K-ε-GG Antibody Enrichment for Mass Spectrometry: A Complete Guide from Principles to Profiling the Ubiquitinome
This article provides a comprehensive guide to the K-ε-GG antibody enrichment protocol for the mass spectrometry-based analysis of protein ubiquitination.
K-ε-GG Antibody Enrichment for Mass Spectrometry: A Complete Guide from Principles to Profiling the Ubiquitinome
Abstract
This article provides a comprehensive guide to the K-ε-GG antibody enrichment protocol for the mass spectrometry-based analysis of protein ubiquitination. Tailored for researchers and drug development professionals, it covers the foundational principles of ubiquitin biology and the specific role of the K-ε-GG remnant. It delivers detailed, optimized methodological workflows for sample preparation, peptide immunoprecipitation, and LC-MS/MS analysis, including automated high-throughput platforms. The content also addresses common troubleshooting scenarios and offers strategies for protocol optimization to enhance sensitivity and reproducibility. Finally, it explores validation techniques and compares the K-ε-GG method to alternative approaches, providing a holistic resource for perturbational studies and the characterization of disease-related ubiquitination signatures.
Ubiquitin Biology and the Revolution of K-ε-GG Antibody Enrichment
The Ubiquitin-Proteasome System (UPS) is a highly complex, temporally controlled, and evolutionarily conserved pathway that serves as the major intracellular, non-lysosomal mechanism for protein degradation in eukaryotic cells [1] [2]. By selectively targeting old, damaged, misfolded, and regulatory proteins for destruction, the UPS plays an indispensable role in maintaining cellular protein homeostasis (proteostasis) and is involved in virtually all cellular processes, from cell cycle progression and differentiation to apoptosis and stress response [3] [1]. The critical importance of this system is underscored by the fact that its dysfunction is linked to the pathogenesis of numerous diseases, including cancer, neurodegenerative disorders, and auto-inflammatory conditions [3] [1] [2].
The UPS operates through a sophisticated enzymatic cascade that conjugates the small protein modifier ubiquitin to specific substrate proteins, marking them for degradation by the proteasome or altering their cellular localization and function [1] [4]. This tagging process provides the cell with a powerful mechanism to precisely control protein half-lives and rapidly modulate signaling pathways in response to environmental stimuli, a capability particularly crucial for immune cells which undergo profound functional remodeling upon pathogen recognition [1].
The Biochemical Pathway of Ubiquitination
The E1-E2-E3 Enzymatic Cascade
The process of ubiquitination occurs through a three-step, ATP-dependent enzymatic cascade often referred to as the E1-E2-E3 pathway [2].
E1: Activation - The pathway initiates with a ubiquitin-activating enzyme (E1), which activates ubiquitin in an ATP-hydrolyzing reaction. This step forms a thioester linkage between a cysteine residue in the E1 active site and the C-terminal carboxyl group of ubiquitin, essentially serving as the "alarm clock" that alerts ubiquitin to begin the degradation process [2] [4].
E2: Conjugation - The activated ubiquitin is then transferred to a cysteine residue of a ubiquitin-conjugating enzyme (E2), creating an E2-ubiquitin intermediate. This step represents the "baton passer" of the UPS, preparing to hand off ubiquitin to the final step in the cascade [2].
E3: Ligation - The final step involves a ubiquitin ligase (E3), which takes ubiquitin from the E2-ubiquitin intermediate and catalyzes its covalent attachment to the target substrate. The E3 enzyme provides substrate specificity, recognizing specific degradation signals on target proteins. With approximately 10 E1, 40 E2, and over 600 E3 enzymes encoded in the human genome, this system offers tremendous specificity and regulatory complexity [2] [4].
Ubiquitin Chain Topologies and Their Functions
Ubiquitin itself contains eight potential ubiquitination sites (M1, K6, K11, K27, K29, K33, K48, and K63), enabling the formation of various polyubiquitin chains with distinct biological functions [2]. The specific topology of the ubiquitin chain determines the fate of the modified protein:
- K48-linked chains: Represent the canonical signal for proteasomal degradation [1] [2].
- K63-linked chains: Primarily function in non-proteolytic processes including DNA repair, endocytosis, and signal transduction, and can target proteins for clearance via the autophagy-lysosome pathway [1] [4].
- Other linkages (M1, K6, K11, K27, K29, K33): Serve diverse regulatory functions in cellular signaling, with chain-specific functions still being elucidated [1] [4].
Additionally, proteins can be modified by a single ubiquitin moiety (monoubiquitination), which regulates processes such as chromatin remodeling, protein sorting, and trafficking without targeting proteins for degradation [1].
Figure 1: The E1-E2-E3 Ubiquitination Cascade. This diagram illustrates the three-step enzymatic process that conjugates ubiquitin to target proteins.
The Proteasome: Architecture and Function
The 26S proteasome is a massive 2.5 MDa multi-subunit complex that serves as the executioner of the UPS, responsible for recognizing, unfolding, and proteolytically degrading ubiquitinated proteins [1] [4]. Its structure consists of two main subcomplexes:
Core Particle (20S CP)
The 20S core particle (CP) exhibits a barrel-like structure composed of four stacked heptameric rings: two identical outer α-rings and two identical inner β-rings [1]. The proteolytic activity resides in the β-rings, which contain three pairs of catalytically active threonine protease subunits:
- β1 subunit: Exhibits caspase-like activity
- β2 subunit: Exhibits trypsin-like activity
- β5 subunit: Exhibits chymotrypsin-like activity [1]
This enclosed chamber architecture ensures that protein degradation occurs in a controlled manner, preventing uncontrolled proteolysis of cellular proteins [1].
Regulatory Particle (19S RP)
The 19S regulatory particle (RP) caps one or both ends of the 20S CP and performs multiple critical functions: recognizing ubiquitin-modified proteins, removing ubiquitin chains, unfolding target proteins, and gating entry into the degradation chamber [1]. The 19S RP contains approximately 19 subunits, including ubiquitin receptors, deubiquitinating enzymes (DUBs), and ATPases that drive substrate unfolding and translocation [1] [4].
Proteasome Variants and Specialized Functions
Cells can assemble specialized proteasome variants with distinct functional properties:
- Immunoproteasomes (IP): Incorporate inducible catalytic subunits (β1i, β2i, β5i) instead of the standard subunits, exhibiting altered cleavage preferences and enhanced capacity to generate antigenic peptides for MHC class I presentation [1].
- Hybrid Proteasomes: Feature different regulators at each end of the 20S CP, such as a 19S RP at one end and an 11S regulator (PA28αβ) at the other, potentially enabling more efficient processing of specific substrates [1].
Table 1: Proteasome Types and Their Characteristics
| Proteasome Type | Catalytic Subunits | Distribution | Primary Functions |
|---|---|---|---|
| Standard Proteasome | β1, β2, β5 | Ubiquitous in most tissues | General protein turnover, homeostasis |
| Immunoproteasome | β1i, β2i, β5i | Constitutive in immune cells; induced by IFN in other tissues | Antigen processing, cytokine signaling |
| Mixed-type Proteasome | Combination of standard and inducible subunits | Tissues with high protein turnover (e.g., liver) | Specialized substrate processing |
K-ε-GG Antibody Enrichment: A Revolutionary Proteomic Technique
Principle of K-ε-GG Remnant Detection
The development of antibodies specific for the di-glycine (GG) remnant left on ubiquitinated peptides after trypsin digestion has revolutionized the large-scale identification of ubiquitination sites by mass spectrometry [5] [4] [6]. When ubiquitinated proteins are digested with trypsin, the enzyme cleaves after lysine and arginine residues, but leaves the two C-terminal glycine residues of ubiquitin attached to the modified lysine (K-ε-GG) in substrate proteins [5]. This GG remnant serves as a specific "fingerprint" of ubiquitination that can be recognized and enriched using highly specific antibodies, enabling comprehensive ubiquitinome profiling [7] [5].
It is important to note that this method also detects modifications by ubiquitin-like proteins NEDD8 and ISG15, as they produce an identical GG remnant after trypsin digestion. However, control experiments have demonstrated that >94% of K-ε-GG sites result from ubiquitination rather than these related modifications [5].
Technical Workflow and Optimization
The standard workflow for K-ε-GG enrichment involves multiple critical steps that have been progressively refined to dramatically improve sensitivity and specificity [5] [6]:
Figure 2: K-ε-GG Enrichment Workflow for Ubiquitinome Analysis. This diagram outlines the key steps in the proteomic profiling of ubiquitination sites.
Key methodological improvements that have enabled the routine identification of >20,000 distinct ubiquitination sites from single experiments include [5] [6]:
- Antibody cross-linking: Chemical cross-linking of anti-K-ε-GG antibodies to solid supports significantly reduces contamination from antibody fragments in final samples.
- Enhanced fractionation: Implementation of basic pH reversed-phase chromatography (bRP) prior to immunoaffinity enrichment dramatically increases the depth of ubiquitinome coverage.
- Optimized input requirements: Refinement of antibody-to-peptide ratios enables efficient enrichment from moderate protein amounts (5-10 mg).
- Integration with quantitative methods: Combination with SILAC (Stable Isotope Labeling by Amino Acids in Cell Culture) allows precise quantification of ubiquitination dynamics in response to cellular perturbations.
Table 2: Evolution of K-ε-GG Enrichment Performance
| Methodological Improvement | Typical Identified Sites (Pre-improvement) | Typical Identified Sites (Post-improvement) | Key Reference |
|---|---|---|---|
| Initial K-ε-GG antibody | ~500-1,000 sites | ~3,300 sites | [4] |
| Basic pH fractionation | ~3,300 sites | ~10,000+ sites | [5] |
| Antibody cross-linking | ~10,000 sites | ~20,000+ sites | [6] |
| Current optimized workflow | ~10,000 sites | ~20,000-30,000 sites | [6] |
Detailed Experimental Protocol: K-ε-GG Enrichment for Ubiquitinome Analysis
Sample Preparation and Protein Digestion
Cell Lysis: Lyse cells or tissue samples in freshly prepared urea lysis buffer (8 M urea, 50 mM Tris HCl pH 8.0, 150 mM NaCl, 1 mM EDTA) supplemented with protease inhibitors (2 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mM PMSF) and deubiquitinase inhibitors (50 μM PR-619) [5] [4]. Critical: Prepare urea lysis buffer fresh to prevent protein carbamylation.
Protein Reduction and Alkylation: Reduce disulfide bonds with 5 mM dithiothreitol (45 min, room temperature), then alkylate with 10 mM iodoacetamide or chloroacetamide (45 min, room temperature in the dark) [5] [4].
Protein Digestion: Dilute the sample to 2 M urea with 50 mM Tris/HCl pH 7.5 and digest first with LysC (Wako, 1:100 enzyme-to-substrate ratio) for 3-4 hours, followed by overnight digestion with sequencing-grade trypsin (Promega, 1:100 ratio) at room temperature [5].
Peptide Cleanup: Acidify peptide mixtures with trifluoroacetic acid (TFA) and desalt using C18 solid-phase extraction cartridges. Lyophilize desalted peptides for storage or further processing [5].
Peptide Fractionation and Enrichment
Basic pH Reversed-Phase Fractionation: Resuspend peptides in basic pH solvent A (5 mM ammonium formate pH 10/2% acetonitrile) and fractionate using a C18 column with a shallow gradient of increasing acetonitrile in basic conditions. Collect 8-12 fractions across the elution profile [5] [6].
Antibody Cross-Linking: Cross-link anti-K-ε-GG antibody to protein A agarose beads using dimethyl pimelimidate (DMP) to prevent antibody leakage during enrichment. Wash beads sequentially with cross-linking buffer (20 mM DMP in 100 mM sodium borate pH 9.0) and quenching buffer (100 mM ethanolamine pH 8.0) [5].
Immunoaffinity Enrichment: Resuspend fractionated peptides in immunoaffinity purification (IAP) buffer and incubate with cross-linked antibody beads for 1.5-2 hours at 4°C with gentle agitation. Wash beads extensively with IAP buffer and cold PBS before eluting bound peptides with 0.1-0.2% TFA [7] [5].
Mass Spectrometric Analysis
LC-MS/MS Preparation: Desalt and concentrate enriched peptides using C18 StageTips or micro-columns. Elute peptides in 50% acetonitrile/0.1% formic acid for LC-MS/MS analysis [5].
Instrumental Analysis: Analyze peptides by nanoflow liquid chromatography coupled to a high-resolution tandem mass spectrometer. Use data-dependent acquisition methods with dynamic exclusion to maximize ubiquitination site identifications [5] [4].
Data Processing: Search MS/MS spectra against appropriate protein databases using search engines that include the K-ε-GG modification (+114.04293 Da) as a variable modification on lysine residues. Apply strict false discovery rate thresholds (typically <1%) to identify high-confidence ubiquitination sites [5] [4].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Ubiquitin-Proteasome System Studies
| Reagent / Kit | Supplier Examples | Primary Application | Key Features |
|---|---|---|---|
| PTMScan Ubiquitin Remnant Motif (K-ε-GG) Kit | Cell Signaling Technology (#5562) | Immunoaffinity enrichment of ubiquitinated peptides | Includes bead-conjugated antibody, protocols for enrichment; also available in magnetic bead format (#59322) [7] |
| Ubiquitin-Activating Enzyme (E1) Inhibitors | Multiple suppliers | Investigation of E1 function, upstream UPS inhibition | Blocks entire ubiquitination cascade |
| Proteasome Inhibitors (MG-132) | Calbiochem, MilliporeSigma | Proteasome function studies, protein stabilization research | Reversible inhibitor of chymotryptic-like activity; widely used in UPS research [4] |
| Deubiquitinase Inhibitors (PR-619) | LifeSensors | DUB inhibition studies, ubiquitination dynamics | Broad-spectrum, reversible DUB inhibitor; targets multiple DUB families [4] |
| Tandem Ubiquitin-Binding Entities (TUBEs) | LifeSensors | Protection of polyubiquitin chains from DUBs, purification of ubiquitinated proteins | High-affinity reagents with multiple UBA domains; pan-selective or linkage-specific versions [2] |
| SILAC Amino Acid Kits | Multiple suppliers | Quantitative proteomics, ubiquitination dynamics | Enable metabolic labeling for accurate quantification of ubiquitination changes [5] [4] |
Applications in Drug Discovery and Therapeutic Development
The UPS has emerged as a promising target for therapeutic intervention, particularly in oncology and inflammatory diseases. Several key approaches have been developed:
PROTAC Technology
PROteolysis TArgeting Chimeras (PROTACs) represent a revolutionary approach in drug discovery that hijacks the UPS to selectively degrade target proteins [2]. These bifunctional molecules consist of:
- A ligand that binds to the target protein of interest
- A second ligand that recruits an E3 ubiquitin ligase
- A linker connecting these two elements
This configuration brings the target protein into proximity with the E3 ligase, leading to its ubiquitination and subsequent degradation by the proteasome. The exceptional selectivity of PROTACs, along with their ability to target proteins previously considered "undruggable," makes them particularly attractive therapeutic modalities [2].
Proteasome Inhibitors in Clinical Use
Several proteasome inhibitors have been successfully translated to clinical practice, particularly for the treatment of hematological malignancies:
- Bortezomib (Velcade): First-in-class proteasome inhibitor approved for multiple myeloma and mantle cell lymphoma
- Carfilzomib (Kyprolis): Second-generation epoxyketone inhibitor with improved specificity and reduced peripheral neuropathy
- Ixazomib (Ninlaro): First oral proteasome inhibitor with convenient dosing
These agents work primarily by inhibiting the chymotryptic-like activity of the proteasome, leading to accumulation of polyubiquitinated proteins and ultimately inducing apoptosis in rapidly dividing cancer cells [3] [1].
The Ubiquitin-Proteasome System represents one of the most sophisticated and crucial regulatory pathways in eukaryotic cells, governing protein turnover with exceptional specificity and temporal control. The development of K-ε-GG antibody enrichment techniques has revolutionized our ability to study this system at unprecedented depth and scale, enabling researchers to quantitatively monitor dynamic changes in thousands of ubiquitination sites in response to cellular perturbations. As our understanding of UPS complexity continues to grow, coupled with advanced proteomic methodologies and innovative therapeutic approaches like PROTACs, this field promises to yield profound insights into cellular physiology and novel treatments for human diseases. The continued refinement of ubiquitinome profiling techniques will undoubtedly uncover new regulatory mechanisms and therapeutic opportunities in the coming years.
Protein ubiquitination is an essential post-translational modification (PTM) that regulates a vast array of cellular processes, including protein degradation, signaling, trafficking, and DNA repair [8]. This versatility stems from the complexity of ubiquitin conjugates, which can range from a single ubiquitin monomer to polymers of various lengths and linkage types [8]. The conventional method for detecting ubiquitination involved immunoblotting with anti-ubiquitin antibodies, a low-throughput technique that requires mutation of putative lysine residues for validation [8]. However, the discovery that tryptic digestion of ubiquitinated proteins leaves a characteristic di-glycine remnant (K-ε-GG) covalently attached to the modified lysine residue revolutionized the field [9]. This 114.04 Da mass shift on the modified lysine serves as a mass spectrometry-detectable "signature" for the original modification site [8]. The commercialization of highly specific anti-di-glycine remnant (K-ε-GG) antibodies dramatically improved the ability to enrich and detect endogenous ubiquitination sites, enabling the identification of thousands of sites in single experiments [10] [11].
The Biochemical Basis of the K-ε-GG Signature
From Ubiquitination to a Mass Spectrometry-Detectable Signature
The ubiquitination process begins with the covalent attachment of the C-terminal glycine (G76) of ubiquitin to an epsilon-amino group of a lysine residue in the substrate protein via an isopeptide bond, a process catalyzed by the sequential action of E1, E2, and E3 enzymes [8]. When ubiquitinated proteins undergo tryptic digestion, the C-terminal region of ubiquitin is cleaved, leaving a di-glycine (Gly-Gly) moiety derived from residues G75-G76 of ubiquitin attached to the modified lysine side chain of the substrate peptide [9]. This constitutes the K-ε-GG remnant. The same signature is produced regardless of whether the modification is monoubiquitination or polyubiquitination, as the Gly-Gly remnant originates from the most C-terminal ubiquitin molecule in the chain [9]. This K-ε-GG motif is therefore a universal indicator of ubiquitination that can be targeted for enrichment and detection.
Visualizing the K-ε-GG Signature Formation
The following diagram illustrates the biochemical process that generates the mass spectrometry-detectable K-ε-GG signature from a ubiquitinated protein:
K-ε-GG Antibody Enrichment: Principles and Advancements
Antibody Development and Specificity
The critical advancement that enabled routine large-scale ubiquitinome analysis was the development and commercialization of high-affinity antibodies specifically recognizing the K-ε-GG remnant [10] [11]. These antibodies are typically conjugated to protein A agarose beads for immunoprecipitation purposes [9]. The refined preparation and use of these antibodies, including optimization of cross-linking techniques to prevent antibody leakage during enrichment, has been fundamental to achieving high-specificity enrichment [10] [11]. The specificity of these antibodies is crucial as it minimizes non-specific binding and maximizes the enrichment efficiency of low-abundance ubiquitinated peptides from complex biological samples.
Quantitative Advancements in Ubiquitinome Profiling
The table below summarizes the dramatic improvements in ubiquitination site identification achieved through optimized K-ε-GG antibody enrichment protocols:
Table 1: Evolution of Ubiquitination Site Identification Using K-ε-GG Antibody Enrichment
| Study/Protocol | Sample Type | Key Methodological Improvements | Number of Ubiquitination Sites Identified |
|---|---|---|---|
| Peng et al. (2003) [8] | S. cerevisiae (His-tagged Ub) | First proteomic approach using tagged ubiquitin | 110 sites on 72 proteins |
| Denis et al. [8] | MCF-7 breast cancer cells | FK2 antibody enrichment | 96 sites |
| Udeshi et al. (2013) [10] | HeLa cells | Refined antibody preparation, cross-linking, offline fractionation | ~20,000 sites in single SILAC experiment |
| Current Advanced Protocols [9] | HeLa cells (with proteasome inhibition) | Offline high-pH fractionation, advanced fragmentation, filter-based cleanup | >23,000 distinct diGly peptides |
Comprehensive K-ε-GG Enrichment Protocol
Sample Preparation and Lysis
Proper sample preparation is critical for successful ubiquitinome profiling. For cultured cells, lysis is typically performed using ice-cold 50 mM Tris-HCl (pH 8.2) with 0.5% sodium deoxycholate (DOC), followed by boiling at 95°C for 5 minutes and sonication [9]. For tissue samples such as mouse brain, a lysis buffer containing 100 mM Tris-HCl (pH 8.5), 12 mM sodium DOC, and 12 mM sodium N-lauroylsarcosinate is recommended [9]. The boiling step is essential for denaturing proteins and inactivating deubiquitinases. Notably, some protocols explicitly recommend against using deubiquitinase inhibitors such as N-ethylmaleimide (NEM) as they may introduce unwanted protein modifications that complicate peptide identification [9]. After lysis, total protein amount should be quantified using a colorimetric absorbance BCA protein assay, with several milligrams of total protein typically required for successful diGly peptide immunoprecipitation [9].
Protein Digestion and Peptide Preparation
Following protein quantification, reduction is performed using 5 mM 1,4-dithiothreitol for 30 minutes at 50°C, followed by alkylation with 10 mM iodoacetamide for 15 minutes in the dark [9]. Protein digestion is then carried out first with Lys-C (1:200 enzyme-to-substrate ratio) for 4 hours, followed by overnight digestion with trypsin (1:50 enzyme-to-substrate ratio) at 30°C or room temperature [9]. The use of sequential Lys-C/trypsin digestion has been shown to provide superior cleavage efficiency over trypsin digestion alone [11]. After digestion, trifluoroacetic acid (TFA) is added to a final concentration of 0.5% to precipitate and remove detergents via centrifugation at 10,000 × g for 10 minutes [9].
Peptide Fractionation for Depth Enhancement
To dramatically increase the depth of ubiquitinome coverage, offline high-pH reverse-phase fractionation is highly recommended prior to immunoenrichment [10] [9]. This involves using high pH RP C18 chromatography with polymeric stationary phase material (300 Å, 50 µM) loaded into an empty column cartridge [9]. The stationary phase bed size should be adjusted to the amount of protein digest, with approximately 0.5 g of stationary phase material recommended for ~10 mg of protein digest (1:50 w/w ratio) [9]. After loading, peptides are typically eluted in multiple fractions (e.g., three fractions with 10 mM ammonium formate at pH 10 containing 7%, 13.5%, and 50% acetonitrile, respectively) [9]. This fractionation step significantly reduces sample complexity and enables the identification of over 20,000 ubiquitination sites in a single experiment [10].
Immunoaffinity Enrichment and Mass Spectrometry Analysis
For the enrichment itself, K-ε-GG antibodies conjugated to protein A agarose beads are used [9]. The beads are washed with PBS before use, and the exact amount of antibody per batch should follow manufacturer recommendations as this information is often proprietary [9]. The enrichment is typically performed by incubating the fractionated peptides with the antibody-bound beads. After extensive washing to remove non-specifically bound peptides, the enriched K-ε-GG-containing peptides are eluted and analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) [12] [9]. For quantification, stable isotope labeling by amino acids in cell culture (SILAC) can be incorporated during cell culture prior to lysis [9] [11].
Visualizing the Complete K-ε-GG Enrichment Workflow
The following diagram outlines the comprehensive experimental workflow for K-ε-GG-based ubiquitinome profiling:
Essential Research Reagent Solutions
The table below outlines key reagents and materials required for implementing the K-ε-GG enrichment protocol:
Table 2: Essential Research Reagents for K-ε-GG Enrichment Experiments
| Reagent/Material | Function/Purpose | Examples/Specifications |
|---|---|---|
| Anti-K-ε-GG Antibody | Immunoaffinity enrichment of diGly-containing peptides | Commercial kits available; cross-linked to protein A beads for reduced antibody leakage [10] [11] |
| Cell Lysis Buffer | Protein extraction while maintaining ubiquitination status | 50 mM Tris-HCl (pH 8.2) with 0.5% sodium deoxycholate; boiling for protease inactivation [9] |
| Digestion Enzymes | Protein cleavage into peptides | Sequential Lys-C (1:200) and trypsin (1:50) digestion for optimal coverage [9] [11] |
| Fractionation Material | Offline peptide separation to reduce complexity | High-pH RP C18 chromatography material (300 Å, 50 µM) [9] |
| LC-MS/MS System | Peptide separation and identification | High-resolution mass spectrometer (e.g., Orbitrap) with nanoflow LC system [9] [13] |
| Proteasome Inhibitors | Increasing ubiquitinated protein abundance (optional) | Bortezomib (10 µM, 8h treatment) to enhance signal for ubiquitinated peptides [9] |
Applications and Integration with Broader Research Goals
The K-ε-GG enrichment methodology has become the gold standard for ubiquitinome profiling, enabling researchers to investigate ubiquitination dynamics in diverse biological contexts. It has been successfully applied to quantify changes in ubiquitination following proteasome and deubiquitinase inhibition [10], profile tissue-specific ubiquitination patterns [9], and identify substrates of specific E3 ubiquitin ligases [11]. When integrated with other proteomic approaches, such as phosphoproteomics or acetylation profiling, this technology provides unprecedented insights into the complex interplay between different PTMs [11]. Furthermore, the ability to profile ubiquitination sites in clinical samples and animal tissues without genetic manipulation makes it particularly valuable for translational research and drug development [8]. As mass spectrometry instrumentation continues to advance, with improvements in electron transfer dissociation (ETD) and ultraviolet photodissociation (UVPD) enhancing sequence coverage [13], the depth and precision of ubiquitinome mapping using K-ε-GG enrichment will continue to expand, further illuminating this critical regulatory layer in cellular physiology and disease.
The comprehensive profiling of protein ubiquitination, known as the ubiquitinome, has presented a formidable challenge to proteomics researchers for decades. As a versatile post-translational modification (PTM), ubiquitination regulates diverse fundamental features of protein substrates, including stability, activity, and localization [8]. The complexity of ubiquitin conjugates ranges from single ubiquitin monomers to polymers with different lengths and linkage types, creating a sophisticated regulatory system that controls numerous cellular processes [8]. Before the development of K-ε-GG antibodies, ubiquitination studies were severely constrained, limited to identifying only several hundred ubiquitination sites, which restricted the scope of global ubiquitination studies and our understanding of ubiquitin biology [14]. The commercialization of highly specific antibodies recognizing lysine residues modified with a di-glycine remnant (K-ε-GG) has fundamentally transformed this landscape, enabling researchers to routinely identify and quantify thousands of endogenous ubiquitination sites by mass spectrometry [14] [10]. This technological breakthrough has opened new avenues for understanding the molecular mechanisms of ubiquitination signaling in both normal physiology and disease states, including cancer and neurodegenerative disorders [8].
Historical Challenges in Ubiquitinome Research
Technical Limitations in Pre-K-ε-GG Antibody Era
Prior to the advent of K-ε-GG antibody-based enrichment, researchers faced significant technical hurdles in ubiquitination detection. Traditional biochemical approaches, such as immunoblotting with general anti-ubiquitin antibodies followed by site-directed mutagenesis of putative ubiquitinated lysine residues, were time-consuming and low-throughput [8]. These methods relied on testing individual proteins and their mutants, making system-wide ubiquitinome profiling practically impossible. Early mass spectrometry-based methods struggled with multiple inherent challenges: the large size of the modification (8.6 kDa), the presence of polyubiquitinated modifications, and the characteristically low stoichiometry of ubiquitination under normal physiological conditions [15]. Additionally, the tryptic digestion process used to generate peptides for mass spectrometry analysis presented a unique challenge—cleavage at arginine and lysine residues in both the substrate protein and the attached ubiquitin created complex peptide mixtures where modified peptides were vastly outnumbered by their unmodified counterparts [15]. This combination of factors rendered comprehensive ubiquitinome profiling an elusive goal for the proteomics community, limiting our understanding of this crucial regulatory mechanism.
Key Technological Limitations Before K-ε-GG Antibodies
Table 1: Major Technical Challenges in Historical Ubiquitinome Profiling
| Limitation Category | Specific Challenge | Impact on Research |
|---|---|---|
| Detection Sensitivity | Low stoichiometry of endogenous ubiquitination | Limited identification to abundant modifications |
| Sample Throughput | Requirement for large protein input (up to 35 mg) | Restricted application to samples with limited material |
| Identification Scale | Maximum of several hundred ubiquitination sites per study | Incomplete understanding of ubiquitination scope |
| Quantification Capability | Lack of robust multiplexed quantification | Difficulty monitoring dynamic ubiquitination changes |
| Technical Complexity | Need for numerous experimental replicates | Increased time, cost, and analytical complexity |
The K-ε-GG Antibody Breakthrough: Mechanism and Advantages
Fundamental Principle of K-ε-GG Recognition
The K-ε-GG antibody technology leverages a fundamental aspect of trypsin digestion biochemistry. When trypsin digests ubiquitinated proteins, it cleaves after arginine and lysine residues in both the substrate protein and the attached ubiquitin molecule. This process results in a characteristic signature—the C-terminal glycine-glycine (Gly-Gly) dipeptide of ubiquitin remains attached to the epsilon-amino group of the modified lysine residue in the substrate protein, creating the K-ε-GG motif [15]. This tryptic remnant serves as a specific "molecular handle" that the K-ε-GG antibody recognizes with high specificity and affinity. The commercial development of antibodies targeting this di-glycine remnant represented a watershed moment in ubiquitinomics, transforming the field by enabling direct immunoaffinity enrichment of formerly ubiquitinated peptides from complex tryptic digests [14] [15] [10]. This approach effectively bypassed the previous limitations of detecting the intact ubiquitin modification or relying on overexpression of tagged ubiquitin variants, allowing researchers to probe endogenous ubiquitination events at an unprecedented scale.
Comparative Advantages Over Previous Methods
The K-ε-GG antibody approach offers several distinct advantages over previous methodologies for ubiquitination detection. Unlike ubiquitin-tagging strategies that require genetic manipulation and expression of tagged ubiquitin in cells, the K-ε-GG antibody workflow can be applied to any biological sample, including primary tissues and clinical specimens, without the need for genetic engineering [8] [15]. This capability is particularly valuable for translational research investigating ubiquitination alterations in human diseases. Additionally, whereas traditional ubiquitin antibodies (such as P4D1 and FK1/FK2) enriched ubiquitinated proteins before digestion, the K-ε-GG approach enriches at the peptide level after digestion, providing precise site-specific identification of modified lysine residues [8]. This site-specific information is crucial for understanding regulatory mechanisms and designing functional experiments. The technology also demonstrates remarkable specificity, with optimized workflows achieving enrichment specificities of 85.7% for K-ε-GG peptides compared to 44.2% for in-solution TMT labeling methods [15]. This high specificity minimizes false positives and reduces interference from non-ubiquitinated peptides during mass spectrometry analysis.
Evolution of K-ε-GG Workflows: Key Methodological Advances
Refined Enrichment and Cross-linking Protocols
Substantial improvements to the original K-ε-GG enrichment workflow have dramatically enhanced its performance and reliability. A critical refinement involves antibody cross-linking using dimethyl pimelimidate (DMP), which covalently attaches the antibody to protein A agarose beads, preventing antibody leaching during enrichment and subsequent contamination of mass spectrometry instruments [14]. The cross-linking protocol involves washing anti-K-ε-GG antibody beads with sodium borate buffer (pH 9.0), resuspending in DMP solution, incubating at room temperature for 30 minutes with rotation, followed by blocking with ethanolamine and extensive washing with IAP buffer before storage [14]. Additionally, systematic optimization of antibody and peptide input requirements has identified that approximately 31μg of antibody provides efficient enrichment for most applications, significantly reducing reagent costs while maintaining high performance [14]. Off-line basic reversed-phase fractionation prior to enrichment has also been improved through non-contiguous pooling strategies, where fractions are combined in a staggered manner (e.g., combining original fractions 1, 9, 17, 25, etc.) to create eight pooled fractions that reduce sample complexity and increase ubiquitinome coverage [14]. These practical refinements have collectively enabled routine identification and quantification of approximately 20,000 distinct endogenous ubiquitination sites in single SILAC experiments using moderate protein input (5mg per SILAC channel) [14] [10].
Advanced Quantification and Throughput Innovations
Further methodological innovations have addressed the challenge of multiplexed quantification, particularly for tissue samples and primary cells where metabolic labeling is not feasible. The UbiFast method represents a significant advancement by enabling TMT labeling while K-ε-GG peptides are still bound to the antibody [15]. This approach protects the di-glycine remnant from derivatization, overcoming the previous limitation where commercial antibodies failed to recognize TMT-labeled K-ε-GG peptides. The optimized UbiFast protocol uses 0.4mg of TMT reagent for 10 minutes, followed by quenching with 5% hydroxylamine, resulting in >92% labeling efficiency and identifying approximately 6,087 K-ε-GG peptide-spectrum matches with 85.7% relative yield [15]. This method enables quantification of ~10,000 ubiquitylation sites from just 500μg of peptide input per sample in a TMT10plex experiment completed in approximately 5 hours, making it suitable for large-scale studies in primary tissue samples [15]. Parallel advances in data-independent acquisition mass spectrometry (DIA-MS) coupled with neural network-based data processing (DIA-NN) have further boosted ubiquitinome coverage, quantifying over 68,000 ubiquitinated peptides in single MS runs while significantly improving robustness and quantification precision compared to traditional data-dependent acquisition [16].
Performance Metrics and Comparative Analysis
Quantitative Assessment of Methodology Evolution
The dramatic improvement in ubiquitinome profiling capabilities enabled by K-ε-GG antibodies is clearly demonstrated through comparative performance metrics across methodological generations. Early studies using the initial K-ε-GG antibody technology typically identified several hundred to a few thousand ubiquitination sites, already representing a substantial advancement over pre-antibody methods [14]. The implementation of systematic workflow optimizations, including antibody cross-linking, optimized input requirements, and improved fractionation, pushed these limits further, enabling routine identification of approximately 20,000 ubiquitination sites from moderate protein inputs (5mg) [14] [10]. The most recent technological innovations, including UbiFast TMT labeling and DIA-MS with neural network processing, have achieved unprecedented depths of coverage, quantifying up to 68,000-70,000 ubiquitinated peptides in single experiments with high quantitative precision [15] [16]. This represents more than a 100-fold improvement in identification capability compared to pre-antibody era methodologies and a 3-fold improvement over early K-ε-GG implementations.
Table 2: Evolution of Ubiquitinome Profiling Performance Metrics
| Methodology Generation | Typical Sites Identified | Protein Input | Quantification Approach | Key Advantages |
|---|---|---|---|---|
| Pre-Antibody Methods | Hundreds | Large (tens of mg) | Limited or none | Established foundational knowledge |
| Early K-ε-GG Workflows | 1,000-5,000 | 10-35 mg | SILAC (3-plex) | First site-specific profiling at scale |
| Optimized K-ε-GG | ~20,000 | 5 mg | SILAC (3-plex) | Routine deep coverage with moderate input |
| UbiFast (TMT On-Bead) | ~10,000 | 0.5 mg | TMT (10-plex) | High multiplexing with minimal input |
| DIA-MS Ubiquitinomics | 68,000-70,000 | 2 mg | Label-free or TMT | Maximum depth and quantitative precision |
Sample Preparation and Lysis Buffer Innovations
Recent advancements in sample preparation have further enhanced the performance of K-ε-GG based ubiquitinomics. Comparative studies have demonstrated that sodium deoxycholate (SDC)-based lysis buffer supplemented with chloroacetamide (CAA) provides significant advantages over traditional urea-based buffers [16]. The SDC-based approach yields approximately 38% more K-ε-GG peptides (26,756 vs. 19,403) while maintaining high enrichment specificity and improving quantitative reproducibility [16]. Immediate sample boiling after lysis with high concentrations of CAA rapidly inactivates cysteine ubiquitin proteases, preserving the endogenous ubiquitination state and increasing ubiquitin site coverage. Additionally, CAA avoids the di-carbamidomethylation artifact that can occur with iodoacetamide, which creates a modification (114.0249 Da) that mimics the ubiquitin remnant mass tag and can lead to false positive identifications [16]. These sample preparation optimizations complement the enrichment improvements, contributing to the overall enhanced performance of contemporary ubiquitinomics workflows.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Research Reagents for K-ε-GG Ubiquitinomics
| Reagent / Kit | Supplier | Primary Function | Application Notes |
|---|---|---|---|
| PTMScan Ubiquitin Remnant Motif Kit | Cell Signaling Technology | Immunoaffinity enrichment of K-ε-GG peptides | Higher sensitivity magnetic bead version available (#59322, #19089) [17] |
| Anti-K-ε-GG Antibody | Multiple suppliers | Recognition and binding to di-glycine remnant | Cross-linking recommended to prevent antibody leaching [14] |
| Tandem Mass Tag (TMT) Reagents | Thermo Fisher Scientific | Multiplexed peptide quantification | UbiFast protocol enables on-antibody labeling [15] |
| IAP Buffer | Cell Signaling Technology | Immunoaffinity purification buffer | Optimized for antibody-peptide binding interactions [17] |
| SDC Lysis Buffer | Laboratory-prepared | Protein extraction with protease inhibition | Superior to urea for ubiquitinome coverage [16] |
Application Protocols: Current Best Practices
Optimized K-ε-GG Enrichment Workflow
Based on the collective advancements in the field, the following protocol represents current best practices for K-ε-GG-based ubiquitinome profiling:
Cell Lysis and Protein Extraction:
- Lyse cells or tissue in SDC lysis buffer (4% SDC, 100 mM Tris-HCl, pH 8.5) supplemented with 40 mM chloroacetamide to rapidly alkylate cysteine residues and inhibit deubiquitinases [16].
- Immediately boil samples at 95°C for 10 minutes to further inactivate enzymes.
- Cool to room temperature and dilute SDC concentration to 1% with 50 mM ammonium bicarbonate.
- Determine protein concentration using bicinchoninic acid (BCA) assay.
Protein Digestion and Peptide Cleanup:
- Digest proteins with sequencing-grade trypsin at 1:50 enzyme-to-substrate ratio overnight at 37°C.
- Acidify with trifluoroacetic acid (TFA) to a final concentration of 1% to precipitate SDC.
- Centrifuge at 10,000 × g for 10 minutes to remove precipitated SDC.
- Desalt supernatant containing peptides using C18 solid-phase extraction cartridges.
Basic Reversed-Phase Fractionation:
- Separate desalted peptides using basic reversed-phase chromatography (pH 10) with a Zorbax 300 Extend-C18 column.
- Collect 80 fractions and pool in a non-contiguous manner into 8 super-fractions (e.g., combine fractions 1, 9, 17, 25, etc.) to reduce sample complexity.
K-ε-GG Peptide Enrichment:
- Cross-link anti-K-ε-GG antibody to protein A agarose beads using 20 mM dimethyl pimelimidate in 100 mM sodium borate (pH 9.0) for 30 minutes at room temperature [14].
- Block cross-linked beads with 200 mM ethanolamine (pH 8.0) for 2 hours at 4°C.
- Incubate peptide fractions with cross-linked antibody beads (31μg antibody per fraction) in IAP buffer for 1 hour at 4°C with rotation.
- Wash beads four times with ice-cold PBS.
- Elute K-ε-GG peptides with 0.15% TFA.
Mass Spectrometry Analysis:
- For maximum depth: Analyze using DIA-MS with 75-120 minute gradients and process with DIA-NN software in library-free mode against an appropriate sequence database [16].
- For multiplexed quantification: Implement UbiFast protocol with on-antibody TMT labeling using 0.4mg TMT reagent for 10 minutes, followed by quenching with 5% hydroxylamine [15].
Quality Control and Data Interpretation Considerations
Implement robust quality control measures including:
- Process negative control samples (without antibody) to assess non-specific binding.
- Include positive controls from proteasome inhibitor-treated cells (e.g., MG-132) to verify enrichment efficiency.
- Monitor enrichment specificity by calculating the percentage of K-ε-GG peptides relative to total identified peptides (should exceed 85% for optimized workflows) [15].
- For DIA data, use DIA-NN with the cross-run normalization and robust LC (high accuracy) alignment options enabled [16].
- Apply stringent false discovery rate (FDR) thresholds (<1%) at both peptide and protein levels.
The development and continuous refinement of K-ε-GG antibody technology has fundamentally transformed ubiquitinome research, enabling the systematic profiling of ubiquitination sites at an unprecedented scale and precision. This transformation has moved the field from struggling to identify hundreds of sites to routinely quantifying tens of thousands of ubiquitination events in single experiments. The methodological innovations surrounding this technology—including antibody cross-linking, optimized fractionation strategies, on-antibody TMT labeling, and advanced DIA-MS acquisition—have collectively addressed the historical limitations of sensitivity, specificity, and throughput that previously constrained ubiquitinomics. These advances have opened new avenues for understanding the intricate regulatory networks controlled by ubiquitination in both physiological and pathological contexts, particularly in cancer and neurodegenerative diseases where ubiquitination plays central roles [8]. As the technology continues to evolve, integration with complementary approaches such as proximity-dependent labeling [18] and further improvements in mass spectrometry instrumentation and computational analysis promise to deepen our understanding of this crucial regulatory system, potentially unlocking new therapeutic opportunities targeting the ubiquitin-proteasome system.
In mass spectrometry-based proteomics, the specific enrichment of ubiquitinated peptides is foundational for understanding the role of this post-translational modification (PTM) in cellular processes. The K-ε-GG antibody enrichment protocol is a cornerstone technique for this purpose, leveraging an antibody that recognizes the di-glycine ("K-ε-GG") remnant left on trypsinized peptides following ubiquitination [14] [19]. However, the specificity of this antibody is a critical concern, as the human proteome encodes several ubiquitin-like proteins (UBLs) that, upon tryptic digestion, can generate a similar or identical di-glycine motif [20]. This application note details the sources of cross-reactivity and provides validated protocols to distinguish true ubiquitination from UBL modifications.
Ubiquitin and Ubiquitin-Like Modifications: A Comparative Analysis
Ubiquitin and UBLs share a common structural fold but are functionally distinct. The table below summarizes key characteristics of major UBLs, highlighting those that pose a potential risk for cross-reactivity in K-ε-GG enrichment protocols.
Table 1: Comparative Analysis of Ubiquitin and Select Ubiquitin-Like Modifiers
| Modification Type | Molecular Weight (kDa) | C-terminal Diglycine Motif After Trypsinization? | Primary Cellular Functions | Potential for K-ε-GG Cross-reactivity |
|---|---|---|---|---|
| Ubiquitin (Ub) | ~8.5 | Yes (definitive) | Protein degradation, signaling, trafficking [19]. | Reference standard. |
| SUMO (1-5) | ~11 | Yes (with homologous motif) [20]. | Nuclear transport, transcription, stress response [20]. | High - C-terminal motif is very similar. |
| ISG15 | ~15 | Yes (identical) [20]. | Antiviral defense, immune modulation [20]. | High - Leaves identical K-ε-GG remnant. |
| NEDD8 | ~9 | Yes (identical in humans) [20]. | Regulation of cullin-RING ligases (CRLs) [20]. | High - Leaves identical K-ε-GG remnant. |
| FAT10 | ~18 | No (constitutively exposed Gly) [20]. | Immune homeostasis, proteasomal degradation [20]. | Low - Does not require proteolytic activation. |
| ATG8/LC3 | ~14-16 | Yes (after propeptide cleavage) [20]. | Autophagy, membrane trafficking [20]. | Moderate - Requires cleavage; context-dependent. |
Experimental Protocol for Specific Ubiquitin Peptide Enrichment
This section provides a refined workflow for the specific enrichment of ubiquitinated peptides using the K-ε-GG antibody, incorporating steps to mitigate UBL cross-reactivity.
Cell Lysis and Protein Digestion
- Cell Lysis: Harvest and lyse cells in a denaturing buffer (8 M Urea, 50 mM Tris-HCl pH 7.5, 150 mM NaCl) supplemented with protease inhibitors (e.g., 2 μg/mL aprotinin, 10 μg/mL leupeptin) and 1 mM PMSF. Include 50 μM PR-619, a broad-spectrum deubiquitinase (DUB) inhibitor, to preserve ubiquitin and UBL conjugates [14].
- Protein Handling: Centrifuge lysates at 20,000 × g for 15 minutes at 4°C to remove insoluble material. Determine protein concentration using a BCA assay.
- Reduction and Alkylation: Reduce proteins with 5 mM dithiothreitol (DTT) for 45 minutes at room temperature. Alkylate with 10 mM iodoacetamide for 30 minutes in the dark.
- Trypsin Digestion: Dilute the lysate to 2 M urea with 50 mM Tris-HCl, pH 7.5. Digest with sequencing-grade trypsin (1:50 enzyme-to-substrate ratio) overnight at 25°C [14]. Desalt the resulting peptides using a C18 solid-phase extraction cartridge.
Peptide Fractionation and Immunoaffinity Enrichment
- Off-line Basic pH Reversed-Phase Fractionation: To reduce sample complexity and increase depth of coverage, fractionate the desalted peptide sample using basic pH reversed-phase chromatography. Pool fractions in a non-contiguous manner into 8-12 final fractions for subsequent enrichment. This step helps resolve ubiquitinated peptides from UBL-modified peptides [14].
- Anti-K-ε-GG Antibody Enrichment:
- Antibody Cross-linking (Recommended): To reduce antibody leaching and background, cross-link the anti-K-ε-GG antibody to protein A agarose beads using 20 mM dimethyl pimelimidate (DMP) in 100 mM sodium borate (pH 9.0) for 30 minutes at room temperature. Block the reaction with 200 mM ethanolamine (pH 8.0) [14].
- Immunoprecipitation: Resuspend each dried peptide fraction in 1.5 mL of IAP Buffer (50 mM MOPS pH 7.2, 10 mM Sodium Phosphate, 50 mM NaCl). Incubate with the cross-linked antibody beads (e.g., 31 μg antibody per fraction) for 1 hour at 4°C with rotation [14].
- Washing and Elution: Wash beads four times with 1.5 mL of ice-cold PBS. Elute bound peptides with two applications of 50 μL of 0.15% trifluoroacetic acid (TFA). Desalt the eluates using C18 StageTips prior to LC-MS/MS analysis [14].
Validation and Distinction Strategies
Relying solely on K-ε-GG enrichment is insufficient for definitive ubiquitin assignment. The following orthogonal approaches are required.
Bioinformatics and MS/MS Data Interrogation
The primary method for distinction is the analysis of MS/MS spectra. The diagram below outlines the bioinformatic logic for differentiating ubiquitin from UBL modifications.
- Ubiquitin Modification: The peptide sequence will originate from a host protein substrate, and the K-ε-GG modification will be on a lysine within that sequence.
- UBL Modification: The peptide sequence itself will be derived from the UBL protein (e.g., SUMO, ISG15, NEDD8). The C-terminal glycine of the UBL will be modified with a glycine, but this will be part of the UBL's own sequence, not the substrate's.
Table 2: Diagnostic Peptide Signatures for UBLs in MS/MS Data
| Modifier | Diagnostic Peptide Sequence (C-terminal) | Monoisotopic Mass [M+H]+ | Interpretation |
|---|---|---|---|
| Ubiquitin | TLSDYNIQK*ESTLHLVLR | 2271.192 | K* is the modified lysine on a substrate peptide. |
| ISG15 | LRLRGG* | 829.525 | The GG* is the C-terminus of ISG15 itself. |
| NEDD8 | AQGG* | 431.214 | The GG* is the C-terminus of NEDD8 itself. |
| SUMO1 | QTGG* | 460.215 | The GG* is the C-terminus of SUMO1 itself. |
Genetic and Pharmacological Validation
- Enzyme Knockdown: Use siRNA or CRISPR/Cas9 to knock down specific E1 enzymes for UBLs (e.g., UBA6 for ISG15, UBE1C for SUMO). A significant reduction in a specific K-ε-GG signal upon knockdown indicates it was derived from that UBL [20].
- Mutagenesis: Mutate the lysine residue identified in the substrate to arginine. The disappearance of the K-ε-GG signal confirms the modification site.
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for K-ε-GG Enrichment and Validation Studies
| Reagent / Kit | Function / Application | Key Features |
|---|---|---|
| PTMScan Ubiquitin Remnant Motif (K-ε-GG) Kit [19] | Immunoaffinity enrichment of K-ε-GG peptides from complex digests. | Includes motif-specific antibody, protocol, and buffers; optimized for MS. |
| Anti-K-ε-GG Antibody | Core component for enrichment of ubiquitin and UBL remnants. | Highly specific for the diglycine lysine remnant; commercial availability ensures reproducibility [14]. |
| PR-619 | Broad-spectrum DUB inhibitor. | Preserves ubiquitin/UBL conjugates during cell lysis by inhibiting deconjugating enzymes [14]. |
| MG-132 / Bortezomib | Proteasome inhibitor. | Stabilizes polyubiquitinated proteins targeted for degradation, increasing yield for analysis. |
| Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC) | Quantitative proteomics. | Allows accurate quantification of changes in ubiquitination/UBLylation sites upon cellular perturbations [14]. |
The K-ε-GG antibody is a powerful tool for profiling the ubiquitin and UBL-modified proteome. However, its inherent potential for cross-reactivity with UBL-derived di-glycine motifs necessitates rigorous experimental and bioinformatic validation. By implementing the refined enrichment protocol, orthogonal genetic controls, and careful MS/MS data analysis detailed herein, researchers can confidently distinguish ubiquitination from other UBL modifications, ensuring accurate data interpretation in their research.
A Step-by-Step Protocol for K-ε-GG Peptide Enrichment and LC-MS/MS Analysis
In mass spectrometry-based proteomics, the reliability of any downstream analysis, including the profiling of ubiquitination sites via K-ε-GG antibody enrichment, is fundamentally dependent upon the initial steps of sample preparation. Inefficient or inconsistent cell lysis, protein digestion, and peptide cleanup can irrevocably compromise data quality, leading to poor ubiquitination site coverage and unreliable quantification. This protocol details the optimized, critical first steps for preparing samples intended for deep-scale ubiquitinome analysis. When integrated with the highly specific immunoaffinity enrichment of diglycine remnant peptides [10] [12], these methods enable the routine identification and quantification of >10,000 endogenous ubiquitination sites from a single experiment [10]. The following sections provide a structured comparison of methodological choices, detailed executable protocols, and a catalog of essential reagents to ensure robust and reproducible results for researchers and drug development professionals.
Method Selection: Optimizing Lysis and Digestion for Ubiquitinome Analysis
The selection of appropriate methods for cell lysis and protein digestion significantly impacts protein recovery and the depth of proteomic coverage. The tables below summarize key findings from comparative studies to guide protocol selection.
Table 1: Comparison of Cell Lysis and Homogenization Methods
| Method | Key Characteristics | Typical Protein Recovery/Performance | Compatibility Notes |
|---|---|---|---|
| Sonication | Physical disruption using sound waves; common lab method [21]. | Comparable protein recovery to BeatBox; effective for standard lysis [21]. | Compatible with SDS, urea, and SDC buffers [21]. |
| BeatBox | Homogenization using high-speed magnetic beads; a recent innovation [21]. | Comparable protein recovery and coverage to sonication [21]. | Compatible with SDS, urea, and SDC buffers; offers standardization [21]. |
| SDS-based Lysis | Uses sodium dodecyl sulfate; strong denaturant [22] [23]. | High protein recovery; enables digestion via S-Trap for complex workflows like MONTE [23]. | Ideal for difficult-to-lyse samples or workflows requiring serial multi-omic analysis [22] [23]. |
Table 2: Comparison of Protein Digestion Methods
| Method | Key Characteristics | Unique Proteins Identified (Mean ± SD) | Key Advantages |
|---|---|---|---|
| SDC-based | Uses sodium deoxycholate; detergent-based [21]. | ~4,900 ± 68 (Sonication), ~4,800 ± 60 (BeatBox) [21]. | Highest protein and peptide yields; excellent for global profiling [21]. |
| S-Trap | Commercial kit; uses suspension trapping [21] [23]. | High and consistent peptide recovery [21]. | Efficient detergent removal without extra columns; ideal for SDS-lysed samples [21] [23]. |
| Urea-based | Classical denaturant; widely used [21]. | ~4,200 ± 120 (Sonication), ~4,300 ± 110 (BeatBox) [21]. | Well-established; requires dilution before digestion [21]. |
| EasyPep | Commercial kit; all-in-one buffers and columns [21]. | Higher variability in peptide recovery (±10%) [21]. | Convenience; streamlined workflow for fast processing [21]. |
Experimental Protocols
Protocol 1: SDC-Based Lysis and Digestion for Deep Proteome Coverage
This reagent-based method is ideal for experiments prioritizing maximum protein and ubiquitination site identification [21].
Step 1: Cell Lysis and Homogenization
- Resuspend cell pellets in SDC lysis buffer (1% SDC, 100 mM Tris-HCl, pH 8.5).
- Add universal nuclease to digest DNA and reduce viscosity.
- Homogenize using either sonication (10 cycles of 5-second pulses at 25% power on ice) or BeatBox (high speed for 10 minutes, twice).
- Centrifuge the lysate at 13,000g for 10 minutes and collect the supernatant.
- Determine protein concentration using a BCA assay [21].
Step 2: Protein Digestion
- Aliquot 100 µg of protein lysate.
- Reduce proteins with 1 µL of 500 mM TCEP (20 min at 37°C, shaking).
- Alkylate proteins with 3 µL of 500 mM chloroacetamide (CAA, 15 min in the dark).
- Add 16.67 µL of trypsin/Lys-C protease mix (1:30 enzyme-to-protein ratio).
- Digest overnight at 37°C with shaking.
- Stop digestion with 20% trifluoroacetic acid (TFA) to a final concentration of ~1%. SDC will precipitate. Centrifuge at 13,000g for 10 minutes and collect the supernatant for desalting [21].
Step 3: Peptide Desalting
- Desalt acidified peptides using a C18 solid-phase extraction column (e.g., MonoSpin C18).
- Equilibrate the column with methanol, then 0.2% TFA.
- Load the acidified peptide supernatant.
- Wash with 0.2% TFA to remove salts and contaminants.
- Elute peptides with 70% acetonitrile (ACN), 0.2% formic acid (FA).
- Dry peptides completely using a refrigerated SpeedVac concentrator [21].
Protocol 2: SDS-Based Lysis and S-Trap Digestion for Complex Workflows
This method is superior for samples requiring robust denaturation or originating from detergent-containing buffers, such as in the MONTE serial multi-omic workflow [23].
Step 1: Cell Lysis
Step 2: Protein Digestion on S-Trap Microcolumns
- Dilute the protein lysate with S-Trap dilution buffer (100 mM TEAB, pH 8.5) to reduce SDS concentration to <0.1%.
- Reduce and alkylate proteins using TCEP and CAA as in Protocol 1.
- Acidify the sample with phosphoric acid to a final concentration of 1.2%.
- Add 6-7 volumes of S-Trap binding buffer (90% methanol, 100 mM TEAB, pH 8.5) to the sample and mix.
- Load the mixture onto the S-Trap column and centrifuge gently.
- Wash the column 3-4 times with S-Trap binding buffer to remove detergents and impurities.
- Add trypsin/Lys-C in 50 mM TEAB and incubate for 1-4 hours at 47°C or overnight at 37°C.
- Centrifuge to elute digested peptides, followed by two additional elutions with 0.2% FA and 50% ACN/0.2% FA. Pool eluents and dry [21] [23].
Protocol 3: Peptide Desalting for Mass Spectrometry Analysis
Proper desalting is critical for maintaining mass spectrometer performance and ionization efficiency.
- Procedure:
- Conditioning: Activate a C18 desalting column or tip with 100% ACN.
- Equilibration: Equilibrate with 0.2% TFA or 0.2% FA in water.
- Sample Loading: Acidify the peptide sample to pH <3 and load it onto the column.
- Washing: Wash with 0.2% TFA or 0.2% FA to remove non-hydrophobic contaminants.
- Elution: Elute purified peptides with 50-80% ACN containing 0.2% FA.
- Drying: Dry the eluted peptides in a SpeedVac concentrator. The peptides are now ready for K-ε-GG antibody enrichment or direct LC-MS/MS analysis [21] [12].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Sample Preparation
| Item | Function/Application | Example Products |
|---|---|---|
| Anti-K-ε-GG Antibody | Immunoaffinity enrichment of ubiquitinated peptides [10] [12]. | Cell Signaling Technology Antibody [10] [12]. |
| S-Trap Micro Columns | Efficient digestion and cleanup of SDS-lysed samples; removes detergents without extra steps [21] [23]. | Protifi S-Trap [21] [23]. |
| C18 Desalting Columns | Solid-phase extraction for desalting and cleaning up peptides prior to MS [21]. | GL Sciences MonoSpin C18 [21]. |
| Trypsin/Lys-C Mix | High-efficiency protease for protein digestion, providing specific cleavages for MS identification [21]. | Thermo Scientific Trypsin/Lys-C Mix [21]. |
| SDS & SDC | Ionic detergents for effective cell lysis and protein denaturation [21] [23]. | Sodium Dodecyl Sulfate, Sodium Deoxycholate [21]. |
| TCEP & CAA | Reducing and alkylating agents for breaking disulfide bonds and cysteine alkylation [21]. | Tris(2-carboxyethyl)phosphine, Chloroacetamide [21]. |
Workflow Integration and Visualization
The sample preparation steps detailed herein form the foundational part of a larger workflow that culminates in the identification of ubiquitination sites. The following diagram illustrates the logical progression from sample to data, highlighting how the initial steps feed into the critical K-ε-GG enrichment.
Diagram 1: A simplified overview of the complete ubiquitinome analysis workflow, from cell lysis to ubiquitination site identification.
The selection of the initial lysis and digestion path determines the compatibility with downstream steps. The diagram below contrasts two primary pathways, showing their key differentiators and convergence points.
Diagram 2: A comparison of two primary sample preparation pathways, highlighting the key differentiators in handling detergents (SDC vs. SDS) and their convergence prior to ubiquitinome enrichment.
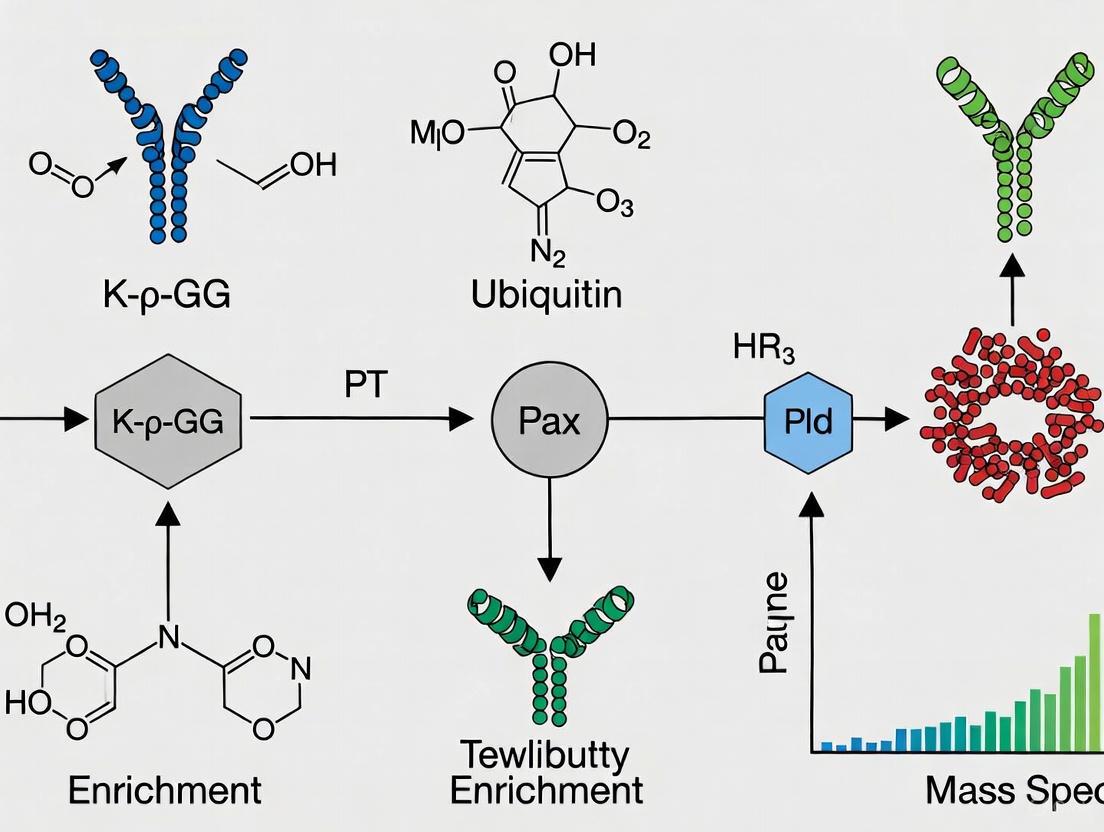
Immunoaffinity chromatography (IAC) is a powerful separation technique that utilizes the specific binding interaction between an antibody and its antigen [24] [25]. When applied to the study of post-translational modifications (PTMs) in proteomics, this method enables the highly selective purification of modified peptides from complex biological samples. The K-ε-GG antibody-based enrichment protocol represents a cornerstone technique for the systematic analysis of ubiquitination, a critical regulatory PTM involved in numerous cellular processes including protein degradation, signal transduction, and cell cycle progression [26] [27].
The core principle of this method relies on the use of a specific antibody that recognizes the di-glycine (GG) remnant left attached to the epsilon-amino group of lysine residues (K-ε-GG) after tryptic digestion of ubiquitinated proteins [6] [10] [27]. This signature serves as a universal marker for ubiquitination sites, allowing researchers to isolate thousands of modified peptides simultaneously for subsequent identification and quantification by liquid chromatography-tandem mass spectrometry (LC-MS/MS) [10] [26]. The refined preparation and use of this antibody enables the routine quantification of >10,000 distinct endogenous ubiquitination sites in a single experiment, providing unprecedented depth in ubiquitinome profiling [6] [10].
Table 1: Key Characteristics of K-ε-GG Immunoaffinity Enrichment
| Characteristic | Description | Significance |
|---|---|---|
| Target Epitope | Di-glycine remnant on lysine (K-ε-GG) | Specific recognition of tryptic ubiquitin signature |
| Antibody Type | Monoclonal anti-K-ε-GG [6] | High specificity and consistency between experiments |
| Application Scale | Single SILAC experiments with moderate protein input [10] | Enables system-wide ubiquitinome profiling |
| Typical Yield | ~20,000 distinct ubiquitination sites [6] [10] | Provides comprehensive coverage of ubiquitination |
Core Principles of the Method
Biochemical Basis of K-ε-GG Recognition
Ubiquitination involves the covalent attachment of the C-terminus of ubiquitin to lysine residues on substrate proteins via an isopeptide bond [26] [27]. During sample preparation for mass spectrometry-based proteomics, proteins are digested with trypsin, which cleaves ubiquitin after arginine residues but leaves the di-glycine signature of ubiquitin attached to the modified lysine on the target peptide [27]. This K-ε-GG motif serves as a specific "handle" that is recognized by the anti-K-ε-GG antibody with high affinity and selectivity [6] [26]. The same principle applies to the enrichment of peptides modified by SUMO (Small Ubiquitin-like Modifier) proteins, which also leave a di-glycine remnant after digestion with specific proteases like wild-type alpha-lytic protease (WaLP) [26].
The exceptional utility of this approach stems from the universal nature of the di-glycine remnant as a marker for ubiquitination. Rather than requiring antibodies against specific protein targets, a single antibody preparation can potentially isolate all ubiquitinated peptides in a proteome, making it possible to study system-wide ubiquitination events without prior knowledge of the modified proteins [26] [27].
Immunoaffinity Chromatography Fundamentals
Immunoaffinity chromatography separates molecules based on a highly specific biological interaction rather than general physicochemical properties [24] [25]. In the K-ε-GG enrichment workflow, antibodies are immobilized onto a solid support, typically agarose or magnetic beads, creating the stationary phase for affinity purification [26] [27] [24]. When the complex peptide mixture from digested cell lysates is applied to the antibody-conjugated beads, peptides containing the K-ε-GG motif are selectively retained while non-modified peptides are washed away [27].
The fundamental components of any IAC system include:
- Stationary Phase: A solid support matrix with immobilized antibodies [24]
- Binding Phase: Application of the sample under conditions favoring antigen-antibody interaction [25]
- Washing Phase: Removal of non-specifically bound contaminants [27]
- Elution Phase: Recovery of target antigens using conditions that disrupt antibody-antigen binding [24] [25]
The strong, specific binding of antibodies for their targets results in association equilibrium constants typically ranging from 10^5 to 10^12 M^-1, enabling exceptional selectivity and enrichment efficiency [24].
Detailed K-ε-GG Bead Incubation Protocol
Reagent and Solution Preparation
Proper preparation of buffers and solutions is critical for successful immunoaffinity enrichment. The following reagents are essential:
- Lysis Buffer: Urea-containing buffer (e.g., 6-8M urea) with protease inhibitors and deubiquitinase inhibitors to preserve ubiquitination sites [27]
- IAP Buffer: Immunoaffinity purification buffer, typically provided in commercial kits or prepared as a tris-buffered saline solution at appropriate pH [27]
- Antibody-Bead Conjugate: Anti-K-ε-GG antibody covalently coupled to protein A agarose or magnetic beads [6] [27]
- Elution Buffer: Dilute acid solution (typically 0.1-0.5% trifluoroacetic acid) for peptide elution [27]
- Wash Buffers: Multiple buffers of varying stringency to remove non-specifically bound peptides
Table 2: Essential Research Reagent Solutions for K-ε-GG Enrichment
| Reagent | Composition/Type | Function in Protocol |
|---|---|---|
| Anti-K-ε-GG Antibody | Monoclonal anti-di-glycine remnant antibody [6] [10] | Specific capture of ubiquitinated peptides |
| Support Matrix | Protein A agarose or magnetic beads [27] [24] | Solid phase for antibody immobilization |
| IAP Buffer | Tris-buffered saline with potential additives [27] | Optimal binding condition maintenance |
| Urea Lysis Buffer | 6-8M urea with protease inhibitors [27] | Protein extraction and denaturation |
| Trypsin/Lys-C Mix | Protease enzymes | Protein digestion to generate peptides |
| WaLP Protease | Serine endopeptidase (for SUMO) [26] | Digestion of SUMOylated proteins |
| Desalting Columns | C18 solid-phase extraction tips [27] | Peptide cleanup before and after enrichment |
Step-by-Step Bead Incubation Procedure
The bead incubation and enrichment process follows a systematic workflow:
Peptide Preparation: Extract proteins from cell or tissue samples using urea-containing lysis buffer. Reduce, alkylate, and digest proteins with trypsin to generate peptides. Desalt the resulting peptide mixture using reversed-phase solid-phase extraction [27].
Bead Equilibration: Resuspend the anti-K-ε-GG antibody bead conjugate by gentle vortexing. Wash beads with IAP buffer to remove storage solution and equilibrate them for optimal binding [27].
Peptide-Bead Incubation:
- Add the appropriate amount of peptide input (typically 10-30 mg for deep ubiquitinome analysis) to the equilibrated beads [6] [10].
- Adjust the volume with IAP buffer to ensure consistent binding conditions.
- Incubate the bead-peptide mixture with continuous gentle mixing (e.g., on a rotator or end-over-end mixer) for the recommended duration, typically 2-4 hours at 4°C [6] [27].
Washing Steps:
- After incubation, pellet the beads by gentle centrifugation or using a magnetic separator.
- Carefully remove the supernatant containing unbound peptides.
- Wash beads sequentially with multiple wash buffers of increasing stringency to remove non-specifically bound peptides while retaining true K-ε-GG-modified peptides [27].
Peptide Elution:
- Add dilute acid solution (e.g., 0.1-0.5% TFA) to the washed beads.
- Incubate briefly with agitation to dissociate antibodies from K-ε-GG peptides.
- Collect the eluate containing enriched ubiquitinated peptides.
- Repeat elution once or twice and pool eluates [27].
Sample Cleanup:
- Desalt the enriched peptides using reversed-phase microtip columns or StageTips.
- Concentrate and dry the peptides in a vacuum concentrator.
- Reconstitute in an appropriate solvent for LC-MS/MS analysis [27].
Critical Parameters for Optimization
Several factors significantly impact the efficiency and specificity of K-ε-GG enrichment:
Antibody Cross-linking: Covalently cross-linking antibodies to beads prevents antibody leaching and co-elution with target peptides, significantly reducing background interference in MS analysis [6] [10].
Peptide-to-Antibody Ratio: Maintaining the optimal peptide input to antibody amount is crucial for achieving maximum enrichment efficiency while avoiding saturation. The refined protocol enables comprehensive analysis with moderate protein input amounts [6] [10].
Incubation Duration: Sufficient incubation time (typically 2-4 hours) ensures adequate binding equilibrium, but excessively long incubations may increase non-specific binding.
Temperature Control: Performing incubations at 4°C enhances binding specificity and reduces protease activity that might degrade samples.
Off-line Fractionation: Implementing fractionation prior to immunoaffinity enrichment dramatically increases the depth of ubiquitinome coverage by reducing sample complexity [6] [10].
Troubleshooting and Quality Control
Common Technical Challenges
High Background: Resulting from insufficient washing, antibody leaching, or non-specific binding. Mitigate by optimizing wash stringency, implementing cross-linked antibodies, and including detergent in wash buffers [6].
Low Yield: Caused by insufficient peptide input, suboptimal antibody activity, or inefficient elution. Ensure proper antibody storage, validate input peptide quantity, and optimize elution conditions [27].
Incomplete Elution: Leads to carryover between experiments. Use fresh elution buffer and consider performing multiple sequential elutions.
Quantitative Performance Metrics
The refined K-ε-GG enrichment protocol enables the identification and quantification of approximately 20,000 distinct endogenous ubiquitination sites in a single Stable Isotope Labeling with Amino acids in Cell culture (SILAC) experiment [6] [10]. This represents a significant advancement over earlier methods, making large-scale ubiquitinome profiling routinely accessible.
Table 3: Quantitative Performance of Refined K-ε-GG Enrichment
| Performance Metric | Typical Result | Technical Basis |
|---|---|---|
| Ubiquitination Sites Identified | ~20,000 sites/single experiment [6] [10] | Antibody cross-linking and optimized fractionation |
| Sample Input Requirement | Moderate amounts [6] [10] | Refined antibody efficiency and enrichment protocol |
| Applications | Single SILAC experiments [10] | Compatibility with quantitative proteomics |
| Commercial Availability | PTMScan HS Ubiquitin/SUMO Remnant Motif Kit [26] | Standardized reagents for reproducibility |
Applications in Mass Spectrometry Research
The K-ε-GG immunoaffinity enrichment method serves as a critical sample preparation step for downstream LC-MS/MS analysis, enabling comprehensive ubiquitinome profiling across diverse research areas [26] [27]. Key applications include:
Global Ubiquitination Dynamics: Monitoring system-wide changes in ubiquitination in response to cellular stimuli, perturbations, or disease states [27].
Proteasome Substrate Identification: Discovering novel substrates of the ubiquitin-proteasome system by monitoring protein abundance changes following proteasome inhibition [26].
E3 Ligase Specificity: Defining substrate specificity of individual E3 ubiquitin ligases through comparative ubiquitinome profiling [27].
Cross-talk with Other PTMs: Investigating functional relationships between ubiquitination and other post-translational modifications such as phosphorylation and acetylation [22].
Drug Mechanism Studies: Identifying ubiquitination-dependent mechanisms of pharmacological agents and therapeutic candidates [26].
The integration of K-ε-GG immunoaffinity purification with modern mass spectrometry represents a powerful technological platform that continues to drive discoveries in ubiquitin biology, with implications for understanding fundamental cellular processes and developing novel therapeutic strategies for human diseases including cancer, neurodegenerative disorders, and immune dysfunction.
In mass spectrometry-based proteomics, the depth of analysis for complex samples is often limited by a wide dynamic range of protein concentrations. This is particularly true for ubiquitination site profiling using K-ε-GG antibody enrichment, where target peptides are of low stoichiometry and masked by abundant unmodified peptides. To overcome this challenge, fractionation strategies that reduce sample complexity prior to LC-MS/MS analysis are essential. Basic reversed-phase (bRP) chromatography and strong cation exchange (SCX) represent two powerful, orthogonal separation techniques that significantly increase proteome coverage when integrated into ubiquitination analysis workflows. By systematically implementing these fractionation methods, researchers can achieve unparalleled depth in identifying and quantifying ubiquitination sites, thereby unlocking deeper insights into ubiquitin signaling pathways in health and disease.
The Role of Fractionation in Ubiquitin Remnant Enrichment
The Complexity Challenge in Ubiquitinomics
Protein ubiquitination is a reversible post-translational modification that regulates diverse cellular functions including protein degradation, activity modulation, and cell cycle progression [8]. The analysis of ubiquitination sites by mass spectrometry relies on the recognition of a di-glycine remnant (K-ε-GG) left on modified lysine residues after tryptic digestion [28] [15]. Despite the commercialization of highly specific anti-K-ε-GG antibodies, the comprehensive profiling of ubiquitination sites remains challenging due to the low stoichiometry of modified peptides and interference from the complex background of unmodified peptides [8] [15].
Orthogonality in Separation Science
The effectiveness of multidimensional separation strategies depends on the orthogonality of the separation mechanisms—the degree to they exploit different physicochemical properties of peptides. Research has demonstrated that methods with high orthogonality significantly enhance proteome coverage. A recent evaluation of six different fractionation sorbents found that quaternary methyl-ammonium (QMA) and mixed strong anion exchange/reversed-phase (MAX) exhibited particularly high orthogonality when paired with standard low-pH reversed-phase separations [29]. Understanding these orthogonal relationships allows researchers to design efficient fractionation strategies that maximize peptide separation while minimizing redundancy.
Comparative Performance of Fractionation Methods
Systematic Evaluation of Separation Techniques
A comprehensive comparison of fractionation methods for plasma proteome analysis demonstrated that high-pH reverse-phase HPLC (hpRP-HPLC) exhibited superior peptide resolution and enabled detection of the largest number of known low-abundant proteins compared to 1-D SDS-PAGE and peptide isoelectrofocusing (OFFGEL electrophoresis) [30]. The advantages of peptide-level fractionation methods like bRP and SCX include better compatibility with quantitative biomarker validation methods such as stable isotope dilution multiple reaction monitoring, higher reproducibility, and increased throughput [30].
Table 1: Performance Comparison of Fractionation Methods in Proteomic Analysis
| Fractionation Method | Separation Mechanism | Advantages | Limitations | Typical Number of Fractions |
|---|---|---|---|---|
| Basic Reversed-Phase (bRP) | Hydrophobicity at high pH | High resolution, salt-free, compatible with LC-MS | Less orthogonal to low-pH RP | 6-8 [31] |
| Strong Cation Exchange (SCX) | Electrostatic interaction | Orthogonal to RP, good for charged peptides | Requires desalting, salt gradients | 6-8 [29] |
| Size Exclusion (SEC) | Molecular size | Effective for cross-linked peptide separation | Limited resolution | 2 [31] |
| 1-D SDS-PAGE | Molecular weight | Visual quality control, distinguishes protein forms | Low throughput, not quantitation friendly | 10-40 [30] |
Impact on Ubiquitination Site Identification
The implementation of optimized fractionation strategies has dramatically improved the sensitivity and coverage of ubiquitination site profiling. Research demonstrates that through systematic optimization of pre-analytical variables including off-line fractionation, researchers can achieve routine quantification of approximately 20,000 distinct endogenous ubiquitination sites in a single SILAC experiment [14]. Furthermore, the integration of bRP fractionation with advanced instrumentation such as high-field asymmetric waveform ion mobility spectrometry (FAIMS) has enabled identification of up to 50% more peptides from limited sample inputs [32], a critical advancement for clinical and translational research where sample amounts are often restricted.
Detailed Experimental Protocols
Protocol 1: Basic Reversed-Phase Fractionation for Ubiquitinated Peptides
Materials and Reagents
- Column: Zorbax 300 Extend-C18 column (9.4 × 250 mm, 300 Å, 5 μm) or equivalent [14]
- Solvent A: 2% acetonitrile, 5 mM ammonium formate, pH 10 [14]
- Solvent B: 90% acetonitrile, 5 mM ammonium formate, pH 10 [14]
- Equipment: HPLC system capable of handling high-pH solvents
Step-by-Step Procedure
- Sample Preparation: Resuspend desalted peptide samples in 1.8 mL of basic RP solvent A [14].
- Column Equilibration: Equilibrate the column with 100% solvent A at a flow rate of 3 mL/min for at least 10 minutes [14].
- Gradient Elution:
- 0-8 minutes: Increase to 8% solvent B (1.1% B/min)
- 8-46 minutes: Linear gradient from 8% to 27% B (0.5% B/min)
- 46-50 minutes: Increase to 31% B (1% B/min)
- 50-58 minutes: Increase to 39% B (0.5% B/min)
- 58-60 minutes: Increase to 60% B (3% B/min) [14]
- Fraction Collection: Collect 80 fractions across the elution profile in a 96-well polypropylene plate [14].
- Fraction Pooling: Pool fractions in a non-contiguous manner to create 8 final fractions (e.g., combine fractions 1, 9, 17, 25, 33, 41, 49, 57, 65, and 73 for fraction 1) [14].
- Sample Concentration: Dry pooled fractions completely in a SpeedVac concentrator before K-ε-GG enrichment [14].
Protocol 2: Strong Cation Exchange Fractionation
Materials and Reagents
- SCX Material: Quaternary methyl-ammonium (QMA) sorbent [29]
- Buffer A: 5 mM ammonium formate, 25% acetonitrile, pH 2.5-3 [29]
- Buffer B: 500 mM ammonium formate, 25% acetonitrile, pH 2.5-3 [29]
- Equipment: Positive pressure μSPE system or HPLC system with SCX column
Step-by-Step Procedure
- Sorbent Preparation: Condition QMA sorbent with methanol followed by Buffer A [29].
- Sample Loading: Acidify peptide sample to pH <3 and load onto SCX cartridge [29].
- Step Gradient Elution:
- 0-5% B: 5 column volumes
- 5-15% B: 5 column volumes
- 15-30% B: 5 column volumes
- 30-50% B: 5 column volumes
- 50-100% B: 5 column volumes [29]
- Fraction Collection: Collect 6 fractions across the step gradient [29].
- Desalting: Desalt fractions using C18 StageTips before further analysis [29].
Protocol 3: Integrated SEC-bRP 2D Fractionation for Complex Samples
Materials and Reagents
- SEC Column: Superdex Peptide 3.2/300 [31]
- bRP Material: C18 tips for high-pH fractionation [31]
- Solvents: Same as bRP protocol above
Step-by-Step Procedure
- SEC Separation: Load 250 μg desalted peptides in 30% ACN/0.1% FA onto SEC column [31].
- Cross-linked Peptide Collection: Collect two SEC fractions containing cross-linked peptides [31].
- bRP Fractionation: Dissolve each SEC fraction in 160 μL of ammonia water (pH 10) and subject to bRP fractionation [31].
- Fraction Analysis: Generate 12 total fractions (2 SEC × 6 bRP) for LC-MS/MS analysis [31].
Workflow Integration and Visualization
Diagram 1: Integrated workflow for ubiquitination site analysis incorporating multidimensional fractionation. The workflow shows how fractionation can be implemented at different stages, either before or after K-ε-GG antibody enrichment, to significantly improve depth of coverage.
Quantitative Impact of Fractionation on Coverage
The implementation of strategic fractionation consistently demonstrates substantial improvements in detection sensitivity and proteome coverage. These enhancements are particularly critical for ubiquitination site mapping, where modified peptides typically represent a small fraction of the total peptide population.
Table 2: Quantitative Benefits of Fractionation in Proteomic Studies
| Study Application | Fraction Method | Input Material | Identifications Without Fractionation | Identifications With Fractionation | Improvement |
|---|---|---|---|---|---|
| Ubiquitination Site Profiling [14] | bRP (8 fractions) | 5 mg protein | Not specified | ~20,000 ubiquitination sites | Established routine deep coverage |
| XL-MS Analysis [31] | SEC-HpHt (12 fractions) | 250 μg peptides | 818 cross-linked peptides | 10,932 unique cross-linked peptides | >12-fold increase |
| HLA Immunopeptidomics [32] | bRP + FAIMS | 100 million cells | Not specified | 8,107 distinct peptides | 20-50% increase |
| Plasma Proteome Profiling [30] | hpRP-HPLC | Immunodepleted plasma | Not specified | Best performance for low-abundant proteins | Superior to SDS-PAGE/OFFGEL |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Fractionation and Ubiquitin Enrichment
| Reagent / Kit | Supplier | Function | Application Notes |
|---|---|---|---|
| PTMScan Ubiquitin Remnant Motif Kit | Cell Signaling Technology | Immunoaffinity enrichment of K-ε-GG peptides | Core enrichment technology; compatible with fractionated samples [28] |
| Anti-K-ε-GG Antibody | Cell Signaling Technology | Recognition of di-glycine remnant on lysine | Critical for specific ubiquitination site enrichment [14] [15] |
| Zorbax 300 Extend-C18 Column | Agilent | Basic reversed-phase separation | 300 Å pore size optimal for peptide separation [14] |
| Superdex Peptide SEC Column | Cytiva | Size-based peptide separation | Effective first dimension for 2D workflows [31] |
| ProteoPrep20 Immunodepletion Column | Sigma-Aldrich | Abundant protein removal | Reduces dynamic range in plasma/serum samples [30] |
| TMT10plex/Isobaric Labels | Thermo Fisher | Multiplexed quantification | Enables comparison of multiple conditions [15] |
Advanced Applications and Future Perspectives
Integration with Novel Proteomic Platforms
Emerging technologies are creating new opportunities for enhanced fractionation strategies in ubiquitination research. The development of multiphasic liquid chromatography chips that integrate reversed-phase and strong cation exchange chromatography upstream of mass spectrometry offers improved reproducibility and detection limits [33]. Similarly, the incorporation of ion mobility separation (FAIMS) with fractionation techniques has demonstrated identification improvements of up to 58% for immunopeptidomics applications [32], suggesting similar benefits could be realized in ubiquitination studies.
Translational and Clinical Applications
The optimization of fractionation strategies has direct implications for translational research and drug development. The ability to profile ubiquitination sites from limited clinical samples enables the identification of ubiquitination signatures associated with disease states and treatment responses [15]. Furthermore, the integration of bRP and SCX fractionation with highly multiplexed quantification methods such as the UbiFast protocol enables quantification of ~10,000 ubiquitylation sites from as little as 500 μg of peptide material from tissues or primary cells [15], opening new possibilities for biomarker discovery and therapeutic target identification.
Basic reversed-phase and strong cation exchange fractionation represent powerful tools for dramatically increasing the depth of coverage in K-ε-GG antibody-based ubiquitination profiling. When strategically implemented in multidimensional separation workflows, these techniques reduce sample complexity, enhance detection of low-abundance ubiquitinated peptides, and ultimately expand the dynamic range of mass spectrometry analysis. As proteomic technologies continue to advance, the integration of orthogonal fractionation methods with improved enrichment protocols and sensitive instrumentation will further accelerate discoveries in ubiquitin biology and their translation to therapeutic applications.
This application note details the instrumental setup and data acquisition protocols for the analysis of ubiquitinated peptides following their enrichment via the K-ε-GG antibody-based protocol. The identification and quantification of ubiquitination sites are critical for understanding their roles in cellular regulation and disease pathogenesis, such as in breast cancer subtypes [34]. The methodology described herein is designed to be integrated with the sample preparation steps outlined in the broader thesis, ensuring a seamless transition from peptide enrichment to high-quality data generation using liquid chromatography tandem mass spectrometry (LC-MS/MS).
Experimental Protocols and Workflows
K-ε-GG Ubiquitinated Peptide Enrichment Protocol
The following protocol is adapted from established procedures for immunoaffinity enrichment, which can be performed manually or using an automated platform like AUTO-SP to enhance reproducibility [35] [34].
- Protein Digestion and Peptide Cleanup: Following protein extraction and reduction/alkylation, digest proteins with Lys-C and trypsin. Acidify the digested peptides to approximately pH 2.0 and desalt them using a C18 solid-phase extraction plate [34].
- Immunoaffinity Enrichment (IP): Resuspend the purified peptides in PTMScan IAP Buffer. Incubate the peptide solution with the K-ε-GG motif-specific antibody conjugated to protein A agarose or magnetic beads. The proprietary antibody specifically recognizes the di-glycine remnant (K-ε-GG) left on trypsinized peptides [35].
- Washing and Elution: After incubation, wash the beads thoroughly to remove non-specifically bound peptides. Elute the captured ubiquitinated peptides using a dilute acid solution.
- Final Cleanup and Concentration: Desalt the eluted peptides using reversed-phase purification on microtips or StageTips to remove salts and contaminants. Concentrate the peptides before LC-MS/MS analysis [35].
LC-MS/MS Instrumentation and Data Acquisition
For optimal results, the following setup is recommended based on recent literature [34].
Liquid Chromatography (LC):
- System: Evosep One or equivalent nanoflow LC system.
- Column: PepSep C18 column (15 cm × 150 μm, 1.5 μm particle size).
- Gradient: 44 minutes (30 samples per day method), with a gradient from 3% to 35% solvent B.
- Flow Rate: 0.5 μL/min.
- Temperature: 50 °C.
Mass Spectrometry (MS) - DIA Mode:
- System: timsTOF HT or other high-sensitivity instrument capable of data-independent acquisition (DIA) or dia-PASEF.
- Ion Mobility: Utilize dia-PASEF for enhanced peak capacity and confidence in identifications.
- MS1 Scan Range: 100–1700 m/z.
- MS2 Scan Range for Ubiquitinated Peptides: 341.6–1216.6 m/z [34].
- Ion Mobility (1/K0) Range: 0.70 to 1.45 V/s/cm².
Table 1: Optimal LC-MS/MS Parameters for Ubiquitinated Peptide Analysis
| Parameter | Recommended Setting |
|---|---|
| LC System | Evosep One or equivalent nanoflow system |
| Column | 15 cm PepSep C18, 1.5 μm |
| Gradient | 44 min (3-35% B) |
| Flow Rate | 0.5 μL/min |
| MS Acquisition | DIA / dia-PASEF |
| MS1 Range | 100-1700 m/z |
| MS2 Range (K-ε-GG) | 341.6-1216.6 m/z |
| Ion Mobility Range | 0.70-1.45 V/s/cm² |
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents and Kits for K-ε-GG Enrichment and Analysis
| Item | Function / Application |
|---|---|
| PTMScan Ubiquitin Remnant Motif (K-ε-GG) Kit [35] | Immunoaffinity enrichment of tryptic peptides containing the K-ε-GG remnant motif. |
| PTMScan IAP Buffer [35] | Optimized buffer for the immunoprecipitation reaction. |
| Magnetic Fe-NTA Beads [34] | For phosphopeptide enrichment in parallel multi-PTM studies. |
| Urea Lysis Buffer (8 M Urea, protease/phosphatase inhibitors) [34] | For efficient protein extraction and denaturation from tissues or cells. |
| Sequencing-Grade Modified Trypsin [34] | For specific protein digestion to generate peptides compatible with K-ε-GG antibody recognition. |
| C18 Solid-Phase Extraction Plates/Tips | For desalting and cleaning up peptide samples before enrichment and MS analysis. |
Data Analysis and Bioinformatics
- Database Search: Process raw DIA data using software such as Spectronaut (Biognosys) or DIA-NN. Search data against a relevant UniProt database.
- Search Parameters:
- Fixed Modification: Carbamidomethyl (C).
- Variable Modifications: Acetyl (Protein N-term), Oxidation (M), and GlyGly (K) for the ubiquitination remnant [34].
- False Discovery Rate (FDR): Set to 1% at the precursor level (Q value cutoff of 0.01).
- Quantification and Statistics: Quantify proteins and modified peptides using the top 3 peptides or precursors. Perform differential analysis using log₂ fold changes and statistical tests (e.g., t-test with Benjamini-Hochberg correction). Pathway analysis can be conducted via tools like WebGestalt for over-representation analysis (e.g., KEGG pathways) [34].
Workflow and Data Analysis Diagrams
Protein ubiquitylation, a fundamental post-translational modification, regulates a vast array of cellular processes including protein degradation, cell signaling, and progression through the cell cycle [15]. This modification involves the covalent attachment of ubiquitin to substrate proteins via a coordinated cascade of E1 (activating), E2 (conjugating), and E3 (ligating) enzymes [15]. For mass spectrometry (MS)-based analysis, tryptic digestion of ubiquitylated proteins generates peptides bearing a di-glycine remnant (K-ε-GG) on modified lysine residues, which serves as a signature for the prior ubiquitylation event [15]. The development and commercialization of highly specific anti-K-ε-GG antibodies revolutionized the field by enabling immunoaffinity enrichment of these low-abundance peptides, transforming our ability to monitor endogenous ubiquitination sites proteome-wide [14] [15].
Despite this breakthrough, early implementations required substantial sample input (up to ~35 mg) and multiple experimental replicates to identify thousands of ubiquitylation sites, limiting practical applications [14]. The UbiFast method, introduced by Udeshi et al., represents a significant advancement by combining on-antibody tandem mass tag (TMT) labeling with streamlined automation, achieving unprecedented sensitivity and throughput [15]. This protocol enables quantification of approximately 10,000 ubiquitylation sites from just 500 μg of peptide input per sample and reduces processing time to approximately five hours, making it suitable for large-scale studies involving primary cells and tissue samples [15] [36]. This application note details the implementation of both manual and automated UbiFast workflows within the context of K-ε-GG antibody enrichment for mass spectrometry research.
Key Methodological Comparisons and Performance Metrics
Evolution of Ubiquitylation Profiling Workflows
The progression from early ubiquitin enrichment methods to the current UbiFast protocol demonstrates remarkable improvements in efficiency, sensitivity, and throughput. Initial workflows identified only several hundred ubiquitination sites, which severely constrained global ubiquitination studies [14]. Refinements in antibody cross-linking, peptide input optimization, and off-line fractionation subsequently enabled identification of approximately 20,000 distinct endogenous ubiquitination sites in single proteomics experiments [14] [10]. The introduction of the UbiFast methodology with its innovative on-antibody TMT labeling marked another substantial leap forward, particularly for working with limited sample quantities [15].
Table 1: Comparative Performance of Ubiquitylation Profiling Methods
| Method | Sample Input | Number of Sites Identified | Processing Time | Multiplexing Capacity | Key Innovation |
|---|---|---|---|---|---|
| Early K-ε-GG Workflows | ~35 mg protein | Up to 5,000 sites | ~24+ hours | Limited (SILAC: 3-plex) | Anti-K-ε-GG antibody enrichment [14] |
| Refined K-ε-GG Enrichment | 5 mg protein per SILAC channel | ~20,000 sites | ~20 hours | SILAC (3-plex) | Antibody cross-linking, optimized fractionation [14] [10] |
| UbiFast (Manual) | 1 mg peptide | ~6,000 K-ε-GG PSMs | ~5 hours | TMT10-plex | On-antibody TMT labeling [15] |
| UbiFast (Automated) | 500 μg peptide per sample | ~20,000 sites | ~2 hours for 10-plex | TMT10-plex (96 samples/day) | Magnetic bead conjugation, robotic automation [37] |
Quantitative Advantages of Automated UbiFast
Automation of the UbiFast method using magnetic bead-conjugated K-ε-GG antibody (mK-ε-GG) and a magnetic particle processor delivers substantial practical benefits for research pipelines [37]. This automated approach not only maintains the high sensitivity of the manual UbiFast protocol but also significantly enhances reproducibility across process replicates while drastically reducing hands-on time [37]. The system enables processing of up to 96 samples in a single day, transforming the scale at which ubiquitylation studies can be designed and implemented [37]. This level of throughput is particularly valuable for pharmaceutical research and translational studies where consistent processing of numerous samples under identical conditions is essential for reliable data interpretation [37] [15].
Table 2: Technical Advantages of Automated UbiFast Workflow
| Parameter | Manual UbiFast | Automated UbiFast | Practical Implication |
|---|---|---|---|
| Processing Time | ~5 hours for TMT10-plex [15] | ~2 hours for TMT10-plex [37] | 60% reduction in processing time |
| Sample Throughput | Limited by manual processing | Up to 96 samples per day [37] | Enables large-scale cohort studies |
| Reproducibility | Subject to manual variation | Significantly reduced variability [37] | Improved data quality and statistical power |
| Operator Dependency | High technical expertise required | Standardized protocol execution | Reduced training requirements |
| Input Requirement | 500 μg - 1 mg peptide [15] | 500 μg peptide per sample [37] | Compatible with biopsy-scale samples |
Experimental Protocols
Antibody Preparation and Cross-Linking
Proper preparation of the anti-K-ε-GG antibody is fundamental to successful ubiquitylation enrichment. The cross-linking procedure prevents antibody co-elution with target peptides, thereby reducing background interference and improving MS detection sensitivity [14] [38].
- Antibody Bead Preparation: Transfer anti-K-ε-GG antibody beads (from PTMScan Ubiquitin Remnant Motif Kit, Cell Signaling Technology, #5562) to a suitable microcentrifuge tube [38].
- Washing: Wash beads twice with 1 ml of 100 mM sodium borate (pH 9.0) at 4°C using gentle centrifugation between washes [14] [38].
- Cross-linking: Resuspend beads in 1 ml of 20 mM dimethyl pimelimidate (DMP) and incubate for 30 minutes at room temperature on a rotating wheel [14] [38].
- Blocking: Wash cross-linked beads twice with 1 ml of 200 mM ethanolamine (pH 8.0) at 4°C, then resuspend in 1 ml of 200 mM ethanolamine and incubate for 2 hours at 4°C with rotation [38].
- Storage Preparation: Wash beads twice with 1 ml of IAP Buffer (50 mM MOPS, pH 7.2, 10 mM sodium phosphate, 50 mM NaCl) at 4°C, resuspend in IAP buffer, and store at 4°C until use [14] [38].
Sample Preparation and Digestion
Proper sample preparation ensures optimal protein extraction and digestion efficiency, which is critical for comprehensive ubiquitylation site identification.
- Cell Lysis: Resuspend cell pellets in denaturing lysis buffer (8 M urea, 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA) supplemented with protease inhibitors (2 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mM PMSF) and deubiquitinase inhibitor (50 μM PR-619) [14].
- Protein Extraction: Centrifuge lysates at 20,000 × g for 15 minutes at 4°C to remove insoluble material. Determine protein concentration using bicinchoninic acid (BCA) assay [14].
- Reduction and Alkylation: Reduce proteins with 5 mM dithiothreitol (DTT) for 45 minutes at room temperature, then alkylate with 10 mM iodoacetamide for 30 minutes in the dark [14].
- Trypsin Digestion: Dilute lysates to 2 M urea with 50 mM Tris-HCl (pH 7.5) and digest overnight at 25°C with sequencing-grade trypsin at an enzyme-to-substrate ratio of 1:50 [14].
- Peptide Desalting: Acidify digested peptides with formic acid (FA) and desalt using C18 solid-phase extraction cartridges. Condition cartridges with 5 ml 100% MeCN, 5 ml 50% MeCN/0.1% FA, and 20 ml 0.1% TFA. Load sample, wash with 15 ml 0.1% TFA, and elute with 6 ml 50% MeCN/0.1% FA. Dry eluents completely by SpeedVac centrifugation [14].
Basic Reversed-Phase Fractionation
Off-line fractionation prior to enrichment significantly improves depth of analysis by reducing sample complexity.
- Sample Resuspension: Resuspend dried peptide samples in 1.8 ml basic reversed-phase solvent A (2% MeCN, 5 mM ammonium formate, pH 10) [14].
- Chromatographic Separation: Perform fractionation using a Zorbax 300 Extend-C18 column (9.4 × 250 mm, 300 Å, 5 μm) with a 64-minute gradient from 2% to 60% solvent B (90% MeCN, 5 mM ammonium formate, pH 10) at a flow rate of 3 ml/min [14].
- Fraction Collection: Collect 80 fractions in a 96-well plate, then pool in a non-contiguous manner into eight super-fractions (e.g., combine fractions 1, 9, 17, 25, 33, 41, 49, 57, 65, and 73 for fraction 1) to reduce quantitative bias while maintaining resolution [14].
- Sample Drying: Dry pooled fractions completely by SpeedVac centrifugation before K-ε-GG enrichment [14].
UbiFast Enrichment with On-Antibody TMT Labeling
The core innovation of UbiFast is the on-antibody TMT labeling, which protects the di-glycine remnant from derivatization while enabling sample multiplexing.
- K-ε-GG Enrichment: Resuspend dried peptide fractions in 1.5 ml IAP buffer and incubate with cross-linked anti-K-ε-GG antibody beads for 1-2 hours on a rotating unit at 4°C. Use approximately 31 μg antibody per enrichment [14] [38].
- Bead Washing: Wash antibody beads four times with 1.5 ml ice-cold PBS to remove non-specifically bound peptides [14].
- On-Antibody TMT Labeling: Resuspend beads with bound K-ε-GG peptides in 100 μl 50 mM HEPES (pH 8.5). Add 0.4 mg TMT reagent dissolved in anhydrous acetonitrile and incubate for 10 minutes with occasional agitation [15].
- Reaction Quenching: Add 5% hydroxylamine to a final concentration of 0.3% (v/v) and incubate for 15 minutes to quench the TMT reaction [15].
- Sample Combining: For multiplexed experiments, combine equal amounts of TMT-labeled samples from different conditions [15].
- Peptide Elution: Elute combined TMT-labeled K-ε-GG peptides with 2 × 50 μl of 0.15% trifluoroacetic acid (TFA) [14].
- Sample Cleanup: Desalt eluted peptides using C18 StageTips or similar micro-solid phase extraction tips before LC-MS/MS analysis [14].
Automated UbiFast Implementation
Automation significantly enhances reproducibility and throughput while reducing manual labor requirements.
- Magnetic Bead Preparation: Use magnetic bead-conjugated K-ε-GG antibody (mK-ε-GG) instead of traditional resin-based antibodies [37].
- Robotic Processing: Implement the enrichment and labeling workflow on a magnetic particle processor capable of handling 96-well plates [37].
- Liquid Handling: Program automated systems for all washing, labeling, and elution steps to ensure consistency across samples [37].
- Parallel Processing: Process up to 96 samples simultaneously using the automated platform, completing a TMT10-plex experiment in approximately 2 hours [37].
The Scientist's Toolkit: Essential Research Reagents and Equipment
Successful implementation of the UbiFast method requires specific reagents and instrumentation optimized for ubiquitin remnant enrichment and analysis.
Table 3: Essential Research Reagent Solutions for UbiFast Implementation
| Item | Specification/Function | Application Notes |
|---|---|---|
| Anti-K-ε-GG Antibody | PTMScan Ubiquitin Remnant Motif Kit (Cell Signaling Technology, #5562) [38] | Highly specific antibody for K-ε-GG peptide enrichment; requires cross-linking |
| Magnetic Bead-conjugated Antibody | mK-ε-GG for automated workflows [37] | Enables high-throughput processing on magnetic particle processors |
| TMT Reagents | Tandem Mass Tags (10-plex or 11-plex) | For sample multiplexing; use 0.4 mg reagent per labeling reaction [15] |
| Digestion Enzymes | Sequencing-grade trypsin | Ensure complete protein digestion with minimal autolysis [14] |
| Chromatography Column | Zorbax 300 Extend-C18 (9.4 × 250 mm, 300 Å, 5 μm) [14] | For basic pH reversed-phase fractionation prior to enrichment |
| LC-MS/MS System | High-performance nanoLC coupled to Orbitrap mass spectrometer | Essential for detection and quantification of enriched peptides |
| FAIMS Device | High-field asymmetric waveform ion mobility spectrometry | Improves quantitative accuracy for TMT-based PTM analysis [15] |
| Magnetic Particle Processor | Automated system for magnetic bead handling | Enables processing of 96 samples per day [37] |
Analytical Considerations and Data Acquisition
Mass Spectrometry Configuration
Optimal MS configuration is critical for comprehensive ubiquitylation site identification and quantification.
- Chromatographic Separation: Use nanoflow liquid chromatography with C18 reversed-phase columns (75 μm × 25 cm, 2.2 μm particles) with a 180-minute gradient from 2% to 30% acetonitrile in 0.1% formic acid [15].
- Ion Mobility Separation: Implement High-field Asymmetric Waveform Ion Mobility Spectrometry (FAIMS) with compensation voltages of -50 V, -65 V, and -80 V to improve peptide isolation and quantitative accuracy [15].
- Mass Analysis: Operate mass spectrometer in data-dependent acquisition mode with MS1 spectra collected at 120,000 resolution and MS2 spectra at 50,000 resolution. Use higher-energy collisional dissociation (HCD) fragmentation at normalized collision energy of 32-35% [15].
- Dynamic Exclusion: Set dynamic exclusion to 60 seconds to maximize peptide identifications [15].
Data Processing and Analysis
- Database Searching: Process raw files using search engines (e.g., Sequest, MaxQuant) against appropriate protein databases. Search parameters should include fixed carbamidomethylation of cysteine (+57.021 Da), variable TMT modification on peptide N-termini and lysine (+229.163 Da), and variable di-glycine remnant on lysine (+114.042 Da) [15].
- False Discovery Control: Apply strict false discovery rate (FDR) thresholds (typically ≤1%) at the peptide and protein levels using target-decoy strategies [15].
- Quantification Analysis: Extract TMT reporter ion intensities from MS2 or MS3 spectra for relative quantification across samples. Normalize data to account for variations in sample loading and instrument response [15].
Troubleshooting and Optimization Guidelines
Successful implementation requires attention to potential challenges throughout the workflow.
- Low Enrichment Efficiency: Ensure proper antibody cross-linking and use fresh DMP prepared in sodium borate buffer (pH 9.0). Verify that peptide input falls within the linear range of the antibody capacity (typically 0.5-2 mg peptide per 31 μg antibody) [14] [38].
- Incomplete TMT Labeling: Use fresh TMT reagents stored under anhydrous conditions and ensure HEPES buffer is at pH 8.5 for optimal labeling efficiency. The 10-minute labeling time with 0.4 mg TMT reagent typically achieves >92% labeling completeness [15].
- High Background in MS: Increase number of wash steps after enrichment and ensure complete cross-linking of antibody to prevent antibody leakage. Implement basic reversed-phase fractionation to reduce sample complexity before enrichment [14].
- Low Ubiquitylation Site Identifications: Treat cells with proteasome inhibitors (e.g., 2-5 μM MG-132 for 4 hours) or deubiquitinase inhibitors (e.g., 5 μM PR-619) prior to lysis to stabilize ubiquitylated proteins [14].
- Poor Quantitative Reproducibility: Utilize the automated UbiFast platform to minimize manual processing variability. Incorporate FAIMS separation to improve quantitative accuracy in TMT-based experiments [37] [15].
The UbiFast method, particularly in its automated implementation, represents the current state-of-the-art in high-throughput ubiquitylation profiling. By enabling comprehensive analysis of thousands of ubiquitylation sites from sub-milligram amounts of sample, this workflow opens new possibilities for investigating ubiquitin biology in physiologically relevant systems including primary cells, patient-derived xenografts, and clinical specimens [37] [15]. The continued refinement of these approaches will further accelerate our understanding of ubiquitin signaling in health and disease, potentially revealing new therapeutic opportunities targeting the ubiquitin-proteasome system.
Optimizing Sensitivity and Reproducibility: Troubleshooting the K-ε-GG Workflow
Within K-ε-GG antibody enrichment protocols for mass spectrometry research, antibody cross-linking is a critical laboratory technique that significantly enhances experimental outcomes. This process involves chemically immobilizing the capture antibody onto solid support beads, preventing its leaching during the immunoprecipitation workflow. By covalently attaching antibodies to beads, researchers can minimize co-elution of antibody fragments that would otherwise contribute to significant background noise and mask low-abundance target peptides during mass spectrometry analysis. This application note details standardized protocols and analytical data demonstrating how proper antibody cross-linking dramatically reduces contamination and improves the yield of ubiquitinated peptides, enabling more sensitive and reproducible proteomic research.
Experimental Protocols & Workflows
Core Antibody Cross-Linking Protocol for K-ε-GG Enrichment
The following step-by-step methodology is adapted from refined preparation techniques that enable routine quantification of >20,000 ubiquitination sites in single proteomics experiments [14] [10].
Materials Required:
- Anti-K-ε-GG antibody (PTMScan Ubiquitin Remnant Motif Kit)
- Dimethyl pimelimidate (DMP)
- Sodium borate (100 mM, pH 9.0)
- Ethanolamine (200 mM, pH 8.0)
- IAP Buffer (50 mM MOPS, pH 7.2, 10 mM sodium phosphate, 50 mM NaCl)
- PBS (ice-cold)
- Purified peptide fractions from digested samples
Step-by-Step Procedure:
- Antibody Bead Preparation: Wash anti-K-ε-GG antibody beads three times with 1 ml of 100 mM sodium borate (pH 9.0) [14] [38].
- Cross-Linking Reaction: Resuspend washed antibody beads in 1 ml of 20 mM dimethyl pimelimidate (DMP). Incubate at room temperature for 30 minutes with rotation [14] [38].
- Quenching: Wash cross-linked beads twice with 1 ml of 200 mM ethanolamine (pH 8.0). Subsequently, incubate beads in 1 ml of 200 mM ethanolamine for 2 hours at 4°C with rotation to block any remaining active groups [14].
- Buffer Exchange: Wash the cross-linked antibody beads three times with 1.5 ml of ice-cold IAP buffer. Resuspend in IAP buffer and store at 4°C until use [14].
- Peptide Enrichment: Resuspend dried peptide fractions in 1.5 ml IAP buffer and incubate with cross-linked anti-K-ε-GG antibody beads for 1-2 hours at 4°C with rotation [14] [38].
- Wash and Elution: Wash beads four times with 1.5 ml ice-cold PBS. Elute bound K-ε-GG peptides with two applications of 50-100 μl of 0.15% trifluoroacetic acid (TFA) [14] [38].
- Sample Purification: Desalt eluted peptides using C18 StageTips or similar micro-purification methods prior to LC-MS/MS analysis [14].
Workflow Visualization
The following diagram illustrates the key decision points and procedural flow in the antibody cross-linking process:
Performance Data and Optimization
Quantitative Impact of Cross-Linking on Ubiquitin Site Identification
The implementation of antibody cross-linking in the K-ε-GG enrichment workflow has demonstrated substantial improvements in both the number of identifications and the reduction of contaminating antibodies in eluates.
Table 1: Performance Metrics of Cross-Linked vs. Non-Cross-Linked Antibody Enrichment
| Parameter | Non-Cross-Linked Antibody | Cross-Linked Antibody | Improvement Factor |
|---|---|---|---|
| Unique Ubiquitination Sites Identified | ~5,000 sites [14] | ~20,000 sites [14] [10] | 4x |
| Antibody Peptides in Eluate | High | Minimal to non-detectable | Significant reduction |
| Required Protein Input | ~35 mg [14] | 5 mg [14] | 7x less material |
| Inter-experimental Reproducibility | Lower due to antibody leakage | Higher consistency | Substantially improved |
Optimization Guidelines for Maximum Yield
Based on systematic studies, the following optimization parameters have been established for the cross-linking protocol [14]:
- Antibody Amount: 31 μg of antibody per enrichment provides optimal results with minimal nonspecific binding.
- Peptide Input: 5 mg of protein input per SILAC channel enables identification of thousands of ubiquitination sites.
- Fractionation Scheme: Basic reversed-phase chromatography with non-contiguous pooling into 8 fractions significantly enhances depth of coverage.
- Cross-linking Efficiency: The use of dimethyl pimelimidate (DMP) at 20 mM concentration for 30 minutes provides consistent cross-linking without compromising antibody affinity.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for Antibody Cross-Linking and K-ε-GG Enrichment
| Reagent / Material | Function / Application | Specifications / Alternatives |
|---|---|---|
| Anti-K-ε-GG Antibody | Specific enrichment of ubiquitinated peptides containing diglycine remnant | PTMScan Ubiquitin Remnant Motif Kit; specific for K-ε-GG motif [14] [38] |
| Dimethyl Pimelimidate (DMP) | Homobifunctional cross-linker for immobilizing antibodies to beads | Amine-reactive; spacer arm length ~9.2Å [14] |
| Sodium Borate Buffer | Provides optimal alkaline conditions for efficient DMP cross-linking | 100 mM, pH 9.0 [14] [38] |
| IAP Buffer | Immunoaffinity purification buffer for peptide-antibody incubation | 50 mM MOPS, 10 mM Na phosphate, 50 mM NaCl, pH 7.2 [14] |
| Ethanolamine | Quenches unreacted DMP after cross-linking completion | 200 mM, pH 8.0 [14] [38] |
| C18 Purification Media | Desalting and concentration of eluted peptides prior to MS analysis | StageTips, Spin Columns, or HLB Cartridges [14] |
Integration with Mass Spectrometry Quality Control
The benefits of antibody cross-linking extend beyond the enrichment process to significantly improve downstream mass spectrometry analysis:
- Reduction of MS Contamination: By preventing antibody leaching, cross-linking minimizes the introduction of non-target peptides into the mass spectrometer, reducing instrument downtime and maintenance [39].
- Improved MS Detection Sensitivity: With reduced background contamination, the MS instrument can allocate more sequencing time to low-abundance ubiquitinated peptides rather than contaminant proteins [40].
- Enhanced Quantitative Accuracy: Cross-linking improves reproducibility between technical and biological replicates, leading to more reliable quantification of ubiquitination dynamics in response to cellular perturbations [14].
Antibody cross-linking represents a fundamental advancement in K-ε-GG enrichment protocols for ubiquitin proteomics. The implementation of this straightforward chemical modification to immobilize capture antibodies directly addresses two critical challenges in proteomics research: contamination from reagent proteins and variable enrichment efficiency. Through the optimized protocols and conditions detailed in this application note, researchers can achieve dramatic improvements in both the depth of ubiquitinome coverage and the reproducibility of their mass spectrometry results, thereby accelerating discoveries in protein regulation and drug development.
In mass spectrometry-based ubiquitinome research, the K-ε-GG antibody enrichment protocol has revolutionized the detection of endogenous ubiquitination sites. The commercialization of highly specific anti-di-glycine remnant (K-ε-GG) antibodies has dramatically improved identification capabilities, transforming our understanding of ubiquitin biology. However, achieving comprehensive coverage of the ubiquitinome requires precise optimization of critical pre-analytical variables, particularly protein input amounts and antibody quantities. This application note details a refined and practical workflow that enables routine identification and quantification of approximately 20,000 distinct endogenous ubiquitination sites in a single SILAC experiment using moderate protein input, representing a significant advancement over previous methodologies.
Key Optimization Data
The relationship between antibody amount, peptide input, and ubiquitination site identification is fundamental to experimental design. The following tables summarize optimized parameters derived from systematic studies.
Table 1: Antibody and Peptide Input Optimization for K-ε-GG Enrichment
| Parameter | Recommended Amount | Identification Yield | Key Findings |
|---|---|---|---|
| Antibody Input | 31 µg (1/8 vial) | ~35,000 diGly sites (DIA) | Saturation occurs beyond this point; optimal for 1mg peptide input [41] |
| Peptide Input | 1 mg | Maximum peptide yield | Optimal balance for depth of coverage in single DIA experiments [41] |
| Protein Starting Material | 5 mg per SILAC channel | ~20,000 ubiquitination sites | 10-fold improvement over previous methods [14] |
| Cell Input for Selection | 1 × 10⁷ target cells | Antibodies to low-expressed receptors | Enables enrichment of antibodies to low-abundance targets [42] |
Table 2: Impact of Acquisition Method on Identification Numbers
| Acquisition Method | Peptide Identifications | Quantitative Precision (CV) | Key Advantages |
|---|---|---|---|
| Data-Independent Acquisition (DIA) | 35,111 ± 682 diGly sites | 45% of sites with CV < 20% | Superior completeness, sensitivity, quantitative accuracy [41] |
| Data-Dependent Acquisition (DDA) | ~20,000 diGly sites | 15% of sites with CV < 20% | Established method but with greater variability [41] |
Experimental Protocols
Protocol 1: Optimized K-ε-GG Enrichment for Ubiquitinome Analysis
Sample Preparation and Digestion
- Cell Lysis: Lysate cells in 8 M urea buffer containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, and protease inhibitors [14].
- Protein Quantification: Determine protein concentration using BCA assay [14].
- Reduction and Alkylation: Reduce proteins with 5 mM DTT (45 min, room temperature) and alkylate with 10 mM iodoacetamide (30 min, room temperature in dark) [14].
- Digestion: Dilute lysates to 2 M urea with 50 mM Tris-HCl (pH 7.5) and digest overnight at 25°C with trypsin (1:50 enzyme-to-substrate ratio) [14].
- Desalting: Desalt peptides using C18 SPE cartridges and dry completely [14].
Basic Reversed-Phase Fractionation
- Column: Zorbax 300 Extend-C18 column (9.4 × 250 mm, 300 Å, 5 μm) [14].
- Solvent System: Solvent A (2% MeCN, 5 mM ammonium formate, pH 10) and Solvent B (90% MeCN, 5 mM ammonium formate, pH 10) [14].
- Gradient: 64-min linear gradient from 8% to 60% Solvent B [14].
- Fraction Pooling: Collect 80 fractions and pool in a noncontiguous manner into 8 fractions to reduce complexity [14].
Antibody Cross-Linking and Enrichment
- Antibody Cross-Linking:
- Wash anti-K-ε-GG antibody beads with 100 mM sodium borate (pH 9.0) [14].
- Resuspend in 20 mM dimethyl pimelimidate (DMP) and incubate 30 min at room temperature with rotation [14].
- Wash with 200 mM ethanolamine (pH 8.0) and incubate in ethanolamine for 2 h at 4°C [14].
- Wash with ice-cold IAP buffer (50 mM MOPS, pH 7.2, 10 mM sodium phosphate, 50 mM NaCl) and store at 4°C [14].
- Peptide Enrichment:
- Desalting: Desalt eluted peptides using C18 StageTips [14].
Protocol 2: Data-Independent Acquisition for Ubiquitinome Analysis
Spectral Library Generation
- Treatment: Treat HEK293 and U2OS cells with 10 μM MG132 (proteasome inhibitor) for 4 hours [41].
- Fractionation: Separate peptides by basic reversed-phase chromatography into 96 fractions, concatenate into 8 fractions [41].
- K48-Peptide Handling: Process fractions containing abundant K48-linked ubiquitin-chain derived diGly peptides separately to reduce competition during enrichment [41].
- Enrichment: Enrich diGly peptides from each pool and analyze using DDA to create comprehensive spectral library [41].
DIA Method Optimization
- Window Layout: Optimize DIA window widths based on empirical precursor distributions [41].
- Scan Settings: Use 46 precursor isolation windows with MS2 resolution of 30,000 [41].
- Sample Loading: Inject only 25% of total enriched material due to improved DIA sensitivity [41].
- Data Analysis: Employ hybrid spectral library generated by merging DDA library with direct DIA search [41].
Workflow Visualization
Optimized Ubiquitinome Analysis Workflow
This workflow illustrates the optimized path from sample preparation to data analysis, highlighting critical optimization points.
Antibody Input Optimization Impact
This diagram illustrates the direct relationship between antibody input and identification yield, demonstrating the saturation point at 31μg.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for K-ε-GG Enrichment Protocols
| Reagent/Resource | Function/Application | Specifications |
|---|---|---|
| Anti-di-glycine remnant (K-ε-GG) antibody | Enrichment of ubiquitinated peptides | Commercial PTMScan kit; cross-link for improved performance [14] |
| Protein A/G beads | Antibody immobilization | Agarose or magnetic formats; UV-treated polypropylene plates alternative [43] |
| Dimethyl pimelimidate (DMP) | Antibody cross-linking | Improves antibody reusability and consistency [14] |
| Stable Isotope Labeling (SILAC) | Quantitative proteomics | Arg-0/Lys-0, Arg-6/Lys-4, Arg-10/Lys-8 for triple encoding [14] |
| Protease Inhibitors | Sample integrity | Include aprotonin, leupeptin, PMSF, chloroacetamide [14] |
| Basic RP Chromatography | Peptide fractionation | Zorbax 300 Extend-C18 column; pH 10 solvent system [14] |
| Data Analysis Software | DIA data processing | Spectral library generation; hybrid search capabilities [41] |
| Spectral Libraries | DIA ubiquitinome analysis | >90,000 diGly peptides; cell line-specific [41] |
Discussion
The optimization of protein and antibody inputs represents a critical advancement in ubiquitinome research. The precise balance of 31 μg antibody with 1 mg peptide input from 5 mg protein starting material achieves maximum identification capability while conserving valuable reagents. This refined protocol demonstrates that dramatic improvements in ubiquitination site coverage are achievable through systematic optimization rather than simply increasing sample inputs.
The implementation of DIA methodology further enhances these benefits, providing superior quantification accuracy and data completeness compared to traditional DDA approaches. The combination of optimized sample preparation with advanced acquisition methods enables researchers to explore ubiquitin signaling pathways with unprecedented depth and precision, opening new possibilities for understanding the role of ubiquitination in cellular regulation and disease pathogenesis.
These protocols provide a robust foundation for ubiquitinome studies while remaining adaptable to specific research needs. As mass spectrometry technologies continue to evolve, further refinements to these methods will undoubtedly emerge, but the fundamental principles of balanced input optimization will remain essential for achieving comprehensive ubiquitinome coverage.
In mass spectrometry-based proteomics, particularly in post-translational modification (PTM) analysis such as K-ε-GG antibody enrichment for ubiquitination studies, low peptide recovery during desalting and elution steps represents a critical bottleneck. Sample loss significantly compromises detection sensitivity, leading to incomplete PTM mapping and potentially misleading biological conclusions. This challenge is especially pronounced when working with limited sample amounts, such as in clinical proteomics or single-cell analysis, where maximizing recovery is paramount [44]. The stoichiometry of most ubiquitination sites is inherently low, making efficient sample preparation not merely an optimization concern but a fundamental requirement for successful identification and quantification [45]. This protocol details established and innovative strategies to overcome recovery limitations, focusing on practical improvements to elution chemistry and solid-phase extraction materials.
Background: The Recovery Challenge in PTM Enrichment
The K-ε-GG antibody enrichment protocol is a powerful tool for profiling ubiquitination sites. Following tryptic digestion of protein samples, ubiquitinated peptides are recognized and isolated using antibodies specific to the di-glycine remnant (K-ε-GG) left on the modified lysine residues [46] [45]. While this enrichment is highly specific, the subsequent steps of desalting and peptide elution are often where significant sample loss occurs. Traditional reversed-phase materials can lead to irreversible peptide adsorption, a problem exacerbated for hydrophobic peptides or when using suboptimal elution buffers [44]. These losses directly impact the number of PTM sites identified and the reliability of quantitative measurements, ultimately affecting data quality in downstream mass spectrometry analysis.
Technical Strategies and Comparative Data
Advanced Desalting Stationary Phases
The choice of desalting material profoundly impacts peptide recovery. Conventional StageTips, which use chromatographic particles entangled in a Teflon mesh, can suffer from significant and irreversible peptide adsorption.
Table 1: Comparison of Desalting Tip Performance
| Desalting Tip Type | Stationary Phase Composition | Key Feature | Peptide Recovery Performance |
|---|---|---|---|
| Traditional StageTip | SDB-XC particles in Teflon mesh | Standard method, widely used | Baseline recovery; significant loss of hydrophobic peptides [44] |
| ChocoTip | Thermoplastic polymer-coated St-DVB particles | Polymer coating inhibits irreversible adsorption | >2x more peptides identified from 20 ng HeLa digest; superior recovery of long-retention-time (hydrophobic) peptides [44] |
| C18-Membrane Micro Spin Column | C18-derivatized membrane | Large surface area, low bed volume | Successful desalting and concentration of peptides from 1 fmol/µL solution [47] |
Innovative materials like ChocoTip address this by using a unique hybrid polymer. The thermoplastic polymer coating covers the pores of the chromatographic particles, thereby inhibiting irreversible peptide adsorption into the mesopores. This design maintains strong hydrophobic interactions for binding during loading and washing but allows for more complete release during elution, dramatically improving recovery, especially for hydrophobic peptides [44].
Optimized Elution Buffer Strategies
The composition of the elution buffer is a critical, yet often overlooked, factor in maximizing peptide yield. Standard elution buffers may not efficiently disrupt all interactions between peptides and the stationary phase.
Recent research in immunoaffinity enrichment for biotinylated peptides demonstrates that introducing a highly organic solvent in the elution buffer can substantially boost the identification of modified peptides [48]. While demonstrated for biotinylation, this principle is directly transferable to the elution of peptides from reversed-phase desalting tips and other enrichment resins. A buffer containing elevated concentrations of acetonitrile (ACN) more effectively competes with hydrophobic interactions, leading to a more complete and efficient elution of tightly bound peptides.
Integrated Protocol for High-Recovery Desalting Post K-ε-GG Enrichment
This protocol assumes you have already performed K-ε-GG immunoaffinity enrichment and have your enriched ubiquitinated peptides in a collection tube.
Materials and Reagents
Table 2: Research Reagent Solutions
| Item | Function/Description |
|---|---|
| ChocoTip or equivalent high-recovery tips | Pipette-tip based microcolumn for solid-phase extraction, minimizing sample loss [44]. |
| Trifluoroacetic Acid (TFA), Mass Spec Grade | Ion-pairing agent used in loading and washing buffers to promote peptide binding. |
| Acetonitrile (ACN), LC-MS Grade | Organic solvent for eluting peptides from the stationary phase. |
| Formic Acid (FA), LC-MS Grade | Volatile acid for neutralizing TFA in the final eluate, making it MS-compatible. |
| Water, LC-MS Grade | Base for all aqueous solutions. |
| Thermoplastic polymer-coated particles | The core material in ChocoTip that minimizes irreversible adsorption [44]. |
Step-by-Step Procedure
- Conditioning: Attach the ChocoTip to a pipette or a centrifugation device. Pipette 200 µL of 80% ACN / 0.1% TFA into the tip and centrifuge at 1,000 × g for 2 minutes. Discard the flow-through. Repeat this step once [44].
- Equilibration: Add 200 µL of 0.1% TFA in water to the tip. Centrifuge at 1,000 × g for 2 minutes. Discard the flow-through. Repeat this step to ensure the stationary phase is fully equilibrated to the loading conditions [44].
- Sample Loading: Acidify the entire volume of your K-ε-GG enriched peptide sample with TFA to a final concentration of 0.1-1%. Slowly load the sample onto the conditioned tip via pipetting or centrifugation. For centrifugation, use 400 × g for 2 minutes. Collect the flow-through and reload it once to maximize binding efficiency [47].
- Washing: Add 200 µL of 0.1% TFA in water to the tip. Centrifuge at 1,000 × g for 1 minute. Discard the flow-through. This step removes residual salts and contaminants. For samples originally in strong denaturants like urea, repeat this wash 3-4 times to ensure complete removal [47].
- Reducing Aqueous Residue: Perform a quick spin (e.g., 30 seconds at 13,000 × g) to remove any residual aqueous solution from the membrane or packing, which would otherwise dilute the elution buffer [47].
- High-Efficiency Elution: Place a clean, low-binding microcentrifuge tube under the tip for collection. Pipette 30-50 µL of 50-80% ACN / 0.1% FA directly onto the center of the stationary phase. The high ACN concentration ensures efficient elution, while formic acid replaces TFA, making the final eluate ready for MS analysis. Let it sit for 1 minute, then centrifuge at 400 × g for 1 minute, followed by a final spin at 13,000 × g for 30 seconds to collect the entire volume [47] [48].
- Sample Preparation for MS: The eluted peptides can now be concentrated in a vacuum centrifuge (if necessary) and reconstituted in a small volume (e.g., 10-20 µL) of 0.1% FA / 2-5% ACN for LC-MS/MS injection.
Workflow Visualization
The following diagram illustrates the complete high-recovery desalting workflow integrated with the preceding K-ε-GG enrichment.
Concluding Remarks
Achieving high peptide recovery is not a single-step fix but the result of optimized materials and techniques. By integrating advanced stationary phases like ChocoTip, which minimizes irreversible adsorption, with a robust elution strategy employing high concentrations of acetonitrile, researchers can significantly boost the yield of precious K-ε-GG enriched ubiquitinated peptides. These protocols provide a practical path to enhancing sensitivity and coverage in PTM studies, ensuring that the data generated more accurately reflects the biological reality of the ubiquitinated proteome.
Utilizing Proteasome and Deubiquitinase Inhibitors to Stabilize the Ubiquitinome
The ubiquitin-proteasome system (UPS) represents a crucial regulatory network responsible for maintaining cellular protein homeostasis, with dysfunction linked to cancer, autoimmune disorders, and neurodegenerative conditions [49] [50]. Within this system, deubiquitinases (DUBs) perform the reverse reaction, cleaving ubiquitin from substrate proteins to modulate their stability, activity, and localization [50] [4]. The strategic application of proteasome and deubiquitinase inhibitors provides researchers with powerful chemical tools to perturb this system, leading to the accumulation of ubiquitinated proteins and enabling comprehensive mapping of ubiquitination events.
The development of antibodies specific to the di-glycine (K-ε-GG) remnant left on trypsinized ubiquitinated peptides has revolutionized ubiquitinome studies by enabling immunoaffinity enrichment prior to mass spectrometry analysis [51] [4]. When combined with targeted inhibition strategies, this approach allows for deep-scale profiling of ubiquitination dynamics under various physiological and pathological conditions, offering unprecedented insights into cellular signaling networks and protein turnover mechanisms that are particularly valuable for drug discovery research [49] [15].
Key Research Reagent Solutions
Table 1: Essential Research Reagents for Ubiquitinome Stabilization Studies
| Reagent Category | Specific Examples | Primary Function | Research Application |
|---|---|---|---|
| Proteasome Inhibitors | MG-132 [51] [4] | Inhibits chymotryptic activity of the proteasome | Blocks degradation of polyubiquitinated proteins, causing ubiquitome accumulation [4] |
| Broad-Spectrum DUB Inhibitors | PR-619 [4] | Reversible inhibitor targeting multiple DUB families | Induces widespread ubiquitination changes; useful for initial screening studies [4] |
| Specific DUB Inhibitors | IU1 (USP14 inhibitor) [52] | Selective inhibition of proteasome-associated USP14 | Increases degradation of specific substrates like tau; enhances proteasome activity [52] |
| USP7 Inhibitors | Multiple compounds in development [51] [50] | Target oncology-relevant deubiquitinase | Modulates stability of p53 and other cancer-relevant substrates [51] |
| Cell Lysis Additives | Chloroacetamide (CAA) [51] | Rapid alkylation of cysteine proteases | Immediate DUB inactivation during lysis preserves native ubiquitination states [51] |
Experimental Protocol: Ubiquitinome Stabilization and Enrichment
Cell Treatment and Protein Extraction
Begin by treating cells with selected inhibitors. For proteasome inhibition, use 5 μM MG-132 for 5 hours prior to harvest. For DUB inhibition, 5 μM PR-619 provides broad coverage across multiple DUB families [4]. For more targeted studies, IU1-47 (an optimized USP14 inhibitor) has demonstrated enhanced potency in inducing degradation of specific substrates like tau protein [52].
Upon treatment completion, immediately place cells on ice and aspirate media. Lyse cells using a SDC (sodium deoxycholate)-based lysis buffer (4°C) supplemented with 5 mM chloroacetamide (CAA) for rapid cysteine protease alkylation [51]. The SDC buffer significantly improves ubiquitin site coverage compared to conventional urea buffers, increasing K-ε-GG peptide identification by approximately 38% [51]. Immediately boil samples after lysis to further inactivate DUBs, then quantify protein concentration using a BCA assay.
Protein Digestion and Peptide Cleanup
Reduce and alkylate proteins with 5 mM dithiothreitol (45 minutes, room temperature) followed by 10 mM iodoacetamide (45 minutes, room temperature in dark) [4]. Dilute the urea concentration to 2 M using 50 mM Tris/HCl pH 7.5, then digest with sequencing-grade trypsin overnight at room temperature. Acidify peptide mixtures with trifluoroacetic acid (TFA) and desalt using C18 solid-phase extraction cartridges. Lyophilize desalted peptides for long-term storage or proceed directly to enrichment.
K-ε-GG Peptide Enrichment and Mass Spectrometry Preparation
Reconstitute peptides in immunoaffinity purification (IAP) buffer. For each enrichment, use ~2 mg of peptide input for optimal ubiquitinome coverage [51]. Incubate with anti-K-ε-GG antibody-conjugated beads for 2 hours at 4°C with gentle agitation. Wash beads extensively with IAP buffer followed by water to remove non-specifically bound peptides.
Elute K-ε-GG peptides using 0.2% TFA. For TMT-based multiplexed experiments, the UbiFast protocol recommends on-bead TMT labeling while peptides are still bound to antibodies, which significantly improves specificity and yield compared to in-solution labeling [15]. This approach enables quantification of >10,000 distinct ubiquitination sites from just 500 μg of peptide input per sample.
Mass Spectrometry Analysis and Data Processing
For comprehensive ubiquitinome profiling, Data-Independent Acquisition (DIA) mass spectrometry coupled with neural network-based processing (DIA-NN) significantly outperforms traditional data-dependent acquisition (DDA) methods, tripling identification numbers to >70,000 ubiquitinated peptides in single MS runs while improving quantitative precision [51]. Alternatively, for multiplexed studies, the UbiFast method with on-antibody TMT labeling enables quantification of ~10,000 ubiquitination sites across 10 samples simultaneously with high reproducibility [15].
Data Interpretation and Analysis
Quantitative Profiling of Ubiquitination Changes
Table 2: Representative Ubiquitination Changes Following Inhibitor Treatment
| Experimental Condition | Total K-ε-GG Sites Identified | Significantly Regulated Sites | Key Biological Insights | Reference |
|---|---|---|---|---|
| MG-132 (5μM, 5h) | >5,500 sites quantified | Hundreds increased | Majority of ubiquitination changes do not necessarily lead to protein degradation | [4] |
| PR-619 (5μM, 5h) | >4,900 sites quantified | Widespread regulation | Demonstrates distinct mechanism from proteasome inhibition | [4] |
| USP7 Inhibition | >8,000 proteins monitored | Hundreds increased within minutes | Small fraction of ubiquitinated proteins are ultimately degraded | [51] |
| USP14 Inhibition (IU1-47) | Targeted analysis | Accelerated tau degradation | Identified lysine 174 as critical for proteasomal degradation of tau | [52] |
Effective data analysis requires integrating ubiquitination data with protein abundance measurements to distinguish regulatory ubiquitination events from non-degradative functions [51]. The median coefficient of variation for quantified K-ε-GG peptides in DIA-based methods is approximately 10%, enabling robust detection of significant changes [51]. Contemporary computational tools like DIA-NN provide specialized analysis modes for ubiquitinomics data, significantly enhancing identification confidence and quantitative accuracy [51].
Advanced Applications in Drug Discovery
The integration of proteasome and DUB inhibitors with advanced ubiquitinome profiling techniques has enabled significant advances in drug discovery, particularly in oncology. Inhibitors targeting specific DUBs such as USP1, USP7, USP14, and USP30 have shown promising results in preclinical and clinical studies for cancer therapy [49] [50]. Furthermore, DUB inhibitors are being incorporated into novel therapeutic approaches including PROTACs (Proteolysis-Targeting Chimeras) and DUBTACs (Deubiquitinase-Targeting Chimeras) that represent cutting-edge modalities for targeted protein degradation and stabilization [49] [50].
For researchers investigating specific disease models, the inhibition of USP14 has demonstrated particularly promising effects in neurodegenerative disease models, where IU1-47 was found to accelerate degradation of pathological tau proteins, including the P301L and P301S mutants associated with tauopathies [52]. This targeted approach demonstrates how ubiquitinome stabilization strategies can be translated into therapeutic concepts for challenging disease targets.
Troubleshooting and Technical Considerations
Several technical challenges require consideration when designing ubiquitinome stabilization experiments. The stoichiometry of ubiquitination is typically low, meaning that even significant changes at the ubiquitination site level may not correspond to dramatic changes in total protein abundance [4]. The specificity of DUB inhibitors varies considerably, with PR-619 affecting multiple DUB families while compounds like IU1 demonstrate much greater selectivity for specific DUB family members [52] [4]. Finally, the dynamic range of ubiquitination changes can be extensive, with some substrates responding within minutes of inhibitor treatment while others require longer exposure [51].
Optimization should include titration of inhibitor concentrations and treatment durations, validation of expected pathway engagement through Western blotting for known substrates, and careful consideration of the appropriate mass spectrometry acquisition method based on sample amount and throughput requirements. The continued development of more selective DUB inhibitors and advanced mass spectrometry methods promises to further enhance the resolution and biological insights gained from these powerful stabilization approaches.
Validating Ubiquitination Sites and Comparing Enrichment Methodologies
Post-translational modifications (PTMs) are crucial and dynamic players in a large variety of cellular processes and signaling, regulating protein functions, conformation, protein-protein interactions, and subcellular localization [53]. Ubiquitination, a key PTM, involves the covalent attachment of ubiquitin to lysine residues of target proteins and regulates nearly all aspects of biology, including growth, development, and responses to stresses [54]. However, characterizing PTMs like ubiquitination presents significant challenges due to low PTM stoichiometry, the presence of multiple PTMs per peptide, and neutral losses during fragmentation [53].
This application note details an integrated workflow combining K-ε-GG antibody-based immunoaffinity enrichment with advanced mass spectrometry fragmentation techniques—Collision-Induced Dissociation (CID) and Electron Activated Dissociation (EAD). We demonstrate how this approach enables confident site-specific localization of ubiquitination, even in long-chain peptides or those with multiple candidate modification sites [55].
The Scientist's Toolkit: Essential Research Reagents and Solutions
The following table catalogues the core reagents and instrumentation essential for executing the protocols described in this note.
Table 1: Key Research Reagent Solutions for Ubiquitinome Analysis
| Item Name | Function/Application | Key Characteristics |
|---|---|---|
| PTMScan HS Ubiquitin/SUMO Remnant Motif (K-ε-GG) Kit [55] | Immunoaffinity enrichment of ubiquitinated peptides from digested protein lysates. | Highly specific antibody enrichment for peptides with diGly (GG) remnant on lysine. |
| Anti-GGX Monoclonal Antibodies [56] | Selective enrichment of tryptic peptides with an N-terminal diglycine remnant for profiling N-terminal ubiquitination. | Four unique clones (1C7, 2B12, 2E9, 2H2) with minimal cross-reactivity to K-ε-GG peptides. |
| ZenoTOF 7600 System [53] [55] | High-sensitivity QqTOF mass spectrometer for LC-MS/MS analysis. | Capable of both CID and EAD fragmentation; features Zeno trap for >90% MS/MS duty cycle and enhanced sensitivity. |
| Electron Activated Dissociation (EAD) [53] [55] | Alternative fragmentation method that preserves labile PTMs during MS/MS. | Tunable electron kinetic energy (0-25 eV); maintains labile modifications intact for confident site localization. |
| S-Trap or C18 StageTips [55] | Micro-desalting and cleanup of enriched peptides prior to LC-MS/MS. | Efficient removal of salts and buffers after immunoaffinity enrichment. |
Performance Comparison: EAD vs. CID for PTM Analysis
We evaluated the performance of EAD alongside the conventional CID method for the analysis of ubiquitinated and other labile PTMs. The following table summarizes quantitative data highlighting their complementary strengths.
Table 2: Comparative Performance of CID and EAD Fragmentation for PTM Analysis
| Parameter | CID (Collision-Induced Dissociation) | EAD (Electron Activated Dissociation) |
|---|---|---|
| Fragmentation Mechanism | Collisional activation; cleaves weakest bonds (amide backbone) [53]. | Electron beam; generates c, z• ions via free radical chemistry [53] [57]. |
| PTM Localization Confidence | Lower for labile PTMs; can suffer from neutral losses [53]. | High; preserves labile modifications, enabling confident site assignment [53] [55]. |
| Performance with Long Peptides | Often insufficient backbone fragmentation, especially with proline residues [55]. | Superior; provides high peptide backbone sequence coverage [55] [57]. |
| Isobaric Isomer Differentiation | Limited capability to differentiate Leu/Ile or Asp/isoAsp [57]. | Excellent; produces diagnostic fragments (e.g., w ions) for clear isomer identification [57]. |
| Quantitative Reproducibility | CVs for Ub-PTMs: ~15% with CV <20% in DDA [41]. | High reproducibility; CVs of ~2-7% in targeted PRM assays [53]. |
| Optimal Precursor Charge State | Efficient for low charge states (1+, 2+) [57]. | Effective for all positive charge states, superior for multiply charged (3+) precursors [53] [57]. |
Experimental Protocols
Protocol for Ubiquitinated Peptide Enrichment Using K-ε-GG Antibody
This protocol is adapted from established methods for the immunoaffinity purification of ubiquitinated peptides from complex cell lysates prior to LC-MS/MS analysis [55].
Materials:
- Lysis Buffer: 8 M Urea, 200 mM HEPES, pH 8.5
- Protease and Phosphatase Inhibitors
- Reduction/Alkylation: Dithiothreitol (DTT), Iodoacetamide (IAA)
- Digestion Enzymes: Trypsin, Lys-C
- PTMScan HS Ubiquitin/SUMO Remnant Motif (K-ε-GG) Kit (Cell Signaling Technology, #59322)
- IP Buffer (10X): 500 mM MOPS, 100 mM Na₂HPO₄, 500 mM NaCl, pH 7.2
- Desalting: SEP-PAK C18 columns or S-Trap micro spin columns
- StageTips with C18 material
Procedure:
- Cell Lysis and Protein Preparation:
- Harvest HCT-116 cells (or your cell line of interest) and wash 2x with cold PBS.
- Lyse cells in Urea Lysis Buffer supplemented with 1X Protease and Phosphatase Inhibitors.
- Sonicate the lysate (e.g., three times for 20 seconds each at 15W, with 1-minute cooling intervals).
- Centrifuge the sonicated lysate at 20,000 g for 15 minutes at room temperature to pellet insolubles.
- Collect the supernatant and determine protein concentration using a BCA assay.
Protein Digestion:
- Reduce 3 mg of protein lysate with 4.5 mM DTT for 30 minutes at 55°C.
- Alkylate with 10 mM IAA for 30 minutes at room temperature in the dark.
- Dilute the sample 1:4 with 20 mM HEPES, pH 8.5, containing 1 mM CaCl₂.
- Digest first with trypsin (37.5:1 substrate-to-enzyme ratio) overnight at room temperature, followed by Lys-C digestion for 4 hours at 37°C.
- Acidify the digest to ~1% TFA final concentration to stop digestion.
- Desalt the resulting peptides using a SEP-PAK C18 column. Elute with 50% acetonitrile/0.1% TFA, dry under vacuum, and reconstitute in 1 mL of 1X IP Buffer.
Immunoaffinity Enrichment (IAE):
- Incubate the digested peptides with the pre-washed K-ε-GG antibody beads from the PTMScan kit for 2 hours at 4°C on a rotator.
- Centrifuge the peptide-bead mixture at 2,000 g for 5 seconds and carefully discard the flow-through.
- Wash the beads twice with 1 mL of 1X IP Buffer, followed by three washes with 1 mL of HPLC-grade water.
- Elute the bound ubiquitinated peptides with two aliquots of 50 μL of 0.15% TFA.
Post-Enrichment Cleanup:
- Desalt the enriched peptides using C18 StageTips.
- Dry the purified peptides using a speed vacuum and store at -20°C until LC-MS/MS analysis.
Protocol for LC-MS/MS Analysis with CID and EAD Fragmentation
This protocol describes the liquid chromatography and mass spectrometry parameters for analyzing enriched peptides using a ZenoTOF 7600 system, enabling direct comparison of CID and EAD fragmentation [53] [55].
Materials:
- LC System: Evosep One or similar nanoLC system
- LC Column: C18, 15 cm x 150 μm ID, 1.5 μm particle diameter (e.g., EV-1137)
- Mobile Phase A: 0.1% Formic Acid in water
- Mobile Phase B: 0.1% Formic Acid in acetonitrile
- Mass Spectrometer: ZenoTOF 7600 system with an OptiFlow Turbo V ion source
Liquid Chromatography (LC) Conditions:
- Column Temperature: 40°C
- Flow Rate: 0.5 μL/min (for Evosep One 30 SPD method)
- Gradient: Use a 44-minute gradient (e.g., 30 samples per day method on Evosep One)
- Sample Load: Approximately 20 ng of enriched peptides estimated from total ion current.
Mass Spectrometry (MS) Acquisition: A data-dependent acquisition (DDA) method is built to incorporate both CID and EAD events.
Table 3: Key TOF MS and DDA Parameters for ZenoTOF 7600 System
| Parameter | Setting |
|---|---|
| Ion Source Gas 1 (GS1) | 25 psi |
| Ion Source Gas 2 (GS2) | 25 psi |
| Curtain Gas (CUR) | 25 psi |
| Ion Spray Voltage (ISVF) | 2300 V |
| Source Temperature | 200 °C |
| TOF MS Mass Range | 300-1800 Da |
| Accumulation Time | 0.25 s |
| CID (Collision Energy) | Rolling CE (e.g., 25-45 eV) |
| EAD (Electron Kinetic Energy) | 7 eV [55] |
| EAD Beam Current | 5500 nA [55] |
| EAD Reaction Time | 20 ms [55] |
| Zeno Trap | Enabled for MS/MS |
Workflow Visualization and Data Analysis
Integrated Workflow for Confident PTM Localization
The following diagram illustrates the logical sequence of the complete experimental procedure, from sample preparation to data analysis, as described in the protocols.
Data Processing and Analysis
- Software: Process raw data using PEAKS Studio or similar software (e.g., Fragpipe, Mascot, Byonic) [58] [55].
- Search Parameters:
- Database: Human UniProt FASTA.
- Enzymes: Trypsin (specific).
- Fixed Modification: Carbamidomethylation (C).
- Variable Modifications: Ubiquitination (GlyGly (K)), Oxidation (M), Acetylation (Protein N-term).
- Precursor Mass Tolerance: 20 ppm.
- Fragment Mass Tolerance: 0.1 Da.
- Validation: Apply false discovery rate (FDR) estimation, typically ≤ 1% at the peptide-spectrum match level.
The synergistic combination of high-specificity K-ε-GG antibody enrichment and the complementary fragmentation techniques of CID and EAD on modern high-sensitivity mass spectrometers provides a powerful solution for one of the most challenging aspects of ubiquitinome research: confident site-specific localization. EAD excels in preserving labile modifications and fragmenting long peptides, while CID remains highly effective for standard peptides. This integrated workflow, supported by the detailed protocols and reagent toolkit provided, empowers researchers to achieve a deeper and more confident characterization of the ubiquitinome, thereby advancing our understanding of this critical regulatory PTM in health and disease.
The study of proteome dynamics in response to cellular perturbations is fundamental to understanding biological mechanisms in disease and drug development. Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC) provides a robust metabolic labeling strategy for tracking newly synthesized proteins over time, enabling precise measurement of protein turnover rates [59]. However, traditional SILAC-based kinetic analyses present practical limitations, as each labeling time point requires a separate mass spectrometry run, making large time-course studies expensive and time-consuming [59]. The integration of Tandem Mass Tag (TMT) isobaric labeling with SILAC—a approach sometimes termed "hyperplexing"—overcomes these limitations by enabling multiplexed analysis of multiple time points in a single mass spectrometry run [59]. This combined methodology significantly enhances the efficiency and precision of temporally-resolved dynamic proteomic experiments, particularly when applied to study post-translational modifications such as ubiquitination using K-ε-GG antibody enrichment protocols [59] [14].
Key Research Reagent Solutions
The following table details essential materials and reagents required for implementing integrated SILAC-TMT labeling with K-ε-GG enrichment:
Table 1: Essential Research Reagents for SILAC-TMT-K-ε-GG Workflow
| Reagent/Kit | Function/Application | Key Characteristics |
|---|---|---|
| SILAC Amino Acids ( [59]) | Metabolic labeling of newly synthesized proteins | L-arginine:HCl (13C6, 99%) and L-lysine:2HCl (13C6, 99%) |
| TMT Multiplex Reagents ( [59]) | Isobaric chemical labeling for sample multiplexing | 10-plex or 11-plex kits enabling simultaneous analysis of multiple time points |
| PTMScan Ubiquitin Remnant Motif (K-ε-GG) Kit ( [60]) | Immunoaffinity enrichment of ubiquitinated peptides | Highly specific antibody recognizing di-glycine remnant left after trypsin digestion |
| Anti-K-ε-GG Antibody ( [14]) | Enrichment of endogenous ubiquitination sites | Specific for lysine residues modified with di-glycine remnant (K-ε-GG) |
| Cell Lysis Buffer ( [14]) | Protein extraction and denaturation | 8 M urea, 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, protease inhibitors |
| Protease Inhibitors ( [14]) | Preservation of ubiquitination state | PR-619 (deubiquitinase inhibitor), chloroacetamide, PMSF |
Integrated SILAC-TMT Experimental Protocol
Cell Culture and Metabolic Labeling
- Cell Line Preparation: Utilize human dermal fibroblasts (e.g., HCA2-hTert) or other relevant cell models. Maintain cells in appropriate media (e.g., Eagle's Minimum Essential Medium) supplemented with 15% dialyzed fetal bovine serum, penicillin, and streptomycin at 37°C with 5% CO₂ [59].
- SILAC Labeling Scheme:
- Dividing Cells: Switch adapted cultures to SILAC media containing heavy isotopes of arginine (13C6) and lysine (13C6) at standard concentrations (0.1264 g/L and 0.087 g/L, respectively). Collect cells after 0, 24, 48, and 72 hours of labeling [59].
- Quiescent Cells: Grow cultures to confluency (approximately 8 days) to induce contact inhibition. Switch to SILAC labeling media and collect cells at multiple time points (e.g., 0, 6, 12, 24, 36, 48, 72, 96, 144, 192, 336 hours) to capture slow protein turnover [59].
- Cellular Perturbation: Apply experimental treatments (e.g., drug compounds, growth factors, or environmental stressors) according to research objectives. Include appropriate controls.
Protein Extraction, Digestion, and TMT Labeling
- Cell Lysis: Lyse cell pellets in urea-based buffer (8 M urea, 75 mM NaCl, 50 mM Tris, pH 8.5) using vortexing and sonication (e.g., 5 cycles of 10 seconds with 1-minute ice incubations). Centrifuge at 15,000 × g for 10 minutes and collect supernatant [59].
- Protein Quantification: Determine protein concentration using BCA assay.
- Trypsin Digestion: Digest 100 μg of protein extract with trypsin (1:50 enzyme-to-substrate ratio) overnight at 37°C. Reduce disulfide bonds with TCEP (5 mM, 55°C for 1 hour) and alkylate with iodoacetamide (10 mM, room temperature for 30 minutes in the dark). Add a second aliquot of trypsin for 3 hours to complete digestion [59].
- Peptide Desalting: Desalt digested peptides using C18 solid-phase extraction tips or cartridges to remove interfering primary amines [59].
- TMT Labeling: Reconstitute desalted peptides in 100 mM triethylammonium bicarbonate. Label each SILAC time point sample with a unique TMT channel (e.g., TMT-126, 127N, 127C, etc.). Use 0.8 mg TMT reagent resuspended in 40 μL acetonitrile per 100 μg peptide. Incubate for 1 hour at room temperature, then quench with 5% hydroxylamine for 15 minutes [59].
- Sample Pooling: Combine equal amounts (e.g., 10 μg) of each TMT-labeled sample according to experimental design. The remaining labeled peptides should be kept separate for MS1-level SILAC quantitation [59].
K-ε-GG Peptide Enrichment
- High-pH Fractionation: Fractionate pooled TMT-labeled peptides using high-pH reversed-phase chromatography. Pool fractions in a non-contiguous manner into 8 final fractions to reduce complexity [14].
- Antibody Cross-Linking: Cross-link anti-K-ε-GG antibody to protein A agarose beads using dimethyl pimelimidate (20 mM in 100 mM sodium borate, pH 9.0) for 30 minutes at room temperature. Block with 200 mM ethanolamine (pH 8.0) for 2 hours at 4°C [14].
- Immunoaffinity Enrichment: Incubate each fraction with cross-linked anti-K-ε-GG antibody beads (31 μg antibody per fraction) for 1 hour at 4°C with rotation. Wash beads 4 times with ice-cold PBS, then elute K-ε-GG peptides with 0.15% TFA [14].
- Sample Cleanup: Desalt enriched ubiquitinated peptides using C18 StageTips prior to LC-MS/MS analysis [14].
LC-MS/MS Analysis and Data Processing
- Chromatography: Separate enriched peptides using reverse-phase liquid chromatography with a linear gradient of increasing acetonitrile.
- Mass Spectrometry: Acquire data using a tandem mass spectrometer capable of high-resolution MS1 and MS2 measurements. Implement Synchronous Precursor Selection (SPS) for MS3-level TMT quantitation to minimize co-isolation interference [59].
- Data Analysis:
- Protein Identification: Search MS/MS spectra against appropriate protein sequence databases using search engines that account for SILAC heavy amino acids, TMT modifications, and ubiquitination (K-ε-GG remnant).
- Quantification: Extract SILAC incorporation rates from MS1 spectra for protein turnover kinetics. Extract TMT reporter ions from MS2/MS3 spectra for relative quantitation across time points [59].
- Turnover Kinetics: Determine rate constants for protein degradation and synthesis by least squares regression analysis of time-resolved data fitted to appropriate kinetic models [59].
Workflow Visualization
Figure 1: Integrated SILAC-TMT-K-ε-GG Workflow. This diagram illustrates the comprehensive experimental workflow combining metabolic labeling (SILAC), isobaric tagging (TMT), and ubiquitinated peptide enrichment for perturbational studies of protein turnover dynamics.
Quantitative Data Analysis and Interpretation
Key Quantitative Parameters
The integrated SILAC-TMT approach generates rich quantitative data on protein turnover and abundance changes. The table below summarizes key measurable parameters:
Table 2: Quantitative Parameters from SILAC-TMT Experiments
| Parameter | Description | Measurement Approach | Biological Interpretation |
|---|---|---|---|
| Protein Degradation Rate (kdeg) | Rate constant of protein clearance | SILAC heavy-to-light ratio decay over time | Cellular protein half-life; increased in enhanced degradation |
| Protein Synthesis Rate (ksyn) | Rate constant of new protein production | SILAC heavy label incorporation kinetics | Translational efficiency; regulation of new protein synthesis |
| Relative Abundance Change | Protein expression differences between conditions | TMT reporter ion intensities | Regulatory responses to perturbations |
| Ubiquitination Site Dynamics | Turnover at specific ubiquitination sites | K-ε-GG peptide SILAC kinetics | Targeted degradation via ubiquitin-proteasome system |
| Global Turnover Shifts | System-wide changes in protein half-lives | Correlation of kdeg across conditions | Cellular adaptation states (e.g., quiescence vs. proliferation) |
Data Analysis Workflow
Figure 2: Quantitative Data Analysis Pipeline. Dual data streams from SILAC (protein turnover) and TMT (relative abundance) converge for comprehensive ubiquitination site analysis and biological interpretation.
Expected Results and Validation
In a representative application studying protein turnover in dividing versus quiescent fibroblasts, this methodology simultaneously measured kinetics for over 3,000 proteins [59]. Key findings included:
- Global Regulation: Upon quiescence, fibroblasts compensated for lack of cellular growth by globally downregulating protein synthesis and upregulating protein degradation [59].
- Method Validation: Measurements showed strong correlation with established non-multiplexed approaches, confirming accuracy [59].
- Ubiquitination Coverage: Optimized K-ε-GG enrichment enables quantification of approximately 20,000 distinct ubiquitination sites from moderate protein input (5 mg per SILAC channel) [14].
Technical Considerations and Optimization
Critical Optimization Parameters
- SILAC Labeling Efficiency: Ensure >95% incorporation of heavy amino acids through sufficient cell doublings (typically 5-6 population doublings) in SILAC media before experimental time course.
- TMT Labeling Efficiency: Optimize peptide-to-TMT reagent ratio and reaction conditions to achieve >98% labeling efficiency. Include quality control steps to verify labeling.
- Antibody Capacity: For K-ε-GG enrichment, use appropriate antibody-to-peptide ratios (approximately 31 μg antibody per basic RP fraction) to maintain binding capacity and specificity [14].
- Fractionation Strategy: Non-contiguous pooling of high-pH fractions significantly reduces sample complexity while maintaining comprehensive ubiquitinome coverage [14].
- MS Acquisition Methods: Implement SPS-MS3 quantification to minimize ratio compression from co-isolated peptides, significantly improving TMT quantification accuracy [59].
Troubleshooting Guide
- Low SILAC Incorporation: Verify dialyzed serum quality, ensure proper cell adaptation to SILAC media, and confirm adequate number of cell divisions.
- Poor K-ε-GG Enrichment: Check antibody cross-linking efficiency, optimize incubation time and temperature, and verify peptide input amounts.
- Ratio Compression in TMT: Implement SPS-MS3 methods, optimize collision energy, and verify peptide separation chromatography.
- High Background in Enrichment: Include stringent washing conditions, ensure proper desalting before immunoprecipitation, and verify antibody specificity.
Ubiquitination is a fundamental post-translational modification (PTM) that regulates diverse cellular functions including protein stability, activity, and localization [61]. The dysregulation of ubiquitination signaling pathways is implicated in numerous pathologies, including cancer and neurodegenerative diseases [61]. Research in this field necessitates precise methods for enriching and analyzing ubiquitinated proteins, particularly given the low stoichiometry of this modification under normal physiological conditions and the complexity of ubiquitin chains, which vary in length, linkage type, and architecture [61].
This application note provides a detailed comparative analysis of contemporary methods for enriching ubiquitinated proteins, with particular emphasis on the K-ε-GG antibody-based approach and its alternatives. We place specific focus on the technical aspects relevant to mass spectrometry research, providing structured quantitative comparisons, detailed experimental protocols, and visual workflow representations to guide researchers in selecting and implementing the most appropriate methodology for their specific research objectives.
The core methodologies for ubiquitinated protein enrichment can be broadly categorized into three groups: anti-K-ε-GG remnant antibodies, ubiquitin-binding domains (UBDs), and tagged ubiquitin systems. Each method possesses distinct principles, advantages, and limitations, making them suitable for different experimental scenarios.
Table 1: Core Methodologies for Ubiquitinated Protein Enrichment
| Method Category | Principle | Key Advantages | Inherent Limitations |
|---|---|---|---|
| K-ε-GG Remnant Antibody [14] [62] | Immunoaffinity enrichment of tryptic peptides containing the di-glycine remnant (K-ε-GG) left after trypsin digestion. | - High specificity for ubiquitin-derived GG-signature- Compatible with endogenous ubiquitin- Amenable to high-throughput and automated workflows [37] | - Requires complete tryptic digestion- Cannot distinguish chain topology- Potential epitope masking |
| Ubiquitin-Binding Domains (UBDs) [63] [64] | Engineered tandem hybrid UBDs (ThUBDs) with high affinity for ubiquitin chains. | - Enriches intact ubiquitinated proteins- Unbiased recognition of all linkage types [63]- Compatible with native conditions | - May not efficiently capture monoubiquitination- Potential for non-specific binding |
| Tagged Ubiquitin Systems [61] | Ectopic expression of affinity-tagged ubiquitin (e.g., His, Strep, HA). | - Easy and low-cost enrichment- Can be used in non-human systems | - Cannot study endogenous ubiquitination- Tag may alter ubiquitin structure/function- Genetic manipulation required |
Quantitative Performance Comparison
The selection of an enrichment strategy is critically informed by performance metrics. The following table summarizes key quantitative data for the leading methodologies, providing a basis for evidence-based decision-making.
Table 2: Quantitative Performance Comparison of Enrichment Methods
| Methodology | Reported Scale of Identification | Input Material | Key Performance Metrics | Linkage Specificity/Bias |
|---|---|---|---|---|
| K-ε-GG (Optimized Workflow) [14] | ~20,000 ubiquitination sites | 5 mg protein (SILAC) | 10-fold improvement over earlier K-ε-GG protocols | Linkage-blind; recognizes the GG remnant regardless of chain type [61] |
| Automated UbiFast (K-ε-GG) [37] | ~20,000 ubiquitination sites | 500 μg per sample (TMT10-plex) | Processes 96 samples in a single day; high reproducibility | Linkage-blind |
| ThUBD (Engineered UBD) [63] | 7,487 ubiquitinated proteins (1125 with defined sites) from mammalian cells | Not specified | Markedly higher affinity than natural UBDs; unbiased high affinity to all 7 lysine-linked chains [63] | Unbiased to all major chain types (K6, K11, K27, K29, K33, K48, K63) [63] |
| ThUBD-Coated 96-Well Plate [64] | High-throughput quantification of ubiquitinated proteins | As low as 0.625 μg | 16-fold wider linear range compared to TUBE-based plates | Unbiased capture of all ubiquitin chain types [64] |
| His-Tagged Ubiquitin (Early Method) [61] | 110 ubiquitination sites on 72 proteins (Yeast) | Not specified | Pioneering method for proteomic profiling | Linkage-blind |
Detailed Experimental Protocols
Refined K-ε-GG Immunoprecipitation Protocol
The following protocol, adapted from refined methodologies [14] [65], is designed for high-sensitivity enrichment of ubiquitinated peptides from cell lines or tissues.
Materials & Reagents:
- Lysis Buffer: 8 M Urea, 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, supplemented with protease inhibitors (e.g., 2 μg/mL aprotinin, 10 μg/mL leupeptin, 1 mM PMSF) and deubiquitinase inhibitors (e.g., 50 μM PR-619) [14].
- Anti-K-ε-GG Antibody: PTMScan Ubiquitin Remnant Motif Kit (#5562, Cell Signaling Technology) or its magnetic bead version (#59322, #19089) for higher sensitivity [62] [65].
- Cross-linking Reagent: Dimethyl Pimelimidate (DMP).
- IAP Buffer: 50 mM MOPS-NaOH (pH 7.2), 10 mM Sodium Phosphate, 50 mM NaCl.
- Solid-Phase Extraction: C18 Sep-Pak cartridges (500 mg, Waters) and Oasis HLB μElution Plates.
Procedure:
- Cell Lysis and Protein Digestion: Lyse cells in urea-based lysis buffer. Reduce proteins with 5 mM DTT (45 min, RT) and alkylate with 10 mM iodoacetamide (30 min, RT in the dark). Dilute the lysate to 2 M urea with 50 mM Tris-HCl (pH 7.5) and digest with sequencing-grade trypsin (enzyme-to-substrate ratio 1:50) overnight at 25°C [14].
- Peptide Cleanup and Fractionation: Acidify digested peptides with formic acid (FA) or TFA to pH < 2 and desalt using a C18 Sep-Pak cartridge. For deep coverage, subject the peptide mixture to offline basic reversed-phase (bRP) fractionation using a high-pH stable C18 column (e.g., Zorbax 300 Extend-C18). Pool fractions in a non-contiguous manner (e.g., 8-12 pooled fractions from 80 original fractions) to reduce complexity and improve LC-MS/MS depth [14].
- Antibody Cross-linking: To minimize antibody co-elution and improve MS sensitivity, cross-link the anti-K-ε-GG antibody to its support beads. Wash antibody beads with 100 mM sodium borate (pH 9.0). Resuspend in 20 mM DMP and incubate for 30 min at RT with rotation. Quench the reaction with 200 mM ethanolamine (pH 8.0) for 2 hours at 4°C. Wash and store the cross-linked beads in IAP buffer at 4°C [14] [65].
- Immunoprecipitation (IP): Resuspend the pooled bRP fractions in IAP buffer. For each mg of peptide, incubate with ~31 μg of cross-linked anti-K-ε-GG antibody for 1 hour at 4°C with rotation [14] [65].
- Washing and Elution: Wash the beads stringently to reduce non-specific binding: 2x with IAP buffer, 2x with IAP buffer containing 0.05% RapiGest SF, and 3x with PBS [65]. Elute bound K-ε-GG peptides with 2 x 50 μL of 0.15% TFA.
- Post-IP Cleanup and MS Analysis: Desalt the eluate using C18 StageTips or an Oasis HLB μElution Plate [14] [65]. Concentrate and analyze by LC-MS/MS.
Tandem Hybrid UBD (ThUBD) Enrichment Protocol
This protocol leverages engineered ThUBDs for the enrichment of intact ubiquitinated proteins, preserving information about ubiquitin chain topology [63] [64].
Materials & Reagents:
- Recombinant ThUBD Protein: Purified from E. coli [63] [64].
- Binding Buffer: 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 0.1% NP-40, supplemented with protease and DUB inhibitors.
- Elution Buffer: 1% SDS or 8 M Urea.
Procedure:
- Cell Lysis: Lyse cells in a native lysis buffer (e.g., containing 0.1% NP-40) or a denaturing buffer (e.g., 1% SDS or 8 M Urea) supplemented with inhibitors. For SDS-containing buffers, ensure subsequent dilution or use of cyclodextrins to sequester SDS before binding if required [66].
- Enrichment: Incubate the clarified cell lysate with purified ThUBD protein (immobilized on glutathione resin for GST-tagged ThUBD [63] or pre-coated onto the wells of a 96-well plate for high-throughput assays [64]) for 1-2 hours at 4°C with gentle agitation.
- Washing: Wash the resin or coated wells extensively with the appropriate binding buffer to remove non-specifically bound proteins.
- Elution: Elute the bound ubiquitinated proteins using a denaturing elution buffer (e.g., 1% SDS or 8 M Urea) for subsequent proteomics analysis, or directly with SDS-PAGE loading buffer for western blotting.
- Downstream Processing: For MS analysis, the eluted proteins can be subjected to standard proteomic workflows, including tryptic digestion and peptide cleanup. The resulting peptides will contain the K-ε-GG remnant, allowing site-specific identification.
Integrated Workflows and Advanced Applications
Modern proteomics increasingly relies on multi-omic profiling from single, limited samples. The MONTE (Multi-Omic Native Tissue Enrichment) workflow exemplifies this by serially enriching multiple 'omes, including the ubiquitylome, from the same tissue specimen [23].
- Workflow Integration: MONTE begins with native HLA-I and HLA-II immunopeptidome enrichment from tissue. The flow-through is then denatured with SDS, digested, and subjected to the UbiFast workflow for K-ε-GG peptide enrichment and TMT labeling. The flow-through from the K-ε-GG IP is subsequently used for deep-scale proteome, phosphoproteome, and acetylome analysis [23].
- Value: This serialized approach demonstrates that K-ε-GG enrichment is fully compatible with, and can be seamlessly integrated into, complex multi-omic pipelines without compromising the depth of coverage or quantitative precision of any individual 'ome [23].
Another innovative approach, SCASP-PTM, addresses the challenge of sequential PTM enrichment from a single sample for Data-Independent Acquisition (DIA) MS. It uses SDS for denaturation, which is then sequestered by cyclodextrins prior to trypsin digestion, enabling desalting-free sequential enrichment of ubiquitinated peptides alongside other PTMs like phosphopeptides and acetylated peptides [66].
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for Ubiquitinated Protein Enrichment
| Reagent / Kit | Vendor / Source | Primary Function | Key Feature |
|---|---|---|---|
| PTMScan Ubiquitin Remnant Motif Kit [62] | Cell Signaling Technology | Immunoaffinity enrichment of K-ε-GG peptides | High-specificity antibody for the di-glycine remnant |
| PTMScan HS Ubiquitin/SUMO Remnant Motif Kit [62] | Cell Signaling Technology | High-sensitivity magnetic bead-based K-ε-GG enrichment | Magnetic bead format for improved performance |
| Recombinant ThUBD Protein [63] [64] | In-house laboratory production | Enrichment of intact ubiquitinated proteins | Unbiased high affinity for all ubiquitin linkage types |
| ThUBD-Coated 96-Well Plates [64] | In-house laboratory production | High-throughput ubiquitination assay | Enables screening of ubiquitination status in a plate format |
| S-Trap Micro Spin Columns | Multiple Vendors | Protein digestion and detergent removal | Ideal for processing SDS-lysed samples post-HLA enrichment |
| C18 Sep-Pak Cartridges [14] | Waters | Peptide desalting and cleanup | Standard for sample preparation prior to fractionation or MS |
| Zorbax 300Extend-C18 Column [14] | Agilent | Offline basic pH reversed-phase fractionation | Increases depth of coverage by reducing sample complexity |
The choice between K-ε-GG and alternative enrichment methods is dictated by the specific research question. The K-ε-GG antibody-based approach remains the gold standard for deep, site-specific profiling of ubiquitination and is ideally suited for integration into high-throughput and multi-omic workflows. In contrast, ThUBD-based technologies offer a powerful alternative for applications requiring unbiased capture of ubiquitin chain topology and enrichment of intact ubiquitinated proteins, demonstrating superior performance over older UBD technologies like TUBE. By providing detailed protocols, performance metrics, and context for integration into broader experimental designs, this application note equips researchers to make informed decisions that will advance our understanding of the complex ubiquitin code.
This application note details the integration of the K-ε-GG antibody enrichment protocol with functional assays to transition from merely identifying ubiquitination sites to defining their biological consequences. Within drug discovery, understanding whether ubiquitination at a specific site leads to protein degradation, alters cellular localization, or modulates activity is paramount for target validation. We provide a detailed methodology for ubiquitin remnant profiling coupled with mechanistic follow-up experiments, enabling researchers to systematically classify ubiquitination events as degradative or non-degradative and link them to functional protein outcomes. This structured approach facilitates the identification of novel drug targets within the ubiquitin-proteasome system and provides a framework for assessing compound efficacy.
Protein ubiquitination, a pivotal post-translational modification (PTM), regulates a vast spectrum of cellular processes, with its complexity rivaling that of phosphorylation [67]. The development of ubiquitin remnant profiling using anti-K-ε-GG antibodies has enabled the identification of tens of thousands of ubiquitination sites, moving the field from characterization of individual events to proteome-wide analyses [67] [14]. A critical challenge, however, lies in interpreting these massive datasets; the majority of identified sites may represent quality control ubiquitination of misfolded or nascent proteins rather than targeted regulatory events involved in signaling pathways [67].
The functional consequence of ubiquitination is exquisitely diverse. While K48-linked polyubiquitination typically targets substrates for proteasomal degradation, other chain linkages or monoubiquitination can alter protein-protein interactions, subcellular localization, and activity [67]. Furthermore, ubiquitination often occurs as part of a PTM cascade, being preceded by phosphorylation, acetylation, or methylation, which can act as a switch to promote or inhibit subsequent ubiquitination [68] [69]. Therefore, a protocol that not only identifies sites but also contextualizes them within cellular regulation is essential for advancing therapeutic development. This document provides a detailed roadmap for such functional characterization, framed within the robust methodology of K-ε-GG immunoaffinity enrichment.
Core Experimental Workflow: Ubiquitin Remnant Profiling
The following section outlines the optimized protocol for enriching and identifying ubiquitinated peptides from complex cell lysates, forming the foundation for all subsequent functional analyses.
Optimized K-ε-GG Enrichment Protocol
This protocol is adapted from refined methodologies [14] and commercial kit procedures [70], and is designed for a triple-encoded SILAC experiment starting with 5 mg of protein per SILAC state.
Step 1: Cell Culture and Lysis
- Culture cells in SILAC media for ~6 doublings to ensure complete labeling. Apply experimental treatments (e.g., with proteasome inhibitor, E3 ligase inhibitor MLN4924, or vehicle control).
- Lysis: Pellet cells and lyse in 4°C denaturing lysis buffer (8 M Urea, 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA) supplemented with protease inhibitors (e.g., 2 μg/mL aprotinin, 10 μg/mL leupeptin, 1 mM PMSF) and deubiquitinase inhibitor (e.g., 50 μM PR-619) [14].
- Clarify lysate by centrifugation at 20,000 × g for 15 min at 4°C. Determine protein concentration using a BCA assay.
Step 2: Protein Digestion and Peptide Cleanup
- Reduce proteins with 5 mM dithiothreitol (45 min, RT) and alkylate with 10 mM iodoacetamide (30 min, RT in the dark).
- Dilute lysate to 2 M urea with 50 mM Tris-HCl, pH 7.5. Digest overnight at 25°C with sequencing-grade trypsin at a 1:50 (enzyme-to-substrate) ratio.
- Acidify peptides with formic acid and desalt using a C18 solid-phase extraction cartridge (e.g., a 500-mg tC18 Sep-Pak). Elute peptides with 50% acetonitrile, 0.1% formic acid and dry completely in a SpeedVac [14].
Step 3: Basic Reversed-Phase (bRP) Fractionation
- To reduce sample complexity, resuspend dried peptides in basic RP solvent A (2% MeCN, 5 mM ammonium formate, pH 10).
- Fractionate using a C18 column with a high-pH gradient. Collect 80 fractions and pool them in a non-contiguous manner into 8 final fractions (e.g., combine fractions 1, 9, 17... into pool 1; 2, 10, 18... into pool 2, etc.) [14]. Dry pooled fractions.
Step 4: Immunoaffinity Enrichment with Anti-K-ε-GG Antibody
- Antibody Cross-linking (Recommended): To reduce antibody leaching and improve sample cleanliness, wash anti-K-ε-GG antibody beads (e.g., from PTMScan Kit #5562) with 100 mM sodium borate, pH 9.0. Resuspend in 20 mM dimethyl pimelimidate (DMP) and incubate 30 min at RT. Quench with 200 mM ethanolamine, pH 8.0, for 2 hours at 4°C [14].
- Resuspend each dried bRP fraction in 1.5 mL of IAP Buffer (50 mM MOPS, pH 7.2, 10 mM sodium phosphate, 50 mM NaCl).
- Incubate each fraction with 31 μg of cross-linked anti-K-ε-GG antibody beads for 1 hour at 4°C with rotation.
- Wash beads four times with 1.5 mL of ice-cold PBS.
- Elute captured K-ε-GG peptides with 2 × 50 μL of 0.15% trifluoroacetic acid (TFA).
Step 5: Mass Spectrometry Analysis
- Desalt eluted peptides using C18 StageTips [14].
- Analyze by LC-MS/MS using a nanoflow LC system coupled to a high-resolution tandem mass spectrometer.
- Search data against the appropriate protein sequence database using search engines that can accommodate the K-ε-GG remnant (mass shift of +114.04293 Da on lysine) as a variable modification.
The following workflow diagram illustrates the core protocol.
The Scientist's Toolkit: Essential Research Reagents
Table 1: Key reagents and materials for K-ε-GG enrichment and functional analysis.
| Item | Function / Description | Example/Catalog Number |
|---|---|---|
| Anti-K-ε-GG Antibody | Immunoaffinity enrichment of tryptic peptides containing the diglycine remnant on lysine. | PTMScan Ubiquitin Remnant Motif Kit #5562 [70] |
| SILAC Amino Acids | For metabolic labeling enabling quantitative comparison of ubiquitination sites across conditions. | L-lysine-4,4,5,5-d4 (Lys-4); L-arginine-[13C6]HCl (Arg-6) [14] |
| Proteasome Inhibitor | To stabilize proteins targeted for degradation, increasing detection of degradative ubiquitination sites. | MG-132 [67] [14] |
| cullin-RING Ligase Inhibitor | To identify substrates of cullin-RING E3 ligases by inhibiting their activity. | MLN4924 [67] |
| Deubiquitinase (DUB) Inhibitor | Prevents removal of ubiquitin during cell lysis, preserving the native ubiquitinome. | PR-619 [14] |
| Cross-linking Reagent | Covalently links antibody to beads, reducing contamination in MS analysis. | Dimethyl Pimelimidate (DMP) [14] |
Defining Functional Outcomes: From Sites to Mechanisms
After identifying ubiquitination sites, the next critical step is to determine their biological role. The following assays are designed to classify sites and link them to protein stability and activity.
Classifying Ubiquitination as Degradative or Non-Degradative
A primary functional distinction is whether ubiquitination leads to proteasomal degradation. This can be determined by profiling ubiquitination site abundance in response to proteasome inhibition.
Protocol: Treatment with Proteasome Inhibitors
- Divide SILAC-labeled cells into two pools.
- Treat one pool with a proteasome inhibitor (e.g., 5 μM MG-132) and the other with a vehicle control (e.g., 0.5% DMSO) for 4-6 hours [14].
- Combine the cells in a 1:1 protein ratio and process them through the K-ε-GG enrichment protocol described in Section 2.1.
- Analyze the data by quantifying the SILAC ratios for each identified ubiquitination site (MG-132 / DMSO).
Data Interpretation:
- Increased Abundance (Ratio > 1): Sites that show significantly increased ubiquitination upon proteasome inhibition are likely directly involved in targeting the protein for degradation. The proteasome blockade causes accumulation of polyubiquitinated species [67].
- Decreased Abundance (Ratio < 1): Sites that decrease in abundance are likely involved in non-proteolytic functions. The model proposes that these sites undergo cycles of ubiquitination and deubiquitination, and the free ubiquitin pool is depleted when the proteasome is inhibited, leading to reduced ubiquitination at these sites [67].
Table 2: Quantitative response of ubiquitination sites to proteasome inhibition provides functional classification.
| Ubiquitination Site Response to MG-132 | Proposed Functional Classification | Potential Molecular Role |
|---|---|---|
| Significant Increase | Proteasome-dependent / Degradative | K48-linked polyubiquitination leading to proteasomal degradation. |
| Significant Decrease | Proteasome-independent / Regulatory | Monoubiquitination or alternative chain linkages (K63, K11, etc.) affecting signaling, localization, or activity. |
| No Change | Constitutive / Unaffected | May represent quality control ubiquitination or regulatory events not coupled to the proteasome under the tested conditions. |
Linking Ubiquitination to Protein Stability and Degron Mapping
To directly connect a specific ubiquitination site to changes in protein half-life, and to identify upstream regulatory PTMs, the following approaches are recommended.
Global Protein Stability (GPS) Profiling: This method uses a library of reporters (e.g., ORFs tagged with EGFP) to monitor protein half-lives. Treatment with an E3 ligase inhibitor (e.g., MLN4924) stabilizes its substrates, leading to an increase in the EGFP/DsRed ratio for those specific reporters. This provides direct functional evidence of E3 ligase substrates and their degradation kinetics [67].
Mapping PTM Crosstalk: Ubiquitination is often preceded by other PTMs that serve as degron activation/inactivation signals [68] [69]. To map this crosstalk:
- Perform parallel proteomic analyses for phosphorylation (using TiO2 or IMAC enrichment), acetylation, and methylation on the same samples.
- Integrate the datasets to identify co-occurring PTMs on the same protein or even the same peptide.
- Mutagenesis studies are then critical. Mutate the putative regulatory PTM site (e.g., a phosphorylation site) to a non-modifiable residue (Alanine) and assess the impact on ubiquitination at the nearby site and overall protein stability.
The following diagram illustrates the strategic process for moving from site identification to functional characterization.
The integration of the refined K-ε-GG antibody enrichment protocol with targeted functional assays provides a powerful, systematic framework for moving beyond ubiquitination site catalogs toward a mechanistic understanding of protein regulation. By quantitatively classifying sites as degradative or non-degradative, identifying their governing E3 ligases, and mapping the PTM crosstalk that controls them, researchers can prioritize therapeutically relevant ubiquitination events. This approach significantly de-risks drug discovery efforts aimed at the ubiquitin-proteasome system, whether the goal is to develop E3 ligase inhibitors, molecular glues, or agents that modulate specific ubiquitination pathways.
Conclusion
The K-ε-GG antibody enrichment protocol has unequivocally become the gold standard for the site-specific, large-scale profiling of the ubiquitinome, enabling the routine identification of tens of thousands of ubiquitination sites. By integrating a solid understanding of ubiquitin biology with a robust and optimized methodological workflow, researchers can achieve unprecedented depth and reproducibility in their studies. The ongoing development of automated, high-throughput platforms and advanced fragmentation techniques like EAD promises to further accelerate discovery. The future of this methodology lies in its expanded application to clinical samples, such as patient-derived tissues and biopsies, which will be crucial for deciphering the role of ubiquitination in disease mechanisms and identifying novel therapeutic targets in oncology and neurodegeneration.
References
- 1. The Ubiquitin – Proteasome in Immune Cells - PMC System [https://pmc.ncbi.nlm.nih.gov/articles/PMC7824874/]
- 2. - Ubiquitin - LifeSensors Proteasome System [https://lifesensors.com/ubiquitinproteasomesystem/]
- 3. How Ubiquitin Works — In One Simple Flow ( Proteasome ) 2025 [https://www.linkedin.com/pulse/how-ubiquitin-proteasome-works-one-simple-flow-2025-nbt5e]
- 4. Methods for Quantification of in vivo Changes in Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3418844/]
- 5. Large-Scale Identification of Ubiquitination Sites by Mass ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4725055/]
- 6. Refined Preparation and Use of Anti-diglycine Remnant (K- ... [https://www.sciencedirect.com/science/article/pii/S1535947620330565]
- 7. PTMScan® Ubiquitin Remnant Motif (K-epsilon-GG) Kit [https://www.cellsignal.com/products/proteomic-analysis-products/ubiquitin-remnant-motif-k-epsilon-gg-kit/5562?srsltid=AfmBOooPlGBJnSDz6QQYBXDbNvjvWnGGCC0v8IRtlmbbNywyNG7icSLZ]
- 8. Current methodologies in protein ubiquitination characterization: from... [https://cellandbioscience.biomedcentral.com/articles/10.1186/s13578-022-00870-y]
- 9. Detection of Protein Ubiquitination Sites by Peptide Enrichment and... [https://www.jove.com/t/59079/detection-protein-ubiquitination-sites-peptide-enrichment-mass]
- 10. Refined preparation and use of anti-diglycine remnant ( K - ε - GG )... [https://pubmed.ncbi.nlm.nih.gov/23266961/]
- 11. Large-scale identification of ubiquitination sites by mass spectrometry [https://experiments.springernature.com/articles/10.1038/nprot.2013.120?error=cookies_not_supported&code=4d6f15bf-8726-49f0-a8e0-d866504b7a39]
- 12. Global Mass -Based Analysis of Protein Spectrometry ... Ubiquitination [https://link.springer.com/protocol/10.1007/978-1-0716-1665-9_11]
- 13. Review Top-Down and Middle-Down Mass Spectrometry of ... [https://www.sciencedirect.com/science/article/pii/S1535947625000878]
- 14. Refined Preparation and Use of Anti-diglycine Remnant (K- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3591673/]
- 15. Rapid and deep-scale ubiquitylation profiling for biology ... [https://www.nature.com/articles/s41467-019-14175-1]
- 16. Time-resolved in vivo ubiquitinome profiling by DIA-MS ... [https://www.nature.com/articles/s41467-021-25454-1]
- 17. PTMScan® Ubiquitin Remnant Motif (K-epsilon-GG) Kit [https://www.cellsignal.com/products/proteomic-analysis-products/ubiquitin-remnant-motif-k-epsilon-gg-kit/5562?srsltid=AfmBOooj_yl2GX8cmeuQwSnRkRroLGtmUdxPGmBs1HYzo369Bqn2L7x3]
- 18. Integrative proximal-ubiquitomics profiling for ... [https://www.sciencedirect.com/science/article/pii/S2451945625001278]
- 19. PTMScan® Ubiquitin Remnant Motif (K-epsilon-GG) Kit [https://www.cellsignal.com/products/proteomic-analysis-products/ubiquitin-remnant-motif-k-epsilon-gg-kit/5562?srsltid=AfmBOoqDqRCFgTCpNsyr1WTGtT1JpLWod1V4sCtDdQCeEtmZ8p846__j]
- 20. The emerging roles of ubiquitin-like modifications in ... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2025.1593445/full]
- 21. In-Depth Comparison of Reagent-Based Digestion ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11923677/]
- 22. Protocol for tandem enrichment of ubiquitinated, ... [https://www.sciencedirect.com/science/article/pii/S2666166725003508]
- 23. Workflow enabling deepscale immunopeptidome ... [https://www.nature.com/articles/s41467-023-37547-0]
- 24. Immunoaffinity chromatography: an introduction to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2903764/]
- 25. | SpringerLink Immunoaffinity Chromatography [https://link.springer.com/protocol/10.1385/1-59259-655-X:167]
- 26. PTMScan ® HS Ubiquitin/SUMO Remnant Motif (K-epsilon- ... [https://www.cellsignal.com/products/proteomic-analysis-products/hs-ubiquitin-sumo-remnant-motif-k-epsilon-gg-kit/59322?srsltid=AfmBOor62QnS1obEVCr8BzCRGNzYPCuj5XVcOlR2z6VmNBTrwPmEx0gj]
- 27. PTMScan® Ubiquitin Remnant Motif (K-epsilon-GG) Kit [https://www.cellsignal.com/products/proteomic-analysis-products/ubiquitin-remnant-motif-k-epsilon-gg-kit/5562?srsltid=AfmBOor5QKZ9Z1NpTlY1ER4aIVo3JJotiz05y5IYMTvpciIVK8vWIZxj]
- 28. PTMScan® Ubiquitin Remnant Motif (K-epsilon-GG) Kit [https://www.cellsignal.com/products/proteomic-analysis-products/ubiquitin-remnant-motif-k-epsilon-gg-kit/5562?srsltid=AfmBOorrXOW2DbbLAZdfExkIwNgtDxAxLDxRkfGfiQC7aQAgHC_hqgF_]
- 29. Orthogonality of separation and sorbent evaluation in ... [https://www.sciencedirect.com/science/article/pii/S277239172500060X]
- 30. Systematic Comparison of Fractionation Methods for In- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3430803/]
- 31. Two-Dimensional Fractionation Method for Proteome-Wide ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9056026/]
- 32. Optimized Liquid and Gas Phase Fractionation Increases ... [https://www.sciencedirect.com/science/article/pii/S1535947621001055]
- 33. Online Peptide Fractionation Using a Multiphasic ... [https://www.sciencedirect.com/science/article/pii/S1535947620331790]
- 34. AUTO-SP: automated sample preparation for analyzing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12355475/]
- 35. PTMScan® Ubiquitin Remnant Motif (K-epsilon-GG) Kit [https://www.cellsignal.com/products/proteomic-analysis-products/ubiquitin-remnant-motif-k-epsilon-gg-kit/5562?srsltid=AfmBOoqbk-An4aN0RyW3niY23bSbpud2FWcdApJIYgXsAQmTYHevUTBW]
- 36. Rapid and deep-scale ubiquitylation profiling for ... - PubMed [https://pubmed.ncbi.nlm.nih.gov/31953384/]
- 37. Automating UbiFast for High-throughput and Multiplexed ... [https://www.sciencedirect.com/science/article/pii/S1535947621001262]
- 38. Enrichment of ubiquitinated (K-ε-GG) peptides [https://bio-protocol.org/exchange/minidetail?id=2242819&type=30]
- 39. Exclusion lists to reduce contamination with HPLC-MS/MS [https://pmc.ncbi.nlm.nih.gov/articles/PMC3714598/]
- 40. Affinity Enrichment for MS: Improving the yield of low ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6081742/]
- 41. Data-independent acquisition method for ubiquitinome ... [https://www.nature.com/articles/s41467-020-20509-1]
- 42. Sequence enrichment profiles enable target-agnostic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10261905/]
- 43. Beads-free protein immunoprecipitation for a mass ... [https://proteomesci.biomedcentral.com/articles/10.1186/s12953-015-0079-0]
- 44. High-Recovery Desalting Tip Columns for a Wide Variety of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11696827/]
- 45. Methods for Quantification of in vivo Changes in Protein ... [https://www.sciencedirect.com/science/article/pii/S1535947620304576]
- 46. Site-specific quantification of the in vivo UFMylome reveals ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12146631/]
- 47. Concentration and Desalting of Peptide and Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2291762/]
- 48. Improved elution strategy and new monoclonal anti-biotin ... [https://www.sciencedirect.com/science/article/pii/S240558082400075X]
- 49. Recent advances in small molecule inhibitors of ... [https://pubmed.ncbi.nlm.nih.gov/39908798/]
- 50. Recent advances in small molecule inhibitors of ... [https://www.sciencedirect.com/science/article/abs/pii/S0223523425000893]
- 51. Time-resolved in vivo ubiquitinome profiling by DIA-MS ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8438043/]
- 52. An inhibitor of the proteasomal deubiquitinating enzyme ... [https://www.sciencedirect.com/science/article/pii/S0021925820328933]
- 53. Localization and Quantification of Post-Translational ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11157679/]
- 54. Comprehensive Ubiquitome Analysis of Nicotiana ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12189520/]
- 55. Confident PTM site localization of immunoaffinity-enriched ... [https://sciex.com/tech-notes/life-science-research/proteomics/confident-ptm-site-localization-of-immunoaffinity-enriched-pepti]
- 56. Antibody toolkit reveals N-terminally ubiquitinated ... [https://www.nature.com/articles/s41467-021-24669-6]
- 57. A single-injection workflow for enhanced peptide mapping ... [https://sciex.com/tech-notes/biopharma/a-single-injection-workflow-for-enhanced-peptide-mapping-using-c]
- 58. Enhancing glycopeptide annotation and glycan localization ... [https://www.sciencedirect.com/science/article/pii/S1874391925001733]
- 59. Time-resolved Analysis of Proteome Dynamics by Tandem ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5141271/]
- 60. PTMScan® Ubiquitin Remnant Motif (K-epsilon-GG) Kit [https://www.cellsignal.com/products/proteomic-analysis-products/ubiquitin-remnant-motif-k-epsilon-gg-kit/5562?srsltid=AfmBOop5GdVCNGeWJ3CANkMqMBkJ8V0q_m5gS_A_3GGtm5-EZVxWYQAf]
- 61. Current methodologies in protein ubiquitination ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9373315/]
- 62. PTMScan® Ubiquitin Remnant Motif (K-epsilon-GG) Kit [https://www.cellsignal.com/products/proteomic-analysis-products/ubiquitin-remnant-motif-k-epsilon-gg-kit/5562?srsltid=AfmBOorEt439Ac4VsfIZgPHKPlpRi_NX1LAnytFPqvjLT3TxBot2dVNl]
- 63. Enhanced Purification of Ubiquitinated Proteins by ... [https://www.sciencedirect.com/science/article/pii/S1535947620336288]
- 64. A High-Throughput Method for Specific, Rapid, Precise and ... [https://www.sciencedirect.com/science/article/abs/pii/S0039914025015681]
- 65. K-ε-GG immunoprecipitation (di-glycine remnant enrichment) [https://bio-protocol.org/exchange/minidetail?id=8183631&type=30]
- 66. Comprehensive PTM profiling with SCASP-PTM uncovers ... [https://www.sciencedirect.com/science/article/pii/S2211124725002712]
- 67. The new landscape of protein ubiquitination - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3242807/]
- 68. Control of protein stability by post-translational modifications [https://www.nature.com/articles/s41467-023-35795-8]
- 69. An inventory of crosstalk between ubiquitination and other ... [https://www.sciencedirect.com/science/article/pii/S258900422300353X]
- 70. PTMScan® Ubiquitin Remnant Motif (K-epsilon-GG) Kit [https://www.cellsignal.com/products/proteomic-analysis-products/ubiquitin-remnant-motif-k-epsilon-gg-kit/5562?srsltid=AfmBOoow2aRpQmPg8DdVLV6iZmn-TQ89XjUdaYgEkpoSuYHsyPLyU2jd]