Ubiquitin Tagging vs. Antibody Enrichment: A Strategic Guide for Proteomics and Therapeutic Development
This article provides a comprehensive comparison of two pivotal methods for analyzing protein ubiquitination: ubiquitin tagging and antibody-based enrichment.
Ubiquitin Tagging vs. Antibody Enrichment: A Strategic Guide for Proteomics and Therapeutic Development
Abstract
This article provides a comprehensive comparison of two pivotal methods for analyzing protein ubiquitination: ubiquitin tagging and antibody-based enrichment. Tailored for researchers, scientists, and drug development professionals, we explore the foundational principles, methodological workflows, and specific applications of each technique. The content delves into troubleshooting common challenges, optimizing protocols for superior results, and offers a direct comparative analysis to guide method selection. By synthesizing current methodologies and emerging trends, this guide aims to equip professionals with the knowledge to effectively map ubiquitin signaling in basic research and advance the development of novel therapeutics, such as antibody-drug conjugates.
Decoding the Ubiquitin Code: Why Analysis is Complex and Crucial
The ubiquitin system, once recognized primarily for its role in targeting proteins for proteasomal degradation, is now understood as a sophisticated post-translational language regulating virtually all cellular processes. This complex signaling system employs diverse ubiquitin chain architectures and conjugation sites to control protein stability, activity, localization, and interactions. The versatility of ubiquitin signaling arises from its ability to form different chain linkages—homotypic, heterotypic, or branched—each capable of encoding distinct functional outcomes. Recent methodological advances have significantly expanded our understanding of non-canonical ubiquitination events, including modifications on non-lysine residues and even non-proteinaceous molecules. This guide objectively compares two fundamental methodological approaches—ubi-tagging and antibody-based enrichment—for studying the ubiquitin system, providing researchers with experimental data and protocols to inform their methodological selections.
Methodological Foundations in Ubiquitin Research
Table 1: Core Methodologies for Studying Protein Ubiquitination
| Method Category | Key Principle | Primary Applications | Key Advantages |
|---|---|---|---|
| Ubi-Tagging | Engineered ubiquitin fusion proteins for site-specific conjugation [1] | Generating homogeneous antibody conjugates, bispecific engagers, targeted therapeutics [1] | Rapid reaction time (30 min), high homogeneity, site-specific control [1] |
| Antibody-Based Enrichment | Immunoaffinity capture using ubiquitin-specific antibodies [2] [3] | Proteome-wide ubiquitination profiling, target-specific ubiquitination status [2] [3] | Preservation of endogenous modification, compatibility with clinical samples [3] |
| UBD-Based Approaches | Tandem ubiquitin-binding domains for high-affinity capture [2] [3] | Unbiased enrichment of polyubiquitinated proteins, detection of various linkage types [2] [3] | High affinity for polyubiquitin chains, reduced linkage bias compared to antibodies [2] |
Ubi-Tagging: A Modular Conjugation Platform
Ubi-tagging represents a protein engineering approach that exploits the natural ubiquitination machinery for controlled, site-specific conjugation. This methodology addresses the long-standing challenge of product heterogeneity in antibody-drug conjugates and other ubiquitin-fusion proteins.
Experimental Protocol and Performance Data
The standard ubi-tagging protocol involves several key components: (1) a donor ubi-tag (Ubdon) containing a free C-terminal glycine with the conjugating enzyme-specific lysine mutated to arginine to prevent homodimer formation; (2) an acceptor ubi-tag (Ubacc) carrying the corresponding conjugation lysine residue with an unreactive C-terminus; and (3) specific ubiquitination enzymes (E1 and E2-E3 fusion proteins) [1].
In a representative experiment demonstrating the efficiency of this system, researchers conjugated anti-mouse CD3 Fab-Ub(K48R)don with rhodamine-labeled Ubacc-ΔGG using recombinant E1 and the K48-specific E2-E3 fusion protein gp78RING-Ube2g2 [1]. The reaction achieved complete consumption of the starting Fab material within 30 minutes, forming a single fluorescent product with the expected molecular weight [1]. Conversion efficiency quantified across multiple reactions reached 93-96% for ubi-tagged antibodies, demonstrating exceptional reaction completeness [1].
Table 2: Quantitative Performance Metrics of Ubi-Tagging
| Performance Parameter | Result | Experimental Context |
|---|---|---|
| Reaction Time | 30 minutes | Complete consumption of Fab-Ub(K48R)don observed [1] |
| Conversion Efficiency | 93-96% | Average across ubi-tagging reactions with antibodies [1] |
| Thermostability | ~75°C infliction temperature | No alteration compared to unconjugated Fab-Ub(K48R)don [1] |
| Functional Antigen Binding | Comparable to parental antibody | Flow cytometry on CD3+ mouse splenocytes [1] |
| Multimerization Capacity | Up to 11th order multimers | Formation of higher-order ubiquitin chains with Fab-UbWT [1] |
Antibody-Based Enrichment: Capturing Endogenous Ubiquitination
Antibody-based methodologies enable the study of endogenous ubiquitination events without genetic manipulation of the target system. These approaches utilize antibodies that recognize ubiquitin itself or specific ubiquitin chain linkages to isolate and characterize ubiquitinated proteins from complex biological samples.
Technological Evolution and Performance Benchmarks
Recent advances in antibody-based platforms have focused on improving affinity, specificity, and throughput. A notable development is the ThUBD (Tandem Hybrid Ubiquitin Binding Domain)-coated 96-well plate technology, which demonstrates significant performance improvements over previous generation TUBE (Tandem Ubiquitin Binding Entity)-based platforms [2].
This high-throughput system exhibits a 16-fold wider linear range for capturing polyubiquitinated proteins from complex proteome samples compared to TUBE-coated plates, with detection sensitivity as low as 0.625 μg of sample input [2]. The platform supports flexible analysis of both global ubiquitination profiles and target-specific ubiquitination status, making it particularly valuable for dynamic monitoring of ubiquitination in PROTAC drug development [2].
Experimental Workflow for Ubiquitination Detection
The standard protocol for ThUBD-based ubiquitination detection involves: (1) coating high-binding 96-well plates with purified ThUBD protein (optimized at 1.03 μg per well for Corning 3603 plates); (2) blocking plates to prevent non-specific binding; (3) incubating with complex proteome samples; (4) thorough washing with optimized buffer systems; and (5) detection using HRP-conjugated detection reagents with chemiluminescent or colorimetric readouts [2]. This workflow enables specific binding to approximately 5 pmol of polyubiquitin chains under optimal conditions [2].
Comparative Analysis: Ubi-Tagging vs. Antibody-Based Enrichment
Table 3: Direct Method Comparison for Ubiquitin Research Applications
| Comparison Parameter | Ubi-Tagging Approach | Antibody-Based Enrichment |
|---|---|---|
| Primary Application | Engineering defined protein conjugates [1] | Analytical detection of endogenous ubiquitination [2] [3] |
| Specificity Control | High (engineered site-specificity) [1] | Variable (depends on antibody quality and linkage specificity) [3] |
| Sample Compatibility | Requires recombinant components [1] | Native tissues and clinical samples [4] [3] |
| Throughput Capacity | Moderate (batch conjugation reactions) [1] | High (96-well plate format) [2] |
| Key Limitation | Not suitable for endogenous profiling [1] | Potential linkage bias and affinity limitations [2] [3] |
| Typical Reaction/Processing Time | 30 minutes [1] | Several hours including incubation and washing steps [2] |
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents for Ubiquitination Studies
| Reagent / Tool | Function / Application | Specific Examples / Notes |
|---|---|---|
| Ubiquitination Enzymes | Catalyze the conjugation of ubiquitin to substrates [1] | E1 activating enzyme, E2 conjugating enzymes (e.g., Ube2g2), E3 ligases (e.g., gp78RING) [1] |
| Linkage-Specific Binders | Enrich or detect specific ubiquitin chain types [2] [3] | ThUBD (unbiased recognition), K48-specific antibodies, K63-specific antibodies [2] [3] |
| Anti-diglycine (K-GG) Antibodies | Enrich tryptic peptides with ubiquitin remnant motif [5] | Immunoaffinity purification of K-GG peptides for mass spectrometry [5] |
| Ubiquitin Variants | Engineered ubiquitin for specific applications [1] | Ub(K48R)don, Ubacc-ΔGG, Strep-tagged Ub, His-tagged Ub [1] [3] |
| Activity-Based Probes | Monitor deubiquitinating enzyme activity | Ubiquitin-based probes with warhead groups (not detailed in results) |
| Platform-Specific Tools | High-throughput ubiquitination analysis [2] | ThUBD-coated 96-well plates, PROTAC assay plates [2] |
Visualization of Ubiquitin Signaling and Methodological Workflows
The Ubiquitin Conjugation Cascade
Ubi-Tagging Experimental Workflow
Antibody-Based Enrichment Workflow
The expanding toolkit for ubiquitin research, exemplified by the contrasting strengths of ubi-tagging and antibody-based enrichment methods, reflects the growing appreciation of ubiquitin signaling complexity. Ubi-tagging offers precision engineering of defined conjugates with exceptional homogeneity and efficiency, making it invaluable for therapeutic applications. Conversely, antibody-based platforms provide the sensitivity and throughput needed for analytical profiling of endogenous ubiquitination events in physiological and pathological contexts. The continued refinement of these methodologies—including emerging approaches for studying non-canonical ubiquitination—will undoubtedly uncover additional layers of complexity in the ubiquitin code, further cementing its status as a central regulatory system far beyond a simple degradation signal.
This guide objectively compares the performance of ubiquitin tagging and antibody-based enrichment methods, two principal strategies for ubiquitination analysis. The comparison is framed within the broader research goal of overcoming the inherent analytical challenges of studying ubiquitination: its low natural abundance and the structural complexity of ubiquitin chains.
Methodologies at a Glance
The table below summarizes the core principles and characteristics of each method.
| Feature | Ubiquitin Tagging | Antibody-Based Enrichment |
|---|---|---|
| Core Principle | Genetic fusion of an affinity tag (e.g., His, Strep) to ubiquitin for purification of conjugated substrates [3]. | Immunoaffinity purification using antibodies against ubiquitin or its remnants (e.g., K-ɛ-GG) [3] [6]. |
| Typical Sample Input | Not explicitly stated; requires genetic manipulation of sample. | ~500 μg - 7 mg of peptide digest for mass spectrometry analysis [7]. |
| Key Reagents | Tagged ubiquitin plasmid, affinity resins (Ni-NTA, Strep-Tactin) [3]. | Anti-K-ɛ-GG or anti-ubiquitin antibodies, protein A/G beads [7] [3]. |
| Stoichiometry | Analyzes tagged ubiquitin pool; may not reflect endogenous stoichiometry. | Targets endogenous ubiquitination; better reflects physiological stoichiometry [6]. |
| Chain Architecture | Can be linkage-specific if tagged ubiquitin with lysine mutations is used [3]. | Requires specific antibodies for different linkage types (M1, K48, K63, etc.) [3]. |
Performance Comparison: Key Metrics and Experimental Data
The following table compares the quantitative performance of both methods based on key metrics critical for researchers.
| Performance Metric | Ubiquitin Tagging | Antibody-Based Enrichment | Comparison Insight |
|---|---|---|---|
| Enrichment Efficiency/ Yield | Relatively low identification efficiency; co-purification of non-ubiquitinated proteins [3]. | High relative yield; ~85.7% of identified peptides are K-ɛ-GG peptides [7]. | Antibody-based methods offer superior specificity and purity for enriched ubiquitinated peptides. |
| Throughput & Scalability | Low-throughput due to requirement for genetic manipulation; not feasible for tissue samples [3]. | High-throughput protocols like UbiFast enable multiplexed analysis of 10+ samples in ~5 hours [7]. | Antibody-based methods are more adaptable to high-throughput studies, especially with clinical samples. |
| Sensitivity & Dynamic Range | Detection sensitivity is limited by background from non-specifically bound proteins [3]. | Highly sensitive; ThUBD-coated plates detect ubiquitinated proteins from amounts as low as 0.625 μg, a 16-fold improvement over TUBE-based methods [2]. | Advanced antibody/UBD-based platforms offer significantly higher sensitivity for detecting low-abundance ubiquitination. |
| Linkage-Type Flexibility | Flexible; allows for exploration of specific linkages by using ubiquitin mutants (e.g., K48R) [1] [3]. | Flexible but requires a specific antibody for each linkage type of interest [3]. | Ubiquitin tagging offers more inherent flexibility for probing non-canonical chain architectures. |
Detailed Experimental Protocols
Protocol 1: Ubi-Tagging for Site-Specific Protein Conjugation
Ubi-tagging repurposes the enzymatic ubiquitination cascade for precise protein engineering rather than analysis. However, it exemplifies the use of ubiquitin tags and linkage specificity [1] [8].
- Step 1: Construct Design. Generate two key protein fusions:
- Donor Ubi-tag (Ubdon): A protein (e.g., an antibody fragment) fused to ubiquitin where a specific lysine (e.g., K48) is mutated to arginine (K48R) to prevent homopolymerization.
- Acceptor Ubi-tag (Ubacc): A cargo molecule (e.g., a fluorescent dye or peptide) fused to ubiquitin that contains the corresponding conjugation lysine (K48) but has an unreactive C-terminus (ΔGG or blocked with a His-tag).
- Step 2: Enzymatic Conjugation. Incubate the Ubdon and Ubacc fusions (e.g., at a 1:5 molar ratio) with recombinant E1 enzyme and a linkage-specific E2-E3 fusion enzyme (e.g., gp78RING-Ube2g2 for K48 linkage) at 37°C.
- Step 3: Reaction Completion and Purification. The conjugation reaction is typically complete within 30 minutes with an efficiency of 93–96%. The homogeneous conjugate (e.g., Rho-Ub2-Fab) is then purified via affinity chromatography (e.g., protein G for antibodies) [1].
Protocol 2: UbiFast for Multiplexed Ubiquitylome Profiling
The UbiFast method is a high-throughput, antibody-based approach for profiling thousands of ubiquitination sites by mass spectrometry [7].
- Step 1: Protein Digestion. Complex protein samples (as little as 500 μg) are digested with trypsin. This cleaves after arginine and lysine residues, generating peptides with a di-glycine (K-ɛ-GG) remnant on formerly ubiquitinated lysines.
- Step 2: On-Bead Immunoaffinity Enrichment. The peptide digest is incubated with anti-K-ɛ-GG antibody beads. The K-ɛ-GG peptides are specifically bound, while non-modified peptides are washed away.
- Step 3: On-Antibody Tandem Mass Tag (TMT) Labeling. While the K-ɛ-GG peptides are still bound to the antibody, they are labeled with TMT reagents. Critically, the di-glycyl remnant is protected and does not get labeled, ensuring specificity.
- Step 4: Elution, Pooling, and Analysis. Peptides are eluted from the antibody, and samples from multiple conditions (up to 10 in a TMT10plex) are combined. The pooled sample is then analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) in approximately 5 hours, enabling the quantification of ~10,000 ubiquitylation sites [7].
Essential Research Reagent Solutions
The table below lists key reagents required for implementing these ubiquitination analysis methods.
| Reagent / Tool | Function / Application | Example Use Case |
|---|---|---|
| Linkage-Specific Ubi-Tag | Genetic construct (e.g., Ub-K48R) for controlled multivalent conjugation [1]. | Generating homogeneous bispecific antibody conjugates for therapeutic development [1] [8]. |
| Recombinant Ubiquitination Enzymes (E1, E2-E3) | Catalyzes the formation of a specific isopeptide bond between ubi-tags [1]. | Executing the ubi-tagging conjugation reaction with high efficiency and linkage control [1]. |
| Anti-K-ɛ-GG Antibody | Immunoaffinity enrichment of endogenous ubiquitination sites from digested samples for MS [7] [3]. | High-throughput ubiquitylome profiling in cell lines and primary tissue samples (UbiFast protocol) [7]. |
| Linkage-Specific Ubiquitin Antibodies | Immunoblotting or enrichment of polyubiquitinated proteins with a specific chain topology [3]. | Detecting K48-linked polyubiquitination of tau protein in Alzheimer's disease research [3]. |
| Tandem Hybrid UBD (ThUBD) | High-affinity, linkage-unbiased capture domain for ubiquitinated proteins [2]. | Sensitive, high-throughput detection of global ubiquitination signals in a 96-well plate format [2]. |
| Tandem Mass Tag (TMT) Reagents | Isobaric chemical labels for multiplexed quantitative mass spectrometry [7]. | Comparing ubiquitylation sites across 10 different experimental conditions simultaneously [7]. |
Protein ubiquitination is one of the most prevalent post-translational modifications (PTMs) within eukaryotic cells, exerting critical regulatory control over nearly every cellular, physiological, and pathophysiological process [9]. This versatility stems from the complexity of ubiquitin conjugates, which can range from a single ubiquitin monomer (monoubiquitination) to polymers of various lengths and linkage types [10]. The covalent attachment of ubiquitin, a 76-amino acid protein, to substrate proteins is mediated by an enzymatic cascade involving E1 activating, E2 conjugating, and E3 ligating enzymes [10]. Conversely, deubiquitinating enzymes (DUBs) remove ubiquitin, maintaining dynamic homeostasis [11].
A pivotal breakthrough in detecting this modification came from the discovery that tryptic digestion of ubiquitinated proteins generates peptides containing a characteristic Lys-ϵ-Gly-Gly (diGLY) remnant on modified lysine residues [9] [5]. This diGLY signature serves as a "molecular fingerprint" for prior ubiquitination. However, its low abundance relative to unmodified peptides necessitates highly specific enrichment strategies prior to mass spectrometry analysis [9]. This guide objectively compares the two dominant methodological philosophies for diGLY enrichment: antibody-based immunoaffinity and antibody-free chemical tagging approaches, providing researchers with the experimental data and protocols needed to inform their methodological choices.
Methodological Principles: Core Enrichment Technologies
Antibody-Based Immunoaffinity Enrichment
The most widely used method for ubiquitinome profiling employs antibodies specifically raised against the diGLY remnant motif [9] [12] [13]. In this workflow, proteins are digested into peptides, and anti-K-ε-GG antibodies are used to immunoaffinity purify diGLY-modified peptides from the complex mixture [5]. A significant advantage of this approach is its direct applicability to any eukaryotic organism or tissue without genetic manipulation [9] [10]. However, a key limitation is that the antibody cannot distinguish the diGLY remnant originating from ubiquitin from those generated by the ubiquitin-like modifiers NEDD8 and ISG15, though studies suggest ~95% of enriched diGLY peptides derive from ubiquitination [9]. Antibodies can also exhibit sequence recognition bias, potentially skewing the representation of certain diGLY peptides [11].
Antibody-Free Chemical Tagging Approaches
To overcome the limitations of antibody-based methods, researchers have developed innovative antibody-free strategies. One such method, the Antibody-Free approach for Ubiquitination Profiling (AFUP), employs a selective chemical tagging strategy [11]. This multi-step process involves:
- Blocking all free amine groups on proteins.
- Hydrolyzing ubiquitin from ubiquitinated proteins using deubiquitinases (USP2 and USP21), which regenerates the free ε-amine group specifically at the former ubiquitination sites.
- Labeling these newly exposed ε-amines with a cleavable biotin tag (NHS-SS-Biotin).
- Enriching the tagged peptides using streptavidin beads [11].
This method eliminates antibody bias and cost concerns, though it requires careful optimization of the blocking and enzymatic steps to ensure specificity [11].
Table 1: Core Principle Comparison of diGLY Enrichment Methods
| Feature | Antibody-Based Immunoaffinity | Antibody-Free Chemical Tagging (AFUP) |
|---|---|---|
| Core Principle | Immunoaffinity purification using anti-K-ε-GG antibodies [9] [5] | Chemical labeling of deubiquitinated lysines followed by streptavidin enrichment [11] |
| Key Reagent | Anti-K-ε-GG antibody [9] | Deubiquitinases (USP2/21) & NHS-SS-Biotin [11] |
| Specificity | Also enriches NEDD8/ISG15 diGLY motifs (~5% of identifications) [9] | Specific for sites deconjugated by the DUBs used; independent of diGLY motif |
| Primary Advantage | Direct application to endogenous samples and tissues; well-established [9] [10] | Avoids antibody sequence bias and high cost [11] |
| Primary Disadvantage | Potential for antibody sequence recognition bias [11] | Multi-step protocol requiring optimized reaction conditions [11] |
The following diagram illustrates the fundamental workflows for these two core methodologies.
Performance Comparison: Quantitative Data
The ultimate value of an enrichment method is reflected in its performance. The tables below summarize key quantitative metrics from published studies using various methodologies.
Identification Depth and Reproducibility
Table 2: Identification Depth and Reproducibility Across Methods
| Method | Sample Type | Input Amount | Sites Identified | Reproducibility (CV) | Citation |
|---|---|---|---|---|---|
| Automated UbiFast (magnetic beads) | Jurkat cells | 500 μg per sample (10-plex) | ~20,000 sites | Significantly improved vs. manual | [14] |
| AFUP (single run) | HeLa cells | 0.8 mg | 349 ± 7 sites | Excellent (CV = 2%) | [11] |
| AFUP + pre-fractionation | 293T cells | Not specified | ~4,000 sites | High (Pearson r ≥ 0.91) | [11] |
| Optimized diGLY-IP (DIA) | MG132-treated HEK293 cells | 1 mg | 35,111 ± 682 sites | 45% of peptides with CV < 20% | [12] |
| Optimized diGLY-IP (DDA) | MG132-treated HEK293 cells | 1 mg | ~20,000 sites | 15% of peptides with CV < 20% | [12] |
Technical and Practical Considerations
Table 3: Technical and Practical Method Comparison
| Characteristic | Antibody-Based | Antibody-Free (AFUP) |
|---|---|---|
| Multiplexing Capability | High (e.g., Automated UbiFast with TMT) [14] | Not demonstrated in cited literature |
| Quantitative Accuracy | High (DIA provides superior accuracy vs. DDA) [12] | High quantitative stability reported [11] |
| Sample Throughput | High, especially with automation (96 samples/day) [14] | Likely lower due to multi-step protocol |
| Novel Site Discovery | Identifies well-known and novel sites [12] | ~40% of identified sites were novel [11] |
| Required Instrumentation | Standard LC-MS/MS; magnetic bead processor for automation [14] | Standard LC-MS/MS |
Experimental Protocols: Key Workflows
Detailed Protocol: Antibody-Based diGLY Enrichment with SILAC
This is a foundational protocol for quantitative ubiquitinome analysis using Stable Isotope Labeling with Amino acids in Cell culture (SILAC) [9].
Cell Culture and Lysis:
- SILAC Media: Culture cells in DMEM lacking lysine and arginine, supplemented with dialyzed FBS and either "light" (L-lysine-2HCl, L-arginine-HCl) or "heavy" (13C6,15N2 L-lysine-2HCl, 13C6,15N4 L-arginine-HCl) isotopes for at least six doublings [9].
- Lysis Buffer: Use a denaturing buffer: 8 M Urea, 150 mM NaCl, 50 mM Tris-HCl (pH 8). Immediately before use, add protease inhibitors (e.g., Complete Protease Inhibitor Cocktail) and 5 mM N-Ethylmaleimide (NEM) to inhibit deubiquitinating enzymes [9].
Protein Digestion and Peptide Cleanup:
- Reduction and Alkylation: Reduce proteins with 5 mM dithiothreitol (45 min, room temperature) and alkylate with 10 mM iodoacetamide (30 min, room temperature in the dark) [14].
- Two-Step Digestion: First, digest with LysC (enzyme-to-substrate ratio 1:50) for 2 hours at room temperature. Then, dilute the urea concentration to ~2 M and digest with trypsin (enzyme-to-substrate ratio 1:50) overnight at room temperature [9] [14].
- Desalting: Acidify peptides with trifluoroacetic acid (TFA) to a final concentration of 0.5-1%. Desalt using a C18 solid-phase extraction cartridge (e.g., Sep-Pak). Condition the cartridge with acetonitrile and 0.1% TFA, load peptides, wash with 0.1% TFA, and elute with 50% acetonitrile/0.1% formic acid. Lyophilize the eluate [9] [15].
diGLY Peptide Immunoaffinity Enrichment:
- Antibody Beads: Use an anti-K-ε-GG antibody conjugated to agarose or magnetic beads. For magnetic beads (HS mag anti-K-ε-GG), one batch is typically sufficient for 1-2 mg of peptide input [14].
- Incubation: Reconstitute peptides in IAP buffer (50 mM MOPS/NaOH pH 7.2, 10 mM Na2HPO4, 50 mM NaCl) and incubate with the antibody beads for 1-2 hours at 4°C with gentle agitation [15].
- Washing: Wash beads 3-4 times with IAP buffer and twice with HPLC-grade water to remove non-specifically bound peptides [15].
- Elution: Elute diGLY peptides from the beads twice using 0.15% TFA. Combine the eluates and desalt using C18 StageTips or similar micro-columns prior to LC-MS/MS analysis [15].
Detailed Protocol: Antibody-Free AFUP Strategy
This protocol outlines the key steps for the Antibody-Free approach for Ubiquitination Profiling (AFUP) [11].
Amine Blocking and Deubiquitination:
- Blocking: Block all free amine groups (lysine ε-amines and protein N-terminal α-amines) by incubating the protein sample with 0.5% formaldehyde. This critical step prevents off-target labeling later [11].
- Deubiquitination: Add the catalytic core domains of deubiquitinases USP2 and USP21 to the blocked protein sample. Incubate to hydrolyze ubiquitin chains from the substrate proteins, which regenerates a free ε-amine group specifically at the ubiquitination sites [11].
Chemical Labeling and Enrichment:
- Biotinylation: Label the newly exposed ε-amines by adding NHS-SS-Biotin reagent. This reagent reacts specifically with the primary amines that were unmasked by deubiquitination [11].
- Streptavidin Enrichment: Digest the labeled proteins with trypsin. Incubate the resulting peptides with Streptavidin Sepharose beads to enrich for biotinylated peptides, which correspond to the former ubiquitination sites [11].
- Elution: Elute the enriched peptides by cleaving the disulfide bond in the NHS-SS-Biotin linker using dithiothreitol (DTT). The eluted peptides can then be analyzed by LC-MS/MS [11].
Essential Research Reagent Solutions
A successful ubiquitinome profiling experiment relies on a suite of specific reagents. The following table details key solutions used in the protocols above.
Table 4: Key Research Reagent Solutions for diGLY Proteomics
| Reagent / Kit | Function / Application | Key Features / Considerations |
|---|---|---|
| PTMScan Ubiquitin Remnant Motif Kit [9] | Immunoaffinity enrichment of K-ε-GG peptides. | Contains agarose-conjugated antibody; well-established for manual protocols. |
| HS mag anti-K-ε-GG antibody [14] | Magnetic bead-based enrichment for high-throughput studies. | Enables automation on magnetic particle processors; increases reproducibility. |
| UbiFast Method [14] | Highly multiplexed ubiquitination profiling. | Uses on-antibody TMT labeling for high sensitivity and quantitation from limited input. |
| NHS-SS-Biotin [11] | Chemical tagging of deubiquitinated lysines in AFUP. | Features a cleavable disulfide bond for efficient peptide elution after enrichment. |
| Recombinant USP2/USP21 [11] | Hydrolysis of ubiquitin chains in antibody-free methods. | Non-linkage specific deubiquitinases crucial for generating free ε-amines in AFUP. |
| Tandem Mass Tag (TMT) Reagents [14] | Isobaric labeling for multiplexed quantitative proteomics. | Allows pooling of samples; compatible with UbiFast (on-bead labeling). |
The diGLY remnant remains the cornerstone of mass spectrometry-based ubiquitinome profiling. Antibody-based enrichment, particularly when enhanced with automation [14], DIA acquisition [12], and multiplexed quantitation, currently sets the benchmark for depth of coverage, throughput, and quantitative robustness. It is the most suitable approach for large-scale, systems-wide studies. In contrast, antibody-free methods like AFUP [11] provide a powerful complementary strategy, offering a means to overcome antibody bias and potentially access a different subset of the ubiquitinome, including novel sites.
The choice between these methods depends on the specific research question, available resources, and sample type. For the foreseeable future, both approaches will coexist and continue to evolve. The ongoing development of more sensitive mass spectrometers, smarter acquisition modes like DIA, and novel biochemical tools promises to further illuminate the complex landscape of the ubiquitin code, driving discoveries in basic biology and drug development.
Ubiquitin-like proteins (UBLs) constitute a family of evolutionarily conserved proteins that share structural similarities with ubiquitin and play crucial roles in posttranslational modification of diverse macromolecules [16]. The eukaryotic ubiquitin family encompasses nearly 20 proteins that adopt the characteristic β-grasp fold of ubiquitin yet regulate strikingly diverse cellular processes including nuclear transport, proteolysis, translation, autophagy, and antiviral pathways [16]. As research continues to identify novel UBL substrates that expand our understanding of their functional diversity, the technical challenges in studying these modifications have become increasingly apparent. A primary bottleneck in the field involves the specific enrichment and identification of UBL-modified substrates, particularly distinguishing between different UBL types and their specific linkage patterns.
The specificity concerns in UBL enrichment stem from several inherent challenges. First, the structural similarity among UBLs complicates the development of highly specific enrichment tools. Second, the stoichiometry of UBL modification is typically low under physiological conditions, necessitating highly sensitive enrichment methods. Third, UBLs can form complex chains with different linkage types that dictate functional outcomes, requiring tools that can distinguish these specific architectures. This comparison guide objectively evaluates the performance of two primary technical approaches—ubiquitin tagging and antibody-based enrichment—for studying UBL modifications, providing researchers with experimental data and methodological insights to inform their experimental design.
Methodological Principles and Technical Specifications
Ubiquitin Tagging Approaches
Ubiquitin tagging methodologies involve the genetic engineering of affinity tags onto UBLs, enabling selective purification of modified substrates. This approach typically utilizes epitope tags (Flag, HA, V5, Myc, Strep, His) or protein/domain tags (GST, MBP, SUMO, Halo) fused to the UBL of interest [10]. After cellular expression of the tagged UBL, ubiquitinated substrates are covalently labeled and can be enriched using commercially available resins such as Ni-NTA for His tags or Strep-Tactin for Strep tags [10]. The pioneering work employing 6× His-tagged Ub in yeast identified 110 ubiquitination sites on 72 proteins, demonstrating the utility of this approach for proteomic profiling [10]. More recent innovations include the Stable tagged Ub exchange (StUbEx) cellular system, which replaces endogenous Ub with His-tagged Ub and has identified 277 unique ubiquitination sites on 189 proteins in HeLa cells [10].
A significant advancement in UBL tagging is the recently described "ubi-tagging" technique that exploits the ubiquitination enzymatic cascade for site-directed protein conjugation [1]. This modular approach utilizes ubiquitin fusions with antibodies, antibody fragments, nanobodies, peptides, or small molecules that can be conjugated to target proteins within 30 minutes with remarkable efficiency (93-96% conversion) [1]. The system employs donor ubi-tags (Ubdon) with free C-terminal glycine and mutated conjugating lysine residues to prevent homodimer formation, alongside acceptor ubi-tags (Ubacc) containing the corresponding conjugation lysine but with an unreactive C terminus [1]. This technology demonstrates that large cargo such as 50 kDa Fab' fragments does not hamper ubi-tagging efficiency, enabling generation of defined multimers including bispecific T-cell engagers [1].
Antibody-Based Enrichment Strategies
Antibody-based enrichment utilizes immunoreagents specifically developed to recognize UBLs or their modification signatures. These include pan-specific anti-ubiquitin antibodies (P4D1, FK1/FK2) that recognize all ubiquitin linkages, as well as linkage-specific antibodies targeting particular chain architectures (M1-, K11-, K27-, K48-, K63-linkage specific antibodies) [10]. For UBLs such as SUMO, specific antibodies have been crucial for enrichment despite challenges in identifying modification sites without mutagenesis [17] [18].
A prominent example of antibody-based innovation is the UbiFast method, which employs anti-K-ε-GG antibodies for sensitive ubiquitylation site detection [19]. The recent automation of UbiFast using magnetic bead-conjugated K-ε-GG antibodies (mK-ε-GG) and magnetic particle processing has significantly enhanced reproducibility and throughput, enabling processing of up to 96 samples in a single day [19]. This automated workflow identifies approximately 20,000 ubiquitylation sites from TMT10-plex experiments with 500 μg input per sample processed in approximately 2 hours, representing a substantial improvement over manual methods [19]. The sensitivity of this approach has been demonstrated in profiling patient-derived xenograft tissue samples, highlighting its applicability to clinically relevant material [19].
For UBLs beyond ubiquitin, specialized tools have emerged such as activity-based probes (ABPs) for interferon-inducible Ubl protease USP18, which incorporates unnatural amino acids into the C-terminal tail of ISG15 to enable selective detection of USP18 activity over other ISG15 cross-reactive deubiquitinases [20]. These probes utilize a chemical biology approach with a ubiquitin-like protein recognition element, an electrophilic warhead, and a reporter tag to covalently label active site cysteines, reporting deISGylase activity in complex biological systems [20].
Table 1: Performance Comparison of UBL Enrichment Methods
| Parameter | Ubiquitin Tagging | Antibody-Based Enrichment |
|---|---|---|
| Specificity Control | Genetic manipulation of UBL sequence | Antibody cross-reactivity profile |
| Throughput | Lower (requires genetic modification) | Higher (direct application to samples) |
| Identification Sensitivity | 110-750 ubiquitination sites [10] | ~20,000 ubiquitylation sites with automated UbiFast [19] |
| Linkage Specificity | Limited without additional manipulation | Excellent with linkage-specific antibodies |
| Physiological Relevance | Potential artifacts from tagged UBL expression [10] | Preserves endogenous modification states |
| Application to Tissue Samples | Infeasible for patient/animal tissues without genetic modification [10] | Direct application to clinical specimens [19] |
| Multiplexing Capacity | Limited by genetic manipulability | High with automated platforms [19] |
Experimental Data and Performance Comparison
Sensitivity and Specificity Metrics
Direct comparison of ubiquitin tagging versus antibody-based approaches reveals significant differences in sensitivity and specificity. The development of specialized search engines like pLink-UBL has addressed a critical limitation in UBL modification site identification, particularly for SUMOylation sites that conventional mass spectrometry methods struggle to characterize [17] [18]. pLink-UBL demonstrates superior precision, sensitivity, and speed compared to alternative search engines such as MaxQuant, pFind, and pLink, increasing identified SUMOylation sites by 50-300% from the same datasets [17] [18]. This represents a significant advancement as traditional approaches required UBL mutation to facilitate identification, potentially altering biological behavior.
For ubiquitin specifically, antibody-based approaches have shown remarkable sensitivity when optimized. The automated UbiFast method identifies approximately 20,000 ubiquitylation sites from limited input material (500 μg per sample), far exceeding the 753 lysine ubiquitylation sites on 471 proteins identified through Strep-tagged Ub approaches [19] [10]. This substantial difference highlights the enhanced detection capability of modern antibody-based platforms, particularly for lower-abundance modifications.
Specificity challenges persist in both approaches, though they manifest differently. Ubiquitin tagging methods risk introducing artifacts as tagged UBLs may not perfectly mimic endogenous UBL structure and function [10]. Additionally, histidine-rich and endogenously biotinylated proteins can co-purify with Ni-NTA agarose and Strep-Tactin resins respectively, reducing identification specificity [10]. Antibody-based methods face challenges of cross-reactivity, particularly problematic when studying specific UBL types or linkage architectures. The development of linkage-specific antibodies and TUBEs (tandem ubiquitin binding entities) has significantly addressed these concerns, enabling precise capture of specific polyubiquitination events [21] [10].
Throughput and Reproducibility
Throughput and reproducibility are critical considerations for large-scale proteomic studies and screening applications. Automated antibody-based methods demonstrate significant advantages in these areas, with robotic processing enabling substantially improved reproducibility compared to manual methods [19]. The automation of UbiFast reduced variability across process replicates while dramatically increasing processing throughput to 96 samples in a single day [19]. This level of throughput is particularly valuable for pharmaceutical applications including drug development and profiling, where consistent processing of many samples is essential.
Ubiquitin tagging approaches inherently involve more complex workstreams requiring genetic modification prior to enrichment, limiting their applicability to screen large sample numbers or clinical specimens. However, for focused mechanistic studies in genetically tractable systems, tagging approaches provide valuable specificity controls through genetic manipulation of modification sites.
Table 2: Workflow Characteristics and Applications
| Characteristic | Ubiquitin Tagging | Antibody-Based Enrichment |
|---|---|---|
| Sample Compatibility | Genetically modified systems only | Native biological samples, clinical specimens [19] [10] |
| Processing Time | Days (including genetic manipulation) | ~2 hours for 10 samples, 96 samples in one day (automated) [19] |
| Quantification Capability | Moderate | Excellent with isobaric labeling (TMT) [19] |
| Specialized Equipment Needs | Standard molecular biology equipment | Magnetic particle processor (for automated workflows) [19] |
| Data Analysis Requirements | Standard proteomic workflows | Specialized search engines (pLink-UBL) for certain UBLs [17] [18] |
| Best Applications | Mechanistic studies, validation experiments | Large-scale profiling, clinical samples, drug screening [19] [21] |
Advanced Applications and Specialized Tools
Linkage-Specific Analysis Tools
The functional diversity of UBL signaling is largely encoded in the specific linkage types of polymer chains. Different ubiquitin linkages regulate distinct cellular processes, with K48-linked chains primarily targeting substrates for proteasomal degradation while K63-linked chains regulate signal transduction and protein trafficking [21]. To address the challenge of linkage-specific analysis, tandem ubiquitin binding entities (TUBEs) have been developed with nanomolar affinities for specific polyubiquitin chains [21]. These specialized tools enable investigation of ubiquitination dynamics in specific contexts, such as monitoring K63 ubiquitination of RIPK2 in inflammatory signaling versus K48 ubiquitination induced by PROTACs targeted for degradation [21]. The application of chain-specific TUBEs in high-throughput screening assays provides a platform for quantifying context-dependent linkage-specific ubiquitination of endogenous proteins, advancing both basic research and drug development efforts [21].
Chemical Biology Tools for UBL Proteases
Beyond enrichment of modified substrates, understanding UBL regulation requires tools to study the enzymes that process these modifications, particularly deubiquitinases (DUBs). Activity-based protein profiling (ABPP) using ABPs has emerged as a powerful platform for mapping reactive proteins and evaluating functional states of enzymes in complex biological systems [20]. For the interferon-inducible Ubl protease USP18, specifically designed ABPs incorporate unnatural amino acids into the C-terminal tail of ISG15 to enable selective detection of USP18 activity over other ISG15 cross-reactive DUBs [20]. These tools employ a hybrid combinatorial substrate library (HyCoSuL) screening approach to identify mutations in the LRGG tail of ISG15 that enhance selective binding to USP18, demonstrating how chemical biology approaches are addressing specificity challenges in the UBL field [20].
Unexpected Substrate Discovery
Advanced enrichment methodologies have enabled the discovery of unexpected UBL substrates, expanding our understanding of UBL biology. Recently, researchers developed a method combining antibody enrichment of UBL C-terminal peptides with LC-MS/MS analysis and pFind 3 blind search to identify nonprotein substrates [17] [18]. This approach revealed spermidine as a major small-molecule substrate for fission yeast SUMO Pmt3 and mammalian SUMO proteins [17] [18]. Spermidine conjugates to the C-terminal carboxylate group of Pmt3 through its N1 or N8 amino group in the presence of SUMO E1, E2, and ATP, without requiring E3 enzymes, and can be reversed by SUMO isopeptidase Ulp1 [17] [18]. This surprising finding that spermidine may be a common small molecule substrate of SUMO and possibly ubiquitin across eukaryotic species underscores the importance of continued methodological development in UBL enrichment, as novel tools may reveal unexpected aspects of UBL biology [17] [18].
Research Reagent Solutions
The following table provides key research reagents essential for implementing UBL enrichment methodologies, based on tools featured in the cited research.
Table 3: Essential Research Reagents for UBL Enrichment Studies
| Reagent | Function | Application Examples |
|---|---|---|
| pLink-UBL Software | Specialized search engine for UBL modification site identification | Identifies SUMOylation sites without UBL mutation; increases sites identified by 50-300% [17] [18] |
| Anti-K-ε-GG Antibody | Enriches ubiquitinated peptides for mass spectrometry | Automated UbiFast platform; enables identification of ~20,000 ubiquitylation sites [19] |
| Tandem Ubiquitin Binding Entities (TUBEs) | High-affinity capture of polyubiquitin chains with linkage specificity | Differentiates K48 vs K63 ubiquitination in cellular signaling and PROTAC mechanisms [21] |
| Activity-Based Probes (ABPs) | Chemical tools to monitor enzyme activity in complex systems | Profiles USP18 deISGylating activity in lung cancer cell lines [20] |
| Ubi-Tagging System Components | Modular system for site-directed protein conjugation | Generates bispecific T-cell engagers, nanobody-peptide conjugates (93-96% efficiency) [1] |
| Linkage-Specific Ub Antibodies | Immunoenrichment of specific ubiquitin chain architectures | Characterizes chain-type specific functions in disease models [10] |
Methodological Protocols
Automated UbiFast Protocol
The automated UbiFast protocol represents a state-of-the-art approach for high-throughput ubiquitylation site identification [19]. This method utilizes magnetic bead-conjugated K-ε-GG antibody (mK-ε-GG) and a magnetic particle processor to achieve highly reproducible enrichment with significantly reduced processing time compared to manual methods. Begin with protein extraction and digestion following standard proteomic protocols. For each sample, utilize 500 μg of peptide material as input. Resuspend peptides in immunoaffinity enrichment buffer and incubate with mK-ε-GG beads for 2 hours at room temperature with gentle agitation. Using the magnetic particle processor, wash beads thoroughly to remove non-specifically bound peptides. While still on-bead, label peptides with isobaric tandem mass tag (TMT) reagents for sample multiplexing. Elute bound peptides and pool samples for simultaneous LC-MS/MS analysis. Analyze resulting spectra using appropriate database search algorithms, achieving identification of approximately 20,000 ubiquitylation sites from a TMT10-plex experiment [19].
Ubi-Tagging Conjugation Protocol
The ubi-tagging protocol enables site-specific protein conjugation using ubiquitin biochemistry [1]. This 30-minute procedure efficiently generates defined conjugates including fluorescently labeled Fab' fragments, Fab' multimers, and Fab'-peptide conjugates. Begin by preparing the ubiquitination enzyme mixture containing 0.25 μM E1 enzyme and 20 μM of the appropriate E2-E3 fusion protein (e.g., gp78RING-Ube2g2 for K48-specific linkages). Combine 10 μM of donor ubi-tagged protein (Ubdon, containing K48R mutation to prevent homodimer formation) with 50 μM of acceptor ubi-tag (Ubacc, with ΔGG C-terminal modification to prevent elongation) in reaction buffer. Add the enzyme mixture to initiate conjugation and incubate at room temperature for 30 minutes. Monitor reaction completion by SDS-PAGE, observing complete consumption of the donor ubi-tagged protein. Purify the conjugate using appropriate affinity chromatography (e.g., protein G for antibody fragments). Validate conjugation efficiency by electrospray ionization time-of-flight (ESI-TOF) mass spectrometry, typically achieving 93-96% conversion efficiency [1].
The comparison of ubiquitin tagging and antibody-based enrichment methods reveals a complex landscape where methodological selection must align with specific research objectives. Ubiquitin tagging approaches offer genetic specificity and are invaluable for mechanistic studies in genetically tractable systems, particularly when combined with recent advances in ubi-tagging conjugation technology [1]. Conversely, antibody-based methods provide superior throughput, sensitivity, and applicability to native biological systems and clinical specimens, especially when implemented in automated platforms like UbiFast [19]. The ongoing development of specialized tools including linkage-specific TUBEs [21], activity-based probes for UBL proteases [20], and advanced search algorithms like pLink-UBL [17] [18] continues to address specificity concerns in UBL enrichment. As these methodologies evolve, they will undoubtedly uncover novel aspects of UBL biology and provide increasingly sophisticated tools for researchers and drug development professionals working in this complex field.
Protein ubiquitylation is a fundamental post-translational modification that regulates diverse cellular functions, including protein degradation, signal transduction, and immune response [3]. This process involves the covalent attachment of the 76-amino acid ubiquitin protein to substrate proteins, primarily through isopeptide bonds with lysine residues [3]. The versatility of ubiquitin signaling arises from the complexity of ubiquitin conjugates, which can range from single ubiquitin monomers to polymers with different lengths and linkage types [3]. However, the low stoichiometry of ubiquitylation and the complexity of ubiquitin chains have made comprehensive analysis challenging [3] [22].
To address these challenges, enrichment strategies have evolved significantly, moving from tagged ubiquitin expression systems to sophisticated methods for capturing endogenous ubiquitylation events. This evolution has been driven by the need for more physiologically relevant data and the ability to study ubiquitin signaling in clinical and tissue samples where genetic manipulation is infeasible [3] [22]. The choice between tagged ubiquitin and endogenous capture methods represents a critical decision point for researchers, with each approach offering distinct advantages and limitations that must be carefully considered based on experimental goals and sample availability.
Methodological Foundations: Key Ubiquitin Enrichment Techniques
Tagged Ubiquitin Approaches
Tagged ubiquitin methodologies involve the genetic engineering of cells to express ubiquitin fused to an affinity tag, enabling purification of ubiquitylated substrates. The most commonly used tags include 6×His and Strep-tag II, which allow enrichment using Ni-NTA agarose and Strep-Tactin resins, respectively [3]. In this approach, cells are engineered to express the tagged ubiquitin, which becomes incorporated into the cellular ubiquitination machinery and covalently attached to substrate proteins. Following cell lysis, ubiquitinated proteins are purified using resins that specifically bind the affinity tag, after which they can be identified through mass spectrometric analysis [3].
This method was pioneered by Peng et al. (2003), who first demonstrated the large-scale identification of ubiquitination sites by expressing 6×His-tagged ubiquitin in Saccharomyces cerevisiae, identifying 110 ubiquitination sites on 72 proteins [3]. Subsequent developments, such as the Stable tagged Ub exchange (StUbEx) cellular system, further refined this approach by enabling replacement of endogenous ubiquitin with His-tagged ubiquitin in human cell lines, leading to the identification of 277 unique ubiquitination sites on 189 proteins in HeLa cells [3]. Similarly, researchers using Strep-tagged ubiquitin constructs have identified 753 lysine ubiquitylation sites on 471 proteins in U2OS and HEK293T cells [3].
Antibody-Based Endogenous Capture Methods
Antibody-based approaches directly target endogenous ubiquitin modifications without requiring genetic engineering of the sample source. The breakthrough enabling these methods was the development of antibodies that specifically recognize the di-glycyl (K-ε-GG) remnant left on tryptic peptides after proteolytic digestion of ubiquitylated proteins [22] [13]. These antibodies allow immunoaffinity enrichment of endogenously ubiquitylated peptides from complex biological samples [13].
A significant advancement in this category is the UbiFast method, which combines antibody-based enrichment with on-bead isobaric labeling for multiplexed analysis [22] [14]. This innovative approach addresses a major limitation: traditional K-ε-GG antibodies cannot recognize their targets when the N-terminus of the di-glycyl remnant is derivatized with tandem mass tags (TMT) [22]. In the UbiFast workflow, K-ε-GG peptides are enriched with anti-K-ε-GG antibody, then labeled with TMT reagents while still bound to the antibody, which protects the di-glycyl remnant from derivatization [22]. The labeled peptides from multiple samples are then combined, eluted, and analyzed by LC-MS/MS [22]. Automation of UbiFast using magnetic bead-conjugated antibodies has further enhanced throughput, enabling processing of up to 96 samples per day with identification of approximately 20,000 ubiquitylation sites from just 500 μg of input material per sample [14].
Table 1: Comparison of Fundamental Ubiquitin Enrichment Approaches
| Feature | Tagged Ubiquitin | Antibody-Based Endogenous Capture |
|---|---|---|
| Principle | Expression of affinity-tagged ubiquitin (His, Strep) in cells | Immunoaffinity enrichment using anti-K-ε-GG or linkage-specific antibodies |
| Sample Requirements | Genetically engineered cell lines | Cell lines, tissues, primary cells, clinical samples |
| Key Advantage | Relatively low-cost; friendly for screening | Studies endogenous ubiquitination without genetic manipulation |
| Main Limitation | Cannot be used on tissues/clinical samples; potential artifacts | High antibody cost; potential non-specific binding |
| Typical Identified Sites | 110-753 sites (early studies) | >10,000 sites with advanced methods like UbiFast |
Technical Comparison: Performance and Applications
Efficiency and Coverage
The evolution of enrichment strategies has led to remarkable improvements in the depth and breadth of ubiquitinome coverage. Early tagged ubiquitin approaches typically identified hundreds of ubiquitination sites, which was groundbreaking at the time but limited compared to current capabilities [3]. In contrast, modern antibody-based methods like the automated UbiFast platform can identify approximately 20,000 ubiquitylation sites from just 500 μg of input material per sample in a TMT10-plex experiment [14].
Quantitative comparisons demonstrate the superior performance of advanced endogenous capture methods. In direct comparisons of sample processing techniques, the automated UbiFast method using magnetic beads significantly outperformed manual agarose-based immunoprecipitation, with the magnetic bead approach identifying 9,624 ubiquitination sites compared to 6,821 sites with agarose beads from the same input amount [14]. This represents a 41% increase in coverage and highlights how methodological refinements continue to enhance experimental outcomes.
Specificity and Background Interference
Both tagged ubiquitin and antibody-based approaches face challenges with specificity, though the nature of interference differs between methods. Tagged ubiquitin systems, particularly those using His-tags, can co-purify histidine-rich proteins, while Strep-tag systems may isolate endogenously biotinylated proteins [3]. These non-specific interactions can reduce the sensitivity of ubiquitinated substrate identification and increase background noise.
Antibody-based methods suffer from different limitations, including potential cross-reactivity and non-specific binding of non-ubiquitinated peptides to the antibody or solid support [3]. However, the development of more specific antibody clones and improved blocking conditions has substantially enhanced signal-to-noise ratios. The implementation of tandem enrichment strategies, such as the SCASP-PTM method that simultaneously enriches ubiquitinated, phosphorylated, and glycosylated peptides from a single sample, further demonstrates how specificity challenges are being addressed through innovative workflow design [23].
Experimental Workflows and Practical Considerations
The practical implementation of ubiquitin enrichment methods involves significantly different workflows, time investments, and technical requirements. Tagged ubiquitin approaches require substantial upfront work in cell line development and validation but offer relatively straightforward downstream processing [3]. In contrast, antibody-based methods eliminate the need for genetic engineering but require careful optimization of enrichment conditions and more complex sample processing.
Table 2: Practical Implementation Comparison Between Methods
| Parameter | Tagged Ubiquitin | Antibody-Based Endogenous Capture |
|---|---|---|
| Sample Preparation Time | Days to weeks (cell line development) | Hours to days (no genetic manipulation needed) |
| Enrichment Processing Time | Several hours | ~2 hours for automated UbiFast [14] |
| Multiplexing Capacity | Limited to 2-3 samples with SILAC | Up to 18 samples with TMT isobaric tagging [22] |
| Required Input Material | Not systematically reported | 500 μg - 1 mg per sample for deep coverage [22] [14] |
| Instrumentation Needs | Standard LC-MS/MS | LC-MS/MS with FAIMS for improved quantification [22] |
Specialized Applications and Emerging Innovations
Linkage-Specific Analysis
Beyond general ubiquitination profiling, specialized approaches have emerged for studying specific ubiquitin chain linkages. Linkage-specific antibodies have been developed that recognize M1-, K11-, K27-, K48-, and K63-linked polyubiquitin chains, enabling researchers to investigate the biological functions associated with different ubiquitin signaling architectures [3]. For example, Nakayama et al. utilized a K48-linkage specific antibody to demonstrate abnormal accumulation of K48-linked polyubiquitinated tau proteins in Alzheimer's disease [3].
Ubiquitin-binding domains (UBDs) provide an alternative tool for linkage-specific enrichment. Tandem-repeated UBDs with enhanced affinity for specific chain types have been successfully employed to purify and characterize ubiquitinated proteins with defined chain architectures [3]. These approaches have been particularly valuable for deciphering the complex signaling codes embodied in the ubiquitin code.
Advanced Instrumentation and Computational Tools
The evolution of enrichment strategies has been paralleled by advances in mass spectrometry instrumentation and computational analysis. High-field asymmetric waveform ion mobility spectrometry (FAIMS) has been integrated into ubiquitin profiling workflows to improve quantitative accuracy for post-translational modification analysis [22]. Additionally, specialized search engines like pLink-UBL have been developed specifically for ubiquitin-like protein modification site identification, demonstrating superior precision, sensitivity, and speed compared to general-purpose proteomics software [17].
A particularly innovative application of ubiquitin biochemistry is the ubi-tagging platform for antibody conjugation. This technology repurposes the ubiquitination machinery for site-specific protein engineering, enabling efficient generation of homogeneous antibody conjugates within 30 minutes with an impressive efficiency of 93-96% [1] [8]. The platform allows creation of multimeric antibody formats, including bispecific T-cell engagers, demonstrating how fundamental ubiquitination principles can be harnessed for therapeutic applications [1].
Research Reagent Solutions: Essential Materials for Ubiquitin Enrichment
Table 3: Key Research Reagents for Ubiquitin Enrichment Studies
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| Affinity Tags | 6×His tag, Strep-tag II | Fused to ubiquitin for purification of ubiquitinated substrates from engineered cells [3] |
| Enrichment Antibodies | Anti-K-ε-GG (di-glycyl remnant) | Immunoaffinity enrichment of endogenously ubiquitinated peptides for mass spectrometry [22] [13] |
| Linkage-Specific Reagents | K48-, K63-, M1-linkage specific antibodies | Selective enrichment of ubiquitin chains with specific linkage types [3] |
| Ubiquitination Enzymes | E1 activating, E2 conjugating, E3 ligating enzymes | In vitro ubiquitination assays; ubi-tagging conjugation platform [1] |
| Deubiquitinases (DUBs) | Ulp1, various DUB inhibitors | Control experiments; validation of ubiquitin-dependent signals [17] |
| Mass Spectrometry Tags | Tandem Mass Tag (TMT) reagents | Multiplexed quantitative analysis of ubiquitination sites across multiple samples [22] [14] |
The evolution from tagged ubiquitin to endogenous capture methods represents significant progress in ubiquitin research, with each approach offering distinct advantages for specific research contexts. Tagged ubiquitin methods remain valuable for hypothesis-driven research in genetically tractable systems where controlled expression enables straightforward validation, while antibody-based endogenous capture approaches provide unparalleled access to physiological ubiquitination events in diverse sample types, including clinical specimens.
Future directions in ubiquitin enrichment will likely focus on increasing sensitivity to work with even smaller sample amounts, enhancing linkage-specific analysis capabilities, and integrating ubiquitin profiling with other post-translational modification analyses in multi-omics frameworks. The continued development of innovative technologies like ubi-tagging for therapeutic applications demonstrates how fundamental research into ubiquitin biochemistry continues to yield unexpected translational opportunities. As these methods mature, researchers will be increasingly equipped to decipher the complex ubiquitin code in health and disease, potentially unlocking new diagnostic and therapeutic strategies targeting the ubiquitin-proteasome system.
Diagram 1: Decision framework for selecting appropriate ubiquitin enrichment strategies based on sample type and research objectives.
Methodologies in Action: Protocols and Applications from Basic Research to Therapeutics
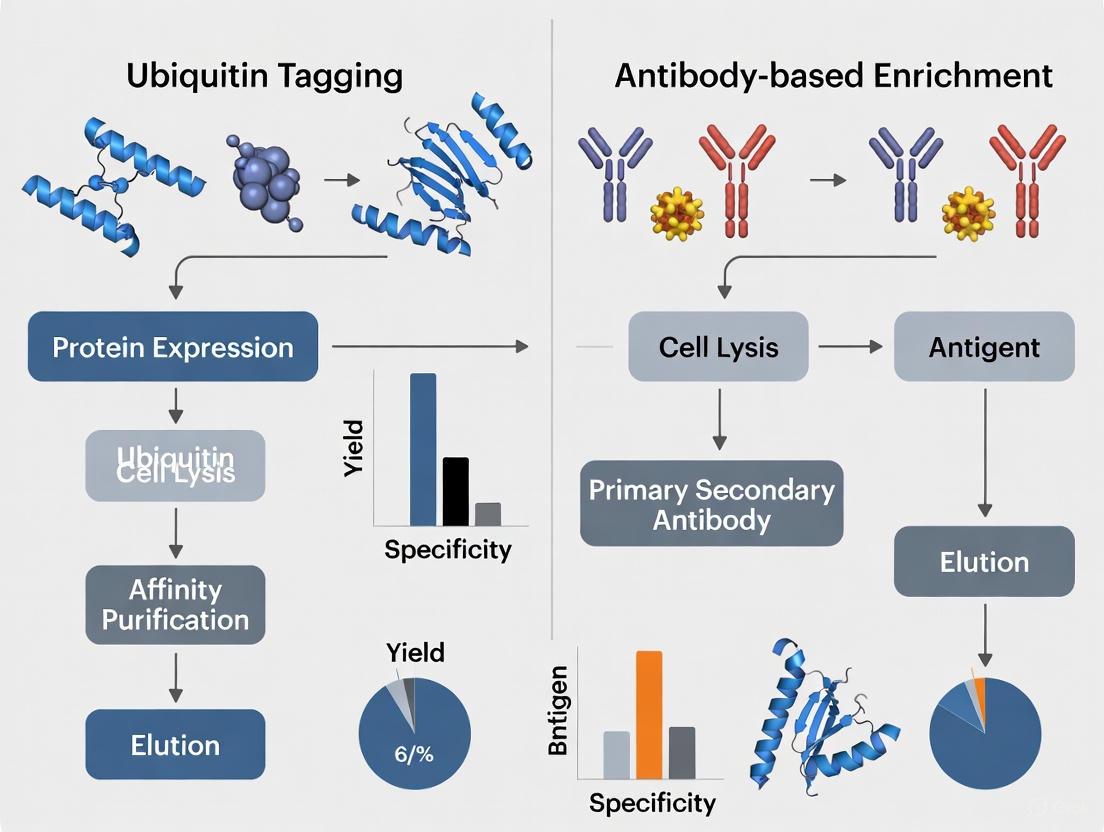
Protein ubiquitination, a crucial post-translational modification, regulates diverse cellular processes from protein degradation to signal transduction. For researchers aiming to study the ubiquitinome, two principal methodological pathways have emerged: ubiquitin tagging (affinity tag approach) and antibody-based enrichment. The ubiquitin tagging approach involves genetically engineering cells to express ubiquitin fused to an affinity tag (such as His or Strep), enabling purification of ubiquitinated proteins directly. In contrast, antibody-based methods utilize antibodies that recognize endogenous ubiquitin signatures, such as the diGly remnant left on trypsinized peptides, to enrich ubiquitinated species from wild-type cells. This guide provides an objective comparison of these approaches, focusing on the principles, workflow, and performance data of the ubiquitin affinity tag method to inform researchers and drug development professionals.
Principle and Components of Ubiquitin Affinity Tagging
The core principle of the ubiquitin affinity tag approach is the genetic replacement or supplementation of endogenous ubiquitin with a tag-modified version. This allows the entire pool of cellular ubiquitinated proteins to be covalently labeled with an affinity handle, facilitating their subsequent purification under denaturing conditions that preserve unstable modifications and protein-complex interactions [3].
- Tag Integration: The DNA sequence for an affinity tag (e.g., His, Strep, FLAG) is fused to the N-terminus of the ubiquitin gene. This construct is then introduced into cells, where the tagged ubiquitin is expressed and processed by the native enzymatic cascade (E1, E2, E3), resulting in its covalent attachment to substrate proteins [3].
- Key Affinity Tags: The most commonly used tags are the 6× His-tag and the Strep-tag. The His-tag binds to immobilized metal ions (e.g., Ni²⁺) on chromatography resins, while the Strep-tag binds with high affinity to Strep-Tactin resins [3] [24].
- System Establishment: In a refined method known as the Stable Tagged Ubiquitin Exchange (StUbEx) system, endogenous wild-type ubiquitin is replaced with His-tagged ubiquitin, ensuring that all cellular ubiquitination events carry the affinity tag [3].
The following diagram illustrates the conceptual workflow and fundamental principle of this approach.
Detailed Workflow for Ubiquitin Affinity Tagging
The experimental workflow for the ubiquitin tagging approach involves a series of defined steps from cell culture to mass spectrometry analysis, as outlined below.
Step-by-Step Protocol:
Cell Line Engineering and Culture:
- Generate a cell line (e.g., HEK293, U2OS) stably expressing N-terminally tagged ubiquitin (e.g., His-Ub or Strep-Ub). The StUbEx system is highly effective for this purpose [3].
- Culture cells under standard conditions. To stabilize ubiquitinated proteins, especially those targeted for proteasomal degradation, treat cells with a proteasome inhibitor such as MG132 (10-25 µM for 3-4 hours) prior to harvesting [25] [26].
Cell Lysis and Protein Extraction:
Affinity Purification of Ubiquitinated Proteins:
- Incubate the clarified, denatured lysate with the appropriate affinity resin:
- For His-tagged Ub: Use Ni-NTA (Nickel-Nitrilotriacetic Acid) agarose resin. Wash with buffers containing decreasing pH (e.g., to pH 6.3) and imidazole (e.g., 20 mM) to reduce non-specific binding. Elute with buffer containing 250-500 mM imidazole [3] [24].
- For Strep-tagged Ub: Use Strep-Tactin resin. Wash and elute under denaturing conditions with a buffer containing desthiobiotin (e.g., 50 mM) or biotin [3].
- Incubate the clarified, denatured lysate with the appropriate affinity resin:
Protein Digestion and Peptide Preparation:
- The purified pool of ubiquitinated proteins is then prepared for mass spectrometry. This typically involves reduction, alkylation, and tryptic digestion. Trypsin cleaves ubiquitin, leaving a signature diGlycine (K-ε-GG) remnant on the modified lysine residue of the substrate peptide, which results in a mass shift of +114.0429 Da [26] [22].
Mass Spectrometry Analysis:
Performance Comparison: Ubiquitin Tagging vs. Antibody-Based Enrichment
The table below summarizes key performance metrics and characteristics of the ubiquitin tagging approach compared to the main alternative, antibody-based diGly remnant enrichment.
| Feature | Ubiquitin Tagging (Affinity Tag) | Antibody-Based (diGly Enrichment) |
|---|---|---|
| Principle | Purification of tagged ubiquitin-protein conjugates at the protein level [3]. | Immunoaffinity enrichment of tryptic peptides containing the K-ε-GG remnant at the peptide level [12] [26]. |
| Required Genetic Manipulation | Yes (stable expression of tagged Ub) [3]. | No (works with wild-type cells and tissues) [12] [22]. |
| Typical Scale of Identified Sites | ~280-750 sites from initial studies [3]. | >35,000 sites in a single experiment (e.g., using DIA) [12]. |
| Compatibility with Tissues/Patients | Infeasible for animal or patient tissues [3]. | Highly suitable for tissue and clinical samples [22]. |
| Linkage-Type Specificity | Can be engineered using ubiquitin mutants for specific linkages [1]. | Requires separate, specific antibodies for different linkage types [3]. |
| Key Advantage | Directly captures the ubiquitinated protein; can study protein complexes. | Extreme depth and sensitivity; high translational potential for clinical samples. |
| Key Disadvantage | Lower identification efficiency; potential for artifacts from tagged Ub expression [3]. | High cost of antibodies; cannot provide information on protein-level conjugates [3]. |
Table: Objective comparison of the ubiquitin affinity tag approach and the antibody-based diGly enrichment method.
Quantitative data from direct comparisons highlights the difference in sensitivity. One study noted that K-GG peptide immunoaffinity enrichment yielded greater than fourfold higher levels of modified peptides than protein-level affinity purification (AP-MS) approaches [26]. Furthermore, while ubiquitin tagging identified hundreds to low-thousands of sites in foundational studies, modern antibody-based diGly methods with Data-Independent Acquisition (DIA) mass spectrometry routinely identify over 35,000 distinct diGly sites in a single measurement [12].
The Scientist's Toolkit: Key Reagents and Solutions
Successful implementation of the ubiquitin affinity tag approach requires the following key reagents:
| Reagent / Solution | Function / Description |
|---|---|
| Plasmid for Tagged-Ub | Expression vector for His-Ub, Strep-Ub, or other tagged ubiquitin constructs. |
| Proteasome Inhibitor (MG132) | Stabilizes ubiquitinated proteins by blocking their degradation [26]. |
| Denaturing Lysis Buffer | Typically contains 6-8 M Urea or Guanidine•HCl; inactivates DUBs to preserve ubiquitination [3]. |
| Affinity Resin | Ni-NTA Agarose (for His-tag) or Strep-Tactin Resin (for Strep-tag) [3] [24]. |
| Imidazole | Used in wash and elution buffers for His-tag purifications to compete with protein binding [24]. |
| Desthiobiotin | A biotin analog used for gentle and efficient elution of Strep-tagged proteins [3]. |
| Trypsin | Protease used to digest purified proteins, generating diGly-modified peptides for MS analysis [22]. |
The ubiquitin affinity tag approach provides a direct method for isolating ubiquitinated proteins and is a powerful tool when studying ubiquitin chain architecture or protein complexes. However, quantitative data clearly shows that antibody-based diGly enrichment methods offer superior depth of coverage, sensitivity, and are indispensable for translational research involving clinical tissues. The choice between these methods should be guided by the specific research question. For hypothesis-driven research on a specific protein or complex where genetic manipulation is feasible, ubiquitin tagging remains valuable. For discovery-phase projects aiming for system-wide coverage of the ubiquitinome in physiologically relevant models, antibody-based diGly enrichment is the unequivocally more powerful and appropriate technology.
Protein ubiquitination is one of the most prevalent post-translational modifications (PTMs), exerting critical regulatory control over virtually every cellular process, from protein degradation to signal transduction and DNA repair [9] [3]. The versatility of ubiquitin signaling arises from its ability to form diverse chain architectures, which encode specific biological functions. However, the low stoichiometry of ubiquitination and the complexity of ubiquitin chains make system-wide analysis challenging [3]. Among the methods developed to characterize the ubiquitinome, the antibody-based enrichment method targeting the ubiquitin remnant (diGLY approach) has emerged as a powerful technique for the identification and quantification of ubiquitination sites with high sensitivity and specificity [9]. This guide provides a comprehensive comparison of the diGLY approach against alternative methods, focusing on its principle, experimental workflow, and performance data to inform researchers and drug development professionals.
The Core Principle of the diGLY Approach
The diGLY approach leverages a defining chemical signature left on proteins after they have been modified by ubiquitin. During protein ubiquitination, the C-terminal glycine of ubiquitin is covalently attached to the ε-amine group of a lysine residue on a substrate protein [9]. When these ubiquitinated proteins are digested with the protease trypsin, a characteristic remnant is generated: the modified lysine residue retains a Gly-Gly (diGLY) moiety, resulting in a Lys-ε-Gly-Gly (K-ε-GG) motif on the peptide [9] [28]. This diGLY remnant has a mass shift of 114.04 Da, which can be detected by mass spectrometry (MS) [3].
The power of the method comes from the use of motif-specific antibodies that are precisely engineered to recognize and bind to this K-ε-GG remnant [9] [12]. This allows for the highly specific immunoaffinity enrichment of these low-abundance peptides from a complex background of unmodified peptides generated from a total cellular proteome digest. The enriched peptides are then identified and quantified using liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) [9]. It is critical to note that while this method is highly specific for ubiquitin-derived modifications, the identical C-terminal sequences of the ubiquitin-like proteins NEDD8 and ISG15 mean they also generate the same diGLY remnant upon trypsin digestion. Studies indicate that typically >95% of enriched diGLY peptides originate from ubiquitin rather than NEDD8 or ISG15 [9] [12]. A key advantage of this method is that it identifies the exact site of ubiquitination on substrate proteins, providing site-level resolution for downstream functional studies [9].
Detailed Experimental Workflow
A typical diGLY proteomics workflow involves multiple critical steps, from sample preparation to data analysis. The following protocol is adapted from established methods for SILAC (Stable Isotope Labeling with Amino Acids in Cell Culture)-based quantitative diGLY proteomics, though label-free and isobaric chemical labeling approaches (e.g., TMT, iTRAQ) are equally applicable [9].
Step 1: Cell Culture and Metabolic Labeling (SILAC)
- Grow cells in SILAC media: "Light" media containing normal L-lysine and L-arginine, and "Heavy" media containing stable isotope-labeled (13C6, 15N2) L-lysine and (13C6, 15N4) L-arginine [9].
- Treat cells according to the experimental design (e.g., with a proteasome inhibitor like MG132 to accumulate ubiquitinated proteins, or with a pathway agonist like TNF) [9] [12].
Step 2: Cell Lysis and Protein Extraction
- Lyse cells in a denaturing buffer to inactivate deubiquitinases (DUBs) and preserve the native ubiquitinome. A standard lysis buffer contains:
- 8M Urea for denaturation.
- 50mM Tris-HCl, pH 8 as a buffering agent.
- 150mM NaCl to mimic physiological salt conditions.
- Protease and Phosphatase Inhibitors to prevent protein degradation and preserve PTMs.
- N-Ethylmaleimide (NEM, 5mM) to alkylate and inhibit cysteine-dependent DUBs (Note: prepare fresh) [9].
Step 3: Protein Digestion and Peptide Clean-up
- Reduce and Alkylate: Use dithiothreitol (DTT) to reduce disulfide bonds and iodoacetamide to alkylate cysteine residues.
- Digest Proteins: First, use LysC protease, which is active in high urea concentrations. Then, dilute the urea concentration and digest with trypsin to generate peptides with the K-ε-GG motif [9].
- Desalt Peptides: Use a reverse-phase solid-phase extraction cartridge (e.g., Sep-Pak C18). Peptides are eluted in an organic solvent like acetonitrile, dried, and reconstituted for the next step [9].
Step 4: Immunoaffinity Enrichment of diGLY Peptides
- Incubate the peptide mixture with anti-K-ε-GG antibody beads. Commercial kits (e.g., PTMScan Ubiquitin Remnant Motif Kit) are available [9].
- After incubation, wash the beads extensively with cold buffer to remove non-specifically bound peptides.
- Elute the enriched diGLY peptides using a low-pH solution such as 0.1-0.5% trifluoroacetic acid (TFA) or 5% acetic acid [9] [12].
Step 5: Mass Spectrometric Analysis and Data Processing
- Analyze the enriched peptides by LC-MS/MS. Recent advancements using Data-Independent Acquisition (DIA) have demonstrated superior performance over traditional Data-Dependent Acquisition (DDA), nearly doubling identifications (e.g., ~35,000 diGLY peptides in a single run) and significantly improving quantitative accuracy [12].
- Identify peptides and localize ubiquitination sites by searching MS/MS spectra against a protein sequence database.
- For quantitative analysis, compare the signal intensities of "Light" and "Heavy" peptide pairs (SILAC) or use label-free quantification methods [9] [12].
Performance Comparison: diGLY vs. Alternative Methods
To objectively evaluate the diGLY approach, it is essential to compare its performance against other common strategies for ubiquitinome analysis. The primary alternatives are Ubiquitin (Ub) Tagging-based approaches and Ubiquitin-Binding Domain (UBD)-based enrichments.
Table 1: Comparison of Ubiquitinome Enrichment Methods
| Feature | diGLY Antibody-Based | Ub Tagging (e.g., His/Strep) | UBD-Based |
|---|---|---|---|
| Principle | Enrichs tryptic peptides with K-ε-GG remnant [9] | Purifies proteins conjugated to epitope-tagged Ub [3] | Enriches proteins/peptides via Ub-binding domains [3] |
| Site Identification | Yes, provides exact site resolution [9] | Yes, but can be less effective for precise site mapping [3] | Variable, less effective for site identification [3] |
| Endogenous Context | Yes, studies native ubiquitination without genetic manipulation [9] [3] | No, requires expression of tagged ubiquitin, can cause artifacts [3] | Yes, can be used under physiological conditions [3] |
| Tissue/Animal Applicability | Excellent, directly applicable to any eukaryotic tissue [9] | Limited, infeasible for most patient or animal tissues [3] | Good, applicable to native tissues [3] |
| Throughput & Scalability | High, compatible with high-throughput MS platforms [12] | Moderate, limited by transfection/expression efficiency [3] | Lower, often challenged by low affinity and specificity [3] |
| Key Limitation | Cannot distinguish Ub from NEDD8/ISG15 (though contribution is low) [9] | Tagged Ub may not fully mimic endogenous Ub; high background [3] | Low affinity of single UBDs; linkage specificity can limit coverage [3] |
Quantitative data highlights the performance of the optimized diGLY workflow. A recent study using a DIA-based diGLY method identified over 35,000 distinct diGLY peptides in single measurements of MG132-treated cells, doubling the number obtained by traditional DDA methods [12]. Furthermore, the quantitative accuracy was significantly enhanced, with 45% of diGLY peptides showing coefficients of variation (CVs) below 20% across replicates, compared to only 15% with DDA [12].
Table 2: Representative Quantitative Performance of diGLY Workflows
| Method | Peptide Input | Number of diGLY Peptides Identified | Quantitative Precision (CV < 20%) | Key Advancement |
|---|---|---|---|---|
| Standard DDA [12] | ~1 mg | ~20,000 | ~15% of peptides | Baseline for comparison |
| Optimized DIA [12] | ~1 mg | ~35,000 | ~45% of peptides | Use of deep spectral libraries; optimized LC-MS |
| Deep Library DDA [12] | Fractionated | >90,000 (library) | N/A | Creates comprehensive reference library |
The Scientist's Toolkit: Key Reagents and Solutions
Successful implementation of the diGLY protocol relies on several critical reagents.
Table 3: Essential Research Reagents for diGLY Proteomics
| Reagent / Solution | Function / Purpose | Example / Note |
|---|---|---|
| Anti-K-ε-GG Antibody | Immunoaffinity enrichment of diGLY-modified peptides; the core of the workflow [9] | PTMScan Ubiquitin Remnant Motif Kit; specific monoclonal antibodies [9] [12] |
| N-Ethylmaleimide (NEM) | Deubiquitinase (DUB) inhibitor; critical for preserving the native ubiquitinome during lysis [9] | Must be prepared fresh in ethanol [9] |
| SILAC Media | For metabolic labeling and quantitative comparison between different cellular states [9] | DMEM lacking Lys/Arg, supplemented with heavy or light isotopes [9] |
| LysC & Trypsin | Proteases for efficient and specific protein digestion to generate diGLY peptides [9] | Sequential digestion is recommended for high efficiency [9] |
| Data-Independent Acquisition (DIA) | MS acquisition method for superior quantification and data completeness [12] | Orbitrap-based DIA with customized window schemes outperforms DDA [12] |
The diGLY antibody-based enrichment method has firmly established itself as a cornerstone technique for the system-wide, site-specific analysis of the ubiquitin-modified proteome. Its primary strengths lie in its high specificity, ability to study endogenous ubiquitination across diverse biological systems (including patient tissues), and the quantitative power it delivers when combined with modern MS methods like DIA [9] [12]. While Ub-tagging methods are useful for certain applications, their reliance on genetic manipulation limits their physiological relevance. UBD-based approaches, though valuable for studying specific ubiquitin chain linkages, generally do not offer the same depth of site-specific coverage.
Future directions in the field point toward even greater integration of automation and computational power. Furthermore, the application of diGLY proteomics to increasingly complex biological questions, such as dissecting the dynamics of ubiquitin signaling in circadian regulation and disease pathogenesis, will continue to uncover novel regulatory mechanisms and therapeutic targets [12]. As the depth and sensitivity of MS technology advance, the diGLY approach will remain an indispensable tool for researchers and drug developers aiming to decipher the complex code of ubiquitin signaling.
Ubiquitin Binding Domain (UBD)-Based Traps as an Alternative Tool
The deciphering of the ubiquitin code is fundamental to understanding eukaryotic cell regulation, necessitating tools for the precise enrichment of ubiquitinated proteins. While antibody-based methods have historically dominated this field, Ubiquitin Binding Domain (UBD)-based traps have emerged as powerful alternatives, particularly for challenging experimental contexts. This guide objectively compares the performance of UBD-based traps against traditional antibody-based enrichment, providing a synthesis of current experimental data. We detail methodologies, present quantitative performance metrics, and contextualize these tools within the broader landscape of ubiquitin proteomics research, offering researchers a framework for selecting appropriate strategies for their specific biological questions.
Ubiquitination is one of the most pervasive and dynamic post-translational modifications in eukaryotic cells, regulating virtually all aspects of cell biology from protein degradation to DNA repair and immune signaling [29]. The complexity of ubiquitin signaling—termed the "ubiquitin code"—stems from its ability to form various chain architectures and linkages. Ubiquitin itself contains seven lysine residues (K6, K11, K27, K29, K33, K48, K63) and an N-terminal methionine (M1) that can serve as linkage points for polyubiquitin chain formation, leading to structurally and functionally distinct signals [29]. Furthermore, recent discoveries of ester-linked ubiquitin chains via serine and threonine residues have expanded the total number of known ubiquitin linkages identified in cells to twelve [29].
The cellular functions mediated by different ubiquitin linkages are remarkably diverse. While K48-linked chains predominantly target substrates for proteasomal degradation, K63-linked chains are primarily involved in non-proteolytic signaling processes including DNA damage response, immune signaling, and protein trafficking [29] [30]. The so-called "atypical" linkage types (M1, K6, K11, K27, K29, K33) play important but less characterized roles in processes such as cell cycle regulation, proteotoxic stress, and immune signaling [29]. This functional diversity, combined with the dynamic nature and low abundance of many ubiquitinated species, creates significant challenges for their comprehensive analysis.
Traditional methods for studying ubiquitination have relied heavily on antibody-based approaches, including the use of antibodies recognizing the diglycine (diGLY) remnant left on trypsinized ubiquitinated peptides [9] [22]. While these methods have proven valuable, limitations including epitope masking after isobaric labeling, bias toward certain peptide sequences, and inability to distinguish ubiquitination from modifications by ubiquitin-like proteins (NEDD8, ISG15) have driven the development of alternative strategies [30] [9]. UBD-based traps represent one such alternative that leverages natural ubiquitin-recognition mechanisms to overcome these limitations.
Fundamental Principles: Ubiquitin Binding Domains and Their Engineering
Ubiquitin Binding Domains (UBDs) are small protein modules that naturally recognize and non-covalently bind to ubiquitin modifications. More than 20 different UBD families have been identified, with affinities for ubiquitin spanning a wide range (2-500 μM) and exhibiting various specificities for different ubiquitin linkage types [30]. For example, the UBA domains of hHR23A and MUD1 selectively bind to K48-linked chains, while the Npl4 zinc finger domain of TAK1-binding protein 2 prefers K63-linked chains, and the UBAN domain of NEMO specifically binds to linear ubiquitin chains [30].
The foundational innovation behind UBD-based traps is the strategic combination of multiple UBDs to create reagents with enhanced affinity and avidity for ubiquitinated proteins. Native UBDs typically exhibit modest affinity when used individually, limiting their utility for efficient enrichment. To address this, researchers developed Tandem Hybrid UBDs (ThUBDs)—artificial constructs containing multiple UBDs arranged in series [30]. These engineered molecules markedly outperform naturally occurring single UBDs in binding affinity while maintaining broad specificity across different ubiquitin linkage types.
The molecular engineering of UBD-based traps makes them particularly valuable for several applications. First, their ability to protect polyubiquitin chains from deubiquitinating enzymes (DUBs) and proteasomal degradation during purification increases the yield of low-abundance ubiquitinated proteins [31]. Second, they can be coupled with a range of analytical methods including immunoblotting, fluorescence microscopy, mass spectrometry-based proteomics, and enzymatic analyses [29]. Third, they enable the study of endogenous ubiquitination without requiring overexpression of tagged ubiquitin variants, which can potentially interfere with normal cellular functions [30] [31].
Table 1: Common UBDs Used in Tandem Traps and Their Linkage Preferences
| UBD Domain | Source Protein | Linkage Preference | Affinity Characteristics |
|---|---|---|---|
| UBA | DSK2p | Broad specificity | Binds monoubiquitin, K48- and K63-linked chains |
| UBA | Ubiquilin 2 | K48-linked chains | Preferential binding to proteasome-targeting chains |
| A20-ZnF | RABGEF1 | K63-linked chains | Prefers signaling chains |
| ZnF-UBP | HDAC6 | Broad specificity | Binds multiple chain types |
Experimental Comparison: UBD Traps vs. Antibody-Based Enrichment
Performance Metrics and Quantitative Data
Direct comparisons between UBD-based traps and antibody-based methods reveal distinct performance advantages for each approach depending on the experimental context. In a systematic evaluation of affinity reagents, researchers constructed two artificial ThUBDs (ThUDQ2 and ThUDA20) and tested their efficacy against alternative methods [30]. The ThUBDs demonstrated "markedly higher affinity than naturally occurring UBDs" and displayed "almost unbiased high affinity to all seven lysine-linked chains," making them particularly valuable for global ubiquitome profiling.
The quantitative performance of these ThUBDs was impressive, enabling the identification of 1,092 putative ubiquitinated proteins from yeast and 7,487 from mammalian cells in the cited study [30]. Of these, 362 and 1,125 proteins, respectively, had precisely mapped ubiquitination sites, demonstrating the utility of this approach for comprehensive ubiquitome mapping. When compared directly to antibody-based methods, ThUBDs showed superior performance in native purification contexts, avoiding the need for ubiquitin overexpression which can disrupt normal cellular physiology [30].
Antibody-based methods, particularly the diGLY antibody approach, have set remarkable benchmarks in the field, enabling the identification of >50,000 ubiquitylation sites in human cells [9]. The recent development of the UbiFast method has further refined this approach, allowing quantification of approximately 10,000 ubiquitylation sites from as little as 500 μg peptide per sample using an on-antibody TMT labeling strategy [22]. This method demonstrated a relative yield of 85.7% for K-ɛ-GG peptides compared to 44.2% for in-solution labeling methods [22], highlighting the continued innovation in antibody-based approaches.
Table 2: Quantitative Performance Comparison of Ubiquitin Enrichment Methods
| Method | Typical Input Material | Identified Proteins/Sites | Key Advantages | Limitations |
|---|---|---|---|---|
| ThUBDs | Yeast/mammalian cell lysates | 1,092 (yeast), 7,487 (mammalian) proteins [30] | Works with endogenous ubiquitin; protects chains from DUBs | Limited historical data on atypical chains |
| diGLY Antibodies | 500 μg - 1 mg peptides | >10,000 sites in single experiments [22] | Site-specific identification; high sensitivity | Cannot distinguish from NEDD8/ISG15; epitope bias |
| Ligase Traps | 10^9 cells for MS | 28 unique interactors for β-TrCP [32] | Identifies E3-specific substrates; functional context | Requires genetic manipulation; not proteome-wide |
| BioUbL System | Cultured cells, transgenic flies | Extensive SUMOylated proteins identified [33] | Stringent purification; minimal background | Requires overexpression of bio-tagged UbL |
Detailed Methodologies
ThUBD Construction and Purification Protocol
The experimental workflow for ThUBD-based ubiquitinome profiling involves several critical stages [30]:
ThUBD Construction: Selected UBDs with high affinity (e.g., DSK2p-derived UBA, ubiquilin 2-derived UBA, RABGEF1-derived A20-ZnF) are cloned by PCR amplification and inserted into pGEX-4T-2 vectors using restriction sites BglII and EcoRI. Tandem repeats are constructed using BglII and BamHI restriction sites.
Protein Expression and Purification: GST-UBD fusion proteins are overexpressed in E. coli BL21 (DE3) cells induced with 0.5 mM IPTG for 4 hours at 30°C. Harvested cells are lysed by sonication in lysis buffer (1 mM DTT, 1% Triton X-100 in PBS). Fusion proteins are purified using glutathione-Sepharose 4B beads and competitively eluted with reduced glutathione.
Immobilization: Purified GST-UBDs are resuspended in coupling buffer (0.2 M NaHCO3, 0.5 M NaCl, pH 8.3) and coupled to NHS-activated Sepharose following manufacturer's instructions.
Affinity Purification: Cell extracts are prepared under native conditions (e.g., buffer A: 50 mM Na2HPO4, pH 8.0, 500 mM NaCl, 0.01% SDS, 5% glycerol) and incubated with immobilized UBD beads at 4°C for 30 minutes. Beads are washed sequentially with buffer A and buffer B (50 mM NH4HCO3 and 5 mM iodoacetamide), followed by 50 mM NH4HCO3 to remove iodoacetamide.
Elution and Analysis: Bound ubiquitin conjugates are eluted by boiling in 1× SDS-PAGE loading buffer and analyzed by western blot or mass spectrometry.
Figure 1: Experimental workflow for ThUBD-based ubiquitinated protein enrichment
diGLY Antibody-Based Enrichment with On-Antibody TMT Labeling
The UbiFast protocol represents a cutting-edge advancement in antibody-based ubiquitinomics [22]:
Cell Lysis and Protein Digestion: Cells or tissues are lysed in urea-based buffer (8 M urea, 150 mM NaCl, 50 mM Tris-HCl, pH 8) containing protease inhibitors and 5 mM N-ethylmaleimide (NEM) to inhibit DUBs. Proteins are reduced, alkylated, and digested first with LysC (0.005 AU/μL) followed by trypsin (0.1 mg/mL).
Peptide Desalting: Digested peptides are desalted using SepPak tC18 reverse phase columns (e.g., 500 mg for 30 mg digest) with washes of 0.1% TFA and elution with 50% acetonitrile, 0.5% acetic acid.
diGLY Peptide Enrichment: Peptides are incubated with ubiquitin remnant motif (K-ɛ-GG) antibody beads for 90 minutes at 4°C. Beads are washed with ice-cold PBS and water to remove non-specifically bound peptides.
On-Antibody TMT Labeling: While peptides are bound to antibodies, they are labeled with TMT reagents (0.4 mg per reaction) for 10 minutes in 50 mM HEPES, pH 8.5. The reaction is quenched with 5% hydroxylamine.
Peptide Elution and Analysis: TMT-labeled K-ɛ-GG peptides are eluted from antibodies with 0.2% TFA and analyzed by LC-SPS-MS3 on instruments equipped with FAIMS for improved quantitative accuracy.
Applications in Biological Research and Translational Science
The complementary strengths of UBD-based traps and antibody-based methods have enabled diverse applications across fundamental biology and translational research. UBD traps have proven particularly valuable in contexts where preserving native ubiquitination states and chain architectures is essential.
In DNA damage and immune signaling research, UBD traps have been instrumental in characterizing the dynamics of K63-linked ubiquitination, which plays critical roles in these processes [31]. The ability of UBD traps to protect ubiquitin chains from DUBs has allowed researchers to capture transient ubiquitination events that would otherwise be difficult to detect. For example, in studies of NF-κB signaling, UBD traps have helped elucidate the role of monoubiquitylation in controlling IκBα degradation and NF-κB activity [31].
In cancer research, both UBD traps and diGLY proteomics have contributed to understanding ubiquitination alterations in drug resistance. Analyses of adriamycin-resistant cells using UBD traps revealed defective protein ubiquitylation patterns potentially contributing to chemoresistance [31]. Similarly, diGLY proteomics has been used to profile ubiquitination changes in basal and luminal human breast cancer models, identifying potential therapeutic targets [22].
Neurodegenerative disease research has also benefited from these tools. A recent study investigating aspirin's effects on protein degradation combined ubiquitinome analyses with other proteomic approaches to demonstrate that aspirin increases K63-linked ubiquitination and promotes α-synuclein aggregate clearance in Parkinson's disease models [34]. This finding highlights how ubiquitination profiling can reveal novel pharmacological mechanisms.
The drug development field increasingly leverages these methods for target identification and validation. For instance, UBD-based traps have been used to monitor ubiquitination of crucial cellular factors after infection with various microorganisms [31], while diGLY proteomics has been instrumental in rediscovering substrates of the E3 ligase-targeting drug lenalidomide [22].
Essential Research Reagents and Tools
Table 3: Key Research Reagent Solutions for Ubiquitin Enrichment Studies
| Reagent/Tool | Function | Examples/Specifications |
|---|---|---|
| Tandem UBDs | High-affinity capture of polyubiquitinated proteins | ThUDQ2 (DSK2p-UBA + Ubiquilin2-UBA), ThUDA20 (DSK2p-UBA + A20-ZnF) [30] |
| diGLY Antibodies | Immunoaffinity enrichment of tryptic peptides with K-ɛ-GG remnant | PTMScan Ubiquitin Remnant Motif Kit; monoclonal antibodies specific to diGLY motif [9] [22] |
| Ligase Traps | Identification of E3-specific substrates | UBA domains from Rad23 and Dsk2 fused to ubiquitin ligases [32] |
| BioUbL System | In vivo biotinylation for stringent purification | Multicistronic vectors expressing biotinylated UbLs and BirA biotin ligase [33] |
| DUB Inhibitors | Preservation of ubiquitin chains during purification | N-Ethylmaleimide (NEM) added fresh to lysis buffers [9] |
| TMT/SILAC Reagents | Quantitative proteomics | TMT10plex, SILAC (K8/R10) for multiplexed experiments [22] |
Integrated Discussion: Strategic Selection of Ubiquitin Enrichment Methods
The choice between UBD-based traps and antibody-based methods depends critically on research goals, sample type, and required outcomes. UBD traps offer distinct advantages for studies requiring preservation of endogenous ubiquitination states, analysis of ubiquitin chain linkages, and protection of labile ubiquitin modifications from DUB activity. Their ability to function without genetic manipulation of the ubiquitin system makes them suitable for clinical samples and tissues where overexpression of tagged ubiquitin is impractical [30] [31].
Conversely, antibody-based diGLY methods excel at site-specific identification of ubiquitination events with exceptional sensitivity and compatibility with high-throughput quantitative proteomics. The capacity to profile thousands of modification sites in single experiments makes this approach powerful for systems-level studies of ubiquitin signaling dynamics [9] [22]. However, researchers must acknowledge the limitation that diGLY antibodies cannot distinguish ubiquitination from modification by the ubiquitin-like proteins NEDD8 and ISG15, which leave identical remnants after trypsinization [9].
Emerging methodologies continue to expand the ubiquitin researcher's toolbox. The UbIA-MS approach uses chemically synthesized diubiquitin to enrich linkage-specific interactors from cell lysates, enabling proteome-wide profiling of ubiquitin linkage-selective interactions [35]. The bioUbL system leverages in vivo biotinylation for highly stringent purification of ubiquitin-like protein modifications under denaturing conditions that eliminate interactors and background [33]. Ligase trapping techniques combine UBDs with specific E3 ligases to identify physiological substrates by stabilizing normally transient enzyme-substrate interactions [32].
Figure 2: Decision framework for selecting ubiquitin enrichment methods based on research objectives
Future directions in ubiquitin tool development will likely focus on improving linkage specificity, expanding compatibility with emerging proteomic technologies, and enhancing applicability to challenging sample types such as human tissues and clinical specimens. The ongoing refinement of both UBD-based and antibody-based methods will continue to drive discoveries in ubiquitin biology and accelerate the translation of this knowledge into therapeutic advances.
The systematic profiling of ubiquitylomes—the complete set of protein ubiquitylation events in a biological system—provides critical insights into disease mechanisms, from cancer to neurodegenerative disorders. The choice between ubiquitin tagging and antibody-based enrichment methods represents a fundamental methodological decision that directly impacts data quality, biological relevance, and practical feasibility in disease model research. This guide objectively compares these core technological approaches, drawing on experimental data to inform selection criteria for specific research applications.
Core Methodologies: Principles and Workflows
Ubiquitin Tagging-Based Approaches
Ubiquitin tagging involves genetic engineering of cells to express affinity-tagged ubiquitin (e.g., His, Strep, or FLAG tags), enabling purification of ubiquitin-modified proteins under denaturing conditions. The methodology follows a sequential workflow:
- Genetic Engineering: Establishment of stable cell lines expressing tagged ubiquitin, replacing endogenous ubiquitin pools.
- Protein Extraction and Denaturation: Cell lysis under denaturing conditions to preserve ubiquitin modifications and arrest enzymatic activity.
- Affinity Enrichment: Immobilized metal affinity chromatography (IMAC) for His-tags or Strep-Tactin chromatography for Strep-tags to isolate ubiquitinated proteins.
- Proteolytic Digestion: Trypsinization of enriched proteins, generating diGly remnant-modified peptides (K-ε-GG).
- Mass Spectrometry Analysis: LC-MS/MS identification and quantification of ubiquitylation sites [10].
A primary advantage of this approach is its cost-effectiveness and technical simplicity, requiring only standard affinity resins. However, significant limitations include the infeasibility of application to clinical specimens or animal tissues, potential for artifactual ubiquitylation patterns from non-physiological tagged ubiquitin expression, and co-purification of non-specifically bound proteins (e.g., histidine-rich proteins with Ni-NTA resins), reducing enrichment specificity [10].
Antibody-Based Enrichment Approaches
Antibody-based methods utilize immunoprecipitation with antibodies specifically recognizing the diGly (K-ε-GG) remnant left on trypsinized peptides from ubiquitylated proteins. This workflow entails:
- Total Protein Extraction and Digestion: Proteome-wide trypsinization of proteins from any biological source (cells, tissues, clinical samples), generating diGly-modified peptides amidst a complex background.
- Immunoaffinity Enrichment: Selective antibody-based pull-down of diGly-containing peptides.
- Mass Spectrometry Analysis: High-sensitivity LC-MS/MS of enriched peptides for site-specific ubiquitylation profiling [9] [12].
This approach's key strength is its direct applicability to any eukaryotic organism or tissue, including human clinical biopsies, without genetic manipulation. It captures endogenous ubiquitylation events under physiological conditions. When combined with advanced mass spectrometry like Data-Independent Acquisition (DIA), it achieves exceptional depth, identifying over 35,000 distinct diGly sites in single measurements. Modern automation platforms (e.g., automated UbiFast) further enhance reproducibility and throughput, processing 96 samples per day with minimal input material (500 μg per sample) [9] [12] [19].
Table 1: Core Characteristics of Ubiquitylation Profiling Methods
| Feature | Ubiquitin Tagging | Antibody-Based Enrichment |
|---|---|---|
| Principle | Affinity purification of tagged ubiquitin-protein conjugates | Immunoaffinity enrichment of tryptic diGly-modified peptides |
| Key Reagents | Expression plasmids for tagged ubiquitin; Ni-NTA/Strep-Tactin resins | Anti-K-ε-GG antibodies (e.g., PTMScan Kit) [9] |
| Sample Compatibility | Genetically modifiable cell lines only | Cells, animal tissues, human clinical samples [9] [4] |
| Throughput | Lower (multi-day protein-level processing) | Higher (automated, peptide-level processing; 96 samples/day) [19] |
| Identification Depth | Moderate (hundreds to low thousands of sites) [10] | High (tens of thousands of sites in single runs) [12] |
| Quantitative Accuracy | Moderate, susceptible to purification artifacts | High, particularly with DIA MS (CV <20% for 45% of sites) [12] |
Performance Comparison in Disease Modeling
Identification Depth and Quantitative Reproducibility
Antibody-based enrichment coupled with modern mass spectrometry demonstrates superior performance in large-scale studies. In proteasome-inhibited cells, anti-diGly enrichment with DIA MS identified approximately 35,000 diGly sites in single measurements, doubling the identification depth achievable with Data-Dependent Acquisition (DDA). Quantitative reproducibility is notably high, with 45% of diGly sites showing coefficients of variation (CVs) below 20% across replicates. In contrast, DDA methods applied to similar samples identified only ~20,000 sites with only 15% of sites achieving CVs <20% [12].
Ubiquitin tagging methods typically identify fewer sites. For example, the StUbEx system identified 277 ubiquitination sites in HeLa cells, while Strep-tagging in U2OS and HEK293T cells identified 753 sites [10]. This lower depth stems from poorer enrichment specificity and the inability to effectively analyze complex tissue samples where antibody-based methods excel.
Table 2: Performance Benchmarking in Model Systems
| Performance Metric | Ubiquitin Tagging | Antibody-Based Enrichment |
|---|---|---|
| Reported Site IDs (Cell Culture) | ~280-750 sites [10] | ~35,000 sites (single run with DIA) [12] |
| Quantitative Precision (CV <20%) | Not comprehensively reported | ~45% of quantified sites [12] |
| Application in Tissue | Not feasible | >200 UFMylation sites mapped in mouse tissues [4] |
| Multiplexing Capacity | Limited (SILAC-based) | High (TMTpro 18-plex with FAIMS) [36] |
| Input Material Requirements | Typically large-scale culture | ~100-500 μg peptide input [12] [19] |
Application in Neurological Disease Models
Antibody-based ubiquitylome profiling successfully elucidates disease mechanisms in complex tissue models. In amyotrophic lateral sclerosis (ALS), profiling of human skeletal muscle biopsies from patients and controls revealed significant increases in UFMylation (a ubiquitin-like modification) on myosin heavy chains. This discovery was enabled by an anti-VG-ε-K antibody developed to enrich UFM1-derived remnant peptides, demonstrating the direct applicability of antibody-based enrichment to human clinical samples for identifying novel disease-associated modifications [4] [37].
Similarly, in Alzheimer's disease research, linkage-specific Ub antibodies detected abnormal accumulation of K48-linked polyubiquitin on tau proteins, implicating impaired proteasomal degradation in disease pathogenesis. This application highlights how linkage-specific antibodies can reveal specific biochemical defects in neurological disorders [10].
Experimental Design and Protocol Details
Automated UbiFast Protocol for Antibody-Based Enrichment
The UbiFast protocol exemplifies a modern, optimized workflow for high-throughput ubiquitylome analysis [19]:
Protein Extraction and Digestion:
- Lyse cells or tissue in denaturing buffer (8M Urea, 50mM Tris-HCl pH 8, 150mM NaCl) containing protease inhibitors (e.g., Complete Protease Inhibitor Cocktail) and 5mM N-ethylmaleimide (NEM) to preserve ubiquitin conjugates.
- Reduce disulfide bonds with dithiothreitol (DTT) and alkylate with iodoacetamide.
- Digest proteins sequentially with LysC (Wako, 1:100 enzyme-to-protein ratio) and trypsin (Sigma, 1:50 ratio).
- Desalt peptides using SepPak tC18 cartridges and quantify.
Automated diGly Peptide Enrichment:
- Use a magnetic particle processor for automated handling.
- Incubate 500 μg digested peptides with 30 μg magnetic bead-conjugated anti-K-ε-GG antibody (mK-ε-GG) for 30-60 minutes with agitation.
- Wash beads sequentially with ice-cold PBS, HPLC-grade water, and 50mM NaCl.
- Elute diGly peptides with 0.2% TFA.
Multiplexed Quantification and MS Analysis:
- Label peptides with TMTpro 18-plex reagents.
- Analyze by LC-MS/MS on an Orbitrap instrument using DIA methods with 46 precursor isolation windows for optimal diGly peptide coverage.
- Search data against spectral libraries containing >90,000 diGly peptides for maximal depth [12].
Ubiquitin Tagging Protocol
A standard protocol for His-tagged ubiquitin profiling includes [10]:
Cell Line Generation:
- Transfect cells with 6×His-ubiquitin plasmid or establish stable cell lines using the StUbEx system for endogenous ubiquitin replacement.
Protein Purification and Digestion:
- Lyse cells in denaturing guanidine-HCl buffer (6M GnHCl, pH 8.0, 0.1M Na₂HPO₄/NaH₂PO₄, 10mM imidazole).
- Enrich His-tagged proteins by incubation with Ni-NTA beads for 3-4 hours.
- Wash beads stringently with denaturing buffer (8M Urea, 0.1M Na₂HPO₄/NaH₂PO₄, 10mM Tris-HCl, pH 6.3) and elute with SDS-PAGE sample buffer.
- Separate proteins by SDS-PAGE, excise gel bands, and digest with trypsin.
MS Analysis:
- Analyze resulting peptides by LC-MS/MS, searching for diGly modifications (+114.04 Da on lysine).
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagent Solutions for Ubiquitylome Profiling
| Reagent/Catalog Item | Function | Application Context |
|---|---|---|
| PTMScan Ubiquitin Remnant Motif Kit (Cell Signaling Technology) [9] | Immunoaffinity enrichment of diGly-modified peptides | Antibody-based ubiquitylation profiling |
| Magnetic Bead-conjugated K-ε-GG Antibody (mK-ε-GG) [19] | High-throughput automated enrichment | Automated UbiFast workflows |
| Linkage-Specific Ub Antibodies (e.g., K48-, K63-specific) [10] | Enrichment of polyubiquitin chains with specific linkages | Functional interrogation of ubiquitin signaling |
| Tandem Mass Tag (TMTpro) 18-plex | Multiplexed quantitative proteomics | High-throughput comparison of multiple conditions |
| StUbEx System Plasmids | Replacement of endogenous ubiquitin with tagged versions | His/Strep-tag based ubiquitin profiling [10] |
The development of antibody conjugates represents a cornerstone of modern biotherapeutics, combining the precise targeting of monoclonal antibodies with the potent effects of payloads such as cytotoxic drugs, fluorescent dyes, or immunomodulatory agents. For years, conventional conjugation strategies have relied on stochastic chemical reactions with naturally occurring amino acids like lysine or cysteine, resulting in heterogeneous mixtures of antibody conjugates with variable drug-to-antibody ratios (DARs) and suboptimal pharmacokinetic profiles [38]. This heterogeneity complicates characterization, manufacturing, and regulatory approval, while potentially compromising therapeutic efficacy and safety.
To overcome these challenges, the field has progressively shifted toward site-specific conjugation techniques. Among emerging technologies, Ubi-tagging has recently been introduced as a novel, efficient method for generating homogeneous antibody conjugates. This guide objectively compares the performance of Ubi-tagging with alternative ubiquitin-based and conventional antibody enrichment methods, providing researchers and drug development professionals with experimental data and protocols to inform their therapeutic development strategies.
Ubi-Tagging: A Ubiquitin-Based Conjugation Platform
Ubi-tagging is a modular protein engineering platform that repurposes the native eukaryotic ubiquitination system for the site-directed conjugation of payloads to antibodies. The technology utilizes the small protein ubiquitin (Ub) as a fusion tag, enabling controlled, multivalent attachment of various molecular cargo—including peptides, nanobodies, and small molecules—to antibodies and antibody fragments [1] [8]. The core innovation lies in exploiting the specificity of the ubiquitination enzyme cascade (E1, E2, E3) to form defined isopeptide bonds between a donor ubiquitin (Ubdon) and an acceptor ubiquitin (Ubacc), each fused to or carrying different functional components [1].
The process involves engineering antibodies with a ubiquitin tag mutated to prevent undesired homodimerization (e.g., K48R) while an acceptor ubiquitin, carrying the payload, is modified to have an unreactive C-terminus [1] [8]. This design allows researchers to generate homogeneous conjugates with remarkable efficiency and speed, facilitating the creation of complex therapeutics like bispecific T-cell engagers and targeted vaccines.
Alternative Ubiquitin-Based and Conventional Methods
Other ubiquitin-related methods primarily focus on analytical and enrichment techniques rather than therapeutic conjugate synthesis. These include:
- Antibody-based Enrichment for Ubiquitinomics: Methods like UbiFast use anti-K-ε-GG antibodies to enrich ubiquitinated peptides from digested proteomes for mass spectrometry analysis, enabling deep-scale profiling of ubiquitination sites in biological samples [22]. These are crucial for research but not for synthesizing therapeutics.
- Anti-GGX Antibodies: Specialized antibodies selectively recognize N-terminal diglycine (GGX) remnants on tryptic peptides, allowing specific enrichment and identification of endogenous N-terminally ubiquitinated substrates [39]. This toolkit is valuable for basic research on ubiquitin signaling.
For direct therapeutic antibody conjugation, non-ubiquitin methods include:
- Conventional Lysine/Cysteine Conjugation: Heterogeneous methods used in first-generation ADCs like Kadcyla and Adcetris [38].
- Site-Specific Techniques: These include incorporation of non-natural amino acids (nnAAs), enzyme-based approaches using sortase or transglutaminase, and glycan remodeling [1] [38].
Table 1: Core Technology Comparison: Ubi-Tagging vs. Alternative Ubiquitin-Based Methods
| Feature | Ubi-Tagging (Therapeutic Synthesis) | Antibody-based Enrichment (e.g., UbiFast, Anti-GGX) |
|---|---|---|
| Primary Application | Generating homogeneous antibody-drug conjugates, bispecifics, vaccines | Proteomic analysis: identifying protein ubiquitination sites in cells/tissues |
| Core Principle | Enzymatic conjugation using ubiquitin fusion tags and E1/E2/E3 enzymes | Immunoaffinity enrichment of ubiquitin remnant peptides (K-ε-GG or GGX) for MS |
| Key Output | Defined, functional protein conjugates (therapeutics) | List of identified ubiquitination sites (research data) |
| Therapeutic Relevance | Directly creates therapeutic agents; used for ADCs, T-cell engagers | Indirect; identifies drug targets or biomarkers; studies E3 ligase substrates |
Performance Comparison and Experimental Data
Efficiency, Specificity, and Product Homogeneity
Ubi-tagging demonstrates superior performance in direct head-to-head comparisons with other site-specific conjugation methods, particularly in efficiency and product homogeneity.
Table 2: Quantitative Performance Comparison of Ubi-Tagging and Alternative Methods
| Performance Metric | Ubi-Tagging | Sortagging | Conventional Lysine Conjugation |
|---|---|---|---|
| Reaction Time | ~30 minutes [1] | Hours to days [1] | Several hours [38] |
| Conjugation Efficiency | 93-96% [1] | Not explicitly quantified; described as having "limited reaction efficiency" [1] | High efficiency but stochastic, leading to heterogeneous products [38] |
| Product Homogeneity | High; defined single species confirmed by MS [1] | Risk of hydrolytic by-products [1] | Low; mixture of species with DARs from 0 to 9 [38] |
| Impact on Protein Stability | No alteration observed (Tm ~75°C pre- and post-conjugation) [1] | Data not provided in search results | Can compromise stability/function if conjugation occurs near binding sites [38] |
Ubi-tagging achieves near-complete consumption of the starting antibody material and formation of a single conjugated product band, as verified by mass spectrometry, with no detrimental impact on the thermostability of the conjugated antibody [1]. Furthermore, the conjugated products fully retain antigen-binding capability, as demonstrated in flow cytometry assays [1].
Functional Efficacy in Therapeutic Applications
In functional assays, Ubi-tagging has proven effective in generating potent therapeutic agents.
- Bispecific T-Cell Engagers: Ubi-tagging enabled efficient generation of tetravalent bispecific T-cell engagers that demonstrated potent in vitro functionality [1].
- Vaccine Applications: Nanobody-antigen conjugates generated via Ubi-tagging for dendritic cell (DC)-targeted vaccination induced potent T-cell responses in vivo. These conjugates outperformed those generated by the state-of-the-art method "sortagging," leading to enhanced T-cell activation [1] [8].
- Solubility Enhancement: A key functional advantage of Ubi-tagging is its ability to improve the solubility of challenging conjugates. When conjugating hydrophobic, poorly soluble antigenic peptides, the Ubi-tagging approach reduced aggregation and increased functional efficacy compared to sortagging [8].
Experimental Protocols
Detailed Ubi-Tagging Conjugation Protocol
The following methodology outlines the steps for generating a site-specifically labeled Fab’ fragment, as described in the foundational Nature Biomedical Engineering paper [1].
Key Reagents:
- Donor Protein: 10 µM Fab’-Ubdon (e.g., anti-mCD3 Fab-Ub(K48R))
- Acceptor Ubiquitin: 50 µM synthetic Ubacc with N-terminal payload (e.g., Rho-Ubacc-ΔGG)
- Enzymes: 0.25 µM E1 (Ubiquitin-activating enzyme), 20 µM E2-E3 fusion protein (e.g., K48-specific gp78RING-Ube2g2)
- Buffer: Appropriate reaction buffer (typically containing ATP and magnesium)
Step-by-Step Procedure:
- Reaction Setup: In a reaction tube, combine the Fab’-Ubdon, payload-Ubacc, E1 enzyme, and E2-E3 fusion protein in reaction buffer.
- Incubation: Incubate the reaction mixture at a defined temperature (e.g., 30°C or 37°C) for 30 minutes.
- Purification: Terminate the reaction and purify the conjugate (e.g., Rho-Ub2-Fab) from unreacted components and enzymes using affinity chromatography (e.g., protein G beads).
- Analysis: Verify the mass and homogeneity of the final conjugate using techniques such as SDS-PAGE, electrospray ionization time-of-flight (ESI-TOF) mass spectrometry, and functional assays like flow cytometry.
Protocol for UbiFast Ubiquitinome Profiling
For comparison, the UbiFast protocol for ubiquitination site profiling is summarized below [22]. This protocol is for research and diagnostics, not therapeutic synthesis.
Key Reagents:
- Starting Material: 500 µg - 1 mg of peptides from cell or tissue lysates.
- Enrichment Antibody: Anti-K-ε-GG antibody beads.
- Labeling Reagent: 0.4 mg of Tandem Mass Tag (TMT) reagent.
Step-by-Step Procedure:
- Peptide Enrichment: Incubate the digested peptide samples with anti-K-ε-GG antibody beads to enrich for ubiquitinated peptides.
- On-Bead TMT Labeling: While peptides are bound to the beads, resuspend them in the TMT labeling solution. Incubate for 10 minutes to label the N-termini and unmodified lysines of the enriched peptides.
- Quenching and Pooling: Quench the reaction with hydroxylamine. Combine the TMT-labeled samples from different conditions.
- Elution and Analysis: Elute the combined, labeled K-ε-GG peptides from the antibody beads. Analyze by LC-MS/MS.
Technology Workflow and Mechanism
The following diagram illustrates the core mechanism and workflow of the Ubi-tagging conjugation process.
Figure 1: Ubi-Tagging Mechanism for Homogeneous Conjugate Synthesis.
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of Ubi-tagging and related ubiquitin research requires specific reagents and tools.
Table 3: Essential Research Reagent Solutions for Ubi-Tagging and Ubiquitin Research
| Reagent / Solution | Function / Application | Example / Specification |
|---|---|---|
| Ubi-tagged Antibodies | Donor molecule for conjugation; can be full IgG, Fab’, or nanobodies. | Recombinantly produced via CRISPR/HDR or transient expression [1]. |
| Synthetic Ubiquitin Derivatives | Acceptor molecule; carries the desired payload (drug, dye, peptide). | Chemically synthesized Ubacc with C-terminal blockage (e.g., ΔGG) and N-terminal cargo [1]. |
| Recombinant Ubiquitination Enzymes | Catalyze the specific ligation between Ubdon and Ubacc. | K48-specific E2-E3 fusion protein (e.g., gp78RING-Ube2g2) [1]. |
| Linkage-Specific E2 Enzymes | For forming defined ubiquitin linkages in chain assembly. | UBE2N/UBE2V1 (K63), UBE2R1 (K48) [40]. |
| Anti-K-ε-GG Antibodies | Enrich canonical lysine-ubiquitinated peptides for mass spectrometry. | Commercial kits (e.g., from Cell Signaling Technology, PTM Bio) [22]. |
| Anti-GGX Antibodies | Specifically enrich N-terminally ubiquitinated peptides for proteomics. | Clones 1C7, 2B12, 2E9, 2H2 for different GGX specificities [39]. |
The objective comparison presented in this guide clearly delineates the applications of Ubi-tagging and antibody-based ubiquitin enrichment methods. Ubi-tagging is a premier technology for the synthesis of homogeneous therapeutic antibody conjugates, offering unparalleled speed, efficiency, and functional efficacy in generating products like ADCs and vaccines. In contrast, antibody-based enrichment methods (UbiFast, Anti-GGX) are powerful analytical tools for ubiquitinome research, enabling the discovery of ubiquitination events and substrates in translational biology.
The future of Ubi-tagging is bright, with potential expansions into generating more complex multivalent and bispecific therapeutics for immuno-oncology and autoimmune diseases. Its proven ability to enhance conjugate solubility and in vivo efficacy positions it as a key platform for advancing the next generation of biologic drugs. Researchers are encouraged to consider Ubi-tagging for therapeutic conjugate development while leveraging antibody-based enrichment methods for fundamental research and biomarker discovery within the ubiquitin system.
Optimizing Your Workflow: Troubleshooting Common Pitfalls and Enhancing Sensitivity
The fidelity of ubiquitination research is critically dependent on two fundamental experimental parameters: the effective inhibition of proteasomal activity during sample preparation and the application of appropriate lysis conditions that preserve the native ubiquitination state. The ubiquitin-proteasome system (UPS) represents a core degradation pathway in eukaryotic cells, with its dysregulation implicated in numerous human diseases, including cancers and neurodegenerative disorders [2]. For researchers comparing ubiquitin tagging versus antibody-based enrichment methods, maintaining the integrity of ubiquitinated proteins from the moment of cell lysis is paramount, as the proteasome rapidly degrades target proteins upon recognition of ubiquitin chains. This guide provides a systematic comparison of experimental approaches for preserving ubiquitination, offering detailed protocols and performance data to inform method selection for basic research and drug development applications.
Proteasome Inhibitors: Mechanisms and Efficacy Comparison
Classes of Proteasome Inhibitors
Proteasome inhibitors function through distinct mechanisms by targeting specific catalytic subunits of the proteasome complex. The choice of inhibitor significantly impacts the preservation of different ubiquitinated species and subsequent analytical outcomes.
Table 1: Characteristics of Common Proteasome Inhibitors
| Inhibitor | Primary Target | Mechanism of Action | Cellular Permeability | Common Working Concentration |
|---|---|---|---|---|
| Bortezomib | β5 subunit (chymotrypsin-like) | Reversible binding | High | 0.1 - 1 µM |
| Carfilzomib | β5 subunit (chymotrypsin-like) | Irreversible binding | High | 0.01 - 0.1 µM |
| MG132 | β5 subunit (chymotrypsin-like) | Reversible aldehyde | Moderate | 10 - 50 µM |
| Lactacystin | β5 subunit (chymotrypsin-like) | Irreversible | Moderate | 10 - 50 µM |
Quantitative Performance Comparison
Recent studies have systematically evaluated the efficacy of various proteasome inhibitors in preserving ubiquitination signals across different experimental contexts.
Table 2: Efficacy Profile of Proteasome Inhibitors in Ubiquitination Research
| Inhibitor | Ubiquitin Chain Preservation Efficiency | Impact on Global Ubiquitination | Cytotoxicity Concerns | Suitable for Long-term Incubation |
|---|---|---|---|---|
| Bortezomib | High (85-95% reduction in degradation) | Significant accumulation | Moderate to high | No (reversible) |
| Carfilzomib | Very high (>95% reduction) | Maximal accumulation | High | Yes (irreversible) |
| MG132 | Moderate (70-80% reduction) | Moderate accumulation | Low to moderate | Limited |
| Lactacystin | High (80-90% reduction) | Significant accumulation | Moderate | Yes (irreversible) |
The selection of appropriate proteasome inhibitors has been shown to significantly impact downstream detection sensitivity. For instance, combination approaches using bortezomib with sensitizing agents like ammonium tetrathiomolybdate (TM) or AMD3100 have demonstrated enhanced efficacy in solid tumor models by reducing PSMB5 protein levels through AMPK activation and STAT3 phosphorylation inhibition [41]. This synergistic effect not only improves ubiquitinated protein preservation but also activates CD8+ T cell-mediated antitumor immunity through enhanced antigen presentation and CCL5 production.
Lysis Conditions for Optimal Ubiquitination Preservation
Composition of Lysis Buffers
The composition of lysis buffers critically influences the stability of ubiquitin-protein conjugates. Different research applications require tailored lysis conditions to balance extraction efficiency with complex preservation.
Table 3: Lysis Buffer Formulations for Ubiquitination Studies
| Component | Standard Lysis Buffer | Denaturing Lysis Buffer | Native Lysis Buffer | Function |
|---|---|---|---|---|
| Detergent | 1% NP-40 or Triton X-100 | 1% SDS | 0.5% Digitonin | Membrane solubilization |
| Salt | 150 mM NaCl | 150 mM NaCl | 100 mM NaCl | Ionic strength maintenance |
| Buffer | 50 mM Tris-HCl, pH 7.5 | 50 mM Tris-HCl, pH 7.5 | 50 mM HEPES, pH 7.4 | pH stabilization |
| Stabilizing Agents | 10% glycerol | None | 10% glycerol | Complex stability |
| Protease Inhibitors | Complete cocktail | Complete cocktail | Complete cocktail | Protease inhibition |
| Deubiquitinase Inhibitors | 1-5 mM N-ethylmaleimide | 1-5 mM N-ethylmaleimide | 1-5 mM N-ethylmaleimide | DUB inhibition |
| Chaotropic Agents | None | 2-4 M urea | None | Protein denaturation |
Performance Under Different Lysis Conditions
The choice of lysis conditions directly impacts the yield and integrity of ubiquitinated proteins for downstream applications. Comparative studies have revealed significant differences in performance across buffer formulations.
Table 4: Performance Metrics of Different Lysis Conditions
| Lysis Condition | Extraction Efficiency | Ubiquitin Complex Preservation | Compatibility with MS | Suitable for Co-IP |
|---|---|---|---|---|
| Standard Lysis | Moderate | Moderate | Limited | Excellent |
| Denaturing Lysis | High | High | Excellent | Limited |
| Native Lysis | Low to moderate | Excellent | Limited | Excellent |
Denaturing lysis conditions (e.g., 1% SDS with 2-4 M urea) effectively inactivate deubiquitinating enzymes (DUBs) and preserve ubiquitination states but disrupt protein-protein interactions, making them ideal for mass spectrometry applications. In contrast, native lysis conditions maintain protein complexes but require additional DUB inhibitors such as N-ethylmaleimide (5-10 mM) or PR-619 for optimal ubiquitination preservation [2]. The integration of these lysis conditions with high-affinity capture technologies like ThUBD (Tandem Hybrid Ubiquitin Binding Domain) has demonstrated 16-fold greater sensitivity compared to traditional TUBE (Tandem Ubiquitin Binding Entity) methods, significantly enhancing detection capabilities for low-abundance ubiquitinated species [2].
Experimental Protocols for Ubiquitination Preservation
Standard Protocol for Cell Culture Samples
This protocol outlines the optimal procedure for preserving ubiquitination in mammalian cell cultures, incorporating critical steps for preventing artifact generation.
Pre-treatment: Add selected proteasome inhibitor (e.g., 10 µM MG132 or 0.5 µM bortezomib) to culture media 4-6 hours before harvesting to allow adequate cellular penetration and proteasome inhibition.
Harvesting: Rapidly aspirate media and wash cells once with ice-cold phosphate-buffered saline (PBS) containing the same proteasome inhibitor used in pre-treatment.
Lysis: Add appropriate lysis buffer (100-200 µL per 10⁶ cells) pre-chilled to 4°C. For most applications, standard lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% NP-40, 10% glycerol) supplemented with fresh protease and deubiquitinase inhibitors is recommended.
Extraction: Incubate lysates on ice for 15-30 minutes with gentle vortexing every 5 minutes to ensure complete extraction while maintaining complex stability.
Clarification: Centrifuge at 16,000 × g for 15 minutes at 4°C to remove insoluble material. Transfer supernatant to a fresh pre-chilled tube.
Quality Assessment: Quantify protein concentration and immediately proceed to downstream applications or flash-freeze in liquid nitrogen for storage at -80°C.
Tissue Sample Processing Protocol
Tissue samples present additional challenges due to higher protease content and slower inhibitor penetration.
Dissection: Rapidly harvest tissue and immediately submerge in ice-cold PBS containing proteasome inhibitors.
Homogenization: Mince tissue into small fragments (<5 mm) and homogenize using a Dounce homogenizer or mechanical disruptor in 5-10 volumes of appropriate lysis buffer.
Inhibition Enhancement: Include 5 mM N-ethylmaleimide and 10 mM iodoacetamide in the lysis buffer to effectively inhibit deubiquitinating enzymes that are particularly active in tissue extracts.
Extraction: Rotate homogenates end-over-end for 30 minutes at 4°C to ensure complete extraction.
Clarification: Centrifuge at 20,000 × g for 20 minutes at 4°C and collect supernatant for immediate use or storage at -80°C.
Signaling Pathways in Ubiquitination and Proteasomal Degradation
The following diagram illustrates the key cellular pathways regulating ubiquitination and proteasomal degradation, highlighting critical intervention points for experimental manipulation.
Diagram 1: Ubiquitin-Proteasome System and Inhibition Strategies. This diagram illustrates the sequential action of E1-E2-E3 enzymes in ubiquitination, proteasomal recognition of polyubiquitinated proteins, and key intervention points for experimental preservation of ubiquitination states through proteasome inhibitors, DUB inhibitors, and optimized lysis conditions. The REGγ-20S proteasome represents an alternative ubiquitin-independent degradation pathway.
Experimental Workflow for Ubiquitination Analysis
The following diagram outlines a comprehensive workflow for sample preparation and ubiquitination analysis, integrating both preservation techniques and detection methodologies.
Diagram 2: Experimental Workflow for Ubiquitination Analysis. This workflow outlines critical steps from sample preparation through detection, highlighting key intervention points for preserving ubiquitination states through inhibitor treatment and optimized lysis conditions before downstream analysis using either ubiquitin tagging or antibody-based enrichment methods.
The Scientist's Toolkit: Essential Research Reagents
Table 5: Essential Research Reagents for Ubiquitination Studies
| Reagent Category | Specific Examples | Function | Key Considerations |
|---|---|---|---|
| Proteasome Inhibitors | Bortezomib, Carfilzomib, MG132, Lactacystin | Block degradation of ubiquitinated proteins | Select based on specificity, reversibility, and cellular permeability |
| Deubiquitinase Inhibitors | N-ethylmaleimide (NEM), PR-619, PYR-41 | Prevent removal of ubiquitin chains | Include fresh in all buffers; NEM is light-sensitive |
| Ubiquitin Affinity Reagents | ThUBD, TUBE, Ubiquitin-binding domains | Enrich ubiquitinated proteins | ThUBD shows 16x higher sensitivity than TUBE [2] |
| Lysis Detergents | NP-40, Triton X-100, SDS, Digitonin | Solubilize proteins while preserving modifications | Match detergent strength to application needs |
| Ubiquitin Ligase Components | E1, E2, E3 enzymes (e.g., gp78RING-Ube2g2) | In vitro ubiquitination assays | Required for ubi-tagging conjugation approaches [1] |
| Mass Spectrometry Reagents | TMT labels, anti-GG-ε-K antibodies, Trypsin | Identify and quantify ubiquitination sites | Anti-VG-ε-K antibodies specific for UFMylation studies [4] |
Comparative Analysis of Ubiquitin Tagging vs. Antibody-Based Enrichment
The preservation methods detailed in this guide directly impact the performance of downstream applications for ubiquitination analysis, particularly when comparing emerging ubiquitin tagging approaches with conventional antibody-based methods.
Performance Metrics Across Methods
Table 6: Method Comparison for Ubiquitination Analysis
| Parameter | Ubi-Tagging Approach | Antibody-Based Enrichment | ThUBD-Based Capture |
|---|---|---|---|
| Sensitivity | High (complete conversion in 30 min) [1] | Variable (depends on antibody affinity) | Very high (16x improvement over TUBE) [2] |
| Linkage Specificity | Controlled (K48-specific demonstrated) [1] | Often linkage-biased | Unbiased recognition of all chain types [2] |
| Multimerization Capacity | Excellent (up to 11th order multimers) [1] | Limited by antibody valency | Designed for polyubiquitin chains |
| Throughput | Moderate (enzymatic steps required) | High (standard immunoassays) | Very high (96-well plate format) [2] |
| Sample Requirements | Moderate (10 µM Fab-Ub demonstrated) [1] | Low to moderate | Low (0.625 µg detection limit) [2] |
Impact of Preservation Methods on Downstream Applications
The choice between ubiquitin tagging and antibody-based approaches significantly influences experimental design, with preservation conditions playing a critical role in each method's success. Ubi-tagging techniques, which enable site-directed multivalent conjugation of antibodies to ubiquitinated payloads, require stringent preservation of ubiquitin chain integrity to maintain enzymatic accessibility for E1, E2, and E3 ubiquitination machinery [1]. These methods achieve complete conversion of substrates within 30 minutes while maintaining protein stability up to 75°C, but depend heavily on initial ubiquitination state preservation.
Antibody-based methods, including the high-throughput ThUBD-coated plate technology, benefit from optimized lysis conditions that maximize ubiquitinated protein yield while minimizing chain dissociation. The ThUBD approach demonstrates a 16-fold wider linear range for capturing polyubiquitinated proteins compared to traditional TUBE-based methods, with sensitivity down to 0.625 μg of input material, making it particularly valuable for limited samples [2]. This method's unbiased recognition of all ubiquitin chain types addresses a critical limitation of conventional antibodies that often display linkage preference.
For mass spectrometry-based ubiquitinome analyses, denaturing lysis conditions with robust proteasome inhibition are essential, as demonstrated in studies identifying over 200 UFMylation sites from mouse tissues [4]. These approaches typically require specialized antibodies for enrichment of modified peptides (e.g., anti-GG-ε-K for ubiquitination, anti-VG-ε-K for UFMylation) following tryptic digestion, with identification efficiency heavily influenced by the initial preservation of modification states.
The comparative analysis presented in this guide demonstrates that effective preservation of ubiquitination states through appropriate proteasome inhibition and lysis conditions is a fundamental prerequisite for successful downstream analysis, regardless of the specific detection methodology employed. While emerging techniques like ubi-tagging offer exceptional specificity and multimerization capabilities, and high-throughput approaches like ThUBD-coated plates provide unprecedented sensitivity, both methodologies depend critically on the initial stabilization of the ubiquitinated proteome. Researchers must carefully match their preservation strategies to their analytical goals, considering factors such as sample availability, required throughput, and specific ubiquitin linkage interests. As the field continues to advance, particularly in drug development applications such as PROTAC development, the precise preservation and detection of ubiquitination states will remain essential for understanding disease mechanisms and developing targeted therapeutics.
In the field of proteomics, achieving deep coverage of the ubiquitylome is a significant challenge due to the low stoichiometry of ubiquitination events within the complex cellular background. This guide objectively compares the performance of the two predominant enrichment strategies—antibody-based enrichment and ubiquitin tagging—by examining experimental data from recent studies. The analysis is framed within the broader thesis that while antibody-based methods are the established workhorse, antibody-free tagging approaches offer a complementary and sometimes superior alternative by mitigating antibody-associated biases.
Core Strategy Comparison: Antibody vs. Tagging Approaches
The following table summarizes the fundamental principles, advantages, and limitations of each strategy.
| Feature | Antibody-Based Enrichment | Ubiquitin Tagging (Genetic) |
|---|---|---|
| Core Principle | Antibodies (e.g., anti-K-ε-GG) enrich for tryptic peptides containing the diGly remnant left after ubiquitination [3] [12]. | Cells are engineered to express affinity-tagged ubiquitin (e.g., His-, Strep-), enabling purification of ubiquitinated proteins prior to digestion [3]. |
| Key Advantage | Applicable to any biological sample, including clinical and animal tissues, without genetic manipulation [3]. | Relatively low-cost and easy enrichment of ubiquitinated substrates [3]. |
| Key Limitations | - Sequence recognition bias [11]- High cost [11] [3]- Cannot distinguish ubiquitination from NEDD8/ISG15 modification [11] [3] | - Infeasible for many clinical samples or animal tissues [11] [3]- Tagged ubiquitin may not perfectly mimic endogenous ubiquitin, potentially creating artifacts [3]- Co-purification of non-ubiquitinated proteins can impair identification sensitivity [3]. |
Performance Benchmarking: Quantitative Data
The transition to advanced mass spectrometry techniques, particularly Data-Independent Acquisition (DIA), has dramatically improved the depth and robustness of ubiquitylome analysis. The table below summarizes key performance metrics from studies utilizing different enrichment strategies coupled with DIA-MS.
| Enrichment Method | MS Acquisition | Sample Input | Unique diGly Peptides Identified (Single Shot) | Key Quantitative Performance | Citation |
|---|---|---|---|---|---|
| anti-K-ε-GG | Optimized DIA (Orbitrap) | 1 mg peptide | ~35,000 sites | Doubled identification vs. DDA; 45% of peptides had CV < 20% [12]. | [12] |
| anti-K-ε-GG | DIA-NN (library-free) | 2 mg protein | ~68,000 peptides | Tripled identification vs. DDA; median CV ~10% [42]. | [42] |
| AFUP (Antibody-Free) | LC-MS/MS | 0.8 mg HeLa lysate | 349 ± 7 sites | Excellent reproducibility (CV = 2%) and high quantitative stability (Pearson, r ≥ 0.91) [11]. | [11] |
The data demonstrates that antibody-based enrichment coupled with DIA-MS is the current state-of-the-art for achieving the deepest possible coverage, with studies consistently identifying tens of thousands of ubiquitination sites in a single experiment. The AFUP method, while identifying fewer sites in the presented experiment, demonstrates exceptional reproducibility and provides a viable antibody-free alternative.
Detailed Experimental Protocols
Protocol 1: Anti-K-ε-GG Enrichment with DIA-MS
This protocol is adapted from studies that achieved identification of over 35,000 ubiquitin sites [12] [42].
- Cell Lysis and Digestion: Lyse cells using a Sodium Deoxycholate (SDC)-based buffer supplemented with Chloroacetamide (CAA) to rapidly alkylate cysteines and inactivate deubiquitinases. This method has been shown to yield ~38% more K-ε-GG peptides than traditional urea buffer [42]. Followed by tryptic digestion.
- Peptide Pre-fractionation: To build a comprehensive spectral library, separate peptides using basic reversed-phase (bRP) chromatography into 96 fractions, which are then concatenated into a smaller number of pools. The highly abundant K48-linked ubiquitin-chain derived diGly peptide is isolated and processed separately to prevent it from dominating the enrichment and MS analysis [12].
- Immunoaffinity Enrichment: Enrich diGly-containing peptides from the fractionated or whole proteome digest using anti-K-ε-GG antibody beads. An optimized ratio is 1 mg of peptide input with 31.25 µg of antibody [12].
- Mass Spectrometry Analysis: Analyze enriched peptides using a DIA method on an Orbitrap mass spectrometer. An optimized method uses 46 precursor isolation windows with a fragment scan resolution of 30,000 [12]. For data processing, software like DIA-NN in library-free mode or with a project-specific spectral library is used [42].
Protocol 2: Antibody-Free Ubiquitination Profiling (AFUP)
This protocol outlines the AFUP strategy, which avoids the use of antibodies [11].
- Blocking Free Amines: At the protein level, block all free amine groups (lysine ε-amines and protein N-terminal) using formaldehyde (dimethylation).
- Deubiquitination and ε-Amine Exposure: Treat the blocked proteins with non-specific deubiquitinases (USP2 and USP21). This hydrolyzes ubiquitin molecules from ubiquitinated proteins, specifically regenerating the free ε-amine groups only at the former ubiquitination sites.
- Biotin Labeling and Enrichment: Label the newly exposed ε-amines with NHS-SS-Biotin. The biotinylated peptides, which correspond to previously ubiquitinated peptides, are then captured using streptavidin beads.
- Elution and Analysis: Elute the enriched peptides by cleaving the disulfide bond in the biotin linker with DTT. The eluted peptides are then analyzed by LC-MS/MS.
The Scientist's Toolkit: Essential Research Reagents
| Reagent / Tool | Function in Ubiquitylomics | Key Consideration |
|---|---|---|
| anti-K-ε-GG Antibody | Immunoaffinity enrichment of ubiquitin remnant peptides after tryptic digestion [12] [43]. | Check for cross-reactivity with NEDD8/ISG15 remnants; batch-to-batch variability can be an issue [11] [3]. |
| Linkage-Specific Ub Antibodies | Enrich for ubiquitinated proteins or peptides with specific chain linkages (e.g., K48, K63) [3]. | Essential for studying the biology of specific ubiquitin signals; availability is limited for some atypical linkages. |
| Tagged Ubiquitin Plasmids | For genetic tagging strategies (e.g., His-, Strep-tag); allows purification of ubiquitinated proteins from engineered cells [3]. | Not suitable for patient samples; potential for artifact generation [3]. |
| Deubiquitinases (DUBs) | Used in antibody-free methods (e.g., AFUP) to expose buried lysines [11]. | USP2 and USP21 are non-specific DUBs used for general ubiquitome profiling [11]. |
| DIA-NN Software | Deep neural network-based software for processing DIA-MS data; significantly boosts ubiquitinome coverage and quantitative accuracy [42]. | Particularly effective in "library-free" mode, circumventing the need for extensive spectral library generation [44] [42]. |
| Proteasome Inhibitors (e.g., MG132) | Increases the abundance of ubiquitinated proteins, particularly K48-linked chains, by blocking their degradation [12]. | Crucial for deep coverage but may alter cellular physiology and skew the ubiquitylome toward proteasomal targets. |
Experimental Workflow Visualization
The following diagrams illustrate the logical steps and key decision points for the two main enrichment strategies.
Antibody-Based Ubiquitylome Profiling
Antibody-Free (AFUP) Ubiquitination Profiling
In the study of the ubiquitin-proteasome system, achieving high specificity is paramount to accurately map ubiquitination events and avoid misleading biological conclusions. The primary challenge researchers face is the inherent cross-reactivity of enrichment methods with ubiquitin-like proteins (UBLs), such as NEDD8 and ISG15, which share structural similarities with ubiquitin and generate identical diGlycine (K-ε-GG) remnants upon tryptic digestion. This methodological limitation can significantly compromise data integrity by introducing false positives. Furthermore, traditional antibody-based approaches often exhibit sequence context bias, while genetic tagging strategies may create artifacts by altering native ubiquitin structure and function. This comparison guide objectively evaluates the performance of modern ubiquitin tagging and antibody-based enrichment methods in addressing these critical specificity challenges, providing researchers with experimental data to inform their methodological selections.
Method Comparison at a Glance
The following table summarizes the core characteristics, advantages, and limitations of the primary methods used to enhance specificity in ubiquitin enrichment.
| Method | Core Principle | Specificity Advantages | Inherent Limitations | Typical Ubiquitin Sites Identified |
|---|---|---|---|---|
| K-ε-GG Antibody (Standard) [45] [3] | Immunoaffinity enrichment of tryptic peptides with diGlycine remnant | High throughput; well-established commercial availability | Cross-reactivity with NEDD8/ISG15; amino acid sequence bias | ~4,000-10,000+ per experiment (highly variable) [45] |
| UbiSite Antibody [45] | Targets longer 13-mer ubiquitin remnant after LysC digestion | Dramatically reduced UBL cross-reactivity; minimized sequence bias | More complex workflow; requires sequential LysC/trypsin digestion | ~30,000 per replicate; 64,000 cumulative with proteasome inhibition [45] |
| Ubi-Fast (On-Bead TMT) [14] [22] | Tandem Mass Tag labeling while peptides are bound to K-ε-GG antibody | High multiplexing capacity (up to 18 samples); improved quantitative accuracy | Does not resolve underlying UBL cross-reactivity of the antibody | ~20,000 sites from 500 μg input per sample (TMT10-plex) [14] |
| Enzymatic Ubi-Tagging [1] | Uses ubiquitination enzyme cascade for site-directed conjugation | Absolute specificity via enzyme linkage control; no UBL cross-reactivity | Primarily for engineered conjugates; not for endogenous site discovery | N/A (Application-specific) |
Detailed Experimental Protocols
UbiSite Protocol for Reduced UBL Cross-Reactivity
The UbiSite method was developed specifically to overcome the cross-reactivity limitations of traditional K-ε-GG antibodies.
- Sample Preparation: Process cell or tissue lysates under denaturing conditions (e.g., 8M urea) to preserve ubiquitination states and inhibit deubiquitinases.
- Protein Digestion: Perform sequential proteolytic digestion:
- Primary Digestion with LysC: Digest proteins with LysC, which cleaves specifically at lysine residues. This generates a longer, ~13-amino acid remnant still attached to the substrate peptide when a ubiquitinated site is encountered.
- Immunoaffinity Enrichment: Use the UbiSite antibody to enrich for peptides containing this specific 13-mer ubiquitin remnant.
- Secondary Digestion with Trypsin: Digest the enriched peptide-antibody complexes with trypsin to reduce the remnant to the standard diGlycine (K-ε-GG) motif, making the peptides amenable to LC-MS/MS analysis [45].
- LC-MS/MS Analysis: Analyze the peptides using standard LC-MS/MS platforms. The initial enrichment for the longer ubiquitin-specific remnant effectively excludes nearly all peptides modified by NEDD8 or ISG15.
Ubi-Fast Protocol for Multiplexed Quantification
The UbiFast approach enhances throughput and quantitative precision while using standard K-ε-GG antibodies.
- Peptide Preparation & Enrichment: Generate a tryptic digest from lysates. Enrich for K-ε-GG peptides using the standard antibody, typically coupled to magnetic beads for automated processing [14].
- On-Antibody TMT Labeling: While the K-ε-GG peptides are still bound to the antibody, resuspend the beads in labeling buffer and add Tandem Mass Tag (TMT) reagents. The antibody protects the ε-amine of the diGlycine remnant from derivatization, allowing the TMT label to react only with the N-terminus and other lysine ε-amines on the peptide. Quench the reaction with hydroxylamine [22].
- Sample Pooling and Analysis: Combine the TMT-labeled samples from multiple conditions (e.g., 10-plex) while they are still on-bead. Elute the combined labeled peptides and analyze them by LC-MS/MS. The single-run analysis reduces missing values and improves quantitative accuracy across samples [14] [22].
Enzymatic Ubi-Tagging for Directed Conjugation
This innovative method bypasses antibodies entirely for generating specific protein conjugates.
- Design of Ubi-Tagged Constructs: Genetically fuse the protein of interest (e.g., an antibody, nanobody, or Fab fragment) to a donor ubiquitin (Ubdon) with a specific lysine-to-arginine mutation (e.g., K48R) to prevent homodimerization. The payload (e.g., a fluorescent dye or peptide) is fused to an acceptor ubiquitin (Ubacc) with a non-reactive C-terminus (ΔGG) [1].
- In Vitro Conjugation Reaction: Incubate the Ubdon and Ubacc constructs with a specific set of recombinant ubiquitination enzymes (E1, and a linkage-specific E2-E3 fusion enzyme) for a short period (e.g., 30 minutes). The enzymes catalyze the formation of a precise ubiquitin linkage between the donor and acceptor, conjugating the payload to the protein [1].
- Purification and Validation: Purify the homogeneous conjugate using standard methods (e.g., protein G affinity). Validate the product using techniques like ESI-TOF mass spectrometry and functional assays to confirm activity is retained [1].
Performance Data and Experimental Evidence
Quantitative Assessment of Specificity and Depth
Empirical data demonstrates the performance trade-offs between different methods.
| Performance Metric | K-ε-GG (DDA) [12] | K-ε-GG (DIA) [12] | UbiSite [45] | UbiFast [22] |
|---|---|---|---|---|
| Sites per Single Run | ~20,000 | ~35,000 | ~30,000 | ~10,000 (from 500 μg input) |
| Quantitative Reproducibility (CV < 20%) | 15% | 45% | Data Not Specified | High (Automation) |
| Estimated UBL Cross-Reactivity | <6% [45] | <6% (inferred) | Dramatically Reduced | <6% (inherent to antibody) |
| Multiplexing Capacity | Low (Label-free) | Low (Label-free) | Low (Label-free) | High (Up to 18-plex with TMT) |
Key Experimental Findings on Specificity
- UBL Cross-Reactivity: Studies quantifying the contribution of UBLs find that traditional K-ε-GG antibodies have a relatively low but non-zero cross-reactivity, with less than 6% of identified sites typically attributable to NEDD8 or ISG15 [45]. While this may be acceptable for some studies, it introduces measurable false positives.
- Impact of DIA on Reproducibility: Data-Independent Acquisition mass spectrometry significantly improves the reproducibility of ubiquitinome studies. One study showed that while DDA quantified 15% of diGly peptides with a coefficient of variation below 20%, DIA increased this figure to 45%, demonstrating superior quantitative accuracy [12].
- Efficiency of Enzymatic Conjugation: The ubi-tagging method demonstrates exceptional efficiency and preservation of function. Conjugation reactions consistently show 93-96% conversion efficiency, and the resulting conjugates, such as fluorescently labeled Fab fragments, maintain their antigen-binding capability without hindrance [1].
The Scientist's Toolkit: Key Research Reagents
Successful implementation of these methods relies on specific, high-quality reagents.
| Reagent / Tool | Function | Example Application |
|---|---|---|
| Linkage-Specific E2-E3 Enzymes [1] | Catalyzes the formation of a defined ubiquitin linkage (e.g., K48) during ubi-tagging. | Generating homogeneous antibody-drug conjugates or bispecific T-cell engagers. |
| HS mag anti-K-ε-GG Antibody [14] | Magnetic bead-conjugated antibody for automated, high-throughput enrichment of diGly peptides. | Automated UbiFast processing of large sample cohorts (e.g., patient-derived xenografts). |
| LysC Protease [45] | Cleaves at lysine residues to generate the long ubiquitin remnant for UbiSite antibody recognition. | Specific workflow for reducing UBL cross-reactivity in endogenous ubiquitinome mapping. |
| Tandem Mass Tag (TMT) Reagents [14] [22] | Isobaric chemical labels for multiplexed quantitative comparison of up to 18 samples in one MS run. | On-bead labeling in UbiFast to compare ubiquitination across multiple time points or conditions. |
| Linkage-Specific Ub Antibodies [3] | Enrich for polyubiquitinated proteins with a specific chain topology (e.g., K48, K63). | Studying the role of a particular ubiquitin chain type in a cellular process like degradation. |
Visualizing Key Workflows
The following diagrams illustrate the core experimental workflows for the primary methods discussed, highlighting the points where specificity is controlled.
K-ε-GG vs. UbiSite Antibody Workflow
Enzymatic Ubi-Tagging Workflow
The choice of an optimal method for ubiquitin enrichment depends heavily on the specific research question and the relative priority of specificity, throughput, and applicability.
- For Mapping Endogenous Ubiquitinomes with High Specificity: The UbiSite method is highly recommended when the primary goal is to minimize UBL cross-reactivity and achieve the most accurate picture of the endogenous ubiquitinome, despite its more complex workflow [45].
- For High-Throughput, Quantitative Profiling: The automated UbiFast platform is unparalleled for large-scale studies involving multiple perturbations, time courses, or patient samples, offering excellent reproducibility and multiplexing capacity, though it does not solve the fundamental UBL cross-reactivity of the underlying antibody [14] [22].
- For Generating Defined Protein Conjugates: Enzymatic ubi-tagging provides absolute specificity and control for engineering therapeutic antibodies or synthetic biology constructs, making it ideal for applications where homogeneity and precise linkage are critical [1].
- For Standard Ubiquitinome Profiling: Traditional K-ε-GG enrichment coupled with DIA-MS remains a robust and sensitive option for many discovery-level studies, especially when the potential for a small percentage of UBL-derived identifications is an acceptable trade-off for the depth of coverage [12].
As the field advances, the combination of these strategies—such as applying UbiSite principles to multiplexed workflows—will further empower researchers to decipher the complex language of ubiquitin signaling with unprecedented accuracy and depth.
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) has become the cornerstone of modern proteomics, enabling high-throughput quantification of complex protein mixtures. Among the various acquisition strategies, Data-Independent Acquisition (DIA) has emerged as a powerful technique that bridges the gap between the broad coverage of data-dependent acquisition (DDA) and the high reproducibility of targeted methods like Selected Reaction Monitoring (SRM). Unlike DDA, which stochastically selects the most intense precursors for fragmentation, DIA systematically fragments all ions within predefined, consecutive mass-to-charge (m/z) windows in a cyclic manner. This fundamental difference allows DIA to overcome the issue of biased sampling inherent in DDA, resulting in significantly improved quantitative accuracy, precision, and reproducibility [46] [47].
The application of DIA is particularly relevant in studies investigating post-translational modifications (PTMs), such as ubiquitination, where accurate quantification under diverse biological conditions is crucial. Ubiquitination, a versatile PTM involving the covalent attachment of ubiquitin to substrate proteins, regulates fundamental cellular processes including protein degradation, cell signaling, and DNA repair. The analysis of ubiquitination presents unique challenges due to the complexity of ubiquitin conjugates, which can range from single monomers to polymers with different lengths and linkage types [3]. Within this context, DIA proteomics provides a robust framework for comparing the performance of different ubiquitin enrichment strategies, notably ubiquitin tagging-based approaches versus antibody-based enrichment methods, enabling researchers to make informed decisions based on empirical data rather than technical limitations.
DIA Fundamentals: Mechanism and Advantages
How DIA Works
The DIA workflow is characterized by its systematic and unbiased approach to data acquisition. A common DIA method is Sequential Windowed acquisition of All Theoretical fragment ion Mass Spectra (SWATH-MS), though the generic term "DIA" is used to refer to all such strategies regardless of instrumentation [47]. In a typical DIA experiment:
- The full m/z range of interest is divided into a series of consecutive, fixed-width isolation windows (e.g., 5-25 Da).
- All precursor ions within each predefined window are simultaneously isolated and fragmented, without intensity-based preselection.
- All resulting fragment ions from each window are recorded by a high-resolution mass analyzer [46] [47].
This process is repeated cyclically throughout the entire LC-MS/MS run, resulting in a complete digital record of all fragment ions across the chromatographic separation. Because multiple peptides are co-fragmented within each window, the resulting MS2 spectra are highly complex. Their deconvolution typically requires a peptide spectral library that contains reference data such as mass-to-charge ratio, retention time, and fragment spectra for peptides expected in the sample. However, recent advances in bioinformatics have enabled the direct analysis of DIA data without traditional libraries [46] [47] [48].
Comparative Advantages of DIA
The design of DIA confers several distinct advantages over other mass spectrometry acquisition methods, as summarized in the table below.
Table 1: Comparison of LC-MS/MS-Based Quantitative Proteomics Strategies
| Feature | Data-Dependent Acquisition (DDA) | Data-Independent Acquisition (DIA) | Targeted Methods (e.g., SRM/PRM) |
|---|---|---|---|
| Acquisition Principle | Intensity-based selection of top precursors for fragmentation | Systematic fragmentation of all precursors in pre-defined m/z windows | Monitoring of pre-selected precursor/fragment ion pairs |
| Protein Coverage | Broad | Very Broad | Limited |
| Quantitative Reproducibility | Moderate (stochastic sampling) | High (consistent sampling) | High |
| Quantitative Accuracy | Moderate | High | High |
| Post-acquisition Analysis Flexibility | Low | High (digital archive allows re-mining) | Low |
| Ideal Application | Discovery proteomics | Biomarker discovery, clinical proteomics, PTM analysis | Validation of predefined targets |
DIA effectively combines the broad protein coverage of DDA with the high reproducibility and accuracy of targeted methods, making it exceptionally well-suited for applications that require large-scale, precise quantification of complex samples, such as in clinical proteomics and drug development [46] [47]. Its comprehensive data recording creates a permanent digital map of the sample, allowing historical data to be re-interrogated as new hypotheses or analysis tools emerge [48].
Experimental Comparison: Ubiquitin Tagging vs. Antibody-Based Enrichment
To objectively evaluate protein ubiquitination analysis methods, we designed an experimental protocol based on DIA quantification, comparing ubiquitin tagging and antibody-based enrichment. The performance of each method was assessed based on key metrics including efficiency, specificity, and practical applicability.
Detailed Experimental Protocols
Ubiquitin Tagging Enrichment Protocol
The ubiquitin (Ub) tagging approach, exemplified by the "ubi-tagging" technique, uses genetic fusion of an affinity tag to ubiquitin for purifying ubiquitinated substrates [1] [3].
- Cell Line Preparation: Generate a stable cell line (e.g., HEK293T or U2OS) expressing His-tagged or Strep-tagged Ubiquitin using lentiviral transduction or similar methods. As a control, a cell line stably expressing only the tag (without Ub fusion) should be generated.
- Cell Lysis and Protein Extraction: Lyse cells in a denaturing buffer (e.g., 6 M Guanidine-HCl, 100 mM NaH₂PO₄, 10 mM Tris-HCl, pH 8.0) supplemented with protease inhibitors and 10 mM N-Ethylmaleimide (NEM) to inhibit deubiquitinases.
- Enrichment of Ubiquitinated Proteins:
- For His-tag purification: Incubate clarified lysates with Ni-NTA agarose beads for 2-4 hours at room temperature. Wash beads sequentially with wash buffer 1 (6 M Guanidine-HCl, 100 mM NaH₂PO₄, 10 mM Tris-HCl, pH 8.0), wash buffer 2 (8 M Urea, 100 mM NaH₂PO₄, 10 mM Tris-HCl, pH 8.0), and wash buffer 3 (same as buffer 2, pH 6.3).
- For Strep-tag purification: Incubate lysates with Strep-Tactin sepharose beads for 1-2 hours. Wash with the manufacturer's recommended buffer.
- Elution and Digestion: Elute bound proteins with Laemmli buffer containing 500 mM Imidazole (for His-tag) or biotin-containing buffer (for Strep-tag). Separate proteins by SDS-PAGE, followed by in-gel tryptic digestion. Alternatively, perform on-bead digestion.
- DIA Mass Spectrometry Analysis: Desalt and concentrate the resulting peptides. Analyze using a established DIA acquisition method on a Q-Orbitrap or Q-TOF instrument [3].
Antibody-Based Enrichment Protocol
This method uses anti-ubiquitin antibodies to immuno-enrich endogenously ubiquitinated proteins directly from biological samples [3].
- Sample Preparation: Lyse tissues or cells in a non-denaturing or mildly denaturing RIPA buffer supplemented with protease inhibitors, 10 mM NEM, and 1 mM PR-619 (a DUB inhibitor).
- Pre-clearing: Incubate the protein lysate with control IgG and Protein A/G beads for 1 hour at 4°C to reduce non-specific binding.
- Immunoprecipitation (IP):
- Incubate the pre-cleared supernatant with a linkage-specific Ub antibody (e.g., anti-K48, anti-K63) or a pan-ubiquitin antibody (e.g., FK1, FK2) cross-linked to Protein A/G beads overnight at 4°C.
- Wash the beads extensively with IP wash buffer (e.g., RIPA buffer) to remove non-specifically bound proteins.
- Elution and Digestion: Elute ubiquitinated proteins using a low-pH glycine solution (pH 2.5-3.0) or by boiling in Laemmli buffer. Proceed with SDS-PAGE separation and in-gel tryptic digestion.
- DIA Mass Spectrometry Analysis: Process and analyze the peptides as described in section 3.1.1.
Performance Data and Comparative Analysis
The following table synthesizes experimental data from published studies to provide a direct comparison of the two enrichment strategies when coupled with DIA analysis.
Table 2: Performance Comparison of Ubiquitin Enrichment Methods Coupled with DIA
| Performance Metric | Ubiquitin Tagging-Based Enrichment | Antibody-Based Enrichment |
|---|---|---|
| Reported Enrichment Efficiency | High; conversion efficiency of conjugation reactions up to 93-96% [1] | Variable; depends on antibody affinity and specificity |
| Key Advantage | Easy, low-cost screening; allows generation of defined conjugates (e.g., bispecific engagers) [1] [3] | Applicable to any biological sample, including patient tissues, without genetic manipulation [3] |
| Primary Limitation | May generate artifacts; tagged Ub may not perfectly mimic endogenous Ub [3] | High cost of quality antibodies; potential for non-specific binding [3] |
| Linkage-Type Specificity | Requires specific E2/E3 enzyme pairs (e.g., gp78RING-Ube2g2 for K48-linkage) [1] | Enabled by linkage-specific antibodies (e.g., K48-, K63-specific) [3] |
| Identification Sensitivity | Can be hampered by co-purification of non-ubiquitinated proteins (e.g., histidine-rich proteins) [3] | Limited by antibody affinity and the stoichiometric scarcity of ubiquitinated peptides |
| Quantitative Accuracy (via DIA) | High quantitative accuracy and reproducibility, characteristic of DIA methods [46] [48] | High quantitative accuracy; enables profiling under physiological conditions [3] |
| Typical Application Context | Cell culture systems for discovery and engineering applications [1] | Profiling ubiquitination in animal tissues or clinical samples for pathological studies [3] |
Optimizing DIA Analysis: Spectral Libraries and Statistical Frameworks
The performance of a DIA-based study is heavily influenced by post-acquisition data processing. Two critical components are the generation of spectral libraries and the choice of statistical frameworks.
Spectral Library Generation
Spectral libraries are crucial for translating complex DIA MS2 spectra into identifiable peptides. Different library generation strategies impact the depth and reliability of results:
- Gas-Phase Fractionated (GPF) Libraries: Generated by repeatedly measuring a representative sample to cover distinct m/z ranges in greater detail. A benchmark study found that all DIA software suites benefit from using a GPF spectral library, which performed best in identifying differentially abundant proteins [48].
- DDA-based Project-Specific Libraries: Created by measuring fractionated samples from the project using DDA. This library type tends to reflect the specific proteome composition and instrumentation used [48].
- In Silico Predicted Libraries: These libraries are generated computationally, for instance using tools like DIA-NN or Prosit, and are gaining momentum. They eliminate the need for additional laboratory work but may carry a higher risk of false discoveries if not properly refined with experimental data [48].
Statistical Analysis for Differential Abundance
Robust statistical analysis is paramount for deriving biological insights from DIA data. A large-scale benchmarking study that evaluated 1428 distinct analysis workflows demonstrated that non-parametric permutation-based statistical tests consistently performed best in correctly identifying differentially abundant proteins, especially when dealing with the heterogeneity typical of clinical samples [48]. Furthermore, the study highlighted that the choice of DIA software, combined with data preprocessing steps like normalization and sparsity reduction, significantly impacts the final list of significant proteins, underscoring the need for careful workflow selection.
The Scientist's Toolkit: Essential Research Reagents
The following table details key reagents and materials essential for conducting DIA-based ubiquitination studies.
Table 3: Essential Research Reagents for DIA-based Ubiquitination Analysis
| Reagent / Material | Function and Importance in the Workflow |
|---|---|
| Tagged Ubiquitin Plasmids | (e.g., 6xHis-Ub, Strep-Ub). For generating stable cell lines for ubiquitin tagging enrichment methods [3]. |
| Ubiquitin-Activating Enzyme (E1) | Essential enzyme for the ubi-tagging conjugation technique, initiates the ubiquitination cascade [1] [3]. |
| Linkage-Specific E2-E3 Fusions | (e.g., gp78RING-Ube2g2 for K48-linkage). Critical for controlled, site-directed multivalent conjugation in ubi-tagging [1]. |
| Pan-Ubiquitin Antibodies | (e.g., P4D1, FK1/FK2). For immuno-enrichment of ubiquitinated proteins regardless of chain linkage type [3]. |
| Linkage-Specific Ub Antibodies | (e.g., K48-, K63-specific). For enriching ubiquitinated proteins with specific chain architectures to study linkage-specific biology [3]. |
| Ni-NTA / Strep-Tactin Beads | Affinity resins for purifying His-tagged or Strep-tagged ubiquitinated proteins, respectively, from complex lysates [3]. |
| Deubiquitinase (DUB) Inhibitors | (e.g., N-Ethylmaleimide (NEM), PR-619). Preserve the native ubiquitinome by preventing the removal of Ub from substrates during sample preparation [3]. |
| Ionic Liquid Surfactants | (e.g., DIA-friendly surfactants). Aid in efficient protein extraction and solubilization while being compatible with MS analysis. |
| Spectral Library | (GPF, DDA-based, or in silico). A mandatory resource for the confident identification and quantification of peptides from DIA data [48]. |
| Retention Time Standard | (e.g., iRT peptides). Used to normalize peptide retention times across different LC-MS/MS runs, improving quantification consistency [48]. |
Workflow and Pathway Diagrams
The following diagrams illustrate the core workflows for the ubiquitin enrichment methods discussed in this guide.
Ubi-Tagging Conjugation and Enrichment
Diagram 1: Ubi-tagging workflow for enriching ubiquitinated proteins. The process involves genetic engineering, affinity-based purification, and DIA analysis.
Antibody-Based Ubiquitin Enrichment
Diagram 2: Antibody-based enrichment workflow. This method uses immunoprecipitation with ubiquitin-specific antibodies to isolate ubiquitinated complexes from native lysates.
This guide has provided a detailed comparison of advanced MS acquisition, specifically leveraging DIA for the superior quantification of ubiquitinated proteins. Through objective experimental data and structured protocols, we have demonstrated that DIA represents a robust and versatile platform for proteomic analysis, effectively combining extensive coverage with high quantitative accuracy. The choice between ubiquitin tagging and antibody-based enrichment is context-dependent; the former is a powerful tool for engineered systems and defined conjugate generation, while the latter is indispensable for studying endogenous ubiquitination in clinical and tissue specimens. Ultimately, the integration of optimized DIA workflows with appropriate enrichment strategies and rigorous statistical analysis, as outlined herein, provides researchers and drug development professionals with a reliable path to uncovering the intricate roles of ubiquitination in health and disease.
Protein ubiquitination, the covalent attachment of a small regulatory protein to substrate proteins, represents one of the most versatile post-translational modifications in eukaryotic cells, governing diverse cellular functions from protein degradation to signal transduction [3] [49]. The identification and quantification of ubiquitination sites has emerged as a critical capability in molecular biology, particularly for drug development professionals seeking to understand disease mechanisms and therapeutic targets. Two primary methodological approaches have dominated the field: ubiquitin tagging (Ub tagging) and antibody-based enrichment, each with distinct advantages, limitations, and sample-specific requirements [3].
Ubiquitin tagging involves the genetic engineering of affinity tags (such as His or Strep tags) onto ubiquitin molecules, enabling purification of ubiquitinated proteins from cell lysates [3]. This method first gained prominence in 2003 when Peng et al. expressed 6× His-tagged ubiquitin in Saccharomyces cerevisiae, identifying 110 ubiquitination sites on 72 proteins [3]. Conversely, antibody-based approaches utilize antibodies that recognize either ubiquitin itself or the characteristic diglycine (K-ε-GG) remnant left on trypsin-digested peptides from ubiquitinated proteins [3] [9]. This method enables researchers to profile endogenously ubiquitinated substrates without genetic manipulation, making it particularly valuable for tissue samples and clinical specimens [3] [22].
The fundamental distinction between these methodologies becomes critically important when designing experiments involving different sample types. While ubiquitin tagging offers an easy, relatively low-cost method for screening ubiquitinated substrates in cultured cells, its application to animal or patient tissues remains infeasible due to the requirement for genetic modification [3]. Antibody-based approaches overcome this limitation but face challenges related to antibody specificity, cost, and potential sequence recognition bias [3] [11]. This comparison guide objectively examines the technical requirements, performance characteristics, and optimal applications of each method, with particular emphasis on their divergent input requirements for cultured cells versus tissue samples.
Technical Foundations and Methodological Principles
Ubiquitin Tagging Methodologies
The ubiquitin tagging approach capitalizes on genetic engineering to facilitate the purification of ubiquitinated proteins. This method involves expressing ubiquitin molecules fused with affinity tags such as polyhistidine (His) or Strep-tag in living cells [3]. The tagged ubiquitin becomes incorporated into the endogenous ubiquitination pathway, ultimately labeling ubiquitinated substrates with the affinity tag. These tagged substrates can then be purified using commercially available resins—Ni-NTA for His tags and Strep-Tactin for Strep-tags—and subsequently identified through mass spectrometry-based proteomics [3].
The technical workflow typically begins with cell culture engineering, where researchers create stable cell lines expressing the tagged ubiquitin constructs. Following experimental treatments, cells are lysed under denaturing conditions to preserve ubiquitination states and disrupt non-covalent protein interactions. The lysate undergoes affinity purification using tag-specific resins, after which the captured proteins are digested with trypsin and analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) [3]. Key to site identification is the characteristic mass shift of +114.04 Da on modified lysine residues, corresponding to the diglycine remnant [3].
A significant advancement in this methodology came with the development of the stable tagged Ub exchange (StUbEx) cellular system, which enables replacement of endogenous ubiquitin with His-tagged ubiquitin [3]. Using this approach, Akimov et al. identified 277 unique ubiquitination sites on 189 proteins in HeLa cells [3]. Similarly, Danielsen et al. constructed a cell line stably expressing Strep-tagged ubiquitin and identified 753 lysine ubiquitylation sites on 471 proteins in U2OS and HEK293T cells [3]. Despite these successes, the method faces limitations including co-purification of histidine-rich or endogenously biotinylated proteins, potential structural alterations to ubiquitin function, and inability to apply the technique to animal tissues or clinical samples [3].
Antibody-Based Enrichment Strategies
Antibody-based enrichment strategies for ubiquitination profiling have evolved along two primary pathways: protein-level immunoprecipitation and peptide-level immunoaffinity enrichment. Protein-level approaches utilize antibodies that recognize ubiquitin itself (such as P4D1, FK1, or FK2) to immunoprecipitate ubiquitinated proteins prior to tryptic digestion and MS analysis [3] [26]. For example, Denis et al. employed FK2 affinity chromatography to enrich ubiquitinated proteins from human MCF-7 breast cancer cells, successfully identifying 96 ubiquitination sites [3].
The more revolutionary advancement came with the development of antibodies specifically recognizing the diglycine (K-ε-GG) remnant left on tryptic peptides derived from ubiquitinated proteins [9] [26]. This peptide-level immunoaffinity enrichment technique enables direct capture of ubiquitinated peptides from complex protein digests, dramatically improving the specificity and sensitivity of ubiquitination site identification [26]. When this approach was initially implemented, it led to the identification of more than 10,000 ubiquitylation sites from cell lysates [9].
The standard workflow for diGLY proteomics involves cell or tissue lysis under denaturing conditions, followed by complete proteolytic digestion using trypsin [9]. The resulting peptides are then incubated with anti-K-ε-GG antibodies immobilized on beads, washed to remove non-specifically bound peptides, and eluted for LC-MS/MS analysis [9]. The diGLY proteomics approach has become an indispensable tool for systematically interrogating protein ubiquitylation with site-level resolution, applicable to any eukaryotic organism or tissue [9]. A recognized limitation is that the diGLY remnant is identical to those generated by the ubiquitin-like proteins NEDD8 and ISG15, though studies indicate that approximately 95% of all diGLY peptides identified using this approach arise from genuine ubiquitylation [9].
Table: Comparative Technical Foundations of Ubiquitination Profiling Methods
| Parameter | Ubiquitin Tagging | Antibody-Based Enrichment |
|---|---|---|
| Core Principle | Genetic fusion of affinity tags to ubiquitin | Immunological recognition of ubiquitin/diGLY remnant |
| Tagging Approach | His, Strep, or other epitope tags | Not applicable |
| Primary Target | Tagged ubiquitin-conjugated proteins | Ubiquitin proteins or K-ε-GG modified peptides |
| Typical Enzymes | Trypsin | Trypsin |
| Key Identifiable Feature | 114.04 Da mass shift on modified lysine | 114.04 Da mass shift on modified lysine |
| Specificity Challenge | Co-purification of endogenous biotinylated/histidine-rich proteins | Cross-reactivity with NEDD8 and ISG15 diGLY remnants |
Input Requirements for Different Sample Types
Cultured Cell Requirements
For proteomic profiling of ubiquitination using cultured cells, significant differences exist in sample input requirements between ubiquitin tagging and antibody-based approaches. Ubiquitin tagging methodologies typically require successful genetic engineering of cell lines, which involves creating stable expressions of tagged ubiquitin constructs. While the exact cell numbers are rarely specified in the literature, the approach necessitates sufficient cell culture scale to generate 1-10 mg of protein lysate for comprehensive analysis [3] [9]. The StUbEx system, for instance, was demonstrated in HeLa cells, requiring complete replacement of endogenous ubiquitin with His-tagged ubiquitin before any ubiquitination profiling could commence [3].
Antibody-based methods offer more flexible and readily quantifiable input requirements for cultured cells. The widely adopted diGLY proteomics approach typically utilizes 1-10 mg of total protein digest from cell lysates as starting material [9] [26]. For example, the UbiFast method enables quantification of approximately 10,000 ubiquitination sites from just 500 μg of peptide per sample when processing cell lysates in a TMT10plex experiment [22]. Standard protocols for diGLY immunoprecipitation from cultured cells recommend beginning with 1-10 mg of total protein, with higher amounts yielding greater depth of ubiquitination site identification [9]. Comparative studies have demonstrated that peptide-level immunoaffinity enrichment consistently yields greater than fourfold higher levels of modified peptides than protein-level affinity purification mass spectrometry (AP-MS) approaches from equivalent starting material [26].
Tissue Sample Requirements
The requirements for tissue samples differ substantially from cultured cells, with antibody-based methods holding a distinct advantage for tissue ubiquitination profiling. Ubiquitin tagging approaches are essentially inapplicable to tissue samples due to the requirement for genetic manipulation of ubiquitin, which cannot be feasibly performed in animal or human tissues [3]. This represents a fundamental limitation of tagging approaches for translational research and clinical applications.
Antibody-based methods have been successfully adapted for tissue ubiquitinome profiling, though typically with increased input requirements compared to cell culture experiments. The diGLY proteomics approach has been applied to various human and murine primary tissues, with recommended starting amounts of 7 mg of tissue protein per sample for TMT-based analyses [22]. However, advanced methodologies like UbiFast have reduced this requirement to 500 μg of peptide per sample from tissues when using TMT10plex multiplexing, making large-scale tissue ubiquitinome profiling more feasible [22]. Tissue processing typically involves mechanical disruption under denaturing conditions, such as 8M urea lysis buffer, to efficiently extract ubiquitinated proteins while preserving the modification state [9]. The UbiFast method has demonstrated particular utility for tissue analysis, completing profiling in approximately 5 hours with enhanced sensitivity, making it suitable for large-scale studies in primary tissue samples where material is often limited [22].
Table: Sample Input Requirements for Ubiquitination Profiling
| Sample Type | Ubiquitin Tagging Requirements | Antibody-Based Enrichment Requirements |
|---|---|---|
| Cultured Cells | • Stable cell line generation• 1-10 mg protein lysate• Complete tag incorporation | • 0.5-10 mg total protein digest• 500 μg for UbiFast TMT10plex• No genetic manipulation needed |
| Tissue Samples | • Not feasible for most applications• Requires genetic manipulation • Limited to transgenic models | • 0.5-7 mg tissue protein• Mechanical disruption essential• Compatible with clinical specimens |
| Primary Cells | • Limited applicability• Challenging genetic manipulation• Variable tag incorporation | • 0.5-2 mg protein digest• Direct application possible• Suitable for patient-derived cells |
Experimental Protocols and Workflows
Ubiquitin Tagging Experimental Protocol
The standard protocol for ubiquitin tagging begins with the creation of a stable cell line expressing tagged ubiquitin. Researchers typically clone ubiquitin cDNA with C-terminal affinity tags (His×6, Strep-tag II, or similar) into mammalian expression vectors, then transfect cells and select stable clones using appropriate antibiotics [3]. For the StUbEx system, endogenous ubiquitin is replaced with His-tagged ubiquitin through a specialized exchange protocol [3].
Once established, cells are typically treated with proteasome inhibitors (e.g., 10-25 μM MG-132 for 1-4 hours) to stabilize ubiquitinated proteins before harvesting [49] [26]. Cells are lysed in denaturing buffers, such as 6M guanidine-HCl or 8M urea, to disrupt non-covalent interactions and preserve ubiquitination states [3]. The lysate is then incubated with tag-specific affinity resin—Ni-NTA for His tags or Strep-Tactin for Strep-tags—for several hours to capture ubiquitinated proteins [3]. After extensive washing with lysis buffer containing decreasing concentrations of denaturant and imidazole (for His tags) or appropriate competitors, bound proteins are eluted using competitive elution (imidazole for His tags, desthiobiotin for Strep-tags) or by boiling in SDS-PAGE buffer [3].
Eluted proteins are digested with trypsin, either in-solution or in-gel after SDS-PAGE separation. For in-solution digestion, proteins are reduced with dithiothreitol, alkylated with iodoacetamide, and digested first with LysC (1:100 enzyme:substrate) followed by trypsin (1:50 enzyme:substrate) overnight at 37°C [9]. The resulting peptides are desalted using C18 solid-phase extraction before LC-MS/MS analysis. Mass spectrometry data acquisition typically uses data-dependent acquisition methods, with MS2 fragmentation targeting peptides with the characteristic +114.04 Da mass shift on lysine residues [3].
Ubiquitin Tagging Experimental Workflow: This diagram illustrates the sequential steps for ubiquitination profiling using ubiquitin tagging approaches, from initial cell line engineering through final data analysis.
Antibody-Based Enrichment Protocol
The antibody-based enrichment protocol for diGLY proteomics begins with sample preparation from cells or tissues. Cells or tissue samples are lysed in denaturing buffer (8M urea, 150mM NaCl, 50mM Tris-HCl, pH 8) supplemented with protease inhibitors and 5mM N-ethylmaleimide (NEM) to preserve ubiquitination by inhibiting deubiquitinases [9]. For tissue samples, mechanical disruption using a Dounce homogenizer or similar equipment is essential for complete lysis [9]. Protein concentration is determined by BCA assay, and typically 1-10 mg of protein is processed for each sample.
Proteins are reduced with dithiothreitol (5mM, 30 minutes, room temperature), alkylated with iodoacetamide (15mM, 30 minutes, room temperature in darkness), and digested first with LysC (1:100 enzyme:substrate, 3 hours) followed by trypsin dilution (1:50 enzyme:substrate, overnight) after diluting urea concentration to 2M [9]. The resulting peptides are desalted using C18 solid-phase extraction cartridges, eluted with 50% acetonitrile/0.5% acetic acid, and vacuum-concentrated.
For diGLY peptide enrichment, desalted peptides are resuspended in immunoaffinity purification (IAP) buffer (50mM MOPS/NaOH, pH 7.3, 10mM Na2HPO4, 50mM NaCl) and incubated with anti-K-ε-GG antibody-coupled beads for 2 hours at 4°C [9]. Beads are washed multiple times with IAP buffer followed by water, and bound peptides are eluted with 0.15% trifluoroacetic acid. For quantitative analyses using isobaric tags (TMT), the UbiFast protocol enables efficient labeling while peptides are still bound to antibodies, improving sensitivity and reducing processing time [22].
Eluted peptides are analyzed by LC-MS/MS using high-resolution mass spectrometers. For maximum depth of coverage, two-dimensional LC separation with high pH reversed-phase fractionation can be employed prior to LC-MS/MS analysis [22]. Data analysis involves database searching with algorithms capable of detecting the +114.0429 Da mass shift on lysine residues, with false discovery rate estimation using target-decoy approaches.
Antibody-Based Enrichment Workflow: This diagram illustrates the key steps in antibody-based ubiquitination profiling, from sample preparation through data analysis, highlighting steps specific to diGLY remnant recognition.
Performance Comparison and Experimental Data
Efficiency and Coverage Metrics
Direct comparison of ubiquitin tagging and antibody-based methods reveals significant differences in efficiency and coverage. Ubiquitin tagging approaches have demonstrated the ability to identify hundreds of ubiquitination sites in single experiments. Peng et al.'s pioneering study identified 110 ubiquitination sites on 72 proteins from Saccharomyces cerevisiae [3]. The StUbEx system improved upon this, identifying 277 unique ubiquitination sites on 189 proteins in HeLa cells [3]. Danielsen et al. further expanded this to 753 lysine ubiquitylation sites on 471 proteins using Strep-tagged ubiquitin in U2OS and HEK293T cells [3].
Antibody-based methods typically achieve substantially higher coverage. Early implementations identified over 5,000 ubiquitination sites from just 1 mg of input material [26]. The UbiFast method enables quantification of approximately 10,000 distinct ubiquitylation sites from as little as 500 μg of peptide per sample from cells or tissue in a TMT10plex experiment [22]. Rose et al. reported identification of 5,000-9,000 ubiquitylated peptides in cells using 1 mg sample per TMT label state and in tissue using 7 mg of sample per TMT label state [22]. The AFUP (Antibody-Free Ubiquitination Profiling) approach identified 349 ± 7 ubiquitination sites from 0.8 mg HeLa lysates with excellent reproducibility (CV = 2%) [11]. When combined with basic C18 pre-fractionation, this method expanded to approximately 4,000 ubiquitination sites in a single run of 293T cells [11].
Quantitative performance also differs between methods. Ubiquitin tagging approaches show variable labeling efficiency and potential artifacts from tag-induced structural changes [3]. Antibody-based methods, particularly the UbiFast approach, demonstrate high quantitative accuracy with >92% completeness of labeling for K-ε-GG peptides bound to anti-K-ε-GG antibody when using on-antibody TMT labeling [22]. Comparative studies show that on-antibody TMT labeling results in 6,087 K-ε-GG peptide-spectrum matches with a relative yield of 85.7%, while conventional in-solution labeling yields only 1,255 PSMs with a relative yield of 44.2% [22].
Application-Specific Performance
The performance characteristics of each method make them differentially suitable for specific research applications. Ubiquitin tagging excels in controlled cell culture systems where genetic manipulation is feasible and the focus is on specific biological questions rather than comprehensive ubiquitinome mapping. Its straightforward purification workflow and relatively low cost make it ideal for hypothesis-driven research in model cell systems [3].
Antibody-based methods demonstrate superior performance in complex samples and translational applications. Their ability to profile endogenous ubiquitination without genetic manipulation makes them indispensable for tissue samples, clinical specimens, and primary cell cultures [3] [22]. The diGLY proteomics approach has been successfully applied to identify and quantify ubiquitination changes in human breast cancer xenograft tissue samples [22], mouse tissues [4], and skeletal muscle biopsies from people living with amyotrophic lateral sclerosis [4].
For specialized applications like identifying deubiquitinase substrates, innovative integrations such as the proximal-ubiquitome workflow combine APEX2-based proximity labeling with K-ε-GG ubiquitin remnant enrichment to define candidate substrates within the native microenvironment of a DUB [50]. When applied to USP30, this method successfully recovered known substrates and identified new ones like LETM1 [50]. Similarly, for studying rare ubiquitin-like modifications such as UFMylation, specialized antibody-based approaches have been developed, identifying over 200 UFMylation sites from various mouse tissues [4].
Table: Performance Metrics for Ubiquitination Profiling Methods
| Performance Metric | Ubiquitin Tagging | Antibody-Based Enrichment |
|---|---|---|
| Typical Site Identification | 110-750 sites | 5,000-10,000+ sites |
| Starting Material Requirements | 1-10 mg protein from engineered cells | 0.5-10 mg protein from any source |
| Reproducibility | Not typically reported | CV = 2% (AFUP method) |
| Quantitative Accuracy | Variable due to tag effects | >92% labeling efficiency (UbiFast) |
| Tissue Compatibility | Not feasible | Excellent (0.5-7 mg input) |
| Multiplexing Capacity | Limited | High (TMT10/11-plex) |
| Specialized Applications | Basic research in model cell systems | DUB substrate identification, clinical samples, tissue profiling |
Research Reagent Solutions
Successful ubiquitination profiling requires specific research reagents optimized for each methodology. The following table details essential materials and their functions for both major approaches.
Table: Essential Research Reagents for Ubiquitination Profiling
| Reagent Category | Specific Examples | Function and Application |
|---|---|---|
| Ubiquitin Tagging System Components | His×6-tagged ubiquitin, Strep-tag II ubiquitin | Affinity tags for purification of ubiquitinated proteins |
| Ni-NTA agarose, Strep-Tactin beads | Affinity resins for capturing tagged ubiquitin conjugates | |
| MG-132, Lactacystin | Proteasome inhibitors to stabilize ubiquitinated proteins | |
| Denaturing lysis buffers (6M Guanidine-HCl, 8M Urea) | Cell lysis while preserving ubiquitination states | |
| Antibody-Based Enrichment Reagents | Anti-K-ε-GG antibodies (diGLY antibodies) | Immunoaffinity enrichment of ubiquitinated peptides |
| Linkage-specific ubiquitin antibodies (K48, K63, etc.) | Enrichment of specific ubiquitin chain linkages | |
| Protein A/G agarose beads | Antibody immobilization for immunoprecipitation | |
| N-Ethylmaleimide (NEM) | Deubiquitinase inhibitor to preserve ubiquitination | |
| General Proteomics Reagents | Trypsin, Lys-C | Proteolytic enzymes for protein digestion |
| TMT10/11-plex, iTRAQ | Isobaric tags for multiplexed quantitative proteomics | |
| C18 solid-phase extraction cartridges | Peptide desalting and cleanup | |
| High-pH reverse-phase chromatography | Peptide fractionation for enhanced depth | |
| Specialized Tools | Ubiquitin-Trap (Nanobody-based) | Commercial system for ubiquitin and ubiquitinated protein pulldown [49] |
| UbiFast reagents | Optimized system for rapid, sensitive ubiquitinome profiling [22] | |
| AFUP reagents | Antibody-free ubiquitination profiling system [11] |
The comprehensive comparison of ubiquitin tagging versus antibody-based enrichment methods reveals a clear division of applications based on sample type and research objectives. Ubiquitin tagging approaches provide a straightforward, cost-effective solution for ubiquitination profiling in genetically tractable cell systems where comprehensive ubiquitinome coverage is not the primary goal. The method's limitations in tissue applications and potential artifacts from tag-induced structural changes restrict its utility in translational research [3].
Antibody-based methods, particularly diGLY proteomics with advanced implementations like UbiFast, offer superior sensitivity, specificity, and breadth of application across diverse sample types [22]. The ability to profile endogenous ubiquitination in clinical specimens, patient tissues, and primary cells makes this approach indispensable for drug development professionals and translational researchers. The ongoing development of improved antibodies, efficient multiplexing strategies, and specialized workflows for specific biological questions continues to expand the applications of antibody-based ubiquitination profiling [22] [50] [4].
For researchers designing ubiquitination profiling studies, the selection between these methodologies should be guided by sample availability, required coverage depth, and specific biological questions. Cell culture studies focused on specific pathways may benefit from ubiquitin tagging's simplicity, while comprehensive ubiquitinome mapping in complex samples necessitates antibody-based approaches. Future methodological developments will likely focus on reducing input requirements further, improving antibody specificity, and enhancing computational tools for data analysis, ultimately making large-scale ubiquitinome profiling accessible to broader research communities.
Head-to-Head Comparison: Selecting the Right Tool for Your Research Goal
Protein ubiquitination, the covalent attachment of a small regulatory protein to substrate proteins, is a fundamental post-translational modification that governs diverse cellular processes including protein degradation, signal transduction, and DNA repair [3]. To decipher the complex ubiquitin code, researchers employ various enrichment strategies, with ubiquitin tagging and antibody-based enrichment representing two foundational approaches. Ubiquitin tagging involves genetic fusion of an affinity tag to ubiquitin itself, enabling purification of ubiquitinated proteins from cellular systems [3]. In contrast, antibody-based methods utilize antibodies that specifically recognize ubiquitin remnants or ubiquitin chains to isolate modified proteins or peptides [9] [3] [51]. This guide provides an objective comparison of these methodologies, supported by experimental data and detailed protocols, to assist researchers in selecting the appropriate technique for their specific applications in drug development and basic research.
Fundamental Principles and Methodologies
Ubiquitin tagging is a genetic approach where an affinity tag (e.g., His, Strep, or FLAG) is fused to ubiquitin and expressed in cells. This tag allows for the purification of ubiquitin-protein conjugates using affinity resins when the tagged ubiquitin is incorporated onto substrate proteins [3]. The method can be applied to study global ubiquitination or specific ubiquitin linkage types when combined with linkage-specific ubiquitin mutants.
Antibody-based enrichment encompasses several techniques that utilize antibodies to capture ubiquitinated proteins or peptides. This includes: (1) Protein-level enrichment using antibodies recognizing ubiquitin or specific polyubiquitin linkages (e.g., FK1, FK2, or linkage-specific antibodies) [3]; (2) Peptide-level immunoaffinity enrichment using antibodies specific for the di-glycine (K-ε-GG) remnant left on trypsinized peptides from ubiquitinated lysine residues [9] [13] [6]; and (3) Antibodies specific for N-terminal ubiquitination that recognize tryptic peptides with an N-terminal diglycine motif (GGX peptides) [51].
Comprehensive Performance Comparison
Table 1: Direct comparison of ubiquitin tagging versus antibody-based enrichment methods
| Feature | Ubiquitin Tagging | Antibody-Based Enrichment (diGLY) |
|---|---|---|
| Basic Principle | Genetic fusion of affinity tags to ubiquitin for purification [3] | Antibodies recognizing di-glycine remnant on lysine after trypsin digestion [9] |
| Specificity | Enriches all proteins modified with tagged ubiquitin | Site-specific identification of modified lysines [13] |
| Sensitivity | Can identify lower abundance ubiquitination events through protein-level enrichment | High sensitivity for detectable sites; limited by antibody affinity and abundance |
| Throughput Capability | Suitable for medium-throughput studies of ubiquitinated proteins | High-throughput capability; identifies thousands of sites in single experiments [6] |
| Quantitative Capability | Compatible with SILAC, label-free, or isobaric tagging methods [9] [3] | Excellent for quantification using SILAC, TMT, or label-free approaches [9] [6] |
| Linkage Type Information | Requires specific ubiquitin mutants to study particular linkages | Limited inherent linkage information; requires additional methods |
| Endogenous Context | Disrupts endogenous ubiquitin system unless complete replacement strategies used | Preserves endogenous ubiquitination patterns [3] |
| Key Advantages | - Tracks newly synthesized ubiquitin conjugates- Can study ubiquitin dynamics- Relatively low cost after initial setup [3] | - No genetic manipulation required- Works with any biological sample- Unbiased site identification [9] [3] |
| Major Limitations | - Tag may alter ubiquitin structure/function- Cannot completely replace endogenous ubiquitin- Co-purification of non-ubiquitinated proteins [3] | - Cannot distinguish ubiquitination from other UBL modifications- Antibody cost- May miss low-stoichiometry sites [9] [3] |
Table 2: Experimental performance metrics from published studies
| Performance Metric | Ubiquitin Tagging | Antibody-Based Enrichment |
|---|---|---|
| Typical Identification Yield | 110-750 ubiquitination sites depending on system [3] | >10,000 ubiquitination sites possible [9] [6] |
| Enrichment Efficiency | Varies based on tag and expression level | 3-4-fold increase in K-ε-GG peptide recovery compared to non-enriched [13] |
| Reproducibility | Moderate to high, depending on expression consistency | Generally high between technical replicates |
| Sample Requirements | Requires genetic manipulation and cell culture | Works with cell lines, tissues, primary samples [9] |
| Compatibility with Clinical Samples | Not applicable to human tissue without genetic modification | Directly applicable to human tissues and clinical samples [3] |
Experimental Protocols
Ubiquitin Tagging Workflow
The following diagram illustrates the key steps in the ubiquitin tagging approach for identifying ubiquitination sites:
Detailed Protocol Steps:
Generation of Cell Lines Expressing Tagged Ubiquitin: Create cell lines stably expressing His- or Strep-tagged ubiquitin using viral transduction or stable transfection. The StUbEx (Stable Tagged Ubiquitin Exchange) system can be used to replace endogenous ubiquitin with tagged variants [3].
Cell Lysis and Protein Extraction: Harvest cells and lyse in denaturing buffer (e.g., 8M urea, 150mM NaCl, 50mM Tris-HCl, pH 8.0) supplemented with protease inhibitors (e.g., Complete EDTA-free protease inhibitor cocktail) and 5mM N-ethylmaleimide (NEM) to preserve ubiquitination by inhibiting deubiquitinases [9].
Affinity Purification of Ubiquitinated Proteins: Incubate lysates with appropriate affinity resin (Ni-NTA for His-tag, Strep-Tactin for Strep-tag) for several hours at 4°C. Wash extensively with lysis buffer followed by wash buffers of increasing stringency (e.g., containing 20mM imidazole for His-tag purifications) to remove non-specifically bound proteins [3].
Elution and Preparation for MS Analysis: Elute bound proteins using competitive elution (250mM imidazole for His-tag, 2.5mM desthiobiotin for Strep-tag) or direct digestion on beads. Reduce, alkylate, and digest proteins with trypsin/Lys-C following standard proteomic protocols [9].
Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS): Analyze resulting peptides using high-resolution LC-MS/MS. Identify ubiquitination sites by searching for the characteristic 114.0429 Da mass shift on lysine residues corresponding to the di-glycine remnant [3].
Antibody-Based Enrichment Workflow
The following diagram illustrates the key steps in the antibody-based diGLY enrichment approach:
Detailed Protocol Steps:
Sample Preparation and Lysis: Lyse cells or tissues in denaturing lysis buffer (8M urea, 150mM NaCl, 50mM Tris-HCl, pH 8.0) supplemented with protease inhibitors and 5mM NEM to preserve ubiquitination sites. Sonicate to shear DNA and reduce viscosity [9].
Protein Digestion and Peptide Cleanup: Reduce proteins with DTT, alkylate with iodoacetamide, and digest first with LysC (1:100 enzyme:protein) for 3-4 hours at room temperature, then dilute to 2M urea and digest with trypsin (1:50 enzyme:protein) overnight at room temperature. Desalt peptides using SepPak C18 cartridges and dry using vacuum centrifugation [9].
Immunoaffinity Enrichment of diGLY Peptides: Resuspend peptides in immunoaffinity purification (IAP) buffer (50mM MOPS/NaOH, pH 7.3, 10mM Na2HPO4, 50mM NaCl). Incubate with anti-K-ε-GG antibody (commercially available from Cell Signaling Technology) conjugated to protein A/G beads for 1.5-2 hours at 4°C [9] [6]. Wash beads sequentially with IAP buffer, followed by water. Elute peptides with 0.15% trifluoroacetic acid.
LC-MS/MS Analysis: Desalt eluted peptides using StageTips or similar micro-scale purification methods. Analyze by LC-MS/MS using high-resolution instruments. Identify ubiquitination sites by searching for the 114.0429 Da mass shift on lysine residues [9].
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential reagents for ubiquitination studies
| Reagent / Tool | Type | Function & Application | Examples & Sources |
|---|---|---|---|
| Tagged Ubiquitin Plasmids | Genetic tool | Expression of affinity-tagged ubiquitin in cells for ubiquitin tagging approaches | His-Ub, Strep-Ub, HA-Ub [3] |
| diGLY Antibody | Antibody reagent | Enrichment of tryptic peptides with K-ε-GG modification for proteomic studies | PTMScan Ubiquitin Remnant Motif Kit [9] |
| Linkage-Specific Ub Antibodies | Antibody reagent | Detection or enrichment of specific ubiquitin linkage types | K48-linkage specific, K63-linkage specific [3] |
| N-terminal GGX Antibodies | Antibody reagent | Specific enrichment of N-terminal ubiquitination sites | Anti-GGX antibodies (1C7, 2B12, 2E9, 2H2 clones) [51] |
| Ubiquitin Binding Domains (UBDs) | Affinity reagent | Enrichment of ubiquitinated proteins based on ubiquitin binding | OtUBD, TUBEs [52] |
| Deubiquitinase Inhibitors | Chemical tool | Preservation of ubiquitination during sample preparation | N-ethylmaleimide (NEM) [9] |
Application Notes and Decision Framework
Method Selection Guide
Choosing between ubiquitin tagging and antibody-based approaches depends on several factors:
Select ubiquitin tagging when: (1) Studying dynamics of newly synthesized ubiquitin conjugates; (2) Working with systems amenable to genetic manipulation; (3) Budget constraints limit antibody purchases; (4) Interested in tracking ubiquitin pool dynamics [3].
Select antibody-based enrichment when: (1) Working with clinical samples or tissues where genetic manipulation is impossible; (2) Requiring site-specific ubiquitination information; (3) Conducting large-scale quantitative studies across multiple conditions; (4) Studying endogenous ubiquitination without tags [9] [3] [6].
Emerging Applications and Advanced Implementations
Recent methodological advances have expanded applications for both techniques. Ubi-tagging has been repurposed beyond proteomics to create a modular platform for site-directed antibody conjugation, demonstrating utility in generating homogeneous antibody-drug conjugates and bispecific T-cell engagers [1] [8]. This application leverages ubiquitin's enzymatic machinery for precise protein engineering, achieving conjugation efficiencies of 93-96% within 30 minutes [1].
For antibody-based approaches, new reagents have enabled the specific detection of unconventional ubiquitination events. Antibodies targeting N-terminal ubiquitination (GGX antibodies) have revealed UBE2W substrates and functional roles for N-terminal ubiquitination in regulating deubiquitinase activity [51]. Additionally, specialized antibodies have been developed for studying other ubiquitin-like modifications, such as UFM1, with the creation of anti-VG-ε-K antibodies for UFMylation site mapping [4].
Integrated approaches using tandem enrichment methods now enable simultaneous analysis of multiple post-translational modifications (ubiquitination, phosphorylation, glycosylation) from single samples, maximizing information from precious clinical specimens [23]. These developments highlight the evolving applications of ubiquitin enrichment technologies beyond traditional proteomic mapping.
Protein ubiquitination, a fundamental post-translational modification, regulates virtually all cellular processes in eukaryotes, including protein degradation, DNA repair, cell signaling, and immune response [3] [12]. This modification involves the covalent attachment of ubiquitin—a small 76-amino acid protein—to substrate proteins via a complex enzymatic cascade. The complexity of ubiquitin signaling arises from its ability to form diverse chain architectures through different linkage types, leading to distinct functional outcomes [3]. To decipher this "ubiquitin code," researchers primarily rely on two powerful enrichment strategies: ubiquitin tagging and antibody-based enrichment. Each method presents a unique balance between throughput—the rate of successful data generation—and the risk of introducing analytical artifacts that can compromise data integrity. This guide objectively compares these methodologies, providing researchers with the experimental data and context needed to select the appropriate approach for their specific investigations in basic research or drug development.
Methodological Foundations: Core Principles and Protocols
Ubiquitin Tagging (Affinity Tag-Based Enrichment)
The ubiquitin tagging method involves genetically engineering cells to express ubiquitin fused to an affinity tag, such as poly-histidine (His) or Strep-tag II [3] [53]. When incorporated into ubiquitinated substrates, these tags enable purification under denaturing conditions using corresponding affinity resins—Ni-NTA for His-tags or Strep-Tactin for Strep-tags [3]. Following enrichment and tryptic digestion, mass spectrometry (MS) analysis identifies ubiquitination sites by detecting the characteristic 114.04 Da mass shift left on modified lysine residues [3].
Detailed Experimental Protocol (His-Tagged Ubiquitin):
- Cell Engineering: Generate a stable cell line expressing N- or C-terminally His-tagged ubiquitin under an appropriate promoter.
- Cell Lysis and Protein Extraction: Lyse cells in a denaturing buffer (e.g., 6 M guanidine-HCl or 8 M urea, pH 8.0) to preserve ubiquitination states and inactivate deubiquitinating enzymes.
- Affinity Purification: Incubate the clarified lysate with Ni-NTA agarose beads. Wash extensively with denaturing buffer containing progressively lower imidazole concentrations (e.g., 20-50 mM) to reduce non-specific binding.
- On-Bead Digestion: Wash beads with a non-denaturing buffer. Digest enriched proteins on-bead with trypsin or Lys-C overnight.
- Peptide Clean-up: Desalt the resulting peptides using C18 solid-phase extraction cartridges.
- LC-MS/MS Analysis: Analyze peptides via liquid chromatography-tandem mass spectrometry (LC-MS/MS) to identify and localize ubiquitination sites.
The Stable Tagged Ub Exchange (StUbEx) system represents an advanced application of this approach, enabling near-complete replacement of endogenous ubiquitin with the tagged version for more comprehensive ubiquitome coverage [3].
Antibody-Based Enrichment (DiGly Antibody Approach)
This methodology leverages a well-characterized antibody that specifically recognizes the di-glycine (diGly) remnant left on tryptic peptides following ubiquitination [12]. Unlike tagging strategies, this approach captures endogenous ubiquitination events without genetic manipulation, making it suitable for clinical samples and tissues.
Detailed Experimental Protocol (DiGly Enrichment):
- Sample Preparation: Lyse cells or tissues in a suitable buffer (e.g., 8 M urea, 50 mM Tris-HCl, pH 8.0) with protease and deubiquitinase inhibitors.
- Protein Digestion: Reduce proteins with dithiothreitol (5 mM, 45 min), alkylate with iodoacetamide (10 mM, 30 min in dark), and digest first with Lys-C (2-4 hours) followed by trypsin (overnight).
- Peptide Clean-up: Desalt digested peptides using C18 Sep-Pak cartridges and dry via vacuum centrifugation.
- Immunoaffinity Enrichment: Reconstitute peptides in immunoaffinity purification (IAP) buffer. Incubate with anti-diGly antibody (e.g., PTMScan Ubiquitin Remnant Motif Kit, Cell Signaling Technology) coupled to protein A/G beads for 1-2 hours at 4°C.
- Wash and Elution: Wash beads extensively with IAP buffer and then with water. Elute diGly-modified peptides with 0.15% trifluoroacetic acid.
- LC-MS/MS Analysis: Analyze the enriched peptides via LC-MS/MS.
Recent automation advances, such as the UbiFast method, utilize magnetic bead-conjugated K-ε-GG antibodies and magnetic particle processors to significantly increase reproducibility and throughput, enabling processing of up to 96 samples in a single day [14].
Comparative Performance Analysis: Throughput and Artifacts
Quantitative Performance Metrics
Table 1: Throughput and Coverage Comparison of Ubiquitin Enrichment Methods
| Performance Metric | Ubiquitin Tagging (His-Tag) | Antibody-Based Enrichment (DiGly) |
|---|---|---|
| Typical Identifications (Sites) | ~277-753 sites [3] | ~20,000-35,000+ sites in single measurements [14] [12] |
| Sample Multiplexing Capacity | Limited by genetic manipulation requirements | High (TMT10-plex demonstrated) [14] |
| Processing Time | Days to weeks (includes cell line generation) | <2 hours for automated enrichment of 10-plex [14] |
| Input Material Requirement | Variable, dependent on expression efficiency | As low as 500 μg per sample [14] |
| Suitability for High-Throughput Screening | Low | High, especially with automated platforms |
| Identification Efficiency | Relatively low [3] | High with optimized DIA methods [12] |
Artifacts and Technical Limitations
Table 2: Artifacts and Limitations of Ubiquitin Enrichment Methods
| Artifact Type | Ubiquitin Tagging | Antibody-Based Enrichment |
|---|---|---|
| Non-Native Structure Impacts | Tagged Ub may not completely mimic endogenous Ub, potentially generating artifacts [3] | Minimal impact on native ubiquitination |
| Co-purifying Contaminants | Histidine-rich or endogenously biotinylated proteins co-purify with His/Strep-tags [3] | Some non-specific antibody binding occurs [3] |
| Sample Compatibility | Limited to genetically manipulable systems; infeasible for patient tissues [3] | Compatible with any biological sample, including clinical specimens [3] |
| Linkage Recognition | Enriches all linkage types non-specifically | Potential linkage bias with some antibodies; newer platforms (ThUBD) address this [2] |
| Stoichiometry Preservation | May alter endogenous ubiquitination dynamics | Preserves native ubiquitination stoichiometry |
| Key Limitations | - Cannot be applied to tissue samples- Potential alteration of Ub structure- Lower identification efficiency [3] | - High-cost antibodies- Non-specific binding- Potential linkage bias with conventional antibodies [3] |
Decision Framework for Ubiquitin Enrichment Method Selection
Advanced Applications and Implementation Tools
Research Reagent Solutions
Table 3: Essential Research Reagents for Ubiquitination Studies
| Reagent / Tool | Function | Application Notes |
|---|---|---|
| HS mag anti-K-ε-GG antibody | Magnetic bead-conjugated antibody for automated ubiquitinated peptide enrichment | Enables processing of 96 samples in <2 hours; 16-fold sensitivity improvement over TUBE technology [14] [2] |
| ThUBD-coated 96-well plates | High-throughput platform with unbiased ubiquitin chain recognition | Captures ~5 pmol of polyubiquitin chains; ideal for PROTAC development and dynamic ubiquitination monitoring [2] |
| Tandem Mass Tag (TMT) Reagents | Isobaric labeling for multiplexed sample analysis | Enables precise relative quantification of up to 18 samples simultaneously; requires on-antibody labeling for diGly peptides [14] [12] |
| UbiFast Methodology | Automated workflow for ubiquitination site profiling | Combines anti-K-ε-GG enrichment with on-antibody TMT labeling; identifies ~20,000 sites from 500 μg input [14] |
| Data-Independent Acquisition (DIA) | MS method for comprehensive peptide fragmentation | Provides greater data completeness; identifies 35,000+ diGly sites in single measurements [12] |
| Linkage-Specific Antibodies | Enrich ubiquitinated proteins with specific chain linkages (M1, K48, K63, etc.) | Essential for studying chain-type specific functions; limited by potential bias and availability [3] |
Technological Advancements Addressing Limitations
Recent innovations have specifically targeted the traditional limitations of both enrichment methods. For antibody-based approaches, the development of Tandem Hybrid Ubiquitin Binding Domain (ThUBD) technology has addressed linkage bias concerns by providing unbiased, high-affinity recognition of all ubiquitin chain types while offering a 16-fold wider linear range for capturing polyubiquitinated proteins compared to previous TUBE technologies [2]. Furthermore, the implementation of data-independent acquisition (DIA) mass spectrometry has dramatically improved the sensitivity and reproducibility of ubiquitinome analyses, enabling identification of over 35,000 distinct diGly sites in single measurements with 45% of peptides showing coefficients of variation below 20% [12]. This represents a substantial improvement over data-dependent acquisition methods, which typically identify approximately 20,000 diGly peptides with only 15% showing comparable reproducibility [12].
For ubiquitin tagging approaches, the StUbEx (Stable Tagged Ub Exchange) system represents a significant advancement, enabling near-complete replacement of endogenous ubiquitin with tagged variants for more comprehensive ubiquitome mapping without competition from native ubiquitin [3]. However, this system still faces the fundamental limitation of not being applicable to clinical or tissue samples where genetic manipulation is infeasible.
High-Throughput Ubiquitinome Analysis Using Automated Antibody-Based Workflow
The choice between ubiquitin tagging and antibody-based enrichment methods represents a critical strategic decision in ubiquitination research that directly impacts data quality, throughput, and biological relevance. Ubiquitin tagging approaches may still offer utility in specialized applications where genetic manipulation is feasible and the research question benefits from controlled expression systems. However, antibody-based enrichment, particularly when implemented with automated platforms like UbiFast and advanced mass spectrometry techniques like DIA, provides substantially superior throughput, reproducibility, and coverage for most research applications [14] [12]. The dramatically higher identification rates (35,000+ vs. <1,000 sites), compatibility with clinical samples, and minimal structural artifacts make antibody-based methods the preferred choice for comprehensive ubiquitinome mapping, particularly in translational research and drug development contexts where monitoring endogenous ubiquitination dynamics is essential. As technologies continue to evolve, particularly with the development of unbiased binding domains like ThUBD and increasingly automated workflows, antibody-based approaches are poised to remain the gold standard for high-throughput ubiquitination analysis while effectively managing artifact-related concerns.
Protein ubiquitination is a crucial post-translational modification that regulates nearly all cellular processes in eukaryotes, with dysregulation contributing to cancer, neurodegenerative disorders, and other human diseases [54] [55]. The ubiquitin-proteasome system (UPS) involves a sophisticated enzymatic cascade that conjugates the 76-amino acid ubiquitin protein to substrate proteins, creating a complex "ubiquitin code" that dictates diverse cellular outcomes [55] [3]. During the past decade, mass spectrometry (MS)-based proteomics has emerged as an indispensable approach for identifying ubiquitinated proteins, modification sites, and ubiquitin chain architectures [54] [3].
Two primary methodological approaches have dominated the field for enriching and detecting ubiquitinated proteins: ubiquitin tagging (Ub tagging) and antibody-based enrichment. Ub tagging involves engineering cells to express affinity-tagged ubiquitin (e.g., His, Strep, or FLAG tags), enabling purification of ubiquitinated substrates [3]. Antibody-based approaches utilize antibodies that recognize either the diglycine (K-ɛ-GG) remnant left on trypsinized ubiquitinated peptides or specific ubiquitin chain linkages [22] [56]. Understanding the quantitative accuracy and reproducibility of these competing methodologies is essential for researchers investigating ubiquitin signaling pathways in both basic biology and drug discovery contexts.
This guide provides an objective comparison of ubiquitin tagging versus antibody-based enrichment methods, focusing on data quality assessment through systematic evaluation of experimental data, protocols, and performance metrics.
Ubiquitin Tagging-Based Approaches
Ubiquitin tagging employs genetic engineering to express affinity-tagged ubiquitin in living cells, enabling covalent labeling and subsequent purification of ubiquitinated substrates [3]. The tagged ubiquitin is incorporated into the endogenous ubiquitination machinery, labeling substrates that can be enriched using commercially available resins such as Ni-NTA for His tags or Strep-Tactin for Strep tags [3]. Following purification and tryptic digestion of ubiquitinated proteins, mass spectrometry identifies ubiquitination sites through detection of a characteristic 114.04 Da mass shift on modified lysine residues [3].
Peng et al. pioneered this approach in 2003 by expressing 6× His-tagged ubiquitin in Saccharomyces cerevisiae, identifying 110 ubiquitination sites on 72 proteins [3]. Subsequent developments include the stable tagged Ub exchange (StUbEx) cellular system, which replaces endogenous ubiquitin with His-tagged ubiquitin, enabling identification of 277 unique ubiquitination sites on 189 proteins in HeLa cells [3]. Similarly, Danielsen et al. constructed a cell line stably expressing Strep-tagged ubiquitin, identifying 753 lysine ubiquitylation sites on 471 proteins in U2OS and HEK293T cells [3].
Antibody-Based Enrichment Approaches
Antibody-based methodologies utilize ubiquitin-specific antibodies to enrich ubiquitinated proteins or peptides without genetic manipulation. Two primary strategies exist: (1) enrichment at the protein level using antibodies that recognize ubiquitin or specific ubiquitin chain linkages, and (2) immunoaffinity purification of tryptic peptides containing the K-ɛ-GG remnant [3] [22] [56].
The breakthrough in antibody-based approaches came with developing antibodies that specifically recognize the diglycyl remnant (K-ɛ-GG) left on lysine residues after tryptic digestion of ubiquitinated proteins [22] [56]. This enabled comprehensive profiling of ubiquitination sites by LC-MS/MS and facilitated the development of highly multiplexed quantification methods like the UbiFast protocol, which can quantify approximately 10,000 ubiquitylation sites from as little as 500 μg peptide per sample [22]. Linkage-specific antibodies have also been generated that recognize particular ubiquitin chain types (M1-, K11-, K27-, K48-, K63-linkage specific antibodies), enabling research into the functional consequences of specific ubiquitin linkages [3] [57].
Ubiquitin-Binding Domain (UBD) Based Approaches
A third category employs ubiquitin-binding domains (UBDs)—protein domains that naturally recognize and bind ubiquitin—as affinity reagents. Tandem-repeated UBDs demonstrate higher affinity than single domains and can be engineered for improved performance [3] [2]. The Tandem Hybrid Ubiquitin Binding Domain (ThUBD) represents an advanced version that combines different UBD types to achieve unbiased, high-affinity capture of all ubiquitin chain types [2].
This approach has been adapted to high-throughput formats, with ThUBD-coated 96-well plates demonstrating 16-fold wider linear range for capturing polyubiquitinated proteins compared to Tandem Ubiquitin Binding Entity (TUBE)-coated plates [2]. The technology enables sensitive detection of global ubiquitination profiles and target-specific ubiquitination status, providing robust technical support for developing Proteolysis-Targeting Chimeras (PROTACs) and other ubiquitin-focused therapeutics [2].
Quantitative Performance Comparison
Table 1: Overall Method Performance Comparison
| Performance Metric | Ubiquitin Tagging | Antibody-Based Enrichment | UBD-Based Approaches |
|---|---|---|---|
| Identification Depth | 110-753 ubiquitination sites [3] | ~10,000 ubiquitylation sites with UbiFast [22] | Not specifically quantified in search results |
| Sample Requirement | Requires genetic manipulation | 500 μg peptide per sample for UbiFast [22] | As low as 0.625 μg for ThUBD plates [2] |
| Quantitative Accuracy | Potential artifacts from tagged Ub expression [3] | >92% labeling efficiency with on-antibody TMT [22] | 16-fold wider linear range vs TUBE [2] |
| Experimental Throughput | Lower throughput due to cell line generation | High-throughput: ~5 hours for TMT10plex [22] | High-throughput: 96-well plate format [2] |
| Linkage Specificity | Limited to general ubiquitination detection | Enabled by linkage-specific antibodies [3] | Unbiased recognition of all chain types [2] |
Table 2: Sensitivity and Specificity Comparison
| Method | Relative Yield | Labeling Efficiency | Dynamic Range | Key Limitations |
|---|---|---|---|---|
| His-Tag Ub Tagging | Not specifically reported | Not applicable | Not specifically reported | Co-purification of histidine-rich proteins; cannot be used in tissues [3] |
| Strep-Tag Ub Tagging | Not specifically reported | Not applicable | Not specifically reported | Co-purification of endogenously biotinylated proteins [3] |
| On-Antibody TMT (UbiFast) | 85.7% [22] | >92% [22] | Not specifically reported | High antibody cost; non-specific binding [3] |
| In-Solution TMT Labeling | 44.2% [22] | 98% (but lower yield) [22] | Not specifically reported | Requires large sample amounts (1-7 mg) [22] |
| ThUBD-Coated Plates | Not specifically reported | Not specifically reported | 16-fold wider than TUBE plates [2] | Specialized reagent requirement [2] |
Experimental Protocols
Ubiquitin Tagging Workflow
The ubiquitin tagging protocol involves multiple stages with critical quality control checkpoints:
- Cell Line Generation: Engineer cells to stably express tagged ubiquitin (His, Strep, or FLAG tags). For the StUbEx system, endogenous ubiquitin is replaced with His-tagged ubiquitin [3].
- Cell Lysis and Protein Extraction: Harvest cells and lyse using appropriate buffers containing protease and deubiquitinase inhibitors to preserve ubiquitination status.
- Affinity Purification: Incubate cell lysates with affinity resin (Ni-NTA for His tags, Strep-Tactin for Strep tags).-
- Stringent Washing: Wash resins with buffers containing low concentrations of imidazole (for His tags) or desthiobiotin (for Strep tags) to reduce non-specific binding.
- Elution: Elute ubiquitinated proteins using high-concentration imidazole (His tags), biotin (Strep tags), or competitive elution.
- Trypsin Digestion: Digest purified proteins with trypsin, which cleaves ubiquitin but leaves the C-terminal Gly-Gly remnant attached to modified lysines.
- LC-MS/MS Analysis: Analyze resulting peptides using capillary liquid chromatography-tandem mass spectrometry.
Critical Quality Controls:
- Verify tagged ubiquitin expression levels by immunoblotting
- Monitor purification efficiency through comparison of input, flow-through, and eluate fractions
- Include negative controls with non-tagged ubiquitin cells
UbiFast Protocol for Antibody-Based Enrichment
The UbiFast method enables highly multiplexed quantification of ubiquitylation sites with minimal sample input:
- Protein Extraction and Digestion: Extract proteins from cells or tissues and digest with trypsin to generate peptides with K-ɛ-GG remnants [22].
- Peptide-Level Enrichment: Incubate peptides with anti-K-ɛ-GG antibody beads to enrich ubiquitinated peptides [22].
- On-Antibody TMT Labeling: While peptides are bound to antibodies, label with Tandem Mass Tag (TMT) reagents (0.4 mg TMT reagent for 10 minutes). The diglycyl remnant is protected from labeling when bound to the antibody [22].
- Reaction Quenching: Add 5% hydroxylamine to quench the TMT reaction [22].
- Peptide Elution and Combination: Elute TMT-labeled K-ɛ-GG peptides from different samples and combine them.
- LC-MS Analysis: Analyze using single-shot, high-performance LC-MS/MS with High-field Asymmetric Waveform Ion Mobility Spectrometry (FAIMS) to improve quantitative accuracy [22].
Optimization Parameters:
- 10-minute labeling with 0.4 mg TMT reagent provides optimal balance of identification and labeling efficiency
- Quenching with 5% hydroxylamine increases K-ɛ-GG peptide identification by almost 10%
- FAIMS interface improves quantitative accuracy for post-translational modification analysis
ThUBD-Coated Plate Assay
The ThUBD-coated plate platform enables high-throughput detection of ubiquitination signals:
- Plate Coating: Coat Corning 3603-type 96-well plates with 1.03 μg ± 0.002 of ThUBD fusion protein [2].
- Sample Application: Apply complex proteome samples to plates and incubate to allow binding of ubiquitinated proteins.
- Stringent Washing: Wash with optimized buffers to remove non-specifically bound proteins.
- Detection: Detect captured ubiquitinated proteins using ThUBD-HRP conjugates or target-specific antibodies [2].
- Quantification: Measure signal intensity and compare to standards for quantification.
Performance Characteristics:
- Specific binding capacity of approximately 5 pmol polyubiquitin chains
- 16-fold wider linear range compared to TUBE-based plates
- Suitable for dynamic monitoring of ubiquitination in PROTAC drug development [2]
Research Reagent Solutions
Table 3: Essential Research Reagents for Ubiquitination Studies
| Reagent Category | Specific Examples | Function and Application | Key Considerations |
|---|---|---|---|
| Affinity Tags | 6× His-tag, Strep-tag, FLAG-tag | Purification of ubiquitinated proteins from engineered cells | His-tag co-purifies histidine-rich proteins; Strep-tag co-purifies biotinylated proteins [3] |
| Ubiquitin Antibodies | Anti-K-ɛ-GG, P4D1, FK1/FK2, linkage-specific antibodies | Enrichment and detection of ubiquitinated proteins/peptides | Linkage-specific antibodies enable research on specific ubiquitin signaling functions [3] [22] |
| UBD-Based Reagents | TUBE, ThUBD, Ubiquitin Binding Entities | Unbiased capture of polyubiquitinated proteins | ThUBD shows no linkage bias and higher affinity than TUBE [2] |
| Enzymatic Tools | E1 activating, E2 conjugating, E3 ligating enzymes | In vitro ubiquitination and ubiquitin chain assembly | E2 and E3 enzymes can be fused to increase ligation activity [1] |
| Mass Spec Standards | SILAC, TMT, iTRAQ reagents | Quantitative comparison of ubiquitination across conditions | On-antibody TMT labeling prevents derivatization of di-glycyl remnant [22] |
| Deubiquitinase Inhibitors | PR-619, N-ethylmaleimide | Preservation of ubiquitination states during extraction | Essential for maintaining ubiquitin signals during sample preparation |
Workflow Visualization
Diagram 1: Experimental workflow comparison of major ubiquitin enrichment methodologies
Diagram 2: UbiFast protocol workflow with critical quality control checkpoints
The comparative analysis of ubiquitin tagging versus antibody-based enrichment methods reveals a complex landscape where methodological selection significantly impacts data quality, reproducibility, and biological relevance. Ubiquitin tagging approaches provide a straightforward mechanism for ubiquitinated protein purification but suffer from limitations including potential structural artifacts from tagged ubiquitin expression, co-purification of non-specifically bound proteins, and incompatibility with native tissue samples [3]. Antibody-based methods, particularly the recently developed UbiFast protocol, offer superior sensitivity, specificity, and throughput, enabling quantification of approximately 10,000 ubiquitylation sites from minimal sample input [22]. UBD-based approaches present a promising alternative with unbiased ubiquitin chain recognition and adaptability to high-throughput screening formats, showing particular utility in drug discovery applications such as PROTAC development [2].
For researchers requiring analysis of engineered cell systems, ubiquitin tagging remains a viable option, though careful controls must be implemented to address potential artifacts. For native tissue samples, clinical specimens, or studies requiring deep ubiquitinome coverage, antibody-based enrichment—particularly the UbiFast method—delivers superior quantitative accuracy and reproducibility. UBD-based technologies offer compelling advantages for screening applications and target validation studies where linkage-independent ubiquitin capture is desirable. As ubiquitin research continues to evolve toward more physiological models and translational applications, methodological selection should be guided by specific research questions, sample availability, and required data quality thresholds.
Protein ubiquitylation is one of the most prevalent post-translational modifications (PTMs), exerting critical regulatory control over nearly every cellular, physiological, and pathophysiological process [9]. This modification involves the covalent attachment of the small protein ubiquitin to substrate proteins, typically marking them for proteasomal degradation or altering their function, localization, and activity [9]. To study and harness this powerful biological system, researchers have developed two principal technological approaches: antibody-based enrichment methods for analytical profiling of endogenous ubiquitylation, and ubiquitin tagging (ubi-tagging) for engineering protein conjugates with therapeutic and diagnostic potential.
Antibody-based enrichment leverages antibodies specific to the diglycine (diGLY) remnant left on trypsinized peptides from ubiquitylated proteins, enabling system-wide identification of ubiquitylation sites through mass spectrometry [9] [22]. This approach has become indispensable for basic research and translational studies, allowing profiling from cells and primary tissues. In contrast, ubi-tagging repurposes the enzymatic ubiquitination cascade—using E1, E2, and E3 enzymes—for the site-directed, multivalent conjugation of antibodies to various payloads such as fluorescent dyes, peptides, and nanobodies [1] [8]. This synthetic biology approach facilitates the rapid generation of homogeneous antibody conjugates for therapeutic and diagnostic applications.
The following comparison guide objectively evaluates the performance of these two methodologies against key operational and application-specific parameters, providing researchers with a framework for selecting the appropriate tool for their experimental or development goals.
Performance Comparison Table
The table below summarizes the core characteristics and performance metrics of antibody-based enrichment and ubi-tagging.
Table 1: Performance comparison of antibody-based enrichment and ubi-tagging methods
| Parameter | Antibody-based Enrichment (diGLY) | Ubi-tagging |
|---|---|---|
| Primary Application | Analytical profiling of endogenous ubiquitylation sites [9] | Engineering defined protein conjugates (e.g., antibodies, nanobodies) [1] |
| Mechanism of Action | Immunoaffinity enrichment of diGLY-modified peptides after trypsinization [9] [22] | Enzymatic conjugation via ubiquitin ligase cascade [1] |
| Typical Sample Input | 500 µg - 1 mg of peptide per sample [22] | ~10 µM of recombinant ubi-tagged protein [1] |
| Throughput / Reaction Time | Several hours to days (including digestion, enrichment, and MS analysis) [9] | ~30 minutes for conjugation [1] [8] |
| Key Performance Metrics | >10,000 ubiquitylation sites identified from 500 µg peptide [22]; ~85% relative yield of diGLY peptides [22] | 93-96% conjugation efficiency; defined product formation [1] [8] |
| Genetic Manipulation Required | No (for endogenous protein analysis) | Yes (for generating ubi-tagged proteins) [1] |
| Compatibility with Clinical/Tissue Samples | Yes (e.g., patient-derived xenografts) [22] | Primarily for production of therapeutics/diagnostics; not for direct tissue analysis |
Experimental Protocols and Workflows
Protocol for Antibody-Based diGLY Enrichment and Mass Spectrometry
The following detailed protocol is adapted from established methods for ubiquitin diGLY proteomics [9].
1. Cell Culture and Lysis
- Culture cells in SILAC (Stable Isotope Labeling by Amino acids in Cell culture) media for quantitative comparisons if desired [9].
- Lyse cells in a buffer containing 8M Urea, 150mM NaCl, 50mM Tris-HCl (pH 8), supplemented with protease inhibitors (e.g., Complete Protease Inhibitor) and phosphatase inhibitors (1mM NaF, 1mM β-glycerophosphate, 1mM Sodium Orthovanadate) [9].
- Critical Step: Include 5mM N-Ethylmaleimide (NEM) in the lysis buffer, prepared fresh in ethanol, to inhibit deubiquitinases and preserve the ubiquitin modification [9].
2. Protein Digestion and Peptide Cleanup
- Digest proteins first with LysC (e.g., 0.005 AU/µL) followed by trypsin (e.g., 0.1 mg/mL) [9].
- Desalt the resulting peptides using a C18 reverse-phase column (e.g., SepPak tC18) [9].
3. diGLY Peptide Immunoaffinity Enrichment
- Incubate the peptide mixture with an anti-K-ɛ-GG antibody (e.g., PTMScan Ubiquitin Remnant Motif Kit) to enrich for ubiquitin remnant peptides [9].
- Wash the beads extensively to remove non-specifically bound peptides.
4. On-Antibody TMT Labeling for Multiplexing (UbiFast Method)
- While peptides are bound to the antibody, label them with Tandem Mass Tag (TMT) reagents. This innovative approach protects the diGLY motif from derivatization, allowing for subsequent enrichment [22].
- Optimized Condition: Use 0.4 mg of TMT reagent and a 10-minute labeling reaction, achieving >92% labeling completeness [22].
- Quench the reaction with 5% hydroxylamine [22].
5. Mass Spectrometry Analysis
- Elute the TMT-labeled diGLY peptides from the antibody.
- Analyze by LC-MS/MS. The use of High-field Asymmetric Waveform Ion Mobility Spectrometry (FAIMS) can improve quantitative accuracy [22].
This workflow enables the identification and quantification of thousands of endogenous ubiquitylation sites from limited sample material, making it suitable for clinical and tissue samples [22].
Protocol for Site-Directed Conjugation via Ubi-Tagging
This protocol outlines the steps for generating site-specific antibody conjugates using the ubi-tagging platform [1].
1. Generation of Ubi-Tagged Proteins
- For Fab' Fragments, Antibodies, or Nanobodies: Genetically fuse a ubiquitin tag to the protein of interest. This can be achieved via CRISPR/HDR genomic engineering in hybridomas or by transient expression [1] [8].
- Design of Ubi-Tags:
- Donor Ubi-tag (Ubdon): This tag should have a free C-terminal glycine but contain a point mutation (e.g., K48R) in the internal lysine used for conjugation to prevent homodimerization and uncontrolled polymerization [1].
- Acceptor Ubi-tag (Ubacc): This tag should contain the corresponding conjugation lysine (e.g., K48) but have an unreactive C-terminus, achieved by removing the di-glycine motif (ΔGG) or blocking it with a His-tag or molecular cargo [1].
2. Conjugation Reaction
- Combine the following components in a reaction mixture:
- Donor protein (e.g., Fab-Ub(K48R)don) at 10 µM.
- Fivefold molar excess of acceptor module (e.g., Rho-Ubacc-ΔGG carrying a fluorescent dye) at 50 µM.
- Ubiquitination enzymes: 0.25 µM E1 (ubiquitin-activating enzyme) and 20 µM of a linkage-specific E2-E3 fusion protein (e.g., gp78RING-Ube2g2 for K48 linkage) [1].
- Incubate the reaction for 30 minutes at room temperature.
3. Purification and Validation
- Purify the conjugate, for instance, using protein G affinity purification for antibody-based constructs [1].
- Validate the product using techniques like SDS-PAGE and ESI-TOF mass spectrometry to confirm homogeneity and correct mass [1].
- Functional Validation: Test the functionality of the conjugate (e.g., antigen binding via flow cytometry) and its stability (e.g., thermal unfolding assays) [1].
This streamlined protocol allows for the rapid and efficient production of homogeneous, site-specifically modified protein conjugates.
Visualized Workflows and Signaling Pathways
The following diagrams illustrate the core workflows and logical relationships for the two methods.
Antibody-based diGLY Proteomics Workflow
Diagram 1: Analytical workflow for diGLY proteomics.
Ubi-tagging Conjugation Mechanism
Diagram 2: Engineering workflow for ubi-tagging.
The Scientist's Toolkit: Key Research Reagent Solutions
The table below lists essential reagents and their functions for implementing the described methodologies.
Table 2: Essential reagents for ubiquitin profiling and conjugation studies
| Reagent / Tool | Function / Application | Key Features / Considerations |
|---|---|---|
| Anti-K-ɛ-GG Antibody [9] [22] | Immunoaffinity enrichment of diGLY-modified peptides for MS. | Specific for ubiquitin remnant; enables identification of >10,000 sites. |
| Tandem Mass Tag (TMT) Reagents [22] | Multiplexed quantitative proteomics; used in UbiFast on-antibody labeling. | Allows comparison of up to 18 samples; increases throughput and reduces missing data. |
| Ubiquitin Remnant Motif (K-ɛ-GG) Kit [9] | All-in-one solution for diGLY peptide enrichment. | Includes antibodies and protocols for standardized enrichment. |
| Recombinant Ubiquitination Enzymes (E1, E2-E3) [1] | Catalyzes site-specific conjugation in ubi-tagging. | Linkage-specific control (e.g., K48-specific gp78RING-Ube2g2). |
| Ubi-tagged Constructs [1] [8] | Serve as substrates for conjugation (donor or acceptor). | Generated via CRISPR/HDR or transient expression; designed to prevent homodimerization. |
| Synthetic Ubiquitin Derivatives [1] [8] | Provide chemically defined acceptor modules (e.g., with dyes, peptides). | Enables attachment of diverse cargo, expanding application scope. |
The analysis of protein ubiquitination is fundamental to advancing our understanding of cellular regulation and disease mechanisms. Researchers primarily rely on two powerful methodologies to capture and study this complex post-translational modification: Ubiquitin (Ub) tagging and antibody-based enrichment. Ub tagging involves genetically engineering cells to express ubiquitin fused to an affinity tag, which allows for the purification of ubiquitinated substrates [3]. In contrast, antibody-based methods utilize specific antibodies, either pan-specific or linkage-specific, to immunopurify endogenous ubiquitinated proteins directly from biological samples [3]. Each approach offers distinct advantages and faces specific challenges regarding specificity, applicability to physiological conditions, and technical artifacts. This guide provides an objective, data-driven comparison of these techniques to inform method selection for proteomic studies aimed at deciphering the ubiquitin code.
Performance Comparison: Ub Tagging vs. Antibody-Based Enrichment
The choice between ubiquitin tagging and antibody-based enrichment involves trade-offs between specificity, physiological relevance, and practical experimental constraints. The table below summarizes a direct comparison of their core performance characteristics based on current methodologies.
Table 1: Comparative Analysis of Ubiquitin Enrichment Techniques
| Feature | Ub Tagging-Based Approaches | Antibody-Based Approaches |
|---|---|---|
| Basic Principle | Expression of affinity-tagged Ub (e.g., His, Strep) in cells to label substrates [3] | Use of anti-Ub antibodies (e.g., P4D1, FK1/FK2) to immunopurify endogenous ubiquitinated proteins [3] |
| Specificity & Background | Can co-purify histidine-rich or endogenously biotinylated proteins, leading to background [3] | Potential for non-specific antibody binding, which can co-enrich non-ubiquitinated proteins [3] |
| Physiological Relevance | May generate artifacts as tagged Ub cannot perfectly mimic endogenous Ub behavior [3] | Captures proteins under native, physiological conditions without genetic manipulation [3] |
| Sample Applicability | Not feasible for use with animal or patient tissue samples [3] | Directly applicable to a wide range of samples, including animal tissues and clinical specimens [3] |
| Linkage-Type Analysis | Requires the use of linkage-specific Ub mutants [1] | Enabled by the availability of linkage-specific antibodies (e.g., for K48, K63) [3] |
| Cost & Accessibility | Relatively low-cost and user-friendly for cellular systems [3] | High cost associated with high-quality antibodies; accessible for most sample types [3] |
Experimental Data and Workflows
Ubi-Tagging Conjugation Efficiency and Validation
The emerging "ubi-tagging" technique demonstrates the capabilities of modern Ub tagging. This method uses an enzymatic cascade (E1, E2, E3) to achieve rapid, site-directed conjugation of payloads to ubi-tagged antibodies or nanobodies. Experimental data shows this reaction achieves a high conjugation efficiency of 93–96% for ubi-tagged antibodies and completes within 30 minutes [1].
Functional validation is critical. In one experiment, a fluorescently labeled Fab fragment generated via ubi-tagging (Rho-Ub2-Fab) was used to stain CD3+ mouse splenocytes. Flow cytometry analysis confirmed that the staining percentage was comparable to that of a fluorescein isothiocyanate (FITC)-labelled parental antibody, demonstrating that the ubi-tagging process did not hinder antigen-binding functionality [1]. Furthermore, thermal unfolding assays showed the conjugated product maintained stability, with an infliction temperature of approximately 75 °C, identical to the unconjugated Fab [1].
Side-by-Side Protocol Comparison
Detailed protocols highlight the practical steps and requirements for each method, from sample preparation to mass spectrometric analysis.
Table 2: Comparison of Experimental Protocols
| Protocol Step | Ub Tagging Method | Antibody-Based Method |
|---|---|---|
| Sample Preparation | Genetic manipulation of cells to stably express tagged-Ub (e.g., His- or Strep-tag) [3] | Protein extraction from native, unmodified cells or tissues [3] |
| Enrichment | Affinity purification using Ni-NTA (for His-tag) or Strep-Tactin (for Strep-tag) resins [3] | Immunoprecipitation using anti-Ub antibodies (e.g., FK2) immobilized on beads [3] |
| Downstream Processing | On-bead or solution digestion of enriched proteins with trypsin for MS analysis [3] | Similar digestion steps; tandem enrichment of other PTMs (e.g., phosphorylation) is possible without intermediate desalting [23] |
| MS Data Acquisition | LC-MS/MS analysis to identify ubiquitination sites via a 114.04 Da mass shift on modified lysine [3] | LC-MS/MS analysis, often coupled with Data-Independent Acquisition (DIA) methods for comprehensive profiling [23] |
Visualizing the Core Workflows
The following diagrams illustrate the logical sequence of steps for each enrichment methodology, providing a clear overview of the experimental workflows.
Ubiquitin Tagging Workflow
Antibody-Based Enrichment Workflow
The Scientist's Toolkit: Essential Research Reagents
Successful ubiquitination profiling requires specific reagents and tools. The following table details key solutions and their functions for experiments in this field.
Table 3: Key Research Reagent Solutions for Ubiquitination Analysis
| Reagent / Tool | Function / Application |
|---|---|
| Affinity Tags (His, Strep) | Genetically fused to Ub for purification of ubiquitinated substrates under denaturing conditions [3]. |
| Pan-Ubiquitin Antibodies (P4D1, FK1/FK2) | Recognize ubiquitin regardless of chain linkage; used for immunoblotting and enrichment of total ubiquitinated proteins [3]. |
| Linkage-Specific Ub Antibodies | Target specific polyUb chain linkages (e.g., K48, K63, M1); enable study of linkage-dependent functions [3]. |
| Ubiquitin-Activating (E1) Enzyme | Initiates the ubi-tagging enzymatic cascade by activating Ub in an ATP-dependent manner [1]. |
| E2-E3 Fusion Enzyme (gp78RING-Ube2g2) | A linkage-specific enzyme (e.g., for K48) that catalyzes the transfer of Ub from E1 to the target protein in ubi-tagging [1]. |
| Tandem Ub-Binding Domains (TUBEs) | High-affinity tools to purify endogenous ubiquitinated proteins and protect them from deubiquitinase (DUB) activity [3]. |
| SCASP-PTM Buffer System | Enables serial enrichment of multiple PTM peptides (Ub, phosphorylation, glycosylation) from a single sample [23]. |
Conclusion
Ubiquitin tagging and antibody-based enrichment are not competing but complementary technologies, each excelling in distinct domains. Antibody-based methods, particularly diGLY proteomics, offer a powerful, direct route for profiling endogenous ubiquitination across diverse biological systems, including patient tissues, and have been revolutionized by advanced mass spectrometry like DIA for unparalleled depth and quantitative accuracy. In contrast, ubiquitin tagging provides a genetically engineered system ideal for substrate identification in model cell lines and has found a unique, high-impact application in the precise engineering of therapeutic antibodies through 'ubi-tagging'. The choice of method is therefore dictated by the research question—whether it is the discovery of endogenous ubiquitination events or the controlled manipulation of proteins for therapeutic design. Future directions will likely see further refinement of linkage-specific tools, the integration of multi-omics data, and the expanded use of these methodologies to crack the ubiquitin code in complex diseases, paving the way for new diagnostic and therapeutic breakthroughs.
References
- 1. Site-directed multivalent conjugation of antibodies to ... [https://www.nature.com/articles/s41551-024-01342-z]
- 2. A High-Throughput Method for Specific, Rapid, Precise and ... [https://www.sciencedirect.com/science/article/abs/pii/S0039914025015681]
- 3. Current methodologies in protein ubiquitination ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9373315/]
- 4. Site-specific quantification of the in vivo UFMylome reveals ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12146631/]
- 5. Characterizing Ubiquitination Sites by Peptide-based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3518114/]
- 6. Methods for Quantification of in vivo Changes in Protein ... [https://www.sciencedirect.com/science/article/pii/S1535947620304576]
- 7. Rapid and deep-scale ubiquitylation profiling for biology ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6969155/]
- 8. Advancing Antibody Conjugates with Ubi-Tagging [https://communities.springernature.com/posts/behind-the-paper-advancing-antibody-conjugates-with-ubi-tagging]
- 9. Ubiquitin diGLY proteomics as an approach to identify and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6791129/]
- 10. Current methodologies in protein ubiquitination characterization [https://cellandbioscience.biomedcentral.com/articles/10.1186/s13578-022-00870-y]
- 11. Antibody-free approach for ubiquitination profiling by ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267023000983]
- 12. Data-independent acquisition method for ubiquitinome ... [https://www.nature.com/articles/s41467-020-20509-1]
- 13. Peptide Level Immunoaffinity Enrichment Enhances ... [https://www.sciencedirect.com/science/article/pii/S1535947620342663]
- 14. Automating UbiFast for High-throughput and Multiplexed ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9357436/]
- 15. Detection of Protein Ubiquitination Sites by Peptide ... [https://www.jove.com/t/59079/detection-protein-ubiquitination-sites-peptide-enrichment-mass]
- 16. Ubiquitin-like proteins [https://pubmed.ncbi.nlm.nih.gov/22404627/]
- 17. Global Analysis of Protein and Small-Molecule Substrates ... [https://www.sciencedirect.com/science/article/pii/S1535947625000738]
- 18. Global analysis of protein and small-molecule substrates ... [https://pubmed.ncbi.nlm.nih.gov/40254064/]
- 19. Automating UbiFast for High-throughput and Multiplexed ... [https://www.sciencedirect.com/science/article/pii/S1535947621001262]
- 20. Chemical tools to define and manipulate interferon ... [https://www.nature.com/articles/s41467-025-56336-5]
- 21. Unraveling chain specific ubiquitination in cells using tandem ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12217038/]
- 22. Rapid and deep-scale ubiquitylation profiling for biology ... [https://www.nature.com/articles/s41467-019-14175-1]
- 23. Protocol for tandem enrichment of ubiquitinated, ... [https://www.sciencedirect.com/science/article/pii/S2666166725003508]
- 24. How to choose between affinity tags for protein purification [https://www.biotage.com/blog/how-to-choose-between-affinity-tags-for-protein-purification]
- 25. Proteomic approaches to study ubiquitinomics - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10612121/]
- 26. Peptide Level Immunoaffinity Enrichment Enhances ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3879610/]
- 27. Overview of Affinity Purification [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-affinity-purification.html]
- 28. Ubiquitin diGLY Proteomics as an Approach to Identify and Quantify the Ubiquitin-Modified Proteome | SpringerLink [https://link.springer.com/protocol/10.1007/978-1-4939-8706-1_23]
- 29. The Molecular Toolbox for Linkage Type‐Specific Analysis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12118340/]
- 30. Enhanced Purification of Ubiquitinated Proteins by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4824862/]
- 31. KIT UBI trap-B for Western-blot [https://bmolecular.eu/product/kit-ubi-trap-b-for-western-blot/]
- 32. Isolation of ubiquitinated substrates by tandem affinity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6499077/]
- 33. A comprehensive platform for the analysis of ubiquitin-like ... [https://www.nature.com/articles/srep40756]
- 34. Aspirin inhibits proteasomal degradation and promotes α ... [https://www.nature.com/articles/s41467-025-56737-6]
- 35. An Interaction Landscape of Ubiquitin Signaling [https://www.sciencedirect.com/science/article/pii/S1097276517300047]
- 36. Recent Advances in Chemical Proteomics for Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12445971/]
- 37. Site-specific quantification of the in vivo UFMylome reveals ... [https://www.sciencedirect.com/science/article/pii/S2667237525000840]
- 38. Methods to Make Homogenous Antibody Drug Conjugates [https://pmc.ncbi.nlm.nih.gov/articles/PMC4596908/]
- 39. Antibody toolkit reveals N-terminally ubiquitinated ... [https://www.nature.com/articles/s41467-021-24669-6]
- 40. Emerging tools and methods to study cell signalling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12224918/]
- 41. Augment proteasome inhibitor efficacy activates CD8+ T ... [https://www.sciencedirect.com/science/article/pii/S2666379125002848]
- 42. Time-resolved in vivo ubiquitinome profiling by DIA-MS ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8438043/]
- 43. What is Ubiquitin ? Uses, How It Works & Top Companies... Antibody [https://www.linkedin.com/pulse/what-ubiquitin-antibody-uses-how-works-top-companies-isoef]
- 44. Benchmarking commonly used software suites and ... [https://www.nature.com/articles/s41467-022-35740-1]
- 45. Ubiquitomics: An Overview and Future - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7603029/]
- 46. Data-Independent Acquisition: A Milestone and Prospect in ... [https://www.sciencedirect.com/science/article/pii/S1535947624000902]
- 47. Data-independent acquisition (DIA) [https://pmc.ncbi.nlm.nih.gov/articles/PMC8674493/]
- 48. Benchmarking of analysis strategies for data-independent ... [https://www.nature.com/articles/s41467-022-30094-0]
- 49. How to Better Study Ubiquitination [https://www.ptglab.com/news/blog/how-to-better-study-ubiquitination/?srsltid=AfmBOoqblD0IDR5V68J8yuM1Ay1Mh87P4fHgkARiuBBEEx1Yi_HJL11V]
- 50. Integrative proximal-ubiquitomics profiling for ... [https://www.sciencedirect.com/science/article/pii/S2451945625001278]
- 51. toolkit reveals N-terminally... | Nature Communications Antibody [https://www.nature.com/articles/s41467-021-24669-6?error=cookies_not_supported&code=4f969dd8-dfb0-4f04-b624-3dd98cc42765]
- 52. Use of a High-Affinity Ubiquitin -Binding Domain to Detect and Purify... [https://bio-protocol.org/en/bpdetail?id=5426&type=0]
- 53. Deciphering the ubiquitin proteome: Limits and advantages ... [https://www.sciencedirect.com/science/article/abs/pii/S1357272513001751]
- 54. Quantitative proteomics to decipher ubiquitin signaling - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3498854/]
- 55. The Ubiquitin Tale: Current Strategies and Future Challenges [https://pmc.ncbi.nlm.nih.gov/articles/PMC11406696/]
- 56. Characterizing Ubiquitination Sites by Peptide-based ... [https://www.sciencedirect.com/science/article/pii/S1535947620334502]
- 57. Strategy for Development of Site-Specific Ubiquitin ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2020.00111/full]