The Ubiquitin Cascade: From E1-E2-E3 Enzymatic Mechanisms to Targeted Drug Development
This article provides a comprehensive analysis of the ubiquitin activation cascade, a crucial three-step enzymatic pathway involving E1, E2, and E3 enzymes that regulates virtually all eukaryotic cellular processes through...
The Ubiquitin Cascade: From E1-E2-E3 Enzymatic Mechanisms to Targeted Drug Development
Abstract
This article provides a comprehensive analysis of the ubiquitin activation cascade, a crucial three-step enzymatic pathway involving E1, E2, and E3 enzymes that regulates virtually all eukaryotic cellular processes through protein ubiquitination. We explore the foundational biochemistry and structural biology governing this system, examine cutting-edge methodologies for studying ubiquitination, address key challenges in targeting this pathway for therapeutic intervention, and evaluate emerging technologies for drug discovery. Aimed at researchers, scientists, and drug development professionals, this review synthesizes current knowledge of ubiquitin signaling with a focus on translating mechanistic understanding into clinical applications for cancer, neurodegenerative disorders, and other human diseases.
Decoding the Ubiquitin Cascade: E1, E2, E3 Enzymatic Architecture and Core Mechanisms
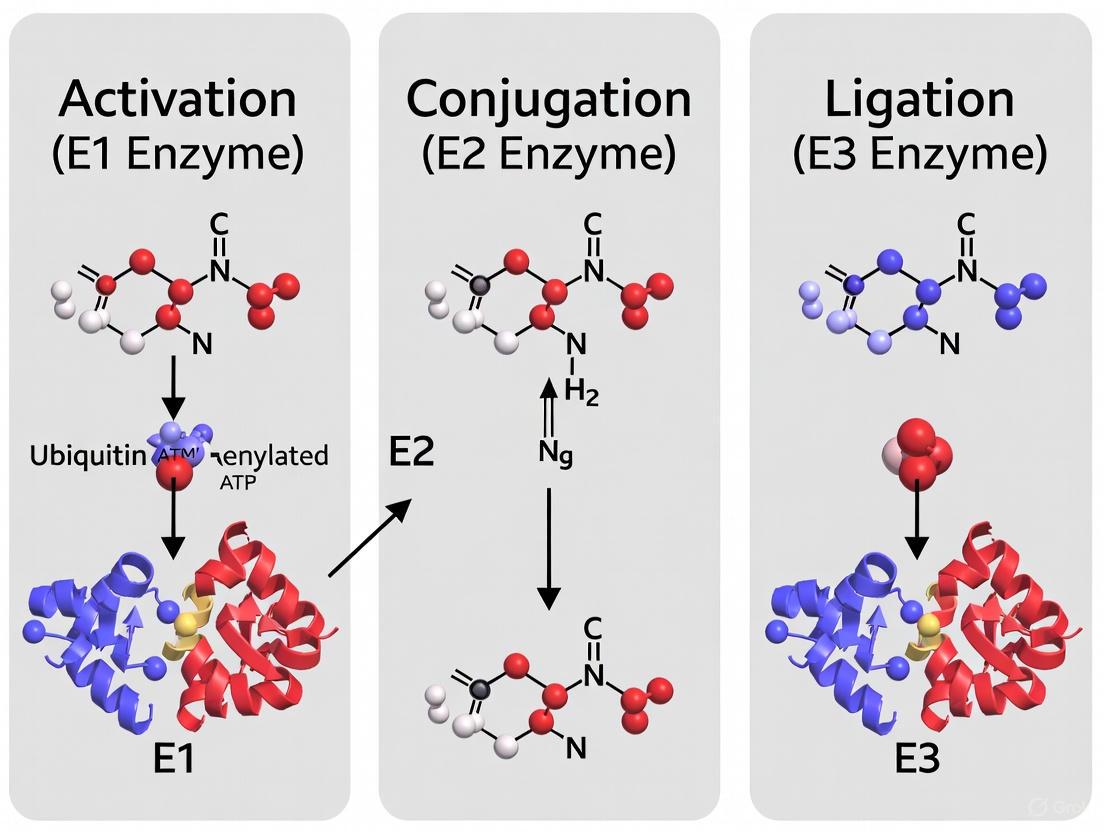
The ubiquitin-proteasome system (UPS) is a master regulator of eukaryotic cell biology, controlling the stability, activity, and localization of a vast array of proteins [1] [2]. At the heart of this system is the ubiquitination cascade, a three-step enzymatic process that covalently attaches the small, highly conserved protein ubiquitin (Ub) to substrate proteins [3] [4]. This process is executed by the sequential action of ubiquitin-activating (E1), ubiquitin-conjugating (E2), and ubiquitin-ligating (E3) enzymes [1] [5]. The outcome of ubiquitination is remarkably diverse, influencing proteasomal degradation, DNA repair, signal transduction, immune response, and autophagy [4] [6]. The specific biological consequence is dictated by factors such as the number of ubiquitin molecules attached (mono- versus polyubiquitination) and the topology of polyubiquitin chains, which can be formed through different lysine residues (K6, K11, K27, K29, K33, K48, K63) or the N-terminal methionine (M1) of ubiquitin itself [4] [6]. Given its central role in cellular homeostasis, dysregulation of the ubiquitination cascade is implicated in numerous diseases, including cancers, neurodegenerative disorders, and autoimmune conditions, making its enzymatic components attractive targets for therapeutic intervention [1] [2].
The Three-Step Enzymatic Mechanism
The ubiquitination cascade is characterized by a relay of enzymatic activities that activate and transfer ubiquitin through a series of high-energy thioester bonds to a final substrate protein.
Step 1: Activation by E1 Enzymes
The cascade initiates with the ATP-dependent activation of ubiquitin by an E1 enzyme [1] [2]. The human genome encodes two ubiquitin E1 enzymes: UBE1 and UBA6 [3]. E1 enzymes are approximately 100 kDa in size and contain a recognizable nucleotide-binding motif for ATP and a conserved catalytic cysteine residue [3]. The mechanism proceeds in two key steps:
- Adenylation: The E1 enzyme binds ATP and ubiquitin, catalyzing the adenylation of the C-terminal glycine of ubiquitin. This reaction results in a ubiquitin-adenylate intermediate and the release of pyrophosphate (PPi) [1] [2].
- Thioester Formation: The adenylated ubiquitin is then attacked by the conserved cysteine thiolate of the E1, forming a high-energy E1~Ub thioester bond and releasing AMP [1] [2] [3].
The E1 enzyme subsequently binds a second molecule of ATP and ubiquitin, forming a ternary complex that is competent for engaging an E2 enzyme [2]. Structural studies of UBA1, the prototypical E1, reveal it is a multi-domain enzyme that undergoes significant conformational changes during its catalytic cycle, transitioning between "open" and "closed" states to facilitate adenylation and thioester transfer [1].
Step 2: Conjugation by E2 Enzymes
The activated ubiquitin is transferred from the E1~Ub thioester to a conserved cysteine residue within the core ubiquitin-conjugating (UBC) domain of an E2 enzyme via a transthiolation reaction, forming an E2~Ub thioester [5] [3]. Humans possess approximately 40 E2 enzymes, which are roughly twice the size of ubiquitin [5]. The core UBC domain, composed of ~150 amino acids forming an α/β-fold, is common to all E2s and is sufficient for catalytic activity with some E3s [5]. Many E2s also feature N- or C-terminal extensions that can regulate their cellular localization, stability, or interactions with specific E3s [5]. E2s are not mere passive carriers; they are critical determinants of the chemistry of ubiquitin transfer. While most E2s facilitate the formation of an isopeptide bond between the C-terminus of ubiquitin and a lysine ε-amino group on a substrate, some E2s exhibit unique reactivities. For instance, UBE2W catalyzes the monoubiquitination of protein N-terminal α-amines, and UBE2J2 has been reported to modify serine and threonine residues [5] [7].
Step 3: Ligation by E3 Enzymes
The final step is the ligation of ubiquitin to a substrate protein, which is facilitated by an E3 ligase. E3s are the most diverse components of the cascade, with hundreds of members in humans, and they are primarily responsible for substrate recognition and specificity [3]. E3s can be divided into three major families based on their mechanism of action:
- RING E3s: These E3s, which include the related U-box proteins, function as scaffolds that simultaneously bind the E2~Ub conjugate and the substrate. They facilitate the direct transfer of ubiquitin from the E2 to the substrate lysine without forming a covalent intermediate [5] [2] [3].
- HECT E3s: HECT E3s contain a conserved ~350-residue HECT domain with an active-site cysteine. They catalyze a two-step transfer: first, ubiquitin is transferred from the E2 to the HECT domain cysteine, forming a transient E3~Ub thioester intermediate; second, ubiquitin is delivered from the E3 to the substrate [2] [3].
- RBR E3s: RING-between-RING (RBR) E3s are functional hybrids. They contain RING domains that bind the E2~Ub, but they also feature a conserved cysteine in a "RING2" domain that forms an obligatory thioester intermediate with ubiquitin, similar to HECT E3s, before final transfer to the substrate [5].
The collaboration between a charged E2 and an E3 results in the formation of an isopeptide bond between the C-terminus of ubiquitin and the substrate, completing the cascade.
Diagram 1: The Three-Step Ubiquitin Cascade
Quantitative Analysis of Cascade Components
The following tables summarize key quantitative data on the enzymes of the ubiquitination cascade and the diversity of ubiquitin signals they generate.
Table 1: Enzymatic Components of the Human Ubiquitination Cascade
| Enzyme Class | Number of Human Genes | Core Functional Domains/Motifs | Key Catalytic Residue | Primary Function |
|---|---|---|---|---|
| E1 (Activating) | 2 (UBE1, UBA6) [3] | Nucleotide-binding motif, Active Cysteine Domain [3] | Cysteine [1] [2] | ATP-dependent ubiquitin activation; E2 charging |
| E2 (Conjugating) | ~40 [5] | UBC domain (~150 residues) [5] | Cysteine [5] [3] | Ubiquitin carrier; influences linkage specificity |
| E3 (Ligating) | Hundreds (RING, HECT, RBR) [3] | RING, U-box, HECT, or RBR domains [2] [3] | Cysteine (HECT, RBR) or none (RING) [5] [2] | Substrate recognition; facilitates final ubiquitin transfer |
Table 2: Ubiquitin Chain Linkages and Their Primary Functions
| Linkage Type | Representative Cellular Functions |
|---|---|
| K48-linked | Proteasomal degradation [1] [4] |
| K63-linked | DNA repair, NF-κB signaling, endocytosis, kinase activation [4] |
| K11-linked | Cell cycle regulation, ER-associated degradation (ERAD) [4] |
| K6-linked | DNA damage response, mitochondrial homeostasis [4] |
| K27-linked | Immune signaling, Wnt/β-catenin signaling [4] |
| K29-linked | Proteasomal degradation, Wnt/β-catenin signaling [4] |
| K33-linked | T-cell receptor signaling, kinase suppression [4] |
| M1-linked (Linear) | NF-κB activation, inflammatory signaling [4] [6] |
| Monoubiquitination | Endocytosis, histone regulation, DNA repair [4] [6] |
Detailed Experimental Protocols
This section provides methodologies for key in vitro experiments used to study the biochemistry of the ubiquitination cascade.
Protocol: E1~Ub Thioester Formation Assay
Objective: To demonstrate the initial activation of ubiquitin by an E1 enzyme through the formation of a high-energy thioester bond.
Principle: This assay exploits the fact that the E1~Ub thioester bond is stable under non-reducing conditions but is rapidly hydrolyzed by reducing agents like DTT or β-mercaptoethanol. Reaction progress can be monitored by a mobility shift on non-reducing SDS-PAGE [7].
Materials:
- Purified E1 enzyme (e.g., UBA1)
- Ubiquitin
- ATP
- MgCl₂
- Reaction Buffer: 50 mM Tris-HCl (pH 7.5), 50 mM NaCl, 2 mM ATP, 5 mM MgCl₂
- Non-reducing SDS-PAGE sample buffer (lacking DTT or β-mercaptoethanol)
- 4x Reducing SDS-PAGE sample buffer (with 200 mM DTT)
Procedure:
- Reaction Setup: In a microcentrifuge tube, combine on ice:
- 2 µL 10x Reaction Buffer
- 1 µM E1 enzyme
- 10 µM ubiquitin
- Nuclease-free water to a final volume of 20 µL.
- Incubation: Incubate the reaction mixture at 30°C for 0, 1, 5, and 10 minutes.
- Termination and Analysis:
- At each time point, remove 5 µL of the reaction and mix with 5 µL of non-reducing SDS-PAGE sample buffer. Do not heat the sample above room temperature to preserve the thioester bond.
- In parallel, take a 5 µL aliquot from the 10-minute time point and quench it with 5 µL of reducing SDS-PAGE sample buffer. Heat this sample at 95°C for 5 minutes.
- Detection: Load all samples onto an SDS-PAGE gel. Perform Western blotting using an anti-ubiquitin antibody. The E1~Ub thioester will appear as a higher molecular weight band in non-reducing lanes that disappears and is replaced by free ubiquitin in the reducing lane [7].
Protocol: Analysis of Lipid-Dependent E2 Activity
Objective: To investigate how membrane lipid composition regulates the activity of the ERAD-associated E2 enzyme, UBE2J2 [7].
Principle: Full-length, membrane-anchored E2s like UBE2J2 are reconstituted into liposomes of defined lipid composition. Ubiquitin loading by E1 is then measured to assess how lipid saturation and packing impact E2 activity.
Materials:
- Purified full-length human UBE2J2 (or UBE2J1 for comparison)
- Purified E1 enzyme
- Ubiquitin
- ATP-Mg²⁺
- Lipids: e.g., POPC (1-palmitoyl-2-oleoyl-glycero-3-phosphocholine), DPPC (dipalmitoylphosphatidylcholine), Cholesterol
- Detergent (for solubilization control, e.g., n-Dodecyl β-D-maltoside)
- Size-exclusion chromatography columns for detergent removal
Procedure:
- Proteoliposome Preparation:
- Create lipid films by evaporating chloroform solutions of lipids under nitrogen gas. Use two distinct compositions:
- Hydrate the lipid films in an appropriate buffer to form multilamellar vesicles. Subject the suspension to extrusion through a membrane with 100 nm pores to form large unilamellar vesicles (LUVs).
- Solubilize the LUVs with a mild detergent and incubate with purified UBE2J2.
- Remove the detergent by dialysis or size-exclusion chromatography to form proteoliposomes with UBE2J2 incorporated into the membrane.
- Ubiquitin Loading Reaction:
- In separate tubes, combine:
- Proteoliposomes (with reconstituted UBE2J2) OR soluble UBE2J2 in detergent buffer (control)
- 100 nM E1
- 5 µM Ubiquitin
- 2 mM ATP
- 5 mM MgCl₂
- in 50 mM Tris pH 7.5, 50 mM NaCl.
- Incubate at 30°C for various time points (e.g., 0, 30 sec, 1 min, 5 min).
- In separate tubes, combine:
- Analysis:
- Quench reactions with non-reducing SDS-PAGE sample buffer.
- Analyze by non-reducing SDS-PAGE and Western blotting with an anti-ubiquitin antibody.
- Expected Result: UBE2J2 loading is inefficient in ER-like membranes but is markedly enhanced in tightly-packed, saturated membranes and in detergent solution [7].
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for Studying the Ubiquitination Cascade
| Reagent / Tool | Function in Research | Example Use-Case |
|---|---|---|
| PYR-41 | Irreversible, small-molecule inhibitor of E1 ubiquitin-activating enzyme [2] | Investigating global effects of ubiquitination inhibition; stabilizes p53 and induces apoptosis in cancer cells [2] [3]. |
| Proteasome Inhibitors (Bortezomib, Carfilzomib) | Inhibit the 26S proteasome, blocking degradation of polyubiquitinated proteins [2] | FDA-approved for multiple myeloma; used experimentally to accumulate ubiquitinated substrates and study protein turnover [2]. |
| Recombinant E1, E2, E3 Enzymes | Highly purified, active enzyme components for in vitro reconstitution assays [7] | Defining minimal components for substrate ubiquitination; studying enzyme mechanisms and kinetics without cellular complexity [7]. |
| ATPγS (Adenosine 5'-O-[γ-thio]triphosphate) | Non-hydrolyzable ATP analog [1] | Traps the ubiquitin-adenylate intermediate, inhibiting the E1 catalytic cycle and subsequent thioester formation [1]. |
| Ubiquitin Variants (UbVs) | Engineered ubiquitin mutants that act as potent and specific inhibitors of UPS enzymes [2] | Targeting specific E2 or E3 enzymes with high selectivity, enabling functional dissection of individual pathways [2]. |
| Defined Lipid Liposomes | Synthetic membranes with controlled lipid composition [7] | Studying the regulation of membrane-associated E2s (e.g., UBE2J2) and E3s by specific lipids and membrane properties like lipid packing [7]. |
Ubiquitin-activating (E1) enzymes stand at the apex of the ubiquitination cascade, initiating a sophisticated pathway that regulates critical cellular processes ranging from protein degradation to DNA repair. This application note delves into the structural biology of E1 enzymes, elucidating their multi-domain architecture and the remarkable conformational changes that underpin their catalytic mechanism. Through detailed experimental protocols and structural analyses, we provide a framework for investigating E1 function, its interaction with E2 conjugating enzymes, and the implications for targeted drug discovery. The insights herein are framed within the broader context of ubiquitin activation cascade research, offering methodologies applicable to both academic and pharmaceutical development settings.
The ubiquitin-proteasome system (UPS) represents a crucial regulatory mechanism for maintaining cellular homeostasis through the targeted degradation of specific proteins and the clearance of misfolded proteins [8]. At the heart of this system lies a sequential enzymatic cascade involving three key enzymes: E1 (activating), E2 (conjugating), and E3 (ligase) enzymes [8] [9]. Ubiquitin, a highly conserved 76-amino acid protein, is covalently attached to substrate proteins via a process that requires ATP and involves three sequential enzymatic steps [9] [10]. This covalent modification acts as a molecular tag, primarily directing proteins to the 26S proteasome for degradation, though it also regulates non-proteolytic processes including cell cycle progression, DNA repair, and receptor endocytosis [8] [10].
The enzymatic cascade begins with E1 enzymes, which activate ubiquitin in an ATP-dependent reaction [11]. The activated ubiquitin is then transferred to an E2 conjugating enzyme, and finally, an E3 ligase facilitates the transfer of ubiquitin to the target substrate, determining specificity within the pathway [8]. The human genome encodes approximately 40 E2 enzymes and over 600 E3 ligases, creating a hierarchical system that allows for precise regulation of thousands of substrate proteins [9] [10] [5]. Dysregulation of this pathway is associated with numerous difficult-to-treat diseases, including cancer, neurodegenerative disorders, and viral infections, highlighting its significance as a therapeutic target area [10].
E1 Enzyme Domain Architecture and Catalytic Mechanism
Multi-domain Structure of E1 Enzymes
Canonical E1 enzymes exhibit a multi-domain architecture that facilitates their unique catalytic functions. Structural studies, primarily through X-ray crystallography, have revealed that E1s consist of several distinct domains:
- Adenylation Domain: This pseudo-dimeric domain is responsible for binding ATP·Mg²⁺ and ubiquitin, catalyzing the first step of ubiquitin C-terminal adenylation [12] [11]. It forms the core structural scaffold to which other domains are attached.
- Catalytic Cysteine Domain (Cys Domain): This domain harbors the active-site cysteine residue that forms a high-energy thioester bond with the C-terminus of ubiquitin following adenylation [12]. This domain undergoes massive conformational rotations during the catalytic cycle.
- Ubiquitin-Fold Domain (UFD): This domain plays a critical role in recruiting cognate E2 conjugating enzymes and presents them in an orientation where the E1 and E2 active sites face one another [12]. The UFD undergoes conformational "unlocking" to facilitate E2 binding.
Table 1: Core Domains of Ubiquitin-Activating (E1) Enzymes
| Domain Name | Key Structural Features | Catalytic Function |
|---|---|---|
| Adenylation Domain | Pseudo-dimeric structure; ATP-Mg²⁺ binding pocket | Binds ATP and ubiquitin; catalyzes ubiquitin adenylation |
| Cysteine Domain (Cys) | Contains active-site cysteine residue; mobile domain | Forms thioester bond with ubiquitin; rotates ~130° during catalysis |
| Ubiquitin-Fold Domain (UFD) | β-grasp fold similar to ubiquitin; flexible linker region | Recruits E2 conjugating enzymes; presents E2 for thioester transfer |
Catalytic Mechanism of Ubiquitin Activation
The E1 enzyme catalyzes ubiquitin activation through a carefully orchestrated mechanism involving distinct chemical steps:
- Ubiquitin Adenylation: E1 binds ATP·Mg²⁺ and ubiquitin, catalyzing the formation of a ubiquitin-adenylate intermediate (Ub-AMP) with the release of pyrophosphate [11].
- Thioester Bond Formation: The catalytic cysteine residue within the Cys domain attacks the Ub-AMP complex, forming a high-energy E1~Ub thioester linkage while releasing AMP [12] [11].
- E2 Recruitment and Ubiquitin Transfer: The E1~Ub complex recruits a cognate E2 conjugating enzyme, and ubiquitin is transferred from the E1 catalytic cysteine to the E2 catalytic cysteine via a transthioesterification reaction [12] [5].
Throughout this mechanism, the E1 enzyme maintains binding to two ubiquitin molecules simultaneously—one forming the thioester bond and a second that is adenylated but does not form a thioester complex. This secondary ubiquitin is believed to facilitate conformational changes during the transthioesterification process [11].
Conformational Dynamics in E1 Enzymes
E1 enzymes undergo remarkable conformational changes to fulfill their catalytic functions, with these dynamics being integral to their reaction cycle [13]. Structural studies comparing E1 structures in different states have revealed several key transitions:
Cys Domain Rotation and Active Site Remodeling
Following ubiquitin adenylation, the E1 Cys domain undergoes a 130-degree rotation (or closing) from an "open" to a "closed" conformation [12]. This substantial movement serves two critical purposes: First, it transits the E1 catalytic cysteine approximately 35 Å into the adenylation active site, bringing it into proximity with the C-terminal carbonyl carbon of ubiquitin. Second, it replaces half of the catalytic residues required for adenylation with residues necessary for thioester bond formation, a process termed active site remodeling [12]. After thioester bond formation and AMP release, the E1 Cys domain rotates back to its open configuration, reforming the adenylation active site to enable a subsequent round of adenylation [12].
UFD Unlocking for E2 Recruitment
The Ubiquitin-Fold Domain (UFD) also undergoes significant conformational changes, particularly a 120-degree rotation (or unlocking) observed in the Nedd8 E1 system [12]. This unlocking event uncovers a cryptic E2 binding surface on the E1 that facilitates contacts between the ubiquitin thioester and the E2 enzyme. Following transfer of ubiquitin from E1 to E2, the UFD is presumed to switch back to the locked conformation to facilitate E2~Ub thioester product release through a steric mechanism [12]. Notably, the UFD of Ub E1 adopts an unlocked configuration even in the absence of E2, likely due to distinct structural elements in its UFD linker region [12].
These coordinated movements ensure the precise spatial and temporal alignment of active sites required for efficient ubiquitin transfer while preventing premature or off-target reactions.
Structural Basis of E1-E2 Complex Formation
The transfer of ubiquitin from E1 to E2 represents a critical juncture in the ubiquitination cascade, requiring precise molecular recognition between these enzymes. Structural studies of a engineered Ub E1-E2(Ubc4)/Ub/ATP·Mg complex have provided unprecedented insights into this process [12].
The structure reveals that E2 recognition occurs through combinatorial binding involving both the E1 UFD and Cys domains [12]. This dual-site interaction brings the E1 and E2 catalytic cysteine residues into proximity for efficient thioester transfer. Mutational analysis coupled with thioester transfer assays demonstrates that both interfaces are essential for the transfer reaction [12].
Comparison of the E1-E2 complex structure with the E1/Ub/ATP·Mg structure alone reveals several key conformational changes in the E1 that enable productive complex formation:
- A 25-degree rotation of the UFD relative to its position in the E1/Ub/ATP·Mg structure.
- Displacement of E1 residues that normally mask the E1 catalytic cysteine, exposing it for interaction with the E2 active site.
- Rearrangements that create complementary surfaces on both the UFD and Cys domains for E2 binding.
This structural arrangement exhibits a degree of plasticity at the E1 UFD/E2 interface while maintaining a high degree of conservation at the E1 Cys domain/E2 interface, allowing a single E1 enzyme to interact with multiple E2 partners while maintaining catalytic fidelity [12].
Figure 1: Ubiquitin Activation and Transfer Cascade. This diagram illustrates the sequential enzymatic steps of the ubiquitination pathway, from initial E1-mediated ubiquitin activation to final substrate modification.
Experimental Protocols for Structural Analysis of E1 Enzymes
Protocol: Crystallographic Analysis of E1-E2 Complexes
This protocol outlines the methodology for determining the crystal structure of an E1-E2 complex, based on approaches used in the structural characterization of a Ub E1-E2(Ubc4)/Ub/ATP·Mg complex [12].
Materials and Reagents
- Purified recombinant E1 enzyme (e.g., S. pombe Uba1)
- Purified recombinant E2 conjugating enzyme (e.g., Ubc4)
- Ubiquitin
- ATP·Mg²⁺ solution
- Crystallization screening kits (e.g., Hampton Research)
- Cryoprotectants (e.g., glycerol, ethylene glycol)
- Liquid nitrogen for flash cooling
Procedure
Complex Stabilization and Preparation:
- To facilitate crystallization, engineer a disulfide bond between the E1 and E2 active sites to stabilize the transient complex [12].
- Incubate E1 enzyme with E2 enzyme, ubiquitin, and ATP·Mg²⁺ in a molar ratio of 1:1.2:1.5:5 for 30 minutes at 4°C.
- Purify the complex using size-exclusion chromatography to isolate homogeneous complex populations.
Crystallization:
- Set up crystallization trials using the sitting-drop vapor-diffusion method at 20°C.
- Mix 0.1 μL of protein complex (10-15 mg/mL) with 0.1 μL of reservoir solution.
- Optimize initial hits by systematic variation of pH, precipitant concentration, and temperature.
Data Collection and Processing:
- Flash-cool crystals in liquid nitrogen using appropriate cryoprotectant.
- Collect X-ray diffraction data at a synchrotron source (e.g., APS 24-ID-E or NSLS X29) [12].
- Process diffraction data using HKL-2000 or XDS, followed by scaling and merging with AIMLESS.
Structure Determination and Refinement:
- Solve the structure by molecular replacement using known E1 structures as search models.
- Conduct iterative cycles of manual building in Coot and refinement in Phenix or Refmac.
- Validate the final model using MolProbity.
Table 2: Crystallography Statistics for E1-E2 Complex Structure Determination
| Parameter | E1/Ub/ATP·Mg | E1-E2/Ub/ATP·Mg |
|---|---|---|
| PDB ID | 4II3 | 4II2 |
| Resolution (Å) | 2.9 | 2.2 |
| Space Group | P2₁2₁2₁ | - |
| R₍w₎ₑₑₑ / R𝒻ᵣₑₑ | 0.239 / 0.283 | - |
| Data Source | APS 24-ID-E | NSLS X29 |
Protocol: Molecular Dynamics Simulations of E1 Conformational Changes
Molecular dynamics (MD) simulations provide insights into the conformational dynamics of E1 enzymes that are difficult to capture through crystallography alone. This protocol is adapted from approaches used in studying PROTAC-induced protein dynamics [14] and structural bioinformatics [15].
Materials and Software
- High-performance computing cluster
- GROMACS MD simulation package [14] [15]
- AMBER force field (e.g., ff14SB) [15]
- Initial E1 enzyme structure from PDB
- Visualization software (e.g., PyMOL, VMD)
Procedure
System Preparation:
- Obtain the initial coordinates from crystallographic studies of E1 enzymes.
- Parameterize the system using the AMBER ff14SB force field for proteins [15].
- Solvate the protein in a cubic box with TIP3P water molecules, maintaining a minimum 1.0 nm distance between the protein and box edges [15].
- Add ions to neutralize the system and achieve physiological salt concentration.
Energy Minimization and Equilibration:
- Perform energy minimization using the steepest descent algorithm until convergence.
- Conduct equilibration in two phases: NVT (constant Number, Volume, Temperature) for 100 ps, followed by NPT (constant Number, Pressure, Temperature) for 100 ps.
Production Simulation:
- Run production MD simulation for 100 ns to 1 μs, depending on the system size and research question.
- Maintain constant temperature and pressure using coupling algorithms (e.g., Berendsen or Parrinello-Rahman).
- Save coordinates every 10 ps for subsequent analysis.
Trajectory Analysis:
- Calculate root-mean-square deviation (RMSD) to assess structural stability.
- Analyze root-mean-square fluctuation (RMSF) to identify flexible regions.
- Use principal component analysis (PCA) to identify essential dynamics and collective motions.
- Monitor specific distances and angles relevant to catalytic activity.
Figure 2: Molecular Dynamics Simulation Workflow. This diagram outlines the key steps in performing MD simulations to study E1 enzyme conformational dynamics.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for E1 Enzyme Structural Biology
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Recombinant Enzymes | S. pombe Uba1, Human UBE1 | Source of E1 for biochemical and structural studies [12] |
| E2 Conjugating Enzymes | Ubc4, Ube2L3 (UbcH7), Ube2W | E2 partners for transthiolation assays and complex formation [12] [5] |
| Chemical Reagents | ATP·Mg²⁺, Ubiquitin | Essential cofactors for E1 catalytic activity [12] |
| Crystallization Kits | Hampton Research Screens | Initial screening for crystal formation [12] |
| MD Simulation Software | GROMACS, AMBER | Studying E1 conformational dynamics [14] [15] |
| Structural Biology Tools | PyMOL, Coot, Phenix | Model building, refinement, and visualization [12] [15] |
Discussion and Research Applications
The structural insights into E1 enzyme mechanism and dynamics have far-reaching implications for both basic research and therapeutic development. Understanding E1 domain architecture and conformational changes provides a foundation for investigating pathological mechanisms in diseases associated with UPS dysfunction, such as cancer and neurodegenerative disorders [9] [10]. Notably, mutations in the UBE1 gene are associated with X-linked infantile spinal muscular atrophy (XL-SMA), likely due to disturbed complex formation with gigaxonin and impaired degradation of microtubule-associated proteins [11].
From a drug discovery perspective, the E1 enzyme represents a potential therapeutic target, though its broad specificity presents challenges for selective inhibition [10]. More promising approaches may involve targeting specific E1-E2 interactions or developing strategies that exploit the conformational dynamics of E1 enzymes. Recent advances in targeted protein degradation, particularly PROTACs (PROteolysis TArgeting Chimeras), rely on recruiting E3 ubiquitin ligases to target proteins, underscoring the therapeutic potential of modulating the ubiquitination cascade [14]. The structural principles governing E1-E2 interactions provide valuable insights for optimizing these novel therapeutic modalities.
Future research directions include further elucidation of E1 dynamics through advanced techniques such as cryo-electron microscopy and single-molecule studies, exploration of E1 interactions with the growing family of ubiquitin-like proteins, and development of small-molecule modulators that specifically target distinct conformational states of E1 enzymes.
Ubiquitin-conjugating (E2) enzymes serve as the crucial central hub in the E1-E2-E3 enzymatic cascade, functioning as more than mere passive carriers of ubiquitin (Ub). In the canonical pathway, the ubiquitin-activating enzyme (E1) activates Ub in an ATP-dependent manner to form a high-energy thioester intermediate. This activated Ub is then transferred to the catalytic cysteine residue of an E2 enzyme via a transthiolation reaction. Finally, working in concert with a ubiquitin ligase (E3), the E2~Ub thioester conjugate facilitates the attachment of Ub to substrate proteins, most commonly forming an isopeptide bond with the ε-amino group of a lysine residue [5] [16]. The human genome encodes approximately 40 E2 enzymes, which interact with a vastly larger repertoire of over 600 E3 ligases [16]. This numerical imbalance highlights a fundamental principle: E2s are highly versatile and conserved factors that must collaborate with diverse, cell-type-specific E3s to govern the ubiquitylation landscape [16]. While E3s are primarily responsible for substrate recognition, E2 enzymes play an indispensable and often underappreciated role in determining the specificity of lysine selection and the topology of polyubiquitin chains, thereby dictating the functional outcome for the modified substrate [16].
Diversity and Structural Determinants of E2 Enzymes
Conserved Core and Functional Adaptations
All E2 enzymes share a conserved core catalytic domain, known as the ubiquitin-conjugating (UBC) domain. This domain typically comprises ~150 amino acids folded into an α/β structure with four α-helices and a four-stranded β-sheet [5] [16]. Despite this common scaffold, E2s have evolved distinct functional adaptations. While most E2s consist solely of the UBC domain, many feature N- or C-terminal extensions that confer enzyme-specific functionality. These extensions can be intrinsically disordered or adopt secondary structures that contact the UBC core. A few E2s, such as Ube2K, even contain additional structured domains or are part of large multi-domain proteins like Ube2O or BIRC6 [5]. Based on these structural features, E2s can be classified into four groups: those with only a UBC domain, those with an additional N-terminal domain, those with a C-terminal extension, and those possessing both N- and C-terminal domains [17].
Classification and Unique Reactivities
The structural diversity of E2 enzymes underlies a remarkable spectrum of chemical reactivities that extend beyond canonical lysine modification, as summarized in Table 1.
Table 1: Diversity of E2 Enzyme Reactivity and Specificity
| E2 Enzyme | Primary Reactivity | Key Feature / Specificity | Bond Formed | E3 Dependence |
|---|---|---|---|---|
| UBE2D3 | Lysine (Canonical) | Works with many RING E3s [5] | Isopeptide (stable) | RING-type [5] |
| UBE2L3 (UbcH7) | Cysteine | Exclusive partner for HECT & RBR E3s [5] | Thioester (E3~Ub) | HECT & RBR-type [5] |
| UBE2W | N-terminal α-amine | Prefers disordered N-termini; monoubiquitylation [5] | Peptide bond (stable) | RING-type (e.g., BRCA1/BARD1) [5] |
| ATG3 | Phosphatidylethanolamine | Ubl (LC3/ATG8) conjugation in autophagy [5] | Amide bond (stable) | E3-like complex [5] |
| UBE2J2 | Serine, Lysine, Sugars | Targets hydroxyl groups; base-sensitive bond [5] [18] | Oxyester (labile) | RING-type (e.g., viral mK3) [5] |
| UBE2Q1/Q2 | Serine, Threonine, Sugars | Lacks canonical HPN triad; RWD domain [18] | Oxyester (labile) | E3-independent activity reported [18] |
These specialized E2s exemplify the adaptability of the UBC fold. For instance, UBE2W's unique ability to modify protein N-terminal is linked to its unusual dynamic C-terminal region, which recognizes and modifies disordered N-termini independently of substrate sequence [5]. The recently characterized UBE2Q family, which lacks the conserved HPN triad found in most E2s and possesses an extended N-terminal RWD domain, can ubiquitylate serine, threonine, and even sugar molecules like glucose, forming labile oxyester bonds [18]. This noncanonical activity was confirmed by the sensitivity of UBE2Q1 autoubiquitylation products to mild alkaline conditions and hydroxylamine treatment, but not to reducing agents, confirming the formation of ester bonds rather than isopeptide or thioester linkages [18].
E2 Enzymes as Determinants of Ubiquitin Chain Topology
The topology of polyubiquitin chains is a primary determinant of a modified protein's fate. E2 enzymes play a critical role in defining this topology, particularly when working with RING-type E3 ligases, which facilitate the direct transfer of ubiquitin from the E2 to the substrate [17]. The specific E2 involved in the reaction heavily influences which lysine residue on the acceptor ubiquitin is used for chain elongation.
Table 2: E2 Enzyme Influence on Ubiquitin Chain Topology and Functional Outcomes
| Ubiquitin Linkage | Role of E2 Enzymes | Primary Biological Function |
|---|---|---|
| Lys48-linked | Specific E2s (e.g., UBE2R1/Cdc34) dictate linkage [16] [17] | Proteasomal degradation; primary degradation signal [16] |
| Lys63-linked | Specific E2s (e.g., UBE2N/Ubc13) dictate linkage [16] | DNA damage response, signaling cascades, endocytosis [16] |
| Lys11-linked | Specific E2s (e.g., UBE2S) dictate linkage [16] | Cell cycle regulation; proteasomal degradation [16] |
| Lys27-linked | E2 guides linkage selection with E3 [16] | Innate immunity, T-cell activation, DNA damage response [16] |
| Lys29-linked | E2 guides linkage selection with E3 [16] | Promotes protein aggregation in NDDs, regulates Wnt/β-catenin signaling, autophagy [16] |
| Lys33-linked | E2 guides linkage selection with E3 [16] | Modulation of T-cell receptor signaling, protein trafficking, autophagy [16] |
| Lys6-linked | E2 guides linkage selection with E3 [16] | Protein stabilization, mitochondrial homeostasis [16] |
| Monoubiquitination | Specific E2s (e.g., UBE2W for N-terminus) [5] | Histone regulation, endocytosis, DNA repair [5] |
In contrast to the mechanism of RING E3s, HECT-domain E3 ligases form a transient thioester intermediate with ubiquitin, which allows them to exert greater control over the topology of the polyubiquitin chain, irrespective of the partnering E2 [17]. This division of labor underscores the intricate partnership between E2s and E3s in shaping the ubiquitin code.
Experimental Protocols for Studying E2 Function
Protocol 1: Assessing Intrinsic E2 Reactivity Using a MALDI-TOF Discharge Assay
Purpose: To directly characterize the intrinsic chemical reactivity of an E2~Ub thioester conjugate toward different nucleophilic amino acids or biomolecules, independent of an E3 ligase [5] [18].
Background: This assay bypasses complications of auto-ubiquitylation or E3-dependent assays to compare the fundamental reactivity of different E2~Ub conjugates. It has been pivotal in redefining the functionality of E2s like UBE2L3 and discovering noncanonical activities in UBE2Q1 and UBE2J2 [5] [18].
Workflow: The following diagram illustrates the key steps and decision points in the MALDI-TOF discharge assay.
Materials:
- Recombinant E2 Enzyme: Purified (e.g., via E. coli expression) [18].
- E1 Activating Enzyme: Recombinant, for E2~Ub charging.
- Ubiquitin: Wild-type and 15N-labeled for internal standard.
- Nucleophiles: Acetyl-lysine (Ac-K), acetyl-serine (Ac-S), acetyl-threonine (Ac-T), glycerol, glucose, etc. [18].
- MALDI Matrix: e.g., α-cyano-4-hydroxycinnamic acid (CHCA).
- MALDI-TOF Mass Spectrometer.
Procedure:
- E2~Ub Conjugate Formation: In a reaction buffer (e.g., 50 mM Tris-HCl, pH 7.5, 50 mM NaCl, 10 mM MgCl₂, 2 mM ATP), incubate E1 (100 nM), the E2 of interest (2.5 µM), and ubiquitin (10 µM) at 30°C for 30-60 minutes to form the E2~Ub thioester [18].
- Discharge Reaction: Add the nucleophile of interest (e.g., 50 mM final concentration) to the E2~Ub mixture. Incubate at 30°C for a defined time (e.g., 1 hour) [18].
- Reaction Quenching and Standard Addition: Stop the reaction by adding trifluoroacetic acid (TFA) to a final concentration of 0.1-0.5%. Add a known amount of 15N-labeled ubiquitin as an internal standard for quantification [18].
- MALDI-TOF MS Analysis: Mix the quenched reaction 1:1 with the MALDI matrix solution. Spot 1-2 µL onto the target plate and allow to dry. Acquire mass spectra in positive ion mode, focusing on the mass range for ubiquitin (≈8.5 kDa) and potential ubiquitin-adducts [18].
- Data Interpretation: Identify peaks corresponding to ubiquitin-adducts (e.g., Ub + Ac-S = 8.5 kDa + 89 Da). The presence of an adduct indicates the E2 can discharge ubiquitin onto that nucleophile. Use the 15N-ubiquitin standard for relative quantification between reactions [18].
Protocol 2: Characterizing Ubiquitin Chain Linkage Specificity
Purpose: To determine the topology of polyubiquitin chains synthesized by a specific E2 enzyme in conjunction with an E3 ligase.
Background: The nature of the polyubiquitin chain (e.g., Lys48 vs. Lys63) dictates the substrate's fate. This protocol uses linkage-specific antibodies and deubiquitinases (DUBs) to characterize chains formed in an in vitro ubiquitylation assay.
Workflow: The experimental setup for determining ubiquitin chain linkage is outlined below.
Materials:
- Enzymes: Recombinant E1, E2, E3 (RING-type for this assay).
- Ubiquitin: Wild-type; mutant ubiquitins (K48R, K63R) can be used for validation.
- Linkage-specific Reagents:
- Antibodies: Anti-K48-linkage specific Ub, Anti-K63-linkage specific Ub, etc.
- DUBs: OTUB1 (preferentially cleaves K48 linkages), AMSH (preferentially cleaves K63 linkages).
- SDS-PAGE and Western Blotting Equipment.
Procedure:
- In Vitro Ubiquitylation Reaction: Assemble reactions in ubiquitylation buffer (e.g., 50 mM Tris-HCl, pH 7.5, 50 mM NaCl, 5 mM MgCl₂, 2 mM ATP, 1 mM DTT) containing E1 (100 nM), E2 (1-5 µM), E3 (1-5 µM), and ubiquitin (50-100 µM). Incubate at 30°C for 1-3 hours.
- Analysis by Immunoblotting:
- Stop the reaction with SDS-PAGE loading buffer.
- Resolve proteins by SDS-PAGE and transfer to a PVDF membrane.
- Probe the membrane with linkage-specific ubiquitin antibodies. A positive signal indicates the formation of that specific chain linkage.
- Analysis by DUB Profiling:
- Split the completed ubiquitylation reaction into separate aliquots.
- To each aliquot, add a specific DUB (e.g., OTUB1 or AMSH) and incubate according to the DUB's optimal conditions.
- Analyze the reactions by anti-ubiquitin immunoblotting. The selective disappearance of high-molecular-weight smears upon treatment with a specific DUB indicates the presence of that linkage type in the synthesized chains.
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for E2 Enzyme Research
| Reagent Category | Specific Example | Function in Research |
|---|---|---|
| Recombinant E2 Enzymes | UBE2D3, UBE2L3, UBE2R1 (Cdc34), UBE2N/Ubc13, UBE2Q1 | Define intrinsic reactivity and linkage specificity; study enzyme mechanics [5] [18]. |
| Activity-Based Probes | E2~Ub thioester conjugates, UBE2Q1 autoubiquitylation complex | Trap and characterize transient enzymatic intermediates for structural studies (e.g., Cryo-EM) [19]. |
| Specialized Ubiquitin Mutants | Lysine-to-Arginine (K48R, K63R), 15N-labeled Ub | Determine chain linkage specificity (K-to-R); quantify ubiquitylation in MS assays (15N-Ub) [18]. |
| Linkage-Specific Antibodies | Anti-K48-Ub, Anti-K63-Ub | Detect specific polyubiquitin chain topologies in Western blotting and cellular imaging [16]. |
| Linkage-Specific DUBs | OTUB1 (K48-specific), AMSH (K63-specific) | Confirm chain linkage identity by selective enzymatic cleavage [16]. |
| Chemical Inhibitors | DHPO (UbcH5c inhibitor) [20] | Tool for probing E2 function in cells and animal models; potential therapeutic lead. |
| Noncanonical Nucleophiles | Acetyl-Serine, Acetyl-Threonine, Glucose | Probe for noncanonical E2 activity targeting hydroxyl groups [5] [18]. |
E2 conjugating enzymes are far from being simple middlemen in the ubiquitin cascade. They are sophisticated enzymes whose diversity, intrinsic reactivity, and partnership with E3 ligases are fundamental to determining the topology and function of the ubiquitin signal. The experimental protocols and tools detailed herein provide a framework for researchers to decipher the specific roles of E2s in biochemical pathways, cellular models, and disease contexts. A deep understanding of E2 enzyme mechanics and specificity is not only crucial for basic science but also opens up promising therapeutic avenues. The successful targeting of UbcH5c with the small molecule inhibitor DHPO, which suppressed pancreatic cancer growth and metastasis in preclinical models, validates E2 enzymes as druggable targets in oncology [20]. As research continues to unveil the complexities of E2 biology, particularly in areas like neurodevelopment and neurodegeneration where E2 dysfunction is increasingly implicated, the strategies outlined in this application note will be vital for driving discovery and therapeutic innovation [16] [17] [9].
Protein ubiquitination is a fundamental post-translational modification that regulates a vast array of cellular processes, including protein degradation, signal transduction, cell cycle progression, and DNA repair [21] [22]. This modification is executed through a sequential enzymatic cascade involving E1 (activating), E2 (conjugating), and E3 (ligating) enzymes [23]. The process initiates when the E1 enzyme activates ubiquitin in an ATP-dependent manner, forming a high-energy thioester bond [24] [25]. The activated ubiquitin is then transferred to an E2 enzyme, before an E3 ligase finally facilitates the attachment of ubiquitin to specific substrate proteins [9] [22].
The human genome encodes approximately 600 E3 ubiquitin ligases, which are categorized into three major families based on their catalytic mechanisms and structural features: RING (Really Interesting New Gene), HECT (Homologous to E6-AP C-Terminus), and RBR (RING-between-RING) [21] [9]. These E3 ligases confer specificity to the ubiquitination system by recognizing and binding to particular protein substrates, thereby enabling the precise regulation of cellular pathways. Dysregulation of E3 ligase function is implicated in numerous diseases, including cancer, neurodegenerative disorders, and neurodevelopmental conditions, making them attractive targets for therapeutic intervention [9] [22].
Catalytic Mechanisms of E3 Ligase Families
RING E3 Ligases: Scaffold-Mediated Direct Transfer
RING E3 ligases represent the largest family of ubiquitin ligases and function primarily as scaffolds that facilitate the direct transfer of ubiquitin from an E2 enzyme to a substrate protein [26] [9]. These enzymes contain a RING domain that binds the E2-ubiquitin conjugate while simultaneously interacting with the target substrate. This spatial positioning enables the direct attack of the E2-ubiquitin thioester bond by the substrate nucleophile (typically a lysine ε-amino group) in an aminolysis reaction [26]. RING domains typically form a cross-brace structure stabilized by Zn²⁺ ions, which is essential for their structural integrity and function [21].
A key mechanistic feature of RING E3 ligases is their ability to induce a closed conformation in the bound E2-ubiquitin conjugate, which activates the thioester bond for nucleophilic attack [26]. Unlike HECT and RBR E3s, RING ligases do not form a covalent thioester intermediate with ubiquitin during the transfer process. The RING family includes both monomeric enzymes and multimeric complexes, such as the SCF (Skp1-Cullin-F-box) complex, which expand the substrate recognition capabilities of the ubiquitin system [22].
HECT E3 Ligases: Two-Step Catalysis with Intermediate
HECT E3 ligases employ a two-step catalytic mechanism that involves a covalent ubiquitin intermediate [23] [21]. In the initial step, the HECT domain accepts ubiquitin from the E2 enzyme through a transthiolation reaction, forming a reactive thioester bond between the C-terminal glycine of ubiquitin and a conserved cysteine residue within the HECT domain [23] [9]. This E3-ubiquitin thioester intermediate is a defining characteristic of the HECT family.
In the second step, the HECT domain catalyzes the transfer of ubiquitin from this covalent intermediate to the target substrate [21]. This often requires a conformational change to properly position the active site cysteine relative to the substrate acceptor site [21]. Some HECT E3s exhibit linkage specificity, preferentially forming particular types of ubiquitin chains on their substrates, which determines the downstream consequences of the modification [22].
RBR E3 Ligases: Hybrid RING-HECT Mechanism
RBR E3 ligases represent a unique family that incorporates mechanistic elements from both RING and HECT-type enzymes, hence their classification as "RING/HECT hybrids" [26] [21]. These enzymes contain three canonical zinc-binding domains: RING1, IBR (In-Between-RING), and RING2, collectively known as the RBR module [26].
Similar to RING E3s, the RING1 domain of RBR ligases binds the E2-ubiquitin conjugate. However, instead of inducing a closed E2-ubiquitin conformation, RBRs stabilize an open conformation and align the active site cysteine in their RING2 domain with the E2-ubiquitin thioester [26]. This facilitates Ub transfer from the E2 to the RBR E3 in a transthiolation reaction (the first step). Subsequently, ubiquitin is transferred from the RING2 active site to the substrate in an aminolysis reaction (the second step), analogous to the mechanism of HECT E3s [26] [21].
Many RBR E3 ligases are subject to complex regulatory mechanisms, including autoinhibition and allosteric activation. For instance, Parkin requires phosphorylation by PINK1 and binding of phospho-ubiquitin for activation, while HOIP is activated by M1-linked di-ubiquitin [26]. This regulatory complexity allows RBR ligases to respond to specific cellular signals and conditions.
Table 1: Comparative Features of Major E3 Ligase Families
| Feature | RING E3 Ligases | HECT E3 Ligases | RBR E3 Ligases |
|---|---|---|---|
| Catalytic Mechanism | Direct transfer from E2 to substrate | Two-step via E3-ubiquitin thioester | Hybrid mechanism with E3-ubiquitin intermediate |
| Covalent Ub Intermediate | No | Yes (on HECT domain cysteine) | Yes (on RING2 domain cysteine) |
| E2-Ub Conformation | Closed conformation induced | Not specified | Open conformation stabilized |
| Representative Members | Cullin-RING ligases (CRLs), HDM2 | E6-AP, NEDD4 | Parkin, HOIP, HHARI |
| Regulatory Mechanisms | Substrate receptor exchange | Autoinhibition, localization | Multistep activation, allosteric regulation |
Quantitative Analysis of E3 Ligase Activation and Function
Understanding the kinetic and functional parameters of E3 ligases provides critical insights into their biological roles and regulatory mechanisms. The following table summarizes key quantitative data related to E3 ligase function and activation.
Table 2: Quantitative Parameters of E3 Ligase Function and Activation
| E3 Ligase/Parameter | Value/Measurement | Experimental Context | Significance |
|---|---|---|---|
| HOIL-1 Activation (M1-diUb) | EC₅₀ = 8 µM | E2-Ub discharge assay with UbcH7 | M1-linked diUb is a potent allosteric activator of HOIL-1 [26] |
| HOIL-1 Activation (K63-diUb) | EC₅₀ = 18 µM | E2-Ub discharge assay with UbcH7 | K63-linked diUb activates HOIL-1 with ~2-fold lower potency than M1-diUb [26] |
| RNF216 Activation | Specific for K63-diUb | E2-Ub discharge assay | Linkage-specific allosteric activation observed with both UbcH5B and UbcH7 [26] |
| Human E3 Ligase Count | ~600 enzymes | Genomic analysis | Vast substrate recognition capacity of the ubiquitin system [21] [22] |
| UBE1-UB Kd Increase | 58-fold | Arg72Leu UB mutation study | Demonstrates critical role of UB C-terminal residues in E1 recognition [24] |
| Phage Selection Enrichment | 350-fold | 8th round selection with Ube1/Uba6 | Indicates successful enrichment of catalytically active UB variants [24] |
Experimental Protocols for E3 Ligase Research
Protocol 1: E2-Ub Discharge Assay for RBR E3 Ligase Activity
Purpose: This assay measures the ability of RBR E3 ligases to catalyze the transfer of ubiquitin from an E2-Ub thioester to the E3 active site cysteine, which is particularly useful for studying RBR activation mechanisms [26].
Materials:
- Purified RBR E3 protein (e.g., HOIL-1 helix-RBR or RNF216 helix-RBR-helix)
- E2 enzyme (e.g., UbcH7 or UbcH5B)
- Ubiquitin
- ATP and Mg²⁺
- Reaction buffer: 50 mM Tris-HCl (pH 7.5), 50 mM NaCl, 1 mM DTT
- Allosteric activators (e.g., M1- or K63-linked di-ubiquitin for HOIL-1)
Procedure:
- E2-Ub Thioester Formation: Pre-form the E2-Ub thioester conjugate by incubating E2 enzyme with ubiquitin, E1 enzyme, and ATP in reaction buffer at 30°C for 10 minutes.
- Reaction Setup: Combine the pre-formed E2-Ub thioester with the RBR E3 ligase in the presence or absence of allosteric activators.
- Incubation: Allow the discharge reaction to proceed at 30°C for specified timepoints (typically 0-60 minutes).
- Termination and Analysis: Stop the reaction by adding non-reducing SDS-PAGE sample buffer. Analyze samples by non-reducing SDS-PAGE and western blotting using anti-ubiquitin antibodies to visualize E2-Ub discharge and E3-Ub formation.
Applications: This protocol can be used to characterize allosteric activation of RBR E3 ligases by different ubiquitin linkages, determine activation kinetics (EC₅₀ values), and investigate regulatory mechanisms [26].
Protocol 2: Phage Display Profiling of Ubiquitin Variants
Purpose: To identify ubiquitin variants with altered C-terminal sequences that maintain reactivity with E1 and E2 enzymes but may be blocked in E3 transfer, useful for studying sequence requirements at different cascade stages [24].
Materials:
- Phage-displayed UB library with randomized C-terminal residues (positions 71-75)
- Biotinylated E1 enzymes (Ube1 or Uba6)
- Streptavidin-coated plates
- Mg-ATP
- Elution buffer (DTT-containing)
- E2 enzymes (e.g., UbcH7, UbcH5a) for secondary screening
Procedure:
- Immobilization: Coat streptavidin plates with biotinylated E1 enzymes.
- Phage Selection: Incubate the UB phage library with immobilized E1 in the presence of Mg-ATP to enable thioester formation.
- Washing: Remove non-specifically bound phage through extensive washing.
- Elution: Release specifically bound phage by cleaving thioester linkages with DTT.
- Amplification and Iteration: Amplify eluted phage and subject to additional rounds of selection with increasing stringency.
- Characterization: Sequence enriched UB variants and test their reactivity with E2 and E3 enzymes.
Applications: This approach has revealed that while E1 enzymes show substantial promiscuity toward UB C-terminal sequences, E3-mediated transfer has stricter requirements [24]. It can also identify UB mutants resistant to deubiquitinating enzymes (DUBs).
Research Reagent Solutions for E3 Ligase Studies
Table 3: Essential Research Reagents for E3 Ligase Investigations
| Reagent/Category | Specific Examples | Function/Application | Research Context |
|---|---|---|---|
| Activity-Based Probes | UbDha [25] | Cascading ABP that irreversibly traps active site cysteines in E1, E2, and E3 enzymes | Monitoring catalysis along Ub/Ubl cascades; proteome-wide profiling |
| E1 Inhibitors | PYR-41, NSC 624206 [27] | Cell-permeable inhibitors of ubiquitin E1 enzymes | General inhibition of ubiquitin cascade; studying E1 function |
| E2-Ub Conjugates | UbcH7(C86K)-Ub [26] | Stable, non-reactive E2-Ub conjugate mimicking Ub-loaded E2 | ITC binding studies with E3 ligases; structural biology |
| Allosteric Activators | M1-linked diUb, K63-linked diUb [26] | Specific ubiquitin linkages that activate RBR E3 ligases | Studying RBR regulation mechanisms (e.g., HOIL-1, RNF216) |
| Engineered UB/E1/E2 Pairs | xUB-xE1, xE1-xE2 [28] | Orthogonal ubiquitin transfer components | Mapping substrate specificity of individual E3s; OUT cascades |
| Stable E3-Ub Intermediates | E3-Ub oxyester/thioster mimics | Structural and biochemical studies of HECT/RBR mechanisms | Circumventing need for active site mutagenesis [25] |
Visualization of E3 Ligase Mechanisms and Experimental Approaches
The Ubiquitin Cascade and E3 Ligase Mechanisms
Experimental Workflow for E3 Ligase Activation Studies
The ubiquitin-proteasome system (UPS) represents a crucial regulatory mechanism that controls virtually all aspects of eukaryotic cell biology through the covalent attachment of a small protein modifier, ubiquitin, to substrate proteins [29] [30]. This post-translational modification operates through a sequential enzymatic cascade involving E1 (activating), E2 (conjugating), and E3 (ligating) enzymes, culminating in the specific transfer of ubiquitin to lysine residues on target proteins [31] [2]. The complexity of ubiquitin signaling emerges from the ability of ubiquitin itself to become ubiquitinated on any of its seven lysine residues (K6, K11, K27, K29, K33, K48, K63) or its N-terminal methionine (M1), giving rise to structurally and functionally distinct polyubiquitin chains that constitute a sophisticated "ubiquitin code" [29]. The specificity of this code is determined by the combinatorial actions of E2 and E3 enzymes, while deubiquitinating enzymes (DUBs) provide reversibility, together enabling dynamic control of protein fate [29] [2]. This application note examines contemporary methodologies for deciphering linkage-specific ubiquitination dynamics, with particular emphasis on quantitative kinetic analyses and regulatory mechanisms governing the ubiquitination cascade.
The Ubiquitin Enzymatic Cascade: Mechanism and Specificity
The ubiquitination pathway initiates with E1 ubiquitin-activating enzymes, which activate ubiquitin in an ATP-dependent manner through the formation of a high-energy thioester bond [31] [2]. The human genome encodes two E1 enzymes that serve as the entry point for the ubiquitination cascade [2]. Activated ubiquitin is subsequently transferred to approximately 38 E2 ubiquitin-conjugating enzymes, which also form thioester linkages with ubiquitin [32] [2]. The final transfer to substrate proteins is facilitated by more than 600 E3 ubiquitin ligases, which provide substrate specificity through direct recognition of target proteins [29] [2].
E3 ubiquitin ligases fall into two major mechanistic classes: RING (Really Interesting New Gene) and HECT (Homologous to E6-AP C-Terminus) ligases [2] [30]. RING E3 ligases function as scaffolds that simultaneously bind E2~Ub complexes and substrates, facilitating direct ubiquitin transfer without forming a covalent intermediate [31]. In contrast, HECT E3 ligases form a transient thioester intermediate with ubiquitin before catalyzing its transfer to the substrate [2]. This enzymatic cascade can result in monoubiquitination, multi-monoubiquitination, or polyubiquitination, with different chain linkages conferring distinct functional outcomes [29] [30]. For instance, K48-linked chains typically target substrates for proteasomal degradation, while K63-linked chains regulate non-proteolytic processes including protein trafficking, DNA repair, and inflammatory signaling [29] [30].
Figure 1: The Ubiquitin Enzymatic Cascade. Ubiquitin is activated by E1 in an ATP-dependent process, transferred to E2, and finally conjugated to substrate proteins by E3 ligases. Different polyubiquitin chain linkages determine distinct functional outcomes for the modified substrate.
Quantitative Analysis of Linkage-Specific Ubiquitination Kinetics
Recent methodological advances have enabled precise quantification of ubiquitination dynamics with high temporal resolution. The development of light-activatable ubiquitin variants has been particularly transformative, allowing researchers to monitor linkage-specific polyubiquitin chain formation kinetics on minute timescales [33].
Light-Activatable Ubiquitin System
The innovative incorporation of photocaged lysine (pcK) at specific positions within ubiquitin enables optical control of ubiquitin chain extension [33]. This approach involves expressing ubiquitin variants bearing a single genetically encoded pcK residue at K11, K48, or K63 sites within a ubiquitin K0 background (which contains lysine-to-arginine substitutions at all other ubiquitination sites) [33]. These variants are expressed at low levels in HEK293T cells to create a minimal, trackable ubiquitin subpopulation that minimally perturbs the endogenous UPS. Upon irradiation with 365 nm light for 4 minutes, the photocaging group is removed, initiating synchronous linkage-specific ubiquitination that can be monitored by SDS-PAGE and anti-myc immunoblotting [33].
Table 1: Kinetic Parameters for Linkage-Specific Polyubiquitin Chain Formation
| Ubiquitin Linkage | Activation Method | Time Scale | Key Experimental Conditions | Functional Significance |
|---|---|---|---|---|
| K48-linked chains | Light activation (365 nm, 4 min) | Minute-scale kinetics | MG132 proteasome inhibition | Primary degradation signal |
| K63-linked chains | Light activation (365 nm, 4 min) | Minute-scale kinetics | MG132 proteasome inhibition | Cell signaling, DNA repair |
| K11-linked chains | Light activation (365 nm, 4 min) | Minute-scale kinetics | MG132 proteasome inhibition | Cell cycle regulation, ERAD |
| ERAD substrates | Reconstituted with purified components | Variable, lipid-dependent | Defined membrane composition | Lipid homeostasis |
Real-Time Monitoring of Ubiquitination Cascades
The UPS-CONFOCAL fluorescence nanoscanning (UPS-CONA) technique provides a complementary approach for monitoring ubiquitination kinetics in real time [32]. This bead-based confocal imaging method immobilizes a substrate or enzyme of interest on polymer micro-beads and incubates them with fluorescently labeled ubiquitin in solution. Upon ubiquitin conjugation, fluorescence emission intensity is detected by confocal imaging through the equatorial cross-section of the beads, appearing as a fluorescent ring [32]. The method enables quantitative tracking of E1, E2, and HECT E3 activities either individually or in integrated cascade reactions with high sensitivity and temporal resolution.
Regulation of Ubiquitination by Membrane Lipid Composition
Emerging research demonstrates that the ubiquitination cascade functions not only as a protein degradation pathway but also as a sensor of cellular membrane properties. Recent findings reveal that the ER-associated degradation (ERAD) pathway integrates multiple lipid signals through regulation of both E2 and E3 enzymes [7] [34].
UBE2J2 as a Lipid Packing Sensor
The membrane-anchored E2 enzyme UBE2J2 exhibits remarkable sensitivity to membrane lipid composition [7]. In loosely packed ER-like membranes characterized by low saturated fatty acyl chain and cholesterol content, UBE2J2 assumes an inactive conformation due to membrane association that impedes ubiquitin loading by E1 [7]. Conversely, in tightly packed membranes with higher saturation levels, UBE2J2 adopts an active conformation that promotes interaction with E1 and subsequent ubiquitin transfer. This lipid packing-dependent regulation of UBE2J2 activity subsequently directs ubiquitin transfer by multiple E3 ligases including RNF145, MARCHF6, and RNF139, affecting both auto-ubiquitination and substrate ubiquitination [7].
Cholesterol Sensing by RNF145 E3 Ligase
The RNF145 E3 ligase directly senses cholesterol levels through a sterol-sensing domain, which modulates its oligomerization state and catalytic activity [7]. Elevated cholesterol concentrations promote RNF145 auto-ubiquitination and destabilization, creating a feedback mechanism that adjusts ERAD activity in response to membrane composition [7]. This dual regulation at both E2 and E3 levels enables the ERAD pathway to integrate multiple lipid signals and maintain ER membrane homeostasis.
Figure 2: Lipid-Dependent Regulation of the ERAD Ubiquitination Cascade. Membrane lipid composition directly modulates UBE2J2 E2 enzyme activity, with tightly packed membranes promoting active conformation. Cholesterol additionally regulates RNF145 E3 ligase stability through direct sensing and auto-ubiquitination.
Experimental Protocols
Protocol 1: Monitoring Linkage-Specific Ubiquitination Kinetics Using Light-Activatable Ubiquitin
Purpose: To quantitatively analyze minute-scale ubiquitination kinetics for specific ubiquitin linkages (K11, K48, K63) following optical activation [33].
Materials:
- HEK293T cell line
- Ubiquitin variant plasmids (Ub K0 background with amber codon at K11, K48, or K63)
- Methanosarcina mazei pyrrolysyl-tRNA-synthetase pair (pcKRS/tRNAPyl)
- Photocaged lysine (pcK, 0.32 mM)
- MG132 proteasome inhibitor (25 µM)
- UV light source (365 nm)
- Anti-myc antibody for immunoblotting
- Lysis buffer (RIPA buffer with protease inhibitors)
Procedure:
- Co-transfect HEK293T cells with pcKRS/tRNAPyl plasmids and ubiquitin variant plasmids containing amber codons at desired positions.
- Culture cells for 24 hours in medium supplemented with 0.32 mM pcK to enable incorporation of photocaged lysine.
- Replace medium with warm DPBS lacking pcK to terminate expression of photocaged ubiquitin.
- Irradiate cells with 365 nm light for 4 minutes to activate ubiquitin chain extension.
- Incubate cells in complete medium containing 25 µM MG132 (to uncouple ubiquitination from proteasomal degradation).
- Harvest cells at specific time points (0, 2, 5, 10, 30, 60, 120, 360 minutes post-irradiation).
- Lyse cells and isolate whole proteomes.
- Analyze ubiquitination by SDS-PAGE and anti-myc immunoblotting.
- Quantify high-molecular-weight smears representing polyubiquitinated proteins using densitometry.
Validation: Include controls expressing non-amber Ub K48 or K0 to confirm light-dependent responses are specific to the photocaged system [33].
Protocol 2: Real-Time Tracking of Ubiquitination Using UPS-CONA
Purpose: To monitor ubiquitination enzyme activities in real time using confocal fluorescence nanoscanning [32].
Materials:
- Ni2+NTA agarose beads (100-120 μm diameter)
- His6-tagged enzyme of interest (E1, E2, or HECT E3)
- Cy5-labeled ubiquitin (Cy5-Ub)
- Untagged partner enzymes (as required for cascade reactions)
- Ubiquitination buffer (50 mM Tris-HCl pH 7.5, 50 mM NaCl, 5 mM MgCl2, 2 mM ATP)
- Confocal fluorescence microscope with scanning stage
- 384-well plate
Procedure:
- Immobilize His6-tagged enzyme on Ni2+NTA agarose beads by incubating for 30 minutes at 4°C.
- Transfer beads to 384-well plate to form a monolayer.
- For E1 activity assays: Incubate immobilized E1 with Cy5-Ub in ubiquitination buffer.
- For E2 activity assays: Incubate immobilized E2 with Cy5-Ub and untagged E1 in ubiquitination buffer.
- For HECT E3 activity assays: Incubate immobilized E3 with Cy5-Ub, untagged E1, and appropriate E2 in ubiquitination buffer.
- For complete cascade: Immobilize substrate protein and incubate with Cy5-Ub, E1, E2, and E3 in solution.
- Monitor Cy5 fluorescence emission at the bead periphery by confocal imaging at regular intervals (e.g., every 2-5 minutes).
- Quantify fluorescence intensity using image analysis software.
- Include control reactions without ATP to account for non-specific ubiquitin binding.
Applications: This protocol can be adapted for inhibitor screening by including small molecule compounds and comparing ubiquitination kinetics to DMSO controls [32].
Table 2: Essential Research Reagents for Ubiquitination Cascade Studies
| Reagent Category | Specific Examples | Function/Application | Key Features |
|---|---|---|---|
| Engineered Ubiquitin Variants | Ubiquitin K0 (lysine-less); Photocaged lysine ubiquitin mutants | Linkage-specific ubiquitination studies; Optical control of ubiquitination | Prevents non-specific chain formation; Enables temporal precision [33] |
| Chemical Inhibitors | MG132 (proteasome inhibitor); PYR-41 (E1 inhibitor); BAY 11-7082 (E2 inhibitor) | Pathway perturbation; Enzyme mechanism studies | Tool compounds for dissecting cascade functions [33] [32] [2] |
| Detection Reagents | Cy5-labeled ubiquitin; Anti-myc antibody; OtUBD enrichment reagent | Visualization and quantification of ubiquitination | Enables real-time monitoring and specific detection [33] [32] |
| Reconstitution Systems | Purified ERAD factors; Liposomes of defined lipid composition | Lipid-protein interaction studies; In vitro ubiquitination | Controlled membrane environment for mechanistic studies [7] |
| Engineered Enzymes | Uba1-VHH05 nanobody fusion; Tagged E2 enzymes | Selective ubiquitin transfer to defined E2s | Dissection of E2-specific functions [35] |
The intricate regulation of the ubiquitin code through linkage-specific chain assembly represents a fundamental mechanism controlling cellular protein homeostasis. The methodologies outlined in this application note—including light-activatable ubiquitin for minute-scale kinetic analyses, real-time UPS-CONA monitoring, and reconstituted lipid-ubiquitination systems—provide powerful tools for deciphering this complex regulatory landscape. These approaches enable researchers to move beyond static observations to dynamic analyses of ubiquitination cascades, revealing how temporal control, subcellular localization, and environmental factors integrate to determine functional outcomes. Continued refinement of these technologies will undoubtedly yield new insights into ubiquitin-mediated regulation and create novel therapeutic opportunities for manipulating the ubiquitin code in disease contexts.
Advanced Methodologies for Ubiquitination Analysis and Therapeutic Targeting
Mass Spectrometry-Based Proteomics for Global Ubiquitination Site Mapping
Protein ubiquitination, a critical post-translational modification, is orchestrated by a sequential enzymatic cascade involving E1 (activating), E2 (conjugating), and E3 (ligating) enzymes [36]. This system regulates diverse cellular processes including protein degradation, apoptosis, and DNA repair. The E1 enzyme initiates the cascade through a molecular choreography involving ubiquitin adenylation and thiolation, forming a E1~Ub thioester intermediate before transthioesterification to a cognate E2 enzyme [36]. E3 ligases then facilitate the transfer of ubiquitin from E2 to specific lysine residues on substrate proteins, creating isopeptide bonds. Understanding the global ubiquitination landscape requires sophisticated proteomic approaches that can capture the spatial and temporal dynamics of this system within native cellular environments.
Recent advances in mass spectrometry (MS) have revolutionized our ability to map ubiquitination sites globally, moving beyond purified systems to in-situ analyses that preserve physiological interactions and compartment-specific specialization [37]. This protocol details comprehensive methodologies for global ubiquitination site mapping, integrating cross-linking MS, top-down fragmentation, and computational analysis to provide unprecedented insights into the ubiquitin code.
Current Methodologies in Ubiquitination Proteomics
In-Situ Cross-Linking Mass Spectrometry (XL-MS)
Workflow Overview: In-situ XL-MS preserves protein complexes in their native cellular environment using cell-permeable cross-linkers, enabling analysis of proteasomal interactions and ubiquitination patterns within intact cells [37].
Detailed Protocol:
- Cross-Linking: Apply cell-permeable, trifunctional cross-linker bis(succinimidyl) with propargyl tag (BSP) to cultured cells at 25-50 μM concentration for 20-30 minutes at 37°C. BSP exhibits low cellular toxicity and diffuses into nuclear and cytoplasmic compartments within minutes [37].
- Subcellular Fractionation: Lyse cells and separate nuclear and cytoplasmic fractions using differential centrifugation. Validate separation efficiency by immunoblotting for compartment-specific markers.
- Affinity Purification: Isubiquitinated proteins or proteasomal complexes using specific antibodies or tagged proteins (e.g., biotin-tagged Rpn11) [37].
- Cross-link Enrichment: Use acid-hydrolyzable click chemistry with streptavidin affinity purification to enrich cross-linked peptides. Remove biotin moiety during liquid chromatography in acidic buffer.
- Mass Spectrometry Analysis: Digest enriched proteins with trypsin/Lys-C and analyze using high-resolution tandem MS (e.g., Q-Exactive Orbitrap). Use stepped collision-induced dissociation (CID) or electron-transfer/higher-energy collision dissociation (EThcD) for cross-linked peptide identification.
- Data Processing: Search MS data against human protein databases using dedicated XL-MS software (e.g., MaxQuant, pLink). Apply false discovery rate (FDR) threshold of <1% at peptide and protein levels.
Table 1: Key Reagents for In-Situ Cross-Linking MS
| Research Reagent | Function in Protocol |
|---|---|
| BSP Cross-linker | Cell-permeable trifunctional reagent (NHS ester) for protein complex stabilization in living cells |
| Cy5 Dye | Fluorescence probe for cross-linker visualization via click chemistry |
| Streptavidin Beads | Affinity matrix for biotinylated peptide enrichment |
| Acid-hydrolyzable Click Reagent | Reversible biotin tag for cross-linked peptide purification |
| Rpn11-Biotin Expressing Cell Line | Stable cell line for proteasomal complex affinity purification |
Computational Top-Down Mass Spectrometry
Workflow Overview: Top-down MS analyzes intact ubiquitinated proteins, preserving information on ubiquitin chain topology and modification sites simultaneously [38].
Detailed Protocol:
- Sample Preparation: Isubiquitinated proteins under non-denaturing conditions. For complex substrates, use selective Asp-N proteolysis to digest substrate proteins while preserving intact ubiquitin chains [38].
- Intact Protein Separation: Separate ubiquitinated proteoforms using reversed-phase liquid chromatography (e.g., C4 column) with shallow acetonitrile gradient (0.5-1.0%/min) in 0.1% formic acid.
- Top-Down MS Analysis: Introduce intact proteins into high-resolution mass spectrometer (e.g., Orbitrap Eclipse Tribrid) using nano-electrospray ionization. Use intact protein mode (m/z 600-2000) for MS1 detection.
- Fragmentation: Isolate specific charge states for tandem MS using higher-energy collisional dissociation (HCD) at 20-25% normalized collision energy. Use ultraviolet photodissociation (UVPD) for improved ubiquitin chain topology analysis.
- Computational Analysis: Process data with UbqTop platform utilizing Bayesian-like scoring algorithm to predict ubiquitin chain topology from fragmentation data [38]. Map modification sites and chain architecture simultaneously.
Table 2: Performance Metrics of Ubiquitination Mapping Methods
| Method Parameter | In-Situ XL-MS | Computational Top-Down MS |
|---|---|---|
| Ubiquitination Sites Identified | 993 unique cross-links within proteasomal subunits [37] | Simultaneous site and chain architecture determination [38] |
| Spatial Resolution | Compartment-specific (nuclear vs. cytoplasmic) | Whole-cell lysates |
| Key Advantage | Preserves native interactions and structural heterogeneity | Resolves isomeric chains and branched architectures |
| Throughput | Moderate (requires fractionation) | High (automated computational pipeline) |
| Structural Information | Distance restraints (20-30 Å) for modeling | Complete proteoform characterization |
Key Applications and Research Insights
Compartment-Specific Proteasomal Specialization
In-situ XL-MS with subcellular fractionation reveals extensive compositional and conformational heterogeneity between nuclear and cytoplasmic proteasomes. Nuclear proteasomes exhibit distinct interactomes including specialization for processes like transcriptional regulation and DNA repair, while cytoplasmic proteasomes show different ubiquitin-binding patterns and dynamic states [37]. This compartment-specific architecture fundamentally regulates proteasome function in maintaining proteostasis.
Novel Proteasome-Interacting Proteins
Recent in-situ studies have identified previously unreported proteasome-interacting proteins, including deubiquitinase USP15 and a hybrid proteasome variant where translation initiation factor EIF3M substitutes for subunit Rpn9 [37]. These findings expand our understanding of proteasomal regulation and its connections to other cellular processes.
Non-Canonical Ubiquitination
Emerging evidence reveals that ubiquitination extends beyond proteins to include drug-like molecules, suggesting a previously unrecognized regulatory mechanism with potential therapeutic applications [39]. This non-canonical ubiquitination offers versatile chemical tools for probing protein regulation and developing new therapeutics.
Visualization of Experimental Workflows
In-Situ XL-MS Workflow for Ubiquitination Mapping
Ubiquitin Cascade and Top-Down MS Analysis
Mass spectrometry-based proteomics has transformed our understanding of the ubiquitin system, moving from simplified in vitro models to comprehensive analyses of its native architecture and dynamics. The integration of in-situ cross-linking MS and computational top-down approaches provides powerful tools for mapping ubiquitination sites globally while preserving critical structural information about chain topology and cellular compartmentalization. These methodologies, framed within the context of the E1-E2-E3 enzymatic cascade, offer unprecedented insights into proteasomal heterogeneity, novel interacting partners, and the complex regulation of cellular proteostasis. As these technologies continue to evolve, they will undoubtedly yield new discoveries in ubiquitin biology and create opportunities for therapeutic intervention in ubiquitination-related diseases.
Ubiquitin Tagging and Antibody-Based Enrichment Strategies for Substrate Identification
Protein ubiquitination is an essential post-translational modification that regulates a vast array of cellular processes, including protein homeostasis, cell cycle progression, DNA damage response, and immune signaling [40] [41]. This modification is executed through a well-defined enzymatic cascade involving E1 (activating), E2 (conjugating), and E3 (ligating) enzymes [23]. The human genome encodes approximately 2 E1s, 50 E2s, and over 600 E3s, which together create a complex network of ubiquitination pathways with immense substrate specificity [40] [42]. The canonical ubiquitination mechanism involves the formation of a thioester bond between the catalytic cysteine residue of E1 and the C-terminal glycine of ubiquitin, followed by transfer to a catalytic cysteine on E2, and finally conjugation to the ε-amino group of a lysine residue on the substrate protein, typically facilitated by an E3 ligase [23] [42]. Understanding the specific substrates of individual E2-E3 pairs remains a significant challenge in the field, necessitating the development of sophisticated enrichment and identification strategies.
Methodological Challenges in Substrate Identification
Identifying specific ubiquitin ligase-substrate relationships has proven challenging due to several intrinsic biological and technical hurdles. Most ubiquitinated substrates are rapidly degraded by the proteasome or processed by deubiquitinating enzymes (DUBs), resulting in transient signal that is difficult to capture [40]. Additionally, many E3-substrate interactions are weak and transient, making them difficult to isolate by conventional immunoprecipitation methods [40]. The tremendous cross-reactivity within the ubiquitin system further complicates assignment of specific relationships, as E2 enzymes can interact with multiple E3s, and E3s can recognize numerous substrates [42]. Finally, the abundance of endogenous ubiquitinated substrates is often low compared to the total cellular proteome, requiring highly sensitive enrichment and detection methods [41]. These challenges have driven the development of innovative antibody-based and protein engineering approaches to stabilize, enrich, and identify ubiquitinated substrates.
Antibody-Based Enrichment Strategies
DiGly Antibody for Lysine Ubiquitination
The development of antibodies recognizing the diglycine (Gly-Gly) remnant left on trypsinized lysine residues has revolutionized the study of ubiquitination. After trypsin digestion of ubiquitinated proteins, a Gly-Gly moiety remains attached to the modified lysine residue, which serves as a signature for ubiquitination sites. Antibodies specifically recognizing this K-ε-GG motif have enabled global profiling of ubiquitination sites through mass spectrometry-based proteomics [40] [41]. This approach has identified thousands of ubiquitination sites across the proteome, but it has limitations for studying specific E3 ligase substrates, as overexpression of a particular ubiquitin ligase combined with proteasome inhibition often does not sufficiently increase diGly peptide levels above background due to ongoing deubiquitination [40].
GGX Antibodies for N-Terminal Ubiquitination
Recent work has expanded antibody tools to include reagents that specifically recognize N-terminal ubiquitination. Unlike canonical ubiquitination, N-terminal ubiquitination involves conjugation of ubiquitin to the α-amino group of a protein's N-terminus, which is catalyzed by specific E2/E3 pairs such as UBE2W [41]. Upon trypsin digestion, N-terminally ubiquitinated proteins generate peptides with a diglycine sequence at their N-terminus (GGX peptides). Researchers have developed monoclonal antibodies (1C7, 2B12, 2E9, and 2H2) that selectively recognize these linear GGX peptides without cross-reacting with isopeptide-linked K-ε-GG peptides [41]. Structural studies of the 1C7 Fab bound to a Gly-Gly-Met peptide revealed the molecular basis for this exquisite selectivity, showing binding in a pocket at the interface of the heavy and light chain complementarity-determining regions [41]. This antibody toolkit enables specific enrichment and global profiling of endogenous N-terminal ubiquitination sites, revealing previously uncharacterized UBE2W substrates including UCHL1 and UCHL5, where N-terminal ubiquitination modulates deubiquitinase activity rather than targeting proteins for degradation [41].
Table: Comparison of Antibody-Based Enrichment Strategies
| Antibody Type | Target Epitope | Ubiquitination Type | Key Applications | Limitations |
|---|---|---|---|---|
| K-ε-GG Antibody | Isopeptide-linked diglycine on lysine | Canonical lysine ubiquitination | Global ubiquitome profiling; identification of degradation signals | Cannot distinguish E3-specific substrates; high background |
| GGX Antibodies | Linear N-terminal diglycine (GGX) | N-terminal ubiquitination | Identification of UBE2W substrates; study of non-degradative ubiquitination | Limited to specific E2/E3 pairs; lower abundance |
| TR-TUBE | Native polyubiquitin chains | All ubiquitin linkages | Stabilization of ubiquitinated substrates; E3 activity assessment | Requires genetic manipulation; potential cellular toxicity |
Engineered Protein Systems for Substrate Identification
TR-TUBE for Substrate Stabilization
The tandem ubiquitin-binding entity (TUBE) system represents a protein engineering approach to overcome challenges in substrate identification. TUBEs are engineered proteins containing multiple ubiquitin-associated domains that exhibit high affinity for polyubiquitin chains. Researchers have developed a trypsin-resistant TR-TUBE for expression in mammalian cells, where it protects polyubiquitin chains on substrates from deubiquitinating enzymes and proteasomal degradation by physically masking the ubiquitin chains [40]. When co-expressed with a specific ubiquitin ligase, TR-TUBE stabilizes ubiquitinated substrates, allowing their accumulation to detectable levels. This system enables quantitative detection of ubiquitin ligase activity toward specific substrates without requiring proteasome inhibition [40]. For example, co-expression of TR-TUBE with the F-box protein Skp2 increased detectable ubiquitination of its substrate p27, even without exogenous p27 overexpression or MG132 treatment [40].
Orthogonal Ubiquitin Transfer (OUT) System
The orthogonal ubiquitin transfer (OUT) cascade represents an ambitious protein engineering strategy to completely isolate specific ubiquitination pathways from the endogenous network. This system involves engineering mutually specific pairs of ubiquitin (xUB), E1 (xE1), E2 (xE2), and E3 (xE3) enzymes that function exclusively with each other without cross-talk with native components [42]. Using structure-based design and phage display, researchers have successfully engineered xUB-xE1 pairs based on the "bump-and-hole" strategy, where mutations are introduced into the E1 adenylation domain (e.g., Q576R, D591R, E594R in yeast Uba1) to disrupt binding to wild-type ubiquitin, followed by complementary mutations in ubiquitin (e.g., R42E, R72E) to restore binding to the engineered E1 [42]. Similarly, specific xE1-xE2 pairs were created by engineering the E1 ubiquitin fold domain (UFD) to block interaction with native E2s, followed by phage selection to identify compatible E2 mutations that restore binding to xE1 [42]. The OUT system allows exclusive transfer of affinity-tagged xUB to the substrate proteins of a specific xE3, enabling unambiguous identification of E3 substrates through affinity purification and mass spectrometry.
Table: Engineered Components for Orthogonal Ubiquitin Transfer
| Component | Engineering Strategy | Key Mutations | Function |
|---|---|---|---|
| xUB | Reverse bump-and-hole | R42E, R72E | Activated only by xE1; contains affinity tag for purification |
| xE1(A) | Adenylation domain mutations | Q576R, D591R, E594R | Specifically activates xUB but not wild-type UB |
| xE1(UFD) | Ubiquitin fold domain mutations | E1004K, D1014K, E1016K | Specifically transfers UB to xE2 but not wild-type E2s |
| xE2 | Phage selection from libraries | H1 helix modifications | Accepts UB only from xE1; transfers specifically to xE3 |
Integrated Experimental Protocols
Protocol 1: Substrate Identification Using TR-TUBE and diGly Antibody
This integrated methodology combines the stabilization power of TR-TUBE with the specificity of diGly antibody enrichment to identify substrates of specific ubiquitin ligases.
- Plasmid Transfection: Co-transfect cells with plasmids encoding FLAG-tagged TR-TUBE and the ubiquitin ligase of interest (e.g., FBXO21) using standard transfection methods. Include controls expressing ubiquitin-binding-deficient TR-TUBE mutant.
- Cell Harvest and Lysis: Harvest cells 48 hours post-transfection. Lyse cells in NP-40 lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% NP-40) supplemented with protease inhibitors and N-ethylmaleimide to inhibit deubiquitinating enzymes.
- Immunoprecipitation: Incubate cell lysates with anti-FLAG M2 affinity gel for 2 hours at 4°C. Wash beads extensively with lysis buffer.
- Elution and Denaturation: Elute ubiquitinated proteins with 3×FLAG peptide or SDS sample buffer. Denature proteins with 1% SDS and dilute to 0.1% SDS.
- Trypsin Digestion: Reduce with 5 mM DTT, alkylate with 10 mM iodoacetamide, and digest with trypsin overnight at 37°C.
- diGly Peptide Enrichment: Desalt peptides and incubate with anti-K-ε-GG antibody conjugated to beads for 2 hours at 4°C.
- Mass Spectrometry Analysis: Wash beads, elute peptides, and analyze by LC-MS/MS. Identify ubiquitination sites by searching for the GG remnant (114.0429 Da) on lysine residues.
Protocol 2: Identification of N-Terminal Ubiquitination Sites
This protocol specifically identifies N-terminal ubiquitination sites using anti-GGX antibodies.
- Cell Culture and Treatment: Culture cells expressing the E2 enzyme UBE2W or empty vector control. Treat with proteasome inhibitor (e.g., MG132) for 4-6 hours before harvesting if studying stabilized substrates.
- Protein Extraction and Digestion: Lyse cells in urea lysis buffer (8 M urea, 50 mM Tris-HCl pH 8.0). Reduce, alkylate, and digest proteins with trypsin overnight.
- Peptide Immunoprecipitation: Desalt peptides and incubate with anti-GGX antibody (1C7, 2B12, 2E9, or 2H2) conjugated to protein A/G beads for 4 hours at 4°C.
- Peptide Washing and Elution: Wash beads sequentially with ice-cold IP buffer, high-salt buffer, and LC-MS grade water. Elute peptides with 0.1% trifluoroacetic acid.
- LC-MS/MS Analysis: Analyze peptides by LC-MS/MS using a high-resolution mass spectrometer. Search data against appropriate database, specifying GG modification on peptide N-termini.
Research Reagent Solutions
Table: Essential Research Reagents for Ubiquitin Substrate Identification
| Reagent | Type | Function | Example Sources |
|---|---|---|---|
| Anti-K-ε-GG Antibody | Monoclonal antibody | Enrichment of canonical ubiquitination sites | Commercial vendors |
| Anti-GGX Antibodies | Monoclonal antibodies (1C7, 2B12, 2E9, 2H2) | Specific enrichment of N-terminal ubiquitination sites | Available from research community [41] |
| TR-TUBE | Engineered ubiquitin-binding protein | Stabilization of ubiquitinated substrates in cells | Custom generation [40] |
| Orthogonal E1/E2 Pairs | Engineered enzymes | Isolation of specific ubiquitination pathways | Custom engineering required [42] |
| Proteasome Inhibitors | Small molecules (MG132, bortezomib) | Stabilization of degradation-targeted ubiquitinated substrates | Commercial vendors |
| DUB Inhibitors | Small molecules (N-ethylmaleimide, PR-619) | Prevention of deubiquitination during processing | Commercial vendors |
Visualizing Experimental Workflows and Signaling Pathways
Ubiquitination Cascade and Stabilization
Orthogonal Ubiquitin Transfer System
The integration of antibody-based enrichment strategies with engineered protein systems provides a powerful toolkit for deciphering the complex landscape of ubiquitin ligase-substrate relationships. The development of diGly antibodies revolutionized global ubiquitome profiling, while newly emerging GGX antibodies enable specific investigation of non-canonical N-terminal ubiquitination. Meanwhile, protein engineering approaches like TR-TUBE and orthogonal ubiquitin transfer systems offer innovative solutions to stabilize and isolate specific ubiquitination events. As these methodologies continue to evolve and become more accessible, they will undoubtedly accelerate our understanding of ubiquitin signaling networks and facilitate the development of targeted therapeutics for diseases characterized by dysregulated protein ubiquitination.
High-Throughput Screening Platforms for Ubiquitin System Modulators
The ubiquitin-proteasome system (UPS) represents a crucial pathway for targeted protein degradation and the regulation of essential cellular processes, including cell cycle progression, DNA repair, and immune signaling [43] [44]. This system operates through a sequential enzymatic cascade involving E1 (activating), E2 (conjugating), and E3 (ligase) enzymes that collectively mediate the attachment of ubiquitin chains to substrate proteins, marking them for proteasomal degradation or functional modification [36] [43]. Dysregulation of this pathway is implicated in numerous disease states, particularly cancer, neurodegenerative disorders, and immune diseases, making it an attractive target for therapeutic intervention [45] [46].
The complexity of the ubiquitin system, with over 600 E3 ligases and approximately 100 deubiquitinating enzymes (DUBs) in humans, presents both challenges and opportunities for drug discovery [46]. High-throughput screening (HTS) platforms have emerged as powerful tools for identifying small molecule modulators of these enzymes, enabling the rapid evaluation of compound libraries against specific targets within the UPS [44] [47]. This application note details established HTS methodologies for identifying modulators of ubiquitin system components, providing researchers with validated protocols and analytical frameworks to advance drug discovery efforts in this rapidly evolving field.
HTS Platform Technologies and Applications
Various HTS platforms have been developed to target different components of the ubiquitin system, each with distinct advantages and applications. The table below summarizes the major HTS platforms currently employed in ubiquitin research.
Table 1: High-Throughput Screening Platforms for Ubiquitin System Modulators
| Platform Technology | Target Enzymes | Readout Method | Key Applications | Throughput Capacity |
|---|---|---|---|---|
| High-Content Imaging | DUBs, E3 Ligases | Fluorescence quantification of intracellular bacteria | Host-pathogen interactions, antimicrobial resistance [48] [49] | 96-384 well plates |
| Homogeneous Time-Resolved Fluorescence (HTRF) | E3 Ligases (Skp2-Cks1) | FRET-based fluorescence ratio (665/620 nm) | Protein-protein interaction inhibitors, cancer therapeutics [44] | 384-well plates |
| Chain-Selective TUBE Assays | Polyubiquitin linkage-specific | ELISA-based detection | Molecular glue degraders, PROTAC characterization [50] | 96-384 well plates |
| Cellular Degradation Screening | E3 Ligases, Molecular Glues | Quantitative proteomics, viability assays | Targeted protein degradation, monovalent degrader discovery [47] | 384-1536 well plates |
The diversity of available screening technologies enables researchers to select platforms based on their specific experimental needs, from in vitro biochemical assays to complex phenotypic cellular screens. High-content imaging approaches have proven particularly valuable for studying host-pathogen interactions, as demonstrated in screens identifying DUB inhibitors that enhance bacterial clearance in Salmonella-infected macrophages [48] [49]. In contrast, HTRF platforms offer robust solutions for quantifying specific protein-protein interactions within the ubiquitination machinery, such as the critical Skp2-Cks1 interaction required for p27 ubiquitination [44].
Recent advances in screening methodologies have also enabled the discovery of monovalent degraders that operate through diverse mechanisms, including direct molecular glues, adaptor-based degraders, and allosteric modulators of E3 ligase activity [47]. These cellular screening approaches, coupled with mechanistic deconvolution using CRISPR and proteomic technologies, have expanded the toolkit for identifying novel modulators of the ubiquitin system with potential therapeutic applications.
Detailed Experimental Protocols
High-Content Imaging Screen for DUB Inhibitors in Bacterial Clearance
This protocol describes a high-content imaging approach for identifying DUB inhibitors that enhance macrophage-mediated clearance of intracellular bacteria, adapted from studies on Salmonella-infected macrophages [48] [49].
Materials and Reagents
- Cell Line: Murine or human macrophage cell line (e.g., RAW 264.7 or THP-1)
- Bacterial Strain: GFP-labeled Salmonella enterica serovar Typhimurium UK-1
- Staining Reagents:
- Hoechst stain (nuclear labeling)
- HCS CellMask Red (cytoplasmic staining)
- Compound Library: 257 small-molecule modulators targeting UPS components
- Equipment: High-content imaging system with automated analysis capabilities
Procedure
Cell Preparation and Infection:
- Seed macrophages into 96-well imaging plates at 2×10^4 cells/well and culture overnight.
- Infect macrophages with GFP-labeled Salmonella at a multiplicity of infection (MOI) of 10:1 (bacteria:cells) for 30 minutes.
- Remove extracellular bacteria by washing 3× with PBS containing gentamicin (100 μg/mL).
Compound Treatment:
- Add UPS-targeting compounds at recommended screening concentrations (typically 1-10 μM).
- Include appropriate controls: DMSO vehicle control, ampicillin (positive control for bacterial clearance).
- Incubate plates for 12-16 hours at 37°C with 5% CO₂.
Staining and Imaging:
- Add Hoechst stain (1 μg/mL) and HCS CellMask Red (1:2000 dilution) to each well.
- Incubate for 30 minutes at 37°C.
- Acquire images using a high-content imaging system with 20× objective.
- Capture multiple fields per well to ensure statistical robustness (minimum 500 cells/well).
Image Analysis and Quantification:
- Use imaging software to identify host cell nuclei (Hoechst channel) and cytoplasm (CellMask Red channel).
- Quantify intracellular GFP-positive bacteria within the masked cytoplasmic regions.
- Normalize bacterial counts to cell number and calculate fold-change relative to DMSO controls.
Table 2: Key Parameters for High-Content Imaging Screen Validation
| Parameter | Optimized Condition | Quality Control Metric |
|---|---|---|
| Cell Density | 2×10^4 cells/well | >80% confluency at time of infection |
| Infection MOI | 10:1 | 20-30% infection rate in controls |
| Compound Incubation | 16 hours | Z' factor >0.5 |
| Image Acquisition | 9 fields/well | >500 cells analyzed per well |
| Hit Selection Criteria | ≥1.5 log10 reduction in bacterial load | p-value <0.01 |
This screening approach successfully identified 59 compounds that significantly reduced intracellular bacterial counts, including the dual USP25/USP28 inhibitor AZ-1, which demonstrated broad-spectrum activity against multiple drug-resistant pathogens [48] [49].
HTRF Assay for Skp2-Cks1 Interaction Inhibitors
This protocol details the establishment of an HTRF-based screening platform for inhibitors targeting the Skp2-Cks1 protein-protein interaction, a critical node in cell cycle regulation [44].
Protein Expression and Purification
Plasmid Transformation:
- Transform pGEX plasmid encoding GST-Skp2/Skp1 into E. coli BL21 (DE3) cells.
- Transform pGEX-4T-1 vector containing GST-Thrombin-His₆-Cks1 into separate BL21 (DE3) cells.
Protein Expression:
- Grow transformed cells in LB medium with appropriate antibiotics at 37°C to OD₆₀₀ = 0.6-0.8.
- Induce protein expression with 0.5 mM IPTG at 16°C overnight.
Protein Purification:
- Lyse cells by sonication in 20 mM Tris-HCl (pH 7.5), 300 mM NaCl, 2.5 mM PMSF, 5 mM β-mercaptoethanol.
- Purify GST-tagged proteins using Glutathione resin with elution buffer (20 mM Tris pH 8.0, 300 mM NaCl, 10 mM glutathione).
- Further purify by anion exchange chromatography (HiTrap Q HP column) with 0-1 M NaCl gradient.
- Apply final gel filtration step (Superdex 200 for Skp2/Skp1; Superdex 75 for His₆-Cks1).
HTRF Binding Assay
Assay Configuration:
- Perform assays in OptiPlate-384 white plates in 20 μL total volume.
- Use anti-GST-Eu cryptate (Cisbio, 61GSTKLA) and anti-His₆-d2 (Cisbio, 61HISDLA) detection antibodies.
- Maintain constant antibody concentrations at 1:1 ratio with target proteins.
Binding Reaction:
- Incubate 5 μL GST-Skp2/Skp1 (2 nM final) with 5 μL His₆-Cks1 (2 nM final) for 1 hour at room temperature.
- Add 5 μL anti-GST-Eu and 5 μL anti-His₆-d2 simultaneously.
- Incubate for 2-4 hours protected from light.
Signal Detection:
- Read plates using HTRF-compatible reader with 337 nm excitation.
- Measure emission at 620 nm and 665 nm.
- Calculate HTRF ratio as (665 nm emission / 620 nm emission) × 10,000.
This optimized HTRF assay demonstrated robust performance with a high signal-to-background ratio, enabling the identification of small molecule inhibitors disrupting the Skp2-Cks1 interaction critical for p27 ubiquitination and degradation [44].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for Ubiquitin System Screening
| Reagent/Library | Supplier Examples | Application | Key Features |
|---|---|---|---|
| K48 Linkage ELISA Kit | LifeSensors (PA480) | Detection of K48-linked polyubiquitin chains | Specific for proteasomal degradation signals [50] |
| K63 Linkage ELISA Kit | LifeSensors (PA630) | Detection of K63-linked polyubiquitin chains | Specific for signaling and trafficking pathways [50] |
| PROTAC In vitro Ubiquitination Assay Kit | LifeSensors (PA770) | Evaluation of engineered PROTAC molecules | Enables E3 ligase discovery and degrader evaluation [50] |
| UPS-Targeted Compound Library | Custom collections | Screening for ubiquitin system modulators | 257+ compounds targeting E1/E2/E3/DUB enzymes [48] [49] |
| Tandem Ubiquitin Binding Entities (TUBEs) | LifeSensors | Linkage-specific ubiquitin chain detection | Sub-nanomolar affinity for specific polyubiquitin chains [50] |
| Anti-GST-Eu Cryptate | Cisbio Bioassays (61GSTKLA) | HTRF-based PPI assays | FRET donor for GST-tagged proteins [44] |
| Anti-His₆-d2 Acceptor | Cisbio Bioassays (61HISDLA) | HTRF-based PPI assays | FRET acceptor for His-tagged proteins [44] |
Signaling Pathways and Experimental Workflows
Ubiquitin Screening Workflow
Ubiquitin Cascade & Screening Targets
The HTS platforms detailed in this application note provide robust and reproducible methods for identifying novel modulators of the ubiquitin system. The integration of biochemical, cellular, and phenotypic screening approaches enables comprehensive interrogation of this complex biological pathway, from specific enzyme activities to system-wide functional outcomes. As research in targeted protein degradation continues to advance, these screening methodologies will play an increasingly important role in the development of next-generation therapeutics for cancer, infectious diseases, and other disorders linked to ubiquitin pathway dysregulation. The protocols and reagents described herein offer researchers a solid foundation for establishing these critical capabilities within their own drug discovery programs.
Targeted protein degradation via Proteolysis-Targeting Chimeras (PROTACs) represents a paradigm shift in therapeutic intervention, building directly upon the foundational ubiquitin activation E1-E2-E3 enzymatic cascade [51]. This technology harnesses the cell's natural protein quality-control machinery—the ubiquitin-proteasome system (UPS)—to achieve catalytic removal of specific disease-associated proteins [52]. Unlike traditional occupancy-based inhibitors, PROTACs operate via an event-driven mechanism, inducing rapid and sustained protein knockdown without requiring continuous high drug concentrations [53].
The core mechanism involves a heterobifunctional molecule that creates a synthetic bridge between a target protein and an E3 ubiquitin ligase, fundamentally reprogramming the final step of the ubiquitination cascade [54] [51]. This induced proximity leads to polyubiquitination of the target protein, primarily through K48-linked chains, marking it for recognition and degradation by the 26S proteasome [51]. A single PROTAC molecule can catalyze multiple rounds of degradation, offering significant pharmacological advantages over conventional inhibitors [55] [53].
Table 1: Core Components of the Ubiquitin-Proteasome System and PROTAC Intervention Point
| System Component | Primary Function | Role in PROTAC Mechanism |
|---|---|---|
| E1 Activating Enzyme | Activates ubiquitin in an ATP-dependent manner [51] | Unmodified by PROTACs; provides activated ubiquitin |
| E2 Conjugating Enzyme | Accepts ubiquitin from E1 and collaborates with E3 for transfer [51] | Unmodified by PROTACs; recruited by the E3 ligase |
| E3 Ubiquitin Ligase | Confers substrate specificity; catalyzes ubiquitin transfer to target [54] [51] | Recruited by PROTAC ligand; determines efficiency and selectivity |
| 26S Proteasome | Recognizes polyubiquitinated proteins and degrades them [51] | Executes final degradation step |
| PROTAC Molecule | N/A | Induces novel E3-Target proximity, enabling ubiquitination of non-native substrates |
The Expanding Universe of E3 Ligases for PROTAC Development
The human genome encodes over 600 E3 ubiquitin ligases, yet current PROTAC development heavily relies on only a handful, notably Cereblon (CRBN) and Von Hippel-Lindau (VHL) [56] [57]. This limited repertoire poses constraints regarding potential resistance mechanisms, tissue-specific targeting, and the range of degradable targets [56] [58]. Systematic analysis of E3 ligases has identified numerous underutilized ligases with high potential for PROTAC development based on criteria such as ligandability, expression patterns, and protein-protein interaction profiles [56].
Table 2: Characterization of Key and Emerging E3 Ligases in TPD
| E3 Ligase | Ligandability & Known Ligands | Expression & Therapeutic Rationale | Representative Degraded Targets |
|---|---|---|---|
| CRBN | High (Thalidomide, Lenalidomide, Pomalidomide) [51] [59] | Broadly expressed; widely validated but potential for resistance [56] | IKZF1/3, BRD4, CDK9, Sirt2 [59] |
| VHL | High (VH032 derivatives) [60] [59] | Broadly expressed; high essentiality may reduce resistance [60] | ERRα, BRD4, RIPK2, HIF-1α [59] |
| MDM2 | Moderate (Nutlins) [51] | -- | AR, p53 [51] [59] |
| IAP (cIAP) | Moderate (Bestatin analogs) [51] | -- | RIPK2, CDK4/6 [59] |
| RNF114 | Emerging [59] | -- | BRD4, BCR-ABL [59] |
| DCAF16 | Emerging (Covalent ligands) [57] | -- | -- |
| KEAP1 | Emerging [57] | -- | -- |
| FEM1B | Emerging [57] | -- | -- |
Expanding the E3 ligase toolbox is critical for overcoming current limitations. New E3 ligases can help circumvent acquired resistance resulting from genomic alterations in common E3s like CRBN, reduce on-target toxicities by leveraging tissue-specific E3 expression, and access a broader target space due to unique ternary complex geometries and subcellular localization [56]. For instance, the poor expression of VHL in platelets was exploited by the PROTAC DT2216 to mitigate the thrombocytopenia side effect associated with BCL-XL inhibition [56].
Experimental Protocols for PROTAC Development and Validation
Protocol 1: Workflow for Validating Novel E3 Ligase Ligands
This protocol outlines a comprehensive workflow for evaluating the utility of a novel E3 ligase ligand in PROTAC development, using promiscuous kinase inhibitors to broadly assess the "degradable" target space [60].
Step 1: Chemical Design and Synthesis
- Design PROTACs by linking promiscuous kinase inhibitors (e.g., compounds 1 or 3 with solvent-exposed moieties like a central pyrimidine ring or terminal piperazine) to the E3 ligase ligand (e.g., VH032) via PEG or alkyl linkers [60].
- Incorporate alternative exit vectors on the E3 ligase ligand (e.g., VH032-NH₂ or VH032-OH) to explore different geometries [60].
- Synthesize negative control compounds, including the E3 ligand alone, the POI ligand-linker conjugate, and matched stereoisomers if applicable [60].
Step 2: Target Engagement and Selectivity Profiling
- Perform Differential Scanning Fluorimetry (DSF): Screen the parent ligands and their linker conjugates against a panel of purified kinases (e.g., 100 kinases). A significant thermal shift (ΔTm > 10°C indicates nanomolar affinity and validates the chosen exit vector does not disrupt binding [60].
- Conduct Kinobeads Pull-Down: Incubate linker-conjugated kinase ligands with cell lysates and Kinobeads to capture kinome-wide interaction profiles and confirm target engagement in a more complex, native-like environment [60].
Step 3: Cellular Target Engagement
- Utilize the NanoBRET Assay: Transfert cells with Nanoluc-fused model kinases. Treat cells with a tracer compound and increasing concentrations of the PROTAC. Measure energy transfer to quantify cellular target engagement and permeability [60].
Step 4: Cytotoxicity and Degradation Assessment
- Viability Assay: Treat cells with PROTACs and measure ATP levels using CellTiter-Glo to identify general cytotoxicity, a major confounder for degradation readouts [60].
- Proteomic Analysis: Use quantitative mass spectrometry (MS)-based proteomics to assess kinome-wide degradation efficacy and selectivity after PROTAC treatment [60].
Step 5: Validation and Mechanism Confirmation
- Western Blotting: Validate the degradation of selected hits from MS data.
- HiBiT Assay: Utilize the split-luciferase system for time-resolved degradation kinetics.
- Pathway Inhibition: Use neddylation inhibitors (e.g., MLN4924) and proteasome inhibitors (e.g., MG132) to confirm that degradation is VHL- and proteasome-dependent [60].
Protocol 2: Rapamycin-Induced Proximity Assay (RiPA) for E3/Target Pairing
This genetic assay identifies which E3 ligases can effectively degrade a specific protein of interest (POI) before undertaking chemical PROTAC synthesis, thus focusing medicinal chemistry efforts [57].
Step 1: Plasmid Construction
- Clone the FRB domain into a lentiviral vector downstream of a strong promoter (e.g., SFFV).
- Clone the POI, fused to FKBP12 and a minimal luciferase (e.g., Oplophorus gracilirostris Luc), into a separate vector.
- Clone candidate E3 ligases, fused to FRB, into expression vectors [57].
Step 2: Cell Line Generation and Transfection
- Co-transfect HEK293 cells with the POI-FKBP12-Luc construct and individual E3-FRB constructs at varying DNA ratios (e.g., 1:1, 1:10, 1:100). Generate stable cell lines via lentiviral transduction if required for long-term studies [57].
Step 3: Rapamycin Induction and Readout
- Treat transfected cells with 0.1 µM rapamycin for 6-24 hours to induce dimerization.
- Measure luciferase activity in living cells at various time points. A significant decrease (>50%) in luminescence indicates successful POI degradation [57].
- Confirm degradation via immunoblotting analysis of cell lysates [57].
Key Controls:
- Transfect with POI-FKBP12-Luc and FRB domain alone (without E3) to ensure degradation is E3-dependent.
- Include well-characterized pairs (e.g., WDR5 and VHL) as a positive control [57].
Advanced Methodologies and Emerging Technologies
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Reagents and Tools for PROTAC Research and Development
| Reagent / Tool | Function & Application | Example & Notes |
|---|---|---|
| E3 Ligase Ligands | Recruit specific E3 ligases to form ternary complex [51] [59] | VH032 (for VHL), Pomalidomide (for CRBN) [60] [59] |
| Linker Conjugates | Evaluate exit vector suitability before full PROTAC synthesis [60] | Kinase inhibitor-PEG-COOH; critical for validating maintained target engagement |
| Neddylation Inhibitor | Inhibits cullin-RING ligase activation; confirms on-mechanism degradation [60] | MLN4924; lack of degradation confirms E3 dependence |
| Proteasome Inhibitor | Blocks final degradation step; confirms UPS involvement [60] | MG132; used to confirm proteasome-dependent degradation |
| NanoBRET Tracers | Measure target engagement in live cells [60] | e.g., Broad-spectrum kinase tracer; assesses cell permeability and engagement |
| HiBiT System | Quantifies real-time protein degradation kinetics [60] | Nanoluc-based split luciferase; high-throughput compatible |
| Caged PROTACs | Enable spatiotemporal control of degradation with light [55] [59] | e.g., DMNB-caged thalidomide; inactive until UV (365 nm) irradiation |
| RiPA System | Genetically identify functional E3/POI pairs [57] | FKBP12-POI and FRB-E3 plasmids with rapamycin inducer |
Visualization of Core PROTAC Mechanism
The following diagram illustrates the fundamental mechanism of PROTAC-induced degradation, from ternary complex formation to proteasomal degradation, within the context of the E1-E2-E3 cascade.
Emerging Frontiers: PROTAC 2.0 and Novel Modalities
The field is rapidly evolving beyond conventional PROTACs to address pharmacokinetic and selectivity challenges. Key emerging modalities include:
Activatable PROTACs: These are designed as prodrugs (pro-PROTACs) that remain inert until activated by specific physiological or external stimuli. Photo-caged PROTACs (opto-PROTACs) use photolabile groups (e.g., DMNB) on critical E3-binding motifs (e.g., the glutarimide -NH of CRBN ligands or the hydroxy group of VHL ligands) to block activity. Upon irradiation with UV light (365 nm), the cage is removed, releasing the active PROTAC for spatiotemporally controlled degradation [55] [59] [53].
Nanotechnology-Enabled Delivery: Nano-PROTACs utilize lipid nanoparticles, polymeric micelles, or other nanostructures to improve the solubility, bioavailability, and targeted tissue delivery of PROTACs, overcoming inherent challenges related to their high molecular weight [53].
Dual-Targeting and Macrocyclic PROTACs: Bivalent or trivalent degraders simultaneously engage two different POIs, potentially addressing pathway redundancy in cancer. Macrocyclization constrains the PROTAC linker into a bioactive conformation, potentially improving degradation efficiency and physicochemical properties [53].
These advanced technologies represent the cutting edge of TPD, aiming to translate the profound therapeutic potential of PROTACs into viable clinical strategies beyond the current ~30 candidates in clinical trials [55] [53].
The ubiquitin-proteasome system is a crucial regulatory pathway in eukaryotic cells, controlling protein degradation, signal transduction, and cellular localization. This system operates through a sequential enzymatic cascade involving E1 (activating), E2 (conjugating), and E3 (ligating) enzymes [24] [43]. The E1 enzyme initiates the pathway by activating ubiquitin in an ATP-dependent manner, forming a ubiquitin-AMP intermediate. The activated ubiquitin is then transferred to the E1 catalytic cysteine residue, forming a thioester bond. Subsequently, ubiquitin is shuttled to an E2 enzyme via transthioesterification. Finally, an E3 ligase facilitates the transfer of ubiquitin from the E2 to a lysine residue on the target protein, determining substrate specificity [24] [61].
The complexity of this system—with 2 E1s, approximately 50 E2s, and over 1,000 E3s in humans—creates a challenging landscape for researchers attempting to dissect specific ubiquitination pathways [24] [28]. This review explores three emerging technologies—Ubiquitin Variants (UbVs), Phage Display, and DNA-Encoded Libraries (DELs)—that are revolutionizing our ability to probe, manipulate, and target the ubiquitin cascade for both fundamental research and therapeutic development.
Ubiquitin Variants (UbVs) for Cascade Modulation
Principles and Applications
Ubiquitin Variants (UbVs) are engineered forms of ubiquitin designed to modulate the activity of specific components within the ubiquitination cascade. Using phage display technology, researchers have developed UbVs that can either activate or inhibit particular E3 ligases or other enzymes in the pathway, offering unprecedented precision for functional studies [24] [28].
Key applications of UbVs include:
- Orthogonal Ubiquitin Transfer Cascades: Engineered UbVs (xUB) can be paired with engineered E1 (xE1) and E2 (xE2) enzymes to create orthogonal transfer pathways that operate independently of native cellular machinery, enabling specific ubiquitination of target proteins without cross-talk [28].
- E1 Enzyme Profiling: Phage display of UB libraries with randomized C-terminal sequences has revealed substantial promiscuity in E1 enzyme specificity, with UB mutants at positions 71, 73, and 74 retaining efficient E1 activation capability [24].
- DUB-Resistant Ubiquitin Chains: Specific UB mutants such as Leu73Phe and Leu73Tyr can form poly-UB chains through the E1-E2-E3 cascade while exhibiting resistance to cleavage by deubiquitinating enzymes (DUBs), providing stable UB polymers for studying biological signals encoded in UB chains [24].
Quantitative Profiling of UbV Specificity
Table 1: Phage Display Selection Results for UB C-terminal Sequence Specificity with E1 Enzymes
| UB Residue Position | Wild-type Amino Acid | Permissive Mutations | Effect on E1 Activation | Downstream Processing |
|---|---|---|---|---|
| Arg72 | Arginine (R) | None | Absolutely required | Essential for E1 recognition |
| Leu71 | Leucine (L) | Bulky aromatic side chains | Efficient activation | Transfer to E2 preserved |
| Leu73 | Leucine (L) | Bulky aromatic side chains (Phe, Tyr) | Efficient activation | DUB resistance; blocks E2 to E3 transfer |
| Arg74 | Arginine (R) | Bulky aromatic side chains | Efficient activation | Not specified |
| Gly75 | Glycine (G) | Ser, Asp, Asn | Efficient activation | Affects discharge from E2 |
Protocol: Phage Display Selection for E1-Specific UbVs
Objective: Identify Ubiquitin Variants with specific reactivity toward E1 enzymes Ube1 and Uba6.
Materials:
- Phage-displayed UB library (randomized residues 71-75, Gly76 unchanged)
- Biotin-labeled PCP-E1 fusions (Ube1 and Uba6)
- Streptavidin-coated plates
- Reaction buffer: 1 mM Mg-ATP, standard buffer conditions
- Elution buffer: 100 mM DTT (dithiothreitol)
- E. coli host strains for phage amplification
Procedure:
- Immobilization: Bind biotin-labeled PCP-E1 fusions to streptavidin plates.
- Library Incubation: Add phage-displayed UB library (1×10^11 pfu) to plates containing 100 pmol immobilized E1.
- Catalytic Selection: Incubate for 1 hour with 1 mM Mg-ATP to allow formation of UB~E1 thioester conjugates.
- Washing: Remove non-specific and unbound phage particles.
- Elution: Release specifically bound phage by cleavage of thioester linkages with 100 mM DTT.
- Amplification: Infect E. coli with eluted phage and amplify for subsequent selection rounds.
- Stringency Increase: Repeat selection with decreasing amounts of E1 (down to 1 pmol) and shorter reaction times (down to 10 minutes) over 8 rounds.
- Clone Analysis: Sequence enriched phage clones after 8 rounds of selection.
Technical Notes:
- Monitor phage enrichment at each round compared to controls without E1 or Mg-ATP.
- The library diversity of 1×10^8 clones adequately covers the sequence space of 5 randomized residues (3.2×10^6 possible sequences).
- Correct phage orientation in proteoliposomes is essential for E1 accessibility [24].
Diagram 1: Phage display biopanning workflow for UbV selection.
Phage Display for Ubiquitin Cascade Engineering
Phage display is a powerful screening method that enables the selection of peptides, proteins, or antibody fragments with desired binding properties from large combinatorial libraries. The technology leverages the direct genotype-phenotype linkage where bacteriophages are engineered to display protein variants on their surface while carrying the encoding DNA internally [62] [63] [64].
The M13 filamentous phage is the most commonly used system, where foreign DNA sequences are fused to genes encoding coat proteins (typically pIII or pVIII). The displayed protein variants can then be screened against targets of interest through an iterative process called biopanning [62] [63].
Applications in Ubiquitin Research
In ubiquitin cascade research, phage display has been instrumental in:
- Profiling E1-E2-UB Interactions: Identifying key residues in UB C-terminal recognition by E1 enzymes, revealing that while Arg72 is absolutely required, other positions show unexpected flexibility [24].
- Engineering Orthogonal Cascades: Developing specialized E1-E2 pairs that operate independently of native cellular machinery for precise ubiquitination studies [28].
- Mapping Enzyme Specificity: Comprehensive profiling of E3 ligase specificity and deubiquitinase (DUB) recognition patterns using UB variant libraries [24].
Protocol: Biopanning for E3 Ligase Binders
Objective: Identify peptide or antibody fragments that bind specifically to target E3 ligases.
Materials:
- Phage display library (naïve, immune, or synthetic)
- Immobilized E3 ligase target (purified protein or whole cells)
- Coating buffers (appropriate for target immobilization)
- Washing buffers: PBS with 0.1% Tween-20, PBS alone
- Elution buffers: 0.1 M glycine-HCl (pH 2.2), 0.5 mg/mL trypsin, or target-specific competitive eluents
- E. coli culture (log-phase) for infection
- PEG/NaCl for phage precipitation
Procedure:
- Target Immobilization: Coat solid support (immunotube, microplate, or beads) with purified E3 ligase (10-100 μg/mL) overnight at 4°C.
- Blocking: Incubate with blocking buffer (2-5% BSA or milk in PBS) for 1-2 hours at room temperature.
- Phage Incubation: Add phage library (10^10-10^12 pfu) in blocking buffer, incubate 1-2 hours with gentle agitation.
- Washing: Remove unbound phage with 10-20 washes using PBS-Tween (0.1%), followed by 10-20 washes with PBS alone.
- Elution: Recover specifically bound phage using:
- Acid elution: 0.1 M glycine-HCl (pH 2.2), 10-minute incubation, neutralize with 1 M Tris-HCl (pH 9.1)
- Enzymatic elution: 0.5 mg/mL trypsin in PBS, 30-minute incubation at 37°C
- Amplification: Infect log-phase E. coli with eluted phage, culture overnight with helper phage if using phagemid system.
- Phage Precipitation: Purify amplified phage from culture supernatant using PEG/NaCl precipitation.
- Iterative Selection: Repeat steps 3-7 for 3-5 rounds with increasing wash stringency.
Technical Notes:
- Monitor enrichment by titering input, output, and wash fractions at each round.
- Include negative selection steps against unrelated proteins or empty solid support to reduce non-specific binders.
- For membrane-protein E3 ligases, consider using whole-cell panning approaches [62] [63] [64].
DNA-Encoded Libraries (DELs) for Ubiquitin System Targeting
Technology Foundation
DNA-Encoded Library technology represents a paradigm shift in screening methodologies, combining combinatorial chemistry with DNA barcoding to create and screen enormous chemical diversity (10^6-10^12 compounds) in a single-tube format [65] [66]. Each small molecule in a DEL is conjugated to a unique DNA tag that serves as an amplifiable identification barcode, enabling pooled screening of billions of compounds simultaneously through affinity selection [65].
DEL construction typically follows a split-and-pool approach where chemical building blocks are conjugated to DNA fragments encoding their identity. Multiple cycles of chemical transformation and DNA tag elongation yield large combinatorial libraries where the final DNA tag records the synthetic history of each compound [65].
Applications in Ubiquitin Drug Discovery
DEL technology has been particularly valuable for targeting components of the ubiquitin system:
- E1 Enzyme Inhibitors: Identification of small molecules that block the initial ubiquitin activation step.
- E2-E3 Interaction Disruptors: Finding compounds that interfere with specific E2-E3 partnerships.
- DUB Inhibitors: Developing selective inhibitors of deubiquitinating enzymes.
- Molecular Glues and PROTACs: Discovering compounds that induce proximity between E3 ligases and target proteins for degradation [66].
Protocol: DEL Screening for Ubiquitin Enzyme Inhibitors
Objective: Identify small-molecule ligands against E1, E2, E3, or DUB targets from DNA-encoded libraries.
Materials:
- DNA-encoded chemical library (typically 10^8-10^10 diversity)
- Target protein (biotinylated or with appropriate tag for immobilization)
- Streptavidin-coated magnetic beads
- Selection buffer: PBS with 0.01-0.1% Tween-20 and BSA (1 mg/mL)
- Washing buffers: Selection buffer, PBS, 10 mM Tris-HCl
- Elution buffers: Denaturing conditions (95°C, 8 M urea) or specific competitors
- PCR reagents for DNA tag amplification
- Next-generation sequencing platform
Procedure:
- Target Immobilization: Incubate biotinylated target protein with streptavidin magnetic beads (1-2 hours, 4°C).
- Blocking: Block beads with selection buffer containing BSA (1-2 hours, 4°C).
- Library Incubation: Incubate DEL with immobilized target (10 nM-1 μM target concentration, >100-fold library excess) in selection buffer (2-16 hours, 4°C or room temperature).
- Washing: Remove unbound library members with multiple washes (5-10 washes with selection buffer, 5-10 washes with PBS, 1-2 washes with Tris buffer).
- Elution: Recover bound ligands using:
- Denaturing elution: 8 M urea or guanidinium HCl at 95°C for 15 minutes
- Competitive elution: Excess of known binder (if available) for 1-2 hours
- DNA Recovery: Purify DNA tags from eluates (phenol-chloroform extraction or commercial kits).
- PCR Amplification: Amplify recovered DNA tags with indexing primers for sequencing.
- Sequencing and Analysis: Perform high-throughput sequencing and analyze enrichment patterns compared to control selections.
Technical Notes:
- Include control selections with irrelevant proteins or no protein to identify non-specific binders.
- For challenging membrane-associated targets (e.g., ERAD components like UBE2J2), optimize membrane mimetics in selection buffer [7].
- Consider multiplexed screening against multiple targets or conditions simultaneously to assess selectivity [65] [66].
Diagram 2: DNA-encoded library selection and analysis workflow.
Research Reagent Solutions
Table 2: Essential Research Reagents for Ubiquitin Cascade Studies
| Reagent Category | Specific Examples | Function/Application |
|---|---|---|
| Engineered Enzymes | xE1 (engineered E1), xE2 (engineered E2) | Orthogonal ubiquitin transfer cascades; specific substrate targeting [28] |
| Ubiquitin Variants | UbV clones (Leu73Phe, Leu73Tyr, Arg72 mutants) | E1 profiling; DUB-resistant chain formation; cascade specificity studies [24] |
| Phage Display Systems | M13 phage libraries (peptide, scFv, Fab) | E3 ligase binder identification; epitope mapping; protein interaction studies [62] [63] |
| DNA-Encoded Libraries | DELs with privileged scaffolds | Small-molecule inhibitor discovery; PROTAC development; molecular glue identification [65] [66] |
| Lipid Membrane Systems | ER-like liposomes (low SFA, low cholesterol) | Studying membrane-sensitive ubiquitination (e.g., UBE2J2 regulation in ERAD) [7] |
| Detection Tools | Biotinylated PCP-E1 fusions, Streptavidin plates | Phage selection setup; monitoring thioester conjugate formation [24] |
Integrated Workflow for Ubiquitin Cascade Research
The synergistic application of UbVs, phage display, and DEL technologies creates a powerful toolkit for comprehensive ubiquitin cascade analysis. Below is an integrated workflow for identifying and characterizing modulators of specific ubiquitination pathways:
Diagram 3: Integrated workflow combining multiple technologies for ubiquitin research.
This integrated approach enables researchers to:
- Identify Key Targets: Use phage display to profile E1-E2-E3 specificities and identify critical interaction interfaces [24] [28].
- Develop Specific Modulators: Engineer UbVs for precise cascade intervention or utilize DELs for small-molecule inhibitor discovery [24] [65].
- Validate in Physiologically Relevant Systems: Test candidates in reconstituted systems with defined lipid compositions to account for membrane sensitivity of enzymes like UBE2J2 [7].
- Establish Orthogonal Cascades: Implement engineered xUB-xE1-xE2 pairs for specific substrate targeting without cellular cross-talk [28].
The combination of these technologies provides an unprecedented ability to dissect the complexity of the ubiquitin system, enabling both fundamental mechanistic studies and the development of targeted therapeutic interventions for diseases involving ubiquitination dysregulation, including cancer, neurodegenerative disorders, and acute kidney injury [43].
Challenges in Ubiquitin System Targeting: Specificity, Resistance, and Technical Limitations
Overcoming Specificity Hurdles in E1, E2, and E3 Enzyme Targeting
The ubiquitin-proteasome system (UPS) is a crucial regulatory mechanism that controls nearly every biological process, from cell cycle progression to DNA repair, by directing proteins for degradation and modulating their function [10]. At the heart of this system operates a sequential enzymatic cascade comprising ubiquitin-activating (E1), conjugating (E2), and ligase (E3) enzymes. This hierarchical system presents both challenges and opportunities for therapeutic intervention. The human genome encodes approximately 2 E1s, 35-40 E2s, and over 600 E3s, creating a complex network of potential interactions that ultimately governs the specificity of ubiquitination [67] [16] [5]. While this elaborate architecture enables precise control of protein fate, it also creates significant hurdles for targeted therapeutic development, as the cross-reactivities among these enzymes make it difficult to isolate specific pathways for modulation [28].
The pursuit of specificity in targeting ubiquitination pathways represents one of the most promising frontiers in drug discovery, particularly for complex diseases such as cancer, neurodegenerative disorders, and viral infections [16] [10]. This application note examines the molecular basis of specificity within the E1-E2-E3 cascade and provides detailed methodologies for profiling and engineering selective components, enabling researchers to overcome the inherent challenges in this pathway. By leveraging recent structural and biochemical insights, we outline practical experimental approaches to develop targeted modulators of ubiquitination with therapeutic potential.
Understanding Specificity Determinants in the Ubiquitin Cascade
Structural and Functional Basis of Enzyme Specificity
The ubiquitination process initiates when the E1 enzyme activates ubiquitin in an ATP-dependent reaction, forming a ubiquitin-adenylate intermediate before transferring ubiquitin to its catalytic cysteine residue to generate a E1~ubiquitin thioester conjugate [24] [67]. This activated ubiquitin is then transferred to a catalytic cysteine of an E2 conjugating enzyme, finally being delivered to substrate proteins with the assistance of E3 ligases that provide substrate specificity [24] [5].
Table 1: Key Specificity Determinants in Ubiquitin Cascade Enzymes
| Enzyme Class | Key Specificity Determinants | Functional Role | Therapeutic Implications |
|---|---|---|---|
| E1 Activating Enzymes | Ubiquitin C-terminal sequence recognition [24]; Crossover loop for ubiquitin/UBL discrimination [67]; UFD domain for E2 selection [67] | Controls initial ubiquitin activation and E2 charging | Broad inhibition affects entire pathway; opportunities for engineered orthogonal systems [28] |
| E2 Conjugating Enzymes | Active site environment and flanking loops [5]; N-terminal helix for E1/E3 binding [67]; β-sheet "backside" for non-covalent ubiquitin binding [67] | Determines ubiquitin transfer reactivity and linkage specificity [16] [5] | E2-specific inhibitors potentially more selective than E1-targeting approaches [16] |
| E3 Ligases | Substrate recognition domains; RING domains for E2 recruitment [67]; HECT/RBR catalytic cysteine residues [5] | Provides ultimate substrate specificity through direct recognition | Most promising therapeutic targets due to high specificity; >600 E3s offer precision targeting [10] |
The E1 enzyme exhibits remarkable specificity for the C-terminal sequence of ubiquitin, with phage display studies revealing that while Arg72 is absolutely required for E1 recognition, residues at positions 71, 73, and 74 can accommodate bulky aromatic side chains, and Gly75 can be substituted with Ser, Asp, or Asn while maintaining efficient E1 activation [24] [68]. This specificity profile has been leveraged to engineer orthogonal ubiquitin transfer systems that operate independently of native cellular machinery [28].
E2 enzymes, historically viewed as simple carriers, are now recognized as central specificity determinants that influence which lysine residue is modified and what type of ubiquitin linkage is formed [16] [5]. The intrinsic reactivity of different E2~ubiquitin conjugates varies significantly, with some E2s showing preference for aminolysis (transfer to lysine) while others favor transthiolation (transfer to cysteine) [5]. For example, Ube2L3 (UbcH7) exhibits reactivity exclusively toward cysteine residues, explaining its functional specialization with HECT and RBR E3 ligases rather than typical RING E3s [5].
E3 ligases constitute the most diverse enzyme class in the cascade and provide the ultimate substrate specificity. RING-type E3s function as scaffolds that simultaneously bind E2~ubiquitin conjugates and substrates, facilitating direct ubiquitin transfer, while HECT and RBR E3s form obligate E3~ubiquitin thioester intermediates before modifying substrates [24] [5]. The therapeutic appeal of E3s lies in this precise substrate recognition capability, which theoretically enables targeting of specific pathogenic proteins without global disruption of ubiquitination [10].
Quantitative Profiling of Ubiquitin C-terminal Sequence Specificity
Phage display has emerged as a powerful methodology for comprehensively profiling the specificity of E1 enzymes toward ubiquitin C-terminal sequences. This approach enables the identification of ubiquitin variants (xUB) with alternative C-terminal sequences that maintain efficient E1 activation while becoming orthogonal to native enzymes [24] [28].
Table 2: Experimentally Determined E1 Specificity for Ubiquitin C-terminal Residues
| Ubiquitin Position | Wild-type Residue | Permissible Mutations | Functional Consequences | Experimental Validation |
|---|---|---|---|---|
| 71 (Leu) | Leu | Phe, Tyr, other bulky aromatic residues [24] | Tolerated without significant E1 activation loss [24] | Phage display selection with Ube1 and Uba6 [24] |
| 72 (Arg) | Arg | No substitutions tolerated [24] | Absolute requirement for E1 recognition; Arg72Leu increases Kd by 58-fold [24] | Site-directed mutagenesis and binding assays [24] |
| 73 (Leu) | Leu | Phe, Tyr, other bulky aromatic residues [24] | Single mutants (Leu73Phe, Leu73Tyr) form polyUB chains but resist DUB cleavage [24] [68] | Phage display; DUB resistance assays [24] |
| 74 (Arg) | Arg | Phe, Tyr, other bulky aromatic residues [24] | Tolerated without significant E1 activation loss [24] | Phage display selection with Ube1 and Uba6 [24] |
| 75 (Gly) | Gly | Ser, Asp, Asn [24] | Efficient E1 activation maintained; impacts downstream E3 transfer [24] | Phage display and biochemical characterization [24] |
| 76 (Gly) | Gly | No functional substitutions [24] | Gly76Ala mutant has very low E1 activity; inhibits UB-AMP formation [24] | Site-directed mutagenesis and activity assays [24] |
The experimental data reveals several key insights: First, E1 enzymes display substantial promiscuity toward ubiquitin C-terminal sequences, with only Arg72 and Gly76 being absolutely required for efficient activation. Second, engineered ubiquitin variants can be efficiently transferred from E1 to E2 enzymes but often encounter blocks in subsequent transfer to E3 enzymes, indicating stricter sequence requirements at later cascade stages. Third, specific mutations (particularly Leu73Phe and Leu73Tyr) confer resistance to deubiquitinating enzymes (DUBs) while maintaining compatibility with the E1-E2-E3 cascade, providing valuable tools for stabilizing ubiquitin signals in cellular contexts [24] [68].
Experimental Protocols and Methodologies
Protocol 1: Phage Display Selection for Engineering E1-Specific Ubiquitin Variants
This protocol describes a method for identifying ubiquitin variants with tailored specificity for engineered E1 enzymes using phage display, based on established methodologies [24] [28].
Materials and Reagents
- M13 phage library displaying ubiquitin variants with randomized C-terminal sequences (residues 71-75)
- Recombinant E1 enzymes (Ube1 or Uba6) fused to N-terminal peptidyl carrier protein (PCP) domain
- Sfp phosphopantetheinyl transferase
- Biotin-CoA conjugate
- Streptavidin-coated plates
- Binding buffer: 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.1% Tween-20, 1 mg/mL BSA
- Reaction buffer: 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 5 mM MgCl₂, 1 mM ATP
- Elution buffer: 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 50 mM DTT
- Escherichia coli strain for phage amplification
Procedure
- E1 Enzyme Biotinylation: Label 100 pmol of PCP-E1 fusion protein with biotin using Sfp phosphopantetheinyl transferase and biotin-CoA in 100 μL reaction for 1 hour at room temperature.
- Enzyme Immobilization: Immobilize biotinylated PCP-E1 on streptavidin-coated plates by incubating for 1 hour at room temperature, followed by three washes with binding buffer.
- Phage Selection: Incubate 1 × 10^11 phage particles displaying the ubiquitin library with immobilized E1 in reaction buffer containing 1 mM Mg-ATP for 1 hour at room temperature.
- Washing: Remove unbound phage by washing ten times with binding buffer containing 0.5 M NaCl to reduce nonspecific binding.
- Elution of Active Phage: Elute specifically bound phage by incubating with elution buffer for 30 minutes at room temperature to cleave the thioester linkage between ubiquitin variants and E1.
- Phage Amplification: Infect eluted phage into log-phase E. coli culture and amplify overnight for subsequent selection rounds.
- Iterative Selection: Repeat steps 3-6 for 6-8 rounds with increasing stringency (reduced E1 amount, shorter reaction time, increased salt concentration in washes).
- Clone Isolation and Sequencing: After final selection round, isolate individual clones and sequence ubiquitin genes to identify selected variants.
Technical Notes
- Include control selections without E1 or without Mg-ATP to monitor background binding
- Monitor phage enrichment through each round by titering input and output phage
- For initial rounds, use 100 pmol E1 and 1-hour reaction time; for later rounds, increase stringency to 1 pmol E1 and 10-minute reaction
- The crossover loop connecting adenylation and SCCH domains contributes significantly to ubiquitin/UBL discrimination and should be considered when engineering specificity [67]
Protocol 2: Orthogonal Ubiquitin Transfer Cascade Assembly
This protocol enables the reconstitution of an orthogonal ubiquitin transfer cascade using engineered components, allowing specific ubiquitination without cross-talk with endogenous systems [28].
Materials and Reagents
- Engineered ubiquitin variant (xUB) with modified C-terminal sequence
- Engineered E1 enzyme (xE1) with mutations in ubiquitin-binding pocket
- Engineered E2 enzyme (xE2) with complementary mutations for xE1 recognition
- Recombinant E3 ligase of interest
- Target substrate protein
- Reaction buffer: 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 5 mM MgCl₂, 2 mM ATP
- SDS-PAGE reagents and immunoblotting equipment
- Anti-ubiquitin and anti-substrate specific antibodies
Procedure
- E1 Activation Assay: Incubate 1 μM xE1 with 10 μM xUB in reaction buffer for 30 minutes at 30°C to form xE1~xUB thioester complex.
- E2 Charging Reaction: Add 5 μM xE2 to the xE1~xUB complex and incubate for additional 30 minutes at 30°C to facilitate transthiolation and xE2~xUB formation.
- E3-Substrate Complex Formation: Pre-incubate 2 μM E3 ligase with 5 μM target substrate in reaction buffer for 15 minutes at room temperature.
- Ubiquitination Reaction: Combine E2 charging reaction with E3-substrate complex and incubate for 1-2 hours at 30°C.
- Reaction Termination: Stop the reaction by adding SDS-PAGE loading buffer with or without reducing agent (DTT or β-mercaptoethanol).
- Analysis: Analyze samples by SDS-PAGE followed by immunoblotting with anti-ubiquitin and anti-substrate antibodies to detect ubiquitinated products.
- Specificity Validation: Perform parallel reactions with wild-type ubiquitin and enzymes to confirm orthogonal system functionality.
Technical Notes
- Include controls with individual component omissions to verify cascade dependency
- For time-course experiments, take aliquots at various time points (0, 15, 30, 60, 120 min)
- Engineered E1-E2 pairs should show no cross-reactivity with native ubiquitin or E2 enzymes [28]
- The UFD domain of E1 enzymes is critical for E2 recruitment and should be considered in engineering efforts [67]
Protocol 3: E2 Intrinsic Reactivity Profiling
This protocol characterizes the intrinsic reactivity of E2~ubiquitin conjugates toward different nucleophiles, revealing fundamental specificity properties that determine E2 functional specialization [5].
Materials and Reagents
- Purified E2 enzymes
- E1 activating enzyme
- Ubiquitin
- Reaction buffer: 50 mM HEPES (pH 7.5), 150 mM NaCl, 5 mM MgCl₂, 2 mM ATP
- Nucleophile substrates: free lysine (50 mM), cysteine (50 mM), or small lysine-less peptides (1 mM)
- SDS-PAGE reagents and staining solutions
- Anti-ubiquitin antibodies for immunoblotting
Procedure
- E2~Ub Thioester Formation: Incubate 5 μM E1, 10 μM E2, and 50 μM ubiquitin in reaction buffer for 30 minutes at 30°C to generate E2~Ub conjugates.
- Nucleophile Reactivity Assay: Add selected nucleophile (lysine, cysteine, or peptide) to the E2~Ub conjugate and incubate for various time points at 30°C.
- Reaction Termination: At each time point, remove aliquots and mix with non-reducing SDS-PAGE loading buffer to preserve thioester linkages.
- Product Analysis: Resolve samples by non-reducing SDS-PAGE followed by Coomassie staining or immunoblotting with anti-ubiquitin antibodies.
- Quantification: Measure the decrease in E2~Ub thioester and appearance of ubiquitin-nucleophile adducts over time.
- Kinetic Analysis: Plot reaction progress curves and calculate rates of ubiquitin transfer for different nucleophiles.
Technical Notes
- Maintain consistent pH as aminolysis rates are pH-dependent
- Include control reactions without nucleophiles to assess background hydrolysis
- Ube2W shows unique specificity for N-terminal α-amino groups over lysine side chains [5]
- Ube2L3 (UbcH7) exhibits exclusive reactivity toward cysteine, explaining its preference for HECT/RBR E3s [5]
Visualization of Ubiquitin Cascade and Specificity Engineering
Ubiquitin Cascade Specificity Nodes
Phage Display Engineering Workflow
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Research Reagents for Ubiquitin Specificity Studies
| Reagent Category | Specific Examples | Key Applications | Technical Considerations |
|---|---|---|---|
| Engineered Ubiquitin Variants | xUB with C-terminal mutations (L71F, R72L, L73F, L73Y, G75S) [24] [68] | E1 specificity profiling; Orthogonal cascade engineering; DUB-resistant signaling | Leu73Phe/Tyr mutants resist DUB cleavage while maintaining E1-E2-E3 compatibility [24] |
| Phage Display Systems | M13 ubiquitin phage library (randomized residues 71-75) [24] | Comprehensive E1 specificity mapping; xUB discovery | Library size of 1×10^8 recommended to cover sequence diversity of 5 randomized residues [24] |
| E1 Enzyme Variants | Ube1 (UBA1), Uba6 (UBA6) [24]; PCP-E1 fusions for immobilization [24] | E1-E2 interaction studies; Activation assays; Structural biology | Uba6 has distinct E2 specificity compared to Ube1, specializing with Ube2Z [67] |
| E2 Enzyme Panels | Ube2L3 (cysteine-specific) [5]; Ube2W (N-terminal specific) [5]; Ube2R1 (Cdc34) | Reactivity profiling; Linkage specificity studies; Cascade reconstitution | Intrinsic reactivity assays reveal specialized functions (e.g., Ube2W for N-terminal ubiquitination) [5] |
| Specialized E3 Ligases | RING-types (Mdm2, BRCA1/BARD1) [10]; HECT-types (NEDD4L) [5]; RBR-types (Parkin, HHARI) [5] | Substrate targeting studies; Therapeutic development | RING E3s facilitate direct transfer; HECT/RBR form E3~Ub intermediates [24] [5] |
| Activity Detection Tools | Biotin-CoA/Sfp for E1 labeling [24]; Non-reducing SDS-PAGE for thioesters [5]; Linkage-specific antibodies [43] | Reaction monitoring; Product characterization | Non-reducing conditions essential for preserving E2~Ub thioester conjugates in gels [5] |
The systematic engineering of specificity within the ubiquitin cascade represents a powerful approach for both basic research and therapeutic development. By leveraging the structural and mechanistic insights summarized in this application note, researchers can design targeted strategies to overcome the inherent complexity of this system. The methodologies outlined—from phage display profiling of ubiquitin variants to orthogonal cascade engineering—provide practical roadmaps for creating selective modulators of ubiquitination pathways.
Future directions in this field will likely focus on expanding the toolkit of orthogonal enzyme pairs, developing small-molecule inhibitors that exploit specificity nodes within E1-E2-E3 interactions, and applying these engineered systems to map the physiological functions of specific ubiquitination events in disease contexts. As our understanding of ubiquitin cascade specificity continues to deepen, so too will our ability to precisely manipulate this fundamental regulatory system for therapeutic benefit.
Addressing Off-Target Effects in Proteasome and Ubiquitin System Inhibition
The ubiquitin-proteasome system (UPS) represents a master regulatory network for intracellular protein degradation, controlling virtually all cellular processes through the orchestrated action of E1 (activating), E2 (conjugating), and E3 (ligating) enzymes [29]. This enzymatic cascade culminates in the polyubiquitination of specific substrate proteins, marking them for destruction by the 26S proteasome [69] [70]. While therapeutic intervention in the UPS holds immense promise, particularly in oncology, a fundamental challenge persists: achieving sufficient specificity to minimize off-target effects that can compromise therapeutic efficacy and cause dose-limiting toxicities [71] [72]. This Application Note delineates strategic approaches and detailed protocols for identifying, quantifying, and mitigating these off-target effects within the context of E1-E2-E3 enzymatic cascade research.
Core Mechanisms and Quantitative Profiling of Off-Target Effects
Inhibitors targeting the proteasome or the ubiquitination cascade can exhibit off-target effects through several mechanisms. Proteasome inhibitors, like bortezomib, simultaneously inhibit multiple catalytic subunits (β5/β1/β2 and their immunoproteasome counterparts) within the 20S core particle, disrupting a wide array of normal cellular protein turnover processes [71]. This non-selective inhibition can lead to the accumulation of misfolded proteins and endoplasmic reticulum (ER) stress, not only in malignant cells but also in healthy tissues, contributing to side effects such as peripheral neuropathy [70]. Furthermore, the constitutive inhibition of immunoproteasome subunits (e.g., LMP2, LMP7) can inadvertently modulate antigen presentation and immune responses [71].
Beyond the proteasome, interventions upstream at the level of E1, E2, or E3 enzymes face a combinatorial specificity challenge. The human genome encodes two E1s, approximately 50 E2s, and over 600 E3s [28] [29]. A single E3 ligase interacts with multiple E2s and hundreds of substrates. Therefore, inhibiting or modulating one component of this network can have ripple effects, leading to the unintended stabilization or degradation of non-target proteins. For instance, the stabilization of oncoproteins is a known risk, while the unintended degradation of tumor suppressors could potentially exacerbate disease progression [72].
Experimental Profiling of Inhibitor Selectivity
A critical first step in mitigating off-target effects is the comprehensive profiling of compound selectivity. The following protocol provides a methodology for quantifying proteasome inhibitor activity against all six catalytic subunits.
Protocol 1: Profiling Proteasome Inhibitor Specificity Using Fluorogenic Assays
Objective: To determine the IC₅₀ values of a proteasome inhibitor against the three constitutive and three immunoproteasome catalytic subunits.
Principle: This assay utilizes fluorogenic peptide substrates that are specifically cleaved by each catalytic subunit. Upon proteolytic cleavage, the fluorophore is released, generating a fluorescent signal that is proportional to enzyme activity [71] [70].
Materials:
- Purified 20S constitutive proteasome and 20S immunoproteasome.
- Proteasome inhibitor (e.g., Bortezomib, dissolved in DMSO).
- Fluorogenic substrates:
- Suc-LLVY-AMC (for β5/caspase-like and β5i)
- Z-LLE-AMC (for β1/caspase-like and β1i)
- Bz-VGR-AMC (for β2/trypsin-like and β2i)
- Assay Buffer (e.g., 50 mM HEPES, pH 7.5, 5 mM EDTA, 0.05% PEG).
- Black 96-well or 384-well microplates.
- Fluorescence plate reader capable of excitation ~380 nm and emission ~460 nm.
Procedure:
- Enzyme Pre-incubation: In a microplate, serially dilute the inhibitor (e.g., 0.1 nM - 10 µM) in assay buffer. Include a DMSO-only control for 100% activity. Pre-incubate the proteasome (0.1-1 nM) with the inhibitor dilutions for 30-60 minutes at 37°C.
- Reaction Initiation: Initiate the reaction by adding the appropriate fluorogenic substrate (final concentration 10-50 µM) to each well.
- Kinetic Measurement: Immediately place the plate in the fluorometer and measure the fluorescence continuously or at 2-5 minute intervals for 60-120 minutes at 37°C.
- Data Analysis: Calculate the initial velocity (V₀) of the reaction for each inhibitor concentration from the linear range of the fluorescence vs. time curve. Normalize V₀ values as a percentage of the DMSO control activity. Plot % activity vs. inhibitor concentration and fit the data to a sigmoidal dose-response model to calculate the IC₅₀ for each subunit.
Table 1: Example Selectivity Profile of Clinical Proteasome Inhibitors [71]
| Compound | Chemical Nature | Primary Target (IC₅₀) | Secondary Targets (IC₅₀) | Reported Off-Target Liabilities |
|---|---|---|---|---|
| Bortezomib | Peptide boronate | β5 / β5i | β1 / β1i > β2i | Peripheral neuropathy, thrombocytopenia |
| Carfilzomib | Peptide epoxyketone | β5 / β5i | >> β2i ~ β1i | Cardiovascular events, acute renal failure |
| Marizomib | β-lactone | β5 / β5i | β2 / β2i > β1 / β1i | Psychiatric effects (e.g., euphoria, somnolence) |
Strategic Mitigation: From Inhibition to Targeted Degradation
A paradigm shift in addressing off-target effects is to move from inhibiting protein function to specifically directing protein destruction. Proteolysis-Targeting Chimeras (PROTACs) exemplify this strategy by hijacking the ubiquitin-proteasome system with much greater precision [73] [74].
The PROTAC Mechanism
PROTACs are heterobifunctional molecules consisting of a ligand for a protein of interest (POI) connected via a chemical linker to a ligand for an E3 ubiquitin ligase. This structure creates a ternary complex where the E3 ligase is brought into proximity with the POI, leading to its polyubiquitination and subsequent degradation by the proteasome [73]. The catalytic nature of PROTACs allows for sub-stoichiometric activity, potentially reducing the required drug exposure and associated off-target effects [74].
Diagram: Mechanism of Action of a PROTAC Molecule
Optimizing PROTAC Specificity
The specificity of a PROTAC is not solely determined by its POI-binding warhead but is critically dependent on the formation of a productive ternary complex. The following protocol outlines key steps for optimizing and validating PROTAC specificity.
Protocol 2: Assessing Ternary Complex Formation and Degradation Specificity
Objective: To confirm the formation of a specific POI-PROTAC-E3 ligase complex and identify the resulting degradome.
Materials:
- PROTAC molecule and matching inactive control (e.g., linker-deficient analog).
- Cell line expressing the POI and the recruited E3 ligase (e.g., VHL, CRBN).
- Control cell line lacking the E3 ligase (CRISPR knockout recommended).
- Antibodies for immunoprecipitation (against POI or E3 ligase) and western blotting (against POI, ubiquitin, and loading controls).
- Proteasome inhibitor (e.g., MG-132).
- Cycloheximide.
Procedure: Part A: Co-immunoprecipitation (Co-IP) of Ternary Complex
- Treat cells (E3+ and E3-) with PROTAC or inactive control (e.g., 1 µM for 1-2 hours) in the presence of MG-132 (10 µM) to stabilize the complex.
- Lyse cells with a mild, non-denaturing lysis buffer.
- Incubate the lysate with an antibody against the POI or the E3 ligase, coupled to protein A/G beads.
- Wash beads extensively to remove non-specifically bound proteins.
- Elute bound proteins and analyze by western blotting. Probe for the POI, the E3 ligase, and the other component of the complex. Successful complex formation is indicated by the co-precipitation of all three components specifically in E3+ cells treated with the active PROTAC.
Part B: Degradation Kinetics and Specificity (Degradome) Analysis
- Treat cells (E3+ and E3-) with a range of PROTAC concentrations (e.g., 1 nM - 1 µM) over a time course (e.g., 0, 2, 4, 8, 24 hours) in the presence of cycloheximide to block new protein synthesis.
- Harvest cells and prepare lysates for western blotting.
- Probe for the POI to establish DC₅₀ and Dmax values. Confirm on-target mechanism by pre-treating with a proteasome inhibitor (MG-132) or an E1 inhibitor, which should block degradation.
- For global degradome analysis, use tandem mass tag (TMT) proteomics. Compare the protein abundance profiles of cells treated with active PROTAC vs. inactive control. This identifies proteins whose levels change significantly, revealing on-target degradation and potential off-target degradants.
Table 2: Key Research Reagent Solutions for UPS and TPD Research
| Reagent / Tool | Function / Application | Key Consideration for Specificity |
|---|---|---|
| Activity-Based Probes (ABPs) | Label active-site residues of proteasome subunits or DUBs. | Enables monitoring of target engagement and occupancy in cell and animal models [70]. |
| Orthogonal E1-E2 Pairs | Engineered E1 and E2 enzymes that function exclusively with engineered ubiquitin [28]. | Isolates the activity of a specific E2-dependent cascade, eliminating cross-talk with native systems. |
| Inactive PROTAC Control | A PROTAC analog with a broken linker or inactive E3 ligand. | Critical control to distinguish on-target degradation from warhead- or linker-mediated off-target effects [73]. |
| E3 Ligase Knockout Cells | Isogenic cell lines lacking a specific E3 ligase (e.g., CRBN, VHL). | Confirms that PROTAC activity is mechanistically dependent on the intended E3 ligase. |
| Cellular Thermal Shift Assay (CETSA) | Measures drug-induced thermal stabilization of target proteins. | Validates direct binding of an inhibitor to its intended protein target in a cellular context. |
Addressing off-target effects in proteasome and ubiquitin system inhibition is a multifaceted challenge that requires a combination of rigorous selectivity profiling and the adoption of next-generation therapeutic modalities. By employing detailed enzymatic profiling and leveraging catalytic, event-driven technologies like PROTACs, researchers can significantly enhance the specificity of their interventions in the E1-E2-E3 cascade. The protocols and strategies outlined herein provide a framework for systematically evaluating and improving the specificity of UPS-targeting compounds, thereby de-risking their path toward clinical translation.
Technical Barriers in Ubiquitin Chain Analysis and Substrate Identification
The ubiquitin-proteasome system (UPS) is a central regulator of protein turnover and signaling, with E3 ubiquitin ligases conferring substrate specificity and chain-type control [54]. The process of ubiquitination involves a sophisticated enzymatic cascade commencing with ubiquitin activation by an E1 enzyme, followed by transfer to an E2 conjugating enzyme, and finally delivered to a substrate via an E3 ligase [75]. This system regulates diverse cellular functions including proteasomal degradation, signal transduction, DNA repair, and immune responses [76] [75].
The versatility of ubiquitin signaling stems from the complexity of ubiquitin conjugates, which can range from a single ubiquitin monomer to polymers with different lengths and linkage types [77]. Ubiquitin itself contains seven lysine residues (K6, K11, K27, K29, K33, K48, K63) and an N-terminal methionine (M1) that can be ubiquitinated, enabling formation of ubiquitin chains with varying topology and linkages [6]. Additionally, recent discoveries have revealed that ubiquitin can modify non-proteinaceous substrates including phospholipids, carbohydrates, glycolipids, metabolites, and nucleic acids, further expanding the functional scope of ubiquitination [78] [6].
Table 1: Key Components of the Ubiquitin Enzymatic Cascade
| Component | Number in Humans | Primary Function | Notable Characteristics |
|---|---|---|---|
| E1 Activasing Enzymes | 2 [77] | Activates ubiquitin in an ATP-dependent manner | Initiates the entire ubiquitination cascade |
| E2 Conjugating Enzymes | ~40 [77] | Accepts ubiquitin from E1 and cooperates with E3 for substrate transfer | Determines chemoselectivity and linkage specificity [7] |
| E3 Ligases | >600 [76] | Confers substrate specificity | Over 1000 estimated in human genome [77]; RING, HECT, RBR types |
| Deubiquitinases (DUBs) | ~100 [77] | Removes ubiquitin modifications | Cleaves ubiquitin from substrates and disassembles chains |
Technical Barriers in Ubiquitin Analysis
Key Analytical Challenges
The structural and functional diversity of ubiquitin signals presents substantial technical hurdles for comprehensive analysis. Several interconnected barriers impede progress in this field:
Low Stoichiometry and Dynamic Nature: The stoichiometry of protein ubiquitination is typically very low under normal physiological conditions, dramatically increasing the difficulty of identifying ubiquitinated substrates [77]. Furthermore, ubiquitination is a highly dynamic modification due to the constant activity of deubiquitinating enzymes (DUBs), making capture of transient modifications particularly challenging.
Structural Complexity of Ubiquitin Chains: Ubiquitin can modify substrates at one or several lysine residues simultaneously, significantly complicating the localization of specific ubiquitination sites [77]. Additionally, the capacity of ubiquitin to form chains of varying length, linkage type (homotypic, mixed, or branched), and architecture creates an enormous diversity of potential signals that are difficult to deconvolute [6] [77].
Limitations of Current Analytical Tools: Traditional methods for ubiquitination analysis often lack the sensitivity, specificity, or throughput required to fully characterize the ubiquitin code. Antibody-based approaches may exhibit linkage bias or limited affinity, while mass spectrometry-based methods struggle with identifying rare modifications and distinguishing isopeptide linkages amidst complex backgrounds [77].
Specific Methodological Limitations
Table 2: Technical Barriers in Ubiquitin Chain Analysis
| Barrier Category | Specific Challenges | Impact on Research |
|---|---|---|
| Enrichment and Capture | Non-specific binding with affinity tags [77]; Low abundance of endogenous ubiquitination events; Inability to preserve labile ubiquitin linkages (oxyester, thioester) [78] | Reduced sensitivity and potential artifacts in ubiquitin profiling |
| Linkage-Specific Analysis | Limited availability of high-quality linkage-specific reagents; Difficulty in analyzing atypical chains (K6, K11, K27, K29, K33) [77]; Inability to detect branched ubiquitin chains | Incomplete understanding of ubiquitin chain functions |
| Technology Access | Sophisticated instrumentation requirements (e.g., high-resolution mass spectrometry); Labor-intensive protocols; High cost of specific antibodies and affinity matrices | Limited accessibility for many research laboratories |
| Non-Proteinaceous Ubiquitination | Lack of standardized tools and reference materials for studying ubiquitination of lipids, carbohydrates, and nucleic acids [78] [79] | Emerging field with limited methodological foundation |
Current Methodologies and Applications
Established Workflows for Ubiquitin Analysis
Several strategic approaches have been developed to overcome the technical barriers in ubiquitin analysis, each with distinct advantages and limitations:
Ubiquitin Tagging-Based Approaches: These methods involve engineering epitope tags (Flag, HA, V5, Myc, Strep, His) or protein/domain tags (GST, MBP, SUMO) onto ubiquitin to facilitate purification of ubiquitinated substrates [77]. The tagged ubiquitin is expressed in cells, where it becomes incorporated into endogenous ubiquitination pathways, enabling subsequent affinity purification and identification of ubiquitinated proteins. While this approach is relatively accessible and cost-effective, concerns remain about potential structural perturbations and the inability to apply this method to clinical or animal tissue samples without genetic manipulation [77].
Antibody-Based Enrichment Strategies: This methodology utilizes anti-ubiquitin antibodies (e.g., P4D1, FK1/FK2) that recognize all ubiquitin linkages or linkage-specific antibodies (M1-, K11-, K27-, K48-, K63-linkage specific) to enrich endogenously ubiquitinated substrates from complex biological samples [77]. This approach preserves physiological relevance by studying endogenous ubiquitination without genetic manipulation and enables investigation of specific chain types. However, it is limited by the high cost of high-quality antibodies, potential linkage bias, and non-specific binding issues that can reduce sensitivity [77].
Ubiquitin-Binding Domain (UBD)-Based Approaches: This technique exploits natural ubiquitin recognition modules, such as tandem-repeated ubiquitin-binding entities (TUBEs), to capture ubiquitinated proteins with high affinity and protect them from deubiquitination during processing [76] [77]. TUBEs offer enhanced affinity compared to single UBDs and can be engineered for linkage specificity, making them particularly valuable for studying specific chain types and preserving labile ubiquitin modifications.
The following workflow diagram illustrates a comprehensive approach for linkage-specific ubiquitination analysis integrating these methodologies:
Specialized Protocol: TUBE-Based Linkage-Specific Ubiquitination Analysis
This protocol details the methodology for using Tandem Ubiquitin Binding Entities (TUBEs) to capture and analyze linkage-specific ubiquitination of endogenous proteins, adapted from studies on RIPK2 ubiquitination [76].
Principle
Chain-specific TUBEs with nanomolar affinities for particular polyubiquitin chains (e.g., K48 or K63-linked) are utilized to selectively capture endogenous ubiquitinated proteins, enabling differentiation between degradative and signaling ubiquitination events in response to specific cellular stimuli [76].
Materials and Reagents
- Cell Line: THP-1 human monocytic cells (or other relevant cell type)
- Stimuli: L18-MDP (200-500 ng/mL) for K63 ubiquitination induction; PROTAC compound for K48 ubiquitination induction
- Inhibitors: Ponatinib (100 nM) for RIPK2 inhibition; protease and deubiquitinase inhibitor cocktails
- TUBE Reagents: K48-specific TUBEs, K63-specific TUBEs, Pan-selective TUBEs
- Antibodies: Anti-target protein (e.g., anti-RIPK2), anti-ubiquitin, secondary antibodies
- Lysis Buffer: Optimized to preserve polyubiquitination (e.g., 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% NP-40, 1 mM EDTA, plus fresh protease and deubiquitinase inhibitors)
- Coating Buffer: For immobilizing TUBEs in 96-well plates (e.g., PBS or carbonate-bicarbonate buffer)
Procedure
Cell Treatment and Stimulation:
- Culture THP-1 cells in appropriate medium and maintain at optimal density.
- Pre-treat cells with inhibitors (e.g., 100 nM Ponatinib) or DMSO control for 30 minutes.
- Stimulate cells with L18-MDP (200 ng/mL) for 30 minutes to induce K63-linked ubiquitination or with PROTAC compounds to induce K48-linked ubiquitination.
Cell Lysis and Protein Extraction:
- Lyse cells in optimized lysis buffer (200-500 μL per 10⁶ cells) with constant agitation at 4°C for 30 minutes.
- Clarify lysates by centrifugation at 14,000 × g for 15 minutes at 4°C.
- Transfer supernatant to fresh tubes and perform protein quantification.
TUBE-Based Capture of Ubiquitinated Proteins:
- Immobilize chain-specific TUBEs (K48-TUBE, K63-TUBE, or Pan-TUBE) in wells of a 96-well plate by incubating with coating buffer overnight at 4°C.
- Block plates with appropriate blocking buffer (e.g., 3% BSA in TBST) for 2 hours at room temperature.
- Apply 50-100 μg of clarified cell lysate per well and incubate for 2-4 hours at 4°C with gentle shaking.
- Wash wells extensively with wash buffer to remove non-specifically bound proteins.
Detection and Analysis:
- Elute bound proteins with SDS-PAGE sample buffer or directly detect ubiquitinated targets by immunoblotting.
- For immunoblotting: Resolve proteins by SDS-PAGE, transfer to PVDF membranes, and probe with anti-target protein (e.g., anti-RIPK2) and appropriate secondary antibodies.
- Quantify band intensities using densitometry software to compare ubiquitination levels across conditions.
Validation and Specificity Controls:
- Include control wells without TUBE coating to assess non-specific binding.
- Use linkage-specific ubiquitin standards to verify TUBE specificity.
- Confirm ubiquitination by reprobing membranes with anti-ubiquitin antibodies.
Applications and Data Interpretation
This TUBE-based methodology enables researchers to:
- Differentiate between K48-linked (proteasomal targeting) and K63-linked (signaling) ubiquitination in response to specific stimuli [76]
- Capture and quantify ubiquitination of endogenous proteins without overexpression artifacts
- Monitor temporal changes in ubiquitination linkage patterns
- Assess efficacy and mechanism of action of PROTAC compounds and DUB inhibitors
Advanced Research Applications
Methodological Innovations
Recent technological advances have expanded the toolbox for ubiquitin research, particularly in the areas of linkage-specific analysis and emerging ubiquitination types:
Chain-Specific TUBE Assays: The development of high-throughput screening assays leveraging chain-specific TUBEs enables precise capture of linkage-specific polyubiquitination events on native target proteins with high sensitivity [76]. This method facilitates analysis of ubiquitin linkage diversity in response to various stimuli and can differentiate between inflammatory signaling (K63-linked) and targeted protein degradation (K48-linked) events, as demonstrated in studies of RIPK2 ubiquitination dynamics [76].
Analysis of Non-Proteinaceous Ubiquitination: The discovery that ubiquitin can modify non-proteinaceous substrates including phospholipids, carbohydrates, and nucleic acids has created new frontiers in ubiquitin research [78]. For example, the RBR E3 ligase HOIL-1 can ubiquitinate serine and threonine residues as well as various di- and monosaccharides in vitro [79]. Specialized methodologies are being developed to study these atypical modifications, including engineered E3 ligases for generating ubiquitinated tool compounds and standards [79].
E3-Independent Ubiquitination Strategies: Structure-guided engineering has enabled the development of E3 ligase-free protein ubiquitination using engineered UBE2E1 variants [80]. This sequence-dependent ubiquitination using UBE2E1 (SUE1) approach efficiently generates ubiquitinated proteins with customized modification sites, ubiquitin chain linkages, and lengths, bypassing the challenge of identifying specific E3 ligases for target proteins [80].
Research Reagent Solutions
Table 3: Essential Research Reagents for Ubiquitin Analysis
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| Affinity Capture Reagents | Tandem Ubiquitin Binding Entities (TUBEs) [76]; Linkage-specific antibodies (K48, K63, M1) [77]; His/Strep-tagged ubiquitin constructs [77] | Enrichment of ubiquitinated proteins from complex mixtures with linkage specificity |
| Activity-Based Probes | Ubiquitin vinyl sulfones; HA-Ub-VS; Biotin-labeled ubiquitin probes | Profiling deubiquitinase activity and ubiquitin interactions |
| Engineered Enzymes | Constitutively active HOIL-1 variants [79]; UBE2E1 mutants for E3-free ubiquitination [80]; Ubch5c P121G/E122S mutant [80] | Generation of defined ubiquitinated compounds and tool development |
| Specialized Substrates | Ubiquitinated saccharides [79]; Ubiquitinated peptide libraries; Defined ubiquitin chain linkages | Standards for assay development and method validation |
| Cell-Based Tools | StUbEx cellular system [77]; Reporter cell lines (NanoLuc fusions) [76]; DUB knockout cell lines | Functional validation of ubiquitination events in physiological contexts |
The field of ubiquitin research continues to face significant technical challenges in the comprehensive analysis of ubiquitin chains and substrate identification. The structural complexity and dynamic nature of ubiquitin signaling, coupled with the expanding repertoire of ubiquitination targets beyond proteins, demands increasingly sophisticated methodological approaches. Current strategies employing TUBE-based enrichment, linkage-specific reagents, and advanced mass spectrometry techniques have substantially advanced our capacity to decipher the ubiquitin code.
The ongoing development of novel research tools, including engineered ubiquitination enzymes, defined ubiquitin chain standards, and high-throughput compatible assays, promises to accelerate progress in this field. These methodological advances are particularly crucial for drug discovery efforts targeting the ubiquitin-proteasome system, such as PROTAC development and DUB inhibitor screening, where precise understanding of ubiquitin linkage specificity and substrate selection is essential for therapeutic efficacy. As these technologies mature and become more accessible, they will undoubtedly unlock new opportunities for fundamental biological insights and therapeutic interventions targeting the ubiquitin system.
Mechanisms of Therapeutic Resistance to Proteasome Inhibitors and UPS-Targeted Agents
The ubiquitin-proteasome system (UPS) serves as a critical pathway for regulated intracellular protein degradation in eukaryotic cells, and its proteasome core has been successfully leveraged as a therapeutic target in hematological malignancies, particularly multiple myeloma and mantle cell lymphoma [81] [82]. The enzymatic cascade begins with E1 activating enzymes, which initiate the ATP-dependent activation of ubiquitin, followed by E2 conjugating enzymes and E3 ligases that collectively mediate the specific attachment of ubiquitin chains to protein substrates, marking them for degradation by the 26S proteasome [36] [83] [43]. Proteasome inhibitors (PIs), such as bortezomib and carfilzomib, disrupt this pathway by binding the catalytic β-subunits of the 20S proteasome core particle, leading to the accumulation of polyubiquitinated proteins and ultimately apoptosis in malignant cells [81] [82]. Despite the transformative clinical success of these agents, therapeutic efficacy is invariably limited by the emergence of drug resistance, a multifaceted phenomenon involving alterations in the proteasome itself, adaptive changes in cellular stress response pathways, and rewiring of the broader ubiquitin network [81] [84] [85]. This Application Note delineates the principal molecular mechanisms underlying resistance to UPS-targeted therapies and provides detailed experimental protocols for their investigation within the context of E1-E2-E3 ubiquitin cascade research.
Core Mechanisms of Resistance
Resistance to proteasome inhibitors arises through a complex interplay of tumor-intrinsic adaptations. The major documented mechanisms include direct target modification, compensatory elevation of proteasome activity, and enhanced capacity for protein homeostasis.
Table 1: Major Mechanisms of Resistance to Proteasome Inhibitors
| Resistance Mechanism | Molecular Alteration | Functional Consequence | Supporting Evidence |
|---|---|---|---|
| Target Mutation | Mutations in the PSMB5 gene encoding the β5 proteasome subunit [81] | Reduced drug-binding affinity; maintained chymotrypsin-like (CT-L) activity [81] | Acquired in bortezomib-resistant cell lines [81] |
| Proteasome Subunit Upregulation | Overexpression of PSMB5 (β5) and PSMB6 (β1) subunits [81] | Increased proteasome abundance and capacity; elevated peptidase activity [81] | Observed in vitro in PI-resistant cell lines [81] |
| Immunoproteasome Induction | Increased expression of β1i, β2i, and β5i subunits [81] | Altered catalytic site specificity and inhibitor sensitivity [81] | Adaptive response in some resistant models [81] |
| Dysregulation of Deubiquitinases (DUBs) | Overexpression of specific Ubiquitin-Specific Proteases (USPs) [86] | Enhanced deubiquitination, counteracting polyubiquitin accumulation from partial PI inhibition [86] | Associated with resistance to chemotherapy and targeted therapies in various cancers [86] |
The table above summarizes key tumor-intrinsic resistance mechanisms. Beyond these, resistance is also fueled by cellular adaptations that reduce dependence on proteasomal degradation. Malignant cells can activate alternative protein clearance pathways, such as autophagy, and upregulate anti-apoptotic proteins to survive the proteotoxic stress induced by PIs [84]. Furthermore, the tumor microenvironment contributes to resistance by providing pro-survival signals and promoting the selection of resistant subclones [84] [85]. A comprehensive understanding of these intertwined mechanisms is essential for developing effective strategies to overcome resistance.
Quantitative Profiling of Resistance Phenotypes
Characterizing the functional and molecular profile of resistant cells is a critical first step. The following tables provide a framework for quantifying key resistance parameters.
Table 2: Quantified Proteasome Activity Profiles in Resistant vs. Sensitive Cells
| Proteasome Activity Type | Sensitive Cell IC₅₀ (nM) | Resistant Cell IC₅₀ (nM) | Fold Change | Assay Method |
|---|---|---|---|---|
| Chymotrypsin-like (β5) | 7.9 [81] | >100 [81] | >12.7x | Fluorogenic peptide substrate (e.g., Suc-LLVY-AMC) |
| Caspase-like (β1) | 53 [81] | ~500 (estimated) | ~9.4x | Fluorogenic peptide substrate (e.g., Z-LLE-AMC) |
| Trypsin-like (β2) | 590 [81] | >5000 [81] | >8.5x | Fluorogenic peptide substrate (e.g., Boc-LRR-AMC) |
Table 3: Expression Level Changes of UPS Components in Resistance
| Gene/Protein Target | Function | Fold Change in Resistant Cells | Detection Method |
|---|---|---|---|
| PSMB5 | β5 proteasome subunit (CT-L activity) | ↑ 2-5x [81] | qPCR, Western Blot |
| PSMB8 (β5i) | Immunoproteasome subunit | ↑ 3-8x [81] | qPCR, Western Blot |
| USP14 | Proteasome-associated DUB | ↑ 2-4x [86] | qPCR, Western Blot, Activity Probe |
| E1 (UBA1) | Ubiquitin-activating enzyme | Variable / Context-dependent | qPCR, Western Blot |
Experimental Protocols
This section provides detailed methodologies for investigating resistance mechanisms, with a focus on the ubiquitin cascade.
Protocol 1: Profiling the E1-E2-E3 Ubiquitin Cascade in PI-Resistant Cells
Objective: To systematically analyze the activity and expression of E1 activating, E2 conjugating, and E3 ligase enzymes in PI-resistant versus sensitive cell lines.
Materials:
- Cell Lines: Isogenic pairs of PI-sensitive and PI-resistant cell lines (e.g., MM.1S vs. ANBL-6.V10R).
- Lysis Buffer: RIPA buffer supplemented with 50mM N-Ethylmaleimide (NEM) to inhibit DUBs, and complete protease inhibitors.
- Antibodies: Anti-Ubiquitin (P4D1), Anti-UBA1 (E1), various E2s (e.g., UbcH5a, Ubc13), E3s (e.g., HOIL-1 [87]), and GAPDH (loading control).
- ATP-Regeneration System: 50mM Tris-HCl (pH 7.5), 5mM MgCl₂, 0.2mM DTT, 10mM Phosphocreatine, 10U Creatine Phosphokinase, 50μM ATP.
Procedure:
- Cell Lysis and Quantification: Harvest and lyse 1x10⁷ cells in 1 mL of ice-cold NEM-supplemented lysis buffer. Clarify lysates by centrifugation at 16,000 x g for 15 minutes at 4°C. Determine protein concentration.
- Global Ubiquitination Assessment (Western Blot): Resolve 30 μg of total protein per lane on a 4-12% Bis-Tris gel. Transfer to PVDF membrane and probe with anti-Ubiquitin antibody. High-molecular-weight smearing indicates accumulated polyubiquitinated proteins.
- E1 Enzyme Activity Assay (Thioester Formation):
- Incubate 100 μg of cell lysate with 2 μg of recombinant His-Ubiquitin and the ATP-regeneration system in a total volume of 50 μL for 30 minutes at 37°C.
- Stop the reaction by adding non-reducing Laemmli sample buffer (without DTT or β-mercaptoethanol).
- Resolve proteins on a non-reducing 4-12% Bis-Tris gel and Western blot with anti-Ubiquitin antibody. Covalent E1~Ub thioester intermediates will appear as a band shift that disappears under reducing conditions.
- E2 and E3 Activity Profiling:
- In vitro Ubiquitin Discharge: Pre-charge 100 ng of recombinant E1 and 200 ng of recombinant E2 (e.g., UbcH5a) with His-Ubiquitin in the presence of ATP for 10 minutes. Add 200 ng of recombinant E3 (e.g., HOIL-1 [87]) and take aliquots at 0, 5, 15, and 30 minutes. Stop with non-reducing sample buffer and analyze by non-reducing Western blot with anti-Ubiquitin to monitor the disappearance of the E2~Ub band.
- Auto-ubiquitination: Incubate recombinant E1, E2, E3, and His-Ubiquitin with ATP for 1 hour. Analyze by reducing Western blot with an antibody against the E3 to detect higher molecular weight auto-ubiquitination products.
Data Analysis: Compare the intensity of E1~Ub and E2~Ub bands, the rate of E2~Ub discharge, and the extent of E3 auto-ubiquitination between sensitive and resistant cell lysates. A decreased rate of ubiquitin transfer may indicate dysregulation at specific cascade steps contributing to resistance.
Protocol 2: Functional Reconstitution of a Minimal Ubiquitin Cascade
Objective: To biochemically characterize mutations in UPS components identified from resistant cells, using a synthetic biology approach inspired by archaeal systems [83].
Materials:
- Recombinant Proteins: Purified E1 (UBA1), E2 (UbcH5a), E3 (e.g., HOIL-1 [87]), and Ubiquitin, produced from bacterial or insect cell expression systems.
- Reaction Buffer: 50mM Tris-HCl (pH 7.5), 5mM MgCl₂, 0.2mM DTT, 2mM ATP.
- Substrate: Recombinant protein substrate of the chosen E3 (e.g., a LUBAC component for HOIL-1).
Procedure:
- Cascade Assembly: In a 50 μL reaction volume, combine 100nM E1, 500nM E2, 1μM E3, 50μM Ubiquitin, and 5μM substrate protein in reaction buffer.
- Time-Course Incubation: Incubate at 30°C. Remove 10 μL aliquots at 0, 10, 30, and 60 minutes.
- Reaction Termination: Stop each aliquot by adding SDS-PAGE loading buffer containing DTT.
- Analysis:
- Western Blot: Resolve proteins by SDS-PAGE and probe with anti-Ubiquitin and anti-substrate antibodies to monitor the formation of ubiquitinated products.
- Mass Spectrometry: For precise mapping of ubiquitination sites, scale up the reaction, resolve proteins by gel electrophoresis, excise the modified substrate band, and analyze via LC-MS/MS.
Data Analysis: Compare the efficiency of ubiquitin chain formation and the specific lysine (or non-canonical serine/threonine [87]) linkage types generated by wild-type versus mutant E3 enzymes. Impaired catalysis or altered linkage specificity can reveal mechanistic insights into resistance.
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents for Investigating PI Resistance Mechanisms
| Reagent / Tool | Specific Example | Research Application | Key Function |
|---|---|---|---|
| Activity-Based Probes | Ubiquitin-VS (Vinyl Sulfone) [86] | Profiling active deubiquitinating enzymes (DUBs) | Covalently labels active site cysteine of DUBs for detection and enrichment |
| Fluorogenic Peptide Substrates | Suc-LLVY-AMC, Z-LLE-AMC, Boc-LRR-AMC [81] [82] | Measuring β5, β1, and β2 proteasome activities | Proteasome cleavage releases fluorescent AMC, quantified by fluorometry |
| E1 Inhibitors | TAK-243 (MLN7243) [36] | Blocking global ubiquitin activation | Inhibits UBA1, used to test synthetic lethality with PIs |
| Recombinant E3 Ligases | HOIL-1 (RBR E3) [87] | In vitro ubiquitination assays | Ubiquitinates protein Ser/Thr and non-protein substrates (e.g., sugars) [87] |
| DUB Inhibitors | USP14/UCHL5 inhibitors (e.g., b-AP15) [86] | Targeting proteasome-associated DUBs | Induces ubiquitin accumulation; overcomes PI resistance in some models |
Signaling Pathway and Experimental Workflow Diagrams
The following diagrams visualize the core ubiquitin-proteasome pathway and a key experimental workflow for profiling E1-E2-E3 activity.
Optimizing Screening Strategies and Compound Validation for UPS Drug Discovery
The ubiquitin-proteasome system (UPS) represents a high-value but challenging target class for drug discovery. The enzymatic cascade, involving E1 activating, E2 conjugating, and E3 ligase enzymes, regulates virtually all aspects of cellular physiology through targeted protein degradation and signaling [88]. Table 1 summarizes the core enzymatic components of the ubiquitin cascade and their characteristics as drug targets. Recent advances have revealed that ubiquitination extends beyond proteins to include non-protein biomolecules and even drug-like small molecules, expanding both the potential applications and complexities of UPS drug discovery [89] [90].
Table 1: Core Enzymes of the Ubiquitin-Proteasome System
| Enzyme Class | Human Genes | Key Function | Targetability |
|---|---|---|---|
| E1 Activating Enzymes | 2 (UBA1, UBA6) | Ubiquitin activation; apex of cascade | Challenging; limited specificity |
| E2 Conjugating Enzymes | ~38 | Ubiquitin chain topology determination | Improved specificity potential |
| E3 Ligases | ~600-700 | Substrate specificity | Highest theoretical specificity |
| Deubiquitinases (DUBs) | ~100 | Ubiquitin removal | Diverse mechanisms |
The discovery that the human E3 ligase HUWE1 can ubiquitinate drug-like small molecules containing primary amino groups represents a paradigm shift, revealing that compounds previously characterized as inhibitors may actually serve as substrates for their target ligases [89]. This finding necessitates more sophisticated validation strategies in UPS drug discovery programs.
Advanced Screening Methodologies
MALDI-TOF Mass Spectrometry Screening
The MALDI-TOF E2/E3 assay provides a universal, label-free high-throughput screening (HTS) platform for ubiquitin E2 conjugating enzymes and E3 ligases [91]. This methodology enables testing of E2/E3 active pairs, inhibitor potency and specificity, and library screening without requiring chemical or fluorescent probes.
Key Advantages:
- Label-free detection: Uses unmodified mono-ubiquitin as substrate
- Universal application: Suitable for all E3 ligase families (RING, HECT, RBR)
- Reduced reagent consumption: 5μL reaction volumes
- High reproducibility: Z'-factor >0.7 in 384-well format
Experimental Protocol: MALDI-TOF E2/E3 Assay
Reaction Setup:
- Prepare ubiquitination reaction in 5μL total volume:
- 1 mM ATP
- 3.125-12.5 μM ubiquitin
- 50 nM E1 activating enzyme
- 250 nM E2 conjugating enzyme
- 250-500 nM E3 ligase
- Compound library members (typically 10-50 μM)
Incubate at 37°C for 30 minutes
Terminate reaction with 2.5 μL of 10% (v/v) trifluoroacetic acid
MS Analysis:
- Spot 0.5 μL reaction mixture with 0.5 μL sinapinic acid matrix
- Acquire mass spectra in linear positive mode
- Monitor depletion of mono-ubiquitin (8565 Da) as primary readout
- Quantify inhibition percentage relative to DMSO controls
Validation: This platform has been validated against diverse E3 ligases including MDM2 (RING), ITCH (HECT), and HOIP (RBR), demonstrating its broad applicability across E3 mechanisms [91].
Covalent Fragment-Based Screening
For challenging targets like bacterial E3 ligases, covalent fragment-based drug discovery provides an alternative strategy. This approach is particularly valuable for targeting catalytic cysteines in HECT-type and RBR-type E3 ligases [92].
Experimental Protocol: Covalent Fragment Screening
Library Design:
- Curate 227-compound library with chloroacetamide warheads
- Maintain molecular weight range: 162-321 Da
- Maintain clogP range: -1.4 to 3.4
- Include diversity of chemical scaffolds
Screening Process:
- Incubate recombinant E3 ligases (0.5 μM) with fragments (50 μM)
- Maintain temperature at 4°C for 24 hours
- Analyze by intact protein LC-MS
- Calculate labeling percentage by comparing protein and protein-fragment molecular weights
Hit Elaboration via HTC-D2B:
- Design amine libraries based on Tanimoto similarity to hits
- Perform in-situ amide coupling with N-(chloroacetoxy)succinimide
- Direct screening of crude reaction mixtures
- Rapid SAR establishment without purification steps
This approach successfully identified the first inhibitors of Salmonella SspH1 and SspH2 bacterial E3 ligases, demonstrating its utility for challenging targets [92].
Specialized Screening Considerations
Membrane-Associated Ubiquitination Cascades
ER-associated degradation (ERAD) components present unique screening challenges due to their membrane association. Research reveals that the E2 enzyme UBE2J2 exhibits sensitivity to membrane lipid saturation, with activity modulated by lipid packing density [7].
Key Findings:
- UBE2J2 is inactive in ER-like membranes (33% saturated fatty acids)
- Activity increases markedly in membranes with 50% saturated fatty acids
- This lipid sensitivity extends downstream to multiple E3 ligases (RNF145, MARCHF6, RNF139)
Screening Implications:
- Screening campaigns for membrane-associated UPS components must incorporate physiologically relevant lipid environments
- Detergent-based assays may misrepresent compound effects
- Consider lipid composition as a variable in assay optimization
Orthogonal E1-E2 Tools
Recent engineering of the Uba1-VHH05 fusion enzyme enables selective ubiquitin transfer to tagged E2 enzymes, creating orthogonal ubiquitination cascades for specific screening applications [35]. This tool allows precise dissection of E2-specific functions without altering other cascade components.
Compound Validation and Mechanistic Studies
Distinguishing Inhibitors from Substrates
The discovery that purported HUWE1 inhibitors (BI8622, BI8626) are actually substrates highlights the critical importance of rigorous mechanistic validation [89].
Experimental Protocol: Substrate vs. Inhibitor Discrimination
Ubiquitination Detection:
- Conduct full ubiquitination cascade reactions with test compounds
- Separate by SDS-PAGE and excise ~9 kDa Ub-containing band
- Perform LysC protease digestion of Ub
- Analyze by MS/MS for modified Ub peptides (+408.21 Da for BI8622; +422.23 Da for BI8626)
- Confirm sequence coverage and modification sites
Specificity Profiling:
- Test compounds against multiple E3 ligase families
- Employ cellular detection methods to confirm target engagement
- Monitor proteomic effects to assess specificity
Key Structural Determinants: Primary amino groups in compound structures can serve as ubiquitination sites, necessitating particular scrutiny of compounds containing this functionality [89].
Cellular Validation Approaches
While in vitro screening provides initial hits, cellular validation remains essential due to compartmentalization and regulatory networks.
Experimental Protocol: Cellular Ubiquitination Monitoring
Methods:
- Implement ubiquitin remnant immunoaffinity enrichment
- Utilize tandem ubiquitin-binding entities (TUBEs) for ubiquitin chain capture
- Apply linkage-specific ubiquitin antibodies (K48, K63, M1)
- Monitor endogenous substrate stabilization (e.g., MCL1 for HUWE1)
Functional Assessment:
- Evaluate cytotoxic effects in disease-relevant cell lines
- Measure proteome-wide ubiquitination changes
- Assess effects on pathway activation (NF-κB, NLRP3 inflammasome)
Research Reagent Solutions
Table 2: Essential Research Reagents for UPS Drug Discovery
| Reagent Category | Specific Examples | Function/Application |
|---|---|---|
| E1 Enzymes | UBA1, UBA6, NAE | Ubiquitin/NEDD8 activation; cascade initiation |
| E2 Enzymes | UBE2L3, UBE2D3, UBE2N/V1, UBE2J2 | Ubiquitin conjugation; chain topology determination |
| E3 Ligases | HUWE1 (HECT), MDM2 (RING), HOIP (RBR) | Substrate recognition; specificity determination |
| Specialized Tools | Uba1-VHH05 fusion, Ub-Dha probe | Orthogonal cascades; activity-based profiling |
| Detection Reagents | Linkage-specific Ub antibodies, TUBEs | Ubiquitination readout; signal quantification |
Optimizing screening strategies for UPS drug discovery requires integrated approaches that address the complexity and context-dependence of ubiquitination cascades. The combination of label-free biochemical screening, covalent fragment-based approaches, and physiologically relevant assay conditions provides a robust foundation for identifying and validating UPS-targeting compounds. Future directions will likely include increased emphasis on membrane-environment recapitulation, expanded use of structural biology in compound optimization, and development of more sophisticated cellular models for validation.
Therapeutic Validation and Comparative Analysis of Ubiquitin-Targeting Agents
The ubiquitin-proteasome system (UPS) is the primary pathway for regulated intracellular protein degradation in eukaryotic cells, playing a critical role in maintaining cellular homeostasis. This system involves a sequential E1-E2-E3 enzymatic cascade that tags target proteins with ubiquitin for destruction by the 26S proteasome [29] [4]. The discovery that cancer cells, particularly multiple myeloma (MM) cells, are more sensitive to proteasome inhibition than normal cells transformed this basic biological understanding into a revolutionary therapeutic strategy [93]. This application note details the clinical translation of proteasome inhibitors (PIs) from fundamental ubiquitin cascade research to established oncology treatments, providing experimental protocols and analytical frameworks for researchers and drug development professionals.
The Ubiquitin-Proteasome Pathway: Mechanism and Therapeutic Rationale
The Enzymatic Cascade
Protein ubiquitination begins with a three-step enzymatic cascade. The E1 (ubiquitin-activating) enzyme activates ubiquitin in an ATP-dependent process, forming a high-energy thioester bond. The activated ubiquitin is then transferred to an E2 (ubiquitin-conjugating) enzyme. Finally, an E3 (ubiquitin ligase) enzyme facilitates the transfer of ubiquitin from the E2 to a specific substrate protein, forming an isopeptide bond between the C-terminal glycine of ubiquitin and the ε-amino group of a lysine residue on the substrate [29] [94]. Polyubiquitin chains linked through Lys48 of ubiquitin typically target proteins for degradation by the 26S proteasome [4].
Therapeutic Targeting Rationale
The therapeutic rationale for targeting the proteasome in multiple myeloma is multifaceted. MM cells produce massive quantities of monoclonal immunoglobulins, creating significant endoplasmic reticulum stress and dependence on the UPS to clear misfolded proteins [93]. PIs disrupt this delicate balance, leading to the accumulation of toxic proteins and initiation of apoptosis through multiple mechanisms, including NF-κB pathway inhibition, disruption of cytokine signaling, and induction of the unfolded protein response [93]. The heightened sensitivity of MM cells to proteasome inhibition compared to normal cells provides the critical therapeutic window exploited by PI-based therapies.
Clinically Approved Proteasome Inhibitors: Mechanism and Evolution
Since the initial approval of bortezomib in 2003, the PI class has expanded to include second-generation agents with distinct pharmacological properties. The table below summarizes the key characteristics of major clinically approved PIs.
Table 1: Clinically Approved Proteasome Inhibitors for Multiple Myeloma
| Characteristic | Bortezomib | Carfilzomib | Ixazomib |
|---|---|---|---|
| Chemical Class | Boronate | Epoxyketone | Boronate |
| Binding Mechanism | Reversible | Irreversible | Reversible |
| Primary Target | β5 subunit (Chymotrypsin-like) | β5 subunit (Chymotrypsin-like) | β5 subunit (Chymotrypsin-like) |
| Administration Route | Intravenous/Subcutaneous | Intravenous | Oral |
| Key Metabolic Pathway | Hepatic (CYP450) | Peptidase cleavage | Hepatic (CYP450) |
| Common Toxicities | Peripheral neuropathy, thrombocytopenia | Cardiotoxicity, renal dysfunction | Gastrointestinal, rash |
The first-generation PI bortezomib is a reversible boronate inhibitor that primarily targets the chymotrypsin-like activity of the β5 proteasome subunit [93]. Its initial approval for relapsed/refractory MM was based on the phase 2 SUMMIT trial, which demonstrated a 27% response rate in heavily pretreated patients [93]. The subsequent second-generation agents addressed specific bortezomib limitations: carfilzomib's irreversible epoxyketone binding reduces off-target effects, while ixazomib's oral bioavailability improves patient convenience [93] [95].
Clinical Trial Data and Therapeutic Efficacy
Evolution of Treatment Paradigms
The therapeutic application of PIs has evolved substantially from single-agent use in late-line settings to combination regimens in newly diagnosed patients. Bortezomib is now an established component of induction therapy for both transplant-eligible and ineligible patients, and has been incorporated into conditioning regimens before autologous stem cell transplantation (ASCT), post-ASCT consolidation, and maintenance therapy [93]. The recent meta-analysis of PI maintenance therapy demonstrated significant benefits in both progression-free survival (PFS) and overall survival (OS) compared to placebo [95].
Quantitative Clinical Outcomes
Table 2: Clinical Efficacy of Proteasome Inhibitors in Key Multiple Myeloma Trials
| Regimen | Trial Phase/Name | Patient Population | Key Efficacy Outcomes | Reference |
|---|---|---|---|---|
| Bortezomib Single Agent | Phase 2 SUMMIT | Relapsed/Refractory MM | ORR: 27% (CR+nCR: 10%); Median TTP: 7 months | [93] |
| Bortezomib Maintenance | Meta-analysis | MM after induction | Improved PFS (OR: 1.98; 95% CI: 1.35-2.92; P<0.001) | [95] |
| Carfilzomib Combinations | Multiple trials | Relapsed/Refractory MM | Activity in bortezomib-resistant disease | [93] |
| Ixazomib Maintenance | Meta-analysis | MM after induction | Improved PFS vs. placebo | [95] |
The subcutaneous administration of bortezomib has demonstrated equivalent efficacy to intravenous delivery with reduced peripheral neuropathy incidence, significantly improving the therapeutic index [93]. The recent meta-analysis of 8 randomized controlled trials confirmed that PI maintenance therapy significantly prolongs PFS (OR: 1.98; 95% CI: 1.35-2.92; P<0.001) and OS compared to placebo or observation, establishing a new standard in MM management [95].
Experimental Protocols for Profiling Ubiquitin-Enzyme Interactions
Phage Display Profiling of E1 Enzyme Specificity
Purpose: To identify ubiquitin C-terminal sequences reactive with E1 activating enzymes and profile E1 specificity. Methodology:
- Library Construction: Create a phage-displayed ubiquitin library with randomized C-terminal residues (positions 71-75), preserving Gly76.
- Enzyme Immobilization: Express E1 enzymes (Ube1 or Uba6) as N-terminal peptidyl carrier protein (PCP) fusions. Label with biotin using Sfp phosphopantetheinyl transferase and immobilize on streptavidin-coated plates.
- Selection Rounds: Incubate phage library with immobilized E1 in reaction buffer (50 mM Tris-HCl, pH 7.5, 5 mM MgCl₂, 1 mM ATP) for 1 hour at room temperature.
- Stringency Enhancement: Through successive selection rounds, decrease reaction time (to 10 minutes), E1 concentration (to 1 pmol), and phage input (to 1×10^10 pfu).
- Elution and Amplification: Cleave covalently bound phage with 20 mM DTT, elute, and amplify in E. coli for subsequent rounds.
- Sequence Analysis: After 8 selection rounds, sequence enriched phage clones to identify reactive ubiquitin variants [24].
Key Insight: This approach revealed that while Arg72 is essential for E1 recognition, positions 71, 73, and 74 tolerate bulky aromatic substitutions, and Gly75 can be substituted with Ser, Asp, or Asn while maintaining E1 activation capability [24].
Cascading Activity-Based Probe for Monitoring Enzyme Cascades
Purpose: To simultaneously monitor E1, E2, and E3 enzymatic activities throughout the ubiquitin cascade. Methodology:
- Probe Design: Synthesize UbDha, a ubiquitin probe with C-terminal glycine replaced by dehydroalanine (Dha), a Michael acceptor.
- In Vitro Reactions: Incubate UbDha (1-5 µM) with E1, E2, and/or E3 enzymes in reaction buffer (50 mM HEPES, pH 7.5, 10 mM MgCl₂, 1 mM ATP) for 30 minutes at 30°C.
- ATP Dependence Control: Include reactions without ATP to confirm mechanism-specific labeling.
- Detection: Resolve reactions by SDS-PAGE under reducing and non-reducing conditions. Detect UbDha-enzyme adducts via Western blot using anti-ubiquitin antibodies or through fluorescent tags incorporated into the probe [25].
Applications: This mechanism-based probe enables profiling of enzymatic activities in cell lysates, monitoring ligand-induced changes in living cells, and identifying specific E3 substrates through orthogonal transfer systems [25].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Studying the Ubiquitin-Proteasome Pathway
| Reagent Category | Specific Examples | Research Application | Experimental Utility |
|---|---|---|---|
| Activity-Based Probes | UbDha, Ub-AMP analogues | Monitoring enzymatic activity in cascades | Mechanism-based trapping of E1, E2, and HECT/RBR E3 enzymes; proteome-wide profiling [25] |
| Engineered Enzyme Pairs | xUB-xE1, xE1-xE2 orthogonal pairs | Substrate identification for specific E3s | Orthogonal UB transfer (OUT) to map E3-specific substrates without cross-reactivity [28] |
| Proteasome Inhibitors | Bortezomib, Carfilzomib, MG132 | Functional validation of UPS dependence | Confirm UPS role in cellular processes; study protein turnover dynamics [93] |
| E3 Ligase Inhibitors | Nutlins (MDM2), SM-406 (IAP), SCF inhibitors | Targeted protein stabilization | Investigate specific ubiquitination pathways; potential therapeutic development [94] |
| DUB Inhibitors | PR-619, VLX1570 | Probing deubiquitination functions | Study ubiquitin chain dynamics and recycling; investigate DUB roles in disease [29] |
Future Directions and Emerging Applications
The clinical success of PIs in multiple myeloma has catalyzed exploration of UPS-targeting therapies in other malignancies and therapeutic contexts. Emerging research focuses on E1 enzyme inhibitors (e.g., TAK-243) currently under investigation for solid tumors and hematologic malignancies, representing a strategic shift upstream in the ubiquitin cascade [94]. The development of E3 ligase-specific inhibitors leverages the substrate specificity of E3s to achieve targeted protein stabilization with potentially reduced off-target effects [94]. Additionally, PROTAC (Proteolysis Targeting Chimeras) technology harnesses the ubiquitin-proteasome system for targeted protein degradation by recruiting E3 ligases to non-native substrates, representing one of the most promising applications of ubiquitin cascade research [29].
Combination strategies represent another frontier, with PI + IMiD (immunomodulatory drug) regimens demonstrating synergistic activity in multiple myeloma through concurrent targeting of proteasome and cereblon E3 ligase activity [96]. The ongoing clinical evaluation of PIs in non-oncologic indications, including autoimmune disorders and neurodegenerative diseases, further illustrates the expanding therapeutic landscape emanating from fundamental ubiquitin-proteasome pathway research [29].
The ubiquitin-proteasome system (UPS) represents a crucial pathway for regulated intracellular protein degradation, operating through a coordinated three-enzyme cascade. This system begins with ubiquitin-activating enzymes (E1), proceeds through ubiquitin-conjugating enzymes (E2), and culminates with ubiquitin ligases (E3), which ultimately transfer ubiquitin to substrate proteins. The E1 enzyme catalyzes the first and committed step in this pathway by activating ubiquitin in an ATP-dependent manner, forming a high-energy thioester bond between its catalytic cysteine residue and the C-terminal glycine of ubiquitin. This activated ubiquitin is then transferred to a cysteine residue on an E2 enzyme via a trans-thiolation reaction. Finally, E3 ligases facilitate the transfer of ubiquitin from the E2 to a lysine residue on the target protein, with different E3 families employing distinct mechanisms—HECT E3s form an intermediate thioester with ubiquitin, while RING E3s act as scaffolds to directly transfer ubiquitin from E2 to substrate [97] [3] [23].
Targeting different levels of this enzymatic cascade presents unique therapeutic opportunities and challenges. E1 inhibition offers a broad approach affecting global ubiquitination, while E2 and E3 targeting provides increasingly specific intervention points. This application note systematically compares these strategic approaches, providing quantitative comparisons, detailed experimental protocols, and essential resource information to guide research in ubiquitination cascade modulation.
Comparative Targeting Strategies
E1-Targeted Inhibition Strategies
E1 inhibition represents the most upstream approach to disrupt the ubiquitination cascade, affecting all downstream ubiquitin-dependent processes. The human genome encodes two primary ubiquitin E1 enzymes: UBA1 (the predominant form) and UBA6, both essential proteins that utilize distinct spectra of E2 enzymes [98] [3]. Small molecule E1 inhibitors typically function through several mechanisms: blocking ATP binding to the E1 enzyme, preventing formation of the covalent E1~Ub complex by targeting the active-site thiol, or disrupting E1-E2 enzyme interactions [2].
PYR-41, one of the first identified cell-permeable ubiquitin E1 inhibitors, irreversibly blocks the catalytic cysteine of E1Ub, demonstrating an IC50 of <10 μM without significant activity against E2 or E3 enzymes at this concentration [2] [99]. This compound inhibits MDM2-dependent p53 ubiquitination, stabilizes p53 protein levels, and induces p53-dependent transcription while preferentially killing transformed cells. More recently developed inhibitors such as TAK-243 (MLN7243) show substantially improved potency, with an IC50 of 1 nM against UAE (UBA1), effectively blocking ubiquitin conjugation and disrupting both monoubiquitin signaling and global protein ubiquitination [99]. This compound induces endoplasmic reticulum (ER) stress, abrogates NF-κB pathway activation, and promotes apoptosis, demonstrating strong therapeutic potential.
The table below summarizes key characteristics of representative E1 inhibitors:
Table 1: Comparative Analysis of E1 Ubiquitin-Activating Enzyme Inhibitors
| Compound Name | Molecular Target | IC50/ Potency | Mechanism of Action | Key Cellular Phenotypes | Therapeutic Context |
|---|---|---|---|---|---|
| TAK-243 (MLN7243) | UBA1 | 1 nM | Selective UAE inhibitor; blocks ubiquitin conjugation | Disrupts global protein ubiquitination; induces ER stress and apoptosis | Anti-cancer applications; in clinical development |
| PYR-41 | E1Ub | <10 μM | Irreversibly blocks catalytic cysteine of E1 | Stabilizes p53; inhibits NF-κB activation; preferential killing of transformed cells | Preclinical cancer models |
| PYZD-4409 | UBA1 | Not specified | 3,5-dioxopyrazolidine compound; inhibits E1 enzymatic activity | Induces ER stress; increases expression of E1 stress markers | Mouse leukemia models; decreased tumor weight and volume |
E2-Targeted Inhibition Strategies
E2 conjugating enzymes represent the middle layer of the ubiquitination cascade, with approximately 37 members encoded in the human genome that determine the specificity of ubiquitin chain topology and collaborate with E3 ligases to define substrate selection [3] [100]. Unlike E1 inhibition, targeting E2 enzymes offers the potential for more selective pathway disruption while still affecting multiple E3 ligases that share common E2 partners. E2 enzymes contain a conserved core domain of approximately 150 residues with an invariant catalytic cysteine residue that accepts ubiquitin from E1 through a thioester linkage [100].
Chemical biology approaches have identified E2 inhibitors such as compounds targeting the UBE2N/Ubc13 enzyme, which plays a critical role in error-free DNA damage tolerance and NF-κB signaling. Inhibition of UBE2N induces neuroblastoma cell death through activation of p53 and JNK pathways [101]. Another approach involves developing inhibitors that disrupt specific E2-E3 protein-protein interactions, such as CC-0651 and its analogs that target the E2 enzyme Cdc34 [101]. The selectivity of E2 inhibition derives from the diversity of E2 enzymes and their specific partnerships with different E3 ligases, allowing for more precise intervention in specific ubiquitination pathways compared to broad E1 inhibition.
E3-Targeted Modulation Strategies
E3 ubiquitin ligases represent the most diverse and specific layer of the ubiquitination cascade, with over 600 members in the human genome that confer substrate specificity through direct recognition of target proteins [3] [101]. E3-targeted approaches offer the highest potential for selectivity, enabling intervention against specific disease-relevant pathways without disrupting global protein ubiquitination. E3 modulators include both inhibitors that disrupt the activity of specific E3 ligases and molecular glues that redirect E3 activity toward novel substrates.
Notable E3-targeting compounds include Nutlin-3a, which inhibits the MDM2-p53 interaction with an IC50 of 90 nM, stabilizing p53 protein and inducing cell cycle arrest and apoptosis [99]. The clinical success of immunomodulatory drugs (IMiDs) such as thalidomide derivatives demonstrates the therapeutic potential of E3 modulation—these compounds function by altering substrate specificity of the CRL4CRBN E3 ligase, resulting in targeted degradation of specific pathogenic proteins [101]. More recently, developed CELMoD agents like mezigdomide (CC-92480) show high affinity for cereblon and potent antimyeloma activity through enhanced degradation of specific substrates [99].
Table 2: Comparison of E2 and E3-Targeting Compounds in the Ubiquitin-Proteasome System
| Compound/Target | Molecular Class | Primary Target | Mechanism of Action | Therapeutic Application |
|---|---|---|---|---|
| UBE2N Inhibitors | E2 enzyme inhibitor | UBE2N/Ubc13 | Blocks E2 activity in NF-κB signaling; induces p53/JNK pathway activation | Neuroblastoma; inflammatory conditions |
| Cdc34 Inhibitors (CC-0651 analogs) | E2 enzyme inhibitor | Cdc34 | Disrupts E2-E3 interaction; inhibits SCF ligase activity | Potential anti-cancer applications |
| Nutlin-3a | E3 interaction inhibitor | MDM2-p53 interaction | Inhibits MDM2-p53 binding (IC50=90 nM); stabilizes p53 protein | TP53 wild-type cancers |
| IMiDs (Thalidomide derivatives) | E3 molecular glue | CRL4CRBN E3 ligase | Alters E3 substrate specificity; induces degradation of novel substrates | Multiple myeloma; myelodysplastic syndromes |
| Mezigdomide (CC-92480) | CELMoD agent | Cereblon E3 ligase | Molecular glue enhancing substrate degradation | Relapsed/refractory multiple myeloma |
Experimental Protocols for Ubiquitination Cascade Analysis
Protocol 1: In Vitro E1 Enzyme Activity Assay
Purpose: To quantitatively measure E1 enzyme activity and inhibitor efficacy in a cell-free system.
Principle: This assay measures E1-mediated ubiquitin activation through detection of inorganic pyrophosphate (PPi) released during ubiquitin adenylation, using a coupled fluorescent detection system.
Reagents and Solutions:
- Recombinant His6-tagged E1 enzyme (commercially available or purified in-house)
- E2 conjugating enzyme (e.g., UbcH7, UbcH5a)
- Ubiquitin (20 μM final concentration)
- ATP (250 μM final concentration)
- Assay buffer: 50 mM Tris-HCl (pH 7.0), 5 mM MgCl2
- Fluorogenic pyrophosphate detection reagent
- Test compounds dissolved in DMSO (final DMSO concentration <1%)
Procedure:
- Prepare reaction mixtures in a 96-well plate format with a final volume of 50 μL per well.
- Add assay buffer containing 50 mM Tris-HCl (pH 7.0) and 5 mM MgCl2 to each well.
- Add ATP to a final concentration of 250 μM.
- Add recombinant E1 enzyme (1 μM final concentration).
- Add E2 enzyme (10 μM final concentration) and ubiquitin (20 μM final concentration).
- Add test compounds at varying concentrations or DMSO vehicle control.
- Initiate the reaction by transferring the plate to a pre-warmed (37°C) microplate reader.
- Immediately add 50 μL of inorganic pyrophosphate detection reagent to each well.
- Monitor fluorescence continuously over 30 minutes (excitation 530 nm, emission 567 nm).
- Calculate IC50 values by plotting inhibitor concentration against percentage inhibition of fluorescence increase relative to DMSO control.
Technical Notes: Include appropriate controls without E1 enzyme to account for non-specific signal. For kinetic analyses, vary substrate concentrations while maintaining inhibitor concentrations constant. For irreversible inhibitors like PYR-41, pre-incubate E1 with inhibitor for 15 minutes before adding other reaction components [98] [2].
Protocol 2: Cellular Ubiquitination Status Assessment
Purpose: To evaluate the effects of E1, E2, or E3 inhibitors on global protein ubiquitination in cells.
Principle: This protocol uses Western blot analysis with anti-ubiquitin antibodies to detect changes in high-molecular-weight ubiquitin conjugates following inhibitor treatment.
Reagents and Solutions:
- Cell line of interest (e.g., K562 leukemia cells, HeLa cells)
- Complete culture medium appropriate for cell line
- Inhibitors of interest (e.g., TAK-243 for E1, specific E2 or E3 inhibitors)
- DMSO vehicle control
- Lysis buffer: RIPA buffer supplemented with protease inhibitors and 10 mM N-ethylmaleimide (to inhibit deubiquitinating enzymes)
- Anti-ubiquitin antibody
- HRP-conjugated secondary antibody
- ECL detection reagents
Procedure:
- Seed cells at appropriate density (e.g., 5 × 10^5 cells/well in 6-well plates) and culture for 24 hours.
- Treat cells with inhibitors at varying concentrations or DMSO vehicle control for desired time periods (typically 4-24 hours).
- Wash cells with ice-cold PBS and lyse in RIPA buffer containing protease inhibitors and N-ethylmaleimide.
- Clarify lysates by centrifugation at 14,000 × g for 15 minutes at 4°C.
- Determine protein concentration of supernatants using BCA assay.
- Separate equal protein amounts (20-40 μg) by SDS-PAGE (4-12% gradient gels recommended).
- Transfer proteins to PVDF membranes using standard Western blotting protocols.
- Block membranes with 5% non-fat milk in TBST for 1 hour at room temperature.
- Incubate with primary anti-ubiquitin antibody overnight at 4°C.
- Wash membranes and incubate with HRP-conjugated secondary antibody for 1 hour at room temperature.
- Develop blots using ECL detection reagents and image using a digital imaging system.
- Assess changes in high-molecular-weight ubiquitin conjugates (appearing as a smear above 50 kDa).
Technical Notes: Include loading controls using antibodies against housekeeping proteins (e.g., GAPDH, actin). For assessment of specific protein ubiquitination, perform immunoprecipitation of the target protein followed by ubiquitin Western blotting [98].
Pathway Diagrams and Visualization
Ubiquitination Cascade and Inhibitor Targeting Sites
Diagram 1: Ubiquitination Cascade with Inhibitor Targeting Sites. This diagram illustrates the sequential E1-E2-E3 enzymatic cascade and the specific intervention points for different inhibitor classes. E1 inhibitors block the initial ubiquitin activation step; E2 inhibitors target the conjugating enzymes; E3 inhibitors/modulators affect the substrate recognition and final transfer steps.
Experimental Workflow for Inhibitor Characterization
Diagram 2: Experimental Workflow for Comprehensive Inhibitor Characterization. This workflow outlines a systematic approach for evaluating inhibitors targeting different components of the ubiquitination cascade, from initial in vitro enzyme assays through cellular validation to detailed mechanistic studies.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Ubiquitination Cascade Studies
| Reagent Category | Specific Examples | Key Applications | Commercial Sources |
|---|---|---|---|
| E1 Enzyme Inhibitors | TAK-243 (MLN7243), PYR-41, NSC 624206 | Global ubiquitination blockade, ER stress induction, apoptosis studies | MedChemExpress, Tocris Bioscience |
| E2 Enzyme Reagents | Recombinant E2 enzymes (UbcH5, UbcH7, Ubc13), UBE2N inhibitors | E2-E3 interaction studies, chain topology determination, DNA damage response | Boston Biochem, Sigma-Aldrich |
| E3-Targeting Compounds | Nutlin-3a (MDM2-p53), Idasanutlin (RG7388), IMiDs (thalidomide derivatives) | Targeted protein stabilization/degradation, molecular glue mechanisms | MedChemExpress, Selleck Chemicals |
| Activity Assay Systems | Fluorogenic pyrophosphate assay kits, Ubiquitin conjugation kits | High-throughput screening, enzyme kinetics, inhibitor potency determination | Invitrogen, Boston Biochem, Enzo Life Sciences |
| Detection Reagents | Anti-ubiquitin antibodies, linkage-specific ubiquitin antibodies | Western blotting, immunoprecipitation, ubiquitin chain typing | Cell Signaling Technology, Santa Cruz Biotechnology |
| Cellular Models | Temperature-sensitive E1 mutant cell lines, E3 knockout cells | Functional validation, pathway analysis, synthetic lethality studies | ATCC, academic repositories |
The strategic targeting of different levels in the ubiquitination cascade presents a spectrum of opportunities for basic research and therapeutic development. E1 inhibition offers the most comprehensive approach to disrupt global protein ubiquitination, potentially beneficial in hematologic malignancies as demonstrated by PYZD-4409 in leukemia models [98]. E2 targeting provides intermediate specificity, potentially disrupting subsets of ubiquitination events mediated by specific E2 enzymes. E3 modulation represents the most precise approach, enabling intervention against specific disease-driving pathways while sparing global protein homeostasis, as exemplified by clinical successes with IMiDs in multiple myeloma [101].
Future directions in this field include developing increasingly selective E2 inhibitors, expanding the repertoire of targeted protein degradation approaches using molecular glues and PROTACs, and exploring combination therapies that simultaneously target multiple nodes in the ubiquitination cascade. The continued refinement of experimental approaches and research tools summarized in this application note will support these advances, ultimately enabling more effective therapeutic modulation of the ubiquitin-proteasome system in human disease.
The ubiquitin-proteasome system (UPS) represents a sophisticated enzymatic cascade fundamental to eukaryotic cellular regulation, governing protein stability, localization, and function. This cascade involves the sequential action of E1 (activating), E2 (conjugating), and E3 (ligating) enzymes that collectively mediate the covalent attachment of ubiquitin (Ub) or ubiquitin-like (UBL) modifiers, such as NEDD8, to specific substrate proteins [102] [16]. The E1-E2-E3 enzymatic hierarchy provides a rich source of novel therapeutic targets, particularly for conditions like cancer and neurodegenerative diseases where proteostasis is dysregulated. While E3 ligases, numbering over 600 in humans, have received significant attention for their substrate specificity, recent research highlights the critical and often overlooked roles of E2 enzymes and UBL-specific pathways like neddylation [16] [103] [104]. This application note details standardized protocols and presents case studies for validating novel targets within the NEDD8 activation pathway and for modulating emergent E3 ligases, providing a methodological framework for researchers in drug discovery.
Case Study 1: Validating UBC12 (UBE2M) in the NEDD8 Activation Pathway
Biological Context and Rationale
The neddylation pathway, which conjugates NEDD8 to cullin proteins, is a crucial regulator of Cullin-RING Ligase (CRL) activity. CRLs, the largest family of E3 ubiquitin ligases, control the degradation of approximately 20% of cellular proteins via the proteasome, making their master regulators attractive drug targets [102] [105]. The NEDD8 conjugation cascade mirrors the ubiquitin pathway, utilizing a dedicated E1 (NAE), E2s (primarily UBC12/UBE2M and UBE2F), and E3s (like RBX1/ROC1) [105] [106]. UBC12 serves as the central NEDD8-conjugating enzyme (E2) for multiple cullins, positioning it as a key node for pharmacological intervention. Evidence of neddylation pathway overactivation in numerous cancers further underscores its therapeutic relevance [105].
Experimental Protocol: Genetic and Functional Validation of UBC12
Objective: To validate UBC12 as a critical dependency in lung cancer models by assessing its loss-of-function effects on neddylation, CRL activity, and malignant phenotypes.
Materials:
- Cell Lines: Human lung cancer cell lines (e.g., A549, H1299) and a non-malignant control line.
- Reagents:
- Lentiviral vectors for CRISPR/Cas9-mediated UBC12 knockdown (e.g., lenti-guide-puro with gRNAs against UBC12).
- Packaging plasmids (e.g., AGP091, AGP090).
- Antibodies: Anti-UBC12, Anti-NEDD8, Anti-Cullin (to assess neddylation), Anti-p21, Anti-p27, Anti-Wee1.
- MLN4924 (Pevonedistat), a reference NAE (E1) inhibitor.
Methodology:
- In Silico Expression Analysis:
- Interrogate public datasets (e.g., TCGA, GEO) to compare UBC12 mRNA levels between human lung cancer tissues and normal lung tissues. Analyze correlation between UBC12 and NEDD8 expression and assess association with clinical stages.
Genetic Knockdown:
- Generate stable UBC12-knockdown cells via lentiviral transduction of CRISPR/Cas9 constructs. Use a non-targeting guide RNA as a negative control.
- Validate knockdown efficiency by Western blotting.
Phenotypic Assays:
- Cell Proliferation: Perform Cell Counting Kit-8 (CCK-8) assays. Seed cells in 96-well plates and measure absorbance at 450nm daily.
- Clonogenic Survival: Plate a low density of cells and culture for 14 days. Fix and stain colonies with crystal violet, then count.
- Cell Cycle Analysis: Fix cells and stain DNA with Propidium Iodide (PI). Analyze cell cycle distribution via Fluorescence-Activated Cell Sorting (FACS).
Mechanistic Biochemical Assays:
- Global Neddylation Status: Assess by Western blotting using an anti-NEDD8 antibody. UBC12 knockdown should reduce global protein neddylation.
- CRL Substrate Accumulation: Monitor levels of well-characterized CRL substrates (p21, p27, Wee1) by Western blot upon UBC12 knockdown.
- Rescue Experiments: Test if UBC12 knockdown inhibits growth in MLN4924-resistant cell lines to validate its potential to overcome drug resistance.
Visualization of the NEDD8 Activation Cascade and Experimental Workflow:
Key Data and Interpretation
Table 1: Summary of Key Quantitative Findings from UBC12 Validation Study [105]
| Experimental Readout | Control Cells | UBC12-Knockdown Cells | Interpretation |
|---|---|---|---|
| UBC12 mRNA in Lung Cancer | Elevated in tumors vs. normal tissue | N/A | Supports pathological relevance of target |
| Global Protein Neddylation | High | Significantly Reduced | Confirms on-target effect on pathway |
| CRL Substrate p21 Level | Low | Accumulated | Induces cell cycle arrest |
| CRL Substrate p27 Level | Low | Accumulated | Induces cell cycle arrest |
| Cell Cycle Profile (G2 Phase) | Baseline | Increased by ~2.5-fold | Mechanism for growth suppression |
| Clonogenic Survival | High colonies | Drastically Reduced colonies | Demonstrates loss of malignant potential |
| Growth of MLN4924-Resistant Cells | Unaffected by MLN4924 | Suppressed by UBC12 knockdown | Validates UBC12 as alternative target |
The data from this validation study demonstrate that UBC12 is not merely a passive carrier but a critical regulator of neddylation. Its knockdown phenocopies the effect of the E1 inhibitor MLN4924, inducing G2 cell cycle arrest and suppressing oncogenic phenotypes by inactivating CRLs [105]. The success in inhibiting MLN4924-resistant cells underscores UBC12's potential as a novel anticancer target when resistance to upstream inhibitors emerges.
Case Study 2: Modulating Novel E3 Ligases for Targeted Protein Degradation
Expanding the E3 Ligase Toolbox
The field of targeted protein degradation (TPD), exemplified by Proteolysis-Targeting Chimeras (PROTACs), has historically relied on a handful of E3 ligases, chiefly CRBN and VHL [104]. This limited repertoire restricts the scope of degradable proteins and poses a risk for acquired resistance. There is a strong impetus to identify and validate novel E3 ligases for TPD applications. These emerging E3s often exhibit tissue-specific expression or unique substrate profiles, which can enable degradation of previously inaccessible targets and improve therapeutic windows [104].
Experimental Protocol: Ligand Discovery and Functional Deployment for RNF4
Objective: To discover a covalent ligand for the RNF4 E3 ligase and functionally validate its utility in a BRD4-degrading PROTAC.
Materials:
- Platform: Activity-based protein profiling (ABPP) covalent ligand screening platform.
- Chemical Library: A diverse library of cysteine-reactive covalent ligands.
- Proteins: Recombinant RNF4 E3 ligase.
- Cell Lines: Relevant cancer cell lines (e.g., triple-negative breast cancer).
- Reagents:
- Hit compound (TRH 1-23) and optimized ligand (CCW 16).
- PROTAC component: JQ1 (BET bromodomain ligand).
- Antibodies: Anti-BRD4, Anti-RNF4, Anti-Ubiquitin.
Methodology:
- Ligand Discovery via ABPP Screening:
- Incubate the covalent ligand library with recombinant RNF4 protein.
- Use a broad-spectrum cysteine-reactive probe to compete for binding sites. Ligands that bind RNF4 will block subsequent probe labeling.
- Identify hits by tandem mass spectrometry (LC-MS/MS) to determine the specific cysteine residue (e.g., C132/C135 in the RING domain) modified by the ligand.
Ligand Validation and Optimization:
- Binding Affinity: Perform competitive ABPP dose-response curves to calculate IC50 values (e.g., IC50 for CCW 16 = 1.8 µM).
- Functional Impact: Assess if ligand binding affects RNF4's autoubiquitination activity via in vitro ubiquitination assays. A "non-functional" binder that does not inhibit E3 activity is ideal for recruitment.
PROTAC Assembly and Cellular Validation:
- Synthesize a heterobifunctional PROTAC (e.g., CCW 28-3) by linking the optimized RNF4 ligand (CCW 16) to a target protein ligand (JQ1 for BRD4) via a chemical linker.
- Degradation Assay: Treat cells with the PROTAC and measure BRD4 protein levels over time (e.g., 6-24 hours) by Western blotting.
- Mechanism Validation:
- Use proteasome inhibitors (e.g., MG132) to confirm degradation is proteasome-dependent.
- Use CRISPR/Cas9 to generate RNF4-knockout cells; the PROTAC should be ineffective in this background, confirming RNF4-dependence.
- Specificity Assessment: Perform proteomic analysis to ensure no off-target degradation occurs.
Visualization of Novel E3 Ligase Recruitment via PROTACs:
Key Data and Interpretation
Table 2: Research Reagent Solutions for Ubiquitin Cascade Target Validation
| Reagent / Tool | Function / Target | Application in Validation |
|---|---|---|
| MLN4924 (Pevonedistat) | Inhibitor of NEDD8 Activating Enzyme (E1, NAE) | Positive control for neddylation pathway inhibition; benchmark for E2-targeting effects [105]. |
| CRISPR/Cas9 Knockdown | Gene-specific knockout (e.g., UBC12, RNF4) | Establish genetic dependency and confirm on-target mechanism of small molecules/PROTACs [105] [104]. |
| Covalent Ligand Screens (ABPP) | Discovery of binders to E3 ligases (e.g., RNF4, RNF114) | Identify starting points for developing E3 recruiters, especially for ligases lacking native small-molecule ligands [104]. |
| Nimbolide & Analogues | Covalent ligand for RNF114 E3 ligase | Tool molecule for recruiting RNF114 in PROTACs to degrade targets like BRD4 [104]. |
| Engineered E1/E2 Pairs | Orthogonal Ubiquitin Transfer (OUT) | Isolate and study specific ubiquitination cascades without cross-talk from endogenous systems [42]. |
The successful development of CCW 28-3, which degraded BRD4 in an RNF4- and proteasome-dependent manner, provides critical proof-of-concept for expanding the E3 ligase landscape through chemical biology approaches [104]. While initial degradation efficiency might be modest, this protocol establishes a pipeline for targeting the "undruggable" proteome by leveraging novel E3 ligases.
The following table catalogs key reagents essential for research in NEDD8 activation and E3 ligase modulation.
Table 3: Key Research Reagents for NEDD8 and E3 Ligase Research
| Reagent / Tool | Function / Target | Application in Validation |
|---|---|---|
| MLN4924 (Pevonedistat) | Inhibitor of NEDD8 Activating Enzyme (E1, NAE) | Positive control for neddylation pathway inhibition; benchmark for E2-targeting effects [105]. |
| CRISPR/Cas9 Knockdown | Gene-specific knockout (e.g., UBC12, RNF4) | Establish genetic dependency and confirm on-target mechanism of small molecules/PROTACs [105] [104]. |
| Covalent Ligand Screens (ABPP) | Discovery of binders to E3 ligases (e.g., RNF4, RNF114) | Identify starting points for developing E3 recruiters, especially for ligases lacking native small-molecule ligands [104]. |
| Nimbolide & Analogues | Covalent ligand for RNF114 E3 ligase | Tool molecule for recruiting RNF114 in PROTACs to degrade targets like BRD4 [104]. |
| Engineered E1/E2 Pairs | Orthogonal Ubiquitin Transfer (OUT) | Isolate and study specific ubiquitination cascades without cross-talk from endogenous systems [42]. |
The systematic validation of novel targets within the ubiquitin-proteasome system, as illustrated by the cases of UBC12 in the NEDD8 pathway and RNF4 in TPD, is paramount for advancing therapeutic discovery. The protocols outlined here provide a robust framework for establishing the pathological relevance, functional necessity, and mechanistic action of emerging targets from the E1-E2-E3 cascade. As the field moves beyond the canonical E3 ligases CRBN and VHL, the strategies of covalent ligand screening and genetic dependency testing will become increasingly vital. The continued expansion of the E3 ligase toolbox and the deepening understanding of E2 enzymes like UBC12 promise to unlock new opportunities for targeting previously intractable diseases, culminating in the development of more precise and effective therapeutics.
The ubiquitin-proteasome system (UPS) represents a crucial regulatory pathway for post-translational protein modification, governing essential cellular processes including protein degradation, cell cycle progression, and DNA damage repair [107] [2]. This enzymatic cascade begins with ubiquitin activation by E1 enzymes, proceeds through ubiquitin conjugation to E2 enzymes, and culminates in substrate-specific ubiquitination mediated by E3 ligases [2] [108]. The dynamic reversal of this process is orchestrated by deubiquitinating enzymes (DUBs), which cleave ubiquitin from modified substrates, thereby opposing the action of E3 ligases [107] [109]. As the therapeutic targeting of E3 ligases advances, DUB inhibitors are emerging as sophisticated tools for pharmacological intervention, offering novel opportunities to modulate protein stability and function with precision [110] [111]. This application note examines the current landscape of DUB inhibitor development, provides validated experimental protocols, and contextualizes their role within ubiquitin cascade research.
DUB Biology and Therapeutic Rationale
Classification and Mechanisms of DUBs
Deubiquitinating enzymes constitute a diverse family of approximately 100 proteases in humans, systematically categorized into seven subfamilies based on their catalytic mechanisms and structural domains [110] [109]. The majority are cysteine proteases, including the ubiquitin-specific proteases (USPs), ubiquitin C-terminal hydrolases (UCHs), ovarian tumor proteases (OTUs), Machado-Josephin domain proteases (MJDs), motif interacting with ubiquitin-containing novel DUB family (MINDYs), and zinc finger-containing ubiquitin peptidases (ZUP1). In contrast, the JAB1/MPN/Mov34 metalloenzymes (JAMM) represent the only metal-dependent DUB family [109]. These enzymes employ three primary mechanisms: generating free ubiquitin from precursor molecules, editing ubiquitin chains through selective cleavage, and completely removing ubiquitin chains from substrate proteins to prevent degradation and alter cellular localization [107].
DUBs as Therapeutic Targets in Human Disease
DUBs regulate critical signaling pathways—including NF-κB, PI3K/Akt/mTOR, and MAPK—with their dysregulation implicated in oncogenesis, neurodegenerative disorders, inflammatory conditions, and infectious diseases [112] [111] [109]. The fundamental role of DUBs in maintaining cellular homeostasis, coupled with their substrate specificity, presents a compelling therapeutic rationale. While proteasome inhibitors like bortezomib and carfilzomib have demonstrated clinical efficacy in hematological malignancies, their use is limited by resistance development and toxicity [107] [2]. Targeting specific DUBs upstream in the UPS pathway offers the potential for enhanced selectivity, reduced off-target effects, and applicability to solid tumors [107] [110]. Furthermore, in infectious diseases, pathogens often hijack host DUB machinery to evade immune responses, positioning DUB inhibitors as promising host-directed therapeutics [112] [49].
Current Landscape of DUB Inhibitor Development
Table 1: Promising DUB Inhibitors in Preclinical and Clinical Development
| Target | Inhibitor | Key Characteristics | Therapeutic Application | Development Stage |
|---|---|---|---|---|
| USP1 | Multiple chemotypes [110] | Targets USP1/UAF1 complex; reverses cisplatin resistance | Oncology (e.g., non-small cell lung cancer) | Preclinical |
| USP7 | XL177A, P5091, others [110] [113] | Stabilizes p53; induces apoptosis in cancer cells | Oncology (leukemia, ovarian cancer) | Preclinical |
| USP14 | Multiple inhibitors [110] | Enhances proteasome activity; reduces intracellular bacteria | Oncology, antimicrobial host-directed therapy | Preclinical |
| USP25/USP28 | AZ-1 [49] | Dual inhibitor; suppresses NF-κB signaling | Antimicrobial host-directed therapy | Preclinical |
| USP30 | Multiple inhibitors [110] | Mitochondrial DUB; potential for neurodegenerative diseases | Oncology, neurodegenerative disorders | Preclinical |
| VCPIP1 | Optimized azetidine probe [113] | 70 nM potency; demonstrates in-family selectivity | Chemical probe for target validation | Preclinical |
The development of selective DUB inhibitors has been accelerated by advanced screening technologies and structural biology insights. Recent platforms utilizing activity-based protein profiling (ABPP) coupled with quantitative mass spectrometry have successfully identified selective hits against 23 endogenous DUBs spanning four subfamilies from a purpose-built library of 178 compounds [113]. This approach demonstrates the feasibility of targeting DUBs with small molecules despite historical challenges with inhibitor selectivity.
Experimental Protocols for DUB Inhibitor Evaluation
Protocol 1: High-Content Screening for Host-Directed Antimicrobial Therapeutics
Purpose: Identify DUB inhibitors that enhance intracellular bacterial clearance without host cell toxicity [49].
Workflow:
- Cell Preparation: Seed macrophages (e.g., RAW 264.7 or primary human macrophages) in 96-well imaging plates and culture overnight.
- Bacterial Infection: Infect cells with GFP-expressing Salmonella enterica serovar Typhimurium (UK-1 strain) at a predetermined MOI (e.g., 1:10 to 1:50 cell-to-bacterium ratio).
- Compound Treatment: Add DUB inhibitors from screening library (257 compounds recommended) at optimal concentrations (typically 1-10 µM) post-infection.
- Staining: Fix cells and stain with Hoechst (nuclei) and HCS CellMask Red (cytoplasm) to differentiate intracellular bacteria.
- Image Acquisition: Utilize high-content imaging system with automated microscopy (e.g., ImageXpress Micro Confocal) capturing multiple fields per well.
- Quantitative Analysis: Employ image analysis software (e.g., CellProfiler) to quantify intracellular bacteria per cell, normalized to control wells.
Validation: Confirm host cell viability through nucleus count and exclude compounds with direct antibacterial activity using axenic culture growth assays [49].
Diagram 1: Host-directed therapy screening workflow
Protocol 2: Chemoproteomic ABPP Screening for DUB Inhibitor Discovery
Purpose: Simultaneously assess compound potency and selectivity across multiple endogenous DUBs in native cellular environment [113].
Workflow:
- Cellular Extract Preparation: Generate protein extracts from HEK293 cells or relevant cell lines using non-denaturing lysis buffer.
- Compound Incubation: Treat extracts with DUB-focused covalent library compounds (50 µM recommended) for 1 hour at room temperature.
- ABPP Probe Labeling: Add biotinylated ubiquitin-based active-site probes (biotin-Ub-VME and biotin-Ub-PA in 1:1 combination) to covalently label active DUBs.
- Streptavidin Enrichment: Capture probe-labeled DUBs using streptavidin beads, followed by extensive washing to remove non-specific binders.
- On-Bead Digestion: Digest enriched proteins with trypsin/Lys-C directly on beads.
- TMT Multiplexing: Label resulting peptides with isobaric tandem mass tag (TMT) reagents for quantitative comparison.
- LC-MS/MS Analysis: Analyze peptides using high-resolution mass spectrometry with true nanoflow LC columns.
- Data Analysis: Process raw data using proteomics software (e.g., MaxQuant) and normalize to DMSO controls to calculate percentage inhibition.
Key Insight: This platform enables screening 178 compounds against 65 endogenous DUBs, providing both hit identification and structure-activity relationships across the DUB gene family [113].
Diagram 2: ABPP screening for DUB inhibitor discovery
Protocol 3: Global Ubiquitinome Profiling for DUB Inhibitor Mechanisms
Purpose: Elucidate proteome-wide changes in ubiquitination in response to DUB inhibition to identify substrates and mechanisms [114].
Workflow:
- Cell Treatment: Treat U2OS cells (or cell line of interest) with DUB inhibitor (e.g., PR619), proteasome inhibitor (MG132), or E1 inhibitor (TAK243) for 3 hours.
- Ubiquitinated Protein Enrichment:
- Option A (His10-Ub): Use U2OS cells stably expressing His10-tagged ubiquitin. Lyse cells under denaturing conditions (6 M guanidine-HCl) and enrich ubiquitinated proteins using Ni-NTA beads.
- Option B (UbiSite): For endogenous ubiquitination sites, digest proteins with Lys-C, then immunopurify using UbiSite antibody specific for Lys-C ubiquitin fragment.
- Sample Preparation: Digest enriched proteins with trypsin, desalt peptides, and concentrate for MS analysis.
- Mass Spectrometry: Analyze peptides on high-resolution mass spectrometer (e.g., Orbitrap Fusion Lumos) using data-dependent acquisition.
- Bioinformatics: Identify significantly changed ubiquitination sites using MaxQuant and Perseus software, applying thresholds of FDR=0.05 and S0=0.1.
Application: This protocol identified over 55,000 ubiquitination sites, revealing distinct substrates preferentially regulated by DUBs versus the proteasome, including PARP1 hyperubiquitination that enhances its enzymatic activity [114].
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagents for DUB Inhibitor Studies
| Reagent Category | Specific Examples | Function/Application | Key Features |
|---|---|---|---|
| DUB Inhibitors | PR-619, AZ-1 (USP25/28), XL177A (USP7), VCPIP1 probe [49] [113] | Pan-DUB inhibition; selective DUB targeting; chemical probes | Varying selectivity profiles; covalent vs. non-covalent mechanisms |
| Activity-Based Probes | Biotin-Ub-VME, Biotin-Ub-PA [113] | Covalent labeling of active DUBs for enrichment and detection | Enable chemoproteomic screening; assess target engagement |
| Proteasome Inhibitors | MG132, Bortezomib, Carfilzomib [2] [114] | Control for UPS disruption; compare DUB vs. proteasome inhibition | Establish differential ubiquitinome regulation |
| E1 Inhibitor | TAK243 [114] | Blocks ubiquitin activation; controls for new ubiquitination | Essential for turnover kinetics studies |
| Cell Lines | U2OS His10-Ub, HEK293 [114] [113] | Ubiquitinome profiling; DUB inhibitor screening | Enable specific enrichment strategies |
| Antibodies | UbiSite antibody, K48- and K63-linkage specific antibodies [114] | Endogenous ubiquitin site enrichment; chain-type analysis | Overcome NEDD8/ISG15 cross-reactivity |
DUB inhibitors represent a promising frontier in targeted therapeutics, building upon the foundation of ubiquitin cascade research. The experimental approaches outlined herein provide robust frameworks for evaluating DUB inhibitor efficacy, selectivity, and mechanisms of action. As screening technologies advance and our understanding of DUB biology deepens, the clinical translation of DUB inhibitors appears increasingly feasible. Future directions include developing heterobifunctional molecules such as DUB-targeting chimeras (DUBTACs), optimizing inhibitor pharmacokinetics for in vivo application, and identifying biomarkers for patient stratification. The continued integration of DUB inhibitors into the ubiquitin pharmacopeia holds significant potential for addressing unmet medical needs across oncology, infectious diseases, and neurodegenerative disorders.
Within the ubiquitin-proteasome system (UPS), the E1-E2-E3 enzymatic cascade represents a primary conduit for selective intracellular protein degradation, regulating most cellular processes [2]. Malfunction of UPS components is implicated in human diseases, including numerous cancers, making this system a rich source of attractive molecular targets for pharmacological intervention [2]. The clinical success of proteasome inhibitors like Bortezomib demonstrated the therapeutic potential of targeting the UPS [2]. However, bridging the gap between preclinical promise and clinical utility remains challenging, with less than 1% of published cancer biomarkers ultimately entering clinical practice [115]. This Application Note provides a structured framework for translating therapeutics targeting the E1-E2-E3 cascade from preclinical discovery to clinical application, with specific protocols for assessing efficacy and toxicity.
The Ubiquitin Activation Cascade: Therapeutic Targeting Landscape
Protein ubiquitination involves a highly coordinated multi-step enzymatic cascade [2]. The process initiates with ubiquitin activation by ubiquitin-activating enzymes (E1), which employ ATP to form a high-energy thioester bond with ubiquitin [2] [116]. The activated ubiquitin is then transferred to ubiquitin-conjugating enzymes (E2) through a similar thioester linkage [2]. Finally, ubiquitin ligases (E3) recruit charged E2 enzymes and facilitate specific transfer of ubiquitin to protein substrates [2]. E3 ligases constitute the most diverse component, with approximately 600 identified in the human genome, and serve as primary regulators of substrate specificity [116]. This cascade is reversible through deubiquitinating enzymes (DUBs) that cleave ubiquitin from modified proteins [2].
Table 1: Key Components of the Ubiquitin-Proteasome System and Their Therapeutic Implications
| Component | Number in Human Genome | Function | Therapeutic Significance |
|---|---|---|---|
| E1 (Ubiquitin-activating enzyme) | 2 [24] | Activates ubiquitin using ATP; initiates cascade [2] | Inhibition provides broad suppression of ubiquitination; two main enzymes: Ube1 and Uba6 [2] [24] |
| E2 (Ubiquitin-conjugating enzyme) | ~40-50 [24] [116] | Accepts activated ubiquitin from E1 [2] | Intermediate specificity; potential targets for selective modulation |
| E3 (Ubiquitin ligase) | ~600-1000 [24] [116] | Recognizes specific substrates; facilitates ubiquitin transfer [116] | High substrate specificity makes attractive drug targets; main families: RING, HECT, U-box [2] [116] |
| DUBs (Deubiquitinating enzymes) | Multiple families | Reverses ubiquitination; cleaves ubiquitin from substrates [2] | Emerging therapeutic targets; regulates ubiquitin homeostasis |
E1 Enzyme Specificity and C-terminal Recognition
Research reveals substantial promiscuity in E1 enzymes regarding ubiquitin C-terminal sequences. Phage display profiling demonstrated that while Arg72 of ubiquitin is absolutely required for E1 recognition, residues at positions 71, 73, and 74 can be replaced with bulky aromatic side chains, and Gly75 can be mutated to Ser, Asp, or Asn while maintaining efficient E1 activation [24] [68]. This flexibility contrasts with the stricter specificities of downstream E2 and E3 enzymes, suggesting strategic considerations for inhibitor design [24].
Figure 1: Ubiquitin-Proteasome System Cascade with Drug Inhibition Points. The diagram illustrates the sequential E1-E2-E3 enzymatic cascade and key inhibition targets (dashed red lines) for therapeutic development.
Quantitative Assessment of UPS-Targeting Compounds
Clinically Evaluated UPS Inhibitors
Several compounds targeting different components of the UPS have advanced to clinical development, with varying success rates across therapeutic areas. Recent analyses indicate great variations in clinical trial success rates (ClinSRs) among various diseases, developmental strategies, and drug modalities [117].
Table 2: Selected UPS-Targeting Compounds in Clinical Development
| Compound | Target | Clinical Status | Indication Context | Key Findings |
|---|---|---|---|---|
| Bortezomib (Velcade) | Proteasome (20S) | FDA-approved (2003) [2] | Multiple myeloma, lymphoma [2] | First FDA-approved proteasome inhibitor; reversible inhibitor of chymotrypsin-like activity [2] |
| Carfilzomib (Kyprolis) | Proteasome (20S) | FDA-approved (2012) [2] | Multiple myeloma (post-Bortezomib) [2] | Irreversible proteasome inhibitor; more potent/selective than Bortezomib [2] |
| TAK-243 (MLN7243) | E1 (UBA1) | Phase I/II trials [118] | Advanced solid tumors, leukemia [118] | First-in-class specific E1 inhibitor; also inhibits UBA6, NAE, SAE with less efficacy [118] |
| PYR-41 | E1 (Ubiquitin-activating enzyme) | Preclinical [2] | Research tool for p53 ubiquitination | Irreversible pyrazone derivative; blocks catalytic cysteine of E1; specificity concerns [2] |
| JS-K | E1 (Ubiquitin-activating enzyme) | Preclinical [2] | Research tool | NO-producing prodrug; inhibits E1~Ub thioester formation [2] |
| SM-406 | IAP E3 ligases | Preclinical [116] | Cancer research | Small molecule IAP inhibitor [116] |
| Nutlins | MDM2 E3 ligase | Preclinical [116] | Cancer research | MDM2-p53 interaction inhibitors [116] |
Clinical Translation Success Rates
The dynamic clinical trial success rate (ClinSR) for drugs has been declining since the early 21st century, though it has recently plateaued and begun to increase [117]. A comprehensive analysis of 20,398 clinical development programs involving 9,682 molecule entities revealed substantial variation in success rates across different therapeutic areas and drug modalities [117]. These findings underscore the importance of robust preclinical efficacy and toxicity assessment to improve clinical success rates.
Experimental Protocols for Efficacy and Toxicity Assessment
Protocol 1: Phage Display Profiling of E1 Enzyme Specificity
4.1.1 Purpose To profile the specificity of human E1 enzymes (Ube1 and Uba6) toward ubiquitin C-terminal sequences and identify ubiquitin variants with altered enzymatic properties [24].
4.1.2 Materials and Reagents
- UB phage display library (complexity: 1×10^8) with randomized C-terminal residues 71-75 [24]
- Human E1 enzymes (Ube1 and Uba6)
- Sfp phosphopantetheinyl transferase
- Biotin-coenzyme A (CoA) conjugate
- Streptavidin-coated plates
- Mg-ATP (1 mM)
- Elution buffer (containing dithiothreitol)
- Blocking buffer (PBS with BSA)
4.1.3 Procedure
- E1 Enzyme Biotinylation: Express E1 enzymes as fusions with N-terminal peptidyl carrier protein (PCP) domain. Label with biotin using Sfp phosphopantetheinyl transferase that catalyzes biotin transfer from biotin-CoA conjugate [24].
- Enzyme Immobilization: Bind biotin-labeled PCP-E1 fusions to streptavidin plates. Block non-specific sites with blocking buffer.
- Phage Selection: Add phage-displayed UB library to plates with Mg-ATP to initiate UB~E1 thioester formation. Incubate for 10 minutes to 1 hour depending on selection round [24].
- Washing: Remove non-specifically bound phage with extensive washing.
- Phage Elution: Release catalytically active UB phage bound to plates by treatment with DTT to cleave thioester linkages.
- Amplification and Iteration: Amplify eluted phage and repeat selection process for 8 rounds with increasing stringency (reduced phage amount, E1 concentration, and reaction time) [24].
- Sequence Analysis: Sequence enriched UB clones after final selection round to identify reactive C-terminal sequences.
4.1.4 Data Analysis
- Calculate phage enrichment from selection reaction (with E1 and Mg-ATP) versus controls (without E1 or Mg-ATP)
- After 8 selection rounds, enrichment should be ≥350-fold over controls [24]
- Analyze sequence logos of enriched UB variants to determine permissible substitutions at each C-terminal position
Protocol 2: Functional Validation of E1 Inhibitors in Disease Models
4.2.1 Purpose To evaluate the efficacy of E1 inhibitors in improving the rescue of misfolded proteins in conjunction with corrector compounds, using cystic fibrosis (F508del-CFTR) as a model system [118].
4.2.2 Materials and Reagents
- CFBE41o- cells expressing F508del-CFTR
- Differentiated human primary airway epithelial cells
- E1 inhibitor (TAK-243, PYR-41, or analogs)
- CFTR correctors (VX-445/elexacaftor, VX-661/tezacaftor)
- CFTR potentiator (VX-770/ivacaftor)
- Anti-CFTR antibodies (#596, #570, Clone 24-1)
- Western blot equipment and reagents
- Using chamber apparatus for short-circuit current measurements
4.2.3 Procedure
- Cell Culture and Treatment: Culture CFBE41o- cells and human primary airway epithelial cells under standard conditions. Differentiate primary cells at air-liquid interface for 4-6 weeks [118].
- Compound Administration: Treat cells with E1 inhibitor (TAK-243) alone or in combination with corrector compounds (VX-445/VX-661) and potentiator (VX-770). Include DMSO vehicle controls.
- Protein Analysis:
- Lyse cells and perform Western blotting using anti-CFTR antibodies to assess mature (band C) and immature (band B) CFTR protein levels.
- Immunoprecipitate CFTR with Clone 24-1 antibody to evaluate ubiquitination status [118].
- Functional Assessment:
- Mount differentiated primary epithelial cells in Using chambers.
- Measure short-circuit current (Isc) to quantify CFTR-dependent chloride transport.
- Activate CFTR with forskolin (10 μM) and genistein (50 μM).
- Inhibit CFTR with CFTRinh-172 (10 μM) to confirm specificity [118].
- Toxicity Assessment:
- Monitor cell viability using MTT or Alamar Blue assays.
- Assess general protein synthesis inhibition by E1 inhibitors via metabolic labeling.
4.2.4 Data Analysis
- Quantify band intensity from Western blots to determine fold-increase in mature CFTR
- Calculate normalized short-circuit current to assess functional correction
- Determine synergy between E1 inhibitors and corrector compounds
- Establish therapeutic index based on efficacy versus toxicity metrics
Figure 2: Integrated Workflow for Ubiquitin Cascade Analysis. The diagram outlines the key experimental protocols for phage display profiling of E1 specificity and functional validation of inhibitors in disease models.
Biomarker Development for Clinical Translation
Strategies for Predictive Biomarker Identification
The successful translation of UPS-targeting therapies requires development of robust biomarkers to guide patient selection and monitor therapeutic response. Several approaches show promise for identifying predictive biomarkers early in drug development:
5.1.1 In Vitro Screening Platforms
- Utilize large panels of well-characterized cancer cell lines or tumor organoids
- Correlate pharmacology screening data with genomic baseline information (gene expression, mutations, copy number variation) [119]
- Employ single gene analysis, composite biomarker generation, and pathway/network analyses to identify signatures of response [119]
5.1.2 Patient-Derived Xenograft (PDX) Models
- Leverage PDX models that retain parental tumor histopathology and genetics [115]
- Conduct Mouse Clinical Trials (MCTs) as surrogate clinical trials using PDX models to capture clinical heterogeneity [119]
- Generate response data correlated with comprehensive characterization data to identify biomarker panels [119]
5.1.3 Multi-Omics Integration
- Combine genomics, transcriptomics, and proteomics to identify context-specific, clinically actionable biomarkers [115]
- Implement cross-species transcriptomic analysis to bridge animal and human biomarker data [115]
- Apply AI and machine learning to identify patterns in large datasets that predict clinical outcomes [115]
Biomarker Qualification Framework
Regulatory agencies have established pathways for biomarker qualification to facilitate drug development:
- FDA Biomarker Qualification Program: Provides a framework for CDER to perform rigorous review and formally qualify biomarkers for specific contexts of use [120]
- EMA Qualification of Novel Methodologies: Established formal procedure for qualification of novel methodologies, primarily biomarkers, for drug development [120]
- Predictive Safety Testing Consortium (PSTC): Successful example of FDA/EMA collaboration that qualified seven preclinical kidney toxicity biomarkers [120]
Research Reagent Solutions
Table 3: Essential Research Reagents for Ubiquitin Cascade Studies
| Reagent/Category | Specific Examples | Function/Application | Key Characteristics |
|---|---|---|---|
| E1 Enzyme Inhibitors | TAK-243 (MLN7243), PYR-41, JS-K [2] [118] | Inhibit ubiquitin activation; block entire cascade [2] [118] | TAK-243: specific UBA1 inhibitor; in clinical trials [118] |
| E3 Ligase Inhibitors | Nutlins, SM-406, MI-63, NSC689857 [116] | Target specific E3-substrate interactions; higher specificity [116] | Nutlins: MDM2-p53 interaction inhibitors [116] |
| Ubiquitin Variants | Phage-displayed UB library, Leu73Phe UB, Leu73Tyr UB [24] | Study enzyme specificity; create DUB-resistant chains [24] | Leu73Phe/Tyr: resistant to DUB cleavage but form poly-UB chains [24] |
| Cell-Based Models | PDX models, CFBE41o- cells, primary airway epithelial cells [118] [115] | Evaluate therapeutic efficacy in physiologically relevant systems [118] | PDX: retain patient tumor characteristics; better clinical predictivity [115] |
| Detection Tools | Anti-ubiquitin antibodies, anti-CFTR antibodies, biotin-CoA [24] [118] | Monitor ubiquitination status and substrate processing | Anti-CFTR #596, #570: for Western blot; Clone 24-1: for IP [118] |
Targeting the E1-E2-E3 ubiquitin cascade represents a promising therapeutic strategy for numerous diseases, particularly cancers and protein-misfolding disorders. The protocols outlined in this Application Note provide a structured framework for evaluating the efficacy and toxicity of compounds modulating this system. Critical to success is the implementation of physiologically relevant models, robust biomarker strategies, and comprehensive assessment across the preclinical-clinical continuum. As our understanding of ubiquitin cascade biology expands, coupled with improved translational methodologies, the potential for developing effective therapies targeting this fundamental regulatory system continues to grow.
Conclusion
The ubiquitin E1-E2-E3 enzymatic cascade represents a master regulatory system with profound implications for human health and disease. Understanding its intricate mechanisms provides not only fundamental biological insights but also unprecedented opportunities for therapeutic intervention. While significant challenges remain in achieving specificity and overcoming technical limitations, emerging technologies in target validation, screening methodologies, and modality development are rapidly advancing the field. The continued elucidation of ubiquitin cascade regulation, coupled with innovative drug development platforms, promises to yield novel treatments for cancer, neurodegenerative disorders, and other conditions linked to ubiquitin system dysregulation. Future directions will likely focus on enhancing target specificity, exploiting neosubstrate relationships, and developing combinatorial approaches that leverage the full potential of the ubiquitin-proteasome system for precision medicine.
References
- 1. Molecular choreography of E1 enzymes in ubiquitin-like ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12355090/]
- 2. Development of inhibitors in the ubiquitination cascade - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7094371/]
- 3. Ubiquitin-Activating Enzyme - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/ubiquitin-activating-enzyme]
- 4. Ubiquitination Cascade Pathway [https://www.rndsystems.com/pathways/ubiquitination-cascade-pathway]
- 5. E2 enzymes: more than just middle men | Cell Research [https://www.nature.com/articles/cr201635]
- 6. Review Ubiquitin—A structural perspective [https://www.sciencedirect.com/science/article/abs/pii/S1097276524010104]
- 7. UBE2J2 sensitizes the ERAD ubiquitination cascade to ... [https://www.nature.com/articles/s41467-025-64777-1]
- 8. E3 ubiquitin ligases: styles, structures and functions - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8607428/]
- 9. The essential role of E3 ubiquitin ligases in ... [https://www.sciencedirect.com/science/article/abs/pii/S0006291X25005121]
- 10. Ubiquitination, E3 Ligases and Drug Discovery – Novel ... [https://www.ddw-online.com/ubiquitination-e3-ligases-and-drug-discovery-novel-technologies-for-a-challenging-pathway-757-201208/]
- 11. Ubiquitin-activating enzyme [https://en.wikipedia.org/wiki/Ubiquitin-activating_enzyme]
- 12. Structure of a ubiquitin E1-E2 complex [https://pmc.ncbi.nlm.nih.gov/articles/PMC3625138/]
- 13. E1 on the Move [https://www.sciencedirect.com/science/article/pii/S1097276505010786#!]
- 14. PROTAC-induced protein structural dynamics in targeted ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11867615/]
- 15. Structural bioinformatics approaches for predicting novel ... [https://www.nature.com/articles/s41598-025-12563-w]
- 16. E2 conjugating enzymes: A silent but crucial player in ... [https://www.sciencedirect.com/science/article/abs/pii/S1568163725000868]
- 17. Ubiquitin Conjugating Enzyme E2 - an overview [https://www.sciencedirect.com/topics/neuroscience/ubiquitin-conjugating-enzyme-e2]
- 18. Discovery and characterization of noncanonical E2 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10971424/]
- 19. Cryo-EM structures reveal the molecular mechanism of ... [https://www.nature.com/articles/s41594-025-01681-8]
- 20. Targeting E2 ubiquitin-conjugating enzyme UbcH5c by small ... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-022-01538-4]
- 21. Emerging roles for E3 ubiquitin ligases in neural development ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12149146/]
- 22. Targeting the ubiquitin-proteasome system [https://www.drugtargetreview.com/article/34078/targeting-the-ubiquitin-proteasome-system/]
- 23. Protein ubiquitination involving an E1–E2–E3 enzyme ... [https://www.nature.com/articles/373081a0]
- 24. Specificity of the E1-E2-E3 Enzymatic Cascade for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4652587/]
- 25. A cascading activity-based probe sequentially targets E1 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5108872/]
- 26. The unifying catalytic mechanism of the RING-between- ... [https://www.nature.com/articles/s41467-023-35871-z]
- 27. Ubiquitin-activating Enzyme E1 | Ligases [https://www.tocris.com/pharmacology/ubiquitin-activating-enzyme-e1]
- 28. Orthogonal Ubiquitin Transfer through Engineered E1-E2 ... [https://www.sciencedirect.com/science/article/pii/S1074552112002888]
- 29. The ubiquitin system: from cell signalling to disease ... [https://www.nature.com/articles/s41418-020-00703-w]
- 30. Ubiquitination - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/ubiquitination]
- 31. E1, E2, E3 Enzyme and Their Regulators [https://www.bocsci.com/resources/e1-e2-e3-enzyme-and-their-regulators.html?srsltid=AfmBOorJWbxhsNTsxBxx_-fuMZim2Ee-70Im1UI5YgZo8F5G7lqgQN6Q]
- 32. Real-time tracking of complex ubiquitination cascades ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6086040/]
- 33. Light‐Activatable Ubiquitin for Studying Linkage‐Specific ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11809417/]
- 34. UBE2J2 sensitizes the ERAD ubiquitination cascade to ... [https://pubmed.ncbi.nlm.nih.gov/41068091/]
- 35. A Modular Uba1-Nanobody Fusion Enables Selective ... [https://www.sciencedirect.com/science/article/pii/S0021925825027620]
- 36. Molecular choreography of E1 enzymes in ubiquitin-like ... [https://pubmed.ncbi.nlm.nih.gov/40570956/]
- 37. In-situ cross-linking mass spectrometry reveals ... [https://www.nature.com/articles/s41467-025-65752-6]
- 38. Computationally Driven Top-Down Mass Spectrometry of ... [https://pubmed.ncbi.nlm.nih.gov/40766653/]
- 39. Drug ubiquitination: an unwelcome mode of action or a ... [https://www.sciencedirect.com/science/article/abs/pii/S0968000425002452]
- 40. A comprehensive method for detecting ubiquitinated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4403176/]
- 41. Antibody toolkit reveals N-terminally ubiquitinated ... [https://www.nature.com/articles/s41467-021-24669-6]
- 42. Orthogonal Ubiquitin Transfer through Engineered E1-E2 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4104569/]
- 43. The Role of Ubiquitination and Deubiquitination in ... [https://www.mdpi.com/2227-9059/13/12/2873]
- 44. Establishment of high-throughput screening HTRF assay ... [https://www.nature.com/articles/s41598-021-00646-3]
- 45. chemical structures and their potential for drug discovery [https://pubs.rsc.org/en/content/articlelanding/2025/np/d5np00004a]
- 46. Targeting the ubiquitin system by fragment-based drug ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2022.1019636/full]
- 47. Mechanisms that matter: unbiased cellular screening for ... [https://www.nature.com/articles/s44386-025-00028-z]
- 48. Targeting deubiquitinating enzymes (DUBs) and ubiquitin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11838268/]
- 49. Targeting deubiquitinating enzymes and ubiquitin pathway ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12506015/]
- 50. HTS Screening of Poly-Ubiquitin Linkage-Specific ... [https://lifesensors.com/hts-screening-of-poly-ubiquitin-linkage-specific-molecular-glues-and-protein-degraders/]
- 51. Targeted protein degradation: mechanisms, strategies and ... [https://www.nature.com/articles/s41392-022-00966-4]
- 52. Targeted Protein Degradation with PROTACs and ... [https://blog.crownbio.com/targeted-protein-degradation-with-protacs-and-molecular-glues]
- 53. PROTAC 2.0: Expanding the frontiers of targeted protein ... [https://www.sciencedirect.com/science/article/abs/pii/S1359644625000893]
- 54. E3 ubiquitin ligases in signaling, disease, and therapeutics [https://www.sciencedirect.com/science/article/pii/S0968000425001860]
- 55. Recent Advances in the Development of Pro-PROTAC for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12473374/]
- 56. Expanding PROTACtable genome universe of E3 ligases [https://www.nature.com/articles/s41467-023-42233-2]
- 57. Identification of Suitable Target/E3 Ligase Pairs for ... [https://elifesciences.org/reviewed-preprints/98450]
- 58. Expanding the landscape of E3 ligases for targeted protein ... [https://www.sciencedirect.com/science/article/pii/S2666246922000027]
- 59. Recent Advances of Degradation Technologies Based on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9496103/]
- 60. Workflow for E3 Ligase Ligand Validation for PROTAC ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11851430/]
- 61. Molecular choreography of E1 enzymes in ubiquitin-like ... [https://www.sciencedirect.com/science/article/pii/S0021925825022653]
- 62. Basics of Antibody Phage Display Technology - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC6024766/]
- 63. Principles, Libraries, and Applications in Drug Discovery [https://www.vipergen.com/molecular-display-systems/]
- 64. Overview of Phage Display Technology [https://www.creative-biolabs.com/overview-of-phage-display-technology.html]
- 65. DNA-Encoded Chemical Libraries: A Comprehensive Review ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8369695/]
- 66. High-power screening (HPS) empowered by DNA- ... [https://www.sciencedirect.com/science/article/pii/S0165614721001978]
- 67. Specificity and disease in the ubiquitin system - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5264512/]
- 68. Specificity of the E1-E2-E3 enzymatic cascade for ubiquitin C ... [https://pubmed.ncbi.nlm.nih.gov/23003343/]
- 69. Mechanism and disease association of E2-conjugating enzymes [https://pmc.ncbi.nlm.nih.gov/articles/PMC5095918/]
- 70. A Practical Review of Proteasome Pharmacology [https://www.sciencedirect.com/science/article/pii/S0031699724012857]
- 71. Proteasome Inhibitors: An Expanding Army Attacking a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3503453/]
- 72. Targeting the ubiquitin-proteasome system - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC11464458/]
- 73. PROTAC Technology as a New Tool for Modern ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12114078/]
- 74. Design and development of PROTACs: A new paradigm in ... [https://www.sciencedirect.com/science/article/pii/S2590098625000181]
- 75. The Ubiquitin Tale: Current Strategies and Future Challenges [https://pmc.ncbi.nlm.nih.gov/articles/PMC11406696/]
- 76. Unraveling chain specific ubiquitination in cells using tandem ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12217038/]
- 77. Current methodologies in protein ubiquitination characterization [https://cellandbioscience.biomedcentral.com/articles/10.1186/s13578-022-00870-y]
- 78. Insights into non-proteinaceous ubiquitination - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12203941/]
- 79. The RBR E3 ubiquitin ligase HOIL-1 can ... - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC11962058/]
- 80. Structure-guided engineering enables E3 ligase-free and ... [https://www.nature.com/articles/s41467-024-45635-y]
- 81. The molecular mechanisms of acquired proteasome inhibitor ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3521846/]
- 82. Comparative Mechanisms of Action of Proteasome Inhibitors [https://www.cancernetwork.com/view/comparative-mechanisms-action-proteasome-inhibitors]
- 83. Functional reconstruction of a eukaryotic-like E1/E2/(RING) ... [https://www.nature.com/articles/s41467-017-01162-7]
- 84. Overcoming proteasome inhibitor resistance in the ... [https://www.sciencedirect.com/science/article/pii/S0165614723001116]
- 85. Resistance to Proteasome Inhibitors and Other Targeted ... [https://pubmed.ncbi.nlm.nih.gov/29676460/]
- 86. Drug resistance mechanisms and treatment strategies ... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-024-02005-y]
- 87. The RBR E3 ubiquitin ligase HOIL-1 can ... [https://www.life-science-alliance.org/content/8/6/e202503243]
- 88. exploring the ubiquitin system for drug development [https://www.nature.com/articles/cr201631]
- 89. Selective ubiquitination of drug-like small molecules by the ... [https://www.nature.com/articles/s41467-025-63442-x]
- 90. The Role of Protein Ubiquitination in the Onset and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12248740/]
- 91. The MALDI-TOF E2/E3 Ligase Assay as Universal Tool for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6162346/]
- 92. Covalent fragment screening to inhibit the E3 ligase activity of ... [https://pubs.rsc.org/en/content/articlehtml/2025/cb/d5cb00177c]
- 93. Proteasome inhibitors in multiple myeloma: 10 years later [https://pmc.ncbi.nlm.nih.gov/articles/PMC4123429/]
- 94. E1, E2, E3 Enzyme and Their Regulators [https://www.bocsci.com/resources/e1-e2-e3-enzyme-and-their-regulators.html?srsltid=AfmBOoqp6D4XU1WJz5upPX0HN7dNkH76ge0U2gE3-Yr9GYC7d_5QgmmQ]
- 95. Meta-analysis of effectiveness and safety of proteasome ... [https://www.sciencedirect.com/science/article/pii/S2405844024123420]
- 96. October - November 2025: What's New in Myeloma? [https://www.myeloma.org/blog/october-november-2025-whats-new-myeloma]
- 97. Protein ubiquitination involving an E1-E2-E3 enzyme ... [https://pubmed.ncbi.nlm.nih.gov/7800044/]
- 98. The ubiquitin-activating enzyme E1 as a therapeutic target ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2920204/]
- 99. E1/E2/E3 Enzyme | Inhibitors [https://www.medchemexpress.com/Targets/E1_E2_E3%20Enzyme.html?srsltid=AfmBOorS4RTAdTwBSqFDzZZe_WIqtuANquJDF2QvoEssb9hlZsBu3H7z]
- 100. Ubiquitin Conjugating Enzyme - an overview [https://www.sciencedirect.com/topics/neuroscience/ubiquitin-conjugating-enzyme]
- 101. Lead discovery and chemical biology approaches targeting ... [https://www.sciencedirect.com/science/article/abs/pii/S0960894X17308648]
- 102. NEDD8 and ubiquitin ligation by cullin-RING E3 ligases - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8096640/]
- 103. Drug Discovery Approaches to Target E3 Ligases [https://pubmed.ncbi.nlm.nih.gov/39686906/]
- 104. The Expanding E3 Ligase-Ligand Landscape for PROTAC ... [https://www.mdpi.com/2813-3137/3/4/30]
- 105. Validation of NEDD8-conjugating enzyme UBC12 as a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6642072/]
- 106. RNF187 neddylation in pancreatic cancer activates ... [https://www.nature.com/articles/s41388-025-03639-y]
- 107. Deubiquitinases (DUBs) and DUB inhibitors: a patent review [https://pmc.ncbi.nlm.nih.gov/articles/PMC4834700/]
- 108. E1, E2, E3 Enzyme and Their Regulators [https://www.bocsci.com/resources/e1-e2-e3-enzyme-and-their-regulators.html?srsltid=AfmBOoqt9Ha-37ua5sa2YTowXc9gHB9T2POfbo-pCUFX-2nCWsPcoeHO]
- 109. Deubiquitinases as novel therapeutic targets for diseases - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11645450/]
- 110. Recent advances in small molecule inhibitors of ... [https://www.sciencedirect.com/science/article/abs/pii/S0223523425000893]
- 111. Deubiquitylating enzymes and drug discovery - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC7097658/]
- 112. DEUBIQUITINATING ENZYMES AS PROMISING DRUG ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5485257/]
- 113. Accelerating inhibitor discovery for deubiquitinating enzymes [https://www.nature.com/articles/s41467-023-36246-0]
- 114. Deubiquitinating enzymes and the proteasome regulate ... [https://www.nature.com/articles/s41467-022-30376-7]
- 115. Bridging the Gap: Translating Preclinical Biomarkers to ... [https://blog.crownbio.com/bridging-the-gap-translating-preclinical-biomarkers-to-clinical-success]
- 116. E1, E2, E3 Enzyme and Their Regulators [https://www.bocsci.com/resources/e1-e2-e3-enzyme-and-their-regulators.html?srsltid=AfmBOoo-DkdTko2u9IOhdtTW6rFFoZ-BhiizDX4G6dpbwIip99eAcBkH]
- 117. Dynamic clinical trial success rates for drugs in the 21st ... [https://www.nature.com/articles/s41467-025-64552-2]
- 118. Targeting the E1 ubiquitin-activating enzyme (UBA1) ... [https://link.springer.com/article/10.1007/s00018-022-04215-3]
- 119. Identify Predictive Biomarkers Using Translational ... [https://blog.crownbio.com/identify-predictive-biomarkers]
- 120. Translational Biomarkers: from Preclinical to Clinical a Report ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3085704/]