Ubiquitin Code: Decoding the Molecular Nexus of DNA Repair and Immune Signaling in Disease and Therapy
This article synthesizes current research on ubiquitination, an essential post-translational modification that acts as a central regulatory node connecting DNA damage repair with immune response pathways.
Ubiquitin Code: Decoding the Molecular Nexus of DNA Repair and Immune Signaling in Disease and Therapy
Abstract
This article synthesizes current research on ubiquitination, an essential post-translational modification that acts as a central regulatory node connecting DNA damage repair with immune response pathways. We explore the foundational mechanisms by which diverse ubiquitin chain topologies—including K48-linked proteolysis, K63-linked signaling scaffolds, and linear ubiquitination—orchestrate cellular homeostasis, from maintaining genomic integrity to directing innate and adaptive immunity. For researchers and drug development professionals, this review further examines cutting-edge methodological approaches for investigating ubiquitin networks, discusses challenges in therapeutic targeting of ubiquitin system enzymes, and validates key targets through comparative analysis of preclinical and clinical data. The integration of these insights highlights the immense potential of targeting the ubiquitin system to develop novel treatments for cancer, inflammatory diseases, and immune disorders.
The Ubiquitin Code: Molecular Mechanisms Linking Genome Integrity and Immune Homeostasis
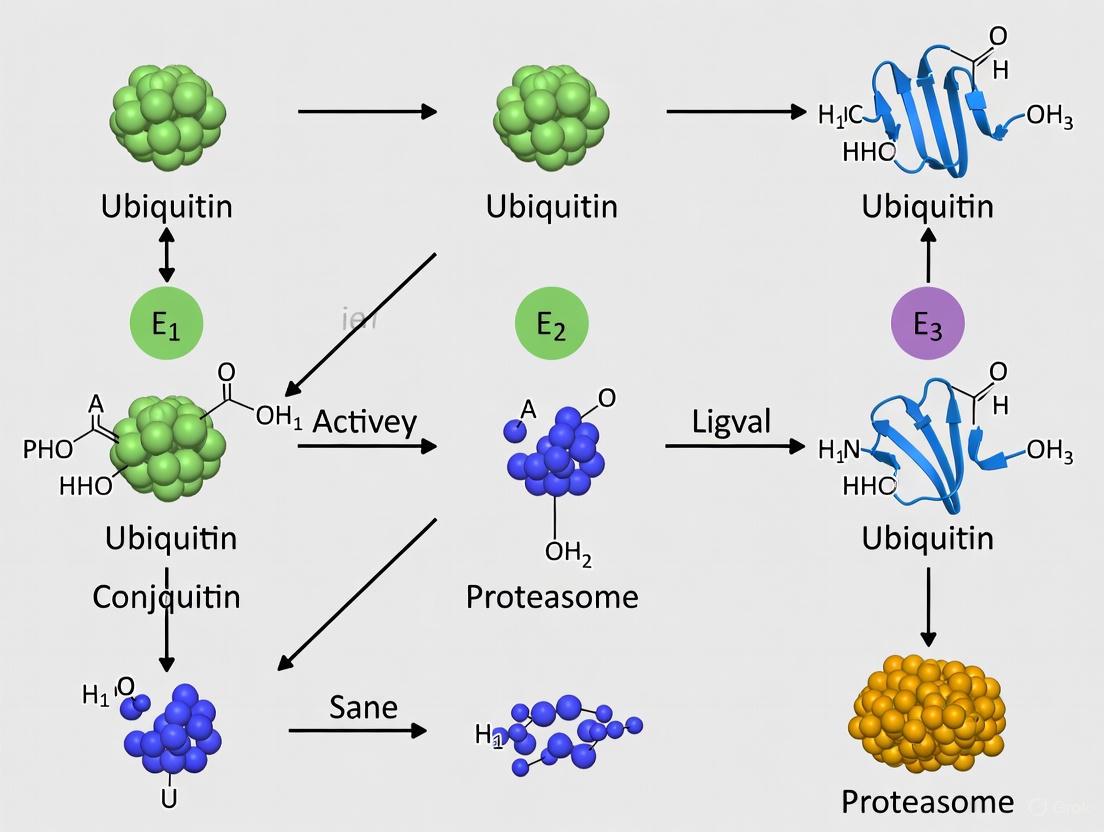
Ubiquitination represents a crucial post-translational modification that regulates virtually all cellular processes in eukaryotes, with particular significance in DNA repair and immune response pathways. This enzymatic process involves a precise cascade mediated by ubiquitin-activating (E1), conjugating (E2), and ligating (E3) enzymes that collectively coordinate the attachment of ubiquitin to substrate proteins. The complexity of ubiquitination extends beyond simple protein degradation, as different ubiquitin chain topologies—linked through specific lysine residues or the N-terminus of ubiquitin—create distinct molecular codes that determine diverse functional outcomes. This technical review examines the core mechanisms of the E1-E2-E3 enzymatic cascade, details the structural and functional diversity of ubiquitin chain architectures, and discusses experimental methodologies for investigating ubiquitination, with special emphasis on its roles in maintaining genome integrity and regulating immune signaling. The growing understanding of ubiquitination networks continues to reveal novel therapeutic targets for cancer and other diseases, particularly through the development of targeted inhibitors against specific components of the ubiquitin-proteasome system.
Ubiquitin is a small, 76-amino acid regulatory protein that is highly conserved across eukaryotic organisms and is expressed in most tissues [1]. The ubiquitination process involves the covalent attachment of ubiquitin to substrate proteins, which subsequently influences their stability, activity, localization, or interactions [1] [2]. This post-translational modification employs a hierarchical enzymatic cascade consisting of E1 (ubiquitin-activating), E2 (ubiquitin-conjugating), and E3 (ubiquitin-ligating) enzymes that work sequentially to transfer ubiquitin to specific substrate proteins [3] [2]. The human genome encodes two E1 enzymes, approximately 40 E2 enzymes, and an estimated 500-1000 E3 enzymes, providing both specificity and versatility to the ubiquitination system [4].
The functional consequences of ubiquitination are determined by the pattern of ubiquitin modification. Monoubiquitination (attachment of a single ubiquitin molecule) can alter protein localization and activity, while polyubiquitination (formation of ubiquitin chains) can lead to diverse outcomes depending on the linkage type between ubiquitin molecules [2] [5]. The ubiquitin code is further complicated by the fact that ubiquitin itself contains seven lysine residues (K6, K11, K27, K29, K33, K48, K63) and an N-terminal methionine (M1), all of which can serve as linkage points for polyubiquitin chain formation [1] [2]. This diversity of chain topologies enables ubiquitination to regulate a vast array of cellular processes, from protein degradation to DNA repair, immune signaling, and beyond [6] [2] [7].
The E1-E2-E3 Enzymatic Cascade
The ubiquitination process proceeds through a three-step enzymatic cascade that ensures precise regulation and substrate specificity:
Step 1: Ubiquitin Activation by E1 Enzymes
The cascade initiates with ATP-dependent activation of ubiquitin by E1 ubiquitin-activating enzymes. During this process, E1 forms a thioester bond between its active-site cysteine residue and the C-terminal carboxyl group of ubiquitin [3] [2] [4]. This activation step first involves the formation of a ubiquitin-adenylate intermediate, followed by transfer of ubiquitin to the E1 active site cysteine, releasing AMP [1]. The human genome encodes only two E1 enzymes with ubiquitin-activating capability: UBA1 (the primary E1) and UBA6, highlighting the convergence of ubiquitin activation at this initial step [4].
Step 2: Ubiquitin Conjugation by E2 Enzymes
Following activation, ubiquitin is transferred from E1 to the active-site cysteine of an E2 ubiquitin-conjugating enzyme through a trans-thioesterification reaction [1] [2]. This step requires the E2 enzyme to bind both the activated ubiquitin and the E1 enzyme [4]. Humans possess approximately 35 E2 enzymes, each characterized by a highly conserved ubiquitin-conjugating catalytic (UBC) fold [1]. While all E2s share this conserved structural domain, different E2 enzymes exhibit significant specificity in their interactions with E3 ligases [2].
Step 3: Ubiquitin Ligation by E3 Enzymes
The final step involves E3 ubiquitin ligases catalyzing the transfer of ubiquitin from E2 to a specific substrate protein [3] [2]. Most commonly, E3s facilitate the formation of an isopeptide bond between the C-terminal glycine of ubiquitin and the ε-amino group of a lysine residue on the substrate protein [1] [2]. E3 enzymes function as the substrate recognition modules of the system and are capable of interacting with both E2 and substrate [2]. With an estimated 500-1000 members in the human genome, E3 ligases provide the remarkable specificity of the ubiquitination system [4].
E3 ubiquitin ligases are categorized based on their structural domains and mechanisms. The two primary classes are HECT (Homologous to E6-AP C-Terminus) domain E3s and RING (Really Interesting New Gene) domain E3s (and the closely related U-box domain) [1]. HECT domain E3s form an obligate thioester intermediate with ubiquitin before transferring it to the substrate, while RING domain E3s catalyze direct transfer from the E2 enzyme to the substrate [1]. Additionally, multi-subunit E3 complexes such as the anaphase-promoting complex (APC) and the SCF (Skp1-Cullin-F-box protein) complex provide further regulatory complexity [1].
Table 1: Key Enzymes in the Ubiquitination Cascade
| Enzyme Type | Number in Humans | Primary Function | Key Features |
|---|---|---|---|
| E1 (Activating) | 2 | Ubiquitin activation via ATP hydrolysis | Forms thioester bond with ubiquitin; initial convergence point |
| E2 (Conjugating) | ~35 | Ubiquitin transfer from E1 to E3/substrate | Contains conserved UBC fold; determines chain topology |
| E3 (Ligating) | 500-1000 | Substrate recognition and ubiquitin transfer | Provides specificity; HECT vs RING mechanistic classes |
Figure 1: The E1-E2-E3 Ubiquitination Enzymatic Cascade. Ubiquitin is activated by E1 in an ATP-dependent process, transferred to E2, and finally conjugated to substrate proteins via E3 ligases that provide substrate specificity.
Ubiquitin Chain Topologies and Functional Diversity
The structural diversity of ubiquitin chains arises from the ability of ubiquitin itself to be modified at any of its seven lysine residues or its N-terminal methionine, creating polyubiquitin chains with distinct structures and functions [1] [2]. The specific linkage type determines the three-dimensional architecture of the polyubiquitin chain and consequently its biological function.
Major Ubiquitin Linkage Types
K48-linked polyubiquitination represents the best-characterized ubiquitin linkage and typically targets substrate proteins for proteasomal degradation [6] [2]. This linkage forms compact structures that are efficiently recognized by the proteasome [6]. In contrast, K63-linked polyubiquitination adopts an extended "beads-on-a-string" conformation that facilitates protein-protein interactions and functions in various signaling pathways, including DNA damage repair, endocytosis, and immune response activation [6] [4].
The other linkage types have more specialized functions: K6-linked chains participate in DNA damage repair [2]; K11-linked chains regulate cell cycle progression and trafficking events [2]; K27-linked chains mediate mitochondrial autophagy [2]; K29-linked chains are involved in proteasomal degradation under specific conditions [2]; K33-linked chains function in protein trafficking [2]; and M1-linked linear chains play crucial roles in NF-κB signaling and inflammatory responses [1] [2].
Table 2: Ubiquitin Chain Topologies and Their Biological Functions
| Linkage Type | Structural Features | Primary Functions | Cellular Processes |
|---|---|---|---|
| K48 | Compact structure | Proteasomal degradation | Cell cycle, protein turnover |
| K63 | Extended, flexible chain | Signaling scaffold | DNA repair, inflammation, endocytosis |
| K11 | Mixed compact/extended | Proteasomal degradation | Cell cycle regulation, ERAD |
| K27 | Not fully characterized | Mitochondrial autophagy | Quality control, metabolism |
| K29 | Not fully characterized | Proteasomal degradation | Stress response |
| K33 | Not fully characterized | Protein trafficking | Vesicular transport |
| K6 | Not fully characterized | DNA damage response | Genome maintenance |
| M1 (Linear) | Linear chain assembly | NF-κB signaling | Immune and inflammatory responses |
Functional Consequences in DNA Repair and Immune Signaling
In the context of DNA damage response, ubiquitination plays critical roles in coordinating repair pathway choice and efficiency. For example, K63-linked ubiquitination creates platforms that recruit DNA repair proteins to sites of damage [6] [7]. Additionally, monoubiquitination of histone H2AX (γH2AX) by UBE2T/RNF8 accelerates damage detection in hepatocellular carcinoma, while RNF40-generated H2Bub1 recruits the FACT complex to relax nucleosomes, facilitating DNA repair [7].
The ubiquitin system also profoundly regulates immune responses through multiple mechanisms. K63-linked ubiquitination of key signaling molecules in pattern recognition receptor pathways (such as TLR, RLR, and STING-dependent signaling) promotes the activation of NF-κB and interferon regulatory factors, thereby initiating anti-viral and inflammatory responses [2] [7]. Furthermore, ubiquitination regulates immune checkpoint molecules such as PD-L1, whose stability is controlled by ubiquitin-mediated degradation [8].
The plasticity of ubiquitin chain configurations allows for dynamic regulation of these processes. Radiation, for instance, dynamically reprograms ubiquitin signaling by altering chain formation, enabling cancer cells to strategically manipulate K63-linked chains to stabilize DNA repair factors while concurrently inhibiting K48-mediated degradation of survival proteins [7].
Experimental Methods for Investigating Ubiquitination
Advancements in methodological approaches have been crucial for elucidating the complexity of ubiquitination pathways. Several key techniques have enabled researchers to identify ubiquitination sites, quantify changes in ubiquitination, and characterize ubiquitin chain architectures.
Ubiquitin Remnant Profiling (Di-Glycine Capture)
This mass spectrometry-based approach has emerged as the gold standard for proteome-wide identification of ubiquitination sites [5] [9]. The method exploits the fact that tryptic digestion of ubiquitinated proteins leaves a di-glycine remnant (∼114 Da mass shift) from the C-terminus of ubiquitin covalently attached to the previously modified lysine [9]. Monoclonal antibodies specifically recognizing this di-glycine adduct (such as the GX41 antibody) are used to enrich for modified peptides from complex protein digests, followed by LC-MS/MS analysis for identification and quantification [9].
The ubiquitin remnant profiling workflow typically involves: (1) protein extraction from cells or tissues; (2) proteolytic digestion with trypsin; (3) immunoaffinity enrichment of di-glycine-modified peptides; (4) LC-MS/MS analysis; and (5) database searching with inclusion of the di-glycine modification (∼114.0429 Da) as a variable modification on lysine [9]. This approach can be combined with quantitative proteomics methods such as SILAC (stable isotope labeling with amino acids in cell culture) or TMT (tandem mass tag) labeling to investigate dynamic changes in ubiquitination in response to cellular perturbations like DNA damage [9].
One limitation of this method is that it cannot distinguish between ubiquitination and modification by other ubiquitin-like proteins (such as NEDD8 and ISG15) that also generate di-glycine remnants after tryptic digestion [9]. However, control experiments have demonstrated that >94% of di-glycine-modified peptides identified in standard cell lines are genuine ubiquitin remnants [9].
Activity-Based Probes for Ubiquitin Enzymes
Cascading activity-based probes such as UbDha have been developed to monitor catalysis along the E1-E2-E3 reaction pathway [10]. These probes function similarly to native ubiquitin—upon ATP-dependent activation by E1, they travel downstream to E2 and subsequently E3 enzymes through sequential trans-thioesterifications [10]. Unlike native ubiquitin, however, these probes contain electrophilic traps that react irreversibly with active-site cysteine residues of target enzymes, enabling their detection and characterization [10].
This methodology allows for: (1) profiling of active ubiquitin-modifying enzymes under different physiological conditions; (2) monitoring enzymatic activity in living cells; and (3) structural studies of enzyme-probe interactions [10]. The approach is diversifiable to ubiquitin-like modifiers and provides novel tools to interrogate ubiquitin and Ubl cascades.
Chain Linkage-Specific Analysis
To decipher the complexity of ubiquitin chain architectures, researchers have developed linkage-specific binders including antibodies, affimers, and TUBEs (tandem ubiquitin-binding entities) that recognize particular ubiquitin linkage types [7] [9]. These tools enable enrichment of proteins modified by specific types of ubiquitin chains, followed by proteomic analysis to identify the modified proteins and their biological contexts [9].
Figure 2: Experimental Workflow for Ubiquitin Remnant Profiling. This mass spectrometry-based approach enables proteome-wide identification and quantification of ubiquitination sites through specific enrichment of di-glycine-modified peptides.
Research Reagent Solutions
The investigation of ubiquitination pathways relies on specialized reagents and tools designed to address the unique challenges of studying this complex post-translational modification.
Table 3: Essential Research Reagents for Ubiquitination Studies
| Reagent/Tool | Primary Function | Key Applications |
|---|---|---|
| Di-Glycine Remnant Antibodies (e.g., GX41) | Immunoaffinity enrichment of ubiquitin remnant peptides | Ubiquitin remnant profiling; site identification |
| Activity-Based Probes (e.g., UbDha) | Irreversible trapping of active ubiquitin enzymes | Profiling E1-E2-E3 activities; mechanistic studies |
| Linkage-Specific Binders (TUBEs, antibodies) | Selective recognition of specific ubiquitin chain types | Chain topology analysis; linkage-specific enrichment |
| Proteasome Inhibitors (e.g., Bortezomib, MG132) | Inhibition of proteasomal degradation | Stabilization of ubiquitinated proteins; pathway analysis |
| DUB Inhibitors | Selective inhibition of deubiquitinating enzymes | Investigation of ubiquitination dynamics; therapeutic探索 |
| Recombinant E1/E2/E3 Enzymes | Reconstitution of ubiquitination cascades | In vitro ubiquitination assays; mechanistic studies |
Therapeutic Targeting of Ubiquitination Pathways
The central role of ubiquitination in regulating cellular processes, combined with its frequent dysregulation in diseases, has made it an attractive target for therapeutic intervention. Several classes of drugs targeting different components of the ubiquitin-proteasome system have been developed, with some achieving clinical success.
Proteasome inhibitors represent the most clinically advanced class of UPS-targeting therapeutics. Bortezomib (Velcade) was the first proteasome inhibitor approved by the FDA for the treatment of multiple myeloma and mantle cell lymphoma [4]. This boronic acid derivative functions by reversibly binding to the catalytic site of the 26S proteasome [4]. Carfilzomib (Kyprolis), a second-generation proteasome inhibitor, derives from the natural product epoxomicin and has shown efficacy in bortezomib-resistant multiple myeloma [4]. Additional proteasome inhibitors in clinical development include marizomib, ixazomib, and CEP-18770 [4].
E1 enzyme inhibitors have also been explored, though their development is more challenging due to the limited number of E1 enzymes and potential toxicity concerns. Nevertheless, MLN7243 (targeting UBA1) has entered Phase I/II clinical trials [4]. The related compound MLN4924 inhibits the NEDD8-activating enzyme (NAE), thereby specifically affecting the activity of Cullin-RING ligases, a major class of E3 ubiquitin ligases [4].
E2 enzyme inhibitors represent an emerging area, with compounds such as Leucettamol A (targeting the Ubc13-Uev1A interaction) and CC0651 (inhibiting Cdc34) showing promise in preclinical studies [4]. These inhibitors typically target protein-protein interactions required for E2 function or specific catalytic activities.
E3 ligase modulators have gained significant attention due to the potential for greater specificity. Notable examples include nutlin-3a and RG7112, which disrupt the interaction between the E3 ligase MDM2 and the tumor suppressor p53, leading to p53 stabilization and activation of apoptosis in cancer cells [4]. Several MDM2 inhibitors have advanced to clinical trials. Additionally, immunomodulatory drugs such as thalidomide, lenalidomide, and pomalidomide have been shown to target the CRL4CRBN E3 ubiquitin ligase, leading to selective degradation of specific substrate proteins in multiple myeloma [4].
The growing understanding of ubiquitination mechanisms has also enabled the development of novel therapeutic modalities such as PROTACs (Proteolysis-Targeting Chimeras) that harness the ubiquitin system to selectively degrade target proteins [7]. These bifunctional molecules simultaneously bind to a target protein and an E3 ubiquitin ligase, resulting in ubiquitination and degradation of the target [7]. Radiation-responsive PROTAC platforms are also emerging, including radiotherapy-triggered PROTAC (RT-PROTAC) prodrugs that are activated by tumor-localized X-rays to degrade specific oncoproteins [7].
The E1-E2-E3 enzymatic cascade and the diverse topology of ubiquitin chains constitute a sophisticated regulatory system that governs virtually all aspects of cellular function. The hierarchical nature of the enzyme cascade—with two E1s, approximately 35 E2s, and hundreds of E3s—provides both efficiency and remarkable specificity in substrate selection. The structural diversity of ubiquitin chains, linked through different lysine residues or the N-terminal methionine, creates a complex ubiquitin code that determines functional outcomes ranging from proteasomal degradation to activation of signaling pathways.
In the context of DNA repair and immune response pathways, ubiquitination serves as a master regulator that coordinates complex cellular decisions. Through mechanisms such as K63-linked ubiquitination in DNA damage signaling and M1-linear ubiquitination in NF-κB activation, the ubiquitin system ensures appropriate cellular responses to genomic and environmental challenges. The development of sophisticated experimental methods, particularly ubiquitin remnant profiling and activity-based probes, has dramatically advanced our understanding of these processes.
The therapeutic targeting of ubiquitination pathways has already yielded clinically successful drugs, particularly proteasome inhibitors for hematological malignancies. The ongoing development of more specific agents targeting E2 and E3 enzymes, along with innovative approaches such as PROTACs, promises to expand the therapeutic applications of ubiquitin manipulation. As research continues to decipher the complexity of the ubiquitin code, new opportunities will emerge for precisely modulating ubiquitination pathways in disease contexts, particularly in cancer and disorders of immune regulation.
The faithful repair of DNA double-strand breaks (DSBs) is fundamental to maintaining genomic integrity and preventing cellular transformation. The cellular response to these lethal lesions is orchestrated by a sophisticated network of post-translational modifications, among which ubiquitin signaling has emerged as a central regulatory mechanism. Similar to phosphorylation, ubiquitination creates a diverse array of molecular signals that control the recruitment, activity, and stability of DNA repair factors at damage sites [6] [11]. This review examines how the ubiquitin system—comprising writers (E3 ligases), erasers (deubiquitinating enzymes), and readers (ubiquitin-binding domains)—orchestrates the complex decision-making processes that govern DSB repair pathway choice, functionality, and termination.
The strategic importance of ubiquitin signaling in the DNA damage response (DDR) is underscored by its involvement in human diseases linked to genomic instability, including various cancer syndromes [12] [13]. Understanding the mechanistic basis of ubiquitin-dependent regulation in DSB repair not only advances fundamental knowledge but also offers promising therapeutic opportunities for cancers characterized by defective DNA repair pathways [12] [14].
The Ubiquitin and Ubiquitin-Like Conjugation Systems
Principles of Ubiquitin Conjugation
Ubiquitylation is a multistep enzymatic process that involves the sequential action of E1 (activating), E2 (conjugating), and E3 (ligase) enzymes [11] [13]. This cascade results in the covalent attachment of ubiquitin—a highly conserved 76-amino acid protein—to target substrates via an isopeptide bond between the C-terminal glycine of ubiquitin and the ε-amino group of a lysine residue on the substrate [11]. The human genome encodes approximately 600 E3 ligases, which provide substrate specificity and are categorized into two main classes: RING (really interesting new gene) and HECT (homologous to E6AP carboxyl terminus) domain-containing enzymes [15] [16].
The remarkable diversity of ubiquitin signaling stems from the ability to form different ubiquitin chain architectures. Ubiquitin itself contains seven lysine residues (K6, K11, K27, K29, K33, K48, K63) and an N-terminal methionine, all of which can participate in chain formation [11] [15]. These distinct linkage types confer different structures and functions—K48-linked chains primarily target substrates for proteasomal degradation, while K63-linked chains typically serve non-proteolytic roles in signaling and protein recruitment [6] [11].
Table 1: Major Ubiquitin Linkage Types and Their Functions in DNA Damage Response
| Linkage Type | Chain Structure | Primary Functions | Key Roles in DSB Repair |
|---|---|---|---|
| K48-linked | Compact conformation [6] | Proteasomal degradation [11] | Regulation of protein stability (e.g., p53) [13] |
| K63-linked | Extended, "beads-on-a-string" [6] | Non-degradative signaling [11] | Recruitment of repair factors (BRCA1, 53BP1) [12] [15] |
| K6-linked | Not well characterized | DNA repair [11] | Involved in Fanconi anemia pathway [11] |
| K11-linked | Unique conformation [11] | Cell cycle regulation [11] | Potential role in cell cycle checkpoints |
| K27-linked | Not well characterized | Non-canonical signaling [12] | DNA damage signaling [12] |
| Linear/M1-linked | Extended structure | NF-κB signaling, inflammation | Emerging roles in genome stability |
SUMO and Other Ubiquitin-Like Modifiers
Beyond ubiquitin, cells employ additional ubiquitin-like modifiers (UBLs) including SUMO (small ubiquitin-like modifier), which follows a similar enzymatic conjugation cascade but utilizes distinct E1 and E2 enzymes [11] [16]. Mammals express three major SUMO isoforms (SUMO1, SUMO2, and SUMO3) that can be conjugated to substrates through a consensus motif (ΨKXE/D) [11]. SUMOylation has been implicated in various aspects of genome maintenance, with recent evidence highlighting extensive crosstalk between ubiquitin and SUMO in the DSB response [12] [17].
Ubiquitin-Dependent Signaling at DNA Double-Strand Breaks
Hierarchical Assembly of the Ubiquitin Signaling Cascade
The initiation of ubiquitin signaling at DSBs begins with the activation of the ataxia telangiectasia mutated (ATM) kinase, which phosphorylates the histone variant H2AX on serine 139 (generating γ-H2AX) within minutes of damage detection [6] [15]. This phosphorylation event spreads over megabase domains flanking the break and serves as a binding platform for the mediator protein MDC1, which in turn recruits the E3 ubiquitin ligase RNF8 through phospho-dependent interactions [12] [15].
RNF8 catalyzes the K63-linked ubiquitination of histone H1 and L3MBTL2, creating a landing platform for a second E3 ligase, RNF168 [12] [15]. RNF168 then monoubiquitinates histones H2A and H2AX at lysine 13/15, which is subsequently extended into K63-linked chains [15]. This RNF8/RNF168-dependent ubiquitin platform serves as a critical recruitment signal for downstream DNA repair factors including 53BP1 and the BRCA1-A complex [12] [18].
Diagram 1: Hierarchical ubiquitin signaling cascade at DNA double-strand breaks. The pathway initiates with ATM activation and progresses through sequential recruitment of E3 ubiquitin ligases that modify chromatin to create binding platforms for downstream effectors.
Ubiquitin-Binding Domains as Decoders of the Ubiquitin Code
The information encoded in ubiquitin modifications is decoded by specialized ubiquitin-binding domains (UBDs) present in DNA repair proteins [6] [14]. These domains recognize specific features of ubiquitin chains, including linkage type and length. For instance, the tandem UIM domains of RAP80 exhibit remarkable specificity for K63-linked ubiquitin chains, enabling recruitment of the BRCA1-A complex to DSB sites [12]. Similarly, 53BP1 utilizes a ubiquitination-dependent recruitment (UDR) motif that directly binds RNF168-ubiquitylated H2A, facilitating its accumulation at breaks [12].
The low affinity of individual UBDs for ubiquitin is strategically overcome through multiple mechanisms, including the presence of tandem UBDs within a single protein and the clustering of UBD-containing proteins within multi-protein complexes [14]. This avidity-based recognition system allows for sensitive detection of ubiquitin signals while maintaining the reversibility essential for dynamic regulation.
Regulation of DSB Repair Pathway Choice by Ubiquitin
The Balance Between NHEJ and Homologous Recombination
One of the most critical decisions following DSB formation is the choice between two major repair pathways: non-homologous end joining (NHEJ) and homologous recombination (HR) [18] [15]. NHEJ promotes direct ligation of broken DNA ends throughout the cell cycle but is error-prone, while HR requires a sister chromatid template and is restricted to S/G2 phases but is largely error-free [12] [14]. Ubiquitin signaling plays a pivotal role in regulating this pathway choice, primarily through the opposing actions of 53BP1 (promoting NHEJ) and BRCA1 (promoting HR) [15].
The recruitment of 53BP1 to DSBs requires recognition of RNF168-dependent H2A ubiquitination (K15ub) combined with H4K20 methylation [12] [15]. Once recruited, 53BP1 interacts with RIF1 and the shieldin complex to inhibit DNA end resection—the nucleolytic processing of DNA ends that represents the committed step for HR [15]. In contrast, BRCA1 promotes end resection through the recruitment and activation of nucleases such as CtIP, effectively opposing 53BP1 function and directing repair toward HR [15].
Diagram 2: Ubiquitin-dependent regulation of DSB repair pathway choice. The balance between NHEJ and HR is controlled by opposing ubiquitin-mediated recruitment of 53BP1 (promoting NHEJ) and BRCA1 (promoting HR), which differentially regulate the critical step of DNA end resection.
Regulation by Deubiquitinating Enzymes (DUBs)
The ubiquitin landscape at DSBs is dynamically regulated by deubiquitinating enzymes (DUBs) that remove ubiquitin modifications, providing an essential counterbalance to E3 ligase activity [18] [17]. Multiple DUB families have been implicated in the DSB response, including ubiquitin-specific proteases (USPs), ovarian tumor proteases (OTUs), and JAMM motif zinc metalloproteases [15].
Specific DUBs such as USP51, USP3, and BRCC36 have been shown to target RNF168-dependent H2A ubiquitination at lysine 13/15, thereby attenuating 53BP1 recruitment and influencing repair pathway choice [15]. The opposing actions of E3 ligases and DUBs create a dynamic equilibrium that allows precise control over the timing and extent of ubiquitin signaling, ensuring appropriate repair outcomes.
Table 2: Key Deubiquitinating Enzymes in the DNA Double-Strand Break Response
| DUB | Family | Target(s) | Functional Consequences |
|---|---|---|---|
| USP51 | Ubiquitin-specific protease | H2A K13/15 ub [15] | Removes RNF168-dependent ubiquitin, regulates 53BP1 recruitment |
| USP3 | Ubiquitin-specific protease | H2A K13/15 ub [15] | Counteracts RNF8/RNF168 signaling, loss increases IR sensitivity |
| BRCC36 | JAMM metalloprotease | H2AX ubiquitination [15] | Opposes RNF8-mediated ubiquitination, enhances radiosensitivity |
| USP11 | Ubiquitin-specific protease | PALB2, H2A [15] | Promotes HR by stabilizing BRCA1-PALB2 interaction |
| USP44 | Ubiquitin-specific protease | H2A ubiquitination [15] | Regulates histone ubiquitination at DSBs |
Experimental Approaches and Research Tools
Methodologies for Studying Ubiquitin in DNA Repair
Advances in our understanding of ubiquitin signaling in the DDR have been driven by the development of sophisticated experimental methodologies spanning genetics, proteomics, and biochemistry [14]. Genetic screens using RNA interference (RNAi) and, more recently, CRISPR-Cas9 have identified numerous components of the ubiquitin system as regulators of DNA repair [14]. However, technical challenges such as off-target effects in RNAi screens have necessitated rigorous validation approaches [14].
Proteomic techniques have been particularly valuable for mapping ubiquitination sites and identifying ubiquitin-dependent protein interactions. Quantitative mass spectrometry approaches have enabled system-wide analyses of ubiquitylation dynamics in response to DNA damage, revealing the astonishing complexity of the ubiquitin code [12] [14]. Additionally, biochemical reconstitution of ubiquitination events using purified components has provided mechanistic insights into the specificity and regulation of E3 ligases and DUBs [14].
Table 3: Essential Research Reagents and Methodologies for Studying Ubiquitin in DNA Repair
| Tool Category | Specific Examples | Applications and Functions |
|---|---|---|
| Genetic Approaches | Genome-wide RNAi/CRISPR screens [14] | Identification of novel regulators of ubiquit-dependent repair |
| Proteomic Methods | Quantitative mass spectrometry [12] [14] | System-wide analysis of ubiquitination sites and dynamics |
| Biochemical Tools | Activity-based probes for DUBs [14] | Profiling deubiquitinating enzyme activities and specificities |
| Visualization Techniques | Immunofluorescence (IRIF) [6] | Spatial and temporal analysis of repair factor recruitment |
| Structural Biology | NMR, X-ray crystallography [6] | Determination of ubiquitin chain structures and UBD interactions |
| Chemical Biology | Proteasome inhibitors, DUB inhibitors [13] | Dissecting specific ubiquitin pathway functions |
Visualization and Monitoring Techniques
The visualization of DNA repair factors at DSBs through immunofluorescence microscopy (detecting ionizing radiation-induced foci, IRIF) has been instrumental in characterizing the ubiquitin-dependent recruitment cascade [6]. This approach revealed the suprastoichiometric accumulation of repair proteins at break sites, with individual DSBs recruiting up to 1,000 molecules of each repair protein [6]. Live-cell imaging techniques have further enhanced our ability to monitor the dynamic assembly and disassembly of repair complexes in real time, providing insights into the kinetics of ubiquitin signaling events.
Therapeutic Implications and Future Perspectives
Dysregulation of ubiquitin signaling factors involved in DSB repair is tightly linked to severe human disorders and cancer predisposition syndromes [12] [13]. For example, epigenetic inactivation of the E3 ligase CHFR has been documented in multiple cancer types, including breast, colorectal, and lung cancers [13]. Similarly, mutations in DUBs such as BRCC36 have been associated with altered DNA damage sensitivity and genomic instability [15].
The mechanistic insights into ubiquitin-dependent DSB repair offer promising therapeutic opportunities for cancers characterized by genetic instability [12] [14]. Several strategies are being explored, including the development of small molecule inhibitors targeting specific E3 ligases or DUBs, as well as synthetic lethal approaches that exploit the differential dependency of cancer cells on specific DNA repair pathways [13] [14]. For instance, cancers deficient in HR repair (such as BRCA-mutant cancers) show heightened sensitivity to PARP inhibitors, and combining these with modulation of ubiquitin pathway components may enhance therapeutic efficacy or overcome resistance [14].
Future research directions will likely focus on deciphering the more complex aspects of the ubiquitin code, including the functions of atypical ubiquitin chains and the extensive crosstalk between ubiquitin and other UBLs [14] [17]. Additionally, understanding how the ubiquitin system integrates with other cellular processes, such as cell cycle regulation and innate immune signaling, will provide a more comprehensive view of its role in maintaining genome stability and cellular homeostasis [14].
Ubiquitin signaling has emerged as a central regulatory mechanism that orchestrates multiple aspects of the DNA double-strand break response, from initial damage recognition to repair pathway choice and termination. The dynamic and reversible nature of ubiquitination, combined with the astounding diversity of ubiquitin chain architectures, provides a sophisticated signaling system that can be precisely tuned to ensure appropriate repair outcomes. Continued elucidation of the complexities of ubiquitin signaling in DNA repair will not only advance our fundamental understanding of genome maintenance mechanisms but also open new avenues for therapeutic intervention in cancer and other diseases associated with genomic instability.
The cellular response to DNA double-strand breaks (DSBs) represents one of the most critical defense mechanisms for maintaining genomic integrity. DSBs are highly deleterious lesions that can lead to chromosomal translocations, genome instability, and malignant transformation if improperly repaired [19]. The DNA damage response (DDR) operates in the context of chromatin, where elaborate signaling pathways coordinate the ordered recruitment of specific factors to damage sites to facilitate repair and activate cell cycle checkpoints [20]. Within this sophisticated network, ubiquitin signaling—particularly histone ubiquitination—has emerged as a central regulatory mechanism that orchestrates the DDR.
Recent advances have illuminated the pivotal role of RNF168, a RING-type E3 ubiquitin ligase, as a master regulator of histone ubiquitination at DSB sites [21]. This technical guide examines the molecular mechanisms through which RNF168-mediated ubiquitination of H2A and H2AX governs DDR pathway activation and chromatin remodeling. Understanding these processes provides critical insights for therapeutic interventions targeting DNA repair pathways in cancer and other diseases.
The RNF168 Ubiquitin Ligase: Mechanism and Regulation
Molecular Architecture and Functional Domains
RNF168 possesses a modular structure containing distinct functional domains that enable its specific role in the DDR cascade. A key discovery is that RNF168 undergoes liquid-liquid phase separation (LLPS) through an intrinsically disordered region (IDR) spanning amino acids 460-550 [21]. This condensation behavior significantly enhances RNF168's catalytic activity and facilitates its rapid accumulation at DNA break sites.
Mechanism of LLPS-Enhanced Function: The phase separation capacity of RNF168 creates a concentrated microenvironment that promotes efficient ubiquitin transfer. This LLPS is significantly enhanced by K63-linked polyubiquitin chains, establishing a positive feedback loop that amplifies RNF168-mediated signaling at DSB sites [21]. Functionally, LLPS deficiency in RNF168 results in delayed recruitment of critical repair factors 53BP1 and BRCA1, ultimately impairing DSB repair efficiency [21].
The Ubiquitination Cascade
RNF168 operates within a hierarchical ubiquitination cascade initiated by the E3 ligase RNF8, which recruits RNF168 to damage sites [19]. RNF168 then catalyzes the monoubiquitination of histone H2A and its variant H2AX on specific lysine residues, including K13, K15, and K119 [20]. This ubiquitin signaling creates docking platforms for the recruitment of downstream DNA repair factors.
Table 1: Key Histone Ubiquitination Targets in the DNA Damage Response
| Histone Target | Ubiquitination Site | E3 Ubiquitin Ligase | Functional Consequence |
|---|---|---|---|
| H2A | K13, K15 | RNF168 | Recruitment of 53BP1 and BRCA1 |
| H2AX | K13, K15 | RNF168 | Amplification of damage signaling |
| H2A | K119 | RNF168/BMI1 | Transcriptional silencing at breaks |
| H2A | K127/129 | BRCA1-BARD1 | Promotion of homologous recombination |
Chromatin Remodeling and Repair Pathway Choice
Histone Ubiquitination in the Epigenetic Landscape of DSBs
The pre-existing chromatin conformation significantly influences both DNA damage induction and repair pathway selection. Compacted heterochromatin may physically shield DNA from damage, while simultaneously presenting challenges for repair machinery accessibility [19]. The "prime-repair-restore" model describes how the initial chromatin landscape is remodeled to facilitate damage signaling, followed by restoration of the original epigenetic state after repair completion [19].
RNF168-generated ubiquitin marks function as central components of this repair-associated epigenetic landscape. The RNF8-RNF168 ubiquitination axis promotes two critical chromatin modifications that 53BP1 recognizes in a bivalent mode: H2AK15ub and H4K20me2 [19]. This dual recognition mechanism ensures specific targeting of 53BP1 to DSB sites, where it promotes non-homologous end joining (NHEJ) by limiting DNA end resection.
Crosstalk with Other Post-Translational Modifications
Ubiquitination does not function in isolation but engages in sophisticated crosstalk with other histone modifications to regulate DDR pathway choice:
Ubiquitination and Acetylation: The histone acetyltransferase TIP60 acetylates H2AK15, directly blocking RNF168-mediated ubiquitination at this site and impairing 53BP1 binding [19]. Additionally, TIP60-mediated acetylation of H4K16 physically inhibits 53BP1 binding to H4K20me2, thereby promoting homologous recombination (HR) [20].
Ubiquitination and Methylation: Histone H4K20 methylation provides a binding platform for 53BP1 recruitment [20]. The removal or absence of methylation at H4K20 (H4K20me0) guides repair pathway choice toward HR by creating a binding site for the BRCA1-BARD1 complex [19].
Ubiquitination and Phosphorylation: Phosphorylation of the histone variant H2AX (forming γ-H2AX) by ATM, ATR, and DNA-PKcs represents one of the earliest DDR signaling events [19]. This phosphorylation cascade promotes the recruitment of RNF8, which initiates the ubiquitination signaling that recruits RNF168, creating a feedback loop that amplifies DDR activation [20].
Diagram 1: RNF168-Driven Ubiquitin Signaling Cascade at DSB Sites. The pathway illustrates the sequential recruitment and activation of key DDR factors following DNA damage, culminating in RNF168-mediated histone ubiquitination that directs repair pathway choice.
Advanced Experimental Approaches
Methodologies for Studying RNF168 Function
LLPS Analysis in RNF168 Condensation: To investigate RNF168 phase separation, researchers employ purified RNF168 protein subjected to in vitro condensation assays [21]. The experimental workflow involves:
- Protein Purification: Recombinant RNF168 is expressed and purified using affinity chromatography.
- LLPS Induction: Purified RNF168 is exposed to K63-linked polyubiquitin chains in physiological buffers.
- Imaging and Quantification: Liquid-like condensate formation is visualized by microscopy and quantified for number, size, and dynamics.
- Functional Validation: H2A.X ubiquitination assays are performed to correlate LLPS with catalytic activity enhancement.
DNA Repair Factor Recruitment Kinetics: To assess the functional consequences of RNF168 activity, time-course experiments monitor the recruitment of 53BP1 and BRCA1 to DSB sites [21]:
- DSB Induction: Cells are irradiated or treated with DSB-inducing agents.
- Immunofluorescence Staining: Fixed cells are stained with antibodies against γ-H2AX, 53BP1, and BRCA1 at various time points.
- Image Analysis: Confocal microscopy quantifies fluorescence intensity at damage sites.
- Statistical Comparison: Recruitment kinetics are compared between wild-type and LLPS-deficient RNF168 mutants.
Techniques for Monitoring Histone Ubiquitination
Chromatin Immunoprecipitation (ChIP) Sequencing: This approach maps the genomic distribution of ubiquitinated histones and repair factors:
- Crosslink cells and isolate chromatin
- Immunoprecipitate with antibodies specific for H2AK15ub
- Sequence bound DNA fragments
- Analyze enrichment patterns at DSB sites and genome-wide
Comet Assay for DNA Break Detection: The neutral comet assay quantifies genomic DSBs in single cells [22]:
- Embed cells in low-melting-point agarose on microscope slides
- Lyse cells to remove membranes and proteins
- Perform electrophoresis under neutral conditions
- Stain with DNA-binding dye and image by fluorescence microscopy
- Quantify DNA damage using Olive Tail Moment analysis
Table 2: Quantitative Analysis of RNF168 LLPS Impact on DSB Repair
| Experimental Parameter | Wild-type RNF168 | LLPS-Deficient RNF168 | Measurement Technique |
|---|---|---|---|
| H2A.X Ubiquitination | Enhanced by K63-ubiquitin | Significantly reduced | In vitro ubiquitination assay |
| 53BP1 Recruitment | Rapid accumulation (<2h) | Delayed (>4h) | Immunofluorescence microscopy |
| BRCA1 Recruitment | Efficient foci formation | Impaired foci formation | Confocal microscopy quantification |
| DSB Repair Efficiency | High survival rate | Reduced survival | Clonogenic survival assay |
| Phase Separation | Robust condensates | Minimal condensation | In vitro LLPS assay |
Research Reagent Solutions
Table 3: Essential Research Tools for Investigating Histone Ubiquitination in DDR
| Reagent/Category | Specific Examples | Research Application | Technical Function |
|---|---|---|---|
| E3 Ligase Inhibitors | RNF168 small molecule inhibitors | Pathway disruption studies | Block specific ubiquitin transfer |
| Ubiquitin Variants | K63-linked ubiquitin chains | LLPS enhancement assays | Promote RNF168 condensation |
| Specific Antibodies | Anti-H2AK15ub, Anti-γH2AX | Damage site visualization | Immunofluorescence detection |
| Cell Line Models | RNF168 knockout cells | Functional rescue experiments | Define pathway requirements |
| Chemical Inhibitors | ATM inhibitors (KU-55933), DNA-PKcs inhibitors (NU7026) [23] | Kinase pathway analysis | Dissect signaling hierarchies |
| LLPS Reporters | RNF168-IDR fusion constructs | Phase separation dynamics | Live imaging of condensates |
Therapeutic Implications and Future Perspectives
The central role of RNF168-mediated ubiquitin signaling in DDR pathway choice presents compelling therapeutic opportunities. Cancer cells with defective HR repair (such as those with BRCA1/2 mutations) depend on NHEJ and alternative repair pathways for survival. Therapeutic strategies targeting RNF168 could potentially sensitize such cancers to DNA-damaging agents by disrupting this compensatory repair mechanism.
Emerging evidence also links RNF168 and histone ubiquitination to immune signaling pathways. In macrophages, DSBs activate a genetic program that promotes inflammasome activation and production of IL-1β and IL-18, with this response being regulated by DDR kinases [23]. This intersection between DNA damage and immune signaling may have significant implications for cancer immunotherapy, particularly in understanding the mechanistic basis of how genomic instability influences antitumor immunity.
Future research directions should focus on:
- Developing specific pharmacological inhibitors of RNF168 catalytic activity
- Elucidating the structural basis of RNF168 LLPS and its regulation
- Investigating the crosstalk between histone ubiquitination and other PTMs in different chromatin contexts
- Exploring the role of RNF168 in immune cell function and tumor microenvironment regulation
The sophisticated experimental approaches outlined in this technical guide provide the foundation for advancing our understanding of histone ubiquitination in DNA repair and developing novel therapeutic strategies that target these pathways in human disease.
Diagram 2: Experimental Workflow for Analyzing RNF168 Function. The diagram outlines key methodological approaches for investigating RNF168-mediated histone ubiquitination and its functional consequences in DNA double-strand break repair.
The ubiquitin system, a pervasive post-translational modification mechanism, is a critical regulator of innate immune responses. It orchestrates the detection of pathogens by pattern-recognition receptors (PRRs) and the subsequent induction of inflammatory and antiviral responses [24] [25]. Ubiquitination involves the covalent attachment of the 76-amino-acid protein ubiquitin to substrate proteins via a sequential enzymatic cascade involving E1 (activating), E2 (conjugating), and E3 (ligating) enzymes [26] [27]. The process is reversible through the action of deubiquitinases (DUBs), allowing for dynamic control of signaling pathways [24] [25]. The functional outcome of ubiquitination is determined by the topology of the ubiquitin chain. K63-linked and linear (Met1-linked) polyubiquitin chains typically serve as non-degradative scaffolds that facilitate the assembly of signaling complexes, whereas K48-linked polyubiquitin chains predominantly target proteins for proteasomal degradation [26] [25] [28]. This "ubiquitin code" enables precise regulation of PRR pathways, including Toll-like receptors (TLRs), RIG-I-like receptors (RLRs), the cytosolic DNA sensor cGAS-STING, and inflammasome complexes, ensuring an effective yet balanced immune response to infection [29] [30] [25].
Ubiquitination in Toll-like Receptor (TLR) Signaling
Toll-like receptors are membrane-bound PRRs that sense pathogen-associated molecular patterns (PAMPs) either at the cell surface or within endosomal compartments [25]. Their signaling primarily transduces signals through the adaptor proteins MyD88 (all TLRs except TLR3) and TRIF (TLR3 and TLR4), culminating in the activation of transcription factors NF-κB and IRF3/7 to induce proinflammatory cytokines and type I interferons (IFNs) [25].
- Key Regulatory Ubiquitination Events: The signaling cascades downstream of TLRs are heavily governed by ubiquitination. The E3 ligase TRAF6 plays a central role: upon recruitment to activated receptors, it catalyzes the formation of K63-linked ubiquitin chains on itself and other proteins, such as NEMO [25]. These chains serve as a platform to recruit and activate the TAK1 kinase complex via ubiquitin-binding domains (UBDs) present in its subunits. TAK1 then phosphorylates and activates the IKK complex, leading to NF-κB activation [25]. Similarly, the adaptor TRIF recruits RIP1 via RHIM-domain interaction, and K63-linked ubiquitination of RIP1 facilitates the recruitment of TAK1 and NEMO for NF-κB activation [25].
- Negative Regulation: Deubiquitinases provide crucial negative feedback to prevent excessive TLR signaling. For example, A20 (also known as TNFAIP3) dampens NF-κB activation by removing K63-linked ubiquitin chains from RIP1 and other signaling molecules [25] [31]. The Met1-linkage-specific DUB OTULIN hydrolyzes linear ubiquitin chains assembled by the LUBAC complex, thereby fine-tuning inflammatory signaling and cell fate decisions [24].
Table 1: Key Ubiquitin-Related Enzymes in TLR Signaling
| Enzyme Name | Type | Target Protein | Ubiquitin Linkage | Function in Pathway |
|---|---|---|---|---|
| TRAF6 | RING E3 Ligase | Itself, NEMO | K63 | Positively regulates NF-κB and MAPK activation [25]. |
| LUBAC | RBR E3 Ligase | NEMO, RIP1 | Linear (Met1) | Positively regulates NF-κB activation [24] [25]. |
| A20 (TNFAIP3) | DUB & E3 Ligase | RIP1 | K63 (DUB activity) | Negatively regulates NF-κB; dual enzyme activity [25] [31]. |
| OTULIN | DUB | Linear ubiquitin chains | Linear (Met1) | Negatively regulates NF-κB by hydrolyzing linear chains [24]. |
Figure 1: Ubiquitin Regulation in TLR Signaling Pathways. K63-linked ubiquitination events are central to the activation of downstream NF-κB and MAPK pathways, while DUBs like A20 and OTULIN provide negative feedback.
Ubiquitination in RIG-I-like Receptor (RLR) Signaling
RLRs, including RIG-I and MDA5, are cytosolic sensors of viral RNA that initiate antiviral innate immunity [29] [25]. Upon ligand binding, they undergo conformational changes and interact with the mitochondrial adaptor protein MAVS (also known as IPS-1, VISA, Cardif), triggering a signaling cascade that leads to the production of type I IFNs [29].
- Sequential Ubiquitination of RIG-I: The activation of RIG-I is a tightly controlled process involving sequential ubiquitination. The E3 ligase Riplet (RNF135) first ubiquitylates RIG-I at K788, which releases its CARD domains from an autorepressed state [29]. Subsequently, TRIM25 and TRIM4 synthesize K63-linked ubiquitin chains on the RIG-I CARD domains (at K172 and K164/172, respectively), facilitating the interaction and aggregation of RIG-I with the downstream adaptor MAVS [29]. Another E3, MEX3C (RNF194), also promotes RIG-I activation through K63-linked ubiquitylation [29].
- Regulation of MDA5 and MAVS: The related RLR MDA5 is similarly regulated by ubiquitin. TRIM65 catalyzes K63-linked polyubiquitylation of MDA5 at K743, promoting its activation [29]. At the level of the mitochondrial adaptor MAVS, TRIM31 and TRIM21 positively regulate signaling. TRIM31 promotes K63-linked polyubiquitylation and aggregation of MAVS, while TRIM21 mediates K27-linked ubiquitylation to enhance TBK1 binding [29]. Negative regulators include RNF5, which targets MAVS for K48-linked degradative ubiquitylation [29].
Table 2: Key Ubiquitin-Related Enzymes in RLR Signaling
| Enzyme Name | Type | Target Protein | Ubiquitin Linkage | Function in Pathway |
|---|---|---|---|---|
| Riplet (RNF135) | RING E3 Ligase | RIG-I | K63 | Initiates RIG-I activation by releasing CARDs [29]. |
| TRIM25 | RING E3 Ligase | RIG-I | K63 | Promotes RIG-I CARD oligomerization and MAVS binding [29]. |
| TRIM65 | RING E3 Ligase | MDA5 | K63 | Positively regulates MDA5 activation [29]. |
| TRIM31 | RING E3 Ligase | MAVS | K63 | Promotes MAVS aggregation and signal transduction [29]. |
| RNF5 | RING E3 Ligase | MAVS | K48 | Negatively regulates signaling by targeting MAVS for degradation [29]. |
Figure 2: Ubiquitin Regulation in RLR Antiviral Signaling. K63-linked ubiquitination is essential for the activation of RIG-I/MDA5 and the MAVS signalosome, while E3 ligases like RNF5 attenuate signaling via K48-linked degradation.
Ubiquitination in the cGAS-STING Signaling Pathway
The cGAS-STING pathway is a key defender against cytosolic DNA from viruses, intracellular bacteria, and self-DNA in autoimmunity [30]. cGAS binds DNA and synthesizes the second messenger 2'3'-cGAMP, which activates the endoplasmic reticulum-resident adaptor STING. Activated STING then traffics to the Golgi, recruiting TBK1 and IRF3 to induce type I IFNs [29] [30].
- Regulation of cGAS: Multiple E3 ligases and DUBs control cGAS stability and activity. TRIM56 induces monoubiquitination of cGAS at Lys335, enhancing its dimerization, DNA-binding capacity, and cGAMP production [30]. RNF185 mediates K27-linked polyubiquitination of cGAS to enhance its enzymatic activity [30]. To prevent excessive activation, the ubiquitin-proteasome system (UPS) also targets nuclear cGAS for degradation, a process mediated by the CRL5-SPSB3 E3 complex [30]. The deubiquitinase USP14 counteracts ubiquitination to stabilize cGAS by recruiting TRIM14 under IFN-I stimulation [30].
- Regulation of STING: STING activation and turnover are critically controlled by ubiquitination. Positive regulators include TRIM56, TRIM32, and RNF115, which promote K63-linked polyubiquitination of STING, facilitating its dimerization, Golgi translocation, and recruitment of TBK1 [30]. The ER-resident E3 complex AMFR/INSIG1 catalyzes K27-linked polyubiquitination of STING, which is also required for TBK1 recruitment and IFN production [30]. Negative regulation is achieved through K48-linked ubiquitination. RNF5 and TRIM30α promote K48-linked polyubiquitination and proteasomal degradation of STING, terminating the signal [30]. Furthermore, activated STING is eventually targeted for lysosomal degradation via a microautophagy mechanism that requires K63-linked ubiquitination at Lys288, a process involving the ESCRT complex [30].
Table 3: Key Ubiquitin-Related Enzymes in the cGAS-STING Pathway
| Enzyme Name | Type | Target Protein | Ubiquitin Linkage | Function in Pathway |
|---|---|---|---|---|
| TRIM56 | RING E3 Ligase | cGAS, STING | Monoubiquitin, K63 | Enhances cGAS activity & STING trafficking [30]. |
| RNF185 | RING E3 Ligase | cGAS | K27 | Enhances cGAS enzymatic activity [30]. |
| AMFR/INSIG1 | RING E3 Complex | STING | K27 | Promotes STING aggregation and TBK1 recruitment [30]. |
| RNF5 | RING E3 Ligase | STING | K48 | Negatively regulates signaling by targeting STING for degradation [30]. |
| USP14 | DUB | cGAS | K48 (removal) | Stabilizes cGAS by cleaving ubiquitin chains [30]. |
Figure 3: Ubiquitin Regulation of the cGAS-STING DNA Sensing Pathway. Activating ubiquitin modifications promote cGAS and STING function, while degradative ubiquitination and lysosomal turnover ensure pathway termination.
Ubiquitination in Inflammasome Activation
Inflammasomes are multiprotein complexes (e.g., NLRP3, AIM2) that assemble in response to infection or cellular stress, leading to the activation of caspase-1 and the maturation and secretion of pro-inflammatory cytokines IL-1β and IL-18 [25] [32]. Ubiquitination serves as a critical regulatory switch for both the priming and activation steps of inflammasome formation.
- Regulation of NLRP3 Inflammasome: The NLRP3 sensor is tightly controlled by ubiquitination. In its inactive state, NLRP3 is constitutively modified by K48-linked ubiquitin chains, maintaining low protein levels [25] [32]. During the priming phase (e.g., via TLR signaling), deubiquitination of NLRP3 by DUBs such as BRCC3 is required for its activation and inflammasome assembly [25]. Conversely, E3 ligases like A20 and TRIM31 can negatively regulate NLRP3 by promoting its ubiquitination and degradation [32].
- Regulation of Inflammatory Cell Death (Pyroptosis): Ubiquitination also controls inflammatory cell death pathways like pyroptosis and necroptosis, which are closely linked to inflammasome activity [24] [32]. For instance, in the necroptosis pathway, the kinase RIPK3 and its substrate MLKL are regulated by ubiquitination. E3 ligases such as cIAP1/2 promote K63-linked ubiquitination of RIPK1, which can suppress necroptotic cell death, while DUBs can promote it [32]. In sepsis, the ubiquitin-dependent modification of RIPK1 and NLRP3 is a key factor in controlling the intensity of the inflammatory response [32].
The Scientist's Toolkit: Research Reagent Solutions
Studying ubiquitination in innate immunity requires a specialized set of molecular tools and reagents. The following table details key resources for probing these pathways.
Table 4: Essential Research Reagents for Studying Ubiquitin in Innate Immunity
| Reagent / Tool | Category | Key Function & Application | Example Use-Case |
|---|---|---|---|
| Ubiquitin Mutants (K-only, K0, ΔGG) | Molecular Biology | Determine ubiquitin linkage specificity; K0 (all lysines mutated to arginine) blocks polyubiquitination; ΔGG (C-terminal glycine deletion) prevents conjugation and serves as a negative control [26]. | Identify if a protein modification is K63-linked by co-expressing a ubiquitin mutant where only K63 is available [26]. |
| Proteasome Inhibitors (Bortezomib, MG132) | Small Molecule Inhibitor | Inhibit the 26S proteasome, causing accumulation of K48-linked polyubiquitinated proteins; used to test if a protein's degradation is proteasome-dependent [27] [31]. | Stabilize a protein of interest (e.g., STING) to investigate if its turnover is mediated by the ubiquitin-proteasome system [30] [27]. |
| Specific E3 Ligase & DUB Inhibitors | Small Molecule Inhibitor | Chemically perturb the activity of specific enzymes to assess their function in a pathway (e.g., HBX 41,108 for USP7; PR-619 as a broad-spectrum DUB inhibitor) [31]. | Test the role of a specific DUB in regulating RLR signaling by treating cells with an inhibitor and measuring IFN production. |
| siRNA/shRNA Libraries | Genetic Tool | Perform loss-of-function screens to identify novel E3s/DUBs involved in PRR signaling; genome-wide libraries enable unbiased discovery [14]. | Identify regulators of the cGAS-STING pathway by screening an E3 ligase-focused siRNA library for modulators of IFN-β promoter activity. |
| Linkage-Specific Ubiquitin Antibodies | Immunological Reagent | Detect and quantify specific ubiquitin chain types (e.g., K48-linkage, K63-linkage, linear) in Western blot or immunofluorescence [14]. | Confirm that an immune stimulus induces K63-linked ubiquitination of MAVS by immunoprecipitation followed by Western blot with anti-K63-linkage specific antibody. |
| Tandem Ubiquitin-Binding Entities (TUBEs) | Affinity Reagents | Isolate and enrich polyubiquitinated proteins from cell lysates while protecting them from DUB activity, facilitating proteomic analysis [14]. | Enrich and identify novel ubiquitinated proteins in the TLR4 signaling complex by mass spectrometry after LPS stimulation. |
Detailed Experimental Protocols
This section provides standardized methodologies for key experiments used to dissect ubiquitination in immune signaling pathways.
Co-Immunoprecipitation (Co-IP) to Assess Protein-Protein Interactions and Ubiquitination
Objective: To determine if a protein of interest (POI) interacts with a specific E3 ligase or DUB, or to confirm its ubiquitination status in a physiological context.
Protocol:
- Cell Transfection & Stimulation: Transfect cells (e.g., HEK293T, THP-1) with plasmids expressing your POI, relevant ubiquitin constructs (e.g., HA-Ub, Myc-Ub), and/or the E3/DUB of interest. Alternatively, stimulate innate immune cells (e.g., bone marrow-derived macrophages) with relevant ligands (LPS, Poly(I:C), cGAMP).
- Cell Lysis: Harvest cells and lyse in a non-denaturing IP lysis buffer (e.g., 1% NP-40, 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA) supplemented with protease inhibitors (e.g., PMSF, aprotinin) and 10-20 mM N-Ethylmaleimide (NEM) to inhibit endogenous DUBs.
- Immunoprecipitation: Pre-clear the lysate with Protein A/G beads. Incubate the supernatant with an antibody against your POI or a tag (e.g., anti-FLAG, anti-HA) for 2-4 hours at 4°C. Add Protein A/G beads and incubate for an additional 1-2 hours.
- Washing and Elution: Wash beads 3-5 times with lysis buffer. Elute the immunoprecipitated complexes by boiling in 2X Laemmli SDS sample buffer.
- Immunoblotting: Resolve the eluted proteins by SDS-PAGE and transfer to a PVDF membrane. Probe the membrane with antibodies against ubiquitin, specific ubiquitin linkages, or the interacting E3/DUB to assess interaction or ubiquitination.
2In VitroUbiquitination Assay
Objective: To reconstitute the ubiquitination reaction using purified components and directly test if an E3 ligase can ubiquitinate a candidate substrate.
Protocol:
- Purification of Components: Purify recombinant E1 enzyme, E2 enzyme, E3 ligase, substrate protein, and ubiquitin from E. coli or using an in vitro transcription/translation system.
- Reaction Setup: In a reaction tube, combine the following in ubiquitination assay buffer (50 mM Tris-HCl pH 7.5, 5 mM MgCl2, 2 mM ATP):
- E1 enzyme (100 nM)
- E2 enzyme (1-5 µM)
- E3 ligase (0.5-2 µM)
- Substrate protein (2-5 µM)
- Ubiquitin (50-100 µM)
- Incubation: Incubate the reaction at 30°C for 1-2 hours.
- Termination and Analysis: Stop the reaction by adding SDS sample buffer and boiling. Analyze the products by SDS-PAGE and Western blotting, using an antibody against the substrate or a tag on the substrate to detect upward molecular weight shifts indicative of ubiquitination.
RNAi-based Functional Screen for Ubiquitin Pathway Regulators
Objective: To systematically identify E3 ligases or DUBs that regulate a specific innate immune pathway.
Protocol:
- Reporter Cell Line Generation: Stably transduce a relevant cell line (e.g., HEK293, monocytic cell line) with a reporter construct (e.g., IFN-β promoter driving luciferase or GFP).
- Screen Execution: Reverse-transfect the reporter cells with a genome-wide or focused siRNA/shRNA library targeting E3s and DUBs. Include non-targeting siRNA controls.
- Stimulation and Readout: After 48-72 hours to allow for gene knockdown, stimulate the cells with a pathway-specific agonist (e.g., Sendai virus for RLRs, HT-DNA for cGAS-STING). Measure the reporter activity (luminescence/fluorescence) 6-24 hours post-stimulation.
- Hit Validation: Identify siRNAs that significantly enhance or suppress the reporter response. Validate primary hits using individual siRNAs and measure endogenous mRNA levels of immune genes (e.g., IFNB1, CXCL10) by qRT-PCR. Confirm the physical interaction between the validated hit and pathway components by Co-IP [14].
The tumor necrosis factor-alpha (TNF-α) signaling pathway represents a paradigm for understanding how cells process extracellular inflammatory signals into decisive fate determinations. This process is governed by an intricate balance between competing ubiquitination and deubiquitination events that ultimately determine whether a cell survives, undergoes apoptosis, or proceeds to necroptosis. Ubiquitination, once primarily recognized as a mere tag for protein degradation, has emerged as a sophisticated signaling mechanism that regulates diverse cellular processes including inflammatory response, immune activation, and cell death. Within the context of a broader thesis on ubiquitination in DNA repair and immune response pathways, the TNF-α system offers a compelling model of how reversible post-translational modifications coordinate complex biological outcomes through dynamic protein regulation.
The ubiquitin system employs a cascade of enzymatic reactions involving E1 activating enzymes, E2 conjugating enzymes, and E3 ligases that collectively conjugate ubiquitin to target proteins [33]. This process is reversible through the action of deubiquitinating enzymes (DUBs) that remove ubiquitin modifications, providing a sophisticated regulatory switch for signaling pathways [34]. The complexity of ubiquitin signaling is magnified by the ability to form different polyubiquitin chain linkages—including K48-linked, K63-linked, and linear chains—each encoding distinct functional outcomes for the modified protein [33]. In TNF-α signaling, this ubiquitin code determines the assembly and disassembly of signaling complexes that dictate cellular fate, making it a quintessential model for understanding how competing ubiquitination and deubiquitination events govern fundamental biological processes.
Molecular Players in the TNF-α Signaling Pathway
Core Signaling Complexes
The TNF-α signaling cascade initiates when the cytokine binds to its cognate receptor TNFR1, leading to the assembly of a primary signaling complex known as Complex I [35]. This membrane-associated complex serves as the platform for the initial ubiquitination events that determine subsequent signaling trajectories. The core components of Complex I include:
- TRADD (TNFRSF1A-associated via death domain): Functions as a scaffold protein that recruits other signaling molecules to the activated receptor [35].
- RIPK1 (Receptor-interacting serine/threonine-protein kinase 1): Serves as a critical signaling hub whose ubiquitination status dictates pathway orientation [33].
- TRAF2 (TNF receptor-associated factor 2): Recruits cellular inhibitor of apoptosis proteins (c-IAPs) to the complex [35].
- c-IAP1/2 (Cellular inhibitor of apoptosis proteins 1 and 2): E3 ubiquitin ligases that mediate ubiquitination of RIPK1 and other complex components [33].
Following Complex I formation, a series of orchestrated ubiquitination events determine whether the cell will proceed toward NF-κB-mediated survival or initiate cell death pathways. The ubiquitination status of RIPK1 represents a critical decision point, with different ubiquitin linkages producing diametrically opposed cellular outcomes.
E3 Ubiquitin Ligases and Their Functions
E3 ubiquitin ligases confer substrate specificity to the ubiquitination system and play decisive roles in TNF-α signaling. The key E3 ligases involved include:
- c-IAP1/2: These RING domain-containing E3 ligases mediate K63-linked ubiquitination of RIPK1, particularly on lysine 377 in humans, which is critical for NF-κB activation [33]. They also promote K48-linked ubiquitination of RIPK1 under certain conditions, leading to its proteasomal degradation and inhibition of cell death [33].
- LUBAC (Linear ubiquitin chain assembly complex): Composed of HOIP, HOIL-1, and SHARPIN, this multi-subunit E3 complex generates linear ubiquitin chains on components including NEMO, RIP1, and TNFR1 itself [33]. LUBAC stabilizes signaling complexes and facilitates MAPK and NF-κB activation [33]. Genetic studies demonstrate that HOIP deficiency induces embryonic lethality that can be rescued by TNFR1 deletion, underscoring LUBAC's critical role in TNF signaling [33].
Table 1: Key E3 Ubiquitin Ligases in TNF-α Signaling
| E3 Ligase | Components | Ubiquitin Linkage | Key Substrates | Biological Function |
|---|---|---|---|---|
| c-IAP1/2 | Single subunit RING ligases | K63, K48, K11 | RIPK1, themselves | Promotes NF-κB activation; regulates cell death |
| LUBAC | HOIP, HOIL-1, SHARPIN | Linear | NEMO, RIPK1, TNFR1 | Stabilizes signaling complexes; enhances NF-κB signaling |
| TRAF2 | Single subunit RING ligase | K63 | RIPK1 | Recruits c-IAP1/2; initiates ubiquitination cascade |
Deubiquitinating Enzymes and Their Regulatory Roles
Deubiquitinating enzymes (DUBs) provide counter-regulatory functions that terminate or modulate ubiquitin-dependent signaling. The major DUBs operating in the TNF-α pathway include:
- CYLD (Cylindromatosis): A tumor suppressor DUB that removes K63-linked ubiquitin chains from RIPK1, thereby attenuating NF-κB activation and promoting cell death [36] [37]. CYLD-mediated deubiquitination of RIPK1 facilitates the transition from Complex I to death-inducing complexes [36].
- A20 (TNFAIP3): This DUB exhibits dual functionality, removing K63-linked ubiquitin chains from RIPK1 while subsequently promoting its K48-linked ubiquitination, thereby terminating NF-κB signaling and promoting RIPK1 degradation [35]. This sequential activity represents a process called "ubiquitin editing" that ensures signal termination [38].
- OTUD7B: Recruited to activated TNFR1 where it promotes RIPK1 deubiquitination, attenuating NF-κB activation [35].
- USP21: Inhibits TNF-α-induced NF-κB signaling by promoting deubiquitination of RIPK1 [39].
- USP4: Targets TRAF2 and TRAF6 for deubiquitination and inhibits TNFα-induced cancer cell migration [40].
Table 2: Major Deubiquitinating Enzymes in TNF-α Signaling
| DUB | Family | Key Substrates | Ubiquitin Linkage Specificity | Biological Function |
|---|---|---|---|---|
| CYLD | USP | RIPK1, TRAF2, TRAF6 | K63 | Attenuates NF-κB; promotes cell death |
| A20 (TNFAIP3) | OTU | RIPK1, TRAF6 | K63 (removal), K48 (addition) | Terminates NF-κB via "ubiquitin editing" |
| USP21 | USP | RIPK1 | K63 | Inhibits NF-κB activation |
| USP4 | USP | TRAF2, TRAF6 | K63 | Negatively regulates TNFα-induced signaling |
| OTUD7B | OTU | RIPK1 | K63 | Attenuates NF-κB activation |
The Ubiquitin-Driven Fate Decision Mechanism
NF-κB Activation Pathway
When pro-survival signaling predominates, the ubiquitination events in Complex I create a platform for IKK (IκB kinase) and TAK1 (TGF-β-activated kinase 1) complex activation, leading to NF-κB-mediated gene transcription. This process involves:
- K63-linked and linear ubiquitin chains on RIPK1, generated by c-IAP1/2 and LUBAC, serve as scaffolding platforms [33].
- TAK1 complex (TAK1-TAB2-TAB3) is recruited through binding of TAB2/3 to K63-linked ubiquitin chains via their C-terminal zinc finger domains [33].
- The IKK complex (IKKα-IKKβ-NEMO) is recruited through binding of NEMO to both K63-linked and linear ubiquitin chains via its UBAN (ubiquitin binding in ABIN and NEMO proteins) domain [33].
- TAK1 phosphorylates and activates IKKβ, which then phosphorylates IκBα, targeting it for K48-linked ubiquitination and proteasomal degradation [37].
- This releases the NF-κB dimer (typically p50-RelA) to translocate to the nucleus and activate transcription of anti-apoptotic and inflammatory genes [35].
The resulting gene expression promotes cell survival through upregulation of anti-apoptotic proteins (BIRC2, BIRC3, BCL2L1) and inflammatory mediators (IL-6), rendering the cell resistant to death signals [35].
Cell Death Pathway Activation
When deubiquitination events prevail, particularly through the actions of CYLD and A20, RIPK1 undergoes deubiquitination and dissociates from Complex I, leading to the formation of cytoplasmic death-inducing complexes:
- Complex IIa (also known as the apoptosome) forms when deubiquitinated RIPK1 associates with FADD and caspase-8, initiating extrinsic apoptosis through caspase-8 activation [35].
- Complex IIb (necrosome) forms when RIPK1 associates with RIPK3 and MLKL, initiating necroptosis, a programmed necrotic cell death pathway [33].
- Activation of caspase-8 within these complexes triggers a proteolytic cascade that executes apoptosis, while phosphorylation of MLKL by RIPK3 initiates necroptosis through membrane disruption [35].
The balance between these opposing pathways is precisely regulated by the ubiquitination status of RIPK1, which serves as a molecular switch determining cellular fate.
Figure 1: The TNF-α Fate Decision Paradigm - Competing ubiquitination and deubiquitination of RIPK1 determine whether cells survive via NF-κB signaling or die through apoptosis or necroptosis.
Experimental Approaches and Methodologies
Key Experimental Models and Reagents
Research into TNF-α signaling ubiquitination has employed sophisticated experimental systems ranging from cell culture models to genetically engineered mice. The following research reagents represent essential tools for investigating this pathway:
Table 3: Essential Research Reagents for TNF-α Ubiquitination Studies
| Reagent/Category | Specific Examples | Experimental Function | Research Application |
|---|---|---|---|
| Cell Lines | Human FLS cells, HeLa, HEK293, Mouse embryonic fibroblasts | Model cellular systems for signaling studies | Pathway manipulation; knockout/knockdown studies |
| Animal Models | ACLT mouse model, TIA mouse model, CYLD-/- mice, HOIP-/- mice | In vivo validation of pathway mechanisms | Osteoarthritis studies, embryonic development, inflammation models |
| Cytokines/Activators | Recombinant TNF-α, IL-1β, TNF-α inducing agents | Pathway stimulation | Signal activation under controlled conditions |
| E3 Ligase Reagents | c-IAP1/2 expression vectors, LUBAC components, TRAF2 plasmids | E3 ligase function manipulation | Overexpression and functional studies |
| DUB Reagents | CYLD plasmids, USP4 siRNA, A20 knockout cells, OTUD7B inhibitors | DUB function manipulation | Deubiquitination mechanism studies |
| Ubiquitin Probes | K63-linked ubiquitin chains, linear ubiquitin chains, ubiquitin binding domain fusions | Ubiquitin chain interaction studies | Binding assays; complex purification |
| Signaling Inhibitors | SMAC mimetics, IAP antagonists, TAK1 inhibitors, IKK inhibitors | Pathway inhibition | Functional dissection of specific components |
Methodological Framework for Investigating Ubiquitination in TNF-α Signaling
The experimental delineation of ubiquitination mechanisms in TNF-α signaling employs a multidisciplinary approach combining biochemical, genetic, and pharmacological techniques:
1. Complex Isolation and Composition Analysis:
- Co-immunoprecipitation (Co-IP): Protein complexes are immunoprecipitated from TNF-α-stimulated cells using antibodies against specific complex components (TNFR1, RIPK1, or TRADD) [39]. The composition of these complexes is then analyzed by Western blotting for associated proteins.
- Tandem Affinity Purification: For higher purity, sequential purification tags allow isolation of native complexes from stimulated cells, followed by mass spectrometric analysis to identify all components.
2. Ubiquitination Status Assessment:
- Denaturing Immunoprecipitation: Cells are lysed in denaturing buffers (containing SDS) to disrupt non-covalent interactions before immunoprecipitation of the protein of interest (e.g., RIPK1). This approach specifically detects covalently attached ubiquitin modifications rather than associated proteins [36].
- Ubiquitin Chain Linkage Determination:
- Linkage-Specific Antibodies: Antibodies specific for K63-linked, K48-linked, or linear ubiquitin chains distinguish chain topology in Western blot analyses [33].
- Ubiquitin Mutants: Expression of ubiquitin mutants (K63R, K48R) identifies chain linkages required for specific signaling outcomes.
3. Functional Manipulation of Ubiquitination Machinery:
- Genetic Approaches:
- RNA Interference: siRNA or shRNA-mediated knockdown of specific E3 ligases (c-IAP1/2, LUBAC components) or DUBs (CYLD, A20, USP21) tests their necessity in pathway regulation [40] [39].
- CRISPR-Cas9 Knockout: Complete gene disruption provides definitive evidence of protein function in ubiquitination signaling.
- Pharmacological Approaches:
- Proteasome Inhibitors: MG132 or bortezomib treatment distinguishes between proteasomal degradation-dependent and -independent ubiquitin signaling.
- IAP Antagonists: SMAC mimetics promote c-IAP1/2 degradation, specifically testing their contributions to pathway regulation.
- DUB Inhibitors: Emerging selective DUB inhibitors enable acute pharmacological inhibition of specific deubiquitinating enzymes.
4. In Vivo Validation:
- Animal Disease Models: The ACLT (anterior cruciate ligament transection) and TIA (TNF-α induced arthritis) mouse models evaluate the pathophysiological relevance of ubiquitination mechanisms in inflammatory diseases [36].
- Genetic Mouse Models: Tissue-specific knockout mice conditionally eliminate ubiquitination pathway components in relevant cell types.
Figure 2: Experimental Workflow for Investigating Ubiquitination in TNF-α Signaling - A multidisciplinary approach combining biochemical, genetic, and in vivo methods.
Quantitative Assessment of Ubiquitination Effects
Rigorous quantification of ubiquitination effects on TNF-α signaling outcomes has yielded critical insights into pathway regulation:
Table 4: Quantitative Experimental Findings in TNF-α Ubiquitination Studies
| Experimental Manipulation | System | Quantitative Outcome | Biological Impact | Reference |
|---|---|---|---|---|
| Spermidine treatment | ACLT mouse OA model | ↓ OARSI scores; ↓ TNF-α scores | Attenuated synovitis, cartilage degeneration | [36] |
| Spermidine (9μM) | Human FLS cells | Activated CYLD-mediated RIP1 deubiquitination | Inhibition of TNF-α-induced NF-κB/p65 signaling | [36] |
| USP4 knockdown | Cancer cell lines | Significantly ↑ TNF-α-induced cytokine expression | Enhanced inflammatory response | [40] |
| RIP1 K376R mutation | Mouse embryonic cells | ↓ NF-κB activation; ↑ cell death | Embryonic lethality; defective development | [33] |
| HOIP deficiency | Mouse model | Embryonic lethality at E10.5 | Rescued by TNFR1 loss until E17.5 | [33] |
Therapeutic Implications and Research Applications
The intricate balance between ubiquitination and deubiquitination in TNF-α signaling presents numerous therapeutic opportunities for inflammatory diseases, cancer, and degenerative conditions. Several strategic approaches have emerged:
1. Targeted Protein Degradation:
- PROTACs (Proteolysis Targeting Chimeras): These bifunctional molecules recruit E3 ubiquitin ligases to target proteins of interest, inducing their degradation. In TNF-α signaling, this approach could selectively eliminate hyperactive signaling components.
- Molecular Glues: Small molecules that enhance interaction between E3 ligases and target proteins, promoting degradation of pathological signaling molecules.
2. DUB-Targeted Therapeutics:
- CYLD Activation: Compounds like spermidine that enhance CYLD-mediated deubiquitination of RIPK1 show promise in osteoarthritis models by inhibiting NF-κB activation and reducing inflammation [36].
- USP Inhibition: Selective inhibitors of specific DUBs could modulate TNF-α signaling outcomes in cancer and inflammatory diseases.
3. IAP-Targeted Strategies:
- SMAC Mimetics: These compounds antagonize IAP proteins, promoting their auto-ubiquitination and degradation, thereby shifting the balance toward apoptosis in cancer cells [33].
The therapeutic manipulation of the ubiquitin system in TNF-α signaling requires precise targeting to avoid disrupting beneficial inflammatory responses while selectively modulating pathological signaling. As our understanding of the structural biology of ubiquitin ligases and DUBs advances, so too does the potential for developing highly specific therapeutics that can precisely tune this critical signaling pathway for clinical benefit.
The TNF-α signaling paradigm exemplifies how competing ubiquitination and deubiquitination events govern fundamental cellular fate decisions through dynamic post-translational modifications. The precise balance between E3 ligase-mediated ubiquitination and DUB-mediated deubiquitination of key signaling nodes like RIPK1 creates a sophisticated regulatory switch that determines whether cells survive, undergo apoptosis, or proceed to necroptosis. This system demonstrates remarkable plasticity, employing different ubiquitin chain linkages to encode distinct functional outcomes—a ubiquitin code that is read by specific binding domains within signaling complexes.
Within the broader context of ubiquitination in DNA repair and immune response pathways, the TNF-α system illustrates how reversible ubiquitin modifications serve as a universal regulatory mechanism across diverse biological processes. The experimental approaches and therapeutic strategies developed for manipulating this balance in TNF-α signaling may find application in other ubiquitin-regulated pathways, advancing our overall understanding of how post-translational modifications coordinate cellular responses to maintain homeostasis. As research continues to unravel the complexities of this system, we move closer to harnessing the ubiquitin machinery for precise therapeutic intervention in cancer, inflammatory diseases, and degenerative conditions.
Abstract The linear ubiquitin chain assembly complex (LUBAC), the only known E3 ubiquitin ligase that generates methionine 1 (M1)-linked linear ubiquitin chains, is a critical regulator of inflammatory signaling, immune cell activation, and cell death. This whitepaper delineates the unique structure and mechanism of LUBAC, its non-redundant role in activating the canonical NF-κB pathway via key substrates like NEMO and RIP1, and its emerging functions in immune regulation, autophagy, and host defense. Furthermore, we detail experimental methodologies for studying linear ubiquitination and discuss the implications of LUBAC dysregulation in human disease, framing these insights within the broader context of ubiquitination in cellular signaling. The content is supported by structured data tables, signaling pathway diagrams, and a catalog of essential research reagents.
Ubiquitination is a versatile post-translational modification involving the covalent attachment of ubiquitin to target proteins. Conventionally, ubiquitin chains are formed via isopeptide bonds between the C-terminal glycine of one ubiquitin and a lysine residue (e.g., K48, K63) on another. The discovery of a linear (M1-linked) ubiquitin chain, connected through a peptide bond between the N-terminal methionine of one ubiquitin and the C-terminal glycine of the next, represented a paradigm shift in the field [41]. LUBAC is the sole E3 ligase complex responsible for writing this "linear ubiquitin code," a crucial process for coordinating inflammatory and immune responses [42] [43]. This whitepaper explores how this unique modification, situated at the nexus of ubiquitination in DNA repair and immune signaling pathways, exerts precise control over cellular fate.
The LUBAC Complex: Structure and Catalytic Mechanism
LUBAC is a multi-subunit ~600 kDa complex composed of three essential components: HOIP (RNF31), HOIL-1L (RBCK1), and SHARPIN [41] [42] [43].
- HOIP: The catalytic core of LUBAC, HOIP, is an RING-in-between-RING (RBR) E3 ligase. Its C-terminal Linear ubiquitin chain-Determining Domain (LDD) is unique and essential for specifically catalyzing the formation of linear ubiquitin chains [42] [43]. The RING2 domain of HOIP contains an active-site cysteine (Cys885) that forms a thioester intermediate with ubiquitin before transferring it to the N-terminus of the acceptor ubiquitin [43].
- HOIL-1L and SHARPIN: These subunits are crucial for stabilizing the complex. Both contain ubiquitin-like (UBL) domains that interact with the ubiquitin-associated (UBA) domains of HOIP, relieving HOIP's autoinhibition and enabling its catalytic activity [42] [43]. HOIL-1L itself is an RBR E3 ligase, but its primary role within LUBAC is to conjugate monoubiquitin to LUBAC subunits, which then serves as a priming event for HOIP-mediated linear chain elongation—a key regulatory mechanism that can attenuate LUBAC function [44].
Table 1: Core Components of the Linear Ubiquitin Assembly Complex (LUBAC)
| Component | Alternative Names | Domain Architecture | Primary Function |
|---|---|---|---|
| HOIP | RNF31, PAUL | RBR, UBA, LDD, NZF | Catalytic subunit; sole enzyme that synthesizes linear ubiquitin chains. |
| HOIL-1L | RBCK1 | RBR, UBL, NZF | Regulatory subunit; stabilizes complex, mediates monoubiquitination. |
| SHARPIN | --- | UBL, NZF | Adaptor subunit; stabilizes complex, recruits LUBAC to substrates. |
The following diagram illustrates the core LUBAC structure and its regulatory interactions with deubiquitinases (DUBs).
Diagram 1: LUBAC complex and DUB regulation. HOIP is the catalytic core, stabilized by HOIL-1L and SHARPIN. The deubiquitinases OTULIN and CYLD (via SPATA2) bind HOIP's PUB domain to regulate linear chain levels.
LUBAC in NF-κB Activation and Immune Regulation
LUBAC is a master regulator of the canonical NF-κB pathway, which is pivotal in inflammation, immunity, and cell survival.
3.1 Mechanism of NF-κB Activation In the canonical pathway, receptors like TNFR1 and IL-1R are activated, leading to the recruitment of signaling complexes. LUBAC is recruited to these complexes, often via interactions with K63-linked ubiquitin chains. Once recruited, LUBAC conjugates linear ubiquitin chains to key signaling molecules, including NEMO (IKKγ) and RIP1 [45] [43]. The linear ubiquitin chains act as scaffolds, with the UBAN domain in NEMO displaying high affinity for them. This binding recruits the IKK complex (IKKα/IKKβ/NEMO) to the activation platform, promoting IKK trans-autophosphorylation and activation [42] [43]. The activated IKK complex then phosphorylates IκBα, targeting it for K48-linked ubiquitination and proteasomal degradation. This releases the NF-κB dimer (e.g., p65/p50), allowing its translocation to the nucleus to drive the expression of pro-survival and inflammatory genes [45].
3.2 Context-Dependent Signaling and Immune Regulation The role of LUBAC extends beyond cytokine signaling:
- T Cell Receptor (TCR) Signaling: In T cells, LUBAC is essential for TCR-mediated NF-κB activation. It linearly ubiquitinates components of the CBM complex (CARMA1-BCL10-MALT1), facilitating IKK recruitment and activation [46]. Mathematical modeling suggests linear ubiquitination of the CBM complex accelerates IKK activation more efficiently than NEMO modification alone [46].
- Antibacterial Autophagy (Xenophagy): LUBAC is recruited to cytosol-invading bacteria (e.g., Salmonella). It decorates the bacterial surface with linear ubiquitin patches, which serve as platforms for recruiting autophagy receptors like Optineurin and NEMO, thereby activating both xenophagy and local NF-κB to restrict bacterial proliferation [47].
- Regulation of Cell Death: By promoting NF-κB-mediated survival gene expression, LUBAC protects against programmed cell death. Dysregulation of LUBAC can lead to aberrant cell death, contributing to inflammatory pathologies [43] [44].
The diagram below integrates LUBAC into the broader NF-κB activation pathway.
Diagram 2: LUBAC-mediated canonical NF-κB activation. Receptor engagement recruits LUBAC, which linearly ubiquitinates NEMO/IKK complex, leading to IκBα phosphorylation, NF-κB release, and nuclear gene transcription.
Experimental Analysis of Linear Ubiquitination
Studying linear ubiquitination requires specific reagents and methodological approaches.
4.1 Key Research Reagents and Tools A well-equipped toolkit is essential for probing LUBAC function.
Table 2: Research Reagent Solutions for Linear Ubiquitination Studies
| Reagent / Tool | Specific Example | Function & Application |
|---|---|---|
| Linear Ubiquitin-Specific Antibodies | LUB9, 1F11/3F5/Y102L | Detect endogenous linear ubiquitin chains in immunoblotting (IB) and immunofluorescence (IF) [48] [46]. |
| LUBAC Component Antibodies | Anti-HOIP, Anti-HOIL-1L, Anti-SHARPIN | Detect protein expression, complex formation (Co-IP), and localization [48] [46]. |
| Plasmids for Expression | Flag-/HA-tagged HOIP, HOIL-1L, SHARPIN, Ub | For overexpression, mutagenesis, and reconstitution experiments. Catalytically inactive HOIP (C699/702/871/874S) is a critical control [48]. |
| Cell Lines | HOIP-/- HCT116, Sharpincpdm/cpdm MEFs, A20.2J (Rnf31+/+) vs H294.10 (Rnf31-/-) | Genetically defined models to study LUBAC loss-of-function [48] [47] [46]. |
| Deubiquitinase (DUB) Inhibitors | --- | Selective inhibitors for OTULIN or CYLD can probe the dynamics of linear ubiquitination. |
| LUBAC Inhibitors | HOIPINs (α,β-unsaturated carbonyls), Gliotoxin | Pharmacological tools to acutely inhibit LUBAC catalytic activity for functional studies [43]. |
4.2 Detailed Experimental Protocol: Co-Immunoprecipitation and Linear Ubiquitination Assay This protocol outlines a method to detect LUBAC-dependent linear ubiquitination of a substrate (e.g., IRF7 or NEMO) in cells, as employed in [48].
- Objective: To demonstrate that LMP1 potentiates LUBAC-mediated linear ubiquitination of IRF7.
Materials:
- 293T cells (high transfection efficiency)
- Expression plasmids: Flag-LMP1, Myc-IRF7, Ub, Flag-RNF31 (HOIP), HA-HOIL-1L, Flag-Sharpin (for active LUBAC)
- Control: Catalytically dead LUBAC (LUBACcs: HOIP C699/702/871/874S, HOIL-1L C282/285/447/450S, Sharpin T358L F359V)
- Antibodies: Anti-Myc (for IP), Anti-linear ubiquitin (LUB9 for IB), Anti-Myc (for IB)
- Lysis Buffer (with protease inhibitors and 10mM N-Ethylmaleimide to inhibit DUBs)
- Protein A/G beads
Methodology:
- Transfection: Seed 293T cells and transfect with the appropriate plasmid combinations. A critical set includes: Myc-IRF7 + Ub + Vector, Myc-IRF7 + Ub + LUBAC, Myc-IRF7 + Ub + LMP1 + LUBAC, and Myc-IRF7 + Ub + LMP1 + LUBACcs.
- Cell Lysis: 48 hours post-transfection, lyse cells in denaturing lysis buffer (e.g., containing 1% SDS) and immediately boil samples to preserve ubiquitination states.
- Immunoprecipitation: Dilute the lysates to 0.1% SDS with non-denaturing buffer. Immunoprecipitate the target protein using an anti-Myc antibody conjugated to beads. Incubate overnight at 4°C.
- Washing and Elution: Wash beads extensively with wash buffer to remove non-specifically bound proteins. Elute the immunoprecipitated complexes by boiling in SDS sample buffer.
- Immunoblotting: Resolve the eluted proteins and total cell lysates by SDS-PAGE. Transfer to a membrane and perform immunoblotting.
- Probe the IP membrane with anti-linear ubiquitin antibody to detect linearly ubiquitinated IRF7.
- Re-probe the membrane with anti-Myc antibody to confirm equal precipitation of IRF7.
- Analyze total lysates with antibodies against LUBAC components and LMP1 to verify expression.
Expected Results: A robust linear ubiquitin signal should be detected in the Myc-IRF7 immunoprecipitate only in the presence of active LUBAC (lane 2), which is potentiated by LMP1 co-expression (lane 3). The catalytically dead LUBAC complex (LUBACcs) should fail to generate this signal (lane 4), confirming the specificity of the reaction [48].
LUBAC in Disease and Therapeutic Targeting
Dysregulation of LUBAC-mediated linear ubiquitination is implicated in various human diseases.
- Cancer: LUBAC components can act as oncogenes. In Epstein-Barr virus (EBV)-transformed cells, LUBAC interacts with the viral oncoprotein LMP1, is required for full NF-κB activation, and supports cell proliferation [48]. The protein levels of RNF31 and LMP1 are correlated in these cells, and RNF31 knockdown decreases proliferation [48]. More broadly, alterations in LUBAC activity are linked to lymphoma, liver cancer, and breast cancer [42].
- Inflammatory and Autoimmune Diseases: Excessive LUBAC activity can drive chronic inflammation. Mutations in LUBAC components or regulators like OTULIN are associated with autoinflammatory syndromes and immunodeficiency [43] [44].
- Therapeutic Implications: Targeting LUBAC is a promising therapeutic strategy. Small-molecule inhibitors of HOIP's catalytic activity, such as the HOIPIN series, have been developed and shown to suppress NF-κB activation in cellular models [43]. These inhibitors represent valuable tools for probing LUBAC biology and potential starting points for drug development against cancers and inflammatory diseases.
Table 3: Quantitative Insights into LUBAC Function and Dysregulation
| Context / Assay | Key Quantitative Finding | Biological Implication |
|---|---|---|
| EBV-Transformed B Cells | Knockdown of RNF31 results in a decrease in cell proliferation [48]. | LUBAC activity is crucial for the survival and growth of virus-transformed cells. |
| TCR Signaling (Mathematical Model) | Linear ubiquitination of the CBM complex accelerates IKK activation compared to NEMO ubiquitination alone [46]. | Substrate selection (CBM vs. NEMO) fine-tunes the dynamics of immune signaling. |
| OTULIN Null Cells | Loss of OTULIN DUB activity markedly increases the amount of linear ubiquitin chains, primarily on LUBAC itself [41] [42]. | OTULIN is the primary DUB preventing LUBAC auto-ubiquitination, maintaining its function. |
| HOIL-1L E3 Mutant Cells | Introduction of E3-defective HOIL-1L mutants augments cellular linear ubiquitination [44]. | HOIL-1L-mediated monoubiquitination is a key negative feedback mechanism for LUBAC. |
LUBAC and the linear ubiquitin code it writes constitute a sophisticated and essential regulatory system within the broader ubiquitin landscape. Its unique ability to control NF-κB activation and other pathways like xenophagy and TCR signaling underscores its centrality in health and disease. Future research will need to fully elucidate the complete repertoire of LUBAC substrates, the mechanisms of its recruitment to diverse signaling platforms, and the complex cross-talk between linear and other ubiquitin linkage types. The continued development of specific pharmacological modulators, including inhibitors and proteolysis-targeting chimeras (PROTACs), will not only serve as powerful research tools but also pave the way for novel therapeutics targeting LUBAC in oncology and inflammatory disorders.
The ubiquitin-proteasome system (UPS) is a master regulator of cellular homeostasis, wielding precise control over protein stability, localization, and activity. Central to this system are E3 ubiquitin ligases, which confer specificity by targeting key signaling proteins for post-translational modification. Emerging research reveals that a select group of E3 ligases and their substrates operate at the critical interface of two fundamental biological processes: DNA damage repair and immune signaling. This review synthesizes current evidence on the shared molecular machinery between these pathways, focusing on the mechanistic role of E3 ligases like RNF8, RNF168, and SPOP in coordinating the cellular response to genomic instability and immune activation. We provide a detailed analysis of experimental methodologies for studying these processes, present key reagent solutions for researchers, and discuss the profound therapeutic implications of targeting this ubiquitin-mediated crosstalk in diseases such as cancer.
Protein ubiquitination is a vital post-translational modification that regulates virtually all cellular processes, including protein degradation, DNA repair, and immune responses [49] [50]. This modification is orchestrated through a sequential enzymatic cascade involving an E1 activating enzyme, E2 conjugating enzyme, and E3 ubiquitin ligase, which collectively coordinate the attachment of ubiquitin to substrate proteins [51] [50]. The human genome encodes over 600 E3 ubiquitin ligases, which are categorized into distinct families such as RING, HECT, and RBR types, each employing different mechanisms for ubiquitin transfer [15] [50]. These E3 ligases provide exquisite specificity to the ubiquitination process by recognizing particular substrate proteins, thereby determining the physiological outcome of the modification [50].
The versatility of ubiquitination lies in its ability to generate diverse signaling outcomes based on the type of ubiquitin chain formed. Whereas K48-linked polyubiquitin chains typically target substrates for proteasomal degradation, K63-linked chains are primarily involved in non-proteolytic processes such as signal transduction, DNA repair, and immune activation [51] [15]. This regulatory complexity enables the UPS to function as a sophisticated communication network that integrates signals from various cellular pathways. Recent investigations have uncovered remarkable crosstalk between DNA damage response (DDR) and immune signaling pathways, with shared E3 ubiquitin ligases and substrates forming critical nodes of intersection [51] [52]. Understanding this molecular crosstalk provides valuable insights for developing novel therapeutic strategies, particularly in oncology, where dysregulation of both DNA repair and immune surveillance drives disease progression.
Shared E3 Ubiquitin Ligases in DNA Repair and Immune Signaling
The RNF8-RNF168 Ubiquitination Cascade
The RNF8-RNF168 axis represents a paradigm of ubiquitin-mediated signaling that coordinates both DNA repair and immune pathway regulation. Following DNA double-strand break (DSB) formation, the master kinase ATM is activated and phosphorylates the histone variant H2AX (creating γH2AX) [15] [52]. The adapter protein MDC1 recognizes γH2AX and recruits the E3 ligase RNF8 to break sites [15]. RNF8 then catalyzes the ubiquitination of L3MBTL2, which recruits a second E3 ligase, RNF168 [15] [52]. RNF168 subsequently ubiquitinates histone H2A at lysine 13/15 (H2AK13/15ub), creating a binding platform for repair proteins including 53BP1 and BRCA1, which determine DSB repair pathway choice [15] [52].
Beyond its established role in DDR, emerging evidence indicates that the RNF8-RNF168 axis similarly regulates immune signaling pathways. RNF168-mediated histone ubiquitination at DSB sites modulates the expression of immune-related genes and cytokine production [52]. Furthermore, RNF168 has been implicated in the regulation of nuclear factor kappa B (NF-κB) signaling, a central pathway in inflammatory and immune responses, though the precise mechanisms remain under investigation. This dual functionality positions the RNF8-RNF168 cascade as a critical integrator of genomic stability and immune homeostasis.
SPOP in Immune Checkpoint Regulation and Genome Stability
Speckle-type POZ protein (SPOP), a substrate recognition component of the Cullin3-RING ubiquitin ligase complex, exemplifies how individual E3 ligases can govern both DNA repair and immune signaling. In cancer biology, SPOP plays a crucial role in mediating the ubiquitination and degradation of programmed death-ligand 1 (PD-L1), a key immune checkpoint protein [51]. In colorectal cancer cells, SPOP directly targets PD-L1 for ubiquitin-mediated degradation, thereby potentially enhancing antitumor immune responses [51]. This degradation can be inhibited by competitive binding of other proteins, such as ALDH2 in hepatocellular carcinoma or SGLT2 in various cancers, which stabilize PD-L1 and promote tumor immune evasion [51].
Simultaneously, SPOP functions in maintaining genome stability through the regulation of DNA repair factors. Although not explicitly detailed in the provided search results, SPOP's role as a multitasking E3 ligase includes the regulation of proteins involved in DDR. The molecular interplay between SPOP's functions in immune checkpoint regulation and DNA repair represents a compelling area for future investigation, particularly in the context of cancer therapy resistance.
Table 1: Key E3 Ubiquitin Ligases Operating in DNA Repair and Immune Pathways
| E3 Ligase | Role in DNA Repair | Role in Immune Signaling | Key Substrates | Ubiquitin Linkage |
|---|---|---|---|---|
| RNF8 | Initiates ubiquitin signaling at DSBs; recruits RNF168 [15] [52] | Emerging role in immune gene regulation [52] | L3MBTL2, H2A [15] | K63-linked chains [15] |
| RNF168 | Amplifies ubiquitin signaling; promotes 53BP1/BRCA1 recruitment [15] [52] | Regulates NF-κB signaling; modulates cytokine production [52] | H2A/H2AX (K13/15) [15] [52] | K13/K15 monoUb and K63 chains [15] |
| SPOP | Regulates DNA repair factors (implied) [51] | Targets PD-L1 for degradation [51] | PD-L1, various DDR proteins [51] | K48-linked chains (degradative) [51] |
| BRCA1/BARD1 | Promotes HR repair; resects DNA ends [15] [52] | Regulates immune signaling pathways | H2A (K127/128) [15] | K6, K63-linked chains [15] |
BRCA1/BARD1 Complex in HR Repair and Immune Regulation
The BRCA1/BARD1 heterodimeric complex represents another E3 ligase with dual functions in DNA repair and immune homeostasis. In DSB repair, BRCA1/BARD1 catalyzes the ubiquitination of histone H2A at lysine 127-129, which promotes the recruitment of DNA end resection factors like SMARCAD1, thereby facilitating homologous recombination (HR) repair [15] [52]. This activity positions BRCA1 as a key determinant in the choice between HR and non-homologous end joining (NHEJ) repair pathways.
Beyond its canonical role in DNA repair, BRCA1 has been increasingly implicated in immune system regulation. While the specific mechanisms were not elaborated in the provided search results, evidence suggests that BRCA1 deficiency alters immune signaling and cytokine production, potentially contributing to the inflammatory microenvironment observed in BRCA1-mutated cancers. The intersection between BRCA1-mediated DNA repair and immune regulation represents a critical area for future research, particularly in understanding how BRCA1 mutations influence both genomic stability and antitumor immunity.
Shared Substrates and Signaling Nodes
Histone Ubiquitination in DDR and Immune Gene Regulation
Histone proteins serve as central substrates ubiquitinated by E3 ligases in both DNA repair and immune contexts. Following DSB induction, RNF168-mediated ubiquitination of H2A at K13/K15 creates a binding platform for 53BP1, which promotes NHEJ and inhibits DNA end resection [15] [52]. Conversely, BRCA1/BARD1-dependent ubiquitination of H2A at K127/K128 recruits SMARCAD1 to facilitate end resection and HR repair [15]. This histone ubiquitination code thus represents a critical mechanism for determining DSB repair pathway choice.
Simultaneously, histone ubiquitination plays a pivotal role in regulating immune gene expression. The ubiquitination status of histones at promoter regions of immune-related genes directly influences their transcriptional activity [52]. For instance, RNF168-mediated H2A ubiquitination can repress the expression of certain immune regulators, thereby fine-tuning immune responses. This dual function of histone ubiquitination in both DDR and immune regulation establishes histones as key shared substrates that enable crosstalk between these pathways.
PD-L1 Ubiquitination in Immune Evasion
Programmed death-ligand 1 (PD-L1) has emerged as a critical shared node in the intersection of ubiquitin-mediated immune and stress response pathways. As an immune checkpoint protein, PD-L1 expressed on tumor cells engages with PD-1 on T cells to suppress antitumor immunity [51]. Multiple E3 ubiquitin ligases, including SPOP, regulate PD-L1 stability through ubiquitin-mediated degradation [51]. In colorectal cancer, SPOP directly binds to PD-L1 and promotes its ubiquitination and proteasomal degradation, thereby potentially enhancing T cell-mediated tumor killing [51].
The regulation of PD-L1 by ubiquitination represents a convergence point where cellular stress responses, including DNA damage, may interface with immune regulation. DNA damage can influence PD-L1 expression through various mechanisms, potentially involving ubiquitin-mediated regulation. This interconnection suggests that ubiquitin-dependent control of PD-L1 stability may serve as an important mechanism linking genomic instability to immune evasion in cancer.
Table 2: Shared Substrates in DNA Repair and Immune Pathways
| Substrate | Role in DNA Repair | Role in Immune Signaling | Regulating E3 Ligases | Functional Outcome |
|---|---|---|---|---|
| Histone H2A | DSB signaling; repair factor recruitment [15] [52] | Immune gene regulation [52] | RNF168, BRCA1/BARD1 [15] [52] | Pathway choice; gene expression |
| PD-L1 | Potential response to genomic instability [51] | Immune checkpoint inhibition [51] | SPOP [51] | T cell suppression; immune evasion |
| 53BP1 | Promotes NHEJ; inhibits resection [15] [52] | Not well characterized | RNF168 (indirect) [15] | Repair pathway choice |
| BRCA1 | HR-mediated repair [15] [52] | Immune modulation | Auto-ubiquitination? | HR repair; immune regulation |
Experimental Approaches for Studying Ubiquitin-Mediated Crosstalk
Methodologies for Analyzing Protein Ubiquitination
Investigating the crosstalk between DNA repair and immune pathways requires sophisticated methodologies to detect and quantify protein ubiquitination. The following experimental protocols represent state-of-the-art approaches for studying ubiquitin modifications:
Co-immunoprecipitation (Co-IP) and Ubiquitination Assays: To detect endogenous protein ubiquitination, cells are lysed in RIPA buffer containing deubiquitinase inhibitors (e.g., N-ethylmaleimide) and proteasome inhibitors (e.g., MG132). Target proteins are immunoprecipitated using specific antibodies, and ubiquitination is detected via immunoblotting with anti-ubiquitin antibodies. For assessing specific E3-substrate relationships, co-IP experiments can be performed under denaturing conditions to disrupt non-covalent interactions [51].
In Vitro Ubiquitination Assays: Recombinant E1, E2, E3 enzymes, ubiquitin, and substrate proteins are incubated in reaction buffer (50 mM Tris-HCl pH 7.5, 5 mM MgCl₂, 2 mM ATP) at 30°C for 1-2 hours. Reactions are terminated with SDS sample buffer and analyzed by immunoblotting to detect ubiquitinated species. This approach allows direct assessment of E3 ligase activity toward specific substrates without confounding cellular factors [50].
Tandem Ubiquitin Binding Entity (TUBE) Assays: TUBE reagents, comprising multiple ubiquitin-associated domains, exhibit high affinity for polyubiquitin chains and protect ubiquitinated proteins from deubiquitinase activity. Cell lysates are incubated with TUBE beads, followed by pull-down and immunoblot analysis to characterize endogenous ubiquitinated proteins under physiological conditions.
Proteomic and Genomic Approaches
Advanced proteomic and genomic technologies provide comprehensive insights into ubiquitin-mediated pathway crosstalk:
Quantitative Mass Spectrometry (MS): Stable Isotope Labeling with Amino acids in Cell culture (SILAC)-based proteomics enables quantification of changes in the ubiquitinome in response to DNA damage or immune activation. Following enrichment of ubiquitinated peptides using di-glycine remnant antibodies, MS analysis identifies specific ubiquitination sites and their regulation under different conditions [50].
Chromatin Immunoprecipitation (ChIP) Sequencing: ChIP-seq with antibodies specific for ubiquitinated histones (e.g., H2AK15ub) or DNA repair factors (53BP1, BRCA1) maps their genomic localization in response to DNA damage. This approach reveals how ubiquitin modifications influence chromatin landscape and gene expression programs at the intersection of DNA repair and immune regulation [52].
Crispr-Cas9 Screening: Genome-wide knockout screens identify novel regulators of ubiquitin-dependent crosstalk. Libraries of sgRNAs targeting E3 ubiquitin ligases, deubiquitinases, and known DNA repair/immune factors are transduced into reporter cell lines to identify genes essential for maintaining pathway coordination.
Research Reagent Solutions
Table 3: Essential Research Reagents for Studying Ubiquitin-Mediated Crosstalk
| Reagent/Category | Specific Examples | Research Application | Key Function |
|---|---|---|---|
| E3 Ligase Inhibitors | PROTACs (Proteolysis-Targeting Chimeras) [49] | Target validation; therapeutic exploration | Selective degradation of pathogenic proteins by recruiting E3 ligases |
| Activity-Based Probes | Covalent fragments for bacterial NEL enzymes [50] | Chemical biology; mechanism studies | Monitor E3 activity in cellular contexts; evaluate therapeutic targeting |
| Deubiquitinase Inhibitors | b-AP15, PR-619 | Pathway modulation studies | Block deubiquitination; stabilize ubiquitinated substrates |
| Ubiquitin Binding Reagents | TUBE (Tandem Ubiquitin Binding Entities) | Ubiquitome analysis | Enrich and protect polyubiquitinated proteins from DUBs |
| DNA Damage Inducers | Etoposide, Bleomycin, Ionizing radiation [15] [52] | DNA repair pathway studies | Induce controlled DNA damage; activate DDR |
| Immune Activation Agents | IFN-γ, anti-CD3/CD28 beads | Immune signaling studies | Activate T cells; induce PD-L1 expression |
| Proteasome Inhibitors | MG132, Bortezomib, Carfilzomib [53] [54] | Ubiquitination assays | Stabilize ubiquitinated proteins; block degradation |
| Specific Antibodies | Anti-H2AK15ub, Anti-PD-L1, Anti-γH2AX [51] [15] [52] | Detection and quantification | Detect specific ubiquitination events and pathway activation |
Visualization of Key Signaling Pathways
Therapeutic Implications and Future Perspectives
The convergence of DNA repair and immune signaling pathways through shared ubiquitin components presents compelling therapeutic opportunities, particularly in oncology. Several strategic approaches are emerging:
Targeted Protein Degradation: Proteolysis-targeting chimeras (PROTACs) represent a groundbreaking therapeutic modality that leverages the ubiquitin system to selectively degrade disease-causing proteins [49]. These bifunctional molecules simultaneously bind to E3 ubiquitin ligases and target proteins, facilitating ubiquitination and degradation of previously "undruggable" targets. The shared E3 ligases discussed in this review, including SPOP and components of the RNF8-RNF168 axis, offer promising platforms for PROTAC development aimed at modulating both DNA repair and immune signaling in cancer.
Combination Therapies: Strategic combination of ubiquitin-targeting agents with existing treatments holds significant potential. Proteasome inhibitors (e.g., bortezomib, carfilzomib) have demonstrated efficacy in hematological malignancies and are being explored in combination with DNA-damaging agents and immune checkpoint inhibitors [53] [54]. Additionally, combining E3 ligase modulators with PD-1/PD-L1 blockade may overcome resistance mechanisms in solid tumors by simultaneously enhancing DNA damage and antitumor immunity [51].
Biomarker-Driven Applications: Understanding ubiquitin-mediated crosstalk enables biomarker development for treatment selection. Tumors with deficiencies in specific DNA repair pathways (e.g., BRCA mutations) may exhibit heightened sensitivity to immune checkpoint blockade when combined with ubiquitin pathway modulators. Similarly, the expression levels of specific E3 ligases like SPOP may predict response to therapies targeting the ubiquitin system.
Future research directions should focus on elucidating the complete network of shared components between DNA repair and immune pathways, developing isoform-specific inhibitors for E3 ligases with minimal toxicity, and exploring the role of atypical ubiquitin chain linkages in pathway crosstalk. As our understanding of this sophisticated regulatory interface deepens, so too will our ability to harness it for therapeutic benefit across a spectrum of human diseases.
From Bench to Bedside: Research Tools and Therapeutic Targeting of the Ubiquitin System
Ubiquitination is a versatile post-translational modification that regulates diverse fundamental aspects of cellular function, including protein stability, activity, localization, and interactions [55]. This modification involves the covalent attachment of ubiquitin, a small 76-residue protein, to substrate proteins via a cascade of E1 (activating), E2 (conjugating), and E3 (ligating) enzymes [56] [55]. The versatility of ubiquitin signaling stems from the complexity of ubiquitin conjugates, which can range from single ubiquitin monomers to polymers (polyubiquitin chains) with different lengths and linkage types [55]. The seven internal lysine residues (K6, K11, K27, K29, K33, K48, K63) and the N-terminal methionine (M1) in ubiquitin provide eight potential linkage sites for polyubiquitin chain formation, creating homotypic chains (same linkage type), heterotypic chains (mixed linkages), and branched chains [56] [55]. This complex repertoire of ubiquitin structures, referred to as the "ubiquitin code," is decoded by cellular proteins containing ubiquitin-binding domains (UBDs) that recognize specific ubiquitin chain architectures [56].
The ubiquitination process is reversible through the action of deubiquitinating enzymes (DUBs), which catalyze the removal or trimming of ubiquitin from substrates, making ubiquitination a highly dynamic process [56] [55]. In humans, the ubiquitin system comprises approximately 2 E1 enzymes, 40 E2 enzymes, over 600 E3 ligases, and nearly 100 DUBs, highlighting the complexity and precision of this regulatory system [55]. Ubiquitin-like proteins (Ubls), which share structural similarities with ubiquitin but serve distinct functions, further expand this complexity [57]. Understanding how to decode this ubiquitin code is technically challenging but essential for unraveling its roles in critical cellular processes, particularly in DNA damage response (DDR) and immune signaling pathways where ubiquitination serves as a key regulatory mechanism [56].
Proteomic Methodologies for Ubiquitin Signaling
Enrichment Strategies for Ubiquitinated Substrates
The identification of ubiquitination sites and ubiquitin chain architectures requires specialized proteomic approaches due to the low stoichiometry of ubiquitination and the complexity of ubiquitin chains. Several enrichment strategies have been developed to isolate ubiquitinated proteins for mass spectrometry-based analysis, each with distinct advantages and limitations (Table 1) [55].
Table 1: Enrichment Strategies for Ubiquitinated Proteins
| Method | Principle | Advantages | Limitations |
|---|---|---|---|
| Ubiquitin Tagging | Expression of affinity-tagged ubiquitin (e.g., His, Strep) in cells enables purification of ubiquitinated proteins [55]. | Relatively easy and low-cost; enables screening of ubiquitinated substrates [55]. | Tag may alter ubiquitin structure; cannot be used in patient tissues; potential co-purification of non-ubiquitinated proteins [55]. |
| Antibody-Based Enrichment | Use of anti-ubiquitin antibodies (e.g., P4D1, FK1/FK2) or linkage-specific antibodies to enrich endogenous ubiquitinated proteins [55]. | Works under physiological conditions without genetic manipulation; applicable to clinical samples; linkage-specific information [55]. | High cost; potential non-specific binding; limited availability of high-quality linkage-specific antibodies [55]. |
| UBD-Based Approaches | Tandem ubiquitin-binding domains (UBDs) with higher avidity used to capture ubiquitinated proteins [55]. | Captures endogenous ubiquitination; can provide linkage specificity based on UBD selectivity [55]. | Optimization required for binding conditions; potential bias toward specific chain types [55]. |
The ubiquitin tagging approach was pioneered by Peng et al. in 2003, who expressed 6×His-tagged ubiquitin in Saccharomyces cerevisiae and identified 110 ubiquitination sites on 72 proteins by detecting the characteristic 114.04 Da mass shift on modified lysine residues after tryptic digestion [55]. This method was further refined through the development of the Stable tagged Ub exchange (StUbEx) cellular system, which enables replacement of endogenous ubiquitin with His-tagged ubiquitin, leading to the identification of 277 unique ubiquitination sites on 189 proteins in HeLa cells [55]. Similarly, expression of Strep-tagged ubiquitin has allowed identification of 753 lysine ubiquitination sites on 471 proteins in human cell lines [55].
For endogenous ubiquitination studies, antibody-based approaches using pan-specific ubiquitin antibodies (e.g., P4D1, FK1/FK2) or linkage-specific antibodies have proven valuable. For instance, FK2 affinity chromatography enabled the identification of 96 ubiquitination sites from human MCF-7 breast cancer cells [55]. Linkage-specific antibodies have been particularly useful for studying the functions of specific chain types, such as the association of K48-linked polyubiquitination of tau protein with abnormal accumulation in Alzheimer's disease [55].
Experimental Protocol: Ubiquitin Proteomics Using Antibody-Based Enrichment
Sample Preparation:
- Lyse cells or tissue in denaturing buffer (e.g., 6 M guanidine hydrochloride, 100 mM Na2HPO4/NaH2PO4, 10 mM Tris-HCl, pH 8.0) to preserve ubiquitination and inhibit DUBs and proteases.
- Reduce disulfide bonds with 5 mM dithiothreitol (DTT) at 56°C for 30 minutes, then alkylate with 15 mM iodoacetamide at room temperature for 20 minutes in the dark.
- Dilute the lysate with dilution buffer (25 mM Tris-HCl, pH 8.0) to reduce denaturant concentration to <1 M.
- Digest proteins with trypsin (1:50 enzyme-to-substrate ratio) at 37°C overnight.
Enrichment of Ubiquitinated Peptides:
- Incubate digested peptides with anti-K-ε-GG antibody beads (e.g., PTMScan ubiquitin/SUMO remnant motif kit) for 2 hours at 4°C with gentle rotation.
- Wash beads sequentially with ice-cold IAP buffer (50 mM MOPS/NaOH, pH 7.2, 10 mM Na2HPO4, 50 mM NaCl), followed by water.
- Elute ubiquitinated peptides with 0.15% trifluoroacetic acid (TFA).
Mass Spectrometry Analysis:
- Desalt eluted peptides using C18 StageTips.
- Analyze peptides by liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) using a high-resolution instrument (e.g., Q-Exactive Orbitrap).
- Generate MS/MS spectra using higher-energy collisional dissociation (HCD) with stepped normalized collision energies (25, 30, 35%).
- Identify ubiquitination sites by searching MS/MS spectra against appropriate protein databases using search algorithms (e.g., MaxQuant, Andromeda) with the following parameters: carbamidomethylation of cysteine as fixed modification; oxidation of methionine, protein N-terminal acetylation, and lysine ubiquitination (GlyGly remnant, +114.042 Da) as variable modifications.
Figure 1: Workflow for Ubiquitin Proteomics Using Antibody-Based Enrichment
CRISPR-Cas Screens in Ubiquitin Research
Genetic Screening for Ubiquitin Pathway Components
CRISPR-Cas9-based genetic screening has revolutionized the identification of genes involved in ubiquitin signaling and DNA damage response pathways. This approach enables genome-scale analyses of gene-gene and gene-drug interactions in distinct DNA damage response deficiency genetic backgrounds [58]. The technology leverages a Cas9 nuclease and single-guide RNAs (sgRNAs) to create targeted double-strand breaks in the genome, resulting in gene knockouts when repaired by error-prone non-homologous end joining [58].
Early genetic approaches to understanding the DNA damage response and ubiquitin signaling relied on RNA interference (RNAi) technologies, including genome-wide libraries of short interfering RNA (siRNA) or short hairpin RNA (shRNA) [56]. However, these methods suffered from major drawbacks including partial knockdowns and non-specific off-target effects [56]. For instance, in a screen designed to identify regulators of homologous recombination, RAD51 was identified as a common off-target hit, complicating data interpretation [56].
CRISPR-Cas9 screens have overcome many of these limitations, providing a more specific and comprehensive tool for functional genomics. Pooled CRISPR screens enable the assessment of complex cellular phenotypes such as viability, drug sensitivity, and synthetic lethality in a high-throughput manner [58]. This has been particularly valuable for identifying synthetic lethal interactions in DNA repair-deficient cancers, leading to new therapeutic strategies [58].
Experimental Protocol: Genome-wide CRISPR-Cas9 Screen for DUBs
sgRNA Library Design and Cloning:
- Design sgRNA target sequences (typically 20 bp) using online applications (e.g., Broad Institute GPP CRISPR Portal).
- Synthesize and clone oligonucleotides into lentiviral sgRNA vectors (e.g., pRG2-CT sgRNA vector) using BsaI restriction sites.
- Validate library representation by next-generation sequencing to ensure adequate coverage (typically >500x per sgRNA).
Virus Production and Cell Transduction:
- Produce lentivirus by transfecting HEK293T cells with sgRNA library, psPAX2 (packaging plasmid), and pMD2.G (envelope plasmid) using polyethylenimine (PEI).
- Harvest virus-containing supernatant at 48 and 72 hours post-transfection, concentrate by ultracentrifugation.
- Transduce target cells at a low multiplicity of infection (MOI ~0.3) to ensure single integration events.
- Select transduced cells with puromycin (1-2 μg/mL) for 7 days.
Screen Execution and Analysis:
- Split cells into experimental arms (e.g., treatment vs. control) with sufficient representation (>1000 cells per sgRNA).
- Harvest cells at multiple time points (e.g., 7, 14, 21 days) for genomic DNA extraction.
- Amplify integrated sgRNA sequences by PCR using primers with Illumina adapters and barcodes.
- Sequence amplified products on Illumina platform and quantify sgRNA abundance using dedicated analysis pipelines (e.g., MAGeCK, CERES).
Figure 2: Workflow for Genome-wide CRISPR-Cas9 Screening
Application Example: Identifying DUBs Regulating REST in Neuroblastoma
A compelling example of CRISPR screening in ubiquitin research comes from a study that performed genome-wide knockout of ubiquitin-specific proteases (USPs) to identify DUBs regulating REST protein abundance in neuroblastoma [59]. The researchers used a CRISPR/Cas9 system with sgRNA vectors and performed western blot analysis to screen for DUBs that control REST protein stability [59]. This approach identified USP3 as a key DUB that interacts with, stabilizes, and increases the half-life of REST protein by counteracting its ubiquitination [59]. Further validation showed that USP3 knockout promotes retinoic acid-induced neuronal differentiation and attenuates REST-mediated neuroblastoma tumorigenesis in mouse xenograft models [59].
Structural Biology and Chemical Biology Approaches
Chemical Synthesis of Ubiquitin and Ubiquitin-like Proteins
Chemical protein synthesis has emerged as a powerful approach for producing ubiquitin and ubiquitin-like modifiers in both free and conjugated forms, particularly when recombinant or enzymatic strategies are challenging [57]. These methods provide precise control over the assembly of ubiquitin and Ubls, enabling the generation of complex constructs with site-specific modifications that facilitate detailed functional and structural studies [57].
The primary method for chemical synthesis of ubiquitin and Ubls involves solid-phase peptide synthesis (SPPS) combined with chemoselective ligation strategies such as native chemical ligation (NCL) [57]. In NCL, a C-terminal thioester of an unprotected peptide is joined with an N-terminal cysteine/selenocysteine peptide under mild, aqueous conditions to form native amide bonds [57]. This approach has been extended through desulfurization and deselenization reactions, expanding its applicability to proteins lacking native cysteine residues [57]. Additional ligation strategies including serine/threonine ligation and α-ketoacid-hydroxylamine (KAHA) ligation have further broadened the scope of accessible proteins [57].
Semi-synthesis approaches combine chemically synthesized fragments with recombinantly expressed protein domains, enabling the preparation of large proteins with diverse modifications including non-canonical amino acids, isotopic labels, and post-translational modifications [57]. This hybrid approach has been extensively applied to study ubiquitination and deubiquitination, allowing researchers to investigate biochemical, structural, and functional properties that are challenging to address using traditional molecular biology and enzymatic approaches [57].
Experimental Protocol: Chemical Synthesis of Ubiquitin Conjugates
Solid-Phase Peptide Synthesis:
- Synthesize peptide fragments (typically 30-50 amino acids) using Fmoc-based solid-phase peptide synthesis on preloaded Wang resin.
- Use coupling reagents (HBTU/HATU) with tertiary amine bases (DIPEA) in DMF for amino acid coupling (2×5 min).
- Remove Fmoc protecting group with 20% piperidine in DMF (1×2 min + 1×5 min).
- Cleave peptides from resin using TFA:water:triisopropylsilane (95:2.5:2.5) for 2-3 hours, precipitate in cold ether, and purify by reversed-phase HPLC.
Native Chemical Ligation:
- Dissolve peptide thioester and N-terminal cysteine peptide in ligation buffer (6 M guanidine hydrochloride, 0.2 M sodium phosphate, pH 7.0, with 2% v/v thiophenol and 2% v/v benzyl mercaptan).
- Incubate at 37°C with gentle agitation for 12-24 hours.
- Monitor reaction progress by analytical HPLC and MALDI-TOF mass spectrometry.
- Purify full-length protein by reversed-phase HPLC.
Desulfurization:
- For converting cysteine to alanine, prepare desulfurization buffer (6 M guanidine hydrochloride, 0.2 M sodium phosphate, pH 7.0).
- Add VA-044 initiator (20 mM) and glutathione (50 mM) to the ligated protein.
- Incubate at 37°C for 2-4 hours under nitrogen atmosphere.
- Purify desulfurized protein by reversed-phase HPLC and characterize by mass spectrometry.
Table 2: Chemical Synthesis Approaches for Ubiquitin and Ubls
| Target | Synthesis Method | Key Features | Applications |
|---|---|---|---|
| SUMO Proteins | NCL using SEA thioester; KAHA ligation; Direct SPPS with aggregation breaker [57]. | Generated SUMO-1, SUMO-2, SUMO-3, and their dimers; Created defined SUMO conjugates [57]. | Study of SUMOylation in transcriptional regulation, DNA repair, and apoptosis [57]. |
| NEDD8 | NCL using [Pd(allyl)Cl]2; KAHA ligation; Direct SPPS with backbone amide propargylation [57]. | Prepared NEDD8-cullin peptide conjugates; Enabled structural and functional studies [57]. | Investigation of neddylation pathway and its role in protein regulation [57]. |
| ISG15 | NCL; NCL with Acm-NMe2 for ISGylated ubiquitin [57]. | Generated ISG15 and its conjugated forms; Created activity-based probes [57]. | Study of ISGylation in immune response and viral infection [57]. |
| UFM1 | KAHA ligation [57]. | Produced UFM1 and its conjugates for biochemical studies [57]. | Exploration of ufmylation pathway in cellular stress response [57]. |
Structural Insights into Deubiquitinases
Structural biology approaches have been instrumental in understanding the mechanisms of deubiquitinases, which play crucial roles in regulating ubiquitin signaling. For instance, structural studies of ubiquitin carboxyl terminal hydrolase L3 (UCHL3) have revealed it to be a bilobed structure consisting of a six-stranded antiparallel β-sheet in the center with α-helices packing on either side [60]. A distinguishing feature of UCHL3 structure is an active site crossover loop that affects its hydrolytic activity for ubiquitin chains [60]. Structural analyses have shown that UCHL3 recognizes and binds to K27-linked di-ubiquitin, inducing conformational changes from compact to extended forms [60].
UCHL3 exhibits dual enzymatic activity, cleaving both ubiquitin chains and the ubiquitin-like modifier NEDD8 from modified proteins through deneddylation, a rare activity among deubiquitinases [60]. This structural understanding has implications for DNA damage repair, as UCHL3 promotes repair by stabilizing key repair proteins such as RAD51 and Ku80, thereby facilitating homologous recombination and non-homologous end joining pathways [60].
Research Reagent Solutions
Table 3: Essential Research Reagents for Ubiquitin Studies
| Reagent Category | Specific Examples | Function/Application |
|---|---|---|
| Affinity Tags | 6×His tag, Strep-tag, Flag tag, HA tag [55]. | Purification of ubiquitinated proteins; detection of ubiquitin conjugates. |
| Ubiquitin Antibodies | P4D1, FK1/FK2 (pan-specific); Linkage-specific antibodies (K48, K63, etc.) [55]. | Detection and enrichment of ubiquitinated proteins; immunofluorescence; Western blotting. |
| CRISPR Tools | Cas9-2A-mRFP-2A-PAC vector; sgRNA vectors (e.g., pRG2-CT) [59]. | Genome-wide knockout screens; validation of ubiquitin pathway components. |
| Expression Plasmids | Flag-REST (#41903), Flag-USP3 (#22582), HA-ubiquitin (#18712) [59]. | Overexpression studies; protein interaction analyses; ubiquitination assays. |
| Activity-Based Probes | Ubiquitin-based probes with warhead groups (e.g., vinyl sulfone, propargylamide) [57]. | Profiling DUB activity; identification of novel DUBs; screening inhibitors. |
| Chemical Biology Tools | Synthetic ubiquitin variants (e.g., di-ubiquitin chains with defined linkages) [57]. | Structural studies; enzyme mechanism investigations; in vitro reconstitution assays. |
| DUB Inhibitors | Small molecule inhibitors targeting specific DUB families (e.g., USP, UCH) [60]. | Functional studies; therapeutic development; combination therapy with DNA-damaging agents. |
Integration of Methodologies in DNA Repair and Immune Signaling
The integration of proteomic, genetic, and structural approaches has significantly advanced our understanding of ubiquitin signaling in DNA repair and immune response pathways. In DNA damage response, ubiquitination plays a critical role in orchestrating the recruitment and activation of repair proteins at damage sites [56]. For example, RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins, with defects in this process leading to human disorders such as RIDDLE syndrome [58]. CRISPR screens have identified novel factors in these pathways, including CCAR2 as an antagonist of DNA end resection and RBMX as a component of the DNA-damage response [58].
In immune signaling, ubiquitination regulates pathways such as NF-κB activation through K63-linked ubiquitin chains and controls antiviral responses through interferon-stimulated gene 15 (ISG15) conjugation [55] [57]. Chemical biology approaches have enabled the synthesis of ISG15 and its conjugates, facilitating studies of its role in immune regulation [57]. Furthermore, the integration of ubiquitin-like modifiers such as UFM1 in cellular stress responses highlights the expanding complexity of ubiquitin-related signaling in maintaining cellular homeostasis [61] [57].
The continued development and integration of these advanced methodologies will be essential for deciphering the complexity of the ubiquitin code and its roles in human health and disease. As these tools become more sophisticated and accessible, they will undoubtedly lead to new insights into ubiquitin signaling and novel therapeutic strategies for cancer, neurodegenerative diseases, and other conditions linked to ubiquitin pathway dysregulation.
The ubiquitin-proteasome system (UPS) represents a crucial regulatory mechanism for maintaining cellular homeostasis, with E3 ubiquitin ligases serving as the pivotal determinants of substrate specificity within this pathway. These enzymes catalyze the final step in the ubiquitination cascade, transferring ubiquitin from E2 conjugating enzymes to specific target proteins, thereby marking them for proteasomal degradation or functional modification [62]. The human genome encodes an estimated 600-700 E3 ligases, which are classified into several major families based on their structural characteristics and mechanisms of action: RING (Really Interesting New Gene), HECT (Homologous to E6AP C-Terminus), RBR (RING-Between-RING), and U-box types [62] [63]. RING-type E3 ligases, the most abundant class, function as scaffolds that directly facilitate ubiquitin transfer from E2 enzymes to substrates, while HECT-type and RBR-type E3 ligases form catalytic intermediates by accepting ubiquitin before transferring it to target proteins [63].
The therapeutic targeting of E3 ligases has emerged as a promising strategy in drug discovery, particularly for conditions traditionally considered "undruggable" through conventional approaches. Two primary modalities have dominated clinical development: small-molecule inhibitors that directly block E3 ligase activity, and activators that hijack the ubiquitination machinery for targeted protein degradation. The latter category includes proteolysis-targeting chimeras (PROTACs), which represent a paradigm shift from occupancy-based pharmacology to event-driven pharmacology [64]. This whitepaper provides a comprehensive technical overview of the clinical development landscape for E3 ligase-targeting therapeutics, with particular emphasis on their applications in DNA repair and immune response pathways—two biological processes critically regulated by ubiquitination.
E3 Ligase Classification and Functional Mechanisms
Structural Families and Catalytic Mechanisms
E3 ubiquitin ligases employ distinct structural domains and catalytic mechanisms to execute their functions. RING-type E3 ligases, representing over 600 members in humans, characteristicly feature a RING domain that simultaneously binds an E2~Ub thioester and a substrate protein, facilitating direct ubiquitin transfer without forming a covalent E3~Ub intermediate [62]. In contrast, HECT-type E3 ligases (28 members in humans) contain a conserved HECT domain that forms a thioester intermediate with ubiquitin before transferring it to substrates [63]. The RBR-type E3 ligases, such as HOIP and HOIL-1 that constitute the LUBAC complex, utilize a hybrid mechanism with both RING-like and HECT-like characteristics [62].
The NEDD4 family represents the largest subgroup of HECT-type E3 ligases and exemplifies the modular architecture of these enzymes. Family members share a common domain structure consisting of: (1) an N-terminal C2 domain that mediates calcium-dependent membrane binding; (2) multiple WW domains that recognize proline-rich motifs (PPxY) in substrate proteins; and (3) a C-terminal HECT domain that catalyzes ubiquitin transfer [63]. This modular design allows for precise substrate recognition and spatial regulation of ubiquitination events.
Ubiquitin Linkage Diversity and Functional Consequences
E3 ligases catalyze the formation of different ubiquitin linkage types through specific lysine residues (K6, K11, K27, K29, K33, K48, K63) or the N-terminal methionine (M1) of ubiquitin, each generating distinct functional outcomes [62]. Table 1 summarizes the major ubiquitin linkage types and their primary biological functions.
Table 1: Major Ubiquitin Linkage Types and Their Biological Functions
| Linkage Type | Primary Biological Functions | Representative E3 Ligases |
|---|---|---|
| K48-linked | Proteasomal degradation | Majority of E3 ligases |
| K63-linked | DNA damage repair, NF-κB signaling, endocytosis | TRAF6, RNF8, RNF168 |
| K27-linked | Innate immune response, mitochondrial quality control | RNF185, AMFR, Parkin |
| K11-linked | Cell cycle regulation, ER-associated degradation | APC/C, UBR5 |
| K29-linked | Proteasomal degradation, innate immune response | HUWE1, TRAF7 |
| M1-linked (linear) | NF-κB activation, inflammation | LUBAC (HOIP/HOIL-1) |
The diversity of ubiquitin linkages enables E3 ligases to participate in a broad spectrum of cellular processes. K48-linked chains primarily target proteins for proteasomal degradation, while K63-linked chains typically serve non-proteolytic functions in DNA damage response and inflammatory signaling [62]. More atypical linkages such as K27-linked chains have been implicated in innate immune regulation, with RNF185 catalyzing K27-linked ubiquitination of cGAS to enhance antiviral responses [65] [62].
E3 Ligases in DNA Repair and Immune Response Pathways
Regulatory Roles in DNA Damage Response
E3 ligases play critical roles in coordinating the DNA damage response (DDR) through the precise spatiotemporal regulation of key signaling and repair proteins. The NEDD4 family of HECT-type E3 ligases exemplifies this regulatory capacity, with multiple members participating in various DDR pathways [63]. Following DNA damage induction, these ligases modulate the stability, localization, or activity of components in the ATM-ATR-DNA-PK signaling axis, which orchestrates cell cycle arrest, DNA repair, and apoptosis decisions.
NEDD4 itself regulates the stability of PTEN, a tumor suppressor that modulates the DNA damage response through its effects on AKT signaling. Meanwhile, other NEDD4 family members such as SMURF1, SMURF2, and WWP1 participate in the regulation of key DDR transducers including KLF4, RNF20, and TOPOIIα, respectively [63]. These regulatory events influence the choice between different DNA repair mechanisms—base excision repair (BER), mismatch repair (MMR), nucleotide excision repair (NER), homologous recombination (HR), and non-homologous end joining (NHEJ)—each appropriate for specific types of DNA lesions.
The following diagram illustrates the central role of E3 ligases in coordinating DNA damage response pathways:
Modulation of Innate Immune Signaling
In innate immunity, E3 ligases regulate pattern recognition receptor (PRR) signaling pathways that detect microbial nucleic acids and initiate antiviral responses. The cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) pathway represents a crucial DNA sensing mechanism that is tightly controlled by ubiquitination [65]. Recent research has identified RNF185 as the first E3 ligase that directly regulates cGAS activity through K27-linked polyubiquitination during HSV-1 infection [65]. This modification enhances cGAS enzymatic activity and promotes the production of the second messenger 2'3'-cGAMP, which subsequently activates STING and downstream IRF3-mediated interferon gene expression.
Concurrently, the E3 ligase TRAF6 bridges immune and inflammatory signaling to the canonical NF-κB pathway through its role in generating K63-linked ubiquitin chains [66]. TRAF6 functions downstream of multiple receptor systems including TLRs, IL-1R, and antigen receptors, making it a central integrator of inflammatory responses. The critical nature of TRAF6-Ubc13 interaction for signal propagation has been established through structural analyses and point mutations that disrupt this binding, thereby reducing TRAF6 E3 ligase activity and NF-κB activation [66].
The diagram below illustrates E3 ligase regulation in innate immune signaling pathways:
Therapeutic Targeting Strategies and Clinical Development
Small-Molecule Inhibitors of E3 Ligases
The development of direct small-molecule inhibitors represents a conventional approach to targeting E3 ligases, with several candidates demonstrating therapeutic potential in preclinical models. The compound C25-140 exemplifies this strategy as a first-in-class inhibitor of the TRAF6-Ubc13 interaction [66]. Discovered through high-throughput screening, C25-140 reduces TRAF6 E3 ligase activity both in vitro and in cellular models, consequently impeding NF-κB activation across various immune and inflammatory signaling pathways. In preclinical mouse models of autoimmune diseases, C25-140 ameliorated inflammation and improved disease outcomes in both psoriasis and rheumatoid arthritis, validating TRAF6 inhibition as a promising strategy for autoimmune and chronic inflammatory conditions [66].
PROTACs and Targeted Protein Degradation
PROteolysis-TArgeting Chimeras (PROTACs) represent a revolutionary approach that hijacks E3 ubiquitin ligases to induce targeted protein degradation [67] [64]. These heterobifunctional molecules consist of three elements: a ligand that binds the protein of interest (POI), a ligand that recruits an E3 ubiquitin ligase, and a flexible linker connecting both moieties. By bringing the POI and E3 ligase into proximity, PROTACs catalyze POI ubiquitination and subsequent proteasomal degradation, enabling targeting of proteins previously considered "undruggable" [64].
Despite the existence of hundreds of E3 ligases in the human genome, current PROTAC development has predominantly utilized four canonical E3 ligases: CRBN, VHL, MDM2, and IAPs [67] [64]. This limited repertoire presents constraints including restricted degradable proteome coverage, potential for acquired resistance, and limited tissue selectivity. In response, significant efforts are underway to expand the E3 ligase toolbox, with recent success in recruiting novel E3s such as RNF4, RNF114, and others for PROTAC applications [67] [64].
Table 2: Clinically Advanced PROTAC Candidates and Their E3 Ligases
| PROTAC Candidate | Target Protein | E3 Ligase | Development Stage | Primary Indication |
|---|---|---|---|---|
| ARV-471 (Vepdegestrant) | Estrogen Receptor | CRBN | Phase 3 (filed for approval) | ER+/HER2- Advanced Breast Cancer |
| ARV-110 | Androgen Receptor | CRBN | Phase 3 | Metastatic Castration-Resistant Prostate Cancer |
| DT2216 | BCL-XL | VHL | Phase 1 | Hematologic Malignancies |
| Multiple candidates | Various | VHL/CRBN | Phase 1/2 | Various Cancers |
The clinical progression of PROTACs reached a landmark with vepdegestrant (ARV-471), which met its primary endpoint in the pivotal Phase 3 VERITAC-2 trial for ER+/HER2- advanced breast cancer, demonstrating statistically significant improvement in progression-free survival compared to standard-of-care fulvestrant [64]. With regulatory submissions filed, ARV-471 positions itself to potentially become the first approved PROTAC degrader, providing definitive clinical validation for the platform.
Expanding the E3 Ligase Toolbox for PROTACs
The limited diversity of E3 ligases utilized in PROTAC development has prompted systematic efforts to characterize and recruit novel E3s. Recent comprehensive analyses have identified 76 E3 ligases as promising PROTAC-interacting candidates based on confidence score, ligandability, expression pattern, and protein-protein interaction potential [67]. Among these, RNF4 and RNF114 have been successfully co-opted for PROTAC applications through innovative ligand discovery approaches.
RNF4 was recruited using covalent ligands identified through activity-based protein profiling (ABPP), with the optimized ligand CCW 16 (IC₅₀ = 1.8 μM) incorporated into PROTAC CCW 28-3 that demonstrated RNF4-dependent degradation of BRD4 [64]. Similarly, RNF114 was engaged through the natural product nimbolide and its synthetic analogs, enabling degradation of BRD4 with nanomolar potency and selectivity [64]. These examples illustrate the potential of expanding the E3 ligase repertoire to overcome current limitations in TPD therapeutics.
Experimental Methodologies and Research Tools
Key Experimental Protocols
Research in E3 ligase biology and therapeutic development relies on specialized experimental methodologies. High-throughput small-molecule screening represents a foundational approach for identifying direct inhibitors or E3 ligase ligands, as demonstrated by the discovery of C25-140 through screening for TRAF6-Ubc13 interaction inhibitors [66]. Standard protocols involve biochemical assays measuring E3 ligase activity through ubiquitin transfer to substrates, typically employing Western blotting to detect ubiquitination or fluorescence-based readouts for high-throughput applications.
Activity-based protein profiling (ABPP) has emerged as a powerful chemoproteomic approach for identifying covalent E3 ligase binders. The methodology involves: (1) incubation of cell lysates or live cells with broad-spectrum cysteine-reactive probes; (2) conjugation with reporter tags (e.g., biotin-azide via click chemistry); (3) enrichment and proteomic analysis to identify ligand-binding sites; and (4) hit validation through competitive ABPP [64]. This approach successfully identified covalent ligands for RNF4 and RNF114, enabling their subsequent application in PROTAC development.
For assessing functional outcomes of E3 ligase modulation, particularly in DNA damage and immune signaling contexts, established protocols include:
- Immunoblotting for ubiquitination and pathway activation: Detection of K27-linked ubiquitination of cGAS by RNF185 requires specific linkage-selective antibodies [65].
- Quantitative PCR for interferon-stimulated gene expression: Assessment of Ifnb, Ifna4, and Cxcl10 mRNA levels in primary cells following E3 ligase knockdown or inhibition [66] [65].
- Confocal microscopy for DNA damage foci analysis: Visualization of γH2AX, 53BP1, or RAD51 foci in cells with modulated E3 ligase activity [63].
- Co-immunoprecipitation for protein complex analysis: Determination of E3 ligase-substrate interactions and ternary complex formation in PROTAC mechanisms [64].
Essential Research Reagents and Tools
Table 3: Key Research Reagent Solutions for E3 Ligase Studies
| Reagent Category | Specific Examples | Research Applications | Technical Considerations |
|---|---|---|---|
| E3 Ligase Inhibitors | C25-140 (TRAF6-Ubc13 inhibitor) | Study of NF-κB signaling in autoimmune models | Validated in primary human and murine cells |
| Covalent Ligands | CCW 16 (RNF4 ligand), Nimbolide (RNF114 ligand) | PROTAC development, E3 ligase functional studies | Specificity confirmation required through knockout controls |
| Linkage-Specific Antibodies | K27-linkage, K63-linkage selective antibodies | Detection of atypical ubiquitin chains in immune signaling | Validation with linkage-specific standards essential |
| PROTAC Molecules | ARV-471, ARV-110, CCW 28-3 | Targeted protein degradation studies | Require ternary complex formation assessment |
| Expression Constructs | Wild-type and catalytic mutant E3 ligases | Mechanistic studies of ubiquitination | Critical for establishing functional relationships |
| siRNA/shRNA Libraries | RNF185-specific siRNAs, NEDD4 family siRNAs | Loss-of-function studies in DNA repair and immune signaling | Confirmation of knockdown efficiency required |
The therapeutic targeting of E3 ligases through small-molecule inhibitors and activators represents a rapidly advancing frontier in drug discovery, with particular relevance to DNA repair and immune response pathways. Direct inhibitors such as C25-140 demonstrate the feasibility of modulating E3 ligase activity for autoimmune disease treatment, while PROTAC technology has matured from concept to clinical validation with the anticipated approval of vepdegestrant for breast cancer. The expanding toolbox of E3 ligases beyond the canonical CRBN and VHL promises to address current limitations and unlock new therapeutic opportunities. As our understanding of E3 ligase biology in DNA repair and immune signaling deepens, and as chemical proteomic approaches continue to identify novel E3 ligase ligands, the clinical landscape for E3-targeting therapeutics is poised for significant expansion in the coming years.
The ubiquitin-proteasome system (UPS) is a critical regulator of protein homeostasis, coordinating the controlled degradation of cellular proteins fundamental to life processes. Within this system, deubiquitinases (DUBs) have emerged as pivotal regulatory enzymes that counterbalance ubiquitin ligase activity by cleaving ubiquitin chains from substrate proteins, thereby dynamically regulating protein stability, localization, and functional activity [68]. The human genome encodes approximately 100 functionally characterized DUBs, systematically classified based on their catalytic domain architecture into several major families, including ubiquitin-specific proteases (USP), ovarian tumor proteases (OTU), ubiquitin C-terminal hydrolases (UCH), Machado-Joseph disease proteases (MJD), and the zinc-dependent JAMM/MPN family [68]. This evolutionary diversity enables exquisite specialization in cellular regulation, with different DUB families exhibiting distinct substrate specificities and cellular functions.
Mounting evidence firmly establishes DUB dysfunction as a critical mechanism in the development of therapy resistance in cancer. DUBs have been implicated in chemoresistance across various cancers, including breast, lung, liver, gastrointestinal, colorectal, ovarian, prostate, and blood cancers [69]. These enzymes can either promote therapeutic resistance or enhance sensitivity depending on their specific protein targets, positioning them as promising therapeutic targets [69]. Similarly, in radiotherapy resistance, DUBs have been identified as master regulators orchestrating tumor adaptation through manipulation of DNA repair fidelity, metabolic reprogramming, and immune evasion [7]. The development of small-molecule inhibitors targeting specific DUBs has consequently gained considerable attention as a strategic approach to overcome therapy resistance and improve cancer treatment outcomes [69] [70].
Molecular Mechanisms of DUBs in Therapy Resistance
DUB-Mediated Chemoresistance Pathways
Chemotherapy remains a cornerstone in cancer treatment by targeting rapidly dividing cancer cells, but the development of chemoresistance represents a major obstacle to successful treatment, often leading to disease recurrence, metastasis, and high mortality [69]. DUBs contribute to chemoresistance through multiple interconnected mechanisms that stabilize oncoproteins, disrupt cell death pathways, and enhance DNA repair capacity. By removing ubiquitin chains from specific substrate proteins, DUBs prevent their proteasomal degradation, thereby maintaining proteins critical for cancer cell survival despite chemotherapeutic assault.
The molecular pathways through which DUBs confer chemoresistance are highly context-dependent and vary across cancer types. For instance, specific DUBs have been demonstrated to stabilize anti-apoptotic proteins, multidrug resistance transporters, and DNA repair enzymes, effectively creating a cellular environment resistant to conventional chemotherapeutic agents [69]. This stabilization function directly counteracts the intended mechanism of many chemotherapeutics that rely on targeted protein degradation or disruption of essential cellular processes. The intricate involvement of DUBs in chemoresistance mechanisms highlights their potential as targets for combination therapies aimed at re-sensitizing resistant cancers to conventional treatments.
DUB-Mediated Radioresistance Pathways
Radiotherapy exerts its anti-tumor effects through direct DNA damage and indirect immune activation, but radiation resistance remains a significant clinical challenge that frequently leads to local recurrence and metastatic progression [7]. The ubiquitin system, particularly through the action of specific DUBs, has emerged as a master regulator of radioresistance by dynamically controlling DNA repair pathways, metabolic adaptation, and immune evasion mechanisms. DUBs accomplish this through spatiotemporal control of critical protein substrates involved in these resistance pathways.
A key mechanism of DUB-mediated radioresistance involves the regulation of DNA repair protein stability. For example, USP14 stabilizes ALKBH5 to maintain glioblastoma stemness, conferring resistance to radiation in these treatment-resistant cells [7]. Additionally, DUBs interface with metabolic reprogramming pathways that support tumor survival under radiation-induced stress. The manipulation of ubiquitin chain topology represents another sophisticated resistance mechanism, where tumors strategically manipulate K63-linked chains to stabilize DNA repair factors while concurrently inhibiting K48-mediated degradation of survival proteins [7]. This ubiquitin signaling rewiring enables cancer cells to maintain genomic integrity despite radiation exposure, highlighting the complex adaptive responses orchestrated by DUBs in the tumor microenvironment.
DUB Targets in Cancer Therapy Resistance
Table 1: Key DUBs Implicated in Cancer Therapy Resistance and Their Mechanisms
| DUB Target | Cancer Type | Resistance Mechanism | Therapeutic Implication |
|---|---|---|---|
| USP30 | Multiple Cancers | Negative regulator of PINK1/Parkin-mediated mitophagy; overactivity leads to accumulation of dysfunctional mitochondria [68] | Inhibition promotes mitophagy and sensitizes to therapy |
| USP14 | Glioblastoma, Head/Neck Cancers | Stabilizes ALKBH5 to maintain glioblastoma stemness; degrades IκBα to activate NF-κB in head/neck cancers [7] | Context-dependent inhibition strategy required |
| UCH-L1 | Multiple Cancers | Regulates α-synuclein degradation and exerts neuroprotective effects; implicated in chemoresistance [68] | Dual functionality complicates therapeutic targeting |
| USP15 | Multiple Cancers | Interferes with Parkin activity by blocking ubiquitin chain formation, impairing mitochondrial quality control [68] | Inhibition enhances mitophagy and cellular quality control |
| OTUD3 | Parkinson's Disease (Potential Cancer Relevance) | Stabilizes iron regulatory protein 2 (IRP2), ameliorating iron deposition pathology [68] | Potential application in cancers with iron metabolism dysregulation |
The targeting of specific DUBs represents a promising strategic approach to overcome therapy resistance in cancer. As illustrated in Table 1, several DUBs have been mechanistically linked to key aspects of therapy resistance through diverse molecular pathways. For instance, USP30 has been identified as a negative regulator of PINK1/Parkin-mediated mitophagy, with its overactivity leading to pathological accumulation of dysfunctional mitochondria [68]. This mechanism is particularly relevant in the context of chemoresistance, as mitochondrial dysfunction can alter cellular survival pathways and drug metabolism.
The functional outcomes of DUB inhibition are highly context-dependent, varying based on cancer type, genetic background, and therapeutic modality. USP14 demonstrates this contextual duality by stabilizing ALKBH5 to maintain glioblastoma stemness while degrading IκBα to activate NF-κB in head/neck cancers [7]. Similarly, UCH-L1 exhibits dual functionality in regulating both α-synuclein degradation and exerting neuroprotective effects [68]. This functional complexity underscores the critical need for precision medicine approaches that selectively disrupt detrimental DUB activities while preserving protective pathways, necessitating robust biomarker development for patient stratification.
DUB Inhibitors in Combination Therapy Strategies
DUB Inhibitors for Chemosensitization
The development of DUB inhibitors has gained considerable attention in cancer therapeutics, with combination therapies showing significant potential to overcome drug resistance and improve treatment outcomes [69]. The rationale for combining DUB inhibitors with conventional chemotherapy stems from their ability to target resistance pathways that are often upregulated in response to chemotherapeutic agents. By inhibiting specific DUBs that stabilize pro-survival proteins, DNA repair enzymes, or drug efflux transporters, these sensitizing agents can effectively lower the threshold for cancer cell death in response to chemotherapy.
Preclinical evidence supports this combination approach across various cancer types. DUB inhibitors have demonstrated efficacy in reversing chemoresistance by targeting the molecular pathways that cancer cells exploit to evade therapy-induced cell death [69]. The strategic advantage of this approach lies in its ability to simultaneously attack multiple vulnerability nodes within cancer cells—direct cytotoxicity from conventional chemotherapy coupled with disruption of adaptive resistance mechanisms through DUB inhibition. This multi-pronged attack can potentially prevent or delay the emergence of resistance, thereby extending the therapeutic efficacy of existing chemotherapeutic agents.
DUB Inhibitors for Radiosensitization
In radiotherapy, DUB inhibitors present unique opportunities to enhance treatment efficacy by exploiting the critical role of ubiquitin signaling in DNA damage response and repair. The combination of DUB inhibitors with radiotherapy represents a promising frontier for improving therapeutic outcomes across various radiation-resistant malignancies [7]. This combination strategy capitalizes on the ability of DUB inhibitors to disrupt DNA repair pathways, thereby preventing cancer cells from effectively repairing radiation-induced DNA damage.
The molecular mechanisms underlying DUB inhibitor-mediated radiosensitization involve multiple interconnected pathways. For instance, blocking USP14 leads to accumulation of K63-ubiquitinated IRF3, triggering STING-dependent antitumor immunity that synergizes with radiotherapy to overcome immune evasion [7]. Additionally, DUB inhibition can prevent the removal of ubiquitin chains from histones and DNA repair proteins, compromising the chromatin remodeling and repair complex assembly necessary for efficient DNA damage resolution. These mechanisms collectively enhance radiation-induced cytotoxicity while simultaneously activating anti-tumor immune responses, creating a multi-faceted therapeutic approach against radioresistant cancers.
Experimental Approaches for DUB Research
Methodologies for Assessing DUB Activity in Therapy Resistance
Table 2: Key Experimental Protocols for Evaluating DUB Function in Therapy Resistance
| Method Category | Specific Technique | Key Applications in DUB Research | Technical Considerations |
|---|---|---|---|
| Activity Assessment | Ubiquitin-Rhodamine Assay | High-throughput screening of DUB inhibitors; measures cleavage of ubiquitin-rhodamine conjugate [70] | Requires purified DUB proteins; suitable for initial inhibitor screening |
| Functional Validation | CRISPR-Based Screening | Identification of DUBs regulating therapy resistance; validation of drug targets [7] | Enables genome-wide functional assessment; requires careful controls |
| Cellular Localization | Immunofluorescence Microscopy | Subcellular localization of DUBs; recruitment to DNA damage sites [7] | Provides spatial information; requires specific validated antibodies |
| Protein-Protein Interactions | Co-Immunoprecipitation + Mass Spectrometry | Identification of DUB substrates and interacting partners in DNA repair complexes [7] | Distinguishes direct vs. indirect interactions; requires rigorous validation |
| Chain Linkage Specificity | Linkage-Specific Ubiquitin Antibodies | Determination of ubiquitin chain topology in DNA damage response [7] | Antibody specificity critical; can be combined with inhibition studies |
Investigating DUB function in therapy resistance requires sophisticated methodological approaches that can capture the dynamic nature of ubiquitin signaling. As outlined in Table 2, researchers employ diverse techniques to dissect the complex roles of DUBs in chemoresistance and radioresistance. The Ubiquitin-Rhodamine assay represents a fundamental tool for initial DUB inhibitor screening, providing a quantitative measure of DUB enzymatic activity through fluorescence-based detection of cleaved ubiquitin-rhodamine conjugates [70]. This approach enables rapid assessment of inhibitor potency and specificity before progressing to more complex cellular models.
Advanced functional genomics approaches, particularly CRISPR-based screening, have revolutionized the identification of DUBs regulating therapy resistance [7]. These unbiased systematic approaches allow researchers to pinpoint specific DUB family members that constitute vulnerability nodes in resistant cancers. Complementary techniques such as immunofluorescence microscopy provide spatial resolution of DUB recruitment to cellular structures like DNA damage foci, while co-immunoprecipitation combined with mass spectrometry enables comprehensive mapping of DUB interaction networks within DNA repair complexes [7]. The integration of these methodologies provides a multi-dimensional understanding of DUB function in therapy resistance pathways.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for DUB Investigation
| Reagent Category | Specific Examples | Research Application | Functional Role |
|---|---|---|---|
| Activity-Based Probes | HA-Ub-VS, HA-Ub-Br2 | DUB activity profiling; identification of active DUBs in cellular extracts [70] | Covalently labels active site cysteine residues in DUBs |
| DUB Inhibitors | b-AP15 (targets USP14 and UCHL5), VLX1570 | Validation of DUB targets; assessment of therapeutic potential [70] | Selective or pan-DUB inhibitors for functional studies |
| Linkage-Specific Ubiquitin Antibodies | K48-linkage specific, K63-linkage specific, mono-Ub antibodies | Assessment of ubiquitin chain topology changes in response to therapy [7] | Detection of specific ubiquitin chain types in Western blot, IF |
| CRISPR Libraries | Whole-genome KO libraries, DUB-focused sgRNA libraries | Functional screening for DUBs involved in therapy resistance [7] | Identification of DUB genetic dependencies in resistant models |
| Recombinant DUB Proteins | Catalytically active USP30, USP14, UCH-L1 | Biochemical characterization; inhibitor screening [68] [70] | Purified proteins for in vitro assays and structural studies |
The investigation of DUBs in therapy resistance relies on specialized research reagents that enable precise manipulation and measurement of DUB activity. As detailed in Table 3, activity-based probes such as HA-Ub-VS and HA-Ub-Br2 represent essential tools for profiling active DUBs in cellular extracts by covalently labeling catalytic cysteine residues [70]. These probes facilitate the identification of DUBs that are enzymatically active in resistant cancer cells, providing insights into which family members might be most therapeutically relevant.
Small molecule DUB inhibitors like b-AP15 (targeting USP14 and UCHL5) serve both as therapeutic leads and functional tools for validating DUB targets in resistance pathways [70]. These pharmacological agents, when used in combination with linkage-specific ubiquitin antibodies that distinguish between different ubiquitin chain topologies, enable researchers to decipher the complex ubiquitin code rewiring that occurs in therapy-resistant cancers [7]. Furthermore, the availability of comprehensive CRISPR libraries and recombinant DUB proteins supports both genetic and biochemical approaches to dissect DUB function, creating a versatile toolkit for mechanistic studies of DUBs in therapy resistance.
Signaling Pathways in DUB-Mediated Therapy Resistance
DUB Regulation of Therapy Resistance Pathways
The signaling pathways through which DUBs mediate therapy resistance involve complex networks that integrate DNA damage response, protein homeostasis, and cell survival signals. As illustrated in the pathway diagram, chemotherapeutic agents induce multiple cellular stresses including DNA damage, oxidative stress, and protein misfolding, which in turn trigger enhanced DUB activity as an adaptive response [69]. This elevated DUB activity promotes chemoresistance through several mechanisms: stabilization of oncoproteins by preventing their ubiquitin-mediated degradation, enhancement of DNA repair capacity, activation of drug efflux pumps, and induction of anti-apoptotic pathways that collectively enable cancer cells to survive chemotherapy-induced cytotoxicity.
In radiotherapy resistance, ionizing radiation generates DNA double-strand breaks, replication stress, and immune activation, leading to the recruitment of specific DUBs to damage sites [7]. These recruited DUBs then activate DNA repair pathways, facilitate chromatin remodeling to access damage sites, and promote immune evasion mechanisms. The strategic inhibition of DUBs represents a promising therapeutic intervention that can sensitize cancer cells to both chemotherapy and radiotherapy by disrupting these adaptive resistance pathways. The interconnected nature of these signaling networks highlights the potential of DUB inhibitors to simultaneously target multiple resistance mechanisms, providing a rational basis for combination therapy approaches.
Future Perspectives and Clinical Translation
The translation of DUB-targeting agents from preclinical models to clinical applications faces several challenges that must be addressed to realize their full therapeutic potential. Functional redundancy within the DUB family represents a significant obstacle, as inhibition of a single DUB may be compensated by related family members with overlapping substrate specificities [7]. This redundancy necessitates careful patient stratification strategies and the development of combination approaches targeting multiple DUBs or parallel resistance pathways. Additionally, unintended on-target toxicity remains a concern, particularly for DUBs with essential functions in normal cellular physiology, requiring the development of therapeutic windows that maximize efficacy while minimizing adverse effects.
Advancements in biomarker-guided therapeutic strategies are critical for successful clinical translation of DUB inhibitors. The integration of advanced technologies such as single-cell transcriptomics has revealed profound intratumoral heterogeneity in the expression of DUBs, uncovering distinct therapy-resistant subpopulations that may require tailored targeting approaches [7]. Furthermore, artificial intelligence and machine learning are transforming DUB research by dissecting the complex interplay between DNA repair and cancer progression with unprecedented precision, enabling streamlined identification of novel biomarkers and optimized patient stratification [71]. These technological innovations, combined with a deepening understanding of ubiquitin biology, position DUB-targeted therapies as promising components of next-generation cancer treatment protocols that may fundamentally alter the landscape of therapy-resistant malignancies.
The future clinical development of DUB inhibitors will likely focus on rational combination strategies with established modalities such as DNA-damaging agents, targeted therapies, and immunotherapies. The unique ability of DUB inhibitors to simultaneously disrupt multiple resistance pathways—DNA repair, metabolic adaptation, and immune evasion—creates synergistic opportunities with conventional treatments [69] [7]. As the field progresses, the continued elucidation of DUB functions in specific cancer contexts and the development of increasingly selective inhibitors will be essential to overcome the challenges of therapy resistance and improve outcomes for cancer patients.
Ubiquitination, the covalent attachment of ubiquitin proteins to substrate proteins, represents the second most abundant post-translational modification in cells and governs virtually every cellular process, from protein degradation and DNA damage repair to immune response regulation [72]. This modification involves a coordinated enzymatic cascade comprising ubiquitin-activating (E1), ubiquitin-conjugating (E2), and ubiquitin-ligase (E3) enzymes, which collectively dictate the specificity and outcome of ubiquitination events [73] [72]. The diversity of ubiquitin chain linkages—including K48-linked chains that target proteins for proteasomal degradation, K63-linked chains that facilitate signaling complex assembly, and monoubiquitination that alters protein localization and function—creates a sophisticated "ubiquitin code" that cells utilize to respond to physiological and pathological stimuli [72] [7]. The dynamic and reversible nature of this code, regulated by deubiquitinating enzymes (DUBs), enables precise control over protein fate and function [60].
Recent technological advances have revealed that disruptions in the ubiquitin network produce molecular signatures with significant diagnostic, prognostic, and therapeutic potential. These ubiquitination signatures—measurable patterns of ubiquitin pathway component expression, activity, or substrate modification—reflect underlying disease states and treatment responses with remarkable specificity [73] [74] [75]. The development of ubiquitin-based biomarkers represents a paradigm shift in precision medicine, offering unprecedented opportunities to classify disease subtypes, predict therapeutic efficacy, and monitor treatment resistance. This technical guide examines the fundamental principles, methodological approaches, and clinical applications of ubiquitination signatures, with particular emphasis on their roles in DNA repair pathways and immune response regulation—two interconnected processes that critically influence cancer progression and therapeutic outcomes.
Biological Foundation: Ubiquitination in Cellular Homeostasis and Disease
Molecular Architecture of the Ubiquitin System
The ubiquitination machinery constitutes a tiered enzymatic system characterized by growing substrate specificity at each level. The human genome encodes approximately 8 E1 enzymes, over 70 E2 enzymes, and more than 600 E3 ligases, which collectively confer specificity to the ubiquitination of thousands of cellular proteins [72]. E1 enzymes initiate the ubiquitination cascade by activating ubiquitin in an ATP-dependent manner through a two-step mechanism: first, ubiquitin is adenylated, then transferred to the E1 active-site cysteine to form a thioester bond [72]. Mammalian cells express two primary ubiquitin-activating enzymes, UBA1 and UBA6, with UBA1 participating in the majority of cellular ubiquitination events [74] [72]. The activated ubiquitin is subsequently transferred to an E2 conjugating enzyme, which then collaborates with an E3 ubiquitin ligase to directly mediate ubiquitin transfer to specific substrate proteins [72].
The ubiquitin code's complexity arises from the ability of ubiquitin itself to form polymer chains through any of its seven lysine residues (K6, K11, K27, K29, K33, K48, K63) or its N-terminal methionine (M1), with each linkage type generating distinct functional consequences [72]. For instance, K48-linked polyubiquitination primarily targets proteins for proteasomal degradation, while K63-linked chains typically facilitate non-proteolytic signaling events in processes such as DNA damage repair and immune activation [72] [7]. Additionally, monoubiquitination (single ubiquitin modification) and multi-monoubiquitination (multiple single ubiquitin modifications on different lysines within the same substrate) regulate protein activity, interactions, and subcellular localization without triggering degradation [72]. The recent discovery of heterogeneous and branched ubiquitin chains has further expanded the coding potential of this system, enabling exquisite precision in cellular information processing [72].
Ubiquitination in DNA Damage Repair and Genome Stability
The ubiquitin system plays an indispensable role in maintaining genome integrity through its regulation of DNA damage response (DDR) pathways. Following radiation-induced DNA damage, a coordinated ubiquitination cascade mediates the recruitment and activation of repair proteins at damage sites [60] [7]. For example, RNF8 and RNF168 catalyze the histone ubiquitination that establishes a platform for the accumulation of BRCA1 and 53BP1, key mediators of homologous recombination (HR) and non-homologous end joining (NHEJ) repair pathways, respectively [7]. The deubiquitinating enzyme UCHL3 promotes DNA repair by stabilizing critical repair proteins including RAD51 (involved in HR) and Ku80 (essential for NHEJ), with UCHL3 overexpression enhancing DNA damage repair capacity and conferring resistance to chemotherapy and radiotherapy in certain tumors [60].
Table 1: Ubiquitination Enzymes in DNA Damage Repair Pathways
| Enzyme | Type | DNA Repair Pathway | Key Substrates | Biological Effect |
|---|---|---|---|---|
| RNF8 | E3 Ligase | Homologous Recombination (HR) | H2AX, H2A | Initiates repair protein recruitment |
| RNF168 | E3 Ligase | HR/Non-Homologous End Joining (NHEJ) | H2A, H2AX | Amplifies repair signaling |
| UCHL3 | Deubiquitinase | HR/NHEJ | RAD51, Ku80 | Stabilizes repair complexes |
| FBXW7 | E3 Ligase | NHEJ | XRCC4 | Enhances repair fidelity via K63 linkage |
| RNF126 | E3 Ligase | ATM-CHK1 signaling | MRE11 | Activates error-prone repair in TNBC |
The functional outcome of ubiquitin-mediated DNA repair regulation exhibits significant context dependency. For instance, while FBXW7 typically acts as a tumor suppressor by promoting the degradation of oncoproteins, it can enhance radioresistance in p53-wild type colorectal tumors by facilitating p53 degradation, yet increase radiosensitivity in non-small cell lung cancer (NSCLC) with SOX9 overexpression by destabilizing SOX9 and alleviating p21 repression [7]. This duality underscores the importance of understanding tissue-specific and genetic context when evaluating ubiquitination signatures as predictive biomarkers for DNA-damaging therapies.
Ubiquitination in Immune Regulation and Cancer Immunotherapy
Ubiquitination serves as a central regulatory mechanism in both innate and adaptive immunity, modulating pattern recognition receptor signaling, cytokine production, immune cell development, and antigen presentation [73]. Within the tumor microenvironment, ubiquitination dynamically shapes anti-tumor immunity by controlling the stability and activity of immune checkpoint proteins. Programmed death-ligand 1 (PD-L1), a critical immune checkpoint molecule, undergoes precise stability control by multiple ubiquitin-specific proteases (USPs), including USP8, USP22, and USP5 [76]. Inhibition of these DUBs induces PD-L1 ubiquitination and degradation, potentially reversing T-cell exhaustion and enhancing anti-tumor immunity [76].
The ubiquitin system also governs immune cell infiltration and function within the tumor microenvironment. Multi-omics analyses have revealed that UBA1 and UBA6 expression patterns significantly correlate with immune microenvironment infiltration across various cancer types [74]. Specifically, UBA6 deficiency in T cells leads to increased intracellular IFN-γ expression and subsequent multi-organ inflammation in mouse models, highlighting its role in maintaining immune homeostasis [74]. Additionally, ubiquitination regulates the cGAS-STING pathway, a central mediator of innate anti-tumor immunity, through mechanisms such as TRIM21-mediated degradation of VDAC2 (which inhibits mitochondrial DNA release and cGAS-STING activation) and USP14-mediated deubiquitination of IRF3 (which suppresses STING-dependent type I interferon responses) [7]. These findings position ubiquitination enzymes as pivotal regulators of tumor immune landscapes and promising targets for combination immunotherapy strategies.
Methodological Approaches for Ubiquitination Signature Discovery
High-Throughput Omics Technologies
Advanced proteomics technologies form the cornerstone of ubiquitination signature discovery. Ubiquitination proteomics utilizing mass spectrometry-based approaches enables comprehensive identification and quantification of ubiquitination sites across the proteome [77]. Current state-of-the-art platforms employ 4D proteomics technology with timsTOF HT or Orbitrap Astral mass spectrometers, which provide the sensitivity and resolution needed to detect the characteristic 114.1 Da mass shift associated with ubiquitin modification on lysine residues [77]. The standard workflow involves sample preparation (protein extraction and digestion), selective enrichment of ubiquitinated peptides using ubiquitin remnant motifs, high-resolution mass spectrometry analysis, and sophisticated bioinformatics processing for ubiquitination site identification and quantification [77].
Multi-omics integration has emerged as a powerful strategy for ubiquitination signature development. As demonstrated in pan-cancer analyses of the UBA family, researchers can extract RNA sequencing, DNA methylation, copy number variation, and clinical data from public repositories such as The Cancer Genome Atlas (TCGA) to identify ubiquitination-related biomarkers with diagnostic and prognostic utility [74]. These approaches typically involve differential expression analysis between normal and tumor tissues, survival analysis using Cox proportional hazards models, correlation with clinical pathological stages, and assessment of immune cell infiltration patterns using algorithms such as CIBERSORT [74] [75]. The resulting signatures can stratify patients based on disease prognosis and therapeutic susceptibility, providing a foundation for precision medicine approaches.
Bioinformatics and Computational Framework
The development of robust ubiquitination signatures requires sophisticated computational frameworks for pattern recognition and validation. A typical analytical workflow for ubiquitination-based biomarker discovery involves several key stages, as illustrated below:
Ubiquitination Signature Development Workflow
This comprehensive bioinformatics pipeline has been successfully applied to develop ubiquitination signatures across multiple cancer types. In breast cancer, researchers analyzed 763 ubiquitin-related genes (UbRGs) from the iUUCD 2.0 database, identifying an 8-gene prognostic signature through univariate and Lasso-Cox regression analyses on TCGA-BRCA data, with subsequent validation in external GEO datasets [75]. Similarly, in esophageal squamous cell carcinoma (ESCC), an integrated analysis of ubiquitination-related genes (URGs) and deubiquitylation-related genes (DRGs) identified an 11-gene signature that effectively stratified patients into distinct prognostic subgroups and predicted response to potential therapeutic agents including dacomitinib and talazoparib [78]. These studies demonstrate the robust utility of ubiquitination signatures in oncology precision medicine.
Experimental Validation Strategies
Candidate ubiquitination signatures require rigorous experimental validation to establish their biological and clinical relevance. Functional validation typically begins with in vitro models, utilizing gene knockdown or overexpression approaches in relevant cell line systems. For example, in ESCC, loss-of-function studies demonstrated that NEURL3 knockdown significantly inhibited cancer cell proliferation and motility, supporting its role as a potential therapeutic target [78]. Similarly, in breast cancer, functional validation of signature genes included lentivirus-mediated FBXL6 knockdown and PDZRN3 overexpression, followed by assessment of phenotypic effects on tumor development [75].
Advanced preclinical models provide critical platforms for evaluating the therapeutic targeting of ubiquitination signatures. Patient-derived xenografts (PDX), genetically engineered mouse models (GEMMs), and organoid cultures recapitulate the tumor microenvironment and enable assessment of treatment responses in physiologically relevant contexts. For instance, in vivo studies have shown that inhibiting UBA1 expression significantly reduces tumor weight and volume in animal models, while UCHL3 inhibitors have demonstrated promising anti-tumor effects in both in vitro and in vivo settings [74] [60]. Additionally, radiation-responsive PROTAC platforms, such as radiotherapy-triggered PROTAC (RT-PROTAC) prodrugs activated by tumor-localized X-rays, have shown synergistic effects with radiotherapy in breast cancer models, highlighting the therapeutic potential of targeting ubiquitination networks [7].
Current Applications in Clinical Oncology
Diagnostic and Prognostic Biomarkers
Ubiquitination signatures show remarkable utility as diagnostic and prognostic biomarkers across diverse cancer types. Pan-cancer analyses reveal that core ubiquitination enzymes, particularly UBA1 and UBA6, are frequently dysregulated in human malignancies [74]. UBA1 demonstrates significantly elevated expression in multiple cancer types, including lung cancer (LC), liver cancer, and colorectal cancer (CRC), where it correlates with advanced disease stage and poor patient survival [74]. The table below summarizes key ubiquitination-related biomarkers with established clinical relevance:
Table 2: Clinically Relevant Ubiquitination Biomarkers in Cancer
| Biomarker | Cancer Type(s) | Expression Pattern | Clinical Correlation | Potential Utility |
|---|---|---|---|---|
| UBA1 | Lung, Liver, Colorectal | Overexpression | Poor survival, Advanced stage | Diagnostic, Prognostic |
| UBA6 | Multiple solid tumors | Overexpression | Shorter survival, Immune infiltration | Prognostic, Immunotherapy response |
| UCHL3 | Various (except prostate) | Overexpression | Therapy resistance, DNA repair capacity | Predictive for chemo/radiotherapy |
| FBXL6 | Breast cancer | Overexpression | Poor prognosis, Tumor progression | Prognostic, Therapeutic target |
| NEURL3 | Esophageal SCC | Overexpression | Shorter overall survival | Prognostic, Therapeutic target |
In breast cancer, ubiquitination-based molecular subtyping has identified distinct patient subgroups with significant differences in overall survival, tumor microenvironment composition, and therapeutic susceptibility [75]. Similarly, in ESCC, a ubiquitination-deubiquitination signature successfully stratified patients into C1 and C2 molecular subtypes, with C2 subtype exhibiting significantly shorter overall survival time and distinct immune microenvironment characteristics [78]. These classification systems provide frameworks for treatment personalization beyond conventional histopathological grading.
Predictive Biomarkers for Therapy Response
Ubiquitination signatures offer exceptional value as predictive biomarkers for therapy response, particularly in the contexts of DNA-damaging agents and immunotherapy. The capacity of tumors to efficiently repair therapy-induced DNA damage represents a major resistance mechanism, and ubiquitination signatures that reflect DNA repair proficiency can inform treatment selection [60] [7]. For example, UCHL3 overexpression enhances DNA damage repair capacity and confers resistance to chemotherapy and radiotherapy in certain tumors, suggesting that UCHL3 expression levels may help identify patients likely to respond poorly to these modalities [60]. Additionally, ubiquitination signatures related to HR proficiency (e.g., RAD51 foci formation) or NHEJ efficiency may predict sensitivity to PARP inhibitors or radiation therapy, respectively [60] [7].
In the immunotherapy realm, ubiquitination signatures that reflect tumor microenvironment immune status show promise as predictors of checkpoint inhibitor response [73] [76]. Multi-omics analyses have established significant correlations between UBA family expression patterns and immune cell infiltration levels across various cancers [74]. Specifically, ubiquitination-based signatures can distinguish "hot" from "cold" tumors, with the former characterized by enhanced T cell infiltration and increased likelihood of responding to immune checkpoint blockade [78] [76]. Furthermore, ubiquitination-related biomarkers may predict response to novel therapeutic strategies that target ubiquitination enzymes themselves, such as DUB inhibitors or PROTACs [72] [7].
Drug Sensitivity and Resistance Signatures
Ubiquitination signatures provide valuable insights into drug sensitivity and resistance mechanisms, enabling more effective treatment selection. Comprehensive drug sensitivity analyses have revealed significant associations between specific ubiquitination patterns and response to conventional chemotherapeutics, targeted agents, and endocrine therapies [75] [78]. In breast cancer, ubiquitination-based risk scores correlate with differential sensitivity to tamoxifen, fulvestrant, cyclophosphamide, cisplatin, paclitaxel, epirubicin, gefitinib, and lapatinib, providing a rational basis for treatment personalization [75]. Similarly, in ESCC, ubiquitination-deubiquitination signatures identify patients with likely sensitivity to dacomitinib and talazoparib, potentially through targeting MAPK14 [78].
The integration of ubiquitination signatures with drug response data enables the development of predictive models for therapy selection. For instance, in non-small cell lung cancer (NSCLC), ubiquitination-based molecular subtyping has informed strategies to overcome resistance to EGFR tyrosine kinase inhibitors [76]. The discovery that the WDR4-Cul4 complex promotes tumorigenesis by inhibiting PTPN23-mediated EGFR degradation, while the miR-4487/USP37 axis and USP22 regulate EGFR ubiquitination, provides potential biomarkers for predicting and overcoming EGFR-TKI resistance [76]. Similarly, in KRAS-mutant NSCLC, ubiquitination networks involving USP5, RNF185/TRIM21-TRIM4, and OTUD7B sustain tumor metabolic reprogramming and represent promising predictive biomarkers for emerging targeted therapies [76].
The Scientist's Toolkit: Essential Research Reagents and Platforms
The experimental approaches discussed in this guide rely on specialized research reagents and technological platforms that enable precise interrogation of ubiquitination signatures. The following table catalogues essential tools for ubiquitination biomarker research:
Table 3: Essential Research Reagents and Platforms for Ubiquitination Studies
| Reagent/Platform | Type | Key Function | Example Applications |
|---|---|---|---|
| timsTOF HT/Orbitrap Astral | Mass Spectrometer | High-resolution identification of ubiquitinated peptides | Ubiquitinome profiling, Site identification |
| CIBERSORT | Computational Algorithm | Deconvolution of immune cell infiltration from RNA-seq data | Tumor microenvironment analysis |
| TISIDB | Database Portal | Integrative analysis of tumor-immune system interactions | Correlation with immunomodulators |
| LASSO-Cox Regression | Statistical Method | Dimensionality reduction for prognostic signature development | Multi-gene biomarker identification |
| PROTACs | Targeted Protein Degradation | Selective degradation of target proteins via ubiquitin-proteasome system | Therapeutic validation of ubiquitination targets |
| ConsensusClusterPlus | R Package | Unsupervised clustering for molecular subtyping | Patient stratification based on ubiquitination patterns |
| GSCA | Bioinformatics Platform | Gene Set Cancer Analysis for genomic mutational landscape | CNV/SNV analysis of ubiquitination genes |
| Ubiquitin Remnant Motif Antibodies | Affinity Reagents | Selective enrichment of ubiquitinated peptides | Mass spectrometry sample preparation |
These research tools enable the comprehensive characterization of ubiquitination signatures across technological domains. Mass spectrometry platforms form the foundation for ubiquitinome profiling, providing the sensitivity and dynamic range needed to detect and quantify ubiquitination events in complex biological samples [77]. Bioinformatics resources such as TISIDB and GSCA facilitate the integration of ubiquitination data with immune parameters and genomic alterations, enabling systems-level analyses [74]. Meanwhile, advanced statistical approaches including LASSO-Cox regression allow for the development of robust multi-gene signatures from high-dimensional datasets [75] [78]. The continuous refinement of these research tools will undoubtedly accelerate the discovery and validation of clinically actionable ubiquitination biomarkers.
The rapidly evolving field of ubiquitination signature research holds tremendous promise for advancing precision medicine across diverse disease contexts, particularly in oncology. Future developments will likely focus on several key areas: (1) the standardization of ubiquitination signature assays for clinical deployment; (2) the integration of ubiquitination signatures with other molecular biomarkers into comprehensive diagnostic platforms; (3) the development of targeted therapies that specifically modulate aberrant ubiquitination events identified through signature analysis; and (4) the application of ubiquitination signatures to monitor treatment response and emerging resistance mechanisms in real time.
The clinical implementation of ubiquitination signatures faces several challenges, including technical standardization, analytical validation, and demonstration of clinical utility through prospective trials. However, the compelling evidence from preclinical and retrospective studies, coupled with advances in mass spectrometry and computational biology, provides a strong foundation for translation into clinical practice. As our understanding of the ubiquitin code continues to expand, and as technologies for measuring ubiquitination events become increasingly sensitive and accessible, ubiquitination signatures are poised to become integral components of the precision medicine toolkit, enabling more accurate diagnosis, prognostication, and treatment selection for cancer patients and potentially for those with other diseases characterized by ubiquitination dysregulation.
Immunomodulatory drugs (IMiDs) such as thalidomide, lenalidomide, and pomalidomide represent a groundbreaking class of therapeutics that function by reprogramming the specificity of E3 ubiquitin ligases. This whitepaper examines the molecular mechanisms by which IMiDs co-opt the CRL4-CRBN E3 ubiquitin ligase complex to target novel protein substrates for ubiquitination and degradation. Within the broader context of ubiquitination in DNA repair and immune response pathways, we explore how IMiDs mediate their therapeutic effects in hematologic malignancies through targeted protein degradation, provide detailed experimental methodologies for investigating these mechanisms, and discuss emerging opportunities in targeted protein degradation therapeutics.
The ubiquitin-proteasome system (UPS) represents a crucial regulatory mechanism in eukaryotic cells, governing protein stability, function, and degradation through the covalent attachment of ubiquitin molecules to target proteins [79] [80]. This process occurs through a sequential enzymatic cascade involving E1 (activating), E2 (conjugating), and E3 (ligating) enzymes, with E3 ubiquitin ligases conferring substrate specificity by recognizing and binding to target proteins [80]. The human genome encodes approximately 600 E3 ligases, which are categorized into three main classes based on their structural domains and mechanisms of action: Really Interesting New Gene (RING), Homologous to E6AP C-Terminus (HECT), and RING-Between-RING (RBR) ligases [80] [81].
E3 ligases function as critical regulatory nodes in diverse cellular processes, including cell cycle progression, DNA damage response, immune signaling, and cellular metabolism [79] [15]. The UPS is particularly crucial for maintaining genomic integrity through its roles in DNA repair pathways, where E3 ligases such as RNF8, RNF168, and BRCA1/BARD1 facilitate the recruitment of DNA repair factors to damage sites and regulate repair pathway choice between non-homologous end joining (NHEJ) and homologous recombination (HR) [15]. In immune regulation, E3 ligases control the stability of key transcription factors and signaling molecules, thereby modulating immune cell development, activation, and function [82].
Molecular Mechanisms of IMiD Action
Historical Development and Clinical Transition
The discovery of IMiDs represents a paradigm shift in understanding molecular glues and targeted protein degradation. Initially developed as sedatives, thalidomide and its derivatives were later found to possess significant immunomodulatory and anti-cancer properties [83] [82]. The teratogenic effects of thalidomide prompted investigations into its molecular targets, culminating in the seminal discovery that thalidomide directly binds to cereblon (CRBN), a substrate receptor of the CRL4 E3 ubiquitin ligase complex [84] [82]. This finding elucidated the mechanism underlying both thalidomide's therapeutic effects and its teratogenicity, revealing that IMiDs function not as simple inhibitors but as molecular glues that reprogram E3 ligase specificity.
Structural Basis of CRBN-IMiD Interactions
Cereblon (CRBN) serves as a substrate receptor component of the Cullin 4-RING E3 ubiquitin ligase (CRL4) complex, which consists of CUL4, ROC1 (RBX1), DDB1, and CRBN [82]. Structural analyses have revealed that CRBN contains an N-terminal Lon-like domain (LLD) and a C-terminal thalidomide-binding domain (TBD) [82]. The TBD features a hydrophobic pocket composed of three tryptophan residues (W386, W380, W400) and one phenylalanine residue (F402), which accommodates the glutarimide moiety common to all IMiDs [82].
Table 1: Key IMiDs and Their Primary Degradation Targets
| IMiD | CRBN Binding Affinity | Primary Degradation Targets | Clinical Applications |
|---|---|---|---|
| Thalidomide | Moderate | Unknown (teratogenicity) | Multiple Myeloma, Leprosy |
| Lenalidomide | High | IKZF1, IKZF3 | Multiple Myeloma, MDS, CLL |
| Pomalidomide | High | IKZF1, IKZF3 | Relapsed/Refractory Multiple Myeloma |
| CELMoDs (e.g., CC-885) | Very High | IKZF1, IKZF3, GSPT1 | Investigational for Hematologic Malignancies |
Upon IMiD binding, CRBN undergoes conformational changes that create novel surface interfaces capable of recognizing proteins that are not native CRBN substrates [83] [84] [82]. For instance, lenalidomide inserts its succinimide ring into the hydrophobic pocket of CRBN's TBD, while its isoindolinone moiety protrudes and interacts with complementary surfaces on neosubstrates such as IKZF1 and IKZF3 [82]. This drug-induced interaction facilitates the ubiquitination and subsequent proteasomal degradation of these transcription factors, ultimately mediating the therapeutic effects of IMiDs in hematologic malignancies.
Figure 1: IMiD-Induced CRL4-CRBN E3 Ligase Complex Reprogramming
Downstream Effects on Immune Function and DNA Damage Response
The degradation of IKZF1 (Ikaros) and IKZF3 (Aiolos) transcription factors by IMiD-reprogrammed CRL4-CRBN has profound effects on immune cell function and cancer cell survival [83] [82]. IKZF1 and IKZF3 normally function as transcriptional repressors of interleukin-2 (IL-2) and interferon-γ (IFN-γ) [82]. Their degradation leads to derepression of IL-2 and IFN-γ expression, enhancing T-cell and natural killer (NK) cell activation and mediating potent immunomodulatory effects [82]. Additionally, IKZF1 and IKZF3 degradation suppresses the expression of IRF4 and c-MYC, two critical survival factors for multiple myeloma cells, thereby directly inhibiting malignant plasma cell proliferation and survival [83] [82].
Beyond their immunomodulatory effects, IMiDs also influence DNA damage response pathways. Recent evidence suggests that CRBN E3 ligase modulators (CELMoDs), the next-generation IMiDs, can promote the degradation of additional substrates such as GSPT1 (G1 to S phase transition 1), a translation termination factor overexpressed in acute myeloid leukemia (AML) [82]. This expanded substrate repertoire highlights the potential of IMiDs and CELMoDs to simultaneously modulate multiple therapeutic pathways, including immune activation, oncoprotein degradation, and potentially DNA damage response through interconnected ubiquitination networks.
Experimental Approaches for Investigating IMiD Mechanisms
Structural Biology Techniques
X-ray Crystallography has been instrumental in elucidating the structural basis of IMiD-induced neosubstrate recruitment. The protocol involves:
- Protein Complex Preparation: Purify recombinant CRBN (TBD domain), IMiD compound, and candidate substrate proteins (e.g., IKZF1/3 zinc finger domains).
- Crystallization: Co-crystallize the CRBN-IMiD-substrate ternary complex using vapor diffusion methods.
- Data Collection and Structure Determination: Collect X-ray diffraction data at synchrotron facilities and solve structures using molecular replacement with existing CRBN structures (e.g., PDB ID: 4CI2) [82].
These structural studies have revealed that lenalidomide binds within CRBN's hydrophobic pocket, with its succinimide ring engaging residues W380 and H378 through hydrogen bonds (2.7-2.8 Å), while the isoindolinone ring mediates interactions with both CRBN and the neosubstrate [82]. For CELMoDs like CC-885, additional interactions occur through urea portions that form hydrogen bonds with CRBN residues E377 and H353 (2.7-2.9 Å) [82].
Cellular Ubiquitination and Degradation Assays
To validate IMiD-induced substrate ubiquitination and degradation, researchers employ the following methodology:
- Cell Culture and Treatment: Culture multiple myeloma cells (e.g., MM.1S, RPMI8226) or HEK293T cells in appropriate media and treat with IMiDs at varying concentrations (typically 1-10 μM) and timepoints (0-24 hours).
- Western Blot Analysis: Harvest cells, lyse in RIPA buffer, separate proteins by SDS-PAGE, and transfer to PVDF membranes. Probe with antibodies against target proteins (IKZF1, IKZF3, GSPT1), CRBN, and loading controls (β-actin, GAPDH).
- Cycloheximide Chase Assays: Treat cells with protein synthesis inhibitor cycloheximide (100 μg/mL) alongside IMiDs to measure substrate half-life.
- Ubiquitination Assays: Transfect cells with HA- or Myc-tagged ubiquitin, immunoprecipitate target proteins under denaturing conditions, and detect ubiquitination by anti-tag Western blotting.
Table 2: Key Research Reagents for IMiD Mechanism Studies
| Reagent/Cell Line | Application | Key Features | Commercial Sources |
|---|---|---|---|
| MM.1S Cells | IMiD sensitivity studies | Sensitive multiple myeloma cell line | ATCC, DSMZ |
| HEK293T Cells | Transfection, ubiquitination assays | High transfection efficiency | ATCC |
| Anti-IKZF1 Antibody | Western blot, IP | Detects Ikaros degradation | Cell Signaling, Abcam |
| Anti-CRBN Antibody | CRBN expression validation | Confirms CRBN presence | Bethyl Labs, Sigma |
| HA-Ubiquitin Plasmid | Ubiquitination assays | Tags ubiquitin for detection | Addgene |
| Proteasome Inhibitor (MG132) | Degradation pathway validation | Blocks proteasomal degradation | Sigma, Millipore |
Functional Cellular Assays
Immunomodulatory Effects:
- Cytokine Production Assays: Isolate primary T-cells from human peripheral blood mononuclear cells (PBMCs) and culture with IMiDs (0.1-10 μM) for 24-72 hours. Measure IL-2 and IFN-γ secretion using ELISA.
- T-cell Co-stimulation Assays: Assess T-cell proliferation using CFSE dilution or 3H-thymidine incorporation in anti-CD3-stimulated T-cells with IMiD treatment.
Anti-cancer Efficacy:
- Cell Viability Assays: Treat multiple myeloma cells with IMiDs in dose-response format (0.001-100 μM) for 72 hours. Measure viability using MTT, CellTiter-Glo, or Alamar Blue assays.
- Apoptosis Assays: Stain IMiD-treated cells with Annexin V/PI and analyze by flow cytometry to quantify apoptotic cells.
- Clonogenic Assays: Plate cells at low density with IMiD treatment and count colonies after 10-14 days to assess long-term proliferation inhibition.
Figure 2: Experimental Workflow for IMiD Mechanism Investigation
Therapeutic Applications and Clinical Translation
Hematologic Malignancies
IMiDs have revolutionized the treatment of multiple myeloma and other hematologic malignancies. Lenalidomide maintenance therapy following autologous stem cell transplantation has significantly improved progression-free and overall survival in multiple myeloma patients [83]. The therapeutic efficacy of IMiDs in multiple myeloma stems from multiple mechanisms: (1) direct degradation of IKZF1 and IKZF3 in malignant plasma cells, suppressing IRF4 and c-MYC expression; (2) immunomodulatory effects enhancing T-cell and NK-cell-mediated tumor cell killing; and (3) inhibition of tumor microenvironment support through modulation of cytokine production and angiogenesis [83] [82].
Beyond multiple myeloma, lenalidomide demonstrates exceptional efficacy in myelodysplastic syndromes with deletion 5q (del(5q)), where it promotes degradation of casein kinase 1A1 (CK1α), a haploinsufficient tumor suppressor encoded on chromosome 5q [82]. This targeted degradation of CK1α leads to selective elimination of del(5q) malignant clones while sparing normal hematopoietic cells, exemplifying the precision medicine potential of IMiDs.
Next-Generation CELMoDs
Cereblon E3 Ligase Modulators (CELMoDs) such as iberdomide (CC-220) and mezigdomide (CC-92480) represent the next evolution of IMiDs with enhanced potency and expanded substrate profiles [83] [82]. These agents feature structural modifications including additional phenyl rings and cyclohexyl groups that enable more extensive interactions with both CRBN and neosubstrates [82]. CELMoDs induce superior degradation of IKZF1/3 compared to first-generation IMiDs and can degrade additional substrates including GSPT1 and CK1α, broadening their therapeutic potential across diverse hematologic malignancies, particularly in IMiD-resistant disease [82].
Table 3: Clinical Efficacy of IMiDs in Hematologic Malignancies
| Agent | Clinical Setting | Response Rates | Key Toxicities |
|---|---|---|---|
| Lenalidomide | Newly Diagnosed Multiple Myeloma (Maintenance) | 60% 3-year PFS | Myelosuppression, Venous Thromboembolism |
| Lenalidomide | del(5q) MDS | 65% Transfusion Independence | Neutropenia, Thrombocytopenia |
| Pomalidomide | Relapsed/Refractory Multiple Myeloma | 30-35% Overall Response | Neutropenia, Fatigue, Pneumonia |
| CELMoDs (e.g., Iberdomide) | Relapsed/Refractory Multiple Myeloma (Phase 1/2) | 25-50% (depending on prior therapy) | Neutropenia, Infections |
Future Directions and Implications for Ubiquitination Research
The discovery and characterization of IMiDs have catalyzed the emerging field of targeted protein degradation (TPD), validating E3 ligases as druggable targets for therapeutic development [84]. Several promising research directions are emerging from this foundation:
Expanding the E3 Ligase Toolkit
While IMiDs target CRBN, only a small fraction of the approximately 600 human E3 ligases have been exploited therapeutically [84] [81]. Research efforts are now focused on identifying molecular glues for other E3 ligase families, including RING-UIM ligases (RNF114, RNF125, RNF138, RNF166) [85] and HECT-type ligases such as HUWE1 [86]. Each E3 ligase offers unique tissue expression patterns, subcellular localization, and intrinsic substrate preferences that could be harnessed for tissue-specific or pathway-selective protein degradation.
Integrating DNA Repair and Immune Modulation
The intersection of ubiquitination in DNA repair and immune response represents a fertile area for future IMiD research [79] [15]. As E3 ligases like CRBN, RNF8, and RNF168 play critical roles in DNA damage response, opportunities exist to develop IMiD-like compounds that simultaneously enhance genomic stability and immune activation. Such approaches could prove particularly valuable in cancers with DNA repair deficiencies, such as those with BRCA mutations or microsatellite instability, where targeted protein degradation could synthetically lethal interactions.
Addressing Resistance Mechanisms
Despite the efficacy of IMiDs, resistance inevitably develops through various mechanisms, including CRBN mutations, downregulation, and alterations in downstream signaling pathways [83] [82]. Future research must focus on understanding these resistance mechanisms and developing strategies to overcome them, such as developing CELMoDs with alternative binding modes, combining IMiDs with other targeted therapies, or targeting alternative E3 ligases in CRBN-deficient malignancies.
IMiDs exemplify the transformative potential of reprogramming E3 ubiquitin ligase specificity for therapeutic benefit. By co-opting the CRL4-CRBN complex to degrade novel substrates, these molecular glues simultaneously modulate multiple pathological pathways in cancer cells and the immune microenvironment. The continued elucidation of IMiD mechanisms provides both a blueprint for developing next-generation targeted protein degraders and valuable insights into the broader roles of ubiquitination in maintaining cellular homeostasis. As research progresses, the strategic manipulation of E3 ligase specificity promises to yield increasingly precise therapeutic modalities that exploit the ubiquitin-proteasome system for cancer therapy and beyond.
The ubiquitin-proteasome system (UPS) has emerged as a master regulator of cellular processes critical to cancer therapy response, including DNA damage repair, cell cycle control, and immune cell activation. This whitepaper examines the mechanistic basis for combining ubiquitin-targeting agents with conventional chemo-, radio-, and immunotherapies. We explore how strategic inhibition of specific E3 ligases, deubiquitinases (DUBs), or proteasomal components can disrupt tumor survival pathways, overcome resistance mechanisms, and enhance antitumor immunity. By integrating current research findings and experimental approaches, this guide provides a framework for developing rational combination strategies that leverage the ubiquitin system to improve therapeutic outcomes across multiple cancer types.
The ubiquitin-proteasome system represents a sophisticated protein modification and degradation pathway that regulates approximately 80% of cellular proteins through a cascade of enzymatic reactions [27]. This system begins with ubiquitin activation by E1 enzymes, conjugation through E2 enzymes, and substrate-specific ligation by E3 ubiquitin ligases, with the entire process being reversible through the action of deubiquitinating enzymes (DUBs) [4]. The critical role of ubiquitination in maintaining cellular homeostasis positions it as a pivotal determinant of therapy response. In the context of cancer treatment, tumors frequently exploit the UPS to evade therapy-induced cell death through enhanced DNA repair capacity, suppression of apoptotic signaling, and creation of immunosuppressive microenvironments [87] [7].
Mounting evidence demonstrates that ubiquitination regulates all three major cancer therapy modalities. In chemotherapy, ubiquitin enzymes mediate resistance to DNA-damaging agents through repair pathway activation and drug efflux pump regulation [87]. In radiotherapy, ubiquitin signaling controls the fidelity of DNA double-strand break repair and resolution of replication stress [15] [7]. In immunotherapy, ubiquitination modulates immune checkpoint expression, antigen presentation, and T-cell activation [88] [32]. This pervasive involvement makes the UPS an attractive target for rational combination therapy design.
Mechanistic Foundations of Ubiquitin-Targeting Combinations
Ubiquitin Signaling in Therapy Resistance Pathways
DNA Damage Repair Regulation
The ubiquitin system orchestrates DNA damage response (DDR) through spatiotemporal control of repair protein assembly and activity. Following DNA double-strand breaks, the sequential action of E3 ligases RNF8 and RNF168 establishes a ubiquitin signaling platform that recruits BRCA1 and 53BP1 to determine repair pathway choice between homologous recombination (HR) and non-homologous end joining (NHEJ) [15]. K63-linked ubiquitin chains facilitate the assembly of repair complexes, while K48-linked chains regulate protein turnover to control repair pathway activity [7]. Cancer cells frequently upregulate these ubiquitin-dependent repair mechanisms to survive genotoxic therapies, creating a vulnerability that can be targeted to sensitize tumors to DNA-damaging agents.
Table 1: Key Ubiquitin Ligases in DNA Damage Repair and Therapeutic Targeting
| E3 Ligase | Primary Function in DDR | Therapeutic Target | Cancer Type |
|---|---|---|---|
| RNF8 | Initiates ubiquitin signaling at DSBs | Not yet targeted | Various |
| RNF168 | Amplifies H2A ubiquitination for 53BP1/BRCA1 recruitment | Not yet targeted | Various |
| BRCA1/BARD1 | Promotes HR through histone ubiquitination | PARP inhibitor combinations | Breast, ovarian |
| FBXW7 | Regulates NHEJ through XRCC4 ubiquitination | Radio-sensitization | Colorectal, NSCLC |
| UBE2T/RNF8 | Mediates H2AX monoubiquitination for damage detection | Radio-sensitization | Hepatocellular carcinoma |
Immune Regulation and Checkpoint Control
Ubiquitination serves as a fundamental mechanism controlling antitumor immunity through regulation of immune cell function and checkpoint protein stability. E3 ligases such as MARCH family members regulate MHC I and II surface expression on antigen-presenting cells, thereby modulating T-cell activation [88]. The CBL family of E3 ligases controls T-cell receptor signaling intensity and duration, while regulation of PD-L1 stability by membrane-associated RING-CH-type E3 ligases represents an emerging mechanism of immune checkpoint control [88] [32]. These immunoregulatory functions position ubiquitin modifiers as promising targets for enhancing cancer immunotherapy efficacy.
Ubiquitin Chain Topology in Therapy Response
The functional consequences of protein ubiquitination are determined by ubiquitin chain topology, with different linkage types encoding distinct biological signals. Understanding these topological codes is essential for designing targeted combination therapies.
Table 2: Ubiquitin Chain Linkages and Their Roles in Therapy Response
| Linkage Type | Primary Function | Therapy Relevance | Example |
|---|---|---|---|
| K48-linked | Targets proteins for proteasomal degradation | Regulates stability of tumor suppressors (p53) and oncoproteins | FBXW7-mediated p53 degradation in colorectal cancer [7] |
| K63-linked | Mediates non-proteolytic signaling, complex assembly | Promutes DNA repair, cell survival signaling | TRAF4-mediated JNK/c-Jun activation in colorectal cancer [7] |
| K11-linked | Proteasomal degradation, cell cycle regulation | Cell cycle control, mitotic progression | APC/C-mediated cyclin degradation [72] |
| K27/K29-linked | DNA damage response, signaling | Activates DNA repair pathways | RNF126-mediated MRE11 ubiquitination in TNBC [7] |
| Monoubiquitination | Alters protein activity, localization, interactions | Regulates histone function, DNA repair | FANCD2 monoubiquitination in crosslink repair [89] |
Combination Therapy Strategies
Ubiquitin-Targeting Agents with Chemotherapy
Overcoming Platinum Resistance
Platinum-based chemotherapeutics such as cisplatin and oxaliplatin induce DNA crosslinks that are repaired through the Fanconi anemia (FA) pathway, a process critically dependent on monoubiquitination of FANCD2 and FANCI by the FA core complex [89]. Inhibition of USP1, the deubiquitinase that reverses FANCD2 monoubiquitination, sensitizes tumors to platinum agents by stabilizing DNA crosslinks and preventing repair completion. Preclinical models of ovarian and pancreatic cancer demonstrate that USP1 inhibitors synergize with cisplatin to enhance tumor cell apoptosis and suppress tumor growth [89].
Targeting Wnt/β-Catenin-Mediated Chemoresistance
In colorectal cancer, ubiquitination regulates chemotherapy tolerance through control of Wnt/β-catenin signaling, epithelial-mesenchymal transition (EMT), and cell cycle progression [87]. E3 ligases such as β-TrCP target key components of these pathways for degradation, while their downregulation in chemoresistant tumors leads to stabilization of oncogenic proteins. Combination strategies employing β-TrCP-stabilizing agents with 5-fluorouracil or irinotecan have shown promise in overcoming chemoresistance in preclinical CRC models [87].
Ubiquitin-Targeting Agents with Radiotherapy
Disrupting DNA Repair Fidelity
Radiotherapy resistance is frequently mediated through enhanced DNA double-strand break repair, a process extensively regulated by ubiquitination. The sequential action of RNF8, RNF168, and other E3 ligases establishes a ubiquitin signaling platform that recruits repair proteins to damage sites [15] [7]. Inhibition of key enzymes in this cascade, such as through small molecule inhibitors targeting RNF168, sensitizes tumor cells to radiation by impairing both homologous recombination and non-homologous end joining. Recent studies in glioblastoma and non-small cell lung cancer models demonstrate that targeting the ubiquitin-dependent repair machinery can enhance radiation-induced cell killing by 2- to 4-fold compared to radiation alone [7].
Metabolic Reprogramming for Radio-Sensitization
Ubiquitination regulates cellular metabolism in ways that impact radiation response. TRIM26 stabilizes GPX4 via K63 ubiquitination to prevent ferroptosis in gliomas, while SOCS2/Elongin B/C complexes drive SLC7A11 destruction to increase ferroptosis sensitivity in liver cancer [7]. Targeting these ubiquitin-dependent metabolic adaptations represents a promising approach for radio-sensitization. Combination of glutathione peroxidase inhibitors with radiation in TRIM26-high gliomas enhances lipid peroxidation and ferroptotic cell death, overcoming a key resistance mechanism [7].
Ubiquitin-Targeting Agents with Immunotherapy
Enhancing Antigen Presentation and T-cell Activation
The ubiquitin system regulates multiple steps in the cancer immunity cycle, from antigen presentation to immune cell activation. MARCH family E3 ligases control MHC I and II surface expression on antigen-presenting cells, while CBL family ligases regulate T-cell receptor signaling intensity [88]. Strategic inhibition of specific immune-suppressive E3 ligases enhances dendritic cell maturation, antigen cross-presentation, and T-cell-mediated tumor killing. Preclinical studies demonstrate that targeting immune-regulatory E3 ligases in combination with PD-1/PD-L1 checkpoint blockade improves therapeutic responses in immunologically "cold" tumors [88] [32].
Modulating the STING Pathway for Immune Activation
The cGAS-STING pathway represents a critical innate immune signaling axis that can be activated by ubiquitin-targeting agents to enhance cancer immunotherapy. TRIM21 suppresses antitumor immunity by promoting K48-linked degradation of VDAC2, inhibiting mitochondrial DNA release and cGAS/STING activation in nasopharyngeal carcinoma [7]. Conversely, USP14 inhibition stabilizes K63-ubiquitinated IRF3, amplifying STING-dependent type I interferon responses [7]. These findings suggest that combining ubiquitin modulators with immune checkpoint inhibitors can convert immunologically suppressed tumors to immunologically active ones.
Experimental Approaches and Methodologies
Assessing Ubiquitin-Dependent Therapy Interactions
DNA Repair Focus Formation Assay
Purpose: To evaluate the impact of ubiquitin-targeting agents on DNA repair protein recruitment to damage sites.
Methodology:
- Seed tumor cells in chamber slides and pre-treat with ubiquitin-targeting agent or vehicle control for 24 hours
- Induce DNA damage using γ-irradiation (2-8 Gy) or chemotherapeutic agent
- Fix cells at specific timepoints post-treatment (0.5, 2, 8, 24 hours)
- Perform immunofluorescence staining for DNA repair markers (γH2AX, 53BP1, RAD51, BRCA1)
- Quantify focus number and intensity using high-content imaging analysis
- Correlate focus formation kinetics with clonogenic survival outcomes
Interpretation: Effective ubiquitin-targeting agents will delay repair focus resolution and increase persistent damage, correlating with enhanced therapy sensitivity [15] [7].
Ubiquitin Chain Linkage Analysis
Purpose: To characterize changes in ubiquitin chain topology following combination treatments.
Methodology:
- Treat cells with ubiquitin-targeting agent alone or in combination with chemo/radio-therapy
- Harvest cells at optimal timepoints and lyse in denaturing buffer
- Enrich ubiquitinated proteins using TUBE (Tandem Ubiquitin Binding Entity) agarose
- Digest purified proteins with specific linkage-preserving or linkage-cleaving DUBs
- Analyze ubiquitin chain composition by mass spectrometry or immunoblotting with linkage-specific antibodies
- Correlate specific chain type alterations with functional outcomes
Interpretation: Effective combinations will induce specific chain alterations (e.g., reduced K63-linked chains at damage sites) that disrupt pro-survival signaling [72] [7].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Investigating Ubiquitin-Targeting Combinations
| Reagent Category | Specific Examples | Research Application | Key Considerations |
|---|---|---|---|
| E1 Inhibitors | MLN7243, PYR-41 | Block global ubiquitination | High toxicity limits therapeutic use; valuable research tools [4] |
| Proteasome Inhibitors | Bortezomib, Carfilzomib, Ixazomib | Inhibit protein degradation | FDA-approved; combination backbone [4] |
| E3 Ligase Modulators | Nutlin-3a (MDM2), Lenalidomide (CRBN) | Target specific E3 ligases | CRBN modulators in clinical use; emerging specific inhibitors [4] |
| DUB Inhibitors | VLX1570 (USP14), PR-619 (pan-DUB) | Block deubiquitination | Emerging specificity challenges; clinical development ongoing [4] |
| Linkage-Specific Antibodies | K48-linkage, K63-linkage, Mono-Ub | Detect specific ubiquitin signals | Validation critical; lot-to-lot variability possible [72] |
| Ubiquitin Activity Probes | HA-Ub-VS, TAMRA-Ub-PA | Monitor DUB and E1/E2 activity | Cell-permeable versions available for live-cell imaging [27] |
Visualization of Key Signaling Pathways
Ubiquitin Regulation of DNA Damage Repair and Immune Signaling
Diagram 1: Ubiquitin Signaling in Therapy Response Pathways. This diagram illustrates how ubiquitination regulates key cellular processes targeted by cancer therapies, highlighting potential intervention points for combination strategies.
Experimental Workflow for Combination Therapy Development
Diagram 2: Experimental Workflow for Combination Therapy Development. This diagram outlines a systematic approach for developing and validating ubiquitin-targeting combination therapies, from target identification to clinical trial design.
Clinical Translation and Future Directions
The translation of ubiquitin-targeting combinations from preclinical models to clinical application requires careful consideration of several factors. First, the contextual duality of many ubiquitin system components necessitates patient stratification based on tumor genetics, ubiquitin enzyme expression patterns, and functional readouts of pathway activity [7]. Second, the timing and sequencing of combination partners must be optimized based on mechanistic understanding of the targeted processes—for example, ubiquitin inhibitors should typically be administered prior to DNA-damaging therapies to preemptively disable repair capacity [87] [7].
Emerging technologies are expanding the toolkit for ubiquitin-targeted therapies. PROTACs (Proteolysis-Targeting Chimeras) represent a promising class of ubiquitin-dependent degraders that recruit E3 ligases to target proteins of interest [7]. Radiation-responsive PROTAC platforms, such as radiotherapy-triggered PROTAC prodrugs activated by tumor-localized X-rays, enable spatial control of protein degradation to minimize systemic toxicity [7]. Additionally, molecular glues that stabilize interactions between specific E3 ligases and target proteins offer another modality for precise manipulation of the ubiquitin system.
Future research directions should focus on developing more selective inhibitors for specific E2 and E3 enzymes, understanding and overcoming resistance mechanisms to ubiquitin-targeting agents, and identifying predictive biomarkers for patient selection. As our understanding of the ubiquitin code deepens, the potential for highly specific, effective combination therapies that leverage this sophisticated cellular regulatory system will continue to expand.
The strategic integration of ubiquitin-targeting agents with conventional cancer therapies represents a promising approach for overcoming treatment resistance and improving patient outcomes. By disrupting the molecular pathways that tumors exploit to survive therapy-induced stress, these combinations can tip the balance toward tumor cell death while potentially sparing normal tissues. The continued elucidation of ubiquitin-dependent signaling networks, coupled with advances in targeted degradation technologies, will enable increasingly sophisticated combination strategies tailored to the molecular vulnerabilities of individual tumors. As this field advances, ubiquitin-targeting combinations are poised to become fundamental components of precision cancer medicine.
Navigating Complexity: Challenges and Optimization in Ubiquitin-Targeted Therapies
Functional redundancy within the ubiquitin system presents a significant challenge in therapeutic development, particularly for DNA repair and immune response pathways. Redundant actions of E3 ubiquitin ligases and deubiquitinating enzymes (DUBs) allow cells to maintain critical functions despite targeted inhibition, limiting the efficacy of single-target approaches. This technical review synthesizes contemporary strategies to overcome this redundancy, focusing on decoy technologies, complex-level targeting, and allosteric regulation. We provide detailed experimental protocols for implementing these approaches, along with visualization of key pathways and a comprehensive research toolkit. The insights presented herein offer a framework for developing more effective therapeutic interventions against cancer and immune disorders by targeting the ubiquitin system at a network level rather than through individual components.
The ubiquitin-proteasome system represents one of the most sophisticated post-translational regulatory networks in eukaryotic cells, governing virtually all cellular processes through targeted protein modification and degradation. This system employs a cascade of enzymes including E1 activating enzymes, E2 conjugating enzymes, and E3 ubiquitin ligases that work in concert to attach ubiquitin to substrate proteins [90]. The reverse reaction is catalyzed by deubiquitinating enzymes (DUBs), which remove ubiquitin modifications, creating a dynamic equilibrium that allows for precise control of protein function, localization, and stability [91] [92]. What makes this system particularly challenging from a therapeutic perspective is the extensive functional redundancy that has evolved across enzyme families, especially among E3 ligases and DUBs.
In humans, approximately 700 E3 ubiquitin ligases and nearly 100 DUBs regulate the ubiquitin code [24] [91]. This expansion has led to significant functional overlap, where multiple enzymes can target the same substrates or perform similar regulatory functions within biological pathways. This redundancy is particularly pronounced in DNA damage response and immune signaling pathways, where system failure would have catastrophic consequences for cell viability [93] [92]. From a therapeutic standpoint, this means that inhibiting a single E3 ligase or DUB often produces minimal phenotypic effects, as parallel enzymes compensate for the lost function.
The NEDD4 family of HECT E3 ligases exemplifies this challenge, with nine members that share structural domains and often substrate specificity [93]. Similarly, the USP family of DUBs contains multiple members with overlapping functions in DNA repair pathways [92]. This technical guide examines innovative strategies to overcome these limitations, focusing specifically on multi-subunit E3 complexes and DUB families operating within DNA repair and immune response pathways.
Mechanistic Insights into Redundancy
Structural and Functional Basis of Redundancy
Functional redundancy in the ubiquitin system arises from structural similarities and evolutionary conservation within enzyme families. E3 ubiquitin ligases are categorized into four major structural classes: RING-finger type, HECT type, RBR type, and U-box type [62]. Each class employs distinct mechanistic approaches to ubiquitination, with RING-type E3s facilitating direct ubiquitin transfer from E2 to substrate, while HECT-type E3s form an obligate thioester intermediate with ubiquitin before substrate modification [62]. The RING-type E3s represent the largest class, with over 600 members in humans, many of which have arisen through gene duplication events that preserve core structural elements while allowing for functional specialization [62].
The structural organization of multi-subunit E3 complexes creates natural redundancy at multiple levels. For instance, cullin-RING ligases (CRLs) utilize a modular architecture where different substrate recognition subunits can assemble around a common catalytic core, enabling diverse substrate targeting while maintaining similar catalytic mechanisms [62]. This modularity allows for compensation when individual components are inhibited, as alternative recognition subunits can maintain pathway function.
DUB families exhibit similar structural conservation, with six major classes defined by their catalytic domain architecture: USPs, UCHs, OTUs, Josephins, JAMMs, and MINDYs [91] [92]. The USP family alone comprises more than 50 members that share a conserved catalytic core despite diverse biological functions and substrate preferences [94]. This structural conservation underlies the functional compensation observed when individual DUBs are inhibited.
Redundancy in DNA Repair and Immune Pathways
In DNA damage response pathways, multiple E3 ligases and DUBs often converge on the same critical substrates. The repair of DNA double-strand breaks, for instance, is regulated by RNF8, RNF168, and BRCA1, which function in a coordinated cascade to promote the accumulation of repair factors at damage sites [7]. Inhibition of any single component often produces only partial pathway impairment due to compensatory mechanisms.
Similarly, in immune signaling pathways, multiple members of the NEDD4 family regulate key mediators such as MDM2 and p53, creating a network that maintains signaling fidelity even when individual components are compromised [93]. The K63-linked ubiquitin chains central to NF-κB activation can be assembled by several E3 ligases, including TRAF6, cIAP1, and cIAP2, creating a robust system resistant to single-point failure [24].
Table 1: Redundant E3 Ligase Families in DNA Repair and Immune Signaling
| Family | Representative Members | Shared Substrates/Pathways | Type of Redundancy |
|---|---|---|---|
| NEDD4 | NEDD4, NEDD4L, ITCH, WWP1, WWP2 | MDM2/p53 pathway, DNA damage response | Substrate overlap, parallel pathways |
| CRL | FBXW7, β-TrCP, SKP2 | Cell cycle regulation, DNA repair initiation | Modular substrate recognition |
| RING | RNF8, RNF168, BRCA1 | DNA double-strand break repair, histone ubiquitination | Sequential action in cascades |
| TRAF | TRAF2, TRAF6, TRAF3 | NF-κB signaling, immune activation | Shared downstream signaling |
Strategic Approaches to Overcome Redundancy
Decoy Technologies for E3 Ligases
The decoy E3 ligase approach represents a innovative strategy to overcome functional redundancy by targeting multiple related E3s simultaneously. This method involves expressing engineered E3 ligases that retain substrate binding capability but lack catalytic function, effectively sequestering substrates from their endogenous E3 ligases [95]. The decoy acts as a dominant-negative by competing with native E3 ligases for substrate binding, thereby stabilizing target proteins that would normally be ubiquitinated and degraded by redundant family members.
The experimental implementation of this approach involves several key steps. First, researchers must identify the substrate recognition domains within target E3 ligases while mutating critical residues required for E2 recruitment or ubiquitin transfer. For RING-type E3s, this typically involves deleting or mutating the RING domain while preserving substrate-binding motifs. For HECT-type E3s, the catalytic cysteine residue required for the thioester intermediate is mutated to serine or alanine [95].
In a landmark study applying this methodology to the plant circadian clock, researchers generated a library of transgenic Arabidopsis plants expressing dominant-negative 'decoy' E3 ubiquitin ligases representing nearly a quarter of all E3s in the organism [95]. This library enabled identification of dozens of previously unknown regulators of circadian function, including the redundant MAC3A and MAC3B ligases that control circadian period through regulation of splicing. This demonstration highlights the power of decoy approaches to overcome genetic redundancy and identify bona fide pathway regulators.
Diagram 1: Decoy E3 mechanism for overcoming redundancy
Targeting Multi-Subunit Complex Assembly
An alternative to targeting individual E3 ligases is focusing on the assembly and stability of multi-subunit E3 complexes. Many E3s, particularly CRLs, require precise stoichiometric assembly of multiple subunits for functionality. Disrupting the protein-protein interactions necessary for complex formation can simultaneously inhibit multiple related E3s that share common structural components [62].
The cullin-RING ligases exemplify this approach, as they utilize a common architecture consisting of a cullin scaffold protein, a RING protein, and various substrate recognition modules. Targeting the neddylation pathway that activates CRLs or developing inhibitors of the key protein-protein interfaces can disrupt entire classes of E3 ligases simultaneously [62]. Similarly, for DUB complexes, many DUBs require binding partners for full activity, providing opportunities for intervention.
Experimental protocols for this approach include:
- Protein-protein interaction mapping: Using techniques such as yeast two-hybrid screening, co-immunoprecipitation, and BioID to identify essential interactions within multi-subunit complexes.
- Structural analysis: Employing X-ray crystallography or cryo-EM to define critical interfaces at atomic resolution.
- Interface inhibitor screening: Developing high-throughput assays to identify small molecules or peptides that disrupt key protein-protein interactions without affecting individual subunit stability.
This strategy has shown promise in targeting the LUBAC complex, which requires precise assembly of HOIP, HOIL-1L, and SHARPIN subunits for linear ubiquitination in NF-κB signaling [24].
Allosteric Regulation of DUB Enzymes
Many DUBs are regulated by complex allosteric mechanisms that can be exploited to overcome functional redundancy. Unlike active-site inhibitors that often lack specificity due to conserved catalytic domains, allosteric modulators can target unique regulatory sites to achieve greater selectivity [94]. Several DUBs, including USP7 and OTULIN, contain misaligned catalytic residues in their apo state that must undergo substrate-induced rearrangements to become active, providing natural opportunities for allosteric regulation [94].
For instance, structural studies of USP7 revealed that its catalytic cysteine and histidine residues exist in a nonproductive conformation in the absence of substrate [94]. Ubiquitin binding triggers a significant conformational rearrangement that aligns the catalytic triad for efficient hydrolysis. The C-terminal ubiquitin-like (HUBL) domains of USP7 regulate this activation by controlling the conformation of a "switching loop" adjacent to the active site. Small molecules that stabilize the inactive conformation could therefore selectively inhibit USP7 without affecting other USPs.
Experimental approaches for targeting allosteric DUB regulation include:
- Conformational analysis: Using hydrogen-deuterium exchange mass spectrometry (HDX-MS) to map allosteric changes upon ligand binding.
- Fragment-based screening: Employing NMR and X-ray crystallography to identify small molecules binding to regulatory sites distant from the active site.
- Cellular conformation sensors: Developing FRET-based reporters that can detect conformational changes in living cells.
Table 2: DUB Regulatory Mechanisms and Targeting Strategies
| DUB | Regulatory Mechanism | Consequence | Targeting Approach |
|---|---|---|---|
| USP7 | HUBL domain-mediated active site rearrangement | Stimulates activity by aligning catalytic triad | Allosteric inhibitors that block HUBL-catalytic domain interaction |
| OTUB1 | Non-catalytic inhibition of E2 enzymes | Binds and inhibits charged E2~Ub complexes | Competitive peptides that disrupt E2-OTUB1 interaction |
| Ataxin-3 | Regulation of E2 enzyme activity | Modulates E2 ubiquitin charging | Small molecules that stabilize inactive E2 complexes |
| OTULIN | Substrate-assisted catalysis | Specificity for linear ubiquitin chains | Met1-linked ubiquitin analogs that trap inactive state |
Experimental Protocols for Key Methodologies
Decoy E3 Ligase Screening Protocol
Objective: Identify E3 ligases regulating specific pathways using a decoy library screening approach.
Materials:
- Decoy E3 ligase expression library (mutant E3s with intact substrate-binding domains but defective catalytic domains)
- Reporter cell line with pathway-specific readout (e.g., luciferase under pathway-specific promoter)
- Control vectors (empty vector, catalytically active E3s)
Method:
- Library Transduction: Introduce decoy E3 library into reporter cells using lentiviral transduction at low MOI to ensure single-copy integration.
- Selection and Expansion: Select transduced cells with appropriate antibiotics for 7-10 days to establish stable expression.
- Pathway Activation: Stimulate the pathway of interest (DNA damage agents, cytokines, etc.) at optimized concentrations.
- Screening: Measure pathway activity using established reporters (luminescence, fluorescence, etc.).
- Validation: Confirm hits using complementary approaches (RNAi, CRISPR knockout, pharmacological inhibition).
Technical Notes:
- Include controls for decoy expression levels and stability
- Use multiple functional assays to minimize false positives
- Counter-screen against related pathways to assess specificity
Complex Disruption Assay Protocol
Objective: Identify inhibitors of multi-subunit E3 or DUB complex assembly.
Materials:
- Purified complex components with appropriate tags
- FRET pairs or split-luciferase system for interaction monitoring
- Candidate inhibitory compounds or peptides
Method:
- Complex Reconstitution: Express and purify individual subunits of target complex with appropriate tags.
- Interaction Assay Development: Establish robust in vitro interaction assay using FRET or split-luciferase systems.
- High-Throughput Screening: Screen compound or peptide libraries for disruption of complex formation.
- Counter-Screens: Eliminate non-specific aggregators and cytotoxic compounds.
- Cellular Validation: Test hits in cellular models for complex disruption and pathway modulation.
Technical Notes:
- Include controls for compound interference with detection system
- Validate hits using orthogonal methods (SPR, ITC, native PAGE)
- Assess effects on individual subunit stability
Research Reagent Solutions
Table 3: Essential Research Reagents for Redundancy Studies
| Reagent/Category | Specific Examples | Function/Application | Key Features |
|---|---|---|---|
| Decoy E3 Libraries | Dominant-negative RING mutants, HECT Cys-to-Ala mutants | Screen for E3s regulating specific pathways | Preserved substrate binding, defective catalysis |
| Activity-Based Probes | Ubiquitin-vinyl sulfone, HA-Ub-VS | Profiling DUB activities in complex samples | Covalently labels active DUBs for identification and quantification |
| Linkage-Specific Ubiquitin Binders | TUBEs (Tandem Ubiquitin Binding Entities), linkage-specific UBDs | Isolation and analysis of specific ubiquitin linkages | High affinity for particular polyubiquitin chain types |
| Complex Disruption Tools | E3 core subunit mutants, competing peptide motifs | Disrupt specific protein-protein interactions in multi-subunit complexes | Target essential interfaces with high specificity |
| DUB Regulatory Compounds | Allosteric USP7 inhibitors, OTULIN substrate analogs | Selective modulation of DUB activity through non-catalytic sites | Bypass catalytic site conservation for greater specificity |
Therapeutic Implications and Future Directions
The strategies outlined herein have significant implications for therapeutic development, particularly in oncology where DNA repair and immune response pathways are frequently dysregulated. The decoy approach offers a unique method to simultaneously target multiple redundant E3s that regulate key tumor suppressors or oncoproteins. Similarly, targeting multi-subunit complex assembly provides opportunities to disrupt entire networks of E3 ligases that would be difficult to inhibit through conventional single-target approaches [7].
The clinical relevance of these strategies is underscored by the growing number of ubiquitin-targeting agents in development, including PROTACs that exploit E3 ligases for targeted protein degradation and DUB inhibitors that modulate key signaling nodes [7]. Understanding and overcoming functional redundancy will be essential for the successful clinical translation of these approaches, particularly in minimizing compensatory resistance mechanisms.
Future directions in this field include the development of more sophisticated multi-targeting strategies, such as bivalent inhibitors that simultaneously engage multiple E3s or DUBs with complementary functions. Additionally, the integration of structural biology with machine learning approaches promises to identify novel allosteric sites and protein-protein interfaces that can be targeted to overcome redundancy more effectively.
As our understanding of the ubiquitin code continues to evolve, so too will our ability to manipulate this system for therapeutic benefit. The strategies outlined in this technical guide provide a framework for addressing one of the most significant challenges in targeting the ubiquitin system - the extensive functional redundancy that has limited the efficacy of conventional single-target approaches.
Functional redundancy within E3 ubiquitin ligase families and DUB complexes represents both a challenge and an opportunity in therapeutic development. By moving beyond single-target inhibition to approaches that address redundancy at a systems level, researchers can develop more effective strategies for modulating DNA repair and immune signaling pathways. The decoy technologies, complex disruption strategies, and allosteric regulation approaches detailed in this technical guide provide a roadmap for overcoming redundancy in the ubiquitin system. As these methodologies continue to evolve, they promise to unlock new therapeutic possibilities for cancer, inflammatory diseases, and other conditions driven by dysregulation of the ubiquitin network.
Ubiquitination, a crucial post-translational modification, governs virtually all cellular processes in a highly context-dependent manner. Within the specific domains of DNA repair and immune response pathways, the functional outcomes of ubiquitin signaling are precisely dictated by tissue-specific environments, subcellular localization, and dynamic chain topology. This technical review delineates the molecular mechanisms underlying this sophisticated regulation, emphasizing how spatial and temporal factors determine whether ubiquitination promotes genomic integrity or triggers immune activation. We provide a comprehensive analysis of the experimental methodologies enabling the dissection of these complex networks and explore the profound implications for developing targeted therapeutic strategies, particularly in oncology and autoimmune disorders. The nuanced understanding that ubiquitin modulation is not a one-size-fits-all mechanism is paramount for future research and drug development.
The ubiquitin-proteasome system (UPS) represents a sophisticated hierarchical network for post-translational regulation, comprising a cascade of E1 activating, E2 conjugating, and E3 ligase enzymes that conjugate ubiquitin to target proteins, with reversal mediated by deubiquitinases (DUBs) [96] [2]. The human genome encodes approximately two E1 enzymes, 40 E2 enzymes, over 600 E3 ligases, and nearly 100 DUBs, creating an immense regulatory potential [2] [97]. The specificity of ubiquitin signaling is further amplified by the diversity of ubiquitin chain topologies, including monoubiquitination and polyubiquitination chains linked through any of seven lysine residues (K6, K11, K27, K29, K33, K48, K63) or the N-terminal methionine (M1) [2] [7]. Traditionally recognized for targeting proteins for proteasomal degradation via K48-linked chains, ubiquitination is now understood to perform a vast array of non-proteolytic functions, from regulating protein-protein interactions to controlling subcellular trafficking and enzymatic activity [98] [96].
Table 1: Major Ubiquitin Chain Linkages and Their Primary Functions in DNA Repair and Immunity
| Chain Type | Primary Functions | Key Regulatory Roles |
|---|---|---|
| K48-Linked | Proteasomal degradation [2] | Controls turnover of cell cycle regulators (e.g., p27), DNA repair proteins, and immune signaling components [99] [2]. |
| K63-Linked | Signal transduction, scaffolding [2] [97] | Activates NF-κB and MAPK pathways in innate immunity [97]; coordinates DNA damage repair complexes [7]. |
| K27-Linked | Immune regulation, mitophagy [30] [2] | Enhances STING activity in immune defense [30]; regulates mitochondrial autophagy [2]. |
| K11-Linked | Cell cycle regulation [2] | Implicated in control of mitotic regulators and trafficking events [2]. |
| Monoubiquitination | Chromatin remodeling, DNA repair [2] [7] | Regulates histone function (H2A, H2B, H2AX) and key DNA repair proteins like FANCD2 and PCNA [2] [7]. |
This ubiquitin "code" is not interpreted uniformly across the cellular landscape. Instead, its readout is profoundly shaped by contextual factors including cell type, subcellular compartmentalization, and the integration of other post-translational modifications (PTMs) such as phosphorylation, SUMOylation, and acetylation [100] [7]. This review will dissect the mechanisms and consequences of this context-dependency, with a focused analysis of its critical roles in maintaining genome stability through DNA repair and orchestrating controlled inflammatory responses through innate immunity.
Molecular Mechanisms of Context-Dependent Ubiquitin Signaling
Determinants of Specificity in the Ubiquitin System
The specificity of ubiquitin signaling is achieved through several interconnected mechanisms. First, the combinatorial pairing of specific E2 enzymes with specific E3 ligases dictates both substrate selection and the type of ubiquitin chain formed [96] [2]. For instance, the E2 enzyme UBE2N (UBC13) complexed with UBE2V1 (UEV1A) specifically synthesizes K63-linked chains and is recruited by the E3 ligase TRAF6 in immune signaling pathways [97]. Second, the spatial organization of ubiquitination machinery within the cell creates specialized signaling hubs. A prime example is the endoplasmic reticulum, where the E3 ligase complex AMFR-GP78/INSIG1 promotes K27-linked polyubiquitination of STING, a modification essential for its trafficking and the subsequent production of type I interferons [30].
Furthermore, the functional outcome of a ubiquitination event is critically determined by the ubiquitin-binding domains (UBDs) present in downstream effector proteins. Different UBDs exhibit distinct preferences for specific chain topologies. This allows K63-linked chains to typically recruit proteins involved in signal transduction and DNA repair, while K48-linked chains are recognized by proteasomal receptors [97]. Finally, the dynamic and reversible nature of this modification, controlled by the balanced actions of E3 ligases and DUBs, allows for rapid and transient cellular responses. For example, the deubiquitinase USP21 negatively regulates STING-mediated signaling by hydrolyzing K63-linked chains, thereby attenuating the innate immune response to prevent excessive inflammation [30].
Crosstalk with SUMOylation and Other PTMs
Ubiquitin signaling does not operate in isolation; it engages in extensive crosstalk with other PTM networks, most notably the SUMO (Small Ubiquitin-like Modifier) system. SUMOylation often intersects with ubiquitination in the regulation of DNA damage response (DDR). During DDR, proteins at damage sites are frequently modified by SUMO, which can then serve as a platform for the recruitment of ubiquitin E3 ligases that further modify targets with ubiquitin chains to promote repair or signaling [101]. This SUMO-ubiquitin crosstalk is a critical layer in the precise coordination of the DNA repair process.
The subcellular localization of SUMO pathway components themselves is subject to regulation, which in turn influences the ubiquitin network. Research in the testis has demonstrated that retinoic acid signaling can modulate the subcellular localization of SUMO-2/3, shifting it from the nucleus to the cytoplasm in Sertoli cells via the action of retinoic acid receptor alpha (RARA) [102]. This developmental and tissue-specific redistribution highlights a sophisticated mechanism for controlling the spatial activity of an entire PTM system, with direct implications for the regulation of ubiquitination events in male germ cell development.
Tissue and Subcellular Specificity in DNA Repair Pathways
The ubiquitin system's role in DNA repair is a paradigm of spatiotemporal regulation, where specific chain types and E3 ligases are recruited to damage sites with precise timing to coordinate the repair process.
Monoubiquitination in DNA Damage Signaling
Monoubiquitination plays a critical role in the earliest steps of the DNA damage response. Following the induction of double-strand breaks, the E2 enzyme UBE2T and E3 ligase RNF8 catalyze the monoubiquitination of histone H2AX. This modification acts as a beacon for the assembly of downstream DNA repair complexes, a process crucial for maintaining genome stability in tissues with high replicative potential, such as the liver [7]. Similarly, the monoubiquitination of FANCD2, a key effector in the Fanconi Anemia (FA) pathway, is essential for resolving DNA interstrand cross-links. This modification enables FANCD2 to associate with chromatin and collaborate with BRCA1 and BRCA2 proteins to facilitate repair, with defects leading to profound genomic instability and cancer predisposition [98].
Chain Topology and E3 Ligase Specificity in Repair Fidelity
The type of ubiquitin chain formed on a DNA repair protein can dictate the choice of repair pathway and its fidelity. The E3 ligase FBXW7, for example, can modify the DNA repair protein XRCC4 with K63-linked ubiquitin chains, which enhances the accuracy of non-homologous end joining (NHEJ) [7]. In contrast, K48-linked ubiquitination typically targets repair proteins for degradation to terminate the repair signal and prevent excessive DNA end resection. The contextual outcome of FBXW7 activity is further illustrated by its role in cancer therapy response. In p53-wild type colorectal tumors, FBXW7 promotes radioresistance by degrading p53, whereas in non-small cell lung cancer (NSCLC) with SOX9 overexpression, it enhances radiosensitivity by destabilizing SOX9 [7]. This duality underscores that the functional impact of a single E3 ligase is entirely dependent on the tissue and genetic background.
Table 2: Context-Dependent Roles of Selected E3 Ligases in DNA Repair and Disease
| E3 Ligase | Biological Process | Tissue/Cancer Specificity | Outcome |
|---|---|---|---|
| FBXW7 | Cell Cycle, DNA Repair | Colorectal Cancer (p53-wild type) [7] | Radioresistance (degrades p53) [7] |
| Non-Small Cell Lung Cancer (SOX9-high) [7] | Radiosensitivity (degrades SOX9) [7] | ||
| BRCA1 | Homologous Recombination | Breast, Ovarian [101] | Genomic Instability, Cancer Predisposition [101] |
| RNF168 | DNA Damage Signaling | All Tissues (RIDDLE Syndrome) [101] | Radiosensitivity, Immunodeficiency [101] |
| SKP2 | Cell Cycle (G1/S) | Lymphoma, Prostate Cancer [99] | Oncogenesis (degrades p27) [99] |
Spatiotemporal Regulation of Immune Responses
The innate immune system relies on the ubiquitin system to generate robust yet self-limiting responses to pathogenic threats. The context-dependent regulation of this system ensures effective host defense while preventing autoimmunity.
Subcellular Trafficking and Ubiquitination in the cGAS-STING Pathway
The cGAS-STING pathway, a central cytosolic DNA sensing mechanism, is exquisitely regulated by ubiquitination at multiple points and locations. The activity of the DNA sensor cGAS is modulated by various E3 ligases. For instance, TRIM56 induces monoubiquitination of cGAS at Lys335, enhancing its dimerization and capacity to produce the second messenger cGAMP, whereas RNF185 promotes K27-linked polyubiquitination to boost its enzymatic activity [30]. The adapter protein STING, upon activation, undertakes a carefully orchestrated journey from the endoplasmic reticulum (ER) to the Golgi apparatus. This trafficking is controlled by a series of ubiquitination events. E3 ligases like TRIM32 and TRIM56 promote K63-linked ubiquitination of STING, facilitating its aggregation in the Golgi and the recruitment of the downstream kinase TBK1 to induce type I interferons [30]. To prevent sustained and harmful immune activation, STING is subsequently ubiquitinated by E3 ligases such as RNF5 and TRIM30α with K48-linked chains, targeting it for proteasomal degradation [30]. Furthermore, for a more definitive termination of signaling, STING is also tagged with K63-linked chains (at Lys288) that direct it to lysosomes for degradation via an ESCRT-dependent microautophagy process [30].
Tissue and Pathway-Specific Ubiquitin Signaling in Innate Immunity
The molecular components of ubiquitin signaling can perform distinct functions in different immune pathways. The E2 enzyme complex UBE2N-UBE2V1 (UBC13-UEV1A), which synthesizes K63-linked chains, is a central node in multiple innate immune pathways. In the TLR-MyD88 pathway, it works with TRAF6 to create K63 chains that activate the TAK1 and IKK complexes, leading to NF-κB-driven pro-inflammatory cytokine production [97]. The same E2 complex is also involved in the RIG-I pathway for antiviral interferon production. This suggests that while the core machinery is shared, the specific cellular context, including the receptor complex and associated E3 ligases, dictates the precise signaling outcome. The critical role of these modifications is highlighted by the fact that pathogens have evolved specific effector proteins to deconjugate or mimic ubiquitin and interfere with these pathways, thereby evading host immunity [97].
Experimental Approaches for Deconstructing Context-Dependency
Methodologies for Mapping Ubiquitin Networks
Elucidating the context-dependent functions of ubiquitination requires a suite of sophisticated experimental techniques.
- Proteomic Profiling of Ubiquitination Sites: Advanced mass spectrometry (MS) methods, particularly quantitative ubiquitin remnant profiling, are used to identify and quantify ubiquitination sites on a proteome-wide scale under different conditions (e.g., stress vs. homeostasis). This approach can reveal tissue-specific ubiquitin targets and stress-induced changes in the "ubiquitinome" [100].
- Spatial Transcriptomics and Single-Cell Analysis: Techniques like single-cell RNA sequencing (scRNA-seq) can reveal profound intratumoral heterogeneity in the expression of ubiquitin ligases and DUBs, uncovering distinct therapy-resistant cellular subpopulations [7]. Creating a "SUMO Cell Atlas," as done in Arabidopsis root tissues, maps the spatial variation of SUMO components, a strategy directly applicable to mapping the ubiquitin system in mammalian tissues [100].
- Genetic Manipulation and Functional Assays: CRISPR-Cas9-based knockout or knockdown of specific E3 ligases or DUBs in specific cell types, followed by functional assays, is a direct method to determine their context-specific roles. For example, knockout of the Rara gene in testicular cells was used to demonstrate its necessity for the normal subcellular localization of SUMO-2/3 [102]. Organoid models, which better preserve tissue-specific architecture and cell-cell interactions, are increasingly valuable for such studies.
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Research Reagents for Ubiquitin Studies
| Reagent / Method | Function / Utility | Key Contextual Insight Provided |
|---|---|---|
| MLN4924 (Pevonedistat) | NEDD8-Activating Enzyme (NAE) Inhibitor [96] | Blocks cullin-RING ligase (CRL) activity; used to probe CRL-specific substrate turnover in different tissues [96]. |
| NSC697923 | Inhibitor of UBE2N (E2 enzyme for K63 chains) [96] | Dissects role of K63-linked ubiquitination in NF-κB signaling vs. DNA repair in various cell types [96]. |
| Tandem Ubiquitin Binding Entities (TUBEs) | Affinity reagents to purify polyubiquitinated proteins [2] | Protects polyubiquitin chains from DUBs during lysis; enables analysis of tissue-specific ubiquitinated proteomes. |
| PROTACs | Proteolysis-Targeting Chimeras [7] | Induces targeted protein degradation in a tissue-specific manner; demonstrates therapeutic potential of harnessing the UPS. |
| CRISPR Screening | Genome-wide functional genomics [7] | Identifies context-specific essential E3s/DUBs, e.g., TRIM21 in radioresistance [7]. |
Protocol: Assessing Ligase Function in a Tissue-Specific Context
Objective: To determine the context-dependent role of a specific E3 ubiquitin ligase (e.g., FBXW7) in the DNA damage response of two different cancer cell lines.
Workflow Diagram:
Methodology Details:
- Cell Model Selection: Utilize two isogenic cell lines representing different tissue types or genetic backgrounds (e.g., a p53-wild type colorectal cancer line and a p53-null NSCLC line).
- Genetic Manipulation: Generate E3 ligase (FBXW7) knockout (KO) clones in both cell lines using CRISPR-Cas9 technology. Include wild-type (WT) controls.
- Treatment: Subject all cell lines (KO and WT) to a relevant DNA-damaging stimulus, such as ionizing radiation (e.g., 2-8 Gy).
- Functional & Molecular Readouts:
- Clonogenic Survival Assay: Measure long-term cell survival post-irradiation to determine radiosensitivity.
- DNA Damage Foci Staining: Fix cells at various time points (e.g., 1, 6, 24h) post-irradiation and perform immunofluorescence for markers like γH2AX and 53BP1 to quantify the kinetics of DNA damage repair.
- Substrate Analysis: Harvest protein lysates and perform Western blotting to monitor the stabilization levels of known E3 ligase substrates (e.g., p53, SOX9) in KO vs. WT cells.
- Interaction Studies: Conduct co-immunoprecipitation (Co-IP) assays to confirm the physical interaction between the E3 ligase and its substrate in the different cell lines.
- Data Integration: Correlate the survival and DNA repair efficiency with substrate stabilization and interaction data. The expected outcome is that the functional consequence of E3 ligase loss will differ between the two cell lines, directly demonstrating context-dependency.
Therapeutic Implications and Future Directions
The context-dependent nature of ubiquitin signaling presents both a challenge and an opportunity for drug development. The success of proteasome inhibitors like Bortezomib in hematological malignancies proves the UPS is a valid therapeutic target [2]. However, the broad activity of these agents leads to significant off-target effects. The current frontier lies in developing more precise agents that target specific nodes within the ubiquitin network, such as E3 ligases, DUBs, or specific protein-protein interactions.
The emergence of PROTACs (Proteolysis-Targeting Chimeras) represents a paradigm shift, as these molecules are themselves context-dependent. A PROTAC that targets a specific oncoprotein for degradation will only be effective in cells where both the E3 ligase and the oncoprotein are expressed [7]. This inherent specificity is being leveraged in clinical development. For instance, EGFR-directed PROTACs are being designed to selectively degrade oncoproteins in EGFR-dependent tumors like lung and head/neck squamous cell carcinomas, thereby suppressing DNA repair and sensitizing these specific cancers to radiotherapy [7].
Future efforts will focus on exploiting the tissue-specific expression of E3 ligases and DUBs to develop highly selective therapeutics. Furthermore, biomarker-driven strategies will be essential to identify patient populations most likely to benefit from ubiquitin-targeting agents. For example, tumors with overexpression of the E3 ligase SKP2 or loss of the tumor suppressor FBW7 might be uniquely vulnerable to therapies that disrupt their specific ubiquitin signaling axis [99] [2]. As our understanding of the ubiquitin network's complexity deepens, so too will our ability to manipulate it with precision, ushering in a new era of targeted therapies for cancer, autoimmune diseases, and beyond.
The ubiquitin-proteasome system (UPS) represents a master regulatory network controlling cellular protein homeostasis, with fundamental roles in critical processes such as the DNA damage response and immune signaling pathways [99] [103]. This system orchestrates the targeted degradation of specific proteins through a sophisticated enzymatic cascade involving E1 activating enzymes, E2 conjugating enzymes, and E3 ligases, ultimately directing substrates to the 26S proteasome for destruction [104] [99]. The profound influence of the UPS on cell cycle regulation, DNA repair mechanisms, and immune activation has positioned it as an attractive therapeutic target for various malignancies and autoimmune disorders [104] [105]. However, the very nature of its fundamental biological roles means that targeting core UPS components frequently leads to on-target toxicity, where the therapeutic inhibition of a pathway essential for normal cellular function causes adverse effects in healthy tissues [104] [99]. This challenge is particularly acute for ubiquitin system drugs, as the constitutive proteasome inhibitor bortezomib demonstrates significant toxicity profiles despite clinical efficacy [105]. Consequently, developing strategies to enhance the therapeutic window—the balance between efficacy and toxicity—has become a paramount objective in the continued development of ubiquitin-targeted therapeutics, especially within the context of their application in DNA repair and immune pathway modulation.
Molecular Foundations of On-Target Toxicity
Essential Roles of UPS in Cellular Homeostasis
The ubiquitin-proteasome system maintains a delicate balance in protein quality control, with its disruption having profound implications for cellular viability. The system's core components are indispensable for normal physiological functions, particularly in rapidly dividing and specialized cells. The 26S proteasome complex, comprising a 20S core particle and 19S regulatory particles, degrades approximately 80% of intracellular proteins, including those that are oxidized, damaged, or misfolded [104]. This degradation is not merely a waste disposal service but a precise regulatory mechanism for controlling the abundance of key regulatory proteins. For instance, the anaphase-promoting complex/cyclosome (APC/C) and the Skp1-Cullin-F-box protein (SCF) complex—two major E3 ligase complexes—orchestrate irreversible cell cycle transitions by targeting cyclins and cyclin-dependent kinase inhibitors for degradation [99]. Similarly, the UPS plays a pivotal role in the DNA damage response (DDR) by modulating the stability of proteins involved in DNA repair pathways, ensuring genomic integrity [99] [103]. In immune cells, ubiquitination regulates critical signaling pathways, including NF-κB activation, which controls the expression of cytokines and chemokines essential for proper immune function [105]. The non-degradative functions of ubiquitination, particularly through K63-linked and linear ubiquitin chains, further expand the UPS's regulatory scope, encompassing processes such as kinase activation, epigenetic regulation, and protein translocation [104] [105].
Mechanisms of Toxicity in UPS-Targeted Therapies
On-target toxicity emerges when therapeutic inhibition of UPS components disrupts these essential homeostatic mechanisms. The mechanisms underlying this toxicity are multifaceted and often tissue-specific, reflecting the diverse cellular processes regulated by the UPS.
Cell Cycle Disruption: Inhibition of E3 ligases like SCF complexes or APC/C prevents the timely degradation of cell cycle regulators, leading to aberrant cell cycle progression and arrested proliferation in healthy cells [99]. For example, compromised APC/C function causes accumulation of mitotic cyclins, disrupting mitotic exit and potentially triggering apoptosis in non-target tissues.
Impaired DNA Damage Response: The UPS is crucial for proper DNA damage repair through the regulation of repair protein stability and activity [99] [103]. Therapeutic interference with this function can sensitize cells to endogenous DNA damage, resulting in genomic instability and cell death, particularly in tissues with high replicative potential.
Immune Dysregulation: Given the central role of ubiquitination in immune cell signaling and cytokine production, UPS inhibition often leads to immune-related toxicities [105]. This includes both immunosuppression, increasing infection risk, and paradoxical inflammation due to disrupted negative feedback mechanisms in immune signaling pathways.
Proteotoxic Stress: By impairing the clearance of damaged or misfolded proteins, UPS inhibition can lead to proteotoxic stress, resulting in endoplasmic reticulum stress and unfolded protein response activation, particularly affecting secretory cells [104] [103].
Table 1: Common On-Target Toxicities in Ubiquitin System-Targeted Therapies
| Therapeutic Class | Primary Target | Common Toxicities | Underlying Mechanism |
|---|---|---|---|
| Proteasome Inhibitors | 20S/26S Proteasome | Peripheral neuropathy, cytopenias, gastrointestinal toxicity | Impaired protein clearance, disrupted cell cycle, ER stress |
| E1 Enzyme Inhibitors | UBA1 (E1) | Hepatotoxicity, dermatological toxicity | Global disruption of ubiquitination, affecting multiple pathways |
| E2 Enzyme Inhibitors | Specific E2 enzymes (e.g., UBE2C) | Tissue-specific based on E2 expression | Accumulation of specific E2 substrates |
| E3 Ligase Inhibitors | Specific E3 ligases (e.g., MDM2) | Varies by E3 specificity | Dysregulation of E3-specific substrates and pathways |
Strategic Approaches to Enhance Therapeutic Windows
Molecular Specificity: From Broad to Targeted Inhibition
The evolution from broad-spectrum proteasome inhibitors to targeted E3 ligase modulators represents a paradigm shift in addressing on-target toxicity. First-generation proteasome inhibitors such as bortezomib and MG132 demonstrate broad anti-neoplastic activity but produce significant toxicity due to their comprehensive disruption of protein degradation [104] [105]. The strategic refinement towards targeting specific E3 ligases capitalizes on the immense diversity of the approximately 600 human E3 ligases, many of which demonstrate tissue-specific or pathway-restricted functions [104] [103]. For instance, inhibitors targeting the E3 ligase MDM2, which regulates p53 stability, enable selective activation of p53-mediated apoptosis in cancer cells with wild-type p53, while theoretically sparing normal tissues that may better tolerate transient p53 activation [104]. Similarly, modulating E3 ligases like FBW7—a tumor suppressor that targets oncoproteins such as c-Myc, Notch, and cyclin E for degradation—offers potential for tissue-selective effects [104] [99]. The clinical development of E6AP inhibitors for HPV-related malignancies further illustrates the principle of context-dependent targeting, where the therapeutic effect relies on viral oncoprotein expression not present in normal cells [104].
Exploiting Synthetic Lethality in DNA Repair Pathways
Synthetic lethality represents a powerful approach to enhance therapeutic windows by leveraging cancer-specific vulnerabilities. This strategy is particularly relevant in the context of ubiquitin-mediated DNA repair pathways, where cancer cells with pre-existing DNA repair defects become uniquely dependent on specific UPS components [99] [103]. For example, tumors with BRCA mutations deficient in homologous recombination repair become exquisitely sensitive to inhibition of ubiquitin-mediated alternative DNA repair pathways. The development of inhibitors targeting specific E3 ligases involved in DNA damage response, such as those regulating Fanconi anemia pathway components or translesion synthesis polymerases, can selectively target repair-deficient cancers while minimizing toxicity to normal cells with intact DNA repair mechanisms [99]. The differential dependency between normal and malignant cells on specific UPS components for maintaining genomic stability provides a foundational principle for designing therapies with improved therapeutic indices.
Tissue-Selective Delivery and Activation
Advanced delivery systems offer a pharmacological approach to enhance therapeutic specificity by minimizing exposure to sensitive tissues. Antibody-drug conjugates (ADCs) targeting cell surface receptors highly expressed on cancer cells can deliver potent E3 ligase modulators or proteasome inhibitors specifically to malignant cells [106]. Similarly, nanoparticle-based delivery systems can be engineered to exploit the enhanced permeability and retention effect in tumor tissues or to respond to tumor-specific microenvironments (e.g., pH, protease activity) for selective drug release [106]. Prodrug strategies that require tumor-specific enzymes for activation represent another promising approach to limit active drug concentrations in normal tissues, thereby reducing on-target toxicities associated with ubiquitin pathway inhibition.
Table 2: Strategies for Enhancing Therapeutic Windows of Ubiquitin-Targeted Therapies
| Strategy | Mechanism | Examples | Potential Limitations |
|---|---|---|---|
| Isoform-Specific Targeting | Targeting tissue-specific enzyme variants | Selective E2 inhibitors (e.g., UBE2C) | Potential compensatory mechanisms |
| Synthetic Lethality | Exploiting cancer-specific genetic vulnerabilities | Targeting UPS in DNA repair-deficient cancers | Requires biomarker identification |
| Temporal Inhibition | Transient versus sustained target engagement | Intermittent dosing schedules | May reduce efficacy |
| Localized Delivery | Direct administration to disease site | Intra-tumoral injections, inhaled formulations | Limited to accessible diseases |
| Combination Therapy | Lower doses of multiple agents | Proteasome inhibitor + selective E3 inhibitor | Potential for novel toxicity interactions |
Experimental Framework for Evaluating Therapeutic Windows
In Vitro Assessment of Therapeutic Index
A robust experimental framework for evaluating potential therapeutic windows begins with comprehensive in vitro modeling. The following protocol outlines a standardized approach for assessing the differential sensitivity of malignant versus normal cells to ubiquitin system-targeted agents:
Protocol 1: Differential Cytotoxicity and Pathway Modulation Assay
Cell Line Selection: Establish co-cultures of relevant cancer cell lines (e.g., multiple myeloma lines for proteasome inhibitors) and matched non-malignant primary cells (e.g., peripheral blood mononuclear cells, mesenchymal stem cells).
Dose-Response Profiling: Treat co-cultures with serial dilutions of the ubiquitin-targeting compound (e.g., E1/E2/E3 inhibitor, proteasome inhibitor) across a 5-log concentration range. Include positive controls (e.g., bortezomib for proteasome inhibitors) and vehicle controls.
Viability Assessment: At 24, 48, and 72 hours, quantify viability using multiplexed assays:
- ATP content (cell viability)
- Caspase 3/7 activation (apoptosis)
- Proteasome activity (specific target engagement)
- Cell cycle distribution via flow cytometry
Pathway-Specific Readouts: Evaluate compound effects on specific pathways:
- For DNA repair-targeting agents: γH2AX foci formation, RAD51 recruitment, comet assay
- For immune pathway modulators: NF-κB nuclear translocation, cytokine secretion profiles, immune cell activation markers
Therapeutic Index Calculation: Determine IC50 values for both malignant and normal cells across all endpoints. Calculate selectivity indices (SI = IC50 normal/IC50 cancer) for both viability and pathway-specific effects.
This multidimensional assessment provides critical early data on potential therapeutic windows before advancing to in vivo models.
In Vivo Toxicity and Efficacy Modeling
Translational assessment of therapeutic windows requires sophisticated in vivo models that capture both efficacy and toxicity endpoints:
Protocol 2: Integrated Efficacy-Toxicity Murine Model
Model Establishment: Implement both xenograft (human tumor cells in immunocompromised mice) and syngeneic (murine tumor cells in immunocompetent mice) models to assess both direct anti-tumor effects and immune-related toxicities.
Dosing Optimization: Test multiple dosing regimens (continuous, intermittent, pulsatile) to identify schedules that maintain efficacy while minimizing toxicity.
Comprehensive Toxicity Monitoring:
- Hematological: Complete blood counts twice weekly
- Neurological: Rotarod performance, nerve conduction velocity
- Hepatic/Renal: Serum chemistry panels weekly
- Gastrointestinal: Histopathological scoring of intestinal crypts
- Immune: Flow cytometry of lymphoid organs, serum cytokine levels
Efficacy Endpoints: Tumor volume measurements, survival analysis, bioluminescent imaging of tumor burden, and post-mortem histopathological assessment of tumor cell death.
Biomarker Correlation: Collect serial blood samples for pharmacodynamic biomarker assessment (e.g., specific ubiquitinated protein accumulation) to correlate target engagement with both efficacy and toxicity.
The following diagram illustrates the key regulatory nodes in ubiquitin-mediated DNA repair and immune pathways where targeted therapeutic intervention is being explored, highlighting points where specificity can be achieved to minimize on-target toxicity:
Diagram 1: Strategic targeting of specific E3 ligases within DNA repair and immune signaling pathways to achieve therapeutic specificity and reduce on-target toxicity. Red dashed lines indicate inhibitory interventions, while green represents activating interventions.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Investigating Ubiquitin System Therapeutics
| Reagent Category | Specific Examples | Research Application | Toxicity Assessment Utility |
|---|---|---|---|
| E1 Inhibitors | PYR-41, TAK-243 | Pan-UPS inhibition controls, understanding global ubiquitination disruption | Modeling dose-limiting toxicity from broad UPS inhibition |
| E2 Inhibitors | NSC697923 (UBE2N/UBC13) | Specific pathway inhibition (NF-κB, DNA repair) | Assessing tissue-specific toxicity from selective pathway disruption |
| E3 Inhibitors | JNJ-165 (MDM2), MLN4924 (NAE) | Targeted protein stabilization/degradation | Evaluating context-dependent toxicity in normal vs. malignant cells |
| Proteasome Inhibitors | Bortezomib, MG132, Carfilzomib | Baseline proteasome inhibition activity, comparison standards | Establishing toxicity benchmarks for new agents |
| DUB Inhibitors | b-AP15, PR-619 | Understanding ubiquitin chain editing and recycling | Assessing toxicity from impaired substrate editing |
| Activity Probes | Ubiquitin-VS, HA-Ub-VME | Target engagement verification, mechanistic studies | Correlating target occupancy with toxicity endpoints |
| Specialized Assays | Ubiquitin chain linkage-specific antibodies, DiUb probes | Pathway-specific ubiquitination assessment | Differentiating on-target vs. off-target effects |
The strategic targeting of the ubiquitin-proteasome system continues to hold tremendous promise for therapeutic intervention in cancer, autoimmune disorders, and other diseases. However, realizing this potential requires sophisticated approaches to manage the inherent on-target toxicities that arise from disrupting fundamental cellular processes. The field is rapidly evolving beyond broad inhibition of core proteasome function toward precision targeting of specific E3 ligases operating in defined pathological contexts, particularly those involved in DNA repair and immune signaling pathways [104] [99] [105]. Future progress will likely come from several complementary strategies: first, the continued identification and validation of context-specific E3 ligases that operate preferentially in disease states; second, the development of bifunctional molecules such as PROTACs that leverage ubiquitin machinery for targeted protein degradation with enhanced specificity; and third, advanced delivery systems that spatially and temporally restrict drug activity to pathological tissues. As our understanding of the intricate biology of ubiquitin signaling in DNA repair and immune response deepens, so too will our ability to design interventions that selectively modulate these pathways in disease contexts while preserving essential functions in healthy tissues. The integration of robust predictive biomarkers, sophisticated preclinical models, and clever chemical biology approaches will be essential to successfully navigate the challenge of on-target toxicity and fully exploit the therapeutic potential of the ubiquitin-proteasome system.
The following diagram outlines an integrated experimental workflow for evaluating both efficacy and toxicity of ubiquitin system-targeted therapies during preclinical development:
Diagram 2: Integrated preclinical workflow for evaluating the therapeutic window of ubiquitin system-targeted therapies, incorporating parallel assessment of efficacy and toxicity endpoints.
Proteolysis Targeting Chimeras (PROTACs) represent a revolutionary therapeutic paradigm in oncology, leveraging the ubiquitin-proteasome system (UPS) to degrade oncogenic proteins. However, tumor cells employ sophisticated resistance mechanisms, including adaptive changes in the ubiquitin machinery and upregulation of deubiquitinating enzymes (DUBs). This whitepaper examines how tumors evade PROTAC-mediated degradation and DUB inhibition, framing these dynamics within the critical roles of ubiquitination in DNA repair and immune signaling. Understanding these adaptive responses is paramount for developing next-generation targeted protein degradation therapies that overcome treatment resistance.
The Ubiquitin-Proteasome System and PROTAC Mechanism
The ubiquitin-proteasome system (UPS) is the primary pathway for regulated intracellular protein degradation in eukaryotic cells, governing essential processes including cell cycle progression, DNA damage response, and immune signaling [99]. The ubiquitination process involves a sequential enzymatic cascade: a ubiquitin-activating enzyme (E1) activates ubiquitin in an ATP-dependent manner, a ubiquitin-conjugating enzyme (E2) accepts the activated ubiquitin, and a ubiquitin ligase (E3) facilitates the transfer of ubiquitin to specific substrate proteins [107] [99]. Polyubiquitinated substrates are subsequently recognized and degraded by the 26S proteasome [99].
PROTACs (Proteolysis-Targeting Chimeras) are heterobifunctional small molecules that hijack this natural degradation machinery [107]. A PROTAC molecule consists of three key components: a ligand that binds to the protein of interest (POI), a ligand that recruits an E3 ubiquitin ligase, and a linker connecting these two moieties [107] [108]. The PROTAC facilitates the formation of a ternary complex (POI-PROTAC-E3 ligase), bringing the E3 ligase into proximity with the target protein and enabling its polyubiquitination and subsequent proteasomal degradation [107]. Unlike traditional inhibitors that rely on occupancy-driven pharmacology, PROTACs operate via an event-driven catalytic mechanism, degrading multiple target protein molecules per PROTAC molecule [109] [110].
Deubiquitinating Enzymes (DUBs): Regulators of Ubiquitin Signaling
Deubiquitinating enzymes (DUBs) constitute a family of approximately 100 human proteases that counterbalance ubiquitin signaling by removing ubiquitin modifications from substrate proteins [91] [111]. DUBs perform several critical functions within the ubiquitin system: (1) processing ubiquitin precursors to generate mature ubiquitin; (2) removing ubiquitin chains from modified substrates to reverse their fate; and (3) disassembling unanchored polyubiquitin chains to recycle ubiquitin [91] [111]. The human DUB family includes cysteine proteases (ubiquitin-specific proteases/USPs, ubiquitin C-terminal hydrolases/UCHs, Machado-Josephin domain proteases/MJDs, and ovarian tumor proteases/OTUs) and metalloproteases (JAMM/MPN+ domain-containing proteins) [111]. Through their specialized activities, DUBs regulate virtually all ubiquitin-dependent processes, including cell cycle control, DNA damage response, and immune signaling pathways [91].
Resistance to PROTAC Therapy: Molecular Mechanisms
Alterations in the Ubiquitin-Proteasome System Components
Tumor cells develop resistance to PROTACs through multiple adaptations in UPS components essential for PROTAC activity:
- E3 Ligase Downregulation: Reduced expression of the E3 ligase recruited by the PROTAC (e.g., CRBN, VHL) prevents formation of the productive ternary complex, diminishing target degradation efficacy. For instance, tumors may downregulate CRBN expression following treatment with CRBN-recruiting PROTACs [109].
- Proteasome Capacity Limitations: Insufficient proteasome capacity creates a bottleneck where ubiquitinated proteins accumulate without degradation, despite successful ubiquitination by the PROTAC mechanism [110].
- Compensatory Ubiquitin Pathway Activation: Tumors may upregulate alternative E3 ligases or components of the ubiquitination cascade that are not engaged by the therapeutic PROTAC, creating bypass signaling pathways [99].
Table 1: Core Components of the Ubiquitin-Proteasome System and Their Roles in PROTAC Resistance
| UPS Component | Normal Function | Resistance Mechanism | Functional Consequence |
|---|---|---|---|
| E3 Ligases (CRBN, VHL) | Substrate recognition for ubiquitination | Downregulation or mutation | Impaired ternary complex formation |
| 26S Proteasome | Degradation of ubiquitinated proteins | Reduced capacity or expression | Accumulation of ubiquitinated targets |
| E2 Enzymes (~40 types) | Ubiquitin conjugation | Altered expression profiles | Changed ubiquitin chain topology |
| Free Ubiquitin Pool | Ubiquitin supply for conjugation | Depletion through excessive demand | Limited ubiquitin availability |
Target Protein Mutations and Adaptations
PROTAC resistance frequently emerges through specific alterations in the target protein itself:
- Point Mutations in Binding Sites: Single amino acid changes in the target protein's PROTAC-binding domain can reduce binding affinity while preserving the protein's oncogenic function. This has been observed in targets such as Bruton's tyrosine kinase (BTK) and androgen receptor (AR) [109].
- Protein Overexpression: Cancer cells may amplify expression of the target protein, exceeding the degradation capacity of the PROTAC at therapeutic concentrations [109].
- Altered Subcellular Localization: Redirecting the target protein to cellular compartments with reduced PROTAC access or decreased E3 ligase activity represents another resistance strategy [110].
Upregulation of Deubiquitinating Enzymes
The increased expression or activity of specific DUBs constitutes a major resistance mechanism to PROTAC therapy:
- Direct Deubiquitination of Targets: Certain DUBs can remove ubiquitin chains from PROTAC-targeted proteins before they reach the proteasome, effectively rescuing them from degradation [91] [111].
- Stabilization of Oncogenic Proteins: DUBs such as USP28 stabilize key oncoproteins like c-Myc, Notch1, and c-Jun, promoting tumor survival despite PROTAC treatment [111].
- Compensatory Stabilization Pathways: Tumors may activate DUB-mediated stabilization of parallel signaling pathways that compensate for the degraded target [91].
Table 2: DUBs Implicated in Cancer Therapy Resistance
| DUB | Class | Cancer Relevance | Role in Resistance |
|---|---|---|---|
| USP28 | Cysteine protease (USP) | Overexpressed in colon/lung cancer | Stabilizes c-Myc, Notch1; confers chemo-resistance |
| UCH-L1 | Cysteine protease (UCH) | Elevated in various malignancies | Promotes tumor survival; unclear direct PROTAC role |
| A20 | Cysteine protease (OTU) | Regulates NF-κB signaling | Modulates inflammatory response to therapy |
| USP7 | Cysteine protease (USP) | Stabilizes p53 and other regulators | Potential stabilization of alternative survival pathways |
DUB Inhibition Resistance Mechanisms
Compensatory DUB Expression and Activity
When specific DUBs are inhibited, tumor cells employ compensatory mechanisms to maintain protein homeostasis:
- Upregulation of Alternative DUBs: Inhibition of one DUB family member often leads to increased expression of other DUBs with similar substrate specificity, creating redundant deubiquitination capacity [91].
- Activation of Backup Signaling Pathways: Tumors activate alternative survival pathways that do not depend on the inhibited DUB's regulatory functions [111].
Alterations in DUB Substrate Specificity
Cancer cells can evolve mutations that change how DUBs recognize their substrates:
- Substrate Recognition Site Mutations: Genetic alterations in DUB substrate-binding domains can modify target specificity, reducing efficacy of DUB inhibitors designed for specific enzyme-substrate interactions [91].
- Post-Translational Modifications: Phosphorylation, acetylation, or ubiquitination of DUBs can alter their activity, localization, and substrate preference [91].
Experimental Approaches for Studying Resistance Mechanisms
Protocol for Monitoring Ternary Complex Formation
Purpose: To assess the impact of E3 ligase downregulation or target protein mutations on PROTAC efficacy.
Methodology:
- Cell Line Selection: Utilize PROTAC-sensitive and -resistant cancer cell lines (e.g., prostate cancer lines for AR degraders, lymphoma lines for BTK degraders).
- PROTAC Treatment: Treat cells with varying PROTAC concentrations (typically 0.1 nM - 10 µM) for different durations (2-24 hours).
- Co-Immunoprecipitation: Harvest cells and lyse with mild detergent. Immunoprecipitate the E3 ligase component (e.g., VHL or CRBN) using specific antibodies.
- Western Blot Analysis: Probe immunoprecipitates for target protein presence to detect ternary complex formation.
- Quantitative Analysis: Compare ternary complex stability and abundance between sensitive and resistant cells using densitometry.
Applications: This protocol identifies defects in the initial steps of the PROTAC mechanism before ubiquitination occurs [107] [108].
Protocol for DUB Activity Profiling in Resistant Cells
Purpose: To identify DUBs upregulated in PROTAC-resistant tumors and assess the efficacy of DUB inhibitors.
Methodology:
- Activity-Based Protein Profiling (ABPP):
- Prepare cell lysates from resistant and sensitive cells.
- Incubate with HA-Ub-VME or Ub-PA, biotinylated activity-based probes that covalently label active DUBs.
- Capture labeled DUBs using streptavidin beads and identify via mass spectrometry.
- DUB Inhibitor Screening:
- Treat resistant cells with panel of DUB inhibitors (e.g., PR-619 broad-spectrum inhibitor or selective compounds like P5091 for USP7).
- Assess resensitization to PROTAC treatment via cell viability assays and target protein degradation immunoblots.
- Functional Validation:
- Knock down candidate DUBs using siRNA or CRISPRi in resistant cells.
- Measure subsequent changes in PROTAC sensitivity and target protein stability.
Applications: This approach identifies which DUBs contribute most significantly to resistance and tests therapeutic strategies to overcome it [91] [111].
Research Reagent Solutions
Table 3: Essential Research Tools for Investigating PROTAC and DUB Resistance
| Reagent/Category | Specific Examples | Research Application | Key Functions |
|---|---|---|---|
| PROTAC Molecules | ARV-471 (ER degrader), ARV-110 (AR degrader) | Resistance mechanism studies | Induce targeted protein degradation; establish resistant models |
| E3 Ligase Ligands | Thalidomide (CRBN), VH032 (VHL) | Ternary complex studies | Recruit specific E3 ligases; mechanistic studies |
| DUB Inhibitors | PR-619 (pan-DUB), P5091 (USP7-specific) | DUB inhibition studies | Block deubiquitination; test synthetic lethal combinations |
| Activity-Based Probes | HA-Ub-VME, Ub-PA | DUB activity profiling | Label active DUBs in complex proteomes |
| Proteasome Inhibitors | Bortezomib, MG-132 | UPS functionality assays | Block proteasome activity; measure ubiquitinated protein accumulation |
| Ubiquitin Variants | K48- and K63-linked ubiquitin chains | Ubiquitination mechanism studies | Specific chain linkage analysis; DUB substrate specificity |
Visualization of Key Pathways and Mechanisms
Diagram 1: PROTAC mechanism and tumor resistance pathways. The diagram illustrates how PROTACs mediate target protein degradation through ternary complex formation and the key resistance mechanisms tumors employ to evade this process.
Diagram 2: DUB regulation in DNA repair and immune signaling. The diagram shows how DUBs fine-tune critical cellular pathways, with imbalances contributing to therapy resistance.
The evolving understanding of PROTAC and DUB inhibition resistance mechanisms reveals the remarkable adaptability of tumor cells. Overcoming these challenges requires multifaceted strategies: developing PROTACs recruiting alternative E3 ligases, creating DUB inhibitors with enhanced specificity, and rational combination therapies that simultaneously target multiple vulnerability nodes. The intricate connections between ubiquitination, DNA repair, and immune signaling pathways suggest that successful approaches will need to consider the system-wide impact of protein degradation therapies. As the field advances, monitoring for these resistance mechanisms in clinical settings and developing diagnostic tools for early detection of adaptive responses will be crucial for extending the therapeutic benefits of targeted protein degradation in oncology.
Proteolysis-Targeting Chimeras (PROTACs) represent a paradigm shift in therapeutic intervention, moving beyond traditional occupancy-based inhibition to event-driven protein degradation [112] [113]. These heterobifunctional molecules function as molecular bridges, connecting a target Protein of Interest (POI) to an E3 ubiquitin ligase. This induced proximity results in the polyubiquitination of the POI, marking it for recognition and destruction by the 26S proteasome [112] [114]. This process hijacks the body's natural Ubiquitin-Proteasome System (UPS), a critical regulatory network that governs protein stability and function. The UPS is particularly vital in maintaining genomic integrity and immune surveillance, as it tightly controls key players in DNA repair pathways and immune response signaling [26]. The catalytic nature of PROTACs allows for sub-stoichiometric activity, meaning a single degrader molecule can facilitate the destruction of multiple POI copies, offering significant advantages over conventional small-molecule inhibitors [113]. The general structure and mechanism of PROTACs are illustrated below.
The Central Role of Linker Chemistry
The linker is far more than a passive tether connecting the warhead and E3 ligase ligand; it is a critical determinant of PROTAC efficacy. Its composition, length, and rigidity directly influence the molecule's physicochemical properties, its ability to form a productive ternary complex, and ultimately, its degradation efficiency [112] [115] [116].
Linker Length and Composition
The optimal linker length is a delicate balance; it must be long enough to connect the two protein-binding domains without inducing steric clashes, yet short enough to pre-organize the complex and maximize binding entropy [116]. Early PROTAC designs predominantly utilized flexible alkyl or polyethylene glycol (PEG) chains due to their synthetic tractability [112]. However, the field is rapidly evolving towards more sophisticated, functionally enriched linkers. Shortening a flexible PEG5 linker to a PEG0 unit in H-PGDS-targeting PROTACs dramatically improved degradation potency, reducing the DC50 from a micromolar to a picomolar range [115]. Conversely, for certain targets like BTK, increasing linker length was found to alleviate steric clashes and improve degradation [117]. This highlights that optimal linker length is highly context-dependent, influenced by the specific warhead and E3 ligase pair.
Linker Rigidity and Conformational Control
Introducing rigidity into the linker is a powerful strategy to reduce the entropic penalty associated with ternary complex formation. Conformationally constrained linkers can pre-organize the PROTAC in a bioactive orientation, enhancing the stability and specificity of the POI-PROTAC-E3 ligase complex [115]. A comparative study on H-PGDS degraders systematically evaluated linkers with varying rigidity, from flexible methylene chains to rigid spirocyclic structures. While all analogues showed similar binding affinities, their cellular behavior differed significantly. The most rigid derivative, PROTAC-3, exhibited markedly higher intracellular accumulation, a key factor for in vivo efficacy [115]. However, it also formed the least stable ternary complex, revealing a critical trade-off between cell permeability and complex stability that must be optimized for each target [115]. The following table summarizes key findings from linker optimization studies.
Table 1: Impact of Linker Design on PROTAC Efficacy
| Target Protein | Linker Type | Key Finding | Experimental Outcome | Citation |
|---|---|---|---|---|
| H-PGDS | Flexible PEG5 vs. PEG0 | Shorter linker drastically increased potency. | DC50 improved from µM to 17.3 pM. | [115] |
| H-PGDS | Piperazine vs. Spirocycles | Rigid linkers improved cellular accumulation but could reduce complex stability. | PROTAC-3 (rigid) had higher intracellular levels but less stable complex. | [115] |
| BTK | Varying Length (RC-1 series) | Optimal length is context-specific; cooperative complex favored shorter linker. | RC-1 (shorter) was more efficacious than longer-linker analogs. | [117] |
| p38α / p38δ | "Amide" vs. "Phenyl" Series | Linker attachment point dictates E3 orientation and substrate specificity. | Achieved isoform-selective degradation (SJFα for p38α, SJFδ for p38δ). | [118] |
| Androgen Receptor | Rigid Phenyl vs. Flexible PEG | Excessive rigidity led to loss of activity. | Flexible PEG chains retained degradation potency. | [115] |
Strategic Warhead Selection and Engineering
The "warhead" is the moiety that binds the POI, and its selection and engineering are fundamental to achieving potent and selective degradation.
Leveraging Selectivity through Ternary Complex Formation
A profound advantage of the PROTAC modality is its ability to transform a non-selective or promiscuous warhead into a selective degrader. This phenomenon is governed by the unique ternary complex, where protein-protein interactions (PPIs) between the POI and the recruited E3 ligase impart an additional layer of specificity [119] [118]. A landmark study using foretinib—a promiscuous kinase inhibitor binding to over 50 kinases—demonstrated that when converted into PROTACs, only a subset of the bound kinases was degraded [119]. The selectivity was dictated by the stability of the ternary complex, which in turn was influenced by the linker design and E3 ligase orientation [118]. For instance, the BET degrader MZ1, which uses the pan-selective inhibitor JQ1 as a warhead, achieves selectivity for BRD4 over BRD2 and BRD3 due to favorable ternary complex interactions with the VHL E3 ligase [112] [116].
Covalent Warheads for Enhanced Engagement
While most PROTACs rely on reversible, non-covalent (RNC) warheads, incorporating covalent chemistry offers a promising strategy to overcome limitations in binding affinity and cellular permeability. A comparative study of BTK degraders explored reversible non-covalent (RNC), irreversible covalent (IRC), and reversible covalent (RC) warheads [117]. The irreversible covalent PROTAC (IRC-1) performed poorly, likely because it is consumed upon binding, negating the catalytic advantage of PROTACs. In contrast, the reversible covalent PROTAC (RC-1) exhibited superior properties. The RC warhead significantly enhanced intracellular accumulation and target engagement, yielding one of the most potent BTK degraders reported (DC50 = 6.6 nM) and functioning as a dual inhibitor-degrader [117]. This reversible covalent approach provides a generalizable path to boost the cellular efficacy of PROTACs.
Experimental Protocols for PROTAC Evaluation
Rigorous in vitro and cellular assays are essential for evaluating PROTAC efficacy and optimizing design. Below is a detailed protocol for a standard degradation assay.
Protocol: Cellular Protein Degradation Assay by Western Blot
Purpose: To measure the concentration-dependent and time-dependent degradation of the target protein induced by a PROTAC in a cellular model.
Materials & Reagents:
- Cell Line: A relevant cell line expressing the POI and the chosen E3 ligase (e.g., MOLM-14 for BTK degradation [117], KU812 for H-PGDS [115]).
- PROTAC Compounds: Serial dilutions of the PROTAC and control compounds (e.g., warhead alone, E3 ligand alone, combination) in DMSO.
- Lysis Buffer: RIPA buffer or similar, supplemented with protease and phosphatase inhibitors.
- Antibodies: Primary antibodies against the POI and a loading control (e.g., GAPDH, β-Actin), and corresponding HRP-conjugated secondary antibodies.
- Other: SDS-PAGE gel, PVDF or nitrocellulose membrane, chemiluminescence detection kit.
Procedure:
- Cell Seeding and Treatment: Seed cells in an appropriate multi-well plate and allow them to adhere and grow overnight to ~70-80% confluence.
- PROTAC Dosing: Treat cells with a concentration gradient of the PROTAC (e.g., from 1 nM to 10 µM) for a predetermined time (typically 4-24 hours). Include a DMSO vehicle control.
- Cell Lysis: After treatment, wash cells with cold PBS and lyse them in ice-cold lysis buffer for 15-30 minutes on ice. Centrifuge the lysates (14,000 x g, 15 min, 4°C) to remove insoluble debris.
- Protein Quantification: Determine the protein concentration of the supernatant using a BCA or Bradford assay. Normalize all samples to the same concentration.
- Western Blotting:
- Separate equal amounts of protein by SDS-PAGE.
- Transfer the proteins from the gel to a membrane.
- Block the membrane with 5% non-fat milk in TBST for 1 hour.
- Incubate with the primary antibody (diluted in blocking buffer) overnight at 4°C.
- Wash the membrane and incubate with the HRP-conjugated secondary antibody for 1 hour at room temperature.
- Detect the signal using a chemiluminescent substrate and image the membrane.
- Data Analysis: Quantify the band intensities of the POI relative to the loading control. Plot the percentage of POI remaining against the log of the PROTAC concentration to generate a dose-response curve and calculate the DC50 (half-maximal degradation concentration). Be alert for the "Hook effect," where degradation decreases at very high PROTAC concentrations due to the formation of non-productive binary complexes [114].
Table 2: The Scientist's Toolkit: Key Reagents for PROTAC Research
| Reagent / Tool | Function in PROTAC Development | Example Usage | Citation |
|---|---|---|---|
| Fluorescence Polarization (FP) Assay | Measures binding affinity (IC50) of PROTAC to the isolated target protein. | Used to confirm H-PGDS PROTACs maintained binding after linker modification [115]. | [115] |
| Quantitative Western Blot | Gold standard for measuring cellular degradation efficiency (DC50, Dmax). | Used to measure BTK and CRBN absolute concentrations in MOLM-14 cells [117]. | [117] |
| Molecular Docking & Modeling | Predicts ternary complex structure and stability to guide rational linker design. | Used with MOE software to model H-PGDS:PROTAC:CRBN complexes and calculate RMSD [115]. | [115] |
| Crystal Structure of Ternary Complex | Provides atomic-level insight into PPIs and linker positioning for a specific complex. | The BRD4-MZ1-VHL structure revealed key interactions driving selectivity [114]. | [114] |
| Click Chemistry | Facilitates modular synthesis and rapid screening of PROTAC libraries with different linkers. | Used to generate a potent JQ1-based BRD4 PROTAC for rapid evaluation [114]. | [114] |
Integration with Ubiquitination in DNA Repair and Immunity
The therapeutic application of PROTACs is deeply intertwined with the biology of the UPS in DNA repair and immune response pathways. Many oncoproteins and proteins involved in immune checkpoint regulation are controlled by ubiquitination [26] [120]. Dysregulation of E3 ligases (e.g., SPOP in prostate cancer) and deubiquitinating enzymes (DUBs) is a common feature in cancer and can confer resistance to therapies [120]. PROTACs can be strategically designed to target these dysregulated pathways. For example, by degrading an oncogenic transcription factor that is normally stabilized in cancer, or by targeting immune checkpoint proteins for degradation to enhance anti-tumor immunity [120]. The following diagram illustrates how PROTACs interface with these critical cellular systems.
Optimizing PROTAC design requires a holistic and integrated approach, where linker chemistry, warhead selection, and ternary complex dynamics are co-optimized for the specific target and E3 ligase pair. The shift from flexible, synthetically simple linkers to more rigid and spatially defined structures, combined with strategic warhead engineering using reversible covalent chemistry, represents the cutting edge of the field. The future of PROTAC development will be increasingly driven by rational design powered by advanced computational modeling of ternary complexes and a deeper understanding of the ubiquitin code in health and disease. As the structural database of PROTAC-induced complexes expands, and as new E3 ligases are harnessed, the ability to precisely target pathogenic proteins—especially those involved in DNA repair deficiencies and immune dysregulation—will unlock novel and transformative therapeutic modalities.
The immune system operates through a precise balance of activation and contraction, a cyclic process essential for effective pathogen clearance while preventing excessive damage to the host [121]. This dynamic equilibrium, reminiscent of the yin-yang philosophy in traditional Chinese medicine, ensures robust threat response followed by wound healing, immune cell death, and immunological memory establishment [121]. At the molecular heart of this regulation lies ubiquitination, an essential post-translational modification that controls protein fate and function through covalent attachment of ubiquitin molecules [73]. The ubiquitin-proteasome system (UPS) orchestrates diverse cellular processes including cell cycle progression, DNA damage response, and immune signaling by precisely modulating protein stability, activity, and interactions [103]. When this sophisticated regulatory system is disrupted, the consequences can be severe, leading to uncontrolled inflammation, cytokine storm syndromes, and autoimmunity [73] [121]. This technical review examines the intricate mechanisms by which ubiquitination maintains immune homeostasis, with particular focus on its dual roles in DNA repair and immune response pathways, and explores emerging therapeutic strategies targeting the ubiquitin system to prevent or mitigate pathological inflammation.
Molecular Mechanisms of Ubiquitination in Immune Signaling
The Ubiquitin Proteasome System: Components and Catalytic Cascade
The ubiquitination process involves a hierarchical enzymatic cascade comprising E1 (activating), E2 (conjugating), and E3 (ligating) enzymes that work sequentially to tag substrate proteins with ubiquitin [2] [103]. The human genome encodes only two E1 enzymes, approximately 35 E2 enzymes, and over 600 E3 ligases, with the latter conferring substrate specificity [2]. This enzymatic process culminates in the formation of an isopeptide bond between the C-terminal glycine of ubiquitin and lysine residues on target proteins [2]. The complexity of ubiquitin signaling arises from the ability to form various chain topologies through different linkage types (K6, K11, K27, K29, K33, K48, K63, and M1), each associated with distinct functional outcomes [2] [103]. For instance, K48-linked chains primarily target proteins for proteasomal degradation, while K63-linked and linear (M1) chains play crucial roles in immune signaling complex assembly and inflammatory responses [96] [103].
Regulatory Counterbalance: Deubiquitinases (DUBs)
Ubiquitination is a reversible modification countered by approximately 100 deubiquitinating enzymes (DUBs) encoded in the human genome [122] [96]. These proteases, including USP, UCH, OTU, JAMM/MPN, and MJD subfamilies, remove ubiquitin from substrates, thereby fine-tuning signaling pathways or rescuing proteins from degradation [122] [96]. DUBs ensure proper levels of ubiquitin, regulate recruitment of downstream effectors, dictate repair pathway choice, and facilitate appropriate termination of cellular responses, including those to DNA damage and immune activation [122]. The balance between E3 ligases and DUB activities constitutes a critical regulatory node in immune homeostasis, with dysregulation contributing to hyperinflammation or autoimmune manifestations [123].
Table 1: Major Ubiquitin Linkage Types and Their Primary Functions in Immune Regulation
| Linkage Type | Primary Functions | Key Immune Processes |
|---|---|---|
| K48 | Proteasomal degradation | Turnover of signaling proteins (IκB, p27) |
| K63 | Signaling scaffold assembly | NF-κB activation, DNA repair complex formation |
| M1 (Linear) | Inflammatory signaling | NF-κB activation, immune cell recruitment |
| K11 | Cell cycle regulation, protein quality control | Immune cell proliferation |
| K27 | Mitophagy, innate immunity | Mitochondrial quality control during stress |
| K29 | Lysosomal degradation | Alternative protein degradation pathway |
| K33 | Kinase regulation, trafficking | T-cell receptor signaling modulation |
| K6 | DNA damage response | Recruitment of DNA repair factors |
Ubiquitination in DNA Damage Response and Immune Crosstalk
Ubiquitination plays a pivotal role in the cellular response to DNA double-strand breaks (DSBs), one of the most lethal forms of DNA damage [122]. Histone ubiquitination at DSB sites creates recruitment platforms for repair machinery and influences pathway choice between non-homologous end joining (NHEJ) and homologous recombination (HR) [122]. This DNA damage response intersects significantly with immune signaling, as unrepaired DNA damage can trigger inflammatory responses through pattern recognition receptors sensing nuclear nucleic acids [122]. Furthermore, E3 ligases such as TRAF6 and RNF168 coordinate both DNA repair and immune signaling pathways, creating molecular crosstalk that maintains genomic integrity while regulating inflammatory responses [122].
Cytokine Storm Syndromes: Molecular Pathogenesis and Ubiquitination Links
Defining Cytokine Storm Syndromes
Cytokine storm syndrome (CSS) represents a life-threatening condition characterized by uncontrolled, systemic hyperinflammation resulting from excessive cytokine release and immune cell activation [124] [121]. Initially described in graft-versus-host disease and severe infections, CSS manifests clinically through fever, hepatosplenomegaly, liver dysfunction, cytopenias, hyperferritinemia, and potentially progressive multiorgan failure [124]. The core pathophysiological feature is the disruption of the normal yin-yang immune balance, with persistent activation overwhelming contraction mechanisms [121]. CSS can be triggered by diverse stimuli, including pathogens (sepsis, SARS-CoV-2, influenza), autoimmune conditions (MAS, sJIA, SLE), and therapeutic interventions (CAR-T cell therapy, immunotherapies) [124] [125].
The Cytokine-Cell Death Feedback Loop
Recent research has elucidated a critical positive feedback loop between cytokine release and inflammatory cell death pathways that perpetuates CSS [124]. This interplay between pyroptosis, apoptosis, and necroptosis—conceptualized as PANoptosis—creates a self-amplifying cycle where cytokine release triggers inflammatory cell death, which in turn facilitates further cytokine secretion through membrane pore formation and cell lysis [124]. Key molecular switches regulating this process include caspase-8, ZBP1, TAK1, and RIPK1, which integrate signals from various pathways to initiate PANoptosis [124]. In experimental models, combined neutralization of TNF and IFN-γ can block this cytokine-mediated cell death cascade, preventing tissue damage and mortality in CSS contexts such as poly(I:C)-induced HLH, LPS-induced sepsis, and SARS-CoV-2 infection [124].
SARS-CoV-2 as a Model for Ubiquitination-Immunity Interactions
The COVID-19 pandemic provided compelling insights into ubiquitination-CSS interactions, with severe manifestations frequently featuring cytokine storm pathology [125] [126]. SARS-CoV-2 infection triggers massive inflammatory response through various mechanisms, including ORF8-mediated activation of IL-17 signaling pathways [126]. Viral ORF8 protein mimics IL-17 function by interacting with IL17RA, recruiting ACT1, and activating TRAF6-mediated NF-κB signaling, ultimately enhancing pro-inflammatory cytokine production [126]. This subversion of normal ubiquitination-related pathways exemplifies how pathogens can disrupt immune homeostasis, tipping the balance toward pathological hyperinflammation. The UPS also plays a regulatory role in the host antiviral response, with various E3 ligases targeting viral proteins for degradation or modifying innate immune signaling components [103].
Diagram 1: Molecular pathogenesis of cytokine storm syndromes showing the central cytokine-cell death feedback loop and ubiquitination system involvement.
Table 2: Major Cytokine Storm Syndromes: Etiologies and Key Mediators
| CSS Category | Specific Conditions | Primary Trigger(s) | Key Cytokine Drivers |
|---|---|---|---|
| Pathogen-induced | Sepsis, Severe COVID-19, Influenza | Bacteria (GAS, S. aureus), Viruses (SARS-CoV-2, IAV, EBV) | TNF, IFN-γ, IL-6, IL-1 [124] [125] |
| Autoinflammatory/Monogenic | pHLH, sHLH/MAS, CAPS, FMF | Genetic mutations (PRF1, UNC13D, NLRP3, MEFV) | IFN-γ, IL-1β, IL-18 [124] |
| Therapy-induced | CAR T-cell CRS, irAEs, GvHD | Immunotherapies, HSCT, Biologics | IL-6, IL-1, T-cell derived cytokines [124] [121] |
| Autoimmune-associated | MAS, SLE flare, sJIA | Underlying autoimmune dysregulation | IFN-γ, IL-1, IL-6, IL-18 [124] [121] |
Experimental Approaches for Studying Ubiquitination in Immune Regulation
Methodologies for Assessing Ubiquitination in Immune Signaling
Elucidating ubiquitination mechanisms in immune pathways requires specialized experimental approaches:
Ubiquitin Pull-Down and Proteomics: Utilizing tandem ubiquitin-binding entities (TUBEs) or ubiquitin remnant motifs for affinity purification enables system-wide identification of ubiquitination events in immune cells under activation conditions [103]. Cells are typically lysed under denaturing conditions to preserve ubiquitination states, followed by enrichment of ubiquitinated proteins using TUBE agarose beads. After extensive washing, bound proteins are eluted and analyzed by mass spectrometry. For immune applications, this can be performed on macrophages or dendritic cells stimulated with LPS, cytokines, or other immune activators to identify ubiquitination changes in signaling pathways [103].
DNA Damage-Ubiquitination Co-Monitoring: Integrated assessment of DNA damage response and ubiquitination involves inducing DSBs (e.g., with ionizing radiation or etoposide) while monitoring histone ubiquitination (γH2AX, ubiquitin-H2A, H2B) and recruitment of repair factors through chromatin immunoprecipitation and immunofluorescence [122]. This approach reveals how ubiquitination coordinates DNA repair and influences subsequent immune signaling, particularly important for understanding the crosstalk between genomic instability and inflammation [122].
CRISPR Screening for UPS Components: Genome-wide CRISPR-Cas9 screens in immune cells exposed to inflammatory stimuli can identify E3 ligases and DUBs critical for maintaining homeostasis [103]. Cells transduced with sgRNA libraries are stimulated with cytokines (e.g., TNF-α, IL-6) or PAMPs, followed by sequencing to identify enriched/depleted guides. This method has revealed novel regulators of cytokine signaling and cell death pathways relevant to CSS [103].
In Vivo Modeling of Ubiquitination-Immune Interactions
Animal models, particularly genetically modified mice, provide indispensable platforms for studying ubiquitination in immune homeostasis:
CSS Induction Models: Administration of TLR agonists (poly(I:C), LPS) or pathogenic infections in transgenic mice with ubiquitination pathway modifications reveals how specific E3 ligases or DUBs influence CSS development [124]. For example, poly(I:C) injection in mice with defective ubiquitin regulation in myeloid cells produces exacerbated CSS pathology, demonstrating the protective role of proper ubiquitination [124].
PANoptosis Assessment: Multi-parameter analysis of cell death pathways in tissue samples from CSS models, including measurement of caspase activation (Western blotting), membrane integrity, and cytokine release, helps elucidate the ubiquitination-PANoptosis connection [124]. Inhibition studies using ZBP1, RIPK1, or caspase-8 targeting can dissect the contribution of specific pathways to the cytokine-cell death cycle [124].
Diagram 2: Integrated experimental workflow for investigating ubiquitination in immune regulation and cytokine storm pathogenesis.
The Scientist's Toolkit: Key Research Reagents and Solutions
Table 3: Essential Research Reagents for Studying Ubiquitination in Immune Pathways
| Reagent Category | Specific Examples | Research Application | Technical Considerations |
|---|---|---|---|
| E1 Inhibitors | PYR-41, PYZD-4409, MLN7243 | Block global ubiquitination; assess broad UPS impact on immune signaling | High toxicity; limited specificity; useful for proof-of-concept studies [96] |
| E2 Inhibitors | CC0651 (CDC34), NSC697923 (UBE2N), BAY 11-7082 | Target specific ubiquitin-conjugating enzymes; more selective than E1 inhibition | CC0651 acts allosterically; NSC697923 blocks K63 chains; BAY 11-7082 has cysteine-reactive off-targets [96] |
| E3-Targeting Compounds | Nutlin (MDM2), MI-219 (MDM2), PROTACs | Modulate specific substrate degradation (e.g., p53); emerging therapeutic strategy | Nutlin stabilizes p53; PROTACs redirect E3 activity to neo-substrates [2] [96] |
| DUB Inhibitors | Compounds G5, F6, Broad-spectrum DUB inhibitors | Investigate deubiquitination roles in immune signaling; potential CSS therapeutics | Varying specificity; can stabilize ubiquitination on immune signaling components [2] [123] |
| Proteasome Inhibitors | Bortezomib, Carfilzomib, Ixazomib | Block protein degradation; assess impact on immune regulator turnover | FDA-approved for cancer; can induce ER stress and modulate cytokine production [2] |
| Ubiquitin Binding Probes | TUBEs, Ubiquitin remnant antibodies, DiGly antibody (K48-GG) | Enrich and detect ubiquitinated proteins; proteomic applications | TUBEs protect from DUBs; DiGly antibody detects K48 linkages after trypsin digestion [103] |
| CRISPR Libraries | E3/DUB-focused sgRNA libraries, Whole-genome screens | Identify novel regulators of immune signaling and CSS | Reveals genetic dependencies in cytokine production and cell death pathways [103] |
Therapeutic Targeting of Ubiquitination in Immune Dysregulation
Existing Modalities and Clinical Applications
Several therapeutic strategies targeting the ubiquitin system have shown promise for modulating immune responses:
Proteasome Inhibitors: Drugs such as bortezomib, carfilzomib, and ixazomib, originally developed for cancer treatment, have demonstrated immunomodulatory effects by preventing degradation of key regulatory proteins [2]. In immune contexts, proteasome inhibition can alter cytokine production profiles and immune cell activation, with potential applications in autoimmune conditions and CSS [2] [123].
E1/Targeted Inhibition: The NEDD8-activating enzyme (NAE) inhibitor MLN4924 (Pevonedistat) blocks cullin neddylation, thereby attenuating the activity of cullin-RING ligases (CRLs) [96]. This compound shows particular promise in hyperinflammatory conditions by preventing the degradation of negative regulators of immune signaling, such as IκB and various cell cycle inhibitors [96]. MLN4924 treatment induces cell death in rapidly dividing cells due to uncontrolled DNA synthesis during S-phase, making it particularly effective against highly proliferative immune populations contributing to CSS [96].
PROTAC Technology: Proteolysis-Targeting Chimeras (PROTACs) represent a groundbreaking approach that redirects E3 ligase activity to specifically degrade target proteins [123]. These bifunctional molecules simultaneously bind to an E3 ligase and a protein of interest, facilitating ubiquitination and degradation of the latter [123]. In immune applications, PROTACs offer potential for degrading pathological signaling components or transcription factors driving cytokine production in CSS [123].
Emerging Targets and Future Directions
The expanding understanding of ubiquitination in immune regulation reveals numerous promising therapeutic targets:
DUB Inhibition: Specific deubiquitinases that stabilize pro-inflammatory signaling components represent attractive targets for CSS intervention [123]. For example, inhibitors of USP7, which regulates NF-κB signaling, or OTULIN, which controls linear ubiquitination in inflammatory signaling, may provide more precise control over hyperinflammation compared to broad immunosuppressants [123].
E3 Ligase Modulation: Rather than general E1/E2 inhibition, developing compounds that target specific E3 ligases involved in immune regulation offers greater specificity [96]. For instance, modulation of SCFSKP2 activity impacts p27 degradation and cell cycle progression in proliferating immune cells, while TRAF6 inhibition could directly dampen NF-κB activation in CSS [2] [96].
Biomarker Development: Ubiquitination signatures show promise as diagnostic and prognostic biomarkers for CSS susceptibility and treatment response [73] [123]. Measuring specific ubiquitination events in immune cells or circulating ubiquitin-related proteins could enable early identification of individuals at risk for severe hyperinflammation and guide personalized therapeutic interventions [73].
The ubiquitin system serves as a master regulator at the intersection of DNA repair, immune activation, and contraction pathways, making it essential for maintaining the delicate balance required for immune homeostasis. Understanding how ubiquitination processes coordinate these diverse functions provides critical insights into the pathogenesis of cytokine storm syndromes and autoimmunity. As research continues to unravel the complexities of the ubiquitin code in immune regulation, new opportunities emerge for targeted therapeutic interventions that can precisely modulate immune responses without causing broad immunosuppression. The integration of ubiquitin-focused approaches with emerging technologies such as PROTACs, CRISPR screening, and ubiquitinomics holds particular promise for developing novel strategies to prevent or treat pathological inflammation, ultimately restoring the crucial yin-yang balance in immune function.
Pharmacodynamic (PD) biomarkers are measurable indicators that provide real-time insights into a drug's biological effects on its target, reflecting the relationship between drug exposure and pharmacological response. In the context of the ubiquitin-proteasome system (UPS), these biomarkers are indispensable for evaluating therapeutic interventions aimed at modulating protein degradation, DNA repair fidelity, and immune response pathways. The UPS mediates over 80% of protein degradation in eukaryotic organisms and regulates critical processes including cell proliferation, DNA damage response, and immune inflammation [74]. As such, PD biomarkers offer a window into the dynamic reprogramming of ubiquitin networks in response to targeted therapies, enabling researchers to confirm target engagement, optimize dosing regimens, and identify mechanisms of resistance.
The clinical utility of PD biomarkers is particularly evident in oncology, where they enable early efficacy assessment of novel agents. For instance, in the development of ubiquitin-targeting therapies, changes in specific ubiquitin chain topology or substrate stability can indicate whether a treatment is effectively engaging its target, often weeks before traditional imaging results become available [127]. This capability accelerates decision-making in drug development and reduces unnecessary patient exposure to ineffective therapies. Furthermore, the integration of PD biomarkers into clinical trials for ubiquitin-modulating agents allows for precise patient stratification based on biological response potential, moving toward personalized treatment approaches that align with the genetic and molecular profiles of individual tumors [7] [127].
The Ubiquitin-Proteasome System: Framework for PD Biomarker Development
Ubiquitin Cascade and Chain Topology
The ubiquitination process involves a sequential enzymatic cascade comprising ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3), which collectively confer substrate specificity. This system generates diverse ubiquitin chain topologies that function as distinct molecular codes governing protein fate and function [7]. The UBA family of E1 enzymes, including UBA1 and UBA6, initiates this process by activating ubiquitin for transfer, making them fundamental to UPS function and potential therapeutic targets [74].
Ubiquitin chain architectures constitute a sophisticated regulatory system with specific functional consequences. K48-linked polyubiquitin chains primarily target proteins for proteasomal degradation, while K63-linked chains facilitate the assembly of non-proteolytic signaling complexes critical for DNA damage response and immune signaling [7]. Additionally, monoubiquitylation of both histone and non-histone proteins regulates chromatin dynamics and genome stability. The diversity of ubiquitin codes extends to other linkage types, including K27 and K29 chains, which have been implicated in DNA repair pathway activation [7]. This complex landscape of ubiquitin modifications provides a rich source of potential PD biomarkers for assessing the functional effects of UPS-targeting therapeutics.
Ubiquitination in DNA Repair and Immune Regulation
Ubiquitination serves as a master regulator orchestrating cellular responses to genotoxic stress and immune signaling. In DNA repair, the ubiquitin system controls the fidelity of multiple repair pathways through spatiotemporal regulation of repair factor assembly and activity. For example, RNF168-mediated ubiquitination of H2A/H2AX creates a platform for recruitment of BRCA1-A complexes to DNA damage sites, enhancing repair fidelity [7]. Similarly, FBXW7 utilizes K63 chains to modify XRCC4, thereby enhancing the accuracy of non-homologous end joining (NHEJ) repair [7]. These specific ubiquitination events represent potential PD biomarkers for therapies targeting DNA repair pathways.
In immune regulation, ubiquitination critically modulates tumor-immune crosstalk and immune evasion mechanisms. TRIM21 exemplifies this role by promoting K48-linked degradation of VDAC2, thereby inhibiting mitochondrial DNA release and cGAS/STING activation in nasopharyngeal carcinoma [7]. Conversely, inhibition of deubiquitinases such as USP14 leads to accumulation of K63-ubiquitinated IRF3, triggering STING-dependent antitumor immunity [7]. The expression levels of specific E3 ligases and deubiquitinases (DUBs) have been correlated with immune cell infiltration patterns in various cancers, suggesting their utility as biomarkers for predicting response to immunotherapy [128] [74]. Multi-omics analyses have revealed that key E3 ubiquitin ligases are negatively correlated with B cells and dendritic cell infiltration but positively related to neutrophil immune infiltration in lung adenocarcinoma, highlighting the complex relationship between ubiquitination and immune microenvironment composition [128].
Table 1: Ubiquitin Chain Types and Their Functional Roles in DNA Repair and Immune Response
| Ubiquitin Chain Type | Primary Function | Role in DNA Repair | Role in Immune Response | Key Regulators |
|---|---|---|---|---|
| K48-linked polyubiquitin | Proteasomal degradation | Regulates stability of DNA repair proteins (e.g., p53 degradation by FBXW7) | Controls immune checkpoint protein turnover; TRIM21-mediated VDAC2 degradation inhibits cGAS/STING | FBXW7, TRIM21 |
| K63-linked polyubiquitin | Signaling scaffold formation | FBXW7-mediated XRCC4 modification enhances NHEJ accuracy | K63-ubiquitinated IRF3 activates STING-dependent immunity | TRAF4, TRAF6, TRIM26 |
| Monoubiquitination | Protein activity and localization modulation | FANCD2 monoubiquitylation resolves DNA crosslinks; H2AX monoubiquitylation accelerates damage detection | Regulates chromatin accessibility for immune gene expression | UBE2T/RNF8, RNF40 |
| K27/K29-linked chains | Atypical signaling roles | RNF126-mediated MRE11 ubiquitination activates ATM-CHK1 axis | Emerging roles in immune signaling pathways | RNF126 |
Quantitative Assessment of Ubiquitin-Related PD Biomarkers
Molecular and Proteomic Biomarkers
The quantitative measurement of ubiquitin-related PD biomarkers requires sophisticated analytical approaches capable of detecting specific ubiquitin chain topologies and substrate modifications. Mass spectrometry-based ubiquitin remnant profiling enables system-wide mapping of ubiquitination sites, providing a comprehensive view of UPS modulation by therapeutic interventions. For assessing target engagement of ubiquitin pathway inhibitors, direct measurement of substrate accumulation offers a straightforward PD readout. For example, the degradation of specific substrates such as SOX9 in non-small cell lung cancer (NSCLC) with SOX9 overexpression serves as a direct biomarker for FBXW7 activity [7]. Similarly, SMURF2-mediated HIF1α degradation provides a measurable PD endpoint for compounds targeting hypoxic adaptation pathways in tumors [7].
Multi-omics approaches integrating genomic, transcriptomic, and proteomic data have identified specific ubiquitin pathway components as quantitative biomarkers with prognostic significance. In lung adenocarcinoma, elevated expression of E3 ligases such as CDC20 is associated with poor patient prognosis and correlates with activation of the mTORC1 signaling pathway [128]. The UBA family members UBA1 and UBA6 are highly expressed in most cancer types and demonstrate stage-dependent expression patterns, making them potential biomarkers for disease progression and treatment response [74]. Quantitative analysis of these biomarkers in tissue samples through immunohistochemistry or mRNA expression profiling can stratify patients based on their likelihood to respond to ubiquitin-targeting therapies.
Imaging and Functional Biomarkers
Non-invasive imaging biomarkers provide complementary approaches to molecular assays for assessing the PD effects of ubiquitin-targeting therapies. Advanced techniques such as radiomics models, texture visualization, convolutional neural networks, and automated segmentation enable quantitative assessment of tumor heterogeneity and treatment response [129]. These imaging biomarkers can capture functional changes in the tumor microenvironment resulting from modulation of ubiquitin pathways, including alterations in perfusion, metabolism, and cellular density.
Functional biomarkers that reflect pathway activity downstream of ubiquitination events offer dynamic readouts of pharmacological effects. For example, in the context of DNA damage response pathways, monitoring the phosphorylation status of ATM/ATR substrates or the formation of DNA repair foci can serve as surrogate markers for the activity of ubiquitin-modulating agents [7]. Similarly, immune-related functional biomarkers such as T-cell activation markers or cytokine profiles can indicate the immunomodulatory consequences of DUB inhibition [7] [74]. The integration of these functional assays with molecular measurements provides a comprehensive framework for evaluating the PD effects of ubiquitin-targeting therapeutics across multiple biological scales.
Table 2: Experimental Assays for Monitoring Ubiquitin-Related PD Biomarkers
| Assay Category | Specific Methodology | Measurable Parameters | Therapeutic Context | Technical Considerations |
|---|---|---|---|---|
| Ubiquitin Profiling | Mass spectrometry-based ubiquitin remnant profiling | Ubiquitination sites, chain topology | Target engagement for ubiquitin pathway inhibitors | Requires specialized instrumentation; complex data analysis |
| Immunoassay | Quantitative immunohistochemistry | Protein expression, localization, and modification | Prognostic stratification; treatment response monitoring | Affected by preanalytical variables; semi-quantitative |
| Molecular Imaging | Radiomics, PET with specific tracers | Tumor metabolism, proliferation, heterogeneity | Non-invasive treatment response assessment | Limited resolution; indirect measurement of target modulation |
| Functional Assays DNA repair foci formation, immune cell activation | Pathway activity downstream of ubiquitination | Mechanism of action confirmation for DNA repair or immune-targeting agents | May reflect integrated biological responses rather than direct target engagement | |
| Multi-omics Analysis | Integrated genomic, transcriptomic, and proteomic profiling | Expression of ubiquitin pathway components, immune signatures | Patient stratification; biomarker discovery | Computational complexity; data integration challenges |
Experimental Protocols for PD Biomarker Evaluation
Preclinical Assessment of Ubiquitin-Targeting Therapies
Preclinical evaluation of ubiquitin-targeting therapies requires well-validated experimental protocols that can accurately measure target engagement and downstream pharmacological effects. For PROTACs (proteolysis-targeting chimeras) and other ubiquitin-dependent degraders, a standard protocol involves treatment of cancer cell lines followed by measurement of target protein degradation over time. Cells are treated with varying concentrations of the test compound for predetermined intervals (typically 2-24 hours), after which they are lysed and analyzed by Western blotting to quantify residual target protein levels. Parallel assessment of ubiquitin chain formation on the target protein using immunoprecipitation followed by ubiquitin immunoblotting provides confirmation of the intended mechanism of action [7].
For in vivo studies, xenograft mouse models implanted with human cancer cells provide a system for evaluating the PD effects of ubiquitin-targeting agents. Animals receive the test compound at predetermined doses and schedules, with tissue collection at multiple time points for biomarker analysis. Protocol details include: (1) compound administration via appropriate route (oral, intraperitoneal, or intravenous); (2) collection of tumor tissue, blood, and relevant normal tissues at specified intervals post-dose; (3) processing of samples for various analytical endpoints including Western blotting, immunohistochemistry, and RNA sequencing; (4) correlation of biomarker changes with antitumor activity [7]. Innovative approaches such as radiation-responsive PROTAC (RT-PROTAC) prodrugs activated by tumor-localized X-rays have demonstrated synergistic effects with radiotherapy in breast cancer models, requiring specialized protocols that combine drug administration with localized radiation [7].
Clinical Translation and Biomarker Validation
The transition from preclinical to clinical evaluation of ubiquitin-related PD biomarkers requires rigorous validation to ensure reliability and reproducibility. Clinical trial protocols for ubiquitin-targeting agents should incorporate serial biomarker assessments to establish PK/PD relationships and determine biologically effective doses. Recommended procedures include: (1) pre- and post-treatment tumor biopsies for assessment of target protein levels, ubiquitin modifications, and pathway modulation; (2) serial blood collection for measurement of soluble biomarkers including circulating tumor DNA, cytokines, and exosome-associated ubiquitin pathway components; (3) functional imaging studies at baseline and early treatment time points to capture metabolic and physiologic changes [127].
Protocols for biomarker analysis must address preanalytical variables that can significantly impact results, including tissue ischemia time, fixation methods, and sample processing techniques [130]. For immunohistochemical analysis of ubiquitin pathway components, standardized scoring systems should be implemented to ensure consistency across samples and time points. Quantitative image analysis (QIA) approaches can reduce subjectivity in biomarker assessment and enable more precise quantification of expression levels and spatial distribution [130]. When evaluating immune-related biomarkers in response to ubiquitin-modulating agents, multiplex immunohistochemistry or immunofluorescence should be employed to characterize immune cell populations and their spatial relationships within the tumor microenvironment [74] [130].
The Scientist's Toolkit: Research Reagent Solutions
The evaluation of ubiquitin-related PD biomarkers relies on a specialized set of research reagents and tools that enable precise measurement of ubiquitin pathway activity. These reagents form the foundation for experimental protocols across preclinical and clinical development stages. Key components include well-validated antibodies specific for different ubiquitin chain topologies, activity-based probes for deubiquitinases, recombinant ubiquitin enzymes, and standardized assay systems for high-throughput screening of ubiquitin-modulating compounds.
Table 3: Essential Research Reagents for Ubiquitin-Related PD Biomarker Studies
| Reagent Category | Specific Examples | Research Application | Function in Experimental Protocols |
|---|---|---|---|
| Chain-Specific Ubiquitin Antibodies | K48-linkage specific, K63-linkage specific, monoUb antibodies | Discriminating ubiquitin chain topology | Western blotting, immunohistochemistry, immunofluorescence for detecting specific ubiquitin modifications |
| Activity-Based Probes | DUB probes, E1/E2/E3 enzyme probes | Assessing enzyme activity in complex mixtures | In-gel fluorescence, pull-down assays, competitive binding studies for target engagement assessment |
| Ubiquitin Enzyme Panel | Recombinant E1, E2, and E3 enzymes | High-throughput screening, mechanistic studies | Biochemical assays for compound screening, enzyme kinetics, mechanism of action studies |
| PROTAC Molecules | EGFR-directed PROTACs, BRD4-targeting PROTACs | Targeted protein degradation studies | In vitro and in vivo proof-of-concept studies, establishing degradation kinetics and downstream effects |
| Ubiquitin Variants | MonoUb, diUb chains with defined linkages, ubiquitin mutants | Structural and mechanistic studies | X-ray crystallography, NMR, biochemical assays to understand molecular recognition and mechanism |
| Quantitative Mass Spectrometry Reagents | Tandem mass tag (TMT) reagents, ubiquitin remnant motif antibodies | Proteome-wide ubiquitination profiling | Global ubiquitinome analysis, quantification of ubiquitination changes in response to treatments |
Signaling Pathways and Ubiquitin Networks
The ubiquitin system interfaces with multiple signaling pathways that govern DNA damage response and immune regulation, creating complex networks that can be monitored through specific PD biomarkers. Understanding these interconnected pathways is essential for interpreting PD biomarker data and developing effective combination therapies. The following diagram illustrates key ubiquitin-mediated signaling pathways in DNA repair and immune response, highlighting potential PD biomarker nodes:
The intricate relationship between ubiquitin pathways and cellular signaling networks creates multiple nodes for PD biomarker development. In DNA damage response, radiation-induced ATM phosphorylation induces conformational changes in RNF168, facilitating K63-chain assembly at DNA damage sites and modulating repair fidelity [7]. This ATM-RNF168 interaction represents a potential PD biomarker node for therapies targeting DNA repair pathways. Similarly, in immune signaling, the TRIM21-VDAC2-cGAS/STING axis provides multiple measurable parameters for assessing the immunomodulatory effects of ubiquitin-targeting agents [7]. Monitoring the phosphorylation status of ATM and the ubiquitination status of VDAC2 in response to treatment can provide direct evidence of target engagement and pathway modulation.
The integration of ubiquitin-related PD biomarkers with other signaling readouts enables comprehensive assessment of therapeutic effects. For example, combining measurement of ubiquitin chain topology with analysis of downstream pathway activation (e.g., mTORC1 signaling in the case of CDC20) provides a more complete picture of drug activity than either measurement alone [128]. Similarly, correlating changes in ubiquitin pathway components with immune cell infiltration patterns offers insights into the interplay between targeted therapies and the tumor microenvironment [74]. These multidimensional biomarker approaches are increasingly important for understanding the complex pharmacology of ubiquitin-modulating agents and optimizing their clinical development.
Pharmacodynamic biomarkers represent essential tools for the development of therapeutics targeting the ubiquitin-proteasome system, providing critical insights into target engagement, pathway modulation, and mechanistic validation. The diverse roles of ubiquitination in DNA repair and immune response pathways create both challenges and opportunities for PD biomarker development. The complexity of ubiquitin chain topology and the contextual duality of many ubiquitin pathway components necessitate sophisticated biomarker strategies that can capture the nuanced effects of therapeutic interventions.
Future directions in ubiquitin-related PD biomarker research will likely focus on several key areas. First, the development of chain-specific ubiquitin antibodies and activity-based probes will enable more precise assessment of specific ubiquitination events in response to treatment. Second, advances in quantitative imaging and mass spectrometry techniques will provide increasingly comprehensive views of ubiquitin pathway modulation in both preclinical models and clinical samples. Third, the integration of ubiquitin-related PD biomarkers with other molecular profiling data through multi-omics approaches will facilitate patient stratification and personalized treatment strategies. Finally, the application of artificial intelligence and machine learning to ubiquitin biomarker data may reveal novel patterns and relationships that would be difficult to detect through traditional analytical approaches.
As the field continues to evolve, ubiquitin-related PD biomarkers will play an increasingly important role in translating fundamental understanding of ubiquitin biology into effective therapeutics for cancer, neurodegenerative diseases, and other conditions. By providing real-time insights into drug activity and biological response, these biomarkers will help accelerate the development of ubiquitin-targeting therapies and maximize their clinical benefit for patients.
Target Validation and Comparative Analysis: From Preclinical Models to Clinical Efficacy
The ubiquitin-proteasome system (UPS) represents a crucial regulatory mechanism in eukaryotic cells, controlling protein stability, function, and localization through the covalent attachment of ubiquitin molecules. This sophisticated enzymatic cascade involves the sequential action of ubiquitin-activating (E1), conjugating (E2), and ligase (E3) enzymes, which collectively mediate the transfer of ubiquitin to target proteins, determining their fate and function [99] [131]. The reverse reaction, deubiquitination, is carried out by deubiquitinating enzymes (DUBs), adding another layer of regulation to this dynamic system [132]. The biological outcome of ubiquitination depends on both the type of ubiquitin chain formed and the specific lysine residues involved. K48-linked polyubiquitination typically targets substrates for proteasomal degradation, while K63-linked chains and monoubiquitination play key roles in signaling, DNA damage response, and immune regulation [72] [98].
Ubiquitination has emerged as a central regulator of two critical cellular pathways: DNA damage repair and immune response. In DNA repair, ubiquitination controls the activation and coordination of various repair pathways, maintaining genomic integrity [98]. In immunity, the ubiquitin system regulates innate immune signaling cascades, including the cGAS-STING pathway, and adaptive immune processes such as antibody diversification in B cells [30] [133]. Dysregulation of ubiquitination components is increasingly implicated in human diseases, particularly cancer, making this system an attractive target for therapeutic intervention [72] [131]. This technical guide explores contemporary genetic validation approaches, primarily CRISPR-based screens and animal models, for characterizing essential components within the ubiquitin pathway and their functional roles in DNA repair and immune regulation.
CRISPR Screening Methodologies for Ubiquitin Pathway Analysis
Design and Implementation of CRISPR-Cas9 Screens
CRISPR-Cas9 screening has revolutionized the functional characterization of ubiquitin pathway components by enabling systematic, genome-wide interrogation of gene function. The design phase begins with selecting an appropriate sgRNA library that provides comprehensive coverage of the ubiquitin pathway genes, including E1, E2, and E3 enzymes, DUBs, and associated regulatory factors [134]. For focused investigations, custom libraries can be designed to target specific ubiquitin pathway subsets, such as the 35 E2 conjugating enzymes and 85 DUBs screened in B cell studies [133]. For phenotypic screens addressing specific biological processes, library design should prioritize genes with putative roles in the pathway of interest, complemented by robust positive and negative controls.
Successful screen implementation requires careful consideration of cellular models. The selection of cell lines should reflect the biological context under investigation—for instance, CH12F3 cells for immunoglobulin class switch recombination studies [133] or cancer cell lines with specific dependencies on ubiquitin pathways [134]. The transduction process must be optimized to ensure single-copy sgRNA integration, typically achieved through low multiplicity of infection (MOI < 0.3) followed by puromycin selection. The duration of the phenotypic selection phase varies depending on the biological question; for positive selection screens identifying essential genes, 14-21 days is typically sufficient to deplete sgRNAs targeting vital ubiquitin pathway components, while negative selection screens may require longer timeframes to accumulate resistant populations [134].
Post-selection, genomic DNA extraction and next-generation sequencing quantify sgRNA abundance changes. Analytical pipelines like MAGeCK identify significantly enriched or depleted sgRNAs, highlighting ubiquitin pathway genes essential for cell viability, DNA repair, or immune function under the screened conditions [134] [133].
Hit Validation and Mechanistic Follow-up Studies
Initial hit identification must be followed by rigorous validation employing multiple sgRNAs per target gene to minimize off-target effects. Validation strategies include complementation rescue experiments with CRISPR-resistant cDNA constructs and orthogonal approaches like RNA interference to confirm phenotype specificity [133]. Functional validation should employ pathway-specific assays tailored to the biological process under investigation, such as immunoglobulin class switch recombination efficiency measurements for immune function [133], γH2AX focus formation for DNA damage response [98], or interferon production assays for cGAS-STING pathway analysis [30].
Mechanistic follow-up experiments characterize how ubiquitin pathway perturbations affect specific substrates and signaling cascades. Western blotting analyzes substrate protein stability, while immunofluorescence and subcellular fractionation assess protein localization changes. Co-immunoprecipitation and proximity ligation assays can identify altered protein-protein interactions in ubiquitin complexes [30]. For E3 ligases and DUBs, substrate identification remains paramount—ubiquitin remnant profiling, combined with CRISPR screening, can map specific enzyme-substrate relationships within DNA repair and immune signaling pathways [98].
Table 1: Key Ubiquitin Pathway Components Identified Through CRISPR Screening
| Gene/Protein | Ubiquitin Pathway Role | Biological Function | Phenotype from CRISPR Screen | Reference |
|---|---|---|---|---|
| Ube2v1 | E2 ubiquitin-conjugating enzyme variant | Suppressor of class switch recombination in B cells | Enhanced CSR efficiency upon knockdown | [133] |
| FBXW7 | E3 ubiquitin ligase (F-box protein) | Cell cycle regulation, DNA damage response | Sensitive to multiple compounds targeting cell cycle | [134] |
| RNF25 | E3 ubiquitin ligase (RING domain) | DNA damage response | Specific sensitivity to methyl methanesulfonate | [134] |
| FBXO42 | E3 ubiquitin ligase (F-box protein) | Mitotic regulation | Sensitivity to mitotic inhibitors; loss causes aberrant mitoses | [134] |
| TRIM41 | E3 ubiquitin ligase (RING domain) | cGAS-STING pathway regulation | Enhances cGAMP synthesis through cGAS monoubiquitination | [30] |
| USP14 | Deubiquitinating enzyme | cGAS stability regulation | Stabilizes cGAS by cleaving K414 ubiquitin chains | [30] |
Specialized CRISPR Screening Applications
Beyond standard knockout screens, specialized CRISPR approaches offer unique insights into ubiquitin pathway function. CRISPR inhibition (CRISPRi) and activation (CRISPRa) platforms enable tunable modulation of gene expression, allowing investigation of dosage-sensitive ubiquitin components that may be lethal in complete knockouts [134]. Base editing and prime editing screens can introduce specific patient-derived point mutations into ubiquitin pathway genes, modeling cancer-associated variants found in E3 ligases like FBXW7 or BRCA1 [98].
Chemical-genetic CRISPR screens represent a particularly powerful approach for ubiquitin pathway analysis. These screens combine gene knockout with small molecule treatment to identify genetic determinants of drug sensitivity and resistance [134]. For example, CRISPR screens conducted with 41 compounds targeting diverse biological processes revealed 466 gene-compound interactions covering 25% of tested E3s/DUBs, highlighting functional specialization within the ubiquitin pathway [134]. Such approaches can identify biomarkers for emerging ubiquitin-targeted therapies like PROTACs and molecular glues, informing clinical development strategies.
Animal Models for Ubiquitin Pathway Validation
Murine Models in Ubiquitin Research
Genetically engineered mouse models (GEMMs) provide indispensable platforms for validating ubiquitin pathway gene functions in physiological contexts. Conventional knockout mice reveal essential roles for ubiquitin pathway components in development and tissue homeostasis. For example, complete loss of Fbw7 results in embryonic lethality due to impaired vascular development, while postnatal deletion causes chromosomal instability and tumorigenesis, underscoring its tumor suppressor function [99]. Tissue-specific conditional knockouts, typically employing Cre-loxP systems, enable bypassing of embryonic lethality and investigation of tissue-specific functions. This approach has been particularly valuable for studying immune-specific roles of ubiquitin components, such as B cell-intrinsic functions of E3 ligases and DUBs in antibody diversification [133].
Humanized mouse models, engrafted with human immune cells or containing human gene knock-ins, facilitate study of human-specific aspects of ubiquitin pathway biology and evaluation of therapeutic agents targeting human ubiquitin enzymes [72]. Spontaneous and carcinogen-induced tumor models in genetically modified mice (e.g., with altered E3 ligase or DUB expression) reveal cell-autonomous and non-cell-autonomous functions of ubiquitin pathway components in tumor initiation, progression, and metastasis [99]. For example, Skp2 overexpression promotes tumorigenesis in multiple tissues, while its deletion impedes cancer progression, confirming its oncogenic function [99].
Table 2: Animal Models for Studying Ubiquitin Pathway Components
| Model Type | Genetic Manipulation | Key Phenotypes | Research Applications | Reference |
|---|---|---|---|---|
| Conventional knockout | Fbw7 deletion | Embryonic lethality, vascular defects | Tumor suppressor function, cell cycle regulation | [99] |
| Conditional knockout | Ube2v1 deletion in B cells | Enhanced class switch recombination | Antibody diversification, humoral immunity | [133] |
| Transgenic overexpression | Skp2 overexpression | Increased tumor incidence, reduced p27 levels | Oncogene function, cell cycle control | [99] |
| Xenograft models | FBXO45 manipulation in ovarian cancer cells | Altered tumor growth and metastasis | Cancer therapeutic development | [135] |
| Disease-specific models | USP11 manipulation in LPS-induced lung injury | Modulated inflammatory response | Inflammation, innate immunity | [132] |
Phenotypic Analysis and Translational Applications
Comprehensive phenotypic characterization of ubiquitin pathway animal models employs diverse methodologies. Histopathological analysis assesses tissue architecture, proliferation indices, and differentiation status, while immunohistochemistry evaluates protein expression and modification states in situ [135]. Flow cytometric immunophenotyping quantifies immune cell populations in primary and secondary lymphoid organs, revealing roles for ubiquitin components in immune development and function [133]. Functional assays measure specific physiological processes such as antibody responses to immunization, DNA repair capacity following genotoxic insult, or metabolic parameters in response to nutritional challenges [98] [133].
Translational applications include preclinical evaluation of ubiquitin-targeted therapeutics, such as small molecule E1, E2, or E3 inhibitors, DUB inhibitors, and PROTAC degraders [72] [131]. Animal models also facilitate biomarker discovery for ubiquitin pathway-targeted therapies, including identification of predictive sensitivity markers and pharmacodynamic indicators of target engagement [96]. For example, murine models have been instrumental in validating MLN4924, a NEDD8-activating enzyme inhibitor, as an anticancer agent, revealing its mechanism of action through dysregulation of cullin-RING ligase substrates [96].
Experimental Protocols for Key Investigations
CRISPR-Cas9 Screen for Ubiquitin Regulators of DNA Repair
Objective: Identify ubiquitin pathway genes essential for DNA damage response using a chemical-genetic CRISPR screen.
Materials:
- Human cell line (e.g., HAP1, RPE1, or cancer cell lines)
- Genome-wide CRISPR knockout library (e.g., Brunello or GeCKO v2)
- DNA damage agents (e.g., methyl methanesulfonate, cisplatin, olaparib)
- Packaging plasmids (psPAX2, pMD2.G)
- Puromycin for selection
- Cell culture reagents and equipment
Procedure:
- Library Amplification and Virus Production: Amplify the CRISPR library in Escherichia coli to maintain representation. Produce lentivirus by transfecting HEK293T cells with library plasmid and packaging vectors using standard protocols.
- Cell Transduction: Transduce target cells at low MOI (0.3-0.5) to ensure single sgRNA integration. Include non-transduced controls.
- Selection and Expansion: Treat cells with puromycin (1-3 μg/mL) for 5-7 days to select successfully transduced cells. Maintain library representation by keeping at least 500 cells per sgRNA throughout the experiment.
- Treatment Arms: Split cells into untreated control and DNA damage treatment groups (e.g., 100 μM methyl methanesulfonate for 24 hours). Culture cells for 14-21 days to allow phenotypic manifestation.
- Genomic DNA Extraction and Sequencing: Harvest 1×10^8 cells per condition. Extract genomic DNA using a maxiprep kit. Amplify integrated sgRNA sequences by PCR with barcoded primers for multiplexing. Sequence using Illumina platforms.
- Bioinformatic Analysis: Align sequences to the reference library. Calculate fold depletion/enrichment of sgRNAs using MAGeCK or similar tools. Identify significantly depleted sgRNAs in treated versus control samples (FDR < 0.05).
Validation: Confirm hits using 3-5 independent sgRNAs. Assess DNA repair proficiency through γH2AX immunofluorescence, comet assays, and clonogenic survival after DNA damage.
In Vivo Validation of Ubiquitin Genes in Immune Function
Objective: Validate the role of Ube2v1 in B cell antibody class switch recombination using a murine model.
Materials:
- Ube2v1 floxed mice (C57BL/6 background)
- CD19-Cre or Mb1-Cre mice for B cell-specific deletion
- Age-matched control littermates (Ube2v1 floxed without Cre)
- Immunization antigens (e.g., NP-LPS, NP-KLH)
- Flow cytometry antibodies (B220, CD19, IgG1, IgG3, IgA)
- ELISA reagents for antigen-specific antibodies
- Cell culture reagents for B cell isolation
Procedure:
- Mouse Breeding and Genotyping: Cross Ube2v1 floxed mice with CD19-Cre mice to generate B cell-specific knockout animals. Use PCR genotyping to identify experimental and control littermates.
- Immunization: Immunize 8-12 week old mice intraperitoneally with 50 μg NP-LPS (for T-independent responses) or 100 μg NP-KLH in alum (for T-dependent responses).
- Analysis of Humoral Responses: At day 7 (early response) and day 14 (peak response) post-immunization:
- Collect serum and measure NP-specific antibody titers by ELISA.
- Isolate splenic B cells and analyze surface immunoglobulin isotypes by flow cytometry.
- Assess germline transcript expression by RT-qPCR.
- Ex vivo CSR Assay: Isolate naïve B cells from spleen using magnetic bead separation. Culture cells with LPS (10 μg/mL) + IL-4 (10 ng/mL) for IgG1 switching, or LPS + TGF-β (5 ng/mL) for IgA switching. Analyze switched B cells by flow cytometry at day 4.
- Immunohistochemistry: Analyze germinal center formation in spleen sections using PNA and BCL6 staining.
Interpretation: Ube2v1 knockout B cells should exhibit enhanced CSR efficiency without affecting germline transcription or AID expression, indicating a post-transcriptional regulatory role.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Ubiquitin Pathway Studies
| Reagent Category | Specific Examples | Function/Application | Key Characteristics |
|---|---|---|---|
| CRISPR Libraries | Brunello, GeCKO v2, Custom ubiquitin-focused libraries | Genome-wide or targeted gene knockout | High coverage, optimized sgRNA designs, minimal off-target effects |
| E1 Inhibitors | PYR-41, PYZD-4409 | Block ubiquitin activation | Irreversible inhibitors, preferentially target malignant cells |
| E2 Inhibitors | CC0651, NSC697923, BAY 11-7082 | Inhibit specific E2 enzymes (CDC34, UBE2N) | Allosteric mechanisms, covalent modification |
| Proteasome Inhibitors | Bortezomib, Carfilzomib, MG132 | Block proteasomal degradation | Clinical applications, tool compounds for mechanistic studies |
| DUB Inhibitors | PR-619, WP1130, VLX1570 | Pan-DUB inhibition | Broad specificity, useful for pathway analysis |
| PROTACs | ARV-471, ARV-110 | Targeted protein degradation | Bifunctional molecules, high specificity |
| Activity-Based Probes | Ubiquitin-vinylsulfone, HA-Ub-VS | DUB activity profiling | Covalent trapping, mechanism-based detection |
| Antibody Reagents | Anti-ubiquitin, anti-K48/K63 linkage, anti-PCNA Ub | Detection of ubiquitination | Chain linkage specificity, modification-state specific |
Signaling Pathways and Experimental Workflows
Ubiquitin Regulation in DNA Repair and Immune Signaling
Diagram Title: Ubiquitin Control of DNA Repair and Immune Pathways
Integrated CRISPR Screening Workflow
Diagram Title: CRISPR Screen Workflow for Ubiquitin Research
Genetic validation of ubiquitin pathway components through CRISPR screens and animal models has dramatically advanced our understanding of this complex regulatory system in DNA repair and immune response. The integration of these approaches provides complementary insights—CRISPR screens enable systematic, unbiased discovery of pathway components, while animal models establish physiological relevance and therapeutic potential. Future developments will likely focus on enhancing spatial and temporal resolution in both approaches, through inducible CRISPR systems and more sophisticated animal models with conditional and tissue-specific genetic manipulations.
The expanding toolkit of ubiquitin-focused research reagents, particularly selective E3 ligase modulators and PROTAC degraders, promises to accelerate both basic research and therapeutic development. As our understanding of ubiquitin signaling complexity grows, so too does the potential for targeting this system in human diseases, particularly cancer, autoimmune disorders, and neurodegenerative conditions where ubiquitin pathway dysregulation is increasingly implicated. The continued refinement of genetic validation approaches will be essential for realizing the full therapeutic potential of the ubiquitin-proteasome system.
The ubiquitin-proteasome system (UPS) represents a sophisticated enzymatic cascade responsible for regulating intracellular protein degradation, thereby controlling essential cellular processes ranging from cell cycle progression to DNA damage repair and immune response. This system operates through a sequential mechanism involving E1 (ubiquitin-activating), E2 (ubiquitin-conjugating), and E3 (ubiquitin-ligase) enzymes that collectively tag target proteins with ubiquitin molecules, marking them for destruction by the 26S proteasome [136]. The delicate balance of this system ensures cellular homeostasis, with dysregulation frequently contributing to oncogenesis and cancer progression. In recent years, therapeutic interrogation of the UPS has expanded beyond initial proteasome inhibitors to encompass more precise strategies targeting specific components upstream in the ubiquitination cascade, including E1/E2/E3 enzymes and deubiquitinating enzymes (DUBs) that reverse ubiquitination [137] [138]. This evolution reflects a growing understanding of UPS complexity and the quest for therapeutic agents with improved specificity and reduced off-target effects. The clinical development of these agents is intricately connected to their roles in DNA repair and immune signaling pathways, wherein the UPS exerts critical regulatory control over damage response elements, checkpoint activation, and immune cell function, positioning UPS-targeting therapies as potent modulators of genomic integrity and anti-tumor immunity [15] [89].
Proteasome Inhibitors: Established Backbone with Evolving Applications
Mechanism of Action and Clinical Trajectory
Proteasome inhibitors function by blocking the proteolytic activity of the 26S proteasome, leading to the accumulation of polyubiquitinated proteins, disruption of protein homeostasis, and induction of endoplasmic reticulum stress, ultimately triggering apoptosis in malignant cells [139] [136]. This drug class has demonstrated particular efficacy in hematological malignancies, especially multiple myeloma, where the first-generation inhibitor bortezomib established a new standard of care. The subsequent development of second-generation agents like carfilzomib and ixazomib aimed to enhance efficacy, reduce resistance, and improve administration convenience [137]. These inhibitors target the proteasome's chymotrypsin-like activity, disrupting the degradation of key regulatory proteins including pro-apoptotic factors and cell cycle controllers, thereby tilting the cellular balance toward apoptosis [136]. The profound sensitivity of multiple myeloma cells to proteasome inhibition is attributed to their high protein synthesis load and dependency on the UPS for handling immunoglobulin production, although the precise mechanistic determinants of sensitivity across different cancer types continue to be elucidated.
DNA Repair and Immune Pathway Interconnections
Proteasome inhibitors exert significant influence on DNA repair pathways through multiple mechanisms. By preventing the degradation of key DNA damage response proteins, they can disrupt the carefully orchestrated sequence of repair events following genotoxic stress. Specifically, proteasome inhibition interferes with the repair of double-strand breaks (DSBs) by impeding the recruitment of essential repair proteins to damage sites and altering the ubiquitin-dependent signaling cascades orchestrated by E3 ligases like RNF8 and RNF168 [15] [89]. Furthermore, the UPS regulates critical checkpoint proteins including CDC25A, whose degradation is essential for cell cycle arrest following DNA damage [89]. Proteasome inhibition consequently disrupts this regulatory circuit, leading to inappropriate cell cycle progression despite persistent DNA damage. In the context of immune response, proteasome inhibitors modulate antigen presentation by affecting the generation of peptide fragments presented by major histocompatibility complex (MHC) class I molecules, thereby influencing T-cell mediated tumor recognition and destruction [139]. This immunomodulatory activity contributes to their therapeutic efficacy in hematologic malignancies and underpins their investigation in various combination immunotherapy regimens.
Table 1: Clinically Utilized Proteasome Inhibitors
| Drug Name | Molecular Target | Key Indications | Clinical Status | Notable Characteristics |
|---|---|---|---|---|
| Bortezomib | 20S proteasome β5 subunit (chymotrypsin-like activity) | Multiple myeloma, mantle cell lymphoma | FDA-approved | First-in-class; reversible binding |
| Carfilzomib | 20S proteasome β5 subunit | Multiple myeloma | FDA-approved | Irreversible binding; reduced neuropathy risk |
| Ixazomib | 20S proteasome β5 subunit | Multiple myeloma | FDA-approved | First oral proteasome inhibitor |
Experimental Protocols for Proteasome Inhibition Studies
Assessment of Proteasome Inhibitory Activity: To evaluate proteasome function in response to inhibitor treatment, researchers typically employ fluorogenic substrate assays using cell lysates or purified proteasomes. The standard protocol involves incubation of proteasome with substrate derivatives such as Suc-LLVY-AMC (for chymotrypsin-like activity), Z-ARR-AMC (for trypsin-like activity), or Z-nLPnLD-AMC (for caspase-like activity). Following inhibitor treatment, the release of fluorescent 7-amino-4-methylcoumarin (AMC) is quantified using a fluorometer with excitation at 380 nm and emission at 460 nm, with results normalized to protein concentration and compared to untreated controls [139].
Analysis of Protein Ubiquitination Accumulation: To confirm target engagement and functional inhibition, western blot analysis of polyubiquitinated proteins is performed. Cells are treated with inhibitors for varying timepoints (typically 4-24 hours), followed by lysis in RIPA buffer containing protease inhibitors and N-ethylmaleimide (to inhibit endogenous DUBs). Lysates are resolved by SDS-PAGE, transferred to membranes, and probed with anti-polyubiquitin antibodies (e.g., FK1 or FK2 clones). Concurrent probing for specific proteasome substrates (e.g., IκBα, p27) provides additional validation of proteasome inhibition [136].
Evaluation of Apoptotic Response: To assess downstream consequences of proteasome inhibition, apoptosis assays are conducted using Annexin V/propidium iodide staining followed by flow cytometry. Cells are treated with inhibitors for 24-48 hours, harvested, and stained according to manufacturer protocols. Additionally, western blot analysis of caspase cleavage (caspase-3, -8, -9) and PARP cleavage provides complementary data on apoptotic pathway activation [136].
E1/E2/E3-Targeting Agents: Expanding the Precision Arsenal
Strategic Targeting of Ubiquitination Cascade Components
The therapeutic targeting of ubiquitin-activating (E1), ubiquitin-conjugating (E2), and ubiquitin-ligase (E3) enzymes represents a more precise approach to modulate specific UPS functions rather than global protein degradation. While E1 inhibitors such as TAK-243 (also known as MLN7243) have entered clinical evaluation, their development has been challenged by toxicity concerns stemming from the central role of E1 in all ubiquitination pathways [137]. More promising approaches have focused on the remarkable substrate specificity afforded by E3 ligases, with over 600 identified in the human genome offering substantial opportunities for selective intervention. Among the most clinically advanced E3-targeting modalities are proteolysis-targeting chimeras (PROTACs), bifunctional molecules that recruit E3 ligases to specific proteins of interest, inducing their ubiquitination and subsequent degradation [140]. This technology leverages the body's natural degradation machinery but redirects it toward disease-causing proteins, effectively expanding the druggable proteome to include transcription factors, scaffolding proteins, and other non-enzymatic targets previously considered undruggable.
DNA Repair and Immune Pathway Implications
The strategic targeting of E3 ligases holds particular significance in DNA repair pathways, where numerous E3s function as critical regulators of damage response. For instance, the RING-type E3 ligases RNF8 and RNF168 orchestrate the ubiquitin-dependent signaling cascade that recruits repair proteins like BRCA1 and 53BP1 to DSB sites, directly influencing repair pathway choice between homologous recombination and non-homologous end joining [15] [89]. Inhibition of specific E3 ligases can therefore sensitize cancer cells to DNA-damaging therapies by compromising their repair capacity. Similarly, in immune regulation, E3 ligases such as CBL-B regulate T-cell activation thresholds, presenting opportunities for immunomodulatory interventions. PROTACs targeting the androgen receptor (ARV-110, ARV-766) and estrogen receptor (ARV-471) have demonstrated clinical proof-of-concept, validating the therapeutic potential of targeted protein degradation [140]. These agents effectively degrade their targets by recruiting E3 ligases such as cereblon (CRBN) or von Hippel-Lindau (VHL), leading to sustained pathway suppression even after drug clearance, a potential advantage over conventional occupancy-based inhibitors.
Table 2: Selected E3-Targeting Agents in Clinical Development
| Agent | Target | E3 Ligase Recruited | Indication | Development Phase |
|---|---|---|---|---|
| ARV-110 (Bavdegalutamide) | Androgen Receptor | CRBN | Metastatic castration-resistant prostate cancer | Phase II |
| ARV-471 (Vepdegestrant) | Estrogen Receptor | CRBN | ER+/HER2- breast cancer | Phase III |
| ARV-766 | Androgen Receptor | CRBN | Metastatic castration-resistant prostate cancer | Phase II |
| CC-94676 (BMS-986365) | Androgen Receptor | CRBN | Metastatic castration-resistant prostate cancer | Phase III |
| KT-474 (SAR444656) | IRAK4 | Not specified | Hidradenitis suppurativa and atopic dermatitis | Phase II |
Experimental Protocols for E3-Targeting Agent Evaluation
PROTAC Degradation Assays: To assess target degradation by PROTACs, cells are treated with compound for 16-24 hours, followed by lysis and western blot analysis for the protein of interest. Time-course and dose-response experiments establish degradation kinetics and DC50 values (concentration causing 50% degradation). Rescue experiments with proteasome inhibitors (MG132, bortezomib) or E1 inhibitors (TAK-243) confirm UPS-dependent degradation, while nuclease-based assays (CRISPR knockout, siRNA) verify the requirement for specific E3 ligases [140].
Cellular Thermal Shift Assay (CETSA): To confirm target engagement by E3-targeting agents, CETSA is performed. Treated and untreated cells are heated to different temperatures (typically 37-65°C) for 3 minutes, followed by cooling, lysis, and removal of precipitated proteins by centrifugation. The remaining soluble target protein in supernatants is quantified by western blot, with stabilization against thermal denaturation indicating direct ligand binding [140].
Ubiquitination Assays: To demonstrate enhanced target ubiquitination, cells are co-transfected with plasmids expressing the target protein and HA- or FLAG-tagged ubiquitin. Following PROTAC treatment, immunoprecipitation of the target protein under denaturing conditions (to isolate covalently linked ubiquitin) is performed, followed by western blot with anti-tag antibodies to detect ubiquitin conjugates [140].
DUB Inhibitors: Emerging Players with Complex Biology
Therapeutic Targeting of Deubiquitination
Deubiquitinating enzymes (DUBs) constitute a diverse family of approximately 100 proteases that reverse protein ubiquitination, thereby stabilizing substrates and modulating their function. DUBs are categorized into seven primary families based on catalytic mechanism and domain structure: ubiquitin-specific proteases (USPs), ubiquitin C-terminal hydrolases (UCHs), ovarian tumor proteases (OTUs), Machado-Joseph disease protein domain proteases (MJDs), JAMM/MPN domain-associated metallopeptidases (JAMMs), Zinc finger containing ubiquitin peptidase 1 (ZUP1), and motif interacting with ubiquitin-containing novel DUB family (MINDYs) [137] [138]. The development of DUB inhibitors has progressed more slowly than other UPS-targeted therapies, reflecting challenges in achieving selectivity against specific DUB family members and understanding their complex biological roles. Nevertheless, several DUB inhibitors have entered early clinical development, targeting enzymes such as USP7, USP1, and USP14 that play roles in oncogenic signaling, DNA damage repair, and cell cycle regulation [137]. The therapeutic rationale for DUB inhibition centers on preventing the rescue of specific oncoproteins from degradation or disrupting DUB-mediated signaling pathways that promote cancer cell survival.
DNA Repair and Metabolic Interconnections
DUBs play particularly important roles in the DNA damage response, where they fine-tune the ubiquitin signals that control repair protein recruitment and activity. For instance, USP51 and USP3 deubiquitinate histone H2A at lysine 13/15, counteracting the activity of RNF168 and thereby modulating the recruitment of 53BP1 and BRCA1 to DSB sites to influence repair pathway choice [15]. Additionally, USP11 regulates homologous recombination by deubiquitinating PALB2, facilitating its interaction with BRCA1 and promoting RAD51 loading [15]. Beyond DNA repair, DUBs are increasingly recognized as important regulators of cancer metabolism, with enzymes such as JOSD2, CSN5, and USP29 stabilizing key metabolic enzymes or transcription factors to promote aerobic glycolysis, a hallmark of cancer metabolism known as the Warburg effect [138]. This intersection between DUB function, DNA repair, and metabolic reprogramming presents both challenges and opportunities for therapeutic intervention, as DUB inhibition may simultaneously disrupt multiple cancer-promoting pathways while requiring careful management of potential on-target toxicities.
Table 3: Selected DUB Inhibitors in Preclinical and Clinical Development
| Target DUB | Inhibitor Examples | Primary Mechanism and Consequences | Development Status |
|---|---|---|---|
| USP1 | KSQ-4279, ML323 | Impairs DNA damage repair; destabilizes oncogenic proteins | Clinical phases |
| USP7 | P5091, HBX 19818 | Stabilizes p53; disrupts MDM2 interaction | Preclinical/early clinical |
| USP14 | IU1 | Enhances proteasome activity; reduces aggregation-prone proteins | Preclinical |
| UCHL3 | Multiple research compounds | Affects RAD51-BRCA2 interaction; impairs homologous recombination | Preclinical |
Experimental Protocols for DUB Inhibitor Characterization
DUB Activity Assays: To evaluate DUB inhibition, fluorogenic ubiquitin-based substrates are employed wherein ubiquitin is fused to a quenched fluorescent reporter (e.g., Ub-AMC). Recombinant DUBs are incubated with inhibitors across a concentration range followed by substrate addition. Fluorescence development (excitation 380 nm/emission 460 nm for AMC) is monitored kinetically, with IC50 values calculated from the rate of substrate cleavage. Specificity profiling across multiple DUBs is essential given the conservation of active sites [137].
Activity-Based Protein Profiling (ABPP): To assess target engagement in cells, activity-based probes containing electrophilic groups that covalently label active site cysteines of DUBs are used. Cells are treated with inhibitors, followed by lysis and incubation with probes such as HA-Ub-VS or HA-Ub-amide. Labeled DUBs are captured on anti-HA beads and identified by western blot or mass spectrometry, confirming cellular target engagement and selectivity [137].
DNA Repair Functional Assays: To evaluate the impact of DUB inhibition on DNA repair, γH2AX foci formation is assessed by immunofluorescence following ionizing radiation. Cells are treated with inhibitors, irradiated (typically 2-10 Gy), fixed at various timepoints post-irradiation, and stained with anti-γH2AX antibodies. Foci counting provides quantitative assessment of DSB formation and repair kinetics. Complementary comet assays under alkaline conditions evaluate overall DNA damage [15].
Comparative Clinical Landscape and Developmental Challenges
The clinical development of UPS-targeting agents reflects a trajectory from broad proteasome inhibition to increasingly specific interventions targeting individual components of the ubiquitination machinery. Proteasome inhibitors remain the most clinically established class, with multiple approved agents and ongoing development focused on combination regimens, novel formulations, and application to new indications. The PROTAC platform has demonstrated remarkable progress, with several agents advancing to late-stage clinical trials and showing particular promise in targeting hormone receptors in prostate and breast cancers [140]. In contrast, DUB inhibitors largely remain in earlier stages of clinical development, reflecting the complex biology and challenges in achieving selectivity for this enzyme class.
Table 4: Comparative Clinical Landscape of UPS-Targeting Agents
| Therapeutic Class | Representative Agents | Highest Development Phase | Key Advantages | Major Challenges |
|---|---|---|---|---|
| Proteasome Inhibitors | Bortezomib, Carfilzomib, Ixazomib | FDA-approved | Established efficacy, predictable toxicities | Resistance development, peripheral neuropathy |
| E3-Targeting PROTACs | ARV-110, ARV-471, CC-94676 | Phase III | Target previously "undruggable" proteins, event-driven pharmacology | Molecular size may limit bioavailability, resistance mechanisms emerging |
| DUB Inhibitors | KSQ-4279 (USP1-targeted) | Early clinical | High specificity potential, novel mechanisms | Complex biology, potential compensatory mechanisms |
A significant challenge across all UPS-targeted therapies is the emergence of resistance mechanisms, including upregulation of alternative degradation pathways, mutations in drug targets, and activation of compensatory survival signaling. Additionally, the interconnectedness of UPS components creates potential for overlapping toxicities, particularly when targeting central regulators like the proteasome or E1 enzyme. The ongoing clinical trials increasingly incorporate biomarker strategies to identify patient populations most likely to benefit, with particular attention to genetic alterations in specific UPS components or dependencies created by oncogenic drivers.
The Scientist's Toolkit: Essential Research Reagents and Methodologies
Table 5: Key Research Reagents for UPS Pathway Investigation
| Reagent Category | Specific Examples | Primary Research Applications | Technical Considerations |
|---|---|---|---|
| Proteasome Inhibitors | MG132, Bortezomib, Carfilzomib | Validation of UPS-dependent processes, induction of ER stress | Varying selectivity profiles; cell-permeable vs. impermeable variants |
| DUB Inhibitors | PR-619 (pan-DUB inhibitor), P5091 (USP7 inhibitor) | Investigation of deubiquitination pathways, substrate stabilization | Limited selectivity for many tool compounds; requires validation |
| E1 Inhibitor | TAK-243 (MLN7243) | Global ubiquitination blockade, assessment of UPS-dependent processes | High toxicity; primarily for mechanistic studies |
| Ubiquitin Probes | HA-Ub-VS, TAMRA-Ub-amide | DUB activity profiling, target engagement studies | Covalent labeling; requires active enzyme for binding |
| PROTAC Molecules | Research-grade PROTACs (e.g., dBET1) | Targeted protein degradation studies, proof-of-concept investigations | Require specific E3 ligase expression; hook effect at high concentrations |
| Ubiquitin Binding Domains | Tandem ubiquitin-interacting entities (TUBEs) | Isolation and detection of ubiquitinated proteins | Affinity varies with ubiquitin chain linkage and length |
| Klinker Mutants | Ubiquitin mutants (K0, K48-only, K63-only) | Dissection of ubiquitin chain signaling in specific pathways | Overexpression may disrupt endogenous ubiquitin pools |
Signaling Pathways and Experimental Workflows
DNA Damage Response Pathway and Therapeutic Intervention Points
PROTAC-Mediated Protein Degradation Mechanism
Future Perspectives and Concluding Remarks
The clinical landscape of UPS-targeting therapies continues to evolve rapidly, with emerging trends including the development of combination strategies that simultaneously target multiple UPS components, the application of UPS modulators to enhance immunotherapy efficacy, and the pursuit of tissue-specific delivery approaches to improve therapeutic indices. The ongoing elucidation of UPS biology in DNA repair and immune signaling continues to reveal new therapeutic opportunities, particularly in cancers characterized by specific vulnerabilities in these pathways. Furthermore, advances in structural biology and chemical design are enabling more precise targeting of challenging UPS components, including protein-protein interfaces and allosteric regulatory sites. As the field progresses, the successful clinical translation of these agents will increasingly depend on robust biomarker strategies to identify patient populations most likely to benefit, careful management of potential compensatory mechanisms and resistance development, and thoughtful integration with established treatment modalities. The comparative analysis presented herein underscores both the substantial progress achieved and the significant opportunities that remain in targeting the UPS for therapeutic benefit across the spectrum of human malignancies, particularly those characterized by dysregulated DNA repair and immune response pathways.
The ubiquitin-proteasome system (UPS) represents a master regulatory network controlling cellular protein homeostasis, with profound implications for oncogenesis and cancer therapy. Ubiquitination, the reversible post-translational modification of proteins with the 76-amino acid ubiquitin polypeptide, governs virtually all cellular processes, from cell cycle progression and DNA damage repair to immune response activation and metabolic adaptation [27]. The exquisite specificity of this system arises from its enzymatic cascade—comprising E1 activating, E2 conjugating, and E3 ligase enzymes—while deubiquitinases (DUBs) provide reversibility, together creating a dynamic regulatory circuit that maintains cellular homeostasis [27] [11]. In cancer, this system becomes dysregulated, with specific E3 ligases and DUBs often hijacked to promote tumor survival, growth, and therapeutic resistance [27] [7]. The clinical validation of UPS targeting emerged with proteasome inhibitors like bortezomib for hematological malignancies, but this approach represents merely the tip of the therapeutic iceberg [27]. More recently, drug development has expanded to target specific nodes within the ubiquitin network, offering unprecedented opportunities for precision oncology while introducing complex efficacy-toxicity considerations across different cancer types.
This review examines the contemporary landscape of ubiquitin-targeting modalities, with particular emphasis on their therapeutic efficacy and toxicity profiles. We situate this analysis within the broader context of ubiquitination's fundamental roles in DNA repair fidelity and immune response regulation, two interconnected pathways that critically determine anticancer therapy success. As we will explore, the therapeutic window for ubiquitin-targeting agents is profoundly influenced by cancer-type-specific dependencies, the dynamic rewiring of ubiquitin signaling in treatment resistance, and the intricate balance between disrupting malignant processes while preserving normal cellular function.
Ubiquitin Biology in Cancer: DNA Repair and Immune Response Interplay
The Ubiquitin Code in DNA Damage Response and Repair
DNA double-strand breaks (DSBs) represent the most lethal form of DNA damage, and their faithful repair is essential for genomic integrity. The ubiquitin system orchestrates the entire DSB response, from initial lesion sensing to repair pathway choice and final resolution. Following DSB induction, the RNF8-RNF168 ubiquitin signaling cascade establishes a complex ubiquitin landscape at damage sites, primarily through histone H2A and H2AX modifications [15] [141]. This ubiquitin signaling recruits downstream repair factors, with K63-linked polyubiquitin chains serving as non-proteolytic scaffolds for protein assembly, while K48-linked chains often regulate protein turnover to control pathway progression [15] [11].
Critical to therapeutic targeting, the ubiquitin system directly governs the choice between two major DSB repair pathways: non-homologous end joining (NHEJ) and homologous recombination (HR). The recruitment of 53BP1 to RNF168-ubiquitinated chromatin promotes NHEJ by limiting DNA end resection, whereas BRCA1 recruitment favors HR through opposing mechanisms [15] [141]. This competition has profound therapeutic implications, as cancer cells with HR deficiencies (e.g., BRCA1/2 mutations) become dependent on alternative repair pathways, creating synthetic lethal opportunities for ubiquitin-targeting agents [142].
Table 1: Key Ubiquitin Ligases in DNA Damage Response and Their Cancer Relevance
| Ubiquitin Ligase | Ubiquitin Linkage | Substrates/Pathways | Cancer Relevance |
|---|---|---|---|
| RNF8 | K63-linked | L3MBTL2, amplification of RNF168 recruitment | Overexpressed in various cancers; promotes repair and radioresistance |
| RNF168 | K13/K15 on H2A/H2AX | H2A/H2AX, recruits 53BP1/BRCA1 | Mutated in RIDDLE syndrome; targeted for radio-sensitization |
| BRCA1/BARD1 | K6/K29-linked | H2A, CtIP, promotes HR repair | Mutated in hereditary breast/ovarian cancer; synthetic lethal with PARP inhibitors |
| FBXW7 | K48-linked | SOX9, p53, NOTCH; context-dependent degradation | Frequently mutated cancers; dual roles in radioresistance |
| UHRF1 | K63-linked | RIF1, regulates DSB repair pathway choice | Overexpressed in cancers; impacts therapeutic response |
Ubiquitin-Mediated Regulation of Antitumor Immunity
Parallel to its DNA repair functions, the ubiquitin system extensively modulates antitumor immunity through multiple mechanisms. The UPS regulates the stability and function of immune checkpoint proteins such as PD-1 and PD-L1, directly influencing T-cell-mediated antitumor responses [143]. For instance, the E3 ligase FBXO38 targets PD-1 for K48-linked polyubiquitination and degradation, thereby promoting T cell activation, while the deubiquitinase USP8 stabilizes PD-L1, enhancing immune evasion [143]. Beyond checkpoint regulation, ubiquitination controls key immune signaling pathways, including NF-κB activation through K63-linked ubiquitination events that trigger pro-inflammatory gene expression [97]. Additionally, the ubiquitin system shapes the tumor immune microenvironment by influencing the function and differentiation of various immune cells, including regulatory T cells (Tregs), dendritic cells, and macrophages [143]. This immunomodulatory capacity creates both challenges and opportunities for ubiquitin-targeting therapies, as their effects extend beyond cancer cells to the broader tumor microenvironment.
Ubiquitin-Targeting Modalities: Mechanisms and Clinical Applications
Proteasome Inhibitors: The First-Generation UPS Therapeutics
Proteasome inhibitors represented the first clinical validation of UPS targeting in oncology, with bortezomib earning FDA approval in 2003 for relapsed multiple myeloma [27]. These agents function through reversible inhibition of the 26S proteasome's chymotrypsin-like activity, leading to accumulation of polyubiquitinated proteins, disruption of protein homeostasis, and ultimately apoptosis, particularly in secretory cells with high protein production rates [27]. While proteasome inhibitors demonstrate remarkable efficacy in hematological malignancies like multiple myeloma and mantle cell lymphoma, their utility in solid tumors remains limited by insufficient efficacy and peripheral neuropathy, a treatment-limiting toxicity [27]. This differential efficacy highlights the concept of cancer-type-specific vulnerabilities to UPS disruption, a theme that recurs across ubiquitin-targeting modalities.
E3 Ligase Modulators: Expanding the Targeting Landscape
Beyond proteasome inhibition, drug development has focused on modulating specific E3 ligases to achieve more precise targeting. The cullin-RING ligase (CRL) family represents a particularly attractive target class, with multiple agents in clinical development [27]. MDM2 inhibitors, which disrupt the p53-MDM2 interaction to stabilize the tumor suppressor, have shown promise in hematological malignancies with wild-type TP53, though clinical efficacy has been hampered by on-target hematological toxicity [27]. Similarly, IAP antagonists have been developed to promote apoptosis by modulating the ubiquitin-mediated regulation of caspase activity, with several agents evaluated in clinical trials [27]. A key challenge in E3-targeted therapy is the context-dependent duality of many ligases; for example, FBXW7 can function as either an oncogene or tumor suppressor depending on cellular background, potentially activating oncogenic drivers like SOX9 while simultaneously degrading tumor suppressors like p53 [7].
Deubiquitinase (DUB) Inhibitors: Emerging Therapeutic Class
Deubiquitinases represent the other major enzymatic component of the ubiquitin system, with approximately 100 human DUBs reversing ubiquitination signals [142]. The largest DUB family, ubiquitin-specific proteases (USPs), has attracted significant therapeutic interest. USP7 inhibitors have shown preclinical promise by stabilizing p53 and other tumor suppressors, though clinical development has encountered challenges with specificity and toxicity [142]. USP1 inhibitors have advanced based on synthetic lethal approaches in HR-deficient cancers, with multiple agents (KSQ-4279, XL-309, TNG348) entering Phase I/II clinical trials, both as monotherapies and in combination with PARP inhibitors [142]. However, the clinical translation of DUB inhibitors faces substantial hurdles, including the high conservation of catalytic sites across USP family members and the multifunctional scaffolding roles of many DUBs beyond their enzymatic activity [142].
Table 2: Selected Ubiquitin-Targeting Agents in Clinical Development
| Therapeutic Agent | Target | Modality | Cancer Types | Development Phase |
|---|---|---|---|---|
| Bortezomib | 26S Proteasome | Reversible inhibitor | Multiple myeloma, mantle cell lymphoma | FDA-approved |
| KSQ-4279 (Roche) | USP1 | Small molecule inhibitor | Solid tumors, HR-deficient cancers | Phase I/II |
| XL-309 (Exelixis) | USP1 | Small molecule inhibitor | Solid tumors | Phase I/II |
| TNG348 (Tango Therapeutics) | USP1 | Small molecule inhibitor | Solid tumors, BRCA1/2 mutations | Phase I/II (discontinued) |
| FT671 | USP7 | Small molecule inhibitor | Hematological malignancies | Preclinical |
| ML323 | USP1/UAF1 | Small molecule inhibitor | Solid tumors | Preclinical |
Induced-Proximity Modalities: PROTACs and Molecular Glues
The most revolutionary advance in ubiquitin-targeted therapy has been the development of induced-proximity modalities, particularly PROteolysis TArgeting Chimeras (PROTACs) and molecular glue degraders (MGDs). These agents hijack the endogenous ubiquitin system to selectively degrade target proteins of interest. PROTACs are bifunctional molecules comprising a target-binding warhead connected via a linker to an E3 ligase recruiter, forming a ternary complex that directs ubiquitination and proteasomal degradation of the target [142]. This approach offers several advantages over traditional inhibition, including the ability to target "undruggable" proteins, achieve sustained pharmacological effects beyond drug exposure, and potentially overcome resistance mechanisms [7].
Notably, radiation-responsive PROTAC platforms are emerging to combat radioresistance, including radiotherapy-triggered PROTAC (RT-PROTAC) prodrugs activated by tumor-localized X-rays to degrade BRD4/2 in breast cancer models, and X-ray-responsive nanomicelles that selectively release PROTACs within irradiated tumors [7]. These innovations represent promising strategies for spatial and temporal control of protein degradation.
Molecular glue degraders typically function by stabilizing the interaction between an E3 ligase and its natural substrate or by inducing novel protein-protein interactions. Clinically approved immunomodulatory drugs (IMiDs) like lenalidomide represent the most advanced MGDs, redirecting the CRL4CRBN E3 ligase to degrade transcription factors IKZF1/3 in multiple myeloma [142]. Compared to PROTACs, MGDs typically have lower molecular weight and better oral bioavailability, though they can face resistance through unintended stabilization of non-target proteins [142].
A more recent innovation, Deubiquitinase Targeting Chimeras (DUBTACs), aims to stabilize rather than degrade target proteins by recruiting DUBs to remove ubiquitin chains, though this approach faces significant technical challenges and remains in earlier development [142].
Comparative Efficacy and Toxicity Across Cancer Types
Hematological vs. Solid Tumors: Differential Vulnerabilities
The therapeutic efficacy of ubiquitin-targeting modalities varies substantially between hematological malignancies and solid tumors, reflecting fundamental biological differences. Proteasome inhibitors demonstrate remarkable efficacy in multiple myeloma, where they induce apoptosis by disrupting protein homeostasis in antibody-producing cells with high secretory activity [27]. Similarly, molecular glue degraders like lenalidomide show significant activity in hematological cancers by targeting cell lineage-specific transcription factors [142]. In contrast, solid tumors have generally proven less responsive to these approaches, likely due to reduced penetrance, tumor microenvironmental factors, and divergent dependencies on specific ubiquitin pathways [27].
The toxicity profiles also differ across tumor types, influenced by both on-target and off-target effects. Dose-limiting hematological toxicities commonly occur with MDM2 inhibitors in leukemias, while peripheral neuropathy frequently limits proteasome inhibitor dosing in multiple myeloma [27]. In solid tumors, on-target toxicities often manifest in rapidly dividing tissues, though the expanded use of combination regimens complicates toxicity attribution.
DNA Repair-Deficient Cancers: Exploiting Synthetic Lethality
Cancers with deficiencies in DNA repair pathways, particularly HR-deficient tumors with BRCA1/2 mutations, exhibit heightened sensitivity to specific ubiquitin-targeting approaches. USP1 inhibitors have demonstrated synthetic lethality in HR-deficient models, with clinical trials exploring combinations with PARP inhibitors to overcome or prevent resistance [142]. The mechanistic basis involves disrupted regulation of DNA damage response factors like PCNA and FANCD2, creating catastrophic replication stress in already repair-compromised cells [142]. Similarly, PROTACs targeting DNA damage response proteins (e.g., BRCA1, RAD51) show enhanced efficacy in repair-deficient contexts, though clinical validation is ongoing [7].
The therapeutic window in these settings is favorably influenced by cancer cell-specific vulnerability, potentially allowing lower dosing that preserves efficacy while minimizing toxicity. However, the discontinuation of TNG348 due to severe liver toxicity underscores that synthetic lethality does not guarantee an adequate therapeutic index, emphasizing the need for careful safety evaluation [142].
Resistance Mechanisms and Adaptive Responses
The efficacy of ubiquitin-targeting therapies is frequently limited by both intrinsic and acquired resistance. Primary resistance mechanisms include functional redundancy within the ubiquitin system, where related E3 ligases or DUBs compensate for inhibited family members [142]. Tumors also activate adaptive responses, such as ubiquitin code rewiring where cancer cells strategically manipulate K63-linked chains to stabilize DNA repair factors while concurrently inhibiting K48-mediated degradation of survival proteins [7]. Additional resistance pathways include E3 ligase downregulation in response to PROTAC treatment, reducing degradation efficiency, and point mutations in either the target protein or E3 ligase that disrupt ternary complex formation [142].
Understanding these resistance mechanisms is crucial for designing effective combination strategies and sequencing approaches. For instance, combining USP1 inhibitors with PARP inhibitors may prevent or overcome resistance in HR-deficient cancers, while radiotherapy-PROTAC combinations leverage spatial activation to enhance tumor-specific degradation while sparing normal tissues [7] [142].
Experimental Approaches and Research Tools
Methodologies for Evaluating Efficacy and Toxicity
Robust preclinical assessment of ubiquitin-targeting agents requires integrated methodological approaches spanning biochemical, cellular, and in vivo models. Key experimental protocols include:
Ubiquitination Assays: In vitro ubiquitination reactions reconstitute the enzymatic cascade using purified E1, E2, E3 enzymes, ubiquitin, and ATP to demonstrate direct substrate modification. These assays determine linkage specificity using ubiquitin mutants (e.g., K48R, K63R) and identify the specific E2-E3 partnerships governing chain topology [15] [11]. For DUB inhibitors, fluorescently-labeled ubiquitin chains (e.g., Ub-AMC) or di-ubiquitin substrates monitor cleavage inhibition.
DNA Repair Functional Assays: Immunofluorescence-based quantification of γH2AX foci formation and resolution kinetics provides a direct measure of DSB recognition and repair proficiency [15] [141]. Complementary clonogenic survival assays evaluate long-term replication capacity following DNA damage induction in the presence of ubiquitin-targeting agents. To specifically assess repair pathway utilization, DR-GFP and EJ5-GFP reporter systems directly measure HR and NHEJ efficiency, respectively, following DSB induction by sequence-specific endonucleases [15].
Immune Activation Profiling: Coculture systems incorporating tumor cells with peripheral blood mononuclear cells or tumor-infiltrating lymphocytes assess immune checkpoint regulation and T-cell activation following UPS perturbation [143]. Flow cytometric analysis of surface PD-1/PD-L1 expression, combined with cytokine secretion profiling (IFN-γ, TNF-α, IL-2), provides quantitative measures of immune modulation [143].
In Vivo Efficacy and Toxicity Models: Syngeneic mouse models enable evaluation of both direct antitumor effects and immunomodulatory activity within an intact immune system [143]. Patient-derived xenografts maintain human tumor biology more faithfully for efficacy assessment. Critical toxicity evaluations include detailed hematological profiling, neurological assessment for peripheral neuropathy, and hepatic/renal function tests, with particular attention to on-target toxicities predicted by target expression in normal tissues.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Ubiquitin-Targeting Studies
| Research Tool | Application | Utility and Function |
|---|---|---|
| Tandem Ubiquitin Binding Entities (TUBEs) | Affinity purification of polyubiquitinated proteins | Protect ubiquitin chains from DUB activity during extraction; identify ubiquitinated substrates |
| Linkage-Specific Ub Antibodies (K48, K63) | Immunoblot, immunofluorescence | Distinguish chain topology in cellular responses; monitor treatment effects on ubiquitin signaling |
| Proteasome Activity Probes | Functional proteasome assessment | Measure chymotrypsin-, trypsin-, and caspase-like activity in live cells or lysates |
| HA-Ubiquitin, FLAG-Ubiquitin plasmids | Transfection-based ubiquitination studies | Trace ubiquitin conjugation and identify modified substrates via epitope tags |
| CRISPR/Cas9 E3/DUB knockout libraries | Functional genomic screens | Identify synthetic lethal interactions and resistance mechanisms |
| Radiation-responsive PROTACs | Spatiotemporally-controlled degradation | Achieve localized protein degradation in irradiated tumor volumes |
Ubiquitin Targeting Modalities and Outcomes
Ubiquitin in DNA Repair and Therapeutic Targeting
The therapeutic targeting of the ubiquitin system has evolved dramatically from broad proteasome inhibition to highly precise modalities that exploit specific nodes within the ubiquitin network. The efficacy-toxicity balance for these agents is profoundly influenced by cancer-type-specific dependencies, the molecular context of tumors, and the dynamic adaptability of ubiquitin signaling in response to therapeutic pressure. Future progress will require deeper understanding of ubiquitin code complexity, including the functional roles of atypical chain linkages and their interplay with other post-translational modifications in determining cancer cell behavior.
Promising directions include the development of biomarker-guided patient selection strategies to identify tumors most vulnerable to specific ubiquitin-targeting approaches, the rational design of combination therapies that simultaneously target multiple UPS nodes or co-target complementary pathways, and advancing novel delivery platforms that enhance tumor-specific activity while minimizing on-target toxicity in normal tissues. The integration of ubiquitin-targeting agents with immunotherapy, radiotherapy, and targeted therapies represents a particularly compelling opportunity to leverage the dual roles of the UPS in DNA repair and immune regulation for enhanced anticancer efficacy.
As the field continues to mature, the ongoing challenge will be to translate our growing mechanistic understanding of ubiquitin biology into clinical strategies that maximize therapeutic index across the spectrum of human malignancies. The expanding toolkit of ubiquitin-targeting modalities, from PROTACs to molecular glues and beyond, offers unprecedented opportunities to manipulate cancer-relevant pathways with increasing precision, potentially ushering in a new era of targeted protein regulation for cancer therapy.
The ubiquitin-proteasome system (UPS) represents a complex post-translational modification network that governs critical cellular processes, including DNA repair and immune response. This technical guide examines current methodologies for identifying and validating ubiquitination-related biomarkers and their correlation with clinical outcomes. We explore experimental protocols spanning bioinformatic discovery to clinical validation, focusing on ubiquitin's role in cancer therapy resistance and immune regulation. The document provides a comprehensive framework for researchers seeking to establish robust ubiquitin-related signatures for prognostic assessment and therapeutic targeting, with particular emphasis on overcoming challenges in biomarker harmonization across diverse patient cohorts.
Ubiquitination constitutes a pivotal post-translational modification mechanism that orchestrates nearly all biological processes through a coordinated enzymatic cascade involving E1 activating, E2 conjugating, and E3 ligase enzymes [28]. The ubiquitin code—comprising diverse chain topologies and linkages—generates a sophisticated signaling network that regulates protein stability, localization, activity, and interactions [14]. Beyond its canonical role in proteasomal degradation, ubiquitination dynamically controls DNA damage repair pathways and immune response modulation, positioning the ubiquitin system as a critical regulator of disease pathogenesis and treatment response [7].
The reversible nature of ubiquitination, governed by the opposing actions of E3 ligases and deubiquitinating enzymes (DUBs), creates a dynamic regulatory network that tumors exploit to develop therapy resistance [7]. Understanding the complex interplay between ubiquitin signaling and clinical outcomes requires sophisticated biomarker discovery and validation approaches that can decode this complexity into clinically actionable signatures. This guide outlines the methodologies and frameworks for establishing such ubiquitin-related biomarker correlations in patient cohorts.
Molecular Mechanisms: Ubiquitination in DNA Repair and Immune Regulation
Ubiquitin Chain Typology and Functional Specificity
The ubiquitin code encompasses diverse chain architectures that dictate functional outcomes in DNA repair and immune signaling:
- K48-linked ubiquitination: Primarily targets proteins for proteasomal degradation, functioning as a proteostatic gatekeeper with contextual duality in radiation response [7]. For instance, FBXW7 exemplifies this duality by promoting radioresistance in p53-wild type colorectal tumors while enhancing radiosensitivity in non-small cell lung cancer with SOX9 overexpression [7].
- K63-linked ubiquitination: Facilitates non-proteolytic signaling cascades in DNA repair and immune activation [7]. These chains act as scaffolds for protein complex assembly, with TRAF4 utilizing K63 modifications to activate the JNK/c-Jun pathway, driving overexpression of anti-apoptotic Bcl-xL in colorectal cancer [7].
- Monoubiquitination: Regulates chromatin dynamics and genome stability through histone and non-histone protein modification [7]. UBE2T/RNF8-mediated H2AX monoubiquitylation accelerates DNA damage detection in hepatocellular carcinoma, while FANCD2 monoubiquitylation resolves carbon ion-induced DNA crosslinks [7].
Table 1: Ubiquitin Chain Linkages and Their Functional Roles in Disease Contexts
| Linkage Type | Primary Function | DNA Repair Role | Immune Regulation | Exemplary Proteins |
|---|---|---|---|---|
| K48-linked | Proteasomal targeting | Context-dependent regulation of repair factors | Degradation of immune signaling components | FBXW7, SMURF2 |
| K63-linked | Signaling scaffold | DNA repair complex assembly | Immune pathway activation | TRAF4, TRAF6, TRIM26 |
| K11/K27-linked | Specialized processes | MRE11 activation in TNBC | Inflammatory signaling | RNF126 |
| Monoubiquitination | Protein activity & localization | Histone modification at damage sites | Immune cell signaling | UBE2T, RNF8, FANCD2 |
Ubiquitin Signaling in DNA Damage Response
The ubiquitin system orchestrates DNA repair fidelity through spatiotemporal control of repair factor recruitment and activity. Following radiation-induced DNA damage, RNF168 activation—amplified by ZNF451-dependent SUMOylation—ubiquitinates H2A/H2AX to open chromatin and recruit BRCA1-A complexes, enhancing repair fidelity but potentially promoting radioresistance [7]. The functional interdependency between ubiquitination and other post-translational modifications creates a sophisticated regulatory network that tumors manipulate to survive genotoxic stress.
Radiation dynamically reprograms ubiquitin signaling by altering chain formation, creating vulnerabilities exploited by tumors. Cancer cells strategically manipulate K63-linked chains to stabilize DNA repair factors (e.g., BRCA1) while concurrently inhibiting K48-mediated degradation of survival proteins like GPX4 [7]. This ubiquitin signaling rewiring represents a key resistance mechanism that biomarker strategies must capture.
Ubiquitin-Mediated Immune Regulation
Ubiquitination critically regulates tumor-immune crosstalk through multiple mechanisms. TRIM21 suppresses antitumor immunity by promoting K48-linked degradation of VDAC2, inhibiting mitochondrial DNA release and cGAS/STING activation in nasopharyngeal carcinoma [7]. Conversely, blocking USP14 stabilizes K63-ubiquitinated IRF3, amplifying STING-dependent type I interferon responses and synergizing with radiotherapy to overcome immune evasion [7]. These immune-editing functions position ubiquitin enzymes as pivotal regulators of tumor microenvironments, suggesting combinatory strategies with immune checkpoint inhibitors.
Experimental Workflows for Ubiquitin Biomarker Discovery
Bioinformatic Identification of Ubiquitination-Related Biomarkers
The initial discovery phase for ubiquitination-related biomarkers employs integrated bioinformatic analyses of multi-omics data. A representative workflow for cervical cancer research illustrates this approach [144]:
Diagram 1: Ubiquitin Biomarker Discovery Workflow
This bioinformatic pipeline identified five key ubiquitination-related biomarkers (MMP1, RNF2, TFRC, SPP1, and CXCL8) in cervical cancer that demonstrated significant prognostic value [144]. The risk score model constructed from these biomarkers effectively predicted patient survival rates (AUC >0.6 for 1/3/5 years) and revealed significant differences in immune cell infiltration patterns between high-risk and low-risk groups [144].
Correlation-Based Network Analysis
Correlation-based network analysis (CNA) provides a powerful data-mining method for visualizing disease-related perturbations of molecular interactions in ubiquitin signaling networks [145]. In this approach, biomarkers are represented as nodes and the statistical correlations between them as edges, allowing identification of highly connected hubs that may drive disease pathogenesis.
Key network properties for interpretation include:
- Node degree: The number of other nodes to which a given node is significantly correlated
- Betweenness centrality: The measure of shortest paths between any two nodes that passes through the node in question
- Network density: The ratio of existing edges to the total number of possible edges in a network [145]
Application of CNA in obesity with metabolic syndrome research revealed a denser network (total edges, n=369) compared to healthy weight individuals (n=299), with three key hubs identified in the immune cell abundance group: Treg cells, neutrophils, and cytotoxic cells [145]. This approach demonstrated superiority over traditional univariate analysis by revealing complex interaction patterns that would otherwise remain undetected.
Methodological Protocols for Biomarker Validation
Laboratory Techniques for Ubiquitin Biomarker Verification
RNA Extraction and Quantitative PCR (qPCR) Total RNA is extracted and purified from tissue samples using TRIzol reagent following manufacturer's instructions [144]. RNA quantity and purity are evaluated using a NanoDrop ND-1000 spectrophotometer, with integrity confirmed through agarose gel electrophoresis. For qPCR validation, RNA is reverse-transcribed into cDNA, which is then amplified using gene-specific primers for target ubiquitination-related genes [144] [146]. The comparative Ct method (2^-ΔΔCt) is used for relative quantification, with normalization to housekeeping genes.
Western Blot Analysis Protein extracts from patient tissues or cell lines are separated by SDS-PAGE and transferred to membranes [146]. After blocking, membranes are incubated with primary antibodies against target ubiquitination-related proteins (e.g., USP15, CUL2), followed by appropriate secondary antibodies. Signal detection is performed using enhanced chemiluminescence, with quantification relative to loading controls.
Immunohistochemical Validation Formalin-fixed, paraffin-embedded tissue sections are deparaffinized, rehydrated, and subjected to antigen retrieval. After peroxidase quenching and blocking, sections are incubated with primary antibodies against ubiquitination-related biomarkers, followed by detection with biotinylated secondary antibodies and streptavidin-HRP complexes. Staining intensity and distribution are scored by experienced pathologists.
Statistical Approaches for Multi-Cohort Harmonization
A major challenge in biomarker validation arises when combining data across multiple studies where biomarker data may be generated using different assay platforms, scanner types, or processing protocols [147]. Bridging studies that involve re-processing a subset of samples across platforms provide a solution for harmonizing biomarker data.
Latent Variable Model for Biomarker Harmonization This approach conceptualizes that a latent biomarker underlies the observed biomarkers across studies and integrates data from bridging studies with study-specific biomarker data for estimating biological correlations between biomarkers and clinical outcomes [147]. The model accounts for measurement error and platform-specific effects, enabling unbiased correlation estimates.
For two studies measuring biomarker B with different platforms, the model structure is:
- Let B represent the true concentration of a protein that cannot be perfectly measured
- Let B_k be the measurement of B from study k (k=1,2)
- A bridging cohort of size n provides paired measurements (B1j, B2j) for j=1,...,n
- Additional unpaired measurements are available from each study (n1 from Study 1, n2 from Study 2)
- The model assumes subjects from both studies are independent random samples from the same population [147]
Table 2: Statistical Methods for Biomarker Correlation Analysis
| Method | Application Context | Key Assumptions | Advantages | Limitations |
|---|---|---|---|---|
| Latent Variable Model | Multi-study harmonization with bridging samples | Random sampling from same population | Accounts for measurement error | Complex implementation |
| Spearman Correlation | Non-parametric relationship assessment | Monotonic relationship | Robust to outliers | Less powerful than parametric |
| Cox Proportional Hazards | Survival outcome analysis | Proportional hazards | Handles censored data | Requires linearity assumption |
| LASSO Cox Regression | High-dimensional prognostic model | Sparse true signal | Automatic feature selection | Requires careful tuning |
| Correlation-Based Network Analysis | Multi-analyte interaction mapping | Linear relationships | Visualizes complex interactions | Correlation ≠ causation |
Case Studies: Ubiquitin Biomarkers in Disease Contexts
Cervical Cancer: Prognostic Ubiquitination Signature
A comprehensive study identified and validated a 5-gene ubiquitination-related signature for predicting cervical cancer outcomes [144]. The research utilized self-collected transcriptomic data from 8 human cervical cancer tissue samples and their adjacent non-cancerous tissues, combined with public data from TCGA-GTEx-CESC dataset (304 tumor, 13 normal samples) for discovery, and GSE52903 (55 tumor, 17 normal samples) for validation.
The analytical process included:
- Identification of differentially expressed genes (DEGs) between tumor and normal samples
- Intersection of DEGs with 465 ubiquitination-related genes from GeneCards
- Functional enrichment analysis of ubiquitination-related DEGs using GO, KEGG, and GSEA
- Prognostic model construction via univariate Cox regression and LASSO algorithms
- Immune infiltration analysis between risk subgroups
- Experimental validation using RT-qPCR
This approach identified five key biomarkers (MMP1, RNF2, TFRC, SPP1, and CXCL8) that formed a prognostic signature significantly associated with patient survival [144]. The risk score model effectively stratified patients into high-risk and low-risk groups with distinct clinical outcomes, demonstrating the clinical potential of ubiquitination-related signatures.
Chronic Obstructive Pulmonary Disease: USP15 and CUL2
In chronic obstructive pulmonary disease (COPD), bioinformatic analysis identified USP15 and CUL2 as ubiquitination-related biomarkers involved in disease progression [146]. The study analyzed gene microarray datasets from COPD patients, identifying 2,932 differentially expressed genes and 96 differentially expressed ubiquitination-related genes.
Functional enrichment analysis revealed that these ubiquitination-related genes were primarily involved in post-translational protein modification and the ubiquitin ligase complex, with KEGG pathway analysis showing enrichment in ubiquitin-mediated proteolysis and TNF signaling pathway [146]. Experimental validation through qPCR and Western blot confirmed elevated expression of USP15 and CUL2 in COPD patients compared to controls, consistent with the bioinformatic predictions.
Breast Cancer: Soluble VEGFR2 as Prognostic Indicator
In a sub-study of the Zamboney trial investigating fulvestrant with vandetanib or placebo in bone-predominant metastatic breast cancer, baseline serum biomarker analysis revealed soluble VEGFR2 (sVEGFR2) as a prognostic factor for overall survival (HR=0.77, 95% CI=0.61-0.96, p=0.020) [148]. Although the primary trial showed no benefit from adding vandetanib to fulvestrant, biomarker analysis demonstrated that increased baseline sVEGFR2 was associated with improved clinical outcomes, supporting future studies on its role in bone metastasis progression.
Table 3: Essential Research Reagents for Ubiquitin Biomarker Studies
| Reagent/Resource | Specific Examples | Application | Key Features |
|---|---|---|---|
| Ubiquitin-Related Gene Sets | GeneCards ubiquitin-like modifiers (465 genes) [144] | Biomarker discovery | Comprehensive ubiquitination-related gene collection |
| Bioinformatics Tools | DESeq2, clusterProfiler, pheatmap, ggplot2 [144] | Differential expression and enrichment analysis | Specialized for omics data analysis |
| ELISA Kits | Quantikine VEGF-A, sVEGFR2, sVEGFR3 [148] | Serum biomarker quantification | High sensitivity and specificity |
| Cell Line Models | COPD cell models [146] | Functional validation | Disease-relevant experimental systems |
| Clinical Cohorts | TCGA-CESC, GTEx, GEO datasets [144] | Validation across populations | Multi-center patient data |
| Statistical Environment | R with Shiny, SIMCor platform [149] | In-silico trial validation | Open-source, menu-driven interface |
The validation of ubiquitination-related biomarker signatures represents a promising frontier in personalized medicine, particularly for cancer therapy and chronic disease management. The complex nature of ubiquitin signaling—with its diverse chain topologies, functional pleiotropy, and context-dependent outcomes—requires sophisticated correlation-based approaches that can decode this complexity into clinically actionable information.
Successful implementation of ubiquitin-related biomarkers depends on robust analytical frameworks that integrate multi-omics data, account for technical variability across platforms, and validate findings in independent cohorts. The methodologies outlined in this guide provide a roadmap for researchers seeking to establish such biomarkers for prognostic assessment, therapeutic stratification, and treatment monitoring.
As our understanding of the ubiquitin network deepens, and as technological advances continue to enhance our ability to measure and interpret ubiquitin-related signatures, these biomarkers are poised to become fundamental components of next-generation diagnostic and therapeutic strategies, particularly in the context of DNA damage response and immune modulation.
The evolution of targeted cancer therapeutics has transformed the treatment landscape for hematological malignancies, with proteasome inhibitors, immunomodulatory drugs, and next-generation engineered agents representing pivotal advances. This review delineates the mechanistic frameworks of bortezomib, lenalidomide, and emerging therapeutic classes, contextualizing their activity within ubiquitination pathways and associated DNA repair and immune response systems. Understanding these intricate mechanisms provides a foundational framework for developing novel agents that exploit ubiquitin-proteasome system (UPS) vulnerabilities and modulate immune surveillance, offering enhanced therapeutic precision against malignant cells.
Core Mechanisms of Established Agents
Bortezomib: Proteasome Inhibition and Immunogenic Cell Death
Bortezomib, a first-in-class proteasome inhibitor, exerts its primary antineoplastic effect through reversible inhibition of the 26S proteasome's chymotrypsin-like activity, disrupting the ubiquitin-proteasome system (UPS) and leading to the accumulation of polyubiquitinated proteins, proteotoxic stress, and apoptosis [150] [151]. The average growth inhibition value (GI₅₀) of bortezomib across 60 NCI cancer cell lines is 7 nM, demonstrating considerable potency [151]. Beyond this direct cytotoxicity, bortezomib triggers immunogenic cell death (ICD), characterized by the pre-apoptotic exposure of calreticulin on the myeloma cell surface, which facilitates phagocytosis by dendritic cells and subsequent activation of myeloma-specific immunity [152].
A pivotal study demonstrated that bortezomib-induced ICD is mediated via activation of the cGAS/STING pathway, culminating in type-I interferon production [152]. This pathway activation enhances the tumor microenvironment's immunogenicity and promotes antitumor immune responses. The clinical relevance of this mechanism is underscored by the identification of a bortezomib-triggered ICD gene signature that correlates with improved patient outcomes in multiple myeloma cohorts [152]. Furthermore, combining bortezomib with STING agonists synergistically potentiates this immunogenic cell death, providing a rational basis for novel combination strategies [152].
Lenalidomide: Immunomodulation and Ubiquitin Ligase Modulation
Lenalidomide, a cornerstone immunomodulatory drug (IMiD), exhibits pleiotropic mechanisms encompassing immunomodulation, anti-angiogenesis, and direct antitumor effects, with its predominant activity varying across pathological contexts [153] [154]. In multiple myeloma, lenalidomide directly inhibits malignant plasma cell proliferation and disrupts protective interactions with bone marrow stromal cells [153] [155]. Its immunomodulatory potency is evidenced by its capacity to downregulate pro-inflammatory cytokines, including TNF-α, with an efficacy up to 50,000-fold greater than its predecessor, thalidomide [153].
A fundamental mechanism involves the cereblon (CRBN)-dependent ubiquitination pathway. Lenalidomide binds to CRBN, a component of the CRL4ᶜᴿᴮᴺ E3 ubiquitin ligase complex, altering its substrate specificity [153]. This molecular interaction leads to the targeted ubiquitination and subsequent proteasomal degradation of specific lymphoid transcription factors, namely Ikaros (IKZF1) and Aiolos (IKZF3) [155]. The depletion of these proteins directly induces cytotoxicity in malignant B cells and concurrently enhances IL-2 production, thereby augmenting T and NK cell-mediated antitumor immunity [153] [155]. Lenalidomide also costimulates T cells via CD28 tyrosine phosphorylation, leading to enhanced NF-κB activation and increased production of IFN-γ and IL-2, which further amplifies immune effector functions [153].
Table 1: Primary Mechanisms of Action of Bortezomib and Lenalidomide
| Feature | Bortezomib | Lenalidomide |
|---|---|---|
| Molecular Target | 26S proteasome (chymotrypsin-like activity) | Cereblon (CRBN) E3 ubiquitin ligase adapter |
| Primary Effect | Accumulation of polyubiquitinated proteins, proteotoxic stress | Altered substrate specificity of CRL4ᶜᴿᴮᴺ ligase |
| Key Downstream Events | cGAS/STING pathway activation, type-I interferon production | Ubiquitination & degradation of Ikaros (IKZF1) & Aiolos (IKZF3) |
| Immunological Consequences | Immunogenic Cell Death (ICD); calreticulin exposure; dendritic cell phagocytosis | T-cell co-stimulation; NK cell activation; cytokine modulation (↓TNF-α, ↑IL-2) |
| Experimental GI₅₀/IC₅₀ | 7 nM (average growth inhibition in NCI cell lines) [151] | Varies by cellular context; pleiotropic immunomodulatory effects |
Next-Generation Therapeutic Platforms
Antibody-Drug Conjugates (ADCs)
Antibody-Drug Conjugates (ADCs) represent a paradigm of precision oncology, comprising a monoclonal antibody covalently linked to a potent cytotoxic payload via a bifunctional linker [156]. This design enables targeted delivery of chemotherapeutic agents to tumors expressing specific surface antigens, minimizing systemic exposure. The drug-to-antibody ratio (DAR) is a critical quality attribute, typically optimized between 2-4 to balance efficacy with pharmacokinetic stability and toxicity [156]. Modern ADCs employ potent payloads with IC₅₀ values in the picomolar to nanomolar range, including DNA-damaging agents (e.g., pyrrolobenzodiazepines), microtubule inhibitors, and topoisomerase inhibitors [156]. Emerging ADC platforms are addressing challenges like tumor heterogeneity and drug resistance through innovative designs such as bispecific ADCs, probody-drug conjugates, and immunostimulatory ADCs (ISACs), which actively engage the immune system alongside targeted payload delivery [156].
Immune Cell Engagers (ICEs)
Immune Cell Engagers (ICEs) are bispecific or trispecific molecules designed to bridge immune effector cells directly to tumor cells, facilitating a cytotoxic immune synapse independent of MHC recognition [157]. Bispecific T-cell Engagers (BiTEs) represent a prominent ICE class, with one arm binding a tumor-associated antigen (TAA) and the other engaging the CD3 complex on T cells. This redirects T cells to lyse tumor cells through perforin and granzyme release, bypassing traditional TCR-MHC interactions [157]. Since the 2014 FDA approval of blinatumomab for acute lymphoblastic leukemia, the ICE landscape has expanded significantly. Newer platforms include NK Cell Engagers (NKCEs), which activate natural killer cells via receptors like CD16a, and trispecific antibodies that incorporate co-stimulatory signals or cytokine domains (e.g., IL-15) to enhance potency and persistence [157]. These agents effectively overcome tumor immune evasion mechanisms, such as MHC downregulation, and can induce bystander killing of antigen-negative tumor cells, mitigating escape heterogeneity.
Table 2: Key Features of Next-Generation Agent Classes
| Feature | Antibody-Drug Conjugates (ADCs) | Immune Cell Engagers (ICEs) |
|---|---|---|
| Core Structure | mAb + Cytotoxic Payload + Linker | Bispecific/Trispecific antibody (e.g., scFv constructs) |
| Mechanism of Action | Antigen-specific internalization; intracellular payload release | Forced immune synapse formation; direct cell-mediated cytotoxicity |
| Primary Effector Cell | N/A (Direct payload effect) | T-cells (BiTEs), NK cells (NKCEs), Phagocytes (PCEs) |
| Key Design Parameter | Drug-to-Antibody Ratio (DAR), linker stability, payload potency | Affinity for immune vs. tumor target, valency, incorporation of co-stimulatory domains |
| Advantages | Targeted delivery of potent cytotoxics; "bystander effect" possible with membrane-permeable payloads | MHC-independent killing; redirection of pre-existing immune effectors; potential for memory responses |
Experimental Analysis of Drug Mechanisms
Protocol 1: Assessing Immunogenic Cell Death (ICD) In Vitro
Objective: To evaluate bortezomib-induced ICD by measuring calreticulin exposure, phagocytosis, and type-I interferon pathway activation.
Methodology:
- Cell Culture: Culture human multiple myeloma cell lines (e.g., MM.1S, RPMI8226) in complete RPMI-1640 medium.
- Drug Treatment: Treat cells with bortezomib (e.g., 10-50 nM) for 12-48 hours. Include a vehicle control (DMSO) and a positive control for ICD (e.g., mitoxantrone).
- Calreticulin Exposure:
- Harvest cells, wash with PBS, and stain with anti-calreticulin primary antibody.
- Incubate with a fluorochrome-conjugated secondary antibody.
- Analyze using flow cytometry. A significant increase in mean fluorescence intensity (MFI) indicates surface calreticulin exposure [152].
- Phagocytosis Assay:
- Label bortezomib-treated myeloma cells with CFSE.
- Co-culture labeled cells with immature dendritic cells (DCs) derived from human monocytes.
- After 4-6 hours, stain DCs with anti-CD11c-APC and analyze by flow cytometry.
- Phagocytosis is quantified as the percentage of CD11c⁺/CFSE⁺ double-positive cells [152].
- cGAS/STING Pathway Activation:
- Isolate total RNA from treated cells and perform RT-qPCR for interferon-stimulated genes (ISGs) like
ISG15andMX1. - Alternatively, measure phospho-STING and phospho-IRF3 levels by western blotting.
- Confirm functional output by quantifying IFN-β secretion in cell culture supernatants using ELISA [152].
- Isolate total RNA from treated cells and perform RT-qPCR for interferon-stimulated genes (ISGs) like
Protocol 2: Evaluating T-cell Co-stimulation by Lenalidomide
Objective: To investigate the effects of lenalidomide on CD28-mediated T-cell co-stimulation and subsequent cytokine production.
Methodology:
- PBMC Isolation: Isolate peripheral blood mononuclear cells (PBMCs) from healthy donors using Ficoll density gradient centrifugation.
- T-cell Stimulation: Seed PBMCs in culture plates coated with sub-optimal concentrations of anti-CD3 antibody (to simulate TCR signal).
- Drug Treatment: Add lenalidomide (1-10 µM) or DMSO control. For some experiments, include a blocking agent like CTLA-4-Ig to assess the ability of lenalidomide to overcome co-stimulatory blockade [153].
- Proliferation Assay:
- Label PBMCs with CFSE prior to culture.
- After 3-5 days, stain cells with anti-CD3 and anti-CD4 or anti-CD8 antibodies.
- Analyze CFSE dilution in T-cell subsets using flow cytometry to determine proliferation indices [158].
- Cytokine Analysis:
- Collect cell culture supernatants after 48-72 hours.
- Use a multiplex bead-based immunoassay (e.g., Luminex) or ELISA to quantify key cytokines, including IL-2, IFN-γ, and TNF-α [153].
- Phospho-protein Analysis:
- After short-term stimulation (15-60 mins), fix and permeabilize T cells.
- Stain intracellularly with antibodies against phosphorylated CD28, NF-κB, and other downstream signaling molecules.
- Analyze by flow cytometry to detect enhanced phosphorylation signaling [153].
Signaling Pathway Visualizations
Diagram 1: Bortezomib triggers ICD via cGAS/STING pathway activation and type-I interferon production, leading to an anti-myeloma immune response [152].
Diagram 2: Lenalidomide binds cereblon, altering E3 ligase specificity to degrade transcription factors and produce immunomodulatory effects [153] [155].
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for Investigating Drug Mechanisms
| Research Reagent | Primary Function/Catalyzed Reaction | Experimental Application |
|---|---|---|
| Anti-Calreticulin Antibody | Binds to calreticulin protein exposed on the cell surface. | Detection and quantification of immunogenic cell death (ICD) by flow cytometry [152]. |
| CFSE (Carboxyfluorescein succinimidyl ester) | Fluorescent cell staining dye that dilutes with each cell division. | Tracking cell proliferation in T-cell or phagocytosis assays [158]. |
| Recombinant CTLA-4-Ig | Recombinant fusion protein that blocks B7-CD28 co-stimulation. | Investigating T-cell co-stimulation mechanisms and ability of lenalidomide to overcome this blockade [153]. |
| Anti-Phospho-STING / IRF3 Antibodies | Specifically bind phosphorylated (active) forms of STING and IRF3. | Confirming activation of the cGAS/STING pathway via western blot or flow cytometry [152]. |
| ELISA Kits (e.g., IFN-β, IL-2) | Enzyme-linked immunosorbent assay for specific cytokine quantification. | Measuring cytokine secretion in cell culture supernatants to assess immune activation [152] [153]. |
| CD138+ Magnetic Beads | Bind and isolate CD138-positive plasma cells from bone marrow. | Purification of primary multiple myeloma cells for FISH analysis or ex vivo drug testing [159]. |
Clinical Translation and Combination Strategies
The mechanistic understanding of these agents directly informs their clinical application and the development of rational combination therapies. Real-world evidence from a multi-centered study in China demonstrated that both lenalidomide maintenance (L-MT) and bortezomib maintenance (B-MT) effectively remedied the negative prognostic impact of high-risk cytogenetic abnormalities in non-transplant multiple myeloma patients, whereas thalidomide maintenance did not [159]. This highlights the clinical relevance of their distinct and potent mechanisms.
The immunogenic effects of bortezomib provide a strong rationale for its combination with STING agonists to enhance anti-myeloma immunity, a strategy supported by preclinical data showing significant potentiation of ICD [152]. Similarly, the ability of lenalidomide to enhance NK cell function and antibody-dependent cellular cytotoxicity (ADCC) underpins its synergistic combination with monoclonal antibodies like rituximab [158] [154]. Furthermore, the convergence of these drug classes on the ubiquitin system suggests potential for synergistic combinations, as well as with next-generation agents such as CELMoDs (degrader-antibody conjugates) [156], which represent the logical evolution of leveraging ubiquitination pathways for therapeutic gain.
Bortezomib and lenalidomide, despite their established roles, continue to reveal complex mechanisms centered on ubiquitination, DNA damage response, and immune activation. Bortezomib extends beyond proteasome inhibition to activate the cGAS/STING pathway, inducing immunogenic cell death. Lenalidomide masterfully co-opts the CRL4ᶜᴿᴮᴺ E3 ubiquitin ligase to achieve targeted protein degradation and multifaceted immunomodulation. Next-generation ADCs and ICEs represent a paradigm shift towards exquisite precision, enabling direct targeting of tumor cells or strategic engagement of immune effectors. The continued dissection of these mechanisms, particularly their interplay with the ubiquitin system, DNA repair, and tumor immunology, will undoubtedly fuel the development of more effective, personalized, and durable cancer therapies.
The translation of preclinical findings into successful clinical outcomes remains a significant challenge in biomedical research, particularly in complex fields such as ubiquitination pathways governing DNA repair and immune responses. This technical review examines the molecular basis of species-specific differences that complicate the extrapolation of data from model systems to human biology. Through detailed analysis of comparative studies and emerging technologies, we provide a framework for enhancing translational predictability in ubiquitination research, with direct implications for drug development targeting DNA repair mechanisms and immune checkpoint regulation.
Protein ubiquitination, a crucial post-translational modification, regulates diverse cellular processes including DNA damage response and immune function. While model organisms have provided fundamental insights into ubiquitination pathways, significant species-specific differences often limit their predictive value for human biology. The ubiquitin system comprises hundreds of enzymes including E1 activating enzymes, E2 conjugating enzymes, and E3 ligases, alongside deubiquitinating enzymes (DUBs) that reverse these modifications [160]. This complexity is further amplified by the diversity of ubiquitin chain linkages that encode distinct functional consequences [161]. Understanding how these components differ between species is critical for rational therapeutic development.
Molecular profiling technologies have revealed fundamental divergences between model systems and human pathology. For instance, comparative analysis of tauopathy models demonstrated that while mouse models recapitulate early disease-associated phosphorylation events, they lack the ubiquitination and acetylation patterns characteristic of late-stage human Alzheimer's disease [162]. Such discrepancies may explain the repeated failure of therapeutics that showed efficacy in preclinical models. This review systematically addresses these translational challenges and provides methodological frameworks for enhancing species-specific research validity.
Molecular Divergence in Ubiquitination Pathways Between Species
Sequence and Structural Determinants of Ubiquitination
Ubiquitination site specificity varies significantly across species due to differences in both protein sequences and structural features. Computational analyses reveal distinct amino acid preferences around ubiquitination sites between humans and common model organisms (Figure 1). These sequence differences directly impact enzyme-substrate recognition and modification patterns.
Table 1: Species-Specific Features in Ubiquitination Pathways
| Species | Key Ubiquitination Features | Divergence from Human | Functional Consequences |
|---|---|---|---|
| Homo sapiens | Complex PTM patterns including phosphorylation, ubiquitination, acetylation in pathological protein aggregates | Reference standard | Late-stage disease signatures |
| Mus musculus (P301S/P301L tau models) | Phosphorylation-dominated tau pathology; minimal ubiquitination | Lacks comprehensive ubiquitination and acetylation profiles | Models early but not late human disease [162] |
| Salmo salar (Atlantic salmon) | Virus-specific ubiquitination patterns in immune response | Differentially regulated E3 ligases | Distinct antiviral response mechanisms [163] |
| Rattus norvegicus | Limited ubiquitination site data (<1000 sites in PLMD) | Sparse annotation compared to human | Reduced predictive utility [164] |
The structural context of ubiquitination sites significantly influences recognition by ubiquitinating enzymes. Protein secondary structure elements, including helices, strands, and coils, affect accessibility to catalytic domains of E3 ligases and DUBs [164]. Species-specific variations in these structural features further complicate cross-species predictions, necessitating specialized computational tools that integrate both sequence and structural information.
Functional Consequences in DNA Repair Pathways
The DNA damage response (DDR) relies heavily on ubiquitin-mediated signaling and protein regulation, with notable species-specific variations in component functionality. Deubiquitinating enzymes like UCHL3 play crucial roles in regulating DNA repair processes through stabilization of key repair proteins such as RAD51 and Ku80, facilitating homologous recombination and non-homologous end joining pathways [60].
UCHL3 demonstrates dual enzymatic activity, cleaving both ubiquitin and the ubiquitin-like modifier NEDD8—a rare capability among DUBs [60]. This functional complexity exhibits species-specific variations that impact therapeutic targeting. For instance, UCHL3 overexpression enhances DNA damage repair capacity and confers resistance to chemotherapy and radiotherapy in certain tumor types, but the molecular mechanisms differ between murine models and human cancers [60]. These differences extend to multiple DDR components, including variations in the ubiquitin-dependent regulation of BRCA1, BRCA2, and RAD51 complexes.
Comparative Analysis of Experimental Models and Human Biology
Limitations of Murine Models in Recapitulating Human Ubiquitination
Murine models expressing human transgenes have provided valuable insights into disease mechanisms but exhibit critical limitations in replicating the full spectrum of human ubiquitination pathology. Comparative proteomic analysis of tauopathy models revealed that while P301S and P301L transgenic mice successfully replicate Tau phosphorylation patterns observed in early human Alzheimer's disease, they lack the ubiquitination and acetylation profiles characteristic of advanced human pathology [162].
This molecular disparity may explain the poor translational record of therapeutic candidates developed in these models. The incomplete recapitulation of human post-translational modification landscapes in mouse models underscores the necessity of validating findings across multiple systems and directly comparing model and human ubiquitination patterns using mass spectrometry-based approaches.
Immune Response Regulation Across Species
Ubiquitination regulates immune responses through species-specific mechanisms that impact translational research. In Atlantic salmon, viral infections trigger dramatic changes in global ubiquitination states, with Infectious Salmon Anaemia Virus (ISAV) and Infectious Pancreatic Necrosis Virus (IPNV) inducing distinct ubiquitination profiles that differentially activate immune pathways [163]. These virus-specific ubiquitination patterns reflect either pathogen-specific post-translational regulation or viral antagonism of immune responses.
In humans, immune checkpoint regulation involves ubiquitination mechanisms that differ from murine systems. Lymphocyte activation gene 3 (LAG3), an emerging immunotherapy target, undergoes robust non-K48-linked polyubiquitination upon ligand engagement, which promotes its inhibitory function instead of targeting it for degradation [165]. This regulatory mechanism, mediated by E3 ligases c-Cbl and Cbl-b, represents a human-specific aspect of immune checkpoint control with direct implications for cancer immunotherapy development.
Table 2: Species-Specific Immune Ubiquitination Patterns
| Immune Process | Human Mechanism | Murine Model Divergence | Therapeutic Implications |
|---|---|---|---|
| LAG3 immune checkpoint function | Ligand-induced non-degradative ubiquitination [165] | Differential ubiquitination site recognition | Altered response to checkpoint inhibitors |
| RIG-I-like receptor signaling | K63-linked ubiquitin chain formation [163] | Variant E2/E3 enzyme expression | Distinct antiviral response kinetics |
| NF-κB pathway activation | Linear ubiquitin chain assembly complex activity | Differential upstream regulation | Altered inflammatory response thresholds |
| TRIM E3 ligase antiviral activity | Conserved and novel family members [163] | Species-specific isoform expression | Virus-specific resistance mechanisms |
Methodological Frameworks for Species-Specific Ubiquitination Research
Advanced Proteomic Workflows for Cross-Species Comparison
Comprehensive characterization of species-specific ubiquitination requires sophisticated proteomic approaches. The following workflow details a standardized method for comparative ubiquitinome analysis:
Protocol 1: Cross-Species Ubiquitin Enrichment and Quantification
Sample Preparation: Homogenize tissue or cell samples in TBS buffer containing protease inhibitors, phosphatase inhibitors, and deubiquitinase inhibitors to preserve ubiquitination states.
Fractionation: Perform sequential extraction using sarkosyl detergent to separate soluble and insoluble protein fractions, enabling analysis of aggregated ubiquitinated species [162].
Ubiquitin Enrichment: Utilize ubiquitin-binding domains (e.g., UBA domains) or di-glycine remnant immunoaffinity purification for selective isolation of ubiquitinated peptides.
Mass Spectrometry Analysis: Employ label-free or tandem mass tag (TMT) quantification for cross-species comparison. Use heavy isotope-labeled protein standards for absolute quantification [162].
Data Processing: Identify ubiquitination sites using sequence databases for respective species. Apply statistical correction for multiple comparisons in cross-species analyses.
This protocol enables direct comparison of ubiquitination patterns between model organisms and human tissues, facilitating identification of species-specific modifications.
Computational Prediction of Species-Specific Ubiquitination
Computational approaches provide complementary tools for predicting ubiquitination sites across species. The SSUbi (Species-Specific Ubiquitination) model integrates both protein sequence and structural information to enhance prediction accuracy [164]:
Algorithm 1: SSUbi Prediction Framework
Data Acquisition: Compile species-specific ubiquitination sites from databases such as the Protein Lysine Modification Database (PLMD).
Feature Extraction:
- Sequence features: Position-specific scoring matrices, amino acid composition
- Structural features: Secondary structure probabilities (helix, strand, coil) predicted using NetSurfP-3.0
Multi-dimensional Feature Integration: Utilize convolutional neural networks with channel attention mechanisms to extract both sequence and structural features.
Species-Specific Classification: Apply capsule networks trained on individual species datasets to predict ubiquitination sites.
This framework outperforms general prediction models by accounting for species-specific sequence variations and structural contexts that influence ubiquitination site accessibility [164].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Species-Specific Ubiquitination Studies
| Reagent/Category | Specific Examples | Function/Application | Species Considerations |
|---|---|---|---|
| Ubiquitin Enrichment Tools | Di-glycine remnant antibodies, TUBE reagents (Tandem Ubiquitin Binding Entities) | Affinity purification of ubiquitinated peptides/proteins | Antibody cross-reactivity varies between species |
| Activity-Based Probes | HA-Ub-VS, Ub-PA (Ubiquitin propargylamide) | Detection of active deubiquitinases | Probe specificity must be verified for each species |
| Mass Spectrometry Standards | Heavy isotope-labeled ubiquitin, SILAC (Stable Isotope Labeling with Amino Acids in Cell Culture) | Absolute quantification of ubiquitination | Requires species-specific protein sequences |
| Computational Resources | SSUbi prediction model [164], PLMD database | In silico prediction of ubiquitination sites | Species-specific training datasets required |
| Genetic Tools | CRISPR/Cas9 for HITI (Homology-Independent Targeted Integration) [166] | Endogenous tagging of ubiquitin system components | Optimization needed for each model organism |
| DUB Inhibitors | LDN-57444 (UCHL1 inhibitor), b-AP15 (proteasome-associated DUB inhibitor) | Functional validation of ubiquitination mechanisms | Potency and specificity vary between species |
Visualization of Key Ubiquitination Pathways with Species-Specific Nodes
The systematic identification of species-specific differences in ubiquitination pathways represents both a challenge and opportunity for translational research. As detailed in this review, disparities between model organisms and humans occur at multiple levels, including ubiquitination site specificity, enzyme-substrate relationships, and functional outcomes in DNA repair and immune regulation. Moving forward, the field requires increased emphasis on direct comparative studies that profile ubiquitination landscapes across species using standardized proteomic workflows.
Promising approaches include the development of humanized mouse models expressing key human ubiquitination enzymes, advanced computational prediction tools that account for structural determinants of ubiquitination, and microphysiological systems that better recapitulate human tissue environments. Furthermore, the integration of multi-omics data with machine learning algorithms will enhance our ability to extrapolate findings from model systems to human biology. By acknowledging and systematically addressing species-specific differences, researchers can enhance the predictive validity of preclinical studies and accelerate the development of therapeutics targeting the ubiquitin system in cancer, neurodegenerative diseases, and immune disorders.
The ubiquitin-proteasome system (UPS) and related signaling pathways have emerged as promising therapeutic targets in oncology, particularly within the contexts of DNA damage response and immune checkpoint regulation. However, the development of resistance to ubiquitin-targeting therapies poses a significant clinical challenge. This technical review provides a comprehensive cross-comparison of resistance mechanisms across different classes of ubiquitin-targeting therapies, including proteasome inhibitors, E3 ligase-targeting agents, and deubiquitinase (DUB) inhibitors. We examine common adaptive responses and synthesize experimental approaches for profiling and overcoming these resistance mechanisms, providing a foundational resource for researchers and drug development professionals working within the broader context of ubiquitination in DNA repair and immune response pathways.
Protein ubiquitination represents a critical post-translational modification that regulates diverse cellular processes through a sophisticated enzymatic cascade involving E1 activating enzymes, E2 conjugating enzymes, and E3 ligases, balanced by deubiquitinating enzymes (DUBs) that reverse this modification [11] [167]. The ubiquitin-proteasome system (UPS) controls protein stability, localization, and activity, making it an attractive therapeutic target, particularly in oncology where it intersects with DNA damage repair and antitumor immunity [168] [167].
Ubiquitin-targeting therapies encompass several mechanistic classes:
- Proteasome inhibitors (e.g., bortezomib) that globally disrupt protein degradation
- E3 ligase-targeting agents that modulate specific substrate degradation
- DUB inhibitors that prevent ubiquitin removal and enhance degradation of specific substrates
- Ubiquitin pathway modulators that target E1/E2 enzymes or ubiquitin-binding domains
Despite initial efficacy across various malignancies, resistance to these agents frequently develops through diverse molecular adaptations. Understanding the commonalities and distinctions between resistance mechanisms across these therapeutic classes is essential for developing effective combination strategies and next-generation agents.
Resistance Mechanisms to Ubiquitin-Targeting Therapies
Proteasome Inhibitor Resistance
Table 1: Key Resistance Mechanisms to Proteasome Inhibitors
| Resistance Mechanism | Molecular Components | Functional Consequences |
|---|---|---|
| Proteasome subunit mutations | PSMB5 mutations | Reduced drug binding affinity |
| Proteasome composition alterations | Immunoproteasome upregulation | Altered catalytic specificity and inhibitor sensitivity |
| Efflux pump upregulation | P-glycoprotein/MDR1 overexpression | Enhanced drug extrusion from cells |
| Unfolded protein response (UPR) activation | XBP1, ATF6 signaling pathways | Enhanced protein folding capacity and ER stress survival |
| Antioxidant response induction | NRF2 pathway activation | Reduced oxidative stress from accumulated proteins |
E3 Ligase-Targeter Therapy Resistance
Resistance to E3 ligase-targeting therapies manifests through several adaptive mechanisms:
- Compensatory ubiquitin ligase upregulation: Inhibition of specific E3 ligases frequently leads to overexpression of alternative E3s that can perform overlapping functions, maintaining substrate turnover through redundant pathways [169].
- Substrate protein mutations: Genetic alterations in substrate proteins can disrupt their recognition by E3 ligases or targeted therapeutics, preventing their degradation even with effective ligase inhibition.
- DUB counter-regulation: Increased expression of specific deubiquitinases, particularly ubiquitin-specific proteases (USPs), can antagonize E3-targeting agents by stabilizing intended substrate proteins [168].
For example, in chronic myeloid leukemia (CML), resistance to tyrosine kinase inhibitors (TKIs) can involve UPS components that directly or indirectly affect BCR-ABL ubiquitination, with specific E3 ligases and DUBs cooperating or antagonizing TKI efficacy [169].
DUB Inhibitor Resistance
Table 2: Documented Resistance Mechanisms to DUB-Targeted Therapies
| Resistance Mechanism | Representative Examples | Experimental Evidence |
|---|---|---|
| Alternative DUB upregulation | USP9X, USP24 overexpression | Compensates for inhibited DUB activity; stabilizes target substrates |
| Altered substrate recognition | Mutation of DUB-binding domains on substrates | Prevents DUB-substrate interaction despite effective DUB inhibition |
| Bypass signaling pathway activation | AKT, ERK, NF-κB pathway activation | Activates survival signals independent of DUB function |
| Epigenetic adaptations | Histone modification changes | Alters expression of DUB-regulated genes and survival pathways |
| Redox homeostasis alterations | Increased glutathione production | Counters oxidative stress induced by DUB inhibition |
The USP family represents the largest class of DUBs, with over 50 members, and USP inhibitors have demonstrated potential for overcoming resistance to conventional therapies [168]. However, cancer cells can develop resistance to USP-targeting agents through multiple compensatory mechanisms. For instance, USP inhibition in some models leads to rapid upregulation of alternative DUB family members that assume stabilization of critical survival proteins [168] [120].
Common Adaptive Responses Across Ubiquitin-Targeting Therapies
Transcriptional Reprogramming
Across different ubiquitin-targeting therapeutic classes, cancer cells demonstrate remarkable plasticity through transcriptional reprogramming. This includes coordinated upregulation of alternative components of the ubiquitination machinery, particularly following prolonged treatment with targeted agents [168]. Resistance to multiple ubiquitin-targeting agents correlates with enhanced expression of genes encoding compensatory E2 conjugating enzymes, E3 ligases with overlapping substrate specificity, and DUBs that stabilize proteins targeted for degradation.
Proteostatic Remodeling
The interconnected nature of cellular protein homeostasis (proteostasis) networks enables resistance development through proteostatic remodeling. When specific UPS nodes are inhibited, cancer cells can activate alternative protein degradation pathways, including autophagy-lysosomal system components, to maintain protein homeostasis [169]. Additionally, increased expression of molecular chaperones, particularly heat shock proteins (HSP70, HSP90), helps stabilize proteins that would otherwise be degraded via the inhibited ubiquitin-dependent pathway.
DNA Damage Response Adaptation
As ubiquitination plays crucial roles in DNA damage response (DDR), therapies targeting ubiquitin pathways frequently engage DNA repair mechanisms, and resistance can develop through DDR adaptations [11] [15]. For example, inhibition of certain DUBs can initially sensitize cells to DNA-damaging agents, but resistant cells often exhibit enhanced alternative DDR pathway activity. Specific adaptations include upregulation of non-homologous end joining (NHEJ) components when homologous recombination (HR) is compromised by ubiquitin-targeting therapies, and vice versa [15].
Experimental Approaches for Resistance Profiling
Methodologies for Identifying Resistance Mechanisms
- Comprehensive Ubiquitin Proteomics: Quantitative mass spectrometry-based ubiquitin remnant profiling enables system-wide assessment of changes in ubiquitination patterns following treatment with ubiquitin-targeting therapies. This approach can identify specific substrates with altered ubiquitination in resistant cells, revealing bypass signaling pathways and compensatory mechanisms.
- CRISPR-Cas9 Synthetic Lethality Screens: Genome-wide knockout screens in the presence of sublethal concentrations of ubiquitin-targeting agents can identify genes whose loss confers resistance or sensitivity. This approach has revealed multiple chromatin regulators and DNA repair factors that modulate sensitivity to DUB inhibitors [168].
- Functional DNA Repair Assays: Direct assessment of DNA repair capacity through comet assays, γH2AX focus formation, and RAD51 recruitment assays can determine how resistance to ubiquitin-targeting therapies correlates with specific DNA repair pathway enhancements [168] [15].
Protocol: Ubiquitin Remnant Profiling for Resistance Marker Identification
This mass spectrometry-based protocol identifies changes in global ubiquitination patterns associated with resistance to ubiquitin-targeting therapies.
Materials:
- DiGly remnant enrichment kit
- Stable isotope labeling by amino acids in cell culture (SILAC) reagents
- Anti-K-ε-GG antibody for ubiquitinated peptide enrichment
- LC-MS/MS system with appropriate analytical columns
Procedure:
- Culture resistant and parental cell lines in SILAC heavy and light media, respectively
- Treat both cell lines with the ubiquitin-targeting therapeutic agent for 4-6 hours
- Harvest cells and lyse in urea-based lysis buffer containing protease and phosphatase inhibitors
- Digest proteins with trypsin and Lys-C overnight at 37°C
- Enrich ubiquitinated peptides using anti-K-ε-GG antibody
- Analyze peptides by LC-MS/MS using a 2-hour gradient
- Process data using MaxQuant or similar software, searching against appropriate databases
- Validate significantly altered ubiquitination sites by immunoblotting
Data Analysis:
- Identify peptides with significant (>2-fold) ubiquitination changes in resistant versus sensitive cells
- Perform pathway enrichment analysis on proteins with altered ubiquitination
- Correlate ubiquitination changes with transcriptomic and proteomic data
- Validate candidate resistance markers using orthogonal approaches
Research Reagent Solutions for Resistance Studies
Table 3: Essential Research Reagents for Investigating Resistance to Ubiquitin-Targeting Therapies
| Reagent Category | Specific Examples | Research Applications |
|---|---|---|
| UPS-targeting inhibitors | Bortezomib, MLN4924, PR-619, VLX1570 | Tool compounds for probing UPS function and inducing resistance |
| DUB-specific inhibitors | P5091 (USP7 inhibitor), G9 (USP9X inhibitor), EOAI3402143 (USP1 inhibitor) | Selective targeting of specific DUB family members |
| Antibodies for ubiquitin signaling | Anti-polyubiquitin (K48, K63-specific), anti-γH2AX, anti-RAD51, anti-K-ε-GG | Detection of ubiquitin linkages and DNA damage markers |
| CRISPR libraries | Whole-genome knockout, DUB-focused, E3 ligase-focused libraries | Systematic identification of resistance genes and synthetic lethal interactions |
| Proteasome activity probes | MV151, BodipyFL-Ahx3Leu3VS | Direct measurement of proteasome function in live cells |
| DNA repair reporters | DR-GFP (HR), EJ5-GFP (NHEJ) | Functional assessment of specific DNA repair pathways |
| Ubiquitin variant (UbV) inhibitors | UbV.4.2 (USP8 inhibitor), UbV.10.1 (USP15 inhibitor) | Highly specific DUB inhibitors for mechanistic studies |
Emerging Therapeutic Strategies to Overcome Resistance
Rational Combination Approaches
Promising combination strategies that address common resistance mechanisms include:
- Sequential DUB and E3 ligase targeting: Simultaneous or sequential inhibition of complementary nodes in the ubiquitination machinery can prevent compensatory stabilization of oncoproteins, overcoming a primary resistance mechanism to single-agent therapy [168].
- Ubiquitin-targeting agents with DNA damage response inhibitors: Combining DUB or E3 ligase inhibitors with PARP, ATR, or ATM inhibitors can exploit DDR adaptations in resistant cells, creating synthetic lethal interactions [15].
- Dual targeting of ubiquitin and immune checkpoint pathways: Simultaneously targeting USPs and immune checkpoints like PD-1/PD-L1 can overcome immune evasion mechanisms that contribute to therapy resistance [167].
Next-Generation Ubiquitin-Targeting Modalities
Emerging approaches to circumvent established resistance mechanisms include:
- Bifunctional PROTAC molecules: Proteolysis-targeting chimeras (PROTACs) that simultaneously bind E3 ligases and target proteins offer enhanced specificity and the potential to overcome resistance to conventional inhibitors through novel degradation mechanisms.
- Allosteric DUB inhibitors: Targeting regulatory domains rather than catalytic sites of DUBs may prevent common resistance mutations in the active site while maintaining selectivity.
- Context-specific UPS targeting: Exploiting differential UPS dependency in resistant cells, such as heightened dependence on specific E3 ligases or DUBs that emerge as resistance adaptations.
The resistance landscape for ubiquitin-targeting therapies reveals both class-specific adaptations and common protective responses that span different therapeutic modalities. Key convergent resistance mechanisms include transcriptional reprogramming of ubiquitination machinery, proteostatic network remodeling, and DNA damage response adaptations. Successful navigation of this complex resistance landscape will require sophisticated profiling approaches, rational combination strategies that anticipate compensatory responses, and next-generation agents designed to circumvent common resistance mechanisms.
Future research directions should prioritize comprehensive resistance profiling across ubiquitin-targeting therapeutic classes, development of predictive biomarkers for specific resistance mechanisms, and innovative clinical trial designs that incorporate adaptive treatment strategies based on emerging resistance patterns. By leveraging our growing understanding of the intricate connections between ubiquitin signaling, DNA repair, and immune regulation, the next generation of ubiquitin-targeting therapies can potentially overcome the limitations of current approaches and deliver more durable clinical responses.
Conclusion
The ubiquitin system emerges as a master regulatory network that intricately connects the fundamental cellular processes of DNA repair and immune response. The decoding of specific ubiquitin chain topologies and their respective writers, erasers, and readers has revealed precise molecular mechanisms that maintain cellular homeostasis, while their dysregulation drives pathogenesis in cancer, inflammatory, and immune disorders. The development of targeted therapeutic strategies—from PROTACs to specific E3 ligase and DUB modulators—demonstrates the immense clinical potential of manipulating this system. However, challenges remain in navigating context-dependent functions, therapeutic resistance, and toxicity. Future research must focus on mapping the complete ubiquitin interactome in different disease states, developing more selective agents, and identifying robust predictive biomarkers. The integration of ubiquitin-targeting agents with conventional and immuno-therapies represents a promising frontier for precision medicine, potentially offering new hope for patients with treatment-resistant diseases. The continued elucidation of the ubiquitin code will undoubtedly unlock novel therapeutic paradigms across biomedical research.
References
- 1. Ubiquitin - Wikipedia [https://en.wikipedia.org/wiki/Ubiquitin]
- 2. The role of ubiquitination in tumorigenesis and targeted drug discovery | Signal Transduction and Targeted Therapy [https://www.nature.com/articles/s41392-020-0107-0]
- 3. Protein ubiquitination involving an E - 1 - E enzyme 2 ... E 3 ubiquitin [https://pubmed.ncbi.nlm.nih.gov/7800044/]
- 4. Targeting ubiquitination for cancer therapies - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4976843/]
- 5. Proteomic Identification of Protein Ubiquitination Events - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3937853/]
- 6. The ubiquitin landscape at DNA double-strand breaks - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2779242/]
- 7. Decoding the ubiquitin network: molecular mechanisms and... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-025-02433-4]
- 8. Frontiers | Ubiquitination and ALL: Identifying FBXO8 as a prognostic... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1554231/full]
- 9. Frontiers | Mass Spectrometry-Based Proteomics for Investigating DNA ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2016.00109/full]
- 10. A cascading activity-based probe... | Nature Chemical Biology [https://www.nature.com/articles/nchembio.2084?error=cookies_not_supported&code=7e63b51d-d6d7-4a2e-9970-04e9e3e12103]
- 11. The ubiquitous role of ubiquitin in the DNA damage response - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7105183/]
- 12. Regulation of DNA double-strand break repair by ubiquitin and... [https://www.nature.com/articles/nrm.2016.58?error=cookies_not_supported]
- 13. Regulation of the DNA damage response by ubiquitin conjugation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4354423/]
- 14. Frontiers | Tools for Decoding Ubiquitin Signaling in DNA Repair [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2021.760226/full]
- 15. and Ubiquitin -like molecules in ubiquitin double strand break... DNA [https://cellandbioscience.biomedcentral.com/articles/10.1186/s13578-020-0380-1]
- 16. Reading, writing, and repair: the role of ubiquitin and the ubiquitin-like proteins in DNA damage signaling and repair - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3612592/]
- 17. Beyond reversal: ubiquitin and ubiquitin -like proteases and the... [https://eprints.whiterose.ac.uk/200096/]
- 18. Opposing roles of RNF8/RNF168 and deubiquitinating enzymes in... [https://pubmed.ncbi.nlm.nih.gov/26983989/]
- 19. Chromatin Ubiquitination Guides DNA Double Strand Break Signaling and Repair - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9294282/]
- 20. Crosstalk between ubiquitin and other post-translational modifications on chromatin during double-strand break repair - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S0962892414000166]
- 21. -induced Ubiquitin condensation promotes RNF ... 168 DNA [https://pubmed.ncbi.nlm.nih.gov/38968116/]
- 22. Microbial Pathogens Trigger Host DNA... | PLOS Pathogens [https://journals.plos.org/plospathogens/article?id=10.1371/journal.ppat.1004030]
- 23. A type I IFN-dependent DNA damage response regulates the genetic... [https://elifesciences.org/articles/24655]
- 24. The ubiquitin system: from cell signalling to disease biology and new therapeutic opportunities | Cell Death & Differentiation [https://www.nature.com/articles/s41418-020-00703-w]
- 25. Ubiquitin signaling in immune responses | Cell Research [https://www.nature.com/articles/cr201640]
- 26. Enzymes in Cancer, Cancer Ubiquitination Evasion, and... Immune [https://pmc.ncbi.nlm.nih.gov/articles/PMC11763706/]
- 27. Targeting the Ubiquitin–Proteasome System and Recent Advances in Cancer Therapy - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10778545/]
- 28. Mechanisms and Pathways of Protein Ubiquitination [https://www.creative-proteomics.com/resource/mechanisms-pathways-protein-ubiquitination-cellular-regulation.htm]
- 29. E3 Ubiquitin Ligases: The Operators of the Ubiquitin Code That... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9740615/]
- 30. Frontiers | The role of cGAS-STING pathway ubiquitination in innate immunity and multiple diseases [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1522200/full]
- 31. Pharmacological targets in the ubiquitin system offer new ways of treating cancer, neurodegenerative disorders and infectious diseases - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3219211/]
- 32. Frontiers | Editorial: The role of ubiquitination in disease development, progression, and prognosis [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1595755/full]
- 33. Ubiquitination in the regulation of inflammatory cell death and cancer | Cell Death & Differentiation [https://www.nature.com/articles/s41418-020-00708-5]
- 34. Frontiers | Elucidating the role of ubiquitination and deubiquitination ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2023.1217466/full]
- 35. -alpha TNF (WP231) - Homo sapiens | WikiPathways signaling [https://www.wikipathways.org/pathways/WP231.html]
- 36. Spermidine activates RIP1 deubiquitination to inhibit TNF-α-induced NF-κB/p65 signaling pathway in osteoarthritis | Cell Death & Disease [https://www.nature.com/articles/s41419-020-2710-y]
- 37. Expanding role of ubiquitination in NF-κB signaling | Cell Research [https://www.nature.com/articles/cr2010170]
- 38. Regulation of death receptor signaling by the ubiquitin system | Cell Death & Differentiation [https://www.nature.com/articles/cdd2009168]
- 39. Ubiquitin specific protease 21 upregulation in breast cancer promotes cell tumorigenic capability and is associated with the NOD-like receptor signaling pathway - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5228564/]
- 40. Ubiquitin-specific protease 4 (USP4) targets TRAF2 and TRAF6 for deubiquitination and inhibits TNFα-induced cancer cell migration - PubMed [https://pubmed.ncbi.nlm.nih.gov/22029577/]
- 41. -mediated linear LUBAC : a crucial ubiquitination of regulator ... immune [https://pmc.ncbi.nlm.nih.gov/articles/PMC8019854/]
- 42. Mechanisms underlying linear ubiquitination and implications in tumorigenesis and drug discovery | Cell Communication and Signaling | Full Text [https://biosignaling.biomedcentral.com/articles/10.1186/s12964-023-01239-5]
- 43. Linear Ubiquitin Code: Its Writer, Erasers, Decoders, Inhibitors, and Implications in Disorders - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7246992/]
- 44. The HOIL-1L ligase modulates immune signalling... | Nature Cell Biology [https://www.nature.com/articles/s41556-020-0517-9?error=cookies_not_supported]
- 45. NF-κB in biology and targeted therapy: new insights and translational implications | Signal Transduction and Targeted Therapy [https://www.nature.com/articles/s41392-024-01757-9]
- 46. Frontiers | Cellular and Mathematical Analyses of LUBAC Involvement... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2020.601926/full]
- 47. -synthesized LUBAC chains... | Nature Microbiology linear ubiquitin [https://www.nature.com/articles/nmicrobiol201763?error=cookies_not_supported&code=45ee4342-dde1-49d7-82be-3da30acd344c]
- 48. The Linear Assembly Complex Modulates Latent Membrane... Ubiquitin [https://pmc.ncbi.nlm.nih.gov/articles/PMC5286870/]
- 49. E3 ubiquitin and their therapeutic potential in disease... ligases [https://pubmed.ncbi.nlm.nih.gov/40120724/]
- 50. Rittinger Lab | Exploring the mechanism of E3 ubiquitin ... | Crick ligases [https://www.crick.ac.uk/careers-study/vacancies/2025-09-23-rittinger-lab-exploring-the-mechanism-of-e3-ubiquitin-ligases-and-their-role-in-infection-and-disease]
- 51. Molecular mechanisms and potential targeting strategies of... [https://www.spandidos-publications.com/10.3892/or.2025.9000?text=fulltext]
- 52. Frontiers | Chromatin Ubiquitination Guides DNA Double Strand Break Signaling and Repair [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2022.928113/full]
- 53. plays a key role in cancer stem cell function and... Ubiquitination [https://www.news-medical.net/news/20250308/Ubiquitination-Plays-a-Key-Role-in-Cancer-Stem-Cell-Function-and-Treatment-Resistance.aspx]
- 54. Delineating Crosstalk Mechanisms of the Ubiquitin Proteasome System That Regulate Apoptosis - PubMed [https://pubmed.ncbi.nlm.nih.gov/29479529/]
- 55. Current methodologies in protein ubiquitination characterization: from ubiquitinated protein to ubiquitin chain architecture - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9373315/]
- 56. Tools for Decoding Ubiquitin Signaling in DNA - PMC Repair [https://pmc.ncbi.nlm.nih.gov/articles/PMC8690248/]
- 57. Chemical approaches to explore ubiquitin-like proteins - RSC Chemical Biology (RSC Publishing) DOI:10.1039/D4CB00220B [https://pubs.rsc.org/en/content/articlehtml/2025/cb/d4cb00220b]
- 58. Revolutionizing DNA research and cancer therapy with... repair [https://www.nature.com/articles/s41580-022-00571-x?error=cookies_not_supported]
- 59. /Cas9-based genome-wide CRISPR of the deubiquitinase... screening [https://jeccr.biomedcentral.com/articles/10.1186/s13046-023-02694-1]
- 60. UCHL3: a crucial deubiquitinase in DNA damage repair and tumor... [https://cancerci.biomedcentral.com/articles/10.1186/s12935-025-03884-x]
- 61. – Silva Lab Research [https://sites.duke.edu/silvalab/research/]
- 62. : styles, structures and functions | Molecular... E 3 ubiquitin ligases [https://link.springer.com/article/10.1186/s43556-021-00043-2]
- 63. Frontiers | The regulatory roles of the E ... 3 ubiquitin ligase [https://www.frontiersin.org/journals/physiology/articles/10.3389/fphys.2022.968927/full]
- 64. The Expanding E3 Ligase-Ligand Landscape for PROTAC Technology [https://www.mdpi.com/2813-3137/3/4/30]
- 65. The E RNF185 facilitates the... | PLOS Pathogens 3 ubiquitin ligase [https://journals.plos.org/plospathogens/article?id=10.1371/journal.ppat.1006264]
- 66. TRAF6 Targeting activity with a E - 3 ... ligase small molecule inhibitor [https://pubmed.ncbi.nlm.nih.gov/29950522/]
- 67. Expanding PROTACtable genome universe of E3 ligases | Nature Communications [https://www.nature.com/articles/s41467-023-42233-2]
- 68. Deubiquitinating enzymes in parkinson’s disease: molecular... [https://molmed.biomedcentral.com/articles/10.1186/s10020-025-01389-x]
- 69. Emerging role of deubiquitinases in modulating cancer chemoresistance [https://pubmed.ncbi.nlm.nih.gov/40118446/]
- 70. Deubiquitinase inhibition as a cancer therapeutic strategy [https://pubmed.ncbi.nlm.nih.gov/25444757/]
- 71. Why DNA Damage Response deserves renewed focus in cancer therapy [https://www.drugtargetreview.com/article/157355/why-dna-damage-response-deserves-renewed-focus-in-cancer-therapy/]
- 72. Ubiquitination Enzymes in Cancer, Cancer Immune Evasion, and Potential Therapeutic Opportunities [https://www.mdpi.com/2073-4409/14/2/69]
- 73. The Role of Ubiquitination in Disease , Progression... Development [https://www.frontiersin.org/research-topics/64036/the-role-of-ubiquitination-in-disease-development-progression-and-prognosis/magazine]
- 74. Frontiers | Multi-omics analysis identifies UBA family as potential... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1510503/full]
- 75. The Clinical Prediction Value of the Ubiquitination Model Reflecting the Microenvironment Infiltration and Drug Sensitivity in Breast Cancer [https://www.jcancer.org/v16p0784.htm]
- 76. Mechanistic Insights and Therapeutic Potentials of Ubiquitin‐Proteasome System in Non‐Small Cell Lung Cancer - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12240642/]
- 77. Ubiquitination Proteomics | Complete Omics Inc. [https://www.completeomics.com/ubiquitination-proteomics/]
- 78. Frontiers | Comprehensive analysis based on the ubiquitination- and deubiquitylation-related genes reveals the function of NEURL3 in esophageal squamous cell carcinoma [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1632090/full]
- 79. Links Ubiquitination Damage and DNA Signaling to Cancer... Repair [https://pmc.ncbi.nlm.nih.gov/articles/PMC10179089/]
- 80. Regulatory role of E3 ubiquitin ligases in multiple myeloma: from molecular mechanisms to therapeutic strategies - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12343653/]
- 81. Multi-omics analysis of core E3 ubiquitin ligase identifies prognostic biomarkers associated with immune infiltration and drug sensitivity in lung adenocarcinoma - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11786024/]
- 82. Frontiers | Molecular glue degrader for tumor treatment [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2024.1512666/full]
- 83. Rapid Progress in the Use of Immunomodulatory and... Drugs [https://pubmed.ncbi.nlm.nih.gov/34572892/]
- 84. Advancing targeted protein degradation for cancer therapy | Nature Reviews Cancer [https://www.nature.com/articles/s41568-021-00365-x]
- 85. The enigma of the RING-UIM E : its transformative impact on... 3 ligases [https://link.springer.com/article/10.1186/s12967-025-07194-8]
- 86. Selective ubiquitination of drug-like small molecules by the ubiquitin ligase HUWE1 | Nature Communications [https://www.nature.com/articles/s41467-025-63442-x]
- 87. The cross talk of ubiquitination and chemotherapy tolerance in... [https://link.springer.com/article/10.1007/s00432-024-05659-9]
- 88. Targeting ubiquitin signaling for cancer immunotherapy | Signal Transduction and Targeted Therapy [https://www.nature.com/articles/s41392-020-00421-2]
- 89. Central Role of Ubiquitination in Genome Maintenance: DNA Replication and Damage Repair - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4908256/]
- 90. Biochemistry, Ubiquitination - StatPearls - NCBI Bookshelf [https://www.ncbi.nlm.nih.gov/books/NBK556052/]
- 91. Deubiquitinating enzymes (DUBs): Regulation, homeostasis, and oxidative stress response - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8424594/]
- 92. The emerging role of deubiquitinating enzymes in genomic integrity, diseases, and therapeutics | Cell & Bioscience | Full Text [https://cellandbioscience.biomedcentral.com/articles/10.1186/s13578-016-0127-1]
- 93. The regulatory roles of the E NEDD4 family in 3 ... ubiquitin ligase DNA [https://pmc.ncbi.nlm.nih.gov/articles/PMC9458852/]
- 94. Mechanisms for regulating deubiquitinating enzymes - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3970886/]
- 95. Decoys provide a scalable platform for the identification of plant... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6483598/]
- 96. Drugging the undruggables: exploring the ubiquitin system for drug development | Cell Research [https://www.nature.com/articles/cr201631]
- 97. The role of ubiquitylation in immune defence and pathogen evasion - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3864900/]
- 98. Regulation of DNA by ubiquitylation | Nature Reviews Molecular... repair [https://www.nature.com/articles/nrm1908?error=cookies_not_supported]
- 99. The Ubiquitin Proteasome Pathway (UPP) in the regulation of cell... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3466981/]
- 100. (PDF) Elucidating tissue and subcellular of the entire... specificity [https://www.academia.edu/144377068/Elucidating_tissue_and_subcellular_specificity_of_the_entire_SUMO_network_reveals_how_stress_responses_are_fine_tuned_in_a_eukaryote]
- 101. and Ubiquitin -relative SUMO in Ubiquitin damage response DNA [https://www.frontiersin.org/research-topics/3933/ubiquitin-and-ubiquitin-relative-sumo-in-dna-damage-response/magazine]
- 102. Retinoic acid modulates the subcellular localization of small... [https://pubmed.ncbi.nlm.nih.gov/20075418/]
- 103. Protein Degradation Through Ubiquitination : Insights... | Preprints.org [https://www.preprints.org/manuscript/202504.2585/v1]
- 104. Dysregulation of the Ubiquitin Proteasome System in Human... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8037609/]
- 105. Frontiers | Ubiquitin -proteasome system : a potential participant and... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1523799/full]
- 106. How Ubiquitin Proteasome Works — In One Simple Flow ( 2025 ) [https://www.linkedin.com/pulse/how-ubiquitin-proteasome-works-one-simple-flow-2025-nbt5e/]
- 107. Frontiers | PROTAC : a revolutionary technology propelling small... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2025.1676414/full]
- 108. An overview of PROTACs: a promising drug discovery paradigm - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9763089/]
- 109. Navigating PROTACs in Cancer Therapy: Advancements, Challenges... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11786021/]
- 110. Proteolysis-targeting chimeras (PROTACs) in cancer therapy | Molecular Cancer | Full Text [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-021-01434-3]
- 111. Deubiquitinating enzyme - Wikipedia [https://en.wikipedia.org/wiki/Deubiquitinating_enzyme]
- 112. strategies for the design of Current : a critical review... PROTAC linkers [https://pmc.ncbi.nlm.nih.gov/articles/PMC9400730/]
- 113. Frontiers | From Conception to Development: Investigating PROTACs Features for Improved Cell Permeability and Successful Protein Degradation [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2021.672267/full]
- 114. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy | Journal of Hematology & Oncology | Full Text [https://jhoonline.biomedcentral.com/articles/10.1186/s13045-020-00885-3]
- 115. rigidity to improve intracellular behavior of... Optimizing linker [https://pubs.rsc.org/en/content/articlehtml/2025/md/d5md00396b]
- 116. Current strategies for the design of PROTAC linkers: a critical review [https://www.explorationpub.com/Journals/etat/Article/100218]
- 117. Enhancing intracellular accumulation and... | Nature Communications [https://www.nature.com/articles/s41467-020-17997-6?error=cookies_not_supported]
- 118. Differential PROTAC substrate specificity... | Nature Communications [https://www.nature.com/articles/s41467-018-08027-7?error=cookies_not_supported&code=576a7166-853c-443c-bd94-05765e53bcfb]
- 119. Lessons in PROTAC Design from Selective Degradation with... [https://pubmed.ncbi.nlm.nih.gov/29129718/]
- 120. Frontiers | The role of ubiquitination and deubiquitination in urological tumours [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1532878/full]
- 121. Frontiers | Into the storm : the imbalance in the yin-yang immune ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2024.1448201/full]
- 122. Regulation of Histone Ubiquitination in Response to DNA Double... [https://pubmed.ncbi.nlm.nih.gov/32708614/]
- 123. Frontiers | Editorial: Ubiquitin-Modifying Enzymes as Drug Targets and Biomarkers in Cancer [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2021.758983/full]
- 124. The ‘ Cytokine ’: molecular Storm and therapeutic... mechanisms [https://pmc.ncbi.nlm.nih.gov/articles/PMC9310545/]
- 125. Coronavirus disease-2019: A review on the disease exacerbation via cytokine storm and concurrent management - PMC [https://ncbi.nlm.nih.gov/pmc/articles/PMC8343371/]
- 126. Elucidation of the underlying mechanism of SARS-CoV-2-mediated cytokine storm syndrome towards enhancing COVID-19 therapeutic modalities [https://www.redalyc.org/journal/2034/203469933001/]
- 127. (PD) Pharmacodynamic in the Real World: 5 Uses You'll... Biomarkers [https://www.linkedin.com/pulse/pharmacodynamic-pd-biomarkers-real-world-imxtf]
- 128. Multi-omics analysis of core E3 ubiquitin ligase identifies prognostic biomarkers associated with immune infiltration and drug sensitivity in lung adenocarcinoma - PubMed [https://pubmed.ncbi.nlm.nih.gov/39895786/]
- 129. The Potential and Emerging Role of Quantitative Imaging Biomarkers for Cancer Characterization - PubMed [https://pubmed.ncbi.nlm.nih.gov/35884409/]
- 130. Quantitative Image Analysis for Tissue Biomarker Use: A White Paper From the Digital Pathology Association - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8354563/]
- 131. Drug Development Targeting the Ubiquitin–Proteasome System (UPS) for the Treatment of Human Cancers - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7226376/]
- 132. Genome-wide identification and characterization of the... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-025-11670-2]
- 133. identifies Ube2v1 as a suppressor of... Genetic screening [https://link.springer.com/article/10.1007/s11033-025-11268-7]
- 134. A comprehensive phenotypic CRISPR -Cas9 screen of the ubiquitin ... [https://pubmed.ncbi.nlm.nih.gov/33539788/]
- 135. Frontiers | Prognostic model of ubiquitination -related genes in... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1654180/full]
- 136. A new era in cancer therapy: targeting the Proteasome -Bcl-2 axis [https://jeccr.biomedcentral.com/articles/10.1186/s13046-025-03505-5]
- 137. Recent advances in small molecule inhibitors of deubiquitinating enzymes - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S0223523425000893]
- 138. The emerging role of deubiquitylating enzymes as therapeutic targets in cancer metabolism | Cancer Cell International | Full Text [https://cancerci.biomedcentral.com/articles/10.1186/s12935-022-02524-y]
- 139. How Proteasome Drug Works — In One Simple Flow ( Inhibitor ) 2025 [https://www.linkedin.com/pulse/how-proteasome-inhibitor-drug-works-one-simple-flow-tt6kc]
- 140. PROTAC Degraders in Clinical Trails: 2025 Update | Biopharma PEG [https://www.biochempeg.com/article/434.html]
- 141. Regulation of DNA double-strand break repair by ubiquitin and ubiquitin-like modifiers | Nature Reviews Molecular Cell Biology [https://www.nature.com/articles/nrm.2016.58]
- 142. Unlocking Cancer : Therapy -Specific Proteases Targeting Ubiquitin [https://www.pharmafocusasia.com/articles/unlocking-new-avenues-in-cancer-therapy-by-targeting-ubiquitin-specific]
- 143. Frontiers | The ubiquitin -proteasome system in the tumor immune... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2024.1436174/full]
- 144. Frontiers | Identification and validation of ubiquitination -related genes... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2025.1578075/full]
- 145. A correlation-based network for biomarker discovery in obesity with metabolic syndrome | BMC Bioinformatics | Full Text [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-3064-2]
- 146. Identification and validation of USP15 and CUL2 as ubiquitination ... [https://pubmed.ncbi.nlm.nih.gov/40413536/]
- 147. Correlational analyses of biomarkers that are harmonized through a bridging study due to measurement errors - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10939855/]
- 148. Correlation of baseline biomarkers with clinical outcomes and response to fulvestrant with vandetanib or placebo in patients with bone predominant metastatic breast cancer: An OCOG ZAMBONEY sub-study - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4620970/]
- 149. An open source statistical web application for validation and analysis of virtual cohorts | Scientific Reports [https://www.nature.com/articles/s41598-025-99720-3]
- 150. The ubiquitin -proteasome system (UPS) and the mechanism ... of [https://pubmed.ncbi.nlm.nih.gov/21504411/]
- 151. (PDF) The ubiquitin -proteasome system (UPS) and the mechanism ... [https://www.academia.edu/3767196/The_ubiquitin_proteasome_system_UPS_and_the_mechanism_of_action_of_bortezomib]
- 152. induces anti-multiple myeloma Bortezomib ... immune response [https://pubmed.ncbi.nlm.nih.gov/34568832/]
- 153. Mechanism of action of lenalidomide in hematological malignancies | Journal of Hematology & Oncology | Full Text [https://jhoonline.biomedcentral.com/articles/10.1186/1756-8722-2-36]
- 154. Lenalidomide and Chronic Lymphocytic Leukemia - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3791640/]
- 155. Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma | Leukemia [https://www.nature.com/articles/leu2009236]
- 156. Frontiers | Antibody-drug conjugates in cancer therapy: applications and future advances [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1516419/full]
- 157. Immune Cell Engagers: Advancing Precision Immunotherapy for Cancer Treatment - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11843982/]
- 158. Lenalidomide Induces Immunomodulation in Chronic Lymphocytic Leukemia and Enhances Antitumor Immune Responses Mediated by NK and CD4 T Cells - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4182694/]
- 159. Frontiers | Lenalidomide or bortezomib as maintenance treatment... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2023.1028571/full]
- 160. Deubiquitylating Enzymes and DNA Damage Response Pathways [https://link.springer.com/article/10.1007/s12013-013-9635-3]
- 161. Ubiquitin chain diversity at a glance - PubMed [https://pubmed.ncbi.nlm.nih.gov/26906419/?dopt=Abstract]
- 162. Common mouse models of tauopathy reflect early but not late human disease | Molecular Neurodegeneration | Full Text [https://molecularneurodegeneration.biomedcentral.com/articles/10.1186/s13024-023-00601-y]
- 163. Frontiers | Characterisation of post- translational and transcriptional... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1532917/full]
- 164. Species-specific model based on sequence and structural information for ubiquitination sites prediction - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S0022283624004017]
- 165. Ligand-induced ubiquitination unleashes LAG3 immune checkpoint... [https://pubmed.ncbi.nlm.nih.gov/40101708/]
- 166. Proximity-dependent biotinylation reveals an interaction between... [https://pubmed.ncbi.nlm.nih.gov/39482856/]
- 167. the Targeting /deubiquitination process to regulate... ubiquitination [https://www.nature.com/articles/s41392-020-00418-x?error=cookies_not_supported&code=c17d2806-6c73-483d-89ab-f0101c8ea551]
- 168. Drug resistance and treatment strategies mediated by... mechanisms [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-024-02005-y]
- 169. Exploiting the potential of the ubiquitin -proteasome system in... [https://www.sciopen.com/article/10.1016/j.gendis.2023.101150]