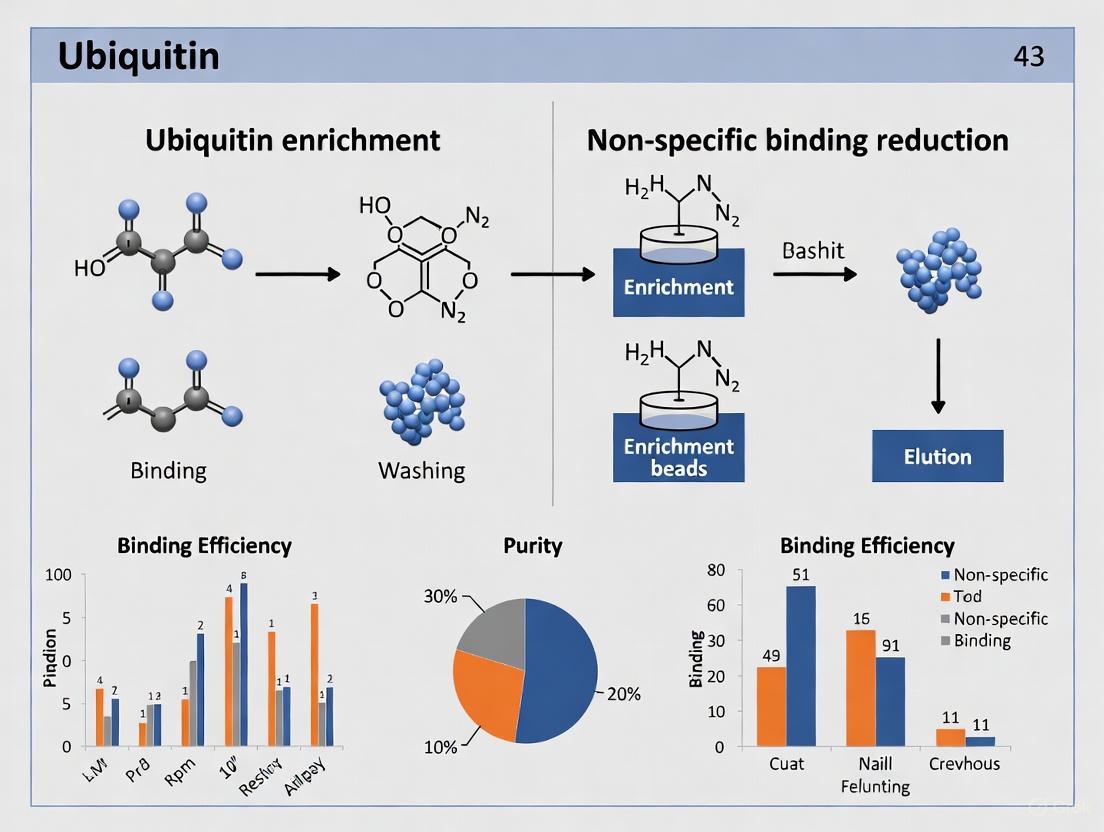
Optimizing Ubiquitin Enrichment: Advanced Strategies to Minimize Non-Specific Binding for Cleaner Results
This article provides a comprehensive guide for researchers, scientists, and drug development professionals on overcoming the critical challenge of non-specific binding in ubiquitin enrichment workflows.
Optimizing Ubiquitin Enrichment: Advanced Strategies to Minimize Non-Specific Binding for Cleaner Results
Abstract
This article provides a comprehensive guide for researchers, scientists, and drug development professionals on overcoming the critical challenge of non-specific binding in ubiquitin enrichment workflows. Covering foundational principles to advanced applications, we detail specific methodologies including Tandem Ubiquitin Binding Entities (TUBEs), chemical biology tools, and optimized commercial kits that significantly enhance specificity. The content further addresses systematic troubleshooting, protocol optimization for various sample types, and rigorous validation techniques using mass spectrometry and functional assays. By synthesizing current methodologies and emerging technologies, this resource aims to empower scientists with practical strategies for obtaining highly specific ubiquitination data, ultimately accelerating research in proteomics, biomarker discovery, and targeted protein degradation therapeutics.
Understanding the Ubiquitin Enrichment Challenge: Why Non-Specific Binding Occurs
The Critical Impact of Non-Specific Binding on Data Quality and Interpretation
In ubiquitin enrichment research, non-specific binding (NSB) refers to the undesirable adherence of biomolecules (like proteins or contaminants) to your experimental surfaces—such as sensor chips, affinity resins, or antibodies—through interactions that are not related to the specific ubiquitin modification you are trying to study [1]. This phenomenon is a critical technical challenge that can lead to increased background noise, false positive signals in pull-down assays, misinterpretation of mass spectrometry (MS) data, and ultimately, a loss of sensitivity and reliability in detecting genuine ubiquitination events [2] [1]. Effectively controlling for NSB is therefore not merely an optimization step but a fundamental requirement for producing high-quality, interpretable data on the ubiquitin code.
FAQs on Non-Specific Binding in Ubiquitin Research
1. What are the primary causes of non-specific binding in ubiquitin enrichment experiments? NSB in ubiquitin studies arises from several factors:
- Sensor Surface or Resin Chemistry: The nature of the affinity resin (e.g., Ni-NTA, Strep-Tactin, or antibody-coupled beads) can passively interact with non-target proteins. For instance, Ni-NTA agarose can co-purify histidine-rich proteins, and anti-biotin resins can bind endogenously biotinylated proteins when using Strep-tagged ubiquitin systems [2].
- Sample Impurities: Contaminants in your cell or tissue lysates, such as lipids, nucleic acids, or other proteins, can bind to the experimental surfaces or to the capture molecules themselves, generating false-positive signals [1].
- Suboptimal Buffer Conditions: The pH, ionic strength, and composition of your binding and wash buffers can significantly influence electrostatic and hydrophobic interactions. Incorrect conditions can promote NSB [1].
2. How can I distinguish between specific ubiquitination signals and non-specific binding?
- Well-Designed Controls: The most robust method is to include control experiments. For immunoblotting or affinity pull-downs, this involves performing the enrichment in the presence of a saturating concentration of an unlabeled competitive ligand (e.g., free ubiquitin) or using a lysate from cells where the E3 ligase of interest is knocked out. The signal remaining in these control conditions is representative of NSB and should be subtracted from your experimental data [3].
- Kinetic Analysis: In real-time binding studies like Surface Plasmon Resonance (SPR), specific interactions typically show characteristic association and dissociation phases. In contrast, NSB often displays rapid, non-saturable association and slower, less structured dissociation [1].
- Mass Spectrometry Follow-up: After enrichment, specific ubiquitinated peptides can be identified by the signature diglycine (Gly-Gly) remnant left on lysine residues after tryptic digestion and MS analysis. The absence of this signature on bound proteins suggests NSB [2] [4].
3. My western blot for ubiquitin shows a high background smear. Is this non-specific binding? Not necessarily. A characteristic smear on a ubiquitin western blot is often indicative of a successful enrichment of polyubiquitinated proteins, which have varying molecular weights due to different chain lengths and linkages [5]. However, a high background signal between expected bands or a strong signal in your negative control lanes is a clear sign of NSB. This can be caused by non-specific antibody interactions or insufficient blocking of the membrane.
4. Which ubiquitin enrichment method is least prone to non-specific binding? No single method is immune, but some are designed to minimize it. Tandem Ubiquitin Binding Entities (TUBEs) and high-affinity ubiquitin-binding domains (UBDs) like OtUBD offer high specificity for ubiquitin, reducing background [6]. The key is to choose a method appropriate for your question. For example, while tagged ubiquitin systems (e.g., His- or Strep-tag) are easy to use, they are prone to the NSB issues mentioned above. Antibody-based methods work on endogenous proteins but can have lot-to-lot variability and high cost [2] [5].
Troubleshooting Guides
Problem: High Background in Affinity Purification and Western Blot
Symptoms: Strong ubiquitin signal in negative control samples (e.g., no primary antibody, sample from ubiquitin-knockdown cells), or a diffuse, high background across the entire lane obscuring specific bands.
Solutions:
- Optimize Blocking: Ensure you are using an effective blocking agent (e.g., 5% BSA or non-fat milk) for a sufficient time (at least 1 hour at room temperature) before antibody incubation.
- Include Control Experiments: Always run a parallel experiment with a control resin (e.g., bare agarose beads) or in the presence of a competing free ubiquitin to define and subtract the NSB signal [3].
- Increase Wash Stringency: Incorporate additional washes with buffers containing mild detergents (e.g., 0.1% Tween-20) or increasing salt concentrations (e.g., 300-500 mM NaCl) to disrupt non-specific electrostatic interactions [6].
- Use Denaturing Conditions: For pull-downs, using strong denaturants like 1-2% SDS in the lysis buffer can help dissociate non-covalent protein interactors from covalently ubiquitinated proteins, providing a cleaner result [6].
Problem: Excessive Non-Specific Binding in SPR Sensorgrams
Symptoms: A rapid, large response unit (RU) signal that does not saturate and exhibits little dissociation, making kinetic analysis of the specific ubiquitin-ligase interaction impossible.
Solutions:
- Surface Optimization: Use a sensor chip with a low propensity for NSB and ensure the ligand (e.g., ubiquitin or a UBD) is properly oriented and immobilized.
- Buffer Additives: Include additives in the running buffer that reduce NSB, such as surfactant P20 (0.05%), carboxymethyl dextran, or chaotropic agents [1].
- Sample Preparation: Centrifuge or filter your analyte samples just before injection to remove any aggregates or particulate matter that can non-specifically stick to the sensor chip [1].
- Reference Subtraction: Always use a reference flow cell (with no ligand or an irrelevant ligand immobilized) and subtract its signal from the active flow cell to account for bulk refractive index changes and system-specific NSB [1].
Problem: Co-precipitation of Non-Ubiquitinated Proteins in MS Workflows
Symptoms: Mass spectrometry analysis of your ubiquitin pull-down identifies a large number of proteins lacking the diglycine modification, suggesting they are non-specifically bound contaminants.
Solutions:
- Tandem Enrichment (Double Pull-Down): Use a tandem purification strategy. For example, if using tagged ubiquitin, perform a first enrichment with the tag-specific resin, then elute and subject the eluate to a second round of enrichment with a ubiquitin-specific antibody or UBD (OtUBD) [2].
- Work in Denaturing Conditions: To isolate the covalent "ubiquitinome" from the non-covalent "interactome," lyse cells in buffers containing 1% SDS or 6 M Guanidine-HCl. This denatures proteins and disrupts nearly all non-covalent interactions, ensuring that only covalently modified proteins are purified in subsequent steps [6].
- Use Specific Enzymes: Treat your enriched samples with a broad-spectrum deubiquitinase (DUB) as a control. A genuine ubiquitin signal should be sensitive to DUB treatment, while NSB will remain.
Essential Methodologies for Ubiquitin Enrichment
The table below summarizes the primary methods used to enrich ubiquitinated proteins, along with their associated NSB challenges and advantages.
Table 1: Comparison of Ubiquitin Enrichment Methodologies
| Method | Principle | Common Sources of NSB | Key Advantages |
|---|---|---|---|
| Tagged Ubiquitin [2] | Expression of His-, HA-, or Strep-tagged Ub in cells; enrichment via tag-specific resin. | Co-purification of histidine-rich or endogenously biotinylated proteins. | Easy to use, relatively low-cost, high-throughput compatible. |
| Antibody-Based [2] [5] | Use of anti-ubiquitin antibodies (e.g., P4D1, FK2) to immunoprecipitate endogenous ubiquitinated proteins. | Non-specific antibody cross-reactivity and binding to protein A/G beads. | Works on endogenous proteins without genetic manipulation; linkage-specific antibodies available. |
| UBD-Based (e.g., TUBEs, OtUBD) [2] [6] | Use of single or tandem ubiquitin-binding domains to capture ubiquitin conjugates. | Lower affinity single UBDs can have poor recovery; can bind free ubiquitin. | High affinity and specificity; can protect chains from DUBs; works under denaturing conditions (OtUBD). |
| Kits (e.g., Pierce) [7] | Proprietary affinity resin supplied with optimized buffers for enrichment. | Varies by kit chemistry, but similar to general resin-based NSB issues. | Fast, convenient, and complete; includes necessary reagents and protocols. |
The Scientist's Toolkit: Key Reagent Solutions
Table 2: Essential Research Reagents for Ubiquitin Enrichment
| Reagent | Function | Example Use Case |
|---|---|---|
| Proteasome Inhibitors (e.g., MG-132, Epoxomicin) [5] | Blocks degradation of ubiquitinated proteins, leading to their accumulation and increased detection signal. | Treat cells with 5-25 µM MG-132 for 1-2 hours before harvesting to boost ubiquitin levels. |
| Deubiquitinase (DUB) Inhibitors (e.g., N-Ethylmaleimide (NEM), PR-619) [6] | Prevents the cleavage of ubiquitin chains by endogenous DUBs during lysate preparation, preserving the ubiquitination signal. | Add 10-25 mM NEM to cell lysis buffer to inactivate DUBs. |
| High-Affinity UBD Resins (e.g., OtUBD, TUBEs) [6] | Provides a highly specific matrix for pulling down mono- and polyubiquitinated proteins with low background. | Use OtUBD resin with harsh wash buffers (e.g., containing 1 M NaCl) to minimize NSB while retaining target proteins. |
| Linkage-Specific Antibodies [2] [4] | Allows for the specific detection or enrichment of ubiquitin chains with a particular linkage (e.g., K48, K63). | Use a K48-linkage specific antibody to confirm if a protein is targeted for proteasomal degradation. |
| Diglycine (Gly-Gly) Remnant Antibodies [8] | Enables the proteomic-level identification of ubiquitination sites by specifically enriching for tryptic peptides containing the Gly-Gly lysine modification. | Use after tryptic digestion of enriched samples for LC-MS/MS analysis to map ubiquitination sites. |
Experimental Workflow and NSB Control Points
The following diagram illustrates a generalized workflow for ubiquitin enrichment, highlighting key steps where NSB can occur and the corresponding mitigation strategies.
Fundamental Principles of Ubiquitin-Binding Interactions and Affinity Matrices
Ubiquitination is a versatile post-translational modification involving the covalent attachment of ubiquitin (Ub), a 76-amino acid protein, to target substrates. This process regulates diverse cellular functions including protein degradation, signal transduction, DNA repair, and endocytosis. The specificity of ubiquitin signaling is governed by the topology of ubiquitin chains, which can vary in length and linkage type. Eight distinct linkage types exist (M1, K6, K11, K27, K29, K33, K48, K63), each potentially encoding different functional outcomes. Understanding the fundamental principles of ubiquitin-binding interactions and affinity matrices is essential for reducing non-specific binding in ubiquitin enrichment research, enabling more accurate characterization of ubiquitin-mediated processes in health and disease.
FAQs: Ubiquitin-Binding and Enrichment Challenges
1. What are the primary causes of non-specific binding in ubiquitin pull-down assays?
Non-specific binding in ubiquitin enrichment experiments commonly results from antibody cross-reactivity, insufficient blocking, inadequate washing stringency, or protein degradation. Polyclonal antibodies, while useful for detecting multiple epitopes, are particularly prone to promiscuous binding. Additionally, the weak affinities of individual ubiquitin-binding domains (typically in the μM range) can lead to non-specific interactions if not properly optimized in experimental conditions. Using high antibody concentrations can exacerbate this problem, as can the presence of protein multimers or degraded protein fragments that share epitopes with your target.
2. Why might my ubiquitin western blot show unexpected bands or smears?
Ubiquitinated proteins often appear as smears or multiple bands on western blots due to several factors: the natural heterogeneity of ubiquitin chain length and linkage type; the formation of protein multimers; partial protein degradation; or the presence of different ubiquitinated protein species. This pattern is actually characteristic of ubiquitinated samples, as the Ubiquitin-Trap captures monomeric ubiquitin, ubiquitin polymers, and ubiquitinylated proteins of varying lengths. However, discrete non-specific bands may indicate antibody cross-reactivity, insufficient protease inhibition during sample preparation, or protein subtypes/splice variants with similar epitopes.
3. How can I improve the specificity of ubiquitin linkage detection?
To enhance linkage-specific detection, employ multiple complementary approaches: (1) Use linkage-specific reagents such as Tandem Ubiquitin Binding Entities (TUBEs) with nanomolar affinities for particular chain types; (2) Incorporate proteolytically stable ubiquitin variants in pull-down assays using chemical biology approaches like genetic code expansion or click chemistry; (3) Validate findings with multiple detection methods including linkage-specific antibodies when available; (4) Include appropriate controls such as ubiquitin mutants (e.g., lysine-to-arginine mutations) to verify specificity.
Troubleshooting Guides
Problem: High Background and Non-Specific Binding in Ubiquitin Enrichment
Potential Causes and Solutions:
Cause: Inefficient blocking
- Solution: Increase blocking reagent concentration (e.g., from 2% to 5% BSA), extend blocking incubation times, and prepare primary antibody in blocking buffer. Add Tween-20 (0.05%) to blocking buffer if not already present.
Cause: Inadequate washing
- Solution: Ensure sufficient washing buffer volume to cover the blot, wash with gentle agitation, increase washes to 4-5 times for 5 minutes each, and consider increasing Tween-20 concentration to 0.1%.
Cause: Antibody-related issues
- Solution: For polyclonal antibodies showing promiscuous binding, switch to monoclonal antibodies when possible. Titrate antibody to determine optimal concentration that minimizes non-specific binding. Always use fresh aliquots of antibodies to maintain specificity.
Cause: Protein degradation
- Solution: Add protease inhibitors to lysis buffer and maintain samples at 4°C during preparation. Precipitate proteins with acetone or TCA to immediately denature and inactivate proteases. Avoid overexposure to urea which can lead to protein degradation.
Problem: Inconsistent Ubiquitin Enrichment Efficiency
Potential Causes and Solutions:
Cause: Variable ubiquitination levels
- Solution: Treat cells with proteasome inhibitors like MG-132 (5-25 μM for 1-2 hours) prior to harvesting to preserve ubiquitination signals. Optimize concentration and duration for each cell type to avoid cytotoxicity.
Cause: Insufficient binding capacity
- Solution: Note that precise binding capacity for ubiquitin chains is difficult to determine as chains can bind at single or multiple sites. Use fresh resin and avoid overloading the affinity matrix. For Ubiquitin-Trap products, follow manufacturer recommendations for sample-to-resin ratios.
Cause: Interference from endogenous proteins
- Solution: When using His-tag systems, be aware that histidine-rich proteins may co-purify. For Strep-tag systems, endogenously biotinylated proteins may cause interference. Include appropriate controls to identify these contaminants.
Research Reagent Solutions for Ubiquitin Studies
Table: Essential Research Reagents for Ubiquitin Enrichment and Detection
| Reagent Type | Specific Examples | Function and Application |
|---|---|---|
| Affinity Matrices | Ubiquitin-Trap Agarose/Magnetic Agarose (ChromoTek) | Immunoprecipitation of monomeric ubiquitin, ubiquitin polymers, and ubiquitinylated proteins from various cell extracts using anti-ubiquitin nanobody. |
| TUBEs (Tandem Ubiquitin Binding Entities) | K48-TUBE, K63-TUBE, Pan-TUBE (LifeSensors) | High-affinity capture of polyubiquitin chains with linkage specificity (K48, K63) or pan-specificity for general ubiquitin enrichment. |
| Linkage-Specific Antibodies | K48-linkage specific, K63-linkage specific, M1-linkage specific | Detection and enrichment of ubiquitinated proteins with specific chain linkages via western blot or immunoprecipitation. |
| Chemical Biology Tools | Genetic Code Expansion (GCE), Click Chemistry (CuAAC), Thiol Chemistry | Generation of defined, proteolytically stable ubiquitin variants for structural and interactome studies. |
| Proteasome Inhibitors | MG-132 | Preserves ubiquitination signals by inhibiting proteasomal degradation of ubiquitinated proteins. |
| Tagged Ubiquitin Systems | His-tagged Ub, Strep-tagged Ub, HA-tagged Ub | Expression systems for purifying ubiquitinated substrates from cellular environments. |
Quantitative Data on Ubiquitin-Binding Affinities
Table: Binding Affinities of Ubiquitin-Binding Domains to Mono-Ubiquitin
| Ub-Binding Domain | Source Protein | Kd (μM) | Method | Structural Features |
|---|---|---|---|---|
| UBA | Dsk2 | 14.8 ± 5.3 | SPR | Three-helical bundle binding Ile44 hydrophobic patch |
| UBA | hHR23A | 400 ± 100 | NMR | Three-helical bundle (UBA2) |
| CUE | Vps9 | 20 ± 1 | ITC | Structurally homologous to UBA domains |
| UIM | Vps27 (UIM1) | 277 ± 8 | NMR | Single α-helix binding shallow hydrophobic groove on ubiquitin |
| UIM | S5a (UIM2) | 73 | NMR | Single α-helix with conserved alanine residue |
| MIU | Rabex-5 | 29 ± 4.8 | SPR/ITC | Helical domain interacting with Ile44 patch |
| ZnF UBP | Isopeptidase T | 2.8 | ITC | Zinc finger domain with high affinity for ubiquitin |
| GAT | TOM1 | 409 ± 13 | SPR | helical domain with distinct binding mode for Ile44 patch |
Experimental Workflows for Specific Ubiquitin Enrichment
Methodology 1: TUBE-Based Enrichment for Linkage-Specific Ubiquitination
Principle: Tandem Ubiquitin Binding Entities (TUBEs) consist of multiple ubiquitin-binding domains fused together, creating high-affinity reagents with specificity for particular ubiquitin chain linkages. These can discriminate between different biological functions, such as K63-linked chains in inflammatory signaling versus K48-linked chains in proteasomal degradation.
Protocol:
- Cellular Treatment and Lysis: Culture THP-1 cells and treat with either L18-MDP (200-500 ng/mL for 30-60 min) to induce K63 ubiquitination of RIPK2 or with PROTAC degrader to induce K48 ubiquitination. Lyse cells using buffer optimized to preserve polyubiquitination (e.g., containing protease inhibitors and N-ethylmaleimide to inhibit deubiquitinases).
- TUBE Coating: Coat 96-well plates with chain-specific TUBEs (K48-TUBE, K63-TUBE, or Pan-TUBE) according to manufacturer's instructions.
- Sample Incubation: Incubate cell lysates (50-100 μg total protein) in TUBE-coated plates for 2-4 hours at 4°C with gentle agitation.
- Washing: Wash plates extensively with wash buffer containing 0.1% Tween-20 to remove non-specifically bound proteins.
- Target Detection: Detect specifically bound ubiquitinated proteins by immunoblotting with target-specific antibodies (e.g., anti-RIPK2).
- Validation: Include controls such as ubiquitin mutants (K48R or K63R) or specific pathway inhibitors (e.g., Ponatinib for RIPK2) to verify linkage specificity.
Methodology 2: Chemical Biology Approaches for Defined Ubiquitin Variants
Principle: Chemical biology tools enable generation of defined ubiquitin variants with proteolytically stable linkages, which can be used as affinity matrices to identify interacting proteins through mass spectrometry. These approaches overcome the limitations of enzymatic generation of ubiquitin chains and the lability of native isopeptide bonds.
Protocol for Click Chemistry-Generated Diubiquitin:
- Ubiquitin Functionalization: Synthesize ubiquitin monomers functionalized with either azide (using azido-ornithine incorporation at desired positions via SPPS) or alkyne groups (using propargylamine coupled to C-terminus).
- Click Reaction: Perform copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) using CuSO4 and sodium ascorbate to generate diubiquitin linked via triazole bonds. These mimic native isopeptide bonds while being resistant to hydrolysis by deubiquitinases.
- Affinity Matrix Preparation: Immobilize triazole-linked ubiquitin chains on appropriate resin (e.g., agarose beads) following standard coupling procedures.
- Affinity Enrichment: Incubate affinity matrix with cell lysates under near-physiological conditions (2-4 hours at 4°C) to allow binding of ubiquitin-interacting proteins.
- Washing and Elution: Wash beads extensively with buffer containing 0.1% Tween-20, then elute bound proteins with SDS-PAGE sample buffer or low pH elution buffer.
- MS Identification: Separate eluted proteins by SDS-PAGE, perform in-gel tryptic digestion, and identify interacting proteins by liquid chromatography-tandem mass spectrometry (LC-MS/MS) with label-free quantification.
Ubiquitin Signaling Pathways and Technical Challenges
Ubiquitin Linkage Diversity and Functional Consequences:
The ubiquitin code encompasses tremendous complexity, with different linkage types directing distinct functional outcomes. K48-linked chains primarily target substrates for proteasomal degradation, while K63-linked chains regulate non-proteolytic functions including inflammatory signaling and protein trafficking. Less common linkages (K6, K11, K27, K29, K33, M1) play roles in specific processes such as DNA repair, autophagy, and immune signaling. This diversity presents both challenges and opportunities for research and therapeutic development.
Technical Considerations for Reducing Non-Specific Binding:
- Ubiquitin-Binding Domain Affinities: Individual ubiquitin-binding domains typically exhibit weak affinities (Kd > 100 μM), which cells leverage through avidity effects using multiple domains or oligomerization. Researchers can mimic this strategy by using tandem domains (e.g., TUBEs) rather than single domains for enrichment.
- Epitope Exposure: The Ile44 hydrophobic patch on ubiquitin serves as the primary interaction surface for many UBDs. Maintaining the structural integrity of this region during experimental procedures is essential for specific binding interactions.
- Stability Considerations: The native isopeptide bond between ubiquitin and substrates is highly labile due to cellular deubiquitinases. Using DUB inhibitors during sample preparation and proteolytically stable ubiquitin analogs (e.g., triazole linkages) in pull-down assays can significantly reduce false negatives.
- Specificity Validation: Always include critical controls such as competition with free ubiquitin, ubiquitin mutants, and pathway-specific inhibitors to confirm the specificity of observed interactions.
Protein ubiquitination is a crucial post-translational modification regulating diverse cellular functions, but its proteomic analysis faces significant challenges due to low stoichiometry and the transient nature of the modification. A primary technical hurdle in ubiquitin proteomics is non-specific binding during enrichment procedures, which can compromise data quality, reduce sensitivity for genuine ubiquitination events, and lead to false positives. Non-specific binding refers to the unintended co-purification of proteins or peptides that are not the target of interest. This guide addresses the common sources of this interference and provides proven methodologies to mitigate it, enabling cleaner and more reliable results in ubiquitination research.
FAQ 1: Why is my ubiquitin western blot a smear with high background, and how can I resolve this?
Issue: A smeared appearance with high background is a classic symptom of non-specific binding during the immunoprecipitation (IP) step. This often occurs because the affinity beads capture abundant, non-ubiquitinated proteins alongside the target ubiquitinated species.
Primary Sources:
- Abundant Protein Contamination: Ubiquitin affinity reagents can non-specifically interact with highly abundant cellular proteins.
- Endogenous Biotinylated Proteins: When using Strep-tag based Ub purification systems, endogenously biotinylated proteins can bind to the Strep-Tactin resin [2].
- His-Rich Proteins: When using His-tagged ubiquitin and Ni-NTA purification, proteins rich in histidine residues can co-purify [2].
- Incomplete Blocking: The affinity beads or the IP system may have insufficient blocking, leading to nonspecific protein adherence.
Solutions:
- Optimize Wash Stringency: Increase the salt concentration or add mild detergents (e.g., 0.1% Triton X-100) to the wash buffer. Consistently use a wash buffer like BlastR Wash Buffer for multiple rigorous washes [9].
- Include Specific Controls: Always run a parallel IP with control beads (e.g., beads without the ubiquitin-binding entity) to identify proteins that bind non-specifically to the bead matrix itself. Subtract these identifications from your experimental sample [9].
- Use Denaturing Conditions: Perform lysis and IP under denaturing conditions (e.g., using 1% SDS in the lysis buffer) to disrupt non-covalent protein-protein interactions that cause co-purification. This must be compatible with your enrichment reagent [9].
- Pre-clear Lysate: Centrifuge the lysate at high speed (e.g., 10,000-16,000 g) to remove insoluble debris. Some protocols also incubate the lysate with control beads before the actual IP to pre-clear non-specific binders.
FAQ 2: I am using tagged ubiquitin for pull-downs. What are the specific non-binding risks with this approach?
Issue: While expressing tagged ubiquitin (e.g., His, Strep, or HA) is a common enrichment strategy, it introduces several specific artifacts that lead to non-specific binding and misinterpretation of data.
Primary Sources:
- Competition with Endogenous Ubiquitin: The expressed tagged ubiquitin may not fully recapitulate the endogenous ubiquitin dynamics, and the presence of both tagged and untagged ubiquitin pools can lead to incomplete enrichment and complex artifacts [2] [10].
- Tag-Specific Interactions: As noted, His-tags bind histidine-rich proteins, and Strep-tags bind endogenous biotinylated proteins [2].
- Structural Alterations: The tag itself may alter the structure of ubiquitin or its ability to form certain chain types, potentially creating unnatural ubiquitination events or failing to mimic endogenous modification patterns [2].
Solutions:
- Choose the Tag Carefully: For mammalian cells, the Stable Tagged Ubiquitin Exchange (StUbEx) system can help replace the endogenous pool with tagged ubiquitin, reducing competition artifacts [2].
- Validate Findings: Corroborate key findings from tagged-ubiquitin experiments with an orthogonal method, such as using pan-specific or linkage-specific Ub antibodies on endogenous proteins.
- Use Tandem Enrichment: To move beyond tag-based limitations, consider methods like the SCASP-PTM protocol, which allows for the enrichment of endogenously ubiquitinated peptides from complex lysates without genetic manipulation, thereby avoiding tag-related artifacts [11].
FAQ 3: How does antibody quality contribute to non-specific binding, and how can I select the best reagent?
Issue: Antibodies are powerful tools for enriching endogenously ubiquitinated proteins, but their quality is paramount. Low-affinity or non-specific antibodies are a major source of false positives and high background.
Primary Sources:
- Cross-Reactivity: Antibodies may recognize epitopes on non-ubiquitin proteins that share similarity with the immunogen.
- Low Affinity: Low-affinity antibodies require a higher amount of antibody and lysate, increasing the chance of non-specific interactions.
- Impure Antisera: Polyclonal antisera can contain antibodies against various contaminants from the immunogen preparation.
Solutions:
- Use High-Affinity Binders: Employ engineered high-affinity reagents like Tandem Ubiquitin Binding Entities (TUBEs) or Ubiquitin-Trap nanobodies. TUBEs, with their avidity effect, show nanomolar affinity for polyubiquitin chains and can outcompete low-affinity non-specific interactions [12] [13]. The Ubiquitin-Trap is noted for producing clean, low-background IPs due to its high specificity [13].
- Select Linkage-Specific Reagents: For studying specific chain types, use well-validated linkage-specific TUBEs or antibodies. These reagents are precisely characterized to enrich for a particular linkage (e.g., K48 or K63), dramatically reducing background from other chain types [12].
- Verify Antibody Specificity: Always validate the specificity of an antibody using controls such as ubiquitin-deficient cell lines, linkage-specific deubiquitinases (DUBs), or competing with free ubiquitin.
FAQ 4: What sample preparation factors can increase non-specific binding?
Issue: The initial steps of sample preparation are critical. Suboptimal handling of cell lysates can greatly exacerbate problems with non-specific binding downstream.
Primary Sources:
- Incomplete Lysis and Viscosity: Crude lysates can be viscous due to released DNA, which can trap proteins non-specifically and clog columns or hinder bead binding [9].
- Protease and Deubiquitinase Activity: During lysis, active proteases can degrade proteins, while deubiquitinases (DUBs) can remove ubiquitin from substrates, both of which distort the true ubiquitination landscape and can generate fragments that bind non-specifically.
Solutions:
- Employ Robust Lysis Protocols: Use lysis buffers containing benzonase or other nucleases to digest DNA and reduce viscosity. Filter lysates using specialized filters (e.g., BlastR Filters) to remove particulate matter [9].
- Use Protease and DUB Inhibitors: It is essential to add broad-spectrum protease inhibitors and, crucially, DUB inhibitors (e.g., PR-619, N-Ethylmaleimide) to the lysis buffer immediately upon cell disruption. This preserves the native ubiquitination state of proteins [9].
- Stabilize Ubiquitination: Treat cells with proteasome inhibitors (e.g., MG-132) for a few hours before harvesting. This prevents the degradation of polyubiquitinated proteins and increases their abundance for detection, reducing the need for excessive starting material that can increase background [13].
Table 1: Common Sources of Non-Specific Binding and Their Impact
| Source Category | Specific Example | Consequence | Frequency in Literature |
|---|---|---|---|
| Tag-Based Enrichment | Co-purification of histidine-rich proteins with His-tag systems [2] | Masks low-abundance ubiquitinated targets | High |
| Co-purification of endogenous biotinylated proteins with Strep-tag systems [2] | False positives in mass spectrometry | High | |
| Antibody-Based Enrichment | Low-affinity or cross-reactive pan-ubiquitin antibodies [2] | High background, smeared western blots | Very High |
| Sample Preparation | Incomplete lysis and viscous DNA in lysate [9] | Trapping of non-target proteins, clogged columns | Common |
| Inadequate DUB inhibition [9] | Loss of signal, altered ubiquitination profile | Very Common | |
| Cellular Context | Abundant non-target proteins | Saturation of binding capacity, reduced sensitivity | Ubiquitous |
Table 2: Comparison of Ubiquitin Enrichment Reagents and Non-Specific Binding Potential
| Enrichment Reagent | Principle | Advantages | Disadvantages / Non-Specific Binding Risks |
|---|---|---|---|
| His-Tag / Ni-NTA [2] | Affinity purification of His-tagged Ub | Relatively low cost, easy to use | High risk from His-rich proteins; requires genetic manipulation |
| Strep-Tag / Strep-Tactin [2] | Affinity purification of Strep-tagged Ub | High specificity and affinity | Risk from endogenous biotinylated proteins; requires genetic manipulation |
| Pan-Ubiquitin Antibodies (e.g., P4D1, FK2) [2] | Immunoaffinity for ubiquitin | Works on endogenous proteins | High risk of cross-reactivity and low-affinity binding; high cost |
| TUBEs (Tandem Ubiquitin Binding Entities) [12] | High-affinity UBDs in tandem | Nanomolar affinity, protects chains from DUBs, low background | May have linkage preferences; requires characterization |
| Ubiquitin-Trap (Nanobody) [13] | High-affinity VHH binding to Ub | Clean IPs, stable under harsh washes, low background | Not linkage-specific; will bind all ubiquitin conjugates |
| diGly Antibody (K-ε-GG) [10] | Enriches tryptic peptides with GlyGly remnant | Direct site identification, works on any sample | Cannot provide protein-level or chain linkage information |
Detailed Experimental Protocols
Protocol 1: Clean Immunoprecipitation of Ubiquitinated Proteins Using Magnetic Beads
This protocol is adapted from commercial best practices and is designed to minimize non-specific binding through stringent washes and appropriate controls [9].
Materials:
- Lysis Buffer (e.g., BlastR Lysis Buffer with 1% SDS)
- Dilution Buffer (e.g., BlastR Dilution Buffer)
- Protease and Deubiquitinase Inhibitor Cocktails
- Wash Buffer (e.g., BlastR Wash Buffer)
- High-affinity ubiquitin binding beads (e.g., TUBE-magnetic beads or Ubiquitin-Trap Magnetic Agarose)
- Control beads (beads without ubiquitin-binding ligand)
- Cell scraper, BlastR filters, magnetic rack, rotating platform.
Method:
- Inhibitor-Enhanced Lysis: Grow and treat cells as required. Wash cells with PBS. Lyse cells in Lysis Buffer supplemented with protease and DUB inhibitors. Use a cell scraper for efficient lysis. The initial use of an SDS-containing buffer helps denature proteins and disrupt non-covalent interactions.
- Clarification and Dilution: Pass the viscous lysate through a BlastR filter using a plunger to remove DNA and debris. Collect the flow-through. Critically, dilute the lysate 1:5 with Dilution Buffer to reduce the SDS concentration to a level compatible with the affinity beads, while maintaining a denaturing environment.
- Quantification and Pre-clearing (Optional): Quantify protein concentration. A starting point of 1.0 mg of total protein per IP is recommended. To reduce non-specific binding, the lysate can be pre-cleared by incubating with control beads for 30 minutes.
- Binding with Controls: Aliquot washed affinity beads and control beads into separate tubes. Add the diluted lysate to both the IP tube and the Control IP tube. Incubate on a rotating platform at 4°C for 2 hours.
- Stringent Washes: Collect beads using a magnetic rack. Aspirate the supernatant. Wash the beads three times with 1 mL of Wash Buffer, incubating for 5 minutes on a rotator with each wash. This step is crucial for removing loosely bound, non-specific proteins.
- Elution: After the final wash, completely remove the supernatant. Add 30 µL of Bead Elution Buffer (a low-ppH buffer or SDS sample buffer) to the beads, resuspend by flicking, and incubate at room temperature for 5 minutes. Transfer the suspension to a spin column and centrifuge to collect the clean eluate.
- Analysis: Add reducing agent (e.g., β-mercaptoethanol) and boil the samples for 5 minutes. Analyze by SDS-PAGE and western blotting.
Protocol 2: Tandem Enrichment of Ubiquitinated Peptides for Mass Spectrometry (SCASP-PTM)
This modern protocol allows for the sequential enrichment of multiple PTMs, including ubiquitination, from a single sample, improving specificity and throughput [11].
Materials:
- SDS-cyclodextrin-assisted sample preparation (SCASP) reagents
- diGly remnant (K-ε-GG) antibody-conjugated beads
- Trypsin
- C18 desalting tips or columns
- Mass spectrometry-compatible buffers.
Method:
- Protein Extraction and Digestion: Extract proteins using the SCASP method, which utilizes SDS and cyclodextrin to efficiently solubilize proteins while maintaining compatibility with downstream enzymatic steps. Digest the extracted proteins with trypsin.
- Primary Enrichment (Ubiquitinated Peptides): Without an intermediate desalting step (which can cause peptide loss), subject the protein digest to enrichment using anti-diGly antibody beads. This directly captures peptides containing the GlyGly remnant left after tryptic digestion of ubiquitinated proteins.
- Secondary Enrichment from Flow-Through: Collect the flow-through from the first enrichment. This flow-through, now largely devoid of ubiquitinated peptides, can be subsequently used for the serial enrichment of other PTMs, such as phosphorylation or glycosylation, without the need for desalting.
- Cleanup and MS Analysis: Desalt the enriched ubiquitinated peptides using C18 tips. The peptides are then analyzed by liquid chromatography with tandem mass spectrometry (LC-MS/MS). Data-independent acquisition (DIA) methods are recommended for comprehensive and quantitative profiling.
Experimental Workflow Visualization
The following diagram illustrates a recommended workflow that integrates solutions for minimizing non-specific binding, from sample preparation to analysis.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents for Clean Ubiquitin Enrichment
| Reagent | Function | Key Feature for Reducing NSB | Example Product/Type |
|---|---|---|---|
| DUB Inhibitors | Prevents deubiquitination during lysis, preserving signal | Stabilizes the target, reducing degradation-related artifacts | PR-619, N-Ethylmaleimide (NEM) [9] |
| Proteasome Inhibitors | Increases abundance of polyubiquitinated proteins | Allows use of less lysate, reducing co-purified background | MG-132 [13] |
| High-Affinity TUBEs | Enriches polyubiquitin chains with high avidity | Nanomolar affinity outcompetes low-affinity non-specific interactions; protects from DUBs [12] | K48-TUBE, K63-TUBE, Pan-TUBE [12] |
| Ubiquitin-Trap | Nanobody-based IP of ubiquitin conjugates | Engineered for high specificity and stability under harsh wash conditions [13] | ChromoTek Ubiquitin-Trap Agarose/Magnetic [13] |
| diGly Remnant Antibody | Enriches ubiquitinated peptides for MS | Directly targets the covalent modification, highly specific for site identification [10] | Anti-K-ε-GG Antibody |
| Control Beads | Identifies non-specific binders to bead matrix | Essential control for subtracting background in MS and WB | Beads without ubiquitin ligand [9] |
High-Specificity Enrichment Tools: From TUBEs to Chemical Biology
The study of the ubiquitin-proteasome system (UPS) is fundamental to understanding cellular homeostasis, signaling, and the mechanisms of targeted protein degradation. A significant technical challenge in this field is the specific enrichment of ubiquitinated proteins from complex cellular lysates. Non-specific binding during pull-down assays can lead to high background noise, masking genuine ubiquitination signals and producing unreliable data. The development of Tandem Ubiquitin Binding Entities (TUBEs) addresses this issue by providing reagents engineered for high-affinity and linkage-specific binding to polyubiquitin chains, thereby reducing non-specific interactions and protecting ubiquitin modifications from deubiquitinating enzymes (DUBs) during processing [12] [14].
TUBEs are recombinant proteins typically composed of multiple ubiquitin-associated (UBA) domains arranged in tandem. This design confers a nanomolar affinity for polyubiquitin chains, which is significantly stronger than that of single UBA domains. Their high affinity allows TUBEs to effectively compete with cellular DUBs, preserving the labile ubiquitin signal in experiments [12] [14]. A key advancement is the development of chain-selective TUBEs, which are engineered to preferentially recognize specific ubiquitin linkage types, such as the degradation-associated K48-linked chains or the signaling-associated K63-linked chains [12]. This specificity enables researchers to dissect the complex biological functions of different ubiquitin codes.
Table 1: Common Ubiquitin Linkages and Their Primary Functions
| Linkage Type | Primary Known Function(s) |
|---|---|
| K48 | Targets proteins for proteasomal degradation [12] [15] |
| K63 | Regulates signal transduction, protein trafficking, and immune responses [12] [15] |
| K6 | Involved in antiviral responses, autophagy, and DNA repair [15] |
| K11 | Associated with cell cycle progression and proteasome-mediated degradation [15] |
| M1 | Regulates cell death and immune signaling [15] |
Quantitative Performance Data
The performance of affinity reagents is quantifiable by their sensitivity and dynamic range. The following table summarizes key metrics for TUBEs and a next-generation technology, Tandem Hybrid Ubiquitin Binding Domains (ThUBDs), which were developed to address some limitations of first-generation TUBEs.
Table 2: Performance Comparison of Ubiquitin-Binding Reagents
| Technology | Affinity/Sensitivity | Key Feature | Linkage Specificity |
|---|---|---|---|
| Traditional TUBEs | Nanomolar affinity (K_d) [12] | High-affinity capture, protection from DUBs | Available as pan-selective or chain-selective (e.g., K48, K63) [12] |
| ThUBD-coated plates | 16-fold wider linear range and higher sensitivity than TUBE-coated plates [16] | Unbiased recognition of all ubiquitin chain types | Designed for unbiased capture, though specific applications may vary [16] |
Experimental Protocol: Using Chain-Specific TUBEs in a 96-Well Plate Format
This protocol is adapted from a study investigating the linkage-specific ubiquitination of RIPK2, demonstrating the high-throughput application of TUBE technology [12].
Materials:
- Chain-specific TUBE-coated plates: e.g., K48-TUBE, K63-TUBE, or Pan-TUBE coated 96-well plates.
- Cell lysate: Prepared from treated cells using a lysis buffer optimized to preserve polyubiquitination (e.g., containing DUB inhibitors).
- Primary antibody: Specific to your protein of interest (e.g., anti-RIPK2).
- HRP-conjugated secondary antibody: Compatible with your primary antibody host species.
- Wash buffer: Typically a phosphate-buffered saline (PBS) solution with a mild detergent.
- ELISA detection reagents: Chemiluminescent or colorimetric substrate compatible with HRP.
- Microplate reader.
Methodology:
Cell Treatment and Lysis:
- Treat cells according to your experimental design. For example, to induce K63-ubiquitination of RIPK2, treat THP-1 cells with 200-500 ng/mL L18-MDP for 30 minutes. To induce K48-ubiquitination, use a specific PROTAC like RIPK2 degrader-2 [12].
- Lyse cells in an appropriate buffer, clarify the lysate by centrifugation, and determine the protein concentration.
Ubiquitin Capture:
- Apply a standardized amount of cell lysate (e.g., 50 µg) to the wells of the chain-specific TUBE-coated plate.
- Incubate for 1-2 hours at 4°C with gentle agitation to allow ubiquitinated proteins to bind to the TUBEs.
Washing:
- Wash the wells thoroughly with wash buffer multiple times to remove all non-specifically bound proteins.
Target Protein Detection:
- Add a primary antibody against your target protein (e.g., RIPK2) and incubate.
- Wash again to remove unbound primary antibody.
- Add an HRP-conjugated secondary antibody and incubate.
- Perform a final wash.
Signal Detection and Quantification:
- Add the HRP substrate to the wells and measure the resulting signal (chemiluminescence or absorbance) using a microplate reader.
- Quantify the level of captured ubiquitinated target protein by comparing to standards or controls.
Workflow Visualization:
High-Throughput TUBE Assay Workflow
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for TUBE-based Ubiquitin Enrichment
| Reagent / Tool | Function / Description | Example Use Case |
|---|---|---|
| Chain-Selective TUBEs | Recombinant proteins to immunoprecipitate specific ubiquitin linkages (K48, K63, etc.) [12]. | Differentiating degradative (K48) from non-degradative (K63) ubiquitination of a target protein like RIPK2 [12]. |
| Pan-Selective TUBEs | Recombinant proteins that bind all ubiquitin chain linkages with high affinity [12] [14]. | Capturing the total pool of ubiquitinated proteins from a cell lysate for global ubiquitome analysis. |
| TUBE-coated 96-well Plates | Microplates pre-coated with TUBEs for high-throughput, plate-based ubiquitination assays [12] [16]. | Rapid screening of PROTAC-induced target ubiquitination in a quantitative format [12]. |
| DUB Inhibitors | Small molecules (e.g., MG-132) added to cell culture media and lysis buffers. | Preserving the ubiquitin signal by preventing its removal by deubiquitinating enzymes during sample preparation [15]. |
| Ubiquitin-Trap Agarose/Magnetic Beads | An alternative nanobody-based technology for pulldown of mono- and polyubiquitinated proteins [15]. | Immunoprecipitation of ubiquitinated proteins for downstream analysis by western blot or mass spectrometry. |
Troubleshooting Guide and FAQs
FAQ 1: My ubiquitin western blot shows a high background smear. How can I reduce this non-specific signal?
- Solution: This is a common challenge. Ensure you are using a high-affinity capture reagent like TUBEs to enrich for true ubiquitination signals over background. Optimize your wash conditions by increasing salt concentration (e.g., 300-500 mM NaCl) or adding a mild detergent to the wash buffer. Always include a control with a TUBE reagent but no cell lysate to identify signal from the reagent itself. Furthermore, treating cells with a proteasome inhibitor like MG-132 (e.g., 5-25 µM for 1-2 hours) prior to harvesting can help preserve and enrich for ubiquitinated proteins [15].
FAQ 2: Can TUBEs differentiate between K48 and K63-linked ubiquitination on my protein of interest?
- Solution: Yes, this is a primary application for chain-selective TUBEs. In a study on RIPK2, K63-TUBEs specifically captured the protein after inflammatory stimulation (L18-MDP), while K48-TUBEs captured it upon treatment with a PROTAC degrader. Pan-TUBEs captured the protein in both contexts. To perform this analysis, you would run parallel experiments using different chain-specific TUBEs and compare the enrichment of your target [12].
FAQ 3: I am working with a low-abundance target protein. How can I improve the sensitivity of ubiquitination detection?
- Solution: Consider moving to a more sensitive platform. While TUBE-coated plates offer good sensitivity, newer technologies like Tandem Hybrid Ubiquitin Binding Domain (ThUBD)-coated plates have been reported to exhibit a 16-fold wider dynamic range and significantly higher sensitivity for capturing polyubiquitinated proteins from complex proteomes compared to TUBE technology [16]. Maximizing the amount of input protein and using high-sensitivity chemiluminescent substrates can also help.
FAQ 4: Why is my ubiquitinated protein yield low after a TUBE pulldown?
- Solution: The transient nature of ubiquitination is a key factor. To protect ubiquitin chains from DUBs, it is critical to include a broad-spectrum DUB inhibitor cocktail in your lysis buffer and perform all steps at 4°C. Also, verify that your lysis buffer is non-denaturing to maintain the native structure of ubiquitin chains required for TUBE recognition. Finally, ensure you are using an adequate amount of TUBE reagent for the amount of lysate input.
FAQ 5: Are there any specific considerations for using TUBEs in mass spectrometry (IP-MS) workflows?
- Solution: TUBEs are compatible with IP-MS. The primary advantage is their ability to protect ubiquitin chains from DUBs, leading to a more representative profile of the ubiquitome. For MS compatibility, ensure you use harsh wash conditions (e.g., high salt) to minimize non-specific co-purifying proteins that can complicate the analysis. Specific protocols for on-bead digestion have been optimized for technologies like the Ubiquitin-Trap, which can serve as a useful guide [15].
Linkage-Specific Antibodies for Selective Ubiquitin Chain Enrichment
Ubiquitination is a critical post-translational modification that regulates diverse cellular functions, including protein degradation, signal transduction, and DNA repair. The specificity of ubiquitin signaling is largely determined by the type of polyubiquitin chain formed, with eight distinct linkage types (M1, K6, K11, K27, K29, K33, K48, and K63) mediating different functional outcomes. Linkage-specific antibodies have become indispensable tools for selectively enriching and detecting these various ubiquitin chain types, enabling researchers to decipher the complex ubiquitin code. However, a significant challenge in these applications is minimizing non-specific binding, which can compromise data quality and lead to erroneous conclusions. This technical resource center addresses common experimental issues and provides optimized protocols to enhance the specificity and reliability of your ubiquitin enrichment studies.
The Scientist's Toolkit: Key Reagents for Ubiquitin Research
The following table summarizes essential reagents used in linkage-specific ubiquitin research:
| Reagent Type | Examples | Primary Function |
|---|---|---|
| Linkage-Specific Antibodies | K48-specific, K63-specific, K11-specific [2] [17] | Immunoblotting and immunofluorescence to detect specific ubiquitin linkages. |
| Tandem Ubiquitin Binding Entities (TUBEs) | Pan-TUBEs, K48-TUBE, K63-TUBE, M1-TUBE [18] [19] | High-affinity enrichment of polyubiquitinated proteins from cell lysates; protect chains from deubiquitinases. |
| Affimers | K6-specific, K33/K11-specific [20] | Non-antibody binding proteins used for linkage-specific detection in blotting, microscopy, and pull-downs. |
| Ubiquitin-Traps | Ubiquitin-Trap Agarose, Ubiquitin-Trap Magnetic Agarose [21] | Immunoprecipitation of monomeric ubiquitin, ubiquitin chains, and ubiquitinylated proteins from various cell extracts. |
| Epitope-Tagged Ubiquitin | His-Ub, HA-Ub, Strep-Ub [2] | High-throughput purification of ubiquitinated substrates from cultured cells. |
Troubleshooting Guide: Addressing Common Experimental Challenges
Problem 1: High Background and Non-Specific Binding in Enrichment
- Potential Cause: Non-specific interaction between the affinity resin or antibody and non-ubiquitinated proteins in the lysate.
- Solution:
- Optimize Wash Stringency: Increase the salt concentration (e.g., 300-500 mM NaCl) or add mild detergents (e.g., 0.1% Triton X-100) to the wash buffers.
- Use Competitor Proteins: Include inert proteins like bovine serum albumin (BSA) in wash buffers to block non-specific binding sites.
- Pre-clear Lysate: Pre-incubate the cell lysate with bare beads or resin to remove proteins that bind non-specifically.
- Validate with Controls: Always include a control with a non-targeting antibody or bare beads to establish the baseline background signal.
Problem 2: Inefficient Capture of Polyubiquitinated Proteins
- Potential Cause: The abundance of the target polyubiquitinated protein is low, or the ubiquitin chains are being degraded by deubiquitinases (DUBs) during lysis.
- Solution:
- Use Proteasome Inhibitors: Treat cells with inhibitors like MG-132 (e.g., 5-25 µM for 1-2 hours) prior to harvesting to stabilize ubiquitinated proteins [21].
- Incorporate DUB Inhibitors: Add broad-spectrum DUB inhibitors (e.g., N-ethylmaleimide or PR-619) directly to the cell lysis buffer [2].
- Employ High-Affinity Binders: Utilize TUBEs, which have nanomolar affinity for polyubiquitin chains and can effectively shield them from DUB activity [18] [19].
- Increase Input Material: As a starting point, use 20 µL of agarose-TUBE beads or 100 µL of magnetic-TUBE slurry per milligram of cell extract [18].
Problem 3: Inability to Distinguish Between Linkage Types
- Potential Cause: The antibody or enrichment reagent has cross-reactivity with non-cognate ubiquitin linkages.
- Solution:
- Use Validated Linkage-Specific Reagents: Employ well-characterized tools like chain-selective TUBEs (e.g., K48-TUBE vs. K63-TUBE) or affimers that are designed for specific linkages [19] [20].
- Validate with Linkage-Defined Standards: Use well-defined ubiquitin chains (e.g., K48-only or K63-only Ub) in your experiments to confirm the specificity of your detection reagent [20].
- Combine Tools: Use a pan-specific TUBE for initial enrichment, followed by immunoblotting with a linkage-specific antibody to determine the chain type [21].
Problem 4: Smear Instead of Discrete Bands on Western Blot
- Potential Cause: This is often an expected result, as a heterogeneous mixture of ubiquitinated proteins and ubiquitin chains of varying lengths will appear as a smear.
- Solution:
- This is a Feature, Not a Bug: A smear indicates successful enrichment of a diverse population of polyubiquitinated proteins [21].
- Probe for Your Specific Target: After confirming ubiquitin enrichment, re-probe the membrane with an antibody against your protein of interest to identify a specific signal within the smear.
Frequently Asked Questions (FAQs)
Q1: What is the difference between a pan-TUBE and a linkage-specific TUBE? Pan-TUBEs (like TUBE1 and TUBE2) bind to all types of polyubiquitin linkages and are ideal for general enrichment and stabilization of ubiquitinated proteins. Linkage-specific TUBEs (e.g., for K48, K63, or M1) are engineered to bind with high affinity to only one specific linkage type, allowing you to investigate the function of that particular chain [18] [19].
Q2: My linkage-specific antibody isn't working in immunofluorescence. What can I do? Linkage-specific antibodies can sometimes perform differently between applications. Consider alternative reagent types that have been validated for imaging. For example, linkage-specific affimers and TUBEs conjugated to fluorophores (e.g., TAMRA, FITC) have been successfully used for confocal fluorescence microscopy [20].
Q3: How do I elute ubiquitinated proteins from TUBE beads for downstream analysis? For TUBE-based enrichments, it is recommended to use a proprietary elution buffer (e.g., LifeSensors Cat # UM411B) or a standard SDS-PAGE loading buffer for direct analysis by western blot. For subsequent mass spectrometry, gentle elution with a low-pH buffer or a solution of free ubiquitin peptide can be effective [18].
Q4: Can I use these tools to study atypical ubiquitin linkages like K6 or K27? Yes, the field is continuously evolving. While reagents for K48 and K63 are most common, linkage-specific tools for atypical chains are becoming available. For instance, K6-linkage-specific affimers have been developed and used to identify HUWE1 as a major E3 ligase for K6 chains [20]. However, commercial availability for all linkages may still be limited.
Experimental Workflow: Assessing Linkage-Specific Ubiquitination
The following diagram illustrates a robust protocol for using chain-specific TUBEs to analyze endogenous protein ubiquitination in a high-throughput format, such as a 96-well plate.
Workflow for TUBE-Based Ubiquitination Assay
Ubiquitin Signaling Pathways and Functional Outcomes
Understanding the functional context of different ubiquitin linkages is crucial for designing relevant experiments. The diagram below summarizes the primary cellular functions associated with the major ubiquitin chain types.
Primary Functions of Major Ubiquitin Linkages
Troubleshooting Guides
Probe Synthesis and Functionality
Problem: Low Yield or Failed Synthesis of Non-hydrolyzable Diubiquitin Probes
| Problem Area | Possible Cause | Solution | Relevant Citations |
|---|---|---|---|
| Chemical Ligation | Inefficient copper-catalyzed alkyne-azide cycloaddition (CuAAC) reaction. | Ensure proper removal of oxygen from the reaction mixture and use fresh catalysts. Confirm the integrity of azido-ornithine and propargylamide precursors [22]. | |
| Probe Assembly | Incorrect handling of C-terminal thioester intermediates during solid-phase peptide synthesis (SPPS). | Use chlorotrityl resin for SPPS and cleave the Ub1–75 precursor with 20% hexafluoro-isopropanol (HFIP) to expose the C-terminal carboxylic acid for proper activation [22]. | |
| Purification | Inadequate purification leading to side products. | Employ a two-step purification using reverse-phase high performance liquid chromatography (RP-HPLC) followed by size-exclusion chromatography (SEC) to isolate pure probe [22]. |
Problem: Synthesized Ubiquitin Variant Lacks Biological Activity
| Problem Area | Possible Cause | Solution | Relevant Citations |
|---|---|---|---|
| Structural Integrity | The synthetic protein is misfolded or the triazole linkage disrupts native Ub structure. | Refold the protein post-synthesis using standard Ub refolding protocols. Verify folding and stability via circular dichroism (CD) or NMR [23] [22]. | |
| Warhead Reactivity | The C-terminal warhead (e.g., propargylamide) is inactive. | Test warhead reactivity using a control reaction with a known, active DUB. Synthesize a small batch of probe with a fluorescent tag (e.g., TAMRA) to confirm successful labeling [22]. | |
| Linkage Specificity | The designed linkage does not mimic the native isopeptide bond for target interaction. | Validate the probe using a DUB with known linkage specificity (e.g., use K48-linked probe for a proteasome-associated DUB) as a positive control [22]. |
Specificity and Background
Problem: High Non-Specific Binding in Affinity Enrichment Experiments
| Problem Area | Possible Cause | Solution | Relevant Citations |
|---|---|---|---|
| Affinity Matrix | Non-specific interactions with the resin or tag (e.g., GST) used for the Ub variant. | Use a different immobilization chemistry or tag. Incorporate stringent wash steps with high salt (e.g., 300-500 mM NaCl) and non-ionic detergents before elution [23]. | |
| Cell Lysate | Endogenous ubiquitin and high-abundance proteins compete for binding. | Pre-clear lysate with bare resin/beads. Use cell lines treated with proteasome inhibitors (e.g., 5-25 µM MG-132 for 1-2 hours) to enrich for ubiquitinated proteins, but note potential cytotoxicity with overexposure [24]. | |
| Probe Concentration | The concentration of the immobilized Ub variant is too high, saturating specific sites and promoting off-target binding. | Titrate the amount of Ub variant conjugated to the beads. Use the minimal amount required for efficient pull-down to minimize non-specific interactions [23]. |
Problem: Probe Binds Non-Target Deubiquitylating Enzymes (DUBs)
| Problem Area | Possible Cause | Solution | Relevant Citations |
|---|---|---|---|
| Probe Design | The probe's warhead is too reactive and lacks selectivity. | Consider using a less reactive warhead or a full-length diUb probe that requires engagement of both S1 and S2 pockets for binding, which is specific to a smaller subset of DUBs [22]. | |
| Experimental Conditions | The reaction buffer or incubation time allows for non-specific reactivity. | Include control experiments with a probe lacking the warhead to identify non-covalent binders. Optimize incubation time and temperature to favor specific enzymatic turnover [22]. |
Detection and Analysis
Problem: Weak or No Signal in Mass Spectrometry After Enrichment
| Problem Area | Possible Cause | Solution | Relevant Citations |
|---|---|---|---|
| Sample Preparation | Inefficient elution of bound proteins from the affinity matrix. | Use a competitive elution with high concentrations (e.g., 0.5-1 M) of free ubiquitin or a low-pH elution buffer. For downstream MS, use protocols optimized for on-bead digestion to minimize sample loss [24]. | |
| Ubiquitinylation Level | The endogenous levels of the target ubiquitinated proteins are too low. | Amplify the ubiquitination signal by treating cells with a proteasome inhibitor like MG-132 prior to harvesting, which prevents the degradation of ubiquitinated proteins [24]. | |
| MS Sensitivity | The enriched ubiquitinated peptides are suppressed by more abundant non-modified peptides. | Use a tandem enrichment strategy, such as SCASP-PTM, which allows for the sequential enrichment of ubiquitinated, phosphorylated, and glycosylated peptides from a single sample without intermediate desalting, thereby reducing sample loss and complexity [11]. |
Frequently Asked Questions (FAQs)
Q1: What are the key advantages of using synthetic ubiquitin variants over enzymatically generated ones? Synthetic biology approaches, such as solid-phase peptide synthesis (SPPS) and native chemical ligation (NCL), provide atomic-level control. This allows for the generation of homogeneously modified Ub variants that are often difficult or impossible to produce enzymatically in high purity. You can incorporate non-hydrolyzable linkages (e.g., triazole), site-specific post-translational modifications (PTMs), non-canonical amino acids, and stable isotopic labels with precision, which is crucial for detailed functional and structural studies [23] [25].
Q2: When should I choose a non-hydrolyzable diubiquitin probe over a monomeric Ub probe? The choice depends on the DUB's mechanism. Use monomeric Ub probes (targeting the S1 pocket) to identify DUBs that cleave monoUb or the distal end of chains. Use non-hydrolyzable diUb probes when you need to study DUBs that require engagement of additional binding pockets. Probes with a warhead between two Ub units (S1-S1' targeting) are good for DUBs that disassemble chains, while probes with a warhead at the C-terminus of the proximal Ub (S1-S2 targeting) are essential for studying DUBs that cleave at the proximal end of a chain, such as some viral DUBs or editors like OTUD2/3 [22].
Q3: My ubiquitin enrichment shows a smear on a western blot. Is this normal? Yes, this is typically expected and often indicates a successful enrichment. A smear represents the heterogeneous mixture of monomeric ubiquitin, polyubiquitin chains of different lengths and linkages, and ubiquitinated proteins of varying molecular weights captured by your method (e.g., using a Ubiquitin-Trap). To distinguish specific linkages within the smear, you must follow up with linkage-specific ubiquitin antibodies during western blot analysis [24].
Q4: How can I confirm that my activity-based probe is functioning correctly? First, validate its reactivity with a positive control DUB known to be labeled by such probes (e.g., USP14 for diUb probes). Use fluorescently tagged probes (TAMRA) to visualize labeling by SDS-PAGE. For functional validation in a complex mixture, incubate the probe with cell lysate, followed by click chemistry addition of a biotin tag for enrichment and western blotting with streptavidin-HRP or avidin to confirm labeling of the expected protein targets [26] [22].
Q5: Can these chemical biology tools be applied to ubiquitin-like proteins (Ubls)? Absolutely. The same chemical synthesis principles have been successfully applied to study Ubls like SUMO, NEDD8, ISG15, and UFM1. These tools enable the generation of defined Ubl chains and Ubl-protein conjugates, which are equally challenging to obtain homogeneously through enzymatic methods. This allows for parallel exploration of the biology and crosstalk within the entire Ub/Ubl family [25].
Research Reagent Solutions
The following table details key reagents used in the synthesis and application of synthetic ubiquitin variants and non-hydrolyzable probes.
| Reagent Name | Function/Description | Key Application | Citation |
|---|---|---|---|
| Ubiquitin-Trap (Agarose/Magnetic) | Anti-Ubiquitin nanobody (VHH) coupled to beads for immunoprecipitation of monoUb, Ub chains, and ubiquitinated proteins. | Pull-down of ubiquitinated proteins from complex cell lysates for detection or MS analysis. [24] | |
| Non-hydrolyzable DiUb Probes (with PA warhead) | Synthetic diubiquitin linked via triazole with a C-terminal propargylamide warhead. Covalently traps DUBs engaging S1 and S2 pockets. | Identifying and characterizing linkage-specific DUB activity in lysates or with purified enzymes. [22] | |
| Ub-MES / UbFluor | Ubiquitin C-terminus conjugated to mercaptoethanesulfonate. Allows E3~Ub complex formation without E1/E2/ATP. Fluorogenic version for HTS. | High-throughput screening for inhibitors of HECT-family E3 ligases like PARKIN. [26] | |
| Azido-ornithine | A non-canonical amino acid incorporated during SPPS to provide an azide group for click chemistry. | Serves as the "anchor" point in the proximal Ub for copper-catalyzed cycloaddition to form triazole-linked chains. [22] | |
| Propargylamine (PA) | A warhead containing an alkyne group. | Used as the C-terminal reactive group on Ub to covalently modify the catalytic cysteine of DUBs. Also used in click chemistry. [22] |
Experimental Workflow Visualizations
Probe Synthesis Workflow
Affinity Enrichment Logic
DUB Profiling Pathway
Technical Support Center
Troubleshooting Guide: Ubiquitin Enrichment
Problem: High Background and Non-Specific Binding
| Potential Cause | Recommended Solution | Principle |
|---|---|---|
| Incomplete Lysate Clearing | Centrifuge lysate at 20,000g for 10 min at 4°C before enrichment [27]. | Removes particulate debris that can trap proteins non-specifically. |
| Insufficient Washing | Increase number of wash steps; use stringent wash buffers (e.g., containing 0.5% NP-40) [28]. | Removes weakly associated, non-specifically bound proteins. |
| Antibody Leakage (Agarose) | Use covalently cross-linked antibodies or magnetic bead-conjugated reagents [27]. | Prevents antibody heavy/light chains from leaching and appearing in MS samples. |
| Non-Optimal Bead Type | Switch to magnetic bead-based platforms (e.g., HS mag anti-K-ε-GG) for more efficient washing [27]. | Magnetic particle processors reduce handling and improve wash consistency. |
Problem: Low Yield of Ubiquitinated Proteins/Peptides
| Potential Cause | Recommended Solution | Principle |
|---|---|---|
| Rapid Deubiquitination | Add Deubiquitinase (DUB) inhibitors (e.g., 5-50 μM PR-619, 1-5 mM N-Ethylmaleimide) to lysis buffer [27] [28]. | Preserves the labile ubiquitin modification during sample preparation. |
| Proteasomal Degradation | Treat cells with proteasome inhibitors (e.g., 1-25 μM MG-132) for 1-2 hours before harvesting [29] [28]. | Stabilizes ubiquitinated proteins destined for degradation. |
| Insufficient Input Material | Use recommended input (e.g., 500 μg peptide for UbiFast); avoid over-dilution [27]. | Ensures target ubiquitinated species are above the detection limit. |
| Inefficient Elution | Use low-pH elution buffer or directly elute in SDS-PAGE loading buffer for western blot [29]. | Disrupts strong antibody-antigen or UBD-ubiquitin interactions. |
Problem: Inability to Detect Specific Ubiquitin Linkages
| Potential Cause | Recommended Solution | Principle |
|---|---|---|
| Using Pan-Specific Reagents | Employ linkage-specific tools (e.g., K48-TUBE, K63-TUBE) for targeted enrichment [12]. | Specific reagents selectively capture the ubiquitin topology of interest. |
| Lack of Downstream Specificity | Follow pan-specific enrichment with western blot using linkage-specific antibodies [29]. | Confirms the identity of the captured ubiquitin chain linkage. |
Frequently Asked Questions (FAQs)
Q1: What are the major advantages of automated magnetic bead systems over manual agarose-based methods?
Automation with magnetic beads offers significant performance improvements. A study comparing the automated UbiFast method using magnetic beads to the manual method reported a drastic reduction in processing time from a manual protocol to approximately 2 hours for a 10-plex sample, enabling processing of up to 96 samples in a single day. Furthermore, automation led to a major increase in reproducibility and significantly reduced variability across process replicates. Notably, the depth of coverage was also enhanced, with the automated method identifying approximately 20,000 ubiquitylation sites from a single experiment [27].
Q2: My goal is to profile ubiquitination sites by mass spectrometry. Should I enrich at the protein or peptide level?
For mass spectrometry-based ubiquitin site mapping (identifying the specific lysine residue modified), enrichment at the peptide level using K-ε-GG antibodies is the established method. Trypsin digestion cleaves ubiquitin, leaving a diagnostic di-glycine (GG) remnant on the modified lysine of the substrate peptide. Anti-K-ε-GG antibodies specifically enrich these peptides for LC-MS/MS analysis, allowing precise site identification [27] [30]. For studying protein-level interactions or complexes, protein-level enrichment with tools like TUBEs or Ubiquitin-Traps is more appropriate [29].
Q3: How can I specifically study K48-linked or K63-linked polyubiquitination in my sample?
You require tools with linkage specificity. Chain-specific Tandem Ubiquitin Binding Entities (TUBEs) are engineered for this purpose. For example:
- K48-TUBEs selectively capture proteins modified with K48-linked chains, which are primarily associated with proteasomal degradation [12].
- K63-TUBEs selectively capture proteins modified with K63-linked chains, which are involved in inflammatory signaling and other non-degradative functions [12].
A recent study demonstrated this by using K63-TUBEs to capture L18-MDP-induced K63 ubiquitination of RIPK2, while K48-TUBEs captured RIPK2 PROTAC-induced K48 ubiquitination [12].
Q4: What is the difference between TUBEs and the Ubiquitin-Trap?
Both are affinity tools, but they use different capture mechanisms:
- TUBEs (Tandem Ubiquitin Binding Entities): These are engineered proteins containing multiple ubiquitin-binding domains (UBDs) that bind non-covalently to polyubiquitin chains. They can protect chains from deubiquitination and some are available with linkage specificity (e.g., for K48 or K63) [12].
- Ubiquitin-Trap: This reagent uses an anti-ubiquitin nanobody (VHH) coupled to beads to immunoprecipitate ubiquitin and ubiquitinated proteins. It is linkage-independent and can capture monomeric ubiquitin, ubiquitin chains, and ubiquitinated proteins [29].
Q5: Can I study unanchored (free) polyubiquitin chains, and why are they important?
Yes, this requires specific tools. Unanchored polyubiquitin chains (not attached to a substrate protein) are biologically significant in processes like innate immune signaling and proteasome regulation. They can be specifically enriched using a Free Ubiquitin-Binding Entity (FUBE), such as the Znf-UBP domain from USP5, which has high specificity for the free C-terminus of ubiquitin [28]. These chains accumulate when the 26S proteasome is pharmacologically inhibited [28].
Quantitative Performance Comparison of Enrichment Methods
The following table summarizes key performance metrics from recent studies for different enrichment strategies.
Table 1: Performance Metrics of Ubiquitin Enrichment Methods
| Enrichment Method | Reported Scale / Throughput | Key Performance Metrics | Specificity Claims | Application Best Suited For |
|---|---|---|---|---|
| Automated UbiFast (magnetic beads) [27] | ~20,000 ubiquitylation sites from a TMT10-plex; 96 samples/day. | High reproducibility; ~2h processing for 10-plex; Reduced variability vs. manual. | K-ε-GG antibody for ubiquitin remnant motif (site-specific). | Deep-scale ubiquitin site mapping for large sample sets (e.g., PDX tissue). |
| Chain-Specific TUBEs [12] | Applied in 96-well plate HTS format for endogenous target ubiquitination. | Can differentiate context-dependent ubiquitination (e.g., K48 vs. K63 on RIPK2). | High affinity for specific polyubiquitin linkages (e.g., K48 or K63). | Investigating linkage-specific functions in signaling or PROTAC mechanism. |
| Ubiquitin-Trap (Nanobody) [29] | Validated for IP from human, mouse, hamster, dog, plant, and yeast cells. | Fast, easy pulldowns; stable under harsh washing conditions; low background. | Linkage-independent; binds monomeric ubiquitin, polymers, ubiquitinated proteins. | General ubiquitin immunoprecipitation and protein-level interaction studies. |
| Pierce Ubiquitin Enrichment Kit [7] [31] | Processes 1 to 15 samples concurrently. | <45 minutes hands-on time; compatible with standard cell lysis products. | Anti-ubiquitin affinity resin; purifies polyubiquitinated proteins. | Western blot analysis of polyubiquitinated proteins from cells and tissues. |
The Scientist's Toolkit: Key Research Reagent Solutions
Table 2: Essential Reagents for Ubiquitin Enrichment Research
| Reagent / Kit | Core Function | Brief Mechanism of Action |
|---|---|---|
| HS mag anti-K-ε-GG [27] | Peptide-level enrichment for site mapping. | Magnetic bead-conjugated antibody enriches tryptic peptides with di-glycine (GG) remnant on lysine. |
| TUBEs (Pan & Chain-Specific) [12] | Protein-level enrichment of polyubiquitin. | Engineered tandem ubiquitin-binding domains (UBDs) with high affinity for polyubiquitin chains. |
| Ubiquitin-Trap (Agarose/Magnetic) [29] | General-purpose ubiquitin immunoprecipitation. | Anti-ubiquitin nanobody (VHH) coupled to beads captures ubiquitin and ubiquitinated proteins. |
| FUBE (Znf-UBP domain) [28] | Specific isolation of unanchored polyubiquitin. | UBD from USP5 selectively binds the free C-terminus of unconjugated ubiquitin and unanchored chains. |
| DUB & Proteasome Inhibitors [27] [29] | Preservation of ubiquitin signals. | PR-619 (DUB inhibitor) and MG-132 (proteasome inhibitor) prevent loss of ubiquitination during processing. |
Experimental Workflow and Pathway Visualization
Ubiquitin Enrichment Workflow Selection
Linkage Specific Ubiquitin Signaling
The following diagram outlines the core BioE3 strategy, which uses proximity-dependent biotinylation to label and isolate ubiquitinated substrates of a specific E3 ligase with high specificity [32].
Step-by-Step Experimental Protocol
This protocol details the BioE3 method for identifying bona fide substrates of a specific E3 ubiquitin ligase, significantly reducing non-specific background [32].
Step 1: Cell Line Engineering and Preparation
- Generate stable cell line: Create a HEK293FT or U2OS cell line with a doxycycline (DOX)-inducible lentiviral vector expressing the
bioGEFUbconstruct. ThebioGEFtag is a mutated AviTag (WHE sequence mutated to GEF) with lower affinity for BirA, which is crucial for minimizing non-specific biotinylation [32]. - Culture in biotin-depleted media: Grow the stable cells in media supplemented with dialyzed, biotin-depleted serum for at least 24 hours prior to the experiment. This step is critical to deplete endogenous biotin and reduce background [32].
- Introduce BirA-E3 fusion: Transfect the cells with your plasmid encoding the
BirA-E3fusion protein. BirA can be fused to the N- or C-terminus of the E3 ligase, but N-terminal fusions are often used to avoid steric hindrance with the C-terminal RING domain [32].
Step 2: Induction, Labeling, and Substrate Capture
- Induce expression: Add doxycycline (DOX) to the culture medium for 24 hours. This simultaneously induces the expression of both
bioGEFUband theBirA-E3fusion protein [32]. - Pulse with biotin: Add exogenous biotin to the culture medium for a short, defined period (e.g., 2 hours). The limited time window ensures that only
bioGEFUbmolecules in very close proximity to theBirA-E3fusion—as they are being conjugated onto substrates—are biotinylated. This is the key step for achieving high specificity [32]. - Lyse cells and perform streptavidin pulldown: Harvest cells and lyse them under denaturing conditions (e.g., using RIPA buffer with 1% SDS) to disrupt non-covalent interactions. Dilute the lysate to reduce SDS concentration and incubate with streptavidin-coated beads. Perform stringent washing (e.g., with high-salt buffers and detergents) to remove non-specifically bound proteins [2] [32].
- On-bead digestion and LC-MS/MS analysis: After washing, digest the captured proteins directly on the beads with trypsin. Elute the peptides and analyze them by Liquid Chromatography with Tandem Mass Spectrometry (LC-MS/MS) to identify the enriched ubiquitinated substrates and their modification sites [32].
Frequently Asked Questions & Troubleshooting
Q1: I am getting high background and non-specific biotinylation in my negative controls. What could be the cause?
- Cause 1: Using the standard AviTag (bioWHE). The wild-type AviTag (bioWHE) has a very high affinity for BirA, leading to widespread, non-specific biotinylation regardless of proximity [32].
- Solution: Switch to the mutated
bioGEFtag, which has a lower affinity for BirA and is essential for proximity-dependent labeling [32].
- Solution: Switch to the mutated
- Cause 2: Inadequate depletion of endogenous biotin. Residual biotin in the growth medium can cause non-specific labeling.
- Solution: Ensure you use dialyzed, biotin-depleted serum and pre-culture cells in this medium for a sufficient time (≥24 hours) before biotin pulsing [32].
- Cause 3: The biotin pulse time is too long. Extended biotin exposure allows for non-proximal labeling events.
- Solution: Optimize and shorten the biotin pulse duration. A 2-hour pulse is a starting point; test shorter times (e.g., 0.5-1 hour) to minimize background [32].
Q2: My pulldown is not efficient, and I am failing to identify known substrates. How can I improve enrichment?
- Cause 1: The BirA-E3 fusion protein is not functional. The fusion may disrupt the native structure or activity of the E3 ligase [32].
- Solution: Validate the activity of your BirA-E3 fusion in a functional assay (e.g., auto-ubiquitination assay). Try fusing BirA to the alternative terminus of the E3 ligase [32].
- Cause 2: Stringency during pulldown is too high. Overly harsh washing conditions may elute weakly bound, genuine substrates.
- Solution: Titrate the stringency of wash buffers (e.g., salt concentration, detergent type). A step-wise increase in stringency can help balance specificity and yield [2].
- Cause 3: Ubiquitinated substrates are scarce or unstable. The stoichiometry of ubiquitination is often low, and substrates can be rapidly degraded or deubiquitinated [2].
- Solution: Treat cells with a proteasome inhibitor (e.g., MG132) for a few hours before lysis to stabilize ubiquitinated substrates. Consider using deubiquitinase (DUB) inhibitors like N-ethylmaleimide (NEM) or chloroacetamide (CAA) in your lysis buffer. Be aware that the choice of DUB inhibitor can affect the outcome of your pulldown, so this may require optimization [33].
Q3: Can this protocol be adapted for different E3 ligase types or ubiquitin-like proteins (UbLs)?
- Answer: Yes. The BioE3 method has been successfully demonstrated for both RING-type (e.g., RNF4, MIB1) and HECT-type (e.g., NEDD4) E3 ligases [32]. The core principle can also be extended to other UbLs, such as SUMO, by using
bioGEFSUMOand the appropriate E3 ligase fusion [32].
The Scientist's Toolkit: Key Research Reagent Solutions
The following table lists essential reagents for implementing the high-specificity BioE3 protocol.
| Item | Function & Description | Key Consideration for Specificity |
|---|---|---|
| bioGEFUb Plasmid | AviTagged Ubiquitin with GEF mutation for proximity-dependent biotinylation. | The GEF mutation is critical over the standard AviTag (bioWHE) to prevent non-specific labeling by reducing affinity for BirA [32]. |
| BirA-E3 Plasmid | Fusion of biotin ligase (BirA) to your E3 ligase of interest. | Fusion at the N-terminus is often preferred to avoid interfering with the catalytic RING or HECT domains [32]. |
| Streptavidin Beads | High-capacity, ultra-pure beads for capturing biotinylated proteins. | Essential for stringent pulldown; allows washing under denaturing conditions to remove non-specific interactors [2] [32]. |
| Dialyzed/Biotin-Depleted Serum | Serum with low-molecular-weight components (like biotin) removed. | Mandatory to lower endogenous biotin levels and minimize background during the biotin pulse [32]. |
| DUB Inhibitors (NEM/CAA) | Cysteine alkylators that inhibit deubiquitinating enzymes. | Preserves ubiquitin chains on substrates during lysis. Note: Choice of inhibitor (NEM vs. CAA) can affect which proteins bind to ubiquitin, so selection should be optimized [33]. |
| Linkage-Specific Affimers | Engineered binding proteins specific to certain Ub chain linkages (e.g., K6, K33). | An alternative to antibodies for downstream validation; useful for confirming chain topology on identified substrates via techniques like Western blot [34]. |
| Linkage-Specific DUBs (e.g., OTUB1, AMSH) | Enzymes that cleave specific ubiquitin linkages (K48, K63). | Used for validation (e.g., UbiCRest assay) to confirm the linkage type of ubiquitin chains enriched in your experiment [33]. |
Specificity Comparison: BioE3 vs. Traditional Methods
The following diagram contrasts the high-specificity BioE3 mechanism with traditional, lower-specificity affinity tag methods.
Troubleshooting Non-Specific Binding: Optimization Strategies for Complex Samples
In ubiquitin enrichment research, the success of experiments such as immunoprecipitation (IP) and pull-down assays critically depends on the composition of your buffers. Non-specific interactions can obscure results, leading to high background noise and false positives. Proper buffer optimization is not merely a technical step; it is a foundational requirement for generating clean, reliable, and interpretable data on the ubiquitinated proteome. This guide details the key components and methodologies for formulating buffers that minimize non-specific binding while preserving the labile ubiquitin modification.
Key Buffer Components and Their Roles
The following table summarizes the essential additives for optimizing buffers to reduce non-specific interactions in ubiquitin research.
Table 1: Key Buffer Components for Reducing Non-Specific Interactions
| Component | Recommended Concentration | Primary Function | Considerations & Tips |
|---|---|---|---|
| Salts (NaCl, KCl) | 150-500 mM | Reduces non-specific ionic interactions by shielding charges. | Start at 150 mM; higher stringency may require higher concentrations, which could disrupt weak specific interactions [35]. |
| Mild Detergents (e.g., NP-40, Triton X-100) | 0.1 - 1% | Disrupts hydrophobic interactions and solubilizes membranes without denaturing proteins [35]. | Compatible with most immunoaffinity purifications. Harsh detergents like SDS are generally used only in denaturing lysis for direct ubiquitin detection [36]. |
| Carrier Proteins (e.g., BSA) | 0.1 - 1% | Blocks non-specific binding sites on tubes, resin, and the support matrix itself [35]. | Add during antibody binding or sample incubation steps. Ensure it does not interfere with downstream mass spectrometry [35]. |
| Inert Polymers (e.g., PVP) | Varies | Acts as a blocking agent, particularly effective in plant protein extracts [37]. | |
| Protease & Phosphatase Inhibitors | As per manufacturer | Prevents general protein degradation, which can generate sticky protein fragments and increase background [35]. | Use broad-spectrum cocktails. |
| DUB Inhibitors (NEM, IAA) | 5 - 100 mM | Critical for ubiquitin work: Preserves the ubiquitination state by inhibiting deubiquitinases (DUBs) [36]. | NEM is often more effective and is preferred for mass spectrometry workflows [36]. Test concentrations >10 mM for optimal preservation [36]. |
Optimized Experimental Protocol for Ubiquitin Enrichment
This protocol is designed for the immunoaffinity purification of ubiquitinated proteins using an anti-ubiquitin antibody (e.g., FK2) or a ubiquitin-binding domain (UBD)-based trap, with steps integrated to minimize non-specific binding.
Step 1: Cell Lysis with Preservation
- Lyse cells in a modified RIPA or NP-40-based buffer containing the optimized components listed in Table 1.
- Crucially, include DUB inhibitors like 20-50 mM NEM or IAA in the lysis buffer to prevent deubiquitination during and after lysis [36].
- For studies focused on proteasomal degradation, include a proteasome inhibitor such as MG132 (e.g., 5-25 µM for 1-2 hours pre-treatment) to stabilize ubiquitinated substrates [36] [37].
- To preserve weak interactions, use low salt concentrations and mild detergents. For more stringent conditions that remove weakly associated proteins, increase salt concentration or include 0.5% sodium deoxycholate [35].
Step 2: Pre-Clearance (Optional but Recommended)
- Incubate the cell lysate with the bare affinity resin (e.g., protein G or A agarose/sepharose, magnetic beads) for 30-60 minutes at 4°C [35].
- Pellet the resin and collect the supernatant. This step removes proteins that non-specifically bind to the resin matrix.
Step 3: Immunoaffinity Purification
- Incubate the pre-cleared lysate with the antibody-bound resin. The choice of resin (e.g., agarose, sepharose, magnetic beads) impacts the surface area and level of non-specific associations [38] [35].
- Perform the incubation for 1-2 hours at 4°C with gentle agitation.
- Wash the resin extensively with the optimized lysis/wash buffer (3-5 times, 5 minutes each) to remove unbound and non-specifically bound proteins. For greater stringency, you may incrementally increase the salt concentration in the final washes [35].
Step 4: Elution and Analysis
- Elute the bound ubiquitinated proteins using a denaturing buffer suitable for your downstream analysis.
- For western blotting, elute by boiling in SDS-PAGE sample buffer.
- For mass spectrometry, elution can be performed with 2–6 M guanidine•HCl or 0.1 M glycine (pH 2.5-3.0), with immediate neutralization for the latter [38].
- Analyze the eluates by western blotting or mass spectrometry.
The following diagram illustrates the core workflow and the parallel strategy of buffer optimization that underpins it.
The Scientist's Toolkit: Essential Reagents
Table 2: Key Research Reagents for Ubiquitin Enrichment and Detection
| Reagent / Tool | Function | Example Use |
|---|---|---|
| DUB Inhibitors (NEM, IAA) | Alkylates active site cysteine of deubiquitinases to prevent ubiquitin chain hydrolysis [36]. | Added at 20-100 mM to lysis and IP buffers to preserve ubiquitin signals [36]. |
| Tandem-repeated UBDs (TUBEs) | High-affinity reagents to pull down polyubiquitinated proteins; can protect chains from DUBs and proteasomal degradation [36] [2]. | Used as an alternative to antibodies for ubiquitin enrichment; can be immobilized on beads [36] [2]. |
| Linkage-specific Ub Antibodies | Detect or immunoprecipitate a particular ubiquitin chain linkage (e.g., K48, K63, M1-linear) [2] [39]. | Confirm the presence and type of ubiquitin chain on a substrate of interest by western blot or IP [2]. |
| Proteasome Inhibitor (MG132) | Inhibits the 26S proteasome, preventing the degradation of polyubiquitinated proteins and aiding in their accumulation and detection [36] [37]. | Treat cells with 5-25 µM for several hours before lysis to stabilize K48-linked and other proteasome-targeted substrates [36]. |
| Anti-Ubiquitin Antibodies (e.g., FK2, P4D1) | Recognize mono- and polyubiquitin conjugates for western blotting, immunohistochemistry, and immunoprecipitation [2] [40]. | FK2 antibody bound to protein G-agarose used for proteomic analysis of the ubiquitinome [40]. |
Troubleshooting Guide & FAQs
FAQ 1: My western blots for ubiquitin show a high background smear. How can I make the specific ubiquitin signal clearer?
A high background smear often results from non-specific protein binding or incomplete blocking.
- Increase Wash Stringency: Add 0.1 - 0.5% of a mild detergent like Tween-20 to your wash buffers and increase the salt concentration (e.g., to 300-500 mM NaCl) [35].
- Optimize Blocking: Extend the blocking time or use a different blocking agent (e.g., 5% BSA or a commercial protein-free blocker) before adding your primary antibody.
- Pre-clear Lysate: As described in the protocol, pre-clearing your lysate with bare resin can significantly reduce non-specific background [35].
FAQ 2: My ubiquitin immunoprecipitation yields very little target protein. What could be the cause?
Low yield can be due to loss of the ubiquitin modification or inefficient capture.
- Check DUB Inhibition: The most common issue in ubiquitin IPs is inadequate DUB inhibition. Ensure fresh NEM or IAA (at least 20 mM, but test up to 50-100 mM) is added directly to the lysis buffer [36].
- Verify Antibody and Resin Capacity: Ensure you are using a sufficient amount of high-affinity antibody and that the resin is not overloaded with total protein.
- Use a Positive Control: Include a well-established positive control (e.g., a protein known to be ubiquitinated under your conditions) to validate your entire workflow.
FAQ 3: I see discrete bands instead of a ubiquitin smear in my western blot. Does this mean my result is incorrect?
Not necessarily. While a characteristic smear often indicates heterogeneous polyubiquitination, discrete bands can represent:
- Monoubiquitination: A single ubiquitin modification on a protein.
- Multi-monoubiquitination: Several lysines on the same protein each modified by a single ubiquitin.
- Short, Homogeneous Ubiquitin Chains: A substrate with a defined number of ubiquitins. To distinguish this from a non-specific band, perform the same experiment with a DUB inhibitor. True ubiquitin signals should be enhanced when DUBs are inhibited [36] [37].
Sample Preparation Techniques to Preserve Ubiquitination States
Protein ubiquitination is a crucial post-translational modification that regulates diverse cellular processes, including proteasomal degradation, DNA repair, and cell signaling [36] [41]. However, studying this modification presents significant challenges due to its transient nature, low stoichiometry, and the dynamic action of deubiquitinating enzymes (DUBs) that can rapidly reverse the modification [36] [42]. The stability of the ubiquitination state is highly vulnerable during sample preparation, where cellular lysis can artificially activate DUBs and proteases. This article provides a technical guide for preserving protein ubiquitination states during experimental procedures, with a specific focus on minimizing non-specific binding in enrichment protocols.
Technical FAQs and Troubleshooting Guides
FAQ 1: How can I prevent the loss of ubiquitination signals during cell lysis?
Answer: The key is to rapidly inactivate deubiquitinating enzymes (DUBs) the moment lysis occurs.
Recommended Action:
- Use Potent DUB Inhibitors: Supplement your lysis buffer with high concentrations of cysteine protease inhibitors. While 5-10 mM N-ethylmaleimide (NEM) or iodoacetamide (IAA) is common, some targets may require up to 50-100 mM for complete preservation of ubiquitin chains, particularly K63- and M1-linked chains [36].
- Include Chelating Agents: Add EDTA or EGTA to chelate metal ions and inhibit metalloproteinase-type DUBs [36].
- Employ Denaturing Conditions: For the most robust preservation, lyse cells directly in boiling buffer containing 1% SDS to instantly denature all enzymes [36]. For downstream applications like immunoprecipitation, the lysate can be diluted with a non-denaturing buffer to reduce the SDS concentration.
Troubleshooting Guide:
- Problem: Smear or signal for ubiquitinated proteins is weak or absent on western blot.
- Potential Cause 1: Inadequate DUB inhibition.
- Solution: Increase the concentration of NEM or IAA in your lysis buffer. Test different concentrations to find the optimum for your system. Consider using a combination of inhibitors.
- Potential Cause 2: Target proteins are degraded by the proteasome.
FAQ 2: What steps can I take to reduce non-specific binding during the enrichment of ubiquitinated proteins?
Answer: Non-specific binding is a major hurdle that can lead to high background and false positives. Mitigation requires a multi-faceted approach involving stringent buffer conditions and appropriate control experiments.
Recommended Action:
- Increase Wash Stringency: Perform washes with buffers containing high salt (e.g., 300-500 mM NaCl) and detergents like Triton X-100 (e.g., 0.5-1%) after the ubiquitin-binding step [6].
- Use Denaturing Pulldowns: When specifically isolating covalently ubiquitinated proteins, use fully denaturing conditions (e.g., 6 M Guanidine-HCl or 1% SDS) during the binding and wash steps. This disrupts non-covalent protein-protein interactions that are a major source of contaminating non-specific binders [6] [41].
- Include Control Reagents: Always run a parallel experiment using a control resin (e.g., empty bead slurry or a resin with an inactive mutant protein) to identify proteins that bind non-specifically to the matrix or apparatus [43].
Troubleshooting Guide:
- Problem: High background noise in western blot or mass spectrometry analysis after enrichment.
- Potential Cause 1: Non-covalent interactors are co-purifying.
- Solution: Switch from native to denaturing purification protocols. If using antibodies for enrichment, ensure lysates are clarified with high-speed centrifugation before incubation.
- Potential Cause 2: Endogenous biotinylated or histidine-rich proteins are contaminating the sample.
- Solution: This is a common issue with Strep-tag and His-tag purifications [41]. Use a control purification from cells not expressing the tagged ubiquitin to identify these contaminants. For His-tag purifications, include 10-20 mM imidazole in the lysis and wash buffers to compete with weak, non-specific binders.
FAQ 3: How do I choose the right ubiquitin enrichment method for my experiment?
Answer: The choice of enrichment method depends on your experimental goal, the need to study endogenous proteins, and the required specificity.
- Recommended Action: Refer to the table below to compare the advantages and disadvantages of common enrichment methodologies.
Table 1: Comparison of Ubiquitin Enrichment Methods
| Method | Principle | Advantages | Disadvantages | Best for Reducing Non-Specific Binding |
|---|---|---|---|---|
| Tagged Ubiquitin [41] | Expression of epitope-tagged Ub (e.g., His, HA, FLAG) in cells. | High affinity and specificity; works well under denaturing conditions. | Not suitable for clinical samples; overexpression may cause artifacts. | Use double-tag systems (e.g., His-Biotin) for tandem purification [43]. |
| Ubiquitin-Binding Domains (UBDs) [6] [41] | Use of high-affinity UBDs (e.g., OtUBD, TUBEs) to pull down ubiquitinated proteins. | Can work with endogenous ubiquitin; TUBEs offer strong DUB protection. | TUBEs may preferentially bind polyubiquitin over monoubiquitin [6]. | Optimize salt and detergent concentrations in wash buffers [6]. |
| Anti-Ubiquitin Antibodies [41] [44] | Immunoprecipitation using antibodies against ubiquitin or specific linkages. | Applicable to any sample, including tissues; linkage-specific antibodies available. | Can be expensive; potential for non-specific antibody binding. | Use linkage-specific antibodies to narrow targets and reduce background. |
| K-ε-GG Antibodies (DiGly) [45] [46] | Enrichment of tryptic peptides with a diGly remnant left on modified lysines. | Directly identifies modification sites; high specificity proteome-wide. | Only applicable for mass spectrometry; requires protein digestion. | Excellent specificity as it targets a unique, defined chemical motif. |
FAQ 4: What are the best practices for sample preparation for mass spectrometry-based ubiquitinomics?
Answer: Recent advances have significantly improved the depth and precision of ubiquitinome profiling via mass spectrometry (MS).
Recommended Action:
- Optimized Lysis for MS: Use a lysis buffer containing 1% Sodium Deoxycholate (SDC) and alkylate proteins immediately with Chloroacetamide (CAA) during extraction. This combination has been shown to inactivate DUBs effectively and increase ubiquitin site coverage by over 38% compared to traditional urea buffers, without inducing di-carbamidomethylation artifacts that can mimic diGly modifications [46].
- High-Input Protein: For deep ubiquitinome analysis, start with a high protein input (e.g., 2-4 mg) to compensate for the low stoichiometry of ubiquitination [46].
- Advanced MS Acquisition: Utilize Data-Independent Acquisition (DIA) mass spectrometry coupled with neural network-based data processing (e.g., DIA-NN software). This method has been shown to more than triple the number of quantified ubiquitinated peptides compared to standard Data-Dependent Acquisition (DDA), while greatly improving reproducibility and quantitative accuracy [46].
Troubleshooting Guide:
- Problem: Low yields of diGly peptides in MS analysis.
- Potential Cause: Incomplete tryptic digestion or inefficient peptide enrichment.
- Solution: Ensure thorough protein denaturation, reduction, and alkylation. Use high-quality, specific antibodies for the diGly immunoaffinity enrichment step. Consider offline high-pH reverse-phase fractionation of peptides prior to enrichment to reduce sample complexity [45].
Essential Reagents and Tools
Table 2: The Scientist's Toolkit: Key Reagents for Ubiquitination Studies
| Reagent / Tool | Function | Key Considerations |
|---|---|---|
| N-Ethylmaleimide (NEM) [36] [6] | Alkylating agent that inhibits cysteine-based DUBs. | More stable than IAA. Preferred for mass spectrometry to avoid adducts that mimic Gly-Gly remnants [36]. |
| MG-132 / Bortezomib [36] [46] | Proteasome inhibitors. | Prevent degradation of polyubiquitinated proteins, enhancing their detection. Use at optimized concentrations and durations to avoid cellular stress responses. |
| Tandem Ubiquitin-Binding Entities (TUBEs) [36] [41] | Engineered molecules with high affinity for polyubiquitin chains. | Protect ubiquitin chains from DUBs during lysis and purification. May have lower affinity for monoubiquitinated proteins [6] [41]. |
| OtUBD Affinity Resin [6] | High-affinity ubiquitin-binding domain from O. tsutsugamushi for protein enrichment. | Effective for both mono- and polyubiquitinated proteins under both native and denaturing conditions. |
| DiGly (K-ε-GG) Antibodies [45] [46] [44] | Immunoaffinity purification of ubiquitin remnant peptides for MS. | Enables proteome-wide mapping of ubiquitination sites. High specificity but requires digested samples. |
| Sodium Deoxycholate (SDC) [46] | Lysis detergent for MS sample preparation. | Boosts ubiquitin site coverage and reproducibility; precipitates in acid for easy removal from peptides. |
Visual Experimental Workflows
Diagram 1: Optimized Workflow for Preserving Ubiquitination States
This diagram outlines a generalized and optimized workflow for sample preparation, highlighting critical steps for preserving ubiquitination and minimizing non-specific binding.
Diagram 2: Decision Pathway for Ubiquitin Enrichment Method Selection
This chart helps researchers select the most appropriate enrichment strategy based on their specific experimental requirements.
Frequently Asked Questions (FAQs)
1. What is the primary consequence of insufficient washing in ubiquitin enrichment protocols? Insufficient washing is a major source of high background and poor specificity. It fails to remove non-specifically bound proteins and unbound reagents, which can mask the detection of true ubiquitination signals and lead to erroneous conclusions [36] [47] [48].
2. How can I improve washing efficiency to reduce background? Beyond increasing the number of wash cycles, incorporating a 30-second soak step between washes can significantly enhance the removal of non-specifically bound material. Always ensure the plate or beads are drained thoroughly after each wash [47] [48].
3. My ubiquitination signal is weak after stringent washing. What could be wrong? A weak signal can result from excessive stringency (e.g., detergent concentration too high or wash volume too large) which elutes the target ubiquitinated proteins. It can also occur if deubiquitinase (DUB) activity was not properly controlled during cell lysis, degrading the target epitopes before enrichment [36].
4. How does the choice of solid support (e.g., magnetic beads vs. agarose) affect washing? Magnetic beads allow for rapid separation and more consistent washing with less physical disruption, which improves reproducibility and reduces sample loss. Automated processing of magnetic beads can standardize wash conditions, significantly cutting down variability across replicates [27].
5. Why is my assay reproducibility poor even when I follow the protocol? Poor assay-to-assay reproducibility is often linked to inconsistent washing (e.g., variable soak times, wash volumes, or draining between experiments) or fluctuations in incubation temperatures. Adhering to a strict, documented washing protocol and using fresh buffers for each run is crucial [48].
Troubleshooting Guide
| Problem | Possible Cause | Recommended Solution |
|---|---|---|
| High Background | Insufficient washing; non-specific binding [47] [48]. | Increase wash number; add 30-second soak steps; use fresh, correctly formulated wash buffers [36] [47]. |
| Weak or No Signal | Overly stringent washes; target elution; inactive DUB inhibitors [36] [48]. | Titrate down detergent concentration in wash buffer; verify freshness and concentration of DUB inhibitors like NEM (up to 50-100 mM may be needed) [36]. |
| Poor Replicate Data | Inconsistent manual washing; uneven bead handling [27] [48]. | Switch to automated washing systems; standardize soak and drain times; use magnetic beads for more consistent processing [27]. |
| Low Ubiquitin Yield | Washing stringency too high; epitope damage [36]. | Optimize wash buffer composition; avoid harsh denaturants unless required; use linkage-specific tools (e.g., ThUBD) for higher affinity capture [16]. |
Quantitative Wash Stringency Data
The table below summarizes key parameters from published protocols that successfully balance yield and specificity.
| Application / Method | Critical Wash Buffer Component | Stringency Impact & Quantitative Outcome | Reference |
|---|---|---|---|
| Automated UbiFast (magnetic beads) | Not specified, but processed on magnetic particle processor | Result: ~20,000 ubiquitylation sites identified from 500 μg input per sample with high reproducibility. Key: Automated, standardized washing reduced variability versus manual method [27]. | |
| ThUBD-Coated 96-Well Plates | Optimized washing buffers (details proprietary) | Performance: 16-fold wider linear range for capturing polyubiquitinated proteins compared to TUBE-based plates. Key: High-affinity ThUBD allows for effective washing without significant signal loss [16]. | |
| General Ubiquitin Immunoblotting | High-concentration N-ethylmaleimide (NEM) | Recommendation: Use up to 50-100 mM NEM in lysis/wash buffers to alkylate DUBs. Impact: Preserves ubiquitin chains during processing, preventing false-negative results from deubiquitination [36]. |
Experimental Protocols for Optimized Washing
Protocol 1: Standardized Washing for Magnetic Bead-Based Ubiquitin Enrichment
This protocol is adapted from the automated UbiFast method for high-throughput ubiquitylation profiling [27].
- Binding: After allowing your ubiquitinated lysate to bind to the HS mag anti-K-ε-GG antibody beads, place the tube on a magnetic separator until the solution clears.
- First Wash (Low Stringency): Carefully remove and discard the supernatant. Add 1 mL of a mild wash buffer (e.g., PBS-based). Gently resuspend the beads by flicking the tube or slow pipetting. Place on the magnetic separator, wait for clearance, and discard the supernatant.
- Subsequent Washes (Higher Stringency): Perform 3-4 additional washes with 1 mL of a stringent wash buffer (e.g., PBS containing 0.1% Triton X-100 or 500 mM NaCl). For each wash, resuspend the beads thoroughly and allow a 30-second soak on the bench before magnetic separation and supernatant removal.
- Final Rinse: Perform a final wash with 1 mL of a volatile buffer (e.g., 50 mM ammonium bicarbonate) to prepare for downstream mass spectrometric analysis.
- Elution: Elute the ubiquitinated peptides from the beads using a low-pH elution buffer (e.g., 0.1-0.5% TFA) or directly by on-bead digestion.
Protocol 2: Preventing Deubiquitination During Lysis and Washing
A critical aspect of "washing" is preserving the ubiquitinated state of proteins throughout the process [36].
- Prepare Lysis/Wash Buffer with DUB Inhibitors:
- To your standard lysis and wash buffers, add N-ethylmaleimide (NEM) to a final concentration of 50-100 mM. NEM is preferred over IAA for mass spectrometry applications as its adduct does not interfere with di-glycine remnant identification [36].
- Include 5-10 mM EDTA or EGTA to chelate metal ions required by metalloproteinase DUBs.
- Consider adding a broad-spectrum DUB inhibitor like PR-619 (e.g., 50 μM) [27].
- Lysis: Lyse cells or tissue directly in the prepared, chilled lysis buffer. For maximum protection, consider direct lysis into boiling SDS-containing buffer to instantly denature and inactivate DUBs.
- Wash: Use the inhibitor-supplemented buffers for all subsequent wash steps during immunoprecipitation or enrichment to maintain the ubiquitination state.
Research Reagent Solutions
The following reagents are essential for successful and specific ubiquitination enrichment.
| Reagent / Tool | Function in Ubiquitin Enrichment | Application Note |
|---|---|---|
| HS mag anti-K-ε-GG Antibody | Magnetic bead-conjugated antibody for enriching peptides with the di-glycine (K-ε-GG) ubiquitin remnant. | Enables automation, reduces hands-on time, and improves reproducibility. Ideal for high-throughput studies [27]. |
| Tandem Hybrid UBD (ThUBD) | Engineered ubiquitin-binding domain with high affinity and minimal linkage bias for capturing polyubiquitinated proteins. | Coated on 96-well plates for high-throughput, sensitive detection. Shows 16x better linear range than TUBEs [16]. |
| N-Ethylmaleimide (NEM) | Alkylating agent that inhibits cysteine-based deubiquitinases (DUBs) by modifying active site cysteines. | Critical for preserving ubiquitin chains. Use at 50-100 mM in lysis and wash buffers [36]. |
| PROTAC Assay Plates | Commercial 96-well plates pre-coated with ubiquitin-binding entities (e.g., TUBEs) for high-throughput screening. | Useful for monitoring target protein ubiquitination status in drug discovery [16]. |
The Relationship Between Washing Stringency and Experimental Outcomes
The diagram below illustrates the critical balance in washing stringency and its consequences.
Addressing Dynamic Range Challenges in Complex Biological Matrices
In ubiquitin enrichment research, the broad dynamic range of protein abundance in complex biological samples presents a significant analytical challenge. The target ubiquitinated proteins are often of low abundance and can be obscured by high-abundance non-specific binders, leading to reduced sensitivity and accuracy. This guide provides targeted strategies to minimize non-specific binding, thereby improving the specific isolation of ubiquitinated proteins for downstream analysis.
Troubleshooting Guides & FAQs
Why is my ubiquitin enrichment yield low despite a strong initial signal?
Low yield often results from non-specific binding depleting your sample or the ubiquitination signal being lost during processing.
- Solution: Implement a multi-faceted approach to protect the signal and reduce interference.
- Use Proteasome Inhibitors: Treat cells with inhibitors like MG-132 (e.g., 5-25 µM for 1–2 hours before harvesting) to prevent the degradation of ubiquitinated proteins [49].
- Employ High-Affinity Capture Reagents: Switch to tools with demonstrated high affinity and low linkage bias, such as Tandem Hybrid Ubiquitin Binding Domains (ThUBDs), which show a 16-fold wider linear range for capturing polyubiquitinated proteins compared to older methods like TUBEs [16].
- Optimize Lysis Conditions: Use lysis buffers that maintain protein complexes while minimizing non-specific interactions. Include a brief, cold sonication step or use freeze-thaw cycles to improve lysate clarity without promoting aggregation [50].
How can I reduce non-specific background in my pull-down experiments?
High background is frequently caused by non-specific proteins binding to the solid support or the capture reagent itself.
- Solution: Enhance the stringency of your wash steps and the specificity of your capture matrix.
- Increase Wash Stringency: Incorporate mild detergents (e.g., 0.1% Tween-20) and slightly elevated salt concentrations (e.g., 300-500 mM NaCl) into your wash buffers to disrupt weak, non-specific interactions without eluting the target complexes [49].
- Include Specific Competitors: Add a non-biotinylated version of the ubiquitin variant or an inert protein like BSA (1-2 mg/mL) during the binding step to block non-specific sites on the affinity matrix [23].
- Pre-clear the Lysate: Pre-incubate your cell lysate with bare beads (without the immobilized capture reagent) to remove proteins that bind non-specifically to the matrix material.
My western blot shows a smear, but MS identification is poor. What is wrong?
A smear confirms successful enrichment of polyubiquitinated proteins, but poor MS results indicate co-elution of non-specific binders or insufficient sample for detection.
- Solution: Focus on improving sample purity and compatibility with mass spectrometry.
- On-Bead Digestion: For MS analysis, perform tryptic digestion directly on the beads after thorough washing. This avoids elution inefficiencies and minimizes sample loss [49].
- Tandem Enrichment: Consider a two-step purification strategy. For example, perform an initial enrichment with a general ubiquitin trap, elute, and then subject the eluate to a second, more specific enrichment (e.g., using linkage-specific antibodies or diUb probes) [23].
- Validate with Controls: Always include negative controls, such as beads with a scrambled-scFv or GST tag alone, to identify proteins that bind non-specifically. Subtract these background proteins from your final list of putative interactors [23].
Key Reagents & Materials
The selection of appropriate reagents is critical for successful and specific ubiquitin enrichment. The table below summarizes key solutions.
Table 1: Essential Research Reagents for Ubiquitin Enrichment
| Reagent / Tool | Function & Mechanism | Key Features & Applications |
|---|---|---|
| ThUBD-coated Plates [16] | High-throughput capture of polyubiquitinated proteins from complex proteomes via a Tandem Hybrid Ubiquitin Binding Domain. | Unbiased recognition of all ubiquitin chain types; 16-fold wider linear range than TUBEs; ideal for high-throughput screening and PROTAC development. |
| Ubiquitin-Trap (Agarose/Magnetic) [49] | Immunoprecipitation of mono- and polyubiquitinated proteins using a anti-ubiquitin nanobody (VHH) coupled to beads. | High affinity; low background; suitable for pull-downs from a wide range of organisms (mammalian, plant, yeast). |
| Chemically Synthesized Ubiquitin Variants [23] | Serve as defined affinity baits (e.g., for AE-MS) to study interactions with specific ubiquitin chain linkages or modifications. | Generated via click chemistry or SPPS; incorporates non-hydrolyzable bonds (e.g., triazole) resistant to DUB activity in lysates. |
| Proteasome Inhibitor (MG-132) [49] | Preserves ubiquitination signals in cell lysates by inhibiting the 26S proteasome, preventing the degradation of ubiquitinated proteins. | Essential for stabilizing low-abundance ubiquitinated targets before enrichment; used during cell harvesting. |
Experimental Workflow for High-Fidelity Enrichment
The following diagram outlines a robust workflow designed to maximize specific ubiquitin enrichment while minimizing non-specific binding.
Detailed Protocol Steps
Cell Treatment and Lysis:
- Culture HEK293T cells (or your cell line of choice) to 70-80% confluency.
- Add MG-132 proteasome inhibitor to a final concentration of 5-25 µM and incubate for 1-2 hours before harvesting [49].
- Lyse cells using a suitable ice-cold lysis buffer (e.g., RIPA buffer) supplemented with protease and deubiquitinase inhibitors. Use freeze-thaw cycles or brief sonication on ice to ensure complete lysis [50].
Lysate Pre-clearing:
- Centrifuge the lysate at 12,000-16,000 x g for 15 minutes at 4°C to remove insoluble debris.
- Transfer the supernatant to a new tube and incubate with bare agarose or magnetic beads (without the ubiquitin-binding protein) for 30-60 minutes at 4°C with gentle agitation.
- Pellet the beads and collect the pre-cleared supernatant for the enrichment step.
Affinity Enrichment:
- Incubate the pre-cleared lysate with the selected affinity matrix (e.g., ThUBD-coated plates [16] or Ubiquitin-Trap magnetic agarose [49]) according to the manufacturer's instructions. Typically, use 1.03 µg of ThUBD per well of a 96-well plate for high-capacity binding [16].
- Perform the binding for 1-2 hours at 4°C with gentle agitation.
Stringent Washing:
- Pellet the beads or empty the wells of the plate.
- Wash the matrix 3-4 times with 1 mL of wash buffer (e.g., PBS or Tris-buffered saline containing 0.1% Tween-20 and 300-500 mM NaCl) to remove non-specifically bound proteins [49].
Elution and Downstream Analysis:
- Elute the bound ubiquitinated proteins using an appropriate method, such as Laemmli sample buffer for Western blotting or a low-pH glycine buffer, followed by neutralization.
- For mass spectrometry, perform on-bead digestion to maximize protein recovery and identification [49].
- Proceed with your chosen analytical method, such as Western blotting, MS/MS analysis, or ELISA.
Performance Data of Enrichment Tools
Selecting the right enrichment tool is paramount. The following table provides a quantitative comparison of common platforms based on recent literature.
Table 2: Quantitative Comparison of Ubiquitin Enrichment Tools
| Enrichment Tool | Affinity Ligand | Detection Sensitivity | Dynamic Range | Linkage Bias | Primary Application |
|---|---|---|---|---|---|
| ThUBD-coated Plates [16] | Tandem Hybrid UBD | As low as 0.625 μg | 16-fold wider than TUBEs | Unbiased | High-throughput screening, PROTAC development |
| TUBE-coated Plates [16] | Tandem Ubiquitin Binding Entity | Lower than ThUBD | Limited | Biased towards specific chains | General ubiquitin enrichment |
| Click Chemistry-Generated DiUb [23] | Synthetic Ubiquitin Variant | High (for defined chains) | N/A | Defined by synthesis | AE-MS for specific linkage interactions |
| Ubiquitin-Trap Agarose [49] | Anti-Ubiquitin Nanobody (VHH) | High | Broad | Unbiased | Standard IP from various species |
The Ubiquitin Code and Recognition
Understanding the "Ubiquitin Code" is essential for interpreting enrichment results. Different ubiquitin chain linkages can trigger distinct cellular signals. The diagram below illustrates this concept and how specific tools interact with the code.
Cross-Validation Approaches to Confirm Target Specificity
Protein ubiquitination, the covalent attachment of a 76-amino acid ubiquitin protein to substrate proteins, represents a crucial post-translational modification regulating diverse cellular functions including protein degradation, signal transduction, and cell division [30] [2]. The versatility of ubiquitination arises from its complexity—ranging from single ubiquitin monomers to polyubiquitin chains with different linkage types (Lys-6, Lys-11, Lys-27, Lys-29, Lys-33, Lys-48, and Lys-63) that determine functional outcomes [30] [2]. Within pharmaceutical research and drug development, mapping ubiquitination sites with high specificity is essential for understanding disease mechanisms and developing targeted therapies.
A significant challenge in ubiquitin research involves the low stoichiometry of protein ubiquitination under normal physiological conditions and the potential for non-specific binding during enrichment procedures [2]. The multiplicity of ubiquitin ligases acting on individual substrates, combined with the editing function of deubiquitinating enzymes, further complicates precise target identification [30]. Cross-validation approaches have therefore become indispensable for confirming target specificity and reducing false positives in ubiquitin enrichment research. This technical support center provides comprehensive guidance on methodologies, troubleshooting, and experimental design to enhance specificity in ubiquitination studies.
Key Methodologies for Ubiquitin Enrichment
Ubiquitin Tagging-Based Approaches
Ubiquitin tagging methodologies utilize affinity tags such as 6× His-tag or Strep-tag genetically fused to ubiquitin, enabling purification of ubiquitinated substrates from cellular lysates.
Experimental Protocol (His-Tagged Ubiquitin Pull-Down):
- Cell Line Preparation: Generate stable cell lines expressing 6× His-tagged ubiquitin using lentiviral transduction or stable transfection methods.
- Lysis: Harvest cells and lyse using denaturing buffer (e.g., 6 M guanidine-HCl, 0.1 M Na₂HPO₄/NaH₂PO₄, 10 mM imidazole, pH 8.0) to preserve ubiquitination status and inhibit deubiquitinating enzymes.
- Immobilized Metal Affinity Chromatography (IMAC): Incubate lysate with Ni-NTA agarose beads for 2-4 hours at room temperature with gentle rotation.
- Washing: Perform sequential washes with:
- Wash Buffer 1: 6 M guanidine-HCl, 0.1 M Na₂HPO₄/NaH₂PO₄, 10 mM imidazole, pH 8.0
- Wash Buffer 2: 8 M urea, 0.1 M Na₂HPO₄/NaH₂PO₄, 10 mM imidazole, pH 8.0
- Wash Buffer 3: 8 M urea, 0.1 M Na₂HPO₄/NaH₂PO₄, 10 mM imidazole, 0.1% Triton X-100, pH 6.3
- Elution: Elute ubiquitinated proteins with 200-300 mM imidazole or low-pH buffer (e.g., 0.2 M acetic acid, pH 3.0).
- Analysis: Process eluates for SDS-PAGE separation or tryptic digestion for mass spectrometry analysis [2] [51].
Advantages and Limitations: This approach enables screening of ubiquitinated substrates in cells with relatively low cost. However, histidine-rich and endogenously biotinylated proteins can co-purify, while tagged ubiquitin may not completely mimic endogenous ubiquitin, potentially generating artifacts [2].
Antibody-Based Enrichment Approaches
Antibody-based methods utilize ubiquitin-specific antibodies for immunoprecipitation, applicable to both endogenous studies and tissue samples without genetic manipulation.
Experimental Protocol (Anti-Ubiquitin Immunoprecipitation):
- Sample Preparation: Lyse cells or tissue in non-denaturing RIPA buffer (or denaturing buffer if needed) supplemented with protease inhibitors and deubiquitinating enzyme inhibitors (e.g., N-ethylmaleimide).
- Antibody Binding: Incubate cleared lysate with anti-ubiquitin antibody (e.g., P4D1, FK1/FK2 for pan-ubiquitin, or linkage-specific antibodies) for 2-4 hours at 4°C.
- Bead Capture: Add protein A/G agarose or magnetic beads and incubate for an additional 1-2 hours.
- Washing: Wash beads 3-5 times with ice-cold lysis buffer.
- Elution: Elute ubiquitinated proteins with low-pH glycine buffer (pH 2.0-2.5) or Laemmli sample buffer for direct Western blot analysis.
- Analysis: Process for Western blotting or mass spectrometry [2].
Advantages and Limitations: This method identifies protein ubiquitination under physiological conditions and can provide chain-linkage information when using linkage-specific antibodies. Limitations include high antibody costs and potential non-specific binding [2].
Diglycine (K-ε-GG) Peptide Immunoaffinity Enrichment
This method exploits the tryptic digestion signature of ubiquitinated proteins, where the C-terminal diglycine remnant of ubiquitin remains attached to the modified lysine residue of substrates.
Experimental Protocol (K-ε-GG Peptide Immunoaffinity Enrichment):
- Protein Digestion: Denature and digest enriched ubiquitinated proteins or whole cell lysates with trypsin or ArgC protease.
- Peptide Immunoprecipitation: Incubate digested peptides with anti-K-ε-GG antibody-conjugated beads for 2 hours at 4°C.
- Washing: Wash beads with PBS or Tris-buffered saline to remove non-specifically bound peptides.
- Elution: Elute K-ε-GG peptides with 0.1-0.2% trifluoroacetic acid or 0.1% formic acid.
- Desalting: Desalt peptides using C18 StageTips or solid-phase extraction cartridges.
- LC-MS/MS Analysis: Analyze peptides by liquid chromatography tandem mass spectrometry for ubiquitination site identification and quantification [30] [52].
Advantages and Limitations: This approach offers high sensitivity and specificity for direct ubiquitination site mapping, with reduced background from non-ubiquitinated proteins. However, it requires efficient tryptic digestion and may miss ubiquitination on non-lysine residues [30].
Cross-Validation Strategies
Employing orthogonal methodologies provides robust validation of ubiquitination targets and sites, significantly enhancing experimental specificity.
Multi-Level Enrichment Cross-Validation
Combining protein-level and peptide-level enrichment strategies provides complementary validation, as demonstrated in a study investigating HRD1 ubiquitin ligase substrates.
Experimental Design: Researchers applied both protein-level (His-tagged ubiquitin enrichment) and peptide-level (K-ε-GG immunoaffinity) approaches in the presence and absence of HRD1. The protein-level method identified and quantified over 400 ubiquitinated proteins, while peptide immunoprecipitation identified over 1,800 ubiquitinated peptides on more than 900 proteins. Several proteins emerged as sensitive to HRD1 levels in both approaches, providing strong cross-validation [51].
Implementation Strategy:
- Perform parallel enrichments using both protein-level (e.g., tagged ubiquitin pull-down) and peptide-level (K-ε-GG immunoaffinity) methods from the same biological samples.
- Identify candidate ubiquitinated substrates/sites from each dataset.
- Compare overlapping targets between methodologies, prioritizing those identified by both approaches for further validation.
Multi-Level Enrichment Cross-Validation Workflow
Genetic Validation Approaches
Combining ubiquitin enrichment with genetic manipulation of ubiquitin pathway components provides powerful mechanistic validation.
E3 Ligase Modulation: Manipulate expression of specific E3 ligases (overexpression or knockdown) and monitor changes in candidate substrate ubiquitination. True substrates should show corresponding increases or decreases in ubiquitination levels [30] [2].
Deubiquitinase (DUB) Co-Expression: Co-express candidate substrates with relevant deubiquitinases. Authentic ubiquitination should be reduced by active DUBs but not by catalytically inactive mutants [2].
Lysine Mutagenesis: Mutate identified ubiquitination sites (lysine to arginine) in substrate proteins and assess ubiquitination status. True sites should show reduced ubiquitination, though functional redundancy of adjacent lysines can complicate interpretation [30].
Quantitative Proteomics for Dynamic Validation
Incorporating quantitative mass spectrometry approaches enables monitoring of ubiquitination dynamics in response to biological perturbations.
Stable Isotope Labeling with Amino Acids in Cell Culture (SILAC) Protocol:
- Culture cells in "light" (L-arginine and L-lysine), "medium" (13C6-arginine and D4-lysine), and "heavy" (13C615N4-arginine and 13C615N2-lysine) media.
- Treat cells with different conditions (e.g., proteasome inhibition, pathway activation, or genetic manipulation).
- Combine cell lysates in 1:1:1 ratio.
- Perform ubiquitin enrichment using preferred methodology.
- Analyze by LC-MS/MS and quantify ubiquitination changes based on isotopic ratios [30] [51].
Label-Free Quantification: Compare ubiquitination levels across multiple samples using spectral counting or peak intensity-based methods, with statistical analysis to identify significant changes.
Troubleshooting Guide
Low Ubiquitinated Peptide Recovery
- Problem: Low yield of ubiquitinated peptides after K-ε-GG immunoaffinity enrichment.
- Possible Causes:
- Incomplete tryptic digestion
- Insufficient antibody binding capacity
- Sample complexity overwhelming enrichment capacity
- Ineffective elution conditions
- Solutions:
- Optimize trypsin-to-protein ratio (1:20-1:50) and digestion time (6-18 hours)
- Include control peptides to assess enrichment efficiency
- Pre-fractionate samples at protein or peptide level before enrichment
- Test alternative elution buffers (0.1% TFA, 0.1% formic acid, or low-pH glycine)
High Background in Tag-Based Enrichments
- Problem: Excessive non-specific binding in His-tagged ubiquitin pull-down experiments.
- Possible Causes:
- Incomplete washing steps
- Co-purification of endogenous histidine-rich proteins
- Non-specific binding to resin matrix
- Solutions:
- Include imidazole (10-20 mM) in binding and wash buffers to reduce non-specific binding
- Increase stringency of wash buffers (e.g., include 0.1% Triton X-100 or increase urea concentration)
- Use competing agents such as 5-10 mM β-mercaptoethanol to reduce non-specific interactions
- Consider alternative tags (Strep-tag) or tandem purification approaches
Inconsistent Results Between Methodologies
- Problem: Poor overlap between targets identified by different enrichment strategies.
- Possible Causes:
- Method-specific biases in ubiquitin chain type enrichment
- Differences in substrate solubility under various lysis conditions
- Variable efficiency for monoubiquitination versus polyubiquitination
- Solutions:
- Use common lysis buffers when possible across methodologies
- Include linkage-specific antibodies to characterize chain types in each approach
- Analyze specific model substrates with known ubiquitination patterns as controls
- Focus quantitative comparisons on well-detected peptides/proteins
Research Reagent Solutions
*Table: Essential Research Reagents for Ubiquitin Enrichment Studies*| Reagent Category | Specific Examples | Function & Application Notes |
|---|---|---|
| Affinity Tags | 6× His-tag, Strep-tag II, HA-tag, FLAG-tag | Enable purification of ubiquitinated proteins; His-tag works with Ni-NTA resin, Strep-tag with Strep-Tactin resin [2] |
| Ubiquitin Antibodies | P4D1, FK1, FK2 (pan-ubiquitin); Linkage-specific antibodies (K48-, K63-specific) | Immunoprecipitation of endogenous ubiquitinated proteins; linkage-specific antibodies characterize chain architecture [2] |
| K-ε-GG Antibodies | Commercial monoclonal and polyclonal anti-K-ε-GG | Immunoaffinity enrichment of tryptic peptides containing diglycine remnant on modified lysines [30] [52] |
| Enzymes | Trypsin, ArgC protease; Recombinant E1, E2, E3 enzymes | Protein digestion for MS analysis; in vitro ubiquitination assays [30] [53] |
| Deubiquitinase Inhibitors | N-ethylmaleimide (NEM), PR-619, Ubiquitin aldehyde | Preserve ubiquitination status during cell lysis and protein purification [2] |
| Enrichment Resins | Ni-NTA agarose, Strep-Tactin sepharose, Protein A/G agarose | Solid supports for affinity purification of tagged proteins or antibody-antigen complexes [2] [51] |
Frequently Asked Questions
Q: What are the major advantages of peptide-level (K-ε-GG) enrichment over protein-level methods? A: K-ε-GG enrichment offers direct identification of modification sites, reduced complexity by analyzing peptides rather than proteins, and avoidance of artifacts from tagged ubiquitin expression. However, it requires efficient tryptic digestion and may miss ubiquitination on non-lysine residues or in suboptimal digestion contexts [30] [52].
Q: How can I determine if identified ubiquitination sites are functionally relevant? A: Functional relevance requires correlation with biological outcomes. Combine site identification with functional assays monitoring substrate stability, activity, localization, or interactions. Mutagenesis of identified sites with assessment of phenotypic consequences provides strong evidence for functional importance [30].
Q: What controls should be included in ubiquitin enrichment experiments? A: Essential controls include: (1) Negative controls with empty vector or tag-only expressions; (2) Catalytically inactive E3 ligase or DUB mutants; (3) Known ubiquitinated substrates as positive controls; (4) Non-specific IgG for immunoprecipitation backgrounds; (5) Quantitative standards for MS-based quantification [2] [51].
Q: How can I distinguish between monoubiquitination and polyubiquitination? A: Use linkage-specific antibodies that recognize particular polyubiquitin chains. Alternatively, examine molecular weights by Western blot—monoubiquitination typically shows discrete ~8 kDa shifts, while polyubiquitination creates smeared patterns. MS-based approaches can directly identify ubiquitin-ubiquitin linkage sites when present [30] [2].
Q: What are common pitfalls in interpreting lysine-to-arginine mutagenesis results? A: A common misconception is that loss of function with lysine mutagenesis definitively proves ubiquitination at that site. However, this effect may result from disrupted protein folding, altered interactions, or elimination of other lysine-dependent modifications. Always combine mutagenesis with direct ubiquitination detection methods [30].
Validating Enrichment Specificity: MS Analysis and Functional Correlations
Mass Spectrometry Verification of Enrichment Specificity and Efficiency
Frequently Asked Questions (FAQs)
Q1: What are the primary causes of non-specific binding in ubiquitin enrichment protocols, and how can I identify them in my MS data?
Non-specific binding primarily occurs due to the affinity resins co-purifying proteins that are not the target ubiquitinated substrates. When using His-tagged ubiquitin, histidine-rich proteins are common contaminants. With Strep-tagged systems, endogenously biotinylated proteins are frequently co-purified [2]. In your MS data, look for an overabundance of peptides from these protein classes, a low ratio of ubiquitin-derived peptides to total identified peptides, and the absence of the diagnostic Gly-Gly (GG) remnant on lysine residues, which is a hallmark of ubiquitination.
Q2: My negative controls still show peptide spectra. How can I distinguish true ubiquitinated substrates from persistent background?
Persistent background in controls suggests inadequate blocking of the affinity resin or non-specific protein-protein interactions. To distinguish true substrates, use quantitative MS strategies like stable isotope labeling. True ubiquitinated substrates will show significant enrichment in the experimental sample compared to the control. Furthermore, confirm hits by verifying the presence of the ~114 Da mass shift on lysine residues corresponding to the diglycine remnant from tryptic digestion of ubiquitinated proteins [2] [54].
Q3: How can I verify that my enrichment method is not disrupting the native ubiquitination landscape or favoring specific chain linkages?
Expressing tagged ubiquitin can sometimes alter the structure of Ub and not completely mimic endogenous Ub, potentially generating artifacts [2]. To verify minimal disruption, employ a dual-strategy approach:
- Use linkage-specific antibodies (e.g., for K48 or K63 chains) in a western blot to compare linkage profiles before and after enrichment [2].
- Perform a quantitative proteomic analysis comparing samples enriched via tagged ubiquitin with those enriched using generic anti-ubiquitin antibodies (e.g., FK2) or Ub-binding domains (UBDs), which are thought to cause less linkage bias [2].
Q4: What MS-based metrics best quantify enrichment efficiency and specificity?
The key metrics are summarized in the table below. These should be calculated by comparing your experimental enrichment to a suitable negative control (e.g., a sample without the affinity tag or a wild-type cell line) [2] [54].
Table 1: Key MS Metrics for Evaluating Enrichment Performance
| Metric | Calculation | Interpretation |
|---|---|---|
| Enrichment Specificity | (Number of spectra with GG-modified peptides / Total number of spectra) x 100% | Measures the purity of the enriched sample. A higher percentage indicates better specificity and less non-specific binding. |
| Substrate Identification Gain | Number of unique ubiquitination sites identified in experimental sample vs. control | Quantifies the depth of the analysis enabled by enrichment. |
| Quantitative Fold-Change | Ratio of peptide abundance in experimental sample versus control, using stable isotope labels or label-free quantification [54] | Confirms true substrates, which should be highly enriched in the experimental sample. |
Troubleshooting Guides
Issue 1: Low Specificity of Enrichment (High Background)
Problem: Your mass spectrometry data shows a low percentage of GG-modified peptides and a high number of non-ubiquitinated protein identifications.
Solutions:
- Optimize Blocking and Wash Stringency: Increase the concentration of competitive eluents (e.g., imidazole for His-tag, biotin for Strep-tag) in the wash buffers. Alternatively, include mild detergents (e.g., 0.1% Triton X-100) and increase salt concentration (e.g., 300-500 mM NaCl) to disrupt weak, non-specific interactions [2].
- Combine Enrichment with Proximity Labeling (APPLE-MS): For studying ubiquitin-linked protein complexes, integrate proximity labeling. The APPLE-MS workflow combines high-specificity Twin-Strep-tag enrichment with PafA-mediated proximity labeling. This labels proteins in close proximity to your target, allowing you to distinguish true interactors from non-specific background during MS data analysis. This method has been shown to improve specificity by over 4-fold compared to standard AP-MS [55].
- Use Tandem Purification: Perform a two-step purification. For example, first enrich with a His-tag pull-down, then elute and subject the eluate to a second round of enrichment with an anti-ubiquitin antibody. This dramatically increases specificity but may reduce yield.
Issue 2: Low Yield of Ubiquitinated Substrates
Problem: The overall number of identified ubiquitinated peptides or proteins is low, even though specificity is good.
Solutions:
- Pre-enrichment Digestion Strategy: Instead of enriching intact proteins, digest the cell lysate first and then enrich for GG-modified peptides using anti-GG remnant antibodies. This can provide deeper coverage of the ubiquitinome by simplifying the sample [2] [54].
- Inhibit Deubiquitinases (DUBs): Ubiquitination is a dynamic process. Add potent DUB inhibitors (e.g., PR-619, N-ethylmaleimide) to all lysis and wash buffers to prevent the removal of ubiquitin during sample preparation.
- Validate Tag Functionality: Ensure your tagged ubiquitin is being properly expressed and processed by the cell's enzymatic machinery. Perform a western blot against the tag and endogenous ubiquitin to confirm integration.
Issue 3: Inability to Detect Specific Ubiquitin Chain Linkages
Problem: You suspect a biological process involves a specific ubiquitin chain linkage (e.g., K63), but your enrichment method does not capture this information.
Solutions:
- Use Linkage-Specific Reagents: Employ linkage-specific Ub-binding domains (UBDs) or antibodies for enrichment. For example, certain UBDs in proteins like RAP80 specifically bind K63-linked chains, while others may prefer K48-linked chains [2].
- Leverage MS2 Spectral Libraries: For more generalized discovery, tools like MS2Query can be used to search MS2 spectra against libraries of known ubiquitin chains. This machine learning-based tool improves the reliability of identifying both exact matches and chemically similar "analogues," which can aid in characterizing complex chain architectures [56].
Experimental Protocols for Key Verification Experiments
Protocol 1: Verification of Enrichment Specificity Using Label-Free Quantitative MS
This protocol is designed to directly quantify how well your enrichment method isolates ubiquitinated proteins over non-specific binders [2] [54].
- Sample Preparation: Prepare two parallel samples: the experimental sample (expressing tagged ubiquitin) and a negative control (isogenic cell line without the tag, or using a "empty-tag" purification).
- Parallel Enrichment: Perform the ubiquitin enrichment protocol identically on both samples.
- On-Bead Digestion: Digest the proteins directly on the beads with trypsin to minimize losses.
- LC-MS/MS Analysis: Analyze both samples separately using a high-resolution LC-MS/MS system.
- Data Analysis:
- Identify proteins and ubiquitination sites (using GG remnant search).
- Use the label-free quantification (LFQ) intensity for each protein.
- Calculate the fold-change (LFQ Experimental / LFQ Control) for every identified protein.
- True substrates will have high fold-change values (e.g., >10).
- Non-specific binders will have fold-change values close to 1.
Protocol 2: APPLE-MS for Mapping Ubiquitin-Dependent Protein Complexes
This protocol enhances the specificity of interaction studies by combining affinity purification with proximity labeling [55].
- Cell Engineering: Create a cell line expressing your ubiquitin-related protein of interest (POI) fused to both the Twin-Strep-tag and the proximity enzyme PafA.
- Stimulation & Labeling: Stimulate the cells as required for your experiment. Induce proximity labeling by adding the small-molecule substrate (biotin-AMP) for a short period (e.g., 5-15 minutes).
- Cell Lysis and Affinity Purification: Lyse the cells and perform Twin-Strep-tag purification under native conditions to isolate the POI and its proximal proteins.
- Streptavidin Enrichment: Denature the purified sample to dissociate interactions and capture the biotin-labeled (proximal) proteins on streptavidin beads.
- MS Sample Prep and Analysis: Digest the proteins on-bead, and analyze the peptides by LC-MS/MS.
Diagram 1: APPLE-MS workflow for specific PPI mapping.
Research Reagent Solutions
Table 2: Essential Reagents for Ubiquitin Enrichment and Verification
| Reagent | Function | Key Considerations |
|---|---|---|
| His-tag / Strep-tag Resins | Affinity purification of tagged ubiquitin and its substrates. | His-tag: Can bind histidine-rich proteins. Strep-tag: Can bind endogenous biotinylated proteins [2]. |
| Linkage-Specific Ub Antibodies (e.g., α-K48, α-K63) | Immuno-enrichment or Western blot verification of specific polyUb chain types. | Critical for validating that enrichment does not skew the native linkage landscape [2]. |
| Tandem Ub-Binding Entities (TUBEs) | High-affinity enrichment of polyubiquitinated proteins, protects from DUBs. | Useful for preserving labile ubiquitination signals during extraction [2]. |
| Deubiquitinase (DUB) Inhibitors (e.g., PR-619, NEM) | Prevent loss of ubiquitin signal during sample preparation by inhibiting deubiquitinating enzymes. | Essential in all lysis and wash buffers to maintain ubiquitination status. |
| Anti-diGly (GG) Remnant Antibody | Immuno-enrichment of tryptic peptides containing the GG signature of ubiquitination. | Enables highly specific, site-specific ubiquitinome mapping by MS [2] [54]. |
| Stable Isotope Labels (SILAC, TMT) | For accurate quantification of enrichment efficiency and specificity in comparative MS experiments. | Allows precise comparison of experimental vs. control samples to filter out non-specific binders [54]. |
Comparative Analysis of Different Enrichment Methodologies
What are the primary ubiquitin enrichment methodologies available?
The main methodologies for enriching ubiquitinated proteins or peptides for mass spectrometry analysis can be categorized into three principal approaches, each with distinct advantages and limitations.
Table: Comparison of Primary Ubiquitin Enrichment Methodologies
| Methodology | Core Principle | Key Advantages | Key Limitations |
|---|---|---|---|
| Antibody-Based Enrichment [2] [57] | Uses anti-ubiquitin antibodies (e.g., K-ε-GG) to immunoaffinity purify ubiquitinated peptides. | - Applicable to endogenous proteins and clinical/animal tissues [2].- High specificity for the ubiquitin remnant motif.- Linkage-specific antibodies are available [2]. | - High cost of antibodies [2].- Potential for non-specific binding [2]. |
| Ubiquitin-Binding Domain (UBD) Based [2] [12] | Utilizes tandem ubiquitin-binding entities (TUBEs) to capture polyubiquitinated proteins. | - Preserves labile ubiquitin linkages during lysis [12].- Can be linkage-specific (e.g., K48 or K63) [12].- Captures endogenous proteins. | - Lower affinity when using single UBDs [2].- May not efficiently capture monoubiquitination. |
| Ubiquitin Tagging [2] | Cells are engineered to express affinity-tagged ubiquitin (e.g., His, Strep). | - Easy and relatively low-cost [2].- Effective for high-throughput screening in cellular models. | - Cannot be used on patient or animal tissues [2].- Tagged Ub may not perfectly mimic endogenous Ub, risking artifacts [2]. |
How can I reduce non-specific binding during immunoaffinity enrichment?
Non-specific binding is a common challenge in antibody-based enrichment. The following troubleshooting guide addresses specific issues and solutions.
Table: Troubleshooting Guide for Non-Specific Binding
| Problem | Potential Cause | Recommended Solution |
|---|---|---|
| High background of non-ubiquitinated peptides. | Insoluble particulates or protein aggregates in the peptide sample. | - Sonicate peptide samples and centrifuge at 10,000×g for 5 minutes to remove particulates before enrichment [57]. |
| Co-purification of endogenously biotinylated proteins (Strep-tag) or histidine-rich proteins (His-tag). | Inherent limitation of certain affinity tags. | - This is a known drawback of tagged Ub approaches [2]. Consider switching to antibody-based or UBD-based enrichment for endogenous studies. |
| Inconsistent results and high variability between replicates. | Manual bead handling inconsistencies. | - Automate the enrichment process using bead-handling platforms (e.g., KingFisher) to improve reproducibility and reduce handling errors [58] [57]. |
What are the best practices for automating ubiquitin enrichment workflows?
Automation significantly enhances reproducibility, throughput, and quantitative accuracy. The key is to select a platform compatible with your enrichment reagent.
Table: Automation Platforms for Ubiquitin Enrichment
| Platform Type | Example Systems | Compatible Reagents | Performance Gain |
|---|---|---|---|
| Bead-Handler | ThermoFisher KingFisher (Apex, Flex) [57] | Magnetic bead-based PTMScan HS kits (e.g., #59322) [57]. | - Recovers a similar number of PTM peptides as manual methods but with greater handling ease and reproducibility [57].Processes up to 96 samples in a single day [59]. |
| Hybrid System | Agilent AssayMAP Bravo [57] | Custom PTMScan reagent formulations without magnetic beads (e.g., agarose-based antibodies) [57]. | - Peptide identifications can be 30-135% higher compared to manual preparation when using a bidirectional aspirate program [57]. |
| Liquid Handler | Hamilton, Tecan, Beckman Coulter systems [57] | PTMScan HS kits [57]. | - Increases throughput and consistency for large sample sets. |
The following workflow diagram illustrates a robust, automated process for ubiquitin enrichment and analysis.
How do I choose between linkage-specific and pan-specific enrichment tools?
Your choice depends entirely on your research question. The diagram below illustrates the decision-making process for method selection.
Use Linkage-Specific Tools (e.g., K48 or K63-TUBEs) when: Your goal is to investigate a specific ubiquitin-dependent process. For example, K48-linked chains are primarily associated with proteasomal degradation, while K63-linked chains regulate signal transduction [12]. These tools allow you to capture and study these specific events in response to stimuli (e.g., an inflammatory agent) or treatments (e.g., a PROTAC molecule designed to induce degradation) [12].
Use Pan-Selective Tools (e.g., general anti-K-ε-GG antibodies, Pan-TUBEs) when: You aim for a global, unbiased profiling of the ubiquitinome without pre-selecting for a particular chain type [2] [57]. This is ideal for discovery-phase experiments.
Can I enrich multiple PTMs from a single sample?
Yes, recent protocols enable the sequential enrichment of multiple PTMs from a single sample, maximizing the information gained from precious biological samples. The SCASP-PTM (SDS-cyclodextrin-assisted sample preparation-post-translational modification) approach allows for the tandem enrichment of ubiquitinated, phosphorylated, and glycosylated peptides from one protein digest without intermediate desalting steps [11]. This streamlined workflow reduces sample loss and processing time.
The Scientist's Toolkit: Key Research Reagent Solutions
Table: Essential Reagents for Ubiquitin Enrichment Experiments
| Reagent / Kit | Function | Application Context |
|---|---|---|
| PTMScan HS Ubiquitin/SUMO Remnant Motif (K-ε-GG) Kit [58] [57] | Immunoaffinity enrichment of ubiquitinated peptides using a high-sensitivity anti-K-ε-GG antibody conjugated to magnetic beads. | Ideal for automated, high-throughput ubiquitinome profiling on bead-handling platforms. |
| Linkage-Specific TUBEs (K48, K63) [12] | Tandem Ubiquitin Binding Entities with high affinity for specific polyubiquitin chain types. | Capturing and studying linkage-specific ubiquitination of endogenous proteins; protects chains from deubiquitinases. |
| His-Ub or Strep-Ub Plasmids [2] | Genetic constructs for expressing affinity-tagged ubiquitin in cells. | High-throughput screening of ubiquitinated substrates in cultured cells. |
| Automated Liquid Handling System (e.g., KingFisher Apex, AssayMAP Bravo) [58] [57] | Platform for automating bead-based or on-tip immunoaffinity enrichment. | Essential for achieving high reproducibility and throughput in large-scale PTM studies. |
Functional Validation Through Downstream Applications and Assays
This technical support resource is designed for researchers and drug development professionals conducting ubiquitin enrichment studies. A central challenge in this field is the reduction of non-specific binding (NSB), which can compromise data quality and lead to inaccurate biological conclusions. The following guides and FAQs provide targeted troubleshooting and detailed protocols to help you optimize your experiments, minimize artifacts, and validate your findings with confidence.
Troubleshooting Non-Specific Binding in Ubiquitin Enrichment
FAQ: My western blots for ubiquitinated proteins show high background noise. How can I improve the signal-to-noise ratio?
High background is frequently caused by inadequate blocking or antibody non-specificity [60] [61].
- Solution 1: Optimize Your Blocking Buffer. The choice of blocking agent is critical. While non-fat dry milk is common and inexpensive, it contains phosphoproteins and biotin, which can interfere with the detection of phosphorylated proteins or streptavidin-biotin systems. For these applications, Bovine Serum Albumin (BSA) is a superior alternative [60] [61]. See Table 1 for a comparison.
- Solution 2: Increase Blocking Efficiency. Ensure you are using a sufficient concentration of blocking buffer (typically 3-5%) and extend the incubation time to 1 hour at room temperature or overnight at 4°C for difficult samples [61].
- Solution 3: Incorporate Detergents. Adding a mild non-ionic detergent like Tween-20 (0.05%-0.2%) to your wash and blocking buffers can help disrupt hydrophobic interactions that cause NSB. Be cautious, as high concentrations may elute weakly bound antibodies [60] [62].
FAQ: During ubiquitin immunoprecipitation, I co-precipitate many non-ubiquitinated proteins. How can I increase specificity?
This is a common challenge due to the transient nature of ubiquitination and the low abundance of ubiquitinated proteins in total lysate [63] [2].
- Solution 1: Use High-Affinity Enrichment Tools. Replace traditional antibodies with high-affinity nanobody-based traps, such as the ChromoTek Ubiquitin-Trap, which is designed for clean, low-background pulldowns of ubiquitin and ubiquitinylated proteins from various cell extracts [63].
- Solution 2: Preserve Ubiquitination Signals. Treat your cells with proteasome inhibitors (e.g., MG-132 at 5-25 µM for 1–2 hours) prior to harvesting. This prevents the degradation of ubiquitinated proteins, thereby increasing the target pool available for enrichment. Note that overexposure can cause cytotoxicity [63].
- Solution 3: Validate with Proper Controls. Always include control samples from non-treated cells or using an empty trap to identify proteins that bind non-specifically to the resin or beads.
FAQ: How can I reduce non-specific binding in Surface Plasmon Resonance (SPR) experiments studying ubiquitin-protein interactions?
NSB in SPR inflates response units and leads to erroneous kinetic calculations [62].
- Solution 1: Adjust Buffer pH. The pH dictates the charge of your biomolecules. If your analyte is positively charged and interacting with a negative sensor surface, adjust the buffer pH to the isoelectric point of your protein to neutralize its charge [62].
- Solution 2: Use Buffer Additives. Incorporate protein blockers like BSA (typically at 1%) or non-ionic surfactants like Tween 20 (at low concentrations) into your running buffer. These additives shield the analyte from non-specific interactions with the sensor surface and system tubing [62].
- Solution 3: Increase Ionic Strength. For charge-based NSB, increasing the salt concentration (e.g., NaCl) can produce a shielding effect. Adding 200 mM NaCl to the running buffer has been shown to significantly reduce non-specific binding of charged analytes like rabbit IgG [62].
Essential Experimental Protocols
Protocol: Tandem Enrichment of Ubiquitinated Peptides for Mass Spectrometry
This protocol, based on the SCASP-PTM method, allows for the sequential enrichment of ubiquitinated, phosphorylated, and glycosylated peptides from a single sample, maximizing data output from precious samples [11].
Workflow Summary:
- Protein Extraction and Digestion: Prepare samples using the SDS-cyclodextrin-assisted sample preparation (SCASP) method.
- Ubiquitinated Peptide Enrichment: Perform the first enrichment step on the digested peptide mixture to isolate ubiquitinated peptides.
- Sequential PTM Enrichment: Use the flow-through from the first enrichment to subsequently isolate phosphorylated or glycosylated peptides without an intermediate desalting step.
- Cleanup and Analysis: Desalt the enriched PTM peptides and analyze via Data-Independent Acquisition (DIA) Mass Spectrometry [11].
The graphical abstract below illustrates this tandem process.
Protocol: Validating Ubiquitination via Immunoprecipitation and Western Blot
This is a conventional method for validating the ubiquitination of a specific protein of interest [63] [2].
Detailed Methodology:
- Cell Treatment: Treat cells with a proteasome inhibitor (e.g., MG-132) for 1-2 hours before harvesting to stabilize ubiquitinated proteins.
- Lysis: Lyse cells in a suitable lysis buffer.
- Immunoprecipitation (IP):
- Option A (Antibody-based): Incubate the clarified lysate with an antibody against your protein of interest, followed by capture with Protein A/G beads.
- Option B (Ubiquitin-Trap): As a more specific alternative, use ChromoTek's Ubiquitin-Trap (agarose or magnetic beads) to isolate all ubiquitinated proteins from the lysate directly [63].
- Wash: Wash the beads stringently to remove non-specifically bound proteins.
- Elution: Elute the bound proteins by boiling in SDS-PAGE sample buffer.
- Western Blot Analysis:
- Separate proteins by SDS-PAGE and transfer to a membrane.
- Block the membrane with an optimized blocker (e.g., 2-5% BSA or a commercial protein-free block) [60] [61].
- Probe with a primary anti-ubiquitin antibody (e.g., P4D1, FK1/FK2), followed by an HRP-conjugated secondary antibody.
- Expected Result: A characteristic "smear" of higher molecular weight bands indicates poly-ubiquitinated forms of your protein. A linkage-specific ubiquitin antibody can be used in this step to determine the chain type [63] [2].
The Scientist's Toolkit: Key Reagent Solutions
The table below lists essential reagents for ubiquitin research, along with their specific functions in overcoming common experimental challenges.
Table 1: Key Research Reagents for Ubiquitin Enrichment and Detection
| Reagent | Function & Application | Key Consideration |
|---|---|---|
| Ubiquitin-Trap (ChromoTek) | High-affinity nanobody-based reagent for immunoprecipitation of ubiquitin and ubiquitinylated proteins from cell extracts [63]. | Not linkage-specific; binds all ubiquitin chains. Ideal for clean pulldowns with low background [63]. |
| Linkage-Specific Ub Antibodies | Enrich or detect polyUb chains with a specific linkage (e.g., K48, K63) in IP or western blot [2]. | Essential for deciphering the ubiquitin code, as different linkages signal different downstream outcomes [63]. |
| Proteasome Inhibitors (e.g., MG-132) | Increases the pool of ubiquitinated proteins in cells by blocking their degradation by the proteasome [63]. | Critical for preserving ubiquitination signals. Titrate for optimal effect (e.g., 5-25 µM), as overexposure is cytotoxic [63]. |
| BSA Blocking Buffer | Protein-based blocking agent for western blotting. Reduces non-specific antibody binding [60] [61]. | Preferred over milk when detecting phosphoproteins or using streptavidin-biotin systems, as it lacks phosphoproteins and biotin [60]. |
| Tween 20 | Non-ionic detergent added to buffers to reduce hydrophobic interactions and minimize NSB in techniques like western blot, ELISA, and SPR [62] [61]. | Use at low concentrations (0.05%-0.2%); high concentrations can disrupt specific antibody-antigen binding [60]. |
The Ubiquitin Code: A Decision Framework
Understanding the downstream consequences of ubiquitination, known as the "Ubiquitin Code," is crucial for experimental design and data interpretation. The following diagram and table summarize how different ubiquitin signals correlate with specific biological outcomes, guiding your functional validation assays.
Table 2: Guide to Ubiquitin Linkages and Downstream Outcomes
| Ubiquitin Signal | Chain Type | Primary Downstream Signaling Event[s] |
|---|---|---|
| Substrate Lysine | Monomer | Endocytosis, Histone Modification, DNA Damage Responses [63] |
| K48 | Polymeric | Targeted Protein Degradation (most canonical signal for proteasomal degradation) [63] |
| K63 | Polymeric | Immune/Inflammatory Responses, NF-κB Pathway Activation, Autophagy [63] |
| K6, K11, K27, K29, M1 | Polymeric | Diverse functions including antiviral responses, cell cycle progression, DNA replication/repair, and neurodegenerative pathways [63] |
What are PROTACs and why is studying their mechanism important? PROteolysis TArgeting Chimeras (PROTACs) are heterobifunctional molecules that target specific proteins for degradation by hijacking the cell's ubiquitin-proteasome system [64] [65]. A typical PROTAC consists of three elements: a warhead that binds to a protein of interest (POI), an anchor that recruits an E3 ubiquitin ligase, and a linker connecting these two ligands [66] [67]. Understanding the precise mechanism of PROTACs is crucial for drug development because it moves beyond traditional inhibition to complete protein removal, offering therapeutic potential for previously "undruggable" targets [66] [68]. However, this promise is tempered by a significant challenge: non-specific binding during ubiquitin enrichment can compromise data integrity and lead to inaccurate conclusions about degradation efficiency and selectivity [2].
Why is reducing non-specific binding critical in ubiquitin enrichment studies? Non-specific binding occurs when proteins other than the intended ubiquitinated targets are captured during enrichment protocols. This interference can lead to false positives, obscure genuine ubiquitination events, and generate misleading data on parameters like DC50 (concentration for 50% degradation) and Dmax (maximum degradation) [69] [2]. In the context of a drug discovery pipeline, such inaccuracies can misguide medicinal chemistry efforts and hinder the development of effective degraders. Therefore, applying specific enrichment techniques is essential to accurately validate PROTAC efficacy and mechanism [70].
Table 1: Key Challenges from Non-Specific Binding in PROTAC Studies
| Challenge | Impact on Data | Consequence for Research |
|---|---|---|
| Co-enrichment of non-ubiquitinated proteins [2] | High background noise, masking true ubiquitination signals | Inaccurate measurement of degradation efficiency (DC50, Dmax) [69] |
| Co-purification of endogenous biotinylated or histidine-rich proteins [2] | False-positive identification of ubiquitination sites | Misleading conclusions about PROTAC selectivity and off-target effects |
| Inefficient enrichment of low-stoichiometry ubiquitination [2] | Failure to detect genuine, functionally relevant ubiquitination events | Incomplete understanding of PROTAC mechanism and potential resistance |
Frequently Asked Questions (FAQs) on Specific Enrichment
FAQ 1: What are the primary sources of non-specific binding in ubiquitin enrichment for PROTAC studies? The main sources of non-specific binding are tied to the enrichment methodology itself. When using Ub tagging-based approaches (e.g., His- or Strep-tagged ubiquitin), histidine-rich or endogenously biotinylated proteins can bind non-specifically to the Ni-NTA or Strep-Tactin resins, respectively [2]. For antibody-based approaches, despite their ability to recognize endogenous ubiquitin, the antibodies (e.g., P4D1, FK1/FK2) can exhibit off-target binding to non-ubiquitinated proteins, pulling them down alongside the true targets [2].
FAQ 2: How can I select the most specific enrichment method for my PROTAC experiment? The choice of method depends on your experimental model and the question you are asking. The table below compares the core features of the primary enrichment strategies.
Table 2: Comparison of Ubiquitin Enrichment Methods for PROTAC Studies
| Method | Principle | Advantages | Limitations & Specificity Concerns |
|---|---|---|---|
| Ub Tagging [2] | Cells are engineered to express affinity-tagged Ub (e.g., His, Strep). Tagged ubiquitinated proteins are purified. | Easy to use; relatively low cost; good for discovery in cell lines. | High non-specific binding from endogenous proteins [2]; does not reflect endogenous conditions; genetic manipulation required. |
| Pan-Ub Antibody [2] | Antibodies that recognize all ubiquitin linkages are used to immunoprecipitate endogenous ubiquitinated proteins. | Works with any cell or tissue sample (no tags needed); captures native ubiquitination. | Linkage-specific information is lost; antibody cross-reactivity can cause non-specific binding [2]. |
| Linkage-Specific Antibody [2] | Antibodies specific to a Ub chain linkage (e.g., K48, K63) enrich for proteins with that chain type. | Provides mechanistic insight (e.g., K48 for proteasomal degradation); high specificity for chain type. | High cost; may still co-enrich non-specifically bound proteins; limited to characterized linkages. |
| UBD-Based Approach [2] | Tandem Ub-binding domains (UBDs) with high affinity are used as bait to purify ubiquitinated proteins. | Can be highly specific for Ub chains; targets endogenous ubiquitin. | Low affinity of single UBDs limits application; requires careful optimization [2]. |
FAQ 3: What specific experimental strategies can minimize non-specific binding?
- Include Rigorous Controls: Always perform enrichment with an isotype control antibody or bare resin in parallel. This control identifies proteins that bind non-specifically to the solid support or antibody Fc region [2].
- Optimize Wash Stringency: Increase the stringency of wash buffers (e.g., with higher salt concentrations, detergents like CHAPS, or mild denaturants) after the binding step to disrupt weak, non-covalent interactions without eluting genuinely ubiquitinated proteins [2].
- Use Tandem Enrichment Tags: For tagging approaches, using a tandem tag (e.g., Twin-Strep) can significantly enhance specificity over single tags, as it requires two binding events for retention [2].
- Pre-clear Lysate: Incubate your cell lysate with the enrichment resin or beads without the specific antibody beforehand to remove proteins that promiscuously bind to the matrix.
Troubleshooting Guides
Problem 1: High Background Noise in Western Blots After Enrichment
Potential Cause: The most common cause is insufficient washing after the enrichment step, leaving non-specifically bound proteins on the beads.
Solution:
- Step 1: Increase the number of washes from 3 to 5-6.
- Step 2: Modify the wash buffer composition. A common effective recipe is:
- 50 mM Tris-HCl, pH 7.5
- 150 mM NaCl
- 1% NP-40 (or 0.5% Sodium Deoxycholate)
- 0.1% SDS
- Step 3: For a final wash, use a low-salt buffer (e.g., 20 mM Tris-HCl, pH 7.5) or plain PBS to remove detergent residues that can interfere with downstream analysis [2].
Problem 2: Failure to Detect Expected Ubiquitination of the Target Protein
Potential Cause: The ubiquitination event may be transient or of low stoichiometry, making it difficult to capture. Alternatively, the PROTAC may not be forming a productive ternary complex.
Solution:
- Step 1: Treat cells with a proteasome inhibitor (e.g., MG-132, 10 µM for 4-6 hours) before lysis. This blocks the degradation of ubiquitinated proteins, allowing them to accumulate and be more readily detected [69].
- Step 2: Validate that your PROTAC is active. Use a complementary, live-cell assay like NanoBRET to confirm target protein degradation in real-time, which can help you confirm that the failure is in detection and not in PROTAC function [65].
- Step 3: If using antibody-based enrichment, try a different antibody or a UBD-based approach to rule out epitope masking or antibody inefficiency [2].
Problem 3: Identifying Too Many Putative Ubiquitination Sites in MS, Suggesting Non-Specificity
Potential Cause: The enrichment protocol is not specific enough, leading to a dataset contaminated with non-ubiquitinated proteins or non-lysine modifications.
Solution:
- Step 1: Implement a pre-clearing step, as described in the FAQs.
- Step 2: For mass spectrometry analysis, use di-glycine remnant immunoprecipitation. After trypsin digestion, which cleaves ubiquitin but leaves a di-glycine signature on the modified lysine, antibodies specific to this di-glycine remnant can be used for a highly specific second-stage enrichment, drastically reducing false positives [2].
- Step 3: Cross-reference your hit list with common contaminants (e.g., histones, ribosomal proteins, proteins with high isoelectric points) and filter them out during data analysis.
Diagram 1: Optimized Workflow for Specific Ubiquitin Enrichment. This flowchart outlines key steps to minimize non-specific binding, including pre-clearing and stringent washes.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Specific Ubiquitin Enrichment in PROTAC Studies
| Reagent / Tool | Function | Specific Application in PROTAC Research |
|---|---|---|
| Linkage-Specific Ub Antibodies [2] | Immunoprecipitation of proteins with specific Ub chain types (e.g., K48, K63). | Confirm PROTAC-induced, proteasome-targeting K48-linked ubiquitination on the POI. |
| Tandem Ub-Binding Domains (TUBEs) [2] | High-affinity enrichment of endogenous polyubiquitinated proteins; protect chains from DUBs. | Capture and stabilize transient ubiquitination events induced by PROTACs for detection. |
| diGly Remnant Antibodies [2] | Highly specific enrichment of ubiquitinated peptides for mass spectrometry. | Unambiguously map the exact lysine residues on the POI that are ubiquitinated by the PROTAC complex. |
| Epitope-Tagged Ubiquitin (His-Strep) [2] | Allows affinity-based purification of ubiquitinated proteins from engineered cell lines. | Useful for initial, broad discovery of PROTAC-mediated ubiquitination targets in a cellular model. |
| Live-Cell Degradation Reporters (e.g., NanoBRET) [65] | Real-time, kinetic monitoring of target protein degradation in live cells. | Correlate ubiquitination data with functional degradation outcomes and determine DC50 values. |
| Proteasome Inhibitors (e.g., MG-132) [69] | Block the proteasome, causing ubiquitinated proteins to accumulate. | Enhances detection sensitivity of ubiquitinated POI in enrichment assays. |
Diagram 2: Core PROTAC Mechanism and Key Validation Points. The diagram shows how a PROTAC brings the POI and E3 ligase together, leading to ubiquitination and degradation. Enrichment assays are critical for directly validating the ubiquitination step.
Within the broader thesis on reducing non-specific binding in ubiquitin enrichment research, benchmarking the performance of your methods is not just a best practice—it is a necessity. The versatility of ubiquitination, encompassing everything from single Ub monomers to complex polyUb chains with different lengths and linkage types, makes the accurate assessment of enrichment specificity particularly challenging [2]. Non-specific binding can lead to high background noise, co-enrichment of non-target proteins, and ultimately, unreliable data. This technical support guide provides targeted troubleshooting advice and methodologies to help you quantify the effectiveness of your ubiquitin enrichment protocols, identify common pitfalls, and implement solutions that enhance the specificity of your results.
Core Concepts: The Ubiquitin Enrichment Landscape
To effectively benchmark your experiments, it is crucial to understand the common enrichment methodologies and their inherent strengths and weaknesses. The primary challenge stems from the low stoichiometry of protein ubiquitination under normal physiological conditions and the complex architecture of Ub chains [2]. The following table summarizes the three predominant techniques used for enriching ubiquitinated proteins.
| Method | Principle | Advantages | Disadvantages & Specificity Concerns |
|---|---|---|---|
| Ubiquitin Tagging [2] | Expression of affinity-tagged Ub (e.g., His, Strep) in cells; purification of tagged conjugates. | Easy, relatively low-cost, good for high-throughput screening in cell culture. | Tag may alter Ub structure, causing artifacts. Co-purification of histidine-rich/biotinylated proteins causes non-specific binding. Infeasible for patient/animal tissues. |
| Antibody-Based [2] [71] | Use of anti-Ub antibodies (pan-specific or linkage-specific) to immunoprecipitate ubiquitinated proteins. | Enables study of endogenous ubiquitination under physiological conditions; applicable to tissues and clinical samples. | Linkage-specific antibodies are expensive. Non-specific binding to non-ubiquitinated proteins is a common issue, leading to high background. |
| UBD-Based [2] | Use of ubiquitin-binding domains (UBDs) from proteins like E3 ligases or DUBs to bind and enrich ubiquitinated proteins. | Can be engineered for general or linkage-selective binding. | Single UBDs often have low affinity, compromising enrichment efficiency. Tandem-repeated UBDs are often required for effective purification. |
Quantitative Benchmarking Metrics
To move beyond qualitative assessments, you should employ quantitative metrics that directly measure enrichment specificity and efficiency. The following table outlines key metrics that can be derived from your experimental data, such as Western blot analysis or mass spectrometry (MS) results.
| Metric | Definition | Calculation | Interpretation & Ideal Outcome |
|---|---|---|---|
| Enrichment Fold-Change | The degree to which target ubiquitinated proteins are concentrated. | Signal intensity in bound fraction / Signal intensity in input lysate. | A higher value indicates more efficient enrichment. A low value suggests poor binding or excessive washing. |
| Non-Specific Binding Index | A measure of non-target proteins co-purified in the experiment. | Signal intensity of non-target proteins in bound fraction / Total protein in bound fraction. | A lower value indicates higher specificity. A high value points to antibody cross-reactivity or insufficient blocking. |
| Background Signal Ratio | The amount of signal detected in negative controls. | Signal in negative control IP / Signal in experimental IP. | Should be minimal. A high ratio indicates problematic background, often from antibody or bead non-specificity. |
| Ubiquitin Smear Profile | Qualitative-quantitative assessment of the poly-ubiquitinated protein pattern on a gel [71]. | N/A | A characteristic smear indicates successful enrichment of diverse ubiquitinated species. A clean or faint smear suggests low yield or protein degradation. |
Experimental Protocols for Benchmarking
Protocol 1: Benchmarking Antibody Specificity via Immunoprecipitation-Western Blot (IP-WB)
This protocol is fundamental for validating the specificity of your enrichment antibody before proceeding to more complex MS workflows.
- Cell Lysis and Pre-Clearing: Lyse cells in RIPA buffer supplemented with proteasome inhibitors (e.g., 5-25 µM MG-132 for 1-2 hours) to preserve ubiquitination [71]. Pre-clear the lysate by incubating with protein A/G beads for 30 minutes at 4°C to reduce non-specific binding.
- Antibody Immobilization: Incubate your chosen anti-Ub antibody (e.g., linkage-specific or pan-specific) with protein A/G beads for 1-2 hours at 4°C.
- Immunoprecipitation: Incubate the pre-cleared lysate with the antibody-bound beads overnight at 4°C with gentle rotation.
- Washing: Wash the beads stringently with lysis buffer 3-5 times. Troubleshooting Tip: Increasing the salt concentration (e.g., 300-500 mM NaCl) in the wash buffer can help reduce non-specific ionic interactions without disrupting specific antibody binding.
- Elution and Analysis: Elute proteins using 2X Laemmli buffer. Separate the proteins by SDS-PAGE and transfer to a membrane for Western blotting.
- Detection: Probe the membrane with the same or a different anti-Ub antibody to detect the enriched ubiquitinated proteins, which should appear as a characteristic smear [71].
Protocol 2: Assessing Enrichment Specificity via Mass Spectrometry (MS)
This protocol builds on the IP to provide a system-wide, quantitative view of your enrichment specificity.
- Enrichment: Perform ubiquitin enrichment as described in Protocol 1, using a method like Ubiquitin-Trap magnetic agarose beads or antibody-based IP [71].
- On-Bead Digestion: After the final wash, instead of eluting, perform on-bead digestion with trypsin to prepare the enriched proteins for MS analysis. This minimizes sample loss [71].
- Mass Spectrometry Analysis: Analyze the digested peptides using a high-resolution LC-MS/MS system.
- Data Analysis:
- Identify Ubiquitination Sites: Search MS data for the signature mass shift (114.04 Da) on modified lysine residues, which is a remnant of the tryptic digest of ubiquitinated proteins [2].
- Calculate Specificity Metric: A key benchmarking metric is the ratio of ubiquitinated peptides to the total number of identified peptides in the sample. A higher ratio indicates a more specific enrichment with less non-specific carryover.
Visualization of Workflows and Signaling
Ubiquitin Enrichment and Benchmarking Workflow
The following diagram outlines the core experimental pathway for ubiquitin enrichment and the key points for benchmarking and troubleshooting.
The Ubiquitin Code and Downstream Signaling
Understanding the "Ubiquitin Code" is essential, as different ubiquitin linkages signal for different cellular events. Your enrichment strategy should be aligned with the biological question you are investigating.
Research Reagent Solutions
Selecting the right reagents is critical for success. The following table details key tools used in ubiquitin enrichment studies.
| Reagent / Tool | Function / Description | Key Considerations for Specificity |
|---|---|---|
| Ubiquitin-Trap (Agarose/Magnetic) [71] | A nanobody-based reagent for pulldown of mono-Ub, Ub chains, and ubiquitinated proteins. | Provides clean, low-background IPs due to high-affinity VHH. Resists harsh washing conditions. Not linkage-specific. |
| Linkage-Specific Antibodies [2] | Antibodies that recognize a specific Ub chain linkage (e.g., K48, K63). | Critical for studying specific pathways. High cost; must be validated for specificity to avoid cross-reactivity. |
| Pan-Ubiquitin Antibodies (P4D1, FK1/FK2) [2] | Antibodies that recognize all ubiquitinated proteins regardless of linkage. | Useful for global ubiquitination assessment. Prone to non-specific binding; requires rigorous controls. |
| Proteasome Inhibitors (MG-132) [71] | Prevents degradation of ubiquitinated proteins, thereby increasing their abundance for detection. | Preserves ubiquitination signals. Concentration and exposure time must be optimized to avoid cytotoxicity. |
| Tandem Ubiquitin Binding Entities (TUBEs) | Engineered tandem UBDs with high affinity for polyUb chains. | Protects Ub chains from deubiquitinases (DUBs) during lysis. High affinity can sometimes present challenges for elution. |
| Click Chemistry-Generated Diubiquitin [23] | Synthetic diubiquitin with non-hydrolyzable triazole linkage, used as standards or affinity matrix. | Resistant to DUB cleavage, allowing accurate identification of linkage-specific interactors. |
Frequently Asked Questions (FAQs)
Q1: Why do I see a smear instead of discrete bands when I Western blot for ubiquitin after enrichment? A: This is expected and indicates a successful enrichment [71]. The smear represents the vast population of proteins of different molecular weights that are modified by polymeric ubiquitin chains of varying lengths. A clean or faint smear suggests low ubiquitinated protein yield or potential protein degradation during your protocol.
Q2: My mass spectrometry results show a low ratio of ubiquitinated peptides. How can I improve this? A: A low ratio typically indicates significant non-specific binding. To improve specificity:
- Increase Wash Stringency: Incorporate higher salt concentrations (e.g., 300-500 mM NaCl) and detergents like 0.1% SDS in your wash buffers.
- Optimize Antibody Amount: Too much antibody can increase non-specific binding; perform a titration experiment.
- Use a Different Enrichment Matrix: Switch from antibody-based IP to a high-affinity nanobody-based system like the Ubiquitin-Trap, which is designed for low background [71].
- Ensure Proper Pre-clearing: Always pre-clear your lysate with bare beads to remove proteins that bind non-specifically to the resin.
Q3: Can I differentiate between different ubiquitin linkage types in my enriched sample? A: Yes, but not in a single-step enrichment using a standard pan-Ub antibody or Ubiquitin-Trap, as these are not linkage-specific [71]. To study a specific linkage, you must use linkage-specific antibodies for your enrichment [2]. Alternatively, you can perform a general enrichment first, and then use linkage-specific antibodies in a subsequent Western blot analysis to probe for the presence of specific chains in your enriched sample.
Q4: How can I preserve weak or transient ubiquitination events in my cell lysates? A: The key is to inhibit deubiquitinating enzymes (DUBs) and the proteasome. Always:
- Use Fresh Proteasome Inhibitors: Add MG-132 (a common proteasome inhibitor) to your cell culture medium for 1-2 hours before harvesting [71].
- Include DUB Inhibitors: Supplement your cell lysis buffer with a broad-spectrum DUB inhibitor cocktail. This prevents the cleavage of Ub from your protein substrates during and after cell lysis.
- Work Quickly: Keep lysates on ice and process them for enrichment as rapidly as possible.
Conclusion
Reducing non-specific binding in ubiquitin enrichment is not merely a technical optimization but a fundamental requirement for generating reliable biological insights. The integration of high-affinity tools like TUBEs, linkage-specific reagents, and chemical biology approaches provides researchers with an expanding toolkit to achieve unprecedented specificity. As the field advances, these refined methodologies will be crucial for deciphering the complex ubiquitin code in physiological and pathological contexts, particularly for developing targeted degradation therapies like PROTACs. Future directions will likely involve further refinement of chain-selective probes, integration with single-cell proteomics, and development of standardized validation frameworks. By implementing these strategies, researchers can significantly enhance the quality of ubiquitination data, accelerating discoveries in basic biology and therapeutic development.
References
- 1. Understanding and Controlling Non-Specific Binding in ... [https://www.amerigoscientific.com/resource-understanding-and-controlling-non-specific-binding-in-spr-experiments.html?srsltid=AfmBOoow4yFZEGtMsjL2QBH5w6nR2jaGzx0G_dl4ug-MDPppul10Sfvv]
- 2. Current methodologies in protein ubiquitination ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9373315/]
- 3. GraphPad Prism 10 Curve Fitting Guide - Nonspecific binding [https://www.graphpad.com/guides/prism/latest/curve-fitting/nonspecificbinding1.htm]
- 4. Ubiquitin modifications | Cell Research [https://www.nature.com/articles/cr201639]
- 5. How to Better Study Ubiquitination [https://www.ptglab.com/news/blog/how-to-better-study-ubiquitination/?srsltid=AfmBOooZC7Ba3NSrKHY6oPwf3lqu4LC91Trafbif230zxRp4_eSpAXrE]
- 6. Use of a High-Affinity Ubiquitin -Binding Domain to Detect and Purify... [https://bio-protocol.org/en/bpdetail?id=5426&type=0]
- 7. Pierce™ Ubiquitin Kit 15 reactions kit | Contact Us Enrichment [https://www.thermofisher.com/order/catalog/product/89899]
- 8. Deciphering non-canonical ubiquitin signaling: biology and ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2023.1332872/full]
- 9. Signal Seeker Kit Protocols [https://www.cytoskeleton.com/signal-seeker-protocols]
- 10. Proteomic approaches for the profiling of ubiquitylation ... [https://www.sciencedirect.com/science/article/abs/pii/S187439192030364X]
- 11. Protocol for tandem enrichment of ubiquitinated, ... [https://www.sciencedirect.com/science/article/pii/S2666166725003508]
- 12. Unraveling chain specific ubiquitination in cells using ... [https://www.nature.com/articles/s41598-025-07242-9]
- 13. How to Better Study Ubiquitination [https://www.ptglab.com/news/blog/how-to-better-study-ubiquitination/?srsltid=AfmBOormcJBN4aqtIo1RXjhjMQ554n3DMHTSzM8pffMHcYBdWUdR8M2T]
- 14. Unraveling Chain specific Ubiquitination in Cells Using ... [https://lifesensors.com/unraveling-chain-specific-ubiquitination-in-cells-using-tandem-ubiquitin-binding-entities-in-a-high-throughput-assay/]
- 15. How to Better Study Ubiquitination [https://www.ptglab.com/news/blog/how-to-better-study-ubiquitination/?srsltid=AfmBOooEbL3NnUI0oMKKWbJWAuFpiDcbSI97ceG1xWPD79GLMz7Ja6i2]
- 16. A High-Throughput Method for Specific, Rapid, Precise and ... [https://www.sciencedirect.com/science/article/abs/pii/S0039914025015681]
- 17. The Molecular Toolbox for Linkage Type‐Specific Analysis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12118340/]
- 18. FAQs [https://lifesensors.com/faqs/]
- 19. Unraveling chain specific ubiquitination in cells using tandem ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12217038/]
- 20. Ubiquitin Linkage-Specific Affimers Reveal Insights into K6 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5640506/]
- 21. How to Better Study Ubiquitination [https://www.ptglab.com/news/blog/how-to-better-study-ubiquitination/?srsltid=AfmBOooKLikRS_HUbekoQcFAXzjoKk3GIEPHsc9UXnX-XbTm5x-sQfqd]
- 22. Non-hydrolyzable Diubiquitin Probes Reveal Linkage-Specific Reactivity of Deubiquitylating Enzymes Mediated by S2 Pockets - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4850247/]
- 23. Investigating the Interaction Profile of Unconjugated Ubiquitin [https://pmc.ncbi.nlm.nih.gov/articles/PMC12447379/]
- 24. How to Better Study Ubiquitination [https://www.ptglab.com/news/blog/how-to-better-study-ubiquitination/?srsltid=AfmBOoo1yNwdC5axrJdj6_UGZve2JSyhISOBHKMiXn3gXhqgcpsChrOm]
- 25. Chemical approaches to explore ubiquitin-like proteins - RSC Chemical Biology (RSC Publishing) DOI:10.1039/D4CB00220B [https://pubs.rsc.org/en/content/articlehtml/2025/cb/d4cb00220b]
- 26. Decoding the Messaging of the Ubiquitin System Using Chemical and Protein Probes - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7611516/]
- 27. Automating UbiFast for High-throughput and Multiplexed Ubiquitin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9357436/]
- 28. Broad Utility of an Affinity- enrichment Strategy for Unanchored P [https://www.longdom.org/open-access/broad-utility-of-an-affinityenrichment-strategy-for-unanchored-polyubiquitin-chains-33380.html]
- 29. How to Better Study Ubiquitination [https://www.ptglab.com/news/blog/how-to-better-study-ubiquitination/?srsltid=AfmBOopB8miKxpCNbiuKbMbxeH1goNMXBt3PPj_DFLxqHBErBQKlv1aB]
- 30. Characterizing Ubiquitination Sites by Peptide-based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3518114/]
- 31. Thermo Scientific™ Pierce™ Ubiquitin Enrichment Kit [https://www.fishersci.ca/shop/products/pierce-ubiquitin-enrichment-kit/pi89899]
- 32. BioE3 identifies specific substrates of ubiquitin E3 ligases [https://www.nature.com/articles/s41467-023-43326-8]
- 33. K48- and K63-linked ubiquitin chain interactome reveals ... [https://www.life-science-alliance.org/content/7/8/e202402740]
- 34. Ubiquitin Linkage-Specific Affimers Reveal Insights into K6 ... [https://www.sciencedirect.com/science/article/pii/S1097276517306172]
- 35. A mass spectrometry view of stable and transient protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5978421/]
- 36. Optimising methods for the preservation, capture and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4709362/]
- 37. How to Better Study Ubiquitination [https://www.ptglab.com/news/blog/how-to-better-study-ubiquitination/?srsltid=AfmBOoqOGSRL1C3h1BD9kawOkhW5-ZDUP2l9rm6GmvfPDOHZXCyRaAGm]
- 38. Overview of Affinity Purification [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-affinity-purification.html]
- 39. Strategy for Development of Site-Specific Ubiquitin ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2020.00111/full]
- 40. An immunoaffinity purification method for the proteomic ... [https://www.sciencedirect.com/science/article/pii/S0003269713002510]
- 41. Current methodologies in protein ubiquitination characterization [https://cellandbioscience.biomedcentral.com/articles/10.1186/s13578-022-00870-y]
- 42. How to Better Study Ubiquitination [https://www.ptglab.com/news/blog/how-to-better-study-ubiquitination/?srsltid=AfmBOoqMdmLboeUz2u1KOahh7jgwV7w_4r_r66q9ipsZVJbBYRI383GM]
- 43. Dissecting the ubiquitin pathway by mass spectrometry - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC1828906/]
- 44. Proteomic Identification of Protein Ubiquitination Events - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3937853/]
- 45. Detection of Protein Ubiquitination Sites by Peptide ... [https://www.jove.com/t/59079/detection-protein-ubiquitination-sites-peptide-enrichment-mass]
- 46. Time-resolved in vivo ubiquitinome profiling by DIA-MS ... [https://www.nature.com/articles/s41467-021-25454-1]
- 47. Troubleshooting Guide: ELISA [https://www.rndsystems.com/resources/troubleshooting-guide-elisa-development]
- 48. ELISA Troubleshooting Guide [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-elisa/elisa-troubleshooting-guide.html]
- 49. How to Better Study Ubiquitination [https://www.ptglab.com/news/blog/how-to-better-study-ubiquitination/?srsltid=AfmBOorF6RhdOMJ4OeCi_lvBFDdB6Er2QJ-_yaY8qnDH-R6lENUx-DGW]
- 50. Approaches to Sample Preparation of Complex Biological ... [https://rjmseer.com/0044-4502/article/view/684150]
- 51. Ubiquitin Ligase Substrate Identification through ... [https://www.sciencedirect.com/science/article/pii/S002192582087207X]
- 52. Characterizing Ubiquitination Sites by Peptide-based ... [https://www.sciencedirect.com/science/article/pii/S1535947620334502]
- 53. Site-directed multivalent conjugation of antibodies to ... [https://www.nature.com/articles/s41551-024-01342-z]
- 54. Weighing in on Ubiquitin: The Expanding Role of Mass ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1224607/]
- 55. Sensitive and specific affinity purification-mass ... [https://www.sciencedirect.com/science/article/pii/S2667237525002024]
- 56. MS2Query: reliable and scalable MS2 mass spectra-based ... [https://www.nature.com/articles/s41467-023-37446-4]
- 57. Automating PTM Profiling for Mass Spec: Scalable Proteomics ... [https://blog.cellsignal.com/high-throughput-proteomics-automating-ptm-enrichment-for-mass-spec]
- 58. AUTO-SP: automated sample preparation for analyzing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12355475/]
- 59. Automating UbiFast for High-throughput and Multiplexed ... [https://www.sciencedirect.com/science/article/pii/S1535947621001262]
- 60. Blocking Buffers for Western Blot and ELISA [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/blocking-buffers-western-blot-elisa.html]
- 61. Western blot blocking: Best practices [https://www.abcam.com/en-us/knowledge-center/western-blot/western-blot-blocking-methods]
- 62. 4 Ways to Reduce Non-Specific Binding in SPR Experiments [https://nicoyalife.com/blog/4-ways-reduce-non-specific-binding-spr/]
- 63. How to Better Study Ubiquitination [https://www.ptglab.com/news/blog/how-to-better-study-ubiquitination/?srsltid=AfmBOorvgr40_hMT1DbMSURE-Wup1NHk-HOUIFCaD59PAtXuAXLmmTyA]
- 64. PROTAC Drug Discovery [https://lifesensors.com/faqs/protac-drug-discovery-faqs/]
- 65. PROTACs: Just the FAQs [https://www.promegaconnections.com/protacs-just-the-faqs/]
- 66. Proteolysis‐Targeting Chimera (PROTAC) - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC12495453/]
- 67. Targeted protein degradation: mechanisms, strategies and ... [https://www.nature.com/articles/s41392-022-00966-4]
- 68. PROTAC targeted protein degraders: the past is prologue [https://www.nature.com/articles/s41573-021-00371-6]
- 69. Methods to accelerate PROTAC drug discovery - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12312393/]
- 70. Target Validation Using PROTACs: Applying the Four ... [https://www.sciencedirect.com/science/article/pii/S2472555222066977]
- 71. How to Better Study Ubiquitination [https://www.ptglab.com/news/blog/how-to-better-study-ubiquitination/?srsltid=AfmBOopOGk4WUZftdR_5c4P-5sYGIaLcbqF90LTmoHHKOztaA0VJtopm]