therapeutic perspective, HECT E3s represent attractive drug targets due to their involvement in human diseases including cancer, neurological disorders, and autoimmunity [12]. The unique catalytic mechanism involving the HECT~Ub intermediate offers opportunities for selective inhibition. The development of Ub-MES and UbFluor-SH probes enables high-throughput screening for HECT-specific inhibitors that don't affect RING E3s [13]. Furthermore, the recent structural insights into full-length HECT E3s like HACE1 and TRIP12 provide blueprints for structure-based drug design targeting either the catalytic HECT domain or specific substrate-binding interfaces [10] [16].
HECT E3 ligases employ a distinctive two-step catalytic mechanism centered on a covalent E3~Ub thioester intermediate that differentiates them from RING-type E3s. This mechanism, combined with diverse structural arrangements including pincer-like architectures, specialized ubiquitin-binding sites, and conformational flexibility, enables HECT E3s to exhibit remarkable specificity for atypical ubiquitin linkages such as K29-linked and branched chains. Recent methodological advances in cryo-EM, chemical biology, and biochemical assays have provided unprecedented insights into these mechanisms, revealing how HECT E3s achieve linkage specificity through precise positioning of donor and acceptor ubiquitins. These findings not only advance our fundamental understanding of ubiquitin signaling but also open new avenues for therapeutic intervention targeting HECT E3s in human diseases.
Ubiquitin ligases (E3s) are pivotal for conferring specificity within the ubiquitin-proteasome system, with RING-type E3s representing the largest class. This guide provides a comparative analysis of the RING E3 mechanism against the HECT and RBR families, focusing on their role as allosteric scaffolds that facilitate direct ubiquitin transfer. We detail the defining one-step mechanism of RING E3s, supported by key experimental evidence, quantitative data on allosteric activation, and essential protocols. The content is structured to serve as a practical resource for researchers in enzymology and drug development, offering clear comparisons, visual workflows, and a catalog of critical research reagents.
The ubiquitination cascade involves a sequential trio of enzymes: a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2), and a ubiquitin ligase (E3). The E3 ligases are the primary determinants of substrate specificity and are categorized into three major families based on their catalytic mechanisms [11] [17]. RING (Really Interesting New Gene) E3s function as allosteric scaffolds to directly transfer ubiquitin from an E2 to a substrate in a single step. In contrast, HECT (Homologous to the E6AP C-Terminus) E3s and RBR (RING-Between-RING) E3s employ a two-step mechanism involving a covalent E3-ubiquitin thioester intermediate [11] [18] [17]. The mammalian genome encodes over 600 RING-type E3s, which are implicated in virtually every cellular process and are increasingly attractive therapeutic targets [11] [19]. This guide objectively compares the core mechanistic features of RING E3s against alternative E3 families, with a specific emphasis on experimental approaches for studying their allosteric activation and scaffold functions.
Comparative Catalytic Mechanisms: RING vs. HECT vs. RBR
Understanding the fundamental differences in catalytic mechanism is essential for comparing E3 ligase families. The table below summarizes the core characteristics of RING E3s in direct contrast to HECT and RBR E3s.
Table 1: Comparative Catalytic Mechanisms of E3 Ubiquitin Ligase Families
| Feature | RING E3s | HECT E3s | RBR E3s |
|---|---|---|---|
| Catalytic Mechanism | One-step, direct transfer | Two-step, with covalent intermediate | Two-step, with covalent intermediate (RING-HECT hybrid) |
| Ubiquitin Thioester Intermediate | No | Yes, on HECT domain Cys | Yes, on RING2 domain Cys |
| Role of E3 | Allosteric scaffold & activator | Catalytic intermediate | RING1: E2-binding; RING2: Catalytic intermediate |
| Primary Determinant of Linkage Specificity | E2 enzyme and RING-E2 combination | HECT C-lobe domain | RBR E3 itself (e.g., specific RING2 domains) |
| Representative Examples | CBL, BRCA1/BARD1, APC11, Mdm2 | E6AP, NEDD4L, UBR5 | Parkin, HOIP, HHARI |
The core mechanistic divergence lies in the formation of a covalent E3~Ub intermediate. RING E3s notably lack this intermediate. Instead, the RING domain binds an E2~Ub thioester conjugate and acts as a scaffold to position the E2~Ub in close proximity to the substrate, facilitating direct ubiquitin transfer [11] [19]. Furthermore, structural studies indicate that RING domains can allosterically activate the E2~Ub conjugate, enhancing the rate of ubiquitin discharge [20].
The following diagram illustrates the distinct catalytic pathways for each E3 family, highlighting the one-step direct transfer of RING E3s versus the two-step transthiolation mechanisms of HECT and RBR E3s.
Key Experimental Evidence for the RING E3 Allosteric Scaffold Model
The model of RING E3s as allosteric scaffolds is supported by foundational biochemical and structural experiments. Key findings include the stimulation of E2 ubiquitin discharge and the identification of allosteric networks within the E2 enzyme.
Allosteric Activation of E2~Ub Discharge
A critical experiment demonstrated that minimal RING domains, even in the absence of a substrate, can significantly enhance the rate of ubiquitin release from the E2~Ub thioester. This was conclusively shown using E3s like Apc2/11 (a subunit of the Anaphase-Promoting Complex/Cyclosome) and CNOT4 [20]. In this experiment, the formation of the E2~Ub thioester (UbcH5b~Ub) was first allowed, followed by the addition of a RING E3. The decay of the thioester band was monitored over time via non-reducing SDS-PAGE and immunoblotting. The results demonstrated that the RING E3 stimulated the discharge of ubiquitin from UbcH5b, indicating it does more than merely bring partners together—it actively promotes the catalytic readiness of the E2 [20].
Table 2: Quantitative Data on RING E3-Mediated Stimulation of E2~Ub Discharge
| Experimental Component | Key Finding | Experimental System |
|---|---|---|
| UbcH5b ~Ub Thioester + Apc2/11 | Rapid stimulation of Ub discharge | In vitro reconstitution with purified human proteins |
| UbcH5b ~Ub Thioester + CNOT4 | Rapid stimulation of Ub discharge | In vitro reconstitution with purified human proteins |
| UbcH5b Mutants (e.g., I37A) | Defective E3-stimulated discharge despite intact E2~Ub formation and E3 binding | In vitro mutagenesis and activity assays |
| Statistical Coupling Analysis (SCA) | Identified a conserved network of co-evolving residues connecting the E3-binding site to the active site in E2s | Bioinformatics analysis of 345 E2 sequences |
Protocol: E2~Ub Thioester Discharge Assay
This protocol is adapted from Ozkan et al. (2005) and is fundamental for assessing RING E3 allosteric function [20].
Thioester Formation Reaction:
- Combine in a 10 µL reaction: 1 µg (0.9 µM) of human E1 enzyme, 1 µg (5 µM) of E2 (e.g., UbcH5b), and 10 µg (130 µM) of ubiquitin.
- Use a reaction buffer containing 10 mM HEPES (pH 7.5), 100 mM NaCl, 40 µM ATP, and 2 mM MgCl₂.
- Incubate for 10 minutes at room temperature to form the E2~Ub thioester.
- (Optional) To stop further E1 activity and synchronize the reaction, add 0.1 unit/µL of apyrase to deplete ATP or 1-5 mM N-ethylmaleimide (NEM) to alkylate the E1 active site cysteine.
E3 Addition and Discharge:
- Add the purified RING E3 protein (e.g., 2.3 µM Apc2/11 or 44 µM CNOT4 fragment) to the reaction mix.
- Take sample aliquots at various time points (e.g., 0, 2, 5, 10, 20 minutes).
Analysis:
- Immediately stop each aliquot by adding SDS-PAGE sample buffer lacking reducing agents (e.g., β-mercaptoethanol or DTT) to preserve the thioester bond.
- Resolve the samples by SDS-PAGE (4-20% gradient gel) and transfer to a membrane for immunoblotting.
- Probe the blot with an antibody against the E2 (e.g., anti-UbcH5).
- Quantify the intensity of the E2~Ub thioester band over time using a densitometer. The data can be fitted to a single-exponential decay curve to model discharge rates.
Identifying Allosteric Networks in E2 Enzymes
The discovery that RING E3 binding allosterically activates E2s prompted investigations into the communication pathways within the E2 structure. Statistical Coupling Analysis (SCA) of a multiple sequence alignment of 345 E2s identified clusters of co-evolving residues that form a physical network connecting the E3-binding site to the distant active site [20]. This provided evolutionary and bioinformatic support for allosteric communication. Subsequent mutagenesis studies validated this model; for instance, the UbcH5b I37A mutation, which is distant from both the active site and the E3-binding interface, was shown to be defective in E3-stimulated discharge while retaining normal E2~Ub thioester formation and E3-binding capacity [20]. This residue is part of the identified allosteric network, confirming its functional role.
The Scientist's Toolkit: Key Research Reagents and Experimental Solutions
Studying RING E3 mechanisms requires a specific set of biochemical and molecular tools. The following table details essential reagents, their functions, and application notes.
Table 3: Research Reagent Solutions for Studying RING E3 Mechanisms
| Reagent / Solution | Function / Purpose | Key Features & Notes |
|---|---|---|
| E2 Enzymes (e.g., UbcH5b, UbcH7) | Core catalytic component; forms thioester with Ub; subject to allosteric activation. | UbcH5 family is often used for its versatility and strong activity with many RING E3s. UbcH7 is also common, particularly with RBR E3s [20] [18]. |
| Minimal RING Domains (e.g., CNOT41-78, Cbl RING) | Isolated functional unit for probing allosteric scaffolding without confounding effects of full-length protein regulation. | Allows for simplified in vitro biochemistry and structural studies (NMR, X-ray crystallography) [20]. |
| Stable E2~Ub Mimetics (e.g., UbcH7(C86K)-Ub) | Non-hydrolyzable mimic of the E2~Ub thioester conjugate for structural and binding studies. | The E2 active site Cys is mutated to Lys, forming an isopeptide bond with Ub, allowing stable complex purification [18]. |
| Wild-type and Mutant Ubiquitin | To study linkage specificity (e.g., K48R, K63R) or create defined chain types (e.g., K48-diUb, K63-diUb). | Essential for probing the role of specific ubiquitin chain linkages in allosteric activation, especially for HECT and RBR E3s [21] [18]. |
| Active-Site Cysteine Mutants (E3 Cys-to-Ala) | Generates catalytically dead E3 for trapping intermediate complexes or studying non-covalent functions. | Critical for HECT and RBR E3 studies to prevent transthiolation and stabilize E2~Ub/E3 interactions [18]. |
| Non-reducing SDS-PAGE | Electrophoretic method to detect and monitor labile thioester bonds. | Standard technique for E2~Ub and E3~Ub discharge assays; requires omission of reducing agents in sample buffer [20]. |
Visualizing the Allosteric Activation Workflow
The following diagram synthesizes the key concepts and experimental workflow for establishing the allosteric scaffold model of RING E3s, integrating bioinformatic, structural, and biochemical validation steps.
Ubiquitination is a crucial post-translational modification that regulates virtually every cellular process in eukaryotes, from protein degradation to immune signaling [11]. This modification involves the sequential action of E1 (activating), E2 (conjugating), and E3 (ligating) enzymes that attach the 76-amino acid protein ubiquitin to substrate proteins. The complexity of ubiquitin signaling arises from the ability of ubiquitin itself to form chains through its seven internal lysine residues (K6, K11, K27, K29, K33, K48, K63) or its N-terminal methionine (M1) [22] [4]. For decades, research has predominantly focused on the canonical K48-linked chains (targeting proteins for proteasomal degradation) and K63-linked chains (involved in signaling and DNA repair). However, recent advances have revealed the biological significance of the remaining "atypical" ubiquitin linkages, which include K6, K11, K27, K29, and K33 connections [23].
The specificity of ubiquitin chain formation is largely determined by E3 ubiquitin ligases, which are categorized into three major families: RING (Really Interesting New Gene), HECT (Homologous to E6AP C-terminus), and RBR (RING-between-RING) types [24] [25]. With over 600 E3s encoded in the human genome, these enzymes provide the specificity that governs cellular ubiquitination patterns. This review systematically compares how HECT and RING E3 ligases recognize, synthesize, and regulate atypical ubiquitin chains, providing researchers in drug development with a framework for understanding this complex regulatory system and its therapeutic implications.
Structural and Mechanistic Divergence Between HECT and RING E3 Ligases
Fundamental Catalytic Mechanisms
HECT and RING E3 ligases employ fundamentally different catalytic mechanisms for ubiquitin transfer, which significantly influences their approach to atypical chain formation:
HECT E3 Mechanism: HECT ligases form an obligate thioester intermediate with ubiquitin before transferring it to substrates. The conserved ~350 amino acid HECT domain consists of two lobes: an N-lobe that binds the E2~Ub complex and a C-lobe containing the catalytic cysteine that forms the transient thioester bond with ubiquitin [11] [25]. These lobes are connected by a flexible hinge region that enables the C-lobe to reposition during ubiquitin transfer. This two-step mechanism—first accepting ubiquitin from E2, then transferring to substrate—gives HECT E3s direct control over linkage specificity [25].
RING E3 Mechanism: In contrast, RING-type E3s function primarily as scaffolds that bring the E2~Ub complex into close proximity with the substrate, facilitating direct ubiquitin transfer without a covalent intermediate [11] [26]. Canonical RING fingers are Zn²⁺-coordinating domains that simultaneously bind E2 and substrate. Some RING E3s may also allosterically activate E2s for enhanced ubiquitin transfer [11].
Table 1: Fundamental Comparison of HECT vs. RING E3 Catalytic Mechanisms
| Feature | HECT E3 Ligases | RING E3 Ligases |
|---|---|---|
| Catalytic Mechanism | Two-step transthiolation with covalent E3~Ub intermediate | Direct transfer from E2 to substrate without covalent intermediate |
| Intermediate Formation | Obligate thioester with catalytic cysteine in HECT domain | No catalytic intermediate |
| Role in Specificity | Direct control over linkage specificity through HECT domain architecture | Primarily brings E2 and substrate together; specificity influenced by E2 identity |
| Structural Features | Bi-lobal HECT domain with flexible hinge; ~28 members in humans | Zn²⁺-coordinating RING domain; >600 members in humans |
| Representative Members | NEDD4, TRIP12, HUWE1, E6AP | Cbl, TRAF, cIAP, BRCA1/BARD1 |
Structural Basis for Atypical Chain Formation
Recent structural studies have illuminated how specific E3 ligases achieve linkage specificity. For HECT E3s, the C-lobe positioning and specific ubiquitin-binding exosites determine which lysine on the acceptor ubiquitin is presented to the catalytic center. The recent cryo-EM structure of TRIP12 (a HECT E3) in complex with ubiquitin components revealed a "pincer-like" architecture that precisely orients the acceptor ubiquitin to facilitate K29-linked chain formation [10]. The structure shows tandem ubiquitin-binding domains engaging the proximal ubiquitin to direct its K29 toward the active site, while selectively capturing a distal ubiquitin from a K48-linked chain to form branched conjugates.
For RING E3s, linkage specificity is often dictated by the combination of E2 identity and additional substrate recognition domains. For instance, the anaphase-promoting complex/cyclosome (APC/C), a multi-subunit RING E3, collaborates with UBE2S to generate K11-linked chains during cell cycle regulation [23]. Structural studies have shown that the E2 enzyme UBE2S contains a specialized acceptor-binding region that positions K11 of the acceptor ubiquitin for chain elongation.
Comparative Analysis of Atypical Ubiquitin Chain Specificity
K6-Linked Chains
K6-linked ubiquitin chains have emerged as important regulators in DNA damage response and mitophagy, with both HECT and RING E3s contributing to their formation:
HECT E3 Involvement: HUWE1 generates K6-, K11-, and K48-linked polyubiquitin chains, with K6 linkages particularly prominent in DNA damage contexts [25] [23]. HUWE1-generated K6-linked chains can serve as degradation signals for substrates like Mfn2, and also play non-proteolytic roles in regulating mitochondrial integrity.
RING E3 Involvement: The Parkin RBR E3 ligase (functionally similar to RING E3s) decorates damaged outer mitochondrial membrane proteins with K6, K11, K48, and K63-linked chains during mitophagy, with K6 and K63 linkages particularly important for designating mitochondria for destruction [23].
Table 2: E3 Ligases Involved in Atypical Ubiquitin Chain Formation and Their Functional Roles
| Ubiquitin Linkage | E3 Ligase | E3 Family | Substrate/Context | Functional Outcome |
|---|---|---|---|---|
| K6 | HUWE1 | HECT | Mfn2, DDR proteins | Proteasomal degradation; DNA damage response |
| K6 | Parkin | RBR | Mitochondrial proteins | Mitophagy initiation |
| K11 | APC/C (UBE2S) | RING | Cell cycle regulators | Mitotic progression; proteasomal degradation |
| K11 | RNF26 | RING | STING | Inhibits STING degradation, enhancing IFN production |
| K27 | TRIM23 | RING | NEMO | Activates NF-κB and IRF3 pathways |
| K27 | TRIM40 | RING | RIG-I, MDA5 | Proteasome-mediated degradation, inhibiting IFN response |
| K29 | TRIP12 | HECT | Multiple substrates | Proteotoxic stress response; protein degradation |
| K29 | SCF-Fbx21 | RING | ASK1 | Induces IFNβ and IL-6 production |
| K33 | RNF2 | RING | STAT1 | Suppresses ISG transcription |
K11-Linked Chains
K11-linked ubiquitination is particularly associated with cell cycle regulation and immune signaling:
RING E3 Specificity: The multi-subunit RING E3 APC/C, in cooperation with the E2 enzyme UBE2S, specializes in generating K11-linked chains to control mitotic progression [23]. APC/C with UBE2S generates K11-linked branch-offs on ubiquitin chains that target cyclins and other cell cycle regulators for proteasomal degradation.
HECT E3 Capabilities: While less prominent in K11 linkage formation, some HECT E3s like HUWE1 can generate K11-linked chains alongside other linkage types [25].
K27-Linked Chains
K27-linked chains have gained attention for their crucial roles in innate immune signaling, with both RING and HECT E3s participating:
RING E3 Dominance: Multiple RING-type TRIM family E3s generate K27-linked chains to regulate immune signaling pathways. TRIM23 catalyzes K27-linked ubiquitination of NEMO, leading to NF-κB and IRF3 activation [22]. Conversely, TRIM40-mediated K27 ubiquitination of RIG-I and MDA5 induces their proteasomal degradation, thereby inhibiting type I interferon response [22].
HECT E3 Contributions: While less characterized in K27 linkage formation, some HECT E3s may contribute to K27 chain assembly in specific contexts, though the current literature primarily documents RING E3 involvement for this linkage type.
K29-Linked Chains
K29-linked chains are associated with proteotoxic stress responses and targeted protein degradation, with HECT E3s playing prominent roles:
HECT E3 Specialization: TRIP12 exemplifies HECT E3 specialization for K29 linkages, forming both homotypic K29 chains and K29/K48-branched chains [10]. Structural studies reveal that TRIP12's preference for K29 linkage formation stems from precise geometric constraints that position the K29 residue of the acceptor ubiquitin optimally in the active site.
RING E3 Involvement: The SCF-Fbx21 RING E3 complex assembles K29-linked chains on ASK1 to induce IFNβ and IL-6 production [22], demonstrating that multiple E3 families can target this linkage to different substrates.
K33-Linked Chains
K33-linked chains represent one of the least understood atypical linkages, though emerging evidence suggests roles in immune regulation:
- RING E3 Examples: RNF2 catalyzes K33-linked ubiquitination of STAT1 to suppress interferon-stimulated gene (ISG) transcription [22]. This demonstrates how atypical chains can exert inhibitory effects on signaling pathways.
Experimental Approaches for Studying Atypical Ubiquitin Chains
Biochemical and Structural Methods
Understanding E3 specificity for atypical chains requires specialized experimental approaches:
In Vitro Ubiquitination Assays: Pulse-chase assays using fluorescently-labeled donor ubiquitin (lacking lysines to prevent chain formation) with defined acceptor ubiquitins enable precise determination of linkage specificity [10]. For TRIP12 studies, researchers used *Ub(K0) to track specific ubiquitin transfer to various di-Ub acceptors, revealing strong preference for K48-linked di-Ubs as acceptors for K29 branching.
Cryo-EM Structural Analysis: Recent advances in cryo-EM have enabled visualization of E3-ubiquitin complexes. For TRIP12, researchers employed a chemical warhead strategy to trap a stable mimic of the transition state during K29-linked chain formation, revealing the "pincer" architecture that dictates linkage specificity [10].
Linkage-Specific Reagents: The development of linkage-specific antibodies and ubiquitin-binding domains (UBDs) has been crucial for detecting atypical chains in cellular contexts. For example, the UBAN domain of NEMO shows preference for linear chains but can also bind longer K63-linked chains, requiring careful experimental interpretation [22].
Mass Spectrometry-Based Proteomics
Advanced proteomic approaches have become indispensable for mapping atypical ubiquitination:
Absolute Quantification (AQUA) Mass Spectrometry: This approach uses synthetic, stable isotope-labeled ubiquitin peptides as internal standards to precisely quantify different ubiquitin linkage types in cellular extracts. This method revealed the abundance of K48-K63 branched chains in mammalian cells and their regulation in NF-κB signaling [27].
Di-Gly Remnant Profiling: Enrichment of tryptic peptides containing the di-glycine remnant left after trypsin digestion of ubiquitinated lysines enables proteome-wide mapping of ubiquitination sites, though distinguishing chain linkage types remains challenging.
Experimental Workflow for Characterizing Atypical Ubiquitin Chains
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for Studying Atypical Ubiquitin Chains
| Reagent Category | Specific Examples | Research Application | Key Considerations |
|---|---|---|---|
| Ubiquitin Mutants | Ub(K0), Ub(K6-only), Ub(K11-only), etc. | Defining linkage specificity in vitro | K0 ubiquitin (all lysines mutated) prevents chain formation; K-only variants contain single lysine |
| Linkage-Specific Binders | UBAN domain (linear), NZF domain (K63) | Detecting specific chain types in cells | Varying affinities and specificities; some domains recognize multiple linkages |
| Activity-Based Probes | Ub-VS, Ub-AMC, Di-Ub probes | Monitoring DUB activity and specificity | Warhead-based probes trap catalytic intermediates |
| E3 Expression Constructs | TRIP12, HUWE1, TRIM E3s, APC/C subunits | Recombinant protein production and cellular studies | Multi-subunit E3s require co-expression; tags may affect activity |
| Mass Spectrometry Standards | AQUA peptides, SILAC ubiquitin | Quantifying linkage abundance | Synthetic heavy-labeled peptides as internal standards |
| Chemical Biology Tools | TRIP12 warhead complex (cryo-EM trapping) | Structural studies of E3 mechanisms | Creates stable mimics of transition states for structural biology |
Biological Significance and Therapeutic Implications
Atypical Chains in Disease Pathways
The involvement of atypical ubiquitin chains in human diseases highlights their therapeutic relevance:
Neurodegenerative Disorders: TRIP12, which generates K29-linked and K29/K48-branched chains, has been associated with neurodegenerative diseases and autism spectrum disorders [10]. The proper regulation of ubiquitin chain topology appears crucial for neuronal health.
Cancer and Immune Disorders: Multiple E3s generating atypical chains are implicated in cancer pathogenesis. For instance, RNF26-mediated K11-linked ubiquitination of STING inhibits its degradation, amplifying antiviral responses [22], suggesting potential applications in immunotherapy or antiviral drug development.
Metabolic Diseases: Emerging evidence connects E3 ligases and their adaptors to metabolic diseases, positioning them as potential therapeutic targets for conditions like diabetes, NAFLD, and obesity [24].
Branching and Heterotypic Chains
Beyond homotypic chains, the emerging complexity of branched and heterotypic ubiquitin chains adds another layer of regulation:
K48-K63 Branched Chains: The E3 ligase HUWE1 cooperates with TRAF6 to assemble K48 branches on K63 chains, creating K48-K63 branched chains that regulate NF-κB signaling [27]. These branched linkages permit recognition by TAB2 while protecting K63 linkages from CYLD-mediated deubiquitylation, thereby amplifying NF-κB signals.
Functional Cooperation: The formation of branched chains represents a potential point of cooperation between different E3 ligase families, with one E3 establishing the initial chain and another adding branching modifications to create specific topological signals.
Atypical Ubiquitin Chains in Innate Immune Signaling Pathways
The expanding landscape of atypical ubiquitin chains reveals remarkable sophistication in how HECT and RING E3 ligases orchestrate specific cellular signals through controlled ubiquitin chain topology. While RING E3s dominate in number and diversity, HECT E3s often exhibit more direct control over linkage specificity through their catalytic mechanisms. The emerging patterns suggest that RING E3s frequently collaborate with specific E2s to determine chain topology, while HECT E3s intrinsically dictate linkage specificity through their structural architecture.
Future research directions will need to address several key challenges: (1) developing more sensitive tools for detecting and quantifying atypical chains in cellular contexts; (2) understanding the complex interplay between different E3 families in establishing the cellular ubiquitin landscape; and (3) elucidating the full spectrum of readers and erasers that interpret and regulate atypical ubiquitin signals. As our understanding of these non-canonical ubiquitin linkages grows, so too will opportunities for therapeutic intervention in the many diseases characterized by dysregulated ubiquitination.
For drug development professionals, targeting the specific interfaces between E3 ligases and their cognate E2 enzymes or substrates offers potential for highly selective therapeutics with fewer off-target effects than general proteasome inhibitors. The distinct structural features of HECT and RING E3 ligases provide multiple entry points for therapeutic modulation, from small molecules that disrupt specific E3-substrate interactions to bifunctional compounds that redirect E3 activity toward disease-causing proteins.
The specificity of ubiquitin signaling—a process governing virtually every aspect of eukaryotic cell biology—is largely determined by two major classes of E3 ubiquitin ligases: HECT (Homologous to E6AP C Terminus) and RING (Really Interesting New Gene) [28] [29]. While both catalyze the transfer of ubiquitin to target proteins, they employ fundamentally different mechanisms to achieve specificity in substrate selection and ubiquitin chain topology. HECT E3s utilize a two-step catalytic mechanism involving a direct thioester-linked intermediate with ubiquitin, with linkage specificity primarily dictated by structural features within the C-lobe of the catalytic HECT domain [30] [10] [31]. In contrast, RING E3s facilitate direct ubiquitin transfer from E2 enzymes to substrates, with specificity emerging from highly selective RING-E2 partnerships that determine which E2~Ub conjugates a given RING can engage [28] [32] [33]. This comparison guide examines the structural determinants underlying these distinct specificity mechanisms, with particular focus on their implications for generating atypical ubiquitin chain linkages (K6, K27, K29, K33) that represent emerging targets for therapeutic intervention [12].
Comparative Structural Architecture and Catalytic Mechanisms
HECT E3 Ligases: C-lobe Directed Specificity
HECT E3 ligases feature a conserved bilobed catalytic domain where the N-lobe binds the E2~Ub conjugate and the C-lobe contains the catalytic cysteine that forms a transient thioester with ubiquitin before its transfer to substrates [30] [31]. The C-terminal lobe plays the predominant role in determining linkage specificity through several key structural features:
- Acceptor Ub Positioning Elements: The C-lobe contains specific surfaces that orient the acceptor ubiquitin to present a particular lysine residue to the thioester-bonded donor ubiquitin. For example, in TRIP12, which forges K29-linked chains, the C-lobe precisely juxtaposes the donor and acceptor ubiquitins to position K29 for isopeptide bond formation [10].
- Linkage-Specific Binding Pockets: Structural studies reveal that HECT C-lobes contain specialized pockets that accommodate the side chain of the targeted lysine residue. In UBR5, which synthesizes K48-linked chains, an intricate web of interactions between the acceptor ubiquitin, UBR5 elements, and the donor ubiquitin strategically positions K48 in the active site [31].
- C-terminal Tail Regulation: The extreme C-terminal tail of HECT domains plays a critical regulatory role. In AREL1, deletion of the last three amino acids completely abrogated autoubiquitination and reduced substrate ubiquitination activity [34].
Table 1: Structural Elements Determining Linkage Specificity in HECT E3 C-lobes
| Structural Element | Function in Specificity | Example E3 | Linkage Formed |
|---|---|---|---|
| Acceptor Ub binding surfaces | Positions acceptor Ub for specific lysine presentation | TRIP12, UBR5 | K29, K48 |
| Catalytic cleft residues | Directs target lysine side chain orientation | E6AP, NEDD4-2 | K48, K63 |
| C-terminal tail | Regulates catalytic activity and Ub transfer | AREL1 | K33/K11 |
| N-lobe/C-lobe hinge | Enables conformational changes for catalysis | All HECTs | Variable |
| Exosite regions | Binds regulatory Ub molecules | NEDD4 family | K63 |
RING E3 Ligases: E2 Partnership Specificity
RING E3 ligases employ a fundamentally different strategy, functioning as allosteric activators that bridge E2~Ub conjugates with substrates without forming a covalent intermediate [28] [29]. Their specificity derives from selective partnerships with particular E2 enzymes:
- E2 Recognition Surfaces: RING domains interact with E2s primarily through the N-terminal helix (α1) and loops L4 and L7 of the E2's ubiquitin-conjugating (UBC) domain [28] [32]. These interfaces exhibit precise complementarity that enables discrimination among the 38 human E2s.
- E2 Active Site Control: The RING-E2 interaction activates the E2~Ub conjugate for catalysis by promoting closed conformations that position the ubiquitin for transfer [28] [33]. Different E2s have inherent preferences for specific linkage types, which they impart to their RING partners.
- Specificity Hotspots: Key residues at the E2-RING interface serve as specificity determinants. For instance, in the UbcH7-E6AP interaction, four side chains on UbcH7 and six on E6AP contribute more than 1 kcal/mol to the binding free energy, with residues K96 and K100 in UbcH7 being particularly critical [32].
Table 2: Key Determinants of RING-E2 Specificity
| Specificity Determinant | Location | Function | Example |
|---|---|---|---|
| F63 residue | E2 Loop 4 | Hydrophobic interface core | UbcH7, UbcH8 |
| K96/K100 | E2 α1 helix | Electrostatic interactions | UbcH7-E6AP |
| L4 and L7 loops | E2 surface | RING binding specificity | Multiple E2s |
| Zinc coordination sites | RING domain | Structural integrity | All RINGs |
| E1/E3 overlapping interface | E2 surface | Mutual exclusivity | All E2s |
Experimental Approaches for Elucidating Specificity Mechanisms
Structural Biology Techniques
Cryo-Electron Microscopy (Cryo-EM) for HECT E3 Mechanism Visualization Recent advances in cryo-EM have enabled visualization of full-length HECT E3s in action. For UBR5, researchers employed a catalytically inactive mutant (C2768A) to capture stable intermediates along the K48-linked ubiquitin chain formation pathway [31]. The protocol involves:
- Expression and purification of full-length UBR5 from insect cells
- Grid preparation with ubiquitin, E1, and E2 enzymes
- Data collection on modern cryo-EM instruments (e.g., Titan Krios)
- 3D classification and model building into maps refined to 2.7-3.7 Å resolution
This approach revealed UBR5 as a dimeric functional unit with HECT domains in the L-conformation, stabilized by domain-swap dimerization and HECT display domains [31].
Crystallography for RING-E2 Complex Characterization X-ray crystallography has been instrumental in defining RING-E2 interfaces at atomic resolution. The typical workflow includes:
- Generation of E2~Ub thioester mimetics using isopeptide linkages
- Co-crystallization of RING domains with E2~Ub conjugates
- Data collection at synchrotron sources (e.g., 0.9792 Å wavelength)
- Structure determination by molecular replacement
This approach revealed how HHARI recruits UbcH7~Ub in an 'open' conformation that prevents spurious ubiquitin discharge and ensures transfer to the RING2 catalytic cysteine [33].
Biochemical and Biophysical Assays
Fluorescence Polarization Binding Assays Quantitative E2-E3 binding affinity measurements utilize fluorescence polarization with bodipy-labeled E2 enzymes [32]. The protocol involves:
- Site-specific labeling of E2 cysteine mutants with thiol-reactive fluorophores
- Titration of increasing E3 concentrations against fixed E2 concentrations
- Measurement of polarization changes to determine dissociation constants (K_D)
- Validation using isothermal titration calorimetry (ITC)
This method identified that E6AP binds UbcH7 and UbcH8 with low micromolar affinity (KD ≈ 5 μM), while interaction with UbcH5b is significantly weaker (KD > 200 μM) [32].
Pulse-Chase Ubiquitination Assays Linkage specificity is determined using pulse-chase assays that track ubiquitin transfer through distinct steps [10] [31]:
- Pulse: Generation of E2~*Ub thioester with fluorescent-labeled ubiquitin
- Chase: Addition of E3 and unlabeled acceptor ubiquitin
- Quenching at timed intervals with SDS-PAGE loading buffer
- Visualization and quantification of reaction intermediates and products by in-gel fluorescence
For TRIP12, this approach demonstrated a striking preference for K48-linked di-ubiquitin acceptors over mono-ubiquitin or other linkage types [10].
Mechanism Visualization
Visual comparison of HECT versus RING E3 specificity mechanisms
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Studying HECT and RING Specificity
| Reagent/Category | Specific Examples | Function/Application | Experimental Use |
|---|---|---|---|
| E2~Ub Thioester Mimetics | UbcH7~Ub (isopeptide-linked) | Stable intermediate for structural studies | Cryo-EM and crystallography of E2-E3 complexes [33] |
| Linkage-Specific Ubiquitin Mutants | Ub(K29R, K48R, K63R) | Determining linkage specificity | Pulse-chase assays to identify target lysines [34] [10] |
| Activity-Based Probes | Ub-VS, Ub-Br2 | Trapping active E3~Ub intermediates | Mechanism-based profiling and inhibition studies [30] |
| Fluorescent Ubiquitin Variants | Bodipy-FL-Ub, Cy5-Ub | Real-time reaction monitoring | Fluorescence polarization and transfer assays [32] [31] |
| HECT Domain Constructs | AREL1(436-823), E6AP HECT | Structure-function studies | Crystallography and biochemical characterization [34] [30] |
| RING Domain Constructs | HHARI RBR, c-Cbl RING | E2 partnership mapping | Binding affinity and specificity studies [32] [33] |
Implications for Therapeutic Development
The structural insights into HECT and RING specificity mechanisms have significant implications for drug development, particularly for targeting E3 ligases in cancer, neurodegenerative disorders, and infectious diseases [34] [12]. HECT E3s like AREL1 represent promising targets because their anti-apoptotic activity in cancer cells depends on specific structural features, including an additional loop (aa 567-573) not found in other HECT members [34]. The development of AREL1-specific ubiquitin variants that inhibit SMAC ubiquitination demonstrates the potential for targeting HECT E3s with protein-based therapeutics [34].
For RING E3s, the precise mapping of E2-RING interfaces enables strategies to disrupt specific pathogenic interactions without affecting global ubiquitination. The identification of hotspot residues contributing significantly to binding free energy provides critical information for designing small-molecule inhibitors or protein-protein interaction blockers [32] [33]. As our understanding of these specificity determinants grows, so does the potential for developing targeted therapies that modulate specific ubiquitination pathways while minimizing off-target effects.
Ubiquitination is a crucial post-translational modification that regulates virtually all cellular processes in eukaryotes, with specificity encoded in the diverse architectures of ubiquitin chains. HECT (Homologous to E6AP C-Terminus) E3 ligases constitute a major family of ~28 enzymes in humans that directly catalyze ubiquitination through a distinctive catalytic mechanism involving a thioester intermediate [11] [35]. Unlike RING E3 ligases that primarily function as scaffolds, HECT E3s determine their own linkage specificity through features intrinsic to their HECT domains, making them fascinating subjects for understanding how the ubiquitin code is written [36] [11]. This review systematically compares the three major HECT subfamilies—NEDD4, HERC, and "Other"—focusing on their structural characteristics, linkage preferences, and the experimental approaches defining their specificities.
HECT E3 Ligase Architecture and Catalytic Mechanism
Domain Organization and Structural Features
All HECT E3 ligases share a conserved ~350-residue catalytic HECT domain at their C-terminus, preceded by diverse N-terminal domains that confer substrate specificity and regulation [11] [35]. The HECT domain itself exhibits a bilobal architecture: an N-lobe (~250 residues) that binds the E2~ubiquitin complex, and a C-lobe (~100 residues) containing the catalytic cysteine that forms a thioester bond with ubiquitin [36] [11]. These lobes are connected by a flexible hinge region that permits substantial conformational rearrangements essential for catalysis [37] [31].
The family is categorized into three principal subfamilies based on their N-terminal domain architectures:
- NEDD4 Subfamily: Characterized by N-terminal C2 domains for membrane localization and multiple WW domains for substrate recognition [38] [35].
- HERC Subfamily: Feature RCC1-like domains (RLDs) that may regulate nucleotide binding or subcellular localization [35].
- "Other" Subfamily: Comprises diverse members with various protein-protein interaction domains that cannot be classified into the above groups [35].
Table 1: Major HECT E3 Subfamilies and Their Characteristics
| Subfamily | Representative Members | N-terminal Domains | Structural Features |
|---|---|---|---|
| NEDD4 | NEDD4-1, WWP2, SMURF1, ITCH | C2 domain, WW domains | Membrane association via C2 domain; substrate recognition via WW domains |
| HERC | HERC1, HERC2, HERC3, HERC4 | RCC1-like domains (RLDs) | Large proteins (>400 kDa); potential nucleotide/GTase regulation |
| Other | E6AP, HUWE1, UBR5 | Various unique domains | Diverse substrate binding domains; UBR5 forms massive dimers |
Catalytic Mechanism and Chain Formation
HECT E3s catalyze ubiquitination through a two-step mechanism fundamentally distinct from RING E3s:
- Transthiolation: Ubiquitin is transferred from the E2 enzyme to the conserved catalytic cysteine within the HECT C-lobe, forming a reactive E3~Ub thioester intermediate [11].
- Substrate Transfer: Ubiquitin is subsequently transferred from the E3 to a lysine residue on the substrate protein or a growing ubiquitin chain [11].
The flexible hinge between the N- and C-lobes enables the substantial domain movements required to juxtapose the E2 and E3 active sites during transthiolation and to position the E3~Ub conjugate for subsequent substrate modification [37] [31]. Recent structural studies of UBR5 and Ufd4, utilizing cryo-electron microscopy (cryo-EM) and chemically trapped intermediates, have visualized these conformational states along the catalytic cycle, revealing a conserved HECT domain trajectory during ubiquitin transfer [31] [39].
A critical distinction from RING E3s is that linkage specificity is determined by the HECT domain itself, particularly the C-lobe, rather than solely by the cooperating E2 enzyme [36]. This intrinsic specificity arises from how the HECT domain positions the acceptor ubiquitin relative to the donor ubiquitin thioester during chain formation.
Diagram Title: HECT vs RING E3 Catalytic Mechanisms
Comparative Analysis of HECT Subfamily Linkage Specificities
NEDD4 Subfamily: Predominant K63 Linkage with Emerging Complexity
The NEDD4 subfamily represents the most extensively characterized group of HECT E3s, with well-established preferences for synthesizing K63-linked ubiquitin chains. Foundational studies on yeast Rsp5 and human homologs NEDD4-1 and ITCH demonstrated their inherent bias toward K63 linkages, which typically function in proteasome-independent signaling pathways such as membrane trafficking and signal transduction [36].
However, recent research reveals considerable diversity and regulatory complexity within this subfamily:
- WWP1 exhibits a hierarchical linkage preference (K63 > K48 > K11), employing a two-phase mechanism where initial unidirectional chain synthesis occurs exclusively through K63, followed by multidirectional elongation incorporating mixed linkages and branched structures [15].
- NEDD4-1 and WWP2 linkage output is modulated by autoinhibitory mechanisms involving their WW domain linkers; relief of autoinhibition by allosteric activators like NDFIP1 or engineered ubiquitin variants influences both catalytic activity and product distribution, including the ratio of K48 versus K63 linkages [38].
- SMURF1, another NEDD4 member, has been implicated in K48-linked degradation of specific substrates, highlighting context-dependent specificity [35].
"Other" Subfamily: Diverse and Specialized Linkage Profiles
The "Other" subfamily encompasses HECT E3s with considerable mechanistic and linkage diversity, including prominent members like UBR5, E6AP, and HUWE1.
- UBR5 specifically generates K48-linked ubiquitin chains, including through branching of pre-existing K11- or K63-linked chains [31]. Recent cryo-EM structures reveal UBR5 functions as a massive dimeric assembly, with ubiquitin-associated (UBA) domains playing a critical role in positioning the acceptor ubiquitin to lure its K48 residue into the active site for efficient chain formation [31].
- E6AP (UBE3A), the founding HECT member, strongly prefers K48-linked chains, consistent with its role in targeting p53 for proteasomal degradation during human papillomavirus infection [36] [35].
- HUWE1 displays conflicting reported specificities, with evidence for both K48- and K63-linkage formation under different contexts, suggesting sophisticated regulatory mechanisms or substrate-dependent specificity [36].
A specialized mechanism is exemplified by Ufd4, which preferentially catalyzes K29-linked ubiquitination on pre-formed K48-linked chains to generate K29/K48-branched ubiquitin chains [39]. Structural studies show this specificity is achieved through coordinated action of Ufd4's N-terminal ARM region and HECT C-lobe, which together recruit K48-linked diubiquitin and orient the proximal ubiquitin's K29 toward the catalytic cysteine [39].
HERC Subfamily: Less Defined but Functionally Significant
While comprehensive biochemical characterization of HERC subfamily linkage specificity remains limited, functional studies implicate them in critical cellular processes. HERC2 regulates p53 stability and the DNA damage response, though interestingly, some of its functions may occur independently of its catalytic activity [35]. The size and complexity of HERC E3s (often exceeding 400 kDa) have presented challenges for detailed mechanistic studies, representing an important frontier for future research.
Table 2: Experimentally Determined Linkage Specificities of Representative HECT E3s
| HECT E3 | Subfamily | Preferred Linkage(s) | Experimental Evidence | Key Regulatory Features |
|---|---|---|---|---|
| Rsp5 (yeast) | NEDD4 | K63 > K29 | In vitro ubiquitination assays; HECT domain determinacy [36] | C-lobe determinants |
| NEDD4-1 | NEDD4 | K63 (modulable to K48) | In vitro assays with autoinhibition relief [38] [36] | WW linker autoinhibition; NDFIP1 activation |
| WWP1 | NEDD4 | K63 > K48 > K11 | Sequential addition mechanism studies [15] | Two-phase chain synthesis |
| ITCH | NEDD4 | K63 (also K29 in vivo) | In vitro and cellular assays [36] | Unknown |
| E6AP | Other | K48 | In vitro ubiquitination [36] | HECT domain specificity |
| UBR5 | Other | K48 (including branches) | Cryo-EM structures; biochemical assays [31] | UBA domain; dimeric architecture |
| Ufd4 | Other | K29 (on K48 chains) | Cryo-EM; Ub-clipping MS; biochemical assays [39] | ARM region; branched chain specificity |
| HUWE1 | Other | K48 and/or K63 (context-dependent) | Conflicting reports on different substrates [36] | Substrate-dependent regulation |
| HERC2 | HERC | Not fully characterized | Functional studies in DNA damage [35] | Catalytic activity-independent functions |
Experimental Approaches for Determining Linkage Specificity
Biochemical and Enzymatic Assays
Standard in vitro ubiquitination assays form the foundation for determining linkage specificity:
- Protocol: Reactions typically contain E1 enzyme, specific E2 (often UbcH5/Ube2D or UbcH7/Ube2L family), E3 ligase (full-length or HECT domain), ubiquitin, and ATP in appropriate buffer [38] [15].
- Analysis: Products are resolved by SDS-PAGE and analyzed by immunoblotting with linkage-specific anti-ubiquitin antibodies to distinguish chain types [15].
- Variants: Assays may utilize mutant ubiquitins (e.g., K48R, K63R) where only a single lysine is available, or employ diubiquitin substrates of defined linkage to study chain elongation specificity [39].
Structural Biology Techniques
Recent advances in structural biology have revolutionized understanding of HECT E3 mechanisms:
- Cryo-EM has enabled visualization of full-length HECT E3s like UBR5 in multiple catalytic states, revealing how dimeric architecture and auxiliary domains coordinate to achieve linkage specificity [31].
- Chemical Trapping strategies employ engineered ubiquitin probes or disulfide crosslinking to stabilize transient catalytic intermediates, as demonstrated in studies of Ufd4-mediated branched chain formation [39].
- X-ray Crystallography of isolated HECT domains, sometimes in complex with E2s or inhibitors, has revealed conformational flexibility and allosteric regulatory sites, such as the conserved glycine hinge targeted by SMURF1 inhibitors [37].
Mass Spectrometry Approaches
Mass spectrometry techniques provide precise linkage mapping:
- Middle-down MS (Ub-clipping): Utilizes the ubiquitin-specific protease USP2 to cleave ubiquitin chains while preserving branching points, allowing identification of branched linkages through detection of diGly remnants on multiple lysines [39].
- Tandem MS: Fragmentation patterns reveal both linkage type and architecture of polyubiquitin chains synthesized by HECT E3s in vitro or isolated from cellular environments.
Research Reagent Solutions
Table 3: Essential Research Reagents for HECT E3 Ligase Studies
| Reagent Category | Specific Examples | Research Applications | Key Features |
|---|---|---|---|
| Ubiquitin Mutants | K48-only Ub, K63-only Ub, K29R Ub | Linkage specificity assays | Restricts chain formation to specific lysines or prevents specific linkages |
| Activity-Based Probes | DiUb/triUb probes with crosslinkable groups | Trapping catalytic intermediates | Enables structural studies of transient states by cryo-EM [39] |
| Engineered Ub Variants | Allosteric Ub variants (engineered) | Autoinhibition relief studies | Probe regulatory mechanisms in NEDD4 family E3s [38] |
| HECT Domain Constructs | Isolated HECT domains, HECT truncations | Determinancy mapping | Identifies regions controlling linkage specificity [36] |
| Linkage-Specific Antibodies | Anti-K48 Ub, Anti-K63 Ub, Anti-K11 Ub | Western blot detection | Rapid assessment of chain linkage in biochemical assays |
| Allosteric Inhibitors | SMURF1 inhibitors (e.g., Cpd-8) | Mechanistic and therapeutic studies | Targets cryptic pocket; restricts glycine hinge motion [37] |
Implications for Therapeutic Development
The distinct linkage specificities and regulatory mechanisms of HECT subfamilies present unique therapeutic opportunities. Unlike RING E3s, HECT E3s offer two potential targeting strategies: active-site inhibition and allosteric regulation. Recent success in identifying allosteric inhibitors of SMURF1 that bind a cryptic pocket and restrict essential hinge motion demonstrates the feasibility of targeting HECT E3s [37]. Machine-learning approaches leveraging this mechanistic understanding have identified similar inhibitors for E6AP, opening a new druggable space for HECT-directed therapies [37].
The linkage specificity of HECT E3s also presents opportunities for pathway-selective intervention. For example, inhibiting K48-specific HECTs like UBR5 or E6AP could modulate proteasomal degradation pathways without affecting K63-mediated signaling, potentially achieving greater specificity than general proteasome inhibitors.
The three major HECT subfamilies—NEDD4, HERC, and "Other"—exhibit distinctive linkage preferences and regulatory mechanisms that enable their specialized cellular functions. The NEDD4 subfamily predominantly generates K63-linked chains but exhibits remarkable regulatory complexity through autoinhibitory mechanisms. The "Other" subfamily displays diverse specificities, including strong K48-linkage formation by UBR5 and E6AP, and specialized branched chain synthesis by Ufd4. The HERC subfamily, while less characterized, represents an important frontier for future research.
These intrinsic linkage specificities contrast sharply with RING E3s, where chain type is primarily determined by the cooperating E2 enzyme. This fundamental distinction, combined with the direct catalytic mechanism of HECT E3s, positions them as attractive targets for therapeutic intervention aimed at rewriting specific ubiquitin signals in disease. Future research elucidating the structural basis for linkage specificity across all HECT subfamilies will be essential for realizing this therapeutic potential.
The Really Interesting New Gene (RING) family of E3 ubiquitin ligases represents the largest and most diverse class of enzymes responsible for substrate specificity within the ubiquitin-proteasome system [19]. These enzymes function as scaffolds that facilitate the direct transfer of ubiquitin from an E2 conjugating enzyme to a substrate protein, a mechanism distinct from the two-step catalytic process employed by HECT-type E3s [19] [8]. RING E3s primarily exert their biological influence through the synthesis of polyubiquitin chains on target proteins, with chain topology determining the substrate's fate—ranging from proteasomal degradation to altered activity or localization [40] [8]. The RING superfamily is broadly divided into two principal architectural classes: monomeric RING E3s and multi-subunit Cullin-RING Ligases (CRLs). This structural dichotomy fundamentally impacts their mechanisms of chain elongation, regulation, and biological functions, creating a fascinating landscape for comparative analysis with HECT-family E3s, particularly regarding their specificity for atypical ubiquitin chains.
Table 1: Fundamental Characteristics of Major RING E3 Subfamilies
| Subfamily | Representative Members | Core Architecture | Key Regulatory Features |
|---|---|---|---|
| Monomeric RING | BRCA1/BARD1, Mdm2/MdmX, RNF25, RNF220 | Single polypeptide or stable hetero/homodimer [19] | Dimerization, post-translational modifications, cellular localization [19] |
| CRL1 (SCF) | SKP1-CUL1-F-box complexes | CUL1 scaffold, SKP1 adaptor, F-box substrate receptor [41] [42] | NEDD8 activation, CAND1 inhibition, substrate receptor exchange [41] [43] |
| CRL2/5 | VHL-CUL2, SOCS-CUL5 | CUL2/5 scaffold, Elongin B/C adaptor, VHL/SOCS substrate receptor [41] [42] | NEDD8 activation, CAND1 inhibition [43] |
| CRL3 | CUL3-BTB complexes | CUL3 scaffold, BTB proteins (combined adaptor/receptor) [41] [42] | BTB protein homo-dimerization [42] |
| CRL4 | DDB1-CUL4-DCAF complexes | CUL4A/B scaffold, DDB1 adaptor, DCAF substrate receptor [41] [42] | NEDD8 activation, CAND1 inhibition [43] |
Architectural Organization of RING E3 Ligases
Monomeric RING E3s: Dimerization-Driven Function
Monomeric RING E3s, despite their name, often function as homodimers or heterodimers, a configuration essential for their catalytic activity and stability [19]. Structurally, they coordinate two Zn²⁺ ions in a cross-braced arrangement that creates a stable platform for E2 binding [19]. Notable examples include the BRCA1-BARD1 heterodimer, crucial for DNA repair, and the Mdm2-MdmX heterodimer, a key regulator of the tumor suppressor p53 [19]. In these complexes, one RING typically serves as the primary E2 engagement site, while the partner RING enhances catalytic activity or contributes to substrate recognition [19]. This dimeric architecture allows for sophisticated regulatory control, as evidenced by cIAP1, where RING dimerization is sequestered in an inactive conformation until activation by IAP antagonists like SMAC/DIABLO [19]. The recent identification of RNF25's role in DNA replication fork protection—through a mechanism independent of its ubiquitin ligase activity—highlights the functional complexity within this subfamily, suggesting that some monomeric RING E3s may serve non-catalytic scaffolding roles in specific biological contexts [44].
Cullin-RING Ligases (CRLs): Modular Assembly Lines for Ubiquitination
CRLs represent the largest family of multi-subunit RING E3s, responsible for approximately 20% of all ubiquitination events mediated by the ubiquitin-proteasome system [41] [45]. These elaborate complexes are built upon a cullin protein scaffold (CUL1, CUL2, CUL3, CUL4A/B, CUL5, CUL7, or CUL9) that serves as a structural backbone [41] [42]. The architecture follows a consistent pattern: the cullin's N-terminal domain recruits variable substrate recognition modules through adaptor proteins, while its C-terminal domain binds a RING-box protein (RBX1 or RBX2) that recruits the E2 enzyme [41] [42]. This modular design creates an extensive family of E3s capable of targeting countless substrates through combinatorial assembly of different substrate receptors. The CRL family exemplifies the concept of functional specialization through modular organization, with each cullin subclass employing distinct sets of adaptors and substrate receptors [41] [42].
Diagram 1: Architectural comparison highlighting the direct substrate and E2 binding of monomeric RING E3s versus the multi-component, scaffold-based architecture of CRLs where the cullin bridges the substrate receptor and E2-binding RING protein.
Mechanisms of Ubiquitin Chain Elongation
The mechanism of polyubiquitin chain synthesis represents a fundamental point of divergence between RING and HECT E3 ligases. While HECT E3s, such as WWP1 and Ufd4, form a catalytic thioester intermediate with ubiquitin before transferring it to the substrate [40] [39], RING E3s facilitate direct ubiquitin transfer from the E2 enzyme to the substrate [19]. This distinction profoundly influences their chain elongation capabilities and linkage specificity.
Chain Elongation by Monomeric RING E3s
Monomeric RING E3s function primarily as scaffolds that position the E2~Ub thioester in close proximity to a substrate lysine residue, enabling direct nucleophilic attack and isopeptide bond formation [19]. The linkage specificity of chain elongation is determined by a combination of the E2 enzyme and specific residues within the RING domain that influence the orientation of the E2~Ub complex [19]. Some E2s exhibit intrinsic specificity for particular ubiquitin lysines, while others are more promiscuous, allowing the RING E3 to fine-tune linkage selection [19]. For branched or atypical chain formation, monomeric RING E3s may collaborate with other E3s or require specific post-translational modifications on their substrates to expose non-canonical acceptor lysines.
Processive Chain Elongation by CRLs
CRLs employ a highly processive mechanism for polyubiquitin chain synthesis, particularly evident in well-characterized complexes like SCF (CRL1) [43]. The neddylated CRL architecture positions the E2 catalytic site approximately 50-60 Å from the substrate binding site, creating an ideal geometry for rapid, successive ubiquitin transfers [43]. Following initial monoubiquitination, the growing ubiquitin chain remains anchored to the substrate receptor while the E2~Ub is repositioned to target the distal ubiquitin in the chain [43]. This processive mechanism enables CRLs to efficiently build degradation-targeting chains (typically K48-linked) without releasing the substrate during chain elongation. The recent structural visualization of HECT E3 Ufd4 catalyzing K29/K48-branched chain formation provides a striking contrast, demonstrating the more complex catalytic cycle required for branched chain synthesis compared to the relatively straightforward processive elongation by many CRLs [39].
Table 2: Comparative Analysis of Ubiquitin Chain Elongation Mechanisms
| Feature | Monomeric RING E3s | Multi-subunit CRLs | HECT E3s (for Comparison) |
|---|---|---|---|
| Catalytic Mechanism | Direct transfer from E2 to substrate [19] | Direct transfer from E2 to substrate [41] | Two-step via E3-thioester intermediate [40] [8] |
| Chain Elongation Mode | Distributive (typically) | Processive [43] | Sequential addition [40] |
| Linkage Determination | E2 intrinsic specificity + RING influence [19] | Primarily E2-dependent [19] | HECT domain specificity [40] |
| Branched Chain Capability | Limited evidence | Limited evidence | Yes (e.g., Ufd4 creates K29/K48 branches) [39] |
| Regulation of Elongation | Dimerization, PTMs, localization [19] | NEDD8 activation, CAND1, substrate receptor exchange [41] [43] | Non-covalent Ub-binding sites, conformational changes [40] |
Experimental Approaches for Studying RING E3 Mechanisms
In Vitro Reconstitution Assays for Linkage Specificity
A powerful methodology for elucidating RING E3 mechanism and linkage specificity involves in vitro reconstitution using purified components, as exemplified by studies on the HECT E3 WWP1 [40]. This approach provides precise control over reaction components and enables quantitative analysis of ubiquitin chain formation.
Protocol Summary:
- Component Purification: Recombinantly express and purify the RING E3 of interest, relevant E2 enzymes, E1 enzyme, and substrate protein [40].
- Reaction Assembly: Combine E1 (50 nM), E2 (500 nM-1 µM), E3 (50-200 nM), substrate (1-5 µM), ubiquitin (10-50 µM), and ATP in reaction buffer [40].
- Time-Course Sampling: Remove aliquots at specified time points (0-120 minutes) and quench with SDS-PAGE loading buffer [40].
- Product Analysis: Resolve reactions by SDS-PAGE followed by immunoblotting with linkage-specific ubiquitin antibodies (e.g., anti-K48, anti-K63, anti-K11) to determine chain topology [40].
- Mutational Validation: Utilize ubiquitin lysine mutants (K48R, K63R, etc.) to confirm linkage identification through elimination of specific chain types [40].
This methodology directly adapted from WWP1 studies can be applied to RING E3s by substituting the relevant E3 and E2 combinations [40]. The recent application of similar approaches to Ufd4 revealed its unique capacity for K29/K48-branched ubiquitin chain formation, demonstrating the power of these reconstitution assays for uncovering atypical chain specificity [39].
Structural Approaches for Elongation Mechanism Visualization
Structural biology techniques have been instrumental in elucidating the molecular mechanisms of ubiquitin chain elongation, particularly through trapping intermediate states.
Cryo-EM Methodology for Transition State Capture: The recent structural determination of HECT E3 Ufd4 in complex with a K29/K48-branched ubiquitin probe illustrates an innovative approach applicable to RING E3 studies [39]:
- Probe Design: Engineer a disulfide-crosslinked ubiquitin probe mimicking the transition state, with the proximal Ub of K48-linked diUb chemically ligated to a donor Ub [39].
- Complex Trapping: Crosslink the engineered ubiquitin probe with the E3 catalytic cysteine to form a stable complex mimicking the transition state [39].
- Cryo-EM Analysis: Collect single-particle cryo-EM data (5,000+ micrographs) and process through 2D/3D classification to obtain high-resolution structures [39].
- Model Building: Dock AlphaFold-predicted E3 structures and known ubiquitin coordinates into cryo-EM density maps with iterative refinement [39].
While this specific approach was used for a HECT E3, analogous strategies utilizing E2-E3-substrate crosslinking could be employed to visualize RING E3 complexes during chain elongation, particularly for understanding the structural basis of branched chain formation.
Diagram 2: Integrated experimental workflow combining biochemical reconstitution assays with structural biology approaches to comprehensively analyze ubiquitin chain elongation mechanisms.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Studying RING E3 Mechanisms
| Reagent Category | Specific Examples | Research Application | Key Function |
|---|---|---|---|
| Ubiquitin Mutants | Ub-K48R, Ub-K63R, Ub-K29R, Ub-K11R, Ub-K0 (no lysines) [40] [39] | Linkage specificity mapping | Identify specific ubiquitin lysines used for chain elongation through elimination of specific linkages |
| E2 Enzyme Library | Commercial kits with ~35 human E2s [40] | E2-E3 pairing identification | Determine which E2s cooperate with specific RING E3s to catalyze ubiquitination |
| Linkage-Specific Antibodies | Anti-K48-Ub, Anti-K63-Ub, Anti-K11-Ub, Anti-K29-Ub [40] | Ubiquitin chain topology analysis | Detect and quantify specific ubiquitin linkage types in immunoblot experiments |
| Activity-Based Probes | DiUb/probe (disulfide-crosslinked ubiquitin probes) [39] | Structural mechanism studies | Trap and stabilize E3-Ub transition states for structural analysis (e.g., Cryo-EM) |
| CRL Modulators | MLN4924 (NAE inhibitor), CAND1, COP9 signalosome [41] [43] | CRL activity regulation | Specifically inhibit or modulate CRL activity through interference with neddylation cycle |
| E3 Expression Constructs | Full-length and truncated RING domains, catalytic cysteine mutants [40] [39] | Structure-function studies | Express recombinant E3s for biochemical assays and determine essential catalytic residues |
Discussion: Comparative Specificity for Atypical Chains in HECT vs. RING E3s
Within the broader context of E3 ligase specificity for atypical ubiquitin chains, several key distinctions emerge between RING and HECT family enzymes. HECT E3s demonstrate a remarkable capacity for synthesizing heterogeneous and branched ubiquitin chains, as exemplified by WWP1's two-phase ubiquitination mechanism (initial Lys-63-specific chain formation followed by multidirectional elongation with mixed linkages) [40] and Ufd4's specialized ability to generate K29/K48-branched chains [39]. This flexibility stems from their catalytic mechanism, which involves direct thioester bonding with ubiquitin, potentially allowing greater control over linkage specification through specialized ubiquitin-binding domains within the HECT catalytic unit [40].
In contrast, most RING E3s primarily facilitate the formation of homogeneous ubiquitin chains, with linkage specificity largely dictated by their partnered E2 enzymes [19]. While some E2s exhibit intrinsic specificity for particular lysines (e.g., UBE2R1 for K48, UBE2N/UBE2V1 for K63), others are more promiscuous [19]. The modular architecture of CRLs, while excellent for substrate diversification, may limit their capacity for synthesizing complex chain topologies due to their more rigid geometric constraints and direct transfer mechanism. However, the potential for collaborative synthesis between different RING E3s, or between RING and HECT E3s, represents an exciting frontier for understanding how complex ubiquitin signals are constructed in cells.
The functional implications of this mechanistic divergence are significant: HECT E3s appear particularly well-suited for creating sophisticated ubiquitin codes involving branched or mixed linkages that can fine-tune substrate fate, while RING E3s excel at efficient, processive synthesis of homogeneous chains for clear biological outcomes like proteasomal degradation. This division of labor suggests that the ubiquitin system has evolved distinct enzyme architectures optimized for different signaling paradigms—precision signaling versus bulk degradation—with both essential for cellular homeostasis.
Toolkit for Discovery: Methodologies for Profiling Atypical Chain Synthesis
Understanding how E3 ubiquitin ligases achieve linkage specificity, particularly for atypical ubiquitin chains, is a fundamental challenge in ubiquitin research. The HECT and RING E3 families represent two major mechanistic paradigms for ubiquitin transfer, each employing distinct catalytic strategies. HECT-type E3 ligases form a catalytic intermediate with ubiquitin via a thioester bond on their active-site cysteine before transferring it to a substrate. In contrast, RING-type E3 ligases primarily function as scaffolds that facilitate direct ubiquitin transfer from an E2 enzyme to the substrate without forming a E3-ubiquitin intermediate [11] [46]. This fundamental mechanistic difference underlies their distinct approaches to generating specific ubiquitin chain topologies.
Recent structural studies have revealed how certain HECT E3s, such as TRIP12 and UBR5, specialize in forming atypical K29-linked and K29/K48-branched ubiquitin chains through sophisticated substrate recruitment and positioning mechanisms [10] [31] [39]. Meanwhile, RING E3s like RNF38 achieve linkage specificity through a conserved "linchpin" residue that stabilizes the closed conformation of the E2~Ub complex [7]. Dissecting these mechanisms requires specialized biochemical techniques that can capture transient reaction intermediates and quantify enzyme activities. This guide compares the experimental applications of pulse-chase and thioester discharge assays for elucidating these specificity mechanisms, with particular focus on their utility in distinguishing HECT versus RING E3 functions in atypical chain formation.
Comparative Analysis of Key Ubiquitination Assays
Table 1: Technical Comparison of Pulse-Chase and Thioester Discharge Assays
| Parameter | Pulse-Chase Assay | Thioester Discharge Assay |
|---|---|---|
| Primary Application | Monitoring multi-step ubiquitination cascades and intermediate formation [47] | Measuring E1-E2 or E2-E3 ubiquitin thioester transfer efficiency [7] |
| Key Measured Output | Ubiquitin transfer from E2~Ub to E3 and then to substrate/acceptor ubiquitin over time [10] [47] | Rate of ubiquitin discharge from E2~Ub to exogenous nucleophile (e.g., DTT) [7] |
| Temporal Resolution | Sequential reaction steps initiated by chase addition [47] | Single time-point or kinetic measurement of thioester breakdown |
| Information Gained | Identifies specific reaction steps, directionality, and enzyme capabilities in polyubiquitination [10] [31] | Probes E2~Ub activation state and the impact of E3 or mutations on E2 catalytic competency [7] |
| Typical Experimental Setup | Fluorescently-labeled ubiquitin, E1, E2, E3, and acceptor ubiquitin/substrate; non-hydrolyzable nucleotides may be used [47] | E2~Ub thioester pre-formed, then incubated with or without E3; reaction stopped with SDS-loading dye ± DTT to assess thioester stability |
| Advantages | Directly monitors physiological reaction steps; can track multiple ubiquitin species simultaneously with dual-color labeling [47] | Simpler setup; directly interrogates E2~Ub conformational state and activation |
Table 2: Application of Assays to HECT vs. RING E3 Mechanisms
| Research Question | Pulse-Chase Assay Application | Thioester Discharge Application |
|---|---|---|
| Linkage Specificity Determination | TRIP12 preferentially modifies K48-linked diUb at K29 of proximal Ub, forming K29/K48-branched chains [10] | RING E3 binding induces closed E2~Ub conformation; discharge rate changes indicate stabilization [7] |
| Acceptor Ub Context Requirement | UBR5 and Ufd4 show strong preference for K48-linked chains over monoUb as acceptors [31] [39] | Not typically applied to acceptor context questions |
| Geometric Constraints | TRIP12 requires precise lysine side-chain length (4 methylenes) for efficient branching [10] | Can probe how E3 binding positions E2~Ub active site |
| Allosteric Regulation | Not primary application | Ubiquitin binding to E2 backside (UbB) allosterically activates RING E3-UbcH5B~Ub complexes [48] |
Experimental Protocols for Key Methodologies
Dual-Color Pulse-Chase Ubiquitination Assay
The pulse-chase assay enables stepwise monitoring of ubiquitin transfer through the enzymatic cascade, making it ideal for characterizing HECT E3 mechanisms that involve distinct E3~Ub intermediates [47].
Protocol Overview:
Pulse Phase: Form the E2~Ub thioester intermediate by incubating E1, E2, ATP, and fluorescently-labeled "donor" ubiquitin (*Ub). The donor ubiquitin is often a K0 (lysine-less) mutant or carries specific lysine mutations (e.g., K48R) to prevent its use as an acceptor. Stop this phase with EDTA to chelate Mg²⁺ and prevent further E1 activity [10] [47].
Chase Phase: Initiate the downstream reaction by adding the E3 ligase and the "acceptor" ubiquitin or substrate to the pre-formed E2~*Ub complex. The acceptor can be monoUb, linkage-defined diUb (e.g., K48-linked diUb), or a substrate protein [10] [31].
Detection: Resolve reaction time points by non-reducing SDS-PAGE to preserve thioester linkages. Visualize fluorescent ubiquitin species to track the decay of E2~Ub and the appearance of E3~Ub and higher molecular weight polyubiquitin products [10] [47].
Key Technical Considerations:
- Use different fluorescent labels (e.g., Cy3, Cy5) for donor and acceptor ubiquitins to simultaneously monitor priming and extension in the same reaction tube [47].
- For HECT E3s, the E3~Ub intermediate is a definitive hallmark of their catalytic mechanism and can be directly observed in this assay.
- To test linkage specificity, include a panel of different linkage-defined diUb molecules (K48-, K63-, K29-diUb, etc.) as potential acceptors in the chase phase [10] [39].
Thioester Discharge Assay
This assay measures the stability and reactivity of the E2~Ub thioester bond, providing insights into the E2's activation state and the allosteric effects of RING E3 binding [7].
Protocol Overview:
E2~Ub Formation: Generate the E2~Ub thioester complex as in the pulse phase of the pulse-chase assay.
Incubation with E3: Add the RING E3 of interest to the E2~Ub complex. As a control, incubate E2~Ub alone.
Thioester Discharge: Include the small nucleophile DTT (dithiothreitol) in the reaction, which attacks the thioester bond, releasing free ubiquitin.
Analysis: Resolve reactions by non-reducing SDS-PAGE. Quantify the percentage of E2~Ub thioester remaining versus discharged ubiquitin over time. Enhanced discharge rates in the presence of E3 indicate E2~Ub conformational activation [7].
Key Technical Considerations:
- This assay is particularly useful for characterizing RING E3 mutants (e.g., linchpin residue variants) and their impact on E2~Ub conformational dynamics [7].
- The assay can demonstrate allosteric effects, such as how non-covalent backside ubiquitin binding (UbB) to the E2 stimulates donor ubiquitin transfer [48].
Research Reagent Solutions
Table 3: Essential Reagents for Ubiquitination Assays
| Reagent / Tool | Function / Application | Key Examples & Notes |
|---|---|---|
| Linkage-Defined DiUb | Acceptor substrates to probe linkage specificity | K48-diUb preferred by TRIP12, UBR5, Ufd4 for branching [10] [31] [39]; Commercial sources or prepared via chemical synthesis/enzymatic ligation |
| Mutant Ubiquitins | To identify essential lysines or probe mechanism | Ub-K0 (lysine-less): track donor ubiquitin [10]; Ub-K29R: test K29 linkage requirement [10] [39]; Site-specific fluorescent labeling |
| Chemical Probe: triUbprobe | Traps transition state for structural studies (e.g., Cryo-EM) | Covalently links E3 catalytic Cys, donor Ub C-terminus, and acceptor Ub K29; Used to determine TRIP12 and Ufd4 mechanisms [10] [39] |
| E2 Enzyme Panel | Test E2-E3 pairing requirements and linkage specificity | UBE2D (UbcH5) family: common for many RING and HECT E3s; UBE2T, Ubc4: used with bacterial SneRING and yeast Ufd4, respectively [49] [39] |
| Cryo-EM with Chemical Crosslinking | Visualize transient E3-Ub-substrate complexes | Provided structural snapshots of UBR5 and TRIP12 during K48 and K29/K48 chain formation [10] [31] [39] |
HECT vs. RING E3 Catalytic Mechanisms
Experimental Workflow for Pulse-Chase Assay
Pulse-chase and thioester discharge assays provide complementary insights into the distinct mechanisms of HECT and RING E3 ligases. The pulse-chase technique is exceptionally powerful for delineating the multi-step ubiquitin transfer pathway of HECT E3s and for establishing their linkage specificity toward atypical chains, as demonstrated by recent work on TRIP12, UBR5, and Ufd4 [10] [31] [39]. In contrast, the thioester discharge assay offers a more focused approach to probe the allosteric activation of E2~Ub by RING E3s and the role of specific structural elements like the linchpin residue [7] [48].
The selection between these techniques should be guided by the specific research question: pulse-chase for comprehensive pathway analysis and linkage specificity determination, particularly for HECT E3s, and thioester discharge for interrogating E2~Ub conformational dynamics in RING E3 mechanisms. Employing both assays in tandem, alongside advanced structural techniques like cryo-EM with chemical crosslinking, provides the most robust strategy for deciphering the complex mechanisms underlying atypical ubiquitin chain formation.
Ubiquitination is a crucial post-translational modification that governs myriad eukaryotic cellular processes, including protein degradation, cell cycle progression, and signal transduction [11]. This process involves a sequential enzymatic cascade wherein ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3) work in concert to attach ubiquitin to substrate proteins [11]. The remarkable functional diversity of ubiquitin signaling arises from the ability to form different ubiquitin chain architectures—including atypical chains linked through non-K48 residues—which create distinct molecular signals recognized by specific cellular machinery [21]. Understanding the structural basis of ubiquitin chain formation requires high-resolution visualization of the E2/E3/Ub complexes, a challenge that has been addressed through two principal structural biology techniques: X-ray crystallography and cryo-electron microscopy (cryo-EM). This guide objectively compares these methodologies within the context of ongoing research comparing HECT versus RING E3 specificity for atypical chain synthesis.
Technical Comparison: Cryo-EM Versus X-Ray Crystallography
Fundamental Principles and Workflows
X-ray crystallography operates on the principle of Bragg's Law, where X-rays diffract from well-ordered three-dimensional crystals of homogeneous molecules, producing distinctive diffraction patterns that can be transformed into atomic models [50]. The technique requires growing highly ordered crystals, which often demands large amounts of purified macromolecules and extensive molecular engineering to improve crystal quality [50]. The critical challenge in crystallography involves solving the "phase problem," typically addressed through experimental methods like SAD/MAD or molecular replacement [50].
Cryo-electron microscopy utilizes high-energy electrons to image biological samples that are rapidly frozen in vitreous ice, preserving their native structure [50] [51]. Single-particle cryo-EM collects thousands of images of individual macromolecules from different orientations, which are computationally aligned, classified, and averaged to reconstruct a three-dimensional density map [50]. This approach avoids crystallization requirements and maintains molecules in near-physiological states, though it demands significant computational resources for image processing [50] [52].
Comparative Performance and Applications
Table 1: Technical comparison of Cryo-EM and X-ray Crystallography for E2/E3/Ub complex studies
| Parameter | Cryo-EM | X-ray Crystallography |
|---|---|---|
| Optimal Resolution | Typically 2.5-4.0 Å (near-atomic) [52] | Up to 1.0 Å possible (atomic resolution) [52] |
| Sample Requirements | 0.1-0.2 mg; moderate heterogeneity acceptable [52] | >2 mg typically; high homogeneity required [52] |
| Molecular Size Suitability | Optimal >100 kDa; no upper limit [52] | Optimal <100 kDa [52] |
| Sample Preparation | Vitrification optimization; grid preparation [52] | Crystal growth & optimization; cryoprotection [52] |
| Data Collection Time | Hours to days per dataset [52] | Minutes to hours per dataset [52] |
| Advantages for E2/E3/Ub | Captures multiple conformational states; studies dynamic complexes; ideal for large assemblies like CRLs [53] | Atomic precision for small complexes; well-established for discrete states; robust validation methods [52] |
| Limitations for E2/E3/Ub | Lower resolution for flexible regions; intensive computing needed [52] | Crystal packing may constrain conformations; challenging for dynamic complexes [50] |
Table 2: Suitability for specific ubiquitination system studies
| Research Application | Recommended Technique | Rationale |
|---|---|---|
| HECT E3 catalytic cycle | Cryo-EM | Can capture multiple intermediates (TS1, E3~Ub, TS2) in near-native states [31] [39] |
| RING E3 static complexes | X-ray Crystallography | Provides atomic details of E2-E3 interfaces and ubiquitin positioning [11] |
| Large CRL complexes | Cryo-EM | No size limitations; reveals conformational dynamics and allosteric regulation [53] |
| Atypical ubiquitin chain architecture | Both complementary | Crystallography for atomic details; Cryo-EM for chain conformation in solution [21] |
| Time-resolved mechanisms | Cryo-EM | Can capture short-lived states via trapping approaches [31] [39] |
Experimental Approaches for E2/E3/Ub Complexes
Trapping Intermediates in HECT E3 Catalytic Cycles
Recent methodological advances have enabled structural visualization of transient intermediates in ubiquitin transfer cascades. For HECT E3 ligases like UBR5 and Ufd4, researchers have employed engineered ubiquitin probes and catalytic cysteine mutations to trap stable mimics of transition states [31] [39].
UBR5 K48-Linked Chain Formation Protocol:
- Generate E2~UbD intermediate using pulse reaction with UBE2D and fluorescently-labeled ubiquitin [31]
- Incubate E2~*UbD with UBR5 and unlabeled acceptor Ub (UbA) in chase reaction
- Resolve reaction intermediates via SDS-PAGE with fluorescent detection
- For structural studies, use C2768A catalytic mutant to stabilize E3~Ub intermediate [31]
- Apply single-particle cryo-EM to determine 3D structure of UBR5~Ub complex at 2.7-3.7 Å resolution [31]
Ufd4 Branched Chain Formation Protocol:
- Synthesize K29/K48-branched triUb probe (triUbprobe) through chemical ligation [39]
- Cross-link triUbprobe with Ufd4 catalytic residue (C1450) to form stable complex mimicking transition state [39]
- Purify complex via size-exclusion chromatography
- Collect 5,332 cryo-EM micrographs for single-particle analysis
- Reconstruct 3D density map at 3.31 Å global resolution [39]
- Dock AlphaFold-predicted Ufd4 structure and refine atomic model [39]
Crystallographic Approaches for RING E3 Complexes
For RING E3 ligases, crystallography has provided fundamental insights into E2-E3 interfaces and ubiquitin positioning:
- Co-crystallize RING domain (e.g., Cbl) with E2~Ub conjugate [11]
- Optimize crystal packing through surface entropy reduction mutagenesis
- Collect diffraction data at synchrotron sources
- Solve structure using molecular replacement with known RING and E2 components
- Model ubiquitin into residual density to determine positioning residues
Diagram 1: Cryo-EM workflow for HECT E3 intermediate analysis
Key Findings: HECT Versus RING E3 Specificity for Atypical Chains
HECT E3 Mechanisms for Atypical Chain Synthesis
Structural studies of HECT E3s reveal distinctive mechanisms for atypical ubiquitin chain formation. Cryo-EM structures of UBR5 demonstrate a 620 kDa dimeric architecture functioning as the fundamental catalytic unit, with flexibly tethered Ub-associated (UBA) domains that capture acceptor ubiquitin and position K48 for linkage formation [31]. The HECT domain undergoes conformational cycling between "L-shaped" and "inverted T" configurations during ubiquitin transfer, with the C-lobe determining linkage specificity [11] [31].
For branched chain formation, Ufd4 employs a clamp-like architecture where N-terminal ARM regions and the HECT C-lobe collaboratively recruit K48-linked diUb substrates, orienting proximal K29 toward the catalytic cysteine [39]. Biochemical assays demonstrate Ufd4 preferentially catalyzes K29-linked ubiquitination on preassembled K48-linked chains, with ~5.2-fold higher efficiency at proximal versus distal K29 sites [39].
WWP1 HECT E3 exhibits a two-phase ubiquitin chain assembly:
- Phase 1: Unidirectional synthesis of Lys-63-linked chains
- Phase 2: Multidirectional elongation with mixed linkages (Lys-63 > Lys-48 > Lys-11) and branched structures [15]
This mechanism involves a low-affinity, noncovalent ubiquitin-binding site within the HECT domain that facilitates sequential ubiquitin addition [15].
RING E3 Specificity Determinants
In contrast to HECT E3s, RING E3s typically lack direct catalytic activity and function as scaffolds that bring E2 and substrate into proximity [11]. Linkage specificity for RING E3s is primarily determined by their cognate E2 enzymes [11] [31]. For example:
- MMS2-UBC13 complex specifically generates K63-linked chains through structural constraints positioning acceptor Ub K63 toward the E2 active site [21]
- BRCA1 interacts with multiple E2s (UBCH6, UBCM2, UBE2W) for monoubiquitination, while partnering with MMS2-UBC13 and UBE2K for K63- or K48-linked chains, respectively [21]
- CBL mediates multiple ubiquitination types (monoUb, K11, K48, K63) on EGFR, with specificity influenced by E2 partnerships and substrate context [21]
Diagram 2: Specificity determinants for HECT versus RING E3 ligases
Research Reagent Solutions for E2/E3/Ub Structural Studies
Table 3: Essential research reagents for E2/E3/Ub structural biology
| Reagent/Category | Specific Examples | Function in Experimental Design |
|---|---|---|
| Engineered Ubiquitin Variants | K48R Ub (acceptor-deficient) [31]; Fluorescently-labeled Ub (e.g., TAMRA) [31]; K29-only triUb substrates [39] | Trapping specific intermediates; monitoring chain formation; mapping modification sites |
| E2 Enzymes | UBE2D family [31]; Ubc4 [39]; MMS2-UBC13 complex [21] | Partner selection for linkage specificity; intermediate formation |
| Stabilization Reagents | Catalytic cysteine mutants (C2768A in UBR5) [31]; Chemical crosslinkers [39] | Stabilizing transient complexes for structural analysis |
| Cryo-EM Grids | UltrAuFoil [31]; Quantifoil [39] | Optimized sample support for high-resolution data collection |
| Detection Systems | Anti-ubiquitin antibodies; Fluorescence detection systems [31] | Monitoring reaction progress and intermediate formation |
Integrated Structural Approaches Advance Ubiquitin Research
The combination of cryo-EM and X-ray crystallography has proven particularly powerful for elucidating complete mechanisms of E2/E3/Ub complexes. Cryo-EM excels at capturing dynamic processes and large assemblies like the 620 kDa UBR5 dimer and multi-subunit cullin RING ligases (CRLs), providing insights into allosteric regulation and conformational transitions [31] [53]. Meanwhile, crystallography offers atomic-resolution views of discrete states, such as E2-E3 interfaces and ubiquitin positioning within active sites [11].
For the specific question of HECT versus RING E3 specificity toward atypical ubiquitin chains, structural biology reveals fundamentally different mechanistic strategies. HECT E3s employ an intrinsic catalytic mechanism with conformational cycling and specialized ubiquitin-binding sites that directly control linkage specificity [15] [31]. In contrast, RING E3s operate through selective E2 partnerships and combinatorial assembly, creating diverse ubiquitination outcomes through contextual specificity [21] [11]. This mechanistic understanding, derived from complementary structural approaches, provides critical insights for drug discovery efforts targeting specific ubiquitination pathways in disease contexts [52] [53].
Ubiquitination is a versatile post-translational modification that regulates diverse fundamental features of protein substrates, including stability, activity, and localization [54]. The versatility of ubiquitination stems from the complexity of ubiquitin (Ub) conjugates, which range from a single Ub monomer to Ub polymers with different lengths and linkage types [54]. While K48-linked ubiquitin chains (targeting proteins for proteasomal degradation) and K63-linked chains (regulating protein-protein interactions and signaling) are well-characterized, the functions of atypical chain types—including mono-ubiquitination, multi-monoubiquitination, and K6-, K11-, K27-, K29-, K33-, and M1-linked ubiquitin chains—are less well defined and represent an emerging frontier in ubiquitin research [54].
The specificity of ubiquitin signaling is largely determined by E3 ubiquitin ligases, which are classified into three main families: Really Interesting New Gene (RING), Homologous to E6AP C-Terminus (HECT), and RING-Between-RING (RBR) ligases [24]. The distinct catalytic mechanisms of HECT and RING E3 ligases directly impact their ability to generate atypical ubiquitin chains. HECT-family E3s form an obligate thioester intermediate with ubiquitin before transferring it to substrates, while RING-family E3s facilitate direct ubiquitin transfer from E2 enzymes to substrates [11] [25]. This fundamental difference influences their linkage specificity and makes the development of sensitive, linkage-specific detection reagents particularly important for advancing our understanding of ubiquitin signaling in health and disease.
HECT vs. RING E3 Ligases: Structural and Mechanistic Basis for Atypical Chain Formation
Comparative Architecture and Catalytic Mechanisms
E3 ubiquitin ligases confer substrate specificity to the ubiquitination cascade, with humans encoding approximately 600 E3s [11]. The mechanistic differences between HECT and RING E3 ligases fundamentally influence their ubiquitin chain linkage specificity:
HECT Family E3 Ligases: The ~30 human HECT E3s feature a conserved bi-lobed HECT domain consisting of an N-lobe that binds the E2 enzyme and a C-lobe containing the active-site cysteine that forms a thioester intermediate with ubiquitin [11] [25]. This intermediate allows HECT E3s to exert direct control over linkage specificity. The flexible hinge region connecting the lobes enables the conformational changes necessary for ubiquitin transfer [11].
RING Family E3 Ligases: The >600 RING-type E3s contain a Zn²⁺-coordinating RING finger domain that simultaneously binds E2~Ub and substrate, facilitating direct ubiquitin transfer without a E3-ubiquitin intermediate [11] [55]. While this provides less direct control over linkage specificity compared to HECT E3s, certain RING E3s can allosterically activate their cognate E2s to influence chain topology [11].
Table 1: Fundamental Differences Between HECT and RING E3 Ubiquitin Ligases
| Feature | HECT Family E3s | RING Family E3s |
|---|---|---|
| Catalytic Mechanism | Forms thioester intermediate with ubiquitin | Direct transfer from E2 to substrate |
| Representative Members | NEDD4, WWP1, E6AP, HUWE1 | Cbl, MDM2, Cullin-RING ligases (CRLs) |
| Ubiquitin Chain Linkage Control | Direct control via E3 catalytic domain | Primarily determined by E2 enzyme |
| Human Genome Encoding | ~28-30 members [25] | >600 members [11] |
| Characteristic Atypical Linkages | K63, K11, K6 (varies by family) [15] [25] | Mixed linkages, branched structures [15] |
Atypical Chain Synthesis by HECT and RING E3 Ligases
Emerging evidence reveals that both HECT and RING E3 families contribute to atypical ubiquitin chain formation through distinct mechanisms:
HECT E3 Specificity and Mechanisms: The ability to build linkage-specific poly-Ub chains appears to be an intrinsic feature of HECT enzymes [25]. Different HECT subfamilies demonstrate characteristic linkage preferences:
- NEDD4 family members (e.g., WWP1) primarily synthesize K63-linked chains, with some producing K48 and K11 linkages (K63 > K48 > K11) [15] [25]. They employ a sequential addition mechanism with distinct synthesis phases: an initial unidirectional phase producing exclusively K63 linkages, followed by a multidirectional phase generating mixed linkages and branched structures [15].
- E6AP (UBE3A) is a K48-specific enzyme [25].
- HUWE1 generates K6-, K11-, and K48-linked poly-Ub chains [25].
The mechanism often involves specialized ubiquitin-binding sites, such as the "Ub exosite" in the N-lobe of NEDD4 family E3s, which stabilizes and orients the growing ubiquitin chain on the substrate [25].
RING E3 and Atypical Chain Formation: RING E3 ligases demonstrate more diverse linkage capabilities, often dictated by their associated E2 enzymes. For example, RNF31 (HOIP), the catalytic component of the Linear Ubiquitin Chain Assembly Complex (LUBAC), primarily generates M1-linked linear ubiquitin chains but has also been demonstrated to catalyze atypical nonlinear ubiquitination of FOXP3, stabilizing the protein rather than targeting it for degradation [56].
The following diagram illustrates the fundamental differences in catalytic mechanisms between HECT and RING E3 ligases and their implications for atypical chain formation:
Linkage-Specific Reagents for Detecting Atypical Ubiquitin Chains
Antibody-Based Detection Methods
Linkage-specific antibodies represent a powerful tool for identifying atypical ubiquitin chains. These reagents are generated to recognize unique epitopes created when ubiquitin molecules form specific linkages:
Polyclonal and Monoclonal Antibodies: Traditional antibodies such as P4D1 and FK1/FK2 recognize ubiquitinated substrates regardless of linkage type, while linkage-specific antibodies target particular chain architectures [54]. For example, researchers have developed antibodies specifically recognizing M1-, K11-, K27-, K48-, and K63-linkages [54].
Application Workflow: Linkage-specific antibodies are typically used in immuno-blotting, immunofluorescence, and immunoprecipitation applications. Nakayama et al. successfully employed a K48-linkage specific antibody to demonstrate abnormal accumulation of K48-linked polyubiquitination on tau proteins in Alzheimer's disease [54].
Advantages and Limitations: Antibody-based approaches enable characterization of protein ubiquitination under physiological conditions without genetic manipulation and can be applied to animal tissues or clinical samples [54]. However, they suffer from high costs, potential non-specific binding, and limited availability for some atypical linkages [54].
TUBEs (Tandem-Repeated Ubiquitin-Binding Entities)
Tandem-repeated Ubiquitin-Binding Entities (TUBEs) represent an alternative approach that exploits natural ubiquitin-binding domains to enrich ubiquitinated proteins:
Design Principle: TUBEs consist of multiple ubiquitin-associated domains (UABs) connected in tandem, creating avidity effects that significantly enhance affinity for ubiquitin chains compared to single domains [54]. This multi-valent approach allows recognition of multiple linkage types simultaneously while protecting ubiquitin chains from deubiquitinating enzyme (DUB) activity during purification.
Applications and Utility: TUBEs are particularly valuable for proteomic studies aiming to capture a broad spectrum of ubiquitinated proteins without linkage bias. They can be engineered with specificity for particular chain types by incorporating linkage-selective ubiquitin-binding domains [54].
Comparative Advantages: Unlike antibodies, TUBEs can be produced recombinantly at lower cost and offer greater versatility in experimental design. Their ability to shield ubiquitin chains from DUBs during cell lysis and purification helps preserve the native ubiquitome [54].
Table 2: Comparison of Key Reagents for Detecting Atypical Ubiquitin Chains
| Reagent Type | Mechanism of Action | Applications | Advantages | Limitations |
|---|---|---|---|---|
| Linkage-Specific Antibodies | Antigen-antibody recognition of linkage-specific epitopes | Immunoblotting, Immunofluorescence, Immunoprecipitation | High specificity, applicable to clinical samples, well-established protocols | Limited availability for rare linkages, potential non-specific binding, high cost |
| TUBEs (Tandem Ubiquitin Binding Entities) | Tandem ubiquitin-binding domains with avidity effect | Enrichment of ubiquitinated proteins, proteomics, DUB protection | Broad ubiquitin recognition, protects from DUBs, recombinantly producible | Less linkage-specific without engineering, requires recombinant expression |
| Affinity Tags (e.g., His-, Strep-tags) | Purification of tagged ubiquitin conjugates | Enrichment of ubiquitinated proteins and substrates, identification of ubiquitination sites | Easy implementation, relatively low cost, compatible with various detection methods | May not mimic endogenous ubiquitin, potential artifacts, genetic manipulation required |
Experimental Approaches for Studying Atypical Ubiquitination
Workflow for Comprehensive Ubiquitin Chain Analysis
The complexity of ubiquitin signaling necessitates integrated experimental approaches that combine multiple methodologies. The following workflow provides a framework for comprehensive analysis of atypical ubiquitin chains:
Key Methodologies and Protocols
Ubiquitinated Protein Enrichment Using TUBEs:
- Cell Lysis: Prepare lysates in modified RIPA buffer containing 50mM Tris-HCl (pH 7.5), 150mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 1mM EDTA, supplemented with N-ethylmaleimide (NEM) to inhibit deubiquitinases and complete protease inhibitors [54].
- TUBE Immobilization: Couple recombinant TUBEs protein to agarose beads using appropriate chemistry (e.g., amine coupling for GST-tagged TUBEs).
- Affinity Purification: Incubate cell lysates with TUBE-coupled beads for 2-4 hours at 4°C with gentle rotation.
- Washing: Wash beads extensively with lysis buffer to remove non-specifically bound proteins.
- Elution: Elute bound ubiquitinated proteins using SDS-PAGE sample buffer or competitive elution with free ubiquitin.
Linkage-Specific Immunoblotting:
- Protein Separation: Separate enriched ubiquitinated proteins by SDS-PAGE and transfer to PVDF membranes.
- Blocking: Block membranes with 5% non-fat milk in TBST for 1 hour.
- Primary Antibody Incubation: Incubate with linkage-specific primary antibodies (typically at 1:1000 dilution) overnight at 4°C.
- Washing and Detection: Wash membranes and incubate with appropriate HRP-conjugated secondary antibodies (1:5000) for 1 hour at room temperature.
- Signal Development: Develop using enhanced chemiluminescence substrate and image with appropriate detection system.
Mass Spectrometry-Based Linkage Identification:
- Sample Preparation: Digest enriched ubiquitinated proteins with trypsin after reduction and alkylation.
- Peptide Identification: Analyze resulting peptides by LC-MS/MS using high-resolution mass spectrometry.
- Data Analysis: Search MS data against appropriate databases, identifying ubiquitination sites through the detection of GG remnant signature (diglycine lysine modification with 114.04 Da mass shift) on modified lysine residues [54].
- Linkage Determination: Identify linkage types through detection of ubiquitin-derived peptides containing the specific lysine linkage.
The Scientist's Toolkit: Essential Reagents for Atypical Chain Research
Table 3: Essential Research Reagents for Studying Atypical Ubiquitin Chains
| Reagent Category | Specific Examples | Primary Applications | Key Features |
|---|---|---|---|
| Linkage-Specific Antibodies | K11-linkage antibody, K48-linkage antibody, K63-linkage antibody, M1-linear antibody | Immunoblotting, Immunofluorescence, Immunoprecipitation | High specificity for particular ubiquitin linkage types |
| TUBEs (Tandem Ubiquitin-Binding Entities) | Agarose-coupled TUBEs, Recombinant TUBEs | Enrichment of ubiquitinated proteins, Proteomic studies | Pan-specific ubiquitin recognition, Protects from DUBs |
| Affinity-Tagged Ubiquitin | His-tagged Ubiquitin, Strep-tagged Ubiquitin, HA-tagged Ubiquitin | Purification of ubiquitinated substrates, Identification of ubiquitination sites | Enables purification under denaturing conditions |
| Ubiquitin Mutants | K6R, K11R, K27R, K29R, K33R, K48R, K63R ubiquitin mutants | Determination of specific ubiquitin linkage requirements | Eliminates specific lysine residues for linkage formation |
| Mass Spectrometry Standards | Heavy-labeled ubiquitin peptides, AQUA peptides | Absolute quantitation of ubiquitin linkages, Mass spectrometry calibration | Enables precise quantification in proteomic experiments |
The expanding toolkit of linkage-specific reagents, particularly antibodies and TUBEs, has dramatically advanced our ability to detect and characterize atypical ubiquitin chains. These tools have revealed that HECT and RING E3 ligases employ distinct mechanistic strategies for generating atypical ubiquitin chains, with HECT E3s directly controlling linkage specificity through their catalytic domains, while RING E3s often collaborate with specific E2 enzymes to determine chain topology.
The continuing development of more sensitive, specific, and comprehensive detection reagents will be essential for unraveling the complex biological functions of atypical ubiquitin chains. As these tools evolve, they will undoubtedly provide deeper insights into how HECT and RING E3 ligases manipulate the ubiquitin code to regulate critical cellular processes, potentially revealing new therapeutic opportunities for diseases characterized by dysregulated ubiquitination.
Mass Spectrometry-Based Proteomics for Mapping Ubiquitination Sites and Chain Topology
Protein ubiquitination is a crucial post-translational modification that regulates diverse cellular functions, including protein degradation, signal transduction, and DNA repair [57]. This modification is orchestrated by a sequential enzymatic cascade involving E1 activating, E2 conjugating, and E3 ligase enzymes, with E3 ligases providing substrate specificity [8] [57]. The remarkable functional diversity of ubiquitination stems from the structural complexity of ubiquitin conjugates, which can range from single ubiquitin monomers to polymers (polyubiquitin chains) of varying lengths and linkage types [8] [57]. Ubiquitin contains seven lysine residues (K6, K11, K27, K29, K33, K48, K63) and an N-terminal methionine (M1), each capable of forming distinct polyubiquitin chain linkages [21] [57].
The E3 ligase families—primarily RING (Really Interesting New Gene), HECT (Homologous to E6-AP C-Terminus), and RBR (RING-between-RING)—differ fundamentally in their catalytic mechanisms, which directly influences the types of ubiquitin chains they assemble [8] [19] [18]. RING-type E3s function as scaffolds that facilitate the direct transfer of ubiquitin from an E2 enzyme to the substrate, with some linkage specificity determined by the associated E2 [19]. In contrast, HECT-type E3s employ a two-step mechanism involving a catalytic cysteine that forms a thioester intermediate with ubiquitin before transferring it to the substrate, granting the HECT domain more direct control over chain linkage specification [8] [40]. The RBR family represents a hybrid mechanism, combining aspects of both RING and HECT-type ligases [18].
Understanding the specificity of different E3 ligase families, particularly regarding their propensity to form "atypical" ubiquitin chains (non-K48 and non-K63 linkages), represents a critical frontier in ubiquitin biology. Mass spectrometry-based proteomics has emerged as an indispensable tool for mapping ubiquitination sites and deciphering ubiquitin chain topology, providing insights into the specialized functions of HECT and RING E3 ligases in generating specific ubiquitin signals.
E3 Ligase Families: Catalytic Mechanisms and Linkage Specificity
RING-type E3 Ligases
RING-type E3 ligases constitute the largest family of ubiquitin ligases and operate through a direct transfer mechanism. They function primarily as scaffolds that simultaneously bind both the E2~Ub thioester and the substrate, facilitating direct ubiquitin transfer without forming a covalent E3-Ub intermediate [19]. The RING domain typically coordinates two Zn²⁺ ions in a cross-braced arrangement that creates a platform for E2 binding [19]. A notable feature of many RING E3s is their tendency to form homodimers or heterodimers, which can enhance catalytic activity and promote processive ubiquitination [19].
Table 1: Characteristic Features of RING-type E3 Ligases
| Feature | Description | Examples |
|---|---|---|
| Catalytic Mechanism | Direct transfer from E2 to substrate; no E3-Ub intermediate | BRCA1-BARD1, Mdm2, cIAP |
| Structural Domains | RING domain (Zn²⁺ coordination), various substrate-binding domains | RING, U-box |
| Dimerization | Common (homodimers and heterodimers) through RING domains or external sequences | Mdm2-MdmX, BRCA1-BARD1 |
| Linkage Specificity | Largely determined by E2 partnership; some E3s show strong preference | CBL (K48, K63), BRCA1 (varies by E2) |
| Regulation | Multiple mechanisms including autoinhibition, subcellular localization, post-translational modifications | cIAP (SMAC/DIABLO activation) |
For RING E3s, the identity of the partnering E2 enzyme significantly influences linkage specificity. Some E2s, such as the UBC13-UEV1A complex, are dedicated to forming specific linkages (K63-linked chains), while others like UBCH5 exhibit broader specificity [21] [19]. The RING E3 CBL exemplifies this partnership-dependent specificity, as it can mediate multiple monoubiquitination or synthesize K11-, K48-, and K63-linked chains on the epidermal growth factor receptor (EGFR) [21]. This flexibility allows RING E3s to generate diverse ubiquitin signals, though the E3 itself primarily functions as a matchmaker between E2~Ub and substrate rather than directly controlling linkage specificity.
HECT-type E3 Ligases
HECT E3 ligases employ a distinct two-step catalytic mechanism that involves a conserved catalytic cysteine residue. In the first step, ubiquitin is transferred from the E2 enzyme to the HECT domain cysteine, forming a reactive E3~Ub thioester intermediate. Subsequently, ubiquitin is transferred from the HECT domain to the substrate lysine residue [8] [40]. This mechanism grants HECT E3s more direct control over the linkage specificity of the ubiquitin chains they assemble.
Table 2: Characteristic Features of HECT-type E3 Ligases
| Feature | Description | Examples |
|---|---|---|
| Catalytic Mechanism | Two-step mechanism with E3~Ub thioester intermediate | Ufd4, WWP1, E6AP, Nedd4 |
| Structural Domains | HECT domain (conserved C-terminal ~350 residues), various substrate-recognition domains | C-lobe (catalytic), N-lobe (E2 binding) |
| Linkage Specificity | Determined by HECT domain; often strong preferences | E6AP (K48), Nedd4 (K63), Ufd4 (K29) |
| Chain Assembly | Sequential addition mechanism; some form branched chains | WWP1 (mixed linkages), Ufd4 (K29/K48-branched) |
| Regulation | Intra- and intermolecular interactions, allosteric regulation | Ndfip1 activation of Nedd4 family |
Different HECT E3s exhibit characteristic linkage specificities. For instance, E6AP primarily synthesizes K48-linked chains, while Nedd4 family members typically prefer K63-linkages [40]. However, recent research has revealed more complexity, with many HECT E3s capable of forming multiple linkage types. The Nedd4 family E3 WWP1 assembles substrate-linked ubiquitin chains containing K63, K48, and K11 linkages (K63 > K48 > K11) [40]. Strikingly, WWP1 catalyzes ubiquitin chain formation in two distinct phases: an initial phase characterized by unidirectional K63-linked chain elongation, followed by a multidirectional phase featuring mixed linkages and branched structures [40].
A remarkable example of HECT E3 specificity is Ufd4, which preferentially catalyzes K29-linked ubiquitination on pre-existing K48-linked ubiquitin chains to form K29/K48-branched ubiquitin chains [39]. Structural studies have revealed that the N-terminal ARM region and HECT domain C-lobe of Ufd4 work together to recruit K48-linked diUb and orient Lys29 of its proximal ubiquitin toward the active cysteine for K29-linked branched ubiquitination [39]. This specialized activity enhances the degradation signal for proteasomal targeting, illustrating how HECT E3s can create complex ubiquitin architectures with distinct functional consequences.
RBR-type E3 Ligases
RBR E3 ligases represent a hybrid category that combines mechanistic elements from both RING and HECT families. Similar to RING E3s, RBRs contain a RING1 domain that binds the E2~Ub conjugate. However, like HECT E3s, they employ a two-step mechanism in which ubiquitin is first transferred from the E2 to a catalytic cysteine in the RING2 domain before subsequent transfer to the substrate [18]. This family includes well-characterized enzymes such as Parkin, HOIP, and HHARI.
RBR E3s are typically regulated by complex autoinhibition mechanisms that must be relieved for catalytic activity. For instance, Parkin is activated by phosphorylation of its ubiquitin-like (UBL) domain by PINK1 kinase, while HOIP is autoinhibited by its UBA domain and activated through interactions with HOIL-1 or Sharpin in the LUBAC complex [18]. A common regulatory feature among RBRs is allosteric activation by ubiquitin or ubiquitin-like proteins, as observed with Parkin (activated by phospho-Ub), HOIP (activated by M1-linked diUb), and RNF216 (activated by K63-linked diUb) [18]. This suggests RBRs may employ a feed-forward mechanism where the product of the reaction enhances further catalytic activity.
Mass Spectrometry Methodologies for Ubiquitination Mapping
Enrichment Strategies for Ubiquitinated Proteins
The low stoichiometry of protein ubiquitination necessitates efficient enrichment strategies prior to mass spectrometry analysis. Three primary approaches have been developed for this purpose, each with distinct advantages and limitations.
Ubiquitin Tagging-Based Approaches utilize genetically engineered ubiquitin containing affinity tags (e.g., His, Strep, HA) expressed in cells. Following lysis, ubiquitinated proteins are purified using appropriate affinity resins (Ni-NTA for His-tag, Strep-Tactin for Strep-tag) [57]. This approach enabled the first large-scale identification of ubiquitination sites, with Peng et al. identifying 110 ubiquitination sites on 72 proteins from Saccharomyces cerevisiae in 2003 [57]. While relatively simple and cost-effective, this method may co-purify non-ubiquitinated proteins (e.g., histidine-rich proteins with His-tag purification) and cannot be applied to clinical tissue samples where genetic manipulation is infeasible.
Ubiquitin Antibody-Based Approaches employ antibodies that recognize endogenous ubiquitin epitopes for enrichment. Pan-specific ubiquitin antibodies (e.g., P4D1, FK1/FK2) can enrich ubiquitinated proteins regardless of linkage type, while linkage-specific antibodies target particular chain architectures (e.g., M1-, K11-, K27-, K48-, or K63-linkages) [57]. This approach enabled Denis et al. to identify 96 ubiquitination sites from human MCF-7 breast cancer cells [57]. The key advantage of antibody-based methods is their applicability to native tissues and clinical samples, though they suffer from high cost and potential non-specific binding.
Ubiquitin-Binding Domain (UBD)-Based Approaches leverage natural ubiquitin receptors containing ubiquitin-binding domains (UBDs) for enrichment. To enhance binding affinity, tandem-repeated ubiquitin-binding entities (TUBEs) have been developed that exhibit higher affinity for ubiquitinated proteins while protecting ubiquitin chains from deubiquitinase activity [57]. UBD-based approaches can capture endogenous ubiquitination without genetic manipulation and may offer some linkage selectivity based on the UBD specificity.
MS Acquisition Methods and Data Analysis
Following enrichment, mass spectrometry analysis typically involves either Data-Dependent Acquisition (DDA) or Data-Independent Acquisition (DIA) approaches. DDA, the traditional method, selects the most abundant precursor ions for fragmentation, which can be effective for abundant ubiquitination events but may suffer from stochastic sampling and missing low-abundance modifications. DIA, alternatively, fragments all ions within predetermined isolation windows, providing more comprehensive coverage and better reproducibility, making it particularly valuable for large-scale clinical studies [58].
For ubiquitination site identification, tryptic digestion produces a characteristic di-glycine remnant (GG-signature) on modified lysine residues, resulting in a mass shift of 114.04292 Da that can be detected by MS [57]. Advanced approaches like "Ub-clipping" combine limited proteolysis with middle-down MS to determine ubiquitin chain architecture, enabling identification of branched chains [39].
Table 3: Comparison of Ubiquitin Enrichment Methods for MS-Based Proteomics
| Method | Principle | Advantages | Limitations | Typical Applications |
|---|---|---|---|---|
| Ubiquitin Tagging | Affinity-tagged Ub expressed in cells | Easy implementation; relatively low cost; good for cell lines | Cannot be used in tissues; potential artifacts from tagged Ub; co-purification of non-ubiquitinated proteins | Screening studies in cultured cells; identification of ubiquitination sites |
| Ubiquitin Antibody | Antibodies recognizing endogenous Ub | Works with native tissues and clinical samples; linkage-specific antibodies available | High cost; potential non-specific binding; limited antibody efficiency | Clinical samples; pathway-specific studies (with linkage-specific antibodies) |
| UBD/TUBE-Based | Tandem ubiquitin-binding domains | Captures endogenous Ub; protects from DUBs; works with tissues | Limited linkage specificity; optimization required for different UBDs | Studies requiring preservation of ubiquitin chains; analysis of endogenous ubiquitination |
High-throughput methods have been developed to streamline ubiquitin proteomics, such as the 96-well plate format that enables processing of up to 80 samples per day with an 18-minute DIA method, achieving identification of approximately 6000 peptides and 600 protein groups from just 2 μL of serum [58]. Such improvements in throughput and sensitivity are crucial for large-scale studies comparing ubiquitination patterns across multiple conditions or time points.
Comparative Analysis of HECT vs. RING E3 Specificity
Methodologies for Determining Linkage Specificity
Several experimental approaches have been developed to characterize the linkage specificity of E3 ligases:
In vitro reconstitution assays involve incubating purified E3 ligase with specific E2 enzymes, ubiquitin, and ATP, followed by MS analysis of the resulting ubiquitin chains. This approach revealed that WWP1 assembles chains with a preference for K63 > K48 > K11 linkages [40].
Linkage-specific antibodies can immunoprecipitate ubiquitinated proteins or ubiquitin chains of particular architectures, allowing assessment of E3 ligase specificity in cellular contexts. For example, Nakayama et al. generated a K48-linkage specific antibody that revealed abnormal accumulation of K48-linked polyubiquitination on tau proteins in Alzheimer's disease [57].
Enzyme kinetics using defined ubiquitin chain substrates with mutations at specific lysine residues can quantify catalytic efficiency for different linkage types. Studies on Ufd4 demonstrated approximately 5.2-fold higher catalytic efficiency (kcat/Km) for K29-linked ubiquitination at the proximal ubiquitin of K48-linked chains compared to the distal ubiquitin [39].
Structural approaches including X-ray crystallography and cryo-EM provide atomic-level insights into the mechanisms underlying linkage specificity. The cryo-EM structure of Ufd4 in complex with a K29/K48-branched triUb probe revealed how specific structural elements orient the acceptor ubiquitin to facilitate K29-linked branching on K48-linked chains [39].
Experimental Data on E3 Ligase Specificity for Atypical Chains
HECT E3 Ligases demonstrate remarkable diversity in their linkage specificity, with many members showing pronounced preferences for atypical chains:
Ufd4 preferentially synthesizes K29/K48-branched ubiquitin chains by catalyzing K29-linked ubiquitination on pre-existing K48-linked ubiquitin chains. Biochemical assays show Ufd4 has significantly higher activity on K48-linked diUb substrates compared to monoUb or other chain types, with a strong preference for modifying the proximal K29 site of K48-linked chains [39].
WWP1 assembles substrate-linked ubiquitin chains containing K63, K48, and K11 linkages (K63 > K48 > K11) through a two-phase mechanism. The initial phase involves unidirectional K63-linked chain elongation, followed by a multidirectional phase characterized by mixed linkages and branched structures [40].
E6AP primarily synthesizes K48-linked chains, consistent with its role in targeting p53 for proteasomal degradation in HPV-infected cells [40].
Nedd4 family members (including Rsp5, Smurf1, Smurf2, Itch) typically prefer K63-linked chains, though several can also synthesize K29 and K48 linkages depending on context [40].
RING E3 Ligases often exhibit more flexible linkage specificity that depends on E2 partnership:
CBL can mediate multiple monoubiquitination or synthesize K11-, K48-, and K63-linked chains on activated EGFR, demonstrating considerable flexibility in linkage specification [21].
BRCA1 cooperates with different E2s to generate distinct ubiquitin linkages: UBCH6, UBE2E2, UBCM2, and UBE2W support monoubiquitination; MMS2-UBC13 catalyzes K63-linked chains; and UBE2K generates K48-linked chains [21].
RBR-type E3s like HOIL-1 and RNF216 show distinct allosteric activation by specific ubiquitin linkages. HOIL-1 is activated by M1- and K63-linked diUb, while RNF216 is specifically activated by K63-linked diUb [18].
Experimental Protocols for E3 Ligase Specificity Characterization
In vitro Ubiquitination Assay for Linkage Specificity
This protocol enables assessment of E3 ligase linkage preference using purified components:
Reagents and Equipment:
- Purified E1 enzyme, E2 enzymes, E3 ligase
- Wild-type and mutant ubiquitin (e.g., K29R, K48R, K63R)
- ATP regeneration system (ATP, creatine phosphate, creatine kinase)
- Reaction buffer (50 mM Tris-HCl pH 7.5, 50 mM KCl, 5 mM MgCl₂, 0.5 mM DTT)
- SDS-PAGE equipment and ubiquitin antibodies for detection
Procedure:
- Set up 20 μL reactions containing 100 nM E1, 1-5 μM E2, 1-5 μM E3, 50 μM ubiquitin, and ATP regeneration system in reaction buffer.
- Incubate at 30°C for 60 minutes.
- Stop reactions by adding SDS-PAGE loading buffer with DTT.
- Analyze by immunoblotting with ubiquitin antibodies or linkage-specific antibodies.
- For linkage determination, include ubiquitin mutants (e.g., K29R, K48-only) to assess requirement for specific lysines.
- Quantify band intensity to determine relative efficiency with different ubiquitin mutants or chain substrates.
Applications: This assay can determine an E3's inherent linkage preference, as demonstrated in studies of Ufd4 which showed dramatically reduced activity with K29R ubiquitin mutant, confirming its preference for K29-linked chain formation [39].
MS-Based Analysis of Ubiquitin Chain Topology
This protocol details the characterization of ubiquitin chain architecture assembled by specific E3 ligases:
Reagents and Equipment:
- Enrichment reagents (affinity resins, ubiquitin antibodies, or TUBEs)
- Trypsin/Lys-C mix for proteolysis
- Mass spectrometer with nanoLC interface
- Data analysis software (MaxQuant, Skyline, etc.)
Procedure:
- Express affinity-tagged ubiquitin in cells or use antibody-based enrichment from native samples.
- Enrich ubiquitinated proteins using appropriate method (Ni-NTA for His-tag, antibody immobilization).
- Wash extensively to remove non-specifically bound proteins.
- Digest enriched proteins with trypsin/Lys-C.
- Analyze peptides by LC-MS/MS using DDA or DIA methods.
- Search data against protein database with ubiquitination (GG-K) as variable modification.
- For chain topology, use middle-down "Ub-clipping" approach with limited proteolysis to preserve ubiquitin chain architecture.
- Identify branched chains by detecting ubiquitin fragments with double-glycine remnants on multiple lysine residues.
Applications: This approach enabled the identification of K29/K48-branched chains synthesized by Ufd4, detecting ubiquitin fragments with double-glycine remnants on both K29 and K48 residues by MS/MS analysis [39].
Visualization of Experimental Approaches
The following diagrams illustrate key methodologies and relationships discussed in this guide.
Ubiquitinated Protein Enrichment Workflow
E3 Ligase Catalytic Mechanisms
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents for Ubiquitination Studies
| Reagent Category | Specific Examples | Function/Application | Considerations |
|---|---|---|---|
| Affinity Tags | 6×His-tagged Ub, Strep-tagged Ub | Purification of ubiquitinated proteins; identification of ubiquitination sites | Potential artifacts from tagged Ub; cannot use in native tissues |
| Linkage-Specific Antibodies | K48-linkage specific, K63-linkage specific, M1-linkage specific | Immunoprecipitation of specific chain types; assessment of chain linkage in cells | High cost; potential cross-reactivity; validation required |
| Ubiquitin Mutants | K29R, K48R, K63R, K48-only (all other lysines mutated to R) | Determination of linkage specificity in vitro; pathway analysis | May affect ubiquitin structure/function; comprehensive analysis needed |
| UBD-Based Reagents | Tandem Ubiquitin-Binding Entities (TUBEs) | Enrichment of endogenous ubiquitinated proteins; protection from DUBs | Variable affinity for different chain types; optimization required |
| Stable E2~Ub Conjugates | UbcH7(C86K)-Ub, UbcH5b(C85S)-Ub | ITC binding studies; structural biology of E2-E3 interactions | Mimics natural thioester linkage; enables study of first catalytic step |
| Activity-Based Probes | Branched triUb probes (e.g., K29/K48-branched) | Trapping catalytic intermediates; structural studies of E3 mechanism | Complex chemical synthesis required; highly specific applications |
| DUB Inhibitors | PR-619, G5, NSC 632839 | Preservation of ubiquitin chains during purification | Potential off-target effects; optimization of concentration required |
Mass spectrometry-based proteomics has revolutionized our ability to map ubiquitination sites and decipher the complex topology of ubiquitin chains. The comparative analysis of HECT and RING E3 ligases reveals distinct strategies for achieving linkage specificity: RING E3s largely depend on E2 partnership and function as matchmakers, while HECT E3s exert more direct control over linkage choice through their catalytic HECT domains and often exhibit strong preferences for specific atypical chains. The emerging understanding of E3 ligase specificity, particularly for atypical ubiquitin chains, provides crucial insights into the molecular logic of ubiquitin signaling and offers new opportunities for therapeutic intervention in diseases characterized by dysregulated ubiquitination. As MS methodologies continue to advance, particularly in sensitivity, throughput, and ability to characterize complex chain architectures, our understanding of the ubiquitin code and the specialized functions of E3 ligase families will undoubtedly deepen, opening new frontiers in ubiquitin biology and drug discovery.
The precise assembly of ubiquitin chains, particularly atypical linkages like K29/K48-branched chains, is fundamental to cellular regulation, yet visualizing the transient catalytic events behind these processes has remained a formidable challenge. Chemical trapping strategies have emerged as powerful tools to stabilize these fleeting intermediates, enabling high-resolution structural visualization of ubiquitination mechanisms. This guide compares the experimental approaches and outcomes of these strategies as applied to HECT-family E3 ligases, with particular emphasis on their specialization for forming atypical ubiquitin chains versus the distinct mechanisms employed by RING-family E3s.
For researchers investigating ubiquitin signaling, understanding these methodologies is crucial for elucidating how E3 ligases achieve linkage specificity, with implications for drug discovery and targeted protein degradation therapeutics. The following sections provide a detailed comparison of protocols, structural insights, and reagent toolkits essential for advancing research in this field.
Comparative Analysis of HECT vs. RING E3 Specificity
Mechanism and Specificity for Atypical Chains
Table 1: Comparative Mechanisms of HECT and RING E3 Ligases in Atypical Chain Formation
| Feature | HECT E3 Ligases (Ufd4/TRIP12) | RING E3 Ligases (RNF38/XIAP) |
|---|---|---|
| Catalytic Mechanism | Two-step transthiolation: E2→E3→substrate [8] [59] | Direct transfer: E2→substrate [7] [8] |
| Domain Architecture | ARM domains + HECT domain (N-lobe/C-lobe) [39] [60] | RING domain (zinc-coordinating) [7] [8] |
| Chain Linkage Specificity | Preferentially forms K29 linkages on K48-linked chains [39] [60] | Variable linkage specificity dependent on E2 identity [7] |
| Key Structural Elements | ARM region and HECT C-lobe orient acceptor ubiquitin [39] | Linchpin residue stabilizes closed E2~Ub conformation [7] |
| Conformational Changes | Inverted-T to L-shape transition during catalytic cycle [60] | Open to closed E2~Ub conformational shift [7] |
| Branched Chain Formation | Direct specialization for K29/K48-branched chains [39] [60] | Limited evidence for direct branched chain formation |
| Acceptor Ub Positioning | Structured pincer mechanism with dedicated Ub-binding domains [60] | Primarily through E2~Ub conformational stabilization [7] |
Quantitative Biochemical Efficiency Data
Table 2: Quantitative Analysis of HECT E3 Catalytic Efficiency and Specificity
| Parameter | Ufd4 | TRIP12 | RNF38 (RING Reference) |
|---|---|---|---|
| Preferred Acceptor | K48-linked diUb (over monoUb) [39] | K48-linked diUb (5-fold over monoUb) [60] | E2-dependent substrate specificity [7] |
| Catalytic Site | C1450 [39] | C2007 [60] | RING domain (no catalytic cysteine) [7] |
| Linkage Specificity | K29-linked on proximal Ub of K48 chain [39] | K29-linked on proximal Ub of K48 chain [60] | Variable (K48/K63 common) [7] |
| Kinetic Preference | ~5.2-fold higher efficiency for proximal K29 (kcat/Km = 0.11 vs 0.021 μM⁻¹min⁻¹) [39] | Strict geometric constraint for K29 (4-methylene linker optimal) [60] | Modulated by linchpin residue identity [7] |
| Processivity Enhancement | Efficiency escalates with K48-chain length [39] | Associated with K48-chain elongation [60] | Primarily monomeric ubiquitination |
Experimental Protocols for Intermediates Trapping
Chemical Trapping Workflow for HECT E3s
The fundamental principle behind chemical trapping involves creating stable mimics of the transition states that occur during ubiquitin transfer. The following diagram illustrates the core workflow for generating these trapped complexes, adapted from studies on Ufd4 and TRIP12 [39] [60] [59].
Diagram 1: Chemical Trapping Workflow for HECT E3s
Step-by-Step Protocol for Trapped Complex Formation
Phase 1: Probe Synthesis
- Acceptor Ub Preparation: Engineer K29C mutation into the proximal ubiquitin of K48-linked diUb using expressed protein ligation [39] [60].
- Donor Ub Modification: Generate donor ubiquitin with C-terminal functionalization (e.g., bromoethylamine warhead) replacing the native glycine [59].
- Conjugation Reaction: Incubate modified donor Ub with acceptor K48-diUb (K29C) in reaction buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM TCEP) for 16 hours at 4°C [39].
- Product Purification: Isulate the branched triUb probe via size exclusion chromatography (Superdex 75) [39].
Phase 2: Complex Formation with HECT E3
- E3 Preparation: Express and purify full-length HECT E3 (Ufd4/TRIP12) with intact catalytic cysteine residue [39] [60].
- Cross-linking Reaction: Incubate 50 μM HECT E3 with 100 μM branched triUb probe in reaction buffer for 2 hours at 4°C [60].
- Complex Isolation: Purify the covalently trapped complex using affinity chromatography (Ni-NTA if His-tagged E3) followed by size exclusion chromatography [39].
- Validation: Verify complex formation and stoichiometry by SDS-PAGE and mass spectrometry [39] [60].
Phase 3: Structural Analysis
- Cryo-EM Grid Preparation: Apply 3.5 μL of complex at 2-4 mg/mL to glow-discharged Quantifoil grids [39] [60].
- Vitrification: Flash-freeze in liquid ethane using Vitrobot (blot time 3-6 seconds) [39].
- Data Collection: Acquire micrographs using 300 keV cryo-electron microscope (e.g., Titan Krios) with automated acquisition [39] [60].
- Image Processing: Process datasets (typically 3,000-5,000 micrographs) through standard cryo-EM workflow to achieve 3.0-3.5 Å resolution [39].
Structural Visualization of Trapped Intermediates
Architectural Features Revealed by Trapped Complexes
Structural studies of trapped HECT E3 complexes have revealed several conserved architectural principles that enable atypical chain formation. The analysis of Ufd4 and TRIP12 in their transition states demonstrates a remarkable pincer-like mechanism where distinct domains collaborate to position the acceptor ubiquitin with precision [39] [60].
The N-terminal ARM domains function as one arm of the pincer, engaging the proximal ubiquitin of the K48-linked chain and specifically orienting its Lys29 residue toward the active site. Simultaneously, the HECT domain forms the opposite pincer arm, with its C-lobe positioned to present the donor ubiquitin for catalysis. This coordinated action creates a structured environment where the epsilon amino group of K29 is positioned at an optimal distance and geometry for nucleophilic attack on the thioester bond [60].
The HECT domain itself undergoes significant conformational rearrangements during the catalytic cycle, transitioning from an "inverted-T" configuration when receiving ubiquitin from the E2 to an "L-shaped" conformation when transferring ubiquitin to the substrate. The trapped complexes capture this L-shaped state, revealing how the junction between the N-lobe and C-lobe of the HECT domain forms the active site where both donor and acceptor ubiquitins are juxtaposed [60].
Structural Determinants of Linkage Specificity
The exceptional K29-linkage specificity of HECT E3s like Ufd4 and TRIP12 emerges from precise geometric constraints within the active site. Biochemical experiments with ubiquitin variants containing lysine analogs demonstrated that branched chain formation was undetectable for acceptor side chains shorter than lysine and impaired with longer side chains, indicating that the epsilon amino group must be positioned with exact spatial precision [60].
The following diagram illustrates the structural organization of a HECT E3 in its catalytically active state, based on cryo-EM structures of trapped intermediates [39] [60].
Diagram 2: HECT E3 Catalytic Architecture for K29/K48-branched Chain Formation
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Chemical Trapping Experiments
| Reagent/Chemical Tool | Function | Example Application |
|---|---|---|
| Branched triUb Probe | Mimics transition state during K29 linkage formation | Trapping Ufd4/TRIP12 catalytic intermediate [39] [60] |
| Ubiquitin K29C Mutant | Enables site-specific incorporation of chemical warhead | Probe synthesis for covalent complex formation [60] |
| C-terminal Ubiquitin Warheads | (e.g., bromoethylamine, dehydroalanine) Forms stable thioether with E3 catalytic cysteine | Creating non-hydrolyzable E3~Ub mimics [59] |
| Linkage-specific DiUb Substrates | M1, K6, K11, K27, K29, K33, K48, K63-linked diUb | Determining E3 linkage specificity preferences [39] [60] |
| Ubiquitin Lysine Analogs | L-ornithine (3 methylenes), L-homolysine (5 methylenes) | Probing geometric constraints of active site [60] |
| Activity-based E2~Ub Probes | E2 enzymes covalently linked to ubiquitin via DTT | Studying E2~Ub recruitment by HECT E3s [59] |
| Tandem Ubiquitin-Binding Entities (TUBEs) | Protect polyubiquitinated substrates from degradation | Substrate identification and stabilization [61] |
Chemical trapping strategies have fundamentally advanced our understanding of HECT E3 ligase mechanisms, particularly their specialized role in generating K29/K48-branched ubiquitin chains. The structural insights gained from these approaches reveal how HECT E3s employ a pincer-like architecture to achieve precise geometric positioning of acceptor ubiquitins, contrasting sharply with the E2-centric mechanisms of RING E3s.
For researchers exploring ubiquitin signaling in disease contexts, these methodologies provide critical tools for interrogating E3 ligase function. The continued refinement of chemical trapping approaches—including the development of new warhead chemistries and probe designs—promises to illuminate remaining questions in ubiquitin signaling, particularly regarding the formation of complex heterogeneous and branched chains in cellular regulation. As structural biology advances, these techniques will undoubtedly play a central role in developing targeted therapeutic strategies that exploit the unique specificities of different E3 ligase families.
Cell-Based Reporter Assays for Monitoring Pathway-Specific Ubiquitination
The ubiquitin-proteasome system is a sophisticated regulatory mechanism that controls the stability, activity, and localization of countless cellular proteins. At the heart of this system are E3 ubiquitin ligases, which confer substrate specificity and determine the type of ubiquitin modification enacted. The approximately 600 E3 ligases encoded in the mammalian genome are generally categorized into two major families based on their structural and mechanistic differences: RING-type E3s and HECT-type E3s [11]. RING (Really Interesting New Gene) finger E3s function primarily as scaffolds that bring ubiquitin-charged E2 enzymes in close proximity to substrate proteins, facilitating direct ubiquitin transfer. In contrast, HECT (Homologous to the E6AP Carboxyl Terminus) domain E3s form an obligate thioester intermediate with ubiquitin via a catalytic cysteine residue before transferring it to substrates [11]. This fundamental mechanistic difference has profound implications for their specificity in generating atypical ubiquitin chains, which has become a focal point in understanding their distinct cellular functions.
The emerging research landscape reveals that both HECT and RING E3 families demonstrate remarkable specificity for particular ubiquitin chain linkages, including the less-characterized atypical chains such as Lys6-linked polymers. Understanding this specificity is crucial for deciphering the ubiquitin code and developing targeted therapeutic interventions. Cell-based reporter assays represent powerful tools for investigating these specificities in physiologically relevant contexts, allowing researchers to monitor pathway-specific ubiquitination events in live cells. This guide provides a comprehensive comparison of current methodologies, their experimental parameters, and their applications in distinguishing HECT versus RING E3 specificity for atypical ubiquitin chains.
E3 Ligase Families: Structural and Mechanistic Foundations
HECT Family E3 Ligases
The HECT family comprises approximately 30 members in mammals, each characterized by a conserved C-terminal HECT domain (~350 amino acids) that is bi-lobed in structure [11]. The N-lobe interacts with the E2 ubiquitin-conjugating enzyme, while the C-lobe contains the active-site cysteine that forms the thioester intermediate with ubiquitin. Structural studies of NEDD4L and E6AP (UBE3A) reveal that the two lobes are connected through a flexible hinge that allows them to come together during ubiquitin transfer [11]. Notably, the ubiquitin chain linkage specificity appears to be inherently dependent on the last 60 amino acids of the HECT domain C-lobe, providing a structural basis for their specificity in generating particular chain types [11]. Among their many cellular functions, HECT E3s have prominent roles in protein trafficking, immune response, and signaling pathways regulating cellular growth and proliferation.
RING Family E3 Ligases
The RING finger family is substantially larger, with over 600 members in the mammalian genome [11]. A canonical RING finger is a Zn²⁺-coordinating domain that facilitates E2-dependent ubiquitylation without forming a catalytic intermediate with ubiquitin. Unlike HECT E3s, RING finger domains do not form a thioester intermediate with ubiquitin but instead serve as scaffolds that bring E2 and substrate together, with evidence suggesting they can also allosterically activate E2s [11]. RING finger ubiquitin ligases can function as monomers, dimers, or multi-subunit complexes. Multi-subunit RING E3s are exemplified by the cullin RING ligase (CRL) superfamily, which includes well-characterized complexes such as the SCF (SKP1-CUL1-F-box protein) and the anaphase-promoting complex/cyclosome (APC/C) [11].
Table 1: Fundamental Characteristics of HECT and RING E3 Ligase Families
| Characteristic | HECT Family E3s | RING Family E3s |
|---|---|---|
| Representative Members | NEDD4, SMURF1, E6AP/UBE3A | CBL, MDM2, BRCA1/BARD1, APC/C, SCF |
| Catalytic Mechanism | Forms thioester intermediate with ubiquitin via catalytic cysteine | Direct ubiquitin transfer from E2 to substrate; no intermediate |
| Structural Features | Bi-lobed HECT domain with flexible hinge | Zn²⁺-coordinating RING finger domain |
| Chain Formation | Processive (builds chains on E3 before transfer) or distributive | Distributive (single ubiquitin transfers) |
| Family Size in Mammals | ~30 members | >600 members |
| Key Cellular Functions | Protein trafficking, immune response, cellular growth signaling | Cell cycle regulation, DNA repair, transcription control |
Reporter Assay Methodologies for Monitoring Ubiquitination
URT-Dual-Luciferase Reporter System
The Ubiquitin-Reference Technique (URT) integrated with a Dual-Luciferase system represents a sophisticated cell-based approach for monitoring E3 ligase activity and substrate ubiquitination. This system employs a linear fusion protein in which ubiquitin is positioned between a protein of interest and a reference protein moiety [62]. The construct design typically consists of N-terminally triple FLAG-tagged Renilla luciferase (RL) linked to a ubiquitin K48R mutant (UbR48) moiety, which is in turn connected to triple FLAG-tagged firefly luciferase (FL), with the target substrate (e.g., RHOB) fused at the C-terminus (3×FLAG-RL-UbR48-3×FLAG-FL-RHOB) [62].
The UbR48 mutant is used instead of wild-type ubiquitin to prevent potential K48-linked ubiquitin conjugation on RL-Ub, which could function as a degradation signal. The fusion protein is co-translationally cleaved by endogenous ubiquitin-specific processing proteases (Ubps) after ubiquitin, yielding equimolar amounts of the target protein (FL-RHOB) and reference protein (RL-UbR48) [62]. The steady-state levels of FL-RHOB and RL-UbR48 are quantified by measuring the activities of FL and RL, respectively, using the Dual-Glo Luciferase Assay System. The RL-UbR48 serves as a stable internal reference, while FL-RHOB is a substrate of the E3 ligase being studied. Therefore, E3 ligase activity is reflected by the ratio of FL activity to RL activity (FL/RL), with inhibitors increasing the ratio and activators decreasing it [62].
This system has been validated for studying HECT family E3 ligases such as SMURF1, demonstrating that co-expression of wild-type SMURF1 reduces the FL/RL ratio from 10 to 4, while catalytically inactive SMURF1-C699A has no effect. Treatment with the proteasome inhibitor MG-132 prevents SMURF1-dependent decrease of the FL/RL ratio, confirming that the observed effects occur through the ubiquitin-proteasome pathway [62]. The URT normalization provides excellent correction for experimental variation, with Z-factor values improving from -0.12 (using FL activity alone) to 0.69 (using the FL/RL ratio), converting a poor method into an excellent assay for high-throughput screening [62].
TUBE-Based TR-FRET Assay
Tandem Ubiquitin Binding Entities (TUBEs) coupled with Time-Resolved Förster Resonance Energy Transfer (TR-FRET) technology provides a homogeneous assay platform for monitoring polyubiquitin chain formation. TUBEs bind selectively to polyubiquitin chains versus mono-ubiquitin, enabling specific detection of polyubiquitin chains in the presence of mono-ubiquitin [63]. This assay reports on the proximity between the protein substrate and TUBEs as a result of polyubiquitin chain formation by an E3 ligase, making it suitable for characterizing and quantitating E3 ligase activity in a rapid and cost-effective manner [63].
The TR-FRET signal is generated when a terbium-cryptate donor antibody (targeting a tag on the E3 ligase or substrate) transfers energy to a d2 acceptor fluorophore conjugated to the TUBE upon polyubiquitin chain formation. This assay format has been successfully applied to profile ubiquitin E3 ligases such as MuRF1 and TRIM25, demonstrating dose dependency and enabling kinetic analysis of E3 ligase activity in real time [63].
HTRF Binding Assays
Homogeneous Time-Resolved Fluorescence (HTRF) technology offers another robust platform for studying E3 ligase interactions and activities. The HTRF XIAP BIR2 Binding Kit exemplifies this approach, designed to identify, characterize, and profile compounds that bind to the BIR2 domain of the E3 ligase XIAP protein [64]. This competitive assay format uses Streptavidin-d2/XIAP BIR2-biotin ligand, a GST-tagged human XIAP BIR2 binding domain, and an anti-GST terbium cryptate-labeled antibody [64].
XIAP-binding compounds compete with the biotinylated ligand, thereby preventing FRET from occurring. This system has been validated for profiling both orthosteric XIAP compounds (SMAC mimetics like LCL161, GDC-0152, A410099.1, and SM164) and PROTAC compounds, demonstrating accurate determination of compound potencies and pharmacological ranking [64]. The assay performs well across a range of DMSO concentrations (0.4% to 1.5% final), with minimal impact on the pharmacological parameters, making it suitable for screening applications [64].
Table 2: Comparison of Reporter Assay Methodologies for Monitoring Ubiquitination
| Assay Parameter | URT-Dual-Luciferase | TUBE-Based TR-FRET | HTRF Binding Assay |
|---|---|---|---|
| Detection Principle | Luciferase activity ratio | FRET between TUBE-d2 and Tb-antibody | Competitive FRET inhibition |
| Throughput Capacity | High (96-well format) | High (homogeneous format) | High (384-well low volume) |
| Key Reagents | Fusion construct, Dual-Glo reagents | TUBEs, TR-FRET antibodies | Tagged protein domains, HTRF reagents |
| Information Output | Functional degradation activity | Polyubiquitin chain formation | Direct binding interactions |
| E3 Families Applicable | HECT and RING | HECT and RING | Primarily RING (XIAP) |
| Assay Development Time | Moderate (construct design) | Rapid (commercial reagents) | Rapid (commercial kits) |
| Specialized Equipment | Dual-luciferase plate reader | TR-FRET capable reader | HTRF capable reader |
Investigating Atypical Ubiquitin Chain Specificity
Methodologies for Detecting Atypical Chains
The specificities of HECT and RING E3 ligases for atypical ubiquitin chains can be investigated using specialized methodologies that discriminate between different linkage types. The bacterial HECT-family E3 ligase NleL (Non-Lee-encoded effector ligase) from enterohaemorrhagic Escherichia coli O157:H7 has been instrumental in studying Lys6- and Lys48-linked ubiquitin polymers [65]. NleL assembles unanchored Ub chains with specificity for Lys6 and Lys48, as demonstrated using single-lysine Ub mutants, with mutation of either Ub Lys6 or Lys48 to Arg (K6R or K48R) resulting in free Ub chains of the other type [65].
Ubiquitin chain restriction analysis using linkage-specific deubiquitinases (DUBs) as "Ub chain restriction enzymes" provides powerful insights into chain architecture. This approach employs DUBs with specific linkage preferences to reveal building blocks comprising distinct linkages. For instance, OTUB1 shows specificity for Lys48-linkages, while OTUD3 demonstrates strong activity against Lys6-linkages with significantly less activity against Lys48 chains [65]. The non-specific viral OTU domain (vOTU) from Crimean Congo Haemorrhagic Fever Virus hydrolyzes Lys6- and Lys48-linkages similarly, serving as a control [65].
Electrophoretic mobility of Ub chains with three or more Ub molecules varies diagnostically with linkage type, enabling preliminary assessment of chain composition. NleL-assembled chains show different electrophoretic mobility for Lys6 and Lys48 diUb, with longer chains (e.g., pentaUb) of WT polymers displaying distinct mobility from both Ub point mutants, indicating the formation of heterotypic Ub chains comprising both Lys6- and Lys48-linkages in the same polymer [65].
Experimental Evidence for E3 Specificity
Research comparing HECT and RING E3 specificity for atypical chains reveals distinct preferences and capabilities. The HECT-family E3 NleL demonstrates a unique capability to assemble both Lys6- and Lys48-linked Ub chains, with kinetics studies showing that Lys6-linkages are assembled into long polymers within minutes, while Lys48-linkages progress with slower kinetics, generating mainly diUb and small amounts of triUb under identical conditions [65]. This suggests that HECT family E3s may have inherent structural features that facilitate formation of atypical linkages.
For RING family E3s, the BRCA1/BARD1 complex has been reported to assemble polyUb with Lys6-linkages on itself and on substrates, linking this atypical chain type to DNA repair processes [65]. Similarly, the related Ring1B/Bmi1 polycomb E3 ligase complex assembles heterotypic Ub chains with branches at Lys6, Lys27, and Lys48 in vitro [65]. These findings indicate that RING-type E3s also possess capability for generating atypical ubiquitin chains, though potentially through different mechanistic principles.
Structural insights into atypical ubiquitin chains reveal distinctive features that may underlie E3 specificity determinants. Analysis of Lys6-linked ubiquitin chains shows an asymmetric interface between Ile44 and Ile36 hydrophobic patches of neighboring ubiquitin moieties, with interactions via the Ile36 patch capable of displacing Leu8 from the Ile44 patch, leading to marked structural perturbations of ubiquitin [65]. These structural characteristics may create specific binding surfaces that are preferentially recognized by particular E3 ligase families.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for Studying E3 Ligase Specificity
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| E3 Ligase Expression Constructs | SMURF1, NleL, XIAP, BRCA1/BARD1 | Functional studies of specific E3 ligases and their mutants |
| Ubiquitin Variants | Single-lysine mutants (K6R, K48R), Lys-less ubiquitin | Determining linkage specificity and chain formation requirements |
| Specialized Assay Kits | HTRF XIAP BIR2 Binding Kit, TUBE-based assays | High-throughput screening and biochemical characterization |
| Linkage-Specific DUBs | OTUB1 (Lys48-specific), OTUD3 (Lys6-preferring) | Ubiquitin chain restriction analysis and linkage verification |
| Detection Reagents | TUBEs, linkage-specific antibodies, luciferase substrates | Signal generation and quantification in various assay formats |
| Proteasome Inhibitors | MG-132, Bortezomib | Confirming ubiquitin-proteasome pathway involvement |
| Cell Line Models | HEK293T, MDCK, specialized knockout lines | Cellular context for pathway-specific ubiquitination studies |
Cell-based reporter assays provide powerful, physiologically relevant platforms for investigating the specificity of HECT versus RING E3 ligases for atypical ubiquitin chains. The URT-Dual-Luciferase system offers exceptional normalization for high-throughput screening applications, while TUBE-based TR-FRET and HTRF assays enable direct monitoring of ubiquitin chain formation and binding interactions. The emerging evidence suggests that both E3 ligase families possess capabilities for generating atypical ubiquitin chains, though potentially through different structural mechanisms and with distinct kinetic properties. The continued refinement of these reporter assay technologies, coupled with advanced structural and biochemical approaches, will undoubtedly yield deeper insights into the complex specificity determinants governing E3 ligase function and their roles in health and disease.
Ubiquitination, a crucial post-translational modification, regulates vast cellular processes, with linkage specificity defining functional outcomes. Among ubiquitin chains, K48-linked polyubiquitination serves as the principal signal for proteasomal degradation [11] [21]. E3 ubiquitin ligases determine substrate specificity and linkage topology, primarily through two major families: RING-type and HECT-type E3s [11] [21]. While RING E3s often function as multi-subunit complexes that directly transfer ubiquitin from E2 to substrate, HECT E3s are typically single-polypeptide enzymes that form a transient thioester intermediate with ubiquitin on their catalytic cysteine [11]. This fundamental mechanistic difference underpins their distinct approaches to establishing linkage specificity. This case study examines the structural mechanisms of K48-linked ubiquitin chain formation by the human HECT E3 UBR5, providing a comparative framework for understanding HECT versus RING E3 specificity in the context of atypical chain synthesis research [31].
Mechanistic Insights into UBR5 Function
Recent cryo-EM structures reveal UBR5 functions as a 620 kDa dimeric assembly, which further oligomerizes into cage-like tetramers [31] [66] [67]. Each UBR5 protomer comprises an extensive α-solenoid scaffold that flexibly tethers numerous protein-interaction domains, including a Ub-associated (UBA) domain, UBR domain, and the C-terminal HECT domain [31] [66]. The functional dimer assembles in a head-to-tail arrangement, creating an "intermolecular jaw" where the N-terminal region of one subunit positions itself adjacent to the HECT domain of the partner subunit [66]. This architecture is essential for coordinating the ubiquitination cascade.
Table 1: Key Structural Features of UBR5
| Structural Element | Location/Residues | Function | Experimental Evidence |
|---|---|---|---|
| HECT Domain | C-terminal (contains C2768) | Catalytic domain with E2-binding N-lobe and catalytic C-lobe | Cryo-EM structures at 2.7-3.7Å [31] [66] |
| UBA Domain | N-terminal region | Binds acceptor ubiquitin (UbA) | Biochemical pulldown, cryo-EM density [31] |
| UBR Domain | Central region | Putative substrate N-degron recognition | AlphaFold2 model fitting into cryo-EM map [31] |
| Dimer Interface | Helical scaffold (7936 Ų interface) | Forms functional 620 kDa dimer | Cryo-EM with focused refinement [66] |
| MLLE Domain | Insert within HECT N-lobe | Protein-protein interactions (substrate binding) | Not visible in maps (flexibly linked) [31] |
Structural Snapshots Along the Ubiquitination Pathway
Through chemical trapping and cryo-EM, researchers have visualized stable mimics of three key intermediates in K48-linked ubiquitin chain formation: the E2~Ub-bound state (TS1), the UBR5~Ub thioester intermediate, and the E3-Ub-acceptor complex (TS2) [31]. These structural snapshots reveal a conserved HECT domain conformational cycle during ubiquitin transfer. The HECT domain transitions between "L-shaped" and "inverted T" conformations, with the flexible interlobe linker enabling the C-lobe to access both the E2~Ub conjugate and the acceptor ubiquitin [31]. During the TS2 transition, the UBA domain plays a critical role by capturing the acceptor ubiquitin and positioning its K48 residue near the catalytic center through an elaborate network of interactions between the acceptor ubiquitin, UBR5 elements, and the donor ubiquitin [31].
Diagram 1: UBR5 Catalytic Cycle. The two-step ubiquitin transfer mechanism showing key intermediates.
HECT vs. RING E3 Specificity: A Comparative Analysis
The structural data on UBR5 reveals fundamental differences in how HECT and RING E3 families achieve linkage specificity, particularly for K48-linked chains.
Determinants of Linkage Specificity
For HECT E3s like UBR5, linkage specificity is an intrinsic property of the catalytic domain itself, particularly the C-lobe region that positions the acceptor ubiquitin [11]. The UBR5 structural data demonstrates how the UBA domain, along with other structural elements, collaborates to specifically orient K48 of the acceptor ubiquitin for isopeptide bond formation [31]. This represents a substrate-assisted mechanism where the E3 directly controls which lysine residue on ubiquitin is targeted.
In contrast, RING E3s typically derive their linkage specificity from their partnered E2 enzymes [11] [68]. For example, the cullin-RING ligase SCF-Cdc34 complex specifically generates K48-linked chains through an acidic loop in Cdc34 that positions K48 of the acceptor ubiquitin for nucleophilic attack [68]. RING E3s function primarily as scaffolds that bring E2~Ub and substrate together without forming a covalent E3~Ub intermediate [11].
Table 2: HECT vs. RING E3 Mechanism Comparison for K48-Linked Ubiquitination
| Feature | HECT E3 (UBR5) | RING E3 (SCF-Cdc34) |
|---|---|---|
| Catalytic Mechanism | Two-step with E3~Ub intermediate [11] | Direct transfer from E2 to substrate [11] |
| Linkage Specificity Determinant | HECT C-lobe and associated domains (e.g., UBA) [31] [11] | Primarily the E2 enzyme (e.g., Cdc34 acidic loop) [68] |
| Structural Configuration | Bilobal HECT domain with flexible hinge [31] | Multi-subunit complex with RING domain scaffold [11] |
| K48-Specific Positioning | UBA domain captures UbA, positioning K48 via multiple interactions [31] | Acidic loop on E2 (Cdc34) positions K48 of acceptor ubiquitin [68] |
| Chain Formation Process | Processive feed-forward mechanism [31] | Processive synthesis enabled by E2 [68] |
| Representative Structures | Full-length UBR5 dimer (2.7-3.7Å) [31] [66] | Isolated HECT domains; SCF-Cdc34 complex [68] |
Implications for Atypical Chain Synthesis
The UBR5 mechanism has significant implications for understanding branched ubiquitin chain formation. UBR5 can generate K11/K48-branched chains, which are particularly potent degradation signals [69] [66]. The structural flexibility observed in UBR5's HECT domain, with its ability to adopt multiple conformations, may enable it to modify pre-formed ubiquitin chains of different linkages to create these branched topologies [31] [69]. This capability contrasts with many RING E3s that typically generate homotypic chains unless functioning in complexes with multiple E2s.
Diagram 2: HECT vs. RING E3 Specificity Mechanisms. Comparative schematic of the distinct mechanisms determining linkage specificity.
Experimental Protocols for Structural Analysis
Cryo-EM Sample Preparation and Data Collection
The structural analysis of UBR5 employed state-of-the-art cryo-EM methodologies [31] [66] [67]:
Protein Expression and Purification:
- Full-length human UBR5 was recombinantly expressed in insect cells or HEK293T cells with N-terminal FLAG tags
- Catalytically inactive mutant UBR5(C2768A) was used for initial structural studies to stabilize intermediates
- Proteins were purified via affinity chromatography followed by size-exclusion chromatography (SEC)
- SEC revealed two oligomeric states: dimer (620 kDa) and tetramer (1.2 MDa)
Grid Preparation and Data Collection:
- Purified UBR5 complexes were applied to cryo-EM grids and vitrified using liquid ethane
- Multiple datasets were collected, including untilted and 30° tilted samples to overcome preferred orientation bias
- Data processing involved 2D classification, 3D refinement, and focused classification to handle flexible regions
- For the UBR5 dimer, application of C2 symmetry improved resolution to 2.66Å
Biochemical and Functional Assays
Ubiquitination Assays:
- Pulse-chase assays monitored ubiquitin transfer from E2~Ub (UBE2D) to UBR5 and then to acceptor ubiquitin
- Fluorescent labeling enabled tracking of ubiquitin transfer via SDS-PAGE mobility shifts
- K48R ubiquitin mutants prevented use as acceptor, confirming linkage specificity
- Substrate ubiquitination (e.g., PEPCK1) verified catalytic activity of purified complexes
Covalent Intermediate Trapping:
- Stable mimics of catalytic intermediates were generated using chemical approaches
- Transition state analogs allowed structural visualization of TS1 and TS2 states
- This approach provided snapshots along the entire ubiquitination cascade
Research Reagent Solutions
Table 3: Essential Research Reagents for UBR5 and Ubiquitination Studies
| Reagent/Method | Specification/Application | Function in Research |
|---|---|---|
| UBR5 Constructs | Full-length (2799 aa), UBR5(C2768A), UBR5(L710D) dimer mutant | Structural and functional studies; defining oligomerization interfaces [31] [66] |
| E2 Enzymes | UBE2D2 (UBCH5B) family | Primary E2 partner for ubiquitin transfer to UBR5 HECT domain [31] |
| Ubiquitin Mutants | K48R, K63R ubiquitin variants | Linkage specificity determination; acceptor function blocking [31] [69] |
| Cryo-EM Workflow | Vitrification, multi-angle data collection, focused refinement | Overcoming flexibility; determining high-resolution structures of full-length E3 [31] [66] |
| Chemical Trapping | Transition state analogs, ester intermediates | Stabilizing and visualizing catalytic intermediates [31] |
| Mass Photometry | Molecular mass determination in solution | Characterizing oligomeric states and complex assembly [67] |
The structural characterization of UBR5 provides unprecedented insights into the mechanism of K48-linked ubiquitin chain formation by HECT E3 ligases. The findings reveal how UBR5's oligomeric architecture, flexible HECT domain conformations, and integrated UBA domain collaborate to achieve linkage specificity through a feed-forward catalytic cycle [31]. This mechanism contrasts sharply with RING E3s, which delegate linkage specificity primarily to their partnered E2 enzymes [11] [68]. The structural flexibility observed in UBR5 may explain its capability to generate branched ubiquitin chains (K11/K48), representing an important expansion of the ubiquitin code [69] [66]. These structural insights not only advance our fundamental understanding of HECT E3 mechanisms but also provide a framework for targeted therapeutic intervention in cancers characterized by UBR5 dysregulation [66] [67].
Navigating Experimental Hurdles: Troubleshooting Specificity and Activity Assays
Overcoming Challenges in Determining Intrinsic vs. E2-Derived Linkage Specificity
The ubiquitin code, which regulates virtually every aspect of cellular function, is defined by the type of ubiquitin chain linkages attached to substrate proteins. A central question in the ubiquitin field is whether the specificity for assembling atypical chains (non-K48/K63 linkages) is an intrinsic property of the E3 ubiquitin ligase or is primarily dictated by its partnered E2 conjugating enzyme. This challenge is particularly acute when comparing the two major classes of E3 ligases: HECT-type and RING-type ligases. This guide objectively compares the mechanistic strategies and experimental approaches used to disentangle this question, providing a framework for researchers in drug development and biochemical research.
Fundamental Mechanistic Divergence Between HECT and RING E3s
The core catalytic mechanisms of HECT and RING E3 ligases provide the first layer of specificity control. The table below summarizes the key mechanistic differences that influence linkage specificity.
Table 1: Core Mechanistic Comparison of HECT and RING E3 Ligases
| Feature | HECT E3 Ligases | RING E3 Ligases |
|---|---|---|
| Catalytic Mechanism | Two-step transthiolation: E2~Ub → E3~Ub → Substrate [8] [25] | Direct transfer: E2~Ub → Substrate [8] [70] |
| Ubiquitin Transfer Site | Catalytic cysteine within the HECT domain [71] [25] | No E3-Ub thioester; acts as a scaffold [70] |
| Primary Specificity Determinant | E3's intrinsic specificity and conformational dynamics [37] [25] | E2's intrinsic specificity, with E3 providing allosteric guidance [72] [73] |
| Representative Examples | SMURF1, NEDD4, HACE1, E6AP [71] [37] [25] | RNF4, MIB1, CBL-c, TRAF-4 [74] [6] |
The following diagram illustrates these distinct catalytic pathways and the key experimental questions for determining linkage specificity.
Experimental Approaches for Decoupling E2 and E3 Contributions
Disentangling the contributions of E2 and E3 requires a combination of rigorous biochemical, structural, and proteomic methods. The following table compares established and emerging techniques.
Table 2: Key Experimental Methods for Determining Linkage Specificity
| Method | Core Principle | Application to HECT vs. RING | Key Outcome |
|---|---|---|---|
| In Vitro Reconstitution [37] | Purified E1, E2, E3, and substrate are combined to reconstitute ubiquitination without cellular complexity. | Foundation for testing multiple E2/E3 pairwise combinations. | Measures intrinsic activity and linkage formation in a minimal system. |
| Chemical Crosslinking & Cryo-EM [71] | Stabilizes transient E2~Ub/E3 or E3~Ub complexes for high-resolution structural analysis. | Visualizes HECT "L" vs "inverted-T" conformations; RING/E2 interfaces. | Reveals structural constraints that dictate Ub positioning and chain elongation. |
| Mechanism-Based Substrate Labeling (e.g., BioE3) [6] | Uses engineered biotin ligase (BirA)-E3 fusions and bioUb to label and capture native substrates for proteomics. | Identifies bona fide cellular substrates and their modification states for a specific E3. | Provides a native cellular snapshot of E3 output and specificity. |
| Deep Learning & High-Throughput Screening (e.g., COMET) [75] | Uses AI and combinatorial genetics to map E3-substrate relationships at a massive scale. | Systematically tests degradation capacity of many E3s on a library of substrates. | Generates large-scale datasets to uncover complex E3-substrate network relationships. |
The workflow below outlines a logical progression for applying these methods to resolve the E2 vs. E3 specificity question.
Detailed Experimental Protocols
In Vitro Ubiquitination Reaction for Linkage Analysis
This foundational protocol assesses the intrinsic linkage-forming capability of a given E2/E3 pair. [70] [37]
Reagents & Buffers:
- Reaction Buffer: 50 mM Tris-HCl (pH 7.5), 50 mM NaCl, 10 mM MgCl₂, 2 mM ATP.
- Energy Regeneration System: 10 mM Creatine Phosphate, 10 U/mL Creatine Kinase.
- Purified Proteins: E1 (50 nM), E2 (500 nM), E3 (1 µM), Ubiquitin (100 µM). Substrate as required.
- Ubiquitin Variants: Wild-type Ubiquitin (for overall activity); Lysine-less Ub (K0) + single Lysine Ub mutants (e.g., K48-only, K63-only) to probe specific linkage formation.
Procedure:
- Assemble reactions on ice in a total volume of 50 µL.
- Add reaction buffer, energy regeneration system, and ubiquitin.
- Initiate the reaction by adding the enzyme cascade (E1, then E2, then E3).
- Incubate at 30°C for 60-90 minutes.
- Terminate the reaction by adding 4x SDS-PAGE loading buffer (with or without DTT to preserve thioester intermediates).
- Analysis: Resolve products by SDS-PAGE and visualize via immunoblotting with anti-ubiquitin or anti-substrate antibodies. For linkage specificity, use linkage-specific anti-Ub antibodies (e.g., anti-K48, anti-K63) or mass spectrometry.
BioE3 Workflow for Identifying Native E3 Substrates and Modifications
This cellular method identifies bona fide substrates and the ubiquitin modifications conferred by a specific E3 ligase in a native environment. [6]
Key Reagents:
- bioGEFUbnc: A non-cleavable Ubiquitin mutant (L73P) fused to an optimized AviTag (bioGEF) for efficient, proximity-dependent biotinylation.
- BirA-E3 Fusion: The E3 ligase of interest fused to the biotin ligase BirA.
- Stable Cell Line: HEK293FT or U2OS cells with doxycycline-inducible bioGEFUbnc.
- Streptavidin Beads: For capture of biotinylated proteins.
Procedure:
- Cell Preparation: Generate a stable cell line expressing doxycycline-inducible bioGEFUbnc. Introduce the BirA-E3 fusion construct into these cells.
- Biotin Depletion: Culture cells in biotin-depleted media for 24 hours to reduce background.
- Induction & Labeling: Induce bioGEFUbnc and BirA-E3 expression with doxycycline. After 24 hours, add exogenous biotin for a short, timed pulse (e.g., 2 hours).
- Cell Lysis & Capture: Lyse cells under denaturing conditions (to disrupt non-covalent interactions) and incubate with streptavidin beads.
- Stringent Washing: Wash beads extensively with high-salt and detergent-containing buffers to remove non-specifically bound proteins.
- Elution & Identification: Elute bound proteins and identify them via liquid chromatography-mass spectrometry (LC-MS).
The Scientist's Toolkit: Essential Research Reagents
This table catalogs critical reagents for investigating E3 ligase specificity, as featured in the cited research.
Table 3: Key Research Reagent Solutions for E3 Specificity Studies
| Reagent / Tool | Function | Key Application & Rationale |
|---|---|---|
| Ubiquitin Mutant Library (K0, K-only) | To probe specific lysine linkages in poly-Ub chain formation. | Used in in vitro reconstitution assays to determine if an E2/E3 pair has an intrinsic preference for K11, K48, K63, etc. [8] [37] |
| Linkage-Specific Anti-Ub Antibodies | Immunodetection of specific Ub chain linkages. | Enables rapid assessment of linkage types formed in in vitro assays or pulled down from cells, without requiring MS. [8] |
| Engineered Ubiquitin (bioGEFUbnc) | A substrate for proximity-based biotinylation of E3-specific targets. | The core component of the BioE3 system; allows for specific capture of substrates ubiquitinated by the BirA-E3 fusion protein in a cellular context. [6] |
| Recombinant E2 Enzyme Library | A panel of purified E2 conjugating enzymes. | Essential for in vitro screening to determine which E2s support a given E3's activity and how the E2 identity influences linkage specificity. [72] [70] |
| Allosteric HECT Inhibitors (e.g., Cpd-8) | Small molecules that lock HECT E3s in an inactive state. | Tools to probe HECT E3 function and conformational dynamics. Useful for validating E3-specific phenotypes and confirming on-target effects in cells. [37] |
The determination of linkage specificity is not a binary E2- or E3-centric problem but exists on a spectrum. The prevailing model, supported by recent structural and biochemical data, is that RING E3s rely more heavily on their cognate E2s to determine linkage type, as they primarily function to allosterically activate the E2~Ub conjugate and bring it into proximity with the substrate. [72] [73] In contrast, HECT E3s exert a greater degree of intrinsic control, as they form a distinct catalytic intermediate (E3~Ub) and contain specialized domains like ubiquitin exosites that orient the growing chain. [37] [25]
For example, the HECT ligase E6AP is a known K48-specific enzyme, while NEDD4 family members predominantly synthesize K63-linked chains, preferences that are intrinsic to their HECT domains. [25] Furthermore, the discovery of an allosteric inhibitor that locks the HECT ligase SMURF1 in an inactive state by restricting the motion of a conserved glycine hinge underscores the critical role of HECT conformational dynamics in catalysis, a layer of control absent in RING E3s. [37] Overcoming the challenge of defining specificity therefore requires a multi-pronged experimental strategy that moves from simplified in vitro systems to validated cellular contexts, leveraging the tools and methods detailed in this guide to precisely map the contributions of each component in the ubiquitination cascade.
Optimizing Conditions to Preserve Labile Thioester Intermediates in HECT E3 Assays
Within the ubiquitin-proteasome system, E3 ubiquitin ligases confer substrate specificity, with HECT-type and RING-type ligases representing two major mechanistic families. Research into their distinct functions is crucial, particularly for understanding the assembly of atypical ubiquitin chains, which serve as complex regulatory signals beyond simple degradation [73]. A significant technical challenge in this field, however, is the inherent lability of the thioester-bonded intermediates that are essential for the catalytic cycle of HECT E3 ligases. This guide objectively compares contemporary methodological solutions for stabilizing these fleeting intermediates, providing structured experimental data and protocols to advance research on HECT versus RING E3 specificity.
Comparative Analysis of Intermediate Stabilization Strategies
The following table summarizes and compares the core strategies developed to trap thioester intermediates, highlighting their key characteristics and applicability to HECT E3 studies.
Table 1: Comparison of Strategies for Trapping Labile Ubiquitin Intermediates
| Strategy Name | Core Principle | Key Stabilizing Feature | Captured Intermediate | Compatibility with Structural Studies | Key Application in HECT Studies |
|---|---|---|---|---|---|
| PSAN Probe [76] | Traps two cysteines to mimic tetrahedral intermediate | Dithioacetal linkage mimicking native geometry | E1–Ub–E2 & E2–Ub–E3HECT | Excellent (cryo-EM) | Visualized E2–Ub(T)–E3HECT transthiolation |
| EDT Cross-linker [77] | Creates atomic bridge between E2 and substrate | Disulfide-bonded bridge with atom count matching tetrahedral state | E2–Ub–Substrate (with E3) | Good (X-ray crystallography) | Adaptable for HECT E2–Ub–substrate complexes |
| Branched Ubiquitin Probe [39] | Pre-formed K29/K48-branched triUb crosslinked to E3 | Covalent linkage to E3 catalytic cysteine | E3~Ub–Ubsubstrate (product state) | Excellent (cryo-EM) | Visualized Ufd4 HECT domain in branched ubiquitination |
Each strategy offers distinct advantages. The PSAN probe technology stands out for its ability to generate near-native intermediates compatible with high-resolution cryo-EM, directly visualizing the E2–Ub(T)–E3HECT transthiolation step [76]. The EDT cross-linker provides a minimalist, atomic-level mimic of the transition state and has proven effective for crystallographic studies [77]. For investigating later stages of the HECT catalytic cycle, particularly chain elongation and branching, the Branched Ubiquitin Probe allows for the capture of the E3 covalently linked to a native-like ubiquitin chain [39].
Detailed Experimental Protocols
Trapping Transthiolation Intermediates with the PSAN Probe
This protocol, based on the seminal work by Zheng et al., details the steps for generating a stable analogue of the E2–Ub(T)–E3HECT intermediate [76].
Ubiquitin Probe Synthesis:
- Begin with Ubiquitin (residues 1–74) in its acyl azide form (Ub(−2) acyl azide).
- Perform aminolysis by reacting Ub(−2) acyl azide with H2N-Gly–PSAN (3-[phenylsulfonyl]−4-aminobut-2-enenitrile) to generate the key reagent, Ub-PSAN.
E2–Ub Vinyl Thioether Formation:
- Incubate Ub-PSAN with the desired E2 conjugating enzyme (e.g., S. pombe Ubc4, a homologue of human UbcH7/UbcH5).
- To maximize yield, first mutate surface-exposed cysteine residues (e.g., Cys21 and Cys107 in Ubc4) to serine to prevent non-specific crosslinking. The predominant product is a crosslink between Ub-PSAN and the E2 active site cysteine (Cys85).
Forming the E2–Ub(T)–E3HECT Intermediate:
- Purify the resulting E2–Ub vinyl thioether adduct.
- Combine this adduct with the HECT domain of your E3 ligase of interest (e.g., Pub2HECT, a NEDD4 family homologue) to form the final, stable dithioacetal analogue of the E2–Ub(T)–E3HECT transthiolation intermediate.
Diagram: Workflow for trapping E2–Ub–E3HECT intermediate with PSAN probe
Trapping E3-Substrate Complexes for Branching Studies
This protocol outlines the approach used by Wang et al. to visualize the HECT E3 Ufd4 during the synthesis of K29/K48-branched ubiquitin chains [39].
Branched Ubiquitin Probe Synthesis:
- Chemically synthesize a K29/K48-branched triUb probe (triUbprobe). In this probe, a "donor" Ub is chemically ligated to the K29 residue of the proximal Ub within a pre-assembled K48-linked diUb.
Cross-linking to the HECT E3 Active Site:
- Incubate the purified triUbprobe with the HECT E3 ligase (e.g., Ufd4).
- The probe covalently crosslinks with the catalytic cysteine residue (C1450 in Ufd4) of the E3, forming a stable complex that mimics the transition state of ubiquitin transfer.
Structural Analysis:
- Purify the crosslinked Ufd4-triUbK29/K48 complex.
- The complex is now stable enough for structural determination via single-particle cryo-EM, enabling the visualization of mechanistic details such as how the N-terminal ARM region and HECT C-lobe orient the ubiquitin chain for branching.
Essential Research Reagent Solutions
The following table lists key reagents and their critical functions in the methodologies described above, serving as a checklist for experimental setup.
Table 2: Key Reagent Solutions for Trapping HECT E3 Intermediates
| Reagent / Tool | Function / Application | Key Feature / Consideration |
|---|---|---|
| Ub-PSAN Probe [76] | Trapping E1–E2 & E2–E3HECT transthiolation intermediates | Mimics tetrahedral intermediate; works with native enzymes |
| EDT (1,2-Ethanedithiol) [77] | Cross-linking E2 active site to substrate lysine (as Cys) | Creates an atomic bridge; optimal distance/orientation mimic |
| BMOE (Bismaleimidoethane) [77] | Alternative cysteine-to-cysteine cross-linker | Stable to reducing agents; but bulkier than EDT |
| Engineered Branched triUb Probe [39] | Trapping HECT E3 in act of ubiquitin transfer to chain | Pre-formed branched chain crosslinked to E3 active site Cys |
| E2 Ubc4 / UbcH7 [76] | E2 partner for HECT E3 transthiolation | Specialized for transthiolation to HECT/RBR E3 ligases |
| Active-site Cysteine Mutant E3 | Generating catalytically inactive control | Essential for validating specificity of crosslinking reactions |
The development of sophisticated chemical and biochemical tools, particularly the PSAN probe and engineered ubiquitin probes, has fundamentally advanced our ability to preserve and visualize the labile thioester intermediates central to HECT E3 ligase function [39] [76]. These strategies have moved the field from indirect inference to direct structural observation, enabling researchers to answer long-standing mechanistic questions. The choice of method depends on the specific research question—whether it is to visualize the initial transthiolation from E2 to E3 or to capture the final ubiquitin transfer to a substrate or growing chain. By implementing these protocols and reagents, researchers can rigorously dissect the unique specificities of HECT-type ligases, particularly their role in forming the atypical ubiquitin chains that are increasingly recognized as critical players in cell signaling and disease.
E3 ubiquitin ligases are pivotal enzymes within the ubiquitin-proteasome system, responsible for conferring substrate specificity during the process of ubiquitination. Their activity is often tightly regulated by intrinsic autoinhibitory mechanisms to prevent uncontrolled protein degradation and signaling. Understanding and overcoming this autoinhibition is particularly crucial for research aimed at comparing the specificity of HECT-type versus RING-type E3 ligases in synthesizing atypical ubiquitin chains. This guide provides a structured comparison of autoinhibition mechanisms and experimentally validated strategies to activate full-length E3 ligases for in vitro studies, providing researchers with practical methodologies to advance functional and structural studies.
Deciphering E3 Ligase Auto-inhibition: A Structural and Functional Comparison
The autoinhibitory mechanisms employed by HECT and RING E3 ligases, while sharing the common goal of preventing uncontrolled activity, are structurally distinct. The table below summarizes the key features of these mechanisms, which must be understood to develop effective activation strategies.
Table 1: Comparative Auto-inhibition in HECT and RING E3 Ligase Families
| Feature | HECT Family (e.g., Nedd4 subfamily) | RING Family (e.g., BRCA1/BARD1, RBR subfamily) |
|---|---|---|
| Inhibitory Domain(s) | N-terminal WW domains and linker regions (e.g., WW2, L, WW4 in WWP1) [78] | Varied: e.g., Ariadne domain in HHARI, UBA domain in HOIP, or inactive RING partner [18] |
| Structural Mechanism | "Multi-lock" mechanism where WW domains and linkers bind the HECT domain, restricting E2~Ub recruitment and HECT lobe flexibility [78] | Steric hindrance from autoinhibitory domains or subunits that prevent productive E2 binding or allosterically suppress the catalytic domain [18] |
| Impact on Catalysis | Prevents ubiquitin transfer from E2 to the HECT active site cysteine and restricts non-covalent ubiquitin binding [78] | Prevents the stabilization of a closed, active E2~Ub conformation or blocks the E3~Ub intermediate formation in RBRs [18] |
| Conformational Change | Large-scale reorientation of the HECT C-lobe relative to the N-lobe is essential for activity and is restricted by autoinhibition [79] [78] | Displacement of inhibitory domains is required to allow E2 access and, for RBRs, to bring the E2 and RING2 active site into proximity [18] |
The following diagram illustrates the generalized autoinhibition and activation pathways for HECT and RBR-type E3 ligases, highlighting key conformational states.
Diagram 1: Pathways for E3 ligase activation. Activation requires specific triggers to overcome autoinhibition.
Experimentally Validated Strategies for Activating Full-Length E3 Ligases
Activation of full-length E3 ligases for in vitro experiments requires strategic disruption of autoinhibitory interactions. The most common and successful approaches are summarized below.
Table 2: Experimental Strategies to Overcome E3 Ligase Auto-inhibition
| Strategy | Mechanism of Action | Exemplary E3 & Experimental Evidence |
|---|---|---|
| Truncation or Deletion Mutants | Physically removes the autoinhibitory domain(s), resulting in a constitutively active catalytic fragment. | WWP1/Itch/WWP2: Deletion of WW domains and linker regions (L) led to progressive increases in autoubiquitination activity. A L34HECT truncation was partially active, while the HECT domain alone was fully active [78]. |
| Adaptor Protein Binding | Binding of specific proteins to the E3's regulatory domains disrupts intramolecular interactions, relieving autoinhibition. | Nedd4 Family E3s: Binding of activator proteins like Ndfip1 to WW domains displaces them from the HECT domain, activating the ligase [79]. E6AP: The viral E6 protein binds to and alters E6AP's substrate specificity, effectively activating it for p53 ubiquitination [79]. |
| Post-Translational Modifications (PTMs) | Phosphorylation or other PTMs at critical residues can destabilize the autoinhibited conformation. | Itch: JNK1-mediated phosphorylation of the linker region releases WW2-mediated autoinhibition [78]. |
| Oligomerization or Dimerization | Can induce activating conformational changes; however, for some HECT E3s, it may also be part of the regulatory mechanism [11] [79]. | RING E3s: Many RING ligases like cIAP, RNF4, and BRCA1/BARD1 function as dimers or multi-subunit complexes, where dimerization is often essential for activity [11]. |
| Allosteric Activation by Ub/UBLs | Binding of ubiquitin or ubiquitin-like proteins to specific allosteric sites stabilizes an active E3 conformation. | RBR E3s (HOIP, HOIL-1, RNF216): Activation by specific diubiquitin linkages (e.g., M1- or K63-linked). HOIL-1 helix-RBR showed a >20-fold increase in E2-Ub discharge activity with an EC₅₀ of 8 µM for M1-diUb [18]. Parkin: Activated by phospho-Ub binding [18]. |
Core Experimental Protocol: Assessing E3 Activation via In Vitro Autoubiquitination
The autoubiquitination assay is a fundamental method to confirm the successful activation of a purified E3 ligase in vitro. The workflow below outlines a robust protocol adapted from established studies [80] [78].
Diagram 2: Workflow for in vitro autoubiquitination assay.
Detailed Methodology
This protocol provides a step-by-step guide for conducting an autoubiquitination assay [80] [78] [81].
Materials and Reagents
- Purified Proteins: The E3 ligase of interest (e.g., full-length and truncated mutants), E1 activating enzyme, and E2 conjugating enzyme (e.g., UbcH5a/b/c for many RINGs, UBCH7 for some RBRs and HECTs).
- Ubiquitin: Wild-type ubiquitin. For linkage specificity studies, use ubiquitin mutants (e.g., Lys-to-Arg mutants).
- Reaction Buffer: 50 mM Tris-HCl (pH 8.0), 50 mM NaCl, 5 mM MgCl₂, 2 mM ATP, and 1 mM DTT.
- Equipment: Thermostat, SDS-PAGE, and Western blot apparatus.
Procedure
- Setup: In a thin-walled PCR tube, combine the reaction components on ice. A typical 20 µL reaction contains:
- 100 nM E1 enzyme
- 1-5 µM E2 enzyme
- 0.5-2 µM E3 ligase (full-length or mutant)
- 50-100 µM Ubiquitin
- 2 mM ATP
- 1x Reaction Buffer
- Incubation: Transfer the tube to a thermocycler or heat block and incubate at 30°C for 60 minutes.
- Termination: Stop the reaction by adding SDS-PAGE loading buffer containing DTT (to reduce thioester bonds) and immediately heating at 95°C for 5 minutes.
- Analysis: Resolve the proteins by SDS-PAGE and transfer to a membrane. Perform Western blotting using an anti-ubiquitin primary antibody and an appropriate HRP-conjugated secondary antibody. Detect the signal using a chemiluminescent substrate.
Troubleshooting and Validation
- Critical Controls: Always include reactions missing individual components (e.g., -E1, -E2, -E3, -ATP) to confirm the specificity of the ubiquitination signal.
- Catalytic Mutants: Use E3 ligases with catalytic cysteine mutations (Cys to Ala in HECT/RBRs) or RING domain mutants to distinguish between genuine enzymatic activity and non-specific labeling.
- Activity Quantification: The increase in ubiquitination signal (high molecular weight smearing or band shifts) for an activated E3 (e.g., via truncation or adaptor addition) compared to the autoinhibited full-length protein is a direct measure of successful activation [78].
The Scientist's Toolkit: Essential Reagents for E3 Activation Studies
Successful in vitro activation studies depend on a core set of high-quality reagents. The following table details these essential components.
Table 3: Key Research Reagent Solutions for E3 Activation Studies
| Reagent / Material | Function / Purpose | Examples & Notes |
|---|---|---|
| E3 Ligase Constructs | To compare autoinhibited vs. active states and map regulatory domains. | Full-length, Truncation mutants (e.g., ΔWW2L34, HECT-only), Catalytic mutants (Cys-to-Ala for HECT/RBR). Purification tags (His, MBP) aid in isolation [78] [80]. |
| E2 Enzyme Panel | Different E2s have varying affinities for E3s and dictate inherent linkage preference in RINGs. | UbcH5 (a, b, c), UbcH7, UbcH10. Essential for testing E2-E3 pairing and catalytic efficiency [81]. |
| Activator Proteins / Molecules | To experimentally disrupt autoinhibition in a physiological manner. | Recombinant adaptors (e.e., Ndfip1), Allosteric activators (e.g., M1- or K63-linked diubiquitin for RBRs) [18] [78]. |
| Ubiquitin Variants | To study chain linkage specificity and the role of allosteric activation. | Wild-type Ub, Lysine-less Ub (K0), Lys-to-Arg mutants (e.g., K48R, K63R), Di-Ub linkages (for allosteric studies) [18] [15]. |
| In Vitro Ubiquitination Kit | Provides a complete, optimized set of core enzymes and buffers for initial setup and validation. | Commercial kits typically include E1, a panel of E2s, ubiquitin, ATP, and reaction buffer, ensuring reproducibility. |
Data Interpretation and Application in Specificity Research
Activating E3 ligases is not an end in itself but a critical step toward understanding their biological function, particularly their specificity for forming atypical ubiquitin chains.
Quantifying Activation: Researchers should measure the efficiency of different activation strategies. For example, in a study on WWP1, truncation mutants showed a clear gradation of activity: the L34HECT construct was partially active, while the HECT-only domain exhibited robust ligase activity, comparable to the 234HECT mutant where the critical linker (L) was deleted [78]. This quantitative assessment is crucial for selecting the right tool for subsequent experiments.
Linking Activation to Atypical Chain Formation: Once activated, the linkage specificity of an E3 can be determined. For instance, the activated Nedd4 family HECT E3 WWP1 was shown to assemble ubiquitin chains containing Lys-63, Lys-48, and Lys-11 linkages (with a preference Lys-63 > Lys-48 > Lys-11) [15]. Furthermore, its chain synthesis occurs in two phases: an initial, unidirectional phase producing Lys-63 chains, followed by a multidirectional elongation phase that creates mixed and branched chains [15]. This nuanced understanding is only possible after autoinhibition has been overcome.
In contrast, for many RING E3s, the linkage specificity is primarily determined by the cognate E2 enzyme [79]. Activating multi-subunit RING E3s like the APC/C or SCF complex often involves recruiting co-activators (e.g., Cdc20 or Cdh1 for APC/C) that simultaneously relieve autoinhibition and define substrate specificity [11]. The RBR subclass, which includes HOIL-1 and HOIP, is notably activated by specific ubiquitin linkages in a feed-forward mechanism; HOIL-1 is activated by M1-linked diubiquitin, while RNF216 is activated by K63-linked diubiquitin [18]. This linkage-specific allosteric activation provides a direct functional readout in specificity research.
By systematically applying the activation strategies and assays detailed in this guide, researchers can effectively compare the intrinsic catalytic properties of HECT and RING E3 ligases, paving the way for a deeper understanding of their roles in signaling and disease.
Selecting Appropriate E2 Enzymes for Probing Atypical Chain Formation with RING E3s
Within the ubiquitin-proteasome system, the modification of proteins with ubiquitin chains is a sophisticated regulatory mechanism controlling virtually every eukaryotic cellular process. The process is catalyzed by a sequential enzymatic cascade involving ubiquitin-activating (E1), ubiquitin-conjugating (E2), and ubiquitin-ligating (E3) enzymes [82]. While E3 ligases, particularly RING-types, are renowned for their substrate specificity, a paradigm shift has established E2 enzymes as active architects in determining the length and topology of ubiquitin chains [82] [83]. This guide focuses on the critical role of E2 selection for researchers investigating the formation of atypical ubiquitin chains—non-K48 and non-K63 linkages—in the context of RING E3 ligases. The fundamental mechanistic distinction between RING and HECT E3s underpins this experimental approach: RING E3s act as scaffolds that direct ubiquitin transfer directly from the E2~Ub thioester to the substrate, whereas HECT E3s form an obligate thioester intermediate with ubiquitin before substrate modification [11] [25]. Consequently, for RING E3s, the E2 directly controls the chemistry of ubiquitin transfer and linkage specification.
E2 Enzyme Classification and Functional Properties
Core Structural and Functional Features of E2s
All human E2s share a conserved catalytic core, the ubiquitin-conjugating (UBC) domain, an approximately 150-residue α/β-fold that contains the active-site cysteine [28] [83]. Despite moderate sequence identity (~30%), the UBC domains across 38 human E2s are structurally remarkably similar [28]. The surface-exposed residues at the E3-binding interface, particularly on the N-terminal helix (α1) and loops L4 and L7, are key sites of variation that influence E3 partnership and functional outcomes [28]. Some E2s, such as UBE2G and UBE2R family members, possess functionally critical insertions within the catalytic cleft that facilitate linkage specificity and E3-independent ubiquitination [28].
Comparative Analysis of E2s for Atypical Chain Formation
The following table summarizes key E2 enzymes, highlighting their propensities for forming specific atypical ubiquitin chains, a crucial consideration for experimental design.
Table 1: E2 Ubiquitin-Conjugating Enzymes and Their Linkage Propensities with RING E3s
| E2 Enzyme | Known Atypical Linkage Propensity | Key Functional Characteristics | Experimental Considerations |
|---|---|---|---|
| UBE2K (E2-25K) | K48-linked chains [82] | Forms K48 chains processively; has C-terminal UBA domain that may regulate activity [83]. | Useful for generating degradation signals; less focused on "atypical" K48. |
| UBE2S (UBE2S) | K11-linked chains [82] | Specialized for K11-chain elongation; critical for cell cycle regulation via APC/C [82]. | Ideal for studying K11 signals in mitotic processes. |
| UBE2N/UBE2V1 | K63-linked chains [82] | UBE2N (Ubc13) requires non-catalytic co-E2 UBE2V1 (Mms2) for K63 synthesis [82]. | Essential for studying DNA repair and NF-κB signaling pathways. |
| UBE2J2 | Potential for ester linkages [83] | Reported to modify serine/threonine hydroxyl groups with viral RING E3 mK3 [83]. | Use base-sensitive assays (hydroxylamine) to detect oxy-ester bonds. |
| UBE2W | N-terminal ubiquitination [83] | Preferentially modifies protein N-termini; exhibits no intrinsic lysine reactivity [83]. | Probe for monoubiquitylation; confirm N-terminal linkage via mass spectrometry. |
| UBE2L3 (UbcH7) | Low aminolysis reactivity [83] | Binds RINGs but is unreactive towards lysine; functions with HECT/RBR E3s [83]. | A poor choice for most RING-E2 ubiquitination assays. |
Experimental Protocols for Probing E2-E3 Specificity
Assessing Intrinsic E2~Ub Reactivity
A foundational step in characterizing a novel E2-E3 pair is to determine the intrinsic reactivity of the E2~Ub thioester conjugate, independent of the E3's allosteric influence.
Detailed Protocol:
- E2~Ub Conjugate Formation: Generate the E2~Ub thioester by incubating purified E2 with E1, ubiquitin, and ATP in reaction buffer (e.g., 50 mM Tris-HCl pH 7.5, 50 mM NaCl, 10 mM MgCl₂, 2 mM ATP). Use a temperature-controlled environment (e.g., 30°C for 15-60 minutes) [83].
- Nucleophile Challenge: Purify the E2~Ub conjugate to remove E1 and excess ATP. Split the conjugate into separate reactions and challenge with different nucleophiles:
- Free Lysine (e.g., 50-100 mM) to test for aminolysis.
- DTT (e.g., 5-10 mM) or another small thiol to test for transthiolation.
- A lysine-less peptide with a free N-terminus to test for N-terminal ubiquitination (particularly relevant for UBE2W) [83].
- Analysis: Quench reactions at timed intervals with SDS-PAGE loading buffer lacking β-mercaptoethanol. Analyze samples by non-reducing SDS-PAGE and western blotting with anti-ubiquitin antibodies to monitor the decay of the E2~Ub thioester and the formation of ubiquitin-adducts [83].
Interpretation: E2s like UBE2L3 show reactivity only to DTT, not lysine, explaining their preference for HECT/RBR E3s. In contrast, E2s like UBE2D family members are reactive toward both, making them generalists for RING E3s [83].
In Vitro Ubiquitination Assays with RING E3s
This core assay tests the combined activity of an E2 and a RING E3 in modifying a substrate.
Detailed Protocol:
- Reaction Setup: In a total volume of 20-50 µL, combine:
- Reaction Buffer (e.g., 40 mM Tris-HCl pH 7.6, 5 mM MgCl₂, 2 mM ATP)
- E1 enzyme (50-100 nM)
- E2 enzyme (1-5 µM)
- RING E3 ligase (0.1-1 µM)
- Substrate protein (1-5 µM)
- Ubiquitin (50-100 µM)
- Initiation and Incubation: Initiate the reaction by adding ATP. Incubate at 30°C or 37°C for desired time points (e.g., 0, 15, 30, 60 minutes).
- Termination and Analysis: Quench reactions with SDS-PAGE loading buffer. Analyze by:
- SDS-PAGE & Western Blotting: Probe with anti-substrate and anti-ubiquitin antibodies to visualize substrate ubiquitination and auto-ubiquitination patterns.
- Linkage-Specific Antibodies: Use antibodies specific for K11, K48, K63, etc., linkages to determine chain topology.
- Mass Spectrometry: For definitive linkage identification, ubiquitinated products can be purified and analyzed by mass spectrometry [82] [83].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for E2-E3 Specificity Studies
| Reagent / Solution | Function in Experiment | Example / Key Specification |
|---|---|---|
| E1 Activating Enzyme | Activates ubiquitin for transfer to all E2s. | Human UBA1 (UBE1), recombinant, purified. |
| E2 Enzyme Library | Core catalysts for ubiquitin transfer; variable linkage specificity. | Recombinant human E2s (e.g., UBE2D, UBE2N/V1, UBE2S, UBE2W). |
| RING E3 Ligases | Scaffolds providing substrate specificity and E2 allosteric activation. | Recombinant full-length or RING-domain constructs. |
| Wild-Type Ubiquitin | Standard modifier for most ubiquitination assays. | Recombinant, non-tagged for unbiased kinetics. |
| Mutant Ubiquitin (K-only) | Determines specific lysine usage in chain formation. | Ubiquitin mutants where only one lysine is available (e.g., K48-only, K63-only). |
| Linkage-Specific Antibodies | Immunodetection of specific ubiquitin chain linkages. | Commercial anti-K11, anti-K48, anti-K63, anti-M1 (linear) Ub. |
| ATP Regeneration System | Maintains constant ATP levels for sustained E1 activity. | Creatine Phosphate and Creatine Kinase. |
Visualizing the E2 Selection Workflow
The following diagram illustrates a logical workflow for selecting and characterizing E2 enzymes for research on atypical ubiquitin chains with RING E3 ligases.
Diagram 1: A logical workflow for the systematic selection and validation of E2 enzymes for probing atypical ubiquitin chain formation with RING E3 ligases.
The strategic selection of E2 enzymes is a critical determinant of success in research aimed at deciphering the biological signals encoded in atypical ubiquitin chains. Moving beyond the traditional view of E2s as passive carriers to recognizing them as active decision-makers in the ubiquitin system allows for more precise experimental design. The protocols and comparative data provided here offer a framework for researchers to objectively match E2 properties with experimental goals, particularly within the context of RING E3 mechanisms. This targeted approach accelerates the deconvolution of complex ubiquitin signaling pathways and enhances the reproducibility of studies in this rapidly advancing field.
Ubiquitination is a vital post-translational modification that regulates diverse cellular processes, from protein degradation to signal transduction [84]. The functional outcome of ubiquitination is largely determined by the topology of the polyubiquitin chain attached to substrate proteins. While the roles of canonical K48-linked (proteasomal degradation) and K63-linked (signaling) chains are well-established, the biological functions of atypical ubiquitin linkages—including K29, K11, K33, K6, and branched chains—have remained more enigmatic [85]. Understanding how E3 ubiquitin ligases, particularly HECT and RING families, dictate the formation of these atypical chains is crucial for deciphering their roles in cellular physiology and disease pathogenesis.
This guide provides a comprehensive comparison of experimental approaches for validating the functional outcomes associated with atypical ubiquitin chains, focusing on the distinct mechanistic features of HECT versus RING E3 ligases. We present structured data, detailed methodologies, and analytical frameworks to equip researchers with the tools necessary to connect specific chain topologies to their cellular consequences, enabling more targeted therapeutic interventions in ubiquitin-related diseases.
E3 Ligase Families: Architectural Divergence in Chain Formation
E3 ubiquitin ligases are categorized into several families based on their structural domains and catalytic mechanisms, with HECT and RING representing two major classes with distinct approaches to ubiquitin transfer [8] [84].
RING (Really Interesting New Gene) E3 ligases function primarily as scaffolds that facilitate the direct transfer of ubiquitin from an E2 enzyme to the substrate protein. They contain a RING domain that binds the E2~Ub thioester intermediate and positions it for ubiquitin transfer, typically without forming a covalent intermediate with ubiquitin itself [84]. This direct transfer mechanism means that linkage specificity is often dictated by the cooperating E2 enzyme, though recent evidence indicates RING E3s can also influence chain topology through substrate and ubiquitin positioning.
HECT (Homologous to E6AP C-terminus) E3 ligases employ a two-step catalytic mechanism involving a covalent E3~Ub thioester intermediate. The HECT domain first accepts ubiquitin from the E2 enzyme onto a conserved catalytic cysteine residue, then transfers it to the substrate lysine residue [8] [10]. This intermediate step allows HECT E3s greater autonomy in determining chain linkage specificity, as they can reposition the donor ubiquitin relative to the acceptor ubiquitin or substrate independently of the E2 enzyme.
Table 1: Fundamental Characteristics of HECT and RING E3 Ligase Families
| Feature | HECT Family E3s | RING Family E3s |
|---|---|---|
| Catalytic Mechanism | Two-step mechanism with covalent E3~Ub intermediate | Direct transfer from E2 to substrate |
| Linkage Specificity Control | Primarily determined by E3 structural features | Largely dictated by collaborating E2 enzyme |
| Representative Members | TRIP12, UBR5, NEDD4, HUWE1 | Cullin-RING ligases (CRLs), APC/C, TRAF6 |
| Branched Chain Formation | Demonstrated capacity (e.g., TRIP12, UBR5) | Collaborative pairs (e.g., TRAF6/HUWE1) |
| Structural Flexibility | Modular domains with conformational flexibility | Often multi-subunit complexes with rigid scaffolding |
Quantitative Profiling of Atypical Ubiquitin Chains
Systematic analysis of linkage preferences provides critical insights into the specialized functions of different E3 ligases. Quantitative assessment of chain formation efficiency and specificity reveals how structural differences between E3 families translate to distinct biological outputs.
Table 2: Experimentally Determined Linkage Specificities of Selected E3 Ligases
| E3 Ligase | Family | Preferred Linkage | Branched Chain Capability | Cellular Function |
|---|---|---|---|---|
| TRIP12 | HECT | K29-linked, K29/K48-branched | Yes: K29/K48 branches | Proteotoxic stress response, neurodegenerative disorders |
| UBR5 | HECT | K48-linked, K11/K48-branched | Yes: K11/K48 branches | Apoptotic regulation, cell division |
| HUWE1 | HECT | K63-linked, K48/K63-branched | Collaborative: with TRAF6 | NF-κB signaling, DNA damage response |
| TRAF6 | RING | K63-linked | Collaborative: with HUWE1 | Innate immune signaling, NF-κB activation |
| APC/C | RING | K11-linked, K11/K48-branched | With UBE2C/UBE2S E2s | Cell cycle progression, mitotic regulation |
| cIAP1 | RING | K11, K48, K63-linked | K48/K63 branches detected | Apoptosis regulation, TNF signaling |
Recent structural studies have illuminated how HECT E3s achieve precise linkage specification. For TRIP12, cryo-EM analyses reveal a pincer-like architecture where tandem ubiquitin-binding domains engage the proximal ubiquitin to position K29 toward the catalytic center, while simultaneously capturing a distal ubiquitin from K48-linked chains to facilitate branching [10]. This sophisticated mechanism enables single E3s to generate complex chain architectures with high fidelity.
Experimental Methodologies for Functional Validation
Biochemical Assembly of Defined Ubiquitin Chains
Table 3: Experimental Approaches for Branched Ubiquitin Chain Assembly
| Method | Principle | Applications | Advantages | Limitations |
|---|---|---|---|---|
| Sequential Enzymatic Assembly | Use C-terminally blocked proximal Ub with mutant distal Ubs; ligate sequentially with linkage-specific enzymes | Generation of defined branched trimers for binding studies, DUB specificity assays | High fidelity, uses established protocols | Limited to trimers without additional processing |
| Photo-controlled Enzymatic Assembly | UV-mediated deprotection of NVOC-caged lysines between elongation cycles | Assembly of longer branched chains (tetramers+) using wildtype ubiquitin | Enables extended branched structures with native linkages | Requires specialized chemical synthesis expertise |
| Thiol-ene Coupling | Chemical modification of distal Ub C-terminus with allylamine for reaction with proximal Ub containing lysine-to-cysteine mutations | Production of isopeptide-linked chains for structural studies | Near-native linkages cleavable by DUBs | Non-physiological synthesis pathway |
| Genetic Code Expansion | Incorporation of noncanonical amino acids with protected side chains via amber stop codon suppression | Incorporation of specific modifications, tags, or warheads at precise positions | Enables precise chemical functionalization | Technically challenging, lower yields |
Detailed Protocol: Sequential Enzymatic Assembly of K48-K63 Branched Trimer
- Proximal Ubiquitin Preparation: Utilize ubiquitin(1-72) or D77-blocked ubiquitin as the proximal unit that cannot function as a donor in subsequent reactions.
- First Elongation Step: Generate K63-linked dimer using UBE2N/UBE2V1 E2 combination with Ub(K48R,K63R) mutant as distal ubiquitin.
- Second Elongation Step: Attach Ub(K48R,K63R) to K48 of proximal ubiquitin using K48-specific E2 enzymes (UBE2R1 or UBE2K).
- Purification and Validation: Purify branched trimer via size-exclusion chromatography and verify structure by mass spectrometry and DUB cleavage patterns [86].
For extended branched structures, the Ub-capping approach combines enzymatic assembly with specific deubiquitinase treatment:
- Initiate with M1-linked dimer containing wildtype distal ubiquitin and proximal Ub(1-72, K48R, K63R).
- Perform K48 and K63 ligation to the distal ubiquitin.
- Use OTULIN (M1-specific DUB) to remove the proximal cap, exposing the native C-terminus for further chain extension [86].
Structural Analysis of E3-Ubiquitin Complexes
Cryo-EM Analysis of TRIP12 in K29/K48-Branched Chain Formation
- Complex Stabilization: Create a stable mimic of the transition state by linking TRIP12's catalytic Cys2007 to a chemical warhead installed between the donor Ub's C-terminus and K29C of the proximal Ub in a K48-linked di-Ub chain.
- Grid Preparation and Vitrification: Apply complex to cryo-EM grids, blot, and plunge-freeze in liquid ethane.
- Data Collection and Processing: Collect micrographs using high-end cryo-EM, perform 2D and 3D classification to isolate homogeneous complexes.
- Model Building and Refinement: Build atomic model into cryo-EM density, iteratively refine against experimental map [10].
This approach revealed TRIP12's pincer-like architecture, with the ARM domain and HECT domain clamping around the acceptor ubiquitin, precisely juxtaposing donor and acceptor ubiquitins to ensure K29 linkage specificity while accommodating K48-linked chains for branching.
Cellular Functional Assays
Validating Degradation versus Signaling Outcomes
- Substrate Stability Profiling:
- Express substrate proteins with defined ubiquitin chain mutants (e.g., K29-only, K48-only, K29/K48-branched) in cells.
- Monitor protein half-life using cycloheximide chase assays and proteasome inhibition (MG132).
- Quantify degradation kinetics via immunoblotting or fluorescent reporters.
Pathway Activation Readouts:
- For signaling chains (K63, M1, K29?), measure downstream pathway activation (NF-κB translocation, kinase phosphorylation, gene expression).
- Use linkage-specific ubiquitin binding domains as biosensors to monitor chain formation in live cells.
- Employ CRISPRi to knock down specific E3s and assess pathway perturbation.
Branched Chain Functional Mapping:
- Express ubiquitin mutants that restrict branching (e.g., K29R, K48R) in relevant cellular models.
- Monitor processing by molecular machines (proteasome, p97) using pull-down assays and mass spectrometry.
- Assess functional rescue with defined branched chains delivered via electroporation or cell-permeable formats.
Pathway Integration and Functional Specialization
The following diagram illustrates the experimental workflow for connecting specific atypical ubiquitin chains to their functional outcomes, integrating biochemical, structural, and cellular validation approaches:
The molecular mechanisms by which atypical chains direct functional outcomes are becoming increasingly clear. K29/K48-branched chains, synthesized by TRIP12 and other HECT E3s, enhance proteasomal targeting by providing multiple degradation signals within a single modification [10] [85]. Meanwhile, K48/K63-branched chains function as dual-fate signals that can either promote degradation or facilitate activation depending on cellular context and interacting proteins [85] [86].
The following diagram illustrates how different E3 ligase families collaborate to build branched ubiquitin chains with distinct functional outcomes:
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Reagents for Studying Atypical Ubiquitin Chains
| Reagent Category | Specific Examples | Function/Application | Key Features |
|---|---|---|---|
| Linkage-Specific E2 Enzymes | UBE2N/UBE2V1 (K63), UBE2R1 (K48), UBE2S (K11) | Controlled synthesis of specific linkage types | Enable homotypic chain assembly as building blocks for branched chains |
| Branched Chain Assembly Kits | Ub(1-72), Ub(K48R, K63R), Ub(K29R) mutants | Sequential enzymatic assembly of defined branched trimers | Provide controlled approach for generating specific architectures |
| DUB Specificity Profilers | OTULIN (M1-specific), Cezanne (K11-specific), TRABID (K29/K33-specific) | Linkage verification and chain editing | Confirm linkage identity and study chain disassembly kinetics |
| Linkage-Specific Binders | K48-specific TUBE, K63-specific TUBE, K11-specific antibody | Affinity enrichment and detection of specific chain types | Enable isolation and visualization of endogenous atypical chains |
| Activity-Based Probes | Ub-VS, Ub-PA, linkage-specific DUB probes | Profiling deubiquitinase activities and specificities | Identify DUBs that recognize and process atypical chains |
| Cellular Delivery Tools | Cell-permeable ubiquitin analogs, electroporation kits | Intracellular introduction of defined ubiquitin chains | Functional testing of specific chain types in living cells |
| Genetic Code Expansion Systems | Noncanonical amino acids (AzF, BOC-lysine), orthogonal tRNA/tRNA synthetase pairs | Site-specific incorporation of chemical handles | Enable precise modification of ubiquitin for advanced applications |
The systematic validation of functional outcomes for atypical ubiquitin chains represents a critical frontier in ubiquitin research. The experimental frameworks presented here enable researchers to move beyond correlation to causation when linking specific chain topologies to biological functions. The distinction between HECT and RING E3 mechanisms provides a conceptual foundation for understanding how chain specificity is achieved, while the methodologies for branched chain assembly and analysis offer practical tools for experimental interrogation.
Future advances will likely come from improved technologies for tracking specific chain types in living cells, more sophisticated structural biology approaches for visualizing E3 mechanisms in action, and computational methods for predicting linkage-specific outcomes. As these tools mature, our ability to design targeted interventions that manipulate specific ubiquitin chain types for therapeutic benefit will transform treatment strategies for cancer, neurodegenerative diseases, and immune disorders where ubiquitin signaling is disrupted.
Troubleshooting Common Pitfalls in Recombinant E3 Production and Purification
Within the Context of Comparing HECT vs. RING E3 Specificity for Atypical Chains
The ubiquitination process, a crucial post-translational modification, regulates myriad cellular processes, including protein degradation, DNA repair, and signal transduction [11] [87]. This process is mediated by a sequential enzymatic cascade involving ubiquitin-activating (E1), ubiquitin-conjugating (E2), and ubiquitin ligase (E3) enzymes. Among these, E3 ligases are pivotal as they confer substrate specificity, determining which proteins are targeted for modification and with what type of ubiquitin signal [88]. The human genome encodes over 600 E3s, vastly outnumbering the approximately 40 E2s and few E1s, highlighting their role in generating signaling diversity [11] [87].
E3 ligases are primarily categorized into two major families based on their structural features and mechanisms: RING (Really Interesting New Gene) and HECT (Homologous to the E6AP C Terminus) [11]. A key mechanistic difference lies in their catalytic activity. RING E3s act as scaffolds, facilitating the direct transfer of ubiquitin from an E2 enzyme to the substrate. In contrast, HECT E3s form an obligate thioester intermediate with ubiquitin on their catalytic cysteine before transferring it to the substrate [11] [88]. This fundamental difference profoundly impacts their functionality and the technical challenges associated with their recombinant production and biochemical characterization. Understanding these challenges is a critical prerequisite for rigorous comparative research, particularly when investigating their distinct specificities for forming atypical ubiquitin chains, such as K29/K48-branched chains [39] [10].
HECT vs. RING E3 Ligases: A Mechanistic and Structural Comparison
The divergence in catalytic mechanism between HECT and RING E3s is rooted in their domain architecture. A canonical RING finger is a Zn²⁺-coordinating domain that serves as a scaffold to bring the E2~Ub thioester and substrate into proximity, sometimes allosterically activating the E2 [11]. RING E3s can function as monomers, dimers, or large multi-subunit complexes, such as the cullin RING ligases (CRLs) that are essential for cell cycle progression [11].
The HECT domain, a ~350 amino acid C-terminal region, is bi-lobed. The N-lobe interacts with the E2, while the C-lobe contains the active-site cysteine that forms a transient thioester with ubiquitin [11]. Structural studies on various HECT E3s, including NEDD4L, E6AP (UBE3A), and the "other" subfamily member AREL1, reveal a flexible hinge between the lobes that allows them to adopt different conformations—such as "L-shaped" and "Inverted-T-shaped"—during the ubiquitin transfer process [11] [34]. This conformational flexibility is integral to its function but often complicates structural studies and stable protein production. Recent cryo-EM structures of HECT E3s like UBR5, Ufd4, and TRIP12 have visualized this process, showing how specific domains recruit acceptor ubiquitins and orient particular lysine residues (e.g., K29 or K48) toward the active site to form linkage-specific chains [39] [10] [31].
Table 1: Fundamental Characteristics of HECT and RING E3 Ligases
| Feature | HECT E3 Ligases | RING E3 Ligases |
|---|---|---|
| Catalytic Mechanism | Direct catalysis via covalent E3~Ub thioester intermediate [11] [88] | Scaffolding role; direct Ub transfer from E2 to substrate [11] [88] |
| Representative Catalytic Domain Structure | Bi-lobed HECT domain with flexible hinge [11] [34] | Zn²⁺-coordinating cross-brace RING finger domain [11] |
| Ubiquitin Chain Linkage Determination | Primarily determined by the HECT E3 itself [34] [15] | Primarily determined by the cognate E2 enzyme [31] |
| Functional Oligomeric State | Often monomers (e.g., AREL1) [34] | Monomers, dimers, or multi-subunit complexes (e.g., SCF, APC/C) [11] |
Common Pitfalls in Recombinant Production and Purification
Producing high-quality, functional recombinant E3 ligases is a common bottleneck in ubiquitin research. The challenges differ significantly between HECT and RING families, necessitating tailored approaches.
HECT E3-Specific Challenges
A primary challenge with HECT E3s is the instability and insolubility of the isolated HECT domain. Research on AREL1 demonstrated that a construct lacking the N-terminal extended region (aa 436–482) preceding the HECT domain was unstable and poorly soluble, whereas the construct containing this region was soluble, though it required reductive alkylation to achieve crystallization-grade concentration [34]. This underscores that regions outside the canonical HECT domain are often indispensable for stability and activity.
Furthermore, the flexible interlobe linker of the HECT domain, while essential for catalytic conformational changes, can render the protein prone to proteolytic degradation and aggregation during expression and purification [11] [31]. The reactive catalytic cysteine (e.g., C1450 in Ufd4, C2007 in TRIP12) is another vulnerability, as it can be prone to oxidation, leading to loss of activity [39] [10].
RING E3-Specific Challenges
The primary challenge for RING E3s often lies in reconstituting functional multi-subunit complexes. Many RING E3s, such as the SCF complex and the anaphase-promoting complex/cyclosome (APC/C), are large assemblies comprising multiple proteins [11]. Co-expressing and purifying these subunits in correct stoichiometry is technically demanding. The development of robust functional assays has been hampered by the need to reconstitute these multi-unit complexes, which can involve more than ten different proteins [88].
For both families, the large size and multi-domain nature of many full-length E3s (e.g., UBR5 is 2799 residues) can lead to low expression yields in standard systems like E. coli, necessitating the use of more complex expression hosts such as insect or mammalian cells [31].
Table 2: Troubleshooting Guide for Common E3 Production Pitfalls
| Pitfall | Underlying Cause | Recommended Solution | Applicable E3 Type |
|---|---|---|---|
| Low Solubility/Stability | Isolated catalytic domain lacking stabilizing N/C-terminal regions [34] | Include flanking regions in construct; use fusion tags (e.g., GST, MBP) [34] [89] | Primarily HECT |
| Loss of Catalytic Activity | Oxidation of catalytic cysteine; improper folding [39] [10] | Purify in presence of reducing agents (DTT, TCEP); use activity assays to validate batches | Primarily HECT |
| Failure to Form Functional Complex | Incorrect stoichiometry or missing subunits in multi-subunit E3s [11] | Implement co-expression systems (e.g., baculovirus); use affinity tags on one subunit | Primarily RING |
| Low Expression Yield | Toxicity to host or intrinsic protein instability [89] | Switch expression host (e.g., insect, mammalian cells); optimize induction conditions | HECT & RING |
| Protein Aggregation | Exposure of hydrophobic surfaces; flexible regions [34] [31] | Include chaperones; use additives like arginine; size-exclusion chromatography | HECT & RING |
Experimental Protocols for Functional Analysis
To compare the specificity of HECT and RING E3s for atypical ubiquitin chains, robust and reproducible biochemical assays are essential. The following protocols are adapted from recent high-impact studies.
Protocol 1: Pulse-Chase Ubiquitination Assay for Linkage Specificity
This assay is ideal for directly visualizing the transfer of ubiquitin and determining linkage preference [10] [31].
- Pulse Reaction: In a 20 µL volume, combine 500 nM E1, 2 µM E2, 100 µM Fluorescently-labeled Donor Ubiquitin (e.g., Ub(K0) with an N-terminal tag), and 2.5 mM ATP in reaction buffer (50 mM Tris-HCl pH 7.5, 50 mM NaCl, 10 mM MgCl₂). Incubate at 30°C for 10 minutes to form the E2~Ub thioester intermediate.
- Chase Reaction: Add the pre-formed E2~*Ub mix to a tube containing 200 nM of the purified E3 ligase (HECT or RING) and 200 µM of the desired Acceptor Ubiquitin (e.g., mono-Ub, K48-linked diUb, or K29R Ub mutants). The total reaction volume is brought to 40 µL. Incubate at 30°C for 30-60 minutes.
- Quenching and Analysis: Stop the reaction by adding SDS-PAGE loading buffer (without reducing agents to preserve thioester bonds). Analyze the products by SDS-PAGE and visualize the fluorescently-labeled ubiquitin species using a gel imager. The appearance of higher molecular weight bands indicates polyubiquitin chain formation. Using mutant acceptors (e.g., K29R) can confirm linkage specificity.
Protocol 2: Determining Ubiquitin Chain Linkage by Middle-Down Mass Spectrometry (Ub-Clipping)
For definitive identification of branched or atypical chains, mass spectrometry is the gold standard [39].
- In Vitro Ubiquitination: Set up a large-scale ubiquitination reaction as above, but with unlabeled wild-type ubiquitin and the E3 of interest.
- Reaction Clean-up: Terminate the reaction and purify the ubiquitin chains using affinity chromatography or size-exclusion chromatography.
- Proteolytic Digestion (Clipping): Digest the purified polyubiquitin chains with a linkage-specific deubiquitinase (DUB) or the broad-spectrum DUB USP2. Alternatively, use the "Ub-clipping" method, which involves digestion with the 3C-protease from the Tobacco Etch Virus (TEVpro), which cleaves after glutamine in the Gly-Gly sequence of ubiquitin, leaving a di-glycine remnant on the modified lysine [39].
- MS Analysis: Analyze the digested peptides by LC-MS/MS. The presence of peptides with di-glycine modifications on specific lysines (e.g., K29 and K48 in the same peptide) provides direct evidence for K29/K48-branched ubiquitin chains.
The following diagram illustrates the logical workflow for producing, assaying, and validating recombinant E3 ligase activity, incorporating key steps to avoid common pitfalls.
Diagram 1: A workflow for recombinant E3 production and functional validation, highlighting critical decision points and troubleshooting loops.
Visualizing the Catalytic Mechanisms of HECT and RING E3s
Understanding the distinct catalytic architectures of HECT and RING E3s is key to studying their specificity. Recent structural biology breakthroughs, particularly for HECT E3s, provide a visual guide for interpreting biochemical data.
The diagram below illustrates the fundamental mechanistic differences and the specific architecture by which a HECT E3, like TRIP12 or Ufd4, assembles an atypical K29/K48-branched ubiquitin chain.
Diagram 2: A comparison of the core catalytic mechanisms of RING and HECT E3 ligases, highlighting the multi-step, covalent catalysis of HECT E3s in forming branched chains.
Successful research in this field relies on a suite of specialized reagents and tools. The table below lists key solutions for studying E3 ligases and atypical ubiquitin chains.
Table 3: Research Reagent Solutions for E3 Ubiquitin Ligase Studies
| Reagent / Resource | Function / Application | Example Use-Case |
|---|---|---|
| Linkage-Specific Di-Ubiquitin | Defined substrates for ubiquitination assays to determine E3 linkage specificity [39] [10]. | Testing if an E3 preferentially elongates K48-linked chains over K63-linked chains. |
| Mutant Ubiquitin (e.g., K29R, K48R) | To identify specific lysine residues used for chain formation in biochemical assays [39]. | Confirming that K29 is essential for E3-mediated branched chain formation. |
| Stabilizing Fusion Tags (GST, MBP) | Enhances solubility and aids in purification of recalcitrant E3 domains [89]. | Improving yield of a poorly expressed HECT domain. |
| Chemical Crosslinkers / Activity Probes | To trap transient E3~Ub intermediates for structural studies (e.g., Cryo-EM) [39] [10]. | Capturing a structural snapshot of TRIP12 during K29-linkage formation. |
| Robotic Liquid Handling Systems | Automation of complex reaction assembly for high-throughput screening of E3 activity or inhibitors [88]. | Screening a compound library for inhibitors of a specific E3-substrate interaction. |
The recombinant production and functional characterization of E3 ubiquitin ligases present distinct challenges for the HECT and RING families, rooted in their fundamental mechanistic differences. For HECT E3s, the primary hurdles involve managing the intrinsic instability of the catalytic domain and preserving its reactive cysteine. For multi-subunit RING E3s, the challenge lies in the faithful reconstitution of large protein complexes. Overcoming these pitfalls is not merely a technical exercise; it is a critical prerequisite for generating reliable, reproducible data.
A clear understanding of these production challenges empowers researchers to objectively compare the biology of these enzymes. As the field advances, particularly in therapeutically relevant areas like targeted protein degradation, the ability to robustly produce and assay E3s will be paramount. The protocols and troubleshooting guides provided here offer a foundation for researchers to delve into the complex and fascinating world of E3-mediated ubiquitin signaling, especially the emerging paradigm of atypical and branched ubiquitin chain specificity.
Comparative Analysis and Therapeutic Validation of E3 Specificity
Ubiquitination is a crucial post-translational modification that regulates virtually all cellular processes in eukaryotes. The functional outcome of ubiquitination is largely determined by the topology of the ubiquitin chain, which refers to the specific lysine residue (K6, K11, K27, K29, K33, K48, or K63) or the N-terminal methionine (M1) used to link ubiquitin molecules [90]. Among these, the "atypical" ubiquitin linkages—K11, K27, and K33—have emerged as important regulators of diverse cellular pathways, from cell cycle progression to immune signaling [90] [12]. The specificity for generating these distinct chain types lies primarily with E3 ubiquitin ligases, the enzymes that catalyze the final step of ubiquitin transfer to substrate proteins [90].
The two major families of E3 ligases, HECT (Homologous to the E6AP C-terminus) and RING (Really Interesting New Gene), employ fundamentally different catalytic mechanisms [91]. This structural and mechanistic divergence directly impacts their ability to specify particular ubiquitin chain linkages. This guide provides a direct comparison of HECT versus RING E3 ligases, focusing specifically on their specificity for K11, K27, and K33 ubiquitin linkages, with supporting experimental data and methodological details to aid researchers in selecting appropriate experimental systems.
Mechanistic Divergence: Catalytic Architectures of HECT and RING E3 Ligases
Fundamental Catalytic Mechanisms
HECT and RING E3 ligases differ fundamentally in their catalytic mechanisms, which directly influences their linkage specificity:
HECT E3s utilize a two-step catalytic mechanism involving a covalent intermediate. They feature an active-site cysteine residue within the HECT domain that forms a thioester bond with ubiquitin transferred from the E2 enzyme before ultimately transferring it to the substrate [4] [91]. This covalent intermediate allows HECT E3s to override the intrinsic linkage specificity of their partner E2 enzymes [12].
RING E3s employ a single-step mechanism where they function as allosteric activators that position the E2~Ub thioester complex in close proximity to the substrate, enabling direct ubiquitin transfer from E2 to substrate without a covalent E3-Ub intermediate [90] [91]. Consequently, RING E3s largely maintain the linkage specificity inherent to their cognate E2 enzymes.
The following diagram illustrates these fundamental mechanistic differences:
Structural Determinants of Linkage Specificity
The structural features that determine linkage specificity differ significantly between these E3 families:
HECT E3 Linkage Determination: For HECT E3s, linkage specificity is primarily determined by regions within the HECT domain itself, particularly elements that position the acceptor ubiquitin relative to the donor ubiquitin attached to the catalytic cysteine [10]. Recent structural studies of TRIP12 reveal that tandem ubiquitin-binding domains and the HECT domain work together like a pincer to precisely orient the acceptor ubiquitin, bringing K29 into proximity with the active site [10]. Similarly, the N-terminal extended region preceding the HECT domain in AREL1 is indispensable for stability and activity [34].
RING E3 Linkage Determination: For RING E3s, linkage specificity is primarily determined by the cognate E2 enzyme, with the RING domain serving mainly to activate the E2 and position it toward the substrate [90] [91]. Different E2 enzymes have intrinsic preferences for specific linkage types, though the RING domain can influence this specificity through allosteric effects.
Comparative Analysis of Linkage Specificity for Atypical Chains
The table below summarizes documented specificities of representative HECT and RING E3 ligases for K11, K27, and K33 linkages based on experimental evidence:
Table 1: Specificity of Selected HECT and RING E3 Ligases for Atypical Ubiquitin Linkages
| E3 Ligase | Family | K11 Linkage | K27 Linkage | K33 Linkage | Experimental Evidence |
|---|---|---|---|---|---|
| WWP1 | HECT (NEDD4) | Secondary preference (after K63/K48) [15] | Not detected as major product [15] | Not detected as major product [15] | In vitro Ub2 chain synthesis assays [15] |
| AREL1 | HECT ("Other") | Not primary | Not primary | Assembled along with K48/K63 linkages [34] | Crystal structure (2.4 Å) and in vitro ubiquitination [34] |
| HUWE1 | HECT ("Other") | Not primary | Capable of synthesis [4] [90] | Not primary | Multiple functional studies [4] [90] |
| E6AP | HECT ("Other") | Specificity influenced by N-terminal domain [92] | Not well characterized | Not well characterized | In vitro Ub2 chain synthesis with HECT domain vs. full-length [92] |
| ITCH | HECT (NEDD4) | Not primary | Not primary | Cooperates with Cbl-b for TCR-ζ ubiquitination [4] | Cellular studies in T-cell activation [4] |
| APC/C | RING (Multi-subunit) | Primary linkage formed [4] [90] | Not primary | Not primary | Cell cycle studies and in vitro reconstitution [4] [90] |
| RNF168 | RING (Single-subunit) | Not primary | Primary linkage in DNA damage response [4] | Not primary | Cellular DNA damage response studies [4] |
| Cbl-b | RING (Single-subunit) | Not primary | Not primary | Cooperates with ITCH for TCR-ζ ubiquitination [4] | Cellular studies in T-cell activation [4] |
Biological Context of Atypical Linkages
The biological functions associated with these atypical linkages provide context for the physiological relevance of the observed E3 specificities:
K11-linked chains are powerful degradation signals that regulate cell division [4] [90] and are particularly enriched during mitosis when catalyzed by the RING E3 APC/C [4]. The HECT E3 WWP1 also demonstrates K11 linkage formation capability, though as a secondary preference after K63 and K48 linkages [15].
K27-linked chains participate in the DNA damage response, with the RING E3 RNF168 generating K27-linked chains that create important chromatin marks upon DNA damage [4]. K27 linkages are also involved in innate immune signaling and mitochondrial regulation [90] [12].
K33-linked chains function in non-proteolytic processes, notably in regulating T-cell receptor signaling through the cooperative action of the RING E3 Cbl-b and HECT E3 ITCH, which together induce K33-linked polyubiquitination of TCR-ζ [4]. K33 linkages also influence post-Golgi membrane protein trafficking [12].
Experimental Approaches for Determining Linkage Specificity
Key Methodologies and Workflows
Researchers have employed several sophisticated biochemical and structural approaches to determine the linkage specificity of E3 ligases:
Table 2: Key Experimental Methods for Determining E3 Linkage Specificity
| Method | Key Procedure | Applications in HECT vs. RING Studies |
|---|---|---|
| In vitro Ub2 Chain Synthesis Assay | Incubates E1, E2, E3 with ubiquitin without substrate; analyzes free diubiquitin chains [92] [15] | Used for WWP1 (HECT) [15] and E6AP (HECT) [92] to determine intrinsic HECT domain specificity |
| Pulse-Chase Ubiquitination Assays | Uses fluorescently-labeled donor ubiquitin to track transfer to specific acceptor ubiquitins [10] | Employed for TRIP12 (HECT) to demonstrate preference for K48-linked diUb acceptors [10] |
| Mass Spectrometry Analysis | Uses middle-down MS (Ub-clipping) to identify linkage types and branched chains [39] | Used for Ufd4 (HECT) to identify K29/K48-branched chains [39] |
| Chemical Biology Probes | Creates stable mimics of ubiquitylation transition states with cross-linked complexes [10] [39] | Enabled cryo-EM structures of TRIP12 and Ufd4 during K29/K48-branched chain formation [10] [39] |
| Cryo-EM Structural Analysis | Determines structures of E3 ligases trapped with donor and acceptor ubiquitins [10] [39] | Revealed structural basis for K29 specificity in TRIP12 and Ufd4 (HECT) [10] [39] |
The following diagram illustrates a typical experimental workflow for determining E3 linkage specificity:
Critical Experimental Considerations
When designing experiments to evaluate E3 linkage specificity, several critical factors must be considered:
Full-length vs. Catalytic Domain: For HECT E3s, the N-terminal regions significantly influence linkage specificity. The E6AP HECT domain alone shows different linkage specificity compared to full-length E6AP, demonstrating the importance of N-terminal domains in determining chain type [92].
E2 Enzyme Selection: For RING E3 studies, the choice of E2 enzyme is critical as it largely determines linkage specificity. Certain E2s like UBE2C and UBE2D interact with RING domains and can assemble all linkage types [12].
Acceptor Ubiquitin Context: Recent studies reveal that some HECT E3s show strong preference for specific ubiquitin chain acceptors. TRIP12 preferentially targets K48-linked diubiquitin over mono-ubiquitin or other diUb linkages when forming K29-linked branches [10].
Temporal Aspects of Chain Formation: Linkage specificity may change over the course of the ubiquitination reaction. WWP1 demonstrates distinct phases of ubiquitination—an initial phase with exclusive K63 linkage formation followed by a multidirectional phase characterized by mixed linkages including K11 and K48 [15].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for Studying E3 Linkage Specificity
| Reagent/Category | Specific Examples | Function/Application | Considerations for HECT vs. RING Studies |
|---|---|---|---|
| Ubiquitin Mutants | Ubiquitin-K29R, Ubiquitin-K11R, Ubiquitin-K0 (lysine-less) | Identify specific linkage requirements; track ubiquitin transfer | Critical for both HECT and RING studies to determine linkage dependence |
| E2 Enzyme Library | UBE2L3 (UbcH7), UBE2D family, UBE2S, UBE2R1/2 | Determine E2-E3 pairing requirements and intrinsic E2 specificity | More critical for RING E3 studies as E2 largely determines linkage |
| Chemical Biology Probes | Disulfide-linked Ub-E2 conjugates; warhead-containing Ub probes | Trap intermediate states for structural studies | Particularly valuable for HECT E3 mechanistic studies [10] [39] |
| E3 Expression Constructs | Full-length vs. HECT-domain only; catalytically inactive mutants (Cys→Ala) | Structure-function studies; determine domain requirements | For HECT E3s, both full-length and isolated HECT domains should be tested [92] |
| Linkage-Specific Antibodies | Anti-K11-linkage, Anti-K27-linkage, Anti-K33-linkage | Detect specific chain types in cells and in vitro | Validation required as cross-reactivity can occur |
| Mass Spectrometry Standards | Synthetic ubiquitin chains with defined linkages | Reference standards for linkage identification | Essential for confirming atypical linkage identification |
The fundamental mechanistic differences between HECT and RING E3 ligases translate to distinct advantages and limitations for generating specific atypical ubiquitin linkages:
HECT E3 Advantages: Their two-step mechanism with a covalent intermediate allows them to override E2 specificity and create defined, often atypical, ubiquitin linkages. Their modular architecture with specific substrate-binding domains and catalytic HECT domains enables precise targeting of particular lysine residues on substrates [34] [10]. This makes HECT E3s particularly valuable for engineering specific ubiquitin signals.
RING E3 Advantages: Their direct transfer mechanism leverages the intrinsic specificity of E2 enzymes, allowing for more predictable linkage outcomes when appropriate E2 partners are selected. The vast diversity of RING E3s (over 600 members) provides a broader range of substrate specificities [90] [91].
For researchers aiming to target specific ubiquitin linkages, HECT E3s may offer more flexibility for engineering specific chain types, while RING E3 systems provide a more natural context that maintains E2-E3 coordination. The emerging structural information on HECT E3s like TRIP12, Ufd4, and AREL1 provides unprecedented insights into the molecular determinants of linkage specificity [34] [10] [39], opening new avenues for the development of specific E3 modulators for research and therapeutic applications.
Roles of HECT- and RING-Synthesized Atypical Chains in Innate Immunity
Protein ubiquitination, a fundamental post-translational modification, extends beyond its canonical role in targeting proteins for proteasomal degradation via K48-linked chains. The diversity of ubiquitin signaling is largely encoded by the topology of polyubiquitin chains, which can be formed through any of the seven lysine residues (K6, K11, K27, K29, K33, K48, K63) or the N-terminal methionine (M1) of ubiquitin [90] [21]. Among these, the atypical ubiquitin chains—those not linked via K48 or K63—have emerged as crucial regulators of innate immune responses, functioning in the activation, assembly, and modulation of signaling complexes [90] [93]. The specificity of chain linkage formation is principally determined by E3 ubiquitin ligases, which are broadly classified into HECT (Homologous to the E6AP C Terminus) and RING (Really Interesting New Gene) families. These enzyme families employ fundamentally different catalytic mechanisms: RING E3s typically act as scaffolds to directly transfer ubiquitin from E2 enzymes to substrates, whereas HECT E3s form an obligate thioester intermediate with ubiquitin before transferring it to substrates [90] [34]. This mechanistic comparison guide synthesizes current experimental evidence to objectively evaluate how HECT- and RING-type E3 ligases dictate the synthesis of atypical ubiquitin chains and thereby control innate immunity.
Comparative Analysis of E3 Ligase Families: HECT vs. RING
Table 1: Fundamental Characteristics of HECT and RING E3 Ligase Families
| Feature | HECT Family E3s | RING Family E3s |
|---|---|---|
| Catalytic Mechanism | Two-step transfer via E3~Ub thioester intermediate [90] [34] | Direct, single-step transfer from E2 to substrate [90] |
| Chain Linkage Determination | Primarily determined by the E3 ligase itself [39] [34] | Primarily dictated by the partnered E2 enzyme [90] [31] |
| Structural Features | Bilobed HECT domain (N-lobe, C-lobe) with catalytic cysteine in C-lobe [31] | RING domain coordinates zinc ions, facilitates E2 binding [94] |
| Representative Atypical Chain Linkages | K29/K48-branched (Ufd4), K6/K48-heterotypic (NleL), K33-linked (AREL1) [39] [34] [65] | K27-linked (RNF185, AMFR), K11-linked, M1-linear (HOIP) [90] [93] |
| Role in Innate Immunity | Less directly characterized; emerging roles in inflammatory signaling [93] | Well-established roles in NF-κB, STING, and cytokine signaling pathways [90] [93] |
Experimental Evidence: Atypical Chain Formation and Function
HECT E3 Ligases and Atypical Chain Synthesis
Recent structural and biochemical studies have provided unprecedented insight into the mechanisms of atypical chain synthesis by HECT E3s. The following experiments showcase the unique capabilities of this family.
3.1.1 Ufd4-Mediated K29/K48-Branched Chain Formation
- Experimental Objective: To elucidate the mechanism by which the HECT E3 ligase Ufd4 preferentially synthesizes K29-linked branches on pre-existing K48-linked ubiquitin chains to form K29/K48-branched ubiquitin chains, a potent signal for proteasomal degradation [39].
- Key Methodology:
- Ubiquitination Assays: In vitro reconstitution assays using E1 (Uba1), E2 (Ubc4), Ufd4, WT Ub, and various Ub chain substrates (monoUb, K29-diUb, K48-diUb) demonstrated enhanced polyubiquitination on K48-linked diUb substrates [39].
- Mutagenesis and Kinetics: Using K48-linked diUb mutants with K29R mutations in either the proximal or distal Ub revealed that the proximal K29 site is preferred, with ~5.2-fold higher catalytic efficiency (kcat/Km) compared to the distal site [39].
- Middle-down Mass Spectrometry (Ub-clipping): Analysis of polyubiquitination products identified Ub fragments with double-glycine remnants on both K29 and K48 residues, confirming the formation of K29/K48-branched chains [39].
- Cryo-EM Structural Analysis: A stable mimic of the transition state was generated by covalently linking Ufd4's catalytic cysteine (C1450) to a chemically synthesized K29/K48-branched triUb probe. Single-particle cryo-EM revealed a closed-ring structure where Ufd4 clamps the donor Ub and the K48-linked diUb substrate, precisely orienting the proximal K29 for catalysis [39].
- Conclusion: Ufd4's N-terminal ARM region and HECT domain collaboratively recognize K48-linked acceptor chains and specifically orient Lys29 of the proximal Ub for linkage, demonstrating a specialized E3 mechanism for generating a degradative branched ubiquitin signal [39].
3.1.2 AREL1 and Atypical Linkage Capability
- Experimental Objective: To characterize the linkage specificity and structural basis of the "other" subfamily HECT E3 AREL1, which has anti-apoptotic functions in cancer [34].
- Key Methodology:
- In Vitro Ubiquitination Assays: The extended HECT domain of AREL1 (aa 436-823) was incubated with E1, E2 (UBE2L3/UbcH7), and ATP. Reaction products were analyzed by western blot to determine linkage specificity [34].
- Structural Biology: The crystal structure of the extended AREL1 HECT domain was solved at 2.4 Å resolution, revealing an inverted T-shaped conformation and a unique loop (aa 567-573) not found in other HECT E3s [34].
- Mutagenesis: A C-terminal tail deletion mutant and a point mutant (E701A) were generated, revealing the tail's role in limiting autoubiquitination and the E701A mutant's enhanced activity [34].
- Conclusion: The AREL1 HECT domain possesses an intrinsic ability to assemble K33-linked polyubiquitin chains, among other atypical linkages, highlighting the diverse linkage specificity inherent to HECT E3s [34].
Table 2: Experimental Data on HECT E3 Ligases and Atypical Chains
| E3 Ligase | Experimental System | Atypical Linkage Synthesized | Key Functional Readout / Role |
|---|---|---|---|
| Ufd4/TRIP12 | In vitro ubiquitination; Cryo-EM; MS [39] | K29/K48-branched | Enhanced proteasomal targeting; N-degron pathway [39] |
| AREL1 | In vitro ubiquitination; X-ray crystallography [34] | K33-, K48-, K63-linked | Anti-apoptotic activity; SMAC degradation [34] |
| NleL | In vitro ubiquitination; DUB restriction analysis [65] | K6-/K48-heterotypic | Bacterial virulence; host cell modulation [65] |
RING E3 Ligases and Atypical Chains in Innate Immunity
RING E3 ligases are heavily implicated in the direct regulation of innate immune signaling pathways through atypical ubiquitin chains.
3.2.1 K27-Linked Ubiquitination in Antiviral Defense
- Experimental Objective: To identify E3 ligases that catalyze K27-linked ubiquitination and define their role in the activation of the innate immune cGAS-STING pathway [90].
- Key Methodology:
- Functional Genetics: RNAi or CRISPR-Cas9 screens and validation were used to identify E3 ligases required for type I interferon (IFN) production upon DNA viral infection or cyclic dinucleotide stimulation [90].
- Biochemical Assays: In vitro ubiquitination assays with purified RNF185 and AMFR E3s, E2s, and substrates (cGAS or STING) confirmed their activity. Immunoprecipitation followed by western blotting with linkage-specific antibodies detected K27-linked ubiquitination in cells [90].
- Immunofluorescence & Signaling Readouts: Microscopy tracked the subcellular localization of STING. ELISA and qPCR measured the production of type I IFN and IFN-stimulated genes (ISGs) [90].
- Conclusion: The RING E3 ligases RNF185 and AMFR target cGAS and STING, respectively, for K27-linked ubiquitination, promoting the antiviral innate immune response by facilitating the activation of the cGAS-STING pathway [90].
3.2.2 M1-Linear Ubiquitination in NF-κB and Inflammatory Signaling
- Experimental Objective: To define the role of the RBR-type RING ligase complex LUBAC (HOIP, HOIL-1) in regulating inflammatory signaling pathways relevant to sepsis and innate immunity [93].
- Key Methodology:
- Cell Signaling Studies: Immune cells (e.g., macrophages) were stimulated with TNF-α or IL-1. The recruitment of LUBAC to receptor complexes (TNFR1, IL-1R) was assessed by co-immunoprecipitation [93].
- Ubiquitination Mapping: The use of linkage-specific DUBs and antibodies confirmed the presence of M1-linked chains on signaling components like RIPK1, RIPK2, and NEMO [93].
- Genetic Models: Cells deficient in LUBAC components (HOIP knockout) were used to measure downstream outcomes, including IKK phosphorylation, IκBα degradation, NF-κB nuclear translocation, and pro-inflammatory cytokine production (TNF-α, IL-6) [93].
- Conclusion: LUBAC-catalyzed M1-linked ubiquitination of key signaling intermediates serves as a critical scaffolding platform for the recruitment and activation of the IKK complex, leading to NF-κB-mediated transcription of pro-inflammatory cytokines [93]. This pathway must be precisely controlled, as its dysregulation is implicated in septic inflammation [93].
Table 3: Experimental Data on RING E3 Ligases and Atypical Chains in Immunity
| E3 Ligase (Family) | Experimental System | Atypical Linkage | Key Functional Readout in Innate Immunity |
|---|---|---|---|
| RNF185 (RING) | Cell-based signaling; in vitro ubiquitination [90] | K27-linked | Activates cGAS-STING pathway for antiviral IFN production [90] |
| AMFR (RING) | Cell-based signaling; in vitro ubiquitination [90] | K27-linked | Activates cGAS-STING pathway for antiviral IFN production [90] |
| LUBAC (RBR) | Cell signaling; KO models; sepsis models [93] | M1-linear | Activates NF-κB pathway; regulates inflammation and septic shock [93] |
Visualization of Signaling Pathways and Experimental Workflows
Atypical Ubiquitin Chains in Innate Immune Signaling
This diagram illustrates how atypical ubiquitin chains synthesized by specific RING and HECT E3 ligases integrate into key innate immune pathways.
Experimental Workflow for Analyzing Atypical Ubiquitin Chains
This diagram outlines a standard biochemical and structural workflow for characterizing E3 ligase specificity and function, integrating methodologies from the cited studies.
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Reagents for Studying Atypical Ubiquitin Chains
| Reagent Category | Specific Example | Function in Research |
|---|---|---|
| Ubiquitin Mutants | Lysine-to-Arginine (K-to-R, e.g., K29R); "K-only" Ub [39] | To identify essential lysine residues for chain formation and create defined acceptor substrates. |
| Linkage-Specific DUBs | OTUB1 (K48-specific); OTUD3 (K6-preferential) [65] | To act as "restriction enzymes" for deciphering chain topology in ubiquitin restriction analysis. |
| Chemical Trapping Probes | Disulfide-linked Ub-E2 conjugates; Branched Ub probes (e.g., triUbprobe) [39] | To generate stable mimics of catalytic intermediates (e.g., E3~Ub, TS2) for structural studies. |
| Linkage-Specific Tools | Linkage-specific Ub antibodies; TUBE (Tandem Ub-Binding Entity) reagents | To detect, enrich, or purify specific chain types from complex mixtures for downstream analysis. |
| Defined E2 Enzymes | UBE2L3/UbcH7; UbcH5; MMS2-UBC13 complex [21] [65] | To provide the ubiquitin-conjugating enzyme partner, which can influence linkage specificity, especially for RING E3s. |
The synthesis of atypical ubiquitin chains represents a sophisticated layer of regulation in innate immunity. The experimental data compiled in this guide demonstrate a fundamental distinction between HECT and RING E3 ligase families: HECT E3s intrinsically determine linkage specificity, often forming complex branched and heterotypic chains that can amplify degradative signals or regulate apoptosis, while RING/RBR E3s are paramount for direct, rapid signaling in immune pathways, often through K27 and M1 linkages that control the NF-κB and cGAS-STING axes. The choice of experimental system—from minimal in vitro reconstitutions to validate biochemical mechanism, to complex cellular models to establish physiological relevance—is critical for accurate interpretation. The continued development of reagents like linkage-specific DUBs and chemical biology probes, combined with powerful structural techniques like cryo-EM, will be essential to fully decipher the complex ubiquitin code written by these enzymes and to harness this knowledge for therapeutic intervention in immune disorders and cancer.
The ubiquitin system regulates essential cellular processes through the post-translational modification of target proteins, with E3 ubiquitin ligases conferring substrate specificity and controlling the type of ubiquitin linkage formed. Approximately 600 E3 ligases in the human genome are categorized into three major families based on their catalytic mechanisms: Really Interesting New Gene (RING), Homologous to E6-AP C-Terminus (HECT), and RING-between-RING (RBR) ligases [8] [95]. These enzymes catalyze the attachment of ubiquitin chains to substrate proteins, with the linkage type determining the functional consequence. While atypical ubiquitin chains (linked through Met1, Lys6, Lys11, Lys27, Lys29, or Lys33) represent less common topological configurations, they serve as crucial regulatory signals in specific biological pathways [21] [96].
The RING-type E3 ligases function as scaffolds that bring the E2 ubiquitin-conjugating enzyme and substrate into proximity, enabling direct ubiquitin transfer without a covalent intermediate. In contrast, HECT-type E3s utilize a two-step mechanism involving a catalytic cysteine that forms a thioester intermediate with ubiquitin before transferring it to the substrate [8] [97]. This fundamental mechanistic difference underlies their distinct linkage specificities and functional capabilities in cellular signaling. This review systematically examines how disease-associated mutations in E3 ligases disrupt specific ubiquitin linkage formation, providing validation for their biological roles and highlighting their potential as therapeutic targets.
E3 Ligase Families: Structural and Mechanistic Determinants of Linkage Specificity
RING E3 Ligases: Direct Ubiquitin Transfer
RING E3 ligases constitute the largest family, characterized by a zinc-binding RING domain that facilitates direct ubiquitin transfer from E2 to substrate. These ligases function primarily as scaffolds that simultaneously bind the E2~Ub thioester and substrate protein, orienting them for efficient ubiquitin transfer [97]. Structural analyses reveal that RING domains activate E2 enzymes by stabilizing a closed conformation that primes the thioester bond for nucleophilic attack by the substrate lysine residue [18]. The specificity for particular ubiquitin linkages is influenced by both the E2 enzyme and the RING domain itself, with certain E2-E3 pairs preferentially generating specific chain types [21].
Notably, some RING E3s exhibit remarkable flexibility in linkage specification. For instance, the mammalian RING-type ligase CBL mediates multiple monoubiquitination and Lys11-, Lys48-, and Lys63-linked polyubiquitination of activated epidermal growth factor receptor (EGFR) [21]. Similarly, BRCA1 interaction with different E2 enzymes (UBCH6, UBE2E2, UBCM2, UBE2W, MMS2-UBC13, and UBE2K) supports monoubiquitination, Lys63-, or Lys48-linked chain formation [21]. This versatility enables RING E3s to participate in diverse signaling pathways through the generation of functionally distinct ubiquitin signals.
HECT E3 Ligases: Two-Step Catalytic Mechanism
HECT E3 ligases employ a conserved two-step mechanism involving a catalytic cysteine residue that forms a thioester bond with ubiquitin before final transfer to the substrate. This intermediate formation allows HECT E3s to exert greater control over linkage specificity compared to RING E3s [8]. The HECT domain consists of two lobes: an N-lobe that binds the E2~Ub conjugate and a C-lobe containing the catalytic cysteine that accepts ubiquitin before transfer to the substrate [8].
Different HECT E3 subfamilies demonstrate distinct linkage preferences. For example, E6AP primarily forms Lys48-linked chains, while KIAA10 catalyzes both Lys48- and Lys29-linked chains [21]. The yeast HECT ligase Rsp5 can modify specific substrates with monoUb, Lys48-, or Lys63-linked chains, demonstrating context-dependent linkage specificity [21]. This capacity for precise linkage control positions HECT E3s as key regulators of diverse cellular processes, with mutations often resulting in severe pathological consequences.
RBR E3 Ligases: Hybrid Mechanism
RBR E3 ligases represent a unique family that combines features of both RING and HECT mechanisms. These enzymes contain RING1 and RING2 domains separated by an in-between-RING (IBR) domain. The RBR catalytic mechanism involves two steps: RING1 binds the E2~Ub conjugate similarly to RING E3s, while RING2 contains a catalytic cysteine that accepts ubiquitin in a transthiolation reaction before transfer to the substrate, analogous to HECT E3s [8] [18]. This hybrid mechanism enables RBRs to exhibit specialized functions in quality control and signaling pathways.
Table 1: Comparison of E3 Ubiquitin Ligase Families
| Feature | RING E3s | HECT E3s | RBR E3s |
|---|---|---|---|
| Catalytic Mechanism | Direct transfer from E2 to substrate | Two-step via E3-thioester intermediate | Hybrid: RING1 binds E2, RING2 has catalytic cysteine |
| Catalytic Domain | RING domain (Zn²⁺-binding) | HECT domain | RING1-IBR-RING2 domains |
| Linkage Control | Primarily determined by E2, influenced by E3 | Strongly determined by E3 | Determined by E3 and allosteric activators |
| Representative Members | Mdm2, Pirh2, RNF168 | E6AP, KIAA10, Rsp5 | Parkin, HOIL-1, HOIP, RNF216 |
| Chain Types Formed | MonoUb, K48, K63, K11, K29, mixed chains | MonoUb, K48, K63, K29 (varies by HECT) | MonoUb, K63, M1 (varies by RBR) |
Disease-Associated Mutations in E3 Ligases and Linkage Defects
Neurological Disorders and Parkin Mutations
Approximately 13% of known E3 ligase genes harbor mutations associated with neurological disorders, providing compelling genetic evidence for their physiological importance [95]. Parkinson's disease (PD) represents a particularly well-characterized example, with mutations in the RBR E3 ligase Parkin (PARK2) accounting for approximately 50% of familial cases and 10-20% of sporadic early-onset PD [95]. Parkin functions in mitochondrial quality control by generating canonical and non-canonical ubiquitin chains on damaged mitochondria, targeting them for autophagic clearance [95].
Functional studies using ubiquitin Absolute Quantification (UB-AQUA) mass spectrometry have revealed that PD-associated Parkin mutations disrupt its ability to form diverse ubiquitin linkages on mitochondrial proteins, with specific defects in K6, K11, K48, and K63-linked chains [95]. These linkage-specific defects impair the feed-forward mechanism of Parkin activation and recruitment to damaged mitochondria, ultimately leading to mitochondrial dysfunction and selective degeneration of dopaminergic neurons. Beyond Parkin, enrichment analyses have identified rare variants in additional E3 ligase genes (HERC1, IRF2BPL, RNF168, and RNF216) in early-onset PD patients, further supporting the involvement of ubiquitin signaling disruption in PD pathogenesis [98].
DNA Damage Response and RNF168 Mutations
The RING E3 ligase RNF168 plays a critical role in DNA double-strand break (DSB) repair by catalyzing the formation of K63-linked ubiquitin chains on histone H2A, creating a platform for the recruitment of downstream repair factors such as 53BP1 and BRCA1 [99]. Mutations in RNF168 are associated with RIDDLE syndrome, a rare immunodeficiency disorder characterized by radiosensitivity, immunodeficiency, dysmorphic features, and learning difficulties [95] [99].
Cancer genome analyses have revealed that RNF168 is mutated in over 10% of several cancer types, with these mutations disrupting its E3 ligase activity and impairing the DNA damage response [99]. Structural and functional studies demonstrate that RNF168 mutations specifically abolish its ability to generate K63-linked ubiquitin chains at DSB sites, without affecting other linkage types. This linkage-specific defect prevents proper accumulation of 53BP1 and BRCA1 at DNA damage sites, compromising DSB repair pathway choice and leading to genomic instability [99]. The dependency of RNF168 on UBC13 as its cognate E2 enzyme further reinforces its specificity for K63-linked chain formation, highlighting how specific E2-E3 partnerships determine linkage outcomes in biological processes.
Cancer-Associated E3 Mutations and Linkage Imbalances
Comprehensive genomic analyses have identified numerous somatic mutations and amplifications in E3 ligase genes across diverse cancer types, with specific alterations disrupting defined ubiquitin linkages. For instance, the RING E3 ligase FBXW7, a critical tumor suppressor, is frequently mutated in multiple human cancers [99]. FBXW7 normally promotes the proteasomal degradation of oncoproteins such as c-MYC, cyclin E, and NOTCH through K48-linked ubiquitination. Cancer-associated FBXW7 mutations disrupt this K48-linked ubiquitination, leading to stabilization of its oncogenic substrates and accelerated tumor progression [99].
Similarly, the HECT E3 ligase HERC2 mutations are observed in various cancers and are linked to defective DNA damage response [99]. HERC2 facilitates the formation of the HERC2/MDC1/RNF8 complex following DNA damage, promoting RNF8-mediated recruitment of RNF168. Mutations in HERC2 impair this process, resulting in reduced K63-linked ubiquitin signaling at DSB sites and compromised DNA repair [99]. These examples illustrate how cancer-associated E3 mutations disrupt the balance between different ubiquitin linkages, leading to altered protein degradation, defective signaling, and ultimately, malignant transformation.
Table 2: Disease-Associated E3 Ligase Mutations and Linkage Defects
| E3 Ligase | E3 Family | Associated Diseases | Disrupted Ubiquitin Linkages | Functional Consequences |
|---|---|---|---|---|
| Parkin (PARK2) | RBR | Parkinson's Disease | K6, K11, K48, K63 | Impaired mitochondrial quality control, defective mitophagy |
| RNF168 | RING | RIDDLE syndrome, Cancer | K63-linked chains | Defective DNA damage response, genomic instability |
| FBXW7 | RING | Various cancers | K48-linked chains | Oncoprotein stabilization, dysregulated cell cycle |
| HERC2 | HECT | Cancer, Neurological disorders | K63-linked chains | Impaired DNA repair, defective protein complex assembly |
| RNF216 | RBR | Neurological disorders | K63-linked chains | Disrupted protein degradation, signaling defects |
Experimental Approaches for Validating Linkage-Specific Defects
Mass Spectrometry-Based Ubiquitin Profiling
Advanced mass spectrometry techniques have become indispensable for characterizing linkage-specific defects in diseased tissues and model systems. The Ubiquitin Absolute Quantification (UB-AQUA) method utilizes isotopically labeled internal standard peptides to precisely quantify different ubiquitin chain linkages from biological samples [95]. This approach enabled the discovery that Parkin generates both canonical and non-canonical ubiquitin chains on mitochondria in response to damage, and that PD-associated mutations disrupt this linkage diversity [95].
The experimental workflow involves: (1) purification of ubiquitinated proteins from control and disease samples; (2) tryptic digestion to generate ubiquitin-derived peptides; (3) enrichment of ubiquitin remnants using specific antibodies; (4) liquid chromatography separation coupled to tandem mass spectrometry analysis; and (5) quantification using heavy isotope-labeled internal standards for each ubiquitin linkage type [96]. This methodology provides comprehensive linkage profiling and has been instrumental in validating linkage-specific defects in numerous E3 ligase disorders.
In Vitro Ubiquitination Assays
Reconstituted in vitro ubiquitination assays remain a cornerstone for directly establishing the biochemical consequences of disease-associated E3 mutations. These assays typically include: (1) purified E1 activating enzyme; (2) specific E2 conjugating enzyme; (3) wild-type or mutant E3 ligase; (4) ubiquitin; (5) ATP regeneration system; and (6) substrate protein [100] [18]. The reactions are incubated at 30°C for specified times, followed by SDS-PAGE and immunoblotting with linkage-specific ubiquitin antibodies to assess chain formation [100].
For example, this approach demonstrated that cancer-associated mutations in the Mdm2 RING domain specifically impair its E3 ligase activity toward p53 without affecting its ability to bind either p53 or its E2 enzyme [100]. Similarly, in vitro assays with purified RBR E3 ligases (HOIL-1 and RNF216) have revealed their dependency on specific di-ubiquitin species for allosteric activation, with HOIL-1 activated by M1- and K63-linked di-Ub, while RNF216 is specifically activated by K63-linked di-Ub [18]. These assays provide direct biochemical evidence for linkage-specific defects resulting from disease mutations.
Cellular Validation Models
Cell-based models expressing wild-type versus mutant E3 ligases offer crucial physiological validation of linkage-specific defects. The experimental protocol typically involves: (1) generating E3 knockout cell lines using CRISPR/Cas9; (2) reconstituting with wild-type or disease-mutant E3 constructs; (3) stimulating pathway activation (e.g., DNA damage, mitochondrial stress); (4) monitoring substrate ubiquitination using linkage-specific antibodies; and (5) assessing functional readouts (e.g., cell viability, reporter assays) [99].
For RNF168-related studies, cells expressing RIDDLE syndrome-associated mutants show defective focus formation of 53BP1 and BRCA1 at DNA damage sites, specifically due to impaired K63-linked ubiquitination [99]. Similarly, patient-derived cells with Parkin mutations exhibit defective mitophagy and increased sensitivity to mitochondrial stress, correlating with specific ubiquitin linkage defects [95]. These cellular models provide critical functional validation of linkage-specific defects in physiologically relevant contexts.
Research Reagent Solutions for E3 Ligase Studies
Table 3: Essential Research Reagents for Investigating E3 Linkage Specificity
| Reagent Category | Specific Examples | Applications and Functions |
|---|---|---|
| Linkage-Specific Antibodies | Anti-K48-Ub, Anti-K63-Ub, Anti-M1-Ub, Anti-K11-Ub | Detection of specific ubiquitin linkages by immunoblotting, immunofluorescence |
| Activity-Based Probes | Ubiquitin vinyl sulfones, HA-Ub-VS | Profiling deubiquitinase activities, monitoring ubiquitin transfer |
| Stable Ubiquitin Mutants | K0-Ub (all lysines mutated to arginine), K48R-Ub, K63R-Ub | Defining linkage requirements, studying chain assembly mechanisms |
| Mass Spectrometry Standards | AQUA peptides, TMT-labeled ubiquitin peptides | Absolute quantification of ubiquitin linkages from complex samples |
| Recombinant E3 Proteins | Purified wild-type and mutant E3 ligases | In vitro ubiquitination assays, structural studies, screening |
| Specialized Cell Lines | E3 knockout lines, substrate reporter cells | Pathway validation, functional characterization of mutants |
The systematic analysis of disease-associated mutations in E3 ubiquitin ligases provides compelling validation for the biological significance of specific ubiquitin linkages in cellular homeostasis. The distinct mechanistic properties of RING, HECT, and RBR E3 families underlie their unique linkage specificities and functional specializations in physiological processes. Through integrated experimental approaches—including advanced mass spectrometry, in vitro biochemical assays, and cellular models—researchers can precisely map linkage-specific defects resulting from pathological E3 mutations. These insights not only enhance our understanding of disease mechanisms but also reveal novel therapeutic opportunities for targeting the ubiquitin system in various disorders.
Visual Guide: Experimental Workflow for Validating E3 Linkage Defects
The following diagram illustrates the integrated experimental approach for characterizing linkage-specific defects in disease-associated E3 ligase mutations:
Visual Guide: E3 Ligase Mechanisms and Linkage Specificity
The following diagram compares the catalytic mechanisms of major E3 ligase families and their relationship to ubiquitin linkage specificity:
The ubiquitin-proteasome system (UPS) is a central regulator of protein turnover and signaling in eukaryotic cells, with E3 ubiquitin ligases conferring substrate specificity and chain-type control [73]. As the key recognition components in this system, E3 ligases facilitate the transfer of ubiquitin from E2 conjugating enzymes to specific target proteins, marking them for proteasomal degradation or other functional modifications [11]. The therapeutic exploitation of these ligases has gained remarkable momentum with the emergence of proteolysis-targeting chimeras (PROTACs)—heterobifunctional molecules that recruit E3 ligases to target proteins of interest (POIs) for degradation [101] [102]. This technology represents a paradigm shift in drug discovery, enabling the targeting of proteins previously considered "undruggable" by conventional small-molecule approaches [103].
Despite the existence of over 600 E3 ligases in the human genome, current PROTAC development heavily relies on a very small subset, predominantly von Hippel-Lindau (VHL) and cereblon (CRBN) [101] [103]. This limited repertoire raises concerns regarding potential resistance mechanisms and restricts the scope of targetable proteins [103]. Expanding the diversity of utilized E3 ligases represents a critical frontier in the field, promising to enhance tissue specificity, circumvent resistance, and access challenging targets [103]. Within this context, understanding the fundamental mechanistic differences between the two major E3 ligase families—HECT and RING—becomes paramount for rational degrader design, particularly regarding their specificity for assembling atypical ubiquitin chains with non-degradative functions [36] [31].
E3 Ligase Families: HECT vs. RING Fundamental Mechanisms
E3 ubiquitin ligases are primarily categorized into two major families based on their structural features and catalytic mechanisms: RING (Really Interesting New Gene) and HECT (Homologous to E6AP C-Terminus) ligases [11]. A third class, RBR (RING-Between-RING) ligases, exhibits hybrid mechanisms but will not be the focus of this comparison [73].
Table 1: Fundamental Comparison of RING and HECT E3 Ligase Mechanisms
| Feature | RING E3 Ligases | HECT E3 Ligases |
|---|---|---|
| Catalytic Mechanism | Act as scaffolds facilitating direct Ub transfer from E2 to substrate [11] | Form catalytic intermediate with Ub via thioester bond at active-site cysteine [36] [11] |
| Ubiquitin Transfer | Direct transfer from E2~Ub to substrate [36] | Two-step transfer: E2 → E3 (transthiolation), then E3 → substrate [36] [31] |
| Representative Members | VHL, CRBN, MDM2, CBL [101] [11] | UBR5, NEDD4, Rsp5, Itch, HUWE1 [36] [31] |
| Linkage Specificity Determinant | Primarily determined by the cooperating E2 enzyme [36] | Inherent to the HECT domain C-lobe, particularly the last 60 amino acids [36] |
| Structural Functional Unit | Monomers, dimers, or multi-subunit complexes (e.g., CRLs) [11] | Primarily monomers or dimers (e.g., UBR5 functions as a dimer) [31] |
The following diagram illustrates the distinct catalytic cycles of RING and HECT E3 ligases, highlighting the direct versus two-step ubiquitin transfer mechanisms.
Chain Linkage Specificity: A Critical Functional Distinction
A pivotal functional distinction between HECT and RING E3 ligases lies in their control over ubiquitin chain linkage specificity, which determines the fate of the modified substrate [36]. K48-linked chains predominantly target substrates for proteasomal degradation, while K63-linked and other atypical chains (e.g., K11, K29, K33) are involved in non-proteolytic signaling processes, such as DNA repair, inflammation, and kinase activation [36] [31].
RING E3 Specificity: The linkage specificity for RING E3s is primarily determined by the cooperating E2 enzyme [36]. For instance, the Ubc13/Mms2 E2 complex specifically generates K63-linked chains, and RING E3s that partner with this complex (e.g., TRAF6) inherit this specificity [36]. The RING domain itself acts primarily as a scaffold, with the E2 controlling the chemistry of ubiquitin transfer [36] [11].
HECT E3 Specificity: In contrast, HECT E3s inherently dictate the type of polyubiquitin chain formed, with specificity determined by the C-terminal lobe (C-lobe) of the HECT domain [36]. Different HECT ligases demonstrate distinct preferences; for example, human E6AP preferentially catalyzes K48-linked chains, while Saccharomyces cerevisiae Rsp5 and human Itch favor K63-linked chains [36]. Recent structural studies of the human HECT E3 UBR5, which specifically generates K48-linked chains, reveal that an intricate web of interactions between the E3, the donor ubiquitin (UbD), and the acceptor ubiquitin (UbA) precisely positions UbA's K48 residue in the active site for chain formation [31].
Table 2: Experimentally Determined Linkage Specificities of Characterized HECT E3 Ligases
| HECT E3 Ligase | Preferred Linkage | Experimental Evidence | Biological Context/Implication |
|---|---|---|---|
| UBR5 | K48-linked [31] | Cryo-EM structures of intermediates; pulse-chase ubiquitin transfer assays [31] | Proteasomal degradation; roles in stem cell pluripotency and oncogenesis [31] |
| E6AP (UBE3A) | K48-linked [36] | In vitro ubiquitination assays with linkage-specific analysis [36] | Proteasomal degradation of p53 when hijacked by HPV E6; mutations cause Angelman syndrome [36] |
| Rsp5 | K63-linked [36] | In vitro and in vivo chain typing; mutagenesis studies [36] | Non-proteolytic functions in yeast; model for NEDD4-family ligases [36] |
| Itch/AIP4 | K63-linked (in vitro) [36] | In vitro ubiquitination assays [36] | Immune signaling and regulation; also reported to form K29 chains in vivo [36] |
| KIAA10 | K48 and K29-linked [36] | In vitro linkage analysis [36] | Dual specificity suggests diverse regulatory potential [36] |
Experimental Approaches for Profiling E3 Ligase Activity and Inhibition
Understanding E3 ligase mechanism and specificity requires a combination of biochemical, biophysical, and structural techniques. The following section outlines key experimental protocols used in the field.
In Vitro Autoubiquitination Assay (ELISA-based)
This assay measures the self-ubiquitination (autoubiquitination) activity of an E3 ligase, which is a common proxy for its catalytic function, and can be adapted to screen for inhibitors [104].
Protocol Details:
- Immobilization: His-tagged or GST-tagged HECT E3 ligase (e.g., WWP1 or WWP2 constructs) is immobilized on corresponding nickel-coated or glutathione plates for 1 hour [104].
- Reaction Setup: The immobilized E3 is incubated with a reaction mixture containing:
- E1 activating enzyme (e.g., GST-Uba1 or His-Uba1, 3-10 ng/well)
- E2 conjugating enzyme (e.g., UbcH7, 15-150 ng/well)
- FLAG-tagged ubiquitin (60 ng/well)
- ATP (1.25 mM) in reaction buffer (25 mM Tris pH 8.0, 100 mM NaCl, 4 mM MgCl2)
- The test compound (inhibitor) or DMSO control is added to the mixture [104].
- Incubation & Detection: The reaction proceeds for 2 hours. After washing, an anti-FLAG antibody conjugated to horseradish peroxidase (HRP) is added. Following a 1-hour incubation and further washes, a 3,3',5,5'-Tetramethylbenzidine (TMB) substrate is added. The resulting colorimetric signal, which is proportional to the amount of ubiquitin conjugated, is quantified by absorbance at 450 nm [104].
- Data Analysis: IC₅₀ values are calculated by fitting the inhibition data to a non-linear regression curve, with results standardized to 0% (no enzyme) and 100% (DMSO control) activity [104].
Pulse-Chase Ubiquitin Transfer Assay
This assay kinetically tracks the transfer of ubiquitin through the E3 catalytic cycle, providing mechanistic insight into the steps of catalysis [31].
Protocol Details:
- Pulse Phase: An E2~Ub thioester intermediate is pre-formed by incubating the E2 (e.g., UBE2D) with E1, ATP, and fluorescently labeled donor ubiquitin (UbD). A UbD K48R mutant can be used to prevent its use as an acceptor in chain formation [31].
- Chase Phase: The pre-formed E2~*UbD is added to the E3 ligase (e.g., UBR5) and unlabeled acceptor ubiquitin (UbA). The reaction is stopped at various time points [31].
- Visualization & Analysis: Reaction products are separated by SDS-PAGE, and the fluorescently labeled ubiquitin is visualized. The transfer of UbD from the E2 to the E3 (forming E3~UbD) and then to UbA (forming polyubiquitin chains) is tracked over time based on electrophoretic mobility [31]. This confirms the catalytic activity and processivity of the E3.
The workflow for these key assays, from protein preparation to data analysis, is summarized below.
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents and Materials for E3 Ligase Research
| Reagent / Material | Function in Research | Specific Examples / Notes |
|---|---|---|
| Truncated E3 Proteins | Simplify structural studies and in vitro assays by focusing on functional domains. | WWP1-L34H (WW domains + HECT) [104]; UBR5 dimer for cryo-EM studies [31]. |
| E1 Activating Enzymes | Essential component to activate ubiquitin to initiate the enzymatic cascade. | GST- or His-tagged Uba1 for in vitro assays [104]. |
| E2 Conjugating Enzymes | Partner with E3s; critical for determining RING E3 specificity. | UbcH7 (UBE2L3) for HECT E3s [104] [11]; UBE2D for UBR5 assays [31]. |
| Linkage-Specific Ubiquitin Mutants | Probe chain type specificity in assays. | Ubiquitin K48R mutant prevents K48-linked chain formation, used in pulse-chase [31]. |
| Cryo-Electron Microscopy | Visualize high-resolution structures of large E3 complexes and intermediates. | Used to solve structures of UBR5 dimer and ubiquitination intermediates [31]. |
| DNA-Encoded Libraries (DELs) | High-throughput discovery of novel E3 ligase binders/ligands. | Emerging technology to expand the ligandable space of E3s for PROTAC development [101]. |
Expanding the Therapeutic E3 Ligase Landscape
The heavy reliance of the current TPD landscape on VHL and CRBN has prompted intensive efforts to characterize and recruit novel E3 ligases. Systematic analyses have identified numerous promising candidates based on criteria such as confidence score, ligandability, expression pattern, and protein-protein interaction potential [103]. Among over 1,000 unique E3s compiled from databases, 76 have been highlighted as strong candidates for PROTAC engagement, vastly expanding the potential beyond the currently used ~1% [103].
Promising new E3s for TPD include:
- DCAF2: Recently harnessed as a novel E3 for TPD. Its frequent overexpression in various cancers presents an opportunity for tumor-targeted degraders [105].
- HECT Family Members (e.g., HUWE1): Despite challenges, their direct catalytic mechanism and diverse substrate recognition make them attractive targets. Small-molecule inhibitors against WWP1 and WWP2 have been identified, providing a potential starting point for future degrader development [104].
- RNF4: A RING E3 with a high confidence score and well-documented roles in the UPS, suggesting high potential for co-option [103].
This expansion is critical not only to access new target space but also to overcome resistance mechanisms such as E3 ligase downregulation or mutation, which have been observed in the context of CRBN-directed therapies [103].
The strategic exploitation of E3 ligases through PROTACs and small molecule inhibitors represents a transformative advance in therapeutic development. The fundamental mechanistic dichotomy between HECT and RING E3 ligases—with RINGs acting as E2-specifying scaffolds and HECTs as intrinsically specific catalysts—has profound implications for their roles in forming atypical ubiquitin chains and their potential as drug targets. While RING E3s like VHL and CRBN have pioneered the clinical application of PROTACs, the vast and underexplored landscape of E3s, particularly the HECT family, offers a rich resource for overcoming current limitations. Future progress will depend on continued mechanistic studies, the development of robust experimental protocols for profiling new E3s, and the innovative application of structural and screening technologies to unlock the full therapeutic potential of the human E3 ligase repertoire.
The ubiquitin-proteasome system represents a master regulatory network controlling virtually every cellular process in eukaryotic cells, with E3 ubiquitin ligases conferring specificity by targeting hundreds of substrate proteins for post-translational modification [106] [11]. Among the three major classes of E3 ligases—RING (Really Interesting New Gene), HECT (Homologous to E6AP C Terminus), and RBR (RING-between-RING)—the HECT family has emerged as a particularly promising therapeutic target due to its direct catalytic involvement in ubiquitin transfer and implication in numerous human pathologies [106] [37]. Unlike RING E3s which primarily function as scaffolds bringing E2∼Ub complexes in proximity to substrates, HECT E3s form an obligate thioester intermediate with ubiquitin via a conserved catalytic cysteine before transferring ubiquitin to substrate proteins [11] [33]. This unique catalytic mechanism, combined with the disease associations of various HECT family members, has fueled intensive efforts to develop specific HECT inhibitors despite considerable structural and mechanistic challenges [106] [37].
The 28 human HECT E3 ligases regulate diverse cellular signaling pathways and are critically implicated in conditions ranging from cancer and neurodevelopmental disorders to viral pathogenesis [106]. For instance, E6AP (UBE3A) is hijacked by human papillomavirus E6 protein to degrade tumor suppressor p53 in cervical carcinogenesis, while genetic alterations in E6AP are linked to Angelman syndrome and autism-spectrum disorders [106]. Similarly, SMURF1, NEDD4-1, WWP1, and other HECT ligases have been implicated in tumorigenesis, bone disorders, and pulmonary arterial hypertension [106] [37]. This review comprehensively compares the development, mechanistic basis, and experimental characterization of HECT-specific inhibitors, contextualizing these advances within the broader landscape of E3 ligase research and the specialized functions of HECT versus RING E3s in synthesizing atypical ubiquitin chains.
Comparative Mechanisms of HECT and RING E3 Ligases
Fundamental Catalytic Differences
HECT and RING E3 ligases employ fundamentally distinct mechanisms for ubiquitin transfer, with significant implications for inhibitor development strategies. RING E3s function as allosteric activators and scaffolds that position ubiquitin-loaded E2 enzymes (E2∼Ub) in close proximity to substrate proteins, facilitating direct ubiquitin transfer without forming a covalent E3-ubiquitin intermediate [11] [33]. In contrast, HECT E3s utilize a two-step catalytic mechanism involving: (1) transthiolation of ubiquitin from E2∼Ub to a conserved catalytic cysteine within the HECT domain, forming a HECT∼Ub thioester intermediate, followed by (2) transfer of ubiquitin to a lysine residue on the target substrate [106] [25]. This hybrid mechanism shares features with both RING and HECT-type catalysis, as RBR E3s employ a RING domain for E2∼Ub recruitment but then transfer ubiquitin to an intermediate catalytic cysteine before substrate modification [33].
Table 1: Fundamental Mechanistic Differences Between HECT and RING E3 Ligases
| Feature | HECT E3 Ligases | RING E3 Ligases |
|---|---|---|
| Catalytic Mechanism | Two-step mechanism with covalent E3∼Ub intermediate | Direct transfer from E2∼Ub to substrate |
| Ubiquitin Intermediate | Thioester-linked HECT∼Ub complex | No covalent E3-ubiquitin intermediate |
| Domain Architecture | Bilobal HECT domain (N-lobe, C-lobe, flexible hinge) | Zinc-binding RING domain |
| Structural Requirement | Conserved catalytic cysteine in C-lobe | Scaffold function without catalytic cysteine |
| E2∼Ub Conformation | Prefers "open" conformation for transthiolation | Stabilizes "closed" conformation for direct transfer |
| Chain Specificity Determinants | Primarily C-lobe with some N-lobe contributions | Primarily E2 enzyme with E3 modulation |
Structural Basis for Ubiquitin Chain Specificity
The bilobal architecture of HECT domains—consisting of an N-lobe that binds the E2∼Ub complex and a C-lobe containing the catalytic cysteine, connected by a flexible hinge—creates unique structural constraints that influence linkage specificity in ubiquitin chain formation [106] [11]. The C-lobe primarily determines linkage specificity, while the N-lobe contains an ubiquitin exosite that contributes to processive chain elongation by orienting the growing ubiquitin chain [106] [25]. This intrinsic linkage specificity varies among HECT family members: NEDD4 subfamily enzymes primarily synthesize Lys63-linked chains, E6AP generates Lys48-linked chains, and HUWE1 assembles Lys6-, Lys11-, and Lys48-linked chains [25]. In contrast, RING E3s largely depend on their cognate E2 enzymes for linkage specificity, though the RING domain can allosterically activate the E2 and influence chain type determination [11] [107].
The following diagram illustrates the comparative catalytic mechanisms of HECT and RING E3 ligases, highlighting their distinct approaches to ubiquitin transfer:
Development of HECT-Specific Inhibitors
Allosteric Inhibitors Targeting Conserved Structural Motifs
Recent advances in HECT inhibitor development have revealed allosteric targeting opportunities despite the absence of deep active-site pockets characteristic of traditional enzyme targets. A groundbreaking 2025 study discovered inhibitors binding a cryptic cavity in SMURF1 distant from the catalytic cysteine, which restrict essential catalytic motion by extending an α-helix over a conserved glycine hinge (G634) present in all HECT domains [37]. This glycine hinge facilitates the movement between N- and C-lobes necessary for ubiquitin transfer, and its inhibition represents a novel allosteric mechanism applicable across HECT family members. Structural analyses demonstrated that inhibitor binding elongates α-helix 10 by one and a half turns over the conserved glycine, shortening the hinge from 27.0 Å to 15.4 Å and replacing the flexible glycine with lysine (K637) at the hinge stem—an amino acid with lower tolerance for the ϕ/Ψ dihedral angles essential for hinge flexibility [37]. Leveraging this mechanistic understanding, researchers conducted an in silico machine-learning-based screen that successfully identified inhibitors of the prototypic HECT E3 ligase E6AP, confirming glycine-hinge-dependent allosteric activity across different HECT family members [37].
Table 2: Characterized Allosteric HECT Inhibitors and Their Properties
| Target | Compound | Mechanism | IC₅₀ / Kᵢ | Specificity | Cellular Activity |
|---|---|---|---|---|---|
| SMURF1 | Compound-8 (Cpd-8) | Glycine hinge allostery | Not specified | Selective over SMURF2 | Reversed PAH in animal models |
| SMURF1 | Piperidine sulfonamides | Glycine hinge allostery | Not specified | Selective over SMURF2 | Restored BMP signaling |
| NEDD4 | Norclomipramine analogs | Ub exosite competition | 0.12-2.05 µM | Selective within NEDD4 family | Inhibited polyubiquitination |
| E6AP | ML-identified compounds | Glycine hinge allostery | Not specified | Confirmed allosteric mechanism | In vitro confirmation |
Ubiquitin Exosite Targeting Strategies
The ubiquitin exosite—a non-covalent ubiquitin-binding pocket in the N-lobe of HECT domains—represents another attractive target for allosteric inhibition. Structural studies of NEDD4 in complex with Norclomipramine (compound 1), a tricyclic antidepressant identified as a HECT inhibitor, revealed binding to the hydrophobic pocket that overlaps with the ubiquitin exosite [108]. Norclomipramine specifically inhibits ubiquitin chain elongation without affecting the initial trans-thioesterification from E2 to E3 or ubiquitin dimer formation, behavior reminiscent of ubiquitin exosite mutants [108]. The compound's tricyclic moiety inserts deeply into the hydrophobic pocket normally occupied by the C-terminal tail of ubiquitin (residues 71-76), establishing additional interactions with residues M600, F637, and I638 while displacing E554 toward the solvent [108]. Building on this mechanism, researchers developed covalent inhibitors targeting cysteine C627 near the Norclomipramine binding pocket, with compound 15 (IC₅₀ = 0.69 µM) and optimized compound 32 (IC₅₀ = 0.12 µM) demonstrating potent inhibition of NEDD4-mediated polyubiquitination while sparing monoubiquitination events [108].
The experimental workflow below illustrates the comprehensive approach used to identify and characterize allosteric HECT inhibitors:
Experimental Approaches for HECT Inhibitor Characterization
Biochemical Assays for Inhibitor Evaluation
Robust biochemical assays form the foundation of HECT inhibitor characterization, with time-resolved fluorescence resonance energy transfer (TR-FRET)-based assays reporting E3 self-ubiquitylation proving particularly valuable for high-throughput screening [37]. The Ub-TR-FRET assay measures ubiquitin chain formation by monitoring FRET signal increases as chains extend using a mixture of fluorescently labeled and unlabeled monoUb, enabling quantitative evaluation of inhibitor potency during polyubiquitin chain elongation [108]. Additional biochemical approaches include single-turnover kinetic assays to assess effects on trans-thioesterification from E2 to E3, ubiquitin chain elongation assays distinguishing between diubiquitin formation and longer chain assembly, and competition assays evaluating inhibitor binding relative to ubiquitin or ubiquitin chains [108]. For linkage specificity assessment, Ub chain restriction analysis using linkage-specific deubiquitinases (DUBs) as "Ub chain restriction enzymes" reveals building blocks comprising distinct linkages and the predominant linkage types in assembly reactions [65].
Structural Biology Techniques
Structural insights have been instrumental in understanding HECT inhibitor mechanisms, with X-ray crystallography providing atomic-resolution views of inhibitor binding modes. Structural studies of SMURF1 and SMURF2 HECT domains with compound 8 revealed the allosteric glycine hinge mechanism, while structures of NEDD4 HECT domain with Norclomipramine elucidated ubiquitin exosite competition [37] [108]. Solution nuclear magnetic resonance (NMR) spectroscopy has complemented crystallographic approaches by characterizing conformational dynamics and detecting more subtle structural perturbations induced by inhibitor binding [107]. These structural techniques have revealed that HECT inhibitors typically induce one of three mechanistic outcomes: (1) restriction of lobe movement through glycine hinge stabilization, (2) competition with ubiquitin binding at the N-lobe exosite, or (3) direct blockage of the catalytic cysteine in the C-lobe.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for HECT Inhibitor Studies
| Reagent / Tool | Function / Application | Example Use Case |
|---|---|---|
| TR-FRET Ubiquitination Assays | High-throughput inhibitor screening | Primary screening of compound libraries [37] [108] |
| Linkage-Specific DUBs | Ubiquitin chain linkage analysis | OTUD3 (Lys6-specific), OTUB1 (Lys48-specific) [65] |
| E2~Ub Thioester Mimetics | Structural studies of E3-E2~Ub complexes | Isopeptide-linked UbcH7~Ub for crystallography [33] |
| Single-Lysine Ub Mutants | Determining linkage specificity | Ub K6R, K48R for chain type assessment [65] |
| HECT Domain Variants | Mechanistic validation | Glycine hinge mutants, catalytic cysteine mutants [37] [108] |
| Computational Screening Platforms | In silico inhibitor identification | Machine-learning approaches for allosteric inhibitors [37] |
The developing landscape of HECT-specific inhibitors reveals distinctive targeting strategies necessitated by the unique structural and mechanistic features of HECT E3 ligases compared to their RING counterparts. While RING E3 inhibitors often target protein-protein interfaces or leverage the linchpin residue to modulate E2∼Ub conformation [107], HECT inhibitor development has converged on allosteric mechanisms exploiting conserved structural motifs—particularly the flexible glycine hinge and ubiquitin exosite—that control catalytic domain movements essential for ubiquitin transfer [37] [108]. These advances highlight the value of structural biology and mechanistic biochemistry in revealing druggable sites beyond traditional active pockets, with glycine hinge targeting representing a particularly promising approach applicable across multiple HECT family members.
Future directions in HECT inhibitor development will likely focus on enhancing specificity within the HECT family, optimizing pharmacological properties for in vivo application, and exploring therapeutic combinations with other ubiquitin-system targeting agents. The successful application of SMURF1 inhibitors in reversing established pulmonary arterial hypertension in animal models demonstrates the therapeutic potential of this approach [37], while the progression of covalent NEDD4 inhibitors with favorable pharmacokinetic properties underscores the tractability of HECT ligases as drug targets [108]. As our understanding of HECT ligase biology and atypical ubiquitin chain signaling continues to evolve, particularly in contrast to RING E3 mechanisms, so too will opportunities for therapeutic intervention in the numerous diseases driven by HECT ligase dysregulation.
Cullin-RING ligases (CRLs) represent the largest family of E3 ubiquitin ligases, functioning as master regulators of protein turnover that control approximately 20% of cellular protein degradation via the ubiquitin-proteasome system (UPS) [109]. These multi-subunit complexes consist of a cullin scaffold protein, a RING finger protein (such as RBX1 or RBX2) that recruits E2 ubiquitin-conjugating enzymes, and a variable substrate recognition module [110]. The activity of CRLs is critically dependent on the post-translational modification of the cullin subunit with the ubiquitin-like protein NEDD8, a process known as neddylation [110] [111]. This modification activates CRLs by inducing conformational changes that facilitate ubiquitin transfer from E2 enzymes to substrate proteins [110].
MLN4924 (Pevonedistat) is a first-in-class, investigational small molecule inhibitor that specifically targets the NEDD8-activating enzyme (NAE), the E1 enzyme that initiates the neddylation cascade [110] [109]. By forming a covalent NEDD8-MLN4924 adduct, MLN4924 effectively blocks the neddylation of cullins, thereby inactivating the entire CRL family and causing the accumulation of CRL substrates that would normally be targeted for degradation [110]. With promising preclinical efficacy, MLN4924 has advanced to multiple Phase I/II/III clinical trials for various human malignancies, representing a novel targeted approach to cancer therapy that operates through a distinct mechanism from conventional proteasome inhibitors such as bortezomib [110] [109].
Molecular Mechanisms of MLN4924 Action
Direct Target and Biochemical Consequences
MLN4924 exerts its primary effects through specific inhibition of the NEDD8-activating enzyme (NAE), which consists of a heterodimer of NAE1 and UBA3 subunits [109]. The inhibitor functions as a mechanism-based analog of ATP that forms a covalent adduct with NEDD8, creating a dead-end complex that cannot proceed through the enzymatic cascade [110]. This inhibition prevents the transfer of NEDD8 to the E2 conjugating enzymes UBE2M (UBC12) or UBE2F, thereby blocking the subsequent neddylation of cullin proteins [110] [109].
The biochemical consequence of MLN4924 treatment is the rapid accumulation of CRL substrates that would normally be targeted for proteasomal degradation. Among the key substrates that accumulate are various cell cycle regulators (such as p21, p27, and CDT1), pro-apoptotic factors, and other regulatory proteins whose stabilization triggers multiple anti-proliferative responses in cancer cells [110] [112]. The selectivity of MLN4924 for neddylation over ubiquitination provides a therapeutic advantage, as it specifically targets a subset of cellular proteins regulated by CRLs, unlike general proteasome inhibitors that affect global protein degradation [110].
Signaling Pathways and Cellular Responses Modulated by MLN4924
Table 1: Key Signaling Pathways and Cellular Responses to MLN4924 Treatment
| Pathway/Response | Molecular Mechanism | Biological Outcome | Experimental Evidence |
|---|---|---|---|
| DNA Damage Response | Accumulation of DNA replication regulators (CDT1) causing re-replication | DNA damage, cell cycle arrest, senescence | Multiple cancer cell lines; in vivo models [110] |
| Apoptosis Induction | Stabilization of pro-apoptotic CRL substrates (e.g., NOXA, Bim); inhibition of NF-κB via βTrCP accumulation | Caspase-dependent apoptosis | Various human cancer cell lines; caspase processing assays [112] |
| Autophagy Induction | DEPTOR accumulation and HIF1α-REDD1-TSC1 axis activation inhibiting mTORC1 | Protective autophagy promoting cell survival | Multiple cancer lines; LC3-I/II conversion, p62 degradation assays [110] [111] |
| Metabolic Reprogramming | Altered PPAR signaling pathway; reduced lipid metabolites (glycerophosphocholine, arachidic acid, palmitic acid) | Disrupted energy metabolism in granulosa cells | Transcriptomic and metabolomic analyses in rabbit models [113] |
| Necroptosis | Enhanced TNFα-induced cell death when combined with TNFα; RIPK1/MLKL-dependent | Alternative cell death pathway when apoptosis is blocked | Multiple cancer cell lines with zVAD-fmk pretreatment [112] |
The following diagram illustrates the core molecular mechanism of MLN4924 action and its primary consequences on CRL function and downstream cellular processes:
Comparative Analysis of MLN4924 Effects Across Experimental Systems
Cell Line-Specific Responses to MLN4924 Treatment
Table 2: MLN4924 Efficacy Across Cancer Cell Lines and Experimental Models
| Cancer Cell Type | MLN4924 Concentration | Exposure Time | Primary Cellular Response | Key Molecular Markers |
|---|---|---|---|---|
| HeLa (Cervical) | 0.1-1 µM | 24 hours | Autophagy induction | EGFP-LC3 puncta formation, LC3-I to LC3-II conversion [110] |
| SK-BR3 (Breast) | 1 µM | 24 hours | Autophagy & apoptosis | Acridine orange staining, caspase processing [110] [112] |
| HCT116 (Colon) | 1 µM | 24 hours | Autophagy | LC3-I/II conversion, p62 degradation [110] |
| Jurkat A3 (Leukemia) | ≥4 µM | 24 hours | Apoptosis | Caspase processing, reduced viability [112] |
| Mino (B-cell lymphoma) | 0.8 µM | 24 hours | Apoptosis | Significant cell death, caspase activation [112] |
| Rabbit Granulosa Cells | Not specified | 48 hours | Metabolic dysregulation | Altered PPAR signaling, reduced lipid metabolites [113] |
| U87 (Glioblastoma) | 1 µM | 24 hours | Autophagy | Acridine orange staining, LC3-I/II conversion [110] |
Key Methodologies for Assessing MLN4924 Activity
Autophagy Assessment Protocols
The induction of autophagy by MLN4924 is typically demonstrated through multiple complementary techniques. For visualization of autophagosome formation, researchers employ fluorescence microscopy in cells expressing EGFP-LC3, where autophagy induction is quantified by counting EGFP-LC3 punctate structures per cell [110]. Alternatively, acridine orange staining is used to detect the acidic vesicular organelles characteristic of autophagic cells, with flow cytometry analysis providing quantitative assessment [110]. At the biochemical level, western blot analysis of LC3-I to LC3-II conversion and p62 (SQSTM1) degradation serve as well-established markers of autophagic flux, often complemented by use of bafilomycin A1 to block lysosomal degradation and confirm autophagic flux [110]. Transmission electron microscopy provides ultrastructural evidence of autophagosome formation, offering direct morphological confirmation of autophagy [110].
Apoptosis and Cell Death Analysis
For quantification of apoptosis, cell viability assays using MTT, MTS, or ATP-based measurements are performed following MLN4924 treatment, typically across a concentration range of 0.1-10 µM for 24-72 hours [112]. Caspase activation is assessed through western blot analysis of caspase-3, -8, and -9 processing, complemented by fluorogenic substrate cleavage assays [112]. To distinguish between apoptosis and necroptosis, specific inhibitors including zVAD-fmk (pan-caspase inhibitor), necrostatin-1 (RIPK1 inhibitor), and necrosulfonamide (MLKL inhibitor) are employed in combination studies [112]. For senescence assessment, β-galactosidase staining is routinely performed, coupled with analysis of senescence-associated secretory phenotype (SASP) factors [110].
mTOR Pathway Inhibition Assessment
The effect of MLN4924 on mTOR signaling is evaluated through western blot analysis of key phosphorylation sites, including mTOR (S2481), S6K1 (T389), and 4E-BP1 (S65/T70) [110]. Time-course experiments typically show mTORC1 inhibition beginning at 4-8 hours post-treatment, lasting up to 24 hours [110]. Direct mTORC1 kinase activity is measured using in vitro kinase assays with immunopurified mTORC1 complex and recombinant S6K1 as substrate [110]. The accumulation of DEPTOR, a natural mTOR inhibitor and CRL substrate, is confirmed through western blot and siRNA knockdown experiments to establish its contribution to mTOR inhibition [110].
The Double-Edged Effects: Anti-Cancer versus Pro-Cancer Consequences
While MLN4924 demonstrates significant anti-tumor activity across diverse cancer models, recent evidence has revealed a more complex picture of its biological effects, with potential pro-survival consequences that may limit therapeutic efficacy [109]. The dual nature of MLN4924 action stems primarily from the diverse functions of CRL substrates that accumulate upon neddylation inhibition.
On one hand, MLN4924 exerts potent anti-cancer effects through stabilization of tumor suppressor proteins and induction of lethal cellular responses. The accumulation of cell cycle regulators such as p21 and p27 induces cell cycle arrest, while DNA re-replication caused by CDT1 stabilization triggers DNA damage response and senescence [110] [109]. Additionally, stabilization of pro-apoptotic factors including NOXA, BIM, and TNF-related receptors promotes programmed cell death through both intrinsic and extrinsic apoptotic pathways [112].
Conversely, MLN4924 can also promote pro-survival signaling through the stabilization of specific oncogenic substrates. The induction of autophagy via DEPTOR accumulation and mTORC1 inhibition represents a key adaptive resistance mechanism, protecting cancer cells from MLN4924-induced apoptosis [110] [111]. This protective autophagy has been demonstrated in multiple cancer cell lines, where pharmacological or genetic inhibition of autophagy significantly enhances MLN4924-induced cell death [110]. Additionally, MLN4924-mediated accumulation of HIF-1α activates the HIF1-REDD1-TSC1 axis, further contributing to mTORC1 inhibition and promoting metabolic adaptations that may support tumor survival under stress conditions [110].
The following diagram illustrates the dual opposing pathways activated by MLN4924 treatment that determine ultimate cell fate:
Strategic Combination Therapies to Overcome Resistance
The dual nature of MLN4924's cellular effects has prompted investigation into rational combination strategies that enhance its anti-tumor efficacy while counteracting pro-survival responses. The most extensively studied approach combines MLN4924 with autophagy inhibitors, based on compelling evidence that MLN4924-induced autophagy serves as a cytoprotective mechanism [110] [111]. Genetic inhibition of autophagy through siRNA knockdown of essential autophagy genes (such as ATG5 or ATG7) or pharmacological inhibition using chloroquine derivatives significantly enhances MLN4924-induced apoptosis across multiple cancer cell lines [110]. This combination strategy effectively converts the cellular response from cytoprotective autophagy to accelerated apoptotic cell death.
Additional combination approaches leverage MLN4924's ability to sensitize cancer cells to other therapeutic agents. MLN4924 enhances TNFα-induced cell death by blocking NF-κB activation through accumulation of IκBα, the inhibitor of NF-κB signaling [112]. This effect is particularly pronounced in cells where apoptotic pathways are compromised, leading to necroptosis as an alternative cell death mechanism [112]. Furthermore, MLN4924 demonstrates synergistic effects with conventional chemotherapeutic agents, radiation therapy, and other targeted agents across various cancer models, highlighting its potential as a sensitizing agent in multi-modality treatment regimens [109] [112].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Investigating MLN4924 Mechanisms
| Reagent/Category | Specific Examples | Research Application | Mechanistic Insight |
|---|---|---|---|
| Cell Death Inhibitors | zVAD-fmk (pan-caspase inhibitor), Necrostatin-1 (RIPK1 inhibitor), Necrosulfonamide (MLKL inhibitor) | Distinguishing apoptosis from necroptosis | Confirmed MLN4924/TNF-induced necroptosis when apoptosis blocked [112] |
| Autophagy Modulators | Bafilomycin A1 (lysosomal acidification inhibitor), Chloroquine (autophagy flux inhibitor), siRNA against ATG5/ATG7 | Assessing autophagic flux and functional significance | Identified protective autophagy as resistance mechanism [110] |
| Metabolic Assays | Metabolomic profiling, Transcriptomic analysis, Seahorse extracellular flux analysis | Evaluating metabolic reprogramming | Revealed PPAR signaling disruption and reduced lipid metabolites [113] |
| Pathway Reporters | EGFP-LC3 transfection, Acridine orange staining, Antibodies for phospho-S6K1, phospho-4E-BP1, LC3-I/II, p62 | Monitoring autophagy and mTOR pathway inhibition | Demonstrated mTORC1-specific inhibition and autophagosome formation [110] |
| Genetic Tools | siRNA against DEPTOR, HIF1α, REDD1; CRISPR/Cas9 knockout cells | Establishing causal relationships in signaling pathways | Verified DEPTOR and HIF1-REDD1-TSC1 axis role in autophagy induction [110] |
MLN4924 represents a pioneering therapeutic agent that has fundamentally advanced our understanding of targeted protein degradation in cancer therapy. Its precise mechanism of action through selective inhibition of the neddylation pathway distinguishes it from broader proteasome inhibitors and offers valuable insights into the complex biology of CRL networks. The experimental evidence comprehensively demonstrates that MLN4924 exerts multi-faceted effects on cancer cells, including induction of apoptosis, senescence, and DNA damage, while simultaneously activating adaptive survival pathways such as protective autophagy.
The dual nature of MLN4924's cellular effects underscores the importance of context-dependent application and strategic combination therapies. Future research directions should focus on identifying predictive biomarkers that can stratify patients most likely to benefit from neddylation inhibition, optimizing combination regimens that exploit synthetic lethal interactions, and developing next-generation neddylation inhibitors with improved therapeutic indices. As our understanding of the nuanced functions of individual CRL complexes advances, more selective approaches targeting specific CRL components or substrate receptors may emerge, potentially mitigating the "double-edged" effects observed with pan-CRL inhibition. The extensive preclinical data summarized in this guide provides a robust foundation for the continued clinical translation of neddylation pathway inhibition as a promising anticancer strategy.
Ubiquitination, a crucial post-translational modification, regulates virtually every cellular process in eukaryotes, from protein degradation and DNA repair to immune signaling and neural development [90] [8]. The functional outcome of ubiquitination is largely determined by the topology of the ubiquitin chain—specifically, which of the seven lysine residues (K6, K11, K27, K29, K33, K48, K63) or the N-terminal methionine (M1) on ubiquitin is used to form polymeric chains [90]. Among the three-enzyme cascade (E1-E2-E3) that catalyzes ubiquitination, E3 ubiquitin ligases serve as the critical specificity determinants, responsible for both substrate recognition and often the linkage specificity of the ubiquitin chain formed [114] [90]. The ability to engineer E3 ligases with tailored linkage specificity represents a frontier in therapeutic development, offering potential for precision targeting of pathological proteins through technologies like PROTACs (Proteolysis-Targeting Chimeras) [84] [103].
This guide compares the inherent structural and mechanistic properties of the two major E3 ligase families—HECT-type and RING-type—in the context of engineering them for predefined linkage specificity, particularly for atypical, non-degradative ubiquitin chains. We provide objective performance comparisons, supporting experimental data, and detailed methodologies to inform research and drug development efforts.
Comparative Analysis of HECT and RING E3 Ligase Families
Fundamental Mechanisms and Architectural Diversity
E3 ubiquitin ligases are primarily classified by their structural domains and catalytic mechanisms. The table below summarizes the core characteristics of the major E3 families, with emphasis on HECT and RING types.
Table 1: Classification and Characteristics of Major E3 Ubiquitin Ligase Families
| E3 Family | Catalytic Mechanism | Representative Members | Key Structural Features | Linkage Specificity Potential |
|---|---|---|---|---|
| HECT-type | Two-step mechanism: Accepts Ub from E2 via catalytic cysteine, then transfers to substrate [90] [8]. | Ufd4, Nedd4 family, HERC family, HUWE1 [90] [39]. | C-terminal HECT domain with N-lobe and C-lobe; N-terminal substrate recognition domains (e.g., WW, C2, RLD) [90]. | High. Direct control over Ub orientation; demonstrated specificity for K29/K48-branched chains [39]. |
| RING-type | One-step mechanism: Acts as a scaffold to bring E2~Ub and substrate into proximity for direct transfer [90] [8]. | UBR4 (hemiRING), RNF220, Cullin-RING ligases (CRLs) [90] [8] [115]. | RING domain (cross-brace Zn²⁺ coordination) or U-box domain; Multi-subunit complexes common [90] [115]. | E2-Dependent. Primarily determined by the recruited E2 enzyme [90]. |
| RBR-type | Hybrid mechanism: RING1 domain binds E2, ubiquitin transferred to catalytic cysteine in RING2 domain, then to substrate (HECT-like) [8]. | HOIP (of LUBAC), Parkin [90]. | RING1 domain, In-Between-RING (IBR) domain, RING2 domain with catalytic cysteine [90]. | Emerging. Specific for M1-linear chains (HOIP) [90]. |
Structural Visualization of HECT and RING Mechanisms
The following diagrams illustrate the distinct catalytic mechanisms of HECT and RING E3 ligases, highlighting the key structural elements involved in ubiquitin transfer and linkage specification.
Experimental Approaches for Determining and Engineering Linkage Specificity
Key Experimental Protocols and Workflows
Cutting-edge research in E3 ligase engineering employs a multi-faceted approach, combining structural biology, biochemical reconstitution, and chemical biology. The workflow below outlines a general protocol for analyzing E3 linkage specificity, integrating methods from recent seminal studies.
Table 2: Key Research Reagent Solutions for E3 Ligase Engineering
| Reagent / Tool | Function / Application | Example Use Case | Reference |
|---|---|---|---|
| Ubiquitin Mutants (K-to-R) | Identify essential lysines for chain formation by blocking specific linkages. | Ufd4 assays with Ub-K29R revealed K29-linkage specificity [39]. | [39] |
| Disulfide Trapping / Crosslinking | Stabilize transient E2~Ub or E3~Ub intermediates for structural analysis. | Structure of UBE2E1 crosslinked to SETDB1-derived peptide [114]. | [114] |
| Chemical Ubiquitin Probes | Mimic specific ubiquitination states (e.g., branched chains) for mechanistic studies. | Trapped Ufd4~Ub complex with K29/K48-branched triUb probe for Cryo-EM [39]. | [39] |
| SUE1 (Sequence-dependent Ubiquitination using UBE2E1) | E3-free system to generate ubiquitinated proteins with custom sites/linkages. | Engineered E3-free ubiquitination using UBE2E1 and target peptide tags [114]. | [114] |
| Linkage-specific Antibodies | Detect and quantify specific ubiquitin chain types in assays and cell lysates. | Standard method for validating linkage specificity in polyubiquitination assays. | [90] |
Quantitative Comparison of Linkage Specificity
The linkage specificity of an E3 ligase is a quantitative trait. The table below summarizes experimental data on the linkage preferences of native and engineered E3s, highlighting their potential for therapeutic targeting.
Table 3: Experimentally Determined Linkage Specificity of E3 Ligases
| E3 Ligase | Family | Preferred Linkage(s) | Key Experimental Findings | Therapeutic Potential / Note |
|---|---|---|---|---|
| Ufd4 / TRIP12 | HECT | K29-linked on K48 chains (Branched) | ~5.2-fold higher efficiency (kcat/Km) for proximal K29 vs. distal K29 on K48-diUb [39]. | Enhanced degradation signal; implicated in N-end rule pathway [39]. |
| UBR4 | RING (hemiRING) | Specificity determined by UBE2A/B | Autoubiquitination exclusively with UBE2A/UBE2B; high intrinsic lysine reactivity of E2 [115]. | Neuronal N-degron pathway; role in neurodevelopment and cancer [115]. |
| UBE2E1 | E2 (E3-independent) | Monoubiquitination (on specific peptide tag) | Enables SUE1 strategy for E3-free, site-specific ubiquitination [114]. | Protein engineering tool; demonstrates feasibility of re-wiring ubiquitination. |
| LUBAC (HOIP) | RBR | M1-linear (Linear) | Unique mechanism for generating linear ubiquitin chains involved in NF-κB signaling [90]. | Target for immune modulation. |
| APC/C with UBE2S | RING (Multi-subunit) | K11-linked on K48 chains (Branched) | Assembles K11/K48-branched chains to enhance proteasomal recognition of Nek2A [39]. | Cell cycle regulation; cancer relevance. |
Engineering Strategies and Therapeutic Applications
Engineering E3 Ligases for Tailored Specificity
Research has revealed several successful strategies for engineering linkage specificity, targeting different components of the ubiquitination machinery:
E3 Ligase Engineering: Structure-guided mutations can alter linkage specificity. A pivotal study on UBE2E1, which naturally performs E3-independent ubiquitination, revealed that its specific recognition of a substrate-derived hexapeptide ("KEGYES") in an L-shaped conformation is key [114]. Mutating key residues in the E2 (e.g., creating Ubch5c-P121G/E122S) can confer E3-free ubiquitination capability, demonstrating the potential for re-engineering the E2-E3 interface [114].
Substrate Targeting via Fusion Tags: The SUE1 system exploits the natural E3-independent activity of UBE2E1. By fusing the "KEGYES" peptide (or an optimized version, "KEGYEE") to a protein of interest, researchers can direct site-specific monoubiquitination or, when combined with Ub mutants, create defined polyubiquitin chains without a canonical E3 ligase [114]. This provides a highly flexible tool for generating homogenously ubiquitinated proteins for functional studies.
Exploiting E2-E3 Pairing Specificity: For RING E3s, specificity is often dictated by the recruited E2. The recent characterization of UBR4's atypical "hemiRING" domain, which specifically recruits UBE2A and UBE2B, underscores this point [115]. The intrinsically high lysine reactivity of UBE2A complements the hemiRING's modest allosteric activation, creating a specific and efficient module for ubiquitin transfer. Engineering efforts can focus on hijacking these specific E2-E3 pairs for therapeutic purposes.
Application in Targeted Protein Degradation (PROTACs)
The field of PROTACs has immense interest in expanding the repertoire of E3 ligases used [84] [103]. Currently, less than 2% of the ~600 human E3s are utilized in PROTAC development, with a heavy reliance on CRBN and VHL [103]. Engineering E3s for tailored linkage specificity can address several key challenges:
- Expanding the PROTACtable Genome: Systematic analyses have identified 76 E3 ligases as promising candidates for PROTAC engagement based on confidence score, ligandability, expression pattern, and protein-protein interactions [103]. Examples include HUWE1 and FBXO7, which have high confidence scores similar to clinically used E3s but remain underexplored.
- Tissue-Specific Targeting: Selecting E3s with tissue-enriched expression (e.g., neural-specific E3s for neurological disorders) can minimize on-target, off-tissue toxicities [84] [103]. For instance, DT2216, a BCL-XL-targeting PROTAC, leverages the low expression of VHL in platelets to reduce thrombocytopenia, a common side effect of BCL-XL inhibition [103].
- Overcoming Resistance: Cancer cells can develop resistance to CRBN-based PROTACs through mutations in the CRBN gene. Having a diverse toolbox of engineered E3s with distinct linkage specificities provides alternative degradation pathways to circumvent such resistance mechanisms [103].
The strategic engineering of E3 ligases for tailored linkage specificity is a transformative frontier in biomedical science. The comparative analysis presented here reveals that while HECT-type E3s offer a more direct path for engineering specific chain topologies due to their central catalytic role, RING-type E3s provide a modular system where specificity can be manipulated through E2 partnership and complex assembly. The integration of high-resolution structural insights (e.g., from Cryo-EM of trapped intermediates [39] [115]) with robust biochemical assays and novel chemical tools [114] is paving the way for rational design.
The future of this field lies in leveraging these engineering strategies to expand the therapeutic reach of targeted protein degradation. By moving beyond the currently narrow set of E3 ligases used in PROTACs and precisely controlling the ubiquitin signal deposited on target proteins, researchers and drug developers can create more effective, specific, and safer therapeutics for a wide range of diseases, including cancer, neurodegenerative disorders, and immune pathologies [84] [103].
Conclusion
The distinct catalytic mechanisms of HECT and RING E3 ligases underpin their specific roles in generating the atypical ubiquitin code. HECT E3s intrinsically determine linkage specificity through their C-lobe, often producing K63-linked (NEDD4 family) or K48-linked (e.g., UBR5, E6AP) chains. In contrast, RING E3s often rely on specific E2 partnerships to dictate chain topology, though some multi-subunit complexes exhibit strong preferences for atypical linkages. This mechanistic understanding, combined with advanced structural and methodological tools, is paving the way for novel therapeutic strategies. The future of E3-targeted drug discovery lies in exploiting these specificity profiles to develop next-generation PROTACs, molecular glues, and specific inhibitors that can precisely manipulate ubiquitin signaling in cancer, neurodegenerative, and inflammatory diseases.
References
- 1. Ubiquitin proteasome system in immune regulation and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10163937/]
- 2. Ubiquitin-Proteasome System [https://lifesensors.com/ubiquitinproteasomesystem/]
- 3. The ubiquitin-proteasome pathway: The complexity and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC34259/]
- 4. Structure and Function of HECT E3 Ubiquitin Ligases and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7500122/]
- 5. Protein Degradation using the Ubiquitin-Proteasome ... [https://www.thermofisher.com/us/en/home/life-science/cell-analysis/cell-analysis-learning-center/protein-degradation-resource-center/protein-degradation-using-ubiquitin-proteasome-pathway.html]
- 6. BioE3 identifies specific substrates of ubiquitin E3 ligases [https://www.nature.com/articles/s41467-023-43326-8]
- 7. Tuning ubiquitin transfer by RING E3 ubiquitin ligases through ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12326332/]
- 8. Emerging roles for E3 ubiquitin ligases in neural development ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12149146/]
- 9. Mechanism of ubiquitin ligation and lysine prioritization by ... [https://elifesciences.org/articles/00828]
- 10. TRIP12 structures reveal HECT E3 formation of K29 ... [https://www.nature.com/articles/s41594-025-01561-1]
- 11. HECT and RING finger families of E3 ubiquitin ligases at a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3381717/]
- 12. Exploring the “Other” subfamily of HECT E3-ligases for ... [https://www.sciencedirect.com/science/article/abs/pii/S0163725821000103]
- 13. Decoding the Messaging of the Ubiquitin System Using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7611516/]
- 14. Structural ubiquitin contributes to K48 linkage specificity of ... [https://www.sciencedirect.com/science/article/pii/S2211124725004590]
- 15. Mechanism of ubiquitin chain synthesis employed by a ... [https://www.sciencedirect.com/science/article/pii/S0021925820395855]
- 16. Structural mechanisms of autoinhibition and substrate ... [https://www.nature.com/articles/s41594-023-01203-4]
- 17. RING-Between-RING E3s ligases: Emerging themes amid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5675740/]
- 18. The unifying catalytic mechanism of the RING-between- ... [https://www.nature.com/articles/s41467-023-35871-z]
- 19. RING-type E3 ligases: Master manipulators of E2 ubiquitin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4109693/]
- 20. Mechanistic insight into the allosteric activation of a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1316884/]
- 21. Atypical ubiquitin chains: new molecular signals. 'Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2427391/]
- 22. The Role of Atypical Ubiquitin Chains in the Regulation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6987411/]
- 23. Beyond K48 and K63: non-canonical protein ubiquitination [https://cmbl.biomedcentral.com/articles/10.1186/s11658-020-00245-6]
- 24. Targeting E3 ubiquitin ligases and their adaptors as a ... [https://www.nature.com/articles/s12276-023-01087-w]
- 25. HECT E3 Ligases: A Tale With Multiple Facets [https://www.frontiersin.org/journals/physiology/articles/10.3389/fphys.2019.00370/full]
- 26. RING domain E3 ubiquitin ligases - PubMed - NIH [https://pubmed.ncbi.nlm.nih.gov/19489725/]
- 27. The K48-K63 Branched Ubiquitin Chain Regulates NF-κB ... [https://www.sciencedirect.com/science/article/pii/S1097276516305639]
- 28. Structural basis of generic versus specific E2–RING E3 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6739811/]
- 29. E2 enzymes: more than just middle men | Cell Research [https://www.nature.com/articles/cr201635]
- 30. Analysis of ubiquitin recognition by the HECT ligase E6AP ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6463701/]
- 31. Structural snapshots along K48-linked ubiquitin chain ... [https://www.nature.com/articles/s41589-023-01414-2]
- 32. Sequence Determinants of E2-E6AP Binding Affinity and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1945100/]
- 33. Structural insights into the mechanism and E2 specificity of ... [https://www.nature.com/articles/s41467-017-00272-6]
- 34. Structural insights into a HECT-type E3 ligase AREL1 and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6937569/]
- 35. Exploring the Roles of HERC2 and the NEDD4L HECT E3 ... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2021.659049/full]
- 36. Polyubiquitination by HECT E3s and the Determinants of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2698738/]
- 37. Therapeutic potential of allosteric HECT E3 ligase inhibition [https://pmc.ncbi.nlm.nih.gov/articles/PMC12087876/]
- 38. Comparative analysis of the catalytic regulation of NEDD4 ... [https://www.sciencedirect.com/science/article/pii/S0021925820307365]
- 39. Structural visualization of HECT-type E3 ligase Ufd4 ... [https://www.nature.com/articles/s41467-025-59569-6]
- 40. Mechanism of ubiquitin chain synthesis employed by a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5481553/]
- 41. Building and remodelling Cullin–RING E3 ubiquitin ligases [https://pmc.ncbi.nlm.nih.gov/articles/PMC3849489/]
- 42. Frontiers | Cullin Ring Ubiquitin Ligases ( CRLs ) in Cancer: Responses... [https://www.frontiersin.org/journals/physiology/articles/10.3389/fphys.2019.01144/full]
- 43. Structural regulation of cullin-RING ubiquitin ligase ... [https://www.sciencedirect.com/science/article/abs/pii/S0959440X11000054]
- 44. The RING finger E3 ligase RNF25 protects DNA replication ... [https://www.nature.com/articles/s41467-025-62368-8]
- 45. The essential role of E3 ubiquitin ligases in ... [https://www.sciencedirect.com/science/article/abs/pii/S0006291X25005121]
- 46. Emerging roles for E3 ubiquitin ligases in neural ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2025.1557653/full]
- 47. Dual-color pulse-chase ubiquitination assays to ... [https://pubmed.ncbi.nlm.nih.gov/30850057/]
- 48. Activation of a Primed RING E3-E2–Ubiquitin Complex by ... [https://www.sciencedirect.com/science/article/pii/S1097276515001318]
- 49. Identification and characterization of a ubiquitin E3 RING ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12591485/]
- 50. How cryo‐electron microscopy and X‐ray crystallography ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5192981/]
- 51. Cryo Electron Microscopy: Principle, Strengths, Limitations ... [https://www.technologynetworks.com/analysis/articles/cryo-electron-microscopy-principle-strengths-limitations-and-applications-377080]
- 52. X-ray Crystallography vs Cryo-EM: Which is Best for Your ... [https://www.creative-biostructure.com/resource-cryo-em-vs-xray-crystallography-comparison.htm?srsltid=AfmBOopNu43KD2PCq892sUx_4UbOObXR6Y7zunYmhGhrKXEM0dIKsTDO]
- 53. How cryo-EM has shed light on Cullin RING E3 ligase ... [https://www.sciencedirect.com/science/article/pii/S0959440X25000739]
- 54. Current methodologies in protein ubiquitination ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9373315/]
- 55. substrate recognition and catalysis by ubiquitin ligases [https://www.sciencedirect.com/science/article/pii/S0962892400018341]
- 56. Ring finger protein 31–mediated atypical ubiquitination ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6311505/]
- 57. Current methodologies in protein ubiquitination characterization [https://cellandbioscience.biomedcentral.com/articles/10.1186/s13578-022-00870-y]
- 58. Simple and robust high-throughput serum proteomics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11579186/]
- 59. Chemical Tools for Probing the Ub/Ubl Conjugation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11727022/]
- 60. TRIP12 structures reveal HECT E3 formation of K29 linkages ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12440805/]
- 61. A substrate-trapping strategy to find E3 ubiquitin ligase ... [https://www.nature.com/articles/s42003-020-01328-y]
- 62. A cell-based high-throughput screening method ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6393603/]
- 63. Analysis of ubiquitin E3 ligase activity using selective ... [https://www.sciencedirect.com/science/article/pii/S016748891200170X]
- 64. HTRF Human XIAP / BIR2 Binding Kit, 500 Assay Points [https://www.revvity.com/product/htrf-human-xiap-bir2-bind-kit-500-pts-64xiapb2peg]
- 65. Assembly, analysis and architecture of atypical ubiquitin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4176834/]
- 66. Structure of the human UBR5 E3 ubiquitin ligase - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10403316/]
- 67. Cryo‐EM structure of the chain‐elongating E3 ubiquitin ... [https://link.springer.com/article/10.15252/embj.2022113348]
- 68. Mechanism of Lysine 48-Linked Ubiquitin-Chain Synthesis ... [https://www.sciencedirect.com/science/article/pii/S0092867405010391]
- 69. Structural basis of K11/K48-branched ubiquitin chain ... [https://www.nature.com/articles/s41467-025-64719-x]
- 70. Understanding Cullin-RING E3 Biology through Proteomics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3518111/]
- 71. Structural mechanisms of autoinhibition and substrate recognition by... [https://www.nature.com/articles/s41594-023-01203-4?error=cookies_not_supported&code=680183c9-46da-4b4c-85b8-2c0afbba53e7]
- 72. Structural insights into the mechanism and E of the RBR... 2 specificity [https://pubmed.ncbi.nlm.nih.gov/28790309/]
- 73. E3 ubiquitin ligases in signaling, disease, and therapeutics [https://www.sciencedirect.com/science/article/pii/S0968000425001860]
- 74. Identification of ligands for E3 ligases with restricted ... [https://pubs.rsc.org/en/content/articlehtml/2025/cb/d5cb00198f]
- 75. Combinatorial mapping of E3 ubiquitin ligases to their ... [https://www.sciencedirect.com/science/article/pii/S1097276525000516]
- 76. Structural basis for transthiolation intermediates in the ... [https://www.nature.com/articles/s41586-024-07828-9]
- 77. Strategies to trap enzyme-substrate complexes that mimic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6390966/]
- 78. A multi-lock inhibitory mechanism for fine-tuning enzyme ... [https://www.nature.com/articles/s41467-019-11224-7]
- 79. Regulating the human HECT E3 ligases - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6063350/]
- 80. In vitro autoubiquitination activity of E3 ubiquitin ligases ... [https://www.sciencedirect.com/science/article/abs/pii/S0076687923000812]
- 81. Activation of the E3 ligase function of the BRCA1/BARD1 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC139111/]
- 82. Building ubiquitin chains: E2 enzymes at work - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3107738/]
- 83. E2 enzymes: more than just middle men [https://pmc.ncbi.nlm.nih.gov/articles/PMC4822130/]
- 84. E3 ubiquitin ligases and their therapeutic potential in ... [https://www.sciencedirect.com/science/article/abs/pii/S0006295225001376]
- 85. Emerging functions of branched ubiquitin chains [https://www.nature.com/articles/s41421-020-00237-y]
- 86. Emerging tools and methods to study cell signalling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12224918/]
- 87. Structural Diversity of Ubiquitin E3 Ligase [https://www.mdpi.com/1420-3049/26/21/6682]
- 88. Ubiquitination, E3 Ligases and Drug Discovery - Novel ... [https://www.ddw-online.com/ubiquitination-e3-ligases-and-drug-discovery-novel-technologies-for-a-challenging-pathway-757-201208/]
- 89. Challenges and opportunities in the purification of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7125906/]
- 90. E3 ubiquitin ligases: styles, structures and functions - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8607428/]
- 91. Ubiquitin ligase [https://en.wikipedia.org/wiki/Ubiquitin_ligase]
- 92. Ubiquitin chain specificities of E6AP E3 ligase and its ... [https://www.sciencedirect.com/science/article/abs/pii/S0006291X17324701]
- 93. The Role of Protein Ubiquitination in the Onset and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12248740/]
- 94. The RING finger protein family in health and disease [https://www.nature.com/articles/s41392-022-01152-2]
- 95. A Comprehensive Atlas of E3 Ubiquitin Ligase Mutations in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5817383/]
- 96. Ubiquitin modifications | Cell Research [https://www.nature.com/articles/cr201639]
- 97. RING-type E3 ligases: Master manipulators of E2 ubiquitin ... [https://www.sciencedirect.com/science/article/pii/S0167488913002164]
- 98. Enrichment of rare variants in E3 ubiquitin ligase genes ... [https://www.sciencedirect.com/science/article/abs/pii/S0197458021002670]
- 99. Ubiquitin E3 ligases in cancer: somatic mutation and ... [https://www.bmbreports.org/view.html?uid=1819&vmd=Full]
- 100. Structural and Functional Comparison of the RING Domains of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3039342/]
- 101. E3 Ligase Ligands for PROTACs: How They Were Found ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8013866/]
- 102. PROTAC: a revolutionary technology propelling small ... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2025.1676414/full]
- 103. Expanding PROTACtable genome universe of E3 ligases [https://www.nature.com/articles/s41467-023-42233-2]
- 104. Expanding the inhibitor space of the WWP1 and WWP2 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11373361/]
- 105. Study identifies new E3 ligase for targeted protein ... [https://www.ddw-online.com/study-identifies-new-e3-ligase-for-targeted-protein-degradation-37439-202510/]
- 106. Developing Small‐Molecule Inhibitors of HECT‐Type ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6471174/]
- 107. Tuning ubiquitin transfer by RING E3 ubiquitin ligases ... [https://www.life-science-alliance.org/content/8/10/e202503394]
- 108. Structure-based design of potent and selective inhibitors ... [https://www.nature.com/articles/s42004-025-01557-4]
- 109. The Double-Edged Effects of MLN4924: Rethinking Anti ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11274557/]
- 110. Targeting Cullin-RING ligases by MLN4924 induces ... [https://www.nature.com/articles/cddis2012125]
- 111. RING E3 ligase by the NEDD8-activating enzyme inhibitor ... [https://pubmed.ncbi.nlm.nih.gov/22874562/]
- 112. The NEDD8-activating enzyme inhibition with MLN4924 ... [https://www.sciencedirect.com/science/article/abs/pii/S0003986120305221]
- 113. Integrated analysis reveals the regulatory mechanism of the ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-024-10118-3]
- 114. Structure-guided engineering enables E3 ligase-free and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10858943/]
- 115. UBE2A and UBE2B are recruited by an atypical E3 ligase ... [https://www.nature.com/articles/s41594-023-01192-4]