Ubiquitination and Phosphorylation Crosstalk: Integrating Signals for Cellular Control and Therapeutic Innovation
This article provides a comprehensive analysis of the intricate crosstalk between ubiquitination and phosphorylation, two paramount post-translational modifications (PTMs) governing eukaryotic cell signaling.
Ubiquitination and Phosphorylation Crosstalk: Integrating Signals for Cellular Control and Therapeutic Innovation
Abstract
This article provides a comprehensive analysis of the intricate crosstalk between ubiquitination and phosphorylation, two paramount post-translational modifications (PTMs) governing eukaryotic cell signaling. Tailored for researchers, scientists, and drug development professionals, we explore the foundational mechanisms of this interplay, from phosphodegrons to reciprocal enzyme regulation. We delve into cutting-edge methodological approaches, including multi-PTM proteomics and systems biology modeling, that are revolutionizing the study of signaling networks in health and disease. The review further addresses the challenges of targeting this crosstalk therapeutically, highlighting progress in drug discovery against the ubiquitin-proteasome system and kinases. Finally, we validate these concepts through compelling case studies in oncology and immunology, synthesizing key takeaways to illuminate future directions for biomedical and clinical research.
The Fundamental Language of Cellular Signals: Defining Ubiquitination and Phosphorylation Crosstalk
In the intricate landscape of cellular biochemistry, post-translational modifications (PTMs) serve as fundamental regulatory mechanisms that control protein function, localization, and stability. Among these, phosphorylation and ubiquitination represent two of the most crucial and widespread enzymatic cascades governing eukaryotic cell signaling. While historically studied in isolation, emerging research reveals extensive crosstalk between these pathways, creating sophisticated regulatory networks that maintain cellular homeostasis [1]. Phosphorylation, characterized by its rapid, reversible nature, primarily regulates protein activity and signal transduction. In contrast, ubiquitination, with its diverse outcomes ranging from proteasomal degradation to non-proteolytic signaling, provides broader functional consequences. Understanding the distinct characteristics, enzymatic mechanisms, and intersecting dynamics of these cascades is essential for researchers and drug development professionals seeking to modulate pathological signaling pathways in cancer, neurodegeneration, and other diseases.
The Phosphorylation Cascade: Architecture and Mechanism
Fundamental Enzymatic Components
The phosphorylation cascade involves a coordinated sequence of enzymatic reactions that transfer phosphate groups to specific target proteins. This system relies on three key components that work in concert to ensure signaling specificity and precision.
Kinases: These enzymes catalyze the transfer of a gamma-phosphate group from adenosine triphosphate (ATP) to specific amino acid residues on substrate proteins, primarily targeting serine, threonine, and tyrosine residues [1]. The human genome encodes approximately 500 protein kinases, each exhibiting specific substrate recognition patterns. Kinases often function within hierarchical cascades, where one kinase phosphorylates and activates the next kinase in the sequence, resulting in signal amplification.
Phosphatases: These enzymes provide the necessary counter-regulation by removing phosphate groups from phosphorylated proteins, thereby terminating signals or reversing functional states [1]. This opposing activity creates a dynamic, reversible modification system that can rapidly respond to changing cellular conditions. The balanced action of kinases and phosphatases allows for precise temporal control over protein function.
Protein Substrates: The targets of phosphorylation cascades include a diverse array of structural and regulatory proteins. A single phosphorylation event can induce conformational changes that alter protein activity, subcellular localization, or interaction with binding partners [1]. Many proteins contain multiple phosphorylation sites, enabling complex regulatory patterns and integration of multiple signals.
Molecular Recognition and Catalysis
Enzyme-substrate interactions in phosphorylation cascades depend on highly specific molecular recognition mechanisms that ensure signaling fidelity:
Active Site Complementarity: Kinases and phosphatases employ specialized active sites with geometric and chemical complementarity to their specific protein substrates, following either lock-and-key or induced-fit binding models [2] [3]. This complementarity ensures precise substrate selection among thousands of cellular proteins.
Binding Interactions: Multiple non-covalent forces stabilize enzyme-substrate complexes, including electrostatic interactions with charged amino acid side chains, hydrogen bonding with polar residues, and hydrophobic interactions that exclude water from binding interfaces [4]. The cumulative effect of these interactions provides the binding energy necessary for substrate specificity and catalytic efficiency.
Catalytic Mechanisms: Phosphorylation enzymes employ several catalytic strategies, including metal ion catalysis using Mg²⁺ or Mn²⁺ to orient ATP and stabilize negative charges, general acid-base catalysis to facilitate proton transfer, and covalent catalysis through transient phosphoenzyme intermediates [2].
Table 1: Key Characteristics of Phosphorylation and Ubiquitination Cascades
| Characteristic | Phosphorylation Cascade | Ubiquitination Cascade |
|---|---|---|
| Enzyme Classes | Kinases, Phosphatases | E1, E2, E3, Deubiquitinases |
| Energy Source | ATP | ATP |
| Modification Site | Ser, Thr, Tyr (His) | Lys (Ser, Thr, N-terminus) |
| Chemical Bond | Phosphoester | Isopeptide |
| Reversibility | Highly reversible | Reversible (by DUBs) |
| Primary Outcomes | Altered protein activity, signaling | Degradation, signaling, trafficking |
| Signal Amplification | Kinase cascades | E2/E3 combinatorial complexity |
The Ubiquitination Cascade: Architecture and Mechanism
The Three-Step Enzymatic Pathway
Ubiquitination employs a sophisticated three-enzyme cascade that conjugates the small protein modifier ubiquitin to substrate proteins, enabling diverse regulatory outcomes beyond mere protein degradation.
E1 (Ubiquitin-Activating Enzyme): The cascade initiates with E1, which activates ubiquitin in an ATP-dependent reaction, forming a high-energy thioester bond between its catalytic cysteine and the C-terminus of ubiquitin [1]. The human genome encodes only two E1 enzymes, creating an initial bottleneck in the ubiquitination pathway.
E2 (Ubiquitin-Conjugating Enzyme): Activated ubiquitin is transferred from E1 to the catalytic cysteine of an E2 enzyme, maintaining the thioester linkage [1]. With approximately 40 E2s in humans, this tier begins to introduce specificity, with different E2s determining the linkage type and chain topology during ubiquitination [5].
E3 (Ubiquitin Ligase): The final and most diverse tier consists of E3 ligases, which number over 600 in humans and provide substrate specificity by simultaneously binding E2~Ub complexes and target proteins [1]. E3s facilitate the direct transfer of ubiquitin from E2 to substrate lysine residues, forming an isopeptide bond. Some E2/E3 pairs can also modify serine, threonine, or the protein N-terminus, expanding the regulatory potential of this pathway [5].
Diversity of Ubiquitin Signals and Outcomes
The ubiquitination cascade generates remarkably diverse signals that dictate distinct functional consequences for modified proteins:
Monoubiquitination: Single ubiquitin attachments typically regulate protein trafficking, endocytosis, and histone function without targeting substrates for degradation [1].
Multi-monoubiquitination: Multiple single ubiquitin molecules attached to different lysines on the same substrate can serve as signals for endocytic sorting and internalization of cell surface receptors [1].
Polyubiquitination: Ubiquitin chains formed through specific lysine linkages create topological codes recognized by different cellular machineries. K48-linked chains predominantly target proteins for proteasomal degradation, while K63-linked chains mediate non-proteolytic functions in DNA repair, kinase activation, and inflammatory signaling [1].
Atypical Linkages: Mixed or branched chains involving K6, K11, K27, K29, or K33 residues provide additional regulatory complexity with specialized functions in ER-associated degradation, immune regulation, and mitophagy [1].
Comparative Analysis: Key Structural and Functional Distinctions
Enzymatic Architecture and Energetics
The fundamental architectures of phosphorylation and ubiquitination cascades reveal distinct evolutionary strategies for cellular regulation. Phosphorylation employs a binary enzyme system of kinases and phosphatases that directly add and remove modifications in a relatively straightforward manner [1]. This streamlined architecture enables rapid, reversible signaling well-suited for fast-responsive regulatory circuits. In contrast, the ubiquitination cascade operates through a three-tiered enzymatic hierarchy (E1-E2-E3) that provides exceptional combinatorial complexity through the pairing of a limited number of E1 and E2 enzymes with a vast repertoire of E3 ligases [1]. This arrangement allows for exquisite substrate specificity and diverse output signals from a limited genomic investment.
Both systems are ATP-dependent processes, but they utilize this energy currency differently. Phosphorylation consumes one ATP molecule per modification event to directly drive the transfer of a phosphate group to the substrate [2]. Ubiquitination requires ATP at the initial activation step where E1 activates ubiquitin, with this energy subsequently preserved through high-energy thioester intermediates that are passed along the enzymatic cascade [1]. This energy conservation mechanism allows multiple ubiquitin transfers from a single activation event, particularly during chain elongation.
Kinetics and Regulatory Dynamics
The temporal dynamics and regulatory properties of these cascades differ significantly, reflecting their distinct biological roles:
Modification Reversibility: Phosphorylation is highly dynamic, with rapid kinase and phosphatase activities enabling millisecond-to-second timescale regulation ideal for signal transduction and metabolic control [1]. Ubiquitination reversal by deubiquitinating enzymes (DUBs) typically occurs on slower timescales, particularly when regulating irreversible processes like proteasomal degradation [1].
Signal Amplification: Phosphorylation cascades, such as the MAPK pathway, provide strong signal amplification through sequential kinase activation, where each enzyme activates multiple copies of the next component [1]. Ubiquitination achieves signal diversification through combinatorial E2-E3 pairing, with single E3 ligases potentially modifying hundreds of substrate molecules and creating different ubiquitin topologies with distinct functional consequences [1].
Modification Diversity: While phosphorylation is limited to the addition of a single phosphate group per modified residue, ubiquitination creates complex signals through chain elongation and branching, with different linkage types encoding distinct functional instructions [1]. This allows a single modification system to regulate multiple cellular processes through topological variation.
Table 2: Quantitative Comparison of Modification Properties
| Property | Phosphorylation | Ubiquitination |
|---|---|---|
| Modifying Enzymes in Humans | ~500 Kinases | 2 E1, ~40 E2, >600 E3 |
| Demodifying Enzymes in Humans | ~150 Phosphatases | ~100 DUBs |
| Modification Sites | Ser, Thr, Tyr, His | Primarily Lys |
| Modification Size | 80 Da (phosphate) | 8.5 kDa (ubiquitin) |
| Representative Turnover Rates | Seconds to minutes | Minutes to hours |
| Common Chain Types | Single phosphate | K48, K63, K11, K29, etc. |
Experimental Methodologies for Cascade Analysis
Core Techniques for Phosphorylation Studies
Investigating phosphorylation cascades requires specialized methodologies to capture the transient nature of these modifications:
Western Blotting with Phospho-Specific Antibodies: This fundamental technique allows detection of phosphorylation changes at specific sites using antibodies that recognize phosphorylated serine, threonine, or tyrosine residues [6]. When combined with appropriate stimulation conditions and pharmacological inhibitors, this approach provides semiquantitative data on phosphorylation dynamics.
Mass Spectrometry-Based Phosphoproteomics: Global phosphorylation analysis employs phosphopeptide enrichment techniques such as immobilized metal affinity chromatography (IMAC) or titanium dioxide (TiO₂) chromatography, followed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) to identify and quantify thousands of phosphorylation sites simultaneously [7]. Stable isotope labeling methods enable precise temporal tracking of phosphorylation events in response to cellular stimuli.
Kinase Activity Assays: Direct measurement of kinase function utilizes radioactive ATP (³²P-ATP) or ATP analogs in in vitro kinase reactions with recombinant or immunoprecipitated kinases and their substrates. These assays provide direct information about enzymatic activity rather than merely phosphorylation status.
Core Techniques for Ubiquitination Studies
Ubiquitination research employs distinct methodological approaches tailored to the complexity of this modification:
Co-immunoprecipitation and Ubiquitin Detection: This classic approach involves immunoprecipitating a protein of interest under denaturing conditions to preserve ubiquitin conjugates, followed by western blotting with anti-ubiquitin antibodies to detect polyubiquitinated species [6]. Modification with tagged ubiquitin constructs (HA-, His-, or FLAG-ubiquitin) enhances detection specificity and sensitivity.
diGly Proteomics: This global ubiquitinome profiling technique exploits the tryptic digestion signature of ubiquitinated proteins, which generates a characteristic di-glycine remnant on modified lysines. Enrichment with diGly-specific antibodies followed by MS analysis enables system-wide identification of ubiquitination sites [7].
In Vitro Reconstitution Assays: Defined biochemical systems with purified E1, E2, and E3 enzymes allow precise dissection of ubiquitination mechanisms without confounding cellular factors [5]. These approaches have revealed how membrane composition regulates E2 activity and how specific E3 ligases recognize their substrates.
Crosstalk Mechanisms: Integration of Phosphorylation and Ubiquitination
Molecular Mechanisms of PTM Interplay
The extensive crosstalk between phosphorylation and ubiquitination creates sophisticated regulatory circuits that enhance cellular signaling capabilities:
Phosphodegrons: These specific phosphorylation motifs serve as recognition signals for E3 ubiquitin ligases, directly linking kinase activity to substrate ubiquitination and degradation [7] [1]. Well-characterized examples include the β-TrCP recognition motif in IκBα and the SCF ubiquitination signals in cell cycle regulators. Phosphodegron-mediated regulation provides irreversibility to cell cycle transitions and transcriptional responses.
E3 Ligase Regulation by Phosphorylation: Many E3 ubiquitin ligases are themselves regulated by phosphorylation events that control their catalytic activity, substrate specificity, or subcellular localization [1]. For instance, phosphorylation of the E3 ligase Cbl on tyrosine 371 induces a conformational change that exposes its RING domain, enabling E2 binding and activation of its ubiquitination function [1].
Kinase Regulation by Ubiquitination: Conversely, ubiquitination directly regulates kinase activity through both degradative and non-degradative mechanisms [1]. While K48-linked ubiquitination typically terminates kinase signaling through proteasomal degradation, K63-linked ubiquitination can directly activate kinases such as NIK in the NF-κB pathway, demonstrating the bidirectional nature of PTM crosstalk.
Systems-Level Integration and Emerging Concepts
Recent research has revealed additional layers of complexity in phosphorylation-ubiquitination crosstalk:
Conservation Patterns: Global proteomic analyses indicate that phosphorylation sites co-occurring with ubiquitination show significantly higher evolutionary conservation than phosphorylation sites in general, suggesting strong functional selection for these integrated regulatory nodes [7].
Hierarchical Regulation: In the EGFR/MAPK pathway, phosphorylation events precede and direct ubiquitination at multiple levels—receptor activation initiates Cbl-mediated ubiquitination, which in turn regulates downstream signaling dynamics through controlled receptor internalization and degradation [1].
Pathway Switching Mechanisms: Recent findings demonstrate that phosphorylation can redirect E2 enzyme specificity, as shown by RSK1-mediated phosphorylation of UBE2L6, which switches its function from ISG15 conjugation to ubiquitin conjugation, consequently altering its role in immune signaling [8].
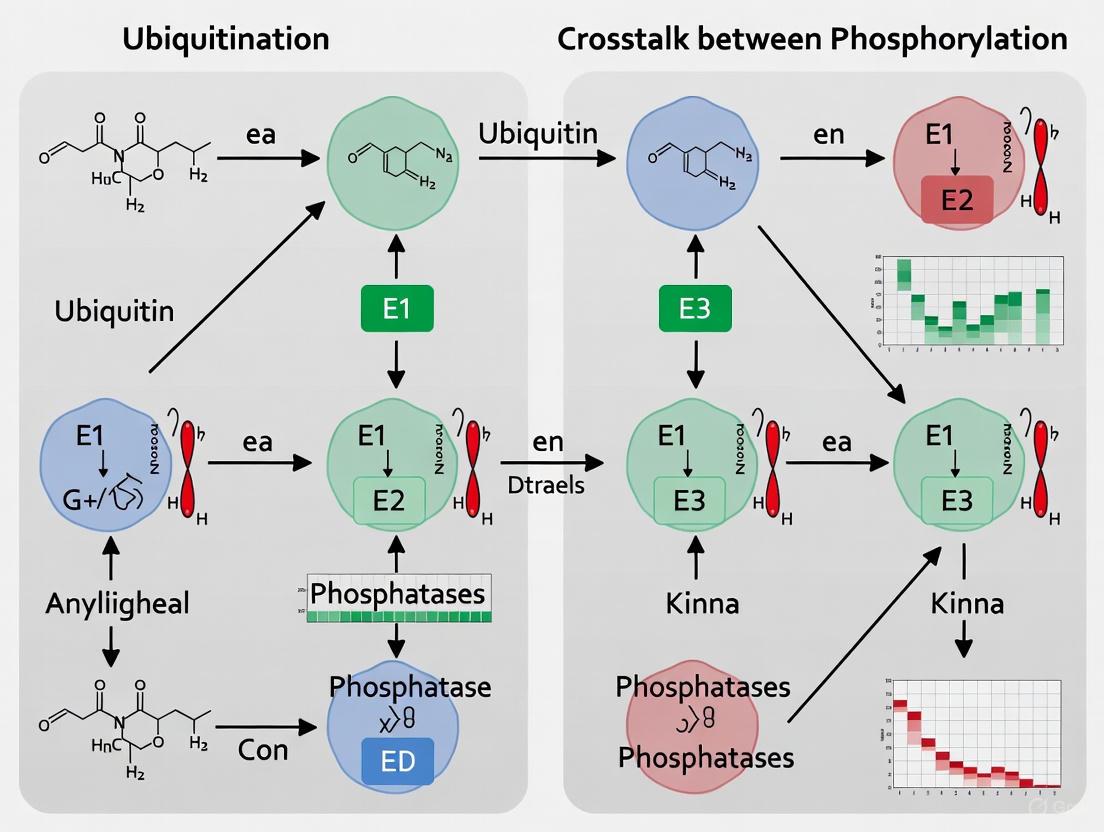
Figure 1: Core Crosstalk Mechanisms Between Phosphorylation and Ubiquitination Cascades. This diagram illustrates how phosphorylation events can create recognition motifs (phosphodegrons) for E3 ubiquitin ligases, while ubiquitination reciprocally regulates kinase activity and stability through both degradative and non-degradative mechanisms.
Table 3: Essential Research Reagents for Studying Phosphorylation-Ubiquitination Crosstalk
| Reagent/Solution | Primary Function | Application Examples | Key Considerations |
|---|---|---|---|
| Phospho-Specific Antibodies | Detect specific phosphorylation events | Western blotting, immunofluorescence | Validation of target specificity required |
| Ubiquitin-Tagging Systems (HA-, His-, FLAG-Ub) | Track protein ubiquitination | Co-IP, pulldown assays | Use under denaturing conditions for direct ubiquitination |
| Proteasome Inhibitors (MG132, Bortezomib) | Block proteasomal degradation | Stabilize ubiquitinated proteins | Can induce cellular stress responses |
| Kinase Inhibitors | Perturb phosphorylation signaling | Functional studies, therapeutic screening | Watch for off-target effects on related kinases |
| Phosphatase Inhibitors | Preserve phosphorylation states | Protein extraction, enzymatic assays | Cocktails often needed for complete inhibition |
| E1/E2/E3 Recombinant Proteins | Reconstitute ubiquitination in vitro | Mechanistic studies, screening | Require optimized buffer conditions |
| diGly-Specific Antibodies | Enrich ubiquitinated peptides | Ubiquitinome profiling by MS | Specific for tryptic digests with Gly-Gly remnant |
| Active Kinases/Phosphatases | Modify phosphorylation in vitro | Enzyme assays, substrate validation | Require proper activation/cofactors |
The comparative analysis of phosphorylation and ubiquitination cascades reveals both striking parallels and fundamental distinctions in their biochemical logic and cellular implementation. While both represent ATP-dependent enzymatic cascades that dynamically modify proteins to regulate their function, they employ markedly different architectural principles—phosphorylation utilizes a direct binary system of opposing enzymes, whereas ubiquitination employs a three-tiered hierarchical cascade with exceptional combinatorial potential [1]. These structural differences underlie their specialized biological roles: phosphorylation excels at rapid, reversible information transfer for signal transduction and metabolic control, while ubiquitination provides diverse functional outcomes ranging from targeted destruction to complex assembly regulation.
The emerging paradigm of extensive crosstalk between these systems demonstrates how eukaryotic cells integrate these pathways to create sophisticated regulatory networks with enhanced information-processing capabilities [1] [9]. The molecular integration through mechanisms such as phosphodegrons, reciprocal enzyme regulation, and pathway switching creates layered control systems that enable precise cellular decision-making. For researchers and drug developers, understanding these interconnected networks provides exciting opportunities for therapeutic intervention, particularly in cancer and immune disorders where both phosphorylation and ubiquitination pathways are frequently dysregulated. Future research will undoubtedly continue to reveal novel aspects of this complex biochemical interplay, expanding our understanding of cellular regulation and creating new avenues for manipulating these fundamental processes in human disease.
{article}
Beyond Degradation: The Diverse Functional Outcomes of Different Ubiquitin Chain Linkages
Ubiquitination, once recognized primarily as a signal for proteasomal degradation, is now understood to govern a vast array of non-degradative cellular processes. The functional outcome of ubiquitination is largely dictated by the topology of the polyubiquitin chain assembled on a substrate protein. This article provides a comparative analysis of the distinct biological functions mediated by different ubiquitin linkage types, with a special emphasis on K48-linked degradative signals versus the non-degradative roles of K63-linked and branched chains. Framed within the context of crosstalk with phosphorylation, we detail the experimental methodologies and reagent toolsets that are enabling researchers to decode the complex logic of ubiquitin signaling in health and disease.
Ubiquitin Chain Linkages: A Functional Spectrum
Ubiquitin can be conjugated to substrate proteins as a monomer or as a polymer, forming chains through one of its seven internal lysine (K) residues (K6, K11, K27, K29, K33, K48, K63) or its N-terminal methionine (M1) [10] [11]. The architecture of these chains dictates their function, creating a specialized signaling system. Homotypic chains, linked uniformly through the same residue, were the first to be characterized. However, the ubiquitin code is further complicated by heterotypic chains, which include mixed chains (more than one linkage type, but each ubiquitin modified at only one site) and branched chains (comprised of ubiquitin subunits simultaneously modified on at least two different sites) [10] [12]. The following table summarizes the well-established functions of major ubiquitin chain types.
Table 1: Comparative Functions of Major Ubiquitin Chain Linkages
| Linkage Type | Primary Functions | Key Biological Processes | Representative E3 Ligases / Complexes |
|---|---|---|---|
| K48-linked | Proteasomal Degradation [11] | Protein turnover, cell cycle progression, metabolic regulation [11] [13] | APC/C, UBR5 [10] |
| K63-linked | Signal Transduction, protein-protein interactions, endocytosis, DNA repair [14] [11] [13] | NF-κB activation, innate and adaptive immunity, autophagy [14] [13] | TRAF6, TRAF2, cIAP1/2 [14] [13] |
| K11-linked | Proteasomal Degradation (especially in concert with K48) [15] | Cell cycle regulation, mitotic progression, proteotoxic stress response [15] | APC/C (with UBE2S) [10] [15] |
| K11/K48-branched | Priority Degradation Signal [15] | Accelerated degradation of cell cycle regulators and misfolded proteins [15] | APC/C (with UBE2C and UBE2S) [10] [15] |
| K29/K48-branched | Proteasomal Degradation [10] | Ubiquitin Fusion Degradation (UFD) pathway [10] | Ufd4 and Ufd2 (in yeast) [10] |
| K48/K63-branched | Degradation & Signaling Switching [10] | NF-κB signaling, apoptotic response, p97/VCP processing [10] | TRAF6 & HUWE1, ITCH & UBR5 [10] |
| M1-linked (Linear) | NF-κB Activation, inflammatory signaling [14] | Innate immunity, cell death regulation [14] | HOIP (LUBAC complex) [14] |
Crosstalk with Phosphorylation: A Central Signaling Nexus
The ubiquitin system does not operate in isolation; it engages in extensive bidirectional crosstalk with other post-translational modifications (PTMs), most notably phosphorylation [7] [16] [17]. This interplay is a fundamental mechanism for increasing the specificity and combinatorial logic of cellular signaling.
One primary mode of crosstalk occurs when phosphorylation directly primes a substrate for ubiquitination. The canonical example is the phosphodegron, a specific motif where phosphorylation creates a binding site for a specialized E3 ubiquitin ligase, leading to the substrate's ubiquitylation and degradation [7]. This mechanism is central to cell cycle control.
Conversely, ubiquitination can regulate components of the phosphorylation machinery. For instance, K63-linked ubiquitination of RIPK2 following NOD2 receptor activation creates a scaffold that facilitates the recruitment and activation of the TAK1 and IKK kinase complexes, which in turn phosphorylate downstream targets to activate NF-κB signaling [13].
Global proteomic studies have quantified this relationship, revealing that phosphorylation sites found on ubiquitylated protein isoforms are significantly more evolutionarily conserved than the broader set of phosphorylation sites, underscoring their functional importance [7]. The following diagram illustrates the core concepts of this PTM crosstalk.
Ubiquitination and Phosphorylation Crosstalk
Methodologies for Decoding Ubiquitin Signaling
Studying ubiquitination presents unique challenges due to its low stoichiometry, the diversity of chain architectures, and its dynamic nature. The field has developed sophisticated methods to overcome these hurdles.
Enrichment and Detection Strategies
A critical first step is the specific enrichment of ubiquitinated proteins or peptides from complex lysates. The table below catalogs key reagent solutions essential for this research.
Table 2: Key Research Reagent Solutions for Ubiquitination Studies
| Reagent / Tool | Function / Principle | Key Features & Applications |
|---|---|---|
| Tandem Ubiquitin Binding Entities (TUBEs) [13] [12] | High-affinity capture of polyubiquitinated proteins; available in pan-specific and linkage-specific (K48, K63) variants. | Protects chains from DUBs; enables detection of endogenous protein ubiquitination in high-throughput assays (e.g., PROTAC screening). |
| Linkage-Specific Antibodies [11] [13] | Immuno-enrichment and detection of ubiquitin chains with defined linkages (e.g., K48, K63, K11). | Useful for Western blotting, immunofluorescence, and immunoprecipitation; can be applied to tissue samples without genetic manipulation. |
| Epitope-Tagged Ubiquitin (e.g., His, Strep, HA) [7] [11] | Affinity purification of ubiquitinated proteins from cell lysates after overexpression of tagged ubiquitin. | Enables system-wide profiling of ubiquitination sites (ubiquitinome) via mass spectrometry. |
| DiGly Antibody (GG-K⁰ antibody) [7] | Enriches for tryptic peptides containing the di-glycine remnant left on modified lysine after trypsin digestion. | The gold standard for mass spectrometry-based mapping of ubiquitination sites on a proteome-wide scale. |
| Linkage-Specific DUBs [12] | Enzymatic tools to confirm linkage identity or to selectively cleave specific chains in vitro. | Used in conjunction with Western blotting or mass spectrometry to validate chain topology. |
| Mutant Ubiquitin Plasmids (K-to-R mutants) [13] | In vivo expression of ubiquitin where a specific lysine is mutated to arginine to prevent formation of that linkage. | Allows functional dissection of the role of a specific ubiquitin linkage in a pathway. |
A prominent application of TUBEs is the investigation of context-dependent ubiquitination. For example, the endogenous kinase RIPK2 can be modified with either K63-linked chains upon inflammatory stimulation (e.g., with L18-MDP) or K48-linked chains when targeted by a PROTAC degrader. As demonstrated in a 2025 study, K63- and K48-TUBEs can selectively capture these distinct ubiquitination events in a high-throughput format, providing a powerful tool for characterizing targeted protein degradation therapeutics [13]. The workflow for such an analysis is detailed below.
TUBE-Based Ubiquitination Assay Workflow
Structural Biology and In Vitro Reconstitution
Understanding how ubiquitin chains are recognized at an atomic level has been revolutionized by structural techniques. A landmark 2025 cryo-EM study of the human 26S proteasome in complex with a K11/K48-branched ubiquitin chain revealed a multivalent recognition mechanism [15]. The structures showed that the proteasome uses a novel binding site formed by RPN2 and RPN10 to engage the K11-linked branch, in addition to the canonical K48-linkage binding site [15]. This explains the "priority degradation signal" conferred by K11/K48-branched chains.
To conduct such studies, the synthesis of defined ubiquitin chains is essential. Branched ubiquitin trimers and tetramers of specific linkages (e.g., K48/K63) can be assembled enzymatically using a combination of linkage-specific E2 enzymes and engineered ubiquitin mutants (e.g., Ub¹⁻⁷²) to control the order of linkage addition [12]. Alternatively, total chemical synthesis and genetic code expansion strategies allow for the incorporation of non-hydrolysable bonds or spectroscopic probes, providing powerful reagents for mechanistic and biophysical studies [12].
Implications for Drug Development and Disease
The intricate relationship between ubiquitin linkages and cellular outcomes has direct translational relevance, particularly in immunology and oncology. In immune signaling, K63-linked and linear ubiquitination are critical for activating NF-κB and MAPK pathways downstream of innate immune receptors [14] [13]. Consequently, inhibiting the enzymes that write (e.g., TRAF6), read, or erase (DUBs) these chains is a promising therapeutic strategy for inflammatory and autoimmune diseases [13].
In cancer, the targeted protein degradation (TPD) paradigm, exemplified by PROTACs, deliberately hijacks the K48-linked ubiquitination machinery to induce the degradation of oncogenic proteins [13]. The efficiency of a PROTAC is fundamentally linked to its ability to induce polyubiquitination with the correct linkage. High-throughput TUBE-based assays are now being deployed to screen for effective PROTAC molecules by directly measuring their ability to induce K48-linked ubiquitination of the target protein [13]. Furthermore, the discovery that branched chains like K11/K48 can serve as superior degradation signals suggests that inducing specific chain topologies could be the next frontier in optimizing TPD therapeutics [15].
{/article}
Post-translational modifications (PTMs) represent a fundamental layer of regulation in cellular signaling, with phosphorylation and ubiquitination standing as two of the most prevalent and functionally interconnected mechanisms. This guide objectively compares the molecular logic by which these PTMs interact to control protein stability through phosphodegrons—specific amino acid motifs where phosphorylation acts as a signal for subsequent ubiquitination and proteasomal degradation. We dissect the experimental evidence for canonical sequential modification, competitive residue mechanisms, and the emerging role of basic residues as electrostatic modulators. Supported by quantitative data and detailed methodologies, this review provides a framework for researchers and drug development professionals to understand and target these regulatory nodes in disease pathways, particularly in cancer and targeted protein degradation therapies.
The functional diversity of the human proteome is vastly expanded by post-translational modifications (PTMs), with phosphorylation and ubiquitination representing two of the most dynamic and interconnected regulatory systems. Ubiquitination-phosphorylation crosstalk is a recurring theme in eukaryotic cell signaling, enabling precise, reversible, and rapid control over protein stability, activity, and localization [1] [17]. The proteome-wide, mass spectrometry-based assessment in budding yeast revealed the scale of this interplay, identifying 466 proteins that were simultaneously modified by both phosphorylation and ubiquitination. Notably, around half of the ~2,000 phosphorylation sites on these proteins were exclusive to the ubiquitin-modified proteoforms, suggesting specialized regulatory roles [7].
At the heart of this crosstalk lies the phosphodegron, a specific motif within a protein that, upon phosphorylation, is recognized by a specialized E3 ubiquitin ligase, leading to the substrate's polyubiquitination and subsequent degradation by the 26S proteasome [18] [19]. These motifs function as critical integration points for cellular signals, translating upstream kinase activity into controlled protein turnover. The mutation of genes encoding E3 ligases like FBXW7 or the phosphodegrons in their substrates, such as NOTCH1, is a frequent occurrence in cancers, underscoring the therapeutic importance of understanding these mechanisms [20] [18]. This guide systematically compares the distinct molecular strategies cells employ to implement phosphodegron-based regulation, providing a side-by-side analysis of their mechanisms, key players, and experimental evidence.
Comparative Mechanisms of Phosphodegron Operation
The term "phosphodegron" encompasses several distinct mechanistic classes of phosphorylation-dependent degradation signals. The following sections and Table 1 provide a structured comparison of these mechanisms, supported by quantitative data from key studies.
Table 1: Comparative Analysis of Phosphodegron Mechanisms
| Mechanism | Molecular Logic | Example E3 Ligase | Example Substrate(s) | Key Regulatory Features |
|---|---|---|---|---|
| Canonical Sequential | Phosphorylation creates a direct binding site for a specific E3 ligase [18]. | FBXW7 (SCF complex) [20] | Cyclin E, c-Myc, KLF5, NOTCH1 [20] | Requires a consensus motif (e.g., FBXW7: Thr-Pro-Pro-Xaa-Ser); often involves multiple kinases in a sequential manner [20]. |
| Kinase-Integrated | The activating kinase is directly recruited to or is a part of the E3 ligase complex [1]. | Cbl (RING-type E3) [1] | EGFR (EGF Receptor) [1] | Kinase activity (EGFR itself) and E3 ligase recruitment are coupled; involves allosteric activation of the E3 ligase [1]. |
| Phospho-Inhibited | Phosphorylation blocks the degron, stabilizing the protein [19]. | Specific E3 complexes for EphB4 [19] | EphB4 Receptor [19] | Phosphorylation and ubiquitylation are alternative fates for the same motif, establishing a biological switch [19]. |
| Electrostatically Modulated | Basic residues near the phosphosite modulate phosphate group properties and E3 binding affinity [21]. | Potentially SCF-Cdc4 [22] | c-Src (Unique domain) [21] | Local electrostatic network fine-tunes phosphodegron function; pKa of phosphate groups is a key parameter [21]. |
Canonical Sequential Phosphodegrons and the FBXW7 Paradigm
The best-characterized mechanism involves a strict sequential process where phosphorylation of a degron motif precedes and is absolutely required for E3 ligase binding. A prime example is the E3 ligase FBXW7 (also known as Cdc4), a critical tumor suppressor. FBXW7 recognizes a consensus phosphodegron sequence, Thr-Pro-Pro-Xaa-Ser, where both the threonine and serine residues must be phosphorylated for high-affinity binding to the WD40 domain of FBXW7 [20]. This mechanism is exemplified by its regulation of oncoproteins like cyclin E (CCNE1) and c-Myc. The structural basis for this interaction was revealed by crystallography studies of the FBXW7-Skp1 complex with a bisphosphorylated cyclin E1 peptide, demonstrating how phosphorylation induces a complementary fit with the ligase [20].
A systems-level study using a ratiometric protein degradation assay discovered 19 novel FBXW7-binding phosphodegrons in proteins involved in transcription (ETV5, KLF4), chromatin regulation (ARID4B, KMT2D), and cytoskeletal functions (MAP2) [20]. This research highlighted the partnership between FBXW7 and Mitogen-Activated Protein Kinases (MAPKs), showing that the degradation of substrates like ARID4B and JAZF1 was diminished upon application of MAPK-specific inhibitors. This demonstrates that MAPKs often serve as the "priming" kinases for FBXG7 substrates, expanding our understanding of how extracellular signals can directly modulate protein stability [20].
Competitive Residues and Phospho-Inhibited Degrons
In a contrasting mechanism, phosphorylation can sometimes inhibit degradation. In these "phospho-inhibited" degrons, modification of a specific residue blocks the interaction with the E3 ligase, thereby stabilizing the protein. This creates a competitive switch where a single residue can be targeted for either stabilization (via phosphorylation) or destabilization (via ubiquitination) [19].
Research on the EphB4 receptor kinase identified a phospho-inhibited degron where phosphorylation at a specific site prevents its own ubiquitination and degradation. This mechanism allows for precise control over the abundance of this angiogenic kinase, and its dysregulation contributes to oncogenesis [19]. This competitive logic is a powerful regulatory switch, enabling cellular signals to directly oppose degradation.
The Role of Basic Residues in Electrostatic Modulation
Beyond the binary switch of phospho-activation/inhibition, the local protein environment can fine-tune degron function. Studies on the Unique domain of c-Src revealed that basic residues (lysine and arginine) located near phosphorylation sites can form specific electrostatic interactions with the introduced phosphate group [21].
These interactions can modulate the acidity (pKa) of the phosphate and introduce local conformational restrictions, effectively coupling various phosphosites into a functional unit. For instance, in c-Src, the phosphorylation sites T37, S43, and S75 all have nearby basic residues that influence their properties. Mutation of arginine 78 (R78) to alanine increased the pKa of the nearby pS75 phosphate group from 6.03 to 6.15, indicating a direct electrostatic interaction that stabilizes the phosphorylated state [21]. This electrostatic network represents a sophisticated layer of regulation that can influence the affinity of a phosphodegron for its cognate E3 ligase, adding a dimension of fine-tuning to the degradation signal.
Experimental Data and Quantitative Analysis
The discovery and validation of phosphodegrons rely on a suite of biochemical and cell-based assays. The quantitative data from these experiments are crucial for comparing the strength and regulation of different degrons.
Table 2: Quantitative Data from Key Phosphodegron and Crosstalk Studies
| Study System / Assay | Key Quantitative Findings | Biological Implication |
|---|---|---|
| Ratiometric Degradation Assay (FBXW7 substrates) [20] | Identified 19 novel FBXW7 phosphodegrons; Degradation of probes (e.g., CycECO) was dependent on FBXW7 overexpression and inhibited by MG132. | Demonstrates partnership between MAPKs and FBXW7; suggests a multitude of substrates may simultaneously mediate FBXW7's tumor suppressor function. |
| Global Yeast PTM Analysis [7] | 466 proteins co-modified by phosphorylation and ubiquitination; 2,100 phosphorylation sites co-occurring with 2,189 ubiquitylation sites. | Phosphorylation sites found co-occurring with ubiquitylation were more highly conserved than the entire set of phosphorylation sites (P = 0.0027). |
| pKa Measurement of c-Src Phosphosites [21] | pKa values of phosphoresidues: pT37 (6.10), pS43 (5.80), pS75 (6.03). R78A mutation increased pS75 pKa to 6.15. | Basic residues form specific electrostatic networks to fine-tune phosphate group properties, potentially affecting E3 ligase binding affinity. |
| Cdk1 Phosphorylation of Ame1 [22] | Ame1 contains a cluster of 4 minimal Cdk1 sites (Thr31, Ser41, Ser45, Ser52/53); Phosphorylation activates SCF-Cdc4 phosphodegrons. | Links core cell cycle machinery (Cdk1) to inner kinetochore assembly, ensuring timely subunit turnover. |
Key Experimental Protocol: PTM-Enhanced (PTMe) Pull-down Assay
To functionally study phosphodegrons and their associated degradation complexes, a novel Post-translational Modification–enhanced (PTMe) Pull-down method has been developed. This protocol integrates kinase and ubiquitination assays into a single pull-down step, offering a significant advantage over traditional, separate co-immunoprecipitation and in vitro modification assays [19].
Workflow Overview:
- Bait Design: A synthetic, biotin-tagged peptide containing the native sequence of the putative phosphodegron is generated.
- PTM-Enhanced Incubation: The bait peptide is incubated with cell extracts, which provide a native source of active kinases, E1/E2/E3 enzymes, and ubiquitin. The reaction is supplemented with ATP (phosphate donor), ubiquitin, and essential cofactors (e.g., Mg²⁺, Mn²⁺) to actively enhance phosphorylation and ubiquitination events on the bait.
- Capture and Analysis: The biotinylated bait and its recruited protein complexes are pulled down using streptavidin-magnetic beads. The complexes can then be analyzed by western blotting (e.g., for specific E3 ligases) or mass spectrometry for proteomic profiling [19].
This method is particularly powerful because it recapitulates the functional dependency between phosphorylation and ubiquitination, allowing for the simultaneous discovery and validation of the degradation complex recruited to a specific phosphodegron under different cellular treatment conditions.
The Scientist's Toolkit: Essential Research Reagents
The following table catalogs key reagents and their applications for studying phosphodegrons and PTM crosstalk, as derived from the cited experimental protocols.
Table 3: Research Reagent Solutions for Phosphodegron Studies
| Reagent / Resource | Specifications & Function | Example Application |
|---|---|---|
| Biotin-tagged pDegron Peptide | HPLC-grade synthetic peptide; serves as the assay bait to recruit degradation complexes. | PTMe pull-down assay for identifying E3 ligases [19]. |
| Recombinant Ubiquitin | Source of ubiquitin moiety for in vitro ubiquitination reactions. | PTMe pull-down assay; standard in vitro ubiquitination assays [19]. |
| SCF Complex Inhibitors | e.g., MLN4924 (NEDD8-activating enzyme inhibitor); blocks activity of cullin-RING ligases (CRLs) like SCF. | Validating the involvement of SCF complexes in substrate degradation [20]. |
| Proteasome Inhibitors | e.g., MG132; inhibits the 26S proteasome, causing accumulation of polyubiquitinated proteins. | Confirming UPS-mediated degradation in cell-based or in vitro assays [20] [19]. |
| Kinase-Specific Inhibitors | Inhibitors for MAPKs (ERK, JNK, p38), Cdk1, AKT, etc.; used to identify the priming kinase. | Determining kinase responsible for phosphodegron activation (e.g., MAPK for FBXW7 substrates) [20] [23]. |
| Phospho-specific Antibodies | Antibodies recognizing phosphorylated degron motifs (e.g., pThr-Pro-Pro-Xaa-pSer). | Validating in vivo phosphorylation of the degron by western blot or immunofluorescence [20]. |
| Ubiquitin Remnant Antibody | Antibody recognizing diglycine (diGly) remnant on lysine after tryptic digest. | Mass spectrometry-based site-specific mapping of ubiquitylation sites [7]. |
Signaling Pathways and Molecular Logic
The molecular mechanisms of phosphodegrons are integrated into larger signaling networks, enabling dynamic cellular responses. The following diagram illustrates the core decision-making pathways that govern protein stability through phosphodegron regulation.
Figure 1: Core Signaling Pathway of a Canonical Phosphodegron. An upstream signal activates a specific kinase, which phosphorylates the degron motif on the substrate protein. This creates a binding site for an E3 ubiquitin ligase, which then polyubiquitinates the substrate, targeting it for proteasomal degradation.
The experimental workflow for discovering and validating these interactions, particularly using modern, integrated methods, can be visualized as a multi-stage process.
Figure 2: Experimental Workflow for Phosphodegron Analysis. The process begins with the identification of a putative degron sequence, followed by the synthesis of a biotin-tagged bait peptide. This bait is used in a PTM-enhanced pull-down with cell extracts to recruit endogenous kinases and E3 ligases. The recruited protein complexes are then analyzed, typically by mass spectrometry for discovery or western blot for confirmation, and finally validated using cellular degradation assays.
The comparative analysis presented in this guide elucidates the sophisticated molecular logic cells employ to control protein stability through phosphodegrons. The canonical sequential model, exemplified by FBXW7, provides a straightforward, kinase-dependent trigger for degradation. In contrast, phospho-inhibited degrons and electrostatic modulation by basic residues demonstrate the nuanced, fine-tuning capabilities of this regulatory system. The experimental data and detailed protocols provided, such as the innovative PTMe pull-down assay, offer a roadmap for researchers to systematically investigate these mechanisms in their systems of interest.
Understanding these interactions is not merely an academic exercise. The frequent dysregulation of phosphodegron pathways in cancer and other diseases highlights their profound therapeutic potential. For drug development professionals, components of these pathways—from the upstream kinases that "write" the phosphodegron signal to the E3 ligases that "read" it—represent promising targets for novel therapies, including molecular glues and PROTACs (Proteolysis-Targeting Chimeras) that aim to hijack the ubiquitin-proteasome system for targeted protein degradation.
Crosstalk between different types of post-translational modifications (PTMs), particularly ubiquitination and phosphorylation, represents a fundamental regulatory layer in eukaryotic cell signaling. This interplay functions as a sophisticated combinatorial logic system that enables cells to process complex information and generate diverse functional outcomes [7] [1]. Rather than operating in isolation, these modification systems engage in intricate cross-regulation that expands the coding potential of the proteome beyond what either PTM could achieve alone. The systems-level properties emerging from this crosstalk—including bistability, oscillations, and precise signal specificity—enable critical cell fate decisions, govern metabolic transitions, and ensure robust homeostatic control [1] [24] [25].
Understanding how ubiquitination-phosphorylation crosstalk generates these sophisticated systems behaviors provides not only fundamental biological insights but also opens new therapeutic avenues. The development of targeted protein degradation technologies, particularly PROTACs (Proteolysis-Targeting Chimeras), exemplifies how mechanistic understanding of ubiquitination machinery can be harnessed for therapeutic intervention [26]. This review synthesizes experimental and computational evidence demonstrating how ubiquitination-phosphorylation crosstalk creates specific systems-level dynamics, with direct implications for drug development and disease treatment.
Quantitative Landscapes of Phosphorylation-Ubiquitination Crosstalk
Global proteomic studies have revealed the extensive co-occurrence of phosphorylation and ubiquitination modifications on cellular proteins. A landmark study in Saccharomyces cerevisiae identified 466 proteins with 2,100 phosphorylation sites co-occurring with 2,189 ubiquitylation sites, demonstrating the remarkable prevalence of this form of crosstalk [7]. The methodological innovation enabling this discovery—sequential enrichment strategies for co-modified proteins—provided the first systematic view of this combinatorial PTM landscape.
Table 1: Global Analysis of Phosphorylation-Ubiquitination Crosstalk in Yeast
| Analytical Method | Ubiquitylated Proteins Identified | Proteins with Both PTMs | Ubiquitylation Sites | Phosphorylation Sites on Co-modified Proteins |
|---|---|---|---|---|
| Ub-protein Enrichment | 891 | 321 | 2,395 | 1,769 |
| SCX-IP | 1,817 | 245 | 4,659 | 437 |
| Total | 1,920 | 466 | 5,629 | 2,100 |
Evolutionary analysis of these co-modification sites revealed striking functional significance. Phosphorylation sites found co-occurring with ubiquitination demonstrated significantly higher conservation than the entire set of phosphorylation sites, suggesting they represent functionally important regulatory nodes under strong selective pressure [7]. This conservation pattern was specific to phosphorylation sites, as ubiquitylation sites on phosphoproteins showed similar conservation levels regardless of phosphorylation status.
The functional domains enriched in ubiquitylated phosphoproteins provide insights into the biological processes most heavily regulated by this crosstalk. Ribosomal proteins, membrane transporters, arrestin domains, and proteins localized to the cell bud showed significant enrichment, pointing to critical roles in protein translation, transmembrane transport, and cell polarity [7].
Systems-Level Dynamics: Bistability and Oscillations
Theoretical Foundations of Bistability
Bistability represents a fundamental systems property where biological networks can exist in two distinct stable states, enabling digital decision-making processes such as cell fate determination. While positive feedback loops have traditionally been considered essential for bistability, recent research demonstrates that multisite phosphorylation alone can generate bistable responses without explicit feedback regulation [25].
The mitogen-activated protein kinase (MAPK) cascade, an essential eukaryotic signaling pathway governing proliferation, differentiation, and programmed cell death, exemplifies this principle. Kholodenko and colleagues established that a single level of the MAPK cascade, involving dual phosphorylation/dephosphorylation cycles, can theoretically exhibit hysteresis and bistability based solely on the intrinsic properties of the pathway architecture [25]. This finding fundamentally altered our understanding of how bistability can emerge from seemingly straightforward biochemical systems.
Metabolic Oscillations Between Growth and Quiescence
The "push-pull" bistability model explains how cells oscillate between quiescent and growth/proliferation states based on the availability of a central metabolic resource [24]. In yeast metabolic cycles (YMCs), synchronized populations exhibit oscillations in oxygen consumption that tightly correlate with distinct physiological states. During high-oxygen-consumption phases, cells activate biosynthetic and growth programs, while low-oxygen-consumption phases are characterized by autophagy, vacuolar function, and quiescence markers [24].
Table 2: Characteristics of Growth-Quiescence Oscillation States in Yeast
| Parameter | Growth/Proliferation State | Quiescent State |
|---|---|---|
| Oxygen Consumption | High | Low |
| Gene Expression | Biosynthesis and growth programs | Autophagy and vacuolar function |
| Metabolic Processes | Anabolic | Catabolic |
| Cell Division | Synchronous division during specific temporal window | Non-dividing |
| Hallmark Features | Commitment to cell cycle progression | Reversibly nondividing state |
This frustrated bistability model requires tripartite communication between the metabolic resource determining oscillations and the quiescent and growth state cells. Cells in each state exhibit hysteresis (memory) of their current state and actively "push-pull" cells from the other state, creating a robust oscillatory system [24]. The identification of specific central metabolites controlling these transitions remains an active research area with profound implications for understanding cellular metabolism in both normal physiology and disease states such as cancer.
Diagram 1: Push-pull bistability model governing growth-quiescence transitions. Metabolic resource availability drives state switching, with each state reinforcing itself and antagonizing the alternative state.
Molecular Mechanisms of Phosphorylation-Ubiquitination Crosstalk
Phosphodegrons and Coordinated Protein Degradation
Perhaps the best-characterized molecular mechanism of phosphorylation-ubiquitination crosstalk is the phosphodegron—a specific phosphorylation pattern that functions in cis to promote subsequent ubiquitylation of a substrate [7]. Phosphodegrons provide temporal control of protein degradation, imparting irreversibility and robustness to critical cellular processes such as cell cycle progression. The identification of 466 co-modified proteins in yeast suggests that phosphodegrons represent a widespread regulatory mechanism rather than a specialized pathway [7].
Beyond substrate recognition, phosphorylation also directly regulates the activity of E3 ubiquitin ligases themselves. The Cbl family of E3 ligases, crucial regulators of receptor tyrosine kinase (RTK) endocytosis, undergoes phosphorylation-induced conformational changes that activate their ubiquitin ligase function [1]. Following EGFR activation and autophosphorylation, Cbl binds to phosphotyrosine residues on the receptor, leading to phosphorylation of a critical tyrosine residue (Y371 in c-Cbl) that enables full rotation of the Cbl linker region. This structural rearrangement exposes the RING domain, allowing E2 binding and stimulating Cbl E3 ligase activity [1].
Reciprocal Regulation and Feedback Loops
The crosstalk between phosphorylation and ubiquitination is notably reciprocal, with ubiquitination events regulating kinase activity and phosphorylation modulating ubiquitin ligase function. This bidirectional regulation creates complex feedback and feedforward loops that enable sophisticated signal processing capabilities [1].
In the EGFR/MAPK pathway, ubiquitination provides crucial negative feedback regulation that shapes both the amplitude and duration of signaling. Cbl-mediated ubiquitination of activated EGFR promotes its endocytosis and subsequent lysosomal degradation, effectively terminating signaling [1]. Additionally, deubiquitinating enzymes (DUBs) such as USP8 can reverse this process, providing another layer of regulation. USP8 itself is regulated by phosphorylation, creating an integrated control system where phosphorylation regulates ubiquitination, which in turn controls receptor trafficking and signaling output [1].
Diagram 2: Phosphorylation-ubiquitination crosstalk in EGFR endocytosis. EGFR autophosphorylation recruits Cbl E3 ligase, leading to receptor ubiquitination and subsequent endocytic sorting for degradation.
Signal Specificity Through Crosstalk and Insulation
The Specificity and Fidelity Problems in Signaling Networks
In complex signaling networks with multiple parallel pathways, maintaining signal specificity presents a fundamental challenge. The problems of "signal fidelity" (a pathway's capacity to prevent activation of its target by non-cognate signals) and "signal specificity" (a pathway's ability to avoid activating non-targets with its own signal) become particularly acute when pathways share components [27]. Single-cell transcriptomic analyses have revealed extensive crosstalk between cell-cell communication pathways, with shared signaling components creating potential interference between signaling channels [27].
Computational methods like SigXTalk leverage hypergraph learning frameworks to quantify pathway fidelity and specificity from single-cell RNA-seq data. This approach encodes higher-order relations among receptors, transcription factors, and target genes within regulatory pathways, allowing systematic analysis of how crosstalk affects signal specificity [27]. By measuring the relative regulatory strength of pathways within crosstalk modules, researchers can identify key shared molecules and their roles in transferring shared signaling information.
Insulating Mechanisms and Combinatorial Control
Cells employ multiple strategies to prevent inappropriate crosstalk and maintain signaling specificity. Spatial compartmentalization represents a powerful insulating mechanism, separating potentially interfering pathways into distinct cellular locations [28]. Additionally, the formation of specific multi-protein complexes creates functional insulation by bringing together dedicated signaling components while excluding others [28].
Combinatorial control represents another key strategy for maintaining specificity. The simultaneous requirement for multiple input signals to activate a pathway reduces the likelihood of accidental activation. In ubiquitination-phosphorylation crosstalk, this often manifests as requirements for specific phosphorylation patterns (rather than single phosphorylation events) to trigger ubiquitination, or the need for multiple ubiquitination events to regulate kinase activity [1]. This combinatorial logic significantly expands the coding capacity of the signaling system while maintaining specificity.
Research Reagent Solutions for Studying PTM Crosstalk
Table 3: Essential Research Tools for Analyzing Ubiquitination-Phosphorylation Crosstalk
| Research Tool | Function/Application | Key Features |
|---|---|---|
| His-tagged Ubiquitin | Enrichment of ubiquitylated proteins | Enables cobalt-NTA affinity purification of modified proteins [7] |
| Phospho-specific Antibodies | Immunoenrichment of phosphopeptides | Isolation of phosphorylated peptides for MS analysis [7] |
| diGly Antibody | Identification of ubiquitylation sites | Recognizes Gly-Gly remnant on modified lysines after tryptic digestion [7] |
| SCX Chromatography | Separation of charged peptides | Enriches for both ubiquitylated peptides and ubiquitylated phosphopeptides [7] |
| SigXTalk Algorithm | Computational crosstalk analysis | Hypergraph learning to quantify pathway fidelity and specificity from scRNA-seq [27] |
| PROTAC Molecules | Targeted protein degradation study | Heterobifunctional molecules recruiting E3 ligases to target proteins [26] |
Experimental Workflows for Global PTM Crosstalk Analysis
The systematic identification of proteins co-modified by phosphorylation and ubiquitination requires sophisticated sequential enrichment strategies. Two complementary methodological approaches have been developed to address the challenges of identifying these typically low-stoichiometry modifications [7].
The first approach employs protein-level enrichment using cobalt-NTA affinity purification to isolate His-tagged ubiquitin-modified proteins. Following digestion, phosphopeptides are enriched from both the ubiquitin-enriched and ubiquitin-depleted fractions, allowing identification of phosphorylation sites specific to ubiquitylated protein isoforms. Additionally, diGly antibody-based enrichment identifies specific ubiquitylation sites in the ubiquitin-enriched population [7].
The second approach utilizes peptide-level sequential enrichment through strong-cation exchange (SCX) chromatography to separate peptides by solution charge, followed by diGly antibody enrichment of ubiquitylated peptides from all SCX fractions. This method specifically identifies peptides concurrently modified by both phosphorylation and ubiquitylation, establishing that both modifications are present on the same protein isoform, though it is limited to modifications in close sequence proximity [7].
Diagram 3: Experimental workflow for identifying co-modified proteins. Sequential enrichment strategies enable identification of phosphorylation and ubiquitination sites on the same protein molecules.
Therapeutic Applications and Future Perspectives
The mechanistic understanding of ubiquitination-phosphorylation crosstalk has enabled groundbreaking therapeutic approaches, particularly in targeted protein degradation. PROTACs (Proteolysis-Targeting Chimeras) represent the most advanced application, utilizing heterobifunctional molecules to recruit E3 ubiquitin ligases to specific target proteins, leading to their degradation [26]. The field has expanded beyond the initial focus on CRBN and VHL ligases to include other therapeutically relevant E3 ligases such as MDM2, which exhibits dual functionality as both a regulator of p53 and an E3 ligase capable of being harnessed for targeted degradation [26].
The expansion of E3 ligases available for PROTAC design has significantly broadened the scope of targetable proteins. Current databases include 6,111 PROTACs targeting 442 distinct protein targets through 20 different E3 ligases [26]. Key targets such as the androgen receptor (AR), epidermal growth factor receptor (EGFR) mutants, and cyclin-dependent kinases (CDKs) have been the focus of extensive PROTAC development efforts, demonstrating the therapeutic potential of manipulating ubiquitination machinery.
Future research directions will likely focus on expanding the repertoire of E3 ligases suitable for therapeutic applications, developing selective ligands for previously untargetable proteins, and exploiting the oscillatory and bistable behaviors generated by PTM crosstalk for chronotherapeutic applications. The integration of computational and experimental approaches will be essential for mapping the complex dynamics of these post-translational regulatory networks and harnessing their therapeutic potential.
Post-translational modifications (PTMs) represent a crucial regulatory layer in cellular signaling, controlling protein function, localization, and stability. While phosphorylation and ubiquitination have established a well-characterized crosstalk paradigm, the network of interactions involving the Small Ubiquitin-like Modifier (SUMO) pathway with acetylation and other PTMs is rapidly emerging as an equally complex and biologically significant area of research. This expansion of the crosstalk network enables cells to implement complex signaling processing modules that function as logical gates in cellular networks, integrating signals from multiple sources to fine-tune biological responses [29] [30]. SUMOylation, the reversible covalent attachment of SUMO proteins to target substrates, regulates diverse cellular processes including transcription, cell cycle progression, DNA repair, and signal transduction [31]. The dynamic interplay between SUMOylation and other PTMs, particularly acetylation, creates sophisticated regulatory circuits that control key cellular functions, with important implications for disease mechanisms and therapeutic development.
Table 1: Key Characteristics of Major PTMs in Signaling Crosstalk
| PTM Type | Enzymes Involved | Target Residues | Primary Functions | Disease Associations |
|---|---|---|---|---|
| SUMOylation | E1 (SAE1/SAE2), E2 (Ubc9), E3 (PIAS, RanBP2), SENPs | Lysine (consensus ψKxE/D) | Transcriptional regulation, DNA repair, protein localization | Cancer, neurodegenerative diseases, autoimmune disorders [31] |
| Acetylation | HATs (p300/CBP), HDACs | Lysine | Chromatin remodeling, transcription activation, metabolic regulation | Cancer, leukemia, neurological disorders [32] |
| Phosphorylation | Kinases, Phosphatases | Serine, Threonine, Tyrosine | Signal transduction, enzyme activation, cell cycle control | Cancer, inflammatory diseases, metabolic disorders [30] [32] |
| Ubiquitination | E1-E2-E3 enzyme cascade, DUBs | Lysine | Protein degradation, endocytosis, DNA repair | Cancer, muscle wasting, viral infection [30] [32] |
Molecular Mechanisms of SUMOylation and Its Crosstalk
The SUMOylation Machinery
SUMOylation involves a well-defined enzymatic cascade that conjugates SUMO proteins to specific lysine residues on target proteins. The process begins with SUMO maturation, where newly synthesized SUMO is cleaved by SUMO-specific proteases (SENPs) to expose a C-terminal diglycine motif. The mature SUMO is then activated in an ATP-dependent manner by the heterodimeric E1 enzyme SAE1/SAE2, transferred to the E2 conjugating enzyme Ubc9, and finally ligated to target proteins with the assistance of E3 ligases such as PIAS family members, RanBP2, and Pc2 [31]. This dynamic modification is reversed through deSUMOylation catalyzed by SENPs, creating a reversible regulatory switch. Mammals express multiple SUMO paralogs (SUMO1-5) with distinct functions and cellular distributions, adding further complexity to this system [31]. SUMOylation can manifest as mono-SUMOylation, multi-SUMOylation (multiple acceptor sites), or poly-SUMOylation (SUMO chains), with SUMO2/3 being primarily responsible for chain formation due to internal consensus sequences [31].
SUMOylation-Acetylation Crosstalk: Molecular Switches and Antagonistic Regulation
The crosstalk between SUMOylation and acetylation represents a sophisticated regulatory mechanism that fine-tunes protein function, particularly for transcription factors and chromatin-associated proteins. Research has revealed that these modifications can engage in both competitive and sequential interactions on the same protein substrates, creating molecular switches that integrate diverse cellular signals.
A paradigm for this crosstalk has been established through studies on p53, where sumoylation at lysine 386 blocks subsequent acetylation by p300, while p300-acetylated p53 remains permissive for ensuing sumoylation [33]. This hierarchical modification directly impacts p53 function, as sumoylation prevents DNA binding, thereby inhibiting transcriptional activation of target genes. Importantly, acetylation can restore the DNA-binding activity of sumoylated p53, demonstrating how this antagonistic relationship creates a dynamic regulatory switch [33]. This switch-like behavior allows cells to rapidly respond to different stimuli by toggling between activating and repressive modifications on a key tumor suppressor.
Beyond direct competition on transcription factors, acetylation can directly regulate the SUMO machinery itself. SUMO proteins undergo acetylation at specific lysine residues (K37 in SUMO1 and K33 in SUMO2) within their basic interface, neutralizing positive charges and preventing interaction with SUMO-interaction motifs (SIMs) in proteins like PML, Daxx, and PIAS family members [34]. This acetyl-dependent switch expands the regulatory repertoire of SUMO signaling by determining the selectivity and dynamics of SUMO-SIM interactions, thereby affecting processes such as PML nuclear body assembly and SUMO-mediated gene silencing [34].
The reverse regulatory direction also occurs, where SUMOylation controls acetylation-dependent processes. The histone reader TRIM24 exemplifies this bidirectional crosstalk, as its binding to acetylated chromatin (H3K4me0/K23ac) via its tandem PHD-bromodomain induces TRIM24 SUMOylation at lysine residues 723 and 741 [35]. Inhibition of histone deacetylation increases TRIM24's interaction with chromatin and its subsequent SUMOylation, establishing a functional connection between histone acetylation and reader protein SUMOylation that regulates oncogenic functions in breast cancer [35].
Figure 1: SUMOylation-Acetylation Crosstalk on p53. Sumoylation at K386 inhibits DNA binding, while acetylation at K373/K382 permits or restores DNA binding capability, creating a molecular switch that regulates p53 transcriptional activity [33].
SUMOylation-Phosphorylation Interplay: Coordinated Regulation
The coordination between SUMOylation and phosphorylation represents another crucial axis in the PTM crosstalk network. These modifications can function sequentially, synergistically, or antagonistically to control protein activity, stability, and interactions. A key mechanism underlying this coordination is the presence of phosphorylation-dependent SUMOylation motifs (PDSMs), which contain a SUMO consensus site adjacent to a proline-directed phosphorylation site (ψ-K-X-D/E-X-X-S-P) [31]. This structural arrangement allows phosphorylation at the serine residue to enhance SUMOylation at the nearby lysine, creating a phospho-switch that integrates kinase signaling with the SUMO pathway.
The interplay between SUMOylation and phosphorylation also extends to their functional outcomes. For instance, SUMOylation can inhibit tyrosine phosphorylation of STAT5, converting it from an active to inactive state and thereby modulating signaling transduction [31]. Additionally, the kinase CK2 phosphorylates serine or threonine residues adjacent to the hydrophobic core of SIMs in proteins like PML and PIAS family members, enhancing their binding to SUMO through electrostatic interactions with basic residues in the SUMO interface [34]. This phosphorylation-dependent enhancement of SUMO binding affects processes such as PML nuclear body dynamics and SUMO-mediated transcriptional regulation.
SUMOylation-Ubiquitination Crosstalk: From Degradation to Stability
SUMOylation and ubiquitination share structural similarities as ubiquitin-like modifiers but often exert opposing effects on protein stability. While ubiquitination typically targets proteins for proteasomal degradation, SUMOylation can counteract this fate or even promote stability under specific contexts. This antagonistic relationship is exemplified by IκBα, where SUMOylation at lysine 21 provides protection against ubiquitin-mediated degradation, thereby modulating NF-κB signaling [31]. Furthermore, phosphorylation of IκBα at serines 32 and 36 inhibits SUMOylation, creating a complex tripartite crosstalk that integrates multiple signaling inputs [31].
Beyond simple antagonism, SUMOylation and ubiquitination can also function cooperatively through sequential modifications. Poly-SUMO chains can serve as platforms for recruitment of SUMO-targeted ubiquitin ligases (STUbLs), which subsequently ubiquitinate the substrate and target it for proteasomal degradation [31]. This SUMO-dependent ubiquitination creates a fail-safe mechanism for eliminating persistently sumoylated proteins and has been implicated in quality control pathways. Advanced proteomic studies have revealed extensive crosstalk between these pathways, with co-regulation observed on deubiquitinase enzymes and SUMOylation of proteasome subunits affecting their recruitment to subnuclear compartments like PML nuclear bodies [36].
Table 2: Experimental Evidence of SUMOylation Crosstalk with Other PTMs
| Crosstalk Type | Experimental System | Key Findings | Functional Outcome |
|---|---|---|---|
| SUMO-Acetylation | In vitro sumoylation system with p53; Cell-based assays [33] | Sumoylation at K386 blocks acetylation by p300; Acetylation restores DNA binding of sumoylated p53 | Fine-tuning of p53 transcriptional activity |
| SUMO-Acetylation | Site-specific acetylation of SUMO paralogs; Biochemical binding assays [34] | Acetylation of SUMO1-K37/SUMO2-K33 prevents binding to SIMs in PML, Daxx, PIAS | Altered PML nuclear body dynamics; Attenuated gene silencing |
| SUMO-Phosphorylation | STAT5 modification studies; CK2 phosphorylation assays [34] [31] | SUMOylation inhibits STAT5 tyrosine phosphorylation; CK2 phosphorylates SIMs to enhance SUMO binding | Modulation of signaling transduction; Enhanced SUMO-SIM interactions |
| SUMO-Ubiquitination | Sequential peptide immunopurification; Proteomic analysis [36] | Co-regulation on deubiquitinase enzymes; SUMOylation of proteasome subunits | Recruitment of proteasome to PML nuclear bodies; Regulation of protein degradation |
Methodological Approaches for Studying PTM Crosstalk
Proteomic Technologies for System-Level Analysis
Advances in mass spectrometry-based proteomics have revolutionized the study of PTM crosstalk by enabling the identification and quantification of thousands of modification sites across the proteome. The development of sequential peptide immunopurification represents a significant methodological innovation, allowing researchers to study both SUMOylation and ubiquitylation from a single biological sample [36]. This approach involves the stable expression of a 6xHis-SUMO3-Q87R/Q88N mutant in HEK293 cells, enrichment of SUMOylated proteins on NiNTA columns, followed by on-bead digestion and immunopurification of SUMO-modified peptides using specific antibodies. Optimization of this method through antibody cross-linking to magnetic beads and refined incubation buffers has achieved remarkable enrichment levels of 62.9%, facilitating the identification of over 10,000 SUMO sites in human cells [36].
Quantitative proteomic analyses using this methodology have revealed dynamic changes in the SUMOylome and ubiquitinome in response to proteasome inhibition, uncovering extensive crosstalk between these modifications in pathways controlling protein degradation. The application of high-sensitivity MS methods with optimized automatic gain control settings and extended injection times has further enhanced the depth of coverage, enabling more comprehensive mapping of the PTM crosstalk network [36]. These technological advances provide unprecedented opportunities to study SUMOylation crosstalk at a systems level, moving beyond candidate-based approaches to discover novel regulatory nodes in the PTM network.
Computational Prediction of PTM Crosstalk
The growing complexity of PTM interactions has spurred the development of computational approaches to predict and prioritize crosstalk events for experimental validation. Recent innovations include WPTCMN/PTCMN, a unified model based on a Multilayer Network structure that simultaneously predicts both intra-protein and inter-protein PTM crosstalk [37]. This integrated deep neural network incorporates evolutionary, structural, and dynamic features of proteins, using random walks to dynamically learn single-layer network features, multilayer network correlation features, and node features within the PTM crosstalk network [37].
These computational approaches address the challenges of limited and imbalanced datasets in PTM crosstalk research, providing valuable insights and guidance for future experimental investigations. The PTMcode web resource represents another valuable tool, compiling approximately 200 manually validated examples of intra-protein PTM crosstalk in the human proteome among ten selected PTM types, while predicting many more potential interactions based on same residue competition, structural proximity, and coevolution [29]. These computational resources complement experimental methods by enabling hypothesis generation and prioritization of functionally relevant crosstalk events for mechanistic validation.
Figure 2: Workflow for Sequential Analysis of SUMOylation and Ubiquitination. This optimized immunoaffinity method enables the study of both modifications from a single sample, facilitating the identification of crosstalk events [36].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for SUMOylation Crosstalk Studies
| Reagent/Category | Specific Examples | Function/Application | Experimental Context |
|---|---|---|---|
| SUMO Mutants | 6xHis-SUMO3-Q87R/Q88N; SUMO2 T91R; ZNF451 tag [36] [38] | Facilitates identification of SUMO sites; induces targeted SUMOylation | Proteomic studies; Functional validation of SUMOylation effects |
| Enzymes & Inhibitors | Proteasome inhibitor MG132; HDAC inhibitors; p300/CBP inhibitors [33] [36] | Modifies PTM pathways; reveals crosstalk dynamics | Perturbation studies to interrogate functional relationships between PTMs |
| Specific Antibodies | Anti-K-(NQTGG); Anti-di-glycine; Anti-acetyllysine; Phospho-specific antibodies [36] | Enrichment and detection of specific PTMs; Immunofluorescence | Immunopurification; Western blotting; Cellular localization studies |
| Expression Systems | HEK293-SUMO3m stable cell line; Site-specific acetylation system [34] [36] | Production of homogenously modified proteins; Controlled PTM expression | Biochemical studies; Functional characterization of specific modifications |
| Computational Tools | WPTCMN/PTCMN; PTMcode; PTM-X [29] [37] | Prediction of PTM crosstalk; Database of known interactions | Hypothesis generation; Prioritization of crosstalk events for validation |
Implications for Disease and Therapeutic Development
The intricate crosstalk between SUMOylation and other PTMs has significant implications for understanding disease mechanisms and developing targeted therapies. Aberrant SUMOylation has been implicated in various pathological conditions, including cancers, cardiovascular diseases, neurodegenerative disorders, and autoimmune diseases [31]. In cancer, the SUMO pathway contributes to malignant transformation through multiple mechanisms, including the regulation of oncogenes and tumor suppressors like p53 [33]. The discovery of crosstalk between histone acetylation and TRIM24 SUMOylation in breast cancer highlights how integrated PTM networks can drive oncogenic processes, suggesting novel therapeutic targets [35].
The therapeutic potential of targeting SUMOylation crosstalk is increasingly recognized, though this field remains relatively immature compared to kinase-targeted therapies. Current strategies include developing inhibitors of SUMO conjugation or deconjugation, as well as approaches to disrupt specific SUMO-SIM interactions [34]. The development of small-molecule inhibitors of the TRIM24 bromodomain (IACS-9571) that disrupt its association with chromatin and subsequent SUMOylation demonstrates the feasibility of targeting reader domains within the PTM crosstalk network [35]. As our understanding of SUMOylation crosstalk deepens, opportunities will emerge to develop more precise therapeutic interventions that modulate specific nodes within these complex regulatory networks rather than broadly inhibiting entire pathways.
The expanding network of crosstalk between SUMOylation, acetylation, and other PTMs represents a sophisticated regulatory layer that fine-tunes cellular signaling with remarkable precision. Through competitive, sequential, and synergistic interactions, these modifications create molecular switches and logic gates that integrate diverse cellular signals to control key biological processes. Methodological advances in proteomics and computational prediction are rapidly accelerating the discovery and characterization of these complex interactions, revealing an increasingly interconnected landscape of PTM crosstalk. As research in this field progresses, a deeper understanding of these regulatory networks will provide new insights into disease mechanisms and unlock novel therapeutic opportunities for targeting specific nodes within the PTM crosstalk network.
Decoding the Interactome: Advanced Methods to Map and Model PTM Crosstalk
In eukaryotic cell signaling, ubiquitination and phosphorylation do not function in isolation. These two major post-translational modifications (PTMs) engage in extensive crosstalk, creating a complex regulatory network that controls protein stability, activity, and interaction partners [30] [17]. This interplay manifests as either positive or negative regulation, where one modification either triggers or blocks the other [29]. A classic example is the "phosphodegron" motif, where phosphorylation of a specific site creates a recognition signal for E3 ubiquitin ligases, marking the protein for ubiquitin-mediated degradation [29] [39]. Understanding this sophisticated crosstalk requires advanced proteomic technologies capable of capturing multiple PTMs simultaneously from the same biological sample. This guide compares the performance and methodologies of sequential enrichment techniques coupled with mass spectrometry that are revolutionizing our ability to decipher this complex PTM language.
Methodological Comparison: Concurrent PTM Analysis Workflows
One-Pot Affinity Enrichment
The one-pot affinity enrichment strategy enables simultaneous extraction of multiple PTMs from a single sample digest without intermediate desalting steps. This approach is particularly valuable for studying crosstalk between acylations such as acetylation and succinylation, which often target the same lysine residues [40].
- Core Principle: A mixture of anti-acetyllysine and anti-succinyllysine antibodies conjugated to beads is incubated with a tryptic digest, allowing concurrent enrichment of both modification types.
- Sample Requirements: Functions effectively with low protein input (100 μg to 1 mg) [40].
- MS Acquisition: Employs Data-Independent Acquisition (DIA) which comprehensively fragments all peptide signals within sampled m/z ranges, overcoming the stochastic sampling limitations of Data-Dependent Acquisition and improving site localization for PTM isoforms [40].
Sequential Enrichment of Post-Translational Modifications (SEPTM)
The SEPTM approach systematically isolates different PTMs from a single protein extract through a series of sequential purification steps, preserving the connectivity between modifications across the proteome [41].
- Core Principle: Beginning with a single protein extract, the method typically enriches for tyrosine-phosphorylated peptides first, followed by sequential enrichment of ubiquitinated, and then acetylated peptides [41].
- Application Power: This methodology has been successfully applied to map coordinated changes in phosphorylation, ubiquitination, and acetylation in lung cancer cell lines treated with tyrosine kinase inhibitors, revealing drug-specific PTM signatures and crosstalk between oncogenic signaling and metabolic pathways [41].
Tandem Enrichment (SCASP-PTM)
The SCASP-PTM protocol represents a streamlined tandem enrichment strategy designed specifically for ubiquitinated, phosphorylated, and glycosylated peptides [42].
- Core Principle: Utilizes the SDS-cyclodextrin-assisted sample preparation method to efficiently extract and digest proteins, followed by serial enrichment of PTMs without intermediate desalting steps [42].
- Workflow Advantage: This "tandem" process reduces sample loss and processing time compared to completely separate enrichments, making it particularly suitable for limited clinical samples [42].
Table 1: Comparative Analysis of Concurrent PTM Proteomic Methods
| Method Characteristic | One-Pot Affinity Enrichment | Sequential Enrichment (SEPTM) | Tandem Enrichment (SCASP-PTM) |
|---|---|---|---|
| PTMs Analyzed | Acetylation, Succinylation [40] | Phosphorylation, Ubiquitination, Acetylation [41] | Ubiquitination, Phosphorylation, Glycosylation [42] |
| Key Advantage | Preserves stoichiometric relationships between co-occurring PTMs; low input requirement [40] | Comprehensive PTM profiling from a single biological source; deep coverage [41] | Streamlined serial processing without desalting; maintains modification connectivity [42] |
| Sample Throughput | High (simplified workflow) | Moderate (multiple enrichment steps) | High (minimal processing steps) |
| MS Method | Data-Independent Acquisition (DIA) [40] | Data-Dependent Acquisition (DDA) or DIA [41] | Data-Independent Acquisition (DIA) [42] |
| Crosstalk Insight | Identifies competitive or cooperative modifications on adjacent residues [40] | Reveals system-wide coordination between different PTM types [41] | Elucidates relationships between degradation, signaling, and structural modifications [42] |
Experimental Protocols for Key Workflows
Detailed Protocol: One-Pot Affinity Enrichment for Acylations
Protein Extraction and Digestion:
- Homogenize tissue or cells in urea-based lysis buffer supplemented with deacetylase inhibitors (e.g., trichostatin A, nicotinamide) and protease/phosphatase inhibitors [40].
- Reduce proteins with dithiothreitol (DTT), alkylate with iodoacetamide (IAA), and dilute urea concentration to <2M.
- Digest with trypsin (1:50 ratio) at 37°C for 14-16 hours and quench with formic acid [40].
Simultaneous PTM Enrichment:
- Desalt the peptide digest using C18 solid-phase extraction.
- Incubate the peptide mixture with a combined bead slurry containing anti-acetyllysine and anti-succinyllysine antibodies (approximately 20-30 μL of bead slurry per mg of protein) for 2 hours at 4°C with gentle agitation [40].
- Wash beads extensively with ice-cold PBS and elute enriched peptides with acidified elution buffer (0.1% TFA).
Mass Spectrometric Analysis:
- Analyze eluted peptides using LC-MS/MS with a DIA method.
- Generate spectral libraries using Data-Dependent Acquisition (DDA) runs of enriched samples.
- Process DIA data with specialized software (e.g., Spectronaut, DIA-NN) for PTM identification and quantification [40].
Detailed Protocol: SEPTM for Phosphorylation, Ubiquitination, and Acetylation
Initial Protein Processing:
- Lyse cells or tissues in a denaturing buffer compatible with subsequent enrichments.
- Digest proteins with trypsin after reduction and alkylation.
Sequential Enrichment Steps:
- First Step: Enrich tyrosine-phosphorylated peptides using anti-phosphotyrosine antibodies.
- Second Step: Subject the flow-through to enrichment for ubiquitinated peptides using anti-diGly remnant antibodies (which recognize the Gly-Ggly remnant left on ubiquitinated peptides after tryptic digestion).
- Third Step: Use the subsequent flow-through for enrichment of acetylated peptides with anti-acetyllysine antibodies [41].
MS Data Acquisition and Analysis:
- Analyze each enriched fraction separately by LC-MS/MS using either DDA or DIA methods.
- Process data to identify and quantify PTM sites, then integrate results to identify coordinated changes across different PTM types [41].
Signaling Pathway Applications: From Cancer to Immunity
Concurrent PTM analysis has revealed sophisticated crosstalk mechanisms in diverse biological contexts:
In the EGFR-MAPK signaling pathway, phosphorylation and ubiquitination engage in intricate interplay. Following EGF stimulation, EGFR undergoes autophosphorylation, creating docking sites for the Cbl E3 ubiquitin ligase, which then mediates receptor ubiquitination [30]. This ubiquitination promotes EGFR endocytosis and degradation, but can be reversed by deubiquitinating enzymes like USP8, creating a dynamic regulatory switch [30].
In plant immunity, the interplay between ubiquitination and phosphorylation provides a regulatory mechanism for rapid response to pathogens. For instance, the immune function of the WRKY45 transcription factor in rice is co-regulated by PUB44-mediated ubiquitination and MPK6-mediated phosphorylation [17] [39].
In lung cancer, SEPTM analysis connected oncogenic signaling with metabolic reprogramming. Network models constructed from phosphorylation, ubiquitination, and acetylation data revealed crosstalk between receptor tyrosine kinase signaling and glycolysis/gluconeogenesis pathways, suggesting new therapeutic targets for combination therapy [41].
The diagram below illustrates the core workflow and biological insight of sequential PTM analysis:
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Research Reagents for Concurrent PTM Analysis
| Reagent / Material | Function in Protocol | Specific Examples |
|---|---|---|
| PTM-Specific Antibodies | Immunoaffinity enrichment of modified peptides | Anti-phosphotyrosine, anti-acetyllysine, anti-succinyllysine, anti-diGly (ubiquitin remnant) antibodies [40] [41] |
| Chromatography Resins | Peptide separation and desalting | C18 solid-phase extraction cartridges, reversed-phase nanoLC columns [43] [40] |
| Protease/Phosphatase Inhibitors | Preservation of PTM states during extraction | Cocktails containing serine/threonine and tyrosine phosphatase inhibitors, deubiquitinase inhibitors [40] |
| Deacetylase Inhibitors | Prevention of acylation loss | Trichostatin A (TSA), nicotinamide [40] |
| Mass Spectrometry Standards | Quantification and quality control | Stable isotope-labeled peptides, tandem mass tag (TMT) reagents [43] |
The advancement of sequential enrichment strategies coupled with sophisticated mass spectrometry has transformed our ability to decipher the complex crosstalk between ubiquitination, phosphorylation, and other PTMs. The one-pot, sequential (SEPTM), and tandem enrichment methods each offer distinct advantages for specific research questions, sample types, and throughput requirements. As these proteomic powerhouses continue to evolve, they will undoubtedly uncover deeper insights into how the dynamic interplay between PTMs orchestrates cellular signaling in health and disease, particularly in therapeutically relevant areas such as cancer and immune signaling where ubiquitination-phosphorylation crosstalk plays a fundamental role.
The integration of multi-layered post-translational modification (PTM) data through computational network biology has emerged as a powerful strategy for elucidating complex cell signaling pathways in cancer research. This guide objectively compares two complementary network modeling approaches—Co-cluster Correlation Networks (CCCNs) and Cluster-Filtered Networks (CFNs)—for investigating crosstalk between ubiquitination and phosphorylation in signaling pathways. We evaluate their performance characteristics, data requirements, and applications in drug discovery research, supported by experimental data and detailed methodologies. By providing a structured comparison of workflows, output interpretability, and practical implementation considerations, this analysis aims to equip researchers with the knowledge to select and implement appropriate network strategies for their specific signaling pathway investigations.
Cell signaling pathways rely on complex interplay between post-translational modifications, particularly ubiquitination and phosphorylation, to regulate protein activity, localization, and degradation. Ubiquitination, once considered primarily a degradation signal, is now recognized as a versatile modification that regulates diverse processes including signal transduction, enzymatic activation, and endocytosis [1]. Phosphorylation, mediated by kinases and phosphatases, serves as the primary switch for activating signaling cascades. The crosstalk between these modifications creates sophisticated regulatory networks that control cellular decisions in health and disease [1]. Computational network biology approaches have become essential for mapping these complex relationships, with CCCNs and CFNs emerging as specialized frameworks for integrating multi-PTM data to reveal functional modules and pathway crosstalk in cancer signaling research [41].
Methodological Frameworks: CCCNs vs. CFNs
Core Definitions and Structural Characteristics
Co-cluster Correlation Networks (CCCNs) represent networks where edges connect proteins whose PTM patterns co-cluster across multiple experimental conditions or treatments. The fundamental premise is that proteins showing coordinated PTM changes likely participate in related functional modules or signaling pathways [41].
Cluster-Filtered Networks (CFNs) are constructed by filtering existing protein-protein interaction (PPI) databases to include only interactions between proteins whose PTMs co-cluster in a given dataset. This approach integrates experimental PTM data with curated biological knowledge [41].
Table 1: Fundamental Characteristics of CCCNs and CFNs
| Feature | CCCNs | CFNs |
|---|---|---|
| Network Basis | Data-driven from PTM correlations | Knowledge-based from curated PPI databases |
| Edge Meaning | Co-clustering relationship between PTM patterns | Physical or functional interaction between proteins |
| Data Sources | Multi-PTM proteomics (e.g., phosphorylation, ubiquitination, acetylation) | STRING, GeneMania, BioPlex, PathwayCommons |
| Construction Approach | Unsupervised clustering of PTM patterns | Filtering of PPI networks using cluster information |
| Primary Application | Discovering novel functional modules | Contextualizing PTM data within known interactions |
Experimental Workflows and Protocols
The implementation of both CCCN and CFN approaches begins with comprehensive PTM data acquisition. The sequential enrichment of post-translational modification (SEPTM) proteomics methodology enables concomitant identification of tyrosine phosphorylated, ubiquitinated, and acetylated proteins from the same samples [41]. This integrated approach is crucial for capturing the interplay between different modification types.
Sample Preparation and PTM Enrichment Protocol:
- Cell Culture and Treatment: Culture appropriate cell lines (e.g., lung cancer cell lines with driver mutations in EGFR, ALK, ERBB2/HER2, ROS1, DDR2) and treat with tyrosine kinase inhibitors (TKIs) such as crizotinib, erlotinib, dasatinib, and afatinib versus DMSO controls [41].
- Protein Extraction and Digestion: Lyse cells in appropriate buffer, quantify protein concentration, and perform tryptic digestion.
- Sequential PTM Enrichment: Implement SEPTM proteomics using:
- Immunoaffinity purification for phosphotyrosine peptides
- Subsequent enrichment for ubiquitinated peptides using diGly remnant antibodies
- Acetylation enrichment using specific antibodies [41]
- Mass Spectrometry Analysis: Analyze samples using nano-reversed-phase liquid chromatography coupled to tandem mass spectrometry (nRPLC-MS/MS) with appropriate replicates.
Computational Analysis Pipeline:
- PTM Identification and Quantification: Process raw MS data using tools like MaxQuant for identification and quantification of PTM sites.
- Data Filtering: Retain PTMs showing significant changes (e.g., ≥2.25-fold up or down compared to controls) [41].
- Clustering Analysis: Apply machine learning algorithms (consensus clustering) to identify groups of PTMs that show coordinated changes across treatments.
- Network Construction:
- For CCCNs: Create edges between proteins whose PTMs belong to the same cluster, representing co-regulated functional modules.
- For CFNs: Filter curated PPI networks to retain only interactions between proteins whose PTMs co-cluster, creating a context-specific network.
Figure 1: Integrated Workflow for CCCN and CFN Construction from Multi-PTM Data
Performance Comparison and Experimental Data
Application in Lung Cancer Signaling
In a comprehensive study analyzing PTM responses to TKIs in ten lung cancer cell lines, both CCCNs and CFNs demonstrated complementary strengths for elucidating signaling pathways [41]. The study identified 12,461 unique PTMs, with 4,987 phosphorylation sites, 3,249 acetylation sites, and 4,452 ubiquitination sites changed by at least 2.25-fold in response to TKI treatment [41].
Table 2: Performance Comparison in Lung Cancer Signaling Study
| Metric | CCCN Performance | CFN Performance |
|---|---|---|
| Nodes Identified | 60 phosphorylation, 21 acetylation,13 ubiquitination sites affected by all TKIs | Core subnetwork of 273 nodeswith 1,060 edges |
| Novel Pathway Insights | Revealed coordinated PTM responsesacross modification types | Identified crosstalk between EGFR/ALKsignaling and metabolic pathways |
| Validation Rate | Functional modules enriched forkinase activity and protein binding | Common core with previous studiesdespite different methodologies |
| Key Finding | Dasatinib and afatinib predominantlyinhibited phosphorylation and acetylation | Revealed connections between RTK signalingand transmembrane transport |
The CCCN approach successfully identified a core set of 60 phosphorylation, 21 acetylation, and 13 ubiquitination sites that were affected by all four TKIs, suggesting these represent central hubs in lung cancer signaling networks [41]. Functional enrichment analysis of these multi-drug-affected PTMs revealed connections from tyrosine kinase signaling to essential cellular processes, demonstrating the ability of CCCNs to identify functionally significant modules.
The CFN constructed from the same data comprised a core subnetwork of 273 nodes with 1,060 edges, highlighting interactions between heat shock/chaperone proteins, metabolic enzymes, cytoskeletal components, and RNA-binding proteins [41]. Comparison with a CFN generated from a previous multi-PTM analysis revealed a common core network despite differences in PTM data acquisition methodology, cell lines, and drugs used, supporting the robustness of this approach.
Analysis of Ubiquitination-Phosphorylation Crosstalk
The integration of ubiquitination and phosphorylation data within network models has revealed intricate crosstalk mechanisms. In a study of WNK4 kinase regulation, researchers identified specific molecular mechanisms where phosphorylation serves as a prerequisite for ubiquitination [6]. Under high-salt conditions, decreased phosphorylation at Ser-1022 led to reduced ubiquitination at Lys-1023, resulting in impaired WNK4 degradation and increased protein abundance—a mechanism relevant to salt-sensitive hypertension [6].
Global analyses in yeast have identified 466 proteins with 2,100 phosphorylation sites co-occurring with 2,189 ubiquitylation sites, demonstrating the extensive overlap between these modification systems [7]. Notably, phosphorylation sites found co-occurring with ubiquitylation showed significantly higher evolutionary conservation than other phosphorylation sites, suggesting functional importance for these coordinated modification sites [7].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for CCCN and CFN Construction
| Reagent/Category | Specification | Application | Example Function |
|---|---|---|---|
| Cell Lines | Lung cancer lines with drivermutations (EGFR, ALK, ROS1) | Pathway perturbation studies | Provide context with specificoncogenic signaling backgrounds |
| Tyrosine Kinase Inhibitors | Crizotinib, Erlotinib,Dasatinib, Afatinib | Experimental perturbation | Inhibit specific kinase targetsto perturb signaling networks |
| PTM Enrichment Antibodies | Anti-phosphotyrosine,Anti-diGly (Ubiquitin remnant),Anti-acetyllysine | SEPTM proteomics | Selective enrichment of modifiedpeptides for MS analysis |
| Mass Spectrometry | nRPLC-MS/MS systems | PTM identification and quantification | High-sensitivity detectionof modified peptides |
| PPI Databases | STRING, GeneMania,BioPlex, PathwayCommons | CFN construction | Source of curated proteininteractions for filtering |
| Clustering Algorithms | Consensus clustering,Machine learning approaches | CCCN construction | Identification of coordinatedPTM patterns across conditions |
Comparative Analysis of Strengths and Limitations
Technical Performance Characteristics
CCCNs excel in discovering novel functional relationships without prior knowledge constraints. The data-driven nature of CCCNs makes them particularly valuable for identifying previously uncharacterized signaling modules and PTM coordinations. However, they may generate connections that lack direct biological interaction mechanisms and require significant computational resources for large datasets.
CFNs provide strong biological context by integrating experimental data with curated knowledge. The reliance on existing PPI databases enhances biological interpretability but may miss novel interactions not present in reference databases. CFNs effectively reduce the complexity of comprehensive PPI networks to context-specific, functionally relevant subsets.
Implementation Considerations for Signaling Studies
For investigating ubiquitination-phosphorylation crosstalk, CCCNs offer advantages in identifying coordinated changes between these modifications across different signaling conditions. The data-driven clusters can reveal how phosphorylation events might trigger subsequent ubiquitination (as in phosphodegron regulation) or how ubiquitination might modulate kinase activity [1].
CFNs provide a structured framework for contextualizing ubiquitination-phosphorylation crosstalk within established pathway architectures. For example, CFN analysis has revealed connections between receptor tyrosine kinase signaling and metabolic pathways through ubiquitination-phosphorylation coordinated networks [41].
Figure 2: Complementary Approaches to Studying Ubiquitination-Phosphorylation Crosstalk
CCCNs and CFNs represent complementary approaches for mapping the complex interplay between ubiquitination and phosphorylation in cell signaling pathways. CCCNs offer a discovery-oriented framework for identifying coordinated PTM patterns without prior knowledge constraints, while CFNs provide biological context by integrating experimental data with curated interaction knowledge.
For research focused on novel pathway discovery in uncharacterized signaling contexts, CCCNs provide superior capability for identifying previously unknown relationships between ubiquitination and phosphorylation events. For studies aiming to contextualize PTM data within established pathway architectures or to identify points of crosstalk between known pathways, CFNs offer enhanced biological interpretability.
The most powerful applications emerge from integrated use of both approaches, leveraging CCCNs to discover novel coordination between ubiquitination and phosphorylation events and CFNs to contextualize these findings within biological pathway frameworks. This combined strategy has proven effective in revealing new connections between oncogenic signaling and metabolic reprogramming in lung cancer, identifying potential targets for combination therapy approaches [41].
As the field advances, the integration of these network approaches with temporal data capturing the dynamics of ubiquitination-phosphorylation crosstalk will further enhance our understanding of how these key post-translational modifications cooperate to control cell signaling decisions in health and disease.
Cell signaling crucially depends on a repertoire of post-translational modification (PTM) mechanisms for its regulation, with ubiquitination and phosphorylation representing two of the most prevalent and functionally significant PTMs in eukaryotic cells [30] [7]. The interplay between these modification systems has emerged as a recurrent theme in cell signalling regulation, adding specificity and combinatorial logic to signal processing that extends far beyond their individual functions [30] [7]. Ubiquitination, once primarily recognized as a tag for proteosomal degradation, is now known to regulate diverse cellular processes including signal transduction, enzymatic activation, endocytosis, trafficking, and DNA repair through both degradative and non-degradative mechanisms [30]. Understanding how these two major PTMs interact to regulate signal transduction represents a critical frontier in cell signaling research, particularly given the implications for human diseases including cancer, neurodegeneration, and metabolic disorders [30] [16].
The complexity of PTM crosstalk arises from several fundamental differences between the modification systems. Phosphorylation involves a relatively straightforward single-step addition of a phosphate group primarily to serine, threonine, tyrosine, or histidine residues, with only a single phosphate group able to be added to a particular residue [30]. In contrast, ubiquitination occurs through a three-step enzymatic cascade requiring E1 (activating), E2 (conjugating), and E3 (ligase) enzymes, targets only lysine residues, but can attach multiple ubiquitin residues in various configurations including monoubiquitination, multi-monoubiquitination, and polyubiquitin chains with diverse linkage types that dictate functional outcomes [30]. This inherent complexity, combined with the extensive cross-regulation between these systems, creates a signaling landscape of overwhelming complexity that necessitates quantitative frameworks based on systems biology and mathematical modeling to efficiently understand their regulatory roles in cell signaling [30].
Molecular Mechanisms of Ubiquitination-Phosphorylation Crosstalk
Fundamental Crosstalk Mechanisms
Crosstalk between ubiquitination and phosphorylation operates through several well-characterized molecular mechanisms that enable sophisticated signal processing capabilities. The most extensively documented form of crosstalk occurs through phosphodegrons - specific phosphorylation motifs that function in a cis-regulatory manner to promote subsequent ubiquitination and degradation of substrates [7]. Global proteomic analyses in Saccharomyces cerevisiae have identified 466 proteins with 2,100 phosphorylation sites co-occurring with 2,189 ubiquitylation sites, demonstrating the remarkable prevalence of this coordination [7]. Evolutionary conservation analysis reveals that phosphorylation sites found co-occurring with ubiquitylation are significantly more conserved than other phosphorylation sites, suggesting strong functional importance for these coordinated modification sites [7].
Beyond phosphodegrons, multiple additional crosstalk mechanisms exist. Phosphorylation can directly regulate the activity of E3 ubiquitin ligases, as exemplified by the Cbl family of E3 ligases that recognize phosphotyrosine residues on activated receptors like EGFR through their tyrosine kinase binding (TKB) domains [30]. Subsequent phosphorylation of Cbl itself on critical tyrosine residues (Y371 in c-Cbl) enables conformational changes that expose the RING domain and stimulate E3 ligase activity [30]. Conversely, ubiquitination can regulate kinase activity, creating bidirectional communication between these modification systems [7]. Additionally, recent research has revealed that ubiquitination can target proteins for degradation through SUMO-primed ubiquitination mechanisms, as exemplified by SUMO-targeted ubiquitin ligases (StUbLs) that mediate ubiquitylation of proteins primed by SUMO modification [44].
Representative Pathway: EGFR-Mediated MAPK Signaling
The epidermal growth factor receptor (EGFR)-mediated MAPK pathway provides a well-characterized example of intricate ubiquitination-phosphorylation crosstalk with implications for cancer biology. Following ligand binding and EGFR activation by autophosphorylation, the E3 ligase Cbl is recruited either directly through phosphotyrosine recognition via its TKB domain or indirectly through adaptor proteins like Grb2 [30]. This recruitment leads to Cbl phosphorylation and activation, resulting in multi-monoubiquitination and polyubiquitination of EGFR [30].
The ubiquitination status of EGFR then determines its trafficking fate and downstream signaling dynamics. Ubiquitinated EGFR associates with adaptor proteins containing ubiquitin-binding domains (e.g., UIM domains), directing receptor internalization through clathrin-coated pits and subsequent sorting toward lysosomal degradation via the ESCRT complexes [30]. This process is dynamically regulated by deubiquitinating enzymes (DUBs) like STAMBP (AMSH) and USP8, which can reverse ubiquitination and redirect EGFR toward recycling pathways back to the plasma membrane [30]. The phosphorylation status of these regulatory DUBs themselves adds another layer of control, as USP8 undergoes tyrosine and serine phosphorylation in an EGFR- and Src-kinase dependent manner, potentially regulating its DUB activity and creating complex feedback regulation [30].
Table 1: Key Molecular Mechanisms of Ubiquitination-Phosphorylation Crosstalk
| Mechanism | Molecular Implementation | Functional Outcome | Representative Examples |
|---|---|---|---|
| Phosphodegron | Phosphorylation creates recognition motif for E3 ligases | Targeted substrate degradation | Cell cycle regulators |
| E3 Ligase Regulation | Phosphorylation activates/deactivates E3 ligases | Control of ubiquitination activity | Cbl family E3 ligases |
| SUMO-Primed Ubiquitination | SUMOylation recruits StUbL complexes | Degradation of SUMOylated targets | PML-RARα, ERα targeting |
| Adaptor Protein Modification | Coupled monoubiquitination of UBD-containing adaptors | Regulation of cargo recognition and trafficking | EPS15, Epsin families |
| DUB Regulation | Phosphorylation of deubiquitinating enzymes | Control of ubiquitination reversal | USP8 phosphorylation |
Mathematical Modeling Approaches for PTM Crosstalk
Modeling Frameworks and Applications
Mathematical modeling provides essential tools for understanding the complex dynamics emerging from ubiquitination-phosphorylation crosstalk. The choice of modeling approach depends on multiple factors including the biological question, extent of mechanistic detail required, and data availability [45]. For the mTOR signaling pathway, which integrates inputs from growth factors, nutrients, energy status, stress, and oxygen, multiple modeling frameworks have been successfully applied [45]. Ordinary differential equation (ODE)-based models have been particularly valuable for capturing the dynamic behavior of mTOR signaling networks, with models ranging from small networks (3-4 components) to comprehensive systems (78 components) simulating processes across timescales from minutes to several hours [45].
These mathematical approaches have revealed that the interplay between feedback regulations and nonlinear PTM cascades can generate rich dynamic behaviors including bistable switches, multistability, and sustained oscillations [30]. For example, both short-period (20-minute) and long-period (4-5 hour) ERK oscillations have been experimentally observed and mathematically modeled [30]. Similarly, GTPase cascades exhibit oscillations that drive periodic protrusion and retraction of lamellipodia during cell migration [30]. The emergence of such complex dynamics from relatively simple network architectures underscores the value of mathematical modeling for elucidating non-intuitive system behaviors that would be difficult to predict through qualitative reasoning alone.
mTOR Pathway Modeling Exemplar
The mTOR signaling pathway exemplifies the successful application of mathematical modeling to understand complex PTM networks. mTOR exists in two distinct complexes (mTORC1 and mTORC2) that integrate signals from multiple upstream pathways including growth factors through PI3K-AKT, energy status through AMPK, nutrients through Rag GTPases, and stress through various sensors [45]. Mathematical models have been particularly instrumental in understanding the functional consequences of multiple feedback loops embedded within this network, including: (1) a positive feedback from AKT to insulin receptor substrate-1 (IRS1) that maintains IRS1 in an active state; (2) a negative feedback from S6K to IRS1 that attenuates PI3K activation; and (3) a positive feedback from AKT to mTORC2 that enhances mTORC2 activity [45].
These models demonstrate how feedback regulation creates complex system dynamics that impact therapeutic interventions. For instance, the limited clinical success of rapamycin and other mTOR-targeted drugs has been partially explained through mathematical modeling revealing that drug effects on mTOR signaling are more complex than initially anticipated, with feedback mechanisms causing counterintuitive system responses [45]. This modeling insight has guided the development of more sophisticated therapeutic strategies that account for these dynamic network properties.
Table 2: Mathematical Models of mTOR Signaling Pathway
| Model Reference | Biological Context | Model Type | Model Size (Components) | Timescale | Key Insights |
|---|---|---|---|---|---|
| Dalle Pezze et al. | mTORC2 regulation mechanisms | ODE | 25-33 | 0-120 min | Dynamics of mTORC2-AKT feedback |
| Sonntag et al. | mTOR-AMPK crosstalk | ODE | 26-28 | 0-120 min | Energy sensing integration |
| Kubota et al. | Insulin signal decoding | ODE | 11 | 0-600 min | Long-term signaling dynamics |
| Borisov et al. | MAPK-mTOR crosstalk | ODE | 78 | 0-30 min | Complex interpathway regulation |
| Toyoshima et al. | Signal transfer mechanisms | ODE | 3-4 | 0-120 min | Core signaling principles |
Experimental Approaches for Studying PTM Crosstalk
Proteomic Methodologies
Global analysis of phosphorylation and ubiquitylation crosstalk requires specialized proteomic methodologies capable of capturing co-modified protein isoforms. Two primary enrichment strategies have been developed to address this challenge [7]. The first approach employs sequential protein-level enrichment, beginning with cobalt-NTA affinity purification to enrich for His-tagged ubiquitin-modified proteins, followed by tryptic digestion and subsequent phosphopeptide enrichment using anti-phosphoantibody-based methods [7]. This method identifies proteins modified by both ubiquitylation and phosphorylation regardless of modification proximity, but cannot establish whether both modifications coexist on the same protein molecule.
The second methodology utilizes sequential peptide-based enrichment through strong-cation exchange (SCX) chromatography to separate peptides by solution charge, followed by antibody-based enrichment of diGly-modified peptides (containing the characteristic ubiquitin remnant) from each fraction [7]. This approach can directly identify peptides concurrently modified by phosphorylation and ubiquitylation, establishing that both PTMs are present on the same protein isoform, but is limited to identifying PTM sites found in close sequence proximity. Application of these complementary methods in Saccharomyces cerevisiae identified 466 co-modified proteins with 2,100 phosphorylation sites co-occurring with 2,189 ubiquitylation sites, demonstrating the extensive coordination between these modification systems [7].
Reconstitution Approaches for Lipid-Dependent Regulation
Recent advances in reconstitution biochemistry have enabled detailed mechanistic studies of how membrane composition modulates ubiquitination cascades, particularly for ER-associated degradation (ERAD) pathways. Studies with purified human ERAD components reconstituted into membranes of defined lipid composition have revealed that the membrane-anchored E2 enzyme UBE2J2 acts as a sensor for lipid packing [5]. In loosely packed membranes resembling the native ER environment, UBE2J2 becomes inactive due to membrane association that impedes ubiquitin loading, while tighter lipid packing promotes its active conformation and interaction with E1 [5].
This reconstitution approach has further demonstrated that UBE2J2 activity directs ubiquitin transfer by multiple E3 ligases including RNF145, MARCHF6, and RNF139, affecting both auto-ubiquitination and substrate ubiquitination [5]. Simultaneously, specific E3 ligases like RNF145 can directly sense cholesterol levels, altering their oligomerization state and activity [5]. These findings reveal a multi-layered regulatory mechanism where ERAD integrates multiple lipid signals through coordinated regulation of both E2 and E3 components, with UBE2J2 serving as a central relay point that extends the effect of lipid packing signals through its cooperation with multiple E3 ligases [5].
Table 3: Essential Research Reagents for Studying Ubiquitination-Phosphorylation Crosstalk
| Reagent/Resource | Category | Function/Application | Representative Examples |
|---|---|---|---|
| His-Tagged Ubiquitin | Protein Modification Tool | Enables affinity purification of ubiquitylated proteins | His-Ub yeast strains [7] |
| diGly-Specific Antibody | Proteomic Reagent | Enrichment of ubiquitylation sites for mass spectrometry | Anti-K-ε-GG antibody [7] |
| Phospho-Specific Antibodies | Proteomic Reagent | Enrichment of phosphopeptides for mass spectrometry | Anti-phosphotyrosine, anti-phosphoserine antibodies [7] |
| Cobalt-NTA Resin | Affinity Purification Matrix | Isolation of His-tagged ubiquitin conjugates | Co2+-NTA chromatography [7] |
| SCX Chromatography | Separation Media | Peptide fractionation by charge prior to PTM enrichment | Strong-cation exchange columns [7] |
| Defined Lipid Compositions | Membrane Biochemistry | Reconstitution of membrane-associated ubiquitination | Synthetic liposomes with controlled saturation [5] |
| Purified E1, E2, E3 Enzymes | Enzymatic Components | Reconstitution of ubiquitination cascades in vitro | UBE2J2, RNF145, MARCHF6 [5] |
| Mathematical Modeling Software | Computational Tools | Dynamic simulation of signaling networks | CellDesigner [46], ODE solvers [45] |
Future Perspectives and Therapeutic Applications
The emerging understanding of ubiquitination-phosphorylation crosstalk has opened new therapeutic avenues, particularly through the development of proteolysis-targeting chimeras (PROTACs) and related modalities that harness cellular degradation machinery. The concept of reprogramming cellular ubiquitination systems for targeted protein degradation represents a promising pharmacological approach, as exemplified by natural products like arsenic trioxide and fulvestrant that leverage SUMOylation-ubiquitylation cascades to inactivate oncogenic fusion proteins PML-RARα and estrogen receptor α, respectively [44]. Recent proof-of-concept studies suggest that proximity-inducing modalities can recruit aggregation-prone proteins to the StUbL machinery, potentially mitigating the formation of neurotoxic inclusions in neurodegenerative diseases [44].
Future research directions will likely focus on expanding our understanding of the versatile dynamics arising from PTM crosstalk in spatial and temporal contexts. As noted in studies of EGFR trafficking and ERAD lipid sensing, the subcellular localization of modification events introduces critical regulatory dimensions that remain incompletely characterized [30] [5]. Similarly, the development of more sophisticated mathematical models that incorporate spatial compartmentalization, stochastic fluctuations, and multi-scale phenomena will enhance our ability to predict emergent behaviors in PTM networks [30] [45]. The continued integration of quantitative experimental approaches with mathematical modeling frameworks promises to transform our understanding of how ubiquitination-phosphorylation crosstalk drives cellular decision-making in health and disease.
The Epidermal Growth Factor Receptor (EGFR) and its downstream Mitogen-Activated Protein Kinase (MAPK) pathway constitute a critical signaling axis governing fundamental cellular processes including proliferation, differentiation, and survival. Dysregulation of this pathway is a hallmark of numerous cancers, driving extensive efforts to develop targeted therapies. Beyond the well-characterized role of phosphorylation in EGFR/MAPK signaling, ubiquitination has emerged as an equally powerful regulatory modification that exhibits intricate crosstalk with phosphorylation events. This interplay creates a sophisticated control system that determines the intensity, duration, and spatial organization of signaling outputs. Understanding the precise mechanisms of ubiquitin-phosphorylation crosstalk is not merely an academic exercise but provides crucial insights into resistance mechanisms for current EGFR-targeted therapies and reveals novel therapeutic vulnerabilities for cancer intervention [1] [47].
The ubiquitin system demonstrates remarkable complexity, rivaling that of phosphorylation. While phosphorylation involves straightforward addition of phosphate groups to serine, threonine, or tyrosine residues, ubiquitination employs a three-enzyme cascade (E1-E2-E3) and can generate diverse ubiquitin chain topologies through its seven lysine residues (K6, K11, K27, K29, K33, K48, K63) or N-terminal methionine (M1), each capable of encoding distinct functional outcomes [1] [48]. This review systematically compares the experimental approaches for investigating ubiquitin-phosphorylation crosstalk within the EGFR/MAPK pathway, providing researchers with methodological frameworks to advance this dynamically evolving field.
Comparative Features of Ubiquitination and Phosphorylation
Table 1: Fundamental characteristics of ubiquitination and phosphorylation modifications
| Feature | Ubiquitination | Phosphorylation |
|---|---|---|
| Chemical Nature | Covalent attachment of 76-amino acid ubiquitin protein | Covalent attachment of phosphate group |
| Amino Acid Targets | Primarily lysine residues | Serine, threonine, tyrosine, histidine |
| Enzyme Machinery | E1 (activating), E2 (conjugating), E3 (ligating) enzymes | Kinases (add phosphate), phosphatases (remove phosphate) |
| Chain Complexity | Monoubiquitination, multimonoubiquitination, homotypic/heterotypic polyubiquitin chains | Single modification per residue |
| Primary Functions | Protein degradation, endocytosis, signal transduction, DNA repair, kinase activation | Protein activation/inactivation, protein-protein interactions, signal transduction |
| Dynamics | Regulated by E3 ligases and deubiquitinases (DUBs) | Regulated by kinases and phosphatases |
| EGFR/MAPK Roles | Receptor endocytosis, degradation, signal modulation | Receptor activation, kinase cascade propagation |
Key Regulatory Nodes of Ubiquitin-Phosphorylation Crosstalk in EGFR/MAPK Signaling
EGFR Receptor Initiation and Endocytosis
The activation of EGFR represents the initial node where ubiquitination and phosphorylation intersect. Upon ligand binding, EGFR undergoes autophosphorylation at specific tyrosine residues within its intracellular domain, creating docking sites for signaling adaptors. Notably, phosphorylation at tyrosine 1045 creates a binding site for Cbl family E3 ubiquitin ligases, which subsequently ubiquitinate the receptor [1]. This phosphorylation-directed ubiquitination serves as a critical regulatory mechanism that controls receptor internalization and endosomal sorting, ultimately determining whether receptors are recycled to the membrane or targeted for lysosomal degradation [1] [47].
The Cbl family E3 ligases exemplify the bidirectional nature of ubiquitin-phosphorylation crosstalk. These ligases are recruited to phosphorylated EGFR through their tyrosine kinase-binding (TKB) domains. Once bound, Cbl itself becomes phosphorylated at tyrosine 371 (in c-Cbl), inducing a conformational change that activates its E3 ligase function and enables ubiquitin transfer to EGFR [1]. This creates a feed-forward loop where phosphorylation activates ubiquitination, which in turn limits phosphorylation by removing receptors from the cell surface. Additional regulatory complexity arises from deubiquitinating enzymes like USP8 and STAMBP, which can reverse ubiquitination and promote receptor recycling, with USP8 itself being regulated by phosphorylation in an EGFR- and Src kinase-dependent manner [1].
RAS/RAF/MEK/ERK Cascade Modulation
Downstream of EGFR, the core MAPK signaling cascade components are similarly subject to coordinated ubiquitin and phosphorylation regulation. RAS activation, triggered by phosphorylation-dependent recruitment of SOS to the membrane, can be modulated by ubiquitination. Similarly, RAF kinases undergo complex regulation by both phosphorylation and ubiquitination, which collectively control their activation state, subcellular localization, and protein stability [49].
The functional consequences of ubiquitination within the MAPK cascade are highly diverse. K48-linked polyubiquitination typically targets proteins for proteasomal degradation, thereby terminating signaling. For instance, the E3 ligase FBXW7 can promote degradation of phosphorylated substrates, with context-dependent outcomes in different cancer types [50]. In contrast, K63-linked ubiquitin chains often serve as non-proteolytic signaling scaffolds, as demonstrated by TRAF4-mediated JNK/c-Jun pathway activation in colorectal cancer [50]. This chain topology diversity enables ubiquitination to exert both negative and positive regulation on MAPK signaling, creating nuanced control mechanisms that extend beyond simple degradation.
Experimental Approaches for Mapping Ubiquitin-Phosphorylation Networks
Proteomic Strategies for System-Wide Mapping
Mass spectrometry-based proteomics provides powerful tools for comprehensively characterizing ubiquitin-phosphorylation crosstalk. Two primary strategies have emerged for system-wide mapping:
Phosphoproteomics with Ubiquitin System Perturbation: This approach involves genetic or pharmacological disruption of specific E3 ligases or deubiquitinases followed by quantitative phosphoproteomic analysis. For example, CRISPR-mediated knockout of Cbl family ligases combined with SILAC (Stable Isotope Labeling with Amino Acids in Cell Culture) enables identification of phosphorylation events dependent on ubiquitin regulation. Experimental workflows typically include: (1) generation of E3 knockout cell lines using CRISPR/Cas9; (2) metabolic labeling with light/heavy amino acids; (3) EGFR stimulation with EGF ligand; (4) protein extraction and phosphopeptide enrichment using TiO2 or IMAC; (5) LC-MS/MS analysis; and (6) bioinformatic analysis to identify differentially regulated phosphosites [50] [1].
Ubiquitin Remnant Profiling with Kinase Inhibition: Complementary to phosphoproteomics, diGly remnant profiling (ubiquitin proteomics) following kinase inhibition identifies ubiquitination events controlled by phosphorylation. The methodology includes: (1) treatment with kinase inhibitors targeting EGFR, MEK, or ERK; (2) cell lysis under denaturing conditions; (3) tryptic digestion; (4) enrichment of K-ε-GG-containing peptides with specific antibodies; (5) high-resolution LC-MS/MS; and (6) computational integration with kinase substrate databases [1] [48].
Table 2: Experimental approaches for studying ubiquitin-phosphorylation crosstalk
| Method | Key Readout | Strengths | Limitations |
|---|---|---|---|
| Quantitative Phosphoproteomics | Changes in phosphorylation sites following E3/DUB perturbation | System-wide identification of phosphorylation dependent on ubiquitination | Cannot distinguish direct vs. indirect effects |
| DiGly Ubiquitin Remnant Profiling | Changes in ubiquitination sites following kinase inhibition | Comprehensive mapping of ubiquitination regulated by phosphorylation | Requires large amounts of protein material |
| Co-immunoprecipitation + Western Blot | Protein-protein interactions and modification status | Accessible, allows examination of specific proteins of interest | Low throughput, antibody-dependent |
| Live-cell FRET/BRET | Real-time dynamics of modification and interaction | Temporal resolution of signaling events | Technically challenging, requires biosensor engineering |
| PROTAC-mediated Degradation | Functional consequences of targeted protein removal | Acute perturbation with high specificity | May induce compensatory mechanisms |
Targeted Mechanistic Studies
While proteomic approaches provide system-wide perspectives, targeted mechanistic studies are essential for validating specific regulatory nodes. Co-immunoprecipitation experiments under carefully controlled conditions can demonstrate physical interactions between ubiquitin system components and phosphorylated signaling proteins. For instance, immunoprecipitation of EGFR followed by Western blotting for Cbl and ubiquitin can reveal ligand-dependent complex formation [1]. Similarly, pulldown assays with ubiquitin-binding domains (UBDs) coupled with phospho-specific antibodies can identify proteins bearing both modifications.
Functional validation typically involves genetic manipulation (siRNA, CRISPR) or pharmacological inhibition of specific E3 ligases, DUBs, or kinases, followed by assessment of MAPK pathway activity using phospho-specific antibodies against ERK or MEK. Rescue experiments with wild-type or mutant forms of the regulated protein (e.g., lysine-to-arginine mutants to prevent ubiquitination, or serine/threonine-to-alanine mutants to prevent phosphorylation) can establish causal relationships [50] [1].
Visualization of EGFR Ubiquitin-Phosphorylation Crosstalk
EGFR Ubiquitin-Phosphorylation Crosstalk
Table 3: Key research reagents for studying ubiquitin-phosphorylation crosstalk
| Reagent Category | Specific Examples | Research Application |
|---|---|---|
| Kinase Inhibitors | Gefitinib (EGFRi), Vemurafenib (BRAFi), Trametinib (MEKi) | Pharmacological disruption of phosphorylation events |
| Ubiquitin System Inhibitors | PYR-41 (E1 inhibitor), MLN7243 (E1 inhibitor), NSC632839 (USP8 inhibitor) | Inhibition of ubiquitin activation or deubiquitination |
| Activation-Specific Antibodies | Anti-phospho-EGFR (Y1068), Anti-phospho-ERK1/2 (T202/Y204), Anti-K63-Ub, Anti-K48-Ub | Detection of specific phosphorylation and ubiquitin linkages |
| PROTACs | EGFR-directed PROTACs, BRD4-targeting PROTACs | Targeted protein degradation to study functional consequences |
| Ubiquitin Mutants | K48R, K63R ubiquitin mutants, Ubiquitin-KO cell lines | Dissection of specific ubiquitin chain functions |
| Mass Spectrometry Standards | TMT/SILAC labeling reagents, DiGly antibody beads, TiO2/IMAC phosphopeptide enrichment | Quantitative proteomics of ubiquitination and phosphorylation |
Therapeutic Implications and Future Perspectives
The therapeutic relevance of understanding ubiquitin-phosphorylation crosstalk is underscored by the emerging resistance mechanisms to EGFR- and MAPK-targeted therapies. Resistance to EGFR inhibitors frequently involves bypass signaling through other receptor tyrosine kinases or downstream pathway reactivation [47]. Simultaneously targeting both phosphorylation and ubiquitination systems represents a promising strategy to overcome such resistance. For example, combination therapies employing EGFR kinase inhibitors alongside ubiquitin system modulators are under investigation [50] [47].
PROTAC (Proteolysis-Targeting Chimera) technology exemplifies the therapeutic translation of ubiquitin-phosphorylation insights. EGFR-directed PROTACs selectively degrade oncogenic proteins in tumor cells by recruiting E3 ubiquitin ligases to target proteins, effectively harnessing the ubiquitin system to counteract dysregulated phosphorylation signaling [50] [51]. Additionally, radiotherapy-triggered PROTAC platforms that degrade radioresistance mediators like BRD4 demonstrate how understanding this crosstalk can inspire innovative treatment modalities [50].
Future research directions should focus on developing more sophisticated tools to manipulate specific ubiquitin chain types and creating computational models that can predict network behavior from ubiquitin and phosphorylation inputs. As our understanding of the ubiquitin-phosphorylation interplay deepens, so too will our ability to therapeutically target this complex regulatory axis in cancer and other diseases.
Post-translational modifications (PTMs) represent a crucial regulatory layer in cellular signaling, fine-tuning protein function, localization, and stability in response to internal and external cues [52] [53]. Among the diverse PTM landscape, ubiquitination and phosphorylation stand out as particularly influential regulators that exhibit extensive crosstalk [54] [1]. While phosphorylation rapidly transmits signals through kinase/phosphatase networks, ubiquitination extends regulatory complexity through diverse outcomes ranging from proteasomal degradation to non-proteolytic signaling [55] [56]. Understanding the molecular logic governing this crosstalk requires identifying functionally relevant modification sites from among thousands of possibilities.
Evolutionary conservation provides a powerful filter for distinguishing functionally significant PTM sites from potentially non-functional ones [57] [58] [59]. The central premise is that functional PTM sites experience evolutionary constraint due to their regulatory importance, while neutral sites accumulate mutations more freely [58]. This review compares experimental and computational approaches for identifying functional PTM sites through evolutionary conservation, with particular emphasis on the crosstalk between ubiquitination and phosphorylation in signaling pathways.
Comparative Analysis of Phosphorylation and Ubiquitination Site Prediction
Fundamental Differences in Site Recognition and Conservation
Phosphorylation and ubiquitination exhibit distinct structural and evolutionary characteristics that influence conservation-based prediction strategies. The table below summarizes key comparative features:
Table 1: Comparative Features of Phosphorylation and Ubiquitination Sites
| Feature | Phosphorylation | Ubiquitination |
|---|---|---|
| Modified residues | Serine, Threonine, Tyrosine [57] | Primarily Lysine [55] [56] |
| Consensus motifs | Kinase-specific motifs [57] | Less defined, often disordered regions [60] |
| Structural context | Ordered domains and disordered regions [59] | Strong preference for intrinsically disordered regions [60] |
| Conservation pattern | Phosphorylation hotspots in protein domains [59] | Limited residue-level conservation [58] |
| Functional prediction | Residue conservation + kinase motif analysis [57] [59] | Random forest classifiers with multiple features [60] |
Performance Metrics of Conservation-Based Prediction Methods
Quantitative assessment of prediction methodologies reveals distinct performance characteristics for phosphorylation versus ubiquitination site identification:
Table 2: Performance Metrics of Conservation-Based Prediction Methods
| Method | Modification Type | Accuracy | Area Under ROC Curve | Dataset Size |
|---|---|---|---|---|
| Domain hotspot analysis [59] | Phosphorylation | N/A | 0.78 (S/T), 0.62 (Y) | 537,321 sites across 40 species |
| UbPred random forest [60] | Ubiquitination | 72% (balanced) | 80% | 141 new sites + known sites |
| Cross-eukaryote conservation [57] | Phosphorylation | Conservation percentage scores | N/A | 20,751 "gold standard" human sites |
Experimental Methodologies for Identifying Functional PTM Sites
Evolutionary Conservation Analysis of Phosphorylation Sites
Large-scale comparative analysis of phosphosites across eukaryotic species represents a powerful methodology for identifying functional sites. A recent conservation-based approach analyzed 20,751 high-confidence human phosphosites across 100 eukaryotic species, calculating percentage conservation scores within specific taxonomic groups [57]. The experimental workflow proceeds through several critical stages:
- "Gold Standard" Phosphosite Curation: Compiling phosphosites with extensive identification evidence from multiple databases to ensure data quality [57].
- Homologous Sequence Identification: Using BLASTp searches against 100 eukaryotic proteomes with stringent E-value thresholds (≤1×10⁻⁵) [57].
- Multiple Sequence Alignment: Employing MUSCLE alignment to positionally map human phosphosites to homologous sequences [57].
- Conservation Scoring: Calculating percentage conservation scores that account for Ser/Threonine substitutions [57].
- Functional Enrichment Analysis: Using clusterProfiler and DAVID to associate conserved phosphosites with biological functions and pathways [57].
This methodology successfully identified highly conserved phosphosites within functional domains including kinase activation loops and Ras effector regions, demonstrating the value of cross-species conservation analysis [57] [59].
Proteomic Identification of Ubiquitination Sites
Ubiquitination site identification employs distinct methodologies that address the technical challenges of capturing this more transient modification. The core protocol involves:
- Affinity Enrichment of Ubiquitinated Proteins: Using tagged ubiquitin systems (His-tag, Strep-tag) or ubiquitin-binding domains (TUBEs) to purify ubiquitinated proteins from complex lysates [56].
- Trypsin Digestion: Cleaving proteins while leaving a recognizable signature on modified lysines (diglycine remnant) [56] [60].
- Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS): Separating and fragmenting peptides for identification [56] [60].
- Database Searching and False Discovery Control: Using tools like SEQUEST and PeptideProphet to identify ubiquitination sites with statistical confidence [60].
- Bioinformatic Analysis: Predicting functional ubiquitination sites using tools like UbPred based on sequence and structural features [60].
These methodologies have revealed that ubiquitination sites demonstrate a strong preference for intrinsically disordered protein regions, contrasting with phosphorylation sites that frequently occur in ordered domains [59] [60].
Integrated Workflow for Identifying Co-Occurring PTM Sites
Table 3: Essential Research Reagents and Computational Resources for PTM Conservation Analysis
| Resource Type | Specific Examples | Function and Application |
|---|---|---|
| PTM Enrichment Reagents | Linkage-specific ubiquitin antibodies (K48, K63) [56] | Selective enrichment of ubiquitin chains with specific linkages |
| Tandem Ubiquitin Binding Entities (TUBEs) [56] | High-affinity capture of ubiquitinated proteins with protection from deubiquitinases | |
| Phosphomotif antibodies [57] | Enrichment of peptides phosphorylated by specific kinase families | |
| Proteomics Tools | LC-MS/MS systems [56] [60] | High-sensitivity identification and quantification of modified peptides |
| Stable isotope labeling (SILAC, TMT) [60] | Quantitative comparison of PTM dynamics across conditions | |
| Computational Resources | PTMNavigator [53] | Pathway-centric visualization of PTM data within signaling networks |
| UbPred [60] | Random forest-based prediction of ubiquitination sites | |
| PhosphoSitePlus [53] | Comprehensive database of experimentally verified PTM sites | |
| ConSurf [57] | Evolutionary conservation analysis of protein sequences | |
| Biological Resources | Mutant yeast strains (e.g., CDC34tm) [60] | Enhanced identification of ubiquitination sites on short-lived proteins |
Case Studies: Conservation of PTM Crosstalk in Signaling Pathways
EGFR/MAPK Signaling Pathway
The EGFR/MAPK pathway exemplifies sophisticated PTM crosstalk, particularly between phosphorylation and ubiquitination [1]. Key conserved regulatory mechanisms include:
Receptor Activation and Ubiquitination: EGFR autophosphorylation creates docking sites for the E3 ubiquitin ligase Cbl, which subsequently ubiquitinates the receptor [1]. This ubiquitination depends on prior phosphorylation of Cbl tyrosine residues (Y371 in c-Cbl), demonstrating sequential PTM dependency [1].
Endocytic Sorting: Ubiquitinated EGFR is recognized by endocytic adaptor proteins containing ubiquitin-binding domains (e.g., EPS15, HRS) [1]. These adaptors themselves undergo "coupled monoubiquitination," creating a cooperative PTM network that regulates receptor trafficking [1].
Signal Modulation: Deubiquitinating enzymes (DUBs) like USP8 and STAMBP provide counter-regulation by removing ubiquitin from EGFR, redirecting it toward recycling rather than degradation [1]. The activity of these DUBs is itself regulated by phosphorylation, creating multi-layered feedback control [1].
NF-κB Signaling Pathway
The NF-κB pathway illustrates competitive crosstalk between ubiquitination and SUMOylation (a ubiquitin-like modification) [54]:
Competitive Modification: IκBα, the NF-κB inhibitor, can be modified either by ubiquitination (at K21/K22) targeting it for degradation, or by SUMOylation (at K21) which stabilizes it [54]. This represents a mutually exclusive PTM arrangement on the same lysine residue.
Phosphorylation Priming: IκBα ubiquitination requires prior phosphorylation at S32/S36, while these same phosphorylation events inhibit SUMOylation [54]. This establishes a hierarchical PTM relationship that determines functional outcome.
Hybrid Chain Formation: IκBα can also undergo simultaneous modification by SUMO and ubiquitin, forming hybrid chains that promote its degradation [54]. This demonstrates the potential for combinatorial PTM signaling.
DNA Damage Response
The PCNA protein exemplifies coordinated PTM crosstalk in DNA damage response, though primarily documented in yeast models [54]. PCNA undergoes sequential modifications - ubiquitination at one site promotes subsequent modifications at other sites, creating a "PTM code" that recruits appropriate repair machinery [54].
Evolutionary conservation provides an powerful framework for identifying functional PTM sites amidst thousands of potential modifications. The distinct conservation patterns of phosphorylation and ubiquitination sites reflect their different roles in cellular signaling - phosphorylation sites are frequently conserved within structured domains where they regulate catalytic activity, while ubiquitination sites often target disordered regions to control protein interactions and stability [59] [60].
Future progress in this field will require improved methodologies for capturing transient PTM events, enhanced computational tools for predicting functional outcomes of co-occurring modifications, and systematic integration of PTM crosstalk data into pathway models [53] [1]. Tools like PTMNavigator represent important steps toward visualizing PTM networks within their biological contexts [53]. As our understanding of the complex interplay between phosphorylation, ubiquitination, and other PTMs deepens, so too will our ability to target these modifications for therapeutic intervention in cancer, neurodegenerative diseases, and other pathologies driven by signaling dysfunction.
Navigating Complexity: Challenges and Strategic Approaches in Crosstalk Research and Drug Discovery
Post-translational modifications (PTMs) represent a crucial regulatory mechanism that expands the functional diversity of the proteome, with over 500 discrete covalent modifications currently identified [61] [62]. Among these, phosphorylation and ubiquitination stand as particularly influential modifications that engage in extensive crosstalk to coordinate cellular signaling pathways [54] [9]. This modification interplay creates a complex regulatory network where phosphorylation can either promote or inhibit ubiquitination, and vice versa, ultimately determining protein stability, activity, and localization [54]. However, the analysis of co-modified proteins presents substantial technical challenges due to their sub-stoichiometric nature—where only a small fraction of a given protein population bears the combination of modifications at any time—and the transient existence of these modified forms within dynamic cellular environments [63] [64] [65].
Enrichment techniques have thus become indispensable tools for overcoming these hurdles, enabling researchers to concentrate these rare molecular species for comprehensive characterization. This guide provides an objective comparison of current enrichment methodologies, evaluates their performance in studying ubiquitination-phosphorylation crosstalk, and outlines detailed experimental protocols to support researchers in advancing signaling pathway research.
Analytical Challenge: The Substoichiometric Nature of PTM Crosstalk
The technical difficulty in analyzing co-modified proteins stems from several intrinsic biological and analytical factors that collectively impede detection without specialized enrichment strategies.
Biological and Technical Barriers
- Low Abundance: Modified proteins and peptides exist at substoichiometric levels within complex biological samples, meaning only a small percentage of any given protein carries the modification of interest at a specific time [63]. This scarcity is particularly pronounced for co-modified species, where the simultaneous presence of multiple modifications on a single protein represents an even smaller fraction of the total proteome.
- Signal Suppression: During mass spectrometry analysis, the signals from modified peptides are often overwhelmed by more abundant unmodified peptides, reducing detection sensitivity [63]. This effect is magnified for co-modified peptides, which may exhibit different ionization efficiencies compared to their singly-modified or unmodified counterparts.
- Dynamic Range Limitations: Mass spectrometers have a finite dynamic range, making it challenging to detect low-abundance modified peptides against a high background of unmodified species [63]. The presence of co-modifications further complicates this analytical landscape.
The Ubiquitination-Phosphorylation Crosstalk Paradigm
The functional significance of PTM crosstalk is exemplified by the intricate relationship between ubiquitination and phosphorylation, which can operate through several distinct mechanistic principles:
- Phosphorylation-Directed Ubiquitination: This represents the most established form of crosstalk, where phosphorylation of a specific site creates a recognition motif for ubiquitin ligases. This mechanism is exemplified by IκBα, where phosphorylation at S32 and S36 creates a recognition site for the β-TrCP ubiquitin ligase, leading to K48-linked polyubiquitination and proteasomal degradation [54]. This process ultimately releases NF-κB for nuclear translocation and activation of inflammatory gene expression.
- Competitive Antagonism: PTMs can compete for modification of the same residue, resulting in functional antagonism. SUMOylation of IκBα at K21, which competes with ubiquitination at the same residue, demonstrates this principle and maintains NF-κB in an inactive state [54].
- Sequential and Cooperative Modification: Some proteins undergo sequential modifications where one PTM primes the protein for subsequent modifications. The coordinated action of multiple PTMs on PCNA during DNA damage response illustrates this sophisticated regulatory mechanism [54].
Comparative Analysis of Enrichment Techniques
Successful analysis of co-modified proteins requires efficient enrichment strategies to overcome the challenges of low stoichiometry and signal suppression. The table below provides a systematic comparison of the most widely used enrichment methods for phosphorylation and ubiquitination research.
Table 1: Performance Comparison of Major Enrichment Techniques for Phosphorylation and Ubiquitination Analysis
| Method | Principle | Target PTMs | Specificity | Recovery Efficiency | Key Limitations | Compatibility with Crosstalk Studies |
|---|---|---|---|---|---|---|
| IMAC | Chelated metal ions (Fe³⁺, Ga³⁺, Ti⁴⁺) bind phosphate groups [63] | Phosphorylation | Moderate to High | Variable (40-85%) [63] | Binds other acidic peptides; metal ion interference [63] | Moderate (sequential enrichment possible) |
| MOAC | Metal oxides (e.g., TiO₂) interact with phosphates [65] | Phosphorylation | Moderate to High | Generally good [65] | Sensitivity to buffer conditions [65] | Moderate (sequential enrichment possible) |
| Immuno-affinity | PTM-specific antibodies (e.g., anti-pTyr, anti-Ub) [64] | Phosphorylation, Ubiquitination, Acetylation | High for known motifs [63] | Antibody-dependent (30-90%) [63] | Limited antibody availability; cost [63] [64] | High (especially with motif-specific antibodies) |
| SCX | Separates peptides based on charge differences [63] [65] | Phosphopeptides (at low pH) | Low | Good for fractionation [63] | Lacks high specificity; pre-fractionation method [63] | Low (mainly for sample simplification) |
| HILIC/ERLIC | Hydrophilic interaction chromatography [65] | Phosphopeptides, Glycopeptides | Moderate | Good for hydrophilic PTMs [65] | Requires optimization [63] | Moderate (for specific PTM combinations) |
Specialized Considerations for Co-modification Analysis
When investigating ubiquitination-phosphorylation crosstalk, researchers must consider several advanced strategies that address the unique challenges of co-modified proteins:
- Sequential Enrichment Approaches: This method involves performing consecutive enrichment steps for different PTMs. For phosphorylation-ubiquitination studies, this typically entails first enriching phosphopeptides using IMAC or MOAC, followed by ubiquitinated peptide enrichment using immunoaffinity purification with anti-ubiquitin antibodies [64] [65]. This sequential approach significantly reduces sample complexity and increases the identification of co-modified peptides.
- Single-Step Co-enrichment: Some specialized resins show affinity for multiple PTM types, potentially allowing simultaneous enrichment. However, this approach remains technically challenging and may require custom-designed capture reagents that specifically recognize co-modified motifs [62].
- Cross-referencing Datasets: As an alternative to physical enrichment, computational integration of datasets from parallel enrichments (performed separately for each PTM) can identify potential crosstalk by revealing proteins that undergo both modifications [66]. While less direct, this approach benefits from optimized individual protocols for each PTM type.
Experimental Design for Crosstalk Analysis
To systematically investigate ubiquitination-phosphorylation crosstalk, researchers should implement carefully designed experimental workflows that incorporate appropriate controls and validation steps. The following workflow diagram illustrates a comprehensive approach for analyzing phosphorylation-directed ubiquitination.
Diagram Title: Experimental Workflow for Phosphorylation-Ubiquitination Crosstalk Analysis
Detailed Protocol: Sequential Enrichment for Phosphorylation-Ubiquitination Crosstalk
This protocol provides a robust method for identifying proteins that undergo both phosphorylation and ubiquitination, with specific emphasis on phosphorylation-directed ubiquitination events.
Sample Preparation and Lysis
- Cell Lysis: Harvest cells and lyse in a denaturing buffer containing 8 M urea, 50 mM Tris-HCl (pH 8.0), 75 mM NaCl, 1x protease inhibitor cocktail, 10 mM N-ethylmaleimide (to preserve ubiquitination), and PhosSTOP phosphatase inhibitors [64] [62].
- Protein Quantification: Determine protein concentration using a compatible assay (e.g., BCA assay). Use 5-10 mg of total protein per experimental condition to ensure sufficient material for sequential enrichment.
- Reduction and Alkylation: Reduce disulfide bonds with 5 mM dithiothreitol (37°C, 45 min) followed by alkylation with 15 mM iodoacetamide (room temperature, 30 min in darkness) [64].
Proteolytic Digestion and Peptide Cleanup
- Trypsin Digestion: Dilute the urea concentration to 2 M using 50 mM Tris-HCl (pH 8.0). Add sequencing-grade modified trypsin at a 1:50 (w/w) enzyme-to-protein ratio and incubate overnight at 37°C [64].
- Peptide Desalting: Acidify digested peptides with 1% trifluoroacetic acid (TFA) and desalt using C18 solid-phase extraction cartridges. Elute peptides with 50% acetonitrile/0.1% TFA and lyophilize for subsequent enrichment steps [64] [62].
Sequential Phosphopeptide and Ubiquitinated Peptide Enrichment
- Phosphopeptide Enrichment: Reconstitute peptides in IMAC binding buffer (80% acetonitrile/0.1% TFA) and incubate with Ti⁴⁺-IMAC beads for 60 min with gentle agitation [63] [65]. Wash beads sequentially with IMAC binding buffer and 1% TFA/30% acetonitrile. Elute phosphopeptides with 1% ammonium hydroxide.
- Ubiquitinated Peptide Enrichment: Acidify the phosphopeptide-enriched eluate and incubate with anti-ubiquitin antibody-conjugated beads overnight at 4°C [64] [65]. Wash beads extensively with PBS and elute ubiquitinated peptides with 0.1% TFA.
- Control Enrichments: In parallel, perform separate phosphopeptide-only and ubiquitinated peptide-only enrichments from the same starting material to establish baseline identifications for each modification type.
Mass Spectrometry Analysis and Data Processing
- LC-MS/MS Analysis: Analyze enriched peptides using a high-resolution tandem mass spectrometer coupled to nanoflow liquid chromatography. Use a 120-min gradient from 2% to 35% acetonitrile in 0.1% formic acid for peptide separation [64] [62].
- Data Acquisition: Employ data-dependent acquisition with dynamic exclusion (30 s) for comprehensive peptide identification. Include both collision-induced dissociation (CID) and higher-energy collisional dissociation (HCD) fragmentation methods to improve PTM localization [62].
- Database Searching and Bioinformatic Analysis: Search MS/MS spectra against the appropriate protein database using search engines such as MaxQuant or Proteome Discoverer. Include phosphorylation (S,T,Y) and GlyGly modification (K, for ubiquitination) as variable modifications [64] [66]. Apply a false discovery rate threshold of 1% at both peptide and protein levels. Use motif analysis tools to identify kinase consensus sequences and potential ubiquitin ligase recognition motifs in co-modified peptides.
Research Reagent Solutions for PTM Enrichment
Successful enrichment of sub-stoichiometric co-modified proteins requires access to high-quality specialized reagents. The following table details essential materials and their specific applications in PTM crosstalk studies.
Table 2: Essential Research Reagents for Co-modification Analysis
| Reagent Category | Specific Examples | Function & Application | Key Considerations |
|---|---|---|---|
| Enrichment Resins | Ti⁴⁺-IMAC beads, TiO₂ beads (MOAC) [63] [65] | Phosphopeptide enrichment | Ti⁴⁺-IMAC shows improved selectivity for phosphopeptides over Fe³⁺-IMAC [63] |
| PTM-Specific Antibodies | Anti-phosphotyrosine, Anti-diGly (ubiquitin remnant) [64] [65] | Immunoaffinity enrichment of specific PTMs | Anti-pTyr antibodies show high specificity; pan-phospho antibodies less specific [64] |
| Protease Inhibitors | N-ethylmaleimide, PR-619 (DUB inhibitor) [54] | Preserve ubiquitin conjugates during preparation | Essential for preventing deubiquitination during cell lysis and protein extraction |
| Phosphatase Inhibitors | PhosSTOP, sodium fluoride, β-glycerophosphate [54] | Maintain phosphorylation status | Critical for preserving labile phosphorylation during sample preparation |
| Chromatography Media | C18 desalting columns, SCX cartridges, HILIC columns [65] | Peptide cleanup and pre-fractionation | Reduces sample complexity before enrichment; improves detection sensitivity |
| Activity-Based Probes | Ubiquitin vinyl sulfone, phosphotyrosine mimetics [54] | Detection and validation of modification sites | Useful for verifying functional consequences of identified modifications |
The field of PTM crosstalk research continues to evolve with emerging technologies promising to overcome current limitations in co-modification analysis. Computational prediction tools utilizing machine learning algorithms are increasingly capable of predicting potential crosstalk sites based on sequence motifs and structural features, providing valuable prioritization for experimental validation [66]. Additionally, new enrichment strategies involving engineered binding proteins and chemical biology approaches are expanding the toolkit available for studying sub-stoichiometric PTM combinations [62].
For researchers investigating ubiquitination-phosphorylation crosstalk, a combined approach utilizing sequential physical enrichment and computational integration of datasets currently provides the most comprehensive strategy. As mass spectrometry sensitivity continues to improve and enrichment methods become more refined, our capacity to decipher the complex language of PTM crosstalk will undoubtedly expand, opening new avenues for understanding cellular signaling and developing targeted therapeutic interventions.
In the intricate world of cellular signaling, post-translational modifications (PTMs) act as master switches, controlling protein function, localization, and stability. Among them, ubiquitination and phosphorylation engage in extensive crosstalk, often in a hierarchical manner where one modification directly influences the occurrence or function of another. However, the vast interconnectedness of PTM networks makes it exceptionally challenging to move beyond observed correlations and establish true causal relationships. This guide objectively compares the performance of leading computational and experimental strategies designed to define these hierarchical PTM relationships, providing researchers with the data needed to select the optimal tools for their investigations into ubiquitination-phosphorylation crosstalk.
Computational & Analytical Approaches for Causal Inference
Computational methods leverage high-throughput data and prior knowledge to infer causal pathways, offering a systems-level view of PTM hierarchies.
CausalPath: Pathway-Centric Causal Analysis
CausalPath evaluates proteomic measurements against a curated database of prior biological knowledge to infer causal relationships between changes in measured features, such as global protein and phosphopeptide levels [67].
- Experimental Protocol: The typical workflow involves:
- Input Data Preparation: Data must be formatted with specific columns: a unique
ID, geneSymbols, modificationSites(e.g., Y401), theEffectof the modification (activating 'a' or inhibiting 'i'), and a numericValue(e.g., signed p-value or fold-change) [67]. - Configuration: A parameters file specifies column names and analysis type (e.g., value-transformation, significance thresholds).
- Execution: The tool is run locally via a Java JAR file or through its web server.
- Visualization & Interpretation: Results are explored as network models using tools like the CausalPath website, Newt, or ChiBE [67].
- Input Data Preparation: Data must be formatted with specific columns: a unique
- Performance Data:
Feature Performance & Capability Analysis Focus Infers causality from proteomic data using prior pathway knowledge Primary Input Phosphoproteomic and global proteomic abundance measurements Key Output Network model of signaling consistent with data and literature Typical Use Case Explaining patterns in experimental data through established mechanisms
Automated Causal Discovery on Mass Cytometry Data
Fundamental causal discovery methods, such as those based on Causal Bayesian Networks (CBNs), use conditional independence tests to learn causal graphs from data. These were applied to mass cytometry data measuring phosphorylated signaling proteins in human immune cells [68].
- Experimental Protocol:
- Perturbation Data Collection: Mass cytometry is used to measure protein phosphorylation under various stimulations and inhibitions.
- Causal Assumptions: Algorithms apply the Causal Markov and Faithfulness assumptions to connect causal structures with observable conditional independence patterns in the data [68].
- Graph Reconstruction: Algorithms test for conditional independencies (e.g., A ⊥ T | S) to identify the most probable causal graph explaining the data [68].
- Performance Data:
Feature Performance & Capability Analysis Focus De novo discovery of causal relationships from multivariate data Primary Input Single-cell protein abundance data under multiple perturbations Key Output Directed causal graph predicting system response to interventions Reproducibility Highly consistent across data from different donors and cell types [68] Validation Often disagrees with known pathway databases (e.g., KEGG); requires experimental validation [68]
Comparative Analysis of Computational Tools
The table below provides a direct comparison of the two featured computational methods.
- Tool Comparison Table:
Tool Methodology Data Input Requirements Key Strength Key Limitation CausalPath Evaluates data against prior pathway knowledge [67] Proteomic data with gene symbols and modification sites Provides mechanistic context; mimics manual literature curation Limited to relationships in its knowledge base Causal Discovery (CBN) Infers structure from conditional independencies in perturbation data [68] Multivariate data under multiple experimental conditions Infers novel, context-specific causal relationships without prior knowledge Sensitive to latent confounders; can produce results that disagree with known biology [68]
Experimental & Proteomic Methods for Elucidating PTM Crosstalk
Mass spectrometry-based proteomics enables the identification and quantification of thousands of PTM sites, providing the data backbone for studying crosstalk.
Proteome-Wide PTM Crosstalk Analysis
Proteomic methods allow for the systematic discovery of PTM crosstalk by identifying modification sites and quantifying their changes across different cellular states [29].
- Experimental Protocol:
- Cell Lysis & Protein Extraction: Under defined experimental conditions (e.g., growth factor stimulation, inhibition of specific enzymes).
- PTM Enrichment: Use of specialized techniques to isolate modified peptides. Immobilized metal ion affinity chromatography (IMAC) is a common method for enriching phosphopeptides [29].
- Mass Spectrometry Analysis: Liquid chromatography-tandem mass spectrometry (LC-MS/MS) to identify PTM sites and quantify their abundance.
- Data Integration and Validation: Computational analysis to identify statistically significant co-occurring or mutually exclusive PTMs, followed by validation using mutagenesis and functional assays [29].
- Performance Data:
Feature Performance & Capability Analysis Scale Proteome-wide, measuring thousands of PTM sites simultaneously Primary Output Catalog of PTM sites and their quantitative changes Crosstalk Identification Infers crosstalk from interdependencies between modified residues, either in proximity or through allostery [29] Validation Requirement Requires downstream biochemical experiments to confirm causal hierarchy
Illustrative Examples of Ubiquitination-Phosphorylation Crosstalk
The following examples from the literature demonstrate established hierarchical relationships where one PTM directly regulates another.
- Phosphorylation-Directed Ubiquitination of IκBα: Upon TNF receptor stimulation, phosphorylation of IκBα on S32 and S36 creates a "phosphodegron," which is directly recognized by the β-TrCP E3 ubiquitin ligase. This triggers K48-linked polyubiquitination of IκBα, leading to its proteasomal degradation and the subsequent activation of NF-κB signaling [54]. This is a clear hierarchical cascade where phosphorylation causes ubiquitination.
- SUMOylation-Directed Ubiquitination of PML: The promyelocytic leukemia (PML) protein is first modified by poly-SUMO chains on K160. This SUMOylation is then recognized by the SUMO-targeted ubiquitin ligase (STUbL) RNF4 via its SUMO-interacting motifs (SIMs). RNF4 subsequently catalyzes the polyubiquitination of PML, targeting it for proteasomal degradation [54]. Here, SUMOylation causes ubiquitination.
- Competitive Crosstalk on IκBα: The same lysine residue (K21) on IκBα can be targeted by either SUMOylation or ubiquitination. SUMOylation at this site directly competes with and antagonizes signal-induced ubiquitination, resulting in the stabilization of IκBα and inhibition of NF-κB transcriptional activity [54]. This represents negative crosstalk through residue competition.
Essential Research Reagent Solutions
The following table details key reagents and their functions in studying PTM crosstalk.
- Research Reagent Solutions Table:
Research Reagent Function in PTM Crosstalk Studies Phospho-specific Antibodies Enable detection and enrichment of specific phosphoproteins or phosphopeptides for western blotting or MS [29]. Ubiquitin Binding Domains (UBDs) Used as affinity tools to pull down and identify ubiquitinated proteins; examples include UIM (Ubiquitin-Interacting Motif) [1]. Active E3 Ligase Complexes (e.g., Cbl, RNF4) Essential for in vitro ubiquitination assays to validate substrates and hierarchy [54] [1]. Deubiquitinases (DUBs) & Phosphatases Used as loss-of-function tools to reverse specific modifications and probe their necessity in a signaling cascade [54] [1]. IMAC & TiO₂ Kits Standard tools for the enrichment of phosphopeptides from complex protein digests prior to MS analysis [29]. Proteasome Inhibitors (e.g., MG132) Block protein degradation, allowing for the accumulation of ubiquitinated proteins for easier detection and analysis [54].
Signaling Pathway and Workflow Visualizations
Hierarchical PTM Crosstalk in Signaling
CausalPath Analysis Workflow
The crosstalk between ubiquitination and phosphorylation represents a fundamental regulatory layer in cellular signaling, enabling precise control over immune responses, cell fate decisions, and homeostatic maintenance. This intricate interplay operates with remarkable context-specificity, where functional outcomes are determined by the convergence of cell-type identities, stimulating signals, and temporal dynamics [16]. The strategic integration of these three dimensions transforms the ubiquitination-phosphorylation crosstalk from a simple binary switch into a sophisticated information processing system capable of directing specific cellular behaviors. Understanding this complexity is paramount for researchers and drug development professionals seeking to target these pathways for therapeutic intervention.
Advances in single-cell technologies have revealed that signaling dynamics contain information that is often lost in traditional bulk population studies [69] [70]. For instance, in immune responses, the temporal patterns of transcription factor activation encode stimulus-specific information that determines functional outcomes [70]. Similarly, phosphorylation-driven ubiquitination switches provide precise temporal control over reactive oxygen species homeostasis during plant immune responses [71]. This review systematically compares experimental approaches for dissecting ubiquitination-phosphorylation crosstalk across varying biological contexts, providing researchers with a methodological framework for investigating this complex regulatory axis.
Comparative Analysis of Methodological Approaches
Single-Cell Trajectory Inference for Temporal Dynamics
Table 1: Comparison of Single-Cell Gene Expression Trajectory (scGET) Methods
| Method Feature | scGET Imputation [69] | Pseudotime Analysis | Live-Cell Imaging [70] |
|---|---|---|---|
| Temporal Resolution | Multiple fixed timepoints (5+ recommended) | Continuous but inferred | Real-time continuous monitoring |
| Technical Basis | scRNA-seq with computational imputation | scRNA-seq with trajectory inference | Fluorescent reporters & microscopy |
| Key Metrics | Peak amplitude, Max LFC, Integral, Speed [69] | Branching points, progression | Oscillation frequency, amplitude, duration [70] |
| Context Preservation | Maintains native cellular environment | Maintains native cellular environment | May require genetic perturbation |
| Multiplex Capacity | Transcriptome-wide (100s-1000s genes) | Transcriptome-wide | Typically 1-2 genes simultaneously |
| Information Capture | Dynamic features substantially contribute to stimulus-response specificity [69] | Identifies branching decisions | Direct observation of signaling dynamics |
The imputation of single-cell gene expression trajectories (scGETs) from time-series scRNA-seq data represents a significant methodological advancement for capturing temporal dynamics. This approach employs principal components analysis across all timepoints, constructs cell archetypes via k-means clustering, and links these archetypes across adjacent timepoints using weighted random walks [69]. The resulting ensembles of trajectories enable quantification of dynamic features such as peak amplitude, maximum log fold change, integral, and activation speed—features that substantially contribute to macrophage stimulus-response specificity and offer clearer discrimination of polarization states than single time-point measurements [69].
Experimental Protocol: scGET Imputation
- Time-series scRNA-seq Data Collection: Collect single-cell RNA sequencing data from at least 5 timepoints, preferably front-weighted to capture early dynamics [69]
- Cell Archetype Construction: Perform PCA on combined temporal data and cluster cells using k-means to create metacells
- Trajectory Linking: Calculate transition probability matrices between adjacent timepoints based on Euclidean distance and perform weighted random walks to connect cell states
- Spline Interpolation: Fit splines across principal components to interpolate between measured timepoints
- Gene Expression Projection: Project cell trajectories back to original gene expression space using PCA loadings
- Feature Extraction: Quantify dynamic metrics (Peak Amp, Max LFC, Integral, Speed) for analysis
Proximity Labeling for Ubiquitination-Phosphorylation Interface Mapping
Table 2: Methods for Mapping Ubiquitination-Phosphorylation Crosstalk
| Method | Application in PTM Crosstalk | Spatiotemporal Resolution | Key Limitations |
|---|---|---|---|
| TurboID Proximity Labeling [71] | Identifies E3 ligase-substrate interactions in resting and activated states | Subcellular, condition-specific | May capture proximal proteins without direct interaction |
| Affinity Purification-MS | Isolates stable protein complexes | Limited to stable interactions | Misses transient interactions |
| Phospho-ubiquitin Mapping | Direct detection of phospho-regulated ubiquitination events | Molecular, but often static | Requires specific antibodies or enrichment strategies |
| Live-cell Imaging [70] | Visualizes dynamics of signaling proteins in real-time | Temporal with subcellular resolution | Limited multiplex capacity and potential perturbation |
Proximity labeling techniques, particularly TurboID, have enabled significant advances in mapping the interface between ubiquitination and phosphorylation. This approach allows for the identification of E3 ubiquitin ligases that interact with phosphorylated transcription factors under different signaling conditions [71]. In studying the Alfin-like 7 (AL7) transcription factor, TurboID-based proximity labeling revealed that the ubiquitin protein ligase E3 component N-recognin 7 (UBR7) interacts with AL7 in both resting and immune-activated states, with phosphorylation at serine-174 enhancing this interaction and subsequent ubiquitination [71]. This phosphorylation-driven ubiquitination switch provides precise temporal control over transcription factor stability and function.
Experimental Protocol: TurboID Proximity Labeling
- Construct Generation: Fuse protein of interest (e.g., transcription factor) to TurboID at C-terminus; validate functional integrity
- Conditional Expression: Express fusion protein in relevant cell types under resting and activated conditions (e.g., ± pathogen effector) [71]
- Biotin Labeling: Administer biotin to allow proximal protein labeling; optimize incubation time to minimize background
- Streptavidin Enrichment: Lyse cells and enrich biotinylated proteins using streptavidin beads
- Protein Identification: Process enriched proteins using on-bead digestion and analyze via LC-MS/MS
- Bioinformatic Analysis: Compare enriched proteins to controls; filter based on statistical thresholds (e.g., P<0.05, fold change ≥1.5); perform pathway enrichment
Cell-Cell Communication Inference for Microenvironmental Context
Understanding ubiquitination-phosphorylation crosstalk requires consideration of the cellular microenvironment, where secreted factors influence intracellular signaling states. Computational methods for inferring cell-cell communication from scRNA-seq data have proliferated, each combining different interaction resources and inference algorithms [72]. Systematic comparison of 16 resources and 7 methods revealed significant variation in predicted interactions, with resources showing biased coverage of specific pathways and tissue-enriched proteins [72]. The choice of resource and method strongly influences biological interpretations, highlighting the importance of context-appropriate tool selection.
Table 3: Performance Comparison of CCC Inference Resources
| Resource | Pathway Coverage Bias | Unique Interactions | Best Application Context |
|---|---|---|---|
| OmniPath | Overrepresents T-cell receptor pathway [72] | Comprehensive (~10% unique) | Broad exploratory analysis |
| Cellinker | Highest uniqueness (39.3% unique interactions) [72] | Highest | Niche signaling events |
| Ramilowski-based | Underrepresents T-cell receptor pathway [72] | Low (<5% unique) | General signaling pathways |
| CellChatDB | Includes protein complexes | Moderate | Multicellular communication |
| Guide to Pharmacology | Manually curated, focused | Limited | Pharmacological studies |
Experimental Protocol: Cell-Cell Communication Inference
- Data Preprocessing: Process scRNA-seq data to identify cell clusters and assign cell type identities
- Resource Selection: Choose interaction resource based on biological context and pathway coverage needs
- Method Application: Apply inference method (e.g., expression product, permutation testing) to estimate communication likelihood
- Integration with Spatial Data: Validate predictions using spatial transcriptomics or protein localization data when available
- Consensus Prediction: Combine multiple methods/resources to increase confidence in predictions
- Functional Validation: Correlate predictions with cytokine activities or receptor protein abundance
Integrated Workflow for Context-Specific Ubiquitination-Phosphorylation Analysis
Figure 1: Phosphorylation-Driven Ubiquitination Switch. This regulatory circuit shows how phosphorylation (green) can trigger ubiquitination (red) to create negative feedback loops, as demonstrated in the AL7-UBR7 system [71].
The integration of multiple methodological approaches provides a comprehensive framework for analyzing ubiquitination-phosphorylation crosstalk. The workflow begins with stimulus application in relevant cellular contexts, followed by time-series monitoring using either live-cell imaging or fixed timepoint sampling. For the AL7-UBR7 system, immune activation leads to phosphorylation of AL7 at serine-174, which enhances its interaction with the E3 ubiquitin ligase UBR7, subsequently promoting AL7 ubiquitination and proteasomal degradation [71]. This phosphorylation-driven ubiquitination switch represents a fundamental regulatory motif that ensures precise temporal control of transcription factor activity and prevents excessive immune responses.
Figure 2: Single-Cell Trajectory Imputation Workflow. This pipeline illustrates the process from multi-timepoint scRNA-seq data to stimulus-response specificity analysis, highlighting how dynamic features provide greater discriminatory power than single timepoints [69].
Table 4: Essential Research Reagents for Investigating PTM Crosstalk
| Reagent/Solution | Function | Example Application |
|---|---|---|
| TurboID System | Proximity-dependent biotin labeling of interacting proteins | Mapping E3 ligase-substrate interactions in different signaling states [71] |
| scRNA-seq Kits | Single-cell transcriptome profiling | Time-series analysis of gene expression trajectories [69] |
| Proteasome Inhibitors | Block ubiquitin-mediated degradation | Stabilizing ubiquitinated proteins for detection (e.g., MG132) [71] |
| Phospho-specific Antibodies | Detect phosphorylation events | Monitoring phosphorylation-dependent ubiquitination |
| Ubiquitin Enrichment Reagents | Isolate ubiquitinated proteins | Identifying ubiquitination substrates |
| Fluorescent Reporter Cell Lines | Live-cell imaging of signaling dynamics | Monitoring NF-κB translocation dynamics [70] |
| LIANA Framework | Cell-cell communication inference from scRNA-seq data | Integrating ligand-receptor interactions with intracellular signaling [72] |
Discussion and Future Perspectives
The comparative analysis of methodologies for investigating ubiquitination-phosphorylation crosstalk reveals a critical dependence on cellular context, stimulus identity, and temporal dynamics. Single-cell trajectory approaches demonstrate that dynamic information substantially contributes to stimulus-response specificity, offering clearer discrimination of cellular states than single time-point measurements [69]. Meanwhile, proximity labeling techniques have uncovered phosphorylation-dependent ubiquitination switches that provide precise temporal control over transcription factor activity and immune response magnitude [71].
Future methodological developments will likely focus on enhancing spatial resolution while maintaining temporal dynamics, possibly through advances in live-cell imaging of ubiquitination events [70]. Additionally, the integration of single-cell proteomics with transcriptomic approaches may provide direct correlation between phosphorylation-ubiquitination events and their transcriptional consequences. For drug development professionals, these methodologies offer pathways to identify context-specific regulatory nodes that could be targeted for therapeutic benefit while minimizing off-target effects. The continued refinement of these tools will undoubtedly uncover further complexity in the ubiquitination-phosphorylation crosstalk, highlighting the importance of context-specificity in understanding cellular signaling networks.
Cell signaling crucially depends on a repertoire of post-translational modification (PTM) mechanisms for its regulation, with ubiquitination and phosphorylation emerging as key players in a complex interplay that controls eukaryotic cell signaling [30]. Unlike the relatively straightforward single-step phosphorylation process, ubiquitination occurs in a three-step reaction requiring three different enzymes: an ubiquitin-activating enzyme (E1), an ubiquitin conjugating enzyme (E2), and an ubiquitin ligase enzyme (E3) [30]. The human genome encodes over 600 E3 ubiquitin ligases, which confer substrate specificity to the ubiquitin-proteasome system (UPS) [73] [74] [75]. These enzymes form a hierarchical system that controls the entire ubiquitination pathway, where several E1 enzymes interact with approximately 30 distinct E2 enzymes, which in turn interact with the hundreds of E3 ligases, leading to the ubiquitination of thousands of substrate proteins [74].
The crosstalk between ubiquitination and phosphorylation represents a recurrent theme in cell signaling regulation [30]. Phosphorylation often serves as a marker that triggers subsequent ubiquitination, particularly where ubiquitination leads to degradation [30]. A prime example of this interplay occurs in the epidermal growth factor (EGF)-mediated extracellular signal-regulated kinase (ERK) signaling pathway, where following ligand binding and activation of EGFR by autophosphorylation, Cbl E3 ligase directly binds to activated EGFR and becomes phosphorylated itself, enabling full activation of its E3 ligase activity and subsequent ubiquitination of EGFR [30]. Understanding how these two major PTMs intersect to regulate signal transduction is fundamental to addressing the challenges of targeting E3 ligases therapeutically.
E3 Ligase Families: Structural and Mechanistic Diversity
E3 ubiquitin ligases are primarily classified into three major families based on their structure and mechanism of action: Really Interesting New Gene (RING), Homologous to E6AP C-Terminus (HECT), and RING-Between-RING (RBR) families [73] [75]. The RING family is the largest and most diverse group, encompassing approximately 270 members, while humans encode around 30 HECT E3 genes [75]. These families employ distinct catalytic mechanisms: RING E3s act as scaffolds that bring the E2~Ub conjugate and substrate into proximity for direct ubiquitin transfer, whereas HECT E3s form an obligate thioester intermediate with ubiquitin before transferring it to the substrate [73] [75]. RBR E3s represent a hybrid mechanism, employing RING domains for E2 binding but utilizing a HECT-like catalytic cysteine for ubiquitin transfer [75].
Table 1: Major E3 Ubiquitin Ligase Families and Their Characteristics
| E3 Family | Catalytic Mechanism | Representative Members | Structural Features | Substrate Recognition |
|---|---|---|---|---|
| RING | Direct transfer from E2 to substrate | MDM2, CBL, SCF complexes | Canonical zinc-binding motif; often multi-subunit complexes | Various substrate recognition domains |
| HECT | Covalent E3~Ub intermediate | SMURF1, NEDD4, E6AP | C-terminal catalytic HECT domain with N- and C-lobes | WW domains for PY motif recognition |
| RBR | Hybrid RING-HECT mechanism | HOIP, HOIL-1, PARC | Two RING domains with central IBR domain | Varied domain architectures |
| CRL (RING subclass) | Direct transfer via RING subunit | SPOP, VHL, CRBN | Cullin scaffold, RING protein, substrate adaptor | Specific adaptor proteins (e.g., BTB proteins for CRL3) |
The Cullin-RING ligases (CRLs) represent the largest known subclass of RING E3s, comprising eight members (CRL1-3, CRL4A-B, CRL5, CRL7, and CRL9) [75]. These multisubunit complexes typically consist of a core cullin scaffold protein, a RING-box protein (RBX1/2) that recruits the E2 enzyme, a substrate receptor protein, and an adaptor protein that connects the substrate receptor to the scaffold [75]. The Speckle-type POZ protein (SPOP) represents a notable example, functioning as a substrate-binding adaptor for the Cullin3 (CUL3)/RBX1 E3 ubiquitin ligase complex [75]. SPOP's role in tumorigenesis demonstrates tissue specificity, acting as a tumor suppressor in prostate cancer, hepatocellular carcinoma, and colorectal cancer, while potentially serving as an oncoprotein in kidney cancer [75].
The Targeting Dilemma: Promiscuity and Compensatory Mechanisms
The therapeutic targeting of E3 ligases faces a fundamental dilemma: their dual nature as both highly specific and potentially promiscuous enzymes. While E3 ligases are renowned for conferring substrate specificity to the ubiquitin system, many recognize multiple substrates and participate in various signaling pathways [75]. This promiscuity creates significant challenges for drug development, as inhibiting a single E3 ligase may have unintended consequences across multiple cellular processes.
Compensatory mechanisms within ubiquitination pathways further complicate therapeutic targeting. Cells maintain proteostasis through intricate networks of E3 ubiquitin ligases that can compensate for each other's functions when specific components are disrupted [75]. For instance, in yeast, dosage compensation mechanisms involve networks of E3 ubiquitin ligases and N-acetyltransferases that collaborate to regulate levels of multiprotein complex subunits [75]. Even more remarkably, compensation extends across different degradation pathways, as demonstrated by the degradation of yeast fatty acid synthase (FASN), which is predominantly degraded through autophagy under nitrogen starvation conditions but can be processed via the UPS through the E3 ligase Ubr1 when autophagy is compromised [75].
The development of resistance to E3-targeted therapies often involves upregulation of alternative degradation pathways or mutation of E3 ligases themselves. In cancer therapy, resistance may occur through various mechanisms, including mutation or overexpression of target proteins, or adaptation to alternative pathways [76]. The tissue-specific functions of E3 ligases like SPOP further complicate therapeutic strategies, as inhibition may produce opposing effects in different tissues [75].
Experimental Approaches and Research Tools
High-Throughput Screening and Allosteric Inhibition
Recent advances in E3 ligase targeting have employed innovative screening approaches to identify novel inhibitors. A large, unbiased biochemical screen of 1.1 million compounds against the HECT E3 ligase SMURF1 utilized a time-resolved fluorescence resonance energy transfer (TR-FRET)-based assay reporting SMURF1 self-ubiquitylation [77]. Primary hits were further screened using biochemical selectivity and cell-based assays to prioritize molecules for specific SMURF1 inhibition, revealing three chemical series with favorable drug-like properties: piperidine sulfonamides, pyrazolones, and pyrroles [77].
Structural analyses through X-ray crystallography of SMURF1 HECT domains with inhibitors revealed an unexpected allosteric mechanism. The inhibitors bind a cryptic cavity in the N-lobe, distant from the catalytic cysteine, and restrict an essential catalytic motion by extending an α helix over a conserved glycine hinge [77]. This elongation shortens the hinge from 27.0 to 15.4 Å and replaces the invariant glycine (G634) with lysine (K637), an amino acid with lower tolerance of ϕ/Ψ dihedral angles, thereby restricting the flexibility necessary for ubiquitin transfer [77].
Table 2: Key Research Reagent Solutions for E3 Ligase Studies
| Research Tool | Function/Application | Key Characteristics | Example Uses |
|---|---|---|---|
| TR-FRET Assays | Reporting E3 self-ubiquitylation | High-throughput compatibility, real-time monitoring | Primary compound screening (e.g., SMURF1 inhibitors) |
| Cullin-RING Ligase Complexes | Structural and biochemical studies | Multi-subunit reconstitution, native architecture | Mechanism of action studies for CRL E3s |
| PROTAC Molecules | Inducible protein degradation | Bifunctional design, catalytic mode of action | Targeted degradation of disease-associated proteins |
| X-ray Crystallography | Structural determination of E3-inhibitor complexes | Atomic-resolution insights, ligand binding modes | Allosteric mechanism elucidation (e.g., SMURF1 glycine hinge) |
| Machine Learning Screening | In silico inhibitor identification | Structure-based prediction, leveraging known mechanisms | E6AP inhibitor discovery based on glycine-hinge understanding |
Targeted Protein Degradation Strategies
Targeted protein degradation (TPD) represents a revolutionary therapeutic strategy that capitalizes on the cell's intrinsic proteolytic systems to selectively eliminate disease-causing proteins [76]. The two primary TPD approaches are proteolysis-targeting chimeras (PROTACs) and molecular glues, both of which utilize the UPS but operate through distinct mechanisms.
PROTACs are heterobifunctional molecules consisting of a target protein-binding ligand connected via a linker to an E3 ligase-recruiting ligand [76]. This design facilitates the formation of a ternary complex that brings the E3 ligase into proximity with the target protein, leading to its ubiquitination and subsequent proteasomal degradation. PROTACs offer several advantages, including their catalytic nature and ability to target proteins without functional binding pockets [76]. However, they face challenges related to their large molecular size and the "hook effect," where high concentrations disrupt ternary complex formation [76].
Molecular glues are typically smaller molecules that induce or stabilize protein-protein interactions between E3 ligases and target substrates [76]. Their smaller size offers advantages for cellular permeability and potential oral administration, but their discovery often occurs serendipitously due to the unpredictable nature of protein-protein interactions [76].
Comparative Analysis of E3 Targeting Modalities
Table 3: Quantitative Comparison of E3 Ligase Targeting Approaches
| Targeting Approach | Molecular Weight Range | Specificity Challenges | Compensation Resistance Risk | Clinical Stage Examples |
|---|---|---|---|---|
| Active-site inhibitors | 300-500 Da | High for conserved catalytic sites | Moderate to High | SMURF1 inhibitors (preclinical) |
| Allosteric inhibitors | 400-600 Da | Medium (targets less conserved regions) | Low to Moderate | SMURF1 Cpd-8 series (preclinical) |
| PROTACs | 700-1200 Da | Medium (depends on ligand specificity) | Low | KT-253 (MDM2-based, Phase I) |
| Molecular Glues | 300-500 Da | Low (induces neo-interactions) | Low | Immunomodulatory imide drugs (approved) |
| PROTAC-based E3 degraders | 900-1300 Da | High (specific E3 elimination) | Low to Medium | Homo-PROTACs (preclinical) |
The diverse approaches to targeting E3 ligases each present distinct advantages and limitations. Traditional active-site inhibitors face challenges due to the absence of confined active-site pockets in many E3s, particularly E2-conjugating enzymes and E3 ligases where active sites protrude from the protein surface [77]. Allosteric inhibitors, such as those discovered for SMURF1, offer an alternative strategy by targeting cryptic cavities distant from catalytic sites, potentially enabling greater specificity [77]. However, their development requires sophisticated screening approaches and detailed structural knowledge.
PROTAC technology has expanded the repertoire of druggable targets, including those previously considered "undruggable" due to the lack of defined active sites [76]. The recyclability of PROTACs increases their efficacy, but their large size and complex structure can reduce stability and cellular permeability [76]. Molecular glues offer advantages in size and pharmacokinetics but present design challenges due to the unpredictable nature of protein-protein interactions [76].
Visualization of Key Concepts
E3 Ligase Mechanisms and Inhibition
E3 Ligase Mechanisms and Inhibition Pathways - This diagram illustrates the distinct catalytic mechanisms of RING and HECT E3 ubiquitin ligases, along with the allosteric inhibition strategy that restricts essential catalytic motions in HECT E3s like SMURF1.
Ubiquitination-Phosphorylation Crosstalk in EGFR Signaling
Ubiquitination-Phosphorylation Crosstalk in EGFR Signaling - This diagram depicts the intricate interplay between phosphorylation and ubiquitination events in EGFR signaling and trafficking, highlighting key regulatory points where phosphorylation controls E3 ligase activity and deubiquitinating enzyme function.
The dilemma of targeting promiscuous E3 ligases while overcoming compensatory mechanisms represents both a formidable challenge and a remarkable opportunity in drug discovery. The extensive complexity of the ubiquitin system, with over 600 E3 ligases regulating diverse cellular processes, necessitates sophisticated approaches that account for tissue specificity, compensatory pathways, and the intricate crosstalk with phosphorylation signaling networks.
Future directions in this field will likely focus on several key areas: First, the expansion of E3 ligases amenable to targeting, moving beyond the current focus on just a few well-characterized E3s like MDM2, IAP, VHL, and CRBN [76]. Second, the development of more sophisticated screening technologies, including machine learning approaches that leverage structural insights like the glycine hinge mechanism discovered in SMURF1 to identify inhibitors for other HECT E3s [77]. Third, addressing the challenges of tissue-specific E3 functions and compensatory mechanisms through more nuanced therapeutic strategies that consider context-dependent roles of E3 ligases like SPOP in different cancer types [75].
The quantitative framework based on systems biology and mathematical modeling will be essential for efficiently understanding the roles of interconnected PTMs in cell signaling [30]. As our comprehension of ubiquitination-phosphorylation crosstalk deepens, and targeting technologies evolve, E3 ligases may indeed fulfill their potential as a transformative therapeutic class, mirroring the trajectory of kinase inhibitors that now represent a $15 billion industry [74].
The intricate crosstalk between ubiquitination and phosphorylation represents a fundamental regulatory layer in cellular signaling pathways, presenting both challenges and opportunities for therapeutic intervention. These two dominant post-translational modifications (PTMs) engage in sophisticated bidirectional communication where phosphorylation often serves as a marker triggering subsequent ubiquitination, while ubiquitination can provide switching mechanisms that turn kinase activity on or off [1]. This dynamic interplay creates robustness in signaling networks that enables cancer cells to develop resistance to single-agent therapies targeting individual nodes. The paradigm of "one genetic abnormality—one drug" demonstrates limitations in patient matching rates (often just 5-10%) and frequently encounters drug resistance due to compensatory signaling activation and tumor heterogeneity [78]. Combination therapies strategically targeting multiple nodes in these interconnected networks offer a promising approach to overcome these limitations by disrupting the resilient feedback loops and cross-regulatory mechanisms that characterize ubiquitination-phosphorylation crosstalk.
Ubiquitination-Phosphorylation Interplay: Molecular Mechanisms and Signaling Dynamics
The crosstalk between ubiquitination and phosphorylation operates through several conserved molecular mechanisms that enable precise control of signal transduction pathways. Understanding these mechanisms is crucial for designing effective combination therapies.
Molecular Basis of PTM Crosstalk
Ubiquitination and phosphorylation, while sharing parallel properties as reversible PTMs, possess distinct characteristics that enable their sophisticated interplay. Phosphorylation involves a single-step addition of phosphate groups primarily to serine, threonine, tyrosine, and histidine residues, whereas ubiquitination requires a three-step enzymatic cascade involving E1 (activating), E2 (conjugating), and E3 (ligating) enzymes that covalently attach ubiquitin to lysine residues [1]. This E3 ubiquitin ligase activity is frequently controlled by phosphorylation events, as exemplified by the Cbl family proteins which regulate EGFR trafficking and degradation. Upon EGFR activation and autophosphorylation, Cbl binds to phosphotyrosine residues on the receptor through its tyrosine kinase binding (TKB) domain, becomes phosphorylated itself on tyrosine 371 (in c-Cbl), and undergoes a structural transformation that exposes its RING domain, subsequently activating its E3 ligase function and leading to EGFR ubiquitination [1].
Phosphodegron Recognition and Coordinated Degradation
A particularly prevalent form of ubiquitination-phosphorylation crosstalk occurs through phosphodegron motifs, where specific phosphorylation events create recognition sites for E3 ubiquitin ligases, thereby targeting proteins for proteasomal degradation. Large-scale proteomic studies in yeast have identified 466 proteins with 2,100 phosphorylation sites co-occurring with 2,189 ubiquitylation sites, with phosphorylation sites found co-occurring with ubiquitylation demonstrating significantly higher evolutionary conservation than other phosphorylation sites [7]. This conservation pattern suggests functional importance for these coordinated modification sites in fundamental biological processes. The degradation of cell cycle regulators through phosphodegron mechanisms provides irreversibility and robustness to cell cycle transitions, illustrating how this form of crosstalk creates unidirectional switches in signaling pathways.
Feedback and Feedforward Loops in Signaling Networks
The EGFR/MAPK pathway exemplifies how ubiquitination-phosphorylation crosstalk creates complex feedback architectures that modulate signal duration and intensity. Following EGF stimulation, Cbl-mediated ubiquitination of EGFR not only targets the receptor for lysosomal degradation but also regulates the strength and duration of downstream ERK signaling [1]. Deubiquitinating enzymes (DUBs) such as STAMBP (AMSH) and USP8 provide counter-regulatory mechanisms that can remove ubiquitin from EGFR, with USP8 itself being regulated by phosphorylation in an EGFR- and Src-kinase dependent manner [1]. This creates a multi-layered regulatory network where phosphorylation regulates ubiquitination, which in turn regulates downstream phosphorylation cascades, generating sophisticated temporal control of signaling outputs. Mathematical modeling of these interconnected PTM networks has revealed their capacity to generate rich dynamical behaviors including bistability, oscillations, and ultrasensitivity [1].
Current Landscape of Combination Therapies: Database Analysis and Clinical Validation
The systematic compilation of drug combination data provides critical insights into current approaches for targeting interconnected signaling nodes. The recently developed OncoDrug+ database represents a comprehensive resource for precision combinatorial therapy, containing 7,895 data entries covering 77 cancer types, 2,201 unique drug combination therapies, 1,200 biomarkers, and 763 published reports [78]. Unlike previous databases that primarily documented synergy scores, OncoDrug+ integrates genetic evidence, pharmacological target information, and evidence scores supporting each combination strategy, enabling evidence-based clinical applications.
Table 1: Evidence Categories for Combination Therapies in OncoDrug+ Database
| Evidence Category | Data Entries | Key Characteristics | Clinical Relevance |
|---|---|---|---|
| FDA-approved Guidelines | 60 | NCI/NCCN recommended regimens | Standard of care |
| Clinical Trials | 349 | Ongoing or recently completed studies | Phase 1-4 investigations |
| Bioinformatics Predictions | 5,066 | REFLECT algorithm predictions | Preclinical candidates |
| Cell Line Screening | 1,577 | Large-scale synergy screening | In vitro validation |
| PDX Models | 30 | Patient-derived xenograft data | In vivo validation |
| Electronic Medical Records | 233 | Real-world patient responses | Clinical outcome data |
Dominant Therapeutic Combinations and Their Molecular Targets
Analysis of combination strategies reveals several prevalent themes in targeting ubiquitination-phosphorylation crosstalk. Dual kinase inhibition strategies, such as combined BRAF and MEK inhibition in BRAF V600E melanoma, demonstrate the principle of targeting consecutive nodes in a linear pathway to enhance efficacy and reduce resistance [78]. This approach reduces skin toxicity associated with BRAF inhibitor monotherapy while extending duration of response by preventing or delaying acquired resistance mechanisms [78]. Another emerging strategy combines kinase inhibitors with protein degradation approaches, such as linking kinase-binding moieties to E3 ligase recruiters to direct targeted protein degradation, effectively harnessing the ubiquitination machinery to eliminate phosphorylated signaling components.
Evidence Grading and Clinical Translation
The OncoDrug+ database implements a prioritization system that ranks combination therapies based on genetic and clinical evidence, including FDA-approval status, evidence type, biomarker reliability, and outcomes in clinical or experimental testing [78]. This evidence-based grading is crucial for clinical decision-making, as it enables clinicians to distinguish between fully validated regimens suitable for standard care and investigational combinations requiring additional validation. The integration of electronic medical records from 233 cancer patients treated with combinatorial therapy at The Second Hospital of Tianjin Medical University provides real-world validation of these approaches [78].
Experimental Approaches: Methodologies for Evaluating Combination Therapies
Rigorous experimental protocols are essential for validating the efficacy of combination therapies targeting ubiquitination-phosphorylation crosstalk. The following section outlines key methodologies cited in the literature.
High-Throughput Combination Screening
Large-scale drug combination screening represents a powerful approach for identifying synergistic interactions. The ALMANAC, AZ-DREAM, and O'Neil et al. studies provide robust platforms for unbiased identification of synergistic drug combinations across hundreds of cell lines [78]. These studies employ standardized protocols where cells are seeded in 384-well plates, treated with compound libraries using liquid handling systems, and assessed for viability using metabolic assays (e.g., CTG, MTS) after 72-120 hours of exposure. Synergy scores are typically calculated using multiple reference models including HSA, Bliss, Loewe, and ZIP, with consistent synergy across all models providing the highest confidence hits [78]. This multi-model assessment is critical as different synergy models can yield varying interpretations of drug interactions.
Proteomic Analysis of Co-Modified Proteins
Advanced proteomic techniques enable systematic identification of proteins undergoing both phosphorylation and ubiquitination, providing insights into direct nodes of crosstalk. Two complementary enrichment strategies have been developed for this purpose [7]:
Table 2: Proteomic Methods for Identifying Co-Modified Proteins
| Method | Principle | Advantages | Limitations |
|---|---|---|---|
| Protein-Level Enrichment | Sequential enrichment of ubiquitylated proteins followed by phosphopeptide enrichment | Identifies co-modified proteins regardless of modification proximity | Cannot establish same-protein isoform |
| Peptide-Level Enrichment | Strong-cation exchange (SCX) chromatography followed by diGly antibody enrichment | Confirms both PTMs on same protein isoform | Limited to adjacent modification sites |
The protein-level enrichment approach involves affinity purification of His-tagged ubiquitin-modified proteins using cobalt-NTA, followed by tryptic digestion and phosphopeptide enrichment using TiO2 or IMAC chromatography [7]. The peptide-level method utilizes SCX chromatography to separate peptides by solution charge, followed by immunoaffinity purification using antibodies specific for the diGly remnant left after tryptic digestion of ubiquitylated peptides [7]. This second method identified 1,008 unique ubiquitylated phosphopeptides compared to only 56 with the first method, demonstrating its superior efficiency for identifying co-modified peptides [7].
Bioinformatics Prediction of Combination Therapies
Computational approaches complement experimental methods for identifying promising combination strategies. The REFLECT (REcurrent Features LEveraged for Combination Therapy) method utilizes multi-omics data to map features that repeatedly and concurrently change in patient cohorts to combination therapy, accurately predicting synergistic effects and survival outcomes [78]. The REFLECT pipeline identifies precision drug combination therapies based on multi-omic co-alteration signatures including mutations, with validation using patient-derived xenografts, in vitro drug screens, and clinical trial data [78]. For implementation, REFLECT signatures define patient sub-cohorts distinguished by recurrent co-alterations, after which the DGIdb database is queried to retrieve potential drugs targeting these alterations, selecting FDA-approved agents with the highest interaction scores [78].
Visualization of Key Signaling Pathways and Experimental Workflows
Visual representation of signaling networks and experimental approaches enhances understanding of the complex relationships in ubiquitination-phosphorylation crosstalk.
EGFR Ubiquitination-Phosphorylation Crosstalk Pathway
The following diagram illustrates the key regulatory steps in EGFR signaling where ubiquitination and phosphorylation intersect:
This pathway visualization captures the essential regulatory circuit where EGFR phosphorylation triggers its own ubiquitination and subsequent degradation, while deubiquitinating enzymes (DUBs) like USP8 can reverse this process. The phosphorylation of both EGFR and regulatory enzymes like Cbl and USP8 creates multiple feedback loops that control signal duration and intensity [1].
Proteomic Identification of Co-Modified Proteins
The experimental workflow for identifying proteins modified by both phosphorylation and ubiquitination involves two complementary approaches:
This workflow illustrates the parallel processing of ubiquitin-enriched and ubiquitin-depleted protein fractions to identify phosphorylation sites unique to ubiquitinated protein isoforms [7]. The identification of such sites is functionally significant as phosphorylation sites found co-occurring with ubiquitylation show higher evolutionary conservation than other phosphorylation sites, suggesting important regulatory roles [7].
The Scientist's Toolkit: Essential Reagents and Research Solutions
Advancing research in ubiquitination-phosphorylation crosstalk requires specialized reagents and tools. The following table catalogues essential research solutions for investigating combination therapies:
Table 3: Essential Research Reagents for Studying PTM Crosstalk and Combination Therapies
| Reagent Category | Specific Examples | Research Application | Key Features |
|---|---|---|---|
| Ubiquitin System Tools | His-tagged ubiquitin, diGly remnant antibodies | Proteomic identification of ubiquitylation sites | Enables affinity purification and MS identification |
| Phosphorylation Tools | Phospho-specific antibodies, TiO2/IMAC beads | Phosphoproteomic enrichment | Selective recognition of phosphorylated residues |
| Bioinformatics Databases | OncoDrug+, DGIdb, REFLECT | Drug combination prediction and prioritization | Evidence-based combination therapy ranking |
| Combination Screening | ALMANAC, AZ-DREAM compound libraries | High-throughput synergy screening | Standardized platforms for unbiased identification |
| Model Systems | PDX models, molecularly characterized cell lines | In vivo and in vitro validation | Preserves tumor heterogeneity and clinical relevance |
| E3 Ligase Modulators | PROTACs, molecular glues | Targeted protein degradation | Harnesses endogenous ubiquitination machinery |
These research tools enable the comprehensive characterization of ubiquitination-phosphorylation crosstalk and the validation of therapeutic combinations that target these interconnected networks. The diGly remnant antibodies have been particularly transformative, enabling proteome-wide identification of ubiquitylation sites through mass spectrometry analysis [7]. Similarly, the development of PROTACs (Proteolysis-Targeting Chimeras) represents a novel therapeutic class that redirects E3 ubiquitin ligase activity to target specific proteins for degradation, effectively harnessing the ubiquitination machinery for therapeutic purposes.
The strategic targeting of ubiquitination-phosphorylation crosstalk through combination therapies represents a paradigm shift in precision oncology, moving beyond single-target approaches to address the robust complexity of signaling networks. The systematic compilation of drug combination data in resources like OncoDrug+, coupled with advanced proteomic methods for identifying co-modified proteins and sophisticated computational prediction tools, provides a powerful foundation for evidence-based combinatorial strategy development [78] [7]. As our understanding of the intricate feedback loops and regulatory dynamics between these PTM networks deepens, so too will our ability to design rational combination therapies that preempt resistance mechanisms and deliver improved patient outcomes. The continued integration of large-scale combination screening, multi-omics profiling, and mechanistic studies of PTM crosstalk will undoubtedly yield next-generation therapeutic strategies that simultaneously target multiple nodes in these interconnected signaling networks.
From Mechanism to Medicine: Validating Crosstalk in Disease Pathways and Therapeutic Interventions
The Epidermal Growth Factor Receptor (EGFR) signaling pathway is a critical regulator of cellular processes such as proliferation, survival, and differentiation. In non-small cell lung cancer (NSCLC), EGFR is frequently dysregulated through overexpression, mutations, or aberrant activation. A sophisticated layer of regulation exists through the crosstalk between different post-translational modifications (PTMs), particularly phosphorylation and ubiquitination. This crosstalk fine-tunes signaling output, influences receptor fate, and represents a critical mechanism that cancer cells exploit for progression and therapy resistance [1]. Understanding this dynamic interplay is not just of academic interest but is essential for developing novel therapeutic strategies that can overcome the limitations of current EGFR-targeted treatments. This case study examines the key nodes of crosstalk within the EGFR network, its functional consequences on lung cancer biology, and the emerging therapeutic paradigms that target these complex interactions.
Key Crosstalk Mechanisms in EGFR Signaling
The interaction between phosphorylation and ubiquitination within the EGFR network can be categorized into several key mechanistic themes, each with distinct functional outcomes.
EGFR transactivation of MET via MAPK signaling
A prime example of receptor crosstalk is the unilateral activation of the MET receptor tyrosine kinase by EGFR signaling. Research using model cell systems has demonstrated that EGFR signaling is sufficient to induce MET phosphorylation, a process enhanced by the co-expression of ERBB3 [79]. This crosstalk is not direct but is mediated through intermediary MAP kinase signaling. Inhibition of EGFR or MAP kinases (MEK/p38MAPK) in NSCLC cells reduces both MET activation and protein levels, establishing a firm link between these pathways [79]. From a therapeutic standpoint, this EGFR-MET axis promotes aggressive behaviors, including migration and invasion. Notably, this signaling is enhanced in highly metastatic subpopulations of EGFR-mutant cells, and attenuation of MET signaling decreases the incidence of brain metastasis, highlighting its role in advanced disease [79].
Phosphorylation-dependent regulation of EGFR ubiquitination and stability
The stability and degradation of EGFR are centrally controlled by the crosstalk between phosphorylation and ubiquitination.
The c-Cbl Mechanism: The E3 ubiquitin ligase c-Cbl is recruited to activated, autophosphorylated EGFR. Upon binding, Cbl itself undergoes phosphorylation, which triggers a conformational change that activates its RING domain, enabling the ubiquitination of EGFR [1]. This classic "phosphodegron" mechanism typically directs the receptor for lysosomal degradation.
The TRIB3-PKCα Axis: The pseudokinase TRIB3 promotes NSCLC progression by interacting with EGFR and recruiting PKCα, which phosphorylates EGFR at threonine 654 (Thr654) in the juxtamembrane region [80]. This phosphorylation, in turn, facilitates WWP1-induced lysine 689 (Lys689) ubiquitination. This unique modification enhances EGFR recycling and stability rather than degradation, leading to sustained downstream signaling and increased cancer stemness [80]. Targeting the TRIB3-EGFR interaction with a stapled peptide accelerates EGFR degradation and sensitizes tumors to chemotherapy, revealing a new therapeutic vulnerability [80].
Mig6 Inactivation in Mutant EGFR: In lung adenocarcinomas harboring EGFR mutations, the negative regulator Mig6 is highly phosphorylated at Ser256 [81]. This phosphorylation inactivates Mig6, leading to reduced ubiquitination and defective degradation of mutant EGFR. This mechanism contributes to the sustained oncogenic signaling characteristic of these cancer cells [81].
Table 1: Key Proteins Regulating EGFR Phosphorylation-Ubiquitination Crosstalk
| Protein | Role in PTM Crosstalk | Functional Outcome in Cancer | Therapeutic Implication |
|---|---|---|---|
| c-Cbl | E3 ligase recruited to phosphorylated EGFR; ubiquitinates receptor [1]. | Promotes EGFR degradation; often dysregulated. | - |
| TRIB3 | Interacts with EGFR, recruits PKCα for Thr654 phosphorylation, enabling WWP1-mediated ubiquitination [80]. | Enhances EGFR recycling/stability, stemness, and progression. | Stapled peptides disrupting TRIB3-EGFR interaction. |
| Mig6 | Tumor suppressor; phosphorylation at S256 inactivates it in mutant EGFR cells [81]. | Reduces ubiquitination, stabilizes mutant EGFR. | Restoring Mig6 function. |
| USP8 | Phosphorylation regulates this deubiquitinase's activity [1]. | Promotes EGFR recycling by deubiquitinating the receptor. | - |
Systems-level dynamics of crosstalk
The interplay between phosphorylation and ubiquitination creates a complex regulatory network capable of rich dynamic behaviors. Quantitative modeling reveals that feedback loops and crosstalk between these PTMs can give rise to bistability, oscillations, and switch-like responses [1]. For instance, oscillations in ERK activity, driven by underlying feedback mechanisms, have been observed with periods ranging from 20 minutes to several hours [1]. These dynamics are not mere curiosities; they can fundamentally influence cellular decisions between proliferation, differentiation, and survival. A systems biology perspective is, therefore, crucial to move beyond a static catalog of interactions and understand the temporal control of signaling that dictates pathological outcomes.
Experimental Approaches for Studying PTM Crosstalk
Investigating the complex relationship between phosphorylation and ubiquitination requires specialized methodologies that can capture their concurrent presence and interaction.
Global Proteomics for Co-modified Proteins
To systematically identify proteins co-modified by both ubiquitination and phosphorylation, large-scale proteomic approaches have been developed. One strategy involves a two-step enrichment: first, proteins are enriched for ubiquitination (e.g., via His-tagged ubiquitin pull-down), and the resulting fractions are then digested and further enriched for phosphopeptides [7]. This protein-level enrichment identifies proteins bearing both modifications, though it cannot confirm if they are on the same molecule. A more precise, peptide-centric approach uses sequential strong-cation exchange (SCX) and diGly remnant immunoaffinity purification to directly identify peptides concurrently modified by both ubiquitination and phosphorylation, confirming their presence on the same protein isoform [7]. These methods revealed that phosphorylation sites found co-occurring with ubiquitination are more highly conserved than other phosphorylation sites, suggesting they serve critical regulatory functions [7].
The 32D Cell Model for Dissecting Receptor Crosstalk
To isolate the specific contributions of individual receptors from the background of promiscuous ERBB family interactions, researchers have employed a 32D mouse myeloid cell model. This system allows for the expression of ERBBs or MET alone and in combination [79]. The key experimental steps are as follows:
- Model Establishment: 32D cells are electroporated with plasmids encoding EGFR, ERBB3, or MET (wild-type or mutant) and selected with antibiotics [79].
- Stimulation & Inhibition: Cells are stimulated with ligands (EGF, HGF) and treated with a panel of kinase inhibitors (e.g., Gefitinib for EGFR, U0126 for MEK) to dissect the signaling pathways [79].
- Analysis: Signaling outputs are analyzed via immunoprecipitation and immunoblotting for phospho-proteins to map activation dependencies [79].
This reductionist system was instrumental in proving that EGFR alone is sufficient to cross-activate MET through MAPK pathways, independent of other ERBBs [79].
Therapeutic Implications and Overcoming Resistance
The crosstalk within the EGFR network is a major driver of resistance to targeted therapies like EGFR tyrosine kinase inhibitors (TKIs). Understanding these mechanisms has opened avenues for novel combination treatments.
Stromal and Microenvironment-Mediated Resistance
The tumor microenvironment, particularly cancer-associated fibroblasts (CAFs), plays an active role in promoting TKI resistance. Direct coculture of EGFR-mutant PC9 lung cancer cells with CAFs induces resistance to the EGFR TKI erlotinib [82]. This resistance is associated with an epithelial-to-mesenchymal transition (EMT) phenotype in the cancer cells and upregulation of the hedgehog signaling pathway component smoothened [82]. Inhibition of smoothened with cyclopamine can reverse this resistance, indicating that stromal crosstalk via hedgehog signaling is a therapeutically targetable mechanism.
Novel Therapeutic Platforms and Strategies
Moving beyond simple kinase inhibition, new strategies are focusing on the broader signaling network and receptor turnover.
Nanotheranostics for Combination Therapy: The H-dot platform is a multifunctional nanoparticle that enables image-guided surgery and targeted drug delivery. Composed of ε-poly-L-lysine and β-cyclodextrin, H-dots can be loaded with drugs like the EGFR-TKI gefitinib and the angiogenesis inhibitor genistein [83] [84]. This allows for simultaneous dual-channel NIR fluorescence imaging and synergistic combination therapy at the tumor site, improving efficacy while reducing off-target effects through ideal biodistribution and rapid renal clearance [83] [84].
Targeting Receptor Stability: Given that aberrant EGFR recycling and stability promote cancer progression, directly promoting EGFR degradation is an emerging strategy. As noted earlier, disturbing the TRIB3-EGFR interaction with a stapled peptide forces EGFR degradation and suppresses tumor growth [80]. This represents a paradigm shift from inhibiting kinase activity to controlling receptor abundance.
Table 2: Mechanisms of Resistance to EGFR-TKIs and Potential Counter-Strategies
| Resistance Mechanism | Description | Therapeutic Counter-Strategy |
|---|---|---|
| Bypass Signaling (e.g., MET) | Activation of alternative RTKs (e.g., MET) sustains pro-survival signaling [79]. | Combination therapy with EGFR + MET inhibitors [79]. |
| T790M Mutation | A secondary mutation in EGFR that impedes TKI binding [82]. | Third-generation EGFR-TKIs (e.g., Osimertinib) [80]. |
| Tumor Microenvironment | CAFs induce EMT and resistance via hedgehog signaling [82]. | Hedgehog pathway inhibitors (e.g., cyclopamine) [82]. |
| Defective EGFR Degradation | Enhanced receptor recycling/stability (e.g., via TRIB3) prolongs signaling [80]. | Agents that promote EGFR degradation (e.g., TRIB3-EGFR disruptor) [80]. |
The Scientist's Toolkit: Key Research Reagents
Studying EGFR signaling crosstalk requires a specific toolkit of reagents and model systems.
Table 3: Essential Research Reagents for Investigating EGFR Crosstalk
| Reagent / Model System | Key Function/Application | Specific Example(s) |
|---|---|---|
| 32D Cell System | Isolated model for expressing individual ERBBs/MET to study specific interactions [79]. | 32D cells expressing EGFR alone or with MET [79]. |
| EGFR-TKIs | Inhibit EGFR tyrosine kinase activity to probe pathway dependency and crosstalk. | Gefitinib, Erlotinib [79] [82]. |
| MAPK Pathway Inhibitors | Inhibit intermediary kinases to dissect indirect crosstalk mechanisms. | MEK inhibitor U0126, p38MAPK inhibitor SB203580 [79]. |
| His-/Strep-Tagged Ubiquitin | Enable affinity-based enrichment and proteomic identification of ubiquitylated proteins [7]. | His-tagged ubiquitin for cobalt-NTA purification [7]. |
| Phospho-specific Antibodies | Detect phosphorylation events key to signaling and regulation. | Antibodies against p-EGFR-Y1068, p-MET-Y1234/1235 [79]. |
| Nanotheranostic Platforms | Enable simultaneous drug delivery and imaging in preclinical models. | H-dots loaded with gefitinib and genistein [83] [84]. |
Visualizing the Crosstalk: Signaling Pathways and Experimental Workflows
MAPK-Mediated EGFR-to-MET Crosstalk
This diagram illustrates the established mechanism by which EGFR signaling activates the MET receptor indirectly through the MAPK pathway, a key crosstalk axis in lung cancer progression.
Ubiquitination-Phosphorylation Regulatory Network
This diagram summarizes the core network motifs involving the key regulators TRIB3, Mig6, and c-Cbl, and how their interplay controls the decision between EGFR recycling and degradation.
The crosstalk within the EGFR signaling network, particularly the intricate interplay between phosphorylation and ubiquitination, extends far beyond a simple linear pathway. It creates a complex, dynamic system that controls critical cell fate decisions, modulates receptor destiny, and drives lung cancer pathogenesis and therapy resistance. The mechanisms involving TRIB3-PKCα-mediated stabilization, MAPK-driven MET transactivation, and phosphorylation-inactivated Mig6 exemplify how this crosstalk is co-opted in cancer. Moving forward, overcoming therapeutic resistance will require strategies that target these network interactions rather than individual nodes. Promising approaches include combination therapies, stromal co-targeting, novel platforms like nanotheranostics, and a new class of drugs that directly promote the degradation of oncogenic proteins like EGFR. A deep, systems-level understanding of this PTM crosstalk will be indispensable for the development of more effective and durable precision medicines for lung cancer patients.
The nuclear factor-kappa B (NF-κB) signaling pathway represents a fundamental regulatory system controlling immune responses, inflammation, and cell survival [85]. At the heart of this pathway lies IκBα (nuclear factor-kappa B inhibitor alpha), a critical regulatory protein that governs NF-κB activity through a sophisticated interplay of post-translational modifications (PTMs) [85]. IκBα serves as a classic model for understanding how phosphorylation, ubiquitination, and SUMOylation compete for the same molecular sites to produce diametrically opposed functional outcomes. This competitive modification system creates a precise regulatory switch that determines whether NF-κB remains sequestered in the cytoplasm or translocates to the nucleus to activate target genes [85] [86]. The study of IκBα regulation provides fundamental insights into the broader thesis of ubiquitination versus phosphorylation crosstalk in signaling pathways, revealing how cells integrate multiple signals to determine functional outcomes in health and disease.
Molecular Anatomy of IκBα and Its Modification Sites
The human IκBα protein consists of 317 amino acids with several critical structural domains that facilitate its regulatory function [85]. The N-terminal signal-receiving domain (SRD; 1-72 aa) contains the primary regulatory sites for phosphorylation (Ser32, Ser36, Tyr42), ubiquitination (Lys21, Lys22), and SUMOylation (Lys21, Lys38) [85]. The ankyrin repeat domain (ARD; 73-280 aa) mediates protein-protein interactions with NF-κB dimers, while the C-terminal PEST domain (281-317 aa) influences protein stability and contains additional phosphorylation sites (Ser283, Ser289, Ser293, Thr291, Thr299) [85]. Two nuclear export sequences (NESs at 45-54 aa and 265-277 aa) enable shuttling between cellular compartments.
Table 1: Key Post-Translational Modification Sites in IκBα
| Modification Type | Specific Sites | Functional Consequences | Regulating Enzymes |
|---|---|---|---|
| Phosphorylation | Ser32, Ser36 | Primes IκBα for ubiquitination | IKK complex (IKKα/IKKβ) |
| Ubiquitination | Lys21, Lys22 | Targets IκBα for proteasomal degradation | E3 ubiquitin ligase (e.g., SCF^β-TrCP) |
| SUMOylation | Lys21 | Competes with ubiquitination, stabilizes IκBα | Ubc9, SUMO E3 ligases |
| Phosphorylation | Tyr42 | Non-canonical regulation; precise function unclear | Unknown kinases |
| Additional Phosphorylation | Ser283, Ser289, Ser293, Thr291, Thr299 | Modulates protein stability and function | Various kinases |
The structural organization of IκBα creates a sophisticated platform for signal integration, where competing PTMs converge on closely spaced residues to determine the protein's fate and function [85]. This molecular competition exemplifies the broader principles of PTM crosstalk that govern cellular signaling networks.
The Competitive PTM Landscape on IκBα
The Phosphorylation-Ubiquitination Axis: Activation of NF-κB Signaling
The canonical NF-κB activation pathway begins with phosphorylation of IκBα at two critical serine residues. Upon stimulation by cytokines, pathogens, or other activators, the IκB kinase (IKK) complex phosphorylates IκBα at Ser32 and Ser36 [85]. This phosphorylation event serves as a molecular "tag" that recruits the SCF^β-TrCP E3 ubiquitin ligase complex, which subsequently catalyzes the attachment of K48-linked polyubiquitin chains to Lys21 and/or Lys22 on IκBα [85]. The ubiquitinated IκBα is then recognized and degraded by the 26S proteasome, thereby releasing NF-κB (typically the p50-p65 heterodimer) to translocate to the nucleus and activate target gene expression [85].
The IKK complex serves as the master regulator for this process, containing two catalytic subunits (IKKα and IKKβ) and a regulatory subunit (IKKγ/NEMO) [87]. Activation of IKK complex is dependent on the phosphorylation of IKKα/β at its activation loop and the K63-linked ubiquitination of NEMO, illustrating additional layers of PTM regulation upstream of IκBα itself [87].
SUMOylation: The Antagonistic Stabilizing Modification
Competing directly with the degradation pathway is SUMOylation, which occurs predominantly at Lys21 of IκBα – the very same residue targeted for ubiquitination [86]. SUMO (Small Ubiquitin-like Modifier) conjugation involves a distinct enzymatic cascade: the E1 activating enzyme (SAE1/SAE2 heterodimer), the E2 conjugating enzyme (Ubc9), and potentially E3 ligases that enhance specificity [88] [89]. SUMO-1 modification of IκBα generates a form that is resistant to signal-induced degradation and consequently inhibits NF-κB activation [86].
The competition between these PTMs occurs through multiple mechanisms. First, SUMOylation and ubiquitination physically compete for the same lysine residue (Lys21), creating mutually exclusive modifications [86]. Second, phosphorylation of IκBα at Ser32 and Ser36 not only promotes ubiquitination but also actively inhibits SUMOylation [86]. This phospho-regulation of SUMOylation creates a bidirectional antagonistic relationship that sharpens the switch-like behavior of the NF-κB pathway.
Table 2: Functional Consequences of Competing IκBα Modifications
| Modification State | IκBα Stability | NF-κB Localization | Transcriptional Outcome | Cellular Response |
|---|---|---|---|---|
| Unmodified IκBα | Stable | Cytoplasmic | Basal repression | Homeostatic state |
| Phospho-IκBα (Ser32/36) | Destabilized | Transition | Primed for activation | Signal responsive |
| Ubiquitinated IκBα (Lys21/22) | Rapid degradation | Nuclear | Gene activation | Inflammatory response |
| SUMOylated IκBα (Lys21) | Stabilized | Cytoplasmic | Enhanced repression | Signal resistance |
Experimental Approaches and Methodologies
Key Techniques for Studying IκBα PTMs
Research elucidating the competitive PTM landscape of IκBα has employed sophisticated methodological approaches. Co-immunoprecipitation (Co-IP) and western blotting have been fundamental for detecting specific modifications under various stimulation conditions. For instance, researchers typically stimulate cells with TNF-α or LPS, followed by lysis in modified RIPA buffer containing protease and phosphatase inhibitors, then immunoprecipitate IκBα using specific antibodies, and finally detect modifications with anti-ubiquitin, anti-SUMO-1, or anti-phospho-IκBα antibodies [86].
Mass spectrometry-based proteomics has provided comprehensive mapping of modification sites. Global analysis of phosphorylation and ubiquitylation crosstalk has been achieved through sequential enrichment strategies – first enriching for ubiquitylated proteins using cobalt-NTA affinity purification (for His-tagged ubiquitin) or diGly remnant antibodies, followed by phosphopeptide enrichment using titanium dioxide (TiO2) or immobilized metal affinity chromatography (IMAC) [7]. For direct identification of co-modified peptides, strong-cation exchange (SCX) chromatography can separate peptides by charge state before sequential diGly and phosphopeptide enrichment [7].
Mutagenesis approaches have been crucial for establishing causal relationships. Site-directed mutagenesis of key residues (e.g., K21R, S32A/S36A) in conjunction with reporter gene assays (e.g., NF-κB luciferase reporters) has demonstrated the functional consequences of disrupting specific modifications [86].
Visualization of the Competitive PTM Network on IκBα
The following diagram illustrates the competitive relationships between phosphorylation, ubiquitination, and SUMOylation on IκBα:
The experimental workflow for comprehensive PTM analysis of IκBα involves multiple enrichment and detection strategies, as shown in the following diagram:
The Scientist's Toolkit: Essential Research Reagents and Methodologies
Table 3: Key Research Reagents for Studying IκBα PTMs
| Reagent Category | Specific Examples | Research Applications | Key Findings Enabled |
|---|---|---|---|
| Phospho-specific Antibodies | Anti-phospho-IκBα (Ser32/36) | Detect activation-specific phosphorylation | IKK-dependent phosphorylation precedes degradation [85] |
| SUMOylation Reagents | Anti-SUMO-1, SUMO expression constructs | Identify and manipulate SUMOylation | SUMO-1 modification inhibits degradation [86] |
| Ubiquitination Tools | Anti-ubiquitin, diGly remnant antibodies | Profile ubiquitination status | Lys21 ubiquitination targets degradation [7] [86] |
| IKK Inhibitors | IKK-16, SC-514, TPCA-1 | Block initial phosphorylation step | Prevents subsequent ubiquitination [87] |
| Proteasome Inhibitors | MG132, lactacystin, bortezomib | Block final degradation step | Accumulation of ubiquitinated IκBα [85] |
| Kinase Expression Constructs | Constitutively active IKK mutants | Enhance phosphorylation | Sufficient to trigger degradation pathway [87] |
| Site-directed Mutants | K21R, S32A/S36A | Disrupt specific modifications | SUMO and ubiquitin compete for Lys21 [86] |
Implications for Drug Development and Therapeutic Intervention
The competitive PTM landscape of IκBα presents multiple attractive targets for therapeutic intervention in inflammatory diseases, cancer, and autoimmune disorders. Most directly, IKK inhibitors have been developed to prevent the initial phosphorylation step, thereby stabilizing IκBα and suppressing NF-κB activation [87]. However, these approaches face challenges due to the pleiotropic functions of NF-κB across multiple tissues. More sophisticated strategies might target the specific E3 ligases responsible for IκBα ubiquitination or develop SUMOylation enhancers that would promote the stabilizing modification at the expense of degradative ubiquitination.
Understanding the competitive relationship between these PTMs has profound implications for drug development. The discovery that phosphorylation not only promotes ubiquitination but also actively inhibits SUMOylation [86] suggests that IKK inhibitors might have dual benefits – both reducing degradation and potentially permitting increased stabilization through SUMOylation. Similarly, developing agents that enhance the SUMOylation machinery could provide an alternative approach to suppressing pathological NF-κB activation.
The principles learned from IκBα regulation extend to numerous other signaling pathways where competing PTMs create sophisticated regulatory networks. This case study exemplifies how understanding PTM crosstalk at the molecular level can reveal novel therapeutic strategies for manipulating cellular signaling in disease contexts.
The intricate crosstalk between post-translational modifications (PTMs) represents a crucial regulatory layer in cancer biology, enabling tumor cells to dynamically rewire signaling networks that connect oncogenic drivers to metabolic reprogramming. Advances in multi-omics technologies and computational biology now allow researchers to move from predictive network modeling to experimental validation of these complex relationships. This guide compares contemporary methodological approaches for elucidating PTM-mediated crosstalk, with a specific focus on the interplay between ubiquitination and phosphorylation in coordinating signaling and metabolic pathways. We objectively evaluate experimental frameworks based on their analytical depth, technical robustness, and translational applicability for researchers and drug development professionals.
Experimental Approaches for PTM Crosstalk Analysis
Multi-PTM Proteomics and Network Modeling
Sequential Enrichment of Post-Translational Modification (SEPTM) Proteomics enables the concurrent identification of tyrosine-phosphorylated, lysine-ubiquitinated, and lysine-acetylated proteins from the same samples. This approach provides a comprehensive view of coordinated PTM regulation in response to perturbations such as tyrosine kinase inhibitors (TKIs). In lung cancer studies, this method has identified over 12,000 unique PTMs, with 4,987 phosphorylation sites, 3,249 acetylation sites, and 4,452 ubiquitination sites showing significant changes following TKI treatment [41].
The experimental workflow involves:
- Treatment of lung cancer cell lines with targeted TKIs (crizotinib, erlotinib, dasatinib, afatinib) matched to specific oncogenic drivers (EGFR, ALK, ERBB2/HER2, ROS1, DDR2)
- Sequential PTM enrichment using modification-specific antibodies and affinity purification
- Mass spectrometry analysis for PTM identification and quantification
- Machine learning clustering to identify functional modules of coordinately regulated PTMs
- Network construction through three complementary approaches:
- Co-cluster Correlation Network (CCCN) based on PTM coordination patterns
- Cluster-Filtered Network (CFN) filtering protein-protein interactions through PTM data
- Pathway Crosstalk Network (PCN) connecting pathways whose proteins show coordinated PTM changes [41]
Integrated Multi-Omics Frameworks
For colorectal cancer research, an integrated multi-omics framework combines bulk RNA-seq (GEO/TCGA, n=1,783), single-cell transcriptomics (41,143 cells), and Mendelian randomization to establish PTM-centric regulatory networks. This approach identified dysregulation in 80% of PTM pathways in colorectal cancer, with ubiquitination sustaining Wnt/β-catenin signaling and GALNT6-mediated glycosylation driving immune evasion [90].
Key methodological components include:
- Differential expression analysis using limma package with FDR correction
- Single-cell RNA sequencing processing via Seurat v4 with Harmony integration
- Machine learning feature selection integrating LASSO, SVM, and Random Forest
- Functional validation using spatial transcriptomics and immune profiling [90]
Comparative Analysis of Experimental Platforms
Table 1: Comparison of PTM Crosstalk Analysis Platforms
| Platform Feature | SEPTM Proteomics [41] | Integrated Multi-Omics [90] | Pan-Cancer PTM Analysis [91] |
|---|---|---|---|
| PTM Types Analyzed | Phosphorylation, Ubiquitination, Acetylation | 20 PTM types including ubiquitination, glycosylation, phosphorylation, SUMOylation, acylations | Multiple modifications via mass spectrometry |
| Primary Data Source | Mass spectrometry proteomics | Transcriptomics (bulk & single-cell) + genomics | Proteomics, genomics, transcriptomics |
| Network Modeling | CCCN, CFN, PCN | PTM Activity Signature | Cross-platform correlation networks |
| Validation Approach | Pathway crosstalk analysis | Mendelian randomization, spatial transcriptomics | Multi-cohort replication |
| Key Applications | TKI response mechanisms, metabolic signaling | Immunotherapy resistance, prognostic stratification | Pan-cancer regulation patterns |
| Technical Throughput | 12,461 unique PTMs identified | 1,783 patients, 41,143 single cells | 100+ samples across cancer types |
| Drug Discovery Utility | TKI combination therapy targets | GALNT6 as immunotherapy target | Biomarker identification |
Signaling Pathway Validation: From Prediction to Mechanism
Ubiquitination-Phosphorylation Interfaces in Lung Cancer
Network models from SEPTM proteomics reveal concrete molecular connections between oncogenic signaling and metabolic reprogramming. The analysis identified specific crosstalk between receptor tyrosine kinase (RTK) signal transduction and metabolic pathways including "Transmembrane transport of small molecules" and "Glycolysis and gluconeogenesis" [41]. These connections were established through:
- PTM Co-clustering: Proteins with coordinated phosphorylation, ubiquitination, and acetylation changes form functional modules
- Cluster-Filtered Networks: Protein-protein interactions are validated through PTM coordination patterns
- Pathway Crosstalk Analysis: BioPlanet pathways show statistically significant connections based on shared PTM regulation
The core network common across multiple lung cancer cell lines and TKI treatments involves heat shock/chaperone proteins, metabolic enzymes, cytoskeletal components, and RNA-binding proteins, suggesting these represent fundamental infrastructure connecting signaling to metabolic reprogramming [41].
Ubiquitination-Mediated Metabolic Reprogramming
Radiotherapy resistance studies provide validated examples of ubiquitination-metabolism crosstalk, with ubiquitin chain architectures (K48-linked, K63-linked, monoubiquitination) creating distinct regulatory outcomes:
- K48-linked ubiquitination: FBXW7 mediates context-dependent radiation responses by degrading p53 in colorectal cancer (radioresistance) versus degrading SOX9 in NSCLC (radiosensitization) [50]
- K63-linked ubiquitination: TRIM26 stabilizes GPX4 via K63 chains to suppress ferroptosis in glioma, connecting ubiquitination to metabolic antioxidant defense [50]
- Metabolic sensor regulation: SMURF2-mediated HIF1α degradation compromises hypoxic adaptation, while SOCS2/Elongin B/C-driven SLC7A11 destruction increases ferroptosis sensitivity in liver cancer [50]
Diagram: PTM Crosstalk Connects Oncogenic Signaling to Metabolic Reprogramming. This network model shows how ubiquitination, phosphorylation, and acetylation form coordinated modules that bridge tyrosine kinase signaling with metabolic pathway regulation.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Essential Research Reagents for PTM Crosstalk Studies
| Reagent/Category | Specific Examples | Function in PTM Studies | Experimental Applications |
|---|---|---|---|
| PTM Enrichment Tools | Phospho-tyrosine antibodies, Ubiquitin remnant motifs, Acetyl-lysine antibodies | Selective isolation of modified peptides for mass spectrometry | SEPTM proteomics, modification-specific proteomics [41] |
| Computational Packages | limma, Seurat v4, glmnet, randomForest, clusterProfiler | Differential expression, single-cell analysis, machine learning feature selection | Multi-omics integration, biomarker identification [90] |
| Network Analysis Platforms | STRING, GeneMania, BioPlex, PathwayCommons | Protein-protein interaction database curation | CCCN, CFN, and PCN construction [41] |
| Pathway Resources | NCATS BioPlanet, KEGG, Gene Ontology | Pathway annotation and enrichment analysis | Pathway crosstalk validation [41] |
| Validation Reagents | Spatial transcriptomics platforms, Immune profiling antibodies | Functional validation of computational predictions | Tumor microenvironment analysis, immune cell infiltration [90] |
The validation of network predictions connecting oncogenic signaling to metabolic reprogramming through PTM crosstalk has progressed from correlative observations to mechanistic insights with therapeutic potential. The comparative approaches outlined here demonstrate that integrated multi-PTM analysis provides a more comprehensive understanding of cancer signaling networks than single-modification studies. The convergence of ubiquitination and phosphorylation regulation emerges as a particularly productive node for therapeutic intervention, with context-dependent outcomes that necessitate precise biomarker-guided strategies. As these methodologies continue to mature, they promise to unlock new opportunities for targeting PTM networks in cancer therapy, particularly for overcoming drug resistance mechanisms that involve metabolic adaptation.
Cell signaling crucially depends on a repertoire of post-translational modification (PTM) mechanisms for its regulation, with phosphorylation and ubiquitination representing two of the most significant modifications [30]. While phosphorylation, catalyzed by kinases, has been successfully targeted for cancer therapy for decades, ubiquitination—the covalent attachment of ubiquitin to target proteins—has emerged as a more recent therapeutic target with expanding clinical potential [30] [92]. The interplay between these PTMs creates a complex regulatory network; phosphorylation often serves as a marker that triggers subsequent ubiquitination, particularly in pathways leading to protein degradation [30]. Understanding this crosstalk is fundamental to appreciating the distinct yet complementary therapeutic approaches of kinase inhibitors and ubiquitin-proteasome system (UPS)-targeting therapies.
The clinical landscape has been dominated by small-molecule kinase inhibitors since the landmark approval of imatinib in 2001, which demonstrated that targeted inhibition of kinase activity could produce transformative outcomes in cancers like chronic myeloid leukemia [93]. Meanwhile, UPS-targeting therapies have evolved from broad proteasome inhibitors to sophisticated targeted protein degradation strategies that exploit the cell's natural ubiquitination machinery [92] [76]. This review provides a comparative analysis of these two therapeutic classes, examining their clinical success, mechanisms of action, and future directions within the framework of phosphorylation-ubiquitination crosstalk.
Clinical Maturity and Approved Agents
Kinase Inhibitors: An Established Therapeutic Class
Kinase inhibitors represent one of the most successful classes of targeted cancer therapies. As of 2025, the FDA has approved 85 small molecule protein kinase inhibitors for clinical use, with the majority (75 drugs) prescribed for neoplastic diseases and seven for inflammatory conditions [94]. This class has not only achieved numerical success but also demonstrated profound clinical impact, particularly in molecularly-defined cancers.
Table 1: Key Milestones in Kinase Inhibitor Development
| Year | Milestone | Clinical Impact |
|---|---|---|
| 2001 | First BCR-ABL inhibitor (imatinib) approved | Revolutionized CML treatment, with 10-year survival rate improved to 83.3% [93] [76] |
| 2003 | First EGFR inhibitor (gefitinib) approved | Paved way for personalized medicine in NSCLC with EGFR mutations [93] |
| 2011 | First ALK inhibitor (crizotinib) approved | Addressed ALK-positive NSCLC; later generations improved progression-free survival to >5 years [93] |
| 2015 | Third-generation EGFR inhibitor (osimertinib) approved | Overcame T790M resistance mutation; became top-selling kinase inhibitor ($6.6B in 2024) [93] |
| 2025 | 100th small-molecule kinase inhibitor approved | Demonstrated steady growth of class with 4 approvals in 2024 and 1 in 2025 [93] [94] |
The success of kinase inhibitors is exemplified by drugs like osimertinib, which demonstrated a median overall survival benefit of 38.6 months versus 31.8 months for earlier-generation EGFR inhibitors in NSCLC, and ibrutinib, which achieved annual sales of $6.4 billion in 2024 [93]. These agents have fundamentally changed treatment paradigms for numerous cancers, establishing kinase inhibition as a validated therapeutic approach.
UPS-Targeting Therapies: An Emerging Frontier
Targeted protein degradation (TPD) represents a revolutionary therapeutic strategy that capitalizes on the cell's intrinsic proteolytic systems, primarily the ubiquitin-proteasome system [76]. Unlike traditional inhibitors that block protein function, TPD aims to completely eliminate disease-causing proteins. The clinical development of UPS-targeting therapies has progressed through several distinct phases:
Table 2: Evolution of UPS-Targeting Therapies
| Therapeutic Class | Key Examples | Mechanism of Action | Clinical Status |
|---|---|---|---|
| Proteasome Inhibitors | Bortezomib, Carfilzomib | Broad inhibition of proteasome function | Approved for multiple myeloma [92] |
| PROTACs | KT-253 (MDM2-recruiting) | Bispecific molecules linking target to E3 ligase | Phase I clinical trials [76] |
| Molecular Glues | Immunomodulatory drugs (e.g., thalidomide derivatives) | Enhance interaction between target and E3 ligase | Approved for hematologic malignancies [76] |
| SNIPERs | LCL-161-based degraders | Simultaneously degrade target and IAP proteins | Preclinical development [76] |
While proteasome inhibitors like bortezomib and carfilzomib have achieved clinical approval, particularly for multiple myeloma, the more targeted approaches of PROTACs and molecular glues represent the cutting edge of UPS-targeted therapy [92] [76]. These novel agents are designed to hijack the cell's ubiquitination machinery to selectively degrade proteins of interest, offering potential advantages for addressing drug resistance and targeting previously "undruggable" proteins [76].
Molecular Mechanisms and Experimental Characterization
Kinase Inhibitor Mechanisms and Signaling Dynamics
Kinase inhibitors primarily function through ATP-competitive binding at the conserved kinase domain, preventing the transfer of phosphate groups to substrate proteins [95]. This inhibition disrupts phosphorylation-mediated signaling cascades, such as the EGFR-mediated MAPK pathway, which is frequently dysregulated in cancer [30]. The specificity of these inhibitors is achieved through strategic molecular design that exploits subtle differences in ATP-binding pockets across kinases.
The experimental characterization of kinase inhibitors typically involves several established methodologies:
- Kinase Activity Assays: Measure inhibition of phosphorylation using biochemical assays with recombinant kinases and substrates, often detecting phosphate transfer using radioactivity or antibodies [95].
- Cellular Proliferation Assays: Determine anti-proliferative effects in cancer cell lines (e.g., MTT, CellTiter-Glo) with IC50 values providing potency metrics [95].
- Target Engagement Validation: Western blotting to confirm reduced phosphorylation of direct kinase targets and downstream pathway components [30].
- Structural Studies: X-ray crystallography of inhibitor-kinase complexes (e.g., PDB: 6MZW for futibatinib-FGFR1) to elucidate binding modes and guide optimization [95].
The crosstalk between phosphorylation and ubiquitination is particularly evident in pathways like EGFR signaling, where receptor autophosphorylation creates docking sites for E3 ligases like Cbl, which then ubiquitinates the receptor, targeting it for endocytosis and degradation [30]. This intricate relationship means that kinase inhibitors can indirectly influence ubiquitination patterns by altering the phosphorylation status of substrates involved in ubiquitination pathways.
UPS-Targeting Therapeutic Mechanisms
UPS-targeting therapies operate through more diverse mechanisms that directly exploit the ubiquitination machinery. PROTACs (Proteolysis-Targeting Chimeras) are bispecific molecules comprising a target-binding ligand, an E3 ligase-recruiting ligand, and a connecting linker [76]. They function by inducing proximity between the target protein and an E3 ubiquitin ligase, leading to polyubiquitination and subsequent proteasomal degradation of the target.
Molecular glues represent a distinct class that typically function by inducing or stabilizing interactions between E3 ligases and target proteins, often by modifying the surface of the E3 ligase complex to create new binding interfaces [76]. Unlike PROTACs, molecular glues are not necessarily composed of two linked pharmacophores and often exhibit more drug-like properties.
Key experimental approaches for characterizing UPS-targeting therapies include:
- Degradation Assays: Western blotting or cellular thermal shift assays to measure reduction in target protein levels over time, with DC50 (half-maximal degradation concentration) and Dmax (maximum degradation) as key metrics [76].
- Ubiquitination Detection: Immunoprecipitation followed by anti-ubiquitin Western blotting to confirm target ubiquitination [76].
- Ternary Complex Formation: Techniques like surface plasmon resonance or analytical ultracentrifugation to assess stabilization of target-PROTAC-E3 ligase complexes [76].
- Functional Consequences: Cell viability assays, cell cycle analysis, and apoptosis assays to determine phenotypic outcomes of protein degradation [92].
Diagram 1: Comparative Mechanisms of Kinase Inhibitors and PROTACs. Kinase inhibitors (top) compete with ATP to block phosphorylation signaling. PROTACs (bottom) recruit E3 ubiquitin ligases to pathological proteins, inducing ubiquitination and degradation.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Studying Kinase and UPS-Targeted Therapies
| Reagent Category | Specific Examples | Research Application | Key Suppliers |
|---|---|---|---|
| Kinase Profiling Panels | Invitrogen SelectScreen, Eurofins KinaseProfiler | Assess inhibitor selectivity across kinase families | Thermo Fisher, Eurofins |
| Phospho-Specific Antibodies | Anti-pEGFR, Anti-pERK, Anti-pAKT | Detect pathway inhibition in cellular models | Cell Signaling Technology |
| ATP-Competitive Assays | ADP-Glo, LANCE TR-FRET | Quantify kinase activity and inhibition | Promega, PerkinElmer |
| E3 Ligase Ligands | VHL ligands (VH032), CRBN ligands (lenalidomide) | PROTAC construction and optimization | MedChemExpress, Sigma |
| DUB Inhibitors | PR-619, P22077 | Study deubiquitination effects on protein stability | Selleckchem, Cayman Chemical |
| Ubiquitination Detection | TUBE2 (Tandem Ubiquitin Binding Entities) | Enrich and detect ubiquitinated proteins | LifeSensors |
| Proteasome Activity Assays | Proteasome-Glo, Suc-LLVY-AMC substrate | Measure proteasome function after treatment | Promega, Boston Biochem |
| Ternary Complex Assays | Time-Resolved FRET, AlphaScreen | Quantify target-PROTAC-E3 ligase interactions | PerkinElmer, Revvity |
These research tools enable comprehensive characterization of both kinase inhibitors and UPS-targeting therapies, from initial mechanistic studies to investigation of resistance mechanisms. The selection of appropriate reagents is critical for generating reproducible and clinically relevant data in both fields.
Resistance Mechanisms and Clinical Limitations
Kinase Inhibitor Resistance
Despite their clinical success, kinase inhibitors face significant challenges with acquired resistance. The primary mechanisms include:
- Gatekeeper Mutations: Single amino acid changes in the kinase ATP-binding pocket (e.g., T790M in EGFR, T315I in BCR-ABL) that sterically hinder drug binding while preserving catalytic activity [93].
- Kinase Amplification: Increased copy number of the target kinase gene, requiring higher drug concentrations for effective inhibition [93].
- Bypass Signaling: Activation of alternative signaling pathways that circumvent the inhibited kinase, such as MET amplification in EGFR-inhibited tumors [93] [96].
- Pharmacological Limitations: The high intracellular ATP concentrations (especially in cancer cells) create a challenging environment for ATP-competitive inhibitors, requiring nanomolar affinity for effective target engagement [95].
These resistance mechanisms have driven the development of successive generations of kinase inhibitors, such as osimertinib for EGFR T790M-mutant NSCLC, which covalently binds to Cys797 in the ATP-binding pocket and shows reduced activity against wild-type EGFR [93].
UPS-Targeting Therapy Limitations
While newer to the clinical landscape, UPS-targeting therapies face their own set of challenges:
- Hook Effect: At high concentrations, PROTACs can form binary complexes with either the target or E3 ligase alone, failing to productively form the ternary complex required for degradation [76].
- E3 Ligase Limitations: Current approaches primarily utilize only a handful of the 600+ human E3 ligases (mainly CRBN, VHL, MDM2, IAP), potentially limiting the scope of degradable targets and enabling resistance through E3 ligase downregulation [76].
- Molecular Properties: PROTACs often have high molecular weights (>700 Da) and poor membrane permeability, presenting challenges for oral bioavailability [76].
- Resistance Mechanisms: Tumors may develop resistance through mutations in the E3 ligase machinery, target protein, or components of the ubiquitin-proteasome pathway [92].
Diagram 2: Phosphorylation-Ubiquitination Crosstalk in Therapeutic Context. The interconnected network of phosphorylation and ubiquitination signals can lead to either oncogenic rerouting (resistance) or enhanced combination response when targeted therapeutically.
Future Directions and Therapeutic Integration
The future of both kinase inhibitors and UPS-targeting therapies lies in their strategic integration, leveraging the natural crosstalk between phosphorylation and ubiquitination. Several promising directions are emerging:
Resistance Overcoming: UPS-targeting therapies offer potential solutions to kinase inhibitor resistance by degrading the target protein completely, thus circumventing mutation-based resistance [76]. KT-253, an MDM2-recruiting PROTAC, has shown 200-fold greater potency than traditional MDM2 inhibitors in preclinical models and has entered Phase I clinical trials [76].
Expanded Target Space: TPD approaches can address "undruggable" targets that lack conventional binding pockets for small-molecule inhibitors, including scaffold proteins and transcription factors [76]. This expands the therapeutic landscape beyond the "druggable" kinome.
Combination Strategies: Rational combinations of kinase inhibitors and UPS-targeted therapies may produce synergistic effects by simultaneously targeting different nodes in interconnected signaling networks [92]. For instance, combining EGFR kinase inhibitors with degraders of resistance proteins could enhance durability of response.
Novel E3 Ligase Exploration: Expanding the repertoire of E3 ligases used for TPD could enhance tissue specificity and reduce adaptation mechanisms [76]. Only about a dozen of the 600+ human E3 ligases are currently utilized in TPD approaches.
Bifunctional Modalities: Advances in bifunctional molecules beyond PROTACs, including lysosome-targeting chimeras and autophagy-targeting chimeras, are further expanding the toolbox for targeted protein degradation [76].
The clinical success of kinase inhibitors has paved the way for these more sophisticated protein-targeting approaches, providing valuable lessons in target validation, resistance management, and patient stratification. As our understanding of phosphorylation-ubiquitination crosstalk deepens, so too will our ability to design increasingly precise therapeutic interventions that exploit this fundamental biological relationship.
The comparative analysis of kinase inhibitors and UPS-targeting therapies reveals a dynamic therapeutic landscape shaped by our evolving understanding of post-translational modification crosstalk. Kinase inhibitors represent a mature, clinically-validated approach with proven efficacy across numerous indications, though limited by resistance mechanisms. UPS-targeting therapies embody an emerging paradigm with distinct advantages for addressing resistance and expanding the druggable proteome, yet face their own developmental challenges. The interplay between phosphorylation and ubiquitination ensures that these therapeutic strategies are not mutually exclusive but potentially complementary. Future advances will likely involve sophisticated combination approaches and novel modalities that increasingly leverage the complex crosstalk between these fundamental regulatory systems to achieve enhanced therapeutic outcomes across human diseases, particularly in oncology.
The intricate crosstalk between ubiquitination and phosphorylation represents a fundamental regulatory mechanism in eukaryotic cell signaling, coordinating virtually all cellular processes from cell cycle progression to stress response [7] [1]. Within this complex interplay, phosphodegrons—short, phosphorylation-dependent recognition motifs that target proteins for ubiquitin-mediated degradation—serve as critical integration points between kinase signaling and proteostatic control [97]. The accurate prediction and validation of these motifs and their associated regulatory circuits have become essential for understanding disease pathogenesis and developing targeted therapies. This review provides a comprehensive comparison of current methodologies for experimental validation of predicted phosphodegrons and kinase-substrate regulatory networks, offering researchers a practical guide for confirming computational predictions through rigorous experimental approaches.
Computational Prediction Tools: Landscape and Limitations
The expanding repertoire of computational tools for predicting phosphodegrons and kinase regulatory circuits has dramatically accelerated discovery, yet each approach carries distinct strengths and limitations that researchers must consider when designing validation strategies. The following table summarizes key computational tools and their primary characteristics:
Table 1: Computational Prediction Tools for Phosphodegrons and Regulatory Circuits
| Tool Name | Prediction Focus | Methodology | Key Features | Known Limitations |
|---|---|---|---|---|
| Degpred [98] | General degrons | BERT-based deep learning | Predicts from sequence alone; captures non-canonical motifs | Limited to sequence features; requires experimental validation |
| Motif_RF [98] | Internal degrons | Random forest classifier | Integrates 11 biochemical features (PTMs, structure, conservation) | Cannot predict terminal degrons; depends on available PTM data |
| deepDegron [98] | N-end & C-end degrons | Deep learning | Trained on GPS high-throughput data | Predicts destabilizing peptides beyond just degrons |
| NetworKIN [99] | Kinase-substrate relationships | Context-aware prediction | Integrates sequence and protein-protein interaction data | Limited to kinases with known recognition motifs |
| Kinase Activity Inference Methods [99] | Kinase activity states | Multiple algorithms (KSEA, PTM-SEA, etc.) | Infers activity from phosphoproteomics data | Performance varies by kinase-substrate library |
These computational approaches face several universal challenges: limited coverage of the full repertoire of E3 ligases and kinases, high false-positive rates for motif-based predictions, and context-dependent functionality that cannot be fully captured by algorithms [99] [98]. Consequently, experimental validation remains indispensable for confirming predicted phosphodegrons and regulatory relationships.
Experimental Validation Strategies for Phosphodegrons
Ratiometric Protein Degradation Assays
The ratiometric protein degradation assay represents a robust methodology for validating phosphodegron functionality, particularly for mapping E3 ligase-specific recognition motifs [20]. This approach employs a dual-fluorescent reporter system where intact DsRed serves as an internal control while EGFP is fused to the putative degron sequence. The core protocol involves:
- Construct Design: EGFP is fused to a probe containing the putative degron sequence alongside a known kinase docking motif to ensure proper phosphorylation, while DsRed is expressed separately as a non-degradable control [20].
- Transfection & Expression: Plasmids encoding both fluorescent proteins are co-transfected into appropriate cell lines (e.g., HEK293T) in equal amounts, establishing a baseline EGFP/DsRed ratio near 1.0 under non-degrading conditions [20].
- Degradation Monitoring: Cells are treated with proteasome inhibitors (e.g., MG132) or co-transfected with E3 ligase expression plasmids (e.g., FBXW7). Functional degrons display significantly reduced EGFP/DsRed ratios due to accelerated EGFP fusion degradation [20].
- Specificity Controls: Critical serine/threonine residues within the degron are mutated to alanine to confirm phosphorylation dependence. Additional controls include cycloheximide chase experiments to measure degradation kinetics [20].
This methodology successfully validated 19 novel FBXW7-binding phosphodegrons in proteins including ETV5, KLF4, JAZF1, and ZMIZ1, demonstrating its utility for systematic phosphodegron discovery [20].
Global Identification of Co-modified Proteins
For proteome-scale analysis of phosphorylation-ubiquitylation crosstalk, two complementary enrichment strategies enable identification of co-modified proteins:
- Ubiquitin Enrichment Followed by Phosphopeptide Isolation: Proteins modified with His-tagged ubiquitin are first enriched via cobalt-NTA affinity purification. Following tryptic digestion, phosphopeptides from both ubiquitin-enriched and ubiquitin-depleted fractions are isolated and analyzed by nRPLC-MS/MS [7].
- Sequential Peptide-based Enrichment: Protein digests are first separated by strong-cation exchange (SCX) chromatography to isolate multiply charged peptides. SCX fractions are subsequently enriched for diGly-modified peptides (ubiquitylation remnants) before MS/MS analysis, directly identifying peptides concurrently modified by phosphorylation and ubiquitylation [7].
These approaches identified 466 co-modified proteins in yeast with 2,100 phosphorylation sites co-occurring with 2,189 ubiquitylation sites, providing a global view of phosphorylation-ubiquitylation crosstalk [7].
Figure 1: Experimental validation workflow for predicted phosphodegrons, integrating computational prediction with multi-layered experimental approaches.
Benchmarking Kinase Regulatory Circuit Predictions
Kinase Activity Inference Methods
Multiple computational methods have been developed to infer kinase activities from phosphoproteomics data, each requiring distinct validation approaches. The benchmarKIN package provides a comprehensive framework for evaluating these methods using both perturbation-based and tumor-based benchmarking [99]:
Table 2: Kinase Activity Inference Methods and Performance Characteristics
| Method Category | Representative Tools | Underlying Algorithm | Key Considerations | Experimental Validation Approach |
|---|---|---|---|---|
| Enrichment-based | PTM-SEA [99] | Single-sample gene set enrichment | Limited by kinase-substrate library coverage | Phosphoproteomics after kinase inhibition |
| Statistical | KSEA [99] | Z-score based aggregation | Requires sufficient substrate coverage | Comparison with direct activity measurements |
| Rank-based | Various [99] | Substrate phosphorylation ranks | Sensitive to background distribution | Correlation with phospho-antibody arrays |
| Linear Models | RoKAI [99] | Multivariate linear modeling | Accounts for kinase promiscuity | Kinase mutagenesis studies |
| Network-based | NetworKIN [99] | Context-aware prediction | Incorporates structural information | Substrate trapping assays |
Experimental Validation of Kinase-Kinase Regulatory Circuits
Machine learning approaches now enable prediction of signed kinase-kinase regulatory relationships (activating/inhibitory), recovering known signaling pathways and suggesting denser inter-kinase regulation than previously appreciated [100]. Key validation methodologies include:
- Perturbation Experiments: Targeted inhibition or genetic ablation of upstream kinases followed by monitoring downstream phosphorylation events. This approach has been systematized in benchmark collections encompassing 230 experiments covering approximately 80 kinases [99].
- Phosphoproteomic Response Profiling: Comprehensive assessment of phosphoproteomic changes following kinase inhibition, with recent studies examining responses to 60 kinase inhibitors across multiple cell lines [99].
- Structural Validation: Crystallographic analysis of kinase-kinase complexes confirms predicted regulatory relationships, particularly when combined with mutational studies of interface residues.
The performance evaluation of these inference methods indicates that most computational approaches perform similarly, with the choice of kinase-substrate library having greater impact than the specific inference algorithm [99]. Combining manually curated libraries with predicted interactions from tools like NetworKIN demonstrates superior performance in recapitulating known kinase activities [99].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for Phosphodegron and Regulatory Circuit Validation
| Reagent Category | Specific Examples | Primary Function | Key Considerations |
|---|---|---|---|
| E3 Ligase Systems | FBXW7, β-TrCP, SPOP [20] [97] [98] | Phosphodegron recognition | Specificity for phosphodegron motifs; cancer mutation hotspots |
| Kinase Inhibitors | MAPK inhibitors, GSK3 inhibitors [20] | Kinase activity modulation | Specificity; off-target effects; concentration optimization |
| Proteasome Inhibitors | MG132 [20] [101] | Block degradation | Reversibility; toxicity with prolonged exposure |
| Tagging Systems | His-tagged ubiquitin, TMTpro [7] [102] | Affinity purification; multiplexing | Tag size impact on function; labeling efficiency |
| Phosphorylation Tools | Phospho-specific antibodies, Phos-tag gels [101] | Phospho-status monitoring | Specificity; validation requirements |
| Cell Line Models | HEK293, HCT116 [102] | Cellular context | Signaling pathway activity; genetic background |
Integrated Workflows for Systematic PTM Analysis
Emerging technologies now enable more comprehensive analysis of the interplay between post-translational modifications and protein assembly states. SEC-MX (Size Exclusion Chromatography fractions MultipleXed) represents a particularly advanced methodology that combines size exclusion chromatography with isobaric tagging to simultaneously characterize PTMs and assembly states from the same sample [102]. The integrated workflow includes:
- Fractionation and Multiplexing: SEC fractions from different conditions are labeled with TMTpro isobaric tags and multiplexed, significantly reducing LC-MS/MS runs while maintaining coverage comparable to label-free methods [102].
- Parallel Global and Phosphoproteomic Analysis: Following multiplexing, aliquots are split for either global proteomic analysis or IMAC-based phosphopeptide enrichment, enabling direct correlation between assembly states and phosphorylation events [102].
- Computational Integration: Data analysis pipelines like SECAT enable identification of high-confidence protein-protein interactions and their phosphorylation-dependent regulation [102].
This approach has revealed intricate relationships between phosphorylation events and assembly states, demonstrating that the same protein can exist in different phosphorylation states across different assembly states, with distinct functional consequences [102].
Figure 2: SEC-MX integrated workflow for simultaneous analysis of protein assembly states and phosphorylation events.
The benchmarking of predictive models for phosphodegrons and regulatory circuits requires multi-layered validation strategies that integrate computational predictions with rigorous experimental testing. The methodologies reviewed here—from targeted ratiometric degradation assays to global co-modification analysis and kinase activity inference benchmarking—provide researchers with a comprehensive toolkit for confirming predicted relationships. As these approaches continue to evolve, particularly with the integration of structural information and deep learning methodologies, the field moves closer to predictive understanding of phosphorylation-ubiquitylation crosstalk in health and disease. The systematic application of these validation frameworks will be essential for translating computational predictions into biologically meaningful insights with potential therapeutic applications.
Conclusion
The crosstalk between ubiquitination and phosphorylation is not merely a secondary phenomenon but a fundamental principle of signal transduction that confers precision, robustness, and dynamic control to cellular networks. The integration of large-scale proteomics with systems biology modeling has been instrumental in moving from studying isolated modifications to understanding the complex, functional PTM code. Validation in specific pathways, such as EGFR and NF-κB, underscores its critical role in human diseases, particularly cancer. Future research must focus on longitudinal studies to capture the temporal dynamics of this crosstalk and expand the exploration into other PTM partnerships. For clinical translation, the development of sophisticated, multi-targeted therapeutic strategies that deliberately manipulate this crosstalk, rather than inhibiting single nodes, holds immense promise for overcoming drug resistance and creating more effective treatments for cancer and other complex diseases.
References
- 1. When ubiquitination meets phosphorylation: a systems ... [https://biosignaling.biomedcentral.com/articles/10.1186/1478-811X-11-52]
- 2. Biochemistry, Proteins Enzymes - StatPearls - NCBI Bookshelf [https://www.ncbi.nlm.nih.gov/books/NBK554481/]
- 3. 8.2: Enzyme Structure and Function [https://bio.libretexts.org/Courses/City_College_of_San_Francisco/Introduction_to_Microbiology_(Liu_et_al.)/08%3A_Microbial_Metabolism_I/8.02%3A_Enzyme_Structure_and_Function]
- 4. Enzyme Structure and Function [https://www.creative-enzymes.com/blog/enzyme-structure-and-function/]
- 5. UBE2J2 sensitizes the ERAD ubiquitination cascade to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12511567/]
- 6. Crosstalk between phosphorylation and ubiquitination is ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7751476/]
- 7. Global analysis of phosphorylation and ubiquitylation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3868471/]
- 8. Article RSK1 reprograms the ubiquitin pathway to promote ... [https://www.sciencedirect.com/science/article/pii/S2211124725010940]
- 9. The Age of Crosstalk: Phosphorylation, Ubiquitination, and ... [https://www.sciencedirect.com/science/article/pii/S1097276507007988]
- 10. Emerging functions of branched ubiquitin chains [https://www.nature.com/articles/s41421-020-00237-y]
- 11. Current methodologies in protein ubiquitination ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9373315/]
- 12. Emerging tools and methods to study cell signalling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12224918/]
- 13. Unraveling chain specific ubiquitination in cells using ... [https://www.nature.com/articles/s41598-025-07242-9]
- 14. Ubiquitin signaling in immune responses - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC4822134/]
- 15. Structural basis of K11/K48-branched ubiquitin chain ... [https://www.nature.com/articles/s41467-025-64719-x]
- 16. An inventory of crosstalk between ubiquitination and other ... [https://www.sciencedirect.com/science/article/pii/S258900422300353X]
- 17. Crosstalk between Ubiquitination and Other Post-translational ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7748009/]
- 18. Control of protein stability by post-translational modifications [https://www.nature.com/articles/s41467-023-35795-8]
- 19. A Post-translational Modification–enhanced Pull-down ... [https://en.bio-protocol.org/en/bpdetail?id=4816&type=0]
- 20. Systematic Discovery of FBXW7-Binding Phosphodegrons ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8955265/]
- 21. Modulation of Functional Phosphorylation Sites by Basic ... [https://www.mdpi.com/1420-3049/28/12/4686]
- 22. Cdc4 phospho-degrons allow differential regulation of ... [https://elifesciences.org/articles/67390]
- 23. An AKT1-and TRIM21-mediated phosphodegron controls ... [https://www.sciencedirect.com/science/article/pii/S258900422300384X]
- 24. A minimal “push–pull” bistability model explains oscillations ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6249812/]
- 25. Uncovering mechanisms of bistability in biological systems [https://www.sciencedirect.com/science/article/abs/pii/S0958166908000797]
- 26. Dual functionality of MDM2 in PROTACs expands the horizons ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12392632/]
- 27. Dissecting crosstalk induced by cell-cell communication ... [https://www.nature.com/articles/s41467-025-61149-7]
- 28. Crosstalk and specificity in signalling. Are we ... [https://pubmed.ncbi.nlm.nih.gov/11516620/]
- 29. Decoding Post-Translational Modification Crosstalk With ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8430371/]
- 30. When ubiquitination meets phosphorylation - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC3734146/]
- 31. Mechanisms and functions of SUMOylation in health and ... [https://jbiomedsci.biomedcentral.com/articles/10.1186/s12929-024-01003-y]
- 32. Protein phosphorylation vs. ubiquitination in drug ... [https://www.genscript.com/protein-phosphorylation-vs-ubiquitination.html]
- 33. Crosstalk between sumoylation and acetylation regulates ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2683057/]
- 34. An Acetylation Switch Regulates SUMO-Dependent Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3614008/]
- 35. Cross-talk between chromatin acetylation and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5950014/]
- 36. Uncovering the SUMOylation and ubiquitylation crosstalk ... [https://www.nature.com/articles/ncomms14109]
- 37. A model for predicting post-translational modification cross ... [https://www.sciencedirect.com/science/article/abs/pii/S0957417424016373]
- 38. A short SUMOylation tag modulates transcription factor ... [https://www.sciencedirect.com/science/article/pii/S0021925825026596]
- 39. Ubiquitination and Phosphorylation: The Dynamic Duo of ... [https://www.metwarebio.com/ubiquitination-phosphorylation-cellular-regulation/]
- 40. Simultaneous Affinity Enrichment of Two Post-Translational ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7275731/]
- 41. Network models of protein phosphorylation, acetylation, and ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1010690]
- 42. Protocol for tandem enrichment of ubiquitinated, ... [https://www.sciencedirect.com/science/article/pii/S2666166725003508]
- 43. Mass spectrometry-based proteomics as an emerging tool in ... [https://clinicalproteomicsjournal.biomedcentral.com/articles/10.1186/s12014-023-09424-x]
- 44. Opinion Reprogramming SUMO-primed ubiquitylation [https://www.sciencedirect.com/science/article/pii/S0165614725002056]
- 45. Understanding the mTOR signaling pathway via mathematical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5573916/]
- 46. The Process Diagram: Rationale and Definition [https://www.celldesigner.org/documents/ProcessDiagram.html]
- 47. Challenges and resistance mechanisms to EGFR targeted ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12196329/]
- 48. Ubiquitination and deubiquitination in cancer [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-024-02046-3]
- 49. Targeting the RAS/RAF/MAPK pathway for cancer therapy [https://www.nature.com/articles/s41392-023-01705-z]
- 50. Decoding the ubiquitin network: molecular mechanisms and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12492556/]
- 51. Targeting the Ubiquitin–Proteasome System for Cancer - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12495026/]
- 52. Evolution and functional cross‐talk of protein post ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4019982/]
- 53. PTMNavigator: interactive visualization of differentially ... [https://www.nature.com/articles/s41467-024-55533-y]
- 54. An inventory of crosstalk between ubiquitination and other ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10165199/]
- 55. Site-specific ubiquitination: Deconstructing the degradation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9208700/]
- 56. Current methodologies in protein ubiquitination characterization [https://cellandbioscience.biomedcentral.com/articles/10.1186/s13578-022-00870-y]
- 57. Applying a Conservation-Based Approach for Predicting ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12418505/]
- 58. Evolutionary Constraint and Disease Associations of Post ... [https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1004919]
- 59. Conserved phosphorylation hotspots in eukaryotic protein ... [https://www.nature.com/articles/s41467-019-09952-x]
- 60. Identification, Analysis and Prediction of Protein Ubiquitination ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3006176/]
- 61. Discovering the Landscape of Protein Modifications - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8106652/]
- 62. Advances in enrichment methods for mass spectrometry ... [https://www.sciencedirect.com/science/article/abs/pii/S0021967322005453]
- 63. 7.3 Enrichment techniques for modified proteins and peptides [https://fiveable.me/proteomics/unit-7/enrichment-techniques-modified-proteins-peptides/study-guide/gRdCkJqac0QXnmIF]
- 64. Modification-specific proteomics: Strategies for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2892724/]
- 65. Peptide Enrichment for Mass Spectrometry-Based Analysis [https://www.creative-proteomics.com/ptms-proteomics/peptide-enrichment-for-mass-spectrometry-based-analysis.htm]
- 66. A Systematic Review of Computational Methods for Protein ... [https://link.springer.com/article/10.1007/s11831-025-10444-z]
- 67. Analyzing causal relationships in proteomic profiles using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8633371/]
- 68. Predicting Causal Relationships from Biological Data [https://www.nature.com/articles/s41598-017-08582-x]
- 69. Single-cell stimulus-response gene expression trajectories ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11560543/]
- 70. Linking signaling dynamics and cell fate decisions through ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2025.1656051/full]
- 71. A phosphorylation-driven ubiquitination switch fine-tunes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12164958/]
- 72. Comparison of methods and resources for cell ... [https://www.nature.com/articles/s41467-022-30755-0]
- 73. Targeting Cullin–RING E3 ubiquitin ligases for drug discovery [https://pmc.ncbi.nlm.nih.gov/articles/PMC4403949/]
- 74. Ubiquitination, E3 Ligases and Drug Discovery - Novel ... [https://www.ddw-online.com/ubiquitination-e3-ligases-and-drug-discovery-novel-technologies-for-a-challenging-pathway-757-201208/]
- 75. Challenges and opportunities for the diverse substrates of ... [https://www.thno.org/v15p6111.htm]
- 76. Targeted protein degradation: advances in drug discovery ... [https://www.nature.com/articles/s41392-024-02004-x]
- 77. Therapeutic potential of allosteric HECT E3 ligase inhibition [https://pmc.ncbi.nlm.nih.gov/articles/PMC12087876/]
- 78. A highly annotated drug combination resource for ... [https://www.nature.com/articles/s41597-025-05630-4]
- 79. EGF Receptor activates MET through MAP kinases to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3745527/]
- 80. TRIB3-EGFR interaction promotes lung cancer progression ... [https://www.nature.com/articles/s41467-020-17385-0]
- 81. Phosphorylation of Mig6 negatively regulates the ... [https://www.sciencedirect.com/science/article/abs/pii/S0898656817303029]
- 82. Crosstalk with cancer-associated fibroblasts induces ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4676617/]
- 83. Image-guided drug delivery of nanotheranostics for ... [https://www.thno.org/v12p4147.htm]
- 84. Image-guided drug delivery of nanotheranostics for targeted ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9169367/]
- 85. Post-translational Modifications of IκBα: The State of the Art [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2020.574706/full]
- 86. SUMO-1 Modification of IκBα Inhibits NF-κB Activation [https://www.sciencedirect.com/science/article/pii/S1097276500801331]
- 87. Phosphorylation and ubiquitination of the IκB kinase complex ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1847656/]
- 88. Mechanisms, regulation and consequences of protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3310159/]
- 89. Regulation of the SUMOylation System in Gene Expression [https://pmc.ncbi.nlm.nih.gov/articles/PMC2495007/]
- 90. Integrated multi-omics analysis reveals PTM networks as key ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12474747/]
- 91. Pan-cancer analysis of post-translational modifications ... [https://www.sciencedirect.com/science/article/pii/S009286742300781X]
- 92. Study unravels the complex interplay of the UPS in cancer ... [https://www.news-medical.net/news/20250224/Study-unravels-the-complex-interplay-of-the-UPS-in-cancer-development-and-treatment.aspx]
- 93. FDA approves 100th small-molecule kinase inhibitor [https://www.nature.com/articles/d41573-025-00188-7]
- 94. Properties of FDA-approved small molecule protein kinase ... [https://pubmed.ncbi.nlm.nih.gov/40252783/]
- 95. ATP-competitive inhibitors for cancer treatment - RSC Publishing [https://pubs.rsc.org/en/content/articlehtml/2025/md/d5md00235d]
- 96. Bispecific antibodies: unleashing a new era in oncology ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12320366/]
- 97. Phosphodegrons in Health and Disease: From Cellular ... [https://www.mdpi.com/2813-3757/3/1/3]
- 98. Systematic prediction of degrons and E3 ubiquitin ligase ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9281121/]
- 99. Comprehensive evaluation of phosphoproteomic-based ... [https://www.nature.com/articles/s41467-025-59779-y]
- 100. Prediction of Signed Protein Kinase Regulatory Circuits [https://www.sciencedirect.com/science/article/pii/S2405471220301484]
- 101. Essential Phosphatases and a Phospho-Degron Are ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2597059/]
- 102. SEC-MX: an approach to systematically study the interplay ... [https://www.nature.com/articles/s41467-025-56303-0]