In Vivo vs. In Vitro Ubiquitination Detection: A Comprehensive Guide for Researchers
This article provides a detailed comparison of in vivo and in vitro ubiquitination detection techniques, tailored for researchers and drug development professionals.
In Vivo vs. In Vitro Ubiquitination Detection: A Comprehensive Guide for Researchers
Abstract
This article provides a detailed comparison of in vivo and in vitro ubiquitination detection techniques, tailored for researchers and drug development professionals. It covers the foundational principles of the ubiquitin-proteasome system, explores established and emerging methodologies like immunoblotting and TUBE-based assays, and offers practical troubleshooting guidance. A critical validation framework is presented to help scientists select the optimal approach for their specific research context, whether studying fundamental biology or developing targeted protein degradation therapies like PROTACs.
Understanding the Ubiquitin Landscape: From Basic Biology to Technical Challenges
The Ubiquitin-Proteasome System (UPS) is the primary pathway for targeted protein degradation in eukaryotic cells, a sophisticated mechanism essential for maintaining cellular homeostasis [1] [2]. This system regulates countless cellular processes, including cell cycle progression, gene expression, responses to oxidative stress, and the removal of damaged or misfolded proteins [2] [3]. The UPS operates through a two-step mechanism: first, proteins destined for degradation are covalently tagged with a ubiquitin chain; second, these tagged proteins are recognized and broken down by the proteasome complex [2]. The discovery of this system was so fundamentally important that it was acknowledged with the award of the 2004 Nobel Prize in Chemistry to Aaron Ciechanover, Avram Hershko, and Irwin Rose [2].
Central to the UPS is ubiquitin, a small, highly conserved protein of 76 amino acids that is universally expressed in eukaryotic cells [1] [4]. Ubiquitin serves as a molecular tag when attached to substrate proteins. This attachment occurs through a sequential enzymatic cascade involving three key enzyme classes: ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3) [1] [4]. The specificity and diversity of ubiquitin signaling are further enhanced by the ability of ubiquitin itself to form polymers (polyubiquitin chains) through any of its seven lysine residues (K6, K11, K27, K29, K33, K48, K63) or its N-terminal methionine (M1) [1] [4]. Different chain linkages create distinct molecular signals, with K48-linked chains being the primary signal for proteasomal degradation, while other linkages (e.g., K63-linked) often regulate non-proteolytic functions like DNA repair and protein trafficking [1] [4] [5].
The Enzymatic Cascade: E1, E2, and E3
The process of ubiquitination is mediated by a well-defined cascade of three enzymes that work in concert to attach ubiquitin to specific substrate proteins.
E1: Ubiquitin-Activating Enzymes
The ubiquitination process initiates with the E1 enzyme, known as the ubiquitin-activating enzyme. The human genome encodes only a few E1 enzymes, highlighting their broad specificity [4] [6]. E1 catalyzes the ATP-dependent activation of ubiquitin. This reaction forms a high-energy thioester bond between the C-terminal glycine (G76) of ubiquitin and a specific cysteine residue in the E1's active site [1] [4]. This initial activation step is ATP-dependent and commits ubiquitin to the conjugation pathway. As the first step in the cascade, E1 regulates all downstream ubiquitination, making it a potential point for therapeutic intervention [5].
E2: Ubiquitin-Conjugating Enzymes
The activated ubiquitin is subsequently transferred from E1 to the active site cysteine of an E2 enzyme (ubiquitin-conjugating enzyme), again via a thioester bond [1] [4]. The human genome encodes more E2s than E1s—over 35 distinct E2s—allowing for greater functional diversification [4] [6]. E2s are not merely passive carriers; they play a critical role in determining the type of ubiquitin chain that will be assembled. For instance, UBE2D3 is an E2 with promiscuous activity that supports the ubiquitination of diverse targets, including ribosomal proteins RPS10 and RPS20, thereby playing a role in protein quality control [7]. The E2 carries the charged ubiquitin to the final enzyme in the cascade, the E3 ligase.
E3: Ubiquitin Ligases
The E3 ubiquitin ligase is responsible for the paramount task of conferring substrate specificity. It simultaneously binds to the E2~ubiquitin complex and a specific target protein, facilitating the direct or indirect transfer of ubiquitin from the E2 to a lysine residue on the substrate [1] [4]. The human genome encodes a vast repertoire of over 600 E3 ligases, which are classified into several families based on their structure and mechanism [4] [6]. The major families include:
- RING (Really Interesting New Gene): These E3s act as scaffolds, bringing the E2~Ub and substrate into close proximity to facilitate direct ubiquitin transfer from the E2 to the substrate [4].
- HECT (Homologous to the E6AP C Terminus): These E3s form a transient thioester intermediate with ubiquitin before transferring it to the substrate [4].
- RBR (RING-Between-RING): These E3s employ a hybrid mechanism, combining aspects of both RING and HECT types [4].
This vast array of E3s allows the UPS to recognize and regulate an immense number of specific protein substrates with high precision.
Table 1: Core Enzymes of the Ubiquitin-Proteasome System
| Enzyme | Number in Humans | Primary Function | Key Mechanism |
|---|---|---|---|
| E1 (Activating) | 2 [6] | Ubiquitin activation | ATP-dependent formation of E1~Ub thioester |
| E2 (Conjugating) | >35 [4] [6] | Ubiquitin carriage | Thioester-linked E2~Ub intermediate |
| E3 (Ligase) | >600 [4] [6] | Substrate recognition | Binds E2~Ub and substrate to facilitate transfer |
The following diagram illustrates the sequential actions of E1, E2, and E3 enzymes in the ubiquitination cascade:
The Proteasome: Structure and Function
The 26S proteasome is the terminal effector of the UPS, responsible for the actual degradation of ubiquitinated proteins. It is a massive, multi-subunit complex with a molecular mass of approximately 2000 kDa [2]. Its structure can be divided into two main components:
The 20S Core Particle (CP)
The 20S core particle is the catalytic heart of the proteasome. It is a barrel-shaped structure composed of four stacked heptameric rings [2] [3]. The two outer rings are made of seven α-subunits each, which function as a gated channel, controlling access to the interior. The two inner rings are composed of seven β-subunits each, which contain the proteolytic active sites. These sites exhibit three distinct cleavage specificities: chymotrypsin-like, trypsin-like, and caspase-like activities, which work in concert to cleave substrate proteins into short peptides [2] [3].
The 19S Regulatory Particle (RP)
The 19S regulatory particle is a cap complex that associates with one or both ends of the 20S core to form the 26S proteasome. The 19S RP is responsible for several critical functions: it recognizes polyubiquitinated proteins, removes the ubiquitin tag for recycling via deubiquitinating enzymes (DUBs), unfolds the target protein in an ATP-dependent manner, and translocates the unfolded polypeptide into the 20S core for degradation [2] [3].
Specialized variants of the proteasome also exist, such as the immunoproteasome, which is induced by inflammatory signals and optimizes the generation of antigenic peptides for immune presentation [3].
Table 2: Proteasome Structure and Key Functions
| Component | Subunits | Key Functions |
|---|---|---|
| 20S Core Particle (CP) | 28 subunits (4 rings of 7) | Proteolytic degradation; contains trypsin-, chymotrypsin-, and caspase-like active sites [2] [3] |
| 19S Regulatory Particle (RP) | ~20 subunits (base and lid) | Substrate recognition, deubiquitination, unfolding, and translocation into the CP [2] [3] |
| Immunoproteasome | Alternative catalytic subunits | Enhanced production of peptides for MHC class I antigen presentation [3] |
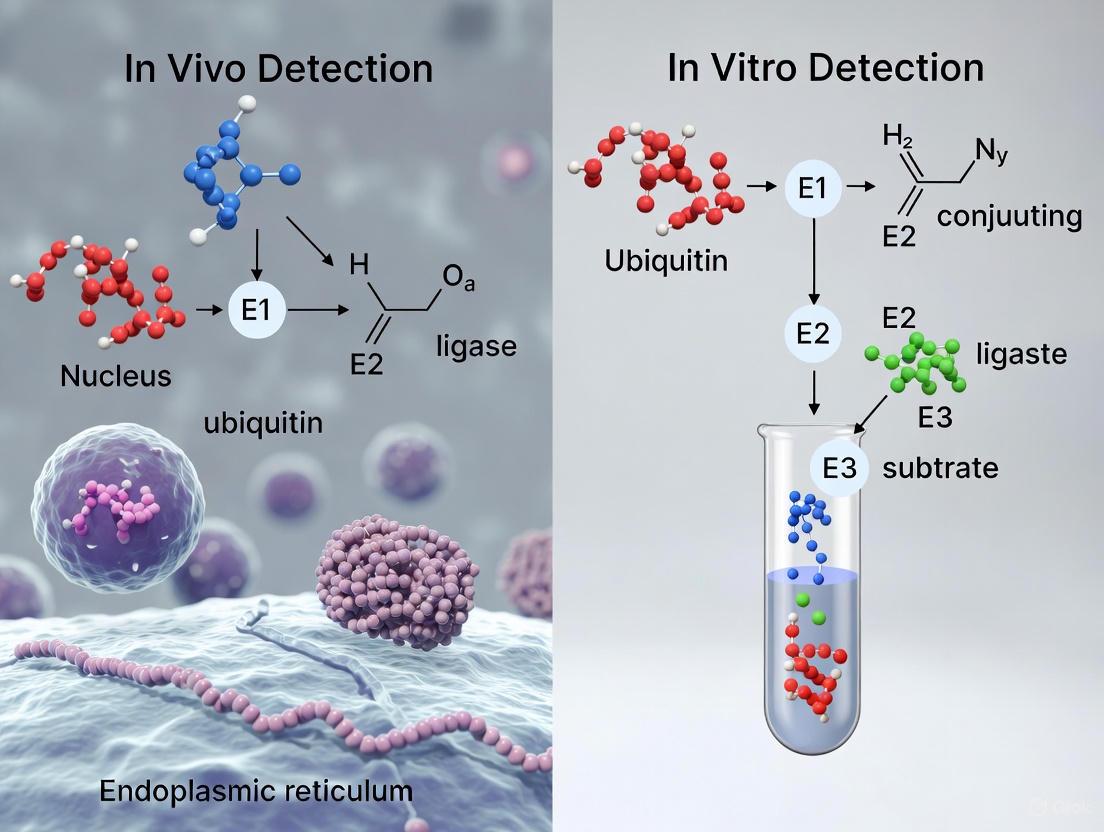
Detection Techniques: In Vivo vs. In Vitro
Characterizing ubiquitination is challenging due to the dynamic nature of the modification, the low stoichiometry of modified proteins, and the complexity of polyubiquitin chains. A wide array of techniques has been developed, falling into two broad categories: in vivo (within living cells) and in vitro (in a controlled cell-free environment).
In Vivo Detection Techniques
In vivo methods aim to capture ubiquitination events within the context of a living cell, preserving physiological relevance.
- Tagged Ubiquitin Systems: A predominant strategy involves genetically engineering cells to express ubiquitin with an affinity tag (e.g., His, HA, or Strep). After lysis, ubiquitinated proteins can be purified en masse using the appropriate resin (e.g., Ni-NTA for His-tags) and identified via mass spectrometry (MS) [6]. While powerful, this approach may introduce artifacts as the tagged ubiquitin does not perfectly mimic endogenous ubiquitin.
- Antibody-Based Enrichment: This method uses antibodies (e.g., P4D1, FK1/FK2) to immunoprecipitate endogenously ubiquitinated proteins directly from cell or tissue lysates without genetic manipulation [6]. Linkage-specific antibodies (e.g., for K48 or K63 chains) can further delineate the type of ubiquitin chain present [6].
- Tandem-Repeated Ub-Binding Entities (TUBEs): TUBEs are engineered proteins with multiple ubiquitin-binding domains, which exhibit high affinity for polyubiquitin chains. They protect ubiquitinated substrates from deubiquitination and proteasomal degradation during lysis, improving the yield of labile ubiquitination events [6].
- Live-Cell Degradation Assays: Techniques like microinjection of fluorescently labeled substrates (e.g., GS-eGFP) followed by live-cell microscopy allow for direct measurement of real-time degradation kinetics in single cells, independent of biosynthesis or uptake variables [8].
In Vitro Detection Techniques
In vitro assays reconstruct the ubiquitination cascade using purified components, offering precise control over reaction conditions.
- Western Blot/Immunoblotting: The most conventional method, where reaction mixtures are probed with anti-ubiquitin antibodies to detect shifts in molecular weight indicative of ubiquitination [4] [6]. It is low-throughput but widely accessible.
- Fluorescence & Luminescence Assays: These include FRET (Förster Resonance Energy Transfer), HTRF (Homogeneous Time-Resolved Fluorescence), and electrochemiluminescence (ECL) assays. They are amenable to high-throughput screening (HTS) for discovering ubiquitination regulators or inhibitors [4] [5].
- Reconstituted Ubiquitination Assays: These assays use purified E1, E2, E3 enzymes, ubiquitin, ATP, and a substrate to recapitulate the entire ubiquitination reaction in a test tube. This allows for detailed mechanistic studies of specific E2-E3 pairs [5].
The following workflow diagram compares the typical steps involved in these two methodological approaches:
Table 3: Comparison of Key Ubiquitination Detection Techniques
| Technique | Key Principle | Throughput | Physiological Context | Key Applications |
|---|---|---|---|---|
| Tagged Ub Pull-Down (in vivo) [6] | Affinity purification of Ub-conjugates from engineered cells | Medium | High (within living cells) | System-wide identification of ubiquitination sites (Ubiquitinome) |
| Antibody-Based IP (in vivo) [6] | Immunoprecipitation using anti-Ub antibodies | Low-Medium | High (native tissue possible) | Validation of substrate ubiquitination; linkage-specific studies |
| Live-Cell Microscopy (in vivo) [8] | Direct visualization of target protein degradation in single cells | Low | High (real-time kinetics) | Measuring precise degradation rates, catalytic efficiency of degraders |
| Reconstituted Assay (in vitro) [5] | Ubiquitination with purified components in a tube | Medium-High | Low (controlled reductionist system) | Mechanistic studies of enzyme activity; HTS for inhibitors |
| FRET/HTRF (in vitro) [5] | Fluorescent signal upon ubiquitination event | High | Low | High-throughput drug screening; kinetic studies |
A Practical Toolkit for UPS Research
To effectively study the UPS, researchers rely on a suite of specialized reagents and tools. Below is a non-exhaustive list of key solutions cited in the literature.
Table 4: Essential Research Reagent Solutions for UPS Studies
| Reagent / Tool | Function / Application | Example Use Case |
|---|---|---|
| Tagged Ubiquitin (His, HA, Strep) [6] | Affinity-based purification of ubiquitinated proteins from cell lysates. | Global ubiquitinome profiling via mass spectrometry [6]. |
| Linkage-Specific Ub Antibodies [6] | Detect or enrich for polyUb chains with a specific linkage (K48, K63, etc.). | Studying K48-linked tau ubiquitination in Alzheimer's disease [6]. |
| TUBEs (Tandem Ubiquitin Binding Entities) [6] | High-affinity enrichment of polyubiquitinated proteins; protect chains from DUBs. | Capturing and stabilizing low-abundance or labile ubiquitinated substrates. |
| Proteasome Activity Assay Kits [3] | Fluorescent-based measurement of proteasome's chymotrypsin-, trypsin-, or caspase-like activity. | Profiling proteasome function in cell lysates under different conditions (e.g., stress, inhibition). |
| Specific E3 Ligase Components (e.g., CHIPΔTPR) [8] | Serves as the recruitment module in bioPROTACs or for in vitro ubiquitination assays. | Targeted protein degradation of a proof-of-concept target like eGFP [8]. |
| Potent Proteasome Inhibitors (e.g., MG-132, Lactacystin) [3] | Block the proteolytic activity of the proteasome, causing accumulation of ubiquitinated proteins. | Validating UPS-dependent degradation of a substrate; studying cell cycle arrest and apoptosis. |
Experimental Protocol: Key Methodologies
This section outlines detailed protocols for two pivotal experiments commonly used to investigate the UPS.
Protocol 1: In Vivo Global Ubiquitinome Profiling using Tagged Ubiquitin
This protocol is adapted from large-scale MS studies and is used to identify ubiquitination sites across the proteome [6] [7].
- Cell Line Engineering: Generate a cell line (e.g., HEK293T) that stably expresses 6xHis-tagged or Strep-tagged ubiquitin using lentiviral transduction or other stable expression methods.
- Cell Lysis and Denaturation: Harvest cells and lyse them in a denaturing buffer (e.g., 6 M Guanidine-HCl, 100 mM NaH₂PO₄, 10 mM Tris-Cl, pH 8.0) to inactivate DUBs and proteases, thereby preserving the ubiquitin-modified proteome.
- Affinity Purification: Incubate the clarified denatured lysate with the appropriate affinity resin:
- Elution and Digestion: Elute the bound ubiquitinated proteins from the beads. A common method is to boil the beads in SDS-PAGE loading buffer. Alternatively, proteins can be on-bead digested with trypsin for MS analysis.
- Mass Spectrometric Analysis: The digested peptides are analyzed by LC-MS/MS. Ubiquitination sites are identified by searching for the signature diGly remnant (a mass shift of +114.04 Da on the modified lysine) that remains after trypsin digestion of ubiquitinated proteins [6] [7].
- Data Analysis and Validation: Use bioinformatic tools to filter and validate the identified ubiquitination sites. Key hits are typically validated by orthogonal methods like immunoblotting.
Protocol 2: In Vitro Ubiquitination Assay
This protocol describes a foundational experiment to study the activity of a specific E2-E3 pair or the ubiquitination of a specific substrate in a controlled environment [5].
- Reagent Preparation: Purify or procure the required recombinant proteins: E1 enzyme, the E2 enzyme of interest (e.g., UBE2D3), the E3 ligase, the substrate protein, and ubiquitin.
- Reaction Setup: Assemble a 20-50 µL reaction mixture containing:
- Energy Regeneration System: 50 mM Tris-HCl (pH 7.5), 5 mM MgCl₂, 2 mM ATP.
- Enzymes and Substrates: 100 nM E1, 1-5 µM E2, 1-5 µM E3, 5-10 µM Substrate protein, and 50-100 µM Ubiquitin.
- Negative Controls: Set up control reactions omitting E1, E2, E3, or ATP to confirm the specificity of the reaction.
- Incubation: Incubate the reaction at 30°C for 1-2 hours.
- Reaction Termination: Stop the reaction by adding SDS-PAGE loading buffer and heating at 95°C for 5 minutes.
- Analysis by Immunoblotting:
- Resolve the proteins by SDS-PAGE.
- Transfer to a nitrocellulose or PVDF membrane.
- Probe the membrane with an antibody against your substrate to observe an upward gel shift (smearing) indicative of ubiquitination.
- Alternatively, probe with an anti-ubiquitin antibody (e.g., P4D1 or FK2) to directly visualize the ubiquitinated species.
Ubiquitination is a crucial post-translational modification that involves the covalent attachment of ubiquitin, a 76-amino acid protein, to target substrate proteins [4]. This modification serves as a versatile cellular signal that regulates diverse fundamental processes, including targeted protein degradation, cell cycle progression, DNA damage repair, and numerous cell signaling pathways [4]. The complexity of ubiquitin signaling arises from its ability to form different types of conjugates—ranging from a single ubiquitin monomer (monoubiquitination) to complex polyubiquitin chains of various lengths and linkage types [6].
The specificity of ubiquitin signals is determined by an enzymatic cascade involving E1 (activating), E2 (conjugating), and E3 (ligase) enzymes [9]. The human genome encodes approximately 40 E2 enzymes and over 600 E3 ligases, which provide remarkable specificity in substrate recognition [10]. This intricate system allows cells to generate precise ubiquitin signals that are decoded by effector proteins containing ubiquitin-binding domains, ultimately leading to specific functional outcomes [10].
Understanding the distinction between monoubiquitination and polyubiquitination is fundamental to deciphering the ubiquitin code. While monoubiquitination typically regulates non-proteolytic processes such as endocytosis, histone modification, and DNA damage responses, different polyubiquitin chain linkages direct distinct cellular outcomes, with K48-linked chains primarily targeting substrates for proteasomal degradation [11]. Recent research has further revealed that ubiquitin chains can form complex branched architectures, adding another layer of complexity to this sophisticated signaling system [12].
Ubiquitin Signal Complexity: Structural and Functional Diversity
Monoubiquitination: Beyond a Simple Signal
Monoubiquitination, the attachment of a single ubiquitin moiety to a substrate, serves as a specific signal that differs functionally from polyubiquitin chains [10]. Contrary to initial assumptions, monoubiquitination is not merely a precursor to chain formation but represents a dedicated signaling event with distinct regulatory consequences. Monoubiquitinated proteins often function in regulatory roles rather than degradation targeting, influencing processes such as gene transcription, protein trafficking, and DNA repair mechanisms [10].
The generation of monoubiquitin signals requires specific cellular strategies to terminate the ubiquitination reaction after addition of a single ubiquitin moiety. Research has revealed several mechanisms that prevent chain elongation, including: (1) the substrate dissociation model, where fast dissociation rates of substrates from E3 enzymes prevent multiple ubiquitin transfers; (2) the self-inactivation model, where intramolecular interactions within monoubiquitinated proteins prevent further E3 binding; and (3) the E2 incompetent model, where specific E2 enzymes lack the capacity for chain extension [10]. These mechanisms ensure that monoubiquitination serves as an independent signal rather than an incomplete polyubiquitination event.
Polyubiquitin Chains: A Complex Language of Linkages
Polyubiquitin chains are classified based on their linkage patterns, which determine their three-dimensional structure and functional specificity [4]. The seven lysine residues (K6, K11, K27, K29, K33, K48, K63) and the N-terminal methionine (M1) of ubiquitin can each form distinct chain linkages with unique functional consequences [4].
Table 1: Polyubiquitin Chain Linkages and Their Cellular Functions
| Linkage Type | Primary Functions | Structural Features |
|---|---|---|
| K48-linked | Targets substrates for proteasomal degradation [4] | Compact structure recognized by proteasomal receptors |
| K63-linked | Regulates protein-protein interactions, DNA repair, NF-κB signaling [4] [6] | More open, extended chain conformation |
| K11-linked | Cell cycle regulation, proteasomal degradation [4] | Shares structural similarities with K48-linked chains |
| K6-linked | DNA damage repair, mitochondrial autophagy [4] | Associated with DNA damage response pathways |
| K27-linked | Controls mitochondrial autophagy [4] | Implicated in mitophagy and cellular stress responses |
| K29-linked | Cell cycle regulation, RNA processing, stress response [4] | Less characterized but linked to neurodegenerative disorders |
| K33-linked | T-cell receptor-mediated signaling [4] | Regulates enzymatic activity in immune signaling |
| M1-linked | Regulates NF-κB inflammatory signaling [4] | Linear chains generated by LUBAC complex |
Branched Ubiquitin Chains: Expanding the Signaling Landscape
Beyond homotypic chains, ubiquitin can form heterotypic chains with mixed or branched architectures [12]. Branched chains contain at least one ubiquitin subunit modified simultaneously at two different acceptor sites, creating complex topological structures that significantly expand the ubiquitin signaling vocabulary [12]. For example, branched K11/K48 chains synthesized by the APC/C complex during mitosis combine the degradative signal of K48 linkages with the cell cycle regulatory function of K11 linkages, potentially enabling more precise control of substrate degradation timing [12].
The synthesis of branched chains often involves collaboration between E3 ligases with distinct linkage specificities. For instance, TRAF6 (which synthesizes K63-linked chains) and HUWE1 (a K48-linkage specialist) collaborate to form branched K48/K63 chains during NF-κB signaling [12]. Similarly, UBR5 recognizes K63-linked chains synthesized by ITCH and attaches K48 linkages to produce branched K48/K63 chains on the pro-apoptotic regulator TXNIP, converting a non-degradative signal into a proteasome-targeting signal [12].
Detection Methodologies: In Vivo vs. In Vitro Approaches
In Vivo Detection Techniques
In vivo ubiquitination detection methods aim to capture ubiquitination events within their native cellular context, preserving physiological enzyme concentrations, subcellular localization, and cellular compartmentalization.
Genetic Manipulation Approaches involve expressing tagged ubiquitin constructs (e.g., His-, Flag-, or HA-tagged ubiquitin) in cells, allowing affinity purification of ubiquitinated proteins under denaturing conditions [6]. The Stable Tagged Ubiquitin Exchange (StUbEx) system enables replacement of endogenous ubiquitin with tagged variants, providing a more physiological approach [6]. These methods allow identification of ubiquitination sites through mass spectrometry analysis by detecting the characteristic 114.04 Da mass shift on modified lysine residues [6].
Proximity Labeling Techniques represent cutting-edge in vivo approaches that capture spatial organization of ubiquitination events. The proximal-ubiquitinome workflow combines APEX2-based proximity labeling with enrichment of ubiquitin remnant motifs (K-ε-GG), allowing spatially resolved detection of ubiquitination events within specific cellular microenvironments [13]. This method has been successfully applied to identify substrates of deubiquitinases like USP30 in their native mitochondrial context [13].
BioPROTAC Degradation Assays provide functional readouts of ubiquitination efficiency in living cells. Recently developed assays use microinjection of fluorescently labeled target proteins and bioPROTACs followed by live-cell microscopy to directly measure degradation kinetics, avoiding confounding factors like biosynthesis and transport variability [8]. This single-cell approach has revealed that bioPROTAC efficiency depends critically on the correct orientation of the ternary complex for target ubiquitination rather than just binding affinity [8].
In Vitro Detection Techniques
In vitro methods offer controlled experimental conditions for mechanistic studies of ubiquitination, enabling precise manipulation of individual components and reaction conditions.
Reconstituted Biochemical Assays use purified E1, E2, and E3 enzymes to study ubiquitination mechanisms in isolation. These systems allow systematic testing of individual enzyme contributions and linkage specificity [14]. For example, studies with HUWE1 demonstrated that certain small-molecule inhibitors are actually substrates that become ubiquitinated, revealing unexpected dimensions of E3 ligase specificity [14].
Western Blotting/Immunoblotting remains widely used for initial ubiquitination detection using ubiquitin-specific antibodies [4] [6]. While this method is accessible and cost-effective, it provides limited information about chain linkage and architecture [4].
Fluorescence and Chemiluminescence Assays offer higher sensitivity and quantitative capabilities for kinetic studies. These assays often use labeled ubiquitin or antibodies to monitor ubiquitination in real-time, enabling determination of kinetic parameters [4].
Nanopore Sensing Assays represent an emerging technology that shows promise for direct detection of ubiquitin chains and their linkages without labeling, potentially enabling analysis of chain architecture [4].
Table 2: Comparison of Major Ubiquitination Detection Techniques
| Technique | Key Features | Applications | Advantages | Limitations |
|---|---|---|---|---|
| Tagged Ubiquitin (in vivo) | Expression of epitope-tagged ubiquitin (His, HA, Flag) | Ubiquitinome profiling, site identification [6] | High-throughput capability, identification of modification sites | May not mimic endogenous ubiquitin, potential artifacts |
| Antibody-Based Enrichment (in vivo) | Use of ubiquitin-specific antibodies (P4D1, FK1/FK2) or linkage-specific antibodies | Endogenous ubiquitination profiling, tissue samples [6] | Works under physiological conditions, no genetic manipulation needed | High cost, non-specific binding, limited antibody specificity |
| Proximity Labeling (in vivo) | APEX2-mediated biotinylation near DUBs of interest | Mapping DUB substrates, spatial ubiquitinomics [13] | Captures microenvironment-specific ubiquitination, identifies direct substrates | Complex workflow, requires genetic engineering |
| Reconstituted Biochemical Assays (in vitro) | Purified E1, E2, E3 enzymes with ubiquitin | Mechanistic studies, enzyme specificity, inhibitor screening [14] | Controlled conditions, precise manipulation of components | May lack cellular context, potential over-simplification |
| TUBEs (Tandem Ubiquitin Binding Entities) | Recombinant proteins with multiple ubiquitin-binding domains | Stabilization of ubiquitin conjugates, proteomics [6] | Protects against DUBs, enhances purification efficiency | Requires recombinant protein production |
| BioPROTAC Kinetic Assays (in vivo) | Microinjection + live-cell microscopy | Degradation rate measurement, degrader optimization [8] | Direct kinetic measurements, single-cell resolution | Technically challenging, low throughput |
Experimental Protocols for Key Methodologies
Proximal-Ubiquitinome Profiling for DUB Substrate Identification [13]:
- Generate cell lines expressing DUB-APEX2 fusion proteins
- Perform proximity labeling with biotin-phenol and H₂O₂ stimulation
- Harvest cells and streptavidin-based enrichment of biotinylated proteins
- Digest enriched proteins with trypsin
- Immunoaffinity enrichment of K-ε-GG-containing peptides
- Analyze by liquid chromatography-tandem mass spectrometry (LC-MS/MS)
- Validate candidate substrates through orthogonal approaches
Reconstituted HUWE1 Ubiquitination Assay [14]:
- Purify E1 (UBA1), E2 (UBE2L3 or UBE2D3), and HUWE1HECT or full-length HUWE1
- Set up reaction buffer: 50 mM Tris-HCl (pH 7.5), 50 mM NaCl, 10 mM MgCl₂, 2 mM ATP
- Combine enzymes with ubiquitin and potential substrates/inhibitors
- Incubate at 30°C for desired time points
- Stop reaction with SDS-PAGE loading buffer containing DTT
- Analyze by SDS-PAGE and western blotting with ubiquitin-specific antibodies
- For inhibitor studies, monitor dose-dependent effects on auto-ubiquitination
BioPROTAC Degradation Kinetic Measurements [8]:
- Purify target protein (e.g., GS-eGFP) and bioPROTAC (e.g., DARPin-CHIPΔTPR fusion)
- Label bioPROTAC with fluorescent dye (TMR5-maleimide)
- Form 1:1 complexes between target and bioPROTAC in vitro
- Microinject pre-formed complexes into HEK293 cell cytosol
- Perform time-lapse fluorescence microscopy to monitor protein levels
- Quantify fluorescence decay in individual cells over time
- Calculate degradation rate constants from single-cell trajectories
Technical Challenges and Methodological Considerations
Key Challenges in Ubiquitination Research
Ubiquitination research faces several technical challenges that complicate data interpretation. The low stoichiometry of ubiquitination under physiological conditions makes detection difficult without enrichment strategies [6]. The transient nature of ubiquitination due to active deubiquitinating enzymes (DUBs) requires use of proteasome inhibitors (e.g., MG-132) or DUB inhibitors to preserve signals [11]. Linkage complexity presents analytical challenges, as polyubiquitin chains can be homotypic, mixed, or branched with different functional consequences [12].
Method-specific limitations include artifacts from tagged ubiquitin expression that may not fully replicate endogenous ubiquitin dynamics [6]. Antibody specificity issues plague the field, with many commercial ubiquitin antibodies showing cross-reactivity or poor linkage specificity [11]. Cellular context loss in in vitro systems may fail to recapitulate physiological regulation, while complexity of intracellular environments in in vivo systems can obscure specific mechanisms [14].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Ubiquitination Studies
| Reagent Category | Specific Examples | Applications and Functions |
|---|---|---|
| Ubiquitin Expression Constructs | His-Ub, HA-Ub, Strep-Ub, GFP-Ub | Affinity purification, microscopy, ubiquitinome profiling [6] |
| Linkage-Specific Antibodies | K48-specific, K63-specific, M1-specific antibodies | Detection of specific chain types, immunohistochemistry [6] |
| Pan-Ubiquitin Antibodies | P4D1, FK1, FK2 | General ubiquitination detection, western blotting [6] |
| Ubiquitin Enrichment Tools | Ubiquitin-Trap (nanobody-based), TUBEs | Pull-down of ubiquitinated proteins, stabilization against DUBs [11] |
| Enzyme Inhibitors | MG-132 (proteasome inhibitor), TAK243 (E1 inhibitor) | Pathway inhibition, stabilization of ubiquitinated species [9] [11] |
| Recombinant Enzymes | E1 (UBA1), E2s (UBE2L3, UBE2D3), E3s (HUWE1, Parkin) | In vitro ubiquitination assays, mechanism studies [14] |
| Activity Probes | Ubiquitin vinyl sulfone, HA-Ub-VS | DUB activity profiling, enzyme characterization |
| Mass Spec Standards | DiGly remnant peptides, SILAC-labeled ubiquitin | Quantitative proteomics, site identification [13] |
Research Applications and Future Directions
Applications in Drug Discovery and Disease Research
Ubiquitination detection methodologies have enabled significant advances in understanding disease mechanisms and developing targeted therapies. In cancer research, detection of aberrant ubiquitination has revealed dysregulated pathways in cell cycle control and DNA damage response [4]. Small molecule inhibitors targeting specific E3 ligases, such as Nutlin against MDM2, have shown promise in clinical trials for treating cancers like multiple myeloma [4]. The proteasome inhibitor bortezomib has become an established treatment for multiple myeloma and mantle cell lymphoma [4].
In neurodegenerative disease research, characterization of ubiquitin chains in pathological protein aggregates has provided insights into disease mechanisms. For example, K48-linked polyubiquitination of tau protein was found to be abnormally accumulated in Alzheimer's disease [6]. Understanding these patterns may lead to novel therapeutic strategies targeting the ubiquitin system.
The emerging field of targeted protein degradation has leveraged insights from ubiquitination mechanisms to develop novel therapeutic modalities. PROTACs (Proteolysis-Targeting Chimeras) and bioPROTACs use the ubiquitin system to selectively degrade disease-causing proteins, with applications in cancer, neurodegenerative diseases, and genetic disorders [8]. These approaches demonstrate how fundamental research on ubiquitination mechanisms can translate into innovative therapeutic strategies.
Emerging Technologies and Future Perspectives
Future developments in ubiquitination research will likely focus on single-cell ubiquitinomics to capture cell-to-cell heterogeneity, spatiotemporally resolved detection to monitor ubiquitination dynamics in real-time, and improved linkage-specific reagents to better discriminate between ubiquitin chain architectures [13] [8].
The integration of chemical biology tools with genetic approaches will enable more precise manipulation and monitoring of ubiquitination events. For example, the development of photo-crosslinkable ubiquitin variants could help capture transient ubiquitination events, while improved mass spectrometry methods may enable comprehensive analysis of branched chain architectures [12].
As our understanding of the complexity of ubiquitin signals grows, so does the appreciation for its role in integrating cellular information. The continued development of sophisticated detection methodologies will be essential for deciphering this complex regulatory code and harnessing its potential for therapeutic intervention.
Visualizing Ubiquitination Pathways and Detection Methods
Ubiquitination Enzymatic Cascade and Signaling Outcomes
Proximal-Ubiquitinome Profiling Workflow
Ubiquitination, the covalent attachment of ubiquitin to target proteins, is a fundamental post-translational modification regulating diverse cellular processes including protein degradation, DNA repair, and cell signaling [4]. The detection and accurate quantification of ubiquitination events are therefore critical for understanding cellular homeostasis and developing therapies for diseases such as cancer and neurodegenerative disorders [4] [15]. However, researchers face significant technical challenges in ubiquitination studies, primarily stemming from the low stoichiometry of modification, transient nature of ubiquitin signaling, and constant interference from deubiquitinases (DUBs) that rapidly reverse these modifications [16] [15]. This guide compares the performance of contemporary methodologies addressing these hurdles, providing experimental data and protocols to inform research design.
The Core Technical Hurdles in Ubiquitin Research
Low Stoichiometry of Modification
Ubiquitination typically occurs at low levels, with only a small fraction of any target protein being modified at a given time [17]. This low stoichiometry presents substantial detection challenges, as the signal from modified species is often obscured by abundant unmodified proteins. Mass spectrometry-based ubiquitinomics reveals that most ubiquitination sites exhibit low occupancy, acting as subtle modulators rather than binary switches [17]. This necessitates highly sensitive enrichment and detection methods to capture meaningful biological signals.
Transient Nature of Ubiquitination
The dynamic balance between ubiquitination by E1-E2-E3 enzymatic cascades and deubiquitination by DUBs creates transient ubiquitination states [4] [15]. This transient nature means ubiquitination events can be brief, making them difficult to capture without methodological interventions. The half-life of ubiquitin-protein conjugates is often short, particularly for substrates targeted to the proteasome, requiring precise temporal control in experimental setups.
Deubiquitinase Interference
Approximately 100 human DUBs constantly oppose ubiquitination by cleaving ubiquitin from modified substrates [15] [18]. This DUB activity presents a major interference factor in ubiquitination studies, as it can rapidly remove ubiquitin signals during cell lysis and sample processing. Different DUB families (USP, UCH, OTU, MJD, MINDY, ZUFSP, and JAMM) exhibit varying linkage specificities and cellular functions, adding complexity to this interference [19] [18]. Bacterial pathogens have even evolved DUB effectors that interfere with host ubiquitination, highlighting the ubiquity of this challenge [20].
Table 1: Major Deubiquitinase Families and Their Characteristics
| DUB Family | Catalytic Mechanism | Representative Members | Key Characteristics |
|---|---|---|---|
| USP (Ubiquitin-Specific Proteases) | Cysteine protease | USP7, USP9X | Largest family (58 members), generally linkage-promiscuous |
| UCH (Ubiquitin C-Terminal Hydrolases) | Cysteine protease | UCH-L1, UCH-L3 | Prefer small adducts or unstructured C-termini |
| OTU (Ovarian Tumor Proteases) | Cysteine protease | OTUD1, A20 | Often linkage-specific (e.g., K63-specific OTUD1) |
| MJD (Machado-Josephin Domain Proteases) | Cysteine protease | Ataxin-3, JosD1 | Josephin domain-containing, involved in neurodegeneration |
| MINDY | Cysteine protease | MINDY-1, MINDY-2 | K48-linkage specific, senses chain length |
| ZUFSP | Cysteine protease | ZUP1 | K63-specific, associated with genome integrity |
| JAMM | Zinc metalloprotease | PSMD14, AMSH | Requires zinc ions, only metalloprotease DUB family |
Methodological Comparisons: Addressing the Key Hurdles
Mass Spectrometry-Based Approaches
Modern mass spectrometry techniques, particularly Data-Independent Acquisition (DIA-MS), have revolutionized ubiquitinome profiling by simultaneously addressing multiple technical challenges.
Performance Comparison: DIA-MS significantly outperforms traditional Data-Dependent Acquisition (DDA) methods, quantifying over 68,000 ubiquitinated peptides in single runs compared to approximately 21,400 with DDA [16]. This enhanced coverage is crucial for detecting low-stoichiometry modifications. DIA-MS also demonstrates superior quantitative precision, with median coefficients of variation (CV) of approximately 10% across replicates, enabling reliable detection of transient dynamics [16].
Experimental Protocol (DIA-MS Ubiquitinomics):
- Cell Lysis: Use sodium deoxycholate (SDC) buffer supplemented with 40mM chloroacetamide (CAA) for immediate cysteine protease inhibition [16].
- Protein Digestion: Perform tryptic digestion to generate ubiquitin remnant peptides containing diglycine (K-ε-GG) signatures.
- Peptide Enrichment: Use immunoaffinity purification with anti-K-ε-GG antibodies to isolate ubiquitinated peptides.
- Mass Spectrometry: Analyze using DIA-MS with 75-minute nanoLC gradients and optimized MS methods.
- Data Processing: Utilize DIA-NN software in "library-free" mode for identification and quantification [16].
Table 2: Comparison of Ubiquitinomics Method Performance
| Method | Sensitivity (Peptides Identified) | Quantitative Precision (Median CV) | Sample Input | Throughput |
|---|---|---|---|---|
| DDA-MS with Urea Lysis | ~19,400 peptides | >20% | 2mg protein | Low (50% missing values in replicates) |
| DDA-MS with SDC Lysis | ~26,750 peptides | 15-20% | 2mg protein | Moderate |
| DIA-MS with SDC Lysis | ~68,400 peptides | ~10% | 2mg protein | High (88% overlap between replicates) |
| UbiSite Method | ~30,000 peptides | Not specified | 40mg protein | Very Low (requires fractionation) |
Biochemical and Cellular Assays
For functional validation of specific ubiquitination events, biochemical approaches provide complementary information to proteomic methods.
DUB Inhibition Studies: Targeted inhibition of specific DUBs combined with ubiquitinomics enables mapping of DUB substrates and distinction between degradative and non-degradative ubiquitination. Upon USP7 inhibition, hundreds of proteins show increased ubiquitination within minutes, but only a small fraction undergo degradation, revealing the complex regulatory landscape [16].
Experimental Protocol (In Vitro Deubiquitination Assay):
- Substrate Preparation: Generate ubiquitinated substrates using recombinant E1, E2, and E3 enzymes or by immunopurification from cells treated with proteasome inhibitors.
- DUB Incubation: Incubate ubiquitinated substrates with purified DUBs or cell lysates expressing DUBs.
- Reaction Conditions: Use appropriate buffers (typically Tris or HEPES, pH 7.5-8.0, with DTT for cysteine DUBs).
- Detection: Monitor deubiquitination by immunoblotting with ubiquitin-specific antibodies or by changes in substrate molecular weight [15] [18].
Advanced Methodologies for Specific Challenges
Addressing Stoichiometry: Site-specific ubiquitination stoichiometry can be quantified using chemical labeling, isotopic tagging, and targeted proteomic approaches [17]. These methods reveal functional thresholds where ubiquitination transitions from subtle modulation to decisive regulatory switch.
Capturing Transient Events: Rapid inhibition techniques using specific DUB inhibitors or crosslinking strategies help preserve transient ubiquitination states. Time-resolved analyses with high temporal resolution enable tracking of ubiquitination dynamics [16].
Countering DUB Interference: Immediate protease inhibition during cell lysis is critical. SDC lysis with CAA alkylation rapidly inactivates DUBs, resulting in 38% more ubiquitinated peptide identifications compared to urea-based methods [16]. Additionally, specific DUB inhibitors and ubiquitin variants (UbVs) can target particular DUB families [18].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Ubiquitination Studies
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Protease Inhibitors | Chloroacetamide (CAA), N-ethylmaleimide | Rapid cysteine protease/DUB inhibition during lysis |
| Lysis Buffers | SDC (Sodium Deoxycholate) buffer | Efficient extraction with concurrent DUB inhibition |
| DUB Inhibitors | MLN4924 (NAE1 inhibitor), P5091 (USP7 inhibitor) | Specific pathway inhibition to study ubiquitination dynamics |
| Proteasome Inhibitors | MG-132, Bortezomib | Prevent degradation of ubiquitinated proteins, enhancing detection |
| Activity-Based Probes | Ubiquitin-based ABPs with warhead groups | Profiling DUB activity and specificity in complex mixtures |
| Ubiquitin Variants | Linkage-specific UbVs (e.g., K48-, K63-specific) | Inhibiting specific DUBs or recognizing particular chain types |
| Enrichment Reagents | Anti-K-ε-GG antibodies, TUBE (Tandem Ubiquitin Binding Entities) | Affinity purification of ubiquitinated proteins/peptides |
| Mass Spec Standards | Heavy-labeled ubiquitin remnant peptides | Quantitative precision and normalization in proteomics |
Visualization of Key Concepts
Ubiquitination-DUB Balance and Methodological Interventions
Experimental Workflow for Comprehensive Ubiquitinomics
The methodological landscape for studying ubiquitination has evolved significantly to address the core challenges of low stoichiometry, transient dynamics, and DUB interference. Integrated approaches combining optimized sample preparation (SDC/CAA lysis), advanced mass spectrometry (DIA-MS), and specific pharmacological tools (DUB inhibitors) now enable comprehensive analysis of ubiquitination events that were previously undetectable. As these technologies continue to advance, particularly through improved quantitative accuracy and temporal resolution, researchers will gain unprecedented insights into the complex regulatory networks governed by ubiquitin signaling, accelerating both basic biological discovery and therapeutic development.
Ubiquitination, the covalent attachment of a small regulatory protein called ubiquitin to target substrates, represents one of the most versatile post-translational modifications in eukaryotic cells. This highly conserved process regulates virtually every cellular pathway, with particular significance for maintaining protein homeostasis through directed proteasomal degradation. The ubiquitination machinery consists of a sequential enzymatic cascade involving E1 activating enzymes, E2 conjugating enzymes, and E3 ligases, with counterbalancing activity provided by deubiquitinating enzymes (DUBs) that remove ubiquitin modifications [21] [22]. The system's importance in human disease stems from its regulatory control over fundamental cellular processes including cell cycle progression, DNA damage repair, signal transduction, and immune responses [22].
Dysregulation of ubiquitination pathways manifests differently across disease states. In cancer, mutations in ubiquitin system components often drive uncontrolled proliferation and evasion of cell death, while in neurodegenerative disorders, impaired ubiquitin-mediated clearance of toxic protein aggregates leads to neuronal dysfunction and death [21] [23]. This review examines the distinct and overlapping roles of ubiquitination in these disease contexts, with particular emphasis on comparing methodologies for detecting ubiquitination events in experimental models—a critical consideration for both basic research and drug development.
Molecular Mechanisms of Ubiquitination in Cancer
The ubiquitin-proteasome system (UPS) contributes to multiple hallmarks of cancer, influencing cellular survival, proliferation, and metabolic reprogramming. Cancer-associated dysregulation occurs through mutations in ubiquitin pathway enzymes or through the hijacking of normal ubiquitination mechanisms to destabilize tumor suppressor proteins [21] [22].
E3 Ligases and DUBs as Oncogenes and Tumor Suppressors
E3 ubiquitin ligases demonstrate remarkable substrate specificity, with many functioning as either oncogenes or tumor suppressors depending on their target proteins. For instance, the E3 ligase MDM2 acts as an oncogene by targeting tumor suppressor p53 for degradation, while BRCA1 functions as a tumor suppressor through its role in DNA damage repair [22]. The following table summarizes key E3 ligases and their roles in cancer:
Table 1: Key E3 Ubiquitin Ligases in Cancer Pathogenesis
| E3 Ligase | Category | Key Substrate(s) | Associated Cancers | Functional Outcome |
|---|---|---|---|---|
| MDM2 | Oncogene | p53 | Sarcomas, breast, lung | Proteasomal degradation of tumor suppressor |
| E6AP | Oncogene | p53 | HPV-associated cancers | Viral hijacking leads to p53 degradation |
| SCFSkp2 | Oncogene | p27 | Lung, glioma, gastric, prostate | Cell cycle progression |
| VHL | Tumor Suppressor | HIF-1α | Renal cell carcinoma | Regulates tumor vascularization |
| BRCA1 | Tumor Suppressor | Multiple DNA repair proteins | Breast, ovarian | DNA damage response |
Similarly, deubiquitinating enzymes (DUBs) counterbalance E3 ligase activity, with several functioning as oncoproteins. For example, ubiquitin-specific protease 2 (USP2) stabilizes the immune checkpoint protein PD-1, thereby promoting tumor immune escape through deubiquitination [21]. The DUB OTUB2 enhances glycolysis and accelerates colorectal cancer progression by inhibiting Parkin-mediated ubiquitination of pyruvate kinase M2 (PKM2) [21].
Emerging Therapeutic Strategies Targeting Ubiquitination
Novel anticancer strategies increasingly leverage the ubiquitin-proteasome system. Proteolysis targeting chimeras (PROTACs) represent a groundbreaking approach that redirects E3 ligase activity to specifically degrade disease-causing proteins. ARV-110 and ARV-471 exemplify this technology, having progressed to phase II clinical trials for metastatic castration-resistant prostate cancer and breast cancer, respectively [21]. Molecular glues offer an alternative degradation strategy with smaller molecular dimensions, simplifying optimization of pharmaceutical properties. CC-90009, which promotes degradation of GSPT1, is currently in phase II trials for leukemia therapy [21].
Ubiquitination Dysregulation in Neurodegenerative Disorders
In contrast to cancer, neurodegenerative diseases typically involve failures in ubiquitin-dependent protein quality control mechanisms. The post-mitotic nature of neurons makes them particularly vulnerable to accumulated protein damage, as they cannot dilute toxic aggregates through cell division [23].
Protein Aggregation and Impaired Clearance Mechanisms
A hallmark of most neurodegenerative disorders is the accumulation of ubiquitin-positive protein aggregates, indicating a failure of normal degradation pathways. Key aggregated proteins include β-amyloid and tau in Alzheimer's disease, α-synuclein in Parkinson's disease, huntingtin in Huntington's disease, and TDP-43 in amyotrophic lateral sclerosis [23] [24]. These aggregates often contain ubiquitin and components of the ubiquitination machinery, suggesting attempted clearance by proteasomal and autophagic pathways.
The two main degradation pathways—the ubiquitin-proteasome system (UPS) and autophagy-lysosomal pathway (ALP)—both depend on ubiquitin signaling. The UPS primarily degrades soluble, misfolded proteins modified with K48-linked ubiquitin chains, while autophagy (particularly aggrephagy) clears larger protein aggregates tagged with K63-linked chains via receptors like p62/SQSTM1 [23] [24]. Aging-associated decline in both pathways contributes to the late onset of most neurodegenerative conditions.
Disease-Linked Mutations in Ubiquitin System Components
Several familial forms of neurodegeneration result from mutations in genes encoding ubiquitin pathway components. Parkinson's disease provides compelling examples, with loss-of-function mutations in the E3 ligase Parkin (PRKN) and its upstream kinase PINK1 causing autosomal recessive juvenile parkinsonism [23]. These proteins coordinate a quality control pathway for damaged mitochondria (mitophagy), with PINK1 accumulating on dysfunctional mitochondria to recruit and activate Parkin, which ubiquitinates mitochondrial outer membrane proteins to trigger autophagic clearance [23].
Table 2: Ubiquitin Pathway Components in Neurodegenerative Diseases
| Disease | Key Aggregated Protein | Relevant Ubiquitin System Component | Functional Consequence |
|---|---|---|---|
| Alzheimer's Disease | β-amyloid, tau | UCHL1, USP14 | Proteasomal impairment |
| Parkinson's Disease | α-synuclein | Parkin (E3), PINK1 | Impaired mitophagy |
| Huntington's Disease | Huntingtin | E3 ligases (CHIP) | Aggregate clearance failure |
| Amyotrophic Lateral Sclerosis | TDP-43, SOD1 | UBQLN2, OPTN | Defective autophagy |
| Angelman Syndrome | - | UBE3A/E6AP (E3) | Neurodevelopmental defects |
Frontotemporal dementia and amyotrophic lateral sclerosis (ALS) have been linked to mutations in UBQLN2, which encodes a proteasome shuttle factor, and OPTN, which encodes an autophagy receptor [23]. These genetic connections underscore the critical importance of efficient ubiquitin-mediated degradation for neuronal survival.
Comparative Analysis: Detection Methods for Ubiquitination
Understanding ubiquitination in disease contexts relies on robust detection methodologies, each with distinct advantages and limitations. The choice between in vivo and in vitro approaches depends on the research question, with considerations for biological relevance versus experimental control.
In Vivo Ubiquitination Detection
The most common method for detecting protein ubiquitination in living cells involves immunoprecipitation followed by immunoblotting under stringent conditions to eliminate non-covalent interactions [25].
Protocol: In Vivo Ubiquitination Assay in Cultured Cells
- Transfection: Introduce plasmids expressing the protein of interest and epitope-tagged ubiquitin into cultured cells.
- Cell Lysis: Lyse cells using complete lysis buffer (2% SDS, 150 mM NaCl, 10 mM Tris-HCl, pH 8.0) containing protease inhibitors and deubiquitinating enzyme inhibitors (N-ethylmaleimide or ubiquitin aldehyde).
- Denaturation: Immediately boil samples for 10 minutes to denature proteins and inactivate DUBs.
- Shearing: Sonicate lysates to shear DNA and reduce viscosity.
- Dilution: Add dilution buffer (10 mM Tris-HCl, pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% Triton) and incubate at 4°C for 30-60 minutes with rotation.
- Immunoprecipitation: Incubate pre-cleared lysates with antibody-conjugated Protein A/G beads targeting the protein of interest overnight at 4°C.
- Washing: Wash beads extensively with high-stringency buffer (10 mM Tris-HCl, pH 8.0, 1 M NaCl, 1 mM EDTA, 1% NP-40).
- Analysis: Elute proteins by boiling in SDS sample buffer, then perform immunoblotting with anti-ubiquitin and target protein antibodies [25].
This approach preserves physiological enzyme-substrate relationships and regulatory mechanisms but may be complicated by endogenous ubiquitin and low abundance of modified species.
In Vitro Ubiquitination Detection
Reconstituted ubiquitination assays using purified components offer precise control over reaction conditions and enzyme composition.
Protocol: In Vitro Ubiquitination Assay
- Reaction Setup: For each 40μL reaction, combine:
- 8μL 5X ubiquitination buffer (100 mM Tris-HCl, pH 7.5, 25 mM MgCl₂, 2.5 mM DTT, 10 mM ATP)
- 250 ng E1 activating enzyme
- 500 ng E2 conjugating enzyme
- 0.5-1 μg E3 ligase (if testing E3-dependent ubiquitination)
- 0.5 μg ubiquitin
- 0.5 μg substrate protein
- Water to 40μL total volume
- Control Reactions: Prepare parallel reactions omitting E1, E2, E3, or ubiquitin to confirm specificity.
- Incubation: Incubate at 37°C for 1-2 hours.
- Termination: Stop reactions by adding SDS-PAGE sample buffer and boiling for 10 minutes.
- Analysis: Resolve proteins by SDS-PAGE and detect ubiquitination by immunoblotting [25].
This method allows direct assessment of specific enzyme requirements and kinetic parameters but lacks cellular context and regulatory complexity.
Table 3: Comparison of Ubiquitination Detection Methods
| Parameter | In Vivo Detection | In Vitro Detection |
|---|---|---|
| Biological relevance | High, maintains cellular context | Limited, reduced complexity |
| Experimental control | Lower, multiple variables | High, defined components |
| Detection sensitivity | May require overexpression | Direct visualization possible |
| Equipment requirements | Standard cell culture and molecular biology | Protein purification capabilities |
| Technical challenges | Non-specific interactions, DUB activity | Enzyme stability, reconstitution efficiency |
| Applications | Disease mechanisms, drug screening | Enzyme mechanics, biochemical characterization |
Experimental Visualization and Research Tools
Key Signaling Pathways in Ubiquitination-Related Diseases
The following diagrams illustrate critical ubiquitin-dependent pathways in cancer and neurodegeneration, created using DOT language with specified color palette:
Diagram 1: PINK1/Parkin-mediated mitophagy pathway relevant to Parkinson's disease.
Diagram 2: HPV E6/E6AP-mediated p53 degradation pathway in cervical cancer.
The Scientist's Toolkit: Essential Research Reagents
Table 4: Essential Reagents for Ubiquitination Research
| Reagent Category | Specific Examples | Research Applications | Technical Considerations |
|---|---|---|---|
| E1 Activating Enzymes | UBA1, UBA6 | Initiates ubiquitin activation | Essential for all in vitro assays |
| E2 Conjugating Enzymes | UBE2D, UBE2R, UBE2N/UEV1 | Ubiquitin chain formation | Determine linkage specificity |
| E3 Ubiquitin Ligases | Parkin, MDM2, BRCA1, E6AP | Substrate recognition | High specificity; often mutated in disease |
| Deubiquitinases (DUBs) | USP14, UCHL1, OTUB1, CYLD | Reverse ubiquitination | Use inhibitors in detection assays |
| Ubiquitin Variants | Wild-type, K48-only, K63-only, HA-tagged, FLAG-tagged | Chain linkage studies | Tags enable detection in assays |
| Proteasome Inhibitors | Bortezomib, MG132 | Stabilize ubiquitinated proteins | Can induce stress responses |
| DUB Inhibitors | PR-619, N-ethylmaleimide | Prevent deubiquitination | Improve detection sensitivity |
| Antibodies | Anti-ubiquitin, anti-K48, anti-K63 linkage-specific | Detection and quantification | Variable specificity between lots |
The investigation of ubiquitination in human disease reveals both shared mechanisms and pathological specializations between cancer and neurodegeneration. In cancer, ubiquitination pathways are typically hyperactive or hijacked to promote growth and survival, while in neurodegenerative conditions, impaired ubiquitin-mediated degradation enables toxic accumulation of misfolded proteins. These distinctions have profound implications for therapeutic development, with cancer strategies often aiming to inhibit specific E3 ligases or proteasomal activity, while neurodegenerative approaches seek to enhance ubiquitin-mediated clearance.
Methodologically, the complementary use of in vivo and in vitro detection techniques provides the most comprehensive understanding of ubiquitination events. In vivo approaches maintain physiological context essential for disease modeling, while in vitro methods offer mechanistic precision for dissecting molecular interactions. The continuing development of targeted protein degradation technologies like PROTACs underscores the therapeutic potential of manipulating ubiquitination pathways, offering promising avenues for addressing previously "undruggable" targets in both cancer and neurodegeneration.
A Practical Guide to Established and Next-Generation Detection Techniques
Immunoprecipitation and Immunoblotting in Cultured Cells
Protein ubiquitination is a crucial post-translational modification that regulates diverse cellular functions, including proteasomal degradation, signal transduction, and immune responses [26] [6]. The detection of ubiquitination in cultured cells provides critical insights into protein regulation under physiological conditions, enabling researchers to study this dynamic process within its native cellular context. Unlike in vitro approaches, in vivo detection preserves the complex regulatory environment of the cell, including the full complement of E1, E2, and E3 enzymes, deubiquitinases (DUBs), and competing post-translational modifications [27] [6]. This guide objectively compares immunoprecipitation (IP) and immunoblotting methodologies for detecting ubiquitination in cultured cells, evaluating their performance against alternative techniques while providing detailed experimental protocols and data for researcher implementation.
Core Methodology: Immunoprecipitation and Immunoblotting Workflow
The standard protocol for detecting protein ubiquitination in cultured cells involves immunoprecipitation of the target protein under denaturing conditions, followed by immunoblotting with ubiquitin-specific antibodies [25]. This approach enables researchers to confirm whether a specific protein is ubiquitinated and to compare ubiquitination levels under different experimental conditions.
Detailed Experimental Protocol
Cell Lysis under Denaturing Conditions:
- Aspirate culture medium and lyse cells directly in 100-200μl of hot SDS-containing lysis buffer (2% SDS, 150mM NaCl, 10mM Tris-HCl, pH 8.0) supplemented with protease inhibitors (e.g., 2mM sodium orthovanadate, 50mM sodium fluoride) and DUB inhibitors (N-ethylmaleimide or ubiquitin aldehyde) to prevent deubiquitination during processing [25] [6].
- Immediately boil cell lysates for 10 minutes to denature proteins and inactivate DUBs [25].
- Sonicate samples to shear genomic DNA and reduce sample viscosity.
Immunoprecipitation:
- Dilute the denatured lysates 10-fold with dilution buffer (10mM Tris-HCl, pH 8.0, 150mM NaCl, 2mM EDTA, 1% Triton X-100) to reduce SDS concentration below its critical micelle concentration (0.1-0.2%) [25].
- Incubate diluted lysates with target protein-specific antibody conjugated to Protein A/G agarose beads (14-20μl of 50% slurry per 500-1500μg total protein) at 4°C overnight with rotation [25].
- Pellet beads by centrifugation at 5,000×g for 5 minutes and wash twice with high-stringency buffer (10mM Tris-HCl, pH 8.0, 1M NaCl, 1mM EDTA, 1% NP-40) to remove non-specifically bound proteins [25].
- After final wash, completely remove residual buffer and elute immunoprecipitated proteins by boiling beads in 2X SDS-PAGE loading buffer for 10 minutes [25].
Immunoblotting:
- Separate immunoprecipitated proteins by SDS-PAGE and transfer to PVDF or nitrocellulose membranes.
- Probe membranes with ubiquitin-specific antibodies (e.g., P4D1, FK1, FK2) to detect ubiquitinated target protein [6].
- Stripping and re-probing membranes with antibody against the target protein confirms equal precipitation across samples [25].
The following diagram illustrates this core experimental workflow:
Performance Comparison with Alternative Methods
Immunoprecipitation and immunoblotting offer distinct advantages and limitations compared to other ubiquitination detection methods. The table below provides a systematic comparison of key performance characteristics:
| Method | Throughput | Sensitivity | Linkage Specificity | Required Resources | Key Applications |
|---|---|---|---|---|---|
| IP + Immunoblotting | Low to moderate | High (detects endogenous proteins) | No (unless linkage-specific antibodies) | Standard molecular biology equipment | Target protein validation, time-course studies [26] [25] |
| TUBE-Based Assays | High (96/384-well format) | Very high (nanomolar affinity) | Yes (chain-specific TUBEs) | Specialized TUBE reagents | High-throughput screening, linkage-specific analysis [26] |
| Ubiquitin Tagging (StUbEx) | Moderate | Moderate | No | Cell lines expressing tagged ubiquitin | Proteomic screening of ubiquitinated proteins [6] |
| Mass Spectrometry | High for discovery | Lower for site mapping | Yes with advanced methods | Specialized instrumentation | Global ubiquitinome profiling, site identification [28] [6] |
| In Vitro Reconstitution | Moderate | High for direct substrates | Controllable | Purified enzyme components | Mechanism dissection, direct substrate validation [27] |
Quantitative Performance Data
Recent studies provide direct comparisons of methodological performance:
Signal Detection Sensitivity: TUBE-based assays demonstrate nanomolar affinities for polyubiquitin chains, significantly enhancing detection sensitivity compared to standard immunoblotting [26]. In studies of RIPK2 ubiquitination, TUBE-based approaches enabled clear differentiation between K48- and K63-linked ubiquitination in response to different stimuli (L18-MDP vs. RIPK2 PROTAC), while standard IP/immunoblotting showed limited linkage discrimination [26].
Throughput Capacity: Traditional IP/immunoblotting is considered low-throughput, requiring 1-2 days for processing limited sample numbers. In contrast, TUBE-based HTS assays have been successfully implemented in 96-well plate formats, enabling rapid quantification of endogenous protein ubiquitination dynamics [26].
Target Specificity: IP/immunoblotting provides high target specificity when quality antibodies are available. However, a comparative study of E3 ligases found that some targets initially identified in cellular studies could not be validated in direct in vitro assays (e.g., CRL3SPOP with PD-L1), highlighting potential false positives in complex cellular environments [27].
Advanced Applications and Modifications
Linkage-Specific Detection
While standard IP/immunoblotting does not inherently distinguish ubiquitin linkage types, researchers can incorporate linkage-specific reagents to gain this information:
Linkage-Specific Antibodies: Antibodies specifically recognizing K48, K63, M1, or other linkage types can be used in immunoblotting steps [6]. For example, K48-linkage specific antibodies have been used to demonstrate abnormal tau ubiquitination in Alzheimer's disease [6].
Tandem Ubiquitin-Binding Entities (TUBEs): TUBEs with specificity for particular chain types can be incorporated into the IP step. In a recent study, K63-TUBEs successfully captured L18-MDP-induced RIPK2 ubiquitination, while K48-TUBEs captured PROTAC-induced ubiquitination [26].
The following diagram illustrates how these advanced reagents enable linkage-specific detection:
Quantitative Dynamic Analysis
IP and immunoblotting can be adapted to monitor ubiquitination dynamics:
Time-Course Studies: Monitoring RIPK2 ubiquitination at 30 and 60 minutes after L18-MDP stimulation revealed higher ubiquitination at the earlier time point, demonstrating the method's applicability for kinetic studies [26].
Inhibitor Characterization: Pre-treatment with Ponatinib (100 nM) completely abrogated L18-MDP-induced RIPK2 ubiquitination, demonstrating how this approach can characterize ubiquitination pathway inhibitors [26].
The Scientist's Toolkit: Essential Research Reagents
Successful detection of ubiquitination in cultured cells requires specific reagents to preserve and detect this labile modification:
| Reagent/Category | Specific Examples | Function and Importance |
|---|---|---|
| Deubiquitinase Inhibitors | N-ethylmaleimide, Ubiquitin aldehyde | Prevent deubiquitination during cell lysis and processing [25] |
| Protease Inhibitors | PMSF, Complete Protease Inhibitor Cocktail | Prevent protein degradation during sample preparation [25] |
| Denaturing Lysis Buffer | 2% SDS, 150mM NaCl, 10mM Tris-HCl | Denatures proteins and disrupts non-covalent interactions [25] |
| Ubiquitin Antibodies | P4D1, FK1, FK2 | Detect ubiquitinated proteins in immunoblotting [6] |
| Linkage-Specific Reagents | K48-TUBEs, K63-TUBEs, linkage-specific antibodies | Enable discrimination of ubiquitin chain types [26] [6] |
| Positive Control Compounds | L18-MDP, PROTACs, MG-132 (proteasome inhibitor) | Induce ubiquitination for protocol validation [26] [28] |
Methodological Limitations and Considerations
Despite its widespread use, researchers must recognize several important limitations of the IP/immunoblotting approach:
Stoichiometry Challenges: Ubiquitination is typically a low-stoichiometry modification, with the majority of ubiquitination sites identified in proteomic studies showing low modification rates [28]. This can make detection challenging without enrichment or signal amplification.
Antibody Specificity Issues: Commercial ubiquitin antibodies vary considerably in their specificity and ability to recognize different ubiquitin chain types. Validation with appropriate controls is essential [6].
Linkage Discrimination Limitations: Standard IP/immunoblotting does not inherently distinguish between ubiquitin linkage types without specialized reagents [26] [6].
Throughput Constraints: The multi-step nature of IP and immunoblotting limits its throughput compared to plate-based or proteomic approaches [26].
Immunoprecipitation combined with immunoblotting remains a foundational methodology for detecting protein ubiquitination in cultured cells, offering robust target-specific detection with accessible laboratory equipment. While newer approaches like TUBE-based assays provide enhanced sensitivity and linkage specificity, and mass spectrometry enables global ubiquitinome profiling, the direct visual confirmation provided by immunoblotting continues to make it a verification standard in the field. By incorporating appropriate controls, DUB inhibitors, and potentially linkage-specific reagents, researchers can reliably employ this technique to investigate ubiquitination dynamics in physiological and pathological contexts.
In vitro reconstitution represents a foundational methodology in the study of the ubiquitin-proteasome system, enabling researchers to deconstruct complex cellular signaling into defined biochemical steps. This approach involves assaying ubiquitination using a complete set of purified components—E1 activating enzymes, E2 conjugating enzymes, E3 ligases, ubiquitin, and a substrate—in a controlled test tube environment [29]. By isolating the enzymatic cascade from the intricate cellular milieu, scientists can dissect precise molecular mechanisms, define minimal requirements for activity, and characterize the specificity of E3 ligases and their downstream effects [30] [27]. As research increasingly highlights the therapeutic potential of targeting ubiquitination pathways, from PROTACs to molecular glues [26], the role of in vitro reconstitution in validating and characterizing these mechanisms has never been more critical. This guide provides a comprehensive comparison of in vitro reconstitution methodologies, detailing experimental protocols, key findings, and strategic applications to empower researchers in selecting the optimal approach for their ubiquitination studies.
Core Principles and Methodologies of In Vitro Reconstitution
The Biochemical Foundation of the Ubiquitination Cascade
The ubiquitination cascade is a sequential process catalyzed by three enzyme families. The E1 ubiquitin-activating enzyme initiates the cycle in an ATP-dependent manner, forming a high-energy thioester bond with ubiquitin. The activated ubiquitin is then transferred to the catalytic cysteine of an E2 conjugating enzyme. Finally, an E3 ubiquitin ligase facilitates the transfer of ubiquitin from the E2 to a lysine ε-amino group on the target protein substrate, forming an isopeptide bond [4] [29]. HECT-type and RBR-type E3 ligases employ a unique catalytic mechanism, forming a transient thioester intermediate with ubiquitin before transferring it to the substrate, whereas RING-type E3s facilitate direct transfer from the E2 to the substrate [31] [32].
In vitro reconstitution replicates this cascade using recombinant proteins, allowing researchers to define the exact composition of the reaction mixture. This reductionist approach enables precise control over variables including enzyme concentrations, reaction conditions, and timing—parameters that are often difficult to manipulate in cellular environments [27]. The typical workflow begins with the preparation of recombinant enzymes and substrates, followed by assembly of the reaction mixture with essential co-factors (notably ATP), incubation under controlled conditions, and finally, termination of the reaction and analysis of products, typically via SDS-PAGE and Western blotting [33] [29].
Standardized Experimental Protocol for In Vitro Ubiquitination
The following protocol outlines the core steps for conducting an in vitro ubiquitination assay, synthesizing methodologies from multiple recent studies [27] [29]:
- Recombinant Protein Preparation: Express and purify the E1 enzyme, E2 enzyme, E3 ligase, and substrate protein. For transmembrane proteins like PD-L1, a recombinant cytoplasmic domain (e.g., amino acids 260-290) is often sufficient and simplifies purification [27].
- Reaction Mixture Assembly: Combine the following components in a reaction buffer:
- ATP-regenerating system (ATP, Mg²⁺)
- Recombinant ubiquitin (wild-type or mutant)
- E1 activating enzyme (e.g., Uba1)
- E2 conjugating enzyme (e.g., Ubc4, UbcH5c, or UBE2L3)
- E3 ubiquitin ligase
- Target substrate protein
- Incubation and Reaction Termination: Incubate the reaction mixture at 30°C for 30-90 minutes. Terminate the reaction by adding SDS-PAGE loading buffer and boiling for 5-10 minutes.
- Product Analysis: Resolve the reaction products by SDS-PAGE. Analyze ubiquitination using Western blotting with antibodies specific for ubiquitin (e.g., P4D1, FK1/FK2) or an epitope tag on the substrate (e.g., anti-HA, anti-biotin) [30] [27].
Table 1: Essential Components for a Standard In Vitro Ubiquitination Assay
| Component | Function | Example Molecules |
|---|---|---|
| E1 Enzyme | Activates ubiquitin in an ATP-dependent manner | Uba1 |
| E2 Enzyme | Carries activated ubiquitin | Ubc4, UbcH5c, CDC34B |
| E3 Ligase | Recognizes substrate and catalyzes ubiquitin transfer | Ufd4, TRIP12, ARIH1, NEDD4, HOIL-1 |
| Ubiquitin | The modifying protein | Wild-type Ub, Ub mutants (K29R, K48R, etc.) |
| Substrate | The target protein for modification | PD-L1 cytoplasmic domain, K48-linked diUb |
| Cofactors | Provides energy for the enzymatic reaction | ATP, Mg²⁺ |
Diagram 1: The core ubiquitination enzyme cascade. E1 activates ubiquitin in an ATP-dependent step, E2 carries the activated ubiquitin, and E3 facilitates final transfer to the substrate.
Comparative Analysis of In Vitro Applications and E3 Ligase Mechanisms
The true power of in vitro reconstitution emerges when comparing activities across different E3 ligase families and substrate types, revealing distinct mechanistic insights that are often obscured in cellular environments.
Recent structural and biochemical studies have meticulously delineated how different E3 ligases dictate ubiquitin chain topology. A prime example is the HECT-type E3 ligase Ufd4, which preferentially synthesizes K29-linked ubiquitin chains onto pre-existing K48-linked diubiquitin to form K29/K48-branched ubiquitin chains [31]. Biochemical assays demonstrated that ubiquitination efficiency escalated with increasing length of the K48-linked ubiquitin chain substrate (tri-, tetra-, penta-Ub). Enzyme kinetics further revealed a ~5.2-fold higher catalytic efficiency ((k{cat}/Km)) for the proximal K29 site (0.11 μM⁻¹ min⁻¹) compared to the distal K29 site (0.021 μM⁻¹ min⁻¹) within a K48-linked diUb substrate [31].
In contrast, studies on RBR-family E3 ligases like HOIL-1 reveal a capacity for non-canonical ubiquitination. HOIL-1 efficiently ubiquitinates serine residues and various saccharides in vitro but shows no activity toward lysine residues. This specificity is governed by a critical catalytic histidine residue (His510) in the flexible active site that enables O-linked ubiquitination while prohibiting ubiquitin discharge onto lysine side chains [32].
Table 2: Comparative Analysis of E3 Ligase Activities via In Vitro Reconstitution
| E3 Ligase | Ligase Type | Substrate | Key Findings | Linkage Preference |
|---|---|---|---|---|
| Ufd4/TRIP12 | HECT | K48-linked Ub chains | Preferentially extends K48 chains with K29 linkages; 5.2-fold preference for proximal K29 site [31] | K29-branched on K48 |
| ARIH1 | RBR | PD-L1 cytoplasmic domain | Directly ubiquitinates PD-L1; activity requires release from autoinhibition (e.g., S427D mutation) [27] | Not Specified |
| NEDD4 Family | HECT | PD-L1 cytoplasmic domain | Biochemically validated E3 activity toward PD-L1; redundant activities among family members [27] | Not Specified |
| HOIL-1 | RBR | Serine, Saccharides | Ubiquitinates hydroxyl groups of Ser/saccharides; no Lys activity; depends on His510 [32] | O-linked (Ser, sugars) |
| CRL3SPOP | RING | PD-L1 cytoplasmic domain | No direct ubiquitination observed in vitro despite cellular evidence; suggests need for co-factors [27] | Inactive in minimal system |
Elucidating Non-Canonical Mechanisms and Cooperative Catalysis
In vitro reconstitution has been instrumental in uncovering non-canonical ubiquitination mechanisms that diverge from the standard E1-E2-E3 paradigm. For instance, the RBR E3 ligase ARIH1 can function not only as an independent E3 but also as a "substrate receptor" that cooperates with Cullin RING ligases (CRLs) to form a super-assembly complex for ubiquitination [27]. This cooperative mechanism was elucidated through systematic in vitro mixing of purified components, demonstrating that ARIH1's autoinhibition could be released either by complex formation with neddylated CRLs or by phosphorylation at Ser427 [27].
Furthermore, in vitro studies with HOIL-1 have expanded the scope of ubiquitination beyond protein substrates. Using purified components, researchers demonstrated HOIL-1's ability to ubiquitinate diverse non-protein molecules including maltose, glycogen, and other physiologically relevant di- and monosaccharides [32]. This surprising activity, which could be enhanced using an engineered, constitutively active HOIL-1 variant, enables the production of ubiquitinated saccharides as tool compounds for studying this emerging field of non-proteinaceous ubiquitination.
Diagram 2: Comparative E3 ligase mechanisms. HECT and RBR E3s form transient thioester intermediates with ubiquitin, while RING E3s facilitate direct transfer from E2 to substrate. Some RBR E3s like HOIL-1 can ubiquitinate non-protein substrates.
The Scientist's Toolkit: Essential Reagents and Methodologies
Successful in vitro ubiquitination studies require careful selection of reagents and methodologies. The following toolkit summarizes critical components and their applications:
Table 3: Essential Research Reagent Solutions for In Vitro Ubiquitination Studies
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| E3 Ligases | Ufd4, TRIP12, ARIH1, NEDD4, HOIL-1 | Catalyze substrate-specific ubiquitination; different types (HECT, RBR, RING) employ distinct mechanisms [31] [27] [32] |
| Ubiquitin Variants | Wild-type Ub, K29R Ub, K48-only Ub | Determine linkage specificity; mutant ubiquitins (e.g., K29R) help identify chain topology [31] |
| Activation Systems | S427D-ARIH1, Engineered HOIL-1 | Constitutively active E3 variants bypass regulatory mechanisms for simplified in vitro studies [27] [32] |
| Substrate Design | PD-L1 cytoplasmic domain, K48-linked diUb | Recombinant substrates (often truncated) maintain functionality while simplifying purification and handling [31] [27] |
| Detection Reagents | Anti-ubiquitin antibodies (P4D1, FK2), Anti-HA, HRP-streptavidin | Enable detection and quantification of ubiquitination products via Western blot [30] [27] |
In vitro reconstitution stands as an indispensable methodology for mechanistically dissecting the ubiquitination cascade, offering unparalleled control and specificity in characterizing E3 ligase activities, substrate preferences, and linkage specificities. The experimental data and comparative analyses presented herein demonstrate how this reductionist approach enables researchers to validate direct E3-substrate relationships, elucidate non-canonical mechanisms of ubiquitination, and characterize the formation of specific ubiquitin chain topologies—findings that often form the foundational validation for subsequent cellular and in vivo studies. While this minimal system approach necessarily excludes the complex regulatory networks present in living cells, its power lies precisely in this simplification, allowing researchers to build mechanistic understanding from the ground up before reintegrating these components into more complex biological contexts. For researchers embarking on ubiquitination studies, particularly in the characterization of novel E3 ligases or the development of targeted degradation technologies, in vitro reconstitution provides the critical biochemical foundation upon which robust and reproducible scientific conclusions can be built.
Ubiquitination, a fundamental post-translational modification, involves the covalent attachment of ubiquitin to substrate proteins, regulating diverse cellular functions from protein degradation to DNA repair and cell signaling [4] [6]. The complexity of ubiquitin signaling arises from the ability to form polyubiquitin chains with different linkage types, each dictating distinct functional outcomes for the modified substrate [4]. For instance, K48-linked chains primarily target proteins for proteasomal degradation, while K63-linked chains regulate non-proteolytic processes like inflammatory signaling and protein trafficking [34] [26]. Dysregulation of ubiquitination pathways contributes to various pathologies, including cancers and neurodegenerative diseases, making accurate detection crucial for both basic research and drug development [4] [6].
The critical challenge in ubiquitination research lies in detecting and characterizing these modifications with specificity, sensitivity, and physiological relevance. Traditional methods like immunoblotting have limitations in throughput and specificity, while mass spectrometry approaches require sophisticated instrumentation and can lack sensitivity for dynamic changes [6] [26]. This comparison guide examines two advanced enrichment technologies—Tandem Ubiquitin Binding Entities (TUBEs) and linkage-specific antibodies—evaluating their performance in both in vivo and in vitro research contexts to inform selection for different experimental needs.
Linkage-Specific Antibodies
Linkage-specific antibodies represent a targeted approach for ubiquitin detection, generating immunological reagents that recognize specific ubiquitin chain architectures. These antibodies are developed to distinguish between different ubiquitin linkage types (M1, K6, K11, K27, K29, K33, K48, K63) through selective binding to unique epitopes presented by each chain configuration [6]. The application scope encompasses both enrichment of ubiquitinated proteins from complex mixtures and detection in techniques like Western blotting and immunofluorescence [6]. For researchers studying specific biological processes known to involve particular ubiquitin linkages, these antibodies provide targeted insight into specialized ubiquitin signaling pathways.
Tandem Ubiquitin Binding Entities (TUBEs)
TUBEs represent an engineered approach to ubiquitin detection, constructed as recombinant proteins comprising multiple ubiquitin-associated (UBA) domains arranged in tandem [6] [34]. This multivalent architecture significantly enhances affinity for polyubiquitin chains through avidity effects, with reported affinities reaching the sub-nanomolar range [34] [26]. TUBEs are available in both pan-selective variants that recognize all ubiquitin chain types and linkage-specific formulations designed to capture particular chain architectures [34] [26]. A key advantage of TUBEs is their ability to protect ubiquitin chains from deubiquitinating enzyme (DUB) activity during cell lysis and processing, preserving the native ubiquitination status of proteins [26]. This feature makes TUBEs particularly valuable for capturing transient or low-abundance ubiquitination events that might otherwise be lost during sample preparation.
Performance Comparison & Experimental Data
Technical Specifications Comparison
Table 1: Direct comparison of key technical parameters between TUBEs and linkage-specific antibodies
| Parameter | TUBEs | Linkage-Specific Antibodies |
|---|---|---|
| Affinity | Sub-nanomolar to nanomolar range [34] [26] | Variable; dependent on antibody generation |
| Throughput Capacity | Adaptable to 96-well plate HTS formats [35] [26] | Limited by conventional immunoassays |
| Linkage Specificity | Available in pan-selective and linkage-specific variants [34] [26] | Highly specific to designated linkage types [6] |
| Detection Universality | Broad detection of polyubiquitin chains [35] | Restricted to recognized linkage type |
| DUB Protection | Yes; protects chains during processing [26] | No inherent protection capability |
| Applicability to Native Systems | Suitable for endogenous proteins in cells and tissues [35] [26] | Requires validation for endogenous detection |
Experimental Performance Data
Recent studies demonstrate the utility of both technologies in specific research contexts. TUBE-based platforms have successfully differentiated context-dependent ubiquitination events, as shown in research on RIPK2, a key regulator of inflammatory signaling. K63-selective TUBEs specifically captured L18-MDP-induced ubiquitination of endogenous RIPK2, while K48-selective TUBEs captured RIPK2 ubiquitination induced by a PROTAC molecule, demonstrating precise linkage discrimination in cellular environments [26]. The technology has been validated for high-throughput applications, with ThUBD (tandem hybrid ubiquitin-binding domain)-coated multi-well plates showing strong universality and specificity in detecting ubiquitination signals across diverse biological samples including cells, tissues, and urine [35].
Linkage-specific antibodies continue to provide critical insights in specialized applications. For example, K48-linkage specific antibodies revealed abnormal accumulation of K48-linked polyubiquitination on tau proteins in Alzheimer's disease models, contributing to understanding disease mechanisms [6]. The technology enables investigation of endogenous protein ubiquitination in native tissue and clinical samples without genetic manipulation, maintaining physiological relevance [6].
Table 2: Summary of quantitative performance data from application studies
| Application Context | Technology Used | Performance Outcome | Reference |
|---|---|---|---|
| RIPK2 Ubiquitination Profiling | K48 & K63 TUBEs | Selective capture of linkage-specific endogenous ubiquitination | [26] |
| Ubiquitination in Biological Samples | ThUBD multi-well plates | Accurate quantification across cells, tissues, urine | [35] |
| Tau Protein Ubiquitination in Alzheimer's | K48-linkage specific antibody | Detection of pathological ubiquitination accumulation | [6] |
| High-Throughput Drug Screening | TUBE-based platform | Enabled HTS for molecular glues and protein degraders | [34] |
Detailed Experimental Protocols
TUBE-Based Enrichment for Endogenous Ubiquitination Analysis
The following protocol adapts methodology from recent studies investigating linkage-specific ubiquitination of endogenous proteins [26]:
Cell Stimulation and Lysis:
- Culture THP-1 cells in appropriate medium and stimulate with desired agonist (e.g., 200-500 ng/mL L18-MDP for RIPK2 ubiquitination) for specified duration (e.g., 30-60 minutes) [26].
- Prepare lysis buffer optimized to preserve polyubiquitination: 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% NP-40, 1 mM EDTA, supplemented with fresh protease inhibitors (e.g., 1 mM PMSF) and deubiquitinase inhibitors (e.g., 5 mM N-ethylmaleimide) [26] [25].
- Lyse cells using 0.5-1 mL buffer per 10⁷ cells, incubate on ice for 15-30 minutes with occasional vortexing, then clarify by centrifugation at 14,000 × g for 15 minutes at 4°C.
Ubiquitin Affinity Enrichment:
- Transfer clarified supernatant to fresh tubes containing chain-specific TUBE-coated magnetic beads (e.g., K48-TUBE or K63-TUBE based on experimental goals) [26].
- Rotate incubation at 4°C for 2-4 hours to facilitate binding.
- Wash beads three times with wash buffer (e.g., 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.1% NP-40) with stringent washing including high-salt conditions if needed (e.g., 1 M NaCl) to reduce non-specific binding [25].
Detection and Analysis:
- Elute bound proteins by boiling in 2× SDS-PAGE sample buffer for 10 minutes [25].
- Separate by SDS-PAGE and transfer to PVDF membrane for immunoblotting.
- Probe with target protein-specific antibody (e.g., anti-RIPK2) to detect ubiquitinated species, typically appearing as higher molecular weight smears or ladders [26] [25].
Experimental workflow for TUBE-based ubiquitin enrichment
Linkage-Specific Antibody-Based Detection
This protocol outlines the application of linkage-specific antibodies for ubiquitination detection, adapted from conventional immunoblotting and immunoprecipitation methods [6] [25]:
Cell Processing and Lysis:
- Transfert cells with plasmids expressing protein of interest and epitope-tagged ubiquitin if studying overexpression systems [25].
- For stringent lysis, use complete cell lysis buffer (2% SDS, 150 mM NaCl, 10 mM Tris-HCl, pH 8.0) with protease inhibitors and DUB inhibitors [25].
- Boil cells immediately for 10 minutes to denature proteins and inactivate DUBs, then shear DNA by sonication.
- Dilute lysate 10-fold with dilution buffer (10 mM Tris-HCl, pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% Triton) to reduce SDS concentration for compatibility with immunoprecipitation [25].
Immunoprecipitation:
- Pre-clear lysate with Protein A/G beads for 30 minutes at 4°C.
- Incubate with linkage-specific ubiquitin antibody (e.g., K48- or K63-specific) or target protein antibody overnight at 4°C with rotation.
- Add Protein A/G-agarose beads (50% slurry) and incubate for additional 2-4 hours.
- Wash beads extensively with washing buffer (10 mM Tris-HCl, pH 8.0, 1 M NaCl, 1 mM EDTA, 1% NP-40) to remove non-specifically bound proteins [25].
Analysis:
- Elute proteins by boiling in 2× SDS loading buffer.
- Resolve by SDS-PAGE and transfer to membrane.
- Probe with appropriate antibodies to detect ubiquitinated proteins or specific targets.
Research Reagent Solutions
Table 3: Essential research reagents for ubiquitination detection studies
| Reagent/Category | Specific Examples | Function & Application |
|---|---|---|
| Chain-Selective TUBEs | K48-TUBE, K63-TUBE, Pan-TUBE [34] [26] | Linkage-specific enrichment of ubiquitinated proteins from complex mixtures |
| Linkage-Specific Antibodies | K48-linkage specific, K63-linkage specific [6] | Detection and immunoprecipitation of specific ubiquitin chain types |
| DUB Inhibitors | N-ethylmaleimide, Ubiquitin aldehyde [25] | Preserve ubiquitination status during sample processing |
| Ubiquitination Assay Kits | PROTAC In vitro Ubiquitination Assay Kit (PA770) [34] | Configured systems for in vitro ubiquitination reactions |
| Positive Control Reagents | L18-MDP (RIPK2 ubiquitination inducer) [26] | Experimental controls for assay validation |
Application Scenarios & Decision Framework
Technology Selection Guide
Choose TUBEs when:
- Studying endogenous ubiquitination without genetic manipulation [35] [26]
- Working with easily degradable ubiquitin chains requiring DUB protection [26]
- Conducting high-throughput screening of ubiquitination modulators [34] [26]
- Analyzing diverse biological samples including tissues and body fluids [35]
- Seeking broad detection of multiple polyubiquitin chain types [35]
Choose Linkage-Specific Antibodies when:
- Investigating specific ubiquitin linkage types with established high-quality antibodies [6]
- Working with fixed samples for immunohistochemistry applications
- Budget constraints limit reagent options
- Studying well-characterized ubiquitination events with known linkage involvement
Emerging Applications & Future Directions
The application of these technologies continues to expand into novel research areas. TUBEs are increasingly employed in drug discovery campaigns targeting the ubiquitin-proteasome system, particularly for characterizing PROTACs (Proteolysis Targeting Chimeras) and molecular glues that redirect E3 ligase activity [34] [26]. The ability to monitor linkage-specific ubiquitination in high-throughput formats enables screening for compounds that induce specific ubiquitin chain types on target proteins [26]. Meanwhile, linkage-specific antibodies remain valuable for diagnostic applications and validating ubiquitination patterns in pathological tissue samples [6]. Emerging engineering applications also leverage ubiquitination machinery, such as the development of "ubi-tagging" for site-specific antibody conjugation, which utilizes ubiquitin's enzymatic machinery for bioconjugation [36].
TUBEs and linkage-specific antibodies represent complementary advanced tools for ubiquitination research, each with distinct advantages for specific research scenarios. TUBEs offer superior capabilities for protecting labile ubiquitin modifications, detecting endogenous proteins across diverse sample types, and adapting to high-throughput screening formats essential for modern drug discovery. Linkage-specific antibodies provide targeted insight into specific ubiquitin chain types and maintain utility for applications requiring immunohistochemistry or benefiting from established immunoassay workflows. The optimal technology selection depends on specific research goals, with TUBEs generally preferable for discovery-phase research and systems-level investigation, while linkage-specific antibodies remain valuable for focused studies of specific ubiquitin linkages. As ubiquitination continues to emerge as a therapeutic target across multiple disease areas, appropriate application of these enrichment tools will remain crucial for advancing both basic understanding and drug development efforts.
Targeted protein degradation (TPD) represents a revolutionary approach in drug discovery, with proteolysis-targeting chimeras (PROTACs) emerging as a promising modality for therapeutic development [37]. Unlike conventional small-molecule inhibitors that merely block protein activity, PROTACs catalyze the complete degradation of target proteins through the ubiquitin-proteasome system, effectively eliminating all functions of the protein, including scaffolding activities [37]. This novel mechanism enables targeting of proteins previously considered "undruggable," expanding the therapeutic landscape [37]. The efficacy of PROTACs is not measured by traditional target occupancy but by degradation parameters such as DC50 (concentration for 50% degradation) and Dmax (maximal degradation achieved) [37]. Evaluating these parameters efficiently requires robust high-throughput and real-time assay systems that can rapidly screen compound libraries and provide quantitative degradation metrics, accelerating the development of this innovative therapeutic class.
High-Throughput Assay Platforms for PROTAC Screening
Cellular Degradation Assays
Advanced cellular assay technologies enable researchers to efficiently quantify PROTAC-mediated degradation in high-throughput formats. These systems move beyond traditional western blotting to provide more rapid and quantitative assessment of degradation efficacy [37]. Luminescence-based assays have been particularly valuable in this context, offering real-time or endpoint measurements of protein levels with sensitivity compatible with automated screening platforms [37]. These assays can be configured to measure the cellular concentration of the protein of interest (POI) directly, allowing determination of key degradation parameters including DC50 and Dmax values that define PROTAC potency and efficacy [37]. The implementation of direct-to-biology (D2B) approaches has further streamlined PROTAC development by enabling high-throughput evaluation of compound activity in cellular contexts without intermediate purification steps [37]. These integrated screening approaches allow researchers to rapidly triage potential PROTAC molecules based on their functional degradation activity rather than mere binding affinity.
Table 1: Comparison of Cellular Assay Technologies for PROTAC Screening
| Assay Technology | Throughput | Quantitative Output | Key Advantages | Limitations |
|---|---|---|---|---|
| Western Blotting | Low | Semi-quantitative | Direct visualization of protein degradation; widely accessible | Low throughput; difficult to quantify precisely |
| Luminescence-Based Assays | High | Quantitative (DC50, Dmax) | High sensitivity; compatible with automation; real-time capability | May require genetic engineering of reporter constructs |
| Direct-to-Biology (D2B) | High | Functional activity ranking | Streamlined workflow; minimal compound manipulation | May have higher variability than purified systems |
Labeling and Detection Strategies
Effective detection of protein degradation requires sophisticated labeling strategies that maintain physiological relevance while enabling quantitative measurement. Tag-targeted protein degrader (tTPD) systems provide a valuable approach for pre-assessing target degradability using fusion proteins such as dTAGs, BromoTAGs, and HaloTAGs [37]. These systems employ a "mutant protein" platform for which TPD-inducing ligands are already available, allowing simulation of degradation and its functional consequences before developing target-specific PROTACs [37]. For endogenous proteins, antibody-based detection methods remain fundamental, with recent advances improving their compatibility with high-throughput platforms. Furthermore, the development of linkage-specific ubiquitin antibodies enables researchers to characterize the specific ubiquitin chain topology involved in PROTAC-mediated degradation, providing mechanistic insights alongside efficacy data [6]. These detection strategies form the technological foundation for high-content screening campaigns aimed at identifying novel degraders with optimal efficacy and selectivity profiles.
Methodologies for Ubiquitination Detection: In Vivo vs. In Vitro
In Vivo Ubiquitination Detection Techniques
In vivo ubiquitination detection methods preserve the physiological context of the ubiquitination process, capturing the complex cellular environment in which PROTACs operate. Tandem ubiquitin-binding entities (TUBEs) represent a powerful approach for isolating polyubiquitinated proteins directly from cellular lysates [6]. These tools, based on ubiquitin-associated (UBA) domains, protect polyubiquitin chains from deubiquitinating enzymes (DUBs) and proteasomal degradation, effectively stabilizing otherwise transient ubiquitination events for detection and analysis [38]. The trypsin-resistant TR-TUBE variant can interact with all eight types of polyubiquitin chain linkages (M1, K6, K11, K27, K29, K33, K48, and K63), providing comprehensive coverage of ubiquitin signaling [38]. When combined with mass spectrometry, TR-TUBE enables identification of endogenous substrates ubiquitinated by specific E3 ligases in their natural cellular environment [38]. Immunoaffinity enrichment using antibodies targeting the diglycine (K-ε-GG) remnant left on trypsinized ubiquitinated peptides provides another powerful in vivo approach, enabling large-scale mapping of ubiquitination sites from cellular lysates [28]. This method has been successfully employed to study global ubiquitination changes in response to proteasome inhibition (MG-132) and deubiquitinase inhibition (PR-619), detecting up to approximately 3300 distinct K-ε-GG peptides from human Jurkat cells [28].
Diagram 1: In vivo ubiquitination detection workflow
In Vitro Ubiquitination Detection Techniques
In vitro ubiquitination detection methods provide controlled environments for mechanistic studies but lack the full complexity of cellular systems. Reconstituted ubiquitination systems utilize purified E1, E2, and E3 enzymes along with ubiquitin, ATP, and candidate substrate proteins to demonstrate direct ubiquitination capability [38]. While this approach offers precision in defining biochemical requirements, it may lack physiological relevance as post-translational modifications or protein complexes present in cells may be necessary for proper ubiquitination [38]. Immunoblotting with ubiquitin antibodies following immunoprecipitation represents a widely used in vitro method, though it can produce artifacts from non-specific ubiquitination or co-precipitating proteins [38]. To address these limitations, researchers have developed tagged ubiquitin systems (e.g., His-, HA-, Flag-tagged Ub) that enable affinity purification of ubiquitinated proteins from overexpression systems, often in the presence of proteasome inhibitors to stabilize ubiquitin conjugates [6]. However, this method may not accurately reflect endogenous ubiquitination dynamics due to tag-induced structural alterations and the potential for artifacts from overexpression [6].
Table 2: Comparison of In Vivo vs. In Vitro Ubiquitination Detection Methods
| Parameter | In Vivo Methods | In Vitro Methods |
|---|---|---|
| Physiological Relevance | High (cellular context preserved) | Limited (purified components) |
| Mechanistic Control | Limited (multiple variables) | High (defined components) |
| Throughput Capacity | Moderate to High | Low to Moderate |
| Technical Complexity | Higher | Lower |
| Identification of Endogenous Substrates | Yes | No |
| Requirement for Genetic Manipulation | Often required | Not required |
| Ability to Capture Post-Translational Regulation | Yes | No |
Diagram 2: Comparison of in vivo and in vitro approaches
Advanced Screening Methodologies for Degrader Discovery
Unbiased Cellular Screening Approaches
Unbiased phenotypic screening represents a powerful strategy for identifying novel monovalent degraders that leverage diverse cellular mechanisms for target protein degradation. Unlike knowledge-based approaches that derivatize known ligands, unbiased screening of diverse compound libraries takes a ligand-agnostic approach, harnessing the broad range of E3 ligases and endogenous cellular pathways capable of inducing degradation [39]. These live-cell screens can uncover compounds that trigger degradation through varied mechanisms, including direct molecular glue degraders (MGDs), adaptor MGDs, and allosteric degraders [39]. For example, the identification of (S)-ACE-OH, a metabolite of acepromazine that functions as a molecular glue degrader of nuclear pore proteins via TRIM21 recruitment, was achieved through phenotypic high-throughput screening using cell viability as a readout [39]. Similarly, CRISPR screening approaches can identify E3 ligases essential for degrader activity, providing mechanistic insights alongside efficacy data [39]. These unbiased methods are particularly valuable for discovering compounds with favorable drug-like properties, as monovalent degraders typically exhibit better pharmacokinetic profiles than their bifunctional PROTAC counterparts [39].
Proteomics and Chemoproteomics in Degrader Screening
Mass spectrometry-based proteomic approaches have revolutionized degrader discovery by enabling global analysis of protein ubiquitination and degradation events. Quantitative proteomics using immunoaffinity enrichment of K-ε-GG peptides allows comprehensive mapping of ubiquitination sites and their dynamics in response to degrader treatment or pathway perturbation [28]. This approach has been successfully applied to study the effects of proteasome and deubiquitinase inhibition, revealing that not all regulated ubiquitination sites are necessarily proteasomal substrates [28]. Chemoproteomics combines phenotypic screening with target identification, as demonstrated in the discovery of EN450, a covalent ligand that modifies the E2 enzyme UBE2D and induces NF-κB degradation [39]. This integrated approach enables simultaneous assessment of compound efficacy and mechanism of action, accelerating the development of novel degraders. Furthermore, advances in sample preparation, including minimal fractionation prior to immunoaffinity enrichment, have significantly improved the yield of K-ε-GG peptides, enabling detection of up to approximately 3300 distinct ubiquitination sites in single experiments [28]. These proteomic technologies provide unprecedented resolution for understanding the molecular mechanisms underlying targeted protein degradation.
Experimental Protocols for Key Ubiquitination Assays
TR-TUBE Protocol for Detection of Endogenous Ubiquitination
The TR-TUBE method enables sensitive detection of ubiquitin ligase activity and identification of endogenous substrates by protecting polyubiquitin chains from deubiquitinating enzymes and proteasomal degradation [38].
Materials Required:
- Plasmid encoding TR-TUBE with affinity tag (e.g., FLAG-tagged TR-TUBE)
- Transfection reagent
- Cell culture materials for chosen cell line
- Lysis buffer (HEPES-Triton buffer with protease inhibitors)
- 1 mM N-ethylmaleimide (NEM, deubiquitinase inhibitor)
- 10 μM MG132 (proteasome inhibitor)
- Anti-FLAG affinity resin
- Wash buffer (lysis buffer with 300-500 mM NaCl)
- Elution buffer (with FLAG peptide or 2× SDS sample buffer)
- SDS-PAGE and western blot equipment
- Ubiquitin antibodies for detection
Procedure:
- Transfect cells with TR-TUBE plasmid using standard transfection protocols.
- At 24-48 hours post-transfection, treat cells with proteasome inhibitor (MG132) if needed for 4-6 hours before harvesting.
- Harvest cells in HEPES-Triton lysis buffer containing 1 mM NEM and 10 μM MG132 to preserve ubiquitination.
- Incubate lysates with anti-FLAG affinity resin for 2-4 hours at 4°C with gentle rotation.
- Wash resin thoroughly with high-salt wash buffer (300-500 mM NaCl) to reduce non-specific binding.
- Elute bound proteins using FLAG peptide competition or direct elution with SDS sample buffer.
- Analyze eluates by SDS-PAGE and western blotting using appropriate ubiquitin or substrate-specific antibodies.
Applications: This protocol effectively detects ubiquitination of endogenous proteins by specific E3 ligases. For example, TR-TUBE has been successfully used to detect ubiquitination of endogenous p27 by overexpressed SKP2, demonstrating superior sensitivity compared to conventional ubiquitin overexpression methods [38].
K-ε-GG Immunoaffinity Enrichment for Ubiquitinomics
This mass spectrometry-based protocol enables large-scale identification and quantification of ubiquitination sites from cell lysates.
Materials Required:
- Cell culture and lysis reagents
- Trypsin for protein digestion
- Anti-K-ε-GG antibody (commercially available)
- Protein A/G agarose beads
- Urea lysis buffer (8M urea in 50 mM Tris pH 8.0)
- Reduction and alkylation reagents (DTT and iodoacetamide)
- Desalting columns (C18 or StageTips)
- High-performance mass spectrometry system
Procedure:
- Lyse cells in urea buffer and quantify protein concentration.
- Reduce disulfide bonds with 5 mM DTT (30 min, 25°C) and alkylate with 15 mM iodoacetamide (30 min, 25°C in dark).
- Dilute urea concentration to 2M and digest proteins with trypsin (1:50 w/w) overnight at 25°C.
- Acidify digests to pH < 3 with trifluoroacetic acid and desalt using C18 columns.
- Immunoaffinity purify K-ε-GG-containing peptides using anti-K-ε-GG antibody coupled to protein A/G beads (2 hours at 4°C).
- Wash beads extensively with ice-cold PBS and elute with 0.1% trifluoroacetic acid.
- Analyze eluted peptides by liquid chromatography coupled to tandem mass spectrometry.
Applications: This approach has been used to identify over 4900 distinct K-ε-GG peptides in SILAC-encoded experiments, enabling quantitative assessment of ubiquitination changes in response to proteasome inhibition (MG-132) and deubiquitinase inhibition (PR-619) [28]. The method provides comprehensive profiling of the ubiquitin landscape under various perturbational conditions.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Ubiquitination and Degradation Studies
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Ubiquitin Enrichment Tools | TR-TUBE [38], Anti-K-ε-GG antibodies [28], Linkage-specific ubiquitin antibodies [6] | Isolation and detection of ubiquitinated proteins from complex mixtures |
| Affinity Tags | His-tag [6], Strep-tag [6], FLAG-tag [38] | Purification of tagged ubiquitin or ubiquitinated proteins |
| Enzyme Inhibitors | MG-132 (proteasome) [28], PR-619 (deubiquitinase) [28], MLN4924 (NAE1) [4] | Pathway perturbation to stabilize ubiquitinated proteins or study mechanism |
| Cellular Reporters | Luminescence-based degradation reporters [37], Tag-targeted systems (dTAG, HaloTag) [37] | Real-time monitoring of protein degradation in live cells |
| Mass Spectrometry | Quantitative proteomics platforms, SILAC encoding [28], TMT labeling | Global identification and quantification of ubiquitination sites |
| E3 Ligase Modulators | Thalidomide derivatives (cereblon) [39], VHL ligands [37], TRIM21 ligands [39] | PROTAC warheads for recruiting specific ubiquitin ligases |
High-throughput and real-time assay technologies have become indispensable tools in the PROTAC drug discovery pipeline, enabling rapid evaluation of degradation efficacy and mechanism. The complementary application of in vivo and in vitro ubiquitination detection methods provides both physiological relevance and mechanistic precision, offering researchers a comprehensive toolkit for degrader development. As the field advances, integration of unbiased cellular screening with sophisticated proteomic technologies will likely yield novel degraders targeting previously inaccessible proteins, expanding the therapeutic potential of targeted protein degradation. The continued refinement of these assay platforms, particularly through enhanced throughput, sensitivity, and real-time monitoring capabilities, will accelerate the development of this promising class of therapeutic agents.
Ubiquitination is a crucial post-translational modification (PTM) that regulates diverse cellular processes, including protein degradation, cell cycle progression, DNA damage repair, and signal transduction [4]. This modification involves the covalent attachment of ubiquitin, a 76-amino acid protein, to substrate proteins via a three-enzyme cascade consisting of ubiquitin-activating (E1), conjugating (E2), and ligating (E3) enzymes [25] [6]. The complexity of ubiquitin signaling arises from its ability to form various chain architectures and linkages, with eight distinct polyubiquitin linkage types identified (K6, K11, K27, K29, K33, K48, K63, and M1), each associated with specific cellular functions [4].
Mass spectrometry-based proteomics has revolutionized the study of protein ubiquitination on a global scale. Traditional methods like immunoblotting have provided valuable insights but are limited by low throughput and an inability to precisely map modification sites [6]. The development of sophisticated enrichment strategies and advanced mass spectrometry techniques now enables researchers to identify thousands of ubiquitination sites in a single experiment, dramatically accelerating our understanding of the "ubiquitinome" [40] [41] [16]. This guide objectively compares the current methodologies for global ubiquitination site profiling, with a particular focus on the critical comparison between in vivo and in vitro approaches, providing researchers with experimental data and protocols to inform their study designs.
Key Methodological Approaches for Ubiquitinome Profiling
The evolution of ubiquitination site profiling has been marked by several technological breakthroughs, with current methods primarily relying on the enrichment of ubiquitinated peptides followed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis [42] [40]. The cornerstone of modern ubiquitinomics is the immunoaffinity enrichment of peptides containing the di-glycine (K-ε-GG) remnant, which remains attached to modified lysine residues after tryptic digestion [42] [40] [43]. This section compares the principal approaches, while subsequent sections will detail their application in in vivo versus in vitro contexts.
Table 1: Comparison of Primary Methods for Ubiquitination Site Profiling
| Method | Principle | Throughput | Sites Identified | Key Advantages | Major Limitations |
|---|---|---|---|---|---|
| K-ε-GG Antibody Enrichment | Immunoaffinity purification of tryptic peptides containing di-glycine remnant | High | >10,000-70,000 sites [40] [16] | High specificity and sensitivity; applicable to any biological sample | Cannot distinguish ubiquitination from other Ub-like modifications (ISG15, NEDD8) [43] |
| Tagged Ubiquitin Expression | Expression of epitope-tagged ubiquitin (His, HA, Strep) in cells | Medium | ~500-1,100 sites [43] | Effective enrichment at protein level; controlled expression possible | May interfere with normal cellular functions; not applicable to clinical samples [43] |
| UBD-Based Enrichment | Use of Ubiquitin-Binding Domains (e.g., TUBEs) to purify ubiquitinated proteins | Medium | ~300 sites [43] | Preserves ubiquitin chain architecture; works under native conditions | Bias toward polyubiquitinated substrates; co-purification of binding partners [43] |
In Vivo Ubiquitination Detection Techniques
In vivo ubiquitination analysis provides a comprehensive view of ubiquitination events within their native cellular context, preserving the physiological relevance of the modifications. The dominant methodology for large-scale in vivo profiling centers on the K-ε-GG antibody enrichment approach, which has been extensively optimized for sensitivity and coverage.
Standard Protocol for Global In Vivo Ubiquitination Site Mapping
The following protocol represents the current state-of-the-art for in vivo ubiquitinome profiling, incorporating key improvements from recent methodological advances [40] [16]:
- Cell Lysis and Protein Extraction: Use SDC (sodium deoxycholate) lysis buffer supplemented with chloroacetamide (CAA) for immediate cysteine protease inactivation. SDC-based lysis has been shown to increase K-ε-GG peptide identification by 38% compared to conventional urea buffers [16].
- Protein Digestion: Digest proteins with LysC and trypsin to generate peptides. Trypsin cleaves after the arginine residue following the two C-terminal glycines of ubiquitin, leaving the diagnostic di-glycine remnant on modified lysines [40].
- Peptide Fractionation: Perform basic pH reversed-phase (bRP) chromatography to reduce sample complexity. This step significantly increases the number of quantified K-ε-GG sites in SILAC-labeled samples [40].
- K-ε-GG Peptide Enrichment: Incubate peptides with anti-K-ε-GG antibody conjugated to beads. Chemical cross-linking of the antibody to the solid support reduces contamination from antibody fragments [40].
- LC-MS/MS Analysis: Analyze enriched peptides using liquid chromatography coupled to tandem mass spectrometry. Data-independent acquisition (DIA) methods more than triple identification numbers compared to data-dependent acquisition (DDA), with demonstrated quantification of up to 70,000 ubiquitinated peptides in single MS runs [16].
Technical Advances in In Vivo Profiling
Recent methodological innovations have substantially improved the depth and precision of in vivo ubiquitinome analyses:
- SDC Lysis Protocol: The introduction of SDC-based lysis with immediate boiling and high concentrations of chloroacetamide (CAA) improves ubiquitin site coverage while avoiding di-carbamidomethylation artifacts that can occur with iodoacetamide [16].
- DIA-MS with Neural Network Processing: Coupling data-independent acquisition mass spectrometry with deep neural network-based data processing (DIA-NN) boosts reproducibility, identification numbers, and quantitative accuracy compared to traditional DDA methods [16].
- Cross-linked Antibody Beads: Chemical cross-linking of the anti-K-ε-GG antibody to beads significantly reduces contamination in final enriched samples [40].
- SILAC Quantification: Incorporation of Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC) enables relative quantification of ubiquitination changes across different cellular states [40].
The following diagram illustrates the optimized workflow for in vivo ubiquitinome profiling:
Figure 1: Workflow for in vivo ubiquitinome profiling incorporating recent optimizations including SDC lysis and DIA-MS analysis.
Performance Metrics and Applications
The performance of in vivo ubiquitination profiling is demonstrated through multiple large-scale studies:
- Wagner et al. identified 11,054 ubiquitination sites on 4,273 proteins in human cells by combining immunoenrichment with peptide fractionation and high-throughput MS [41].
- In rice panicles, researchers identified 1,638 ubiquitination sites on 916 unique proteins, representing the largest ubiquitinome dataset in plants to date [41].
- A time-resolved in vivo ubiquitinome profiling study upon USP7 inhibition simultaneously recorded ubiquitination changes for more than 8,000 proteins at high temporal resolution [16].
In Vitro Ubiquitination Detection Techniques
In vitro ubiquitination assays provide a reductionist approach to study specific aspects of the ubiquitination machinery under controlled conditions, free from the complexity of cellular environment.
Protocol for In Vitro Ubiquitination Assay
The in vitro ubiquitination assay allows researchers to reconstitute ubiquitination using purified components [25]:
Reaction Setup: For each 40 μL reaction, combine:
- 8 μL 5X ubiquitination buffer (100 mM Tris-HCl, pH 7.5, 25 mM MgCl2, 2.5 mM DTT, 10 mM ATP)
- 250 ng ubiquitination E1 enzyme
- 500 ng ubiquitination E2 enzyme
- 0.5-1.0 μg E3 ligase (if testing E3 specificity)
- 0.5 μg ubiquitin
- 0.5 μg protein of interest
- Water to 40 μL total volume
Control Reactions: Prepare control reactions omitting E1, E2, E3, or ubiquitin to confirm specificity.
Incubation: Incubate the reaction mixture at 37°C for 1 hour or longer.
Termination and Analysis: Stop the reaction by adding SDS-PAGE sample buffer and boiling for 10 minutes. Analyze by immunoblotting with anti-ubiquitin and target protein antibodies.
Technical Considerations for In Vitro Assays
- ATP Regeneration: Some protocols add creatine phosphate and creatine kinase to the buffer for ATP regeneration [25].
- Deubiquitination Prevention: Include deubiquitinating enzyme inhibitors such as N-ethylmaleimide or ubiquitin aldehyde in all buffers to prevent deubiquitination [25].
- Stringent Detection: Use harsh conditions for cell lysis, immunoprecipitation, and washing to ensure detected ubiquitination is specific to the target protein rather than non-covalently interacting proteins [25].
The simplified workflow for in vitro ubiquitination analysis is illustrated below:
Figure 2: Workflow for in vitro ubiquitination assays using purified system components.
Comparative Analysis: In Vivo vs. In Vitro Approaches
Understanding the strengths and limitations of each approach is essential for selecting the appropriate methodology for specific research questions. The quantitative comparison below highlights key performance differences.
Table 2: Performance Comparison of In Vivo vs. In Vitro Ubiquitination Detection
| Parameter | In Vivo Profiling | In Vitro Assays |
|---|---|---|
| Physiological Relevance | High - captures modifications in native cellular context | Limited - reduced system lacking cellular regulation |
| Throughput | High - 10,000-70,000 sites per experiment [40] [16] | Low - typically single proteins or simple mixtures |
| Mechanistic Insight | Limited - identifies sites but not direct enzyme-substrate relationships | High - enables precise dissection of enzymatic requirements |
| Technical Complexity | High - requires specialized expertise and instrumentation | Moderate - accessible to most molecular biology laboratories |
| Cost | High - antibodies, MS instrumentation, and maintenance | Low - standard laboratory reagents and equipment |
| Linkage Information | Limited with standard K-ε-GG approach - requires specialized methods | Can be designed to test specific linkage types |
| Dynamic Range | Wide - can detect low stoichiometry modifications in complex mixtures | Narrow - focused on specific interactions |
| Quantification Capability | Excellent - SILAC, label-free, or TMT-based quantification [40] [16] | Limited - typically semi-quantitative immunoblotting |
Strategic Application of Each Approach
The complementary nature of these methodologies means they often serve different research purposes:
In vivo profiling is ideal for:
- Discovery-phase research to identify novel ubiquitination events
- Understanding global changes in ubiquitination in response to cellular perturbations
- Studying ubiquitination in disease models or during developmental processes
- Profiling ubiquitination in clinical samples or animal tissues [41] [16]
In vitro assays are optimal for:
- Validating direct ubiquitination of specific substrates
- Determining enzymatic requirements for ubiquitination (E2/E3 specificity)
- Characterizing ubiquitin chain linkage specificity
- Studying the biochemical properties of ubiquitin machinery components [25]
The Scientist's Toolkit: Essential Research Reagents
Successful ubiquitination profiling requires specific reagents and materials optimized for these specialized applications. The following table details key solutions used in the featured methodologies.
Table 3: Essential Research Reagents for Ubiquitination Studies
| Reagent | Function | Application Notes |
|---|---|---|
| Anti-K-ε-GG Antibody | Immunoaffinity enrichment of ubiquitinated peptides after tryptic digestion | Central to modern ubiquitinomics; multiple commercial sources available [42] [40] |
| SDC Lysis Buffer | Protein extraction while maintaining ubiquitination status | Superior to urea for ubiquitinome studies; 38% increase in identifications [16] |
| Chloroacetamide (CAA) | Alkylating agent for cysteine protection | Preferred over iodoacetamide to avoid di-carbamidomethylation artifacts [16] |
| DUB Inhibitors | Prevent deubiquitination during sample processing | Essential for preserving ubiquitination; examples: N-ethylmaleimide, ubiquitin aldehyde [25] |
| SILAC Amino Acids | Metabolic labeling for quantitative comparisons | Enables precise relative quantification across experimental conditions [40] |
| Recombinant E1, E2, E3 Enzymes | Reconstitute ubiquitination in vitro | Available from multiple commercial sources with varying specificity [25] |
| Linkage-Specific Ub Antibodies | Detect specific polyubiquitin chain types | Examples: K48-, K63-specific antibodies for functional characterization [4] [6] |
Emerging Methods and Future Directions
The field of ubiquitinomics continues to evolve with several promising technological developments:
- Linkage-Specific Profiling: New methods are emerging to characterize ubiquitin chain architecture, moving beyond simple site identification to understanding chain linkage and topology [6].
- Non-Canonical Ubiquitination Detection: Most current methods focus on lysine ubiquitination, but approaches are being developed to capture non-canonical ubiquitination on cysteine, serine, threonine, and protein N-termini [44].
- Single-Cell Ubiquitinomics: While currently challenging due to sensitivity limitations, future methodological improvements may enable ubiquitination analysis at single-cell resolution.
- Integration with Other PTMs: Simultaneous analysis of ubiquitination with other modifications (phosphorylation, acetylation) provides a more comprehensive view of cellular signaling networks [41].
Mass spectrometry-based proteomics has dramatically expanded our ability to profile protein ubiquitination on a global scale. The comparison between in vivo and in vitro approaches reveals their complementary strengths: while in vivo profiling offers unprecedented comprehensiveness and physiological relevance, in vitro assays provide mechanistic clarity and experimental control. The optimal research strategy often involves an iterative process between these approaches—using in vivo methods for discovery and in vitro techniques for validation and mechanistic dissection. As methodologies continue to advance, particularly in the areas of quantification, sensitivity, and linkage specification, our understanding of the complex ubiquitin signaling network will continue to deepen, offering new insights into cellular regulation and opportunities for therapeutic intervention in ubiquitination-related diseases.
Optimizing Your Assay: Critical Pitfalls and Proven Solutions for Reliable Data
The ubiquitin-proteasome system (UPS) represents a fundamental regulatory mechanism in eukaryotic cells, controlling protein stability, localization, and activity through the precise addition and removal of ubiquitin modifications [15]. This dynamic process involves a delicate balance between ubiquitinating enzymes (E1, E2, and E3) that attach ubiquitin to target proteins, and deubiquitinases (DUBs) that catalyze the removal of these ubiquitin modifications [15] [45]. The strategic inhibition of DUBs has emerged as a crucial therapeutic approach, particularly in oncology and neurodegenerative diseases, where disrupted protein homeostasis drives pathogenesis [46] [45].
The complexity of ubiquitination signaling is remarkable—ubiquitin itself contains eight potential linkage sites (M1, K6, K11, K27, K29, K33, K48, and K63), with K48-linked chains primarily targeting proteins for proteasomal degradation, while K63-linked chains typically regulate signal transduction, protein trafficking, and kinase activation [26] [30] [47]. This intricate signaling system is further refined by approximately 100 human DUBs that ensure precise temporal control over ubiquitin-mediated processes [15] [30]. Understanding and strategically disrupting specific DUB activities provides researchers with powerful tools to investigate protein turnover and develop targeted therapies for diseases characterized by ubiquitination dysregulation [46] [45].
Table: Major DUB Families and Their Characteristics
| DUB Family | Catalytic Mechanism | Representative Members | Primary Functions |
|---|---|---|---|
| USP | Cysteine protease | USP7, USP14, USP30 | Cleaves K48-linked chains; regulates protein stability & signaling [15] |
| UCH | Cysteine protease | UCH-L1 | Removes single ubiquitin molecules; maintains free ubiquitin pools [15] |
| OTU | Cysteine protease | OTUD5 | Deubiquitinates K63-linked chains; signaling regulation [15] |
| MJD | Cysteine protease | Ataxin-3 | Processes ubiquitin and non-ubiquitin substrates [15] |
| JAMM | Zinc metalloprotease | AMSH, RPN11 | Requires metal ions; regulates immune responses & protein homeostasis [15] |
Methodological Framework: Comparing Ubiquitination Detection Techniques
In Vivo versus In Vitro Approaches
Investigating deubiquitination requires sophisticated methodological approaches that can be broadly categorized into in vivo (cellular) and in vitro (cell-free) systems, each with distinct advantages and limitations. The choice between these systems fundamentally shapes experimental outcomes and interpretation, particularly when evaluating DUB inhibitor efficacy.
In vivo approaches provide the distinct advantage of preserving native cellular architecture, including subcellular compartmentalization, physiological enzyme concentrations, and competitive binding interactions that maintain the natural equilibrium of ubiquitination dynamics [15]. These systems are indispensable for understanding the biological context of DUB inhibition, as they capture complex regulatory networks and compensatory mechanisms that exist in living cells. However, in vivo systems present significant challenges in controlling variables and establishing direct causal relationships, as cellular complexity often obscures specific DUB-substrate interactions [15].
In vitro approaches offer unparalleled experimental control by reconstituting specific components of the ubiquitination machinery in isolation [15]. These simplified systems enable precise manipulation of reaction conditions, substrate concentrations, and enzyme composition, allowing researchers to establish direct mechanistic relationships and quantify kinetic parameters without confounding cellular factors. The primary limitation of in vitro systems lies in their potential oversimplification, as they may lack critical regulatory components or post-translational modifications present in native cellular environments [15].
Advanced Detection Technologies
Recent technological advancements have significantly enhanced our ability to detect and characterize ubiquitination events in both experimental paradigms:
Tandem Ubiquitin Binding Entities (TUBEs) have emerged as powerful tools for studying endogenous protein ubiquitination with linkage-specific resolution [26] [47] [48]. These engineered affinity reagents consist of multiple ubiquitin-associated domains that exhibit nanomolar affinity for polyubiquitin chains, protecting ubiquitinated substrates from deubiquitination and proteasomal degradation during lysis and analysis [26] [47]. The development of chain-specific TUBEs enables selective capture of distinct ubiquitin linkages, allowing researchers to differentiate between degradative (K48-linked) and non-degradative (K63-linked) ubiquitination signals in high-throughput formats [26] [47]. For example, TUBE-based assays have successfully demonstrated that inflammatory stimuli induce K63-linked ubiquitination of RIPK2, while PROTAC compounds promote K48-linked ubiquitination of the same protein [26].
Mass spectrometry-based proteomics has revolutionized the global analysis of ubiquitination sites and ubiquitin chain architecture [30] [49]. Advanced enrichment strategies coupled with high-resolution mass spectrometry enable system-wide quantification of ubiquitylation site occupancy and turnover rates [49]. Recent studies have revealed that ubiquitylation site occupancy spans over four orders of magnitude, with a median occupancy approximately three orders of magnitude lower than phosphorylation [49]. These sophisticated approaches have identified distinct classes of ubiquitination sites based on their occupancy, turnover rate, and regulation by proteasome inhibitors, providing insights into the specialized functions of ubiquitination in proteasomal degradation versus cellular signaling [49].
Table: Comparison of Key Ubiquitination Detection Methodologies
| Methodology | Key Features | Throughput | Key Applications | Limitations |
|---|---|---|---|---|
| TUBE-Based Assays | Nanomolar affinity; linkage-specific variants; protects ubiquitination during lysis [26] [47] | High (96-well plate format) [26] | Studying endogenous protein ubiquitination; PROTAC characterization [26] | Requires specific TUBE reagents |
| Immunoblotting | Uses anti-ubiquitin antibodies; widely accessible [30] | Low | Initial validation of substrate ubiquitination [30] | Semi-quantitative; low sensitivity [26] |
| Ubiquitin Tagging | Expression of tagged ubiquitin (e.g., His, Strep) [30] | Medium | Proteomic screening of ubiquitination sites [30] | May not mimic endogenous ubiquitin [30] |
| Linkage-Specific Antibodies | Enrich ubiquitinated proteins with specific chain linkages [30] | Medium | Characterizing chain-type specificity [30] | High cost; potential non-specific binding [30] |
Experimental Applications: DUB Inhibitors in Action
DUB Inhibitor Mechanisms and Classes
Deubiquitinase inhibitors represent a diverse class of compounds that target the catalytic activity of DUBs through various mechanisms. N-Ethylmaleimide (NEM) is a broad-spectrum cysteine protease inhibitor that irreversibly alkylates catalytic cysteine residues in multiple DUB families, including USPs, UCHs, OTUs, and MJDs [15]. While non-specific in its action, NEM remains a valuable tool compound for initial investigations of DUB-dependent processes and for preserving ubiquitinated substrates during cell lysis by preventing deubiquitination [15].
The landscape of DUB inhibitors has evolved significantly toward more selective compounds with defined mechanisms of action:
Ubiquitin-based inhibitors such as ubiquitin aldehyde (Ubal) and ubiquitin vinyl sulfone (UbVS) represent early mechanistic tools that form covalent complexes with the active sites of multiple DUBs [46] [45]. Although these compounds lack selectivity, they have been invaluable for structural studies and as positive controls in DUB activity assays [45].
Small-molecule inhibitors targeting specific DUB families have shown promise in preclinical and clinical development. Notable examples include inhibitors targeting USP1 (involved in DNA damage repair), USP7 (a key regulator of tumor suppressors and oncogenes), USP14 (proteasome-associated DUB), and USP30 (mitochondrial DUB) [46]. Several of these inhibitors have demonstrated antitumor efficacy in preclinical models, with some advancing to clinical trials for hematological malignancies and solid tumors [46] [45].
Targeted protein degradation approaches represent an innovative extension of DUB inhibition strategies. Deubiquitinase-Targeting Chimeras (DUBTACs) are heterobifunctional molecules designed to recruit specific DUBs to target proteins to stabilize them by removing degradative ubiquitin signals [46]. Conversely, DUB inhibitors have also been incorporated into PROTACs to target DUBs themselves for degradation, offering an alternative approach to conventional inhibition [45].
Detailed Experimental Protocol: Assessing DUB Inhibition in Cellular Models
The following protocol outlines a comprehensive approach for evaluating DUB inhibitor efficacy using cellular models, incorporating best practices from recent methodological advances:
Cell Culture and Treatment:
- Culture appropriate cell lines (e.g., THP-1 monocytes for inflammatory signaling studies [26] [47]) in optimized medium.
- Pre-treat cells with selected DUB inhibitors (NEM or more specific inhibitors) at varying concentrations for 30 minutes to 4 hours, depending on inhibitor pharmacokinetics.
- Apply relevant stimuli: inflammatory agents (e.g., L18-MDP at 200-500 ng/mL for 30-60 minutes to induce K63-linked ubiquitination [26]) or degradative agents (e.g., PROTACs to induce K48-linked ubiquitination [26]).
- Include appropriate controls: vehicle-only treatments, irreversible inhibitors, and catalytically dead DUB mutants when available.
Cell Lysis and Ubiquitin Enrichment:
- Lyse cells using specialized buffers optimized to preserve polyubiquitination, typically containing protease inhibitors and, when using NEM, additional cysteine protease inhibitors to prevent deubiquitination during processing [26] [47].
- Clarify lysates by centrifugation at 14,000 × g for 15 minutes at 4°C.
- Quantify protein concentration using standardized methods (e.g., BCA assay).
- For ubiquitin enrichment, incubate clarified lysates with selected capture reagents:
- Incubate with rotation for 2-4 hours at 4°C.
- Capture using appropriate magnetic beads or affinity resins, followed by 3-5 washes with lysis buffer.
Detection and Analysis:
- Elute bound proteins using Laemmli buffer with heating at 95°C for 5-10 minutes.
- Separate by SDS-PAGE and transfer to PVDF membranes.
- Probe with primary antibodies specific to:
- Protein of interest (e.g., anti-RIPK2 for inflammatory signaling studies [26])
- Ubiquitin (pan-ubiquitin or linkage-specific antibodies)
- Loading controls
- Visualize using enhanced chemiluminescence and quantify band intensities using densitometry software.
- For higher throughput assessment, consider adapting to 96-well TUBE-coated plate formats enabling more rapid screening of multiple conditions [26] [48].
Diagram 1: Experimental workflow for assessing DUB inhibition, showing both cellular (in vivo) context and subsequent in vitro detection methods that preserve ubiquitination signals.
Comparative Analysis: DUB Inhibitor Profiles
The strategic selection of DUB inhibitors requires careful consideration of their specificity, mechanism of action, and experimental applicability. The following analysis compares key inhibitor classes:
Table: Comprehensive Comparison of DUB Inhibitor Classes
| Inhibitor Class | Representative Compounds | Mechanism of Action | Specificity | Research Applications | Therapeutic Potential |
|---|---|---|---|---|---|
| Broad-Spectrum Cysteine Protease Inhibitors | N-Ethylmaleimide (NEM) | Irreversible alkylation of catalytic cysteine [15] | Low (targets all cysteine proteases) | Preservation of ubiquitination during lysis; initial DUB involvement screening [15] | Limited due to toxicity |
| Ubiquitin-Based Probes | Ubiquitin aldehyde (Ubal), Ubiquitin vinyl sulfone (UbVS) | Covalent modification of DUB active sites [46] [45] | Medium (pan-DUB activity) | Structural studies; DUB activity profiling; positive controls [45] | Research tools only |
| Small Molecule USP Inhibitors | USP1, USP7, USP14, USP30 inhibitors [46] | Competitive or allosteric inhibition of specific DUBs | High (family or isoform-specific) | Target validation; pathway analysis; combination therapy studies [46] [45] | Promising (several in clinical trials) |
| Targeted Degradation Approaches | DUBTACs, DUB-targeting PROTACs [46] [45] | Recruitment to E3 ligases (PROTACs) or stabilization (DUBTACs) | Variable (depends on warhead specificity) | Protein stabilization studies; alternative to catalytic inhibition [46] | Emerging field with high potential |
Research Reagent Solutions
The following table details essential reagents and methodologies for conducting deubiquitination research, compiled from current technological advances:
Table: Essential Research Reagents for Deubiquitination Studies
| Reagent Category | Specific Examples | Key Features | Primary Research Applications |
|---|---|---|---|
| DUB Inhibitors | N-Ethylmaleimide (NEM), USP-specific inhibitors (e.g., USP7, USP14 inhibitors) [15] [46] | Varying specificity; irreversible (NEM) vs. reversible (specific inhibitors) | Pathway analysis; therapeutic target validation [46] [45] |
| Ubiquitin Enrichment Tools | TUBEs (pan-selective and linkage-specific), ubiquitin antibodies (P4D1, FK1/FK2) [26] [30] [47] | High-affinity ubiquitin binding; linkage-specific variants available | Capturing endogenous ubiquitinated proteins; linkage-specific ubiquitination analysis [26] [30] |
| Tagged Ubiquitin Systems | His-tagged Ub, Strep-tagged Ub, HA-tagged Ub [30] | Affinity purification compatible; can be expressed in cells | Proteomic screening of ubiquitination sites; ubiquitination dynamics [30] |
| Mass Spectrometry Tools | TMT/iTRAQ labeling, ubiquitin remnant motif antibodies [30] [49] | System-wide quantification; site-specific identification | Global ubiquitinome analysis; occupancy and turnover rate calculations [49] |
| Functional Screening Systems | DNA-encoded libraries with ubiquitin transfer detection [50] | High-throughput capability; matches small molecules with protein substrates | Identifying novel molecular glue degraders; substrate profiling [50] |
The strategic application of DUB inhibitors, from broad-spectrum tools like N-ethylmaleimide to increasingly specific small molecule compounds, provides powerful approaches for dissecting ubiquitination dynamics in both physiological and pathological contexts. The continuing refinement of detection methodologies, particularly TUBE-based assays and advanced mass spectrometry techniques, has significantly enhanced our ability to capture and quantify ubiquitination events with unprecedented specificity and sensitivity.
Future developments in the DUB inhibition field will likely focus on achieving greater selectivity through structural-guided drug design and innovative approaches such as DUBTACs and targeted degradation strategies. Furthermore, the integration of functional screening platforms like DNA-encoded library selections with ubiquitin transfer assays promises to accelerate the discovery of novel degrader molecules and their optimal protein substrates [50]. As these technologies mature, they will undoubtedly provide deeper insights into the complex landscape of ubiquitination signaling and enable more effective therapeutic interventions for diseases characterized by ubiquitination dysregulation.
The consistent advancement in both inhibitor design and detection methodologies underscores the dynamic nature of this research field and its critical importance for both basic science and drug development. Researchers should maintain awareness of these evolving tools and approaches to ensure their investigations leverage the most appropriate and powerful technologies available.
In the study of protein ubiquitination, the choice of detection technique—conducted either within living cells (in vivo) or in a controlled tube environment (in vitro)—fundamentally shapes the experimental approach. However, a common challenge unites these methodologies: the critical need to distinguish specific, covalent ubiquitin attachments from non-specific, transient protein interactions. The integrity of this distinction rests heavily on the experimental conditions employed, particularly during the steps of cell lysis and immunoprecipitation washing. This guide objectively compares the performance of in vivo and in vitro ubiquitination detection, highlighting how stringent protocols are the decisive factor in ensuring data specificity and reliability for researchers and drug development professionals.
Experimental Protocols for Specific Detection
In Vivo Ubiquitination Detection Protocol
The following protocol for detecting ubiquitination in cultured cells is designed to maximize specificity by disrupting non-covalent interactions [25].
Detailed Step-by-Step Methodology:
- Transfection & Lysis: Culture and transfect cells with plasmids expressing your protein of interest and an epitope-tagged ubiquitin. Prepare a complete cell lysis buffer with the following stringent composition:
- 2% Sodium Dodecyl Sulfate (SDS)
- 150 mM NaCl
- 10 mM Tris-HCl (pH 8.0)
- Supplement with 2mM sodium orthovanadate, 50 mM sodium fluoride, and protease inhibitors immediately before use.
- Immediate Denaturation: Lyse cells directly in a 6 cm dish with 100 µl of the pre-warmed lysis buffer. Immediately scrape the cells and transfer the lysate to a tube to boil for 10 minutes. This rapid and harsh denaturation is crucial for inactivating enzymes and disrupting non-covalent protein complexes.
- Clarification & Dilution: Shear DNA by sonicating the boiled lysate. Add 900 µl of dilution buffer (10 mM Tris-HCl, pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% Triton) to reduce the SDS concentration, and incubate at 4°C for 30-60 minutes with rotation.
- Immunoprecipitation under Stringent Conditions: Centrifuge the diluted sample at 20,000 x g for 30 minutes. Transfer the supernatant and incubate it with an antibody against the target protein that is pre-conjugated to Protein A- or G-agarose beads. Use a cut pipette tip to handle the resin. Incubate this mixture at 4°C overnight with rotation.
- Rigorous Washing: Spin down the beads and aspirate the supernatant. Wash the resin twice with a high-stringency washing buffer (10 mM Tris-HCl, pH 8.0, 1 M NaCl, 1 mM EDTA, 1% NP-40). The high salt concentration (1 M NaCl) is essential for removing electrostatically bound contaminants.
- Elution and Analysis: Perform a final high-speed spin, aspirate all residual wash buffer, and elute the immunoprecipitated proteins by boiling the resin in 2X SDS loading buffer. The samples can then be loaded onto an SDS-PAGE gel for immunoblotting analysis with antibodies against ubiquitin and the target protein.
In Vitro Ubiquitination Assay Protocol
The in vitro assay offers direct control over reaction components but requires purified elements [25].
Detailed Step-by-Step Methodology:
- Reaction Setup: Prepare a 5X ubiquitination buffer (100 mM Tris-HCl, pH 7.5, 25 mM MgCl₂, 2.5 mM DTT, 10 mM ATP). Store in aliquots at -20°C.
- Component Assembly: For a 40 µl reaction, mix the following components:
- 8 µl of 5X ubiquitination buffer
- 250 ng ubiquitin-activating enzyme E1
- 500 ng ubiquitin-conjugating enzyme E2
- 0.5 µg ubiquitin
- 0.5 µg protein of interest (the substrate)
- Water to 40 µl
- Critical Controls: Always include control reactions that omit E1, E2, E3 (if used), or ubiquitin to confirm the specificity of the observed ubiquitination.
- Incubation and Termination: Incubate the reaction mixture at 37°C for 1 hour or longer. Stop the reaction by adding SDS-PAGE sample buffer and boiling for 10 minutes.
- Detection: Analyze the products by SDS-PAGE and immunoblotting for ubiquitin and the target protein.
Comparative Performance Data
The table below summarizes the key characteristics, advantages, and limitations of the in vivo and in vitro ubiquitination detection techniques, with a focus on how they achieve specificity.
Table 1: Performance Comparison of In Vivo and In Vitro Ubiquitination Detection
| Feature | In Vivo Detection | In Vitro Reconstitution |
|---|---|---|
| Physiological Context | Full cellular environment; preserves complex biology [4] | Simplified, controlled system; lacks native cellular regulation [25] |
| Specificity Assurance | Relies on stringent lysis/wash buffers (high SDS, high salt) to eliminate non-covalent interactions [25] | Built-in through defined components; controlled by omitting specific enzymes in reactions [25] |
| Key Advantage | Can identify true physiological substrates and modifications. | Directly demonstrates a protein is a substrate for the ubiquitin machinery. |
| Primary Limitation | Potential for co-precipitating non-specifically bound proteins if conditions are not stringent enough. | May produce non-physiological results due to the absence of regulatory factors or incorrect localization. |
| Technical Complexity | High, due to transfection, optimization of lysis/IP conditions, and risk of enzyme activity (DUBs) during lysis. | Lower, but requires purification of all components (E1, E2, E3, substrate). |
| Throughput | Lower, more suited for targeted studies. | Higher potential for screening, as demonstrated in luminescent assays screening hundreds of proteins [51]. |
The Scientist's Toolkit: Essential Research Reagents
Successful and specific detection of ubiquitination relies on a set of key reagents. The following table details these essential materials and their critical functions in the protocol.
Table 2: Key Research Reagents for Ubiquitination Detection
| Reagent / Solution | Function & Importance |
|---|---|
| Stringent Lysis Buffer (2% SDS) | The foundation of specificity in in vivo work. The strong ionic detergent SDS denatures proteins, dissolves membranes, and most importantly, disrupts nearly all non-covalent interactions, ensuring only covalent ubiquitin conjugates are isolated [25]. |
| High-Salt Wash Buffer (1 M NaCl) | Used during immunoprecipitation to remove contaminants that are bound through electrostatic (charge-based) interactions, which are common sources of non-specific background [25]. |
| Deubiquitinating Enzyme (DUB) Inhibitors | Chemicals like N-ethylmaleimide are added to lysis and wash buffers to prevent the removal of ubiquitin from substrates by endogenous DUBs during sample preparation, thereby preserving the native ubiquitination signal [25]. |
| Epitope-Tagged Ubiquitin | A mainstay of in vivo studies. Tags (e.g., HA, Myc, FLAG) allow for standardized immunodetection and more efficient immunoprecipitation of ubiquitinated proteins than often variable native ubiquitin antibodies. |
| Purified E1, E2, and E3 Enzymes | The core components for in vitro assays. Their purity and activity are prerequisites for a successful reconstitution of the ubiquitination cascade [25] [51]. |
Workflow and Specificity Visualization
The following diagrams illustrate the key experimental workflows and the critical role stringent conditions play in ensuring specificity.
In Vivo Ubiquitination Workflow
How Stringent Conditions Ensure Specificity
The comparative data and protocols presented herein lead to a clear conclusion: while the fundamental question (is a protein ubiquitinated?) can be addressed by either in vivo or in vitro techniques, the specificity of the answer is dictated by the rigor of the experimental conditions.
For in vivo studies, this rigor is embodied by the stringent lysis and wash buffers. The use of high concentrations of SDS and salt is not merely a technical detail; it is the primary defense against the co-purification of proteins that merely associate with the target, ensuring that the final western blot smear represents a true ubiquitination ladder [25]. For in vitro assays, specificity is engineered into the system through the use of purified components and omission controls, providing a direct and unambiguous result, albeit in a non-physiological setting [25].
Emerging technologies, particularly advanced mass spectrometry, are pushing the boundaries of specificity and throughput in ubiquitinomics. These methods rely on sophisticated lysis protocols, such as those using sodium deoxycholate (SDC) with immediate boiling and alkylation, to comprehensively profile ubiquitination sites across the proteome with high precision [16]. This evolution underscores a persistent theme: the initial steps of sample preparation remain the foundation upon which reliable ubiquitination data is built. Therefore, whether employing classical immunoblotting or cutting-edge proteomics, a critical evaluation and stringent application of lysis and wash conditions are indispensable for generating credible, publication-quality results in ubiquitination research.
Choosing the Right Controls for In Vitro and In Vivo Experiments
The detection of protein ubiquitination is a cornerstone of research in cell biology, cancer, and drug development. This post-translational modification, regulated by a cascade of E1, E2, and E3 enzymes, controls diverse cellular functions from protein degradation to signal transduction. A critical challenge researchers face is selecting appropriate experimental controls to accurately distinguish between direct ubiquitination events and indirect effects. This guide provides a systematic comparison of controls for in vitro and in vivo ubiquitination assays, empowering scientists to generate more reliable and interpretable data in their investigation of the ubiquitin-proteasome system.
Core Principles of Ubiquitination Detection
Ubiquitination involves the covalent attachment of ubiquitin to target proteins, primarily through isopeptide bonds to lysine residues. The complexity of ubiquitin signaling—encompassing monoubiquitination, multiple monoubiquitination, and various polyubiquitin chain linkages—necessitates carefully controlled experiments to draw meaningful biological conclusions [6].
The Ubiquitination Cascade and Major Detection Methods
Comparative Analysis: In Vitro vs. In Vivo Ubiquitination Detection
| Parameter | In Vitro Reconstitution | In Vivo Cellular Systems |
|---|---|---|
| Control Requirements | Omit individual E1, E2, E3, ubiquitin, or ATP [25] | Include DUB inhibitors (N-ethylmaleimide), proteasome inhibitors (MG-132) [25] [16] |
| Key Experimental Applications | Direct E3 ligase validation (e.g., ARIH1-PD-L1, HUWE1-small molecules) [27] [14] | Physiological context, PROTAC/MGD mechanism studies [26] [50] |
| Appropriate Negative Controls | Catalytically dead E3 mutants, substrate binding mutants [27] | siRNA/shRNA knockdown of E3, substrate ubiquitination site mutants [52] |
| Appropriate Positive Controls | Known E3-substrate pairs (e.g., IKZF1a-Pomalidomide-CRBN) [50] | Stimuli-induced ubiquitination (e.g., L18-MDP-induced RIPK2 ubiquitination) [26] |
| Technical Advantages | Direct mechanism proof, minimal confounding factors [27] | Physiological relevance, native cellular environment [26] |
| Key Limitations | May miss cellular regulators; false negatives possible (e.g., CRL3SPOP-PD-L1) [27] | Indirect effects hard to exclude; specificity challenges [14] |
| Quantitative Potential | Good for kinetics with purified components [14] | Advanced MS methods enable high-throughput (70,000+ ubiquitin sites) [16] |
| Linkage-Type Analysis | Can control E2 to influence linkage [6] | Requires TUBEs or linkage-specific antibodies [26] [6] |
Detailed Experimental Protocols with Essential Controls
In Vitro Ubiquitination Reconstitution Assay
This protocol establishes a minimal system to test whether an E3 ligase directly ubiquitinates a substrate protein [25] [27].
Step-by-Step Methodology:
- Prepare 5X ubiquitination buffer: 100 mM Tris-HCl (pH 7.5), 25 mM MgCl₂, 2.5 mM DTT, 10 mM ATP. Aliquot and store at -20°C [25].
- Assemble reaction mixture (40 μL total volume):
- 8 μL 5X ubiquitination buffer
- 250 ng E1 activating enzyme
- 500 ng E2 conjugating enzyme
- 0.5-1 μg E3 ligase
- 0.5-1 μg substrate protein
- 0.5-1 μg ubiquitin
- Deionized water to volume [25]
- Critical control reactions: Assemble identical mixtures but omit individually: E1, E2, E3, ubiquitin, or ATP [25]. Include catalytically dead E3 mutant (e.g., cysteine mutant for HECT E3s) [14].
- Incubate at 37°C for 1-3 hours.
- Terminate reaction by adding SDS-PAGE sample buffer and boiling for 10 minutes.
- Analyze by immunoblotting with anti-ubiquitin and anti-substrate antibodies.
In Vivo Ubiquitination Detection Protocol
This protocol detects ubiquitination of a target protein in its cellular context [25] [52].
Step-by-Step Methodology:
- Cell preparation and transfection: Culture appropriate cell lines (e.g., HEK293T, HepG2) and transfect with plasmids expressing:
- Protein of interest
- Epitope-tagged ubiquitin (e.g., His-ubiquitin, HA-ubiquitin)
- E3 ligase (for overexpression studies) [52]
- Treatment: 4-6 hours before harvesting, add 10-20 μM MG-132 proteasome inhibitor to prevent degradation of ubiquitinated proteins [16] [52].
- Cell lysis under denaturing conditions: Use stringent lysis buffer (2% SDS, 150 mM NaCl, 10 mM Tris-HCl, pH 8.0) with protease inhibitors and 2 mM N-ethylmaleimide to inhibit deubiquitinases [25]. Immediately boil lysates for 10 minutes to inactivate enzymes.
- Sonication and dilution: Shear DNA by sonication, then dilute 10-fold with Triton X-100-containing buffer (10 mM Tris-HCl, pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100) [25].
- Immunoprecipitation: Incubate with target protein antibody conjugated to Protein A/G beads overnight at 4°C [25].
- Stringent washing: Wash beads twice with high-salt buffer (10 mM Tris-HCl, pH 8.0, 1 M NaCl, 1 mM EDTA, 1% NP-40) to remove non-covalently associated proteins [25].
- Immunoblot analysis: Detect ubiquitin and target protein with respective antibodies.
Advanced Methodologies and Specialized Applications
Chain-Linkage Specific Ubiquitination Analysis
Different ubiquitin chain linkages serve distinct cellular functions, with K48-linked chains primarily targeting proteins for proteasomal degradation while K63-linked chains regulate signaling and protein interactions [26]. Tandem Ubiquitin Binding Entities (TUBEs) with nanomolar affinities for specific polyubiquitin chains enable researchers to differentiate between these linkage types in high-throughput assays [26].
Application Example: Research on RIPK2 ubiquitination demonstrated that inflammatory agent L18-MDP stimulates K63 ubiquitination captured by K63-TUBEs, while RIPK2 PROTAC-induced K48 ubiquitination is specifically captured by K48-TUBEs [26].
Mass Spectrometry-Based Ubiquitinomics
Advanced proteomic approaches now enable system-wide ubiquitination profiling. Key methodological improvements include:
- SDC-based lysis protocol: Sodium deoxycholate buffer with immediate boiling and chloroacetamide alkylation significantly improves ubiquitin site coverage compared to traditional urea buffer [16].
- Data-Independent Acquisition (DIA-MS): This method more than triples ubiquitinated peptide identifications (to ~70,000 per run) while significantly improving quantitative precision compared to data-dependent acquisition [16].
- False discovery control: Using decoy databases and neural network-based processing (DIA-NN) ensures confident identification of modified peptides [16].
The Scientist's Toolkit: Essential Research Reagents
| Reagent/Category | Specific Examples | Function and Application |
|---|---|---|
| Ubiquitin Tags | His-tagged Ub, Strep-tagged Ub, HA-Ub | Affinity purification of ubiquitinated proteins [6] [52] |
| E3 Ligase Modulators | BI8622/BI8626 (HUWE1), Pomalidomide (CRBN), PROTACs | Investigate E3 function and targeted protein degradation [14] [50] |
| Proteasome Inhibitors | MG-132, Bortezomib | Stabilize ubiquitinated proteins by blocking degradation [16] [52] |
| Deubiquitinase Inhibitors | N-ethylmaleimide, Ubiquitin aldehyde | Prevent removal of ubiquitin during processing [25] |
| Linkage-Specific Binders | K48-TUBEs, K63-TUBEs, linkage-specific antibodies | Enrich and detect specific ubiquitin chain types [26] [6] |
| Activity Detection Assays | CCK-8 assay | Measure functional consequences of ubiquitination [52] |
| Ubiquitination Enrichment Resins | Ni-NTA agarose, Anti-HA beads, Streptavidin beads | Isolate ubiquitinated proteins from complex mixtures [6] [52] |
Choosing appropriate controls for ubiquitination experiments requires careful consideration of the biological question and technical approach. For mechanistic studies seeking to establish direct E3-substrate relationships, in vitro reconstitution with omission controls provides the most definitive evidence. For physiological studies investigating ubiquitination in cellular contexts, in vivo approaches with proper inhibition controls and validation through mutagenesis are essential. The increasing availability of linkage-specific tools and advanced mass spectrometry methods now enables researchers to move beyond simple ubiquitination detection to detailed characterization of ubiquitin chain architecture and function. By implementing the controlled experimental designs outlined in this guide, researchers can generate more reliable data and advance our understanding of the complex ubiquitin signaling network.
The ubiquitin-proteasome system represents a crucial regulatory mechanism governing virtually all aspects of cellular physiology, from DNA repair and cell cycle progression to inflammatory signaling and metabolic reprogramming. However, studying this system presents a fundamental challenge: the low stoichiometry of protein ubiquitination under normal physiological conditions. This low abundance, combined with the staggering complexity of ubiquitin chain architectures and their dynamic regulation, creates significant detection hurdles. Researchers must navigate a landscape where target proteins may be modified at one or several lysine residues simultaneously, while ubiquitin itself can form polymers of varying length, linkage types, and overall architecture. The critical importance of overcoming these sensitivity limitations is underscored by the central role ubiquitination plays in pathological conditions, including cancer radiotherapy resistance and inflammatory disorders, and by the emergence of revolutionary technologies that hijack the ubiquitin system for therapeutic purposes, such as PROTACs (Proteolysis Targeting Chimeras) and molecular glues. This guide systematically compares the predominant methodologies for ubiquitination detection, focusing specifically on their capabilities to address the sensitivity challenge in both in vivo and in vitro research contexts.
Methodological Landscape: Techniques for Ubiquitination Detection
Table 1: Core Methodologies for Detecting Protein Ubiquitination
| Method Category | Key Principle | Typical Context | Primary Sensitivity Challenge | Best Application Scenario |
|---|---|---|---|---|
| Immunoblotting | Uses anti-ubiquitin antibodies to detect shifts in molecular weight via Western blot. [30] | In vivo & in vitro | Low-throughput, semi-quantitative, poor sensitivity for subtle changes. [26] | Initial validation of ubiquitination for a single, abundant protein. |
| TUBE-Based Enrichment | Uses Tandem Ubiquitin Binding Entities (high-affinity UBDs) to capture polyubiquitinated proteins from lysates. [26] | Primarily in vivo | Preserving labile ubiquitination events during cell lysis. [26] | Capturing endogenous, linkage-specific ubiquitination (e.g., K48 vs. K63) for proteomics or downstream analysis. [26] |
| Tagged Ubiquitin | Cells express affinity-tagged Ub (e.g., His, Strep, HA); tagged substrates are purified. [30] | In vivo | Artifacts from overexpressed, non-native Ub; co-purification of non-ubiquitinated proteins. [30] | High-throughput identification of ubiquitination sites from cultured cells via mass spectrometry. |
| Linkage-Specific Antibodies | Antibodies specific to a Ub chain linkage (e.g., K48, K63) enrich or detect that chain type. [30] | In vivo & in vitro | High cost; potential for non-specific binding; availability for atypical linkages. [30] | Studying the biology of a specific, well-defined ubiquitin linkage type. |
| In Vitro Reconstitution | Reconstructs ubiquitination cascade with purified E1, E2, E3, Ub, and substrate. [53] [50] | In vitro | May oversimplify complex cellular environment and regulation. | Mechanistic studies of E3 ligase activity and specificity, including for non-protein substrates. [53] [50] |
Decoding the Ubiquitination Cascade and Detection Strategy
The diagram below illustrates the core enzymatic cascade of ubiquitination and the subsequent point of application for major detection techniques, highlighting the central strategy of enrichment to overcome low abundance.
Deep Dive: Experimental Protocols for Enhanced Sensitivity
Protocol 1: TUBE-Based Enrichment for Linkage-Specific Endogenous Ubiquitination
This protocol leverages Tandem Ubiquitin Binding Entities (TUBEs) with nanomolar affinities for polyubiquitin chains to capture and study endogenous ubiquitination events, such as those on RIPK2, a key inflammatory signaling regulator. [26]
Step 1: Cell Stimulation and Lysis
- Treat cells (e.g., human monocytic THP-1 cells) with a stimulus (e.g., 200-500 ng/ml L18-MDP for 30-60 minutes to induce K63-linked ubiquitination of RIPK2) or a PROTAC (to induce K48-linked ubiquitination). [26]
- Lyse cells using a buffer specifically optimized to preserve labile polyubiquitination events. This typically includes protease inhibitors, deubiquitinase (DUB) inhibitors (e.g., N-ethylmaleimide), and often 1% SDS, which can be diluted for subsequent steps. [26]
Step 2: Affinity Enrichment with TUBEs
- Incubate the clarified cell lysate (e.g., 50 µg to 1 mg total protein) with chain-specific TUBE-coated magnetic beads (e.g., Pan-, K48-, or K63-TUBEs). [26]
- Perform incubation for 2-4 hours at 4°C with gentle agitation to allow high-affinity binding between the TUBEs and polyubiquitin chains.
Step 3: Washing and Elution
- Wash beads stringently with a compatible wash buffer to remove non-specifically bound proteins.
- Elute the captured ubiquitinated proteins by boiling the beads in SDS-PAGE sample buffer, which denatures the proteins and disrupts the TUBE-ubiquitin interaction.
Step 4: Downstream Analysis
- Analyze the eluates by immunoblotting using an antibody against the protein of interest (e.g., anti-RIPK2) to detect its ubiquitinated forms, which appear as higher molecular weight smears or discrete bands. [26]
- Alternatively, the eluates can be used for mass spectrometric analysis to identify the ubiquitinated proteins or specific ubiquitination sites.
Protocol 2: DNA-Encoded In Vitro Ubiquitination Assay
This functional screening approach identifies small molecule/protein pairs susceptible to ubiquitination by a specific E3 ligase (e.g., CRL4CRBN) in a high-throughput, multiplexed format. [50]
Step 1: Preparation of DNA-Encoded Components
- Proteins/Peptides: Fuse the protein or peptide of interest to a self-labelling tag (e.g., SNAP-tag). Incubate with benzylguanine (BnG)-bearing DNA oligonucleotides to create covalent DNA-protein conjugates. [50]
- Small Molecules: Synthesize a DNA-linked version of the small molecule library, such as a derivative of pomalidomide (Pom_N3) coupled to a DNA strand via a linker. [50]
Step 2: Assembly and Reaction
- Hybridize the small molecule DEL (SM_DEL) with the pooled DNA-encoded protein collection via complementary DNA sequences. This self-assembly also induces proximity between the small molecule and its potential protein substrate. [50]
- Add a reconstituted E3 ligase system (e.g., CRL4CRBN, E1, E2, ubiquitin, ATP) to the hybridized complexes. If a small molecule successfully recruits its paired protein to the E3 ligase, ubiquitin will be transferred to the protein. [50]
Step 3: Affinity Capture of Ubiquitinated Substrates
- Use anti-ubiquitin antibody-conjugated beads to selectively capture and purify the DNA sequences that are now linked to ubiquitin-modified proteins. [50]
Step 4: Decoding Active Pairs
- Sequence the enriched pool of DNA tags to identify the small molecule and protein sequences of the active pairs that successfully underwent ubiquitination. [50]
The Scientist's Toolkit: Essential Reagents for Ubiquitination Research
Table 2: Key Research Reagents and Their Functions
| Reagent / Tool | Core Function | Application Context | Considerations for Sensitivity |
|---|---|---|---|
| Chain-Specific TUBEs (e.g., K48, K63) | High-affinity capture of endogenous proteins modified with specific Ub chain linkages. [26] | In vivo enrichment; HTS assays. | Nanomolar affinity allows capture of low-abundance ubiquitinated species; differentiates context-dependent ubiquitination. [26] |
| Linkage-Specific Antibodies (e.g., FK2, P4D1) | Recognize all Ub linkages (pan-specific) or specific linkages (e.g., K48-only) for enrichment/detection. [30] | Immunoblotting, immunofluorescence, enrichment. | Enables study of endogenous proteins without genetic manipulation; potential for non-specific binding. [30] |
| Tagged Ubiquitin (His, HA, Strep, FLAG) | Affinity purification of ubiquitinated substrates from cell lysates after overexpression. [30] | Global ubiquitinome profiling via mass spectrometry. | Can introduce artifacts; co-purification of non-ubiquitinated proteins reduces identification sensitivity. [30] |
| DUB Inhibitors (e.g., NEM, PR-619) | Inhibit deubiquitinating enzymes in cell lysis buffers to prevent loss of Ub signal during processing. [26] | Essential pre-analytical step for all methods. | Critical for preserving low-abundance or labile ubiquitination events, directly enhancing detection sensitivity. [26] |
| Reconstituted E3 System (E1, E2, E3, Ub) | Provides defined components for in vitro ubiquitination reactions. [53] [50] | Mechanistic studies and functional screens. | Removes cellular complexity, allowing clear assessment of direct ubiquitination events on low-abundance substrates. [50] |
Choosing the optimal strategy for detecting ubiquitination hinges on the specific research question and the inherent abundance of the target. For profiling endogenous ubiquitination events in a physiological context, TUBE-based enrichment and linkage-specific antibody approaches offer the most direct path to enhancing sensitivity without genetic manipulation. When the goal is high-throughput discovery of novel ubiquitination substrates or small molecule inducers, DNA-encoded in vitro systems and tagged ubiquitin proteomics provide powerful, multiplexable alternatives, albeit with considerations about their biological translatability. Ultimately, a combination of these techniques—using in vitro reconstitution for mechanistic discovery followed by TUBE or antibody-based validation in cellular models—often constitutes the most robust strategy. As the ubiquitin field continues to evolve, driven by its profound therapeutic implications, the development of even more sensitive and specific detection methodologies will remain a critical frontier for both basic research and drug development.
Ubiquitination is a fundamental post-translational modification that regulates virtually all cellular processes, with linkage specificity dictating fundamentally different biological outcomes. Among the eight possible ubiquitin chain linkages, K48-linked chains primarily target substrates for proteasomal degradation, while K63-linked chains predominantly regulate non-proteolytic functions including signal transduction, protein trafficking, and inflammation [47] [4]. The accurate differentiation between these signals is not merely an academic exercise but a critical requirement for understanding disease mechanisms and developing targeted therapies. This guide provides a comprehensive comparison of contemporary methodologies for validating K48 versus K63 ubiquitination signals, presenting experimental data and protocols to empower researchers in making informed methodological choices. The emerging complexity of heterotypic and branched ubiquitin chains, which contain multiple linkage types, further underscores the necessity for rigorous validation approaches [54] [55].
Technical Comparison of Key Validation Methodologies
Antibody-Based Enrichment and Detection Approaches
Table 1: Comparison of Antibody-Based Ubiquitin Detection Methods
| Method | Principle | Applications | Throughput | Key Advantages | Major Limitations |
|---|---|---|---|---|---|
| Linkage-Specific Immunoblotting | Uses K48- or K63-specific antibodies for detection | Validation of putative substrates, chain type confirmation | Low to medium | Direct visualization, widely accessible | Semi-quantitative, limited multiplexing capability |
| diGly Antibody Enrichment (Pan-Specific) | Enriches tryptic peptides with K-ε-GG remnant | System-wide ubiquitinome profiling by MS | High | Identifies exact modification sites, high sensitivity | Cannot distinguish linkage types on its own |
| Linkage-Specific Immunoprecipitation | Antibodies against specific linkage types enrich particular chain structures | Isolation of proteins modified with specific chain types | Medium | Direct linkage information, works with endogenous proteins | Potential cross-reactivity, antibody availability |
Antibody-based methods form the cornerstone of ubiquitin signal validation. Traditional immunoblotting using linkage-specific antibodies remains the most accessible approach for initial validation. For instance, K48-linkage specific antibodies have been used to demonstrate the abnormal accumulation of K48-linked polyubiquitinated tau proteins in Alzheimer's disease [6]. However, the emergence of more sophisticated enrichment strategies has significantly enhanced our profiling capabilities. The anti-K-ε-GG antibody approach, which recognizes the diglycine remnant left on tryptic peptides from ubiquitinated proteins, has revolutionized large-scale ubiquitinome studies by enabling the identification of thousands of ubiquitination sites via mass spectrometry [56] [57]. When combined with linkage-specific antibodies, this approach provides both site-specific and linkage-type information, offering a more comprehensive view of the ubiquitination landscape.
Ubiquitin-Binding Entity (UBE) Based Approaches
Table 2: TUBE-Based Applications for Linkage-Specific Ubiquitination Analysis
| TUBE Type | Specificity | Reported Affinity | Application Context | Experimental Evidence |
|---|---|---|---|---|
| K63-TUBE | K63-linked chains | Nanomolar range | Monitoring inflammatory signaling (e.g., RIPK2) | Captured L18-MDP-induced RIPK2 ubiquitination; no PROTAC signal |
| K48-TUBE | K48-linked chains | Nanomolar range | Validation of degradation signals | Captured RIPK2 PROTAC-induced ubiquitination; no L18-MDP signal |
| Pan-TUBE | All chain types | Nanomolar range | General ubiquitination assessment | Captured both inflammatory and degradation-induced RIPK2 ubiquitination |
Tandem Ubiquitin Binding Entities (TUBEs) have emerged as powerful tools for linkage-specific ubiquitination analysis, offering significant advantages over traditional antibody-based methods. These engineered reagents consist of multiple ubiquitin-binding domains (UBDs) fused in tandem, resulting in dramatically enhanced affinity for ubiquitin chains compared to single UBDs [6]. The application of chain-specific TUBEs with nanomolar affinities enables highly specific capture of polyubiquitin chains in high-throughput screening formats [47]. In a compelling demonstration of their specificity, K63-TUBEs successfully captured L18-MDP-induced K63 ubiquitination of RIPK2 but showed no appreciable binding to PROTAC-induced K48 ubiquitination of the same protein. Conversely, K48-TUBEs specifically captured the PROTAC-induced degradation signal while ignoring the inflammatory signal [47]. This exquisite specificity makes TUBEs particularly valuable for drug discovery applications, including the characterization of PROTACs (Proteolysis Targeting Chimeras) that specifically induce K48-linked ubiquitination of target proteins [47].
Mass Spectrometry-Based Structural Elucidation
Advanced mass spectrometry approaches provide the most detailed structural information about ubiquitin chains. Data-independent acquisition (DIA) methods have recently been developed that markedly improve the sensitivity and reproducibility of ubiquitinome analysis. One optimized workflow combining diGly antibody-based enrichment with Orbitrap-based DIA mass spectrometry identified approximately 35,000 distinct diGly peptides in single measurements of proteasome inhibitor-treated cells—nearly double the identification rate of traditional data-dependent acquisition methods [57]. This approach demonstrated significantly better quantitative accuracy, with 45% of diGly peptides showing coefficients of variation below 20% across replicates [57].
For precise quantification of specific linkage types, the Ub-AQUA/PRM (Absolute Quantification of Ubiquitin with Parallel Reaction Monitoring) method enables targeted quantification of specific ubiquitin linkages. This approach was used to confirm that OTUD5 is specifically modified with K29-linked chains by TRIP12 E3 ligase, providing linkage-type specificity through mass spectrometric quantification [55]. The method combines stable isotope-labeled ubiquitin internal standards with targeted mass spectrometry for highly precise linkage quantification.
Detailed Experimental Protocols for Key Applications
Protocol 1: TUBE-Based Linkage-Specific Ubiquitination Assessment
Application: Differentiating K48 vs. K63 ubiquitination signals in cellular pathways.
Cell Treatment and Lysis:
- Treat cells with pathway-specific stimuli (e.g., 200-500 ng/mL L18-MDP for K63-signaling or PROTACs for K48-signaling) for 30-60 minutes [47].
- Use optimized lysis buffer (e.g., 50 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA) with fresh protease inhibitors and deubiquitinase inhibitors (5 mM chloroacetamide or 50 μM PR-619) to preserve ubiquitin chains [47] [56].
Linkage-Specific Capture:
- Coat 96-well plates with K48-TUBE, K63-TUBE, or Pan-TUBE (50-100 μL per well).
- Incubate clarified cell lysates (100-500 μg total protein) with TUBE-coated plates for 2 hours at 4°C with gentle agitation.
- Wash extensively to remove non-specifically bound proteins.
Target Detection and Quantification:
- Elute bound proteins or directly detect target protein ubiquitination using specific antibodies.
- Quantify signals using chemiluminescence or fluorescence detection.
- Confirm specificity by comparing signals across different TUBE types—true K48 ubiquitination should be enriched specifically on K48-TUBE, while K63 signals should preferentially bind to K63-TUBE [47].
Protocol 2: diGly Enrichment for System-Wide Ubiquitinome Profiling
Application: Comprehensive identification and quantification of ubiquitination sites.
Sample Preparation and Digestion:
- Lyse cells in 8 M urea buffer with protease and DUB inhibitors (5 mM chloroacetamide, 50 μM PR-619) [56] [57].
- Reduce with 5 mM dithiothreitol (45 minutes, room temperature) and alkylate with 10 mM iodoacetamide (45 minutes, room temperature in dark).
- Digest with trypsin (1:50 enzyme-to-substrate ratio) overnight at room temperature.
diGly Peptide Enrichment:
- Desalt peptides using C18 solid-phase extraction cartridges.
- Enrich diGly peptides using anti-K-ε-GG antibody (31.25 μg antibody per 1 mg peptide input) [57].
- Incubate for 2 hours at 4°C with gentle rotation.
- Wash extensively and elute with 0.1-0.5% trifluoroacetic acid.
LC-MS/MS Analysis:
- Analyze using optimized DIA method with 46 precursor isolation windows and MS2 resolution of 30,000 [57].
- Utilize comprehensive spectral libraries (containing >90,000 diGly peptides) for optimal identification [57].
- For linkage-specific information, combine with linkage-specific antibodies or TUBEs in separate experiments.
Essential Research Reagent Solutions
Table 3: Essential Research Reagents for Ubiquitin Linkage Validation
| Reagent Category | Specific Examples | Key Functions | Considerations for Use |
|---|---|---|---|
| Deubiquitinase Inhibitors | Chloroacetamide (CAA), N-Ethylmaleimide (NEM), PR-619 | Preserve ubiquitin chains during processing | CAA: milder, chain-specific; NEM: potent but potential off-target effects [54] |
| Linkage-Specific Binders | K48-TUBE, K63-TUBE, Pan-TUBE | Selective enrichment of specific chain types | Nanomolar affinity enables high-sensitivity capture [47] |
| Mass Spec Standards | Stable isotope-labeled ubiquitin, TMT/iTRAQ tags | Quantitative precision | Enable absolute quantification of linkage types [58] [55] |
| Activity Probes | Linkage-specific DUB substrates | Validation of chain integrity and identity | Functional assessment of enriched chains |
| Validation Reagents | Single-lysine ubiquitin mutants (K48R, K63R) | Confirm linkage specificity | Critical controls for antibody and TUBE specificity [55] |
The selection of appropriate DUB inhibitors is particularly critical, as this choice can significantly impact experimental outcomes. Recent research has demonstrated that chloroacetamide (CAA) and N-ethylmaleimide (NEM)—two commonly used DUB inhibitors—can yield different interactor profiles in ubiquitin pulldown experiments. NEM provides nearly complete chain stabilization but may have off-target effects on cysteine residues in ubiquitin-binding surfaces, while CAA exhibits fewer side effects but allows partial chain disassembly [54]. For most applications, CAA is recommended when studying specific ubiquitin-binding interactions, while NEM may be preferable when complete preservation of ubiquitin chains is the highest priority.
Pathway Integration and Functional Correlations
The biological significance of ubiquitin linkage validation extends to numerous signaling pathways with profound therapeutic implications. In the NF-κB inflammatory signaling pathway, K63-linked ubiquitination of RIPK2 following L18-MDP stimulation activates downstream signaling, while PROTAC-induced K48-linked ubiquitination of the same protein targets it for degradation [47]. Similarly, the TRIP12-OTUD5 regulatory axis demonstrates how branched ubiquitin chains containing both K29 and K48 linkages can overcome the protective deubiquitinating activity of OTUD5 to promote proteasomal degradation [55]. These examples highlight how linkage-specific ubiquitination governs critical cellular decisions between activation and degradation.
The following diagram illustrates the experimental workflow for comprehensive ubiquitin linkage validation, integrating multiple methodological approaches:
Experimental Workflow for Ubiquitin Linkage Validation
The validation of K48 versus K63 ubiquitination signals requires sophisticated methodological approaches that can distinguish between these structurally similar but functionally distinct modifications. As this comparison demonstrates, methodological selection should be guided by the specific research question—whether it involves validating a single substrate, profiling system-wide ubiquitination changes, or quantifying specific linkage types. Antibody-based methods offer accessibility and direct visualization, TUBE-based approaches provide exceptional specificity and enrichment capability, while mass spectrometry delivers unparalleled structural detail and quantification precision.
The emerging complexity of heterotypic and branched ubiquitin chains presents both challenges and opportunities for future methodology development [54] [55]. The discovery that K48/K63-branched ubiquitin chains comprise approximately 20% of all K63 linkages in cells underscores the limitations of approaches that consider only homotypic chains [54]. Future methodological advances will need to address this complexity, potentially through the development of branch-specific binders or computational tools that can deconvolute mixed chain signals. For now, a combined approach utilizing multiple orthogonal methods provides the most robust validation of linkage specificity, ensuring that observed biological effects can be correctly attributed to specific ubiquitin signals.
Choosing Your Path: A Critical Comparison of In Vivo and In Vitro Methodologies
In the study of the ubiquitin-proteasome system, a central pathway in cellular regulation and drug discovery, researchers face a fundamental choice: to observe ubiquitination in its native cellular environment (in vivo) or to reconstitute the process with purified components in a tube (in vitro). Each approach presents a core trade-off between preserving full biological context and maintaining stringent experimental control. This guide objectively compares the performance, applications, and limitations of these foundational methodologies to inform research and development strategies.
Core Methodological Comparison
The decision between in vivo and in vitro approaches impacts every facet of experimental outcomes, from the complexity of the biological information obtained to the level of control over variables. The table below summarizes the key performance differentiators.
| Feature | In Vivo Ubiquitination Detection | In Vitro Ubiquitination Assay |
|---|---|---|
| Biological Context | Full cellular environment with native physiology, compartmentalization, and competing enzymatic activities [26]. | Minimal context; defined components in a cell-free system [59]. |
| Experimental Control | Low; complex cellular variables can influence results [26]. | High; complete control over reaction components (E1, E2, E3, substrate) [59] [32]. |
| Throughput & Quantitative Ease | Lower throughput; quantification can be challenging [26] [6]. | Higher throughput; easily quantifiable and reproducible reactions [59]. |
| Primary Application | Validating physiological relevance, studying endogenous protein regulation, and profiling linkage-specific signaling [26] [47] [7]. | Direct biochemical validation of E3 ligase activity, mechanistic studies, and screening for ubiquitination regulators [59] [5] [32]. |
| Data Output | Information on endogenous protein ubiquitination status and chain linkage type in a pathological or signaling context [26] [6]. | Direct evidence of ubiquitination, requiring follow-up studies to confirm physiological occurrence [59] [32]. |
Featured Experimental Protocols
To illustrate this trade-off in practice, the following sections detail two robust protocols: one for capturing endogenous ubiquitination in cells and another for a controlled, reconstituted reaction.
Protocol 1: Capturing Endogenous K63 Ubiquitination using TUBEs
This protocol leverages Tandem Ubiquitin Binding Entities (TUBEs) to study stimulus-induced, linkage-specific ubiquitination of an endogenous protein in cells, as demonstrated for RIPK2 [26] [47].
1. Cell Stimulation and Lysis:
- Culture human monocytic THP-1 cells.
- Pre-treat cells with a compound of interest (e.g., 100 nM Ponatinib) or a vehicle control (DMSO) for 30 minutes [26].
- Stimulate cells with an inflammatory agent (e.g., 200 ng/mL L18-MDP) for 30-60 minutes to induce K63-linked ubiquitination of RIPK2 [26].
- Lyse cells using a buffer optimized to preserve polyubiquitination (e.g., containing N-ethylmaleimide to inhibit deubiquitinases) [26] [6].
2. Ubiquitinated Protein Enrichment:
- Incubate the cell lysate with magnetic beads conjugated with K63-TUBEs (e.g., LifeSensors UM401M) [26] [47].
- For comparison, parallel samples can be enriched with Pan-TUBEs (all chain types) or K48-TUBEs.
- Wash the beads thoroughly to remove non-specifically bound proteins.
3. Detection and Analysis:
- Elute the bound ubiquitinated proteins and subject them to SDS-PAGE and immunoblotting [26].
- Probe the blot with an antibody against the protein of interest (e.g., anti-RIPK2) to detect its ubiquitinated forms [26].
- Expected Result: L18-MDP stimulation leads to a RIPK2 smear captured by K63- and Pan-TUBEs, but not K48-TUBEs, confirming linkage-specific endogenous ubiquitination [26].
Protocol 2: In Vitro Ubiquitination of a Purified Cytoplasmic Domain
This protocol details a method to directly test whether a specific E3 ligase can ubiquitinate a substrate, using purified recombinant proteins, as established for the cytoplasmic domain of PD-L1 [59].
1. Protein Expression and Purification:
- Express and purify the recombinant cytoplasmic domain of the immune checkpoint protein (e.g., PD-L1 residues 260-290) from E. coli BL21(DE3) with an N-terminal GST tag, using a pGEX4T-1 vector [59].
- Express and purify the relevant E3 ligases (e.g., CUL3/RBX1 complex from Sf9 insect cells) and other ubiquitination machinery (E1, E2, Ub) as recombinant proteins [59].
2. Liposome Preparation (Optional for Membrane Association Studies):
- Prepare liposomes from DC2.4 cell membranes to mimic the plasma membrane environment [59].
- Incorporate the purified cytoplasmic domain of PD-L1 into these liposomes.
3. In Vitro Ubiquitination Reaction:
- Assemble the reaction mixture containing:
- 50-100 nM E1 activating enzyme
- 1-2 µM E2 conjugating enzyme (e.g., UbCH5B, UbcH7)
- 2-4 µM E3 ligase (e.g., CUL3/RBX1, ARIH1, WWP2)
- 5-10 µM purified cytoplasmic domain substrate (soluble or incorporated into liposomes)
- 4-8 µM Ub (wild-type or mutant)
- 2 mM ATP in an appropriate reaction buffer [59].
- Incubate the reaction at 30°C for 1-2 hours.
4. Reaction Detection and Analysis:
- Terminate the reaction by adding SDS-PAGE loading buffer.
- Analyze the products by immunoblotting using an antibody against the substrate or an epitope tag (e.g., anti-HA) [59].
- Expected Result: A characteristic upward smear on the blot indicates polyubiquitination of the substrate, providing direct evidence of E3 ligase activity [59].
Key Signaling Pathways and Workflows
The diagrams below outline the core logical and experimental relationships central to this field.
Diagram 1: The Ubiquitination Cascade and Key Tools
Diagram 2: Experimental Workflow Comparison
The Scientist's Toolkit: Key Research Reagents
Successful ubiquitination research relies on specific, high-quality reagents. The table below details essential tools for both in vivo and in vitro approaches.
| Reagent | Function | Application Context |
|---|---|---|
| TUBEs (Tandem Ubiquitin Binding Entities) | High-affinity matrices for enriching polyubiquitinated proteins from cell lysates; available in pan-specific or linkage-specific (K48, K63) forms [26] [47] [6]. | In Vivo |
| Recombinant E1, E2, E3 Enzymes | Purified, active components of the ubiquitination cascade for reconstituting the reaction in a cell-free system [59] [32]. | In Vitro |
| Linkage-Specific Ub Antibodies | Antibodies that recognize specific polyubiquitin chain linkages (e.g., K48, K63) for detecting chain topology in immunoblots or enrichment [6]. | Primarily In Vivo |
| Epitope-Tagged Ubiquitin (e.g., His, HA) | Ubiquitin with an affinity tag for purifying and identifying ubiquitinated substrates from cell lysates [6] [7]. | Both |
| NEDD8 | A ubiquitin-like protein that activates cullin-ring E3 ligases (CRLs); essential for many in vitro assays involving CRLs [59]. | In Vitro |
The choice between in vivo and in vitro ubiquitination detection is not about selecting a superior method, but about aligning the experimental approach with the specific research question. For confirming the physiological relevance of a ubiquitination event and its linkage-specific role in signaling, in vivo methods with tools like TUBEs are indispensable. For mechanistic dissection, direct validation of E3 ligase activity, and screening applications, the controlled environment of in vitro assays is paramount. A robust research program often leverages the complementary strengths of both to move from initial discovery to mechanistic understanding.
The detection and characterization of protein ubiquitination are fundamental to understanding a vast array of cellular processes, from protein degradation to DNA repair and immune signaling. Research in this field relies on a diverse toolkit of methodologies, broadly categorized into in vivo (conducted within living cells or organisms) and in vitro (conducted in a controlled laboratory environment outside of a living organism) techniques. Each approach offers distinct advantages and limitations in throughput, cost, physiological relevance, and technical difficulty. This guide provides an objective comparison of these methods, supported by experimental data, to help researchers and drug development professionals select the appropriate strategy for their investigative goals.
Key Ubiquitination Detection Methodologies
The following table summarizes the core characteristics of prominent ubiquitination detection techniques used in modern research.
Table 1: Comparative Analysis of Ubiquitination Detection Methodologies
| Methodology | Category | Throughput | Relative Cost | Physiological Relevance | Technical Difficulty |
|---|---|---|---|---|---|
| TUBE-Based ELISA [48] [26] | In vitro / High-Throughput Screening | High (96/384-well plate format) | Moderate | Moderate (uses endogenous cell lysates) | Low |
| Immunoblotting (Western Blot) [6] | In vitro / Low-Throughput | Low | Low | High (detects endogenous proteins) | Moderate |
| Mass Spectrometry (MS) Proteomics [6] | In vitro / Discovery | Medium (multiplexed samples) | High | High (can profile endogenous ubiquitination) | High |
| In Vitro Reconstituted Assays [14] [50] [60] | In vitro / Targeted | Medium (adaptable to HTS) | Low to Moderate | Low (minimal system components) | Low to Moderate |
| Tagged Ubiquitin Expression [6] | In vivo (genetically engineered) | Low to Medium | Moderate | High, but with potential artifacts | Moderate |
| Genetic-Fusion-Dependent Degradation (e.g., TCD) [61] | In vivo (genetically engineered) | Low (per construct) | Low (after initial development) | High (targets endogenous proteins) | High |
Detailed Experimental Protocols
To ensure reproducibility and provide clarity on how these techniques are implemented, this section outlines standard protocols for key methodologies.
TUBE-Based ELISA for Linkage-Specific Ubiquitination
This protocol is used to study chain-specific ubiquitination of endogenous proteins in a high-throughput format, as demonstrated for RIPK2 [26].
- Step 1: Cell Stimulation and Lysis. Culture cells (e.g., THP-1 monocytic cells) and treat with stimuli (e.g., L18-MDP to induce K63-ubiquitination) or inhibitors (e.g., Ponatinib). Lyse cells using a buffer optimized to preserve polyubiquitination and prevent deubiquitinase (DUB) activity.
- Step 2: Affinity Capture. Coat a 96-well plate with chain-specific Tandem Ubiquitin Binding Entities (TUBEs), such as K63-TUBEs or K48-TUBEs. Incubate the prepared cell lysates in the coated wells to allow linkage-specific ubiquitinated proteins to bind.
- Step 3: Target Detection. After washing, detect the captured target protein (e.g., RIPK2) using a target-specific primary antibody followed by an HRP-conjugated secondary antibody.
- Step 4: Signal Measurement. Add a chemiluminescent or colorimetric detection reagent and measure the signal using a plate reader. The signal intensity corresponds to the level of linkage-specific ubiquitination of the target protein [26].
In Vitro Ubiquitination Assay for PROTAC Screening
This protocol is designed to screen for and characterize PROTAC (Proteolysis Targeting Chimera) efficiency using a reconstituted system [60].
- Step 1: Reconstitute System. Combine the core ubiquitination machinery in a tube or well: E1 activating enzyme, E2 conjugating enzyme, ubiquitin, and ATP in an appropriate reaction buffer.
- Step 2: Introduce Specificity. Add the E3 ligase of interest (e.g., Cereblon, VHL, or HDM2), the target protein, and the heterobifunctional PROTAC molecule. The PROTAC acts as a bridge, bringing the E3 ligase and target protein into proximity.
- Step 3: Incubate. Allow the reaction to proceed at a defined temperature (e.g., 30°C) for a set time to enable ubiquitin transfer onto the target protein.
- Step 4: Detect Ubiquitination. The ubiquitinated target protein can be detected by various methods, including immunoblotting with an anti-ubiquitin or target-specific antibody, or via specialized assay kits that use affinity-based detection in a plate format [60].
Targeted Condensation-Prone-Protein Degradation (TCD) in Plants
This is an in vivo, genetic-fusion-dependent method for targeted protein degradation in plants, which does not require small molecules [61].
- Step 1: Identify an E3 Ligase. Screen for and identify a condensation-prone RING-type E3 ubiquitin ligase (e.g., E3TCD1/E3IDP45) that exhibits the capacity to form condensates and has self-degradation properties.
- Step 2: Create a Genetic Fusion. Genetically fuse the gene of the endogenous target protein (X) with the identified E3 ligase (E3TCD1) to create an X–E3TCD1 fusion construct.
- Step 3: Transgenic Expression. Introduce the X–E3TCD1 fusion construct into the plant (e.g., rice) to generate transgenic lines.
- Step 4: Functional Validation. The X–E3TCD1 fusion protein integrates into condensates containing the native target protein X. Through its E3 ligase activity, it then recruits the cellular degradation machinery to ubiquitinate and degrade the entire condensate, effectively reducing the levels of the target protein and allowing observation of the resulting phenotypic changes [61].
Visualization of Key Concepts and Workflows
The following diagrams illustrate the core ubiquitination process and the experimental workflow for a key methodology.
The Ubiquitination Cascade
TUBE-Based ELISA Workflow
The Scientist's Toolkit: Essential Research Reagents
This table lists key reagents and tools used in ubiquitination research, as cited in the methodologies above.
Table 2: Key Research Reagent Solutions for Ubiquitination Studies
| Reagent / Tool | Function | Example Application |
|---|---|---|
| Tandem Ubiquitin Binding Entities (TUBEs) [48] [26] [6] | High-affinity reagents that bind polyubiquitin chains, protecting them from DUBs and enabling enrichment/detection. | Capturing endogenous ubiquitinated proteins from cell lysates for immunoassays (ELISA) or pulldown. |
| Linkage-Specific Antibodies [6] | Antibodies that recognize a specific ubiquitin chain linkage (e.g., K48-only, K63-only). | Enriching and detecting a particular type of polyubiquitin chain in immunoblotting or immunofluorescence. |
| PROTAC Molecules [60] [26] | Heterobifunctional small molecules that recruit an E3 ligase to a target protein to induce its ubiquitination and degradation. | As an experimental tool to degrade a protein of interest; as a therapeutic modality in drug discovery. |
| Tagged Ubiquitin (e.g., His-, HA-, Strep-tag) [6] | Ubiquitin genetically fused to an affinity tag for purification. | Global profiling of ubiquitinated substrates and identification of ubiquitination sites via mass spectrometry. |
| Reconstituted E1/E2/E3 Systems [14] [60] | Purified components of the ubiquitination enzymatic cascade. | Studying the biochemistry of ubiquitination in a controlled, minimal system, free from cellular complexity. |
Ubiquitination is a versatile post-translational modification that regulates diverse cellular functions, including proteasomal degradation, signal transduction, and immune responses [26]. The functional consequences of ubiquitination are determined by the type of polyubiquitin chain assembled on substrate proteins. Among the eight possible ubiquitin linkage types, K48-linked chains primarily target proteins for proteasomal degradation, while K63-linked chains predominantly regulate non-degradative signaling processes [26] [62]. This linkage specificity presents both a challenge and opportunity for researchers studying disease mechanisms and therapeutic development.
Receptor-Interacting Serine/Threonine-Protein Kinase 2 (RIPK2) serves as an ideal model for studying linkage-specific ubiquitination due to its well-characterized roles in inflammatory signaling. RIPK2 is a critical adaptor kinase in the NOD1/NOD2 signaling pathway, which regulates immune responses to bacterial pathogens [63] [64]. Upon activation by nucleotide-binding oligomerization domain (NOD) receptors, RIPK2 undergoes context-dependent ubiquitination: K63-linked ubiquitination during inflammatory signaling and K48-linked ubiquitination during targeted degradation [26]. The ability to differentiate these distinct ubiquitination events is essential for understanding RIPK2 biology and developing targeted therapies.
Traditional methods for studying ubiquitination, including Western blotting and mass spectrometry, face significant limitations in throughput, sensitivity, and linkage specificity [26]. This case study examines how Tandem Ubiquitin Binding Entities (TUBEs) address these limitations by enabling high-throughput, linkage-specific analysis of RIPK2 ubiquitination, providing insights relevant to both basic research and drug discovery.
Technical Comparison: Ubiquitination Detection Methods
Limitations of Conventional Approaches
Studying endogenous protein ubiquitination has historically presented substantial technical challenges. Conventional methods each carry significant limitations that hinder comprehensive analysis of linkage-specific ubiquitination events.
Table 1: Comparison of Ubiquitination Detection Methodologies
| Method | Key Applications | Throughput | Linkage Specificity | Major Limitations |
|---|---|---|---|---|
| Western Blotting | General ubiquitination detection | Low | Limited | Semi-quantitative, low sensitivity, inability to differentiate linkages [26] |
| Mass Spectrometry | Ubiquitination site mapping | Low to medium | High with enrichment | Labor-intensive, requires sophisticated instrumentation, limited sensitivity for rapid changes [26] |
| Mutant Ubiquitin Expression | Linkage-specific function studies | Medium | High | May not accurately represent wild-type ubiquitin modifications, potential artifacts [26] |
| TUBE-Based Assays | Linkage-specific endogenous ubiquitination | High | High | Requires specialized reagents, relatively new technology [26] [48] |
Mass spectrometry, while powerful for identifying ubiquitination sites, faces challenges in detecting subtle changes in endogenous protein ubiquitination and requires extensive sample preparation [26]. The use of exogenously expressed mutant ubiquitins, where specific lysines are mutated to arginine to prevent certain chain types, may introduce artifacts and not accurately recapitulate modifications involving wild-type ubiquitin [26]. These limitations highlight the need for improved techniques to specifically capture, detect, and study linkage-specific ubiquitination of endogenous proteins.
TUBE Technology Fundamentals
Tandem Ubiquitin Binding Entities (TUBEs) represent a technological advancement for studying ubiquitination. These engineered affinity matrices consist of multiple ubiquitin-associated (UBA) domains arranged in tandem, conferring nanomolar affinities for polyubiquitin chains [26] [48]. This structural configuration enables TUBEs to protect polyubiquitin chains from deubiquitinating enzymes (DUBs) during cell lysis and processing, preserving the native ubiquitination status of proteins [48].
The development of chain-selective TUBEs has been particularly transformative for linkage-specific analyses. These specialized TUBEs are engineered with mutations that favor binding to specific ubiquitin linkage types while discriminating against others [26]. For RIPK2 research, K48- and K63-specific TUBEs enable researchers to differentiate between degradative and non-degradative ubiquitination events in response to different cellular stimuli. When coated on microplates, these TUBEs facilitate high-throughput applications in 96-well format, overcoming the throughput limitations of Western blotting [26] [48].
Experimental Application: TUBE Technology for RIPK2 Ubiquitination
Methodologies and Protocols
Cell Culture and Stimulation
The following protocol has been optimized for studying endogenous RIPK2 ubiquitination in human monocytic THP-1 cells:
- Cell Culture: Maintain THP-1 cells in RPMI-1640 medium supplemented with 10% fetal bovine serum at 37°C with 5% CO₂.
- Stimulation Conditions:
- For K63 ubiquitination: Treat cells with 200-500 ng/ml L18-MDP (Lysine 18-muramyldipeptide) for 30-60 minutes to activate NOD2-RIPK2 signaling [26].
- For K48 ubiquitination: Treat cells with RIPK2 PROTAC degrader (e.g., RIPK degrader-2) to induce degradative ubiquitination [26].
- For inhibition studies: Pre-treat cells with 100 nM Ponatinib (RIPK2 kinase inhibitor) for 30 minutes prior to stimulation [26].
- Cell Lysis: Harvest cells and lyse in ubiquitination-preserving lysis buffer (recommended: 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 10% glycerol, plus protease and deubiquitinase inhibitors) [26].
TUBE-Based Ubiquitin Capture
- TUBE Selection: Choose chain-specific TUBEs based on experimental goals:
- K63-TUBEs for inflammatory signaling studies
- K48-TUBEs for degradation studies
- Pan-TUBEs for total ubiquitination assessment
- Affinity Capture:
- For magnetic bead-based TUBEs (e.g., LifeSensors UM401M): Incubate 50-100 µg of cell lysate with TUBE-conjugated magnetic beads for 2 hours at 4°C with gentle rotation [26].
- For plate-based TUBEs: Coat 96-well plates with chain-specific TUBEs, block with appropriate buffer, then add cell lysates and incubate for 2 hours at room temperature [26] [48].
- Washing and Elution: Wash captured complexes 3-5 times with lysis buffer, then elute bound proteins with 2× Laemmli buffer for Western blot analysis.
Detection and Analysis
- Immunoblotting: Separate eluted proteins by SDS-PAGE, transfer to PVDF membranes, and probe with anti-RIPK2 antibody to detect ubiquitinated RIPK2 species.
- Quantification: Use densitometry software to quantify ubiquitination signals normalized to total input RIPK2.
- Data Interpretation: Compare linkage-specific ubiquitination patterns across experimental conditions.
Diagram 1: RIPK2 Signaling and Ubiquitination Pathways. RIPK2 undergoes K63-linked ubiquitination during NOD2-mediated inflammatory signaling (top) versus K48-linked ubiquitination during PROTAC-induced degradation (bottom).
Key Experimental Findings
Application of TUBE technology to RIPK2 ubiquitination has yielded crucial insights into its regulation and function:
Stimulus-Dependent Ubiquitination Switching
Research demonstrates that RIPK2 undergoes distinct ubiquitination patterns depending on cellular context:
- L18-MDP stimulation induces robust K63-linked ubiquitination of endogenous RIPK2, detectable within 30 minutes and declining by 60 minutes [26]. This ubiquitination is mediated by the E3 ligase XIAP, which builds K63-linked chains on multiple lysine residues of RIPK2 [26] [64].
- RIPK2 PROTAC treatment induces primarily K48-linked ubiquitination, targeting RIPK2 for proteasomal degradation [26].
- Pharmacological inhibition with Ponatinib (100 nM) completely abrogates L18-MDP-induced RIPK2 ubiquitination, demonstrating the requirement for kinase activity in this process [26].
Quantitative Assessment of Linkage Specificity
TUBE-based assays enable precise quantification of linkage-specific ubiquitination:
Table 2: Quantitative Analysis of RIPK2 Ubiquitination Using TUBE Technology
| Experimental Condition | K63-TUBE Signal | K48-TUBE Signal | Pan-TUBE Signal | Biological Outcome |
|---|---|---|---|---|
| Untreated Cells | Baseline | Baseline | Baseline | Homeostatic state |
| L18-MDP (30 min) | ++++ | + | ++++ | Inflammatory signaling [26] |
| L18-MDP (60 min) | ++ | + | ++ | Signal attenuation [26] |
| RIPK2 PROTAC | + | ++++ | ++++ | Targeted degradation [26] |
| Ponatinib + L18-MDP | - | - | - | Signaling inhibition [26] |
The data clearly demonstrate that chain-selective TUBEs can differentiate between context-dependent ubiquitin linkages on endogenous RIPK2. K63-TUBEs specifically capture inflammatory signaling-induced ubiquitination, while K48-TUBEs selectively recognize degradative ubiquitination [26].
Comparative Data: TUBE Technology Versus Traditional Methods
Performance Metrics
The advantages of TUBE-based approaches become evident when directly compared to traditional methodologies:
Table 3: Comprehensive Method Comparison for RIPK2 Ubiquitination Studies
| Performance Parameter | Western Blotting | Mass Spectrometry | TUBE-Based Assays |
|---|---|---|---|
| Detection Sensitivity | Low to moderate | High (with enrichment) | High (nanomolar affinity) [26] [48] |
| Linkage Specificity | Limited without linkage-specific antibodies | High | High (chain-selective TUBEs) [26] |
| Throughput Capacity | Low (1-10 samples/day) | Low to medium | High (96-well format) [26] [48] |
| Endogenous Protein Compatibility | Yes | Yes | Yes [26] |
| Quantitative Capability | Semi-quantitative | Quantitative | Quantitative [26] [48] |
| Equipment Requirements | Standard molecular biology | Specialized instrumentation | Standard plate readers |
| Assay Development Time | Short | Extensive | Moderate |
| Data Reproducibility | Variable | High | High [26] |
Technical Advantages of TUBE-Based Approaches
TUBE technology provides several distinct technical advantages for studying RIPK2 ubiquitination:
- Preservation of Native Ubiquitination: TUBEs protect polyubiquitin chains from deubiquitinating enzymes (DUBs) during cell lysis and processing, maintaining the endogenous ubiquitination state [48].
- Compatibility with Endogenous Proteins: Unlike overexpression systems, TUBEs enable study of ubiquitination on endogenous RIPK2 at physiological expression levels, avoiding artifacts associated with protein overexpression [26] [64].
- High-Throughput Capability: The adaptation of TUBEs to 96-well plate formats enables screening of multiple conditions and compounds in parallel, facilitating drug discovery applications [26] [48].
- Linkage-Specific Resolution: Chain-selective TUBEs provide unambiguous differentiation between ubiquitin linkage types that mediate distinct functional outcomes [26].
Successful application of TUBE technology for RIPK2 ubiquitination studies requires several key reagents and tools:
Table 4: Essential Research Reagents for RIPK2 Ubiquitination Studies
| Reagent/Tool | Specific Example | Function/Application | Technical Notes |
|---|---|---|---|
| Chain-Selective TUBEs | K48-TUBE, K63-TUBE, Pan-TUBE | Linkage-specific ubiquitin capture | Available as magnetic beads or pre-coated plates [26] [48] |
| RIPK2 Activators | L18-MDP, Muramyl Dipeptide (MDP) | Induce K63-linked ubiquitination via NOD2 signaling | Use at 200-500 ng/ml for 30-60 min [26] |
| RIPK2 Degraders | RIPK2 PROTACs (e.g., RIPK degrader-2) | Induce K48-linked ubiquitination and degradation | Concentration varies by specific compound [26] |
| RIPK2 Inhibitors | Ponatinib, GSK2983559 | Block kinase activity and ubiquitination | Ponatinib used at 100 nM for pre-treatment [26] [65] |
| Cell Models | THP-1 cells, BMDMs | Study endogenous RIPK2 ubiquitination | IFNγ priming enhances MDP response in BMDMs [26] [64] |
| Detection Antibodies | Anti-RIPK2, Anti-Ubiquitin | Detect ubiquitinated RIPK2 species | Validate for specific applications |
| DUB Inhibitors | Broad-spectrum DUB inhibitor cocktails | Preserve ubiquitination during processing | Include in lysis buffers [26] |
Research Implications and Future Directions
Application to Drug Discovery and Development
The ability to monitor linkage-specific ubiquitination of RIPK2 has significant implications for pharmaceutical research:
- PROTAC Characterization: TUBE-based assays enable rapid assessment of PROTAC efficiency by quantifying K48-linked ubiquitination of target proteins like RIPK2 [26]. This facilitates optimization of degraders for enhanced potency and selectivity.
- Mechanistic Insights: Recent studies reveal that some kinase inhibitors, including RIPK2 inhibitors, can accelerate target degradation by promoting K48-linked ubiquitination, adding a new dimension to their mechanism of action [66]. TUBE technology provides a direct means to investigate this phenomenon.
- Biomarker Development: Monitoring linkage-specific ubiquitination patterns could serve as pharmacodynamic biomarkers for target engagement in clinical trials of RIPK2-directed therapies.
Integration with Complementary Techniques
While TUBE technology offers significant advantages, optimal experimental design often involves integration with complementary approaches:
- Genetic Validation: Combine TUBE assays with CRISPR/Cas9-mediated gene editing to validate the roles of specific E3 ligases (e.g., XIAP) or ubiquitination sites in RIPK2 regulation [64] [67].
- Deubiquitinase Studies: Investigate DUB regulation of RIPK2 by pairing TUBE assays with DUB inhibition or genetic ablation. For example, OTUB2 has been identified as a DUB that stabilizes RIPK2 by removing K48-linked ubiquitination [67].
- Structural Biology: Correlate ubiquitination findings with structural studies of RIPK2 complexes to understand how ubiquitination interfaces with RIPK2 dimerization and oligomerization [63].
Diagram 2: TUBE Technology Experimental Workflow. The general workflow for studying RIPK2 ubiquitination using TUBE technology, from cell stimulation to downstream analysis applications.
TUBE technology represents a significant advancement in the study of protein ubiquitination, addressing critical limitations of traditional methods while enabling high-throughput, linkage-specific analysis. Applied to RIPK2, this approach has revealed the dynamic regulation of inflammatory signaling and targeted degradation through distinct ubiquitin linkages. The ability to differentiate K48- versus K63-linked ubiquitination on endogenous RIPK2 provides researchers with a powerful tool for investigating basic biological mechanisms and developing targeted therapeutics for inflammatory diseases, cancer, and other conditions involving dysregulated ubiquitination.
As the ubiquitin field continues to evolve, TUBE technology offers a versatile platform for answering fundamental questions about linkage-specific ubiquitination while accelerating drug discovery efforts targeting the ubiquitin-proteasome system.
Ubiquitination, a crucial post-translational modification, regulates diverse cellular functions from protein degradation to signal transduction. The ubiquitination cascade involves E1 activating, E2 conjugating, and E3 ligase enzymes that collectively target proteins for modification, with complexity arising from various ubiquitin chain linkages that determine functional outcomes [6]. For researchers investigating this complex system, a significant challenge lies in effectively correlating findings from controlled in vitro experiments with biologically relevant in vivo validation. This guide objectively compares the performance, applications, and limitations of current ubiquitination detection techniques across experimental settings to help researchers develop robust validation strategies.
Methodological Comparison: In Vitro versus In Vivo Ubiquitination Assays
Fundamental Approaches and Technical Considerations
In vitro ubiquitination assays utilize purified components—E1, E2, and E3 enzymes, ubiquitin, and substrate—in a controlled environment to establish direct mechanistic relationships. These reconstituted systems allow researchers to dissect specific enzymatic activities without confounding cellular factors [27]. For example, Xie et al. systematically compared PD-L1 ubiquitination by CRL3SPOP, ARIH1, and NEDD4 family ligases using purified components, discovering that ARIH1 independently ubiquitinates PD-L1 while CRL3SPOP showed no direct activity despite previous cellular studies implicating it in PD-L1 degradation [27].
In vivo ubiquitination assays investigate ubiquitination within its native cellular context, preserving physiological conditions including competing enzymes, subcellular localization, and post-translational modifications that regulate ubiquitination. These assays typically involve immunoprecipitation of the target protein followed by anti-ubiquitin immunoblotting, often with proteasome inhibition (e.g., MG132) to preserve ubiquitinated species [68]. For instance, studies of maize viral infection responses demonstrated increased global ubiquitination levels during infection, findings that would be difficult to recapitulate in purified systems [69].
Table 1: Core Characteristics of In Vitro and In Vivo Ubiquitination Assays
| Characteristic | In Vitro Assays | In Vivo Assays |
|---|---|---|
| Complexity | Minimal, defined components | High, cellular environment |
| Control | High precision over reaction conditions | Limited control over cellular environment |
| Mechanistic Insight | Direct establishment of enzyme-substrate relationships | Indirect, influenced by cellular context |
| Throughput | Potentially high for screening | Lower, requires transfection/transduction |
| Physiological Relevance | Limited, may lack necessary co-factors | High, native cellular environment |
| Key Applications | Mechanistic studies, enzyme characterization, drug screening | Validation of physiological relevance, disease modeling |
Quantitative Performance Metrics
Recent technological advances have enhanced the detection sensitivity and specificity for both in vitro and in vivo applications. In vitro, the use of liposome-associated assays has revealed how phosphorylation enhances PD-L1 ubiquitination by disrupting its membrane association, a finding that required membrane context to elucidate [27]. In vivo, chain-specific TUBEs (Tandem Ubiquitin Binding Entities) with nanomolar affinities now enable researchers to differentiate context-dependent linkage-specific ubiquitination of endogenous proteins like RIPK2, distinguishing between K48-linked degradative ubiquitination and K63-linked signaling ubiquitination in cellular environments [26].
Table 2: Performance Metrics of Ubiquitination Detection Methods
| Method | Sensitivity | Linkage Specificity | Quantitative Capability | Throughput Potential |
|---|---|---|---|---|
| Traditional In Vivo IP+WB | Low to moderate (microgram protein input) | Limited without specific antibodies | Semi-quantitative | Low |
| TUBE-Based Enrichment | High (captures endogenous ubiquitination) | Yes, with linkage-specific TUBEs | Quantitative with normalization | Medium to high (96-well format) |
| In Vitro Reconstitution | High (precise control of components) | Can be designed with specific Ub mutants | Highly quantitative | High for enzyme characterization |
| Ubiquitinome Profiling | Very high (requires specialized expertise) | Comprehensive linkage analysis | Quantitative with isotopic labeling | Low to medium |
Experimental Protocols for Integrated Workflows
In Vitro Ubiquitination Assay Protocol
The following protocol, adapted from studies of HUWE1 ligase activity, provides a framework for establishing direct enzyme-substrate relationships [53] [14]:
Reaction Setup: Combine purified E1 enzyme (50 nM), E2 enzyme (200 nM-1 μM), E3 ligase (50-500 nM), ubiquitin (10-50 μM), and substrate (1-10 μM) in reaction buffer (50 mM Tris-HCl pH 7.5, 50 mM NaCl, 5 mM MgCl₂, 2 mM ATP, 1 mM DTT)
Incubation: Conduct reactions at 30°C for 1-3 hours
Termination and Analysis: Stop reactions with SDS-PAGE sample buffer, followed by western blotting with anti-ubiquitin antibodies or substrate-specific antibodies
Validation: Confirm ubiquitination sites through mass spectrometry analysis of reaction products, identifying characteristic di-glycine remnants on modified lysines (+114.04 Da mass shift) [6]
This approach enabled researchers to discover that small-molecule HUWE1 inhibitors like BI8622 and BI8626 are themselves ubiquitinated at primary amine groups, revealing an unexpected substrate-competitive inhibition mechanism [53] [14].
Integrated In Vivo Validation Protocol
For validating in vitro findings in cellular contexts, the following protocol provides a standardized approach [68]:
Cell Treatment: Culture appropriate cell lines (HEK293T, HeLa, or specialized lines like THP-1 for immune signaling studies) and transfect with target protein constructs
Proteasome Inhibition: Treat cells with 50 μM MG132 for 4-6 hours before harvesting to stabilize ubiquitinated proteins
Cell Lysis and Denaturation: Lyse cells in buffer containing 1% SDS and boil for 10 minutes to disrupt non-covalent interactions, then dilute 10-fold with standard lysis buffer
Immunoprecipitation: Incubate lysates with target protein antibody-conjugated beads overnight at 4°C
Ubiquitination Detection: Analyze immunoprecipitates by SDS-PAGE and western blotting with anti-ubiquitin antibodies (e.g., P4D1, FK1/FK2) or linkage-specific antibodies
This methodology has been successfully applied to validate inflammatory signaling pathways, demonstrating that L18-MDP stimulation induces K63-linked ubiquitination of endogenous RIPK2 in THP-1 cells, which can be completely abrogated by the RIPK2 inhibitor Ponatinib [26].
Advanced Technical Solutions for Ubiquitination Research
Research Reagent Solutions
Table 3: Essential Research Reagents for Ubiquitination Studies
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| Linkage-Specific Binders | K48-TUBEs, K63-TUBEs, M1-specific antibodies | Selective enrichment and detection of specific ubiquitin chain linkages |
| Activity-Based Probes | Ub-VS, Ub-AMC, Di-Ub probes | Monitoring deubiquitinase activity and ubiquitin chain cleavage |
| Tagged Ubiquitin | His-Ub, HA-Ub, Strep-Ub, GFP-Ub | Affinity purification and visualization of ubiquitinated proteins |
| Proteasome Inhibitors | MG132, Bortezomib, Carfilzomib | Stabilization of ubiquitinated proteins by blocking proteasomal degradation |
| E3 Ligase Modulators | PROTACs, Molecular Glues (e.g., Pomalidomide) | Targeted manipulation of specific ubiquitination pathways |
| Ubiquitination Sensors | Tandem UBD reporters, Ubiquitination-sensitive FRET probes | Live-cell monitoring of ubiquitination events |
Technology Integration Workflows
Emerging technologies are creating new opportunities for bridging the in vitro-in vivo gap. DNA-encoded library (DEL) screening approaches now enable multiplexed functional screens that simultaneously evaluate small molecules and protein targets for ubiquitination susceptibility. This method uses DNA hybridization to pre-associate encoded small molecules with potential protein substrates, allowing identification of optimal ligand/substrate pairs in a single functional experiment [50]. Similarly, the development of the StUbEx (Stable Tagged Ubiquitin Exchange) cellular system enables replacement of endogenous ubiquitin with tagged variants in human cell lines, facilitating high-confidence identification of ubiquitination sites through quantitative proteomics [6].
Visualization of Integrated Experimental Workflows
In Vitro to In Vivo Validation Pathway
Ubiquitination Signaling and Detection Methodology
Bridging the gap between in vitro and in vivo ubiquitination research requires strategic methodological integration rather than sequential application of techniques. The most robust research approaches:
- Establish mechanistic foundations using purified component systems to demonstrate direct enzyme-substrate relationships
- Validate physiological relevance in cellular models that preserve native context and regulation
- Employ linkage-specific tools to decipher the functional consequences of ubiquitination
- Leverage emerging technologies like TUBEs and DEL screens for higher-resolution analysis
This integrated methodology framework enables researchers to navigate the complexities of ubiquitination signaling with greater confidence, ultimately accelerating the translation of basic research findings into therapeutic applications, particularly in cancer immunotherapy and targeted protein degradation.
The ubiquitin-proteasome system (UPS) is a fundamental regulatory mechanism in eukaryotic cells, controlling protein stability, signaling, and degradation. For researchers investigating this system, particularly in the context of drug development for conditions like cancer, neurodegeneration, and autoimmune diseases, selecting the appropriate experimental approach for detecting ubiquitination is critical. The choice between in vivo (within living systems) and in vitro (in controlled laboratory environments) methodologies carries significant implications for data interpretation, physiological relevance, and technical feasibility. This guide provides an objective comparison of these approaches, highlighting their respective strengths and limitations to inform experimental design in ubiquitination research.
Fundamental Principles of Ubiquitination Detection
Ubiquitination involves a sequential enzymatic cascade where E1 activating enzymes, E2 conjugating enzymes, and E3 ligases work together to attach ubiquitin to substrate proteins. This modification can result in various outcomes depending on the ubiquitin chain linkage type, with K48-linked chains primarily targeting substrates for proteasomal degradation, and K63-linked chains typically regulating signal transduction and protein trafficking [26]. Detection methods must therefore consider not only whether ubiquitination occurs but also the specific linkage types and cellular context.
The core challenge in ubiquitination research lies in capturing these often transient modifications while maintaining physiological relevance or achieving precise experimental control. The following diagram illustrates the fundamental workflows distinguishing in vivo from in vitro approaches:
Comparative Analysis: In Vivo vs. In Vitro Ubiquitination Detection
The table below summarizes the core characteristics, strengths, and limitations of in vivo and in vitro approaches for ubiquitination detection:
| Parameter | In Vivo Detection | In Vitro Detection |
|---|---|---|
| Physiological Relevance | High - maintains cellular context, compartmentalization, and native regulation [8] | Reduced - lacks cellular environment and endogenous regulatory mechanisms [59] |
| Experimental Control | Limited - complex cellular environment with competing processes | High - defined components and controlled conditions [59] |
| Technical Complexity | High - requires specialized techniques like microinjection or genetic manipulation [8] | Moderate - utilizes purified components in controlled reactions [59] |
| Throughput Potential | Lower - limited by cellular viability and transduction efficiency | Higher - amenable to multi-well formats and screening approaches [26] |
| Key Applications | Studying endogenous protein regulation, pathway interactions, and therapeutic effects in physiological contexts [8] [70] | Mechanism validation, enzyme characterization, and initial screening under defined conditions [59] [53] |
| Primary Limitations | Complex data interpretation due to cellular complexity; potential interference from parallel pathways [8] | May not recapitulate native cellular regulation; requires protein purification and reconstitution [59] |
| Data Output Examples | Single-cell degradation rates; endogenous protein ubiquitination status; pathway activation in native context [8] [26] | Direct E3 ligase activity; ubiquitination kinetics; minimal component requirements for activity [59] [53] |
Detailed Methodological Approaches
In Vivo Ubiquitination Detection
Microinjection-Based Degradation Assay
This sophisticated approach involves direct introduction of pre-formed complexes into living cells, allowing precise measurement of degradation kinetics without confounding factors from biosynthesis and transport. The methodology includes:
Complex Preparation: Purified target proteins (e.g., GS-eGFP with additional N-terminal Gly-Ser residues for proteasomal engagement) are complexed with fluorescently labeled bioPROTACs at 1:1 molar ratios [8].
Fluorescent Labeling: The bioPROTAC component is labeled with TMR5-maleimide dye coupled to intrinsic cysteines, with labeling efficiency verified by ESI-MS analysis [8].
Microinjection: Pre-formed complexes are microinjected directly into the cytosol of HEK293 cells using specialized equipment [8].
Live-Cell Imaging & Quantification: Fluorescence degradation is monitored in real-time using live-cell microscopy, with approximately 30 single-cell degradation rates measured to calculate mean degradation rates, accounting for natural cell-to-cell variability [8].
This method directly measures actual degradation rates, independent of biosynthesis or uptake kinetics, providing high physiological relevance for assessing ubiquitination efficiency in living cells [8].
TUBE-Based Endogenous Ubiquitination Detection
Tandem Ubiquitin Binding Entities (TUBEs) enable high-throughput analysis of linkage-specific ubiquitination on endogenous proteins:
Cell Stimulation: Cells (e.g., THP-1 human monocytic cells) are treated with pathway-specific stimuli (e.g., L18-MDP for K63 ubiquitination or PROTACs for K48 ubiquitination) [26].
Cell Lysis & Preservation: Cells are lysed using optimized buffers that preserve polyubiquitination states, preventing deubiquitinase activity during processing [26].
Chain-Specific Enrichment: Lysates are incubated with linkage-specific TUBEs (K48-, K63-, or pan-specific) coated on 96-well plates, enabling high-throughput processing [26].
Detection & Quantification: Captured ubiquitinated proteins are detected using target-specific antibodies, allowing quantification of linkage-specific ubiquitination events on endogenous proteins like RIPK2 [26].
This approach faithfully captures context-dependent ubiquitination, distinguishing between inflammatory (K63) and degradative (K48) signaling on the same endogenous protein [26].
In Vitro Ubiquitination Detection
Reconstituted Ubiquitination Assay
This reductionist approach utilizes purified components to establish minimal systems for mechanistic studies:
Protein Purification: Recombinant proteins are expressed and purified, including:
- E1 activating enzyme (UBA1)
- E2 conjugating enzymes (UBE2L3 or UBE2D3)
- E3 ligases (full-length or catalytic domains)
- Substrate proteins (often cytoplasmic domains of target proteins) [59]
Reaction Assembly: Components are combined in optimized buffers containing ATP, ubiquitin, and energy regeneration systems [59].
Incubation & Time-Course: Reactions are incubated at appropriate temperatures, with time points taken to monitor ubiquitination kinetics [59].
Analysis: Products are analyzed by SDS-PAGE and immunoblotting using ubiquitin-specific or tag-specific antibodies [59].
This approach has been successfully applied to characterize ubiquitination of immune checkpoint proteins like PD-L1, demonstrating direct E3 ligase activity on purified cytoplasmic domains [59].
Specialized Applications: Non-Protein Ubiquitination
Recent advances have revealed that E3 ligases can ubiquitinate diverse non-protein substrates:
Small-Molecule Ubiquitination: HUWE1-mediated ubiquitination of drug-like small molecules (BI8622 and BI8626) occurs through canonical catalytic cascades, transferring ubiquitin to primary amino groups on compounds [53].
Carbohydrate Ubiquitination: HOIL-1 efficiently ubiquitinates serine residues and various saccharides in vitro, with minimal activity toward threonine and no activity for lysine residues, mediated by critical active site residue His510 [32].
These specialized applications demonstrate the expanding substrate repertoire of E3 ligases and provide novel platforms for investigating ubiquitination mechanisms.
The Scientist's Toolkit: Essential Research Reagents
The table below outlines key reagents and their applications in ubiquitination research:
| Research Tool | Primary Function | Application Context | Key Features |
|---|---|---|---|
| Chain-Specific TUBEs | High-affinity capture of linkage-specific polyubiquitin chains [26] | In vivo and cell-based assays | Nanomolar affinity; distinguishes K48 (degradation) vs. K63 (signaling) chains [26] |
| Tagged Ubiquitin (His/Strep) | Affinity purification of ubiquitinated substrates [30] | Proteomic identification of ubiquitination sites | Enables enrichment of ubiquitinated proteins from complex mixtures for MS analysis [30] |
| Linkage-Specific Antibodies | Immunodetection of specific ubiquitin linkages [30] | Western blotting, immunofluorescence | Targets specific chain types (K48, K63, M1, etc.); validated for specific applications [30] |
| Recombinant E1/E2/E3 Enzymes | Reconstitution of minimal ubiquitination systems [59] | In vitro ubiquitination assays | Purified components enable mechanistic studies without cellular complexity [59] |
| Active E3 Variants | Enhanced efficiency for challenging substrates [32] | In vitro production of ubiquitinated molecules | Engineered constitutively active variants (e.g., HOIL-1) simplify tool compound generation [32] |
| Proteasome Inhibitors | Stabilization of ubiquitinated proteins [26] | Cell-based ubiquitination assays | Precludes degradation of ubiquitinated targets, enhancing detection sensitivity [26] |
Method Selection Framework
Choosing between in vivo and in vitro approaches requires careful consideration of research objectives, technical constraints, and desired outcomes. The following decision pathway illustrates key considerations in experimental design:
The selection between in vivo and in vitro ubiquitination detection methodologies represents a fundamental strategic decision in experimental design. In vivo approaches offer unparalleled physiological context but face challenges in mechanistic interpretation and throughput. In vitro methods provide precise experimental control and mechanistic insights but may not fully recapitulate native cellular environments. The most robust research programs often employ integrated approaches, using in vitro methods for initial characterization and mechanistic studies, followed by in vivo validation in physiologically relevant contexts. As ubiquitination research continues to evolve, particularly in drug development areas such as PROTAC technology and targeted protein degradation, understanding the complementary strengths and limitations of each approach becomes increasingly critical for generating reliable, translatable findings.
Conclusion
The choice between in vivo and in vitro ubiquitination detection is not a matter of superiority but of strategic alignment with research goals. In vivo methods provide essential physiological context, crucial for understanding disease mechanisms and evaluating drugs in a native cellular environment. In vitro systems offer unparalleled reductionist control for mechanistic dissection and high-throughput screening. The future lies in their integrated use, leveraging emerging tools like chain-specific TUBEs and advanced mass spectrometry to decode the complex language of ubiquitin signaling. This synergy will accelerate the development of targeted therapies, particularly in oncology and neurodegeneration, by providing a more complete picture of protein regulation through ubiquitination.
References
- 1. The Ubiquitin Conjugating Enzyme : An Important Ubiquitin Transfer... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7215765/]
- 2. - Wikipedia Proteasome [https://en.wikipedia.org/wiki/Proteasome]
- 3. Unlocking the proteasome : Structure and function | Abcam [https://www.abcam.com/en-us/knowledge-center/cell-biology/proteasome]
- 4. Ubiquitination detection techniques - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC10625345/]
- 5. Ubiquitination Regulators Discovered by Virtual Screening ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2021.665646/full]
- 6. Current methodologies in protein ubiquitination characterization [https://cellandbioscience.biomedcentral.com/articles/10.1186/s13578-022-00870-y]
- 7. Ubiquitinome Profiling Reveals in Vivo UBE2D3 Targets ... [https://www.sciencedirect.com/science/article/pii/S1535947623000580]
- 8. Molecular features defining the efficiency of bioPROTACs [https://www.nature.com/articles/s42003-025-08352-w]
- 9. The pros and cons of ubiquitination on the formation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10423694/]
- 10. Cellular strategies for making monoubiquitin signals - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3476054/]
- 11. How to Better Study Ubiquitination [https://www.ptglab.com/news/blog/how-to-better-study-ubiquitination/?srsltid=AfmBOopqh-RsyPj-Frdqs1eLea4XN_y3_KF8b5tagAxWHPFbyc3Ec57r]
- 12. Emerging functions of branched ubiquitin chains [https://www.nature.com/articles/s41421-020-00237-y]
- 13. Integrative proximal-ubiquitomics profiling for ... [https://www.sciencedirect.com/science/article/pii/S2451945625001278]
- 14. Selective ubiquitination of drug-like small molecules by the ... [https://www.nature.com/articles/s41467-025-63442-x]
- 15. Deubiquitinase dynamics: methodologies for understanding ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12123204/]
- 16. Time-resolved in vivo ubiquitinome profiling by DIA-MS ... [https://www.nature.com/articles/s41467-021-25454-1]
- 17. Expanding the landscape of lysine acetylation ... [https://www.sciencedirect.com/science/article/pii/S1874391925001496]
- 18. On the Study of Deubiquitinases: Using the Right Tools for ... [https://www.mdpi.com/2218-273X/12/5/703]
- 19. Functional and structural diversity in deubiquitinases of the ... [https://www.nature.com/articles/s41467-023-43144-y]
- 20. Bacterial Interference of Ubiquitination and Deubiquitination [https://pmc.ncbi.nlm.nih.gov/articles/PMC7173291/]
- 21. Ubiquitination and deubiquitination in cancer [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-024-02046-3]
- 22. Ubiquitin becomes ubiquitous in cancer - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC3023568/]
- 23. Ubiquitin signalling in neurodegeneration - Nature [https://www.nature.com/articles/s41418-020-00706-7]
- 24. Ubiquitin signaling in neurodegenerative diseases [https://pmc.ncbi.nlm.nih.gov/articles/PMC7862246/]
- 25. Detection of Protein Ubiquitination - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3149903/]
- 26. Unraveling chain specific ubiquitination in cells using ... [https://www.nature.com/articles/s41598-025-07242-9]
- 27. Biochemical analysis of PD-L1 ubiquitination by CRL3SPOP ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12335357/]
- 28. Methods for Quantification of in vivo Changes in Protein ... [https://www.sciencedirect.com/science/article/pii/S1535947620304576]
- 29. Protein Ubiquitination: Assays, Sites & Proteomics Methods [https://www.creative-proteomics.com/resource/protein-ubiquitination-detection-assays-site-identification-proteomics.htm]
- 30. Current methodologies in protein ubiquitination ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9373315/]
- 31. Structural visualization of HECT-type E3 ligase Ufd4 ... [https://www.nature.com/articles/s41467-025-59569-6]
- 32. The RBR E3 ubiquitin ligase HOIL-1 can ... [https://www.life-science-alliance.org/content/8/6/e202503243]
- 33. An optimized protocol to detect ubiquitination modification ... [https://www.sciencedirect.com/science/article/pii/S2666166723006172]
- 34. HTS Screening of Poly-Ubiquitin Linkage-Specific ... [https://lifesensors.com/hts-screening-of-poly-ubiquitin-linkage-specific-molecular-glues-and-protein-degraders/]
- 35. [Evaluation of high-throughput detection technology for ... [https://pubmed.ncbi.nlm.nih.gov/40873330/]
- 36. Site-directed multivalent conjugation of antibodies to ... [https://www.nature.com/articles/s41551-024-01342-z]
- 37. Methods to accelerate PROTAC drug discovery - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12312393/]
- 38. Detection of ubiquitination activity and identification ... [https://www.sciencedirect.com/science/article/abs/pii/S0076687918305287]
- 39. Mechanisms that matter: unbiased cellular screening for ... [https://www.nature.com/articles/s44386-025-00028-z]
- 40. Large-Scale Identification of Ubiquitination Sites by Mass ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4725055/]
- 41. Ubiquitinome Profiling Reveals the Landscape of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7801245/]
- 42. Global Mass Spectrometry-Based Analysis of Protein ... [https://pubmed.ncbi.nlm.nih.gov/34432246/]
- 43. Ubiquitinated Proteome: Ready for Global? - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3098603/]
- 44. Deciphering non-canonical ubiquitin signaling: biology and ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2023.1332872/full]
- 45. Recent advances in the development of deubiquitinases ... [https://www.sciencedirect.com/science/article/abs/pii/S0223523424000412]
- 46. Recent advances in small molecule inhibitors of ... [https://pubmed.ncbi.nlm.nih.gov/39908798/]
- 47. Unraveling chain specific ubiquitination in cells using tandem ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12217038/]
- 48. Unraveling Chain specific Ubiquitination in Cells Using ... [https://lifesensors.com/unraveling-chain-specific-ubiquitination-in-cells-using-tandem-ubiquitin-binding-entities-in-a-high-throughput-assay/]
- 49. Global, site-resolved analysis of ubiquitylation occupancy ... [https://www.sciencedirect.com/science/article/pii/S0092867424003155]
- 50. A method to identify small molecule/protein pairs susceptible ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc01251a]
- 51. A High Throughput Screen to Identify Substrates for the ... [https://www.sciencedirect.com/science/article/pii/S0021925820563709]
- 52. An optimized protocol to detect protein ubiquitination and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10050764/]
- 53. Selective ubiquitination of drug-like small molecules by the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12405493/]
- 54. K48- and K63-linked ubiquitin chain interactome reveals ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11109483/]
- 55. Combinatorial ubiquitin code degrades deubiquitylation- ... [https://www.nature.com/articles/s41467-025-57873-9]
- 56. Methods for Quantification of in vivo Changes in Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3418844/]
- 57. Data-independent acquisition method for ubiquitinome ... [https://www.nature.com/articles/s41467-020-20509-1]
- 58. Quantifying Ubiquitin Signaling - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC4441763/]
- 59. Protocol to investigate the biochemical details of immune ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12554173/]
- 60. PA770: PROTAC® In Vitro Ubiquitination Assay Kit [https://lifesensors.com/product/pa770-protac-in-vitro-ubiquitination-assay-kit/]
- 61. Genetically-encoded targeted protein degradation ... [https://www.nature.com/articles/s41467-025-56570-x]
- 62. The Role of Ubiquitination and Deubiquitination in ... [https://www.mdpi.com/2227-9059/13/12/2873]
- 63. Current advances on RIPK2 and its inhibitors in ... [https://www.frontiersin.org/journals/molecular-neuroscience/articles/10.3389/fnmol.2025.1492807/full]
- 64. A regulatory region on RIPK2 is required for XIAP binding ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7645253/]
- 65. CMD-OPT model enables the discovery of a potent and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12278411/]
- 66. Kinase inhibitors can accelerate the degradation of target ... [https://www.news-medical.net/news/20251126/Kinase-inhibitors-can-accelerate-the-degradation-of-target-proteins-study-reveals.aspx]
- 67. Deubiquitination of RIPK2 by OTUB2 augments NOD2 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11446981/]
- 68. In vivo and in vitro ubiquitination assay [https://bio-protocol.org/exchange/minidetail?id=2434213&type=30]
- 69. Proteome and Ubiquitinome Analyses Reveal the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12413316/]
- 70. The ubiquitin E3 ligase TRIM21 suppresses type I ... [https://www.nature.com/articles/s12276-025-01490-5]