The β-Grasp Fold: From Ubiquitin's Structure to Versatile Functions and Therapeutic Targeting
This article provides a comprehensive resource for researchers and drug development professionals on the β-grasp fold, a structurally simple yet functionally versatile protein scaffold.
The β-Grasp Fold: From Ubiquitin's Structure to Versatile Functions and Therapeutic Targeting
Abstract
This article provides a comprehensive resource for researchers and drug development professionals on the β-grasp fold, a structurally simple yet functionally versatile protein scaffold. We explore the evolutionary origins and core structural architecture of this fold, best known for its role in ubiquitin and ubiquitin-like proteins (UBLs). The content details the experimental and computational methodologies used to study β-grasp proteins, addresses key challenges in probing their dynamics and interactions, and compares the diverse functional families within this superfamily. By integrating foundational knowledge with current research, we highlight the significant implications of targeting β-grasp fold pathways, particularly the ubiquitin-proteasome system, for developing novel therapeutics in areas such as cancer, neurodegenerative, and infectious diseases.
The Architectural Blueprint and Evolutionary History of the β-Grasp Fold
The β-grasp fold (β-GF) represents a fundamental and versatile structural motif in protein architecture, prototyped by the ubiquitous protein ubiquitin (UB) [1]. This compact fold is characterized by a β-sheet that appears to "grasp" a single α-helical segment, forming a stable scaffold that has been recruited for a strikingly diverse range of biochemical functions across all domains of life [1]. Its discovery in ubiquitin, a key regulator of protein stability and signaling in eukaryotes, initially highlighted its importance. Subsequent structural studies have revealed its presence in a vast array of proteins with functionally distinct roles, including sulfur transfer, RNA binding, enzymatic activity, and adaptor functions in signaling complexes [1]. This in-depth technical guide delineates the core structural features of the β-grasp fold, its evolutionary trajectory, and its functional plasticity, with a specific focus on its implications for ubiquitin and ubiquitin-like protein (Ubl) research. Understanding this fold is paramount for researchers and drug development professionals, as it forms the structural basis for critical cellular processes, and its dysregulation is often implicated in disease.
Core Structural Features of the β-Grasp Fold
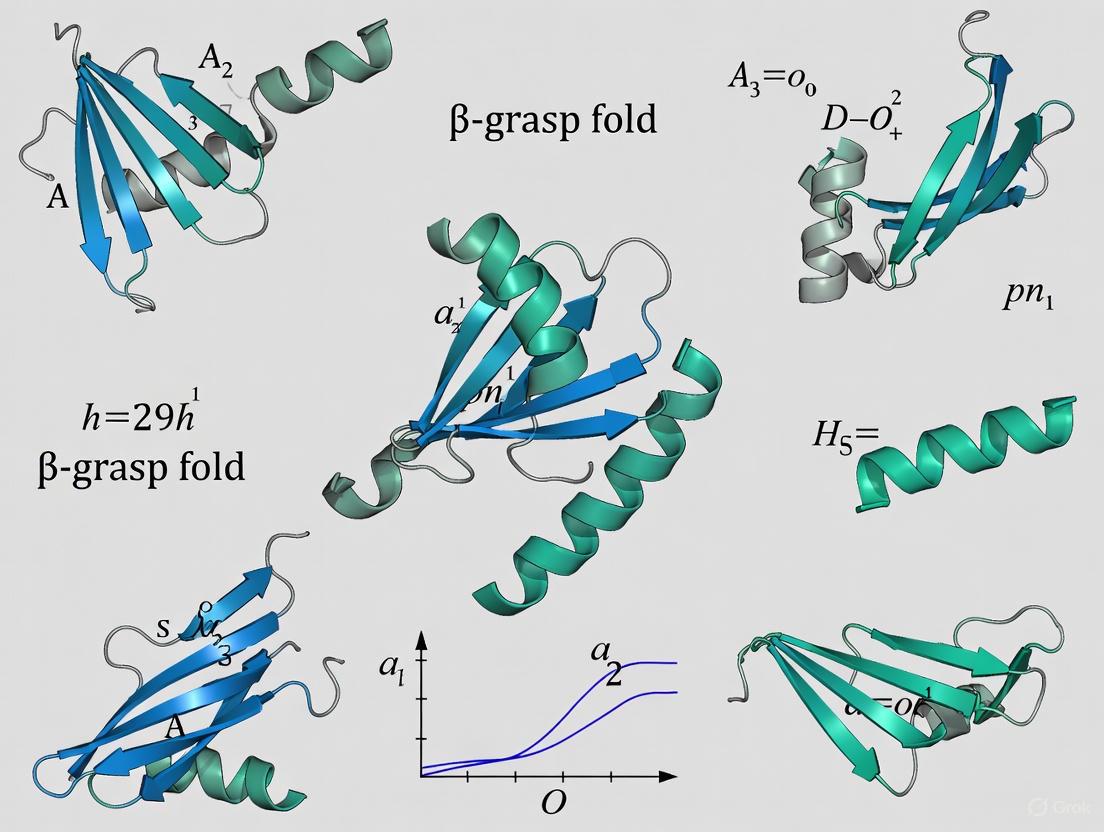
The β-grasp fold is defined by a conserved core structure that serves as a stable platform for functional diversification. The defining characteristic is a β-sheet composed of four to five anti-parallel β-strands that form a twisted, exposed surface. This sheet "grasps" a single α-helical segment that is positioned diagonally across the sheet [1]. The core structural elements are consistently arranged in a specific order, forming the classic β-grasp topology.
Table 1: Core Structural Elements of the β-Grasp Fold
| Structural Element | Description | Functional Role |
|---|---|---|
| β-Sheet | 4-5 anti-parallel strands; provides a large, exposed interaction surface. | Primary site for interactions with diverse partners (proteins, RNA, ligands, co-factors) [1]. |
| α-Helix | Single helical segment; positioned between strands 2 and 3 of the core fold. | Stabilizes the core structure; can participate in specific binding interactions. |
| Loop Regions | Variable connectors between secondary structures; often contain specific inserts. | Major source of functional diversification; can form binding pockets or active sites [2]. |
The structural versatility of the β-GF arises primarily from its prominent β-sheet, which provides an exposed surface for diverse interactions. In some cases, this sheet can also form open barrel-like structures to accommodate other functions [1]. Beyond the core, the fold is subject to numerous elaborations, including inserts of additional secondary structures, such as the β-hairpin found in the transcobalamin-like clade of the SLBB superfamily, which plays a direct role in ligand binding [2]. These structural variations, while adorning the core, do not obscure the fundamental β-grasp topology, which remains readily identifiable.
Diagram 1: The core β-grasp fold structure and its functional versatility.
The β-Grasp Fold in Ubiquitin and Ubiquitin-Like Proteins
The ubiquitin superfamily represents a major radiation of the β-grasp fold within eukaryotes. Ubiquitin itself is a 76-residue polypeptide that adopts the classic β-GF, with a five-stranded β-sheet and a single α-helix [1]. This structural scaffold is not only stable but also serves as the foundation for a vast post-translational modification system. Other Ubiquitin-like proteins (Ubls), such as SUMO, Nedd8, Apg12, and Urm1, share the same core fold and are conjugated to target proteins via a cascade of E1, E2, and E3 enzymes [1]. This system allows for the precise regulation of protein stability, localization, and activity.
The origin of the eukaryotic ubiquitin system is deeply rooted in more ancient bacterial metabolic pathways. Sensitive sequence and structural analyses have revealed that ubiquitin is closely related to bacterial sulfur carrier proteins like ThiS and MoaD, which are involved in thiamine and molybdenum cofactor biosynthesis, respectively [1]. These bacterial proteins also possess a C-terminal glycine that forms a thiocarboxylate, catalyzed by enzymes (ThiF/MoeB) that are structural and mechanistic ancestors of the eukaryotic E1 enzyme [1]. This evolutionary connection highlights a remarkable functional shift: a fold and associated enzymatic machinery originally used for sulfur transfer in core metabolism were co-opted in eukaryotes to form a sophisticated protein-tagging system. The eukaryotic phase of β-GF evolution was marked by a specific expansion of UB-like members, leading to at least 67 distinct families, with 19-20 families already present in the last eukaryotic common ancestor [1].
Functional Diversity and Evolutionary History
The functional repertoire of the β-grasp fold is extraordinarily diverse, extending far beyond the ubiquitin superfamily. Systematic analyses show that this small fold has been independently recruited for multiple distinct biochemical activities throughout evolution [1].
Table 2: Functional Diversity of the β-Grasp Fold
| Functional Category | Example Protein/Domain | Specific Function | Independent Evolutions |
|---|---|---|---|
| Post-translational Modification | Ubiquitin, SUMO, ThiS, MoaD | Protein or sulfur carrier conjugation [1]. | Multiple |
| Enzymatic Activity | NUDIX phosphohydrolases | Hydrolysis of diverse substrates [1]. | ≥ 3 |
| Co-factor Binding | 2Fe-2S Ferredoxin, Molybdopterin-binding | Electron transport, redox reactions [1]. | ≥ 3 (co-factors), ≥ 2 (Fe-S clusters) |
| Soluble Ligand Binding | SLBB Superfamily (e.g., Transcobalamin) | Vitamin B12 binding and uptake [2]. | Multiple |
| RNA Binding | TGS Domain | Binding tRNA and other RNAs [1]. | Multiple |
| Protein-Protein Interaction | RA, PB1, FERM domains | Adaptors in signaling complexes [1]. | Multiple |
Evolutionary reconstruction indicates that the β-grasp fold is ancient, having differentiated into at least seven distinct lineages by the time of the last universal common ancestor (LUCA) [1]. The earliest members were likely involved in RNA metabolism and related functions [1]. Subsequently, the fold radiated into various functional niches. Most of the structural diversification occurred in prokaryotes, while the eukaryotic phase was characterized by a dramatic expansion of Ub-like domains and an increase in the domain architectural complexity of proteins, facilitating their use in numerous adaptor roles [1]. A notable example of ongoing discovery is the identification of the SLBB superfamily, a novel group of β-GF domains that bind soluble ligands like vitamin B12 [2].
Diagram 2: Evolutionary history of the β-grasp fold from LUCA to eukaryotes.
Experimental Protocols for β-Grasp Fold Analysis
Identification of Novel β-Grasp Fold Members
The small size and high divergence of β-GF members make exhaustive identification challenging. A multi-pronged computational strategy is required [1].
Materials:
- Structural Datasets: Protein Data Bank (PDB), SCOP database.
- Sequence Databases: NCBI Non-Redundant (NR) database.
- Software Tools: PSI-BLAST, DALI, HMMER package, T-Coffee multiple alignment tool.
Methodology:
- Seed Collection: Compile a set of known β-GF structures from PDB and SCOP as initial seeds [1].
- Iterative Sequence Profiling: Use seeds to perform PSI-BLAST searches against the NR database. Iterate until convergence (e-value threshold e < 0.01), collecting statistically significant hits [1].
- Structural Similarity Searches: Use programs like DALI to perform structural comparisons with known β-GF domains. Retrieve hits with significant Z-scores (e.g., Z > 5-7) [2].
- Transitive Searches and Model Building: Use newly detected members to initiate further iterative searches. Construct Hidden Markov Models (HMMs) and Position-Specific Scoring Matrices (PSSMs) from alignments to search for more divergent homologs in sequenced genomes [1] [2].
- Multiple Alignment and Classification: Align sequences using a tool like T-Coffee, guided by structural superpositions. Analyze the alignment to identify conserved core features (e.g., glycine residues) and classify sequences into families and superfamilies (e.g., the SLBB superfamily) [2].
Structural and Functional Characterization
Once identified, potential β-GF domains require experimental validation and functional insight.
Materials:
- Cloning and Protein Purification Systems: (e.g., E. coli expression vectors, chromatography equipment).
- Crystallization Trays and X-ray Source: For X-ray crystallography.
- NMR Spectrometer: For solution-state structure determination.
- Functional Assays: (e.g., enzyme activity assays, binding measurements like ITC/SPR).
Methodology:
- Structure Determination:
- X-ray Crystallography: Purify the protein, grow crystals, and solve the structure via molecular replacement or experimental phasing. The structure will confirm the presence of the core β-GF (β-sheet grasping a helix) and reveal any unique inserts (e.g., the β-hairpin in transcobalamin) [3] [2].
- NMR Spectroscopy: For smaller, soluble β-GF domains (like ubiquitin), NMR can be used to determine the solution-state structure and study dynamics.
- Functional Analysis:
- Ligand Binding Studies: Co-crystallize the β-GF domain with its proposed ligand (e.g., vitamin B12 for transcobalamin) or perform binding assays. Analyze the structure to identify contact residues from the core sheet and variable inserts [2].
- Site-Directed Mutagenesis: Mutate conserved residues (e.g., the glycines in the SLBB superfamily or residues in the binding interface) to confirm their role in fold stability and function.
- Structure Determination:
Table 3: The Scientist's Toolkit: Key Research Reagents and Materials
| Reagent/Material | Function/Application |
|---|---|
| PSI-BLAST | Sensitive sequence database searching to identify divergent homologs [1]. |
| DALI Server | Structural similarity searches to detect β-GF folds based on 3D shape [2]. |
| HMMER Suite | Building and searching with probabilistic models (HMMs) for remote homology detection [2]. |
| Protein Data Bank (PDB) | Repository of 3D structural data for use as search seeds and comparative analysis [1]. |
| E. coli Expression Systems | Standard platform for recombinant overexpression of β-GF domain proteins for purification. |
| Crystallization Kits | Sparse matrix screens to identify initial conditions for growing protein crystals. |
| Ubiquitin (Wild-type & Mutants) | Essential control and reference molecule for studies of UB/Ubl structure and function. |
Ubiquitin, a 76-residue regulatory protein, serves as the prototypical member of the β-grasp fold (β-GF), a structural archetype distinguished by its remarkable functional versatility and evolutionary conservation. This fold is characterized by a β-sheet that appears to "grasp" an α-helical segment, forming a compact globular structure [1] [4]. Despite its small size, the β-grasp fold has been recruited for a stunning array of biochemical functions, including post-translational modification, sulfur transfer, RNA binding, enzymatic catalysis, and small molecule coordination [1] [2] [5]. This whitepaper provides a comprehensive technical analysis of the ubiquitin fold, detailing its structural features, evolutionary relationships, and the experimental methodologies central to its study. Framed within ongoing research on ubiquitin and ubiquitin-like proteins (Ubls), this guide aims to equip researchers and drug development professionals with the structural and mechanistic insights necessary to navigate this complex protein family and exploit its therapeutic potential.
The discovery of the ubiquitin fold marked a pivotal advancement in molecular biology. Initially identified as a post-translational modification signal, ubiquitin's structure was first resolved in the 1980s [6]. Structural analyses revealed that ubiquitin's fold was not unique but was shared by functionally disparate proteins, leading to the formal definition of the β-grasp fold [1] [5]. This fold is characterized by a core structure comprising a mixed β-sheet of four to five strands that clutches a single α-helix between its second and third strands [1] [6]. The N and C termini are strategically positioned in close proximity, a feature critical for its function in conjugation [7] [6].
Evolutionary reconstruction indicates that the β-grasp fold had already diversified into at least seven distinct lineages by the time of the last universal common ancestor (LUCA), encompassing much of the structural diversity seen today [1]. The earliest members were likely involved in RNA metabolism and sulfur transfer operations in prokaryotic systems [1]. The eukaryotic lineage witnessed a specific and dramatic expansion of ubiquitin-like (Ubl) members, with the eukaryotic UB superfamily diversifying into at least 67 distinct families [1] [5]. A key innovation in eukaryotes was the integration of Ubl domains into complex multidomain proteins, increasing the architectural complexity of proteins involved in signaling and adaptor roles [1]. This evolutionary history establishes ubiquitin not as an outlier, but as a highly specialized derivative of an ancient and versatile structural scaffold.
Structural Bioinformatics of the β-Grasp Fold
Core Architectural Principles
The β-grasp fold is defined by a conserved core structure that can be elaborated upon through various inserts and extensions, giving rise to its functional diversity. The canonical fold, as prototyped by ubiquitin, includes the following elements [6]:
- A central, mixed β-sheet: Typically composed of five anti-parallel strands arranged with a -1, +3x, +1x, -2x topology [6].
- An α-helix: Positioned between the second and third β-strands, which is "grasped" by the β-sheet.
- A 3₁₀ helix: A shorter helical segment is also commonly present.
The stability of the fold is remarkable, with ubiquitin maintaining its structure across a pH range of 1.18–8.48 and temperatures up to 80°C, exhibiting a melting point near 100°C [6]. This stability is primarily due to extensive intra-hydrogen bonding and a well-packed hydrophobic core, as the fold contains no disulfide bonds, metal ions, or cofactors [6].
Table 1: Secondary Structural Elements of Human Ubiquitin (PDB: 1UBQ)
| Element Type | Start Residue | End Residue | Description | Sequence/Identifier |
|---|---|---|---|---|
| α-Helix | 23 | 34 | 3.5-turn α-helix | IENVKAKIQDKE |
| 3₁₀ Helix | 56 | 59 | Short 3₁₀ helix | LSDY |
| β-Strand 1 | 2 | 7 | N-terminal strand | QIFVKT |
| β-Strand 2 | 12 | 16 | TITLE | |
| β-Strand 3 | 41 | 45 | QRLIF | |
| β-Strand 4 | 48 | 49 | KQ | |
| β-Strand 5 | 66 | 71 | C-terminal strand | TLHLVL |
| β-Turn 1 | 7 | 10 | Type I | TLTG |
| β-Turn 2 | 18 | 21 | Type I | EPSD |
| β-Hairpin 1 | 2-7 | 12-16 | 3:5 hairpin |
Functional Versatility and Structural Elaborations
The manifold functions of the β-grasp fold arise primarily from its prominent β-sheet, which provides an exposed surface for diverse interactions [1]. This surface can mediate protein-protein, protein-RNA, and protein-ligand interactions. In some cases, the sheet can curve to form open barrel-like structures for binding larger ligands or cofactors [1].
Systematic analysis has shown that this small fold has independently evolved to support a wide range of biochemical activities on multiple occasions [1]:
- Enzymatic active sites: Recruited as a scaffold for different enzymes, such as NUDIX phosphohydrolases, on at least three independent occasions.
- Cofactor binding: The binding of diverse cofactors like iron-sulfur clusters and molybdopterin has evolved independently at least three times.
- Soluble ligand binding: A novel superfamily termed the Soluble-Ligand-Binding β-grasp (SLBB) domain was identified, which includes proteins like transcobalamin (vitamin B12 binding), bacterial polysaccharide export proteins, and the Nqo1 subunit of NADPH-quinone oxidoreductase [2].
Table 2: Major Functional Classes of β-Grasp Fold Proteins
| Functional Class | Representative Members | Key Structural Features | Independent Evolutionary Origins |
|---|---|---|---|
| Post-translational Modifiers | Ubiquitin, SUMO, NEDD8, Atg8, Atg12 | Conserved C-terminal glycine for conjugation, exposed hydrophobic patch (Ile44) [1] [6] | Derived from sulfur carrier systems (ThiS/MoaD) [1] |
| Sulfur Carriers | ThiS, MoaD | C-terminal thiocarboxylate, similarity to Ub fold [1] [8] | Ancient, predating Ub |
| Enzymatic Scaffolds | NUDIX hydrolases, Staphylokinases | Active site residues positioned on loops of the β-sheet [1] | At least 3 |
| Iron-Sulfur Cluster Binding | 2Fe-2S Ferredoxins | Cysteine residues ligating the cluster [1] [2] | At least 2 |
| Soluble Ligand Binding (SLBB) | Transcobalamin, Nqo1, ComEA | Inserts for ligand specificity (e.g., β-hairpin in transcobalamin) [2] | At least 2 major clades (Transcobalamin, Nqo1) |
| RNA Binding | TGS domain, IF3, RPB2 subunit | Positive surface patches for nucleic acid interaction [1] | Multiple |
| Protein Interaction Adapters | RA, PB1, FERM-N domains | Surface loops and strands for specific protein binding [1] | Multiple |
The SLBB superfamily exemplifies how structural variations enable new functions. The transcobalamin-like clade is defined by a β-hairpin insert after the core helix, which directly contacts the vitamin B12 ligand [2]. In contrast, the Nqo1-like clade features a distinct insert between strands 4 and 5 of the core fold [2]. Despite different inserts, both clades likely bind their soluble ligands in a similar spatial location relative to the core fold.
Experimental Analysis of Ubiquitin Structure and Folding
Protocol 1: Atomic-Resolution Folding Studies via Molecular Dynamics
Objective: To characterize the folding mechanism, thermodynamics, and kinetics of ubiquitin at an atomic level using equilibrium molecular dynamics (MD) simulations [7].
Methodology:
- System Setup:
- Initial Structures: Simulations are initiated from both the folded state (e.g., PDB 1UBQ) and an extended, unfolded state.
- Solvation: The protein is solvated in a water box (e.g., ~5,581 TIP3P water molecules) with periodic boundary conditions. System size artifacts should be checked using a larger water box.
- Force Field: Use the CHARMM22* force field, modified to correct proline isomerization balance.
- Electrostatics: Employ a Gaussian Split Ewald (GSE) method for long-range electrostatic interactions with a 10.5 Å cutoff. A simple shifted-force truncation is insufficient as it can produce artificially compact unfolded states.
Simulation Execution:
- Equilibration: Equilibrate the system in the NPT ensemble (constant Number of particles, Pressure, and Temperature) for 2 ns.
- Production Run: Perform simulations in the NVT ensemble (constant Number of particles, Volume, and Temperature) at a temperature near the protein's melting point (e.g., 390 K) to observe spontaneous folding and unfolding events. Use a specialized machine like Anton for the required computational throughput.
- Integration: Use a reference system propagator algorithm (RESPA) scheme with a 5 fs inner and 10 fs outer timestep, which can be facilitated by modifying hydrogen and water oxygen masses.
Data Analysis:
- Transition Path Identification: Identify folding/unfolding transition paths using dual cutoffs on the Cα root-mean-square deviation (RMSD) of key secondary structure elements (e.g., residues 2–45 and 65–69).
- Reaction Coordinate Optimization: Optimize a one-dimensional reaction coordinate as a linear combination of the Q-values (native contacts) for individual residues.
- Kinetic and Thermodynamic Analysis: Calculate folding rates and free energy surfaces from the simulations. Φ-values for point mutations can be computed from folding/unfolding rates derived from Langevin dynamics simulations along the optimized reaction coordinate.
- State Clustering: Identify metastable states on the free-energy landscape using kinetic-clustering analysis of Cα–Cα contact autocorrelation functions.
Key Findings: MD simulations reveal that ubiquitin folding is a relatively sequential process following a few dominant paths. The order of formation of native structure is correlated with relative structural stability in the unfolded state. The transition state ensemble (TSE) is characterized by a well-defined folding nucleus in the N-terminal region, involving the α-helix and the first two β-strands, while C-terminal strands are less structured in the TSE [7]. These principles align with those derived from studies of fast-folding proteins.
Protocol 2: Structural Dissection of a Ubiquitin Ligase Complex by Cryo-EM
Objective: To determine the architecture and molecular basis of substrate recognition and ubiquitination by the human HRD1 ubiquitin ligase complex using single-particle cryo-electron microscopy (cryo-EM) [9].
Methodology:
- Complex Preparation:
- Reconstitution: Reconstitute the subcomplex by overexpressing core components (HRD1, SEL1L, and the lectin adapter XTP3B) in HEK293 cells. While native purification is ideal, reconstitution improves yield and homogeneity for structural studies.
- Validation: Conduct functional assays (e.g., monitoring degradation of a known ERAD substrate like CD147) to ensure the reconstituted complex is functional.
- Cryo-EM Workflow:
- Vitrification: Apply purified complex to cryo-EM grids, blot, and plunge-freeze in liquid ethane.
- Data Collection: Collect a large dataset of micrographs using a high-end cryo-electron microscope (e.g., Titan Krios).
- Image Processing:
- Particle picking and 2D classification to select homogeneous particles.
- Ab-initio reconstruction and 3D refinement.
- Perform focused classification and refinement to improve resolution for flexible regions.
- Model Building:
- Dock AlphaFold2-predicted models of components into the cryo-EM density map.
- Manually build and adjust the atomic model in Coot, followed by real-space refinement in Phenix.
Key Findings: The cryo-EM structure of the human HRD1-SEL1L-XTP3B complex revealed that HRD1 forms a dimer, but only one protomer carries the SEL1L-XTP3B complex, forming a 2:1:1 stoichiometry [9]. The structure captured a trimmed N-glycan substrate sandwiched between XTP3B and SEL1L. Furthermore, the engagement of Derlin family proteins was found to induce dramatic conformational changes, breaking the HRD1 dimer and forming a new four-helix bundle from two SEL1L molecules, potentially inducing membrane curvature for retrotranslocation [9].
The following diagram illustrates the key conformational changes in the HRD1 complex induced by Derlin protein binding, as revealed by cryo-EM studies [9].
Table 3: Key Research Reagents for the Study of Ubiquitin and β-Grasp Fold Proteins
| Reagent / Resource | Function / Description | Example Use Case |
|---|---|---|
| CHARMM22* Force Field | An all-atom empirical force field for molecular dynamics simulations, optimized for proteins. | Simulating ubiquitin folding and dynamics at atomic resolution [7]. |
| E1-E2-E3 Enzyme Cascade | The three-enzyme cascade (Activating, Conjugating, and Ligating enzymes) for in vitro ubiquitination. | Reconstituting specific ubiquitin linkage formation on target substrates for biochemical study [8]. |
| MLN4924 (Nedd8-Adenylate Analog) | A mechanism-based inhibitor that forms a covalent adduct with NEDD8, inhibiting the NEDD8 E1 enzyme. | Probing the role of neddylation pathways in cells; a tool for targeted protein stabilization [8]. |
| Cryo-EM with Direct Electron Detectors | High-resolution structural biology technique for visualizing large macromolecular complexes in near-native state. | Determining the architecture of large E3 ligase complexes like HRD1 [9]. |
| Ubiquitin-Binding Domains (UBDs) | Modular protein domains (e.g., UBA, UIM, NZF) that recognize and non-covalently bind ubiquitin motifs. | As pull-down probes to isolate and identify ubiquitylated proteins from cell lysates [6]. |
| Activity-Based Probes (ABPs) for DUBs | Suicide substrates that covalently label the active site of deubiquitinating enzymes (DUBs). | Profiling active DUBs in complex proteomes and inhibitor screening [6]. |
| Tandem Ubiquitin Binding Entities (TUBEs) | Engineered multimeric UBDs with high affinity for polyubiquitin chains, protecting them from DUBs. | Protecting polyubiquitin chains during purification and detecting endogenous ubiquitylation [6]. |
Concluding Perspectives and Future Directions
The ubiquitin prototypical β-grasp fold exemplifies a profound principle in structural biology: a simple, stable scaffold can be evolutionarily co-opted for an extraordinary range of biochemical functions. The functional versatility of this fold stems from its prominent β-sheet, which serves as a versatile interaction surface, and its ability to tolerate structural elaborations like inserts and extensions that confer specificity [1] [2]. From its ancient origins in RNA metabolism and sulfur transfer in prokaryotes, the fold radiated into niches including enzyme catalysis, small molecule binding, and, most notably in eukaryotes, the post-translational regulatory system centered on ubiquitin and Ubls [1].
Future research will focus on several frontiers. First, the full scope of the "ubiquitin code" is still being deciphered, including the physiological functions of atypical ubiquitin linkages and crosstalk with other post-translational modifications like phosphorylation [6]. Second, structural studies on full-length, multi-component complexes like HECT ligases and the HRD1 complex are revealing how domain architecture and conformational dynamics regulate ligase activity and specificity [9] [10]. A key finding is the role of "structural ubiquitin" molecules, which are non-covalently bound and contribute to ligase activity and linkage specificity, as seen in yeast Tom1 [10]. Finally, the continued discovery of new β-grasp families and their functions, particularly in prokaryotes and viruses, promises to uncover novel biology and potential therapeutic targets. A deep understanding of this conserved structural archetype is therefore not only fundamental to cell biology but also crucial for pioneering new therapeutic strategies in disease areas ranging from cancer to neurodegeneration.
The β-grasp fold (β-GF) represents a remarkable evolutionary success story in molecular structural adaptation. Characterized by a five-strand antiparallel β-sheet that appears to grasp a single α-helical segment, this compact fold has been recruited for a strikingly diverse range of biochemical functions throughout the history of cellular life [1] [11]. While best known for its role in eukaryotic ubiquitin and ubiquitin-like proteins (UBLs) that regulate protein degradation and signaling, the deepest origins of this fold predate the emergence of eukaryotes by billions of years [1] [11]. This whitepaper examines the evolutionary journey of the β-grasp fold from its primordial manifestations in prokaryotic systems to its sophisticated regulatory functions in eukaryotic cells, providing researchers with both theoretical frameworks and experimental approaches for investigating these ancient molecular systems.
Evolutionary reconstructions indicate that the β-grasp fold had already differentiated into at least seven distinct lineages by the time of the last universal common ancestor (LUCA) of all extant organisms, encompassing much of the structural diversity observed in modern versions of the fold [1]. The earliest β-grasp members were likely involved in RNA metabolism and subsequently radiated into various functional niches, with most structural diversification occurring in prokaryotes before experiencing specific expansions in eukaryotes [1]. This extensive evolutionary history provides critical context for understanding how simple structural domains can be co-opted for increasingly complex cellular functions across the tree of life.
Evolutionary History and Phylogenetic Distribution
Deep Evolutionary Origins
Molecular clock analyses using pre-LUCA gene duplicates estimate that LUCA lived approximately 4.2 billion years ago (4.09-4.33 Ga), with a genome encoding around 2,600 proteins [12]. This prokaryote-grade anaerobic acetogen possessed an established ecological system, within which early versions of the β-grasp fold likely functioned [12]. The fold appears to have first emerged in the context of translation-related RNA interactions before exploding to occupy various functional niches [11].
The last universal common ancestor contained several β-grasp fold proteins that would subsequently diverge into distinct lineages. Evolutionary reconstruction reveals that the earliest β-grasp members were probably involved in RNA metabolism and subsequently radiated into various functional niches, with most structural diversification occurring in prokaryotes [1]. The eukaryotic phase was mainly marked by a specific expansion of the ubiquitin-like β-grasp members, with the eukaryotic UB superfamily diversifying into at least 67 distinct families, of which at least 19-20 families were already present in the eukaryotic common ancestor [1].
Distribution Across Domains of Life
Table 1: Distribution of β-Grasp Fold Proteins Across Life Domains
| Domain | Representative β-Grasp Proteins | Key Functions | Structural Features |
|---|---|---|---|
| Bacteria | ThiS, MoaD, TtuB, UBact, BilA, Bub | Sulfur transfer, cofactor biosynthesis, antiphage defense | Single or multiple β-grasp domains, filament-forming variants |
| Archaea | SAMPs (Small Archaeal Modifier Proteins) | Protein conjugation, sulfur transfer | Ubiquitin-like β-grasp, forms polysamp chains |
| Eukaryotes | Ubiquitin, SUMO, NEDD8, ISG15, ATG8, ATG12 | Protein degradation, signaling, autophagy, immune response | Classic Ub-fold, UBL domains with conjugation capability |
The β-grasp fold is widely distributed across all domains of life, though its representation and functional specialization vary significantly [13] [11]. In comparison to eukaryotes, prokaryotic proteins with relationships to UBLs are phylogenetically restricted but demonstrate remarkable functional diversity [13]. For example:
- Bacteria possess ubiquitin-like proteins such as Pup in actinobacteria (though structurally distinct from β-GF) and TtuB in Thermus species, which shares the β-grasp fold and has dual functions as both a sulfur carrier and covalently conjugated protein modification [13].
- Archaea encode small archaeal modifier proteins (SAMPs) that share the β-grasp fold and play a ubiquitin-like role in protein degradation, with some lineages possessing seemingly complete sets of genes corresponding to a eukaryote-like ubiquitin pathway [13].
- Eukaryotes have dramatically expanded the UBL clade, with at least 70 distinct UBL families observed, of which nearly 20 families were probably present in the last eukaryotic common ancestor [11].
Structural and Functional Diversity of β-Grasp Fold Proteins
Core Structural Principles
The β-grasp fold is a small, compact protein fold dominated by a β-sheet with 5 anti-parallel β-strands and a single helical segment [1] [11]. The name derives from the characteristic arrangement where the β-sheet appears to grasp the helical segment [11]. Despite its small size, this fold serves as a multifunctional scaffold in diverse biological contexts [11].
Systematic analysis of all known interactions of the fold shows that its manifold functional abilities arise primarily from the prominent β-sheet, which provides an exposed surface for diverse interactions or additionally, by forming open barrel-like structures [1]. This structural versatility has enabled the fold to be recruited for strikingly diverse biochemical functions, including providing scaffolds for different enzymatic active sites, iron-sulfur clusters, RNA-soluble-ligand and co-factor-binding, sulfur transfer, adaptor functions in signaling, assembly of macromolecular complexes, and post-translational protein modification [1].
Functional Spectrum
Table 2: Functional Diversity of β-Grasp Fold Proteins
| Functional Category | Example Proteins | Organismic Domain | Specific Biochemical Role |
|---|---|---|---|
| Sulfur Transfer | ThiS, MoaD, URM1 | Bacteria, Eukarya | Thiamine and molybdenum cofactor biosynthesis |
| Protein Modification | Ubiquitin, SUMO, SAMPs | Eukarya, Archaea | Target proteins for degradation or signaling |
| RNA Binding | TGS domain, IF3 | All domains | Translation regulation, RNA metabolism |
| Enzymatic Activities | NUDIX phosphohydrolases | All domains | Phosphohydrolase activity on diverse substrates |
| Signaling Adaptors | RA, PB1, DCX domains | Eukarya | Mediate protein-protein interactions in signaling |
| Antiphage Defense | BilA, Bub proteins | Bacteria | Conjugate to phage proteins to inhibit virion assembly |
The β-grasp fold demonstrates remarkable functional plasticity, with both enzymatic activities and the binding of diverse co-factors independently evolving on at least three occasions each, and iron-sulfur-cluster-binding on at least two independent occasions [1]. This functional versatility stems from:
- Surface Plasticity: The prominent β-sheet provides an exposed surface for diverse interactions [1].
- Structural Elaborations: Numerous elaborations on the core fold enable functional specialization [1].
- Domain Combinations: Fusion with other domains creates proteins with novel capabilities [14].
In the eukaryotic phase of β-grasp evolution, a key aspect was the dramatic increase in domain architectural complexity of proteins related to the expansion of UB-like domains in numerous adaptor roles [1]. This expansion facilitated the evolution of complex regulatory networks that characterize eukaryotic cellular processes.
Prokaryotic Antecedents of Ubiquitin Signaling
Sulfur Transfer Systems: The Evolutionary Bridge
The evolutionary connection between eukaryotic ubiquitin systems and prokaryotic sulfur transfer machinery represents one of the most compelling examples of molecular exaptation. The first major advances in understanding ubiquitin's origin came with the identification of the sulfur transfer proteins ThiS and MoaD, involved in thiamine and molybdenum cofactor (MoCo) biosynthesis, respectively [11]. These proteins contain β-grasp folds closely related to ubiquitin and form thiocarboxylates at their C-termini, catalyzed by enzymes (ThiF and MoeB) that are strikingly similar to ubiquitin-activating enzymes (E1) [11].
The URM1 protein in eukaryotes represents a molecular fossil bridging sulfur carrier and protein modifier functions [13] [11]. Like ThiS and MoaD, URM1 functions as a sulfur carrier through thiocarboxylate formation in the context of tRNA thiolation, but it also undergoes covalent attachment to target proteins in response to oxidative stress, similar to classical ubiquitin-like modifiers [11]. This dual functionality provides a living snapshot of the evolutionary transition from metabolic to regulatory functions.
Prokaryotic Ubiquitin-like Conjugation Systems
Recent research has revealed that bacteria possess biochemical pathways related to eukaryotic ubiquitination that mediate protein conjugation in contexts such as antiphage immunity [14]. These include:
- Bil (Bacterial ISG15-like) Operons: Encode separate E1, E2, Ubl, and deubiquitinase (DUB) proteins that conjugate their Ubl to phage tail proteins during infection to inhibit virion assembly and infectivity [14].
- Bub (Bacterial Ubiquitination-like) Operons: Previously termed "6E" or "DUF6527" operons, encode E1, E2, Ubl, and peptidase proteins that perform protein conjugation [14].
- Type II CBASS Operons: Encode an E2-E1 fusion protein that conjugates the C-terminus of their cognate CD-NTase to unknown targets to activate antiviral signaling [14].
These bacterial Ubls show high structural diversity, with up to three predicted β-grasp domains and diverse fused N-terminal domains [14]. Many form higher-order oligomers, with a large subset containing three β-grasp domains and forming filamentous assemblies in vitro upon calcium ion binding [14]. This filament formation occurs in diverse Ubls from type II Bil, type I Bub, and type II Bub operons, suggesting this property plays an important role in their function, potentially enabling cells to respond to changes in metal ion concentration during phage infection or other stress conditions [14].
Figure 1: Evolutionary relationships between prokaryotic and eukaryotic β-grasp fold proteins, showing functional transitions from metabolic to regulatory roles.
Experimental Approaches and Methodologies
Key Experimental Protocols
Structural Characterization of Bacterial Ubl Oligomerization
The recent discovery of filament-forming bacterial Ubls requires specialized approaches for structural characterization [14]:
Protocol for Ca²⁺-Induced Filament Analysis
- Protein Purification: Express recombinant bacterial Ubls (BilA or Bub) in E. coli Rosetta2 pLysS strains with N-terminal His-tags using pET-based vectors. Purify using nickel-affinity chromatography followed by TEV protease cleavage to remove tags and subsequent size-exclusion chromatography.
- Calcium Titration: Incubate purified Ubls (5-10 mg/mL) with CaCl₂ across a concentration gradient (0-10 mM) in buffer containing 20 mM HEPES pH 7.5, 150 mM NaCl for 1 hour at 4°C.
- Structural Analysis:
- Cryo-EM Grid Preparation: Apply 3.5 μL samples to glow-discharged gold grids, blot for 3-5 seconds at 100% humidity, and plunge-freeze in liquid ethane.
- Data Collection: Image using a 300 keV cryo-electron microscope with a K3 direct electron detector at 105,000x magnification.
- Image Processing: Process movies with MotionCor2, followed by CTF estimation in CTFFIND4 and particle picking in cryoSPARC.
- X-ray Crystallography: For crystalline specimens, screen crystallization conditions using sitting-drop vapor diffusion with commercial screens. Collect diffraction data at synchrotron beamlines and solve structures by molecular replacement using known β-grasp domains as search models.
Phylogenetic and Genomic Analysis
Protocol for Evolutionary Reconstruction of β-Grasp Fold Families
- Sequence Identification: Perform iterative PSI-BLAST searches of non-redundant databases using known β-grasp domains as queries (E-value threshold < 0.01) [1].
- Multiple Sequence Alignment: Align identified sequences using MAFFT or MUSCLE with structural guidance where available.
- Gene Tree Reconstruction: Construct maximum likelihood trees using IQ-TREE or RAxML with best-fit model selection and 1000 bootstrap replicates.
- Reconciliation Analysis: Use the ALE (Amalgamated Likelihood Estimation) algorithm to reconcile gene trees with species trees, inferring duplications, transfers, and losses [12].
- Ancestral State Reconstruction: Reconstruct ancestral sequences at key nodes (including LUCA) using empirical Bayesian methods as implemented in PAML.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Investigating Prokaryotic Ubl Systems
| Reagent/Category | Specific Examples | Function/Application | Technical Notes |
|---|---|---|---|
| Expression Systems | E. coli Rosetta2 pLysS, pET vectors | Recombinant protein production | Optimized for toxic protein expression |
| Purification Tools | Nickel-NTA resin, TEV protease, Size-exclusion columns | Protein purification and tag removal | Maintain reducing conditions for cysteine mutants |
| Crystallization Screens | Commercial sparse matrix screens (Hampton Research) | Protein crystallization | Screen with/without Ca²⁺ for filamentous Ubls |
| Structural Biology | Cryo-EM facilities, Synchrotron access | High-resolution structure determination | Cryo-EM essential for filamentous assemblies |
| Bioinformatics Tools | PSI-BLAST, MAFFT, IQ-TREE, ALE | Sequence analysis and phylogenetics | Iterative searches crucial for distant homologs |
| Antibodies | Custom anti-Ubl antibodies, His-tag antibodies | Detection and immunoprecipitation | Validate native expression in host systems |
Research Implications and Future Directions
Basic Research Applications
Understanding the evolutionary origins of ubiquitin signaling provides fundamental insights with broad research applications:
- Protein Engineering: The structural plasticity of the β-grasp fold enables engineering of novel protein modifiers with customized specificities [1] [14].
- Evolutionary Analysis: Comparative studies of Ubl systems across domains reveal general principles of molecular evolution and functional diversification [1] [11].
- Systems Biology: Mapping complete Ubl networks in prokaryotes reveals primordial regulatory architectures that evolved into eukaryotic complexity [14] [11].
Therapeutic Opportunities
The emerging understanding of prokaryotic Ubl systems creates novel therapeutic avenues:
- Antimicrobial Strategies: Targeting bacterial Ubl pathways involved in antiphage defense could disrupt bacterial immunity or create new antiviral approaches [14].
- Cancer Therapeutics: Understanding the deep evolutionary origins of ubiquitin signaling may reveal conserved structural features that can be targeted for proteostasis manipulation in cancer cells [15].
- Immunomodulation: As bacterial Ubl systems interact with host ubiquitin pathways during pathogenesis, these interfaces represent potential targets for anti-infective strategies [13].
Unanswered Questions and Research Frontiers
Despite significant advances, key questions remain about prokaryotic antecedents of ubiquitin signaling:
- Mechanistic Details: How do bacterial E1-E2-E3 cascades achieve specificity in the absence of the extensive regulatory networks characteristic of eukaryotic systems? [14] [11]
- Physiological Roles: What are the native functions of bacterial Ubl conjugation systems beyond antiphage defense? [14]
- Evolutionary Transitions: What specific evolutionary steps transformed sulfur-carrier systems into protein modifiers? [13] [11]
- Structural Plasticity: How do the architectural variations in bacterial Ubls (multiple domains, oligomerization) relate to their functional specialization? [14]
Future research should focus on combining comparative genomics with experimental characterization of diverse prokaryotic Ubl systems to build a more complete picture of how this remarkable protein fold has been adapted and readapted throughout evolutionary history.
Figure 2: Integrated workflow for investigating prokaryotic ubiquitin-like systems, combining bioinformatic, structural, and functional approaches.
The investigation of prokaryotic antecedents of ubiquitin signaling has transformed our understanding of one of biology's most important regulatory systems. What was once considered a eukaryotic innovation is now recognized as having deep evolutionary roots extending back to LUCA, with functional precursors in prokaryotic sulfur transfer systems that were progressively co-opted for regulatory functions [1] [11]. The recent discovery of sophisticated bacterial ubiquitination-like pathways involved in antiphage defense demonstrates that protein conjugation systems continue to evolve novel functions in prokaryotic contexts [14].
For researchers and drug development professionals, these evolutionary insights provide valuable perspectives for manipulating ubiquitin signaling in therapeutic contexts. The structural and functional diversity of β-grasp proteins across life domains represents a rich source of mechanistic insights and potential engineering templates. As research continues to unravel the molecular complexities of these ancient systems, our ability to harness their principles for basic research and therapeutic applications will undoubtedly expand, potentially leading to new classes of therapeutics that target the deep evolutionary foundations of cellular regulation.
The last universal common ancestor (LUCA) represents a pivotal stage in early evolution, possessing a complex cellular structure with a genome encoding approximately 2,600 proteins [12] [16]. This organism exhibited sophisticated metabolic capabilities and an early immune system, existing within an established ecological framework. From this ancestral state, a remarkable functional radiation occurred, whereby primitive biomolecules diversified to fulfill specialized roles across biochemistry. This review examines the trajectory of this radiation, with particular focus on the β-grasp fold, a structural scaffold that evolved from fundamental RNA metabolism functions in LUCA to specialized sulfur transfer systems and eventually to the complex regulatory apparatus of ubiquitin and ubiquitin-like proteins in modern eukaryotes.
Functional radiation describes the evolutionary process where a single ancestral structure or molecule diversifies into multiple forms with distinct biological functions. Unlike adaptive radiation typically observed at the organismal level, functional radiation operates at the molecular level, where protein folds and metabolic pathways are recruited for novel functions through gene duplication, divergence, and structural adaptation.
Current research indicates LUCA was a prokaryote-grade, anaerobic acetogen with a genome of at least 2.5 Mb, encoding approximately 2,600 proteins – comparable in complexity to modern prokaryotes [12]. LUCA possessed a functional CRISPR-Cas system, indicating an established evolutionary arms race with viral elements [16]. The β-grasp fold (β-GF), prototyped by modern ubiquitin, represents a prime example of molecular exaptation, where a simple structural scaffold was repeatedly recruited for novel functions throughout evolutionary history.
Table 1: Key Characteristics of the Last Universal Common Ancestor (LUCA)
| Feature | Reconstruction | Methodology | Citation |
|---|---|---|---|
| Age | ~4.2 Ga (4.09-4.33 Ga) | Divergence time analysis of pre-LUCA gene duplicates | [12] |
| Genome Size | ~2.5 Mb (2.49-2.99 Mb) | Phylogenetic reconciliation & comparative genomics | [12] |
| Proteome | ~2,600 proteins | Probabilistic gene-tree species-tree reconciliation | [12] [16] |
| Metabolism | Anaerobic acetogen with Wood-Ljungdahl pathway | Phylogenomic analysis of metabolic proteins | [12] [16] |
| Cellular Features | DNA genome, ribosomes, cell membrane, ion transporters | Universal conserved cellular machinery | [17] [16] |
| Ecological Context | Part of established ecosystem, possibly hydrothermal vents | Metabolic reconstruction & geochemical constraints | [12] [17] |
The β-Grasp Fold: A Versatile Structural Scaffold
The β-grasp fold is a compact protein domain characterized by a β-sheet with 4-5 antiparallel strands that appears to "grasp" an α-helical segment [1]. This simple yet versatile architecture serves as a structural scaffold for an extraordinary diversity of biochemical functions in modern organisms. The fold is defined by its core structural features, which provide stable surfaces for molecular interactions while accommodating extensive sequence variation.
Structural Characteristics and Functional Versatility
The manifold functional abilities of the β-grasp fold arise primarily from its prominent β-sheet, which provides an exposed surface for diverse interactions. In some cases, these sheets form open barrel-like structures that accommodate larger ligands or catalytic centers [1]. This structural plasticity has enabled the fold to be recruited for:
- Enzymatic active sites (e.g., NUDIX phosphohydrolases)
- Iron-sulfur cluster binding
- RNA, soluble ligand, and co-factor binding
- Sulfur transfer reactions
- Adaptor functions in signaling pathways
- Assembly of macromolecular complexes
- Post-translational protein modification
Systematic analysis indicates that both enzymatic activities and co-factor binding have independently evolved on at least three separate occasions within different β-grasp fold lineages, while iron-sulfur-cluster-binding emerged at least twice independently [1].
Evolutionary Trajectory: From LUCA to Modern Systems
β-Grasp Fold Lineages Already Present in LUCA
Evolutionary reconstruction indicates that by the time of LUCA, the β-grasp fold had already differentiated into at least seven distinct lineages, encompassing much of the structural diversity observed in extant versions of the fold [1]. The earliest β-grasp fold members in pre-LUCA evolution were likely involved in fundamental RNA metabolism, with subsequent radiation into various functional niches.
Table 2: Major Evolutionary Transitions in β-Grasp Fold Function
| Evolutionary Stage | Functional Innovations | Key Examples | Evidence | |
|---|---|---|---|---|
| Pre-LUCA RNA World | RNA metabolism, nucleotide binding | Primitive RNA-binding domains | Phylogenetic analysis | [1] [18] |
| LUCA Era | Diversification into 7 distinct lineages | Sulfur transfer systems (ThiS, MoaD) | Universal distribution in Archaea and Bacteria | [1] |
| Early Prokaryotic Radiation | Metabolic specialization, co-factor binding | NUDIX hydrolases, ferredoxins, SLBB domains | Lineage-specific expansions in bacteria and archaea | [1] |
| Eukaryotic Emergence | Protein modification, signaling adaptors | Ubiquitin, Ubls, adaptor domains (UBX, RA, PB1) | Domain architecture complexity increase | [1] |
The Sulfur Transfer Precursor Hypothesis
The evolutionary connection between sulfur transfer systems and ubiquitin-like protein modification represents a particularly illuminating example of functional radiation. The sulfur transfer proteins ThiS and MoaD, involved in thiamine and molybdenum cofactor biosynthesis respectively, contain β-grasp folds closely related to ubiquitin [1]. These systems share remarkable mechanistic similarities with modern ubiquitination:
- C-terminal residues form thiocarboxylates (analogous to ubiquitin's C-terminal glycine)
- Activation by enzymes (ThiF and MoeB) structurally similar to E1 ubiquitin-activating enzymes
- ATP-dependent activation mechanisms
This phylogenetic and mechanistic evidence strongly supports the hypothesis that eukaryotic ubiquitin-conjugation systems evolved from more ancient bacterial precursors involved in sulfur transfer reactions for metabolite biosynthesis [1].
Experimental Approaches for Reconstruction of Ancient Functions
Phylogenomic Analysis and Tree Reconciliation
Methodology: Probabilistic gene-tree species-tree reconciliation using algorithms such as ALE (Amalgamated Likelihood Estimation) enables reconstruction of gene family evolution across deep evolutionary timescales [12].
Protocol:
- Species Tree Construction: Infer a reference species tree from universal marker genes (57 phylogenetic markers recommended) across broadly sampled archaeal and bacterial lineages [12].
- Gene Family Identification: Cluster orthologous groups using KEGG Orthology (KO) or Clusters of Orthologous Genes (COG) databases.
- Gene Tree Reconstruction: Generate bootstrapped phylogenetic trees for each gene family.
- Reconciliation Analysis: Compare gene trees to the species tree to infer evolutionary events (duplications, transfers, losses) using probabilistic models.
- Ancestral State Reconstruction: Calculate presence probabilities for gene families at ancestral nodes, including LUCA.
Application: This approach identified 399 high-probability protein families in LUCA, with estimates of a total proteome of 2,451-2,855 proteins, indicating substantial molecular complexity [12] [16].
Universal Paralog Dating for Molecular Clock Calibration
Methodology: Divergence time estimation using pre-LUCA gene duplicates provides cross-bracing for molecular clock analyses [12].
Protocol:
- Paralog Identification: Identify protein families that duplicated before LUCA (e.g., catalytic and non-catalytic subunits of ATP synthases, elongation factors EF-Tu and EF-G) [12].
- Sequence Alignment and Tree Building: Construct phylogenetic trees for each paralogous family.
- Fossil Calibration: Apply multiple fossil calibrations (e.g., oxygenic photosynthesis evidence ~2.95 Ga) to constrain node ages.
- Cross-bracing Implementation: Use shared nodes across paralog trees to improve divergence time estimates.
- Molecular Clock Analysis: Apply relaxed clock models (GBM and ILN) to estimate divergence times with confidence intervals.
Application: This method dated LUCA to approximately 4.2 Ga (4.09-4.33 Ga), suggesting rapid evolution from life's origin to complex cellular organization [12].
Structural Phylogenetics and Fold Analysis
Methodology: Comprehensive sequence-structure analysis to identify remote homology and evolutionary relationships across diverse β-grasp fold members [1].
Protocol:
- Structure-Based Searches: Use known β-grasp fold structures as queries for similarity searches against structural databases.
- Iterative Sequence Profiling: Apply PSI-BLAST with progressively built hidden Markov models to detect distant homologs.
- Topological Analysis: Compare structural topologies and conservation of core elements.
- Functional Site Mapping: Identify conserved residues and structural motifs associated with specific functions.
- Phylogenetic Tree Construction: Build trees based on structural alignments to reconstruct evolutionary relationships.
Application: This approach revealed previously unrecognized β-grasp fold variants and established evolutionary connections between functionally distinct families [1].
Diagram 1: Evolutionary trajectory of β-grasp fold functions from LUCA to modern systems
The Scientist's Toolkit: Key Research Reagents and Methods
Table 3: Essential Research Tools for Studying β-Grasp Fold Evolution and Function
| Reagent/Method | Specific Application | Function/Utility | Example Use Case | |
|---|---|---|---|---|
| ALE Software | Phylogenetic reconciliation | Infers gene family evolution events (duplication, transfer, loss) | Reconstructing LUCA gene content | [12] |
| PSI-BLAST | Remote homology detection | Identifies distantly related protein sequences | Finding novel β-grasp fold members | [1] |
| Molecular Clock Calibration (Universal Paralogs) | Deep evolutionary dating | Provides cross-bracing for divergence time estimates | Dating LUCA with pre-LUCA duplicates | [12] |
| CRISPR-Cas Systems | Ancient immunity study | Models early host-viral coevolution | Understanding LUCA's defense mechanisms | [12] [16] |
| Twister Ribozymes | RNA world reconstruction | Molecular fossils of early RNA catalysis | Studying pre-LUCA RNA replication | [18] |
| Single-Molecule Techniques (Magnetic Tweezers) | Enzyme mechanism analysis | Characterizes low-probability catalytic events | Studying error-prone RNA polymerases | [18] |
| Structural Phylogenetics | Fold evolution mapping | Establishes evolutionary relationships based on structure | Connecting ubiquitin to sulfur transfer proteins | [1] |
The functional radiation from LUCA represents a fundamental evolutionary process whereby limited molecular components diversified to create biochemical complexity. The β-grasp fold exemplifies this phenomenon, evolving from fundamental RNA metabolism and sulfur transfer functions in LUCA to the sophisticated regulatory systems of modern eukaryotes. This trajectory underscores how ancient metabolic systems can be co-opted for novel signaling and regulatory functions through evolutionary processes.
Future research should focus on: (1) experimental reconstruction of ancestral β-grasp fold proteins to test functional predictions; (2) exploration of potential ubiquitin-like conjugation systems in extant bacteria and archaea; and (3) investigation of how structural plasticity enables functional radiation at the molecular level. Understanding these evolutionary principles provides not only insights into life's history but also frameworks for engineering novel protein functions for therapeutic and biotechnological applications.
Diversification in Prokaryotes vs. UBL Expansion in Eukaryotes
The β-grasp fold (β-GF) is a compact protein structural scaffold characterized by a β-sheet with five anti-parallel strands that appears to "grasp" a single α-helical segment [1] [11]. This ancient fold serves as a remarkable example of evolutionary recruitment, having been utilized for a strikingly diverse range of biochemical functions across all domains of life. While this fold is prototyped by eukaryotic ubiquitin (Ub), its evolutionary history reveals a fundamental divergence in adaptive strategies between prokaryotes and eukaryotes. Research indicates that prokaryotes primarily exploited this fold for structural and functional diversification, leading to its incorporation into various enzymatic and metabolic pathways. In contrast, eukaryotes leveraged this scaffold for a massive expansion of ubiquitin-like protein (UBL) modifiers that regulate cellular physiology through reversible post-translational modifications [1] [19] [11]. This whitepaper examines the comparative evolutionary trajectories of the β-grasp fold in prokaryotic and eukaryotic systems, with implications for understanding fundamental biological mechanisms and developing targeted therapeutic interventions.
Structural Hallmarks and Functional Versatility of the β-Grasp Fold
Core Structural Features
The β-grasp fold comprises several conserved structural elements that contribute to its stability and functional versatility:
- A β-sheet with five anti-parallel strands that forms the core structural scaffold
- A single α-helical segment positioned adjacent to and "grasped" by the β-sheet
- Potential for diverse loop regions and insertions that enable functional specialization
- A compact globular structure that provides a stable platform for molecular interactions [1] [11] [20]
The fold's remarkable functional plasticity arises from its ability to present diverse interaction surfaces, particularly through the prominent β-sheet, which provides an exposed platform for binding various biomolecules or forming open barrel-like structures [1].
Functional Versatility of the Fold
The β-grasp fold has been recruited for an extraordinary diversity of biological functions, including:
Table 1: Functional Diversity of β-Grasp Fold Proteins
| Function Category | Specific Examples | Organismic Distribution |
|---|---|---|
| Post-translational modification | Ubiquitin, SUMO, NEDD8 | Primarily eukaryotic |
| Enzymatic activities | NUDIX phosphohydrolases, staphylokinases | Both prokaryotic and eukaryotic |
| Cofactor binding/scaffolding | Iron-sulfur clusters, molybdopterin | Primarily prokaryotic |
| RNA/soluble ligand binding | TGS domain, SLBB domain, vitamin B12 binding | Both prokaryotic and eukaryotic |
| Sulfur transfer | ThiS, MoaD in thiamine and molybdenum cofactor biosynthesis | Primarily prokaryotic |
| Adaptor functions | RA, PB1, FERM N-terminal domains | Primarily eukaryotic |
| Toxin activities | Staphylococcal enterotoxin B, superantigens | Primarily prokaryotic |
This functional diversity emerged early in evolution, with the β-grasp fold already having differentiated into at least seven distinct lineages by the time of the last universal common ancestor (LUCA) of all extant organisms [1].
Prokaryotic Diversification: Functional and Structural Radiation
Early Evolutionary Radiation in Prokaryotes
Comparative genomic analyses reveal that the most extensive structural and functional diversification of the β-grasp fold occurred in prokaryotes. By the time of LUCA, the fold had already differentiated into multiple distinct lineages that encompassed much of the structural diversity observed in extant versions [1] [11]. The earliest β-grasp fold members were likely involved in RNA metabolism and translation-related RNA interactions, as evidenced by the TGS domain found in aminoacyl tRNA synthetases and other translation regulators [1].
This prokaryotic diversification led to the incorporation of the fold into various metabolic pathways, particularly those involving sulfur transfer and cofactor biosynthesis. Key examples include:
- ThiS: Involved in thiamine biosynthesis, forms a thiocarboxylate at its C-terminus for sulfur transfer
- MoaD: Participates in molybdenum cofactor biosynthesis, similarly employs a C-terminal thiocarboxylate
- Iron-sulfur cluster scaffolding: β-grasp fold proteins serve as scaffolds for iron-sulfur clusters in ferredoxins [1] [21] [22]
Enzymatic Recruitment and Novel Functions
Prokaryotes extensively recruited the β-grasp fold for diverse enzymatic functions, with both enzymatic activities and cofactor-binding having independently evolved on multiple occasions [1]. Notable examples include:
- NUDIX phosphohydrolases: Utilize the β-grasp fold as a scaffold for catalytic activities against diverse substrates
- Staphylokinases and streptokinases: Fibrinolytic enzymes in low GC Gram-positive bacteria
- TmoB: A subunit of the aromatic monooxygenase oxygenase complex with structurally conserved β-grasp fold
- RnfH: A component of Rnf dehydrogenases utilizing the fold for structural integrity [1] [11]
Table 2: Key Prokaryotic β-Grasp Fold Proteins and Their Functions
| Protein/Domain | Function | Biological Context |
|---|---|---|
| ThiS | Sulfur carrier in thiamine biosynthesis | Forms thiocarboxylate, activated by ThiF (E1-like) |
| MoaD | Sulfur carrier in molybdenum cofactor biosynthesis | Forms thiocarboxylate, activated by MoeB (E1-like) |
| TGS domain | RNA binding | Found in aminoacyl tRNA synthetases, translation regulators |
| YukD | Unknown function | Conserved in Bacillus subtilis and related bacteria |
| SLBB domain | Soluble ligand binding | Binds vitamin B12 and other solutes |
| β-grasp ferredoxin | Iron-sulfur cluster binding | Metal chelation via cysteine-containing flaps |
Prokaryotic Ubiquitin-like Conjugation Systems
Despite the absence of canonical ubiquitin in prokaryotes, several ubiquitin-like modification systems have been identified:
- Pup (prokaryotic ubiquitin-like protein): An intrinsically disordered protein in actinobacteria that targets proteins for proteasomal degradation through conjugation (pupylation) [21] [23]
- SAMP (small archaeal modifier proteins): Ub-fold proteins in archaea that form isopeptide bonds with target proteins (sampylation) [21]
- TtuB: Ub-fold protein in Thermus species involved in both protein modification and sulfur-transfer pathways [21]
These systems represent evolutionary innovations that parallel eukaryotic ubiquitination but employ distinct mechanisms and enzymes. The pupylation system, for instance, utilizes a single ligase (PafA) and depupylase (Dop) rather than the multi-enzyme cascade characteristic of eukaryotic ubiquitination [23].
Eukaryotic Expansion: The Ubiquitin-Like Protein Explosion
Massive Diversification of UBL Modifiers
The eukaryotic phase of β-grasp fold evolution was predominantly characterized by a dramatic expansion of ubiquitin-like proteins, with at least 70 distinct UBL families distributed across eukaryotes [19] [11]. Genomic evidence indicates that nearly 20 UBL families were already present in the last eukaryotic common ancestor, including:
- Multiple protein-conjugated forms (Ub, SUMO, NEDD8, URM1, Apg12)
- Lipid-conjugated forms (ATG8)
- Versions functioning as adaptor domains in multi-module polypeptides [1] [19] [11]
This expansion was accompanied by an increase in domain architectural complexity, with UBL domains incorporated into numerous proteins as adaptors in various signaling contexts [1].
The Ubiquitin Signaling System
The eukaryotic ubiquitin system represents one of the most elaborate manifestations of the β-grasp fold, characterized by:
- A conserved three-enzyme cascade (E1-E2-E3) for conjugation
- A dedicated system for deconjugation by deubiquitinating enzymes (DUBs)
- Specialized recognition receptors for ubiquitin signals
- The capacity to form diverse chain architectures through different lysine linkages [1] [20]
The different types of ubiquitin modifications create a sophisticated "ubiquitin code" that regulates practically all aspects of eukaryotic biology, from protein degradation to DNA repair, signaling transduction, and immune responses [20].
Functional Specialization of UBL Families
The expansion of UBL families in eukaryotes enabled functional specialization and compartmentalization:
Table 3: Major Eukaryotic UBL Families and Their Functions
| UBL Family | Primary Functions | Conjugation System |
|---|---|---|
| Ubiquitin | Protein degradation, DNA repair, signaling, endocytosis | E1-E2-E3 cascade |
| SUMO | Nuclear transport, transcriptional regulation, DNA repair | E1-E2-E3 cascade |
| NEDD8 | Regulation of cullin-RING ligases, cell signaling | E1-E2-E3 cascade |
| Apg12 | Autophagy, vesicle trafficking | E1-E2-like system |
| ATG8 | Autophagy, membrane dynamics | E1-E2-like system |
| ISG15 | Immune response, antiviral defense | E1-E2-E3 cascade |
| UFM1 | ER stress response, development | E1-E2-E3 cascade |
| Urm1 | tRNA thiolation, oxidative stress response | E1-like enzyme |
The early diversification of UBL families played a major role in the emergence of characteristic eukaryotic cellular systems, including nucleo-cytoplasmic compartmentalization, vesicular trafficking, lysosomal targeting, protein processing in the endoplasmic reticulum, and chromatin dynamics [19] [11].
Evolutionary Relationships and Transitional Systems
Evolutionary Origins of Eukaryotic UBL Systems
Recent comparative genomics indicates that precursors of the eukaryotic Ub-system were already present in prokaryotes [11] [22]. The simplest versions combine a Ubl and an E1-like enzyme involved in metabolic pathways related to metallopterin, thiamine, cysteine, siderophore, and modified base biosynthesis [11]. Key evolutionary connections include:
- ThiS/MoaD in prokaryotes are closely related to eukaryotic URM1
- ThiF/MoeB enzymes are structural and functional analogs of eukaryotic E1 enzymes
- Similar mechanisms of C-terminal adenylation and sulfur transfer through thiocarboxylate formation [11] [22]
These systems appear to have been recruited in eukaryotes for protein modification, with sampylation in archaea and urmylation in eukaryotes representing direct recruitment of such systems as simple protein-tagging apparatuses [11].
Transitional Systems Bridging the Functional Gap
Several systems represent evolutionary transitions between sulfur carrier and protein modifier functions:
- Urm1: Functions as both a sulfur carrier in tRNA thiolation and a protein modifier in response to oxidative stress
- SAMPs: Archaeal modifiers that function in both sulfur transfer and protein modification, with SAMP2 capable of forming poly-samp chains analogous to polyubiquitin
- TtuB: Functions in both protein modification and tRNA thiolation in Thermus species [21] [11]
These dual-function systems provide fascinating insights into how the eukaryotic ubiquitination system may have evolved from more ancient metabolic sulfur-transfer systems [11].
The following diagram illustrates the evolutionary relationships and functional transitions between prokaryotic and eukaryotic β-grasp fold systems:
Figure 1: Evolutionary Transitions of β-Grasp Fold Function
Experimental Approaches and Research Tools
Key Methodologies for Studying β-Grasp Fold Evolution
Research into the evolution and diversification of β-grasp fold proteins employs several sophisticated methodologies:
Comparative Genomic Analysis
- Sequence profile searches: Using tools like PSI-BLAST to identify distant homologs through iterative searches [1] [22]
- Domain architecture analysis: Examining gene neighborhoods and domain fusions to infer functional relationships [22]
- Phylogenetic profiling: Reconstructing evolutionary relationships across diverse taxa [1]
Structural Analysis
- Structural similarity clustering: Using programs like DALI to calculate pairwise structural alignment Z-scores [22]
- Topological similarity assessment: Identifying structurally conserved regions despite sequence divergence [1]
- Fold recognition: Detecting remote homologs through structural rather than sequence similarity [1]
Functional Characterization
- Enzyme activity assays: Measuring adenylation, conjugation, and deconjugation activities [21] [23]
- Interaction studies: Using bacterial two-hybrid systems, pull-down assays, and co-expression studies [21]
- Mass spectrometry: Identifying covalent modifications and chain architectures [21] [20]
Essential Research Reagents and Tools
The following table outlines key reagents and methodologies essential for research in this field:
Table 4: Research Reagent Solutions for β-Grasp Fold Studies
| Reagent/Methodology | Function/Application | Key Features |
|---|---|---|
| PSI-BLAST | Detection of distant homologs through sequence profile searches | Iterative search strategy, sensitive for remote homology detection |
| DALI Server | Structural comparison and fold recognition | Z-score based structural alignment, database scanning |
| Tandem Mass Spectrometry | Identification of covalent modifications (ubiquitination, pupylation) | Detection of isopeptide linkages, chain architecture determination |
| Bacterial Two-Hybrid System | Protein-protein interaction screening | Particularly useful for identifying prokaryotic interaction networks |
| JAB Domain Proteases | Deconjugation enzyme tools | Study of ubiquitin/UBL removal mechanisms, conservation from prokaryotes to eukaryotes |
| E1-like Adenylating Enzymes | Activation of UBLs for conjugation | Study of initial activation step, conservation across domains of life |
| Polyclonal Anti-Ub/UBL Antibodies | Detection of modified proteins in cellular contexts | Specific recognition of modifier proteins and their conjugates |
Research Implications and Future Directions
The comparative analysis of β-grasp fold evolution between prokaryotes and eukaryotes reveals fundamental principles of molecular evolution, including:
- How simple structural scaffolds can be recruited for diverse functions
- How metabolic systems can evolve into sophisticated signaling pathways
- How domain architecture complexity contributes to functional innovation
From a therapeutic perspective, understanding these evolutionary relationships provides valuable insights for:
- Antibiotic development: Targeting bacterial-specific ubiquitin-like systems such as pupylation in Mycobacteria [23]
- Cancer therapeutics: Exploiting the ubiquitin-proteasome system for targeted protein degradation
- Autoimmune and inflammatory diseases: Modulating ubiquitin-like signaling pathways in immune regulation
Future research directions should focus on:
- Characterizing the numerous uncharacterized β-grasp fold proteins identified in genomic studies
- Elucidating the structural determinants of functional specificity within this fold family
- Developing chemical probes that selectively target specific ubiquitin-like modification pathways
- Exploring the therapeutic potential of modulating prokaryotic ubiquitin-like systems for antimicrobial applications
The evolutionary journey of the β-grasp fold from simple prokaryotic metabolic functions to sophisticated eukaryotic signaling systems exemplifies how nature creatively reuses successful structural templates, providing both fundamental biological insights and practical therapeutic opportunities.
Techniques for Probing Structure, Dynamics, and Drug Discovery Applications
The β-grasp fold is a widespread and evolutionarily ancient protein structural motif characterized by a β-sheet, typically composed of four or five strands, that wraps around a single α-helix [1] [2]. This compact fold serves as a versatile structural scaffold that has been recruited for a strikingly diverse range of biochemical functions throughout evolution. Its most renowned representatives are ubiquitin (UB) and the family of ubiquitin-like proteins (UBLs), which include well-characterized members such as NEDD8, SUMO, and Ufm1 [1] [24] [25]. Despite their low sequence identity—for instance, only 14% between Ufm1 and ubiquitin—these proteins share this conserved tertiary structure [24]. The functional versatility of the β-grasp fold arises primarily from its prominent β-sheet, which provides an exposed surface for diverse interactions, and its ability to form open barrel-like structures, enabling roles in enzyme active sites, binding of iron-sulfur clusters, RNA-soluble-ligand and co-factor-binding, sulfur transfer, and critical adaptor functions in signaling and post-translational modification [1]. The structural elucidation of UBLs is therefore not merely an exercise in structure determination but a fundamental prerequisite for understanding their vast functional roles in cellular regulation, disease mechanisms, and potential therapeutic targeting.
The β-Grasp Fold: A Versatile Structural Scaffold
Core Structural Features and Functional Diversity
The canonical β-grasp fold, as prototyped by ubiquitin, consists of a mixed β-sheet with five anti-parallel strands and a single α-helix that is grasped by the sheet [1]. This core structure is both stable and malleable, allowing for significant topological variations and elaborations that underlie its functional adaptability. Systematic analyses have revealed that by the time of the last universal common ancestor (LUCA), the β-grasp fold had already differentiated into at least seven distinct lineages, encompassing much of the structural diversity seen today [1].
The table below summarizes major functional classes of β-grasp fold proteins and their representative members.
Table 1: Functional Diversity of the β-Grasp Fold
| Functional Class | Representative Members | Key Function |
|---|---|---|
| Post-translational Protein Modifiers | Ubiquitin, NEDD8, SUMO, Ufm1, Apg12 | Covalent modification of target proteins to regulate stability, activity, or localization [1] [24]. |
| Enzymatic Scaffolds | MutT/Nudix phosphohydrolases | Provide a scaffold for enzymatic active sites [1]. |
| Co-factor Binding Proteins | 2Fe-2S Ferredoxins, PduS | Bind iron-sulfur clusters or other co-factors [1] [2]. |
| Soluble Ligand Binders | Transcobalamin, SLBB Superfamily | Bind small molecules like vitamin B12 [2]. |
| RNA Binding Proteins | TGS Domain | Mediate RNA-protein interactions [1]. |
| Protein-Protein Interaction Adaptors | RA, PB1, FERM domains | Act as adaptors in signaling complexes [1]. |
| Toxins & Superantigens | Staphylococcal enterotoxin B | Mediate toxic shock syndrome [1]. |
A key evolutionary innovation within this fold is the ubiquitin superfamily. In eukaryotes, this family diversified into at least 67 distinct families, with 19–20 families already present in the eukaryotic common ancestor [1]. This expansion was coupled with a dramatic increase in domain architectural complexity, facilitating the sophisticated regulatory networks that are characteristic of eukaryotic cells.
Structural Variations and Ligand Binding
The core β-grasp fold is often embellished with secondary structure inserts that confer functional specificity. A prime example is the Soluble-Ligand-Binding β-grasp (SLBB) superfamily. Members of this superfamily, such as the C-terminal domain of transcobalamin (a vitamin B12 uptake protein), are characterized by the insertion of a β-hairpin after the core α-helix [2]. This insert plays a prominent role in contacting the soluble ligand, with additional interactions contributed by residues from the core β-sheet itself. This demonstrates how the robust core scaffold can be adapted through localized elaborations to generate novel ligand-binding surfaces [2].
Principles of Structure Determination for UBLs
The small size (typically 8-20 kDa) and generally high solubility of standalone UBLs make them amenable to high-resolution structural determination by both X-ray crystallography and Nuclear Magnetic Resonance (NMR) spectroscopy. These techniques provide complementary insights into protein structure.
X-ray Crystallography
X-ray crystallography aims to generate a precise, atomic-resolution, three-dimensional model of a protein based on the diffraction pattern of X-rays passing through a crystalline lattice of the molecule.
Table 2: Key Experimental Parameters from UBL Structure Determinations
| Protein | Technique | Resolution (Å) / Comments | Space Group | R-factor / R-free | Citation |
|---|---|---|---|---|---|
| Human NEDD8 | X-ray | 1.6 Å | Information not in search results | 21.9% | [25] |
| Human Ufc1 | X-ray | 2.54 Å | P1 (with 4 protomers in ASU) | Refined with strong NCS restraints | [24] |
| Human Ufc1 | NMR | N/A | High-quality structure determined in solution | Statistics indicate high quality | [24] |
Nuclear Magnetic Resonance (NMR) Spectroscopy
NMR spectroscopy elucidates protein structures in solution, providing a dynamic view that can capture conformational flexibility. The technique relies on measuring interactions between atomic nuclei in a strong magnetic field, generating constraints (e.g., distances, angles) that are used to calculate an ensemble of structures that satisfy these constraints.
For human Ufc1, the NMR structure was determined using a protocol involving uniformly ¹³C, ¹⁵N-enriched protein. The structure was calculated with the program CYANA based on NMR-derived constraints, resulting in a high-quality solution structure [24]. The complementary nature of X-ray and NMR data was crucial for Ufc1, revealing that an N-terminal helix adopted different conformations in the crystal and in solution, suggesting a potential role in mediating specific protein-protein interactions [24].
Integrated Methodologies in Structural Proteomics
Modern structural biology of UBLs increasingly relies on integrative approaches, combining high-resolution techniques with complementary methods to gain a systems-level understanding.
Table 3: Core Experimental Techniques in Structural Proteomics of UBLs
| Technique | Acronym | Key Principle | Application to UBLs |
|---|---|---|---|
| Cross-linking Mass Spectrometry | XL-MS | Uses bifunctional cross-linkers to covalently link spatially close amino acids, providing distance restraints [26]. | Mapping protein-protein interactions and topology of UBL-conjugating enzyme complexes [26]. |
| Hydrogen-Deuterium Exchange Mass Spectrometry | HDX-MS | Measures the rate of hydrogen/deuterium exchange in the protein backbone, revealing solvent accessibility and dynamics [26]. | Probing conformational dynamics and allosteric changes upon UBL binding [26]. |
| Limited Proteolysis Mass Spectrometry | LiP-MS | Uses proteases to probe protein structure; cleavage sites indicate flexible/accessible regions [26]. | Identifying structural changes and binding interfaces in UBL complexes [26]. |
| Native Mass Spectrometry | Native MS | Analyzes intact proteins and non-covalent complexes under non-denaturing conditions [26]. | Studying stoichiometry and stability of UBL-E2-E3 ligase complexes. |
| Molecular Dynamics Simulations | MD | Computationally simulates physical movements of atoms and molecules over time. | Unveiling allosteric mechanisms and conformational dynamics, e.g., activation of SENP1 by SUMO1 [27]. |
The following diagram illustrates how these techniques can be integrated into a cohesive workflow for the structural analysis of UBLs and their complexes.
Experimental Protocols for Key Techniques
Protocol: Cross-linking Mass Spectrometry (XL-MS) for UBL Complex Analysis
This protocol outlines the steps to identify interacting peptides within a UBL-protein complex, providing spatial constraints for molecular modeling [26].
- Sample Preparation: The UBL complex of interest (e.g., Ufc1~Ufm1) can be analyzed either in purified form (1-10 μg per protein) or in a more native context like intact cells (requiring ~100 μg of total protein) [26].
- Cross-linking Reaction: Incubate the sample with a homo- or hetero-bifunctional cross-linker (e.g., DSSO or BS3). The cross-linker's spacer arm length determines the maximum distance between cross-linkable amino acids (e.g., lysines) [26].
- Quenching and Digestion: Quench the reaction to stop cross-linking. Denature the complex and digest it into peptides using a protease like trypsin [26].
- Liquid Chromatography and Tandem Mass Spectrometry (LC-MS/MS): Separate the complex peptide mixture by liquid chromatography and analyze it by tandem mass spectrometry. Data-Dependent Acquisition (DDA) or Data-Independent Acquisition (DIA) methods can be used [26].
- Data Analysis: Use specialized software (e.g., MaxQuant, FragPipe, Spectronaut) to identify the peptides and, crucially, the cross-linked peptide pairs from the MS/MS spectra. These identified pairs provide distance restraints for modeling [26].
Protocol: Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS) for Probing UBL Dynamics
HDX-MS is used to study protein dynamics and binding interfaces by measuring the exchange of backbone amide hydrogens with deuterium from the solvent [26].
- Deuterium Labeling: Dilute the UBL or UBL-complex sample into a deuterated buffer (e.g., D₂O) for defined time periods (e.g., 10 seconds to 1 hour) at a controlled temperature and pH.
- Quenching: After each labeling time, quench the exchange by lowering the pH and temperature, which slows the exchange rate significantly.
- Digestion and LC-MS/MS: Rapidly digest the quenched sample with an acid-stable protease (e.g., pepsin) and immediately inject the peptides onto a LC-MS system under quenched conditions to minimize back-exchange.
- Data Analysis: Monitor the mass increase of the identified peptides due to deuterium incorporation. A region that shows reduced deuterium uptake upon complex formation is likely involved in a binding interface or has become less dynamic. This data provides insights into conformational changes and allostery.
Case Study: Structural Elucidation of the Human Ufm1 System
The structural characterization of the human Ufm1 system provides an excellent example of the power of integrating X-ray crystallography and NMR spectroscopy.
Ufm1 is a UBL that is conjugated to target proteins via a cascade involving the E1-like enzyme Uba5 and the E2-like conjugating enzyme Ufc1 [24]. The structure of human Ufc1 was solved using both techniques, yielding complementary insights [24].
- NMR Structure Determination: The solution structure of the 167-residue Ufc1 was determined using uniformly ¹³C, ¹⁵N-enriched protein. The structure was calculated with the program CYANA, resulting in a high-quality model that confirmed the presence of the canonical E2 catalytic core domain, consisting of a 4-stranded β-sheet and four α-helices. Importantly, NMR revealed that the active site cysteine (Cys116) was located in a highly solvent-accessible, flexible loop [24].
- X-ray Crystal Structure Determination: Ufc1 was crystallized using the hanging drop vapor diffusion method. A SAD (Single-wavelength Anomalous Diffraction) data set was collected at 2.54 Å resolution using a selenomethionine (SeMet)-derivatized crystal. Phasing and model building were performed, and the structure was refined. The crystal structure showed Ufc1's core domain was consistent with the NMR model but provided a precise atomic-resolution view of the fold and crystal packing [24].
- Integrated Model of Ufm1-Ufc1 Complex: The combination of both structures allowed researchers to generate a model for the Ufc1~Ufm1 thioester complex. The model showed that the C-terminal glycine of Ufm1 could be positioned to form the catalytic thioester bond with Cys116 of Ufc1. Furthermore, a comparative analysis showed that an N-terminal α-helix in Ufc1 adopted different conformations in the crystal and in solution, suggesting a role in mediating specific protein-protein interactions, potentially with E3 ligases [24].
The Scientist's Toolkit: Essential Reagents and Materials
Table 4: Key Research Reagent Solutions for UBL Structural Studies
| Reagent / Material | Function | Example Application |
|---|---|---|
| Heterologously Expressed UBLs | Provides a pure, scalable source of the protein for structural and biochemical studies. | Production of human Ufc1 and Ufm1 in E. coli for NMR and crystallography [24]. |
| Isotopically Labeled Compounds (¹⁵N-NH₄Cl, ¹³C-Glucose) | Enables NMR spectroscopy by incorporating detectable NMR-active nuclei (¹⁵N, ¹³C) into the protein. | Production of uniformly ¹³C, ¹⁵N-enriched Ufc1 for multi-dimensional NMR experiments [24]. |
| Selenomethionine (SeMet) | Used for experimental phasing in X-ray crystallography via SAD or MAD methods. | Production of SeMet-labeled Ufc1 for solving the crystal structure [24]. |
| Chemical Cross-linkers (e.g., DSSO, BS3) | Covalently link proximal amino acids in proteins, providing distance restraints for modeling. | Mapping spatial proximity in UBL-enzyme complexes using XL-MS [26]. |
| Crystallization Screening Kits | Contains diverse conditions (precipitants, salts, buffers) to identify initial crystallization leads. | Initial screening for Ufc1 crystallization [24]. |
| Ubiquitin-Activating Enzyme (E1) and Conjugating Enzymes (E2s) | Essential reconstitution components for in vitro ubiquitination/UBLation assays. | Studying HUWE1 ligase activity and inhibitor ubiquitination [28]. |
Visualization of a UBL Signaling Pathway and Activation Mechanism
The following diagram illustrates the canonical UBL conjugation pathway and a specific allosteric activation mechanism, highlighting key structural insights.
Future Directions and Therapeutic Implications
The field of UBL structural biology is rapidly advancing toward systems structural proteomics, which integrates data from XL-MS, HDX-MS, LiP-MS, crystallography, NMR, and computational models to build proteome-wide, context-aware models [26]. The convergence of these experimental methods with AI-driven predictive models like AlphaFold and RoseTTAFold is revolutionizing our ability to model UBL complexes and their dynamic assemblies at high resolution [26].
Structurally guided drug discovery is a major application of this research. For instance, understanding the allosteric activation mechanism of SENP1 by the SUMO1 β-grasp domain opens new avenues for developing non-covalent, allosteric inhibitors for cancers where SENP1 is overexpressed, such as prostate cancer [27]. Furthermore, recent discoveries that ubiquitin ligases like HUWE1 can modify drug-like small molecules themselves reveal a new dimension of Ub system functionality and highlight the potential for harnessing ubiquitination for therapeutic purposes [28]. The continued structural elucidation of UBLs will undoubtedly remain a cornerstone of understanding cellular regulation and designing novel therapeutic strategies.
The β-grasp fold is a fundamental structural motif in molecular biology, prototyped by the ubiquitous protein ubiquitin (UB). This compact fold, characterized by a β-sheet comprising five anti-parallel strands that appears to "grasp" a single α-helical segment, has been recruited by nature for a strikingly diverse range of biochemical functions [1]. These functions extend far beyond ubiquitin's renowned role in post-translational modification and include providing a scaffold for enzymatic active sites, iron-sulfur clusters, RNA-soluble-ligand binding, sulfur transfer, and assembly of macromolecular complexes [1]. The evolutionary success and functional versatility of this small fold are intrinsically linked to its structural dynamics and folding landscape, properties that can be directly probed through Molecular Dynamics (MD) simulations.
Understanding the folding pathways of ubiquitin and ubiquitin-like proteins (UBLs) is not merely an academic pursuit. The proper folding and stability of these proteins are critical for cellular proteostasis—the delicate balance between protein synthesis, folding, modification, and degradation [29]. Disruptions in proteostasis lead to a pathological state known as dysproteostasis, which is implicated in a growing list of human diseases, including neurodegenerative disorders and cancer [29]. For UBLs like FAT10, which directly targets proteins for proteasomal degradation, intrinsic instability and flexible regions are essential for its function [30]. Computational approaches, particularly MD simulations, provide a powerful, high-resolution toolset to unravel the dynamic processes that underlie the folding, stability, and functional mechanics of β-grasp fold proteins, offering insights that are often difficult to capture through experimental means alone.
The β-Grasp Fold: A Structurally Versatile Scaffold
Core Structural Features and Functional Diversity
The β-grasp fold's remarkable functional plasticity arises from its core architecture. Systematic sequence-structure analysis has shown that its manifold functions originate primarily from the prominent β-sheet, which provides an exposed surface for diverse interactions or can form open barrel-like structures [1]. This fold has served as a scaffold for the independent evolution of enzymatic activities and co-factor binding on multiple occasions throughout evolution [1].
The UBL family, which shares this core fold, exhibits significant structural conservation despite functional divergence. For instance, the UBL modifier FAT10 consists of two flexibly linked ubiquit-like domains (UBDs), each adopting the typical β-grasp fold, yet their surface properties are entirely different from each other and from ubiquitin, explaining their unique binding specificities [30]. NMR studies reveal that FAT10's domains are joined by a flexible linker and possess additional flexible regions at the N- and C-termini, features that contribute to its functional mechanism [30].
Evolutionary Context and Ubiquitin-Like Proteins
Evolutionary reconstruction indicates that the β-grasp fold had already differentiated into at least seven distinct lineages by the time of the last universal common ancestor of all extant organisms [1]. The earliest members were likely involved in RNA metabolism, subsequently radiating into various functional niches. The eukaryotic phase of evolution was marked by a specific expansion of ubiquitin-like β-grasp members, with the eukaryotic UB superfamily diversifying into at least 67 distinct families [1]. Key relatives like ThiS and MoaD, involved in thiamine and molybdenum cofactor biosynthesis in prokaryotes, share both structural homology and a conserved C-terminal double-glycine motif with ubiquitin, pointing to a common evolutionary origin [31].
Table 1: Key Ubiquitin-like Proteins with the β-Grasp Fold
| Protein | Organism | Primary Function | Key Structural Features |
|---|---|---|---|
| Ubiquitin (UBQ) | Eukaryotes | Protein tagging for degradation | Classic β-grasp fold; C-terminal GG motif |
| ThiS | Bacteria | Sulfur carrier in thiamine biosynthesis | β-grasp fold related to Ub; C-terminal GG motif |
| MoaD | Bacteria/Beyond | Molybdenum cofactor biosynthesis | Ub-like fold; C-terminal GG motif |
| UBX (from FAF1) | Mammals | Function not fully defined; implicated in Ub-mediated pathways | Structurally similar to Ub; lacks C-terminal GG motif |
| FAT10 | Mammals | Direct targeting for proteasomal degradation | Two flexibly-linked UBDs; disordered N-terminus |
Molecular Dynamics Simulations: Principles and Methodologies
MD simulations solve Newton's equations of motion for a molecular system, generating a trajectory that describes how the positions and velocities of atoms change over time. This provides a powerful framework for studying protein folding at an atomic level of detail.
Theoretical Foundations and Historical Context
The theoretical underpinnings of protein folding research have evolved significantly. Early work by Christian Anfinsen established that a protein's native structure is determined solely by its amino acid sequence and represents the most thermodynamically stable conformation [29]. However, Levinthal's paradox highlighted the impossibility of a protein randomly sampling all possible conformations to find its native state [29]. This led to proposed models like the diffusion-collision model and the nucleation-condensation model, which suggest that folding proceeds through the formation and assembly of local microdomains or a folding nucleus [29]. MD simulations provide a computational means to test these models and directly observe the folding process.
The energy landscape theory frames protein folding as a funnel-guided process where the native state occupies the global energy minimum [29]. The ruggedness of this landscape accounts for partially folded states and kinetic traps. More recently, the "foldon" model has proposed that proteins contain independently folding units that fold in a hierarchical manner before final assembly [29]. MD simulations are uniquely positioned to characterize these landscapes and identify intermediate states.
Essential Dynamics for Analyzing β-Grasp Fold Proteins
For analyzing the dynamics of β-grasp fold proteins, Essential Dynamics (ED), also known as principal component analysis (PCA), is a particularly valuable technique [31]. ED separates the conformational space explored during an MD simulation into an essential subspace (containing large-scale, functionally relevant motions) and a physically constrained subspace (containing smaller, harmonic fluctuations) [31].
The methodology involves:
- Generating a covariance matrix C of atomic positional fluctuations from the MD trajectory.
- Diagonalizing this matrix to obtain eigenvectors and eigenvalues.
- The eigenvectors represent directions of collective motion, while the eigenvalues indicate the magnitude of motion along these directions [31].
Projecting the MD trajectory onto the first few principal components allows for the visualization and analysis of the most significant conformational changes, enabling researchers to identify conserved dynamics potentially linked to function within the ubiquitin family [31].
Diagram 1: MD Simulation and Analysis Workflow. This flowchart outlines the key stages in a molecular dynamics study of protein folding, from initial structure preparation to functional interpretation.
Protocol: Native-State MD Simulation of Ubiquitin-like Proteins
This section provides a detailed methodological protocol for conducting and analyzing native-state MD simulations of ubiquitin-like proteins, based on established practices in the field [31].
System Setup and Simulation Parameters
Initial Structure Preparation:
- Obtain high-resolution structures from the Protein Data Bank (e.g., PDB IDs: 1UBQ for ubiquitin, 1MJD for DCX, or other relevant β-grasp fold proteins).
- For proteins where only NMR structures are available, use an average structure of the ensemble of low-energy conformers.
Solvation and Energy Minimization:
- Solvate the protein in a cubic box with explicit water molecules (e.g., SPC water model). Ensure a minimum distance (e.g., 10 Å) between the protein and the box edges.
- Perform energy minimization using steepest descent or conjugate gradient algorithms to relieve any steric clashes and optimize solvent orientation.
Equilibration and Production Simulation:
- Equilibrate the system in stages, typically first with position restraints on the protein heavy atoms to allow solvent relaxation, followed by an unrestrained equilibration.
- Run production simulations using a force field such as GROMOS 96. Maintain constant temperature (e.g., 298 K) and pressure (e.g., 1 atm) using weak coupling algorithms.
- Handle electrostatics with a cutoff method (e.g., 8 Å Coulombic cutoff) or particle mesh Ewald for better accuracy.
- A simulation length of 2 ns was used in earlier studies [31], but modern simulations for folding studies often require much longer timescales (microseconds to milliseconds), potentially achieved through enhanced sampling methods.
Trajectory Analysis Techniques
Backbone Stability and Fluctuations:
- Calculate the root mean square deviation (RMSD) of C-α atoms over time to monitor structural stability and convergence.
- Compute the root mean square fluctuation (RMSF) per residue to identify flexible and rigid regions within the protein structure.
Global Dynamics via Essential Dynamics:
- As described in Section 3.2, perform PCA on the C-α atomic coordinates from the production trajectory.
- Diagonalize the covariance matrix to extract eigenvectors (principal components) and eigenvalues.
- Project the trajectory onto the first two or three principal components to visualize the dominant motions and explore the essential subspace.
- Quantify the similarity of dynamics between different β-grasp proteins by calculating the root mean square inner product (r.m.s.i.p.) of the first 10 eigenvectors [31].
Chain Compactness:
- Monitor the radius of gyration throughout the simulation to assess the overall compactness of the protein structure and detect potential collapse events relevant to folding.
Table 2: Key Analysis Metrics for MD Trajectories of β-Grasp Proteins
| Metric | Description | Functional Insight |
|---|---|---|
| C-α RMSD | Measures the average change in backbone atom positions over time. | Indicates overall structural stability and convergence of the simulation. |
| C-α RMSF | Measures the fluctuation of each residue around its average position. | Identifies flexible loops/regions crucial for function (e.g., binding interfaces). |
| Radius of Gyration | Measures the compactness of the protein structure. | Can report on folding/unfolding events or conformational expansion/compaction. |
| Principal Components (PCs) | Define the collective directions of largest atomic displacement. | Reveals large-scale, functionally relevant motions (e.g., domain breathing). |
| RMSIP | Quantifies the similarity of essential dynamics between two proteins. | Assesses conservation of dynamic behavior across related β-grasp proteins. |
Case Study: Conserved Dynamics in the Ubiquitin Family
A seminal study [31] employed the aforementioned protocol to investigate the native-state dynamics of four β-grasp fold proteins: ubiquitin (UBQ), UBX, ThiS, and MoaD. The goal was to determine whether functional relationships are reflected in conserved dynamic properties.
Experimental Design and Findings
The researchers conducted 2 ns MD simulations for each protein and analyzed their backbone fluctuations and global dynamics. The analysis revealed that the three proteins with a common evolutionary ancestor and related C-terminal function (Ubiquitin, ThiS, MoaD) showed a slightly higher degree of conservation in their dynamics compared to UBX, which lacks the C-terminal double-glycine motif and has a different function [31].
Notably, the essential dynamics analysis identified conserved motions in residues critical for function, particularly around the C-terminal region essential for the conjugation chemistry shared by ubiquitin, ThiS, and MoaD [31]. This suggests that evolution has not only conserved the structural fold but also optimized and conserved the protein motions necessary for specific biological activity within this family.
Insight into FAT10 Dynamics and Function
Further illustrating the link between dynamics and function, structural studies of FAT10 using NMR spectroscopy revealed that its two UBDs are independently folded and connected by a flexible linker [30]. This intrinsic flexibility, combined with an unstructured N-terminal heptapeptide and the overall poor stability of FAT10, is thought to enable its rapid degradation alongside its substrates by the proteasome. Stabilizing the FAT10 UBDs through point mutations was shown to decelerate degradation, underscoring how conformational dynamics are directly tuned to govern functional outcomes in UBLs [30].
Diagram 2: Essential Dynamics Analysis Methodology. This flowchart outlines the key computational steps in performing an Essential Dynamics (Principal Component) Analysis on an MD trajectory.
Successful application of MD simulations to study β-grasp fold proteins relies on a suite of sophisticated software tools, force fields, and computational resources.
Table 3: Research Reagent Solutions for MD Simulations
| Resource Category | Specific Examples | Function and Application |
|---|---|---|
| Simulation Software | GROMACS, NAMD, AMBER, OpenMM | Software suites for performing high-performance MD simulations, including energy minimization, equilibration, production runs, and basic analysis. |
| Force Fields | GROMOS 96, CHARMM, AMBER force fields | Parameter sets defining interatomic potentials (bonded and non-bonded interactions) crucial for accurate physical modeling of the molecular system. |
| Analysis Tools | MDAnalysis, VMD, PyTraj, GROMACS analysis modules | Specialized software and built-in tools for analyzing MD trajectories to compute metrics like RMSD, RMSF, hydrogen bonding, and PCA. |
| Structure Databases | Protein Data Bank (PDB) | Repository for initial high-resolution experimental structures (X-ray, NMR, Cryo-EM) used as starting points for simulations. |
| Specialized Algorithms | Essential Dynamics (PCA), Molecular Docking (RosettaDock) | Advanced computational methods for extracting meaningful information from complex simulation data or modeling protein interactions. |
Molecular Dynamics simulations have proven to be an indispensable tool for unraveling the folding pathways and dynamic behavior of proteins with the β-grasp fold. By moving beyond static structural snapshots, MD provides a temporal dimension that reveals how the conserved architecture of ubiquitin and UBLs supports their diverse functions through specific dynamic properties. Studies have shown that evolution has conserved not only the fold itself but also key aspects of its molecular motions, which are critical for function [31]. Furthermore, intrinsic flexibility and instability, as seen in FAT10, can be themselves functional features, directly regulating processes like proteasomal degradation [30]. As computational power continues to grow and simulation methodologies become increasingly refined, MD will undoubtedly remain at the forefront of efforts to connect the sequence, structure, dynamics, and function of this extraordinarily versatile protein fold, with profound implications for understanding cellular proteostasis and designing novel therapeutic strategies.
Ubiquitin-binding domains (UBDs) are critical modular elements that enable cellular proteins to recognize and decode the ubiquitin signal, a central post-translational modification regulating virtually all aspects of eukaryotic cell biology [32] [33]. The versatility of ubiquitin signaling arises from the diversity of ubiquitin modifications—including monoubiquitination and various polyubiquitin chain architectures—that dictate distinct functional outcomes such as protein degradation, DNA repair, inflammation, and autophagy [32] [34]. UBDs serve as deciphering modules that specifically interpret these ubiquitin codes by engaging ubiquitin or ubiquitin chains through defined molecular interfaces [32].
Structurally, ubiquitin itself adopts the β-grasp fold (β-GF), a compact domain dominated by a five-stranded β-sheet that grasps a single α-helical segment [1]. This ancient fold has been extensively recruited throughout evolution for diverse biochemical functions beyond ubiquitin signaling, including providing scaffolds for enzymatic active sites, RNA binding, sulfur transfer, and adaptor functions in signal transduction [1]. Understanding how UBDs, many of which themselves contain β-grasp folds or other structural motifs, recognize and interact with the β-grasp fold of ubiquitin is fundamental to elucidating the specificity of ubiquitin signaling pathways.
This technical guide provides a comprehensive framework for characterizing UBD-ubiquitin interactions, integrating current structural insights, quantitative binding profiles, experimental methodologies, and practical tools for researchers investigating ubiquitin signaling mechanisms in both basic research and drug discovery contexts.
Ubiquitin-Binding Domains: Classification and Recognition Principles
Diversity of UBD Families
More than 20 distinct types of UBDs have been identified, ranging in length from approximately 20 to 150 amino acids [33] [34]. Despite their structural diversity, most UBDs share common recognition principles by binding to characteristic surface patches on ubiquitin, most commonly the hydrophobic I44 patch [32] [33]. These domains typically exhibit weak binding affinities for monoubiquitin, commonly in the range of 50–500 μM, which may facilitate reversible interactions and dynamic regulation of signaling pathways [32].
Npl4-type zinc-finger (NZF) domains represent a well-characterized UBD family that illustrates key recognition principles. These compact domains comprise approximately 30 residues stabilized by four cysteine residues coordinating a zinc ion [32]. NZF domains contain a conserved Thr-Phe/Tyr (TF) motif that mediates binding to the I44 patch of ubiquitin [32]. The human proteome encodes 11 proteins containing a total of 15 NZF domains, with members exhibiting varied specificities for different ubiquitin chain linkage types [32].
Structural Mechanisms of Specificity
UBDs achieve signaling specificity through several structural mechanisms:
Linkage specificity: Some UBDs, including certain NZF domains, can distinguish between different ubiquitin chain linkage types by simultaneously engaging two ubiquitin moieties through distinct binding interfaces [32]. For example, the TAB2 and TAB3 NZF domains specifically recognize Lys63-linked chains, while HOIL-1L and Sharpin NZF domains prefer Met1-linked chains [32].
Substrate-assisted recognition: Recent research reveals that some UBDs recognize ubiquitinated substrates through bidentate interactions that engage both the ubiquitin moiety and the modified substrate itself [32]. The NZF1 domain of HOIP exemplifies this mechanism by preferentially binding to site-specifically ubiquitinated forms of NEMO and optineurin [32].
Avidity effects: Proteins containing multiple UBDs can achieve enhanced affinity and specificity through avidity effects, simultaneously engaging multiple ubiquitin moieties within a chain [33] [34]. This principle is exploited in engineered tandem ubiquitin-binding entities (TUBEs) used for efficient ubiquitin enrichment [34].
Table 1: Linkage Specificity and Affinities of Human NZF Domains
| NZF Domain | Preferred Linkage | Affinity for Preferred Linkage (K_D) | Affinity Range for Other Linkages (K_D) |
|---|---|---|---|
| TAB2 | Phosphorylated K6, K63 | Not specified | 113-189 μM (all linkages) |
| HOIL-1L | M1 | 4 μM | ~200 μM (other linkages) |
| Sharpin | M1, K63 | 55 μM (M1), 170 μM (K63) | ~200 μM (other linkages) |
| TRABID NZF1 | K29, K33 | Not specified | 50-fold specificity |
| ZRANB3 | None (promiscuous) | - | 28-48 μM (all linkages) |
| CAPN15 NZF1 | None (promiscuous) | - | 110-190 μM (all linkages) |
| CAPN15 NZF2 | None (promiscuous) | - | 143-296 μM (all linkages) |
| NPL4 | None (promiscuous) | - | 113-189 μM (all linkages) |
| RYBP | None (promiscuous) | - | 255-348 μM (all linkages) |
Quantitative Profiling of UBD Interactions
Methodological Approaches
Comprehensive characterization of UBD specificity requires quantitative assessment of interactions across the full spectrum of ubiquitin chain linkage types. Surface plasmon resonance (SPR) has emerged as a powerful platform for systematically profiling UBD binding specificities and affinities [32]. In this approach, eight types of diubiquitins (K6, K11, K27, K29, K33, K48, K63, and M1) are immobilized on SPR chips, and equilibrium binding is measured across a range of UBD concentrations to derive dissociation constants (K_D) [32]. This methodology enables direct comparison of linkage preferences and identification of specific versus promiscuous binders.
Pull-down assays complement SPR data by validating interactions in a more complex biochemical context. In these experiments, UBDs immobilized on beads are used to capture ubiquitinated proteins or specific ubiquitin chains from cell lysates or purified preparations, followed by Western blot analysis with linkage-specific antibodies [32] [33].
Quantitative Binding Landscapes
Systematic profiling of human NZF domains reveals distinct specificity patterns:
Specific binders: HOIL-1L NZF displays 50-fold specificity for M1-linked diUb (K_D = 4 μM) compared to other linkage types [32]. TRABID NZF1 shows similar specificity for K29 and K33 linkages [32].
Promiscuous binders: The majority of NZF domains, including those from ZRANB3, CAPN15, NPL4, and RYBP, lack strong linkage preference, binding all chain types with affinities ranging from 28 μM to 348 μM [32].
Phosphorylation-dependent recognition: TAB2 NZF preferentially recognizes Lys6-linked chains phosphorylated on Ser65, explaining its specific recruitment to depolarized mitochondria where this modified ubiquitin signal occurs [32].
Table 2: Research Reagent Solutions for UBD Characterization
| Research Tool | Specifications/Composition | Experimental Applications |
|---|---|---|
| SPR Platform with Immobilized diUb | Eight linkage types of diubiquitin (K6, K11, K27, K29, K33, K48, K63, M1) immobilized on sensor chips | Quantitative profiling of UBD binding specificity and affinity [32] |
| UBD-Conjugated Agarose Beads | UBA domains from p62, hHR23B, NBR1, NUB1, UQ1, Dsk2; UIM from S5a; CUE from VPS9 coupled to agarose | Pull-down assays to capture ubiquitinated proteins from complex lysates [33] |
| OtUBD Affinity Resin | High-affinity UBD from O. tsutsugamushi conjugated to SulfoLink coupling resin | Enrichment of both mono- and polyubiquitinated proteins under native or denaturing conditions [34] |
| Linkage-Specific Antibodies | Monoclonal antibodies specific for K48 (Apu2) and K63 (Apu3) linkages; polyclonal anti-ubiquitin (AB1690) | Detection and validation of specific ubiquitin chain types in Western blotting [33] |
| Tandem UBE (TUBE) | Multiple low-affinity UBDs linked in a single polypeptide | High-avidity capture of polyubiquitinated proteins; protection from deubiquitinases [34] |
Experimental Protocols for UBD Characterization
Surface Plasmon Resonance Analysis of Linkage Specificity
Purpose: To quantitatively determine the binding affinity and linkage specificity of a UBD across all eight ubiquitin chain types.
Procedure:
- Immobilization: Covalently immobilize the eight diubiquitin linkage types (K6, K11, K27, K29, K33, K48, K63, and M1) on separate flow cells of a CM5 sensor chip using standard amine coupling chemistry [32].
- Binding measurements: Dilute the purified UBD to a concentration series (typically spanning 0.1-500 μM) in HBS-EP buffer (10 mM HEPES, 150 mM NaCl, 3 mM EDTA, 0.005% surfactant P20, pH 7.4).
- Data acquisition: Inject UBD concentrations over diUb surfaces at a flow rate of 30 μL/min with a contact time of 60-120 seconds and dissociation time of 300-600 seconds.
- Data analysis: Subtract responses from a reference flow cell and blank injections. Calculate equilibrium dissociation constants (K_D) by fitting the equilibrium binding responses to a 1:1 binding model [32].
Technical considerations: Include regeneration steps with mild acidic or basic conditions (10 mM glycine pH 2.5-3.0) to remove bound UBD between cycles. Analyze data from at least two independent experiments to ensure reproducibility.
OtUBD-Based Enrichment of Ubiquitinated Proteins
Purpose: To isolate ubiquitinated proteins from complex biological samples using the high-affinity OtUBD domain.
Procedure: A. OtUBD resin preparation:
- Express recombinant Cys-His6-OtUBD in E. coli BL21(DE3) using pET21a-cys-His6-OtUBD plasmid and induce with 0.5 mM IPTG at 18°C for 16 hours [34].
- Purify the protein under native conditions using Ni-NTA agarose chromatography.
- Couple purified OtUBD to SulfoLink resin via cysteine residues according to manufacturer's instructions (2-5 mg OtUBD per mL resin).
- Block remaining reactive groups with 50 mM L-cysteine and store in PBS with 0.02% sodium azide at 4°C [34].
B. Native enrichment workflow:
- Prepare cell lysates in lysis buffer (50 mM HEPES, 5 mM EDTA, 150 mM NaCl, 1% Triton X-100, pH 7.5) supplemented with 10 mM N-ethylmaleimide (NEM) to inhibit deubiquitinases and complete protease inhibitor cocktail [34].
- Incubate clarified lysate with OtUBD resin for 2 hours at 4°C with end-over-end mixing.
- Wash resin extensively with lysis buffer followed by a final wash with 50 mM ammonium bicarbonate.
- Elute bound proteins with 2× SDS-PAGE loading buffer containing DTT for Western blot analysis, or with 50 mM ammonium bicarbonate for proteomic analysis [34].
C. Denaturing enrichment workflow:
- Lyse cells in denaturing buffer (6 M guanidine hydrochloride, 100 mM NaH2PO4, 10 mM Tris-HCl, 10 mM NEM, pH 8.0) with vigorous vortexing.
- Dilute lysate 1:4 with 50 mM ammonium bicarbonate before incubation with OtUBD resin.
- Proceed with washing and elution as in native workflow [34].
Applications: The native workflow preserves non-covalent protein interactions, enabling co-purification of ubiquitin-interacting proteins, while the denaturing workflow specifically isolates covalently ubiquitinated proteins, reducing background interactions [34].
Structural Insights and β-Grasp Fold Context
The β-grasp fold represents an evolutionarily ancient structural scaffold that has been extensively adapted for diverse functions throughout evolution [1]. Evolutionary reconstruction indicates that this fold had already differentiated into at least seven distinct lineages by the time of the last universal common ancestor, encompassing much of the structural diversity observed in extant versions [1].
Ubiquitin itself exemplifies a highly specialized β-grasp fold protein that has evolved specifically for post-translational modification functions. The versatility of this fold arises primarily from the prominent β-sheet, which provides an exposed surface for diverse interactions or can form open barrel-like structures that accommodate various ligands and interaction partners [1]. The manifold functional adaptations of this fold include enzymatic activities, binding of diverse co-factors, and iron-sulfur-cluster-binding, each having evolved independently on multiple occasions [1].
In the context of UBD-ubiquitin interactions, we frequently observe β-grasp fold domains recognizing other β-grasp fold domains—a fold-fold interaction that underscores the remarkable adaptability of this structural scaffold. The NZF domain, while distinct in its zinc-coordination mechanism, maintains topological relationships to the β-grasp fold through its central β-sheet organization [32]. This structural compatibility may facilitate the evolution of specific recognition interfaces between UBDs and their cognate ubiquitin signals.
The evolutionary trajectory of β-grasp fold proteins reveals that most structural diversification occurred in prokaryotes, while the eukaryotic phase was marked primarily by specific expansion of ubiquitin-like β-grasp fold members [1]. The eukaryotic ubiquitin superfamily diversified into at least 67 distinct families, with at least 19-20 families already present in the eukaryotic common ancestor [1]. This expansion was accompanied by a dramatic increase in domain architectural complexity, with ubiquitin-like domains incorporated into numerous proteins as adaptors in complex signaling networks [1].
Research Tools and Visualization
Diagram 1: Experimental workflow for UBD characterization
Diagram 2: Molecular recognition mechanisms in UBD-ubiquitin interactions
Ubiquitin signalling represents a fundamental regulatory system in eukaryotic cells, controlling diverse cellular functions from protein degradation to DNA repair and immune response [35]. At the heart of this system lies a conserved structural architecture: the β-grasp fold (β-GF). This compact fold, prototyped by ubiquitin (UB) itself, is characterized by a β-sheet with five anti-parallel strands that appears to "grasp" a single α-helical segment [1]. The evolutionary significance of this fold is profound - by the time of the last universal common ancestor, the β-GF had already diversified into at least 7 distinct lineages [1].
The ubiquitin system comprises an enzymatic cascade of E1 (activating), E2 (conjugating), and E3 (ligating) enzymes that work in concert to attach ubiquitin to substrate proteins, with deubiquitinases (DUBs) providing the counterbalancing activity to edit or remove these signals [35]. What makes this system particularly remarkable is that ubiquitin and ubiquitin-like proteins (Ubls) - all sharing the β-grasp architecture - function as protein modifiers in this cascade [1]. The versatility of the β-grasp fold as a structural scaffold has enabled its recruitment for strikingly diverse biochemical functions throughout evolution, with its prominent β-sheet providing an exposed surface for diverse interactions [1].
The Enzymatic Cascade: Mechanism and Disease Relevance
The Ubiquitination Pathway
The ubiquitination process follows a precise enzymatic sequence:
E1 (Ubiquitin-Activating Enzyme): The cascade initiates with E1-mediated activation of ubiquitin in an ATP-dependent manner. The E1 enzyme forms a high-energy thioester bond between its catalytic cysteine and the C-terminal glycine of ubiquitin [36]. Humans possess two major E1 enzymes: UBA1 and UBA6 [35] [37].
E2 (Ubiquitin-Conjugating Enzyme): Activated ubiquitin is transferred from E1 to a catalytic cysteine residue on an E2 enzyme, forming a E2~Ub thioester intermediate [35] [36]. The human genome encodes approximately 35 E2 enzymes, all sharing a core ubiquitin conjugation (UBC) domain of roughly 150 residues [35].
E3 (Ubiquitin Ligase): E3 enzymes function as substrate recognition modules, facilitating the transfer of ubiquitin from E2 to specific target proteins [36]. With over 600 E3s in humans, these enzymes provide the specificity that determines which proteins are ubiquitinated [36]. E3s are categorized into three major families: RING (really interesting new gene), U-box, and HECT (homologous to E6-AP C-terminus) types [36] [37].
DUBs (Deubiquitinating Enzymes): Completing the cycle, DUBs counteract ubiquitination by cleaving ubiquitin from modified proteins, thus editing or erasing the ubiquitin signal [35].
Table 1: Core Enzymes in the Ubiquitin-Proteasome System
| Enzyme Class | Number in Humans | Core Function | Key Structural Features |
|---|---|---|---|
| E1 (Activating) | 2 [37] | Ubiquitin activation via ATP hydrolysis; forms E1~Ub thioester | Binds C-terminal LRLRGG sequence of UB [37] |
| E2 (Conjugating) | ~35 [35] | Accepts Ub from E1; forms E2~Ub thioester; often determines chain topology | Core UBC domain (~150 residues) with active-site cysteine [35] |
| E3 (Ligating) | ~600 [36] | Substrate recognition; facilitates Ub transfer from E2 to substrate | RING, U-box (scaffold), or HECT (catalytic) domains [36] |
| DUBs (Deubiquitinases) | Multiple families | Cleaves Ub from substrates; processes Ub precursors | Diverse folds, often with catalytic cysteine or metalloprotease domain [35] |
Structural and Functional Relationships of the β-Grasp Fold
The β-grasp fold found in ubiquitin and Ubls represents a remarkable example of structural conservation facilitating functional diversity. Structural analyses reveal that this fold is dominated by a β-sheet with five anti-parallel strands and a single helical segment [1]. The versatility of this fold arises primarily from the prominent β-sheet, which provides an exposed surface for diverse interactions or can form open barrel-like structures [1].
Throughout evolution, the β-grasp fold has been recruited for an extraordinary range of biochemical functions, including providing scaffolds for enzymatic active sites, RNA-soluble-ligand and co-factor-binding, sulfur transfer, adaptor functions in signaling, and assembly of macromolecular complexes [1]. The eukaryotic phase of β-GF evolution was marked by a specific expansion of ubiquitin-like members, with the eukaryotic UB superfamily diversifying into at least 67 distinct families [1].
Disease Associations
Dysregulation of ubiquitin signaling is associated with numerous disease states. While E3 ligases have traditionally been the focus of therapeutic development, emerging evidence demonstrates that mutations or impairment of E2 enzymes can lead to severe pathologies including chromosome instability syndromes, cancer predisposition, and immunological disorders [35]. Given their central positioning in the cascade and relevance to diseases, E2s represent an important class of therapeutic targets [35]. The ubiquitin-proteasome system (UPS) has been linked to a variety of human diseases, including cancer, immune disorders, and viral infections, making its enzymatic components attractive targets for therapeutic intervention [36].
Therapeutic Targeting: Inhibitor Strategies and Classifications
The development of inhibitors targeting the ubiquitin cascade has gained significant momentum as the critical role of this system in human diseases becomes increasingly apparent. Different therapeutic strategies have emerged for each class of enzyme in the cascade.
E1 Inhibitors
E1 inhibitors function at the apex of the ubiquitin cascade, preventing the initial activation step and thereby globally disrupting ubiquitin signaling. These compounds typically target the ATP-binding site or the ubiquitin-binding interface of E1 enzymes. While potent E1 inhibitors have been developed, their broad impact on ubiquitin signaling can lead to significant toxicity, limiting their therapeutic application.
E2 Inhibitors
Targeting E2 enzymes represents a promising therapeutic approach that offers more selectivity than E1 inhibition but broader effects than specific E3 targeting [35]. E2 inhibitors may function by:
- Blocking the E1-E2 transthioesterification reaction
- Disrupting E2-E3 interactions through competitive or allosteric mechanisms
- Interfering with E2~Ub conformational states required for ubiquitin transfer
Recent structural studies have revealed key insights into E2 mechanism that can inform inhibitor design, including the dynamic conformations adopted by the E2~Ub thioester and the conserved surfaces involved in E1 and E3 interactions [35].
E3 Inhibitors
E3 ligases represent particularly attractive therapeutic targets due to their substrate specificity. There are three main strategies employed to develop inhibitors for E3 ligases [36]:
- Directly inhibiting their catalytic activity (particularly relevant for HECT-type E3s)
- Targeting the substrate-binding interface
- Affecting the expression of the protein by transcription or translation inhibition
Most small molecule E3 inhibitors developed to date target RING-type E3s, with particular focus on MDM2, IAP, and SCF complexes [36].
Table 2: Examples of E3 Ubiquitin Ligase Inhibitors in Development
| E3 Target | Example Compounds | Therapeutic Context |
|---|---|---|
| MDM2 | Nutlins, MI-63, Mel 23, HL198, TDP521252, TDP665759 [36] | Cancer (reactivating p53) |
| IAP (XIAP, cIAP1, cIAP2) | SM-406, GDC-0152 [36] | Cancer (promoting apoptosis) |
| Skp2 | NSC689857, NSC681152 [36] | Cancer (cell cycle regulation) |
| Itch | Clomipramine [36] | Immune disorders, inflammation |
| E6AP | CM11-1 [36] | HPV-associated cancers |
DUB Inhibitors
DUB inhibitors prevent the removal of ubiquitin signals, potentially stabilizing ubiquitin-modified proteins and altering signaling outcomes. Therapeutic strategies targeting DUBs aim to modulate the stability of specific proteins or enhance the degradation of disease-driving proteins when combined with other UPS-targeting agents.
Experimental Approaches: Methodologies for Studying the Cascade
Phage Display for Profiling Enzyme Specificity
Phage display has emerged as a powerful technique for profiling the specificity of ubiquitin cascade enzymes, particularly for mapping interactions between E1 enzymes and the C-terminus of ubiquitin.
Experimental Protocol for E1 Specificity Profiling [37]:
Library Construction: Create a UB library with randomized C-terminal sequences (typically residues 71-75, while preserving Gly76 which is indispensable for E1 activation).
Phage Selection: Express E1 enzymes as fusions with a N-terminal peptidyl carrier protein (PCP) domain that can be biotinylated using Sfp phosphopantetheinyl transferase and biotin-coenzyme A.
Immobilization: Bind biotin-labeled PCP-E1 fusions to a streptavidin-coated plate.
Reaction: Add phage-displayed UB library to the plate with Mg-ATP to initiate UB~E1 thioester formation.
Selection: Wash away non-specific phage, then release specifically bound phage by cleaving thioester linkages with dithiothreitol (DTT).
Amplification and Iteration: Repeat selection process over multiple rounds with increasing stringency to enrich for UB variants with high reactivity toward E1 enzymes.
This approach has revealed that while Arg72 of UB is absolutely required for E1 recognition, UB residues at positions 71, 73, and 74 can be replaced with bulky aromatic side chains, and Gly75 can be mutated to Ser, Asp, or Asn while maintaining efficient E1 activation [37].
Phage Display Workflow for E1 Specificity Profiling
Structural Biology Approaches
X-ray crystallography and cryo-EM have been instrumental in elucidating the mechanisms of ubiquitin cascade enzymes. Structural studies of E2 enzymes have revealed:
- The conserved UBC fold containing an N-terminal helix, four-stranded β-meander, short 3₁₀-helix, central cross-over helix, and two C-terminal helices [35]
- The catalytic cleft with active-site cysteine preceding the 3₁₀-helix, structurally supported by a conserved His-Pro-Asn motif [35]
- Partially overlapping surfaces for E1 and E3 interactions, ensuring mutually exclusive binding events during ubiquitin transfer [35]
Structures of E2~Ub thioester mimics have revealed dynamic conformations adopted by ubiquitin relative to the E2, with "closed" conformations promoting efficient ubiquitin discharge [35].
Biochemical Assays
Standard biochemical assays for studying ubiquitin cascade enzymes include:
- Thioester Formation Assays: Monitoring formation of E1~Ub or E2~Ub thioesters using non-reducing SDS-PAGE
- Ubiquitin Discharge Assays: Measuring transfer of ubiquitin from E2~Ub to substrates or competing nucleophiles
- Polyubiquitin Chain Formation Assays: Characterizing linkage-specific chain assembly by E2-E3 pairs
- DUB Activity Assays: Using ubiquitin-AMC or ubiquitin-chain substrates to measure DUB activity
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Studying the Ubiquitin Cascade
| Reagent / Tool | Function and Application | Experimental Context |
|---|---|---|
| Biotin-CoA / Sfp PPTase | Site-specific biotinylation of PCP-tagged proteins for immobilization | Phage display selection; pull-down assays [37] |
| UB C-terminal Mutant Library | Profiling enzyme specificity toward ubiquitin C-terminal sequence | Phage display; enzyme specificity studies [37] |
| E1/E2/E3 Recombinant Proteins | Reconstituting ubiquitination cascade in vitro | Biochemical assays; structural studies [35] |
| DUB Inhibitors | Probing DUB function in cellular processes | Cell-based assays; target validation |
| Linkage-specific Ub Antibodies | Detecting specific polyubiquitin chain types | Western blot; immunofluorescence |
| Activity-based DUB Probes | Labeling active DUBs in complex mixtures | Profiling DUB activity; competitive inhibition assays |
| ATPγS (ATP analog) | Studying ATP-dependent steps in ubiquitin activation | E1 mechanism studies |
| Ub-VS (Ubiquitin vinyl sulfone) | Trapping thioester intermediates | Monitoring E1~Ub and E2~Ub formation |
Signaling Pathways and Molecular Interactions
The ubiquitin cascade operates within complex signaling networks that regulate essential cellular processes. Understanding these pathways is crucial for developing targeted therapeutic interventions.
Ubiquitin Cascade Coordination and Inhibition Points
Targeting the E1-E2-E3-DUB enzymatic cascade represents a promising therapeutic strategy for numerous diseases, particularly cancer and immunological disorders. The central role of the β-grasp fold throughout this system highlights the remarkable functional versatility of this ancient structural scaffold. As our understanding of the intricate mechanisms governing ubiquitin signaling deepens, so too does our ability to develop precisely targeted inhibitors that modulate specific pathways with minimal off-target effects.
Future directions in this field will likely focus on developing increasingly selective inhibitors, particularly for E2 enzymes and E3 ligases that offer the ideal balance of specificity and broader pathway modulation. The integration of structural biology, mechanistic enzymology, and chemical biology will continue to drive innovation in therapeutic development targeting the ubiquitin-proteasome system.
The ubiquitin-proteasome system (UPS) represents the primary pathway for controlled intracellular protein degradation in eukaryotic cells, playing an indispensable role in maintaining cellular homeostasis by regulating the concentration of key proteins [38]. This sophisticated system orchestrates the degradation of damaged, misfolded, or short-lived regulatory proteins through an ATP-dependent process, thereby influencing critical cellular processes including cell cycle progression, DNA repair, and stress response [38] [39]. The UPS functions through a coordinated two-step mechanism: first, target proteins are marked for degradation through covalent attachment of ubiquitin chains in an enzymatic cascade involving E1 (activating), E2 (conjugating), and E3 (ligating) enzymes; second, the polyubiquitinated proteins are recognized and degraded by the 26S proteasome complex [38] [39].
The clinical significance of the UPS emerged with the understanding that cancer cells, particularly those with high protein turnover like multiple myeloma, exhibit heightened dependence on proteasomal function, creating a therapeutic window that can be exploited for targeted treatment [38]. This review comprehensively examines the UPS as a drug target, from fundamental structural biology to clinical applications, with particular emphasis on the β-grasp fold that constitutes the structural foundation of ubiquitin and ubiquitin-like proteins central to this system.
Structural Foundations: The β-Grasp Fold in Ubiquitin and Ubiquitin-like Proteins
Ubiquitin and the β-Grasp Fold Architecture
Ubiquitin, a 76-amino acid polypeptide, features a distinctive structural motif known as the β-grasp fold (β-GF) [1] [11]. This compact fold is dominated by a mixed β-sheet consisting of five anti-parallel β-strands that appears to "grasp" a single α-helical segment positioned across the sheet [1]. Despite its small size, the β-grasp fold serves as a remarkably versatile structural scaffold that has been recruited for a strikingly diverse range of biochemical functions throughout evolution [1].
The evolutionary history of the β-grasp fold is profound, with reconstructions indicating that it had already differentiated into at least seven distinct lineages by the time of the last universal common ancestor of all extant organisms [1]. Early β-grasp members were likely involved in RNA metabolism, subsequently radiating into various functional niches including enzymatic active sites, iron-sulfur cluster scaffolding, RNA-soluble-ligand and co-factor-binding, sulfur transfer, and adaptor functions in signaling [1]. The structural versatility of the β-grasp fold arises primarily from its prominent β-sheet, which provides an exposed surface for diverse interactions or can form open barrel-like structures to accommodate various functional adaptations [1].
Ubiquitin-like Proteins and Evolutionary Diversification
The ubiquitin system centers on the conjugation and deconjugation of ubiquitin and ubiquitin-like (Ubl) proteins to cellular targets, regulating virtually all aspects of eukaryotic biology [11]. Eukaryotes possess numerous Ubl proteins that share the β-grasp fold, including SUMO, Nedd8, Urm1, and Apg12, which undergo analogous conjugation pathways despite often serving distinct cellular functions [1] [11]. The eukaryotic phase of β-grasp evolution was marked by a dramatic expansion of Ubl proteins, with the last eukaryotic common ancestor already possessing nearly 20 distinct Ubl families that played crucial roles in the emergence of characteristic eukaryotic cellular systems [1].
Table: Major Functional Classes of β-Grasp Fold Proteins
| Functional Class | Representative Members | Primary Cellular Role |
|---|---|---|
| Protein Degradation Tags | Ubiquitin, SUMO, Nedd8 | Post-translational protein modification targeting proteins for degradation or functional modulation |
| Sulfur Transfer | ThiS, MoaD, Urm1 | Biosynthesis of thiamine, molybdenum cofactor, and tRNA thiolation |
| Enzymatic Catalysis | NUDIX phosphohydrolases | Diverse hydrolase activities on nucleotide substrates |
| Adaptor Signaling | FERM N-terminal domain, RA domain | Mediating protein-protein interactions in signaling pathways |
| RNA Binding | TGS domain | RNA binding in aminoacyl tRNA synthetases and translation regulators |
| Toxins | Staphylococcal enterotoxin B | Superantigen activity in toxic shock syndrome |
Notably, precursors of the eukaryotic ubiquitin system first emerged in prokaryotes, with the simplest systems combining a Ubl and an E1-like enzyme involved in metabolic pathways related to metallopterin, thiamine, cysteine, and siderophore biosynthesis [11]. The evolutionary conservation of this fold across domains of life underscores its fundamental utility in cellular regulation, while its functional diversification highlights its remarkable structural plasticity.
The Ubiquitin-Proteasome Pathway: Mechanism and Components
The Ubiquitination Cascade
Protein degradation via the UPS initiates with the precise tagging of target proteins with ubiquitin through a well-orchestrated enzymatic cascade [38]. This process involves three distinct steps:
- Activation: Ubiquitin is activated in an ATP-dependent reaction catalyzed by the E1 ubiquitin-activating enzyme, forming a thioester bond between E1 and the C-terminus of ubiquitin [38].
- Conjugation: The activated ubiquitin is transferred to an E2 ubiquitin-conjugating enzyme, preserving the high-energy thioester linkage [38].
- Ligation: An E3 ubiquitin ligase facilitates the transfer of ubiquitin from E2 to the ε-amino group of a lysine residue on the target protein, forming an isopeptide bond [38].
This process repeats to form polyubiquitin chains, with specific chain linkages determining the fate of the modified protein. The human genome encodes over 600 E3 ligases, which provide substrate specificity to the UPS and represent attractive intervention points for therapeutic development [39].
The Proteasome Complex
The 26S proteasome is a massive multiprotein complex consisting of a 20S catalytic core particle (CP) capped by one or two 19S regulatory particles (RP) [38]. The 20S core particle forms a barrel-like structure composed of four stacked heptameric rings (α7β7β7α7) [38]. The outer α-rings regulate substrate entry, while the inner β-rings contain the proteolytic active sites facing the interior chamber [38]. Three distinct catalytic activities reside within the β-subunits: chymotrypsin-like (β5), trypsin-like (β2), and caspase-like (β1) activities, which cleave after hydrophobic, basic, and acidic residues, respectively [38]. The chymotrypsin-like site is particularly sensitive to inhibition and represents the primary target of clinical proteasome inhibitors [38].
The 19S regulatory particle recognizes polyubiquitinated substrates, removes the ubiquitin chains, unfolds the target protein, and translocates the unfolded polypeptide into the degradative chamber of the 20S core [38]. A specialized variation exists in Actinobacteria, including Mycobacterium tuberculosis, which employs a prokaryotic ubiquitin-like protein (Pup) as a degradation tag and the mycobacterial proteasome ATPase (Mpa) as the regulatory particle, demonstrating evolutionary convergence in targeted protein degradation mechanisms [40].
Diagram: The Ubiquitin-Proteasome Pathway. The pathway involves sequential enzymatic steps for ubiquitin conjugation followed by recognition and degradation by the proteasome complex.
Clinical Translation: Proteasome Inhibitors in Hematologic Malignancies
Mechanism of Proteasome Inhibitor-Mediated Cytotoxicity
Proteasome inhibitors exert their anti-cancer effects through multiple interconnected mechanisms that collectively induce apoptosis in malignant cells [38]. The primary mechanisms include:
- Inhibition of NF-κB Pathway: Proteasome inhibition prevents the degradation of IκBα, an endogenous inhibitor of NF-κB, thereby sequestering this pro-survival transcription factor in the cytoplasm and blocking its anti-apoptotic signaling [38].
- Induction of Unfolded Protein Response (UPR): Multiple myeloma cells produce large quantities of immunoglobulins, creating exceptional endoplasmic reticulum (ER) stress. Proteasome inhibition prevents clearance of misfolded proteins from the ER, exacerbating ER stress and activating the terminal UPR leading to apoptosis [38].
- Stabilization of Pro-apoptotic Factors: Proteasome inhibitors prevent the degradation of pro-apoptotic proteins such as Bim, Bid, Bik, and NOXA, allowing these factors to accumulate and trigger caspase activation and programmed cell death [38].
- Activation of p53 and JNK Pathways: Proteasome inhibition can stabilize the tumor suppressor p53 and activate c-Jun NH2-terminal kinase (JNK), both of which promote apoptotic signaling cascades [38].
The particular susceptibility of multiple myeloma cells to proteasome inhibitors stems from their high rate of immunoglobulin production, which creates exceptional dependence on proteasomal capacity to manage the resulting protein burden [38]. This dependency creates a therapeutic window where malignant cells are more vulnerable to proteasome inhibition than normal cells.
Approved Proteasome Inhibitors and Clinical Applications
Since 2003, three proteasome inhibitors have received FDA approval for the treatment of hematologic malignancies, fundamentally changing the management of multiple myeloma and mantle cell lymphoma [38].
Table: Clinically Approved Proteasome Inhibitors
| Name (Brand) | Year Approved | Inhibition Kinetics | Active Moiety | Primary Indications | Route of Administration | Common Toxicities |
|---|---|---|---|---|---|---|
| Bortezomib (Velcade) | 2003 | Slowly reversible | Boronate | First-line, relapsed or refractory MM and MCL | IV/SC | Peripheral neuropathy, nausea, vomiting, diarrhea, cytopenias, infection |
| Carfilzomib (Kyprolis) | 2012 | Irreversible | Epoxyketone | Relapsed or refractory MM | IV | Dyspnea, cytopenias, nausea, vomiting, diarrhea, fatigue, headache, peripheral edema |
| Ixazomib (Ninlaro) | 2015 | Reversible | Boronate | MM after one prior therapy | Oral | Diarrhea, constipation, cytopenias, peripheral neuropathy, nausea, vomiting, peripheral edema, back pain |
The efficacy of proteasome inhibitors is enhanced when administered in combination with other agents. In multiple myeloma, triplet therapy combining a proteasome inhibitor (bortezomib or carfilzomib) with dexamethasone and an immunomodulatory drug (lenalidomide, thalidomide, or pomalidomide) has demonstrated superior outcomes compared to single-agent or doublet regimens [38]. The landmark APEX trial established bortezomib's superiority over high-dose dexamethasone, with response rates of 38% versus 18% and significantly improved time to progression (6.22 months vs. 3.49 months) in relapsed multiple myeloma [38].
Emerging Frontiers: Expanding the UPS Therapeutic Landscape
Deubiquitinating Enzyme (DUB) Inhibitors
As resistance mechanisms to conventional proteasome inhibitors emerge and the limitations of broad proteasome inhibition become apparent, research focus has expanded upstream in the UPS to target deubiquitinating enzymes (DUBs) [41]. DUBs comprise seven primary families of proteases (USPs, UCHs, OTUs, MJDs, JAMMs, ZUP1, and MINDYs) that cleave ubiquitin from target proteins, thereby counterregulating ubiquitin signaling and substrate degradation [41]. The therapeutic rationale for DUB inhibition includes:
- Achieving greater selectivity for specific pathways compared to broad proteasome inhibition
- Overcoming resistance to existing proteasome inhibitors
- Targeting oncoproteins stabilized by specific DUBs
Notable DUB targets under investigation include USP1, involved in DNA damage repair; USP7, which regulates p53 and other cancer-relevant substrates; and USP9X, which stabilizes anti-apoptotic proteins like Mcl-1 [41]. While no DUB inhibitors have yet received FDA approval, several candidates have entered clinical and preclinical development, representing a promising new frontier in targeting the UPS for cancer therapy [41].
Targeted Protein Degradation Strategies
Beyond conventional inhibition, revolutionary new modalities have emerged that exploit the UPS to achieve targeted protein degradation, most notably proteolysis-targeting chimeras (PROTACs) and molecular glues [39]. These approaches offer several advantages over traditional small molecule inhibitors:
- Ability to target "undruggable" proteins that lack conventional binding pockets
- Catalytic mode of action allowing sub-stoichiometric activity
- Potential to overcome resistance mutations that impair inhibitor binding
PROTACs are heterobifunctional molecules consisting of a target protein-binding ligand connected via a chemical linker to an E3 ubiquitin ligase recruiter [39]. This structure facilitates the formation of a ternary complex that brings the E3 ligase into proximity with the target protein, leading to its ubiquitination and subsequent proteasomal degradation [39]. The first small-molecule PROTACs reported in 2008 have evolved significantly, with many candidates now advancing through clinical trials [39].
Molecular glues represent a distinct approach comprising small molecules that induce or stabilize protein-protein interactions between E3 ligases and target proteins, leading to target ubiquitination and degradation [39]. Though many molecular glues were discovered serendipitously, rational design approaches are emerging that may expand the applicability of this promising technology [39].
Biomolecular Condensates and Intrinsically Disordered Proteins
Recent research has revealed connections between the UPS and biomolecular condensates—membrane-less organelles formed through liquid-liquid phase separation (LLPS) that compartmentalize cellular processes [42]. Intrinsically disordered proteins (IDPs) and regions (IDRs), which lack stable tertiary structures, frequently drive condensate formation and often serve as scaffolds [42]. Notably, aberrant biomolecular condensates have been implicated in cancer and neurodegenerative diseases, with mutations potentially altering phase separation properties and contributing to pathogenesis [42].
A novel class of "condensate-modifying drugs" (c-mods) has emerged that targets IDPs and biomolecular condensates, potentially offering therapeutic approaches for previously "undruggable" targets like c-Myc and p53 [42]. These agents can be categorized as dissolvers (reverse condensate formation), inducers (trigger condensate formation), localizers (alter subcellular localization), or morphers (modify condensate material properties) [42]. This emerging field represents a promising intersection with UPS-targeted therapies, as many condensate components are regulated through ubiquitination and proteasomal degradation.
Experimental Approaches: Methodologies for UPS Drug Discovery
Key Research Reagent Solutions
Table: Essential Research Reagents for UPS-Targeted Drug Discovery
| Reagent Category | Specific Examples | Research Application | Key Features/Functions |
|---|---|---|---|
| Proteasome Inhibitors | Bortezomib, Carfilzomib, MG132, Lactacystin | Mechanism studies, in vitro and cellular assays | Target chymotrypsin-like (β5) activity primarily; different inhibition kinetics (reversible/irreversible) |
| DUB Inhibitors | P5091 (USP7), ML323 (USP1/UAF1), WP1130 (USP9X) | Pathway dissection, target validation | Varying selectivity profiles; some target specific DUBs while others are pan-DUB inhibitors |
| E3 Ligase Ligands | VHL ligands, CRBN ligands (lenalidomide), MDM2 ligands (Nutlin-3) | PROTAC development, E3 ligase function studies | Enable recruitment of specific E3 ligases for targeted protein degradation |
| Activity Probes | Ubiquitin-AMC, Ubiquitin-Rho110, TAMRA-labeled ubiquitin vinyl sulfone | Enzymatic assays, high-throughput screening | Fluorescent or tagged substrates for monitoring DUB or proteasome activity |
| Pupylation System Components | PupE (Mtb), Mpa, Dop, PafA | Bacterial UPS analog studies | Study evolutionarily related but distinct prokaryotic protein degradation system |
| Model Substrates | Ubiquitinated proteins, PupDHFR fusion, UbG76V-GFP | Degradation assays, substrate engagement studies | Reporter substrates for monitoring UPS activity in cellular or biochemical systems |
Structural Biology Techniques for UPS Mechanism Studies
Advanced structural biology approaches have been instrumental in elucidating the mechanistic details of UPS components and their interactions:
Cryo-Electron Microscopy (Cryo-EM) Methodology for Mpa-Proteasome Complex Analysis [40]:
- Complex Preparation: Generate a substrate-engaged complex using Mpa, 20S core particle (with Δ7PrcA α-subunits for stability), and a model PupDHFR substrate. Assemble in presence of ATP on ice to permit substrate translocation while slowing ATP turnover.
- Sample Vitrification: Quench reaction with ATPγS to stall translocation, apply to EM grid, and vitrify using liquid ethane.
- Data Collection: Acquire micrographs using a cryo-electron microscope equipped with a direct electron detector.
- Image Processing: Pick particles from micrographs (initial set: ~860,000 particles), followed by 2D classification to identify complex views. Use 385,394 particles for 3D classification.
- High-Resolution Reconstruction: Perform local 3D classification and refinement with signal subtraction to resolve distinct conformational states of Mpa (3.8-3.9 Å resolution) and the 20S core particle (2.8 Å resolution).
This approach revealed two distinct conformational states of the Mpa-proteasome complex corresponding to sequential stages of substrate translocation, providing unprecedented insights into the mechanism of bacterial protein degradation [40].
Experimental Workflow for DUB Inhibitor Development [41]:
- Target Selection: Prioritize DUBs with validated disease associations (e.g., USP7 in cancer, USP13 in neurodegeneration).
- Primary Screening: Employ biochemical assays using fluorescent ubiquitin substrates (e.g., Ub-AMC) or activity-based probes against DUB libraries.
- Selectivity Profiling: Counter-screen against panel of related DUBs and deubiquitinating enzymes to assess specificity.
- Cellular Target Engagement: Validate cellular activity using ubiquitin-specific antibodies, substrate accumulation assays, or cellular thermal shift assays (CETSA).
- Mechanistic Studies: Determine mode of inhibition (reversible/irreversible) and conduct structural studies (crystallography/cryo-EM) of inhibitor-DUB complexes.
- Functional Characterization: Assess effects on downstream pathways, protein degradation, and phenotypic outcomes in disease-relevant models.
Diagram: UPS-Targeted Therapeutic Strategies. Multiple modalities engage different components of the ubiquitin-proteasome system to achieve therapeutic effects.
The targeting of the ubiquitin-proteasome system represents one of the most successful translations of basic biological insight into clinical therapy in oncology. From the initial approval of bortezomib in 2003 to the emerging paradigms of targeted protein degradation, our growing understanding of the UPS has continuously expanded the therapeutic landscape. The structural foundation of this system in the versatile β-grasp fold underscores the evolutionary ingenuity of cellular regulation mechanisms, while providing a rich repertoire of targets for therapeutic intervention.
Future directions in UPS-targeted drug discovery will likely focus on enhancing specificity through E3 ligase-and tissue-selective approaches, developing novel modalities that exploit biomolecular condensates and phase separation biology, and addressing challenges in drug delivery and resistance mechanisms. The integration of structural biology, chemical biology, and disease biology will continue to drive innovation in this dynamic field, offering new hope for patients with malignancies and other diseases characterized by proteostatic dysfunction. As our fundamental understanding of the UPS deepens, so too will our ability to precisely manipulate this system for therapeutic benefit across a broadening spectrum of human diseases.
Overcoming Challenges in Studying and Therapeutically Targeting β-Grasp Proteins
Addressing Functional Complexity and Redundancy in UBL Pathways
Ubiquitin-like proteins (UBLs) constitute a family of post-translational modifiers characterized by the conserved β-grasp fold structure, which enables their conjugation to target proteins through parallel yet functionally distinct enzymatic cascades. This technical guide examines the intricate complexity and pervasive redundancy within UBL pathways, highlighting how these features pose both challenges and opportunities for therapeutic intervention. We explore the structural mechanisms governing UBL activation, conjugation, and recognition, with particular emphasis on how UBL-UBA proteins exemplify biological redundancy in proteasomal targeting. The article further synthesizes current methodologies for investigating UBL networks and discusses emerging strategies for targeting redundant pathways in drug development, providing researchers with a comprehensive framework for navigating this complex signaling landscape.
The ubiquitin-like protein system represents a sophisticated regulatory network that controls nearly every aspect of cellular homeostasis through covalent modification of target proteins. Central to this system is the β-grasp fold, a structurally conserved domain consisting of a mixed β-sheet that grasps a central α-helix [43]. This evolutionarily ancient structural motif provides the foundation for a diverse family of protein modifiers that includes ubiquitin itself and at least 18 human UBLs such as SUMO, NEDD8, ISG15, and ATG8 [44]. Despite sharing this common structural core, UBLs have evolved distinct functional specialties through variations in surface features, conjugation machinery, and recognition systems.
The functional complexity of UBL pathways arises from their multi-layered architecture, which encompasses activating enzymes (E1), conjugating enzymes (E2), and ligases (E3) that work in concert to modify specific cellular targets [8]. This enzymatic cascade attaches UBLs to substrate proteins through an isopeptide bond, forming a covalent linkage that can alter the substrate's activity, stability, localization, or interaction partners. The system is further complicated by the ability of UBLs to form chains through different linkage types, creating a diverse "code" that is interpreted by specialized recognition proteins containing ubiquitin-binding domains (UBDs) [43].
Redundancy represents a fundamental design principle within UBL networks, manifesting at multiple levels including enzyme specificity, substrate recognition, and functional compensation between parallel pathways. This redundancy provides robustness to the system but creates significant challenges for therapeutic intervention, as inhibiting single components often fails to disrupt pathway outputs. Understanding the structural and mechanistic basis of this complexity is essential for developing targeted strategies to modulate UBL pathways in disease contexts.
Structural and Mechanistic Foundations of UBL Signaling
The UBL Conjugation Cascade
UBL conjugation follows a conserved three-step enzymatic mechanism that is initiated by E1 activating enzymes, which recognize and activate specific UBLs in an ATP-dependent reaction. Structural studies reveal that canonical E1 enzymes share a conserved domain architecture consisting of two pseudosymmetric adenylation domains that form a composite active site for ATP•Mg2+ and Ubl binding, a catalytic cysteine (CYS) domain that forms the E1~Ubl thioester bond, and a ubiquitin fold domain (UFD) that interacts with E2 proteins [8]. The E1 mechanism involves substantial conformational changes, including a 130° rotation of the CYS domain that transits the catalytic cysteine to a position proximal to the UBL C-terminal adenylate [8].
Following UBL activation, the E1~UBL thioester complex recruits specific E2 conjugating enzymes, with transfer occurring through a mechanism that involves a ~25° rotation of the E1 UFD to bring the E2 active site cysteine into proximity with the E1~UBL thioester [8]. The human genome encodes approximately 40 E2 enzymes for ubiquitin alone, with additional specialized E2s dedicated to specific UBL pathways [45]. E2s serve as central determinants of pathway specificity, dictating aspects such as the efficiency of UBL chain assembly and the configuration of polyUBL chains [45].
The final transfer of the UBL to specific substrate proteins is typically facilitated by E3 ligases, which number in the hundreds for ubiquitin and include specialized counterparts for other UBLs [45]. E3s provide critical substrate specificity through recognition of degradation signals or other modification motifs, such as the SUMO consensus motif (ΨKX(E/D)) that directs SUMO conjugation [44]. Depending on the E3 class, substrate modification occurs either through direct transfer from the E2~UBL complex or after formation of a transient E3~UBL thioester intermediate.
Table 1: Major UBL Types and Their Primary Functions
| UBL | Length (aa) | E1 Enzyme | E2 Enzyme(s) | Primary Functions |
|---|---|---|---|---|
| Ubiquitin | 76 | Uba1 | ~40 different E2s | Proteasomal degradation, signaling |
| SUMO-1 | 97 | SAE1/SAE2 | Ubc9 | Transcription, DNA repair, nuclear transport |
| NEDD8 | 81 | Uba3/NAE1 | Ubc12 | Cullin activation, cell cycle |
| ISG15 | 157 | UBE1L | UBCH8 | Immune response, antiviral defense |
| ATG8 | 117 | ATG7 | ATG3 | Autophagy, membrane trafficking |
Diversity of UBL Modifications and Recognition
UBL modifications exhibit remarkable diversity in both form and function. Modifications range from monoubiquitination (single UBL attachment) that often regulates protein activity and localization, to multi-monoubiquitination (multiple single UBLs at different sites), to polyubiquitination (UBL chains) that can signal for proteasomal degradation or serve as scaffolds for protein complexes [46]. The structural basis for this functional diversity lies in the ability of UBLs to form chains through different linkage types, connecting the C-terminus of one UBL to specific lysine residues or the N-terminal methionine of another UBL.
The eight possible linkage sites (K6, K11, K27, K29, K33, K48, K63, M1) in ubiquitin create an extensive coding system where different chain types signal distinct functional outcomes [43]. For instance, K48-linked chains typically target substrates for proteasomal degradation, while K63-linked chains often function in non-proteolytic processes such as DNA repair, endocytic trafficking, and inflammatory signaling [43]. Similar linkage specificity exists for other UBLs, though the functional consequences are distinct—SUMO chains, for example, regulate protein interactions and localization rather than degradation.
The interpretation of this UBL code is carried out by a diverse family of ubiquitin-binding proteins (UBPs) that contain specialized ubiquitin-binding domains (UBDs) [43]. These recognition proteins function as signal adaptors that transmit UBL signals to downstream effectors, thereby connecting modified substrates to appropriate cellular machinery. Recent systematic approaches have identified numerous selective interactors for different UBL chain types, with one study reporting 46 specific interactors for linear polyubiquitin chains [43], highlighting the sophisticated decoding capacity of the UBL recognition system.
Diagram 1: UBL Conjugation Cascade. The E1-E2-E3 enzymatic pathway activates and transfers UBLs to substrate proteins.
Functional Complexity and Redundancy in UBL Pathways
Molecular Mechanisms of Pathway Redundancy
Redundancy in UBL pathways operates at multiple levels, from genetic duplication and functional overlap between paralogous proteins to compensatory interactions between distinct pathways. A prime example of this redundancy can be found in the UBL-UBA protein family in budding yeast, which includes Rad23, Dsk2, and Ddi1 [47]. These proteins function as ubiquitin receptors that shuttle polyubiquitinated substrates to the proteasome, with each containing an N-terminal UBL domain that interacts with the proteasome and one or more C-terminal UBA domains that bind ubiquitin chains.
Genetic studies demonstrate that while single deletions of RAD23, DSK2, or DDI1 are viable, combination deletions show synthetic phenotypes revealing their functional overlap. The rad23Δdsk2Δ double deletion completely stabilizes a model degradation substrate that is only partially stabilized in single deletions [47]. Similarly, the triple deletion rad23Δdsk2Δddi1Δ exhibits temperature sensitivity and distinct cell cycle defects not observed in any single or double mutant combinations [47]. This genetic evidence indicates partially redundant roles where Rad23 can compensate for the loss of both Dsk2 and Ddi1, while Dsk2 and Ddi1 cannot fully compensate for each other, suggesting Rad23 possesses at least two distinct cell cycle-related functions.
At the molecular level, UBL-UBA proteins can form both homodimers and heterodimers, further expanding their functional versatility. Rad23 and Dsk2 homodimerization occurs through their UBL and/or UBA domains, while Ddi1 homodimerization utilizes neither of these domains [47]. Heterodimerization between different UBL-UBA proteins has been demonstrated through both direct UBL-UBA domain interactions and indirect associations mediated by bridging molecules such as ubiquitin chains [48]. This combinatorial interaction network allows UBL-UBA proteins to form multimeric complexes with polyubiquitin chains, potentially increasing the efficiency of substrate delivery to the proteasome [47].
Structural Basis for Redundant Ubiquitin Chain Recognition
The structural flexibility of polyubiquitin chains contributes significantly to the redundancy in UBL recognition systems. While K48-linked tetraubiquitin adopts a closed conformation in crystal structures with only the most distal ubiquitin moiety available for binding, biochemical studies demonstrate that tetraubiquitin can simultaneously bind two Rad23 molecules or combinations of Rad23 and Ddi1 [48]. This finding indicates that polyubiquitin chains can adopt open conformations when bound to their receptors, allowing multiple recognition proteins to engage the same chain simultaneously.
This structural plasticity enables redundant recognition of ubiquitinated substrates by different UBL-UBA proteins, as demonstrated by the ability of Rad23, Dsk2, and Ddi1 to all bind common tetraubiquitin chain types [48]. The UBA domains of these proteins exhibit complementary binding preferences for different chain configurations, creating a system where multiple receptors can recognize the same substrate under different conditions or in different cellular contexts. This redundancy provides robustness to the protein degradation system while complicating efforts to disrupt specific substrate turnover through targeted inhibition of individual components.
Table 2: Redundancy in Yeast UBL-UBA Proteins
| Protein | Domains | Dimerization Mechanism | Genetic Interactions | Cell Cycle Phenotypes |
|---|---|---|---|---|
| Rad23 | UBL + 2 UBA | UBL/UBA domains | Redundant with Dsk2 and Ddi1 | G2/M and anaphase delay in combinations |
| Dsk2 | UBL + UBA | UBL/UBA domains | Redundant with Rad23 only | SPB duplication defects in rad23Δdsk2Δ |
| Ddi1 | UBL + UBA | Non-UBL/UBA domains | Redundant with Rad23 only | Requires homodimerization for function |
Experimental Approaches for Deconvoluting UBL Complexity
Chemical Biology Tools for UBL Research
Chemical protein synthesis has emerged as a powerful approach for producing homogeneously modified UBL conjugates that are challenging to obtain through enzymatic or recombinant methods. These techniques enable precise control over UBL assembly, facilitating the incorporation of site-specific modifications, non-canonical amino acids, and defined linkage types for structural and functional studies [44]. Key methodologies include:
- Solid-phase peptide synthesis (SPPS): Allows assembly of peptides with defined sequences and modifications, particularly useful for generating UBL domains and short conjugated peptides [44].
- Native chemical ligation (NCL): Enables coupling of unprotected peptide segments through reaction of a C-terminal thioester with an N-terminal cysteine, facilitating the synthesis of full-length UBLs and their conjugates [44].
- Extended ligation strategies: Including desulfurization/deselenization reactions that expand NCL to non-cysteine sites, and serine/threonine ligation that further broadens the synthetic toolbox [44].
- Semi-synthetic approaches: Combine chemically synthesized fragments with recombinantly expressed protein domains, enabling preparation of large proteins with specific modifications [44].
These chemical methods have been successfully applied to study various UBLs including SUMO, NEDD8, UFM1, ISG15, and ATG8, allowing researchers to generate defined chain types and complex conjugates for mechanistic investigations [44]. For example, synthetic SUMO conjugates have been used to elucidate the role of SUMOylation in RanGAP1 targeting to the nuclear pore complex and in promyelocytic leukemia protein nuclear body assembly [44].
Structural and Biophysical Methodologies
Structural biology approaches have been instrumental in elucidating the conformational changes and molecular interactions that underlie UBL pathway complexity and redundancy. Key methodologies include:
- X-ray crystallography: Has revealed the dramatic domain rearrangements in E1 enzymes during UBL activation, including the 130° rotation of the CYS domain that positions the catalytic cysteine for thioester formation [8].
- NMR spectroscopy: Has been used to map protein-protein interactions in UBL pathways, such as the characterization of UBL/UBA domain interactions mediating heterodimerization between Rad23 and Ddi1 [48].
- Targeted disulfide cross-linking: Combined with structural analysis has captured transient E1-E2 interaction states, revealing the ~25° rotation of the E1 UFD domain that facilitates thioester transfer [8].
- Mechanism-based inhibitors: Such as MLN4924 (an adenosine sulfamate analog that inhibits NEDD8 activation) have been used as structural and chemical probes to investigate E1 enzyme mechanisms and conformational dynamics [8].
These structural approaches have demonstrated how UBL pathway components utilize conformational plasticity and composite binding surfaces to achieve specificity while maintaining redundant recognition capabilities. The integration of structural information with biochemical and genetic data has been essential for developing comprehensive models of UBL network organization and regulation.
Diagram 2: UBL-UBA Protein Redundancy. Multiple UBL-UBA proteins can recognize the same ubiquitinated substrates and deliver them to the proteasome.
Research Reagent Solutions for UBL Studies
Table 3: Essential Research Tools for Investigating UBL Pathways
| Reagent Category | Specific Examples | Applications | Key Features |
|---|---|---|---|
| Chemical Synthesis Tools | SPPS, NCL, KAHA ligation | Production of homogeneous UBL conjugates | Precise control over modifications and linkages |
| Mechanism-Based Inhibitors | MLN4924 (NEDD8-E1 inhibitor) | Probing specific UBL pathway functions | Forms covalent adduct with NEDD8, trapping E1 |
| Structural Biology Platforms | X-ray crystallography, NMR spectroscopy | Determining molecular mechanisms | Captures conformational states and interactions |
| Yeast Genetic System | rad23Δ, dsk2Δ, ddi1Δ single and combination mutants | Analyzing functional redundancy | Reveals synthetic phenotypes and genetic interactions |
| UBL Interaction Probes | Synthetic diubiquitins (8 linkage types) | Decoding ubiquitin signaling | Identifies linkage-specific interactors |
The functional complexity and redundancy inherent in UBL pathways represent both a fundamental challenge and untapped opportunity for therapeutic development. The β-grasp fold provides an evolutionarily optimized structural platform that supports diverse signaling functions while enabling compensatory mechanisms through conserved interaction surfaces. As research methodologies advance, particularly in chemical biology and structural analysis, our ability to deconvolute these complex networks continues to improve.
Future research directions should focus on developing multi-target therapeutic strategies that address pathway redundancy while minimizing off-target effects. The success of MLN4924, which specifically inhibits the NEDD8 E1 enzyme, demonstrates the potential of targeting nodal points in UBL cascades for therapeutic benefit [8] [45]. Similarly, exploring the allosteric networks that coordinate conformational changes in E1 and E2 enzymes may reveal new opportunities for selective inhibition.
As our understanding of the UBL code continues to expand, particularly through systematic approaches to identify linkage-specific interactors, new patterns of redundancy and specialization will likely emerge. Integrating this knowledge with chemical biology tools for generating defined UBL conjugates will enable more precise dissection of pathway functions and facilitate the development of targeted interventions for diseases characterized by dysregulated UBL signaling.
Achieving Specificity in Drug Design for Highly Conserved Binding Interfaces
The β-grasp fold (β-GF) is a structurally conserved scaffold prototyped by ubiquitin (UB) and is central to a strikingly diverse range of biochemical functions, from post-translational modification and sulfur transfer to RNA binding and adaptor roles in signaling [1]. Its evolutionary success is attributed to its simple yet versatile architecture, dominated by a β-sheet with five anti-parallel strands that "grasp" a single helical segment. This fold had already diversified into at least seven distinct lineages by the time of the last universal common ancestor, encompassing much of the structural diversity seen today [1]. However, this very structural conservation presents a significant challenge in drug design: targeting a specific member of this fold, such as a particular ubiquitin-like (Ubl) protein, without affecting the myriad of other structurally similar proteins. The problem is exacerbated by the fact that protein-protein interaction (PPI) interfaces, common in β-GF protein functions, typically feature small, shallow pockets with an average volume of only 54 ų, making them difficult targets for traditional small-molecule inhibitors [49]. This guide outlines strategies and detailed methodologies for achieving specificity in drug design against these highly conserved yet functionally critical interfaces.
Structural Basis for Specificity: Lessons from the β-Grasp Fold
Core Features and Functional Adaptations
The manifold functional abilities of the β-GF arise primarily from its prominent β-sheet, which provides an exposed surface for diverse interactions. Systematic analysis reveals that both enzymatic activities and the binding of diverse co-factors have independently evolved on multiple occasions within this fold [1]. Understanding these adaptations is the first step toward designing specific inhibitors.
Key structural elements that can be exploited for specificity include:
- The Core β-Sheet: The primary interaction surface, often used for binding a wide array of partners. Its conservation means that targeting its most central residues will likely lead to cross-reactivity.
- Variable Loops and Inserts: Specific lineages within the β-GF are defined by unique structural elaborations. For example, the Soluble-Ligand-Binding β-grasp (SLBB) superfamily is characterized by inserts of β-hairpins or other segments that often play a direct role in ligand binding [2]. These less-conserved regions are prime targets for achieving specificity.
- Conformational Plasticity: Binding sites are not static. Research on PPIs shows that "the critical components of this adaptivity are largely local, involving primarily low energy side-chain motions within 6 Å of a hot spot" [49]. Designing compounds that exploit the specific conformational dynamics of a target, but not its homologs, is a viable strategy.
The Druggable Hot Spot Paradigm
Despite the conservation, PPI interfaces contain druggable hot spots—small regions that are major contributors to the binding free energy. These hot spots are characterized by a concave topology combined with a pattern of hydrophobic and polar functionality, giving them a general tendency to bind organic compounds with a variety of structures [49].
Table 1: Key Characteristics of Druggable Hot Spots at PPI Interfaces
| Characteristic | Description | Implication for Drug Design |
|---|---|---|
| Topography | Concave surface cavities | Provides a defined region for ligand anchorage, despite the overall flat interface. |
| Composition | Mix of hydrophobic and polar functionality | Binds organic species with hydrophobic scaffolds decorated by polar groups. |
| Prevalence | Multiple small pockets (~54 ų average) | Requires identification of the key cluster of pockets that can be targeted. |
| Adaptivity | Local side-chain flexibility (within ~6 Å) | Ligands can induce pocket expansion to a drug-like volume. |
Computational Strategy: Mapping Druggable Hot Spots
A cornerstone of modern PPI drug discovery is the computational identification of hot spots and the prediction of their ability to bind drug-sized molecules. The following protocol, adapted from a landmark study, provides a robust method for this task [49].
Protocol: Computational Solvent Mapping for Hot Spot Identification
Objective: To identify clusters of binding hot spots at a PPI interface and assess their druggability from the structure of the unliganded protein.
Materials and Software:
- Hardware: Standard high-performance computing workstation.
- Software: A computational solvent mapping suite (e.g., FTMap) or molecular dynamics software with custom analysis scripts.
- Input: High-resolution (e.g., < 2.5 Å) crystal or NMR structure of the target β-GF protein (e.g., a Ubl domain or its binding partner) from the PDB. The unliganded structure is preferred.
Methodology:
- Initial Mapping: Place 16-20 different small molecular "probes" (e.g., ethanol, isopropanol, acetonitrile, acetone) on a dense grid around the protein surface. The probes should vary in size, shape, and polarity.
- Identify Favorable Positions: For each probe, find favorable positions using empirical free energy functions. Clustering is essential for identifying consensus sites.
- Rank Consensus Sites: Rank the clusters based on the average free energy. Regions that bind several different probe clusters are designated consensus sites (CS). The site binding the largest number of probe clusters is the main hot spot (CS1).
- Assess Druggability: A site is considered druggable if the main hot spot (CS1) binds at least 16 probe clusters. The presence of nearby secondary hot spots (CS2, CS3, etc.) indicates a potential site that can accommodate a drug-sized ligand.
Accounting for Conformational Flexibility:
- Select Flexible Side Chains: Identify side chains within 6 Å of the main hot spot that are potentially flexible (e.g., based on B-factors or rotamer libraries).
- Generate Conformers: Use a conformer generator to create a set of energetically accessible alternative side-chain conformations.
- Re-map Alternative Structures: Repeat the computational solvent mapping on each of these alternative protein structures.
- Select the Optimal Conformation: The structure resulting in the highest number of probe clusters in the binding site represents the most druggable conformation.
This workflow is summarized in the diagram below:
Application to β-Grasp Fold Proteins
This methodology has been validated on multiple PPI targets. For instance, mapping the unliganded structure of Mdm2 (a key regulator of the tumor suppressor p53) identified two primary hot spots (CS1 and CS2, each with 21 probe clusters) at its p53-binding interface, confirming its druggability and guiding the development of Nutlin inhibitors [49]. When applied to a β-GF protein, the same process can pinpoint the specific surface patches—potentially involving variable loops or co-factor binding inserts unique to that lineage—that are most amenable to selective inhibition.
Experimental Validation of Hot Spots
Computational predictions require experimental validation. The following techniques are essential for confirming the location and importance of predicted hot spots.
Protocol: Alanine Scanning Mutagenesis
Objective: To experimentally determine the energetic contribution of individual residues to a protein-protein interaction.
Materials:
- Reagents: Site-directed mutagenesis kit, expression and purification systems for the target and partner proteins.
- Equipment: Equipment for quantifying protein interactions (e.g., Surface Plasmon Resonance (SPR) biosensor, Isothermal Titration Calorimetry (ITC), or equipment for Fluorescence Polarization (FP) assays).
Methodology:
- Design Mutants: Select a set of residues located at the predicted hot spot, typically those with significant solvent-accessible surface area and non-zero energy coefficients. Design point mutations to change each of these residues to alanine (or a similar inert amino acid).
- Generate Mutants: Use site-directed mutagenesis to create the alanine mutants of the target β-GF protein.
- Express and Purify: Express and purify the wild-type and all mutant proteins.
- Measure Binding Affinity: Determine the binding affinity (KD) of the wild-type and each mutant protein for its natural binding partner using a technique like SPR or ITC.
- Calculate ΔΔG: For each mutant, calculate the change in binding free energy: ΔΔG = -RT ln(KD,mutant / KD,wild-type). A ΔΔG > 1.0 kcal/mol typically indicates a "hot spot" residue.
Protocol: Fragment-Based Screening
Objective: To experimentally identify small molecular fragments that bind to the predicted hot spots.
Materials:
- Reagents: A library of 500-2000 low molecular weight (< 250 Da) compounds.
- Equipment: High-throughput screening facility equipped with X-ray crystallography, NMR, or Surface Plasmon Resonance (SPR).
Methodology:
- Screen Fragment Library: Screen the library against the target protein using a primary method such as SAR by NMR or a high-throughput SPR assay.
- Identify Hits: Identify fragments that show weak but specific binding (typical KD in the µM to mM range).
- Determine Co-crystal Structures: Soak hits into protein crystals and solve the co-crystal structure. This is crucial, as it confirms the binding location and shows how the fragment engages the hot spot.
- Fragment Growing/Linking: Use the structural information to chemically grow the fragment into a higher-affinity lead compound or link two adjacent fragments that bind to separate but nearby hot spots.
Table 2: Essential Research Reagent Solutions for Experimental Validation
| Research Reagent | Function/Application in Specificity Design |
|---|---|
| Site-Directed Mutagenesis Kit | For creating alanine mutants to validate hot spot residues via alanine scanning. |
| Surface Plasmon Resonance (SPR) Biosensor | For label-free, real-time quantification of binding kinetics (KA, KD) between wild-type/mutant proteins and partners or inhibitors. |
| Crystallization Screening Kits | For obtaining protein crystals of the target β-GF protein, essential for structural determination via X-ray crystallography. |
| Fragment Library (500-2000 compounds) | A collection of small, diverse molecules for experimental identification of weak binders to druggable hot spots. |
| Isothermal Titration Calorimetry (ITC) | For measuring the thermodynamics of binding interactions, providing direct measurement of binding affinity (KD) and enthalpy (ΔH). |
Case Study: Applying the Workflow to a β-Grasp Fold Target
Let us consider a hypothetical target: a bacterial ubiquitin-like β-GF protein involved in a sulfur transfer system, which is structurally similar to eukaryotic Ubls but has a distinct biological function and partner network [1].
- Computational Analysis: Computational solvent mapping of the bacterial Ubl reveals a primary hot spot (CS1) on its β-sheet surface, but also a secondary hot spot (CS2) adjacent to a unique β-hairpin insert not found in human homologs.
- Validation: Alanine scanning confirms that residues in both CS1 and the unique insert contribute significantly to binding its natural partner.
- Fragment Screening: A fragment screen identifies a molecule that binds specifically to the pocket formed by the unique β-hairpin insert (CS2). Co-crystallization confirms the binding mode.
- Lead Development: This fragment is optimized into a lead compound that makes extensive contacts with the variable insert but has minimal interaction with the more conserved core β-sheet. This compound potently inhibits the bacterial Ubl's function but shows no cross-reactivity with human Ubls in selectivity panels.
This targeted approach, which leverages both universal and lineage-specific structural features, provides a clear path to achieving specificity against a highly conserved binding interface.
Achieving specificity in drug design for the highly conserved binding interfaces of the β-grasp fold is a formidable but surmountable challenge. The key lies in moving beyond the conserved core and focusing on the structural nuances—the variable inserts, conformational dynamics, and unique clusters of druggable hot spots—that define each individual member of this ancient and versatile fold. By integrating robust computational mapping with rigorous experimental validation, as outlined in this guide, researchers can develop highly specific inhibitors that modulate the function of a single protein within the vast β-GF family, opening new avenues for therapeutic intervention in diseases driven by these critical players.
Optimizing Strategies for Targeting Protein-Protein Interactions
The β-grasp fold (β-GF) is a remarkable evolutionary scaffold characterized by a β-sheet with five anti-parallel strands that appears to "grasp" a single α-helical segment [1]. This compact structural motif, prototyped by ubiquitin (UB), has been recruited for a strikingly diverse range of biochemical functions, many of which involve critical protein-protein interactions (PPIs) in cellular processes [1]. The fold provides an exposed β-sheet surface that is ideal for diverse interactions and can form open barrel-like structures, enabling its participation in adaptor functions in signaling, assembly of macromolecular complexes, RNA-soluble-ligand binding, and post-translational protein modification [1]. The versatility of this small fold makes it an excellent model system for understanding general principles of PPIs and developing strategies to target them therapeutically.
Evolutionary reconstruction indicates that the β-GF had already differentiated into at least seven distinct lineages by the time of the last universal common ancestor of all extant organisms [1]. While most structural diversification occurred in prokaryotes, the eukaryotic phase was marked by a specific expansion of ubiquitin-like (Ubl) β-GF members, with the eukaryotic UB superfamily diversifying into at least 67 distinct families [1]. This evolutionary history has produced a fold that participates in PPIs central to numerous disease pathways, making it a high-value target for therapeutic intervention. The structural conservation yet functional diversity of the β-GF provides unique insights into targeting PPIs, as strategies developed for this fold can often be generalized to other interaction interfaces.
Structural Characterization of PPI Interfaces and Binding Pockets
Classification and Analysis of PPI Binding Pockets
Systematic analysis of PPI interfaces reveals distinct structural characteristics that influence druggability. A comprehensive dataset of pocket-centric structural data has classified ligand-binding pockets in PPI complexes into three main types based on their relationship with the protein-protein interaction interface [50]:
Table 1: Classification of Ligand-Binding Pockets in PPI Complexes
| Pocket Type | Acronym | Description | Functional Implication | Utility in Drug Discovery |
|---|---|---|---|---|
| Orthosteric Competitive | PLOC | Ligand binds directly at the PPI interface, competing with the protein partner's epitope | Direct inhibition of protein complex formation | Positive dataset for competitive inhibitor design |
| Orthosteric Non-Competitive | PLONC | Ligand occupies orthosteric pockets without direct competition with the protein epitope | May influence partner function or conformation | Training data for nuanced scenarios |
| Allosteric | PLA | Ligand binds near but not overlapping with the orthosteric binding pocket | Induces allosteric effects modulating the PPI | Negative dataset for non-competitive modulation |
This classification is crucial for understanding the functional implications of ligand binding and for training machine learning models to design focused chemical libraries [50]. The PLOC subset serves as a positive dataset for machine learning models targeting competitive inhibition, while PLA pockets represent negative datasets for ligands binding to PPI-involved protein chains without direct interface proximity.
Hot Spot Analysis in PPI Interfaces
PPI binding sites typically encompass specific residue combinations, distinct regions, and unique architectural layouts, resulting in cooperative formations referred to as "hot spots" [51]. These are defined as residues whose substitution results in a substantial decrease in the binding free energy (ΔΔG ≥ 2 kcal/mol) of a PPI [51]. The energetic contributions of hot spots stem from their localized networked arrangement within tightly packed "hot" regions, enabling flexibility and the capacity to bind to multiple different partners [51].
Interfaces rich in aromatic residues like tyrosine or phenylalanine have been shown to be particularly amenable to fragment hit identification [51]. This understanding has motivated the development of various strategies for targeting PPIs, with the β-grasp fold providing an excellent model system for studying these interfacial properties due to its well-characterized interaction surfaces and evolutionary conservation.
Computational Approaches for PPI Modulator Discovery
Predictive Methods and Machine Learning
The computational landscape for identifying and optimizing PPI modulators has expanded significantly, with methods broadly falling into two categories [51]:
Homology-based methods: Leverage the principle of "guilt by association" based on sequence similarity with known interactors. These methods are known for their accuracy and reliability, particularly for well-characterized proteins [51].
Template-free machine learning methods: Identify patterns in vast datasets of known interacting and non-interacting protein pairs using features like amino acid sequences, protein structures, or interaction affinities. Common algorithms include Support Vector Machines (SVMs) and Random Forests (RFs) [51].
The rapid progress and widespread adoption of large language models (LLMs) and machine learning have created a significant paradigm shift in PPI prediction and modulator design. These approaches are particularly valuable for targeting β-GF proteins, where evolutionary relationships can inform interaction predictions.
Virtual Screening and Fragment-Based Approaches
Table 2: Computational Strategies for PPI Modulator Discovery
| Strategy | Principle | Advantages | Limitations | Applicability to β-Grasp Fold |
|---|---|---|---|---|
| Structure-Based Virtual Screening | Utilizes 3D structural information of target protein | Direct physical modeling of interactions | Limited to proteins with well-defined binding pockets | Moderate (varies by specific β-GF protein) |
| Ligand-Based Virtual Screening | Screens compounds fitting pre-built pharmacophore model from known inhibitors | Does not require well-defined binding pocket | Dependent on existing known inhibitors | Limited for novel β-GF interactions |
| Fragment-Based Drug Discovery (FBDD) | Uses small, low molecular weight fragments that bind to discontinuous hot spots | Excellent for targeting PPI interfaces | Fragment linking remains challenging | High (suited for discontinuous epitopes) |
Traditional rational medicinal chemistry approaches are often less effective for PPI interfaces, which are frequently flat and featureless compared to enzyme active sites [51]. Fragment-based drug discovery has emerged as a particularly useful technique for designing PPI modulators because the presence of discontinuous hot spots on many PPI interfaces poses challenges for high-throughput screening but is very amenable to the binding of smaller fragments [51].
Experimental Methodologies for PPI Characterization
Atomic Force Microscopy (AFM) Force Spectroscopy
AFM force spectroscopy provides a powerful method for quantifying protein-protein interactions with pico-Newton (pN) resolution, enabling direct measurement of binding strengths between β-GF proteins and their partners [52].
Experimental Protocol: AFM Force Spectroscopy for PPI Measurement
Materials and Reagents:
- Silicon nitride cantilevers (spring constant ~0.06 N/m)
- APTES ((3-Aminopropyl) triethoxysilane)
- NHS-PEG-MAL linker molecules
- SATP (N-succinimidyl-S-acetylthiopropionate)
- Target proteins (e.g., FAK and Akt1 for β-GF relevant studies)
- PD-10 columns for buffer exchange
- Phosphate buffered saline (PBS) for storage
Probe Functionalization:
- Clean AFM probe in chloroform for one hour, rinse in fresh chloroform, and dry with argon gas.
- Process probes in oxygen plasma cleaner to enhance hydroxyl group density on silicon nitride surface.
- Coat APTES onto plasma-treated AFM probes via gas phase deposition in an argon-filled chamber.
- Functionalize APTES-coated probes with NHS-PEG-MAL linker molecules.
- Incubate PEG linker molecule-coated AFM probes in SATP-functionalized target protein molecules for 2-3 hours.
- Rinse functionalized probes three times with PBS and store at 4°C before use [52].
Substrate Preparation:
- Option 1: Direct deposition via hydrophobic interactions on polystyrene substrate.
- Option 2: APTES-mediated binding to amino-functionalized mica substrates.
- Incubate protein molecules on substrate surface for 2 hours, then rinse with PBS [52].
Measurement and Data Analysis:
- Approach-retract cycles are performed with controlled contact force and dwell time.
- Hundreds of force-distance curves are collected to ensure statistical significance.
- A statistical-based data processing method enhances the contrast between control and experimental samples.
- Binding probability is calculated from the percentage of approach-retract cycles showing specific adhesion events [52].
Optimization of this method has focused on improving the signal-to-noise ratio (SNR) through refined functionalization processes and redesigned probe-substrate contact regimes. This is particularly important for drug screening applications where the method's low false positive rate and label-free nature offer significant advantages over fluorescence-based techniques [52].
Structural Biology and Pocket Detection
High-resolution structural information is critical for understanding PPIs involving the β-grasp fold. The following workflow outlines the process for structural characterization of PPI interfaces:
Experimental Protocol: Structural Characterization of β-GF PPI Interfaces
Protein Selection and Quality Filtering:
- Download metadata of the entire PDB database as a .json file from PDBe.
- Select heterodimer complexes representing protein-protein interactions using PDBe annotations and Uniprot identifiers.
- Apply strict quality filters: resolution ≤ 3.5 Å for X-ray structures, R-free - R-factor ≤ 0.07, and remove structures with atoms having alternative locations at interfaces [50].
Structure Preparation:
- Repair incomplete amino acids using FoldX software (version 5).
- Remove heteroatoms and water molecules from structures.
- Protonate complexes with the OPLS-AA force field of GROMACS (version 2020).
- Convert structures to .mol2 format for pocket detection [50].
Pocket Detection and Characterization:
- Employ VolSite to detect and characterize pockets using adapted parameters suitable for PPI pockets.
- Detect pockets within monomers using ligands as reference for selection of surrounding residues.
- For heterodimer complexes, detect pockets on one protein with the other protein treated as the ligand, then reverse roles.
- Verify that binding pockets reside at the interface to confirm orthosteric nature [50].
This systematic approach has been applied to create comprehensive datasets encompassing diverse protein families and organisms, enabling detailed investigations into molecular interactions at the atomic level [50].
Research Reagent Solutions for PPI Studies
Table 3: Essential Research Reagents for PPI Experimental Characterization
| Reagent/Category | Specific Examples | Function/Application | Considerations for β-Grasp Fold Studies |
|---|---|---|---|
| AFM Consumables | Silicon nitride cantilevers (~0.06 N/m) | Molecular force measurement | Spring constant must be calibrated for each experiment |
| Surface Chemistry | APTES, NHS-PEG-MAL, SATP | Covalent immobilization of proteins | Maintains protein orientation and functionality |
| Chromatography | PD-10 desalting columns | Buffer exchange and purification | Ensures proper protein folding and activity |
| Structural Biology | FoldX software, GROMACS | Protein structure repair and simulation | Essential for modeling β-GF structural variants |
| Pocket Detection | VolSite algorithm | Binding site identification and characterization | Parameters must be adjusted for shallow PPI pockets |
| Expression Systems | E. coli, insect cell, mammalian | Recombinant protein production | Choice affects post-translational modifications relevant to β-GF |
| Validation Tools | Secondary antibodies, fluorescence labels | Functional assay validation | Critical for confirming β-GF interaction specificity |
Integration of Strategies for Therapeutic Development
The development of PPI modulators has transitioned beyond early-stage drug discovery, with several FDA-approved drugs now targeting PPIs, including venetoclax, sotorasib, and adagrasib for various diseases [51]. The lessons learned from these successes provide valuable insights for targeting PPIs involving the β-grasp fold.
The strategic integration of computational prediction, experimental validation, and structural characterization creates a powerful pipeline for advancing PPI-targeted therapeutics. For β-grasp fold proteins, this involves:
Evolutionary Analysis: Leveraging the deep evolutionary conservation of the β-GF to identify conserved interaction surfaces and functional motifs [1].
Interface Characterization: Applying pocket detection and classification methodologies to identify potentially druggable regions on often challenging flat interaction surfaces.
Modulator Screening: Utilizing fragment-based approaches and AFM force spectroscopy to identify and validate lead compounds that target hot spots.
Optimization: Employing structure-guided design to enhance potency and selectivity while addressing the pharmacokinetic challenges often associated with PPI modulators.
This integrated approach demonstrates that PPI modulators have transitioned beyond early-stage drug discovery and now represent a prime opportunity with significant therapeutic potential across cancer, inflammation and immunomodulation, and antiviral applications [51]. The β-grasp fold, with its fundamental role in ubiquitin-mediated processes and other cellular pathways, presents particularly promising targets for this expanding therapeutic paradigm.
Resolving Technical Challenges in Studying Unfolding and Transient States
The β-grasp fold (β-GF) is a fundamental structural scaffold in molecular biology, prototyped by the ubiquitous protein ubiquitin (UB) [1] [53]. This compact fold, characterized by a β-sheet that "grasps" a central α-helix, has been recruited for a strikingly diverse range of biochemical functions, from post-translational modification and sulfur transfer to RNA binding and enzymatic activity [1] [2]. Understanding the dynamics of how proteins with this fold—and their substrates—undergo unfolding and refolding is crucial for deciphering cellular regulation, protein quality control, and the mechanisms of diseases like Alzheimer's, Parkinson's, and Huntington's, which are linked to protein misfolding [54].
A central technical problem in this field is the difficulty of capturing transient intermediates and short-lived conformational states. These states are essential for accurately describing dynamic protein folding/unfolding pathways, but they are inherently difficult to study due to their rapid transitions, sub-nm conformational differences, and the heterogeneity of the folding process itself [54]. This guide details the advanced methodologies overcoming these barriers, providing a framework for their application within the specific context of β-grasp fold research.
Key Methodological Advances for Capturing Transient States
Traditional ensemble techniques, which average the properties of many molecules, often underestimate the complexity of folding mechanisms [54]. The following single-molecule and kinetic methods now allow researchers to dissect these processes in unprecedented detail.
Solid-State Nanopore Confinement for Single-Molecule Peptide Dynamics
This novel method uses the electrochemical confinement effect of a solid-state nanopore to trap a single peptide and monitor its real-time conformational changes.
- Core Principle: A single peptide is confined within a SiNx nanopore using a biotin–monovalent streptavidin (mSA) tether system. As the peptide undergoes spontaneous folding/unfolding, its changing conformation alters the hydrodynamic excluded volume and surface charge distribution within the nanopore, resulting in measurable fluctuations in the ionic current [54].
- Protocol Outline:
- Peptide Design: A β-hairpin peptide (e.g., sequence GEWTYDDATKTFTVTE) from the C-terminal fragment of protein G B1 domain is used as a model system [54].
- Complex Formation: The peptide's C-terminus is tagged with biotin and bound to mSA, forming a stable peptide–mSA complex too large to translocate through the pore.
- Nanopore Fabrication: SiNx nanopores with diameters of 4–5 nm are fabricated using controlled dielectric breakdown, creating a confined space suitable for the peptide (cross-section ~2 nm × 3 nm) [54].
- Data Acquisition: A low voltage (+150 mV) is applied to capture the complex in the nanopore without stretching the peptide. The resulting ionic current is monitored at high temporal resolution.
- Data Analysis: A custom data analysis program classifies current blockage levels (ΔI/I0) into distinct stages, each corresponding to a specific peptide conformation (e.g., folded, partially unfolded, unfolded) [54].
- Application to β-Grasp Fold: This technique could be adapted to study the folding landscape of β-GF domains themselves, or to monitor the unfolding of β-GF protein substrates by AAA+ proteases in real time.
Single-Turnover Transient State Kinetics for Processive Unfolding
This approach uses sequential mixing and fluorescence detection to isolate and quantify the kinetics of enzyme-catalyzed protein unfolding, distinct from translocation of an already unfolded chain [55].
- Core Principle: The method employs engineered protein substrates containing a stable, folded domain (e.g., Titin I27) downstream of a specific motor protein binding site. In a single turnover experiment, the motor protein is pre-bound to the substrate, and the reaction is initiated with ATP. The subsequent unfolding of the folded domain is reported by a change in fluorescence, allowing direct measurement of the unfolding rate [55].
- Protocol Outline:
- Substrate Engineering: Construct a fusion protein such as RepA-TitinX, where X represents 1, 2, or 3 tandem repeats of the stably folded Titin I27 domain. The N-terminal RepA sequence provides a high-affinity binding site for the motor protein (e.g., ClpB), and a C-terminal cysteine allows for site-specific fluorescent labeling [55].
- Motor-Substrate Complex Formation: The motor protein (e.g., ClpB) is incubated with the RepA-TitinX substrate in the absence of nucleotide to form a stable complex.
- Sequential Mixing in Stopped-Flow: The pre-formed complex is rapidly mixed with a solution containing ATP or non-hydrolysable ATP analogs (e.g., ATPγS) in a stopped-flow apparatus.
- Fluorescence Detection: The loss of fluorescence (e.g., from the unfolding of the Titin domain) is monitored over time. The stepwise unfolding of multiple domains in a single construct provides evidence of processivity [55].
- Kinetic Analysis: The resulting time courses are fitted to kinetic models to extract rates of protein unfolding and the kinetic step-size (the number of amino acids unfolded between two rate-limiting steps).
- Application to β-Grasp Fold: This is directly applicable to studying how AAA+ proteases and disaggregases (many of which interact with or are regulated by ubiquitin and other β-GF proteins) unfold and process their substrates. It can reveal how stable folds are mechanically disrupted.
Table 1: Quantitative Insights from Single-Turnover Unfolding Studies
| Parameter Measured | Value for E. coli ClpB | Experimental Context | Significance |
|---|---|---|---|
| Protein Unfolding Rate | ~0.9 - 4 amino acids (aa) s⁻¹ | Unfolding of Titin I27 domains at sub-saturating [ATP] [55] | Reveals that unfolding is slow and rate-limiting compared to translocation |
| Kinetic Step-Size | ~60 aa | Distance unfolded between two rate-limiting steps [55] | Suggests the motor unfolds in substantial increments before encountering another kinetic barrier |
| Translocation Rate (unfolded chain) | >240 aa s⁻¹ (reported by others) | Translocation on a mechanically pre-unfolded polypeptide [55] | Highlights the mechanistic distinction between slow unfolding and fast translocation |
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of these advanced protocols requires carefully selected reagents and tools. The table below catalogs key solutions for studying unfolding and transient states.
Table 2: Key Research Reagent Solutions for Unfolding and Transient State Studies
| Reagent / Material | Function / Application | Specific Examples & Notes |
|---|---|---|
| Engineered Model Peptides | High-resolution model system for studying folding mechanisms at the single-molecule level. | β-hairpin peptide (GEWTYDDATKTFTVTE) from protein G B1 domain [54]. |
| Stable Folded Domain Substrates | Reporters for enzyme-catalyzed unfolding in bulk kinetic assays. | RepA-TitinX constructs (RepA-binding site fused to 1-3 Titin I27 domains) [55]. |
| Monovalent Streptavidin (mSA) | Provides a stable, high-affinity tether for nanopore confinement without inducing dimerization. | Essential for capturing peptide-mSA complex in nanopore without translocation [54]. |
| Non-hydrolysable ATP Analogs | Used to trap enzymatic intermediates or activate specific motor functions. | ATPγS alone or in a 1:1 mix with ATP can activate ClpB unfolding without co-chaperones [55]. |
| Hyperactive Mutant Proteins | Bypass regulatory requirements to simplify mechanistic studies. | ClpB(Y503D) variant functions without the need for the DnaK/J/E co-chaperone system [55]. |
Connecting Methodologies to the β-Grasp Fold Research Context
The techniques described herein are not generic; they provide powerful tools to address specific, unresolved questions in the biology of ubiquitin and β-grasp fold proteins.
- Elucidating Ubiquitin Chain Dynamics: The nanopore confinement method could be deployed to study the conformational dynamics of different ubiquitin chain linkages (e.g., K48, K63, M1). This could provide atomic-level insights into how the distinct structures of these chains are "read" by specific receptor proteins to initiate diverse downstream signals [53].
- Probing the Unfolding of β-Grasp Domains: The single-turnover kinetics approach can be used to investigate how β-GF domains themselves are recognized and unfolded by AAA+ proteases like the proteasome. For example, engineering a Titin-I27 substrate with a C-terminal ubiquitin fusion would allow researchers to dissect the kinetics of deubiquitinase (DUB) processing versus the initiation of substrate unfolding.
- Mapping Functional Versatility: The remarkable functional diversity of the β-grasp fold—from sulfur transfer in ThiS/MoaD to ubiquitin signaling—is rooted in structural variations and interactions [1] [2]. These high-resolution techniques can map how transient conformational states in these proteins enable binding to diverse partners, including E1/E2/E3 enzymes, other UBLs, and target biomolecules.
The challenges of studying protein unfolding and transient states are being met by a new generation of sophisticated biophysical tools. Methodologies like single-molecule nanopore confinement and single-turnover transient state kinetics provide a direct window into the dynamics of these previously elusive processes. By applying these techniques to the rich biological context of the β-grasp fold, researchers can move from static structural snapshots to a dynamic, mechanistic understanding of how this versatile fold and its associated networks control fundamental cellular processes, paving the way for novel therapeutic interventions in cancer, neurodegenerative diseases, and infection.
Navigating Cellular Consequences of UPS and UBL Pathway Modulation
The ubiquitin-proteasome system (UPS) and ubiquitin-like protein (UBL) conjugation pathways represent intricate, ATP-dependent regulatory networks essential for cellular homeostasis. These systems, unified by the common β-grasp fold structure of their modifier proteins, orchestrate a vast array of cellular processes including protein degradation, DNA repair, cell cycle progression, and stress response. Dysregulation of these pathways contributes significantly to pathogenesis, particularly in cancer, neurodegenerative disorders, and infectious diseases. This technical review provides an in-depth examination of UPS and UBL pathway mechanisms, their functional crosstalk, and the consequent cellular adaptations to their modulation. We further detail experimental methodologies for investigating these systems and present a curated research toolkit to facilitate drug discovery efforts targeting these critical regulatory networks.
The β-grasp fold is an evolutionarily conserved structural motif that serves as the fundamental architectural unit for ubiquitin and all UBL proteins. This compact globular fold consists of a mixed β-sheet (typically 4-5 strands) that grasps a central α-helix, creating a stable platform that can be specialized for diverse cellular functions [56]. Despite limited sequence conservation, this structural unity underscores the common evolutionary origin of these modifiers from prokaryotic sulfurtransferase systems, such as MoeB and ThiF, which utilize similar folds and mechanistic principles [56] [8].
The functional diversification of β-grasp proteins represents a remarkable case of evolutionary tinkering. While ubiquitin itself is absent from prokaryotes, its structural antecedents underwent significant specialization in eukaryotes to create a sophisticated post-translational modification system. The conserved C-terminal glycine in all UBLs enables activation and conjugation through a conserved enzymatic cascade, while variations in surface features determine specific interactions with downstream effector proteins [44] [56]. This balance between structural conservation and functional diversification makes the β-grasp fold an ideal framework for understanding how cells have engineered a multitude of regulatory pathways from a common structural blueprint.
The Ubiquitin and UBL Protein Families
Ubiquitin and UBLs constitute a growing family of protein modifiers that share the β-grasp fold yet regulate distinct cellular processes. Table 1 provides a comprehensive overview of the principal UBL family members, their enzymatic machinery, and key functional attributes.
Table 1: The Ubiquitin-like Protein Family and Their Conjugation Machinery
| UBL Modifier | Sequence Identity with Ubiquitin | E1 Activating Enzyme | E2 Conjugating Enzyme | Key Biological Functions |
|---|---|---|---|---|
| Ubiquitin | 100% | Uba1, Uba6 | ~40 E2s | Proteasomal degradation, signaling, endocytosis |
| NEDD8 | 55% | Uba3-NAE1 | Ubc12 | Cullin activation, cell cycle regulation |
| SUMO1-3 | 18% | Uba2-SAE1 | Ubc9 | Transcription, DNA repair, nuclear transport |
| ISG15 | 32-37% | Ube1L, Uba7 | UBCH8 | Innate immunity, antiviral response |
| ATG12 | Not determined | Atg7 | Atg10 | Autophagy initiation |
| ATG8/LC3 | Not determined | Atg7 | Atg3 | Autophagosome formation |
| UFM1 | Not determined | Uba5 | Ufc1 | Endoplasmic reticulum homeostasis |
| FAT10 | 32-40% | Uba6 | USE1 | Immune response, mitosis |
The UBL family exhibits considerable diversity in their conjugation apparatus. While the ubiquitin system utilizes two E1 enzymes (Uba1 and Uba6) and approximately 40 E2 enzymes to achieve substrate specificity, most UBLs operate with more restricted enzymatic cascades [44] [56]. For instance, the SUMO pathway employs a single E2 enzyme (Ubc9) that partners with various E3 ligases to modify target proteins [44]. This variation in enzymatic complexity reflects the specialized functions of each UBL and their positions within the cellular regulatory hierarchy.
Functional specificity is further enhanced by distinct modification patterns. Ubiquitin can form diverse polyubiquitin chains through its seven lysine residues (K6, K11, K27, K29, K33, K48, K63) and N-terminal methionine (M1), with K48-linked chains primarily targeting substrates for proteasomal degradation [57] [58]. In contrast, UBL modifications typically function as monomeric attachments or form limited chains that alter protein activity, interactions, or localization without necessarily marking proteins for degradation [56] [59].
Enzymatic Cascades and Conjugation Mechanisms
The E1-E2-E3 Enzymatic Hierarchy
UBL conjugation follows a conserved three-step enzymatic cascade that activates and transfers the UBL to specific substrate proteins:
E1 Activation: E1 activating enzymes initiate the cascade by catalyzing UBL adenylation, followed by formation of a high-energy thioester bond between the E1 active-site cysteine and the UBL C-terminal glycine. Structural studies reveal that E1 enzymes undergo dramatic domain rotations (up to 130°) to transits the catalytic cysteine between adenylation and E2-binding sites [8].
E2 Conjugation: Activated UBLs are transferred from E1 to the active-site cysteine of an E2 conjugating enzyme, forming an E2~UBL thioester. E2 enzymes employ oxyanion stabilization mechanisms, including C-alpha hydrogen bonding, to facilitate this transfer [60].
E3 Ligation: E3 ligases facilitate the final transfer of the UBL from E2 to specific substrate proteins, typically forming an isopeptide bond with a lysine ε-amino group. E3s achieve substrate specificity through diverse recognition domains and mechanisms [8] [59].
Diagram: UBL Conjugation Enzymatic Cascade
Structural Mechanisms of E1 Enzymes
Canonical E1 enzymes exhibit a conserved domain architecture consisting of:
- Adenylation domains that form a composite active site for ATP•Mg²⁺ and UBL binding
- Catalytic cysteine domain (CYS) that harbors the active-site cysteine for thioester bond formation
- Ubiquitin fold domain (UFD) that interacts with E2 proteins [8]
The E1 mechanism involves sophisticated conformational changes. During UBL activation, the CYS domain rotates approximately 130° from an "open" to a "closed" conformation, positioning the catalytic cysteine proximal to the UBL-adenylate intermediate [8]. This transition involves remodeling of the adenylation active site and disordering of secondary structural elements that cover the catalytic cysteine in the open state. For E1-to-E2 transfer, a ~25° rotation of the UFD domain brings the E2 from a distal position to a proximal orientation suitable for thioester transfer [8].
Experimental Approaches for UBL Pathway Investigation
Chemical Protein Synthesis for UBL Research
Chemical synthesis approaches have revolutionized UBL research by enabling atomic-level control over protein structure and modification. Table 2 outlines key methodologies and their applications in UBL studies.
Table 2: Chemical Biology Approaches for UBL Pathway Investigation
| Methodology | Key Features | Application Examples | Advantages |
|---|---|---|---|
| Native Chemical Ligation (NCL) | Chemoselective ligation of unprotected peptide segments; utilizes C-terminal thioester and N-terminal cysteine | Synthesis of SUMO-1, SUMO-2/3, NEDD8, ISG15 conjugates; preparation of defined ubiquitin-UBL hybrids | Enables site-specific modifications, non-hydrolyzable linkages, incorporation of unnatural amino acids |
| Expressed Protein Ligation (EPL) | Hybrid approach combining recombinant protein expression with chemical synthesis | Production of lipidated LC3 for autophagy studies | Access to larger proteins with specific post-translational modifications |
| KAHA Ligation | α-Ketoacid-hydroxylamine ligation independent of cysteine residues | Synthesis of SUMO-2, SUMO-3, UFM1 | Expands ligation sites beyond cysteine residues |
| Activity-Based Probes (ABPs) | Designed to capture and identify enzyme activities in complex mixtures | Profiling of DUB and UBL protease activities; identification of USP18 substrates | Enables functional proteomics and identification of enzyme-substrate relationships |
Functional Proteomic Profiling
Systematic approaches to identify UBL substrates have revealed the non-random organization of UBL modification networks. A comprehensive profiling study examining seven UBLs during mitosis identified approximately 1,500 potential substrates, with 80-200 protein targets exclusive to each UBL [61]. This indicates highly specialized biological roles for each UBL despite their structural similarities. For instance, FAT10 modification profiles suggested a previously underappreciated role in mitotic regulation, highlighting how systematic profiling can reveal novel UBL functions [61].
Cellular Consequences of Pathway Modulation
Interplay Between UPS and Autophagy
The UPS and autophagy exhibit complex, reciprocal crosstalk that maintains cellular proteostasis. While the UPS primarily degrades short-lived soluble proteins, autophagy eliminates insoluble protein aggregates and damaged organelles [57] [58]. Modulation of one pathway often triggers compensatory adaptation in the other:
- UPS inhibition stimulates autophagy through multiple mechanisms, including accumulation of aggregation-prone proteins that activate autophagy receptors [58]
- Autophagy impairment can either activate or inhibit proteasomal flux depending on cellular context and stress conditions [58]
- Shared regulatory nodes integrate both pathways, exemplified by the p62/SQSTM1 protein, which recognizes ubiquitinated substrates and delivers them to both proteasomal and autophagic degradation systems [57]
Ubiquitin-Mediated Regulation of Autophagy
Ubiquitination serves as a key regulatory mechanism controlling multiple stages of autophagy, from initiation to termination:
- Autophagy Induction: K63-linked ubiquitination of ULK1 by TRAF6 enhances ULK1 stability and kinase activity, creating a positive feedback loop that potentiates autophagy initiation [58]
- VPS34 Complex Regulation: K63 ubiquitination of Beclin-1 by TRAF6 disrupts its interaction with Bcl-2, promoting autophagy induction in response to TLR4 signaling [58]. Conversely, K48/K11 ubiquitination by NEDD4 and RNF216 targets Beclin-1 for proteasomal degradation [58]
- Selective Autophagy: Ubiquitin chains serve as recognition signals for autophagy receptors (e.g., p62, NBR1, OPTN) that bridge ubiquitinated cargoes to the autophagic machinery [58]
Diagram: Ubiquitin-Mediated Regulation of Autophagy
Disease Associations and Therapeutic Targeting
Dysregulation of UPS and UBL pathways contributes to numerous human diseases:
- Neurodegenerative disorders (Alzheimer's, Parkinson's, Huntington's): Impaired clearance of protein aggregates due to dysfunctional UPS or autophagy [57] [59]
- Cancer: Altered ubiquitination of oncoproteins and tumor suppressors; mutations in ubiquitin ligases and deubiquitinases [62] [59]
- Infectious diseases: Pathogen manipulation of host ubiquitination systems; antiviral functions of ISG15 [56] [60]
Therapeutic targeting of these pathways has gained significant momentum, with the proteasome inhibitor bortezomib demonstrating clinical efficacy in multiple myeloma [59]. Current drug discovery efforts focus on developing specific inhibitors for E1 enzymes (e.g., MLN4924 for NEDD8 E1), E2 enzymes, E3 ligases, and deubiquitinating enzymes [62] [59]. Multiomics approaches are increasingly employed to delineate the complexity of ubiquitin and UBL modifications in disease contexts and to identify novel therapeutic targets [62].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for UPS and UBL Pathway Investigation
| Research Tool | Specific Examples | Research Applications | Key Features |
|---|---|---|---|
| Activity-Based Probes | Ubiquitin/UBL vinyl sulfones, suicide inhibitors | Profiling enzyme activities in complex lysates; identification of active enzymes in cellular states | Covalently modifies active sites; enables enrichment and identification of low-abundance enzymes |
| Chemical Cross-linkers | Disulfide cross-linking between E1 and E2 cysteines | Trapping transient enzyme complexes for structural studies; mapping interaction interfaces | Stabilizes weak or transient complexes for structural biology approaches |
| UBL Conjugation Assay Components | Recombinant E1/E2/E3 enzymes; ATP regeneration systems; ubiquitin/UBL mutants | In vitro reconstruction of conjugation cascades; mechanistic studies of enzyme specificity | Enables reductionist approach to study individual pathway components |
| Linkage-Specific Antibodies | K48- and K63-linkage specific ubiquitin antibodies | Detection of specific ubiquitin chain types in cells and tissues; monitoring chain-type dynamics in response to perturbations | Provides insight into functional consequences of ubiquitination |
| DUB Inhibitors | PR-619 (broad-spectrum DUB inhibitor); USP14 inhibitors | Investigating DUB functions; modulating ubiquitin chain stability and dynamics | Tool compounds for probing deubiquitination functions in cellular pathways |
| Synthetic UBL Conjugates | Semisynthetic SUMO-substrate conjugates; defined ubiquitin-UBL hybrids | Structural and functional studies of specific conjugates; identification of UBL-binding domains | Provides homogeneous, precisely defined conjugates for biochemical studies |
The UPS and UBL pathways constitute an elaborate regulatory network that governs virtually all aspects of cellular physiology through the versatile β-grasp fold architecture. Their modulation creates cascading cellular consequences, from compensatory proteostatic adaptations to fundamental changes in signaling pathway outputs. The intricate crosstalk between these systems, particularly evident in the ubiquitin-mediated regulation of autophagy, highlights the sophisticated homeostatic mechanisms cells employ to maintain functional integrity under stress conditions.
Advancements in chemical and proteomic methodologies are rapidly accelerating our understanding of these pathways, enabling researchers to precisely manipulate and monitor UBL modifications with unprecedented specificity. These technical innovations, combined with growing recognition of the therapeutic potential in targeting specific pathway components, position UPS and UBL research at the forefront of molecular medicine. Future efforts will undoubtedly focus on unraveling the complexity of heterologous ubiquitin and UBL chains, mapping the complete network of substrate modifications, and developing increasingly specific modulators for therapeutic intervention in the numerous diseases associated with pathway dysregulation.
Functional Diversity and Validation Across the β-Grasp Superfamily
Ubiquitin-like proteins (UBLs) represent a fundamental group of protein modifiers that share a common evolutionary origin in the β-grasp fold (β-GF), a compact structural scaffold characterized by a β-sheet that grasps a single α-helical segment [1] [11]. This fold is remarkably versatile and has been recruited for a stunning diversity of biochemical functions across all domains of life. The human genome encodes several UBLs, including SUMO (Small Ubiquitin-like Modifier), NEDD8 (Neural precursor cell expressed developmentally down-regulated protein 8), and ISG15 (Interferon-Stimulated Gene 15), which are central to regulating virtually all aspects of cellular biology [63] [64]. Although they share a common structural heritage, these UBLs have evolved distinct functions and regulatory mechanisms. SUMO is a key regulator of nuclear processes, NEDD8 primarily controls the activity of cullin-RING ligases, and ISG15 serves as a critical effector of the innate immune response [63] [65] [64]. This review provides a comparative analysis of these three major UBLs, examining their shared structural features, unique enzymatic cascades, specific biological roles, and the experimental tools used to probe their functions, all within the context of the conserved β-grasp architecture.
Structural and Functional Classification of UBLs
The Ubiquitous β-Grasp Fold
The β-grasp fold is a small, ancient protein fold that appeared prior to the last universal common ancestor and has since undergone extensive functional radiation [1] [11]. Its core structure consists of a five-stranded mixed β-sheet that clutches a single α-helix [11]. Despite their sequence divergence, ubiquitin and all UBLs are variations on this structural theme. The β-GF provides a stable platform that can be functionalized in different ways, primarily through the prominent β-sheet, which offers an exposed surface for diverse interactions with other proteins, nucleic acids, or small molecules [1]. In the case of UBLs, this fold was adapted for covalent conjugation to target proteins, a function that evolved from more ancient systems involved in sulfur transfer and cofactor biosynthesis [11].
Comparative Anatomy of Major UBLs
While SUMO, NEDD8, and ISG15 all possess the β-grasp fold, they exhibit distinct structural characteristics that underlie their specific functions. SUMO and NEDD8 are single-domain proteins, each comprising one β-grasp fold, much like ubiquitin. In contrast, ISG15 is a two-domain protein, consisting of two β-grasp folds in tandem, connected by a short linker region, which gives it a larger molecular size [65] [64]. All UBLs are synthesized as precursor proteins and must be processed by specific proteases to expose a C-terminal di-glycine motif that is essential for their conjugation to target proteins [65]. The sequence and structural homology between these UBLs, while significant, is insufficient for functional cross-talk; each operates within its own dedicated enzymatic pathway, ensuring fidelity in cellular signaling [66].
Table 1: Fundamental Properties of SUMO, NEDD8, and ISG15
| Property | SUMO | NEDD8 | ISG15 |
|---|---|---|---|
| Domains | Single β-grasp | Single β-grasp | Two tandem β-grasp domains |
| Mature Length | ~100 amino acids | 81 amino acids | 157 amino acids (two domains) |
| Sequence Identity to Ub | ~18% | ~58% | ~29% (per domain) |
| C-terminal Motif | Di-glycine | Di-glycine | Di-glycine (LRLRGG) |
| Conjugation | Mono & Poly | Primarily Mono | Mono & Hybrid chains with Ub |
| Major Biological Role | Nuclear processes, stress response | Activation of cullin-RING E3 ligases | Innate immunity, antiviral defense |
Enzymatic Cascades and Molecular Mechanisms
The conjugation of UBLs to target substrates is a ATP-dependent process mediated by a dedicated, three-step enzymatic cascade involving E1 (activating), E2 (conjugating), and E3 (ligating) enzymes [63] [64]. While the overall mechanism is conserved, each UBL utilizes a specific set of enzymes that ensure pathway fidelity.
The Conjugation Cascade
The process begins with E1 activation: The E1 enzyme forms a thioester bond with the C-terminal glycine of the UBL in an ATP-dependent reaction [66] [64]. The activated UBL is then transferred to the catalytic cysteine of the cognate E2 conjugating enzyme via a transthioesterification reaction [66]. Finally, an E3 ligase facilitates the transfer of the UBL from the E2 to a lysine residue on the target protein, forming a stable isopeptide bond [63] [67]. The specificity of this cascade is rigorously maintained. For example, the SUMO E1 is a heterodimer of SAE1 and UBA2, and its dedicated E2 is UBC9 [66] [64]. Recent cryo-EM structures of the human SUMO E1-E2 complex have revealed that dramatic conformational changes, including a ~175° rotation of the ubiquitin-fold domain (UFD), are required to align the active sites for thioester transfer, highlighting the precision of this machinery [66].
Deconjugation and Reversibility
Like ubiquitination, UBL modification is reversible. A family of specialized proteases, akin to deubiquitinases (DUBs), cleaves the isopeptide bond between the UBL and the substrate [65]. This deconjugation is essential for dynamic signaling and homeostasis. For ISG15, the primary deISGylating enzyme in humans is the cysteine protease USP18 (Ubp43 in mice) [65]. Other proteases, such as USP16 and USP24, have also been reported to hydrolyze ISG15, and notably, several viral pathogens encode proteases with deISGylating activity to antagonize the host immune response [65].
Figure 1: Enzymatic Cascades for UBL Conjugation and Deconjugation. Each UBL (SUMO, NEDD8, ISG15) is processed and then activated by its specific E1 enzyme. The activated UBL is transferred to a dedicated E2 enzyme, and finally, an E3 ligase facilitates conjugation to the target substrate. The process is reversed by specific proteases. Note: ISG15's primary E3, HERC5, is known to mediate co-translational ISGylation.
Table 2: Dedicated Enzymatic Machinery for SUMO, NEDD8, and ISG15
| Enzyme Type | SUMO | NEDD8 | ISG15 |
|---|---|---|---|
| E1 Activating Enzyme | SAE1-UBA2 heterodimer | NAE1-UBA3 heterodimer | UBE1L (UBA7) |
| E2 Conjugating Enzyme | UBC9 | UBE2F, UBE2M | UBE2L6 (UbcH8) |
| Representative E3 Ligases | PIAS family, HDAC4, HDAC7 | DCN1, RBR-type E3s | HERC5, ARIH1, TRIM25 (EFP) |
| Major Proteases | SENP family | NEDP1, SENP8 | USP18, USP16, USP24 |
Biological Functions and Pathogen Interactions
Distinct Cellular Roles of UBLs
The functional specialization of SUMO, NEDD8, and ISG15 is evident in their distinct biological roles:
SUMO is a key regulator of nuclear processes, including transcription, DNA repair, chromatin organization, and mitosis [66]. It often modifies transcription factors and core histones, thereby altering protein-protein interactions, subcellular localization, and stability. SUMOylation can have both activating and suppressive roles in immunity; for instance, SUMO1 can inhibit NF-κB signaling by stabilizing IκBα, while SUMO2/3 can activate NF-κB by modifying NEMO [64].
NEDD8's most characterized function is the activation of cullin-RING E3 ubiquitin ligases (CRLs) [63]. NEDD8 modification of cullins induces a conformational change that promotes CRL assembly and activity, thereby regulating the ubiquitylation and degradation of a vast array of substrate proteins involved in cell cycle progression, signal transduction, and development [63] [64].
ISG15 functions as a central player in the innate immune response to bacterial and viral infections [65]. Its expression is strongly induced by type I interferons. ISG15 conjugation (ISGylation) can inhibit viral replication by modifying both viral and host proteins. A key mechanism involves the co-translational ISGylation of newly synthesized viral proteins, which is facilitated by the E3 ligase HERC5's association with polysomes, thereby disrupting virion assembly [65].
Crosstalk and Hybrid Chain Formation
A complex layer of regulation arises from the crosstalk between different UBL pathways. A prominent example is the formation of hybrid chains, where one UBL modifies another. Ubiquitin can be modified by ISG15, forming ISG15-ubiquitin mixed chains. Lysine 29 on ubiquitin has been identified as the major acceptor site for ISG15 [68]. These hybrid chains do not appear to serve as degradation signals. Instead, evidence suggests they can negatively regulate the turnover of ubiquitylated proteins, representing a mechanism by which ISGylation can directly antagonize the ubiquitin-proteasome system to fine-tune protein homeostasis during immune stress [68] [67]. Similarly, ubiquitination of SUMO isoforms has been detected, further expanding the combinatorial complexity of the "Ubiquitin Code" [67].
UBLs as Targets of Bacterial Effectors
Bacterial pathogens have evolved sophisticated mechanisms to manipulate host UBL pathways to promote infection. Although bacteria lack genuine eukaryotic UBI/UBL systems, they secrete effector proteins that specifically interfere with these PTMs [63]. For instance, Shigella flexneri delivers effectors such as OspI (a deamidase that inactivates the E2 enzyme UBC13) and IpaH family members (E3 ubiquitin ligases that target components of the NF-κB pathway) to dampen host inflammatory responses [63]. Similarly, Legionella pneumophila uses the effector SdeA to catalyze a non-canonical, E1/E2-independent ubiquitination of host proteins, thereby subverting membrane trafficking [63]. These strategies highlight the critical importance of UBL pathways in host defense and their vulnerability as targets of microbial sabotage.
Experimental Analysis and Research Tools
Studying UBLs requires a suite of specialized reagents and methodologies to dissect their dynamics, specificity, and functional outcomes.
Key Methodologies and Reagents
- Crosslinking Strategies for Structural Biology: Stabilizing transient enzyme-substrate complexes is crucial for structural studies. A disulfide crosslinking strategy between the catalytic cysteines of SUMO E1 (UBA2 C173) and its E2 (UBC9 C93) was instrumental in capturing the human SUMO E1–E2 complex for cryo-EM analysis, revealing the dramatic conformational changes during thioester transfer [66].
- Proteomic Identification of Conjugation Sites: Mass spectrometry-based proteomics is the primary method for identifying UBL modification sites on target proteins. For example, immunoprecipitation of ubiquitin followed by liquid chromatography and tandem mass spectrometry (LC-MS/MS) unequivocally identified Lys29 of ubiquitin as the major acceptor site for ISG15 [68].
- Site-Directed Mutagenesis of Acceptor Lysines: To confirm the functional significance of a modification site, lysine residues are mutated to arginine (e.g., K29R ubiquitin mutant). This abrogates conjugation and allows comparison with the wild-type protein to determine the biological consequence of the modification [68].
- Activity Assays for Enzymatic Function: In vitro activity assays monitor different steps of the enzymatic cascade. For instance, the effect of point mutations (e.g., E483R;R512A in SUMO E1) on thioester transfer activity can be quantified and compared to wild-type enzyme activity, revealing critical residues for catalysis [66].
- Cell-Based Models with Genetic Ablation: Mouse embryonic fibroblasts (MEFs) from wild-type and Ube1L knockout (KO) mice provide a powerful system to demonstrate the dependency of ISGylation on a specific E1 enzyme, as ISG15-conjugated proteins are absent in the KO cells [68].
Table 3: The Scientist's Toolkit: Essential Reagents and Methods for UBL Research
| Tool/Reagent | Function/Application | Example from Literature |
|---|---|---|
| Disulfide Crosslinking | Traps transient E1-E2 complexes for structural biology. | Used to solve cryo-EM structure of SUMO E1–UBC9 complex [66]. |
| LC-MS/MS Proteomics | Identifies specific lysine residues modified by UBLs. | Identified Lys29 of ubiquitin as the major ISG15 acceptor site [68]. |
| K-to-R Mutants | Abolishes conjugation at a specific lysine to study function. | Ubiquitin K29R mutant shows decreased ISGylation [68]. |
| Activity Assays | Quantifies enzymatic activity of E1, E2, E3, or proteases. | Measured ~10-fold reduction in thioester transfer for SUMO E1 mutants [66]. |
| Genetic Knockout Cells | Determines requirement for a specific enzyme in a pathway. | Ube1L KO MEFs show no ISG15 conjugation [68]. |
| Specific Inhibitors | Pharmacologically inhibits UBL pathways. | TAK-981 (SAE inhibitor) blocks SUMOylation and enhances IFN signaling [64]. |
The comparative analysis of SUMO, NEDD8, and ISG15 reveals a fascinating story of evolutionary divergence from a common β-grasp fold scaffold into highly specialized regulatory systems. While they share a conserved structural core and a common enzymatic logic for conjugation, each has evolved unique enzymes and molecular interfaces that dictate their specific functions: SUMO in nuclear organization and signaling, NEDD8 in controlling cullin-RING ligase activity, and ISG15 in innate immunity. The emerging complexity of crosstalk and hybrid chain formation between these pathways, such as ISG15 modification of ubiquitin, adds a sophisticated layer of regulation that cells utilize to integrate different signals, particularly under stress. The fact that bacterial pathogens actively target these systems underscores their paramount importance in cellular homeostasis and defense. Continued research, powered by the advanced experimental tools outlined in this review, will be essential to fully decipher the complex language of UBL signaling and to harness this knowledge for developing novel therapeutic strategies against cancer, autoimmune diseases, and infectious diseases.
Ubiquitin (Ub), a small 76-amino acid protein, is prototypical of the β-grasp fold, a structural motif characterized by a five-stranded β-sheet cradling a central α-helix [53] [69]. For decades, the primary function of ubiquitination was thought to be the targeting of proteins for degradation via the 26S proteasome, a discovery recognized by the Nobel Prize in Chemistry in 2004 [53]. This perspective, however, has undergone a profound shift. It is now clear that the Ub code, written by a cascade of E1 (activating), E2 (conjugating), and E3 (ligating) enzymes and erased by deubiquitinases (DUBs), regulates a vast array of non-proteolytic processes [70] [71]. The remarkable stability, solubility, and structural conservation of the β-grasp fold are the foundational properties that enable Ub's versatility as a signaling molecule [53] [69]. This guide will delve into the experimental validation of these non-proteolytic roles, focusing on their mechanisms in cellular signaling and adaptive processes, all framed within the unique structural context of the Ub/Ub-like (UBL) family of β-grasp fold proteins.
The Non-Proteolytic Ubiquitin Code: Linkages and Functional Outcomes
The type of ubiquitin chain linkage dictates its functional consequences. While K48-linked chains are the canonical signal for proteasomal degradation, several other linkage types mediate non-proteolytic functions by altering protein interactions, localization, and activity [70] [71] [72]. The table below summarizes the key non-proteolytic linkages and their established roles.
Table 1: Non-Proteolytic Ubiquitin Linkages and Their Cellular Functions
| Ubiquitin Linkage | Chain Conformation | Primary Non-Proteolytic Functions | Key Signaling Pathways/Processes |
|---|---|---|---|
| K63-linked | Open, flexible [72] | Signal transduction, endocytic trafficking, DNA repair, kinase activation, scaffolding [70] [73] [72] | NF-κB activation, innate immune response, AMPA receptor trafficking [73] [72] |
| M1-linked (Linear) | Open, extended [53] | Immune and inflammatory signaling, cell death, protein scaffolding [70] [73] | NF-κB activation (via LUBAC complex) [73] [72] |
| K6-linked | Mixed (can be compact) [71] | Mitophagy, DNA damage response, protein stabilization [70] [71] | Parkin-mediated mitophagy [70] [72] |
| K11-linked | Compact [71] | DNA Damage Response (DDR), cell cycle regulation [70] | TNFα signaling [72] |
| K27-linked | Not well characterized | Innate immunity, epigenetic regulation, mitophagy scaffold [70] [72] | DDR (recruitment of repair proteins) [70] |
| K29-linked | Not well characterized | Wnt/β-catenin signaling, neurodegenerative disorders [70] | Midbody assembly in cell division [70] |
| K33-linked | Not well characterized | Protein trafficking, kinase regulation [70] | Post-Golgi transport [72] |
The diversity of these linkages allows ubiquitin to form a complex "code." The structural basis for this functional diversity lies in the β-grasp fold, which provides a stable platform while displaying different interaction surfaces based on the linkage type. For instance, K63-linked and M1-linked chains adopt open conformations that are ideal for serving as scaffolds in large signaling complexes, as they expose key hydrophobic patches and interaction surfaces for specific Ub-binding domains (UBDs) [53] [72].
Methodologies for Validating Non-Proteolytic Functions
Establishing a non-proteolytic function for ubiquitination requires a multi-faceted experimental approach that goes beyond simply detecting ubiquitination of a substrate.
Genetic and Molecular Perturbation
A foundational strategy involves modulating the enzymes that write, read, and erase the ubiquitin signal.
- E2/E3 Targeting: Using RNAi or CRISPR to knock down specific E2 or E3 ligases and observing the functional consequence is a primary method. For example, knockdown of the E3 ligase TRAF6, which collaborates with the E2 complex Ubc13/Uev1A to build K63 chains, impairs NF-κB signaling without necessarily stabilizing the substrate [73] [72].
- DUB Overexpression: Expressing a deubiquitinase (DUB) that specifically cleaves a non-proteolytic chain (e.g., enzymes that disassemble K63 or M1 chains) should inhibit the signaling event if that chain is required.
- Ubiquitin Mutants: A powerful tool is the expression of ubiquitin mutants in which all lysine residues except one are mutated to arginine (e.g., "K63-only Ub"). Reconstituting Ub-deficient cells with K63-only Ub can rescue K63-linked signaling pathways but not degradation-dependent processes [72].
Biochemical and Proteomic Analysis
Direct biochemical evidence is crucial for validating observations.
- Linkage-Specific Affinity Reagents: The development of linkage-specific antibodies and affimer proteins (e.g., for K6-linked chains) allows for the immunoprecipitation and visualization of specific chain types [53] [70].
- In Vitro Reconstitution: Reconstructing a signaling pathway in vitro using purified components (substrate, E1, specific E2/E3, and ubiquitin) can definitively show that a particular E3 ligase builds a specific chain type (e.g., K63 or M1) on a substrate and that this modification activates the pathway in the absence of a proteasome [73].
- Mass Spectrometry (Ubiquitin Proteomics): Advanced proteomics can precisely map ubiquitination sites and determine the topology of polyubiquitin chains on a substrate, providing unambiguous evidence for a non-degradative linkage [71].
Table 2: Essential Research Reagents for Studying Non-Proteolytic Ubiquitination
| Research Reagent | Function and Utility in Experiments | Example Use Case |
|---|---|---|
| K63-only Ubiquitin Mutant | Allows exclusive formation of K63-linked chains in cells; validates chain-specific function. | Rescuing NF-κB signaling in Ub-deficient cells without triggering degradation [72]. |
| Linkage-Specific Antibodies/Affimers | Immunoprecipitation and detection of endogenous chains with specific linkages (e.g., K63, M1, K6). | Confirming the presence of K63 chains on RIP1 in TNFR signaling [70] [73]. |
| TUBE (Tandem Ubiquitin Binding Entity) | Affinity matrices to enrich for ubiquitinated proteins; protects chains from DUBs during purification. | Isolating endogenous ubiquitinated signaling complexes for proteomic analysis [71]. |
| Proteasome Inhibitors (e.g., MG132) | Blocks proteasomal degradation; used to distinguish proteolytic from non-proteolytic outcomes. | Demonstrating that a ubiquitination event does not stabilize a protein substrate [74]. |
| Recombinant E2/E3 Enzymes | For in vitro reconstitution assays to define enzyme specificity and biochemical function. | Showing that LUBAC complex specifically synthesizes M1-linear chains [73] [72]. |
Functional and Phenotypic Assays
The ultimate validation lies in connecting ubiquitination to a specific cellular outcome.
- Monitoring Protein Stability: A straightforward experiment is to measure the half-life of a substrate after its ubiquitination. If the protein is ubiquitinated but its stability is unchanged, a non-proteolytic function is indicated [74].
- Signaling Outputs: Assess the activation of downstream pathway components. For instance, K63 ubiquitination of RIPK1 in the TNFR pathway leads to IKK and MAPK activation, which can be measured by phospho-specific antibodies [73].
- Subcellular Localization: Immunofluorescence or live-cell imaging can reveal if non-proteolytic ubiquitination (e.g., K63-linked) directs the trafficking of membrane receptors like AMPA or EGFR during endocytosis [70] [72].
Signaling Pathway Deep Dive: NF-κB Activation
The NF-κB pathway is a paradigm for non-proteolytic ubiquitin signaling, involving both K63-linked and M1-linked chains. The following diagram illustrates the key steps in the TNFR1 pathway.
Diagram 1: Non-proteolytic ubiquitin signaling in TNFR-mediated NF-κB activation. K63-linked chains on RIP1, assembled by cIAP1/2 and Ubc13/Uev1A, serve as a scaffold to recruit the TAK1 kinase complex. Subsequently, the LUBAC complex modifies NEMO with M1-linear chains, facilitating full IKK activation. This pathway operates independently of proteasomal degradation [73] [72].
Experimental Workflow for Validating K63 Ubiquitination in NF-κB Signaling:
- Stimulate Cells: Treat cells with TNF-α.
- Inhibit Proteasome: Use MG132 to uncouple signaling from degradation.
- Immunoprecipitate: Pull down the endogenous RIP1 protein.
- Probe for Ubiquitin: Use a K63-linkage specific antibody to detect K63 chain formation on RIP1.
- Functional Knockdown: Use siRNA to deplete Ubc13 or cIAP1/2 and repeat steps 1-4. This should abolish K63 ubiquitination of RIP1 and subsequent NF-κB activation (measured by IκBα phosphorylation and degradation, or nuclear translocation of NF-κB).
Non-Proteolytic Ubiquitination in Disease and Therapeutic Targeting
Dysregulation of non-proteolytic ubiquitination is implicated in numerous diseases, making its machinery a attractive therapeutic target.
- Cancer: K63 and M1 linkages are critical in regulating cell proliferation, apoptosis, and inflammation. For instance, K63-linked ubiquitination of Akt can promote its membrane translocation and oncogenic signaling [70] [71].
- Neurological Disorders: In the brain, K63-linked chains are abundant and regulate synaptic function. They control the internalization of glutamate receptors (e.g., via the E3 ligase Nedd4-1), a process critical for synaptic plasticity. Aberrant K63 signaling is linked to Parkinson's disease and autism spectrum disorders [72].
- Inflammation and Autoimmunity: As the NF-κB pathway is central to inflammation, excessive M1 or K63 ubiquitination can drive autoimmune pathologies [73].
- Therapeutic Strategies: Drug discovery is focusing on developing:
- Specific E3 Ligase Inhibitors: Small molecules to target rogue E3s in cancer.
- DUB Inhibitors: To stabilize protective ubiquitin signals.
- UBD Blockers: Molecules that disrupt the interaction between a ubiquitin chain and its reader protein [71].
The non-proteolytic functions of ubiquitin, enabled by the versatile and stable β-grasp fold, represent a sophisticated regulatory layer controlling immunity, neural communication, and cellular homeostasis. Moving beyond the degradation-centric view is essential for a complete understanding of cell signaling. Future research, leveraging the validated methodologies and reagents detailed in this guide, will continue to decipher the complex ubiquitin code. This will undoubtedly unlock new therapeutic avenues for a wide spectrum of human diseases by targeting the writers, readers, and erasers of non-proteolytic ubiquitin signals.
The β-grasp fold (β-GF) is a small, versatile protein fold prototyped by ubiquitin (UB) and utilized in a strikingly diverse range of biochemical functions [1]. Its manifold functional abilities arise primarily from a prominent β-sheet that provides an exposed surface for diverse interactions or can form open barrel-like structures [1]. This fold has been recruited for functions including providing a scaffold for enzymatic active sites and iron-sulfur clusters, RNA-soluble-ligand and co-factor-binding, sulfur transfer, adaptor functions in signaling, assembly of macromolecular complexes, and post-translational protein modification [1]. Among the numerous lineages of the β-grasp fold, the Soluble-Ligand-Binding β-grasp (SLBB) superfamily represents a fascinating adaptive radiation focused on binding diverse soluble ligands, with vitamin B12 recognition as a central, characterized function [75].
This review details the SLBB superfamily as a case study within the broader context of β-grasp fold research. We examine how a fundamental structural scaffold was adapted for specific solute-binding functions, its evolutionary history, and its implications for metabolic processes across domains of life. The integration of structural, sequence, and genomic context data has been crucial in uncovering the extent and functional diversity of this superfamily.
The β-Grasp Fold: A Versatile Structural Scaffold
Core Structural Features
The β-grasp fold is a compact α/β fold dominated by a β-sheet typically composed of four to five anti-parallel β-strands. This sheet "grasps" a single α-helical segment that packs against its concave surface [1] [76]. The core structural elements include a β-sheet of five strands and a helix between strands 2 and 3, though numerous topological variations and elaborations have evolved through insertions and additions to this basic theme [75] [1].
Table 1: Major Functional Classes of β-Grasp Fold Domains
| Functional Class | Representative Domains | Key Functions |
|---|---|---|
| Post-translational Modifiers | Ubiquitin, SUMO, Nedd8 | Protein tagging for degradation, signaling, and regulation [1] |
| Sulfur Carriers | ThiS, MoaD | Sulfur transfer in cofactor biosynthesis [1] |
| Enzymes | MutT/NUDIX phosphohydrolases | Hydrolysis of diverse substrates [1] |
| Electron Transport | 2Fe-2S Ferredoxins | Iron-sulfur cluster binding [1] |
| Soluble Ligand Receptors | SLBB Superfamily | Binding vitamin B12 and other soluble ligands [75] |
| RNA Bindors | TGS domain | RNA-protein interactions in tRNA synthetases [1] |
| Protein-Protein Interaction Adapters | RA, PB1, FERM domains | Signal transduction complex assembly [1] |
Evolutionary Radiance of the Fold
Evolutionary reconstruction indicates the β-grasp fold had differentiated into at least seven distinct lineages by the time of the last universal common ancestor (LUCA), encompassing much of the structural diversity in extant versions [1]. The earliest members were likely involved in RNA metabolism and core cellular functions, subsequently radiating into various functional niches [1]. Most structural diversification occurred in prokaryotes, while the eukaryotic phase was marked by a specific expansion of ubiquitin-like β-GF members, with the eukaryotic UB superfamily diversifying into at least 67 distinct families [1].
The SLBB Superfamily: Discovery and Defining Characteristics
Identification and Structural Relationships
The SLBB superfamily was identified through sensitive sequence and structure similarity searches that revealed a novel superfamily containing the β-grasp fold [75]. Key evidence came from DALI structure comparison searches, which retrieved the C-terminal domain of transcobalamin with significant Z-scores when initiated with other β-GF domains like MoaD [75]. This C-terminal domain aligns completely with all core structural elements of the β-grasp fold but is distinguished by a unique β-hairpin insert after the conserved helix [75].
Sequence profile and hidden Markov model (HMM) searches subsequently identified numerous prokaryotic proteins containing these homologous β-GF domains, establishing the SLBB superfamily as widespread across bacteria and acquired by animals through horizontal gene transfer [75].
Sequence and Structure Hallmarks
A comprehensive multiple alignment of the SLBB superfamily revealed conserved hydrophobic residues forming the stabilizing core of the fold, plus notable sequence features including two strongly conserved glycine residues [75]. One is located in the turn leading into the horizontal flange preceding the third β-strand, and the other immediately downstream of the second conserved β-strand [75]. This conservation pattern is a unique feature distinguishing SLBB domains from other β-GF domains.
Table 2: Key Structural Features of SLBB Domains and Their Roles
| Structural Element | Description | Functional Role |
|---|---|---|
| Core β-Grasp Fold | β-sheet of 5 strands with a helix between strands 2 and 3 | Provides fundamental structural scaffold |
| Conserved Glycine Residues | Two strongly conserved glycines in specific turns | Enables specific conformational flexibility needed for ligand binding [75] |
| β-Hairpin Insert | Insert after the helix of the β-GF; typifies the transcobalamin-like clade | Directly contacts ligand; contributes solvent-mediated interactions [75] |
| Strand 4-5 Insert | Insert between strands 4 and 5 of the core fold; characterizes Nqo1-like clade | Predicted to play a role in ligand interaction [75] |
| Ascending Connector | Region between strand 4 and 5 | Makes additional contacts with ligand from core β-GF [75] |
Classification and Functional Diversity of the SLBB Superfamily
Major Clades and Representative Members
The SLBB superfamily is divided into two major clades, each with characteristic structural inserts and functional associations.
The Transcobalamin-like Clade
This clade is typified by a β-hairpin insert after the helix of the β-grasp fold [75]. Members include:
- Transcobalamin and Intrinsic Factor: Animal-specific proteins involved in vitamin B12 uptake [75]. These proteins contain an N-terminal α/α toroid domain and a C-terminal SLBB domain that cooperate to sandwich a single B12 molecule [75].
- Bacterial Polysaccharide Export Proteins: Cell-surface/secreted sugar-binding proteins involved in polysaccharide export [75].
- ComEA Family DNA Uptake Receptors: DNA uptake receptors in Gram-positive bacteria [75].
The Nqo1-like Clade
This clade is characterized by an insert between strands 4 and 5 of the core fold [75]. Representative members include:
- PduS-like Cobalamin Reductases: Enzymes involved in generating cob(I)alamin [75].
- Nqo1 Subunit of NADPH-quinone Oxidoreductase: A component of the respiratory electron transport chain complex I [75].
- RnfC Subunit of Bacterial Oxidoreductases: Components of bacterial Rnf operons encoding oxidoreductases [75].
Ligand Recognition Mechanisms
In transcobalamin, the B12 ligand is sandwiched between the N-terminal α/α toroid domain and the C-terminal SLBB domain [75]. The unique β-hairpin insert of the SLBB domain plays a prominent role in binding by contributing several direct or solvent-mediated interactions with the ligand [75]. Additional contacts are made by residues from the core β-GF, including those from strand 3, the end of strand 4, and the "ascending connector" between strand 4 and 5 [75]. This binding strategy demonstrates how inserts to the core fold facilitate functional specialization while maintaining the fundamental structural scaffold.
Diagram: Evolutionary and Functional Diversification of the SLBB Superfamily. The core β-grasp fold gives rise to two major clades distinguished by characteristic structural inserts that enable binding to diverse soluble ligands.
Evolutionary History and Genomic Context
Ancient Bacterial Origins
Both major SLBB clades are widely represented in bacteria, suggesting the superfamily was derived early in bacterial evolution [75]. The presence of SLBB domains in diverse bacterial metabolic pathways, including B12 metabolism, polysaccharide export, and competence development, indicates this superfamily was recruited for multiple solute-binding functions early in prokaryotic history.
Horizontal Gene Transfer to Animals
The animal lineage appears to have acquired transcobalamin-like proteins from low GC Gram-positive bacteria [75]. This horizontal gene transfer event might be correlated with the emergence of the ability to utilize B12 produced by gut bacteria, representing a key adaptation in host-microbe symbiosis [75]. The recruitment of the SLBB domain for specialized B12 transport in animals illustrates how conserved structural domains can be repurposed for novel physiological functions during evolution.
Experimental and Methodological Approaches
Key Techniques for Structural and Functional Characterization
The identification and characterization of the SLBB superfamily relied on complementary bioinformatic and structural biology approaches.
Table 3: Essential Methodologies for SLBB Superfamily Research
| Methodology | Application | Technical Considerations |
|---|---|---|
| Sensitive Sequence Searches (PSI-BLAST, HMMER) | Detection of distant homologs beyond sequence identity thresholds; identification of novel superfamily members [75] | Requires iterative searches with statistical evaluation; multiple sequence alignment construction essential |
| Structural Comparison (DALI) | Recognition of structural similarity despite low sequence conservation; identification of β-grasp fold in diverse proteins [75] | Z-scores >5-7 typically indicate significant structural relationships |
| Genomic Context Analysis | Inference of functional associations through conserved operons and gene neighborhoods [75] | Particularly powerful in prokaryotes where genes of related function are often co-localized |
| Domain Architecture Analysis | Understanding functional integration of SLBB domains with other protein domains [75] | Reveals cooperation between domains (e.g., α/α toroid and SLBB in transcobalamin) |
| Binding Affinity Measurements | Quantitative assessment of ligand-receptor interactions | Various approaches including fluorescence accumulation and single-molecule tracking [77] |
Research Reagent Solutions
Table 4: Essential Research Reagents and Resources for SLBB Studies
| Reagent/Resource | Function/Application | Examples/Specifications |
|---|---|---|
| Structural Templates (PDB IDs) | Reference structures for comparative analysis and modeling | 2BBC (Transcobalamin), 2FUG (Nqo1), 1O06 (UIM-ubiquitin complex) [75] [76] |
| Conserved Domain Database (CDD) | Classification and identification of SLBB domains | CDD family cl22925 provides sequence profiles and domain boundaries [78] |
| Sequence Search Tools | Identification of homologs and sequence analysis | PSI-BLAST, HMMER with statistical correction for compositional bias [75] |
| Cell Culture Systems | Functional assays for ligand binding and uptake | Mammalian cell lines (e.g., MM1S) with appropriate culture media [79] |
| Binding Assay Components | Quantitative measurement of protein-ligand interactions | TMB substrate, formaldehyde fixation, specific antibodies, detergent solutions [79] |
Protocol for Binding Affinity Determination
The following methodology, adapted from current approaches for studying ubiquitin ligase interactions, can be modified to study SLBB-ligand binding affinities [79]:
Diagram: Experimental Workflow for Cellular Binding Affinity Assessment. This protocol can be adapted to study SLBB-ligand interactions using cellular or purified component systems.
Detailed Procedure:
Cell Culture and Preparation: Culture appropriate cell lines expressing the SLBB protein of interest. For soluble SLBB domains, engineered cell lines or purified protein systems may be utilized. Ensure cells are ≥90% confluency at time of assay [79].
Compound/Ligand Preparation: Prepare stock solutions of ligands or compounds in preferred solvent (e.g., DMSO) at 10 mM or higher concentration. Verify purity (≥95%) by LC-MS and NMR analysis [79].
Treatment and Binding Reaction: Treat cells or purified SLBB proteins with serial dilutions of the test ligand. Include appropriate controls (vehicle-only and non-specific binding controls). Incubation time and conditions should be optimized for specific SLBB-ligand pairs.
Fixation and Permeabilization: For cellular assays, fix samples with 4% formaldehyde in PBS or TBS. Permeabilize with 0.1% Triton X-100 in TBS if intracellular detection is required [79].
Ligand Complex Detection: Incubate with primary antibody specific to the SLBB-ligand complex or tagged SLBB domain, followed by HRP-linked secondary antibody. Alternatively, direct labeling of ligands may be employed [79].
Signal Development and Quantification: Develop signal using TMB substrate (1:1 mixture of TMB substrate A and B), stop reaction with 2 N H₂SO₄, and measure optical density with plate reader [79].
Data Analysis: Calculate binding affinity using appropriate models (e.g., Zhu-Golan expression for 2D binding [77]). Nonlinear regression analysis in software such as GraphPad Prism can determine Kd values [79].
Research Applications and Future Directions
The study of SLBB domains extends beyond fundamental structural biology to practical applications in biotechnology and medicine. Understanding the molecular basis of soluble ligand recognition by this superfamily provides insights for:
- Drug Development: Targeting vitamin B12 transport pathways in metabolic disorders and cancers.
- Engineered Binding Proteins: Utilizing the SLBB scaffold for designing novel solute-binding proteins.
- Microbiome Research: Understanding host-microbe interactions mediated by vitamin B12 and other solute exchanges.
- Evolutionary Studies: Tracing the adaptation of structural scaffolds for novel functions across domains of life.
Future research directions include structural characterization of more diverse SLBB family members, detailed mechanistic studies of their ligand binding and release cycles, and exploration of their potential as therapeutic targets in metabolic diseases and infectious processes.
The SLBB superfamily exemplifies the remarkable functional versatility of the β-grasp fold, demonstrating how a fundamental structural scaffold can be adapted through evolution for specific solute-binding functions. Through strategic insertions into the core fold and integration with other protein domains, SLBB proteins have evolved to recognize diverse ligands including vitamin B12, polysaccharides, and DNA. Their early origin in bacteria and subsequent horizontal transfer to animals highlights the dynamic evolutionary history of this superfamily. The continued investigation of SLBB domains will undoubtedly yield further insights into the interplay between protein structure, ligand recognition, and metabolic adaptation across the tree of life.
The β-grasp fold is a small, ancient protein fold characterized by a β-sheet composed of four or five strands that "grasp" an α-helix. This structural motif serves as a versatile scaffold for an extraordinary diversity of biological functions, from regulation of protein stability via ubiquitination to RNA binding and enzyme catalysis [1]. Among its most ancient and functionally critical adaptations is its role in coordinating iron-sulfur (Fe-S) clusters in ferredoxins, proteins essential for electron transfer in fundamental metabolic processes [80] [1]. The independent recruitment of the β-grasp fold for Fe-S cluster binding on multiple evolutionary occasions represents a striking case of convergent evolution [1]. This review examines the structural and functional relationships between ferredoxins and other β-grasp fold proteins, framing Fe-S cluster binding within the broader context of this versatile fold's evolutionary trajectory. We summarize quantitative data on ferredoxin diversity, detail experimental methodologies for studying Fe-S cluster assembly and function, and provide essential resources for researchers investigating these ubiquitous protein cofactors.
The β-Grasp Fold: A Versatile Structural Scaffold
The β-grasp fold is defined by a core structure consisting of a mixed β-sheet (typically 4-5 strands) that grasps a single α-helix positioned parallel to the sheet [1]. Despite their structural similarity, proteins with this fold have diverged to perform remarkably diverse cellular functions. The evolutionary history of this fold is deep, with differentiation into at least seven distinct lineages before the last universal common ancestor (LUCA) [1].
Figure 1: The functional diversity of the β-grasp fold
This functional versatility arises primarily from the prominent β-sheet, which provides an exposed surface for diverse interactions, and the ability to form open barrel-like structures that accommodate different ligands and active sites [1]. Enzymatic activities and cofactor binding have evolved independently within the fold on multiple occasions, with iron-sulfur-cluster-binding arising at least twice independently [1].
Ferredoxins: Classification, Structure, and Function
Structural Classification of Ferredoxins
Ferredoxins are small iron-sulfur (Fe-S) cluster-containing proteins found across all biological domains. They are classified into distinct groups based on the composition and structure of their Fe-S clusters, each with characteristic iron-sulfur sequence binding motifs [80].
Table 1: Structural classification of ferredoxins based on Fe-S cluster type
| Cluster Type | Binding Motif Characteristics | Representative Functions |
|---|---|---|
| 2Fe-2S | Four cysteine residues in binding motif | Electron transfer in photosynthesis, steroid metabolism |
| 3Fe-4S | Three cysteines + proline after third cysteine | Electron transfer in anaerobic metabolism |
| 4Fe-4S | Four cysteines + proline after fourth cysteine | CO₂ fixation, hydrogen production |
| 7Fe-8S | Characteristics of both 3Fe-4S and 4Fe-4S clusters | Bacterial electron transfer chains |
| 2[4Fe-4S] | Varying cysteine spacing; includes small proteins and Alvin (Alv) ferredoxins | Multiple redox potentials in complex systems |
A new subtype classification system based on spacing between amino acids in the Fe-S binding motif has revealed unparalleled diversity between ferredoxins and helped identify evolutionarily linked forms across different organisms [80].
Biological Functions of Ferredoxins
Ferredoxins participate in crucial biological processes by mediating electron transfer:
- Photosynthesis: Ferredoxins accept electrons from photosystem I (PSI) and transfer them to ferredoxin-NADP+ reductase (FNR) for NADPH production [80].
- Hydrogen Production: Reduced ferredoxin transfers electrons to [FeFe]-hydrogenase (HYDA1), which catalyzes proton reduction to H₂ [80].
- Cytochrome P450 Systems: Ferredoxins transfer electrons to cytochrome P450 monooxygenases, modulating their function in oxidative reactions including steroid metabolism [80].
- Nitrogen Fixation: Ferredoxins provide electrons to nitrogenase, which converts N₂ to NH₃ [80].
- Sulfur Metabolism: Ferredoxins transfer electrons to sulfite reductase for conversion of sulfite to hydrogen sulfide [80].
- Iron-Sulfur Cluster Biosynthesis: Human mitochondrial ferredoxins (FDX1 and FDX2) transfer electrons for iron-sulfur cluster assembly on scaffold proteins [80].
Table 2: Key functional roles of ferredoxins in biological systems
| Biological Process | Ferredoxin Role | Electron Donor/Acceptor |
|---|---|---|
| Pyruvate Synthesis/CO₂ Fixation | Direct reduction by hydrogen via hydrogenases | Hydrogenase → Ferredoxin |
| Photosynthesis | Electron acceptor from PSI, donor to FNR | PSI → Ferredoxin → FNR |
| Hydrogen Production | Electron donor to hydrogenase | Multiple donors → Ferredoxin → HYDA1 |
| P450 Monooxygenase Reactions | Electron transfer from reductase to P450 | FdR → Ferredoxin → P450 |
| Nitrogen Fixation | Electron donor to nitrogenase | Reduced ferredoxin → Nitrogenase |
| Iron-Sulfur Cluster Assembly | Electron donation to assembly proteins | FDX1/FDX2 → ISC assembly machinery |
Convergent Evolution of Fe-S Cluster Binding in β-Grasp Proteins
The β-grasp fold has been independently recruited for Fe-S cluster binding on multiple evolutionary occasions, representing a remarkable case of convergent molecular evolution [1]. While 2Fe-2S ferredoxins represent one adaptation of this fold for Fe-S cluster coordination, other functionally distinct β-grasp proteins have independently evolved the capacity to bind similar cofactors.
Structural analyses indicate that the versatile β-sheet surface of the β-grasp fold provides an ideal platform for coordinating Fe-S clusters, with specific amino acid insertions and modifications enabling this function to arise independently in different lineages [1] [2]. The fold's intrinsic stability and ability to display coordination residues in precise spatial arrangements have made it particularly amenable to this function.
The evolutionary trajectory of ferredoxins themselves provides evidence for convergent evolution within this protein family. Phylogenetic analyses suggest that ferredoxins evolved through tandem gene duplications encoding smaller proteins, which may have originated from duplicating even simpler ancestral peptides [80]. This evolutionary mechanism has resulted in the emergence of distinct ferredoxin types with similar Fe-S cluster binding capabilities but different evolutionary origins.
Experimental Approaches for Studying Fe-S Cluster Proteins
Spontaneous Assembly of Fe-S Clusters with Cysteine
Figure 2: Experimental workflow for spontaneous Fe-S cluster assembly
Protocol: Fe-S clusters can spontaneously form through interactions of inorganic Fe²⁺/Fe³⁺ and S²⁻ with cysteine in water at alkaline pH under anaerobic conditions [81].
- Solution Preparation: Prepare anaerobic solutions of FeCl₃ (1 mM), Na₂S (1 mM), and L-cysteine (0.2-5 mM) in deoxygenated water at pH 9-11.
- Mixing: Combine solutions in a 5:1:1 ratio (cysteine:Fe³⁺:S²⁻) under anaerobic conditions (<10 ppm O₂).
- Incubation: Allow the mixture to stand at room temperature for cluster formation.
- Analysis: Monitor cluster formation via UV-Vis spectroscopy (characteristic absorption at ~420 nm), ⁵⁷Fe-Mössbauer spectroscopy, and ¹H-NMR.
- Redox Activity Assessment: Perform cyclic voltammetry to confirm cluster redox activity.
Key Findings: This method demonstrates robust, concentration-dependent formation of [4Fe4S], [2Fe2S], and mononuclear iron clusters at cysteine concentrations as low as 0.2 mM, supporting the prebiotic plausibility of spontaneously formed Fe-S clusters [81]. Bicarbonate ions (>10 mM) stabilize clusters and promote formation through salting-out effects.
Complementation Assay for Heterologous Fe-S Enzyme Activity
Protocol: This assay tests the functional compatibility of heterologous Fe-S enzymes with host cellular networks [82].
- Strain Construction: Create knockout strains of E. coli MG1655 lacking conditionally essential Fe-S enzymes (e.g., ΔnadA for quinolinate synthase, ΔispG for 4-hydroxy-3-methylbut-2-enyl-diphosphate synthase).
- Ortholog Cloning: Clone codon-optimized orthologs of target Fe-S enzymes from diverse bacterial phyla into multicopy expression vectors with inducible promoters.
- Complementation Testing: Transform constructs into corresponding knockout strains and assess functional complementation through growth in selective media.
- Condition Variation: Repeat assays under aerobic (37°C) and anaerobic conditions (28°C, 37°C) to evaluate oxygen sensitivity.
- Phylogenetic Analysis: Correlate functional expression success with phylogenetic distance from host ortholog using maximum likelihood phylogenies.
Key Findings: Fe-S enzymes are significantly less likely to retain activity in heterologous hosts than non-Fe-S enzymes (only 14/47 NadA orthologs functional in E. coli) [82]. Functional compatibility correlates with phylogenetic proximity for some enzymes (NadA) but not others (IspG). Anaerobic conditions recover function for orthologs from obligate anaerobes.
Iron-Sulfur Cluster Biogenesis Analysis
Protocol: Analysis of de novo Fe-S cluster formation using mitochondrial or bacterial ISC assembly components [83] [84].
- Component Isolation: Purify core ISC machinery proteins (NFS1, ISD11, ACP, ISCU2, frataxin, FDX2).
- Persulfide Formation: Incubate NFS1 with PLP cofactor and cysteine substrate to generate enzyme-bound persulfide.
- Cluster Assembly: Combine persulfide-loaded NFS1 with ISCU2 scaffold, iron source, and FDX2/ferredoxin reductase for electron donation.
- Cluster Transfer: Monitor transfer of nascent 2Fe-2S clusters to glutaredoxin 5 (GLRX5) with HSC20/HSPA9 chaperone assistance.
- Cluster Conversion: Assess condensation of two 2Fe-2S clusters into 4Fe-4S centers by ISA complex.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key research reagents for studying Fe-S cluster proteins
| Reagent/Category | Function/Description | Research Application |
|---|---|---|
| Cysteine Desulfurases (NFS1, IscS, SufS) | Pyridoxal phosphate-dependent enzymes that provide sulfur by converting cysteine to alanine | Fe-S cluster biogenesis assays |
| Scaffold Proteins (ISCU, SufB) | Platforms for de novo Fe-S cluster assembly | In vitro reconstitution of cluster formation |
| Cluster Transfer Proteins (GLRX5, NfuA, IscA) | Intermediate carriers that receive clusters from scaffolds and deliver to apo-targets | Studying cluster trafficking and insertion |
| Chaperone Systems (HSC20/HSPA9) | ATP-dependent complex facilitating cluster transfer from ISCU to GLRX5 | Analysis of cluster transfer mechanisms |
| Ferredoxins (FDX1, FDX2, Yah1) | [2Fe-2S] cluster proteins providing reducing equivalents for cluster assembly | Electron donation in cluster synthesis |
| Electron Donors (NADPH/FdR system) | Generation of reduced ferredoxin for cluster assembly | Providing reducing power for in vitro systems |
| Oxygen Control Systems | Anaerobic chambers/chambers with <10 ppm O₂ | Maintaining anoxic conditions for cluster stability |
| Spectroscopic Standards | Reference spectra for [2Fe-2S], [4Fe-4S] clusters | Quantification of cluster types and yields |
The convergent evolution of iron-sulfur cluster binding within the β-grasp fold underscores the remarkable functional plasticity of this ancient protein architecture. Ferredoxins represent one of nature's most successful adaptations of this fold for electron transfer, with their diverse Fe-S cluster types supporting fundamental metabolic processes across all domains of life. The experimental methodologies outlined here—from spontaneous cluster assembly to functional complementation assays—provide powerful approaches for investigating Fe-S protein biogenesis, function, and evolution. As research continues to unravel the complexities of Fe-S cluster biosynthesis and insertion, the principles of convergent evolution observed in ferredoxins offer valuable insights for protein engineering and the development of novel bioinorganic catalysts.
The β-grasp fold represents a remarkable example of evolutionary optimization, where a compact structural scaffold has been recruited for a strikingly diverse range of biochemical functions across all domains of life. This fold, prototyped by ubiquitin (UB), is characterized by a β-sheet with 4-5 strands that appears to "grasp" a central α-helical segment [1]. While ubiquitin and ubiquitin-like proteins (Ubls) represent the most extensively studied members of this fold, their structural versatility extends far beyond post-translational modification systems. The NUDIX hydrolase superfamily exemplifies how this ancient fold has been adapted for sophisticated catalytic functions, utilizing the inherent stability and plasticity of the β-grasp architecture to create active sites for hydrolyzing potentially toxic nucleoside diphosphate derivatives [1] [85].
Evolutionary reconstruction indicates that the β-grasp fold had already differentiated into at least seven distinct lineages by the time of the last universal common ancestor of all extant organisms, encompassing much of the structural diversity observed in extant versions of the fold [1]. The earliest β-grasp members were probably involved in RNA metabolism, with subsequent radiation into various functional niches including enzymatic catalysis, sulfur transfer, iron-sulfur cluster binding, and adaptor functions in signaling pathways [1]. This evolutionary trajectory highlights the remarkable adaptability of this structural framework, with NUDIX hydrolases representing a particularly successful functional radiation within this fold family.
Structural Foundations of the β-Grasp Fold
Core Architectural Features
The β-grasp fold exhibits several distinctive structural characteristics that contribute to its exceptional stability and functional versatility:
- Compact β-sheet core: The fold is dominated by a β-sheet with 4-5 anti-parallel β-strands that provides an exposed surface for diverse interactions [1] [53]
- Central α-helix: A single helical segment is cradled by the β-sheet, creating a stable structural core [53]
- Minimal exposed surface area: The compact arrangement minimizes exposed surface area, contributing to remarkable stability including thermostability up to 95°C [53]
- Stabilizing structural elements: Three disulfide bonds and a series of hydrogen bonds further enhance structural stability [53]
The manifold functional abilities of the β-grasp fold arise primarily from the prominent β-sheet, which provides an exposed surface for diverse interactions or additionally, by forming open barrel-like structures that can accommodate various biochemical activities [1].
Structural Variations and Functional Adaptations
Within the conserved core architecture, the β-grasp fold exhibits significant structural variations that enable functional specialization:
Table: Structural and Functional Diversity in β-Grasp Fold Proteins
| Structural Variation | Functional Adaptation | Representative Examples |
|---|---|---|
| Enzymatic active sites | Scaffold for phosphohydrolase activity | NUDIX hydrolases [1] |
| Open barrel formations | Binding of diverse co-factors | Molybdopterin-binding proteins [1] |
| Iron-sulfur cluster binding | Electron transfer | 2Fe-2S ferredoxins [1] |
| Thiocarboxylate modifications | Sulfur transfer | ThiS, MoaD in cofactor biosynthesis [1] |
| Covalent conjugation systems | Post-translational modification | Ubiquitin, SUMO, UFM1 [1] [86] |
NUDIX Hydrolases: Mechanistic Diversity and Catalytic Strategies
Structural Organization and Conserved Motifs
NUDIX hydrolases constitute a large superfamily of hydrolytic enzymes characterized by a conserved structural framework based on the β-grasp fold. All NUDIX hydrolases share a characteristic α-β-α sandwich structure with a specific NUDIX motif that contains the catalytic site and metal-binding residues [87] [88]. The NUDIX motif comprises 23 amino acids with the consensus sequence: GX₅EX₇REUXEEXGU, where U represents a bulky hydrophobic amino acid (typically isoleucine, leucine, or valine), and X represents any amino acid [89] [87]. This motif forms a short helix that contains the catalytic amino acids essential for substrate hydrolysis.
The NUDIX fold domain typically exists as a single domain, though some family members incorporate additional domains that modulate function or substrate specificity [90] [87]. For instance, NUDT12 and NUDT13 contain the SQPWPFPxS sequence motif common in NADH diphosphatases, while DCP2 incorporates additional domains involved in mRNA decapping functions [90]. This structural diversity within the conserved NUDIX framework enables the remarkable functional range observed across this enzyme family.
Catalytic Mechanisms and Metal Dependence
NUDIX hydrolases employ sophisticated catalytic strategies to achieve their remarkable rate accelerations, which range from 10⁹- to 10¹²-fold [85]. The mechanisms involve multiple catalytic components working in concert:
- General base catalysis: Contributes 10³-10⁵-fold to rate acceleration, mediated by a glutamate residue within or beyond the NUDIX box, or occasionally by a histidine residue [85]
- Lewis acid catalysis: Provides 10³-10⁵-fold rate acceleration through one, two, or three divalent cations (typically Mg²⁺ or Mn²⁺) that stabilize the transition state [85]
- Cationic side chain assistance: Lysine and arginine residues provide additional catalysis (10-10³-fold) through charge neutralization and orientation of catalytic groups [85]
- Hydrogen bond donation: Tyrosine residues assist in orienting the general base or promoting departure of leaving groups [85]
The mechanisms of NUDIX hydrolases are highly diverse in both the position on the substrate at which nucleophilic substitution occurs and the number of required divalent cations [85]. While most NUDIX enzymes proceed by associative nucleophilic substitutions by water at specific internal phosphorus atoms of a diphosphate or polyphosphate chain, members of the GDP-mannose hydrolase sub-family catalyze dissociative nucleophilic substitutions by water at carbon atoms [85]. The specific site of substitution is determined by the positions of the general base and the entering water molecule relative to the substrate orientation in the active site.
Table: Representative Human NUDIX Hydrolases and Their Substrate Preferences
| Enzyme | Primary Substrates | Catalytic Features | Biological Functions |
|---|---|---|---|
| MTH1 (NUDT1) | 8-oxo-dGTP, 2-OH-dATP, N2-me-dGTP | Oxidized nucleotide preference | Sanitization of nucleotide pool [90] |
| NUDT2 | Ap4A, Ap4, Ap4dT, Ap4G, p4G | Dinucleoside polyphosphate hydrolysis | RNA metabolism, signaling [90] |
| NUDT12 | β-NADH, Ap3A, ADP-ribose | Broad substrate range | NADH metabolism [90] |
| NUDT15 | 8-oxo-dGTP, 6-thio-GTP, 5-me-dCTP | Promiscuous activity | Thiopurine metabolism [90] |
| DIPP Enzymes (NUDT3,4,10,11) | 5-PP-InsP5, diphosphoinositol polyphosphates | Inositol pyrophosphate specificity | Phosphate signaling [90] |
Functional Diversity and Biological Roles of NUDIX Hydrolases
Cellular Housekeeping and Metabolic Regulation
NUDIX hydrolases function as crucial "housecleaning" enzymes that maintain metabolic homeostasis by hydrolyzing potentially toxic nucleoside diphosphates and their derivatives [89] [87]. This housekeeping function encompasses several critical cellular activities:
- Nucleotide pool sanitization: Removal of oxidized nucleotides such as 8-oxo-dGTP that would otherwise cause transversion mutations if incorporated into DNA [90]
- Signaling molecule regulation: Hydrolysis of dinucleoside polyphosphates (e.g., Ap4A) and inositol pyrophosphates that function as cellular messengers [89] [90]
- Cofactor metabolism: Processing of nucleotide cofactors including NADH, NAD⁺, and Coenzyme A derivatives [90]
- RNA metabolism: Decapping of mRNA and processing of RNA 5' caps, regulating mRNA stability and turnover [87]
The substrate diversity of NUDIX enzymes is extraordinary, encompassing nucleoside di- and triphosphates, nucleotide sugars, dinucleosides, diphosphoinositol polyphosphates, and RNA caps [87] [88]. This functional range underscores the adaptability of the β-grasp fold in creating specialized active sites within a conserved structural framework.
Pathogenic Manipulation of Host Signaling
Recent research has revealed that pathogenic fungi have evolved secreted NUDIX effector proteins that manipulate host signaling pathways to facilitate infection [89]. These effectors exemplify mechanistic diversification within a single enzyme family and highlight the potential clinical relevance of NUDIX hydrolases:
- Magnaporthe oryzae MoNUDIX: Secreted into the host cytoplasm during rice blast infection, where it hydrolyzes host inositol pyrophosphates (PP-InsPs), triggering a phosphate starvation response that suppresses immunity and enhances nutrient mobilization [89]
- Colletotrichum lentis CtNUDIX: Localizes to the apoplast during lentil anthracnose infection, where it disrupts PP-InsP-dependent clathrin-mediated endocytosis by hydrolyzing PP-InsPs bound to adaptor proteins, leading to loss of plasma membrane integrity and hypersensitive cell death [89]
These fungal effectors demonstrate how the NUDIX fold has been evolutionarily co-opted for pathogenic strategies, with subcellular localization and specific substrate preferences determining distinct infection outcomes. The MoNUDIX effector promotes sustained biotrophic growth by suppressing host immunity, while CtNUDIX mediates the transition from biotrophy to necrotrophy by inducing programmed cell death [89].
Experimental Approaches for NUDIX Hydrolase Characterization
Comprehensive Biochemical Screening
Systematic biochemical characterization of NUDIX hydrolase families requires carefully designed experimental protocols to capture their functional diversity:
Table: Essential Research Reagents for NUDIX Hydrolase Characterization
| Reagent/Category | Specific Examples | Experimental Function |
|---|---|---|
| Recombinant Enzymes | Human NUDIX proteins (NUDT1-22) | Functional assays, structural studies [90] |
| Substrate Libraries | 52 putative substrates (oxidized dNTPs, nucleotide sugars, dinucleotides) | Specificity profiling [90] |
| Detection Assays | Malachite Green phosphate detection | High-throughput activity screening [90] |
| Crystallography Resources | Crystal structures of NUDIX-ligand complexes | Structure-function analysis [90] |
| Bioinformatic Tools | CLANS clustering, phylogenetic analysis, molecular docking | Sequence-structure-function relationships [87] [88] |
Protocol: High-Throughput Biochemical Screening of NUDIX Hydrolases
Protein Expression and Purification: Express recombinant NUDIX hydrolases in E. coli system with appropriate affinity tags (e.g., His-tag, GST-tag). Purify using affinity chromatography followed by size-exclusion chromatography to obtain soluble, monodisperse proteins [90]
Enzyme Activity Assay Setup:
- Prepare reaction buffer with physiological pH conditions (e.g., 50 mM Tris-HCl, pH 7.5, 5-10 mM MgCl₂)
- Use enzyme concentrations of 5 nM (low) and 200 nM (high) to detect both high-affinity and low-affinity substrates
- Incubate with 25-50 μM substrate concentrations for 15-60 minutes at 37°C [90]
Phosphate Detection Using Malachite Green:
- Terminate reactions with Malachite Green reagent
- Measure absorbance at 620-650 nm
- Calculate phosphate release using potassium phosphate standard curve [90]
Data Analysis and Redundancy Mapping:
- Generate heat maps of enzymatic activities across substrate panels
- Perform hierarchical clustering to identify substrate preference patterns
- Construct substrate redundancy maps to visualize functional overlap [90]
Structural and Computational Approaches
Structural characterization of NUDIX hydrolases provides critical insights into their mechanistic diversity and evolutionary relationships:
Protocol: Structural Analysis of NUDIX Hydrolase Diversity
Sequence Identification and Classification:
Phylogenetic and Clustering Analysis:
Molecular Docking for Substrate Prediction:
- Generate 3D structure models using AlphaFold2 or molecular modeling
- Prepare ligand libraries of potential substrates (Ap3A, Ap4A, PP-InsPs)
- Perform molecular docking simulations to predict binding affinities and specificities [88]
Structure-Function Correlation:
- Superimpose available crystal structures to identify conserved structural elements
- Map sequence variations onto structural models to identify determinant residues
- Correlate structural features with substrate preferences and catalytic mechanisms [90]
Visualization of NUDIX Hydrolase Functional Diversity
The functional diversity and evolutionary relationships of NUDIX hydrolases can be visualized through the following pathway diagram:
NUDIX Hydrolase Functional Classification and Evolution
This visualization illustrates the evolutionary trajectory of the β-grasp fold from ancestral RNA metabolism functions to specialized NUDIX hydrolase activities. The diagram highlights how this structural scaffold has been adapted for both cellular housekeeping functions and specialized pathogenic effector roles, demonstrating the remarkable functional plasticity of this protein fold.
The β-grasp fold represents a paradigm of structural efficiency, where a compact architectural framework has been evolutionarily optimized for an extraordinary range of biological functions. NUDIX hydrolases exemplify the catalytic potential of this fold, demonstrating how conserved structural elements can be adapted for diverse hydrolytic activities through strategic variations in active site architecture and substrate recognition features. The mechanistic diversity within the NUDIX superfamily—encompassing different nucleophilic substitution mechanisms, metal coordination schemes, and substrate specificities—highlights the remarkable functional plasticity of this protein fold.
Future research directions in this field should focus on several key areas:
- Comprehensive functional annotation: Systematic characterization of the numerous uncharacterized NUDIX family members across diverse organisms [87] [88]
- Therapeutic targeting: Exploitation of NUDIX enzymes as drug targets, particularly in cancer and infectious diseases, building on their upregulated expression in stressed cells [90]
- Pathogen effector mechanisms: Detailed understanding of how pathogenic NUDIX effectors manipulate host signaling, potentially leading to novel antifungal strategies [89]
- Structural dynamics: Investigation of how conformational flexibility within the conserved β-grasp framework enables functional diversification
The continuing study of NUDIX hydrolases and other β-grasp fold proteins promises to yield fundamental insights into enzyme evolution, structural determinants of catalytic efficiency, and novel approaches for therapeutic intervention across a range of human diseases.
Conclusion
The β-grasp fold exemplifies how a simple structural scaffold has been leveraged through evolution to perform an extraordinary range of biochemical functions, with the eukaryotic ubiquitin-signaling system representing one of its most complex manifestations. Research has firmly established that the core components of this system have deep evolutionary roots in prokaryotic sulfur-transfer machinery. The future of biomedical research in this area lies in deepening our understanding of the specific roles of diverse UBLs and overcoming the challenge of developing targeted therapies that can precisely modulate these pathways. The continued development of small-molecule inhibitors targeting E1/E2/E3 enzymes and deubiquitinases (DUBs), informed by advanced structural and dynamic studies, holds immense promise for treating cancer, neurodegenerative disorders, and infectious diseases. Moving forward, integrating computational predictions with experimental validation will be key to unraveling the remaining mysteries of the β-grasp fold's versatility and fully harnessing its therapeutic potential.
References
- 1. Small but versatile: the extraordinary functional and structural ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1949818/]
- 2. A novel superfamily containing the β-grasp fold involved in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1796856/]
- 3. Fold and Function of the InlB B-repeat [https://www.sciencedirect.com/science/article/pii/S0021925820858335]
- 4. ubiquitin [https://www.swarthmore.edu/NatSci/jvoet1/Int_Ex_JSmol/ubiquitin/ubiquitin.html]
- 5. the extraordinary functional and structural diversity of the beta ... [https://pubmed.ncbi.nlm.nih.gov/17605815/]
- 6. Ubiquitin: Structure and Function [https://www.intechopen.com/chapters/87831]
- 7. Atomic-level description of ubiquitin folding - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3625349/]
- 8. Structural and Functional Insights to Ubiquitin-Like Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4118471/]
- 9. Structural insights into the human HRD1 ubiquitin ligase ... [https://www.nature.com/articles/s41467-025-61143-z]
- 10. Structural ubiquitin contributes to K48 linkage specificity of ... [https://www.sciencedirect.com/science/article/pii/S2211124725004590]
- 11. The natural history of ubiquitin and ubiquitin-related domains [https://pmc.ncbi.nlm.nih.gov/articles/PMC5881585/]
- 12. The nature of the last universal common ancestor and its ... [https://www.nature.com/articles/s41559-024-02461-1]
- 13. Ubiquitin-like protein [https://en.wikipedia.org/wiki/Ubiquitin-like_protein]
- 14. Article Structural diversity and oligomerization of bacterial ... [https://www.sciencedirect.com/science/article/abs/pii/S096921262500108X]
- 15. E3 ubiquitin ligases: styles, structures and functions - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8607428/]
- 16. A New View of the Last Universal Common Ancestor - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11458664/]
- 17. Last universal common ancestor [https://en.wikipedia.org/wiki/Last_universal_common_ancestor]
- 18. The Routes of Emergence of Life from LUCA during ... [https://www.mdpi.com/2075-1729/5/2/1445]
- 19. Structure and evolution of ubiquitin and ... - PubMed - NIH [https://pubmed.ncbi.nlm.nih.gov/22350875/]
- 20. The Ubiquitin Tale: Current Strategies and Future Challenges [https://pmc.ncbi.nlm.nih.gov/articles/PMC11406696/]
- 21. Prokaryotic Ubiquitin-Like Protein Modification - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4757901/]
- 22. The prokaryotic antecedents of the ubiquitin-signaling system ... [https://genomebiology.biomedcentral.com/articles/10.1186/gb-2006-7-7-r60]
- 23. Prokaryotic Ubiquitin-Like Protein and Its Ligase/Deligase ... [https://www.sciencedirect.com/science/article/abs/pii/S0022283617301961]
- 24. NMR and X-RAY structures of human E2-like ubiquitin-fold ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2850604/]
- 25. Crystal Structure of the Human Ubiquitin-like Protein ... [https://www.sciencedirect.com/science/article/pii/S0021925818371722]
- 26. State-of-the-Art and Future Directions in Structural Proteomics [https://pmc.ncbi.nlm.nih.gov/articles/PMC12546877/]
- 27. Allosteric activation of SENP1 by SUMO1 β-grasp domain ... [https://elifesciences.org/articles/18249]
- 28. Selective ubiquitination of drug-like small molecules by the ... [https://www.nature.com/articles/s41467-025-63442-x]
- 29. Navigating the landscape of protein folding and proteostasis [https://pmc.ncbi.nlm.nih.gov/articles/PMC12550075/]
- 30. The structure of the ubiquitin-like modifier FAT10 reveals ... [https://www.nature.com/articles/s41467-018-05776-3]
- 31. Native-state dynamics of the ubiquitin family [https://pmc.ncbi.nlm.nih.gov/articles/PMC1578254/]
- 32. Secondary interactions in ubiquitin-binding domains achieve ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11372445/]
- 33. Evaluation of selected binding for the analysis of... domains [https://pmc.ncbi.nlm.nih.gov/articles/PMC3715598/]
- 34. Use of a High-Affinity Ubiquitin - Binding to Detect and Purify... Domain [https://bio-protocol.org/en/bpdetail?id=5426&type=0]
- 35. Mechanism and disease association of E2-conjugating enzymes [https://pmc.ncbi.nlm.nih.gov/articles/PMC5095918/]
- 36. E1, E2, E3 Enzyme and Their Regulators [https://www.bocsci.com/resources/e1-e2-e3-enzyme-and-their-regulators.html?srsltid=AfmBOorHm2ykeLMX-HZpGXHHROzT-_ToR3PMQZRfRvu57V7UxoqK_vE3]
- 37. Specificity of the E1-E2-E3 Enzymatic Cascade for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4652587/]
- 38. Proteasome Inhibitors: Structure and Function - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6020165/]
- 39. Targeted protein degradation: advances in drug discovery ... [https://www.nature.com/articles/s41392-024-02004-x]
- 40. Structural basis of prokaryotic ubiquitin-like protein ... [https://www.nature.com/articles/s41467-021-27787-3]
- 41. Recent advances in small molecule inhibitors of ... [https://www.sciencedirect.com/science/article/abs/pii/S0223523425000893]
- 42. Current perspectives in drug targeting intrinsically disordered ... [https://bmcbiol.biomedcentral.com/articles/10.1186/s12915-025-02214-x]
- 43. The Ubiquitin System: Functional Complexity and Semiosis ... [https://uncommondescent.com/intelligent-design/the-ubiquitin-system-functional-complexity-and-semiosis-joined-together/]
- 44. Chemical approaches to explore ubiquitin-like proteins [https://pubs.rsc.org/en/content/articlehtml/2025/cb/d4cb00220b]
- 45. Recent progress in ubiquitin and ubiquitin-like protein (Ubl ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4822135/]
- 46. The Ubiquitination Machinery of the Ubiquitin System [https://bioone.org/journals/the-arabidopsis-book/volume-2014/issue-12/tab.0174/The-Ubiquitination-Machinery-of-the-Ubiquitin-System/10.1199/tab.0174.full]
- 47. Yeast UBL-UBA proteins have partially redundant functions in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1697804/]
- 48. UBL/UBA Ubiquitin Receptor Proteins Bind a Common ... [https://www.sciencedirect.com/science/article/abs/pii/S0022283605015408]
- 49. Structural conservation of druggable hot spots in protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3158149/]
- 50. A comprehensive dataset of protein-protein interactions ... [https://www.nature.com/articles/s41597-024-03233-z]
- 51. New insights into protein–protein interaction modulators in ... [https://www.nature.com/articles/s41392-024-02036-3]
- 52. Optimization of Protein-Protein Interaction Measurements for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7015265/]
- 53. Review Ubiquitin—A structural perspective [https://www.sciencedirect.com/science/article/abs/pii/S1097276524010104]
- 54. Snapshotting the transient conformations and tracing the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8179386/]
- 55. Single turnover transient state kinetics reveals processive ... [https://elifesciences.org/articles/99052]
- 56. Origin and Function of Ubiquitin-like Protein Conjugation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2819001/]
- 57. Ubiquitination-Proteasome System (UPS) and Autophagy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8909305/]
- 58. Ubiquitin-mediated regulation of autophagy [https://jbiomedsci.biomedcentral.com/articles/10.1186/s12929-019-0569-y]
- 59. Ubiquitin-like protein conjugation and the ... [https://www.nature.com/articles/nrd3321]
- 60. Ubiquitin-like modifications [https://www.nature.com/collections/iiihdafije]
- 61. Profiling of Ubiquitin-like Modifications Reveals Features ... [https://www.sciencedirect.com/science/article/pii/S0092867413001554]
- 62. Function, mechanism and drug discovery of ubiquitin and ... [https://www.sciencedirect.com/science/article/pii/S2211383523002812]
- 63. Ubiquitin, SUMO, and NEDD8: Key Targets of Bacterial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7028394/]
- 64. Ubiquitin-like Proteins in Autoimmune Diseases [https://immunenetwork.org/DOIx.php?id=10.4110/in.2025.25.e21]
- 65. DIG-DUBs: mechanisms and functions of ISG15 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12410001/]
- 66. Cryo-EM structures reveal the molecular mechanism of ... [https://www.nature.com/articles/s41594-025-01681-8]
- 67. Hybrid Chains: A Collaboration of Ubiquitin and ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2019.00931/full]
- 68. Identification and characterization of a novel ISG15 ... [https://www.nature.com/articles/srep12704]
- 69. Small but versatile: the extraordinary functional and structural ... [https://biologydirect.biomedcentral.com/articles/10.1186/1745-6150-2-18]
- 70. Non-proteolytic ubiquitylation in cellular signaling and human ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8826416/]
- 71. Emerging Roles of Non-proteolytic Ubiquitination in ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2022.944460/full]
- 72. Nonproteolytic Ubiquitin Chains in Neural Function and Brain ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9229342/]
- 73. Ubiquitin signaling in immune responses | Cell Research [https://www.nature.com/articles/cr201640]
- 74. Proteolytic and non-proteolytic roles of ubiquitin and the ... [https://www.sciencedirect.com/science/article/abs/pii/S1874939910001604]
- 75. A novel superfamily containing the β-grasp fold involved in binding diverse soluble ligands - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1796856/]
- 76. Structure and Ubiquitin Binding of the Ubiquitin-interacting ... [https://www.sciencedirect.com/science/article/pii/S0021925820843968]
- 77. Supported Lipid Bilayers and the Study of Two ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2022.833123/full]
- 78. CDD Conserved Protein Domain Family: SLBB [https://www.ncbi.nlm.nih.gov/Structure/cdd/cl22925]
- 79. Evaluation of the binding affinity of E3 ubiquitin ligase ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7829257/]
- 80. Ferredoxins: Functions, Evolution, Potential Applications, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11430716/]
- 81. Spontaneous assembly of redox-active iron-sulfur clusters ... [https://www.nature.com/articles/s41467-021-26158-2]
- 82. Cellular assays identify barriers impeding iron-sulfur ... [https://elifesciences.org/articles/70936]
- 83. Outlining the Complex Pathway of Mammalian Fe-S ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8349188/]
- 84. Biogenesis of Iron–Sulfur Clusters and Their Role in DNA ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2021.735678/full]
- 85. Structures and mechanisms of Nudix hydrolases - PubMed - NIH [https://pubmed.ncbi.nlm.nih.gov/15581572/]
- 86. The UFM1 system: Working principles, cellular functions ... [https://www.sciencedirect.com/science/article/pii/S1097276523009796]
- 87. Analysis of NUDIX enzymes across fungi reveals previously ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12210691/]
- 88. Analysis of NUDIX enzymes across fungi reveals previously ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-11778-5]
- 89. Functional mechanistic diversity of the NUDIX effectors in ... [https://phytopatholres.biomedcentral.com/articles/10.1186/s42483-025-00400-x]
- 90. A comprehensive structural, biochemical and biological ... [https://www.nature.com/articles/s41467-017-01642-w]