Ubiquitination Patterns as Stage-Specific Biomarkers and Therapeutic Targets in Cancer
This article synthesizes the latest evidence establishing a direct correlation between dynamic ubiquitination patterns and cancer stage and grade.
Ubiquitination Patterns as Stage-Specific Biomarkers and Therapeutic Targets in Cancer
Abstract
This article synthesizes the latest evidence establishing a direct correlation between dynamic ubiquitination patterns and cancer stage and grade. For researchers, scientists, and drug development professionals, we explore the foundational role of ubiquitinome alterations in tumorigenesis, detail advanced methodologies for profiling these changes, and analyze their clinical applications. The content covers the development of ubiquitination-based prognostic models, discusses challenges in therapeutic targeting of the ubiquitin-proteasome system, and validates these findings through multi-omics integration and clinical trial data. The review underscores the immense potential of ubiquitinomics in advancing predictive, preventive, and personalized medicine (PPPM) in oncology.
Decoding the Ubiquitinome: How Ubiquitination Patterns Define Cancer Progression and Staging
The Ubiquitin-Proteasome System (UPS) serves as the primary pathway for controlled intracellular protein degradation in eukaryotic cells, functioning as a critical mechanism for maintaining cellular protein homeostasis (proteostasis) [1]. This sophisticated system ensures the precise elimination of damaged, misfolded, or short-lived regulatory proteins through an ATP-dependent process [2]. The UPS represents a highly selective degradation machinery that governs the turnover of approximately 80-90% of intracellular proteins, distinguishing it from the autophagy-lysosome pathway which primarily handles bulk degradation [3]. The clinical importance of the UPS is well-established in oncology, particularly in multiple myeloma, where proteasome inhibitors like bortezomib have become first-line therapeutics, validating the UPS as a viable drug target [1] [4].
Beyond its housekeeping functions, the UPS plays indispensable roles in regulating vital cellular processes including cell cycle progression, signal transduction, gene expression, apoptosis, and immune responses [1] [2]. The system's operation involves a coordinated enzymatic cascade that tags target proteins with ubiquitin molecules, marking them for recognition and degradation by the 26S proteasome complex [1]. Understanding the intricate mechanisms of ubiquitination and the proteasome is fundamental to cancer biology, as dysregulation of UPS components contributes significantly to tumor initiation, progression, and therapeutic resistance across various cancer types [1] [5].
Table 1: Core Components of the Ubiquitin-Proteasome System
| Component | Number in Humans | Primary Function | Key Characteristics |
|---|---|---|---|
| Ubiquitin | 1 gene (multiple copies) | Protein tag for degradation | 76-amino acid, highly conserved heat-resistant protein [1] |
| E1 Enzymes | ~2 | Ubiquitin activation | ATP-dependent, forms thioester bond with ubiquitin [1] [6] |
| E2 Enzymes | ~40 | Ubiquitin conjugation | Transfers ubiquitin from E1 to E3/substrate [6] |
| E3 Ligases | 500-1000 | Substrate recognition | Determines specificity, recognizes degradation signals [2] |
| 26S Proteasome | Multiple complexes | Protein degradation | 2.5 MDa complex with 20S core and 19S regulatory particles [2] |
| DUBs | ~100 | Deubiquitination | Cleaves ubiquitin chains, provides reversibility [7] [4] |
The Ubiquitination Enzymatic Cascade
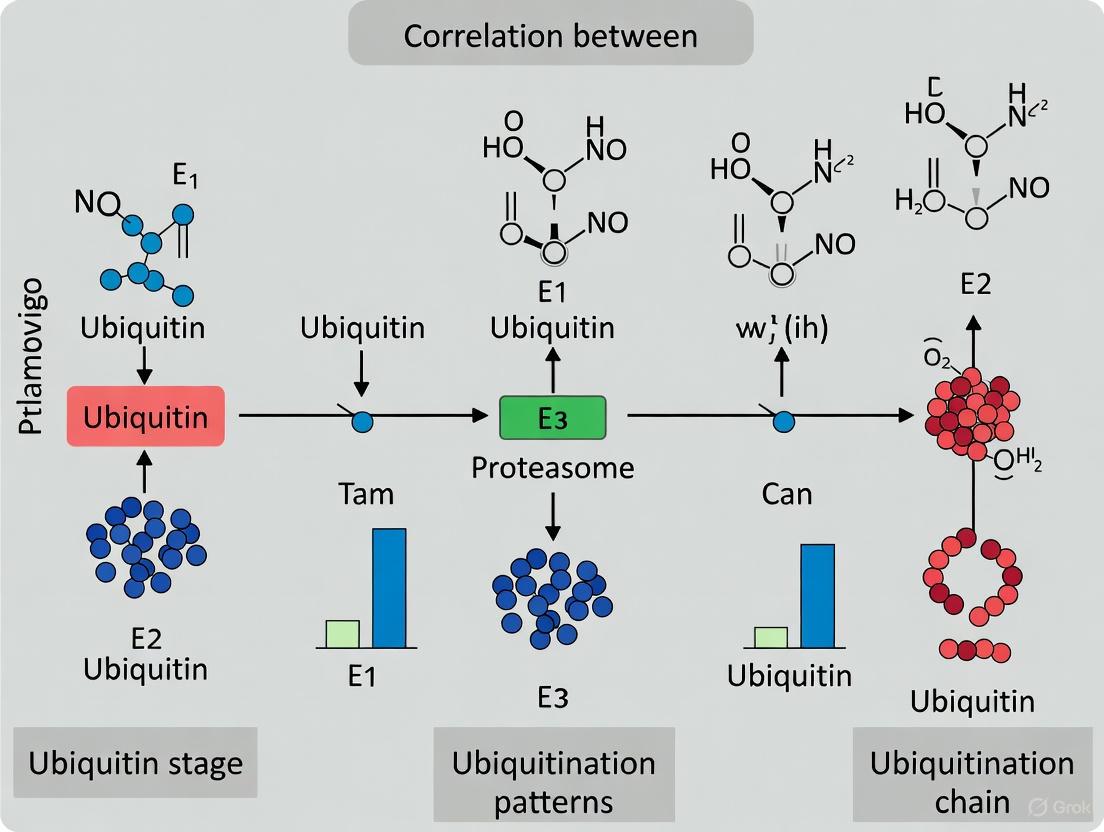
The process of ubiquitination involves a sequential enzymatic cascade that conjugates ubiquitin molecules to specific substrate proteins, thereby marking them for proteasomal degradation. This cascade requires the coordinated action of three distinct enzyme classes: E1 (ubiquitin-activating enzyme), E2 (ubiquitin-conjugating enzyme), and E3 (ubiquitin ligase) [1] [2].
Mechanism of Ubiquitin Transfer
The ubiquitination cascade begins with ubiquitin activation by E1 enzymes through an ATP-dependent reaction. During this initial step, E1 forms a high-energy thioester bond between its active-site cysteine residue and the C-terminal glycine (Gly76) of ubiquitin [1] [4]. The activated ubiquitin is then transferred to the cysteine active site of an E2 conjugating enzyme through a transesterification reaction [1]. The human genome encodes approximately 40 E2 enzymes, each containing a highly conserved catalytic UBC (Ubiquitin-Conjugating) domain [6].
The final step involves E3 ubiquitin ligases, which number between 500-1000 in humans and provide substrate specificity to the system [2]. E3 enzymes facilitate the transfer of ubiquitin from E2 to a lysine residue on the target protein, forming an isopeptide bond between the C-terminus of ubiquitin and the ε-amino group of the substrate lysine [1]. Some E3 ligases directly catalyze ubiquitin transfer, while others act as scaffolds that bring the E2~ubiquitin complex into proximity with the substrate [6]. This enzymatic cascade can be repeated to attach additional ubiquitin molecules to previously conjugated ubiquitin moieties, forming polyubiquitin chains with diverse topologies [2].
Diagram 1: The ubiquitination enzymatic cascade showing sequential E1-E2-E3 action.
Experimental Analysis of Ubiquitination
Investigating the ubiquitination cascade requires specific methodological approaches that can capture these dynamic enzyme-substrate interactions. Co-immunoprecipitation (Co-IP) assays serve as a fundamental technique for validating physical interactions between UPS components. For example, the interaction between UCHL1 (a deubiquitinating enzyme) and CIP2A (an oncoprotein) in gastric cancer was confirmed through endogenous immunoprecipitation assays using anti-CIP2A antibodies, followed by Western blot analysis to detect UCHL1 [7].
Western blotting under denaturing conditions can detect protein ubiquitination by observing characteristic molecular weight shifts, though this approach may not distinguish between mono- and polyubiquitination. More sophisticated methods include in vitro ubiquitination assays that reconstitute the cascade using purified E1, E2, E3 enzymes, ubiquitin, and ATP, allowing researchers to demonstrate direct ubiquitination of substrate proteins [6]. For studying E2 enzymes specifically, covalent inhibitors like NSC697923 that target the catalytic cysteine (Cys85) in UBE2N can be employed to interrogate specific E2 functions, though these inhibitors typically cause loss of function rather than facilitating degradation [6].
Table 2: Experimental Methods for Studying Ubiquitination
| Method | Application | Key Steps | Outcome Measures |
|---|---|---|---|
| Co-immunoprecipitation | Protein-protein interactions | Cell lysis, antibody incubation, precipitation, Western blot | Detection of interacting partners [7] |
| Western Blot | Protein expression/ modification | SDS-PAGE, transfer, antibody probing, detection | Molecular weight shifts, expression levels [7] [8] |
| In vitro Ubiquitination Assay | Direct ubiquitination confirmation | Purified E1/E2/E3, ubiquitin, ATP incubation | Ubiquitinated substrates on Western blot [6] |
| Mass Spectrometry | Ubiquitin chain topology | Protein digestion, LC-MS/MS, data analysis | Identification of linkage types [9] |
| Genetic Knockdown/ Knockout | Functional validation | siRNA/shRNA/CRISPR, phenotypic assays | Changes in proliferation, migration, invasion [7] |
Ubiquitin Chain Topologies and the Ubiquitin Code
Ubiquitin itself contains seven lysine residues (K6, K11, K27, K29, K33, K48, K63) and an N-terminal methionine (M1) that can serve as attachment points for subsequent ubiquitin molecules, enabling the formation of polyubiquitin chains with diverse structures and functions [2]. The specific arrangement of these ubiquitin chains creates a sophisticated "ubiquitin code" that determines the fate of modified proteins [9].
Structural and Functional Diversity of Ubiquitin Chains
The topology of ubiquitin chains directly influences their biological functions. K48-linked polyubiquitin chains represent the canonical signal for proteasomal degradation and serve as the primary topology for marking proteins for destruction [2] [3]. Similarly, K11-linked chains have also been strongly associated with targeting substrates for proteasomal degradation, particularly during cell cycle regulation [9] [3]. In contrast, K63-linked polyubiquitin chains typically function as non-proteolytic signals that regulate various cellular processes including protein trafficking, DNA repair, and kinase activation [2]. These chains also target damaged organelles for clearance via the autophagy-lysosome pathway [3].
Linear ubiquitin chains (linked through M1) play critical roles in immune signaling pathways, particularly in NF-κB activation, where the LUBAC complex (Linear Ubiquitin Chain Assembly Complex) generates these atypical chains to modulate inflammatory responses [2] [4]. Other linkage types, including K27, K29, and K33, participate in various signaling functions, though their roles are less well-characterized [2]. Additionally, mixed or branched chains that incorporate multiple linkage types within a single ubiquitin polymer add further complexity to the ubiquitin code, creating an extensive repertoire of regulatory signals [9].
Diagram 2: Diverse ubiquitin chain topologies and their primary cellular functions.
Analytical Techniques for Ubiquitin Chain Characterization
Deciphering the ubiquitin code requires specialized methodologies that can distinguish between different chain topologies. Mass spectrometry-based approaches have become indispensable for comprehensive ubiquitin chain analysis, particularly tandem mass tagging (TMT) and LC-MS/MS techniques that can identify specific linkage types through characteristic peptide fragments [6]. These proteomic methods enable researchers to map ubiquitination sites and determine chain topology in complex biological samples.
Linkage-specific antibodies that recognize particular ubiquitin chain types provide an accessible alternative for initial screening experiments. These reagents can be employed in Western blotting or immunofluorescence applications to monitor changes in specific ubiquitin signals under different experimental conditions. For functional validation, enzymatic probes including linkage-specific deubiquitinases (DUBs) can be utilized to selectively cleave particular chain types, thereby confirming their presence and importance for specific cellular processes [9].
Recent advances in chemical biology tools have further expanded the methodological toolbox for ubiquitin research. Activity-based probes that covalently label active-site cysteines in E2 enzymes or DUBs can help profile the enzymatic machinery responsible for generating or editing specific ubiquitin chain types [6]. Additionally, di-glycine remnant antibodies that recognize the characteristic signature left after tryptic digestion of ubiquitinated proteins enable proteome-wide identification of ubiquitination sites, though they cannot distinguish between chain topologies [6].
Table 3: Ubiquitin Chain Topologies and Their Functions
| Chain Type | Primary Function | Associated Processes | Key Regulatory Enzymes |
|---|---|---|---|
| K48-linked | Proteasomal degradation | Protein turnover, cell cycle | Most E3 ligases [2] [3] |
| K11-linked | Proteasomal degradation | ERAD, cell cycle | APC/C, UBE2S [9] [3] |
| K63-linked | Signaling, autophagy | DNA repair, inflammation, endocytosis | TRAF6, UBE2N/UBC13 [2] [3] |
| M1-linked (Linear) | Inflammation signaling | NF-κB activation, immunity | LUBAC (HOIP, HOIL-1) [2] [4] |
| K27-linked | Signaling | Immune response, mitophagy | Unknown E3s [2] |
| K29-linked | Signaling, degradation | Proteostasis, Wnt signaling | UBE3A, UBR5 [2] [3] |
| K33-linked | Signaling | Trafficking, metabolism | Unknown E3s [2] |
| Mixed/Branched | Complex signaling | Integrated cellular responses | Multiple E2/E3 combinations [9] |
UPS Dysregulation in Cancer Biology
The ubiquitin-proteasome system plays a dual role in oncogenesis, functioning as both a tumor suppressor mechanism through the degradation of oncoproteins and as a tumor promoter when hijacked to eliminate tumor suppressor proteins [1] [5]. This delicate balance becomes disrupted in numerous cancer types, with specific UPS components demonstrating distinctive dysregulation patterns across different malignancies.
Oncogenic Alterations in UPS Components
Deubiquitinating enzymes (DUBs) frequently exhibit altered expression in cancer, with UCHL1 showing marked upregulation in approximately 70% of gastric cancer cases [7]. Elevated UCHL1 expression correlates with poor patient prognosis and promotes tumor progression through stabilization of oncogenic clients like CIP2A, which in turn enhances c-Myc signaling and drives proliferation [7]. In lung adenocarcinoma, comprehensive bioinformatics analyses have identified a ubiquitination-related risk score (URRS) based on four ubiquitination-related genes (DTL, UBE2S, CISH, and STC1) that effectively stratifies patients into distinct prognostic groups [5].
The ubiquitin-conjugating enzyme UBE2T demonstrates elevated expression across multiple cancer types, including multiple myeloma, breast cancer, renal cell carcinoma, and ovarian cancer [8]. UBE2T upregulation associates with reduced overall and progression-free survival, positioning it as a potential prognostic biomarker and therapeutic target. Functional studies link UBE2T to key oncogenic pathways including cell cycle regulation, p53 signaling, and DNA damage response [8]. Pan-cancer analyses further identify gene amplification as the predominant genetic alteration affecting UBE2T across various malignancies [8].
Tumor-Specific UPS Signatures
The development of ubiquitination-based molecular signatures provides insights into cancer-specific UPS alterations. In lung adenocarcinoma, ubiquitination-related subtypes identified through unsupervised clustering algorithms demonstrate significant differences in tumor mutation burden (TMB) and clinical outcomes [5]. Patients classified into the high-risk URRS group exhibit not only worse prognosis but also distinct tumor microenvironment characteristics, including elevated PD-1/PD-L1 expression, increased TMB, and enhanced tumor neoantigen burden [5].
These UPS alterations directly impact therapeutic responses, as evidenced by differential sensitivity to chemotherapy agents between ubiquitination subtypes. The high URRS group shows lower IC50 values for various chemotherapeutic drugs, suggesting increased susceptibility to specific treatment modalities [5]. Additionally, UBE2T expression correlates with sensitivity to targeted agents like trametinib and selumetinib while showing negative correlations with CD-437 and mitomycin sensitivity [8]. These findings highlight the potential for UPS-based biomarkers to guide treatment selection and predict therapeutic responses.
Diagram 3: UPS component dysregulation in cancer pathogenesis.
Emerging UPS-Targeted Therapeutic Approaches
The clinical validation of proteasome inhibitors for multiple myeloma treatment established the UPS as a legitimate therapeutic target, spurring the development of more precise strategies that target specific UPS components [1] [4]. These innovative approaches aim to achieve enhanced selectivity while mitigating the broader toxicity associated with general proteasome inhibition.
PROTAC Technology and Molecular Glues
Proteolysis-Targeting Chimeras (PROTACs) represent a groundbreaking therapeutic modality that hijacks the UPS for targeted protein degradation [1] [9]. These heterobifunctional molecules consist of two distinct binding moieties connected by a chemical linker: one that engages the target protein of interest, and another that recruits an E3 ubiquitin ligase [9]. This engineered proximity facilitates ubiquitination and subsequent degradation of the target protein, effectively eliminating it from the cell.
The clinical potential of PROTACs is demonstrated by assets currently in advanced development, including ARV-110 and ARV-766 targeting the androgen receptor for prostate cancer, and ARV-471 targeting the estrogen receptor for breast cancer [9]. These degraders offer several advantages over traditional inhibitors, including event-driven catalysis (enabling efficacy at low doses), the ability to target previously "undruggable" proteins like transcription factors and scaffolding proteins, and potentially improved selectivity profiles [9]. Notably, PROTACs have demonstrated success against challenging targets including STAT3 and c-Myc, which have historically resisted conventional inhibition approaches [9].
Molecular glue degraders represent a related approach that induces novel interactions between E3 ligases and target proteins without a physical linker. The immunomodulatory drugs (IMiDs) like thalidomide, lenalidomide, and pomalidomide exemplify this strategy, acting as molecular glues that redirect the CRL4CRBN E3 ligase to degrade specific zinc-finger transcription factors (IKZF1, IKZF3) in multiple myeloma [9]. More recently, covalent molecular glues like EN450 have been discovered that recruit NF-κB to UBE2D, expanding the potential applications of this technology [6].
Expanding the UPS Therapeutic Toolbox
Beyond PROTACs and molecular glues, emerging strategies aim to co-opt additional UPS components for therapeutic purposes. These include approaches that target E2 ubiquitin-conjugating enzymes, non-substrate receptor E3 components, E3-associated proteins, and even the proteasome itself with greater specificity than first-generation proteasome inhibitors [6]. The development of covalent E2-targeted degraders that exploit allosteric cysteines in E2 enzymes represents a particularly innovative direction that bypasses traditional E3 recruitment [6].
The efficacy of UPS-targeted therapies is influenced by various cellular parameters including target protein localization, E3 ligase expression patterns, and the activity of deubiquitinating enzymes [9]. For instance, the subcellular compartmentalization of targets significantly impacts their susceptibility to different PROTACs, with some E3 ligases (like VHL) showing superior activity against endoplasmic reticulum-localized targets, while others (like CRBN) perform better against nuclear and cytoplasmic proteins [9]. Understanding these contextual factors is essential for rational degrader design and patient stratification strategies.
Table 4: Current UPS-Targeted Therapeutic Agents in Clinical Development
| Therapeutic Class | Representative Agents | Molecular Target | Clinical Stage | Primary Indications |
|---|---|---|---|---|
| Proteasome Inhibitors | Bortezomib, Carfilzomib, Ixazomib | 20S proteasome core | Approved (Phase III for newer agents) | Multiple myeloma [1] |
| PROTACs | ARV-110, ARV-766 | Androgen receptor | Phase III | Prostate cancer [9] |
| PROTACs | ARV-471 | Estrogen receptor | Phase III | Breast cancer [9] |
| Molecular Glues | Lenalidomide, Pomalidomide | CRBN (IKZF1/3) | Approved | Multiple myeloma, MDS [9] |
| E1 Inhibitors | TAK-243 | UBA1 (E1 enzyme) | Phase I | Advanced solid tumors [4] |
| DUB Inhibitors | LDN-57444 | UCHL1 | Preclinical | Gastric cancer [7] |
The Scientist's Toolkit: Essential Research Reagents
Investigating the ubiquitin-proteasome system requires specialized research tools that enable precise manipulation and monitoring of its components. The following table summarizes key reagents essential for UPS research, particularly in the context of cancer biology.
Table 5: Essential Research Reagents for UPS Investigation
| Reagent Category | Specific Examples | Research Application | Key Features/Functions |
|---|---|---|---|
| Proteasome Inhibitors | Bortezomib, MG132 | Functional UPS inhibition | Reversible/irreversible proteasome blockade, apoptosis induction [1] |
| E3 Ligase Ligands | VHL ligands, CRBN ligands (thalidomide) | PROTAC development, E3 functional studies | Recruit endogenous E3 machinery for targeted degradation [9] |
| DUB Inhibitors | LDN-57444 (UCHL1 inhibitor) | DUB functional validation | Target-specific DUB inhibition, substrate stabilization studies [7] |
| E2 Inhibitors | NSC697923 (UBE2N inhibitor) | E2 enzymatic studies | Covalent inhibition of specific E2 enzymes, signaling pathway dissection [6] |
| Linkage-Specific Antibodies | K48-linkage, K63-linkage specific | Ubiquitin chain topology analysis | Distinguish chain types in Western blot, immunofluorescence [2] |
| Activity-Based Probes | Ubiquitin-based probes with warheads | DUB/E2 enzymatic activity profiling | Covalently label active enzymes, monitor functional states [6] |
| siRNA/shRNA Libraries | E3/DUB-specific constructs | Functional genetic screening | Targeted knockdown of UPS components, phenotypic characterization [7] |
| Ubiquitin Mutants | K48R, K63R, K0 (no lysines) | Ubiquitination mechanism studies | Define chain linkage requirements, dominant-negative approaches [2] |
The Ubiquitin-Proteasome System represents a sophisticated regulatory network that extends far beyond simple protein disposal, encompassing complex enzymatic cascades and a diverse ubiquitin code that governs virtually all cellular processes. The intricate relationship between ubiquitin chain topologies and their specific biological functions underscores the precision of this system in maintaining cellular homeostasis. In cancer biology, UPS dysregulation emerges as a hallmark of tumorigenesis, with distinct patterns of E3 ligases, deubiquitinating enzymes, and ubiquitin chain dynamics correlating with disease progression, prognosis, and therapeutic responses. The ongoing development of UPS-targeted therapies, particularly PROTACs and molecular glues, highlights the translational potential of understanding these fundamental mechanisms. As research continues to unravel the complexities of the ubiquitin code and its alterations in disease states, the UPS promises to yield novel biomarkers and therapeutic strategies for cancer and beyond.
The ubiquitin-proteasome system (UPS) represents a sophisticated regulatory network that controls approximately 80-90% of intracellular protein degradation in eukaryotic cells, serving as a critical post-translational modification mechanism that governs protein stability, function, and localization [10] [11]. This system employs a precise enzymatic cascade comprising ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3) to tag specific substrate proteins with ubiquitin molecules, ultimately determining their fate through proteasomal degradation or functional alteration [1] [12]. The pathological significance of the UPS becomes particularly evident in oncogenesis, where dysregulation of ubiquitination processes contributes fundamentally to the acquisition of core cancer hallmarks, including sustained proliferation, evasion of growth suppressors, resistance to cell death, and activation of invasion and metastasis programs [11].
Emerging multi-cancer profiling studies have revealed that ubiquitination patterns are not static but undergo dynamic, stage-specific remodeling throughout cancer progression [13] [5]. The ubiquitinome—the complete set of ubiquitinated proteins in a cell or tissue—transforms substantially as tumors advance from early to late stages, reflecting the evolving functional requirements of cancer cells during disease progression. This review synthesizes evidence from recent pan-cancer analyses that delineate how systematic ubiquitinome reprogramming correlates with cancer stage grade, offering insights into prognostic biomarkers and therapeutic vulnerabilities that could inform stage-specific treatment strategies.
Molecular Mechanisms of Ubiquitinome Reprogramming in Cancer Progression
Ubiquitination Machinery and Chain Topology Diversity
The ubiquitination process initiates when E1 enzymes activate ubiquitin in an ATP-dependent manner, subsequently transferring it to E2 conjugating enzymes [1] [12]. E3 ligases then catalyze the transfer of ubiquitin from E2 to specific substrate proteins, with humans encoding only two E1 enzymes, approximately 50 E2 enzymes, and over 600 E3 ligases that provide substrate specificity [10] [12]. This enzymatic cascade can generate diverse ubiquitin modifications, including monoubiquitination (single ubiquitin on one lysine), multi-monoubiquitination (multiple single ubiquitins on different lysines), and polyubiquitination (ubiquitin chains linked through specific lysine residues) [11].
The functional consequences of ubiquitination depend critically on the topology of ubiquitin chain linkage. K48-linked polyubiquitination primarily targets substrates for proteasomal degradation, while K63-linked chains typically facilitate non-proteolytic signaling processes such as DNA repair, inflammation, and protein trafficking [12] [14]. Other linkage types, including K6, K11, K27, K29, and K33, as well as linear M1-linked chains, have been associated with diverse cellular functions from DNA damage repair to metabolic regulation [12] [11]. This "ubiquitin code" complexity allows the UPS to regulate virtually all cellular processes, with distinct chain topologies becoming preferentially enriched at different cancer stages to support stage-specific biological requirements.
Stage-Specific Alterations in Ubiquitination Patterns
Multi-cancer analyses have demonstrated systematic ubiquitinome remodeling during cancer progression. In early-stage tumors, ubiquitination patterns frequently reflect attempts to maintain genomic integrity and cellular homeostasis, with enrichment of K63-linked ubiquitination supporting DNA repair mechanisms and error-free cell division [14]. As tumors advance to higher stages, the ubiquitinome shifts toward degradation of tumor suppressors and enhanced stability of oncoproteins, typically mediated through increased K48-linked ubiquitination of protective factors [13].
Advanced-stage cancers exhibit ubiquitinome signatures characterized by elevated ubiquitination of proteins involved in invasion, metastasis, and treatment resistance pathways [13] [5]. For instance, the ubiquitin-like protein UBD (also known as FAT10) demonstrates progressive overexpression across multiple cancer types, with levels correlating strongly with histological grade and clinical stage [13]. This stage-associated reprogramming creates distinct molecular vulnerabilities that may be exploited for therapeutic intervention, particularly in advanced disease states where conventional therapies often fail.
Table 1: Ubiquitin Chain Linkages and Their Functional Roles in Cancer Progression
| Linkage Type | Primary Functions | Representative Roles in Cancer | Stage Association |
|---|---|---|---|
| K48-linked | Proteasomal degradation | Degradation of tumor suppressors (p53, PTEN) | Late-stage enrichment |
| K63-linked | Signal transduction, DNA repair | NF-κB activation, DNA damage response | Early-stage maintenance |
| K11-linked | Cell cycle regulation, ERAD | Mitotic progression, quality control | Stage-dependent variation |
| K27-linked | Mitophagy, immune signaling | Mitochondrial quality control | Context-dependent |
| K29-linked | Proteasomal degradation, Wnt signaling | Alternative degradation pathway | Limited characterization |
| K33-linked | Kinase regulation, trafficking | AMPK modulation, metabolic adaptation | Emerging evidence |
| K6-linked | DNA damage repair | Fanconi anemia pathway | Damage response |
| M1-linked (Linear) | NF-κB signaling, inflammation | Immune activation, cell survival | Microenvironment interaction |
Pan-Cancer Evidence for Stage-Associated Ubiquitinome Remodeling
Systematic Ubiquitin D (UBD) Overexpression in Advanced Cancers
A comprehensive pan-cancer investigation analyzing UBD across 44 different cancer types revealed compelling evidence of stage-specific ubiquitinome alterations [13]. This study demonstrated that UBD was significantly overexpressed in 29 cancer types compared to corresponding normal tissues, with elevated expression strongly correlating with advanced histological grades and clinical stages [13]. The most prevalent genetic alteration driving UBD overexpression was gene amplification, occurring alongside epigenetic changes characterized by reduced promoter methylation in 16 cancer types [13].
Mechanistically, UBD engages key oncogenic signaling pathways—including NF-κB, Wnt, and SMAD2—and interacts with downstream effectors such as MAD2, p53, and β-catenin to promote tumor survival, proliferation, invasion, and metastatic potential [13]. Patients exhibiting UBD alterations experienced significantly reduced overall survival rates across multiple cancer types, establishing UBD as both a promising prognostic biomarker and a potential predictor of immunotherapy sensitivity [13] [15]. The consistent overexpression pattern across diverse malignancies suggests that UBD upregulation represents a convergent adaptation in advanced cancers, possibly facilitating immune evasion and metastatic progression.
Ubiquitination-Related Gene Signatures in Cancer Staging
Beyond individual ubiquitin-like proteins, systematic analyses of ubiquitination-related gene (URG) signatures have revealed distinct molecular subtypes with profound implications for cancer staging and prognosis. In lung adenocarcinoma (LUAD), ubiquitination-based molecular subtyping successfully stratified patients into groups with significantly different clinical outcomes [5]. A ubiquitination-related risk score (URRS) developed from four prognostic genes (DTL, UBE2S, CISH, and STC1) demonstrated robust predictive capacity, with high-risk patients exhibiting worse prognosis, elevated PD-1/PD-L1 expression, increased tumor mutational burden, and heightened tumor neoantigen load [5].
Similarly, in breast cancer, a six-gene ubiquitination signature (ATG5, FBXL20, DTX4, BIRC3, TRIM45, and WDR78) effectively stratified patients into distinct risk categories with significant survival differences [16]. These ubiquitination-based classifiers consistently outperformed traditional clinical indicators in prognostic accuracy, highlighting the clinical utility of ubiquitinome profiling for cancer staging and outcome prediction. The reproducible performance of URGs across multiple independent validation cohorts underscores the fundamental role of ubiquitination reprogramming in driving cancer progression.
Table 2: Stage-Specific Ubiquitination Alterations Identified in Pan-Cancer Analyses
| Cancer Type | Key Ubiquitination Alterations | Stage Correlation | Functional Consequences |
|---|---|---|---|
| Multiple Cancers (29 types) | UBD/FAT10 overexpression | Higher histological grades and stages | Promotion of chromosomal instability, immune evasion |
| Lung Adenocarcinoma | URRS signature (DTL, UBE2S, CISH, STC1) | Advanced stage, poor prognosis | Immune checkpoint elevation, increased TMB |
| Breast Cancer | 6-gene ubiquitination signature | High-risk disease | Altered tumor microenvironment, metabolic reprogramming |
| Hepatocellular Carcinoma | UBE2T-mediated H2AX monoubiquitination | Radiation resistance | Enhanced DNA damage response, CHK1 activation |
| Glioblastoma | USP14-mediated ALKBH5 stabilization | Stemness maintenance | Preservation of cancer stem cell populations |
| Colorectal Cancer | FBXW7-mediated p53 degradation | Radioresistance | Inhibition of apoptosis |
| B-cell Lymphoma | LUBAC-mediated linear ubiquitination | NF-κB activation | Enhanced survival signaling |
Analytical Frameworks for Ubiquitinome Profiling in Cancer Staging
Multi-Omics Integration for Ubiquitinome Characterization
Comprehensive ubiquitinome profiling in cancer staging employs integrated multi-omics approaches that combine genomic, transcriptomic, epigenomic, and proteomic datasets. The analytical workflow typically begins with data acquisition from large-scale cancer genomics resources such as The Cancer Genome Atlas (TCGA) and Genotype-Tissue Expression (GTEx) projects [13]. Bioinformatics platforms including Gene Expression Profiling Interactive Analysis (GEPIA2.0), cBioPortal, UALCAN, and Sangerbox then facilitate systematic analysis of ubiquitination-related gene expression, prognostic significance, promoter methylation patterns, genetic alterations, and immune infiltration associations [13] [5].
Consensus clustering algorithms applied to URG expression profiles enable identification of distinct molecular subtypes with characteristic clinical outcomes [5]. Subsequent differential expression analysis reveals URGs that are significantly altered between molecular subtypes or cancer stages. Prognostic URGs are identified through integrated application of univariate Cox regression, random survival forests, and least absolute shrinkage and selection operator (LASSO) Cox regression algorithms, ultimately yielding ubiquitination-related risk scores that stratify patients according to clinical outcomes [5] [16].
Diagram 1: Experimental workflow for ubiquitinome profiling in cancer staging research.
Functional Annotation and Pathway Enrichment Analysis
Following identification of stage-associated ubiquitination alterations, functional annotation elucidates the biological processes and pathways impacted by ubiquitinome remodeling. Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database facilitates construction of protein-protein interaction networks for predicted UBD-interacting proteins [13]. Subsequent Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and Gene Ontology (GO) enrichment analyses using platforms like DAVID reveal ubiquitination involvement in critical cancer-relevant processes including neurodegeneration, proteolysis, apoptosis, immune response, and metabolic reprogramming [13].
Advanced ubiquitinome profiling further integrates immune microenvironment assessment, examining correlations between ubiquitination patterns and immune infiltration levels, checkpoint molecule expression, microsatellite instability, tumor mutational burden, and neoantigen load [13] [5]. This comprehensive functional characterization establishes mechanistic links between ubiquitinome alterations and functional consequences across cancer stages, providing insights into how ubiquitination reprogramming drives disease progression.
Research Reagent Solutions for Ubiquitinome Studies
Table 3: Essential Research Reagents for Ubiquitination and Cancer Staging Studies
| Reagent Category | Specific Examples | Research Applications | Key Features |
|---|---|---|---|
| Bioinformatics Platforms | GEPIA2.0, cBioPortal, UALCAN, Sangerbox | Ubiquitination gene expression analysis, survival correlation | Integration of TCGA/GTEx data, multi-omics capability |
| Ubiquitination Databases | iUUCD 2.0, STRING | Ubiquitination-related gene compilation, interaction networks | Comprehensive URG catalogs, PPI network visualization |
| E1 Enzyme Inhibitors | MLN7243, MLN4924 | E1 enzymatic activity blockade | UPS pathway disruption, apoptosis induction |
| E2 Enzyme Inhibitors | Leucettamol A, CC0651 | E2 enzymatic function inhibition | Specific E2 targeting, mechanism studies |
| E3 Ligase Modulators | Nutlin, MI-219, PROTACs | E3 ligase substrate recruitment manipulation | Targeted protein degradation, p53 pathway activation |
| Proteasome Inhibitors | Bortezomib, Carfilzomib, Ixazomib | 20S proteasome catalytic inhibition | Clinical application, protein stabilization |
| Deubiquitinase Inhibitors | Compounds G5, F6 | DUB enzymatic activity suppression | Ubiquitin chain stabilization, signaling modulation |
Therapeutic Implications and Future Perspectives
The systematic characterization of stage-specific ubiquitinome remodeling holds profound implications for cancer diagnostics and therapeutics. Ubiquitination-based biomarkers offer promising tools for cancer staging, prognosis prediction, and treatment selection, particularly in malignancies where conventional staging systems provide limited biological insights [13] [5] [16]. The strong association between specific ubiquitination signatures and immunotherapy response suggests potential applications in predicting checkpoint inhibitor efficacy, possibly through ubiquitination-mediated regulation of PD-1/PD-L1 stability and expression [11].
From a therapeutic perspective, the stage-associated enrichment of specific ubiquitination components reveals actionable vulnerabilities for targeted intervention. Proteasome inhibitors such as bortezomib, carfilzomib, and ixazomib have already demonstrated clinical success, particularly in multiple myeloma, establishing proof-of-concept for UPS-targeting therapies [1] [10]. Emerging strategies including proteolysis-targeting chimeras (PROTACs) and molecular glues represent innovative approaches to leverage the UPS for selective degradation of oncogenic proteins [17] [11]. These technologies exploit the ubiquitination machinery to target previously "undruggable" oncoproteins, with several candidates (ARV-110, ARV-471, CC-90009) advancing through clinical trials [11].
Future research directions should prioritize comprehensive mapping of ubiquitinome dynamics across the entire cancer progression continuum, from preneoplastic lesions to advanced metastatic disease. Single-cell ubiquitinome profiling technologies may reveal intratumoral heterogeneity in ubiquitination patterns and identify rare subpopulations with distinctive ubiquitination signatures driving therapy resistance [14]. The integration of ubiquitinome data with other post-translational modification networks (phosphorylation, acetylation, SUMOylation) will provide a more holistic understanding of the complex regulatory circuitry governing cancer progression [17] [14]. Ultimately, these advances promise to deliver ubiquitination-based precision oncology approaches that align therapeutic strategies with the stage-specific ubiquitinome configuration of individual tumors.
Diagram 2: Ubiquitin signaling pathway in cancer, showing key modifications and functional consequences.
Ubiquitination, a fundamental post-translational modification, has emerged as a critical regulatory mechanism in cancer progression and tumor grading. This process involves the covalent attachment of ubiquitin molecules to target proteins, thereby regulating their stability, activity, localization, and interactions [12] [11]. The ubiquitin-proteasome system (UPS) comprises a sophisticated enzymatic cascade including ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3), which work in concert to tag proteins for proteasomal degradation or functional modification [12] [18]. Beyond its well-established role in protein degradation, ubiquitination participates in nearly all cellular processes, and its dysregulation is now recognized as a hallmark of cancer pathogenesis, influencing tumor initiation, progression, and metastasis [12] [11].
Mounting evidence indicates that specific ubiquitination signatures correlate strongly with tumor grade and stage, offering unprecedented opportunities for prognostic stratification and therapeutic intervention [19] [20] [21]. The ability to decode these ubiquitination patterns provides valuable insights into the molecular drivers of cancer progression, potentially enabling more accurate patient risk assessment and personalized treatment approaches. This review synthesizes current understanding of how ubiquitination signatures reflect and influence tumor grade, with particular emphasis on key regulatory proteins, pathway interactions, and emerging methodological frameworks for clinical translation.
Molecular Mechanisms Linking Ubiquitination to Tumor Grade
The Ubiquitination Machinery in Cancer
The ubiquitination process initiates with E1 enzymes activating ubiquitin in an ATP-dependent manner, followed by transfer to E2 conjugating enzymes, and finally, E3 ligases facilitate the attachment of ubiquitin to specific substrate proteins [12] [18]. The human genome encodes approximately 40 E2 enzymes and over 600 E3 ligases, which confer substrate specificity and determine the biological outcomes of ubiquitination [12] [22]. This enzymatic cascade can generate diverse ubiquitin topologies, including monoubiquitination (single ubiquitin attachment), multimonoubiquitination (multiple single ubiquitins on different lysines), and polyubiquitination (ubiquitin chains linked through specific lysine residues) [11]. The specific linkage type determines the functional consequence for the modified protein, with K48-linked chains typically targeting substrates for proteasomal degradation, while K63-linked chains more often regulate signaling transduction, DNA repair, and endocytic trafficking [12] [22].
The reverse process, deubiquitination, is catalyzed by deubiquitinating enzymes (DUBs) that remove ubiquitin moieties from substrates, thereby counteracting E3 ligase activity and providing an additional layer of regulation [12] [11]. The balanced interplay between ubiquitinating and deubiquitinating enzymes maintains cellular homeostasis, and disruption of this equilibrium frequently drives oncogenic transformation and tumor progression [11]. For instance, the deubiquitinase OTUB2 has been shown to stabilize pyruvate kinase M2 (PKM2) by counteracting Parkin-mediated ubiquitination, thereby enhancing glycolysis and accelerating colorectal cancer progression [11].
Ubiquitination Patterns Across Tumor Grades
Different ubiquitination signatures emerge as tumors progress from low-grade to high-grade malignancies, reflecting the evolving molecular landscape of cancer cells. High-grade tumors often exhibit upregulated ubiquitination of tumor suppressor proteins, leading to their excessive degradation, while oncoproteins may escape ubiquitin-mediated degradation through various mechanisms [12]. A pan-cancer analysis integrating data from 4,709 patients across 26 cohorts and five solid tumor types revealed conserved ubiquitination-related prognostic signatures that effectively stratified patients into distinct risk categories with differential survival outcomes [19]. This study identified that ubiquitination scores positively correlated with squamous or neuroendocrine transdifferentiation in adenocarcinoma, suggesting that ubiquitination patterns may underlie histological fate decisions during tumor progression [19].
Table 1: Ubiquitination Linkage Types and Their Functional Consequences in Cancer
| Linkage Type | Primary Function | Role in Cancer | Associated Tumor Grades |
|---|---|---|---|
| K48-linked chains | Proteasomal degradation | Enhanced degradation of tumor suppressors | High-grade tumors |
| K63-linked chains | Signal transduction | Activation of NF-κB, TGF-β pathways | Advanced stages with metastasis |
| K11-linked chains | Cell cycle regulation | Mitotic control, aneuploidy | High-grade proliferative tumors |
| Linear (M1) chains | NF-κB activation | Inflammation, cell survival | Therapy-resistant cancers |
| K27-linked chains | Mitochondrial autophagy | Metabolic adaptation | Nutrient-deficient tumor microenvironments |
| Monoubiquitination | DNA repair, endocytosis | DNA damage response, receptor trafficking | Varies by cancer type |
At the single-cell resolution, ubiquitination signatures enable more precise classification of distinct cell types within the tumor microenvironment and correlate with immune cell infiltration patterns, particularly macrophages, which vary significantly with tumor grade [19]. The dynamic regulation of ubiquitination across tumor grades underscores its potential as a biomarker for disease progression and therapeutic response prediction.
Prognostic Ubiquitination Signatures Across Cancer Types
Development of Ubiquitination-Related Prognostic Signatures
The establishment of ubiquitination-related prognostic signatures (URPS) represents a significant advancement in molecular cancer classification. These signatures leverage the expression patterns of ubiquitination-related genes (URGs) to stratify patients according to clinical outcomes, often with greater accuracy than traditional histopathological grading alone [19] [20] [21]. Methodologically, URPS development typically involves several standardized steps: (1) comprehensive URG collection from databases like iUUCD or GeneCards; (2) expression analysis in tumor versus normal tissues; (3) identification of URGs with prognostic significance through univariate Cox regression; (4) molecular subtyping of cancers based on URG expression patterns; and (5) construction of multivariable models using machine learning approaches such as LASSO Cox regression [20] [21].
In colon cancer, for instance, researchers have identified distinct molecular subtypes based on URG expression patterns that exhibit significant differences in overall survival, progression-free survival, immune cell infiltration, and pathological staging [21]. A study analyzing 1299 URGs in colon cancer classified patients into two molecular subtypes with markedly different clinical outcomes and immune microenvironment characteristics [21]. Similarly, a pan-cancer study developed a URPS that effectively stratified patients into high-risk and low-risk groups across multiple cancer types, including lung cancer, esophageal cancer, cervical cancer, urothelial cancer, and melanoma [19]. The high-risk group consistently demonstrated worse overall survival, validating the prognostic utility of ubiquitination signatures across diverse malignancies.
Table 2: Experimentally Validated Ubiquitination-Related Prognostic Signatures
| Cancer Type | Key Ubiquitination Regulators | Associated Tumor Grade/Stage | Functional Role | Experimental Validation |
|---|---|---|---|---|
| Colon Cancer | ARHGAP4, MID2, SIAH2, TRIM45, UBE2D2, WDR72 | Advanced stage, poor prognosis | Immune microenvironment modulation, cell proliferation | qRT-PCR, immunohistochemistry, colony formation, EdU staining, xenograft models [21] |
| Pan-Cancer (Multiple) | OTUB1, TRIM28 | High-grade, therapy resistance | MYC pathway modulation, oxidative stress response | In vivo, in vitro, patient cohort analyses [19] |
| Breast Cancer | UBE2N/UBE2V1 complex | Metastatic disease | TGF-β signaling, NF-κB activation, MMP1 expression | ShRNA inhibition, metastasis mouse models [22] |
| Various Cancers | UBE2C | High-grade, aneuploidy | Chromosomal segregation, mitotic regulation | Transgenic mouse models, cell cycle analyses [22] |
Immune Microenvironment and Therapeutic Implications
Ubiquitination signatures not only reflect tumor grade but also shape the immune landscape of tumors, with significant implications for immunotherapy response. In colon cancer, patients classified into the high-risk URPS group exhibit characteristics indicative of enhanced epithelial-mesenchymal transition, immune escape, immunosuppressive myeloid-derived suppressor cell infiltration, regulatory T cell infiltration, and lower immunogenicity [21]. Conversely, the low-risk group demonstrates opposite trends but shows a better response to CTLA-4 checkpoint inhibitors [21]. These findings highlight how ubiquitination patterns influence tumor-immune interactions and may guide immunotherapeutic strategies.
The URPS also demonstrates value in predicting response to conventional therapies. For example, in the pan-cancer study, the ubiquitination score effectively predicted outcomes for patients receiving both surgery and immunotherapy [19]. This suggests that ubiquitination signatures capture fundamental biological features that determine therapeutic vulnerability, potentially guiding treatment selection across the cancer care continuum.
Key Experimental Methodologies for Ubiquitination Signature Analysis
Transcriptomic Profiling and Computational Modeling
The delineation of ubiquitination signatures relies heavily on sophisticated transcriptomic analyses and computational approaches. Bulk RNA sequencing from large patient cohorts like The Cancer Genome Atlas (TCGA) provides the foundational data for identifying ubiquitination-related gene expression patterns associated with tumor grade and clinical outcomes [19] [20]. The standard analytical workflow includes data preprocessing (normalization, batch effect correction), differential expression analysis of URGs between tumor and normal tissues, consensus clustering to define molecular subtypes, and survival analysis to validate prognostic utility [20] [21].
Single-cell RNA sequencing (scRNA-seq) has further enhanced resolution by enabling the dissection of ubiquitination signatures at cellular level within the complex tumor microenvironment [19]. This approach reveals how ubiquitination patterns vary between malignant cells, immune populations, and stromal components, providing insights into cell-type-specific regulatory mechanisms that influence tumor grade and behavior. Gene set variation analysis (GSVA) and single-sample gene set enrichment analysis (ssGSEA) algorithms are then employed to quantify pathway activities and immune cell infiltration based on ubiquitination signatures [19] [20].
Machine learning techniques, particularly least absolute shrinkage and selection operator (LASSO) Cox regression and support vector machine recursive feature elimination (SVM-RFE), are instrumental in refining ubiquitination signatures by identifying the most informative gene subsets for prognostic prediction [20] [21]. These computational methods effectively handle high-dimensional data while minimizing overfitting, yielding robust models that can be validated in independent patient cohorts.
Functional Validation Approaches
While computational analyses identify candidate ubiquitination signatures, functional validation is essential to establish causal relationships with tumor grade and progression. A multi-tiered experimental approach is typically employed, including:
In vitro models: Cell line models enable mechanistic studies of specific ubiquitination regulators. For example, knockdown experiments using siRNA or shRNA demonstrate how modulating specific E3 ligases or DUBs affects cancer cell phenotypes relevant to tumor grade, including proliferation, invasion, stemness, and therapy resistance [19] [22]. Colony formation and EdU staining assays quantitatively assess proliferative capacity, while migration and invasion assays evaluate metastatic potential [21].
In vivo models: Xenograft mouse models provide physiological context for evaluating how ubiquitination regulators influence tumor growth and progression. Orthotopic implantation models particularly recapitulate the tumor-microenvironment interactions that shape cancer behavior [21]. Spontaneous metastasis models further elucidate the role of ubiquitination in advanced disease [22].
Biochemical assays: Co-immunoprecipitation and ubiquitination assays directly demonstrate enzyme-substrate relationships and the effects of specific manipulations on ubiquitin chain formation [19]. For instance, the pan-cancer study validated that OTUB1-TRIM28 ubiquitination regulates MYC pathway activity, establishing a direct molecular link between ubiquitination signatures and oncogenic signaling [19].
Clinical correlation: Immunohistochemistry on patient tissue microarrays correlates protein expression of ubiquitination regulators with tumor grade, stage, and clinical outcomes [21]. Quantitative real-time PCR (qRT-PCR) validates gene expression patterns in independent patient cohorts [21].
Figure 1: Experimental workflow for developing and validating ubiquitination signatures in cancer research, integrating computational analyses with functional studies.
Key Regulatory Proteins and Pathways in Tumor Grading
E3 Ligases and Deubiquitinases as Grade-Associated Regulators
Specific E3 ubiquitin ligases and deubiquitinating enzymes consistently emerge as critical regulators of tumor grade across cancer types. The E3 ligase TRIM28, identified in the pan-cancer ubiquitination analysis, forms a regulatory axis with OTUB1 that modulates MYC pathway activity and influences patient prognosis [19]. This ubiquitination-dependent regulation of MYC signaling represents a particularly significant finding, as MYC activation is a hallmark of aggressive, high-grade malignancies yet has proven notoriously difficult to target therapeutically.
In colon cancer, feature genes including ARHGAP4, MID2, SIAH2, TRIM45, UBE2D2, and WDR72 form the core of a prognostic signature that effectively stratifies patients by risk category [21]. Functional studies demonstrate that WDR72 knockdown significantly inhibits colorectal cancer cell proliferation both in vitro and in vivo, establishing its direct role in promoting aggressive tumor behavior [21]. Similarly, ARHGAP4 and SIAH2 show promising early diagnostic capabilities, suggesting their involvement from initial tumor development through progression to higher grades.
The E2 enzyme UBE2C exemplifies how ubiquitination regulators correlate with tumor grade across diverse cancer types. UBE2C overexpression is detected in breast, colon, prostate, ovarian, and lung cancers, where it causes chromosome missegregation and aneuploidy - cytological features associated with high-grade, poorly differentiated tumors [22]. Transgenic mouse models overexpressing UBE2C develop elevated lung tumor burden and various other malignancies, confirming its oncogenic potential [22].
Ubiquitination in Cancer Stemness and Metabolic Reprogramming
Ubiquitination pathways significantly influence two fundamental determinants of tumor grade: cancer stemness and metabolic reprogramming. The ubiquitin-proteasome system regulates the stability of core stem cell regulators including Nanog, Oct4, and Sox2, thereby modulating the self-renewal capacity and differentiation potential of cancer stem cells (CSCs) [12] [23]. As CSCs are increasingly recognized as drivers of tumor initiation, therapy resistance, and progression to higher grades, ubiquitination-dependent maintenance of stemness represents a crucial mechanism linking ubiquitination signatures to aggressive tumor behavior [23].
Similarly, ubiquitination orchestrates the metabolic adaptations that enable cancer cells to thrive in nutrient-poor environments and support rapid proliferation - characteristics of high-grade malignancies. Key metabolic enzymes including ACLY (ATP-citrate lyase) and FASN (fatty acid synthase) are regulated by ubiquitination, creating a direct molecular connection between the ubiquitin code and cancer-specific metabolic pathways [18]. In lung cancer, the E3 ligase NEDD4 targets ACLY for ubiquitination, thereby influencing lipid synthesis and tumor growth [18]. The deubiquitinase OTUB2 stabilizes pyruvate kinase M2 (PKM2) in colorectal cancer, enhancing glycolysis and accelerating disease progression [11]. These examples illustrate how ubiquitination signatures reflect and contribute to the metabolic reprogramming that underlies tumor progression.
Figure 2: Ubiquitination-dependent regulatory network driving high-grade tumor phenotypes through multiple interconnected mechanisms.
Table 3: Essential Research Reagents for Ubiquitination Signature Studies
| Reagent Category | Specific Examples | Research Application | Technical Considerations |
|---|---|---|---|
| Database Resources | iUUCD 2.0, GeneCards, STRING | Comprehensive URG identification and network analysis | Quality of manual curation, regular updates essential |
| Computational Tools | "ConsensusClusterPlus", "GSVA", "estimate" R packages | Molecular subtyping, pathway analysis, TME scoring | Parameter optimization, multiple algorithm comparison |
| Cell Line Models | Cancer cells with E3/DUB modulation | Functional validation of URGs | Use of CRISPR/Cas9, siRNA, or stable overexpression |
| Animal Models | Xenograft mouse models | In vivo tumorigenesis and metastasis assays | Orthotopic implantation improves physiological relevance |
| Antibodies | Phospho-specific, ubiquitin remnant antibodies | Western blot, immunohistochemistry, immunofluorescence | Validation for specific applications critical |
| Proteasome Inhibitors | Bortezomib, Carfilzomib | UPS functional studies, therapeutic applications | Differential effects on constitutive vs immunoproteasomes |
| E1 Inhibitors | MLN7243, MLN4924 | Investigation of global ubiquitination blockade | Significant toxicity concerns |
| DUB Inhibitors | SIM0501 (USP1 inhibitor) | Targeting specific deubiquitination pathways | Selectivity profile requires verification |
| PROTACs | ARV-110, ARV-471 | Targeted protein degradation applications | Optimization of linker length and E3 recruiter |
The systematic investigation of ubiquitination signatures has fundamentally advanced our understanding of tumor biology and provides a powerful framework for refining cancer classification, prognosis, and treatment. The consistent correlation between specific ubiquitination patterns and tumor grade across diverse malignancies highlights the central role of ubiquitin signaling in cancer progression. As research in this field accelerates, several promising directions emerge for clinical translation and therapeutic development.
Ubiquitination signatures offer particular promise for addressing the challenge of tumor heterogeneity, both between patients and within individual tumors. The development of single-cell ubiquitination signatures could illuminate how distinct cellular subpopulations contribute to therapeutic resistance and disease progression, potentially guiding more effective combination therapies. Additionally, the integration of ubiquitination signatures with other molecular data types - including genomic, epigenomic, and proteomic profiles - may yield multi-dimensional classification systems that surpass the prognostic accuracy of current staging methods.
From a therapeutic perspective, the ubiquitination machinery presents novel opportunities for drug development. While proteasome inhibitors have established efficacy in hematological malignancies, newer approaches targeting specific E3 ligases or deubiquitinases offer the potential for greater selectivity and reduced toxicity [24]. The emergence of PROTAC (Proteolysis-Targeting Chimera) technology exemplifies how understanding ubiquitination mechanisms can enable entirely new therapeutic modalities that target previously "undruggable" oncoproteins [11]. Furthermore, the ability of ubiquitination signatures to predict immunotherapy response suggests their potential utility in guiding immune-based treatments, potentially expanding the benefit of these powerful therapies to more patients.
As methodologies continue to advance, particularly in mass spectrometry-based ubiquitinomics and single-cell technologies, we anticipate that ubiquitination signatures will become increasingly incorporated into clinical cancer diagnostics and therapeutic decision-making. The ongoing decoding of the ubiquitin code in cancer progression represents a frontier of molecular oncology with profound implications for improving patient outcomes across the spectrum of tumor grades and types.
The ubiquitin-proteasome system (UPS) represents a crucial regulatory mechanism for protein degradation and function, playing a paradoxical role in cancer biology as both a guardian against malignant transformation and an accomplice in tumor progression [1]. Ubiquitination, the process whereby ubiquitin molecules are attached to substrate proteins, governs the stability, activity, and localization of virtually all intracellular proteins [11]. This post-translational modification involves a sequential enzymatic cascade comprising ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3), which collectively determine the specificity and outcome of substrate modification [12]. The dynamic reversal of this process is mediated by deubiquitinating enzymes (DUBs), which remove ubiquitin chains and provide an additional layer of regulation [25].
The context-dependent nature of ubiquitination creates a complex landscape in oncology, where specific components can function as either oncogenes or tumor suppressors depending on cellular context, tumor type, and stage of progression [26]. Oncogenic ubiquitination typically promotes the degradation of tumor suppressor proteins or enhances the stability and function of oncoproteins, thereby driving uncontrolled proliferation, evasion of growth suppression, and other cancer hallmarks [12]. In contrast, tumor-suppressive ubiquitination facilitates the elimination of oncoproteins, maintains genomic integrity, and regulates appropriate cell fate decisions [1]. Understanding this delicate balance is paramount for developing targeted therapeutic strategies that specifically modulate the UPS to restore normal cellular homeostasis in cancer cells [11].
Molecular Mechanisms of Ubiquitination
The Ubiquitination Machinery
The ubiquitination process initiates with E1 activating ubiquitin in an ATP-dependent manner, forming a thioester bond between its catalytic cysteine residue and the C-terminal glycine of ubiquitin [1] [12]. This activated ubiquitin is then transferred to an E2 enzyme, which collaborates with an E3 ligase to ultimately conjugate ubiquitin to lysine residues on substrate proteins [18]. The human genome encodes approximately 600 E3 ligases, which provide substrate specificity and determine the fate of modified proteins [12]. Based on their structural characteristics and mechanism of action, E3 ligases are classified into three main families: really interesting new gene (RING) finger domain-containing E3s, homology to E6AP C terminus (HECT) domain-containing E3s, and RING-between-RING (RBR) family E3s [25].
The consequences of ubiquitination are determined by the topology of ubiquitin chains. Monoubiquitination (attachment of a single ubiquitin) and multimonoubiquitination (multiple single ubiquitins on different lysines) typically regulate subcellular localization, protein activity, and endocytosis [11]. Polyubiquitination chains, formed through specific lysine residues (K6, K11, K27, K29, K33, K48, K63) or the N-terminal methionine (M1) of ubiquitin, generate diverse biological signals [12]. K48-linked chains primarily target substrates for proteasomal degradation, while K63-linked chains mediate non-proteolytic functions such as signal transduction, DNA repair, and endocytic trafficking [25]. Other chain types, including K11, K29, and linear M1-linked chains, participate in cell cycle regulation, immune signaling, and other specialized functions [11].
The Deubiquitination Counterbalance
Deubiquitinating enzymes (DUBs) constitute a diverse family of proteases that reverse ubiquitination by cleaving ubiquitin from substrate proteins, thereby opposing the action of E3 ligases [18]. Approximately 100 DUBs are encoded in the human genome, categorized into seven subfamilies based on their catalytic domains: ubiquitin-specific proteases (USPs), ubiquitin C-terminal hydrolases (UCHs), ovarian tumor proteases (OTUs), Machado-Josephin domain proteases (MJDs), JAB1/MPN+/MOV34 (JAMM) domain proteases, monocyte chemotactic protein-induced proteins (MCPIPs), and motif interacting with ubiquitin-containing DUB family (MINDY) [25]. DUBs regulate ubiquitin homeostasis, process ubiquitin precursors, edit ubiquitin chains, and rescue substrates from proteasomal degradation, thereby fine-tuning ubiquitin signaling networks in cancer cells [27].
Figure 1: The Ubiquitin-Proteasome System Cascade. This diagram illustrates the sequential enzymatic process of ubiquitination, beginning with ubiquitin activation by E1 and culminating in substrate degradation by the proteasome following polyubiquitination.
Oncogenic Ubiquitination: Driving Tumor Progression
Enhanced Degradation of Tumor Suppressors
Oncogenic ubiquitination frequently manifests through the elevated activity of specific E3 ligases that target tumor suppressor proteins for destruction. A paradigm of this mechanism is the MDM2-mediated ubiquitination and degradation of p53, a critical tumor suppressor mutated in over 50% of human cancers [26]. In normal cells, p53 induces MDM2 expression as part of a negative feedback loop, but cancer cells often overexpress MDM2, leading to excessive p53 degradation and uncontrolled proliferation [12]. Similarly, the SCFSkp2 E3 ligase complex targets multiple cell cycle inhibitors for degradation, including p27Kip1, p57Kip2, p130, and Tob1 [26]. Overexpression of Skp2 is observed in various human cancers and correlates with poor prognosis, while gene amplification of Skp2 has been identified in gastric cancers, solidifying its role as an oncogene [26].
The ubiquitination-mediated destruction of tumor suppressors extends beyond cell cycle regulators. The E3 ligase ARF-BP1 (Huwe1) ubiquitinates and promotes the degradation of p53 independent of MDM2, providing an alternative mechanism for p53 inactivation in cancer cells [26]. Additionally, BRCA1, a RING finger E3 ligase with crucial functions in DNA damage response, frequently undergoes somatic mutations in breast and ovarian cancers, compromising its tumor suppressor activity and genomic stability maintenance [26]. These examples illustrate how diverse E3 ligases converge on critical tumor suppressor pathways to enable malignant transformation and progression.
Stabilization of Oncoproteins
Oncogenic ubiquitination also operates through the stabilization of oncoproteins, either directly through atypical ubiquitin linkages that enhance protein function rather than degradation, or indirectly through the inactivation of E3 ligases that normally target oncoproteins for destruction [12]. The deubiquitinating enzyme OTUB2 exemplifies this mechanism by interacting with pyruvate kinase M2 (PKM2) and inhibiting its ubiquitination by the E3 ligase Parkin, thereby enhancing glycolysis and accelerating colorectal cancer progression [11]. Similarly, USP2 stabilizes programmed cell death 1 (PD-1) through deubiquitination, promoting tumor immune escape and resistance to immunotherapy [11].
The metabolic reprogramming of cancer cells is particularly dependent on ubiquitination-mediated stabilization of oncoproteins. TRAF6, an E3 ligase upregulated in cancer cells, mediates K63-linked polyubiquitination of mTOR under amino acid stimulation, promoting mTORC1 translocation to lysosomes and subsequent activation [25]. This ubiquitination event enhances mTORC1 signaling, driving metabolic adaptations that support cancer cell growth and proliferation. Additionally, reduced K48-linked ubiquitination of mTOR by E3 ligases FBX8 and FBXW7 decreases proteasome-dependent degradation, further sustaining oncogenic mTOR signaling in cancer cells [25].
Table 1: Key E3 Ubiquitin Ligases with Oncogenic Functions in Cancer
| E3 Ligase | Substrate(s) | Cancer Type(s) | Biological Outcome |
|---|---|---|---|
| MDM2 | p53 | Various (multiple cancer types) | Enhanced proliferation, evasion of apoptosis |
| SCFSkp2 | p27Kip1, p57Kip2, p130, Tob1 | Gastric, various | Cell cycle progression, uncontrolled proliferation |
| ARF-BP1 (Huwe1) | p53 | Various | Apoptosis evasion, genomic instability |
| TRAF6 | mTOR | Various | Metabolic reprogramming, enhanced growth |
| RNF2 | Histone H2A | Hepatocellular carcinoma | Enhanced metastatic potential |
Tumor-Suppressive Ubiquitination: Constraining Malignancy
Degradation of Oncoproteins
Tumor-suppressive ubiquitination primarily functions through the targeted degradation of oncoproteins, preventing their accumulation and subsequent驱动 of malignant phenotypes. The Fbw7 (F-box and WD repeat domain-containing 7) E3 ligase represents a critical tumor suppressor that targets multiple oncoproteins for ubiquitin-mediated degradation, including Cyclin E, c-Myc, c-Jun, c-Myb, Notch, and mTOR [26]. Fbw7 recognizes specific phosphodegron motifs in its substrates and is frequently deleted or mutated in human cancers, leading to stabilization of its oncogenic targets and accelerated tumor development [26]. Conditional inactivation of Fbw7 in mouse models results in thymic hyperplasia due to c-Myc accumulation, eventually progressing to thymic lymphoma, confirming its bona fide tumor suppressor function [26].
The anaphase-promoting complex/cyclosome (APC/C) represents another crucial tumor-suppressive E3 ligase that controls cell cycle progression by targeting key regulators for degradation, including cyclins and securin [28]. Proper APC/C function ensures accurate cell division and prevents chromosomal instability, a hallmark of cancer. The specificity of APC/C is determined by its interactions with coactivators Cdc20 and Cdh1, which recognize distinct substrates during different cell cycle phases. Dysregulation of APC/C activity contributes to uncontrolled proliferation and genomic instability in various cancers, underscoring its importance as a tumor suppressor [28].
Regulation of DNA Repair and Genomic Integrity
Tumor-suppressive ubiquitination plays essential roles in maintaining genomic integrity through the regulation of DNA damage response pathways. BRCA1, in complex with BARD1, forms a RING finger E3 ligase heterodimer that participates in DNA repair processes, cell cycle checkpoint control, and centrosome duplication [26]. The ubiquitin ligase activity of the BRCA1-BARD1 complex is crucial for its tumor suppressor function, as mutations that disrupt this activity are frequently identified in hereditary breast and ovarian cancers [26]. Unlike typical degradation signals, BRCA1-BARD1-mediated ubiquitination of RNA polymerase II and RPB8 in response to DNA damage does not necessarily lead to proteasomal degradation but rather facilitates DNA repair processes through non-proteolytic mechanisms [26].
Monoubiquitination events also contribute to tumor-suppressive functions, particularly in DNA damage response. The monoubiquitination of histone H2AX (γH2AX) by UBE2T regulates the phosphorylation of cell cycle checkpoint kinase 1 (CHK1), influencing DNA repair efficiency and maintaining genomic stability [11]. Additionally, the E3 ligase Rad18 mediates monoubiquitination of proliferating cell nuclear antigen (PCNA) in response to DNA damage, facilitating the recruitment of specialized DNA polymerases that enable error-prone translation synthesis, a double-edged sword that promotes survival but also increases mutation rates [12]. These DNA damage-associated ubiquitination events represent critical tumor-suppressive mechanisms that prevent the accumulation of oncogenic mutations.
Table 2: Key E3 Ubiquitin Ligases with Tumor-Suppressive Functions in Cancer
| E3 Ligase | Substrate(s) | Cancer Type(s) | Biological Outcome |
|---|---|---|---|
| Fbw7 | Cyclin E, c-Myc, c-Jun, c-Myb, Notch | Various (frequently mutated) | Cell cycle control, proliferation inhibition |
| APC/C | Cyclins, Securin | Various | Cell cycle regulation, genomic stability |
| BRCA1-BARD1 | RNA polymerase II, RPB8 | Breast, ovarian | DNA damage repair, genomic integrity |
| Parkin | PKM2 | Colorectal cancer | Metabolic regulation, inhibition of glycolysis |
| CUL3-KLHL25 | ACLY | Lung cancer | Inhibition of lipid synthesis |
Context-Dependent Roles in Cancer Hallmarks
Metabolic Reprogramming
Ubiquitination exerts profound influence on cancer metabolism, regulating key enzymes and signaling pathways that drive metabolic reprogramming. The ubiquitin-mediated regulation of adenosine triphosphate citrate lyase (ACLY), a crucial enzyme linking glycolysis to lipid synthesis, demonstrates the context-dependent nature of ubiquitination in cancer metabolism [18]. The Cullin 3 E3 ligase complex, through its adaptor protein KLHL25, ubiquitinates and degrades ACLY, thereby inhibiting lipid synthesis and suppressing tumor growth in lung cancer [18]. Conversely, ARHGEF3 enhances ACLY stability by reducing its association with the E3 ligase NEDD4, promoting lipogenesis and cancer proliferation [18]. This opposing regulation highlights how different E3 ligases can exert antagonistic effects on the same metabolic enzyme, creating a balance that is disrupted in cancer.
The ubiquitination of pyruvate kinase M2 (PKM2), a key glycolytic enzyme in cancer cells, further illustrates the complex regulation of cancer metabolism. The E3 ligase Parkin facilitates the ubiquitination of PKM2, while the deubiquitinating enzyme OTUB2 inhibits this process by interacting with PKM2, thereby enhancing glycolysis and accelerating colorectal cancer progression [11]. This opposition between ubiquitinating and deubiquitinating enzymes creates a regulatory switch that controls metabolic flux in cancer cells. Similarly, fatty acid synthase (FASN), a critical enzyme in de novo lipogenesis, is regulated by multiple E3 ligases including COP1 and TRIM21, which target FASN for degradation in a context-dependent manner [18]. The tumor suppressor SPOP, an E3 ubiquitin ligase frequently mutated in prostate cancer, regulates lipid metabolism by reducing FASN expression and fatty acid synthesis, highlighting the metabolic dimension of its tumor-suppressive function [18].
Immune Evasion and Tumor Microenvironment
The ubiquitin-proteasome system plays a pivotal role in regulating tumor immune responses, particularly through the modulation of immune checkpoint proteins. The programmed cell death 1/programmed cell death ligand 1 (PD-1/PD-L1) axis, a critical immune checkpoint, is extensively regulated by ubiquitination [11]. USP2, a deubiquitinating enzyme, stabilizes PD-1 through deubiquitination, promoting tumor immune escape and resistance to immunotherapy [11]. Conversely, metastasis suppressor protein 1 (MTSS1) promotes the monoubiquitination of PD-L1 at K263 mediated by the E3 ligase AIP4, leading to PD-L1 internalization, endosomal transport, and lysosomal degradation, thus inhibiting immune escape in lung adenocarcinoma [11]. This opposing regulation of immune checkpoints by ubiquitinating and deubiquitinating enzymes offers promising therapeutic opportunities for enhancing cancer immunotherapy.
The tumor microenvironment (TME) is additionally shaped by ubiquitination through its effects on immune cell function and cytokine signaling. Linear ubiquitination, mediated by the linear ubiquitin chain assembly complex (LUBAC), regulates NF-κB signaling and impacts cancer development and immune responses [11]. HOIP, a component of LUBAC, promotes lymphoma by activating NF-κB signaling, suggesting LUBAC as a therapeutic target for B-cell lymphoma [11]. Additionally, Epsin, a member of the ubiquitin-binding endocytosis adaptor protein family, interacts with LUBAC to facilitate linear ubiquitination of NEMO, contributing to breast cancer progression [11]. These findings highlight the diverse mechanisms through which ubiquitination shapes the tumor immune microenvironment and influences cancer progression.
Figure 2: Oncogenic versus Tumor-Suppressive Ubiquitination in Cancer Hallmarks. This diagram illustrates how different ubiquitination patterns influence key hallmarks of cancer through distinct molecular mechanisms.
Experimental Approaches and Methodologies
Assessing Ubiquitination in Cancer Models
The investigation of ubiquitination pathways in cancer relies on a combination of molecular, cellular, and biochemical techniques that enable the detection and quantification of ubiquitination events, identification of relevant enzymes and substrates, and functional characterization of specific modifications. Co-immunoprecipitation (Co-IP) followed by western blotting represents a fundamental approach for detecting protein-protein interactions and ubiquitination status in cancer cells [28]. Typically, cells are transfected with plasmids expressing ubiquitin and the protein of interest, treated with proteasome inhibitors (e.g., MG132) to accumulate ubiquitinated proteins, and lysed under denaturing or non-denaturing conditions depending on the interaction stability. Immunoprecipitation using specific antibodies against the protein of interest is followed by western blotting with anti-ubiquitin antibodies to detect ubiquitination [25].
In vivo and in vitro ubiquitination assays provide more direct evidence for ubiquitination events and can identify specific E3 ligases responsible for substrate modification. For in vitro ubiquitination assays, purified E1, E2, E3 enzymes, ubiquitin, and the substrate protein are incubated in reaction buffer containing ATP, followed by western blot analysis to detect ubiquitinated substrates [25]. In vivo ubiquitination assays involve transfection of ubiquitin and relevant E3 ligases into cells, treatment with proteasome inhibitors, immunoprecipitation of the substrate, and detection of ubiquitination by western blotting [12]. These approaches are complemented by mass spectrometry-based proteomics, which enables global identification of ubiquitination sites and quantification of ubiquitin chain topology in cancer cells under different conditions [21].
Functional Validation in Cancer Models
Functional validation of ubiquitination pathways in cancer biology employs genetic and pharmacological approaches to modulate specific components of the ubiquitin system and assess resulting phenotypic changes. RNA interference (RNAi) techniques, including small interfering RNA (siRNA) and short hairpin RNA (shRNA), are widely used to knock down expression of specific E3 ligases or DUBs in cancer cells, enabling assessment of their effects on substrate stability, signaling pathways, and malignant phenotypes [28] [21]. CRISPR-Cas9-mediated gene knockout provides a more permanent and complete elimination of target genes, allowing comprehensive analysis of their functions in cancer progression and therapeutic responses [21].
Pharmacological inhibition of UPS components represents another key experimental approach, with proteasome inhibitors such as bortezomib, carfilzomib, and ixazomib being extensively used in both preclinical models and clinical practice, particularly for multiple myeloma [1]. More recently, specific inhibitors targeting E1 enzymes (e.g., MLN7243, MLN4924), E2 enzymes (e.g., Leucettamol A, CC0651), E3 ligases (e.g., nutlin, MI-219), and DUBs (e.g., compounds G5 and F6) have been developed and evaluated in preclinical cancer models [12]. These chemical tools enable acute perturbation of ubiquitination pathways and assessment of therapeutic potential. Functional readouts include cell proliferation assays, colony formation, cell cycle analysis, apoptosis measurement, migration and invasion assays, and in vivo tumor xenograft models [28] [21].
Table 3: Essential Research Reagents for Ubiquitination Studies in Cancer
| Reagent Category | Specific Examples | Research Application |
|---|---|---|
| Proteasome Inhibitors | Bortezomib, Carfilzomib, MG132 | Stabilize ubiquitinated proteins for detection |
| E1 Inhibitors | MLN7243, MLN4924 | Block ubiquitin activation |
| E2 Inhibitors | Leucettamol A, CC0651 | Inhibit ubiquitin conjugation |
| E3 Ligase Modulators | Nutlin (MDM2 inhibitor), MI-219 | Specifically target substrate recognition |
| DUB Inhibitors | Compounds G5, F6, SIM0501 (USP1 inhibitor) | Prevent deubiquitination |
| Ubiquitin Expression Constructs | Wild-type ubiquitin, Lysine mutants | Define chain topology specificity |
| Activity-Based Probes | Ubiquitin-based probes | Monitor DUB activity in cells |
Diagnostic and Therapeutic Applications
Prognostic Biomarkers and Molecular Classification
The expression patterns of ubiquitination-related genes (URGs) show significant promise as prognostic biomarkers and tools for molecular classification of cancers. Multiple studies have demonstrated that URGs can stratify cancer patients into distinct subtypes with different clinical outcomes, therapeutic responses, and tumor microenvironment characteristics [21] [29]. In colon cancer, ubiquitination-related molecular classification has identified subtypes with significant differences in survival, immune cell infiltration, and pathological staging [21]. A six-gene ubiquitination-related signature (ARHGAP4, MID2, SIAH2, TRIM45, UBE2D2, WDR72) effectively categorizes colon cancer patients into high-risk and low-risk groups, with the high-risk group exhibiting enhanced epithelial-mesenchymal transition, immune escape, immunosuppressive cell infiltration, and poorer outcomes [21].
Similar approaches in bladder cancer have identified four ubiquitination-related molecular subtypes with significantly different clinical characteristics, prognosis, PD-L1 expression levels, and tumor microenvironment composition [29]. A prognostic index based on six URGs (HLA-A, TMEM129, UBE2D1, UBE2N, UBE2T, and USP5) successfully predicted overall survival in bladder cancer patients across multiple cohorts, with area under the ROC curve values of 0.736, 0.723, and 0.683 in training, testing, and validation cohorts, respectively [29]. These ubiquitination-based classification systems provide valuable tools for risk stratification and treatment selection, potentially enhancing precision oncology approaches.
The ubiquitin-conjugating enzyme E2 C (UBE2C) exemplifies the prognostic utility of individual URGs in cancer. UBE2C expression is significantly elevated in advanced prostate cancer compared to adjacent non-neoplastic tissues and shows a strong positive correlation with prostate-specific antigen levels, Gleason score, pathological stage, and lymphatic involvement [28]. High UBE2C expression is associated with poor prognosis in various cancers, including ovarian, adrenal gland, breast, colon, liver, and lung cancers, highlighting its potential as a pan-cancer biomarker [28]. Transgenic mice overexpressing UBE2C develop spontaneous tumors and exhibit increased susceptibility to carcinogen-induced tumors with chromosome aneuploidy, confirming its oncogenic character [28].
Targeted Therapeutic Strategies
The therapeutic targeting of ubiquitination pathways has yielded several clinically successful agents, particularly proteasome inhibitors for the treatment of multiple myeloma [1]. Bortezomib, carfilzomib, and ixazomib represent first-line therapeutic agents that disrupt protein degradation in cancer cells, leading to the accumulation of polyubiquitinated proteins, endoplasmic reticulum stress, and apoptosis [1]. These agents demonstrate the clinical viability of targeting the UPS for cancer therapy and have significantly improved outcomes for multiple myeloma patients.
Nover therapeutic strategies focus on more specific modulation of ubiquitination pathways, including the development of molecular glues and proteolysis-targeting chimeras (PROTACs) that redirect E3 ligase activity toward specific oncoproteins [11]. PROTAC technology enables targeted degradation of disease-causing proteins by recruiting them to E3 ubiquitin ligases for ubiquitination and subsequent proteasomal degradation [11]. ARV-110 (bavdegalutamide) and ARV-471 (vepdegestrant) represent pioneering PROTAC candidates that have progressed to phase II clinical trials, targeting the androgen receptor for degradation in metastatic castration-resistant prostate cancer and the estrogen receptor in breast cancer, respectively [11]. Compared to traditional inhibitors, PROTACs offer advantages including increased selectivity, ability to target undruggable proteins, and potential to overcome resistance mechanisms.
Molecular glues represent another innovative approach that induces or stabilizes interactions between E3 ligases and target proteins, leading to selective degradation [11]. CC-90009 facilitates the ubiquitination-mediated degradation of G1-to-S phase transition 1 (GSPT1) by recruiting the CUL4-DDB1-CRBN-RBX1 E3 ligase complex and is currently in phase II clinical trials for leukemia therapy [11]. Additionally, existing drugs have been found to exert part of their anti-cancer effects through ubiquitination pathways. Indomethacin, for instance, diminishes growth and recurrence of esophageal squamous cell carcinoma by enhancing SYVN1-mediated ubiquitination of integrin αv, while honokiol inhibits melanoma growth by inducing keratin 18 ubiquitination and degradation [11]. These findings highlight the expanding repertoire of therapeutic strategies that harness the ubiquitin-proteasome system for cancer treatment.
The dualistic nature of ubiquitination in cancer biology—encompassing both oncogenic and tumor-suppressive functions—reflects the complexity and context-dependency of this essential regulatory system. Oncogenic ubiquitination promotes tumor development through degradation of tumor suppressors, stabilization of oncoproteins, and rewiring of metabolic and signaling pathways, while tumor-suppressive ubiquitination constrains malignancy by eliminating oncoproteins, maintaining genomic integrity, and regulating appropriate cell fate decisions. The balance between these opposing functions is disrupted in cancer, creating therapeutic opportunities for restoring normal ubiquitination patterns.
Advances in understanding ubiquitination pathways have enabled the development of novel diagnostic and therapeutic approaches, including ubiquitination-based molecular classifications, prognostic signatures, and targeted agents such as PROTACs and molecular glues. As research continues to unravel the complexities of ubiquitination in cancer hallmarks, the clinical translation of these findings promises to enhance precision oncology and improve patient outcomes across diverse cancer types.
The tumor microenvironment (TME) represents a complex ecosystem comprising tumor cells, surrounding fibroblasts, immune cells, blood vessels, inflammatory cells, various signal molecules, and the extracellular matrix (ECM) [30]. The interaction network between cancer cells and their microenvironment promotes cell proliferation and angiogenesis, inhibits cell apoptosis and immune detection, and ultimately supports tumor progression and metastasis [30]. The ubiquitin-proteasome system (UPS) has emerged as a crucial regulatory mechanism that controls the delicate crosstalk between tumors and their microenvironment through post-translational modification of protein stability and function [30]. This review comprehensively examines how ubiquitination regulates the TME across cancer stages, with particular focus on its implications for therapeutic development and biomarker discovery.
Molecular Mechanisms of Ubiquitination in TME Regulation
Ubiquitination is a critical post-translational modification process that involves the covalent attachment of ubiquitin, a small 76-amino-acid protein, to target proteins [30] [12]. This process requires a sequential enzymatic cascade involving ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3) [30]. The human genome encodes two E1 enzymes, approximately 35 E2 enzymes, and over 600 E3 ligases that confer substrate specificity [12]. Polyubiquitin chains linked at K48 or K11 sites typically target proteins for proteasomal degradation, while monoubiquitination or K63-linked polyubiquitin chains participate in non-proteolytic functions including signal transduction, autophagy, and DNA damage repair [30]. The process is reversible through deubiquitinases (DUBs), which remove ubiquitin from modified proteins [30].
Ubiquitination in Immune Cell Regulation Within TME
T Cells and Immune Checkpoints
T cells represent critical components of anti-tumor immunity, and their function is extensively regulated by ubiquitination [31]. The FBXO38 E3 ligase specifically targets the programmed death-1 (PD-1) receptor for polyubiquitination at Lys233 residue, marking it for proteasomal degradation [31]. This reduction in PD-1 expression enhances T-cell activity and strengthens anti-tumor immune responses [31]. Additionally, ubiquitination regulates cytotoxic T lymphocytes (CTLs) which recognize and eliminate tumor cells through release of granzymes and perforin, as well as secretion of cytokines like interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α) [31].
Tumor-Associated Macrophages (TAMs)
Macrophages in the TME can be polarized into pro-inflammatory M1 macrophages with antitumor effects or anti-inflammatory M2 macrophages with tumor-promoting effects [30]. The majority of TAMs in tumor tissues are polarized into M2 macrophages, which promote angiogenesis, inhibit antitumor immune responses, and support tumor growth [30]. Ubiquitination pathways regulate this polarization process and subsequent TAM functions through modification of key signaling molecules.
Myeloid-Derived Suppressor Cells (MDSCs) and Dendritic Cells (DCs)
MDSCs inhibit T-cell immune responses through multiple mechanisms including expression of arginase-1, inducible nitric oxide synthase, and anti-inflammatory cytokines such as IL-10 [30]. Ubiquitination regulates the immunosuppressive functions of MDSCs within the TME [30]. Dendritic cells, as potent antigen-presenting cells, participate in immune-mediated cancer elimination via antigen presentation and T-cell activation [30]. The ubiquitin-proteasome system modulates DC function through regulation of co-stimulatory molecules and cytokine production [31].
Table 1: Ubiquitination-Mediated Regulation of Immune Cells in TME
| Immune Cell Type | Regulatory E3 Ligases/DUBs | Molecular Targets | Functional Outcome in TME |
|---|---|---|---|
| T Cells | FBXO38 | PD-1 (degradation) | Enhanced T cell activity, improved antitumor immunity [31] |
| Macrophages | Not specified | Polarization signals | Modulates M1/M2 polarization balance [30] |
| MDSCs | Not specified | Immunosuppressive mediators | Regulates T cell suppression [30] |
| Dendritic Cells | Not specified | Antigen presentation machinery | Affects T cell activation [30] |
| Natural Killer Cells | Not specified | Activating/inhibitory receptors | Modulates cytotoxic function [30] |
Experimental Approaches for Studying Ubiquitination in TME
Ubiquitination-Related Risk Modeling in Cancer
Recent advances in bioinformatics have enabled the development of ubiquitination-related risk scores (URRS) that demonstrate prognostic value across cancer types [5] [32]. In lung adenocarcinoma (LUAD), a URRS based on expression of DTL, UBE2S, CISH, and STC1 effectively stratified patients into high-risk and low-risk groups [5]. Patients with higher URRS had significantly worse prognosis (HR = 0.54, 95% CI: 0.39-0.73, p < 0.001), with validation across six external cohorts confirming these findings (HR = 0.58, 95% CI: 0.36-0.93, p_max = 0.023) [5]. The high URRS group also exhibited higher PD-1/PD-L1 expression, tumor mutation burden, and TME scores, suggesting enhanced immune activation but also potentially greater immunosuppression [5].
Similarly, in cervical cancer, a ubiquitination-based signature comprising MMP1, RNF2, TFRC, SPP1, and CXCL8 demonstrated strong predictive value for patient survival (AUC >0.6 for 1/3/5 years) [32]. Immune microenvironment analysis revealed significant differences in 12 immune cell types between high-risk and low-risk groups, including memory B cells and M0 macrophages [32].
Methodologies for Ubiquitination Analysis
The experimental workflow for studying ubiquitination in TME typically involves multiple coordinated approaches [5] [32]:
Data Acquisition and Processing: RNA sequencing data from cancer tissues (e.g., TCGA, GEO databases) are processed to exclude non-cancerous tissues, formalin-fixed samples, and recurrent tissues. Patients with survival time less than 3 months are typically filtered out [5].
Consensus Clustering Analysis: Unsupervised clustering algorithms applied to ubiquitination-related gene (URG) expression patterns identify distinct molecular subtypes with different clinical outcomes [5].
Differential Expression Analysis: Differentially expressed URGs between molecular subtypes are identified using packages like DESeq2, with adjusted p-value ≤ 0.05 and |log2FC| ≥ 0.8 considered significant [5].
Prognostic Model Construction: Univariate Cox regression, Random Survival Forests, and LASSO Cox regression algorithms identify prognostic URGs. Risk scores are calculated using gene expression and Cox coefficients [5].
Immune Infiltration Analysis: Differences in immune cell composition between risk groups are analyzed using established deconvolution algorithms to estimate abundance of various immune cell types [32].
Table 2: Key Research Reagents and Experimental Solutions for Ubiquitination Studies
| Research Tool | Type | Primary Function | Application Example |
|---|---|---|---|
| ConsensusClusterPlus R Package | Bioinformatics Tool | Identifies molecular subtypes based on URG expression | Unsupervised clustering of TCGA-LUAD cohort [5] |
| DESeq2 Package | Bioinformatics Tool | Identifies differentially expressed genes | Screening DEGs between CC and normal samples [32] |
| LASSO Cox Regression | Statistical Method | Selects most prognostic genes from URGs | Developing ubiquitination-related risk scores [5] |
| IMvigor210CoreBiologies | R Package | Accesses anti-PD-L1 treatment data | Validating URRS in immunotherapy context [5] |
| RT-qPCR Validation | Laboratory Technique | Confirms gene expression trends | Validating MMP1, TFRC, CXCL8 in tumor tissues [32] |
Therapeutic Targeting of Ubiquitination in TME
Current Clinical Applications
The clinical success of proteasome inhibitors (bortezomib, carfilzomib, oprozomib, and ixazomib) in treating multiple myeloma validated the UPS as a therapeutic target in cancer [12]. These inhibitors preferentially induce cell death in malignant cells compared to normal cells, although the precise mechanisms underlying this selectivity are still being elucidated [33]. Beyond proteasome inhibitors, several targeted approaches have emerged:
E1 Enzyme Inhibitors: MLN7243 targets ubiquitin E1 activation, while MLN4924 (Pevonedistat) inhibits NEDD8-activating enzyme, blocking cullin neddylation and subsequent CRL E3 ligase activity [12]. MLN4924 is currently in multiple phase II clinical trials and induces cell death through uncontrolled DNA synthesis during S-phase, leading to DNA damage and apoptosis [34].
E2 Enzyme Inhibitors: Compounds like CC0651 (targeting CDC34) and NSC697923 (inhibiting UBE2N) have shown potential in preclinical studies, though development challenges have limited clinical advancement [34].
E3 Ligase Modulators: Nutlin and MI-219 inhibit MDM2, the primary E3 ligase for p53, stabilizing this critical tumor suppressor [12]. Additionally, proteolysis-targeting chimeras (PROTACs) represent an innovative approach that hijacks E3 ligases to degrade specific target proteins [33].
Emerging Therapeutic Strategies
The combination of UPS-targeted therapies with immunotherapy represents a promising frontier in cancer treatment [31]. By modulating the stability and expression of immune checkpoint proteins like PD-1/PD-L1, ubiquitination regulators can enhance the efficacy of immune checkpoint inhibitors [31]. Additionally, targeting ubiquitination in specific TME components (e.g., TAMs, CAFs, or MDSCs) may reverse immunosuppression and sensitize tumors to existing immunotherapies [30] [31].
Ubiquitination has emerged as a master regulator of the tumor microenvironment, controlling critical processes in immune cell function, stromal activation, and tumor-stroma crosstalk across cancer stages. The development of ubiquitination-related risk models demonstrates clinical utility in prognostic stratification and treatment response prediction. While therapeutic targeting of the UPS has achieved tangible success with proteasome inhibitors, next-generation approaches focusing on specific E3 ligases, DUBs, and combination strategies with immunotherapy hold significant promise. Future research should focus on elucidating stage-specific ubiquitination patterns, developing more selective ubiquitination modulators, and exploring personalized approaches based on individual tumor ubiquitination signatures. As our understanding of the ubiquitin code in TME regulation deepens, so too will our ability to harness this knowledge for improved cancer diagnosis, prognosis, and treatment.
From Bench to Biomarker: Profiling Techniques and Clinical Translation of Ubiquitination Signatures
Ubiquitination, a crucial post-translational modification (PTM), involves the covalent attachment of ubiquitin to lysine residues on target proteins, regulating their stability, activity, and localization. In cancer, ubiquitination patterns are frequently dysregulated, influencing the turnover of key oncoproteins and tumor suppressors. Proteogenomics—the integration of mass spectrometry (MS)-based proteomics with genomic data—is revealing that specific ubiquitination signatures correlate strongly with cancer stage and grade, offering new potential diagnostic and prognostic biomarkers [35]. Quantitative ubiquitinomics using Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) has therefore become an indispensable tool for elucidating these patterns in preclinical and clinical cancer research [36] [35].
Key Ubiquitinomics Workflows and Technologies
The core challenge in ubiquitinomics is the substoichiometric nature of PTMs, requiring specialized methods to enrich low-abundance ubiquitinated peptides from a complex background of unmodified peptides [35]. The primary workflows for this are compared below.
Core Technology Comparison
| Feature | PTMScan Ubiquitinomics | Traditional Immunoprecipitation (IP) | IMAC/TiO₂ for Global Phosphoproteomics |
|---|---|---|---|
| Enrichment Principle | Immunoaffinity purification using antibodies specific for the ubiquitin remnant motif (K-ε-GG) [36] [35]. | Antibodies against full-length ubiquitin or ubiquitin-binding domains. | Immobilized metal affinity chromatography; non-specific for ubiquitin [35]. |
| Specificity | High specificity for the diGly (K-ε-GG) remnant left after tryptic digestion [35]. | Lower specificity; can pull down entire ubiquitinated protein complexes, not just the modified site. | Not applicable for ubiquitination. |
| Compatibility with Automation | High; compatible with bead-handler (e.g., KingFisher) and liquid handling platforms [36]. | Moderate; can be challenging to automate efficiently. | High; easily automated. |
| Typical PTM Sites Identified | Hundreds to tens of thousands of ubiquitination sites per sample [36]. | Limited, often focused on specific proteins or protein families. | N/A |
| Primary Application | System-wide, proteome-wide discovery of ubiquitination sites. | Targeted studies of protein poly-ubiquitination for degradation. | Enrichment of phosphorylated peptides [35]. |
Experimental Performance Data
Quantitative data demonstrates the impact of integrating PTMScan technology with automation for robust ubiquitinomics.
Table 2: Quantitative Performance of PTMScan Ubiquitin K-ε-GG Kit (#59322) [36]
| Performance Metric | Manual Enrichment | Automated Enrichment (KingFisher Apex) |
|---|---|---|
| Number of Unique PTM Peptides Identified | ~1000 (Baseline) | Similar high recovery, with significantly improved reproducibility [36]. |
| Overlap with Manual Workflow | 100% (Baseline) | >90% overlap of randomly selected PTM peptides [36]. |
| Reproducibility (MS1 Peak Area) | Baseline variance | High correlation (R² > 0.9) in label-free quantification of shared peptides [36]. |
| Peptide Identification vs. Manual | Baseline | 30-135% higher identification when using hybrid platforms (e.g., AssayMAP Bravo) [36]. |
Detailed Experimental Protocols
This section outlines the standard and automated protocols for ubiquitin enrichment using PTMScan technology.
Standard Manual Protocol for Ubiquitin Enrichment
- Sample Preparation: Proteins are extracted from tissue or cell lines, reduced, alkylated, and digested into peptides using trypsin. Trypsin cleaves after lysine and arginine, leaving a characteristic K-ε-GG remnant on ubiquitinated lysines [37] [35].
- Peptide Desalting: The resulting peptide mixture is desalted using solid-phase extraction (e.g., C18 columns) to remove detergents and salts.
- Immunoaffinity Enrichment: The desalted peptides are incubated with anti-K-ε-GG antibody conjugated to magnetic beads (PTMScan Ubiquitin/SUMO Remnant Motif Kit). The beads are thoroughly washed to remove non-specifically bound peptides.
- Elution: The enriched K-ε-GG-containing peptides are eluted from the beads using a low-pH buffer.
- LC-MS/MS Analysis: The eluted peptides are separated by liquid chromatography (LC) and analyzed by tandem mass spectrometry (MS/MS). The mass spectrometer fragments the peptides, and the resulting MS/MS spectra are used to identify the peptide sequence and the site of the K-ε-GG modification [37].
Automated Protocol for High-Throughput (KingFisher Apex System)
- Plate Setup: Distribute magnetic beads (from PTMScan HS Kit), peptide samples, and wash/elution buffers across a deep-well plate.
- Bead Handling: Use the KingFisher Apex system's magnetic probes to automatically capture, mix, and transfer the beads through the series of wells for binding, washing, and elution. Author Tip: For consistent results, manually dispense beads with thorough mixing between pipette steps to ensure reproducible aliquots across wells [36].
- Elution and Analysis: The robot elutes the enriched peptides directly into a collection plate, which is then ready for LC-MS/MS analysis. This automated process reduces hands-on time and significantly enhances inter-experimental reproducibility [36].
Diagram 1: Ubiquitinomics workflow for cancer research.
The Scientist's Toolkit: Key Research Reagent Solutions
| Research Reagent | Function in Ubiquitinomics |
|---|---|
| PTMScan HS Ubiquitin/SUMO Remnant Motif (K-ε-GG) Kit | Immunoaffinity beads conjugated to an antibody specific for the K-ε-GG remnant left on ubiquitinated peptides after tryptic digestion; enables specific enrichment of these peptides from complex samples [36] [35]. |
| Cell Lysis Buffer (RIPA or similar) | For efficient extraction of proteins from cancer tissues or cell lines while maintaining the integrity of PTMs. |
| Trypsin (Proteomics Grade) | Enzyme used to digest proteins into peptides. Its specificity cleaves after lysine, revealing the diagnostic K-ε-GG signature for ubiquitination site mapping [37]. |
| PTMScan-Validated Antibodies (Bulk/Custom) | Non-conjugated antibodies available for use on hybrid automation platforms like the Agilent AssayMAP Bravo system, offering experimental flexibility [36]. |
| Iodoacetamide | Alkylating agent used to modify cysteine residues, preventing disulfide bond formation and ensuring complete digestion and accurate protein identification. |
| LC-MS/MS Grade Solvents (Water, Acetonitrile) | High-purity solvents essential for optimal peptide separation in liquid chromatography and ionization in the mass spectrometer, minimizing background noise. |
Integration with Proteogenomics in Cancer Research
The true power of ubiquitinomics is realized through proteogenomic integration. By layering ubiquitination data onto genomic and transcriptomic data from the same tumor, researchers can directly examine the functional consequences of genomic aberrations [35]. For example, a missense mutation might not change protein abundance but could drastically alter its ubiquitination status and stability, a insight only possible through this integrated approach. This strategy is being applied at scale by consortia like the Clinical Proteomic Tumor Analysis Consortium (CPTAC) to generate comprehensive maps of cancer drivers and dependencies [35].
Diagram 2: Proteogenomic integration of ubiquitinomics data.
The ubiquitin-proteasome system (UPS) represents a crucial regulatory mechanism in cellular homeostasis, governing protein degradation and influencing virtually all cancer hallmarks. Ubiquitination involves a sequential enzymatic cascade comprising E1 (activating), E2 (conjugating), and E3 (ligase) enzymes that coordinate to tag target proteins with ubiquitin molecules, marking them for proteasomal degradation or functional modification [38] [39]. Ubiquitination-related genes (URGs) have emerged as valuable biomarkers for cancer prognosis, with their altered expression patterns correlating strongly with disease progression, therapeutic resistance, and survival outcomes across multiple cancer types [40] [5]. The integration of computational approaches with high-throughput genomic data has enabled the construction of sophisticated prognostic models based on URG signatures, offering unprecedented opportunities for personalized cancer management and treatment stratification within the broader context of ubiquitination patterns correlation with cancer stage grade research.
Computational Methodologies for URG Prognostic Model Construction
Data Acquisition and Preprocessing
The construction of URG-based prognostic models begins with comprehensive data acquisition from publicly available repositories. The The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) databases serve as primary sources for transcriptomic data and corresponding clinical information [38] [5] [41]. Normalization procedures such as Fragments Per Kilobase Million (FPKM) or Transcripts Per Million (TPM) are applied to ensure cross-sample comparability. Ubiquitination-related gene sets are typically obtained from specialized databases like the Integrated Ubiquitin and Ubiquitin-like Conjugation Database (iUUCD), which catalogs E1, E2, E3 enzymes, and deubiquitinating enzymes [5] [41] [16]. Differential expression analysis between tumor and normal tissues identifies significantly altered URGs using packages like limma or edgeR with thresholds commonly set at |logFC| ≥ 1 and adjusted p-value < 0.01 [38] [42].
Feature Selection and Model Construction
Multiple statistical and machine learning approaches are employed to select the most prognostically relevant URGs for model construction:
- Univariate Cox Regression Analysis: Identifies URGs significantly associated with overall survival (p < 0.01) [38] [41]
- LASSO (Least Absolute Shrinkage and Selection Operator) Cox Regression: Prevents overfitting by applying a penalty term to regression coefficients, effectively selecting a parsimonious set of prognostic genes [38] [5] [41]
- Random Survival Forests: Non-parametric method that handles complex interactions between variables and provides variable importance metrics [5]
- Multivariate Cox Regression: Finalizes gene coefficients for risk score calculation while adjusting for potential confounders [5]
The resulting risk score formula follows the standard format: Risk score = Σ (Coefficienti × Expressioni) where Coefficienti represents the regression coefficient for each selected URG, and Expressioni denotes its normalized expression value [38] [42] [41].
Model Validation and Performance Assessment
Robust validation strategies are essential for establishing prognostic reliability. Models are typically evaluated using:
- Kaplan-Meier Survival Analysis: Compares overall survival between high-risk and low-risk groups stratified by median risk score [38] [5]
- Time-Dependent Receiver Operating Characteristic (ROC) Curves: Quantifies predictive accuracy at 1, 3, and 5 years using area under the curve (AUC) metrics [38] [41]
- External Validation: Applies the model to independent datasets (e.g., GEO cohorts) to assess generalizability [38] [5]
- Clinical Correlation Analysis: Evaluates whether the risk score associates with established clinical parameters like cancer stage, grade, and molecular subtypes [41]
Table 1: Performance Metrics of URG Prognostic Models Across Cancers
| Cancer Type | Selected URGs | Training Cohort | Validation Cohort(s) | AUC (1/3/5-year) | Reference |
|---|---|---|---|---|---|
| Ovarian Cancer | 17 genes (including FBXO45) | TCGA-OV (n=376) | GSE165808, GSE26712 | 0.703/0.704/0.705 | [38] |
| Lung Adenocarcinoma | DTL, UBE2S, CISH, STC1 | TCGA-LUAD | 6 GEO datasets | HR=0.58 (validation) | [5] |
| Endometrial Cancer | 22 genes | TCGA-UCEC | Internal validation | Satisfactory performance | [41] |
| Breast Cancer | ATG5, FBXL20, DTX4, BIRC3, TRIM45, WDR78 | GSE20685 | 5 external datasets | Significant (p<0.05) | [16] |
Comparative Analysis of URG Models Across Cancer Types
Model Architectures and Gene Signatures
URG prognostic models demonstrate cancer-specific architectures while sharing common computational frameworks. In ovarian cancer, a 17-gene signature was developed through Cox univariate analysis, LASSO regression, and deviance tests, effectively stratifying patients into distinct prognostic subgroups [38] [42]. For lung adenocarcinoma, a more streamlined 4-gene signature (DTL, UBE2S, CISH, STC1) was constructed using univariate Cox regression, Random Survival Forests, and LASSO Cox regression, highlighting the variability in optimal gene set size across malignancies [5]. Breast cancer models identified a 6-gene signature (ATG5, FBXL20, DTX4, BIRC3, TRIM45, and WDR78) through univariate Cox regression followed by non-negative matrix factorization, illustrating how algorithm selection influences final model composition [16].
Biological Insights and Clinical Correlations
The biological relevance of URG models strengthens their clinical utility. In ovarian cancer, the high-risk group exhibited significantly lower overall survival (p < 0.05) and distinct immune microenvironment characteristics, including decreased CD8+ T cells, M1 macrophages, and follicular cells [38]. Experimental validation confirmed FBXO45 as a key E3 ubiquitin ligase promoting ovarian cancer progression through the Wnt/β-catenin pathway [38] [42]. For lung adenocarcinoma, high-risk patients demonstrated elevated PD-1/PD-L1 expression, tumor mutation burden, and tumor neoantigen load, suggesting enhanced responsiveness to immunotherapy [5]. Similarly, breast cancer models revealed distinct tumor microenvironment compositions, with high-risk patients lacking myeloid dendritic cells—a feature potentially contributing to immune evasion [16].
Table 2: Biological and Clinical Correlates of High-Risk URG Signatures
| Cancer Type | Immune Microenvironment Features | Key Pathway Associations | Therapeutic Implications |
|---|---|---|---|
| Ovarian Cancer | Lower CD8+ T cells, M1 macrophages, follicular cells | Wnt/β-catenin signaling | Potential for PROTAC-targeted therapy |
| Lung Adenocarcinoma | Higher PD-1/PD-L1, TMB, TNB | KRAS, HIF-1α, p53 pathways | Enhanced immunotherapy response |
| Low-Grade Glioma | Immune-inflamed vs. immune-exclude phenotypes | CSC stemness, cellular senescence | Immunotherapy stratification |
| Breast Cancer | Absence of myeloid dendritic cells | Apoptosis regulation | Targeted therapy opportunities |
Experimental Protocols for URG Model Development and Validation
In Silico Analysis Pipeline
A standardized computational workflow enables reproducible URG model development:
Data Collection and Curation: Download RNA-seq data (FPKM/TPM normalized) and clinical metadata from TCGA and GEO. Retrieve ubiquitination-related gene sets from iUUCD or UUCD databases [38] [41].
Data Preprocessing: Filter patients with survival time <3 months. Remove non-cancerous, formalin-fixed, and recurrent tissues. Merge expression matrices with clinical data [5].
Differential Expression Analysis: Identify differentially expressed URGs using
limmaoredgeRwith threshold |logFC| ≥ 1 and adjusted p-value < 0.01 [38] [42].Prognostic Gene Selection: Perform univariate Cox regression (p < 0.05). Apply LASSO Cox regression with 10-fold cross-validation to select optimal genes [38] [5] [41].
Risk Score Calculation: Compute risk score using formula: Risk score = Σ (Coefficienti × Expressioni). Stratify patients into high/low-risk groups by median cutoff [38] [41].
Model Validation: Assess prognostic performance via Kaplan-Meier curves and log-rank test. Evaluate predictive accuracy with time-dependent ROC analysis. Validate in external datasets when available [38] [5].
Secondary Analyses: Correlate risk scores with immune infiltration (ESTIMATE, CIBERSORT), tumor mutation burden, pathway enrichment (GSEA, GSVA), and drug sensitivity [5] [41].
Experimental Validation Techniques
Wet-lab validation strengthens computational predictions through mechanistic studies:
Gene Expression Validation: Quantitative RT-PCR and Western blotting confirm differential expression of key URGs (e.g., FBXO45 in ovarian cancer) [38] [42].
Functional Assays: In vitro cell culture models (e.g., A2780, HEY ovarian cancer cells) with gene knockdown/overexpression evaluate effects on proliferation, migration, and invasion [38] [42].
Pathway Analysis: Western blotting and immunohistochemistry assess signaling pathway alterations (e.g., Wnt/β-catenin, AKT, NF-κB) in response to URG manipulation [38] [5].
Drug Sensitivity Testing: Cell viability assays (CCK-8, MTT) measure IC50 values for chemotherapeutic agents and targeted therapies across risk groups [5] [41].
Visualization of Computational Workflows and Biological Mechanisms
Ubiquitination Cascade and Prognostic Model Construction
Figure 1: Integrated workflow depicting ubiquitination biology and computational model development.
URG Model Implementation and Clinical Translation
Figure 2: Implementation pipeline and clinical translation of URG prognostic models.
Table 3: Essential Research Reagents and Computational Resources for URG Prognostic Modeling
| Category | Specific Resource | Function/Application | Source/Reference |
|---|---|---|---|
| Bioinformatics Databases | TCGA (The Cancer Genome Atlas) | Source of cancer genomic data and clinical information | [38] [41] |
| GEO (Gene Expression Omnibus) | Repository of functional genomic datasets | [5] [16] | |
| iUUCD/UUCD Database | Comprehensive ubiquitination-related gene annotations | [5] [41] | |
| Wet-Lab Reagents | RNAiso Reagent | Total RNA extraction from tissues and cells | [38] [42] |
| Real-time PCR kits (e.g., RR064A) | Quantitative gene expression validation | [38] [42] | |
| Lipofectamine Transfection Reagents | Gene knockdown/overexpression in cell lines | [38] [42] | |
| Primary Antibodies (FBXO45, WNT1, β-catenin) | Protein detection and pathway analysis | [38] [42] | |
| Computational Tools | R/Bioconductor Packages (limma, edgeR) | Differential expression analysis | [38] [5] |
| Survival, survminer, timeROC | Survival analysis and ROC curve evaluation | [38] [41] | |
| Maftools | Mutation pattern visualization and analysis | [38] [5] | |
| ESTIMATE, CIBERSORT | Tumor microenvironment and immune infiltration analysis | [38] [40] |
Computational approaches for building prognostic risk models from ubiquitination-related genes represent a powerful paradigm in cancer research, effectively bridging molecular mechanisms with clinical applications. The consistent demonstration that URG signatures stratify patients into distinct prognostic categories across multiple cancer types underscores the fundamental role of protein homeostasis in tumor progression. Future research directions should focus on integrating multi-omics data (proteomics, ubiquitinomics) to capture the full complexity of ubiquitination networks, developing single-cell resolution URG signatures to address tumor heterogeneity, and creating dynamic models that incorporate temporal changes in ubiquitination patterns during disease progression and treatment. Furthermore, the therapeutic implications of these models—particularly their ability to predict response to emerging modalities like PROTACs and immunotherapy—position URG-based prognostication as a cornerstone of personalized oncology. As validation in prospective clinical cohorts accumulates, these computational approaches hold immense promise for refining cancer staging systems and guiding risk-adapted treatment strategies.
The ubiquitin-proteasome system represents a crucial post-translational modification pathway that regulates protein stability, degradation, and function across virtually all biological processes. This system operates through a coordinated enzymatic cascade involving ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3), which collectively target specific proteins for proteasomal degradation or functional modification [32] [43]. In cancer biology, ubiquitination-related genes (UbRGs) have emerged as critical regulators of tumorigenesis, metastasis, and treatment response. However, traditional bulk sequencing approaches have consistently failed to resolve the complex heterogeneity of ubiquitination patterns across different cellular subpopulations within tumors, limiting our understanding of how ubiquitination drives cancer progression [44].
The advent of single-cell RNA sequencing (scRNA-seq) and spatial transcriptomics (ST) has revolutionized our capacity to dissect tumor ecosystems with unprecedented resolution. These technologies now enable researchers to map ubiquitination-related gene expression patterns across individual cells while maintaining critical spatial context within the tumor microenvironment (TME) [44] [45]. This technological advancement is particularly significant for understanding how ubiquitination heterogeneity correlates with cancer stage and grade, as different subclones within the same tumor may exhibit distinct ubiquitination profiles that drive aggressive behavior or therapeutic resistance [46] [47]. By integrating single-cell and spatial approaches, researchers can now resolve the complex spatial architecture of ubiquitination patterns and their functional consequences across the cellular hierarchy of human tumors.
Technological Platforms for Resolution of Ubiquitination Patterns
Single-Cell RNA Sequencing Platforms
Single-cell RNA sequencing technologies have undergone rapid evolution, with several platforms now enabling comprehensive characterization of ubiquitination-related gene expression at cellular resolution. The 10x Genomics Chromium system has emerged as a workhorse for high-throughput scRNA-seq applications, utilizing microfluidic technology to partition individual cells into nanoliter-scale droplets containing barcoded beads [44]. This platform enables profiling of tens of thousands of cells simultaneously, providing sufficient power to detect rare cell populations exhibiting distinct ubiquitination signatures. Alternative platforms include the BD Rhapsody system, which employs microwell-based cell capture with molecular barcoding, and the Smart-seq2 platform, which offers full-length transcript coverage advantageous for detecting splice variants of ubiquitination-related genes [44].
The technical workflow for scRNA-seq begins with tissue dissociation and single-cell suspension preparation, followed by cell capture, reverse transcription, cDNA amplification, and library construction. Critical quality control metrics include cell viability assessment, mitochondrial gene content filtering (typically excluding cells with >15-20% mitochondrial reads), and removal of potential doublets [48] [47]. Unique molecular identifiers (UMIs) are incorporated to correct for amplification biases and enable accurate transcript quantification [44]. For ubiquitination-focused studies, targeted panels can be employed to enrich for ubiquitination-related transcripts, though most current approaches utilize whole-transcriptome profiling to capture ubiquitination genes within broader functional networks.
Spatial Transcriptomics Platforms
Spatial transcriptomics technologies have rapidly advanced from low-resolution spot-based methods to platforms capable of subcellular resolution. These technologies broadly fall into two categories: sequencing-based (sST) and imaging-based (iST) approaches [49] [45]. Sequencing-based platforms like Visium HD (10x Genomics) and Stereo-seq (BGI) utilize spatially barcoded arrays to capture polyadenylated RNAs, with resolutions reaching 2μm and 0.5μm respectively [49]. These approaches offer unbiased whole-transcriptome coverage, enabling discovery of novel ubiquitination-related gene expression patterns within tissue architecture.
Imaging-based platforms such as Xenium (10x Genomics), CosMx (NanoString), and MERFISH employ sequential hybridization and imaging of fluorescently labeled probes to localize transcripts within tissue sections [49] [45]. While these methods typically target predefined gene panels (ranging from hundreds to thousands of genes), they offer superior sensitivity and single-molecule resolution. For ubiquitination studies, custom panels can be designed to include the full complement of ubiquitination-related genes while maintaining spatial context of the tumor microenvironment [45].
Table 1: Performance Comparison of High-Resolution Spatial Transcriptomics Platforms
| Platform | Technology Type | Resolution | Genes Captured | Sensitivity (Transcripts per Cell) | Key Applications in Ubiquitination Research |
|---|---|---|---|---|---|
| Xenium 5K | Imaging-based (iST) | Subcellular | 5001 | High | Ubiquitination zone mapping in tumor niches |
| CosMx 6K | Imaging-based (iST) | Subcellular | 6175 | Moderate-High | Single-cell ubiquitination heterogeneity |
| Visium HD FFPE | Sequencing-based (sST) | 2μm | 18,085 | Moderate | Whole-transcriptome ubiquitination signatures |
| Stereo-seq v1.3 | Sequencing-based (sST) | 0.5μm | Whole transcriptome | Moderate | High-resolution spatial ubiquitination patterns |
Integrated Multi-Omic Approaches
The most powerful applications for resolving ubiquitination heterogeneity combine scRNA-seq and ST with additional molecular modalities. Single-cell multi-omics technologies now enable simultaneous profiling of transcriptome and surface proteins (CITE-seq), chromatin accessibility (scATAC-seq), and even genetic variants (scDNA-seq) from the same cells [44]. Computational integration methods then map these multi-modal single-cell data onto spatial transcriptomics frameworks, creating comprehensive spatial atlases of ubiquitination activity within the tumor ecosystem [48] [45].
For instance, the integration of scRNA-seq with spatial transcriptomics has enabled the identification of specialized tumor subpopulations with distinct ubiquitination signatures localized to specific tissue compartments. In pancreatic cancer, this approach revealed endothelial cells with high ubiquitination scores (High_ubiquitin-Endo) that interact with fibroblasts and macrophages through WNT, NOTCH, and integrin pathways [48]. Similarly, in lung adenocarcinoma, integrated analysis identified malignant epithelial subclusters with distinct ubiquitination-related pathway activities that correlated with patient survival outcomes [50].
Experimental Protocols for Ubiquitination Heterogeneity Mapping
Tissue Processing and Sample Preparation
The fidelity of ubiquitination heterogeneity mapping critically depends on optimal tissue processing and sample preparation. For scRNA-seq, fresh tumor tissues require immediate processing with enzymatic digestion (e.g., collagenase IV, dispase) to generate high-viability single-cell suspensions while preserving RNA integrity [47]. For preservation purposes, tissues can be cryopreserved in appropriate media (e.g., Bambanker, DMSO-containing solutions) or stored in RNAlater for subsequent processing. Fixed tissue approaches are increasingly available, though these may impact RNA quality and recovery [44].
For spatial transcriptomics, optimal cutting temperature (OCT) compound-embedded fresh-frozen tissues generally provide superior RNA preservation compared to formalin-fixed paraffin-embedded (FFPE) tissues, though recent advances have significantly improved FFPE compatibility [49]. Tissue sections of 5-10μm thickness are typically optimal for most spatial transcriptomics platforms, with sectioning performed using cryostats (for frozen tissues) or microtomes (for FFPE tissues) [49] [45]. Consecutive sections should be collected for hematoxylin and eosin (H&E) staining, immunohistochemistry, and spatial transcriptomics to enable integrative analysis.
Single-Cell Library Preparation and Sequencing
The following protocol outlines a standardized workflow for scRNA-seq library preparation focused on capturing ubiquitination-related gene expression patterns:
Cell Viability and Quality Assessment: Use trypan blue or acridine orange/propidium iodide staining to assess viability (>80% recommended). Determine cell concentration and integrity using automated cell counters or flow cytometry.
Single-Cell Capture and Barcoding: Load cells at appropriate concentrations (100-1,000 cells/μL) onto the 10x Genomics Chromium controller or alternative platform to achieve targeted cell recovery (typically 5,000-10,000 cells per sample).
Reverse Transcription and cDNA Amplification: Perform reverse transcription within droplets or wells using template-switching oligonucleotides to incorporate cell barcodes and UMIs. Amplify cDNA with 10-14 PCR cycles using KAPA HiFi HotStart ReadyMix or equivalent.
Library Construction: Fragment amplified cDNA and add sample indices and sequencing adapters via end repair, A-tailing, and ligation or tagmentation approaches. Include UMIs to enable accurate transcript quantification.
Quality Control and Sequencing: Assess library quality using Bioanalyzer or TapeStation, quantifying with qPCR-based methods. Sequence on Illumina platforms with recommended read lengths (28bp Read1, 10bp i7 index, 10bp i5 index, 90bp Read2) to achieve >50,000 reads per cell.
For ubiquitination-focused studies, targeted enrichment can be performed after cDNA amplification using custom panels targeting ubiquitination-related genes, though most current studies prefer whole-transcriptome approaches to capture ubiquitination genes in functional context [48].
Spatial Transcriptomics Library Preparation
The protocol for spatial transcriptomics varies by platform but generally follows these principles:
Tissue Pretreatment: For FFPE sections, perform deparaffinization, rehydration, and antigen retrieval. For frozen sections, fix briefly in methanol or formaldehyde and permeabilize with appropriate enzymes (e.g., pepsin, proteinase K) to expose RNA.
Spatial Barcoding: For sequencing-based approaches, hybridize tissue sections to spatially barcoded oligo-dT arrays. For imaging-based approaches, hybridize with gene-specific probes containing readout sequences.
cDNA Synthesis and Amplification: Reverse transcribe bound RNA using array-bound primers or probe-based templates. Amplify cDNA with appropriate cycle numbers to maintain representation while achieving sufficient yield.
Library Preparation and Sequencing: For sequencing-based approaches, construct libraries from amplified cDNA using tagmentation or fragmentation-based methods. For imaging-based approaches, perform multiple rounds of fluorescent probe hybridization and imaging to decode spatial barcodes.
Image Registration: Align H&E or DAPI-stained tissue images with spatial barcode coordinates to maintain morphological context.
Critical considerations for ubiquitination studies include validation of probe sensitivity for ubiquitination-related genes in imaging-based approaches and confirmation of capture efficiency across expression levels [49].
Computational Analysis and Integration
The analytical workflow for ubiquitination heterogeneity mapping involves several key steps:
Quality Control and Preprocessing: Filter low-quality cells (high mitochondrial percentage, low feature count), remove doublets, and normalize data using standard methods (SCTransform, Seurat).
Cell Type Identification: Cluster cells based on gene expression patterns and annotate using canonical markers. Identify malignant cells using copy number variation inference tools (InferCNV, CaSpER) [47].
Ubiquitination Signature Scoring: Calculate ubiquitination activity scores using gene set variation analysis (GSVA) or additive model approaches based on curated ubiquitination-related gene sets [48] [43].
Spatial Mapping and Niche Identification: For spatial data, identify tissue domains with similar expression patterns using clustering methods (BayesSpace, SpaGCN). Map cell types from scRNA-seq to spatial data using deconvolution approaches (RCTD, Cell2location) [49].
Cell-Cell Communication Analysis: Infer ubiquitination-related signaling interactions between cell types using tools like CellChat, NicheNet, or LIANA, incorporating spatial proximity constraints when available [50] [48].
Diagram Title: Ubiquitination Heterogeneity Mapping Workflow
Comparative Performance of Transcriptomics Platforms
Sensitivity and Specificity Metrics
Systematic benchmarking studies have provided critical insights into the performance characteristics of different transcriptomics platforms for resolving ubiquitination heterogeneity. A comprehensive evaluation of four high-throughput platforms with subcellular resolution—Stereo-seq v1.3, Visium HD FFPE, CosMx 6K, and Xenium 5K—revealed significant differences in sensitivity and specificity [49]. The study utilized serial sections from colon adenocarcinoma, hepatocellular carcinoma, and ovarian cancer samples, with matched single-cell RNA sequencing and CODEX protein profiling as ground truth references.
Xenium 5K demonstrated superior sensitivity for multiple marker genes, including epithelial cell marker EPCAM, which showed well-defined spatial patterns consistent with H&E staining and Pan-Cytokeratin immunostaining on adjacent sections [49]. When analysis was restricted to regions shared across FFPE serial sections to reduce variability, Xenium 5K consistently outperformed other platforms in detection sensitivity. Within carefully selected regions of interest (400 × 400 μm) with similar cancer cell morphology and density, Visium HD FFPE showed better performance than Stereo-seq v1.3, while Xenium 5K maintained higher sensitivity than CosMx 6K [49].
Table 2: Platform Performance Metrics for Ubiquitination-Related Gene Detection
| Performance Metric | Xenium 5K | CosMx 6K | Visium HD FFPE | Stereo-seq v1.3 |
|---|---|---|---|---|
| Gene Detection Correlation with scRNA-seq | High | Moderate | High | High |
| Transcript Diffusion Control | Excellent | Good | Moderate | Moderate |
| Cell Segmentation Accuracy | High | High | Moderate | Moderate |
| Spatial Clustering Resolution | High | High | High | High |
| Concordance with Protein Reference (CODEX) | High | Moderate | High | Moderate |
Technical Considerations for Ubiquitination Studies
The selection of appropriate platforms for ubiquitination heterogeneity studies depends on several technical considerations. For discovery-phase studies aiming to identify novel ubiquitination patterns, sequencing-based approaches like Visium HD and Stereo-seq offer unbiased whole-transcriptome coverage, enabling comprehensive profiling of all ubiquitination-related genes without prior knowledge of specific targets [49] [45]. However, for validation studies or clinical applications focusing on predefined ubiquitination signatures, imaging-based platforms like Xenium and CosMx provide superior sensitivity and single-cell resolution within tissue architecture.
An critical consideration is the trade-off between gene panel size and spatial resolution. While whole-transcriptome approaches capture the full complexity of ubiquitination networks, their resolution is typically limited to multi-cellular spots (55μm in standard Visium, 2μm in Visium HD). In contrast, imaging-based platforms offer subcellular resolution but are restricted to predefined gene panels (5001 genes for Xenium, 6175 for CosMx) [49]. For ubiquitination-focused studies, custom panels can be designed to include the complete set of ubiquitination-related genes (approximately 400-700 genes) while reserving additional space for cell type markers and pathway indicators.
Ubiquitination Patterns Across Cancer Types and Stages
Breast Cancer Ubiquitination Heterogeneity
In breast cancer, integrated single-cell and spatial analyses have revealed striking heterogeneity in ubiquitination patterns across molecular subtypes and disease stages. A comprehensive ubiquitination-related gene signature developed from TCGA-BRCA and validation cohorts identified eight UbRGs with significant prognostic value: FBXL6, PDZRN3, and six others that stratified patients into distinct risk categories [43]. High-risk patients exhibited upregulated ubiquitination pathways associated with immune suppression and therapeutic resistance, confirmed through in vitro and in vivo experiments demonstrating that FBXL6 promoted breast carcinogenesis while PDZRN3 exerted tumor-suppressive effects [43].
Spatial transcriptomics analysis of breast cancer tissues further revealed that ubiquitination heterogeneity was non-randomly distributed across the tumor ecosystem. Specifically, aggressive tumor subregions characterized by hypoxic signatures and epithelial-mesenchymal transition displayed distinct ubiquitination patterns centered around immune-privileged niches [45]. These spatial patterns correlated with clinical outcomes, as patients whose tumors contained >10% of these high-ubiquitination niches experienced significantly shorter progression-free survival compared to those with more homogeneous ubiquitination distributions [45].
Pancreatic Cancer TRIM9-HNRNPU Axis
In pancreatic cancer, integrated single-cell and spatial approaches uncovered TRIM9 as a key ubiquitination-related tumor suppressor that exhibited stage-dependent expression patterns [48]. scRNA-seq of pancreatic cancer samples (GSE155698) identified 12 distinct cell types, with endothelial cells showing particularly high ubiquitination scores (High_ubiquitin-Endo) and enriched interactions with fibroblasts and macrophages through WNT, NOTCH, and integrin pathways [48]. Spatial transcriptomics (GSE235315) validated the preferential localization of TRIM9-expressing cells to well-differentiated tumor regions, with loss of TRIM9 occurring at the transition from pre-malignant lesions to invasive carcinoma.
Mechanistic investigations revealed that TRIM9 mediated K11-linked ubiquitination and proteasomal degradation of HNRNPU, an RNA-binding protein promoting tumor progression [48]. This degradation was dependent on TRIM9's RING domain, establishing a direct molecular link between ubiquitination dysfunction and pancreatic cancer pathogenesis. Clinically, TRIM9 downregulation correlated with advanced tumor stage, metastatic progression, and reduced overall survival, highlighting the prognostic significance of ubiquitination patterning in this aggressive malignancy [48].
Lung Adenocarcinoma MDK-NCL Signaling Axis
In lung adenocarcinoma (LUAD), integrated single-cell and spatial transcriptomics analysis revealed how the MDK-NCL signaling axis shapes the immunosuppressive microenvironment through ubiquitination-related mechanisms [50]. scRNA-seq of LUAD samples identified six malignant epithelial subpopulations with distinct ubiquitination signatures, where clusters with aggressive phenotypes (clusters 0, 1, and 5) exhibited higher copy number variation scores and enriched pathways associated with immune evasion [50].
Spatial transcriptomics analysis further delineated nine distinct tumor niches, with MDK-NCL signaling particularly upregulated at the tumor-immune interface [50]. This spatial patterning created an immunosuppressive microenvironment characterized by reduced T-cell infiltration and increased expression of checkpoint molecules including PD-1 and CTLA-4. Patients with high MDK-NCL expression showed significantly poorer survival outcomes, establishing this ubiquitination-related pathway as both a prognostic biomarker and potential therapeutic target [50].
Diagram Title: Ubiquitination Pathway in Cancer Signaling
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table 3: Essential Research Reagents and Platforms for Ubiquitination Heterogeneity Studies
| Category | Product/Platform | Specification | Application in Ubiquitination Research |
|---|---|---|---|
| Single-Cell Platform | 10x Genomics Chromium X | Up to 1 million cells per run | High-throughput ubiquitination profiling across cell populations |
| Spatial Transcriptomics | 10x Genomics Xenium | 5001-plex gene panel, subcellular resolution | Spatial mapping of ubiquitination patterns in tissue context |
| Spatial Transcriptomics | NanoString CosMx 6K | 6175-plex gene panel, single-cell resolution | Targeted ubiquitination signature validation |
| Cell Isolation | Fluorescence-Activated Cell Sorting (FACS) | High-purity cell population isolation | Ubiquitination analysis in specific cell subtypes |
| Cell Isolation | Magnetic-Activated Cell Sorting (MACS) | Rapid magnetic separation | Preparation of defined cell populations for ubiquitination studies |
| Library Preparation | 10x Genomics Single Cell 3' Reagent Kits | Cell multiplexing and barcoding | Efficient ubiquitination transcript capture |
| Analysis Software | Seurat Toolkit | Single-cell analysis pipeline | Ubiquitination signature identification and visualization |
| Spatial Analysis | Spacexr RCTD Deconvolution | Cell type mapping in spatial data | Ubiquitination pattern assignment to specific cell types |
| Cell Communication | CellChat Package | Ligand-receptor interaction analysis | Ubiquitination-mediated signaling network inference |
The integration of single-cell and spatial transcriptomics has fundamentally transformed our understanding of ubiquitination heterogeneity in human tumors. These technologies have revealed that ubiquitination patterns are not uniformly distributed across tumor ecosystems but instead form specialized spatial domains that correlate with disease progression, therapeutic resistance, and clinical outcomes [44] [48] [45]. The resolution afforded by these approaches has identified novel ubiquitination-related biomarkers and therapeutic targets across multiple cancer types, including TRIM9 in pancreatic cancer, the MDK-NCL axis in lung adenocarcinoma, and FBXL6/PDZRN3 in breast cancer [50] [48] [43].
Looking forward, several technological advancements promise to further enhance our ability to resolve ubiquitination heterogeneity. Multi-omic single-cell platforms that simultaneously profile transcriptome, proteome, and epigenome from the same cells will provide unprecedented insights into the regulatory networks controlling ubiquitination patterns [44]. Similarly, the ongoing improvement in spatial transcriptomics resolution and sensitivity will enable mapping of ubiquitination heterogeneity at subcellular levels, potentially revealing subcellular compartment-specific ubiquitination activities. The integration of these advanced profiling approaches with functional screening technologies will ultimately establish causal relationships between specific ubiquitination patterns and tumor phenotypes, paving the way for targeted interventions that exploit ubiquitination heterogeneity for therapeutic benefit.
For the research community, the current evidence strongly supports the adoption of integrated single-cell and spatial approaches for comprehensive ubiquitination mapping in cancer studies. The consistent correlation between ubiquitination heterogeneity and cancer stage across multiple tumor types suggests that spatial ubiquitination patterns may serve as valuable biomarkers for disease stratification and treatment selection. As these technologies continue to mature and become more accessible, ubiquitination heterogeneity mapping may eventually become a standard component of cancer molecular profiling, enabling truly personalized therapeutic approaches based on the unique ubiquitination architecture of individual tumors.
Ubiquitination, a major post-translational modification, plays a crucial role in regulating protein degradation, localization, and activity, significantly impacting tumorigenesis, progression, and prognosis across various cancers [51]. The malfunction of the ubiquitin system has been associated with numerous malignancies, creating an emerging field for biomarker discovery and molecular classification [21]. Ubiquitination-related gene (URG) signatures demonstrate particular value in stratifying cancer patients based on disease aggressiveness, therapeutic response, and survival outcomes, offering promising avenues for predictive, preventive, and personalized medicine (PPPM) in oncology [39]. This review comprehensively compares URG-based classifier applications, experimental methodologies, and performance metrics across three cancer types—lung adenocarcinoma, colon cancer, and pancreatic cancer—within the broader thesis that ubiquitination patterns correlate strongly with cancer stage and grade, providing critical insights for researchers, scientists, and drug development professionals.
Ubiquitination Machinery in Cancer: Core Components
The ubiquitin-proteasome system (UPS) comprises a sophisticated enzymatic cascade that regulates fundamental cellular processes. Understanding these core components is essential for contextualizing URG-based classifier development and function.
Diagram 1: Ubiquitination machinery core components showing the enzymatic cascade from ubiquitin activation to substrate degradation or deubiquitination.
The ubiquitination process involves a coordinated enzymatic cascade where ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3) work sequentially to tag substrate proteins with ubiquitin molecules [39]. Human cells express approximately two E1 enzymes, more than 50 E2 enzymes, and over 600 E3 ligases, highlighting the system's remarkable specificity and regulatory complexity [39]. Deubiquitinating enzymes (DUBs) counterbalance this process by removing ubiquitin chains, creating a dynamic equilibrium that controls protein stability and function [52]. In cancer cells, aberrations in this system—whether in ubiquitin-activating enzymes, ubiquitin-conjugating enzymes, ubiquitin-protein ligases, or deubiquitinating enzymes—can disrupt critical pathways controlling cell cycle progression, DNA repair, and apoptosis, contributing to malignant transformation and progression [51] [21].
Comparative Analysis of URG-Based Classifiers Across Cancer Types
Colon Cancer URG Classifiers
Colon cancer research has pioneered URG-based classification, with multiple studies demonstrating robust prognostic and predictive capabilities through comprehensive molecular subtyping approaches.
Table 1: URG-Based Classifiers in Colon Cancer
| Study | URG Signature | Cancer Stage | Methodology | Key Findings | Performance Metrics |
|---|---|---|---|---|---|
| Xu et al. [51] | 6-gene signature (ARHGAP4, MID2, SIAH2, TRIM45, UBE2D2, WDR72) | Stages I-IV | NMF clustering, Lasso Cox regression | Stratified patients into high/low-risk with distinct immune microenvironments | Better response to CTLA4 inhibitors in low-risk group |
| Exploration Study [21] | 1299 URGs from iUUCD database | TCGA-COAD cohort (n=424) | ssGSEA, ESTIMATE algorithm | Revealed differences in immune cell infiltration and pathological staging | Predictive of survival and therapy response |
| Ubiquitinomics Study [39] | 1249 ubiquitinated sites within 608 DUPs | Sigmoid colon cancer | Label-free quantitative proteomics, LC-MS/MS | Identified 35 significant signaling pathways including glycolysis and ferroptosis | 46 overall survival-related DUPs identified |
Experimental protocols for colon cancer URG classifier development typically begin with data acquisition from sources like TCGA-COAD and GEO databases, followed by preprocessing and normalization [21]. Researchers then extract URGs from specialized databases such as the Integrated Annotations for Ubiquitin and Ubiquitin-like Conjugation Database (iUUCD), which comprehensively documents ubiquitin-activating enzymes, ubiquitin-conjugating enzymes, ubiquitin-protein ligases, deubiquitinating enzymes, and ubiquitin-binding domain-containing proteins [51]. Molecular subtyping employs non-negative matrix factorization (NMF) to categorize patients based on URG expression patterns, with the optimal cluster number determined by cophenetic coefficient analysis [21]. Feature selection utilizes machine learning techniques including Lasso logistic regression and Support Vector Machine-Recursive Feature Elimination (SVM-RFE) to identify the most prognostically relevant URGs [51]. Finally, validation encompasses survival analysis, receiver operating characteristic (ROC) analysis, decision curve analysis (DCA), and external dataset validation to confirm clinical utility [21].
Lung Adenocarcinoma Classifiers
While lung adenocarcinoma classifier development has advanced significantly, current approaches have primarily focused on broader transcriptional subtypes rather than specifically ubiquitination-related signatures.
Table 2: Gene Expression-Based Classifiers in Lung Adenocarcinoma
| Study | Classifier Type | Cancer Stage | Methodology | Key Findings | Performance Metrics |
|---|---|---|---|---|---|
| Single Sample Predictor Study [53] | 36-gene SSP (TRU/nonTRU) | Stage I | AIMS algorithm, NanoString assay | TRU subtype shows better OS and DMFS | Accuracy: 0.85 (SSP2), 0.81 (SSP3) |
| TCGA Network [53] | Transcriptional subtypes (TRU, PI, PP) | Stages I-IV | Nearest Centroid Classification | TRU subtype associated with EGFR mutations, lower stage | Improved prognosis for TRU subtype |
The experimental workflow for lung adenocarcinoma classifier development involves distinct phases. Training cohorts assemble from multiple public gene expression datasets (e.g., n=1,655 samples across 17 datasets) encompassing diverse technical platforms to ensure robustness [53]. The AIMS algorithm then generates single sample predictors (SSPs) capable of classifying individual samples without requiring cohort normalization, overcoming critical limitations in previous classification approaches [53]. These SSPs are designed to capture established transcriptional subtypes—terminal-respiratory unit (TRU), proximal-inflammatory (PI), and proximal-proliferative (PP)—with the TRU subtype consistently demonstrating association with improved patient outcomes [53]. For clinical translation, researchers transform SSPs into clinically applicable assays like the NanoString nCounter platform, enabling analysis of formalin-fixed paraffin-embedded (FFPE) tissue samples and demonstrating prognostic stratification in Stage I patients [53].
Pancreatic Cancer Detection Approaches
Pancreatic cancer detection has incorporated machine learning and liquid biopsy approaches, though URG-specific classifiers remain less developed compared to colon cancer.
Table 3: Machine Learning Approaches in Pancreatic Cancer Detection
| Study | Classifier Type | Cancer Stage | Methodology | Key Findings | Performance Metrics |
|---|---|---|---|---|---|
| Deep Learning Study [54] | Disease trajectory model | Early-stage | Transformer and GRU models | Sequence of disease codes predicts cancer risk | AUROC=0.88 (36-month prediction) |
| EV-Based Detection [55] | EV protein classifier | Stages I-II | ACE-based EV isolation, machine learning | EV proteins enable early detection | Sensitivity=90%, Specificity=92.8% |
| Radiomics Study [56] | Clinical-radiomics model | PDAC | 7 ML algorithms, 43 combinations | Combined model superior to clinical or radiomics alone | C-index: 0.746 (validation) |
Pancreatic cancer detection methodologies have employed diverse technological approaches. The deep learning algorithm analyzing disease trajectories utilized clinical data from 6 million patients (24,000 pancreatic cancer cases) in the Danish National Patient Registry and 3 million patients (3,900 cases) from the US Veterans Affairs database [54]. This approach embedded diagnosis codes and timestamps into real-number vectors, encoded trajectories into lower-dimensional latent space using Transformer and gated recurrent unit (GRU) models, and predicted time-dependent cancer risk within incremental intervals (3, 6, 12, 36, or 60 months) [54]. Alternatively, extracellular vesicle (EV)-based detection employed alternating current electrokinetics (ACE) technology to isolate EVs from plasma samples, followed by multiplex immunoassays to quantify protein biomarkers [55]. Machine learning analysis then identified the most informative EV proteins for early-stage PDAC detection, with the final model validated on an independent cohort [55]. Radiomics approaches extracted high-throughput features from contrast-enhanced CT images, with seven machine learning algorithms (including Supervised Principal Components, Random Survival Forest, Lasso, and Elastic Network) integrated into 43 combinations to build prognostic models [56]. The most effective radiomics features were combined with clinical variables like TNM stage and systemic inflammation response index (SIRI) to enhance predictive performance [56].
Experimental Workflows and Methodologies
The development of URG-based classifiers follows systematic experimental workflows that integrate multi-omics data, bioinformatics analysis, and machine learning approaches.
Diagram 2: Experimental workflow for URG-based classifier development showing key stages from data acquisition to validation.
The experimental workflow for URG-based classifier development begins with comprehensive data acquisition from public repositories such as The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) [51] [21]. For colon cancer research, this typically involves RNA sequencing or microarray data from hundreds of patients (e.g., TCGA-COAD n=424; GSE39582 n=573) [21]. Data preprocessing follows, including normalization of Fragments Per Kilobase Million (FPKM) to Transcripts Per Kilobase Million (TPM), log2 transformation, and batch effect correction using algorithms like Combat in the SVA R package [21]. Ubiquitination-related genes are then extracted from specialized databases such as the iUUCD, which provides a comprehensive summary of ubiquitin-activating enzymes, ubiquitin-conjugating enzymes, ubiquitin-protein ligases, deubiquitinating enzymes, and ubiquitin-binding domain-containing proteins [51].
Bioinformatics analysis represents the core phase, employing non-negative matrix factorization (NMF) for molecular subtyping based on URG expression patterns [21]. The optimal number of clusters is determined by the rank value corresponding to the maximum change in cophenetic coefficient [21]. Feature selection utilizes machine learning techniques including Lasso logistic regression with 10-fold cross-validation and SVM-RFE to identify the most discriminative genes between subtypes [51]. For prognostic model development, univariate Cox regression identifies survival-associated URGs, followed by Lasso and stepwise regression to construct multi-gene signatures [51]. Validation encompasses multiple approaches: survival analysis using Kaplan-Meier curves and log-rank tests; time-dependent receiver operating characteristic (ROC) analysis; decision curve analysis (DCA) to evaluate clinical utility; and external validation on independent datasets [51] [21]. Additional validation may include experimental assessment of key URGs through quantitative real-time polymerase chain reaction (qRT-PCR), immunohistochemistry, and functional studies using colony formation, EdU staining, and xenograft tumorigenesis assays [51].
Table 4: Essential Research Reagents and Resources for URG-Based Classifier Development
| Resource Type | Specific Examples | Function/Application | Key Features |
|---|---|---|---|
| Data Resources | TCGA (The Cancer Genome Atlas) | Provides genomic, transcriptomic, and clinical data | Multi-dimensional data across cancer types |
| GEO (Gene Expression Omnibus) | Repository of gene expression profiles | Diverse datasets from independent studies | |
| iUUCD 2.0 (Integrated Annotations for Ubiquitin and Ubiquitin-like Conjugation Database) | Comprehensive URG database | 1,360 URGs with functional annotations | |
| Computational Tools | NMF R Package | Molecular subtyping via non-negative matrix factorization | Identifies latent patterns in gene expression |
| glmnet R Package | Lasso logistic regression for feature selection | Regularization prevents overfitting | |
| SVM-RFE Algorithm | Support Vector Machine with recursive feature elimination | Ranks genes by importance | |
| ESTIMATE Algorithm | Scores tumor microenvironment | Evaluates immune and stromal components | |
| ssGSEA Method | Single-sample gene set enrichment analysis | Quantifies immune cell infiltration | |
| Experimental Platforms | Verita Platform (ACE technology) | Extracellular vesicle isolation from plasma | Efficient capture of 50-200nm EVs |
| NanoString nCounter | Gene expression analysis from FFPE tissue | Platform-independent single sample prediction | |
| Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) | Ubiquitinated protein identification and quantification | Label-free quantitative proteomics | |
| Multiplex Immunoassays (Luminex) | High-throughput protein biomarker quantification | Simultaneous measurement of multiple EV proteins |
Discussion: Implications for Cancer Staging and Personalized Therapy
The development of URG-based classifiers represents a significant advancement in cancer stratification, moving beyond traditional histopathological staging to incorporate molecular features that more accurately reflect tumor biology and behavior. In colon cancer, URG signatures have demonstrated remarkable utility in classifying patients into distinct subgroups with differential survival outcomes, immune microenvironment characteristics, and therapeutic responses [51]. Specifically, the 6-gene URG signature (ARHGAP4, MID2, SIAH2, TRIM45, UBE2D2, WDR72) identifies high-risk patients exhibiting enhanced epithelial-mesenchymal transition, immune escape mechanisms, and immunosuppressive cell infiltration, while low-risk patients show better response to CTLA4 checkpoint inhibitors [51]. These findings align with the broader thesis that ubiquitination patterns correlate with cancer stage and grade, as evidenced by the identification of 46 overall survival-related differentially ubiquitinated proteins (DUPs) in sigmoid colon cancer, which are strongly associated with disease progression and patient outcomes [39].
In lung adenocarcinoma, while URG-specific classifiers remain less developed compared to colon cancer, existing transcriptional subtype classifiers (TRU, PI, PP) capture biologically distinct entities with clear prognostic implications [53]. The terminal-respiratory unit (TRU) subtype demonstrates association with better outcomes, lower stage disease, and specific molecular features such as EGFR mutations, reinforcing the connection between molecular signatures and disease behavior [53]. The successful translation of these classifiers into platform-independent single sample predictors (SSPs) and their validation in archival FFPE tissue specimens highlights the clinical feasibility of implementing such approaches in routine practice [53].
Pancreatic cancer detection has leveraged alternative approaches including disease trajectory analysis and extracellular vesicle biomarkers, achieving impressive performance for early-stage detection [55] [54]. The deep learning algorithm analyzing sequences of diagnosis codes in clinical histories represents a particularly innovative approach, achieving AUROC values of 0.88 for 36-month prediction in the Danish dataset [54]. While not exclusively focused on ubiquitination, these models demonstrate the power of machine learning to integrate complex patterns from diverse data sources for risk stratification.
The correlation between ubiquitination patterns and cancer stage emerges consistently across these studies, supporting the central thesis that ubiquitination modifications reflect and potentially drive tumor progression. In colon cancer, comprehensive ubiquitinome analysis revealed 1249 ubiquitinated sites within 608 differentially ubiquitinated proteins between cancer and control tissues, with specific involvement in critical pathways including salmonella infection, glycolysis/gluconeogenesis, and ferroptosis [39]. Relationship analyses between DUPs and their corresponding gene expression patterns identified four distinct models of regulation, suggesting complex feedback mechanisms linking ubiquitination to transcriptional and translational control in cancer cells [39].
From a clinical implementation perspective, URG-based classifiers offer particular promise for patient stratification in the framework of predictive, preventive, and personalized medicine (PPPM) [39]. By identifying high-risk patients who may benefit from more aggressive treatment or specific therapeutic approaches, these classifiers can guide clinical decision-making and resource allocation. Additionally, the identification of key URGs provides potential targets for drug development, as evidenced by studies showing that knockdown of WDR72 significantly inhibited colorectal cancer cell proliferation both in vitro and in vivo [51]. The integration of URG-based risk scores with traditional clinical variables like stage and age in nomograms further enhances predictive performance, offering improved tools for prognostic assessment and treatment planning [51].
Future research directions should focus on validating these classifiers in prospective clinical trials, standardizing analytical approaches across institutions, and further elucidating the functional roles of specific URGs in cancer pathogenesis. The expanding availability of multi-omics data and advances in computational biology will likely accelerate the development and refinement of URG-based classifiers, potentially enabling their integration into routine clinical practice across multiple cancer types.
Integrating Ubiquitination Profiles with TCGA and ICGC Data for Enhanced Patient Stratification
The integration of ubiquitination profiles with genomic data from The Cancer Genome Atlas (TCGA) and International Cancer Genome Consortium (ICGC) represents a transformative approach for cancer patient stratification. Ubiquitination, a dynamic post-translational modification process involving E1 activating enzymes, E2 conjugating enzymes, E3 ligases, and deubiquitinating enzymes (DUBs), regulates virtually all cellular processes and exhibits widespread dysregulation across malignancies. This guide provides a comprehensive comparison of methodologies, experimental protocols, and analytical frameworks for leveraging ubiquitination signatures to identify molecular subtypes, predict clinical outcomes, and inform therapeutic development, with direct implications for researchers and drug development professionals investigating ubiquitination patterns in cancer.
Protein ubiquitination encompasses a sophisticated regulatory network that controls substrate specificity, protein degradation, and signaling outcomes through complex chain topology diversity. The ubiquitin-proteasome system (UPS) maintains cellular homeostasis through a coordinated enzymatic cascade involving E1, E2, and E3 enzymes, while DUBs provide reversibility to this process [57] [58]. Recent pan-cancer analyses reveal that ubiquitination pathway genes frequently harbor genetic alterations, expression perturbations, and pathway activities that correlate with cancer development and progression [58] [59]. The integration of ubiquitination-related genes (URGs) with multi-omics data from consortia like TCGA and ICGC has enabled researchers to stratify patients into subgroups with distinct clinical outcomes, thereby facilitating biomarker discovery and targeted therapeutic development [5] [59] [16].
The clinical significance of ubiquitination profiling stems from its pervasive role in oncogenic processes. Ubiquitination regulates key cancer hallmarks including cell cycle progression, DNA damage repair, apoptosis, metabolic reprogramming, and immune evasion [60] [59]. For instance, the E3 ligase FBXW7, a well-characterized tumor suppressor, demonstrates cancer-type-specific mutation patterns that influence patient survival across multiple cancer types [58]. Similarly, DUBs such as USP4, USP5, and USP7 show elevated expression in various cancers where they stabilize oncoproteins and promote malignant phenotypes [57]. These findings underscore the prognostic and therapeutic potential of ubiquitination networks in precision oncology.
Comparative Analysis of Ubiquitination-Based Stratification Approaches
Pan-Cancer Ubiquitination Signatures
Comprehensive molecular characterization of ubiquitination pathways across 33 cancer types from TCGA has identified distinct clusters of tumors with significantly different survival outcomes [58]. This analysis of 9,125 tumor samples revealed that ubiquitination pathway genes tend to be upregulated in cancer through diverse mechanisms, with specific driver mutations showing cancer-type-specific patterns. For example, mutated FBXW7 exhibits tissue-specific mutation distributions, while amplified MDM2 shows a mutually exclusive pattern with BRAF mutations [58]. A separate pan-cancer investigation of gastrointestinal cancers identified 9 clusters through multi-omics integration—5 single-type-dominant clusters and 4 mixed clusters—revealing both homogeneity and heterogeneity across cancer types [61]. This stratification demonstrated more significant prognosis differences than single-omics approaches, highlighting the power of integrated ubiquitination profiling.
Table 1: Comparison of Ubiquitination-Based Stratification Approaches Across Cancers
| Cancer Type | Stratification Method | Key Ubiquitination Genes | Clinical Utility | Validation Cohort |
|---|---|---|---|---|
| Lung Adenocarcinoma [5] | URRS with 4 genes (DTL, UBE2S, CISH, STC1) | DTL, UBE2S, CISH, STC1 | Prognostic prediction, immune infiltration assessment, drug response | 6 external GEO datasets |
| Breast Cancer [16] | Risk score with 6 genes (ATG5, FBXL20, DTX4, BIRC3, TRIM45, WDR78) | ATG5, FBXL20, DTX4, BIRC3, TRIM45, WDR78 | Prognostic classification, microenvironment analysis | TCGA-BRAC, 5 GEO datasets |
| Hepatocellular Carcinoma [62] | FBXO42 expression and interaction analysis | FBXO42, p57Kip2, YY1 | Prognostic biomarker, therapeutic target | HCCDB, TCGA, ICGC |
| Esophageal Squamous Cell Carcinoma [63] | 5-gene signature (BUB1B, CHEK1, DNMT1, IRAK1, PRKDC) | BUB1B, CHEK1, DNMT1, IRAK1, PRKDC | Prognostic prediction, therapeutic targeting | TCGA-ESCC, GEO datasets |
| Pancreatic Cancer [64] | Machine learning model (CoxBoost+RSF) with 7 genes | TSPAN6, TSC1, RNF167, PBXIP1, LRRC49, KATNAL2, IGF2BP2 | Survival stratification, immune infiltration analysis | TCGA, ICGC, 6 GEO datasets |
Performance Metrics of Ubiquitination-Based Models
Ubiquitination-related risk scores (URRS) demonstrate robust prognostic performance across multiple cancer types. In lung adenocarcinoma, a 4-gene URRS signature (DTL, UBE2S, CISH, STC1) effectively stratified patients into high-risk and low-risk groups with significantly different survival outcomes (hazard ratio [HR] = 0.54, 95% confidence interval [CI]: 0.39–0.73, p < 0.001), which was validated across six external cohorts (HR = 0.58, 95% CI: 0.36–0.93, pmax = 0.023) [5]. The high-risk group exhibited elevated PD1/L1 expression, increased tumor mutation burden (TMB), higher tumor neoantigen load (TNB), and distinct tumor microenvironment scores (p < 0.001) [5]. Similarly, in breast cancer, a 6-gene ubiquitination signature showed superior predictive ability compared to traditional clinical indicators across multiple validation datasets [16].
Table 2: Performance Metrics of Ubiquitination-Based Prognostic Models
| Cancer Type | Model Characteristics | Statistical Significance | Association with Clinical Features | Validation Approach |
|---|---|---|---|---|
| Lung Adenocarcinoma [5] | 4-gene URRS | HR = 0.54, 95% CI: 0.39–0.73, p < 0.001 | Higher TMB, TNB, PD1/L1 in high-risk group | 6 external GEO datasets |
| Breast Cancer [16] | 6-gene risk score | Significant survival difference (p < 0.05) | Superior to traditional clinical indicators | TCGA-BRAC, 5 GEO datasets |
| Hepatocellular Carcinoma [62] | FBXO42 expression | Correlation with poor prognosis | Association with immune infiltration, cancer stemness | HCCDB, TCGA, ICGC |
| Pancreatic Cancer [64] | 7-gene machine learning model | Significant survival stratification | Correlation with mutation burden, immune infiltration | TCGA, ICGC, 6 GEO datasets |
| Esophageal Squamous Cell Carcinoma [63] | 5-gene signature | Significant prognostic value (p < 0.05) | Association with tumor stage and progression | TCGA-ESCC, GSE20347 |
Experimental Protocols for Ubiquitination Profiling
Data Acquisition and Preprocessing
The foundational step in ubiquitination profiling involves comprehensive data acquisition from publicly available repositories. The standard protocol includes downloading RNA-seq data, somatic mutations, copy number variations (CNVs), and clinical data from TCGA and ICGC portals [5] [63]. For ubiquitination-specific analysis, researchers should curate URGs from specialized databases such as iUUCD 2.0 (containing 966 URGs), GeneCards (filtering with relevance score >5-10), or published literature [5] [63] [64]. Quality control measures must include removal of samples with survival time less than 3 months, exclusion of formalin-fixed and recurrent tissues, and normalization of count data to fragments per kilobase per million mapped fragments (FPKM) or transcripts per million (TPM) [5] [63].
For single-cell RNA sequencing (scRNA-seq) analysis of ubiquitination patterns, as performed in pancreatic cancer research [64], the protocol includes: (1) downloading data from GEO database (e.g., GSE155698); (2) quality control excluding genes expressed in <3 cells, cells with <200 or >7,000 genes, cells with >15% mitochondrial gene expression; (3) data normalization using the "NormalizeData" function in Seurat; (4) identification of highly variable genes (3,000 genes using "vst" method); (5) data integration using "CCA" method; and (6) dimensionality reduction with "PCA," "tSNE," and "UMAP" methods (dims = 20) [64]. This approach enables identification of cell-type-specific ubiquitination patterns and cell-cell communication networks.
Ubiquitination-Related Risk Score Calculation
The calculation of ubiquitination-related risk scores (URRS) follows a standardized analytical workflow:
Differential Expression Analysis: Identify differentially expressed URGs between tumor and normal tissues using the "limma" R package with thresholds of |log fold change (FC)| >0.5 and adjusted p-value <0.05 [63].
Prognostic Gene Selection: Apply multiple algorithms to identify prognostic URGs:
Risk Score Calculation: Compute URRS using the formula: [ Risk\ score = \sum {\beta}{RNA} * {Exp}{RNA} ] where βRNA represents the coefficient from multivariate Cox regression analysis and ExpRNA represents the expression of differentially expressed URGs [5].
Stratification: Divide patients into high-risk and low-risk groups based on the median risk score.
Validation: Validate the prognostic model using external datasets and time-dependent ROC curves to evaluate predictive efficiency at 1-, 3-, and 5-year endpoints [5].
Functional Validation of Ubiquitination Mechanisms
For experimental validation of ubiquitination mechanisms, the following protocols are essential:
Cell Culture and Transfection: HCC cell lines (HepG2 and Hep3B) are maintained in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% antibiotic solution (streptomycin/penicillin) at 37°C with 5% CO2 [62]. Lentiviral vectors encoding target genes (e.g., LV-FBXO42, LV-CDKN1C) and short hairpin RNAs (shRNAs) targeting genes of interest (e.g., LV-shFBXO42) are designed and introduced through polybrene-mediated transduction followed by puromycin selection [62].
Functional Assays:
- CCK-8 Assay: Seed stably transfected cells into 96-well plates (500 cells/well), add 10μL CCK-8 solution diluted with 90μL culture medium at designated timepoints, incubate at 37°C for 2 hours, and measure absorbance at 450nm [62].
- Clone Formation Assay: Plate cells into 6-well plates (500 cells/well) and incubate for 14 days, then fix colonies with 4% paraformaldehyde and stain with crystal violet [62].
- Transwell Migration Assay: Seed 1×10^4 cells resuspended in 200μL serum-free DMEM into the upper chamber, with 800μL DMEM containing 10% FBS in the lower chamber. After 24 hours incubation, fix cells with 4% paraformaldehyde for 30 minutes, remove non-migrated cells from the upper surface, and count migrated cells [62].
Ubiquitination Assays: Co-immunoprecipitation (Co-IP) and ubiquitination assays are performed to validate direct interactions. For instance, to demonstrate FBXO42-mediated ubiquitination of p57Kip2, researchers co-transfect cells with FBXO42 and p57Kip2 constructs, treat with proteasome inhibitor MG132, immunoprecipitate p57Kip2, and detect ubiquitination via Western blotting with anti-ubiquitin antibodies [62].
Ubiquitination Signaling Networks in Cancer
Ubiquitination regulates multiple oncogenic signaling pathways through complex networks. The FBXO42-p57Kip2 axis exemplifies a clinically relevant ubiquitination pathway in hepatocellular carcinoma. In this mechanism, the transcription factor YY1 upregulates FBXO42 expression, which then interacts with p57Kip2 to promote its ubiquitination and degradation, ultimately driving HCC progression [62]. This pathway highlights the therapeutic potential of targeting ubiquitination networks, as FBXO42 inhibition stabilizes p57Kip2 and suppresses malignant behaviors.
Another crucial ubiquitination network involves the regulation of radiotherapy resistance. Ubiquitination controls DNA repair fidelity, metabolic reprogramming, and immune evasion through diverse chain topologies, with K48-linked ubiquitination primarily targeting proteins for proteasomal degradation while K63-linked ubiquitination modifies signaling outcomes [60]. The complex crosstalk between ubiquitination and other post-translational modifications (phosphorylation, SUMOylation, acetylation) creates both challenges and opportunities for therapeutic intervention, particularly through targeted protein degradation approaches such as PROTACs [60].
Table 3: Essential Research Reagents and Databases for Ubiquitination Profiling
| Resource | Type | Function | Application Example |
|---|---|---|---|
| TCGA/ICGC Data Portals | Database | Provide multi-omics data for pan-cancer analysis | Ubiquitination alteration identification across 33 cancer types [58] |
| cBioPortal | Analysis Tool | Explore genetic alterations of URGs | Mutation profile analysis of FBXO42 in HCC [62] |
| UbiBrowser 2.0 | Database | Predict ubiquitin ligase-substrate interactions | Identification of FBXO42 interaction with p57Kip2 [62] |
| CCK-8 Assay Kit | Research Reagent | Assess cell proliferation and viability | Measurement of HCC cell proliferation after FBXO42 modulation [62] |
| Lentiviral Vectors | Research Tool | Enable gene overexpression or knockdown | FBXO42 functional validation in HCC models [62] |
| TIMER 2.0 | Analysis Tool | Analyze immune cell infiltration | Correlation of FBXO42 with immune microenvironment [62] |
| ConsensusClusterPlus | R Package | Perform molecular subtyping | Identification of ubiquitination subtypes in LUAD [5] |
| iUUCD 2.0 | Database | Access curated ubiquitination-related genes | Compilation of URGs for risk model construction [5] |
The integration of ubiquitination profiles with TCGA and ICGC data provides a powerful framework for enhanced cancer patient stratification. This approach has demonstrated robust prognostic value across multiple cancer types, including lung adenocarcinoma, breast cancer, hepatocellular carcinoma, and pancreatic cancer. The consistent findings across independent validation cohorts underscore the reliability of ubiquitination-based stratification models for predicting clinical outcomes and therapeutic responses.
Future research directions should focus on several key areas: (1) expanding the integration of multi-omics data to capture the complexity of ubiquitination networks; (2) developing standardized analytical pipelines for ubiquitination profiling across different cancer types; (3) exploring the therapeutic potential of targeting specific ubiquitination pathways in stratified patient populations; and (4) investigating the crosstalk between ubiquitination and other post-translational modifications in driving cancer progression. As our understanding of ubiquitination networks continues to evolve, these approaches will increasingly inform precision oncology strategies and drug development efforts, ultimately improving patient outcomes across diverse cancer types.
Navigating Complexity: Challenges and Solutions in Targeting Ubiquitination for Cancer Therapy
Overcoming Tumor Heterogeneity and Context-Specific Ubiquitination Effects
The ubiquitin-proteasome system (UPS) represents a crucial regulatory pathway in eukaryotic cells, governing protein degradation and a wide array of cellular processes including cell cycle progression, DNA repair, and immune responses [33] [65]. This system employs a precise enzymatic cascade involving E1 activating enzymes, E2 conjugating enzymes, and E3 ubiquitin ligases to tag target proteins with ubiquitin, while deubiquitinating enzymes (DUBs) reverse this process [33]. The dysregulation of ubiquitination is now recognized as a hallmark of cancer, influencing tumor initiation, progression, and therapeutic resistance [39] [66]. However, the profound heterogeneity of tumors and the context-specific nature of ubiquitination events present significant challenges for developing targeted therapies.
Tumor heterogeneity manifests at multiple levels, including molecular, cellular, and spatial dimensions, creating diverse microenvironments that influence ubiquitination patterns [67] [68]. Single-cell RNA sequencing technologies have revolutionized our understanding of this complexity, revealing distinct cellular subtypes within the tumor microenvironment (TME) that exhibit unique transcriptional programs and functional states [68] [69]. This heterogeneity directly impacts ubiquitination pathways, as demonstrated by recent studies showing that ubiquitination-related genes (URGs) can stratify colon cancer patients into molecular subtypes with distinct survival outcomes and immune microenvironments [21]. The development of effective therapeutic strategies requires navigating this complexity through advanced profiling technologies and context-specific analysis of ubiquitination networks.
Ubiquitination Patterns Across Cancer Types and Stages
Pan-Cancer Ubiquitination Signatures
Comprehensive pan-cancer analyses have revealed that specific components of the ubiquitination machinery exhibit consistent dysregulation across multiple cancer types. Ubiquitin D (UBD), also known as HLA-F adjacent transcript 10 (FAT10), has emerged as a particularly significant oncogenic factor that demonstrates elevated expression in 29 different cancer types according to data from The Cancer Genome Atlas (TCGA) and Genotype-Tissue Expression (GTEx) projects [66]. This overexpression correlates strongly with poor prognosis and advanced histological grades, with gene amplification representing the most common genetic alteration [66]. Ubiquitin D engages key oncogenic pathways including NF-κB, Wnt, and SMAD2 signaling, while interacting with downstream effectors such as MAD2, p53, and β-catenin to promote tumor survival, proliferation, and metastatic potential [66].
Table 1: Ubiquitin D (UBD) Expression and Clinical Correlations Across Selected Cancers
| Cancer Type | Expression in Tumor vs Normal | Correlation with Prognosis | Common Genetic Alterations | Associated Pathways |
|---|---|---|---|---|
| Hepatocellular Carcinoma | Significantly upregulated | Reduced overall survival | Gene amplification | PD-L1 upregulation, Immune evasion |
| Colorectal Cancer | Significantly upregulated | Reduced disease-specific survival | Gene amplification | NF-κB, Wnt signaling |
| Breast Cancer | Significantly upregulated | Higher histological grade | Shallow deletions | SMAD2, β-catenin interaction |
| Glioblastoma | Significantly upregulated | Reduced overall survival | Gene amplification | Chromosomal instability |
| Non-Small Cell Lung Cancer | Significantly upregulated | Advanced clinical stage | Not specified | Mitotic non-disjunction |
The clinical relevance of UBD extends beyond mere expression patterns, as its overexpression induces mitotic non-disjunction and chromosomal instability, directly contributing to tumor development [66]. Epigenetically, reduced UBD promoter methylation has been observed in 16 cancer types, suggesting a potential mechanism for its dysregulation [66]. Furthermore, UBD expression significantly correlates with features of the tumor immune microenvironment, including immune infiltration patterns, checkpoint expression, microsatellite instability (MSI), tumor mutational burden (TMB), and neoantigen load, positioning it as a potential predictor of immunotherapy sensitivity [66].
Cancer-Type Specific Ubiquitination Profiles
Beyond pan-cancer patterns, specific cancer types exhibit distinct ubiquitination signatures with clinical implications. In sigmoid colorectal cancer, ubiquitinome analysis has identified 1,249 ubiquitinated sites within 608 differentially ubiquitinated proteins (DUPs) between cancerous and para-carcinoma tissues [39]. These DUPs participate in 35 statistically significant signaling pathways, including salmonella infection, glycolysis/gluconeogenesis, and ferroptosis [39]. Gene Ontology analysis revealed involvement in 98 biological processes, 64 cellular components, 51 molecular functions, and 26 immune system processes, highlighting the functional diversity of ubiquitination in this cancer type [39].
In breast cancer, a novel two-gene signature focusing on the ubiquitination regulation of SKP2, an established oncoprotein, has demonstrated significant prognostic value [52]. This signature consists of the copy number of FZR1 (an E3 ligase component that ubiquitinates SKP2) compared to USP10 (a deubiquitinase that reverses SKP2 ubiquitination) [52]. Patients were stratified into "high SKP2 ubiquitination" (higher FZR1 copy number than USP10) and "low SKP2 ubiquitination" (higher USP10 copy number than FZR1) groups, with the signature showing strong association with clinical outcome specifically in luminal breast cancer but not other subtypes [52]. The signature also correlated significantly with tumor grade, stage, and the number of positive lymph nodes [52].
Table 2: Experimentally Validated Ubiquitination-Related Biomarkers in Specific Cancers
| Cancer Type | Biomarker/Signature | Components | Clinical Utility | Validation Status |
|---|---|---|---|---|
| Colon Cancer | 6-Gene Ubiquitination Signature | ARHGAP4, MID2, SIAH2, TRIM45, UBE2D2, WDR72 | Prognostic stratification, immune microenvironment assessment | qRT-PCR, IHC, functional assays in vitro and in vivo |
| Breast Cancer | SKP2 Ubiquitination Signature | FZR1 vs USP10 copy number | Prognosis in luminal BC, association with grade/stage | TCGA data analysis, correlation with p27 levels |
| Sigmoid Colon Cancer | Differential Ubiquitinated Proteins | 608 DUPs with 1249 ubiquitination sites | Patient stratification, predictive diagnosis | Label-free quantitative ubiquitinomics |
| Multiple Cancers | Ubiquitin D (UBD) | UBD expression levels | Prognostic biomarker, immunotherapy response predictor | Pan-cancer analysis of TCGA and GTEx data |
For colon cancer, researchers have developed a ubiquitination-related signature based on six genes (ARHGAP4, MID2, SIAH2, TRIM45, UBE2D2, and WDR72) that effectively stratifies patients into distinct risk categories [21]. This signature demonstrates that patients in high-risk groups exhibit characteristics indicative of enhanced epithelial-mesenchymal transition, immune escape, immunosuppressive myeloid-derived suppressor cell infiltration, and lower immunogenicity [21]. In contrast, low-risk patients show better responses to CTLA4 checkpoint inhibitors, providing clinical guidance for immunotherapy selection [21]. Experimental validation confirmed the early diagnostic capabilities of ARHGAP4 and SIAH2, while WDR72 knockdown significantly inhibited colorectal cancer cell proliferation both in vitro and in vivo [21].
Methodological Approaches for Ubiquitination Research
Experimental Workflows for Ubiquitination Analysis
The investigation of ubiquitination patterns in the context of tumor heterogeneity requires sophisticated methodological approaches. Single-cell multi-omics technologies have emerged as powerful tools for dissecting tumor heterogeneity at unprecedented resolution, enabling simultaneous analysis of genomics, transcriptomics, epigenomics, proteomics, and spatial information from individual cells [67]. These technologies employ various cell isolation strategies, including micromanipulation, laser capture microdissection (LCM), fluorescence-activated cell sorting (FACS), magnetic-activated cell sorting (MACS), and microfluidic technologies, each with distinct advantages and limitations for specific research applications [67].
Ubiquitinomics workflows typically employ anti-K-ε-GG antibody beads (PTMScan ubiquitin remnant motif)-based enrichment of ubiquitinated peptides followed by label-free quantitative proteomics and bioinformatics analysis [39]. This approach allows for large-scale identification and quantification of ubiquitination sites across the proteome. The typical workflow involves tissue sample preparation, protein extraction and digestion, ubiquitinated peptide enrichment via immunoaffinity purification, liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis, and bioinformatic processing for ubiquitination site identification and quantification [39]. For cellular studies, detection methods for compound ubiquitination include SDS-PAGE separation of ubiquitination reactions followed by excision of Ub-containing bands and MS/MS analyses, with LysC protease digestion of Ub yielding C-terminal peptides identifiable in both unmodified and compound-linked forms [70].
The Scientist's Toolkit: Key Research Reagents and Platforms
Table 3: Essential Research Reagents and Platforms for Ubiquitination Studies
| Reagent/Platform | Function | Application Examples | References |
|---|---|---|---|
| Anti-K-ε-GG antibody beads | Immunoaffinity enrichment of ubiquitinated peptides | Ubiquitinome profiling in sigmoid colon cancer | [39] |
| 10x Genomics Chromium X | Single-cell RNA sequencing | Pan-cancer TME analysis, cell subtype identification | [67] [68] |
| PTMScan Ubiquitin Remnant Motif Kit | Ubiquitinated peptide enrichment | Identification of 1249 ubiquitinated sites in colon cancer | [39] |
| Fragment-based compound libraries | Drug discovery targeting UPS | Identification of HUWE1 inhibitors/substrates | [65] [70] |
| TCGA, GTEx, cBioPortal databases | Bioinformatics analysis of ubiquitination genes | Pan-cancer analysis of UBD expression and alterations | [66] |
| STRING database | Protein-protein interaction network analysis | Mapping interactions of ubiquitination-related proteins | [66] |
Advanced screening technologies have been developed to identify modulators of ubiquitination enzymes. These include target-based high throughput screening (HTS), fragment-based drug discovery (FBDD), proteolysis-targeting chimeric molecules (PROTACs), and protein design engineering approaches [33]. Each technology offers distinct advantages and limitations in sampling chemical space, cost-effectiveness, precision of target degradation, and ease of manipulation [33]. Fragment-based screening has gained prominence due to its efficient coverage of chemical space with small libraries, with both covalent and non-covalent approaches proving valuable for targeting ubiquitination enzymes [65].
For computational analysis, researchers rely on multiple bioinformatics platforms and databases. The Integrated Annotations for Ubiquitin and Ubiquitin-like Conjugation Database (iUUCD) provides comprehensive information on ubiquitination-related genes, including E1, E2, E3 enzymes, deubiquitinating enzymes, and ubiquitin-binding domains [21]. Gene Expression Profiling Interactive Analysis (GEPIA2.0), CBio Cancer Genomics Portal (cBioPortal), University of Alabama at Birmingham CANcer data analysis Portal (UALCAN), and Sangerbox offer specialized tools for analyzing ubiquitination gene expression, genetic alterations, methylation patterns, and clinical correlations across cancer types [66].
Signaling Pathways and Ubiquitination Networks
Key Ubiquitination-Regulated Pathways in Cancer
Ubiquitination modulates numerous critical cancer-relevant signaling pathways through targeted protein degradation and regulation of protein activity. The SKP2-p27 axis represents a well-characterized ubiquitination pathway with significant implications for cancer progression [52]. In this pathway, SKP2 functions as part of the Skp1-Cullin-F-box (SCF) complex E3 ubiquitin ligase that targets cell cycle inhibitors including p27 and p21 for degradation, facilitating cell cycle progression [52]. The ubiquitination of SKP2 itself is controlled by the antagonistic actions of the APC/CFZR1 E3 ligase complex (which promotes SKP2 ubiquitination and degradation) and deubiquitinases such as USP10 (which stabilize SKP2) [52]. The balance between these regulators determines SKP2 protein levels and consequently influences cell cycle progression and proliferation.
The UBD-mediated pathways represent another significant ubiquitination network in cancer biology. UBD promotes oncogenesis through multiple mechanisms, including induction of mitotic non-disjunction and chromosomal instability [66]. It engages key oncogenic pathways including NF-κB, Wnt, and SMAD2 signaling, while interacting with downstream effectors such as MAD2, p53, and β-catenin [66]. These interactions collectively promote tumor survival, proliferation, invasion, and metastatic potential. In hepatocellular carcinoma, UBD (FAT10) specifically drives immune evasion by upregulating PD-L1, fostering an immunosuppressive tumor microenvironment [66].
Analysis of ubiquitination pathways in sigmoid colon cancer has revealed enrichment in 35 statistically significant signaling pathways, including salmonella infection, glycolysis/gluconeogenesis, and ferroptosis [39]. These findings connect ubiquitination to diverse cellular processes beyond conventional protein degradation, including metabolic reprogramming and cell death mechanisms that are increasingly recognized as important therapeutic targets in cancer. The identification of these pathway associations provides insights for developing context-specific therapeutic strategies targeting ubiquitination in different cancer types and stages.
Tumor Microenvironment and Ubiquitination
Single-cell RNA sequencing analyses have revealed extensive heterogeneity in ubiquitination patterns across different cell types within the tumor microenvironment. A recent pan-cancer single-cell atlas that simultaneously considered heterogeneity in five cell types across 230 treatment-naive samples from nine cancer types identified 70 pan-cancer single-cell subtypes with distinct co-occurrence patterns [68]. This study revealed two TME hubs of strongly co-occurring subtypes: one resembling tertiary lymphoid structures (TLS), and another consisting of immune-reactive PD1+/PD-L1+ immune-regulatory T cells and B cells, dendritic cells, and inflammatory macrophages [68]. Subtypes belonging to each hub demonstrated spatial co-localization, with their abundance associating with early and long-term checkpoint immunotherapy response [68].
In breast cancer, integrated single-cell RNA sequencing and spatial transcriptomics analysis identified 15 major cell clusters within the TME, including neoplastic epithelial, immune, stromal, and endothelial populations [69]. Notably, low-grade tumors showed enriched subtypes such as CXCR4+ fibroblasts, IGKC+ myeloid cells, and CLU+ endothelial cells with distinct spatial localization and immune-modulatory functions [69]. These subtypes were paradoxically linked to reduced immunotherapy responsiveness despite their association with favorable clinical features, highlighting the complex relationship between ubiquitination patterns, TME composition, and therapeutic outcomes.
The investigation of ubiquitination in the context of tumor heterogeneity has revealed both challenges and opportunities for cancer diagnosis and treatment. The context-specific nature of ubiquitination effects necessitates sophisticated analytical approaches that account for tumor type, stage, cellular composition, and spatial organization. Single-cell multi-omics technologies, spatial transcriptomics, and advanced ubiquitinomics workflows provide powerful tools for deciphering this complexity, enabling researchers to identify clinically relevant ubiquitination patterns and signatures across diverse cancer contexts.
Future directions in this field will likely focus on developing increasingly precise ubiquitination-targeting therapies that account for tumor heterogeneity. Fragment-based drug discovery approaches show particular promise for targeting challenging ubiquitination enzymes [65], while PROTAC technology offers opportunities for targeted protein degradation [33]. The discovery that ubiquitin ligases can modify drug-like small molecules [70] opens new avenues for harnessing the ubiquitin system to transform exogenous compounds into novel chemical modalities within cells. Furthermore, the integration of ubiquitination signatures with other molecular profiling data may enable more precise patient stratification and personalized treatment selection.
As our understanding of ubiquitination patterns in cancer continues to evolve, the translation of these insights into clinical practice will require validation in large patient cohorts and development of standardized assays for ubiquitination marker assessment. The ongoing refinement of single-cell and spatial analysis technologies will further enhance our ability to map ubiquitination networks within the complex architecture of heterogeneous tumors, ultimately contributing to more effective strategies for overcoming the challenges posed by tumor heterogeneity and context-specific ubiquitination effects.
Resistance Mechanisms to UPS-Targeted Therapies and Combinatorial Strategies
The Ubiquitin-Proteasome System (UPS) is a master regulator of cellular protein homeostasis, controlling the degradation of key proteins involved in cell cycle progression, apoptosis, and DNA repair [39] [71]. Given its pivotal role in cellular regulation, the UPS has emerged as a valuable target for cancer therapy, with proteasome inhibitors (PIs) like bortezomib, carfilzomib, and ixazomib achieving clinical success, particularly for hematological malignancies such as multiple myeloma and mantle cell lymphoma [71]. However, the development of resistance to these targeted therapies remains a significant clinical obstacle, often leading to treatment failure and disease relapse [72] [73]. This resistance is a multifaceted phenomenon, driven by genetic, epigenetic, and microenvironmental factors that allow cancer cells to evade therapy-induced death [72] [74]. Understanding these resistance mechanisms is crucial for developing more effective treatment strategies. This guide objectively compares the performance of UPS-targeted therapies, analyzes the resistance mechanisms that limit their efficacy, and evaluates combinatorial strategies designed to overcome this resistance, all within the context of the growing understanding that ubiquitination patterns correlate with cancer stage and grade [39] [21].
The Ubiquitin-Proteasome System (UPS) and its Therapeutic Targeting
Fundamentals of the UPS Pathway
The UPS is a highly coordinated enzymatic cascade responsible for the targeted degradation of ubiquitinated proteins. The process begins with ubiquitin activation by an E1 enzyme, followed by its transfer to an E2 conjugating enzyme. Finally, an E3 ubiquitin ligase recognizes the specific substrate and facilitates the transfer of ubiquitin from the E2 to the target protein [39] [73]. Polyubiquitinated proteins are then recognized and degraded by the 26S proteasome, a multi-subunit complex comprising a 20S catalytic core and 19S regulatory particles [71]. The 20S core possesses three primary proteolytic activities: caspase-like (β1), trypsin-like (β2), and chymotrypsin-like (β5) [71]. Beyond its role in protein turnover, ubiquitination is a versatile signaling mechanism that regulates protein localization, activity, and interactions, with different polyubiquitin chain linkages (e.g., Lys48 vs. Lys63) dictating distinct functional outcomes [39] [73].
Figure 1: The Ubiquitin-Proteasome System Pathway. This diagram illustrates the sequential enzymatic cascade of ubiquitination and subsequent proteasomal degradation.
Clinically Approved UPS-Targeted Therapies
Table 1: Clinically Approved Proteasome Inhibitors
| Drug Name | Type of Inhibitor | Primary Target | Approved Cancers | Key Limitations |
|---|---|---|---|---|
| Bortezomib | Reversible | Chymotrypsin-like (β5) activity [71] | Multiple Myeloma, Mantle Cell Lymphoma [71] | Peripheral neuropathy, drug resistance [71] |
| Carfilzomib | Irreversible | Chymotrypsin-like (β5) activity [71] | Multiple Myeloma [71] | Cardiotoxicity, limited efficacy in solid tumors [71] |
| Ixazomib | Reversible, Oral | Chymotrypsin-like (β5) activity [71] | Multiple Myeloma [71] | Gastrointestinal side effects, emerging resistance [71] |
Resistance Mechanisms to UPS-Targeted Therapies
Resistance to UPS-targeted therapies can be intrinsic or acquired and involves a complex interplay of mechanisms within cancer cells and their surrounding microenvironment.
Molecular and Cellular Resistance Mechanisms
Table 2: Key Resistance Mechanisms to UPS-Targeted Therapies
| Resistance Mechanism | Description | Impact on Therapy |
|---|---|---|
| UPS Component Alterations | Mutations or overexpression of proteasome subunits (e.g., PSMB5) and upregulation of immunoproteasomes, reducing drug binding and efficacy [71]. | Decreased sensitivity to PIs, requires development of next-generation inhibitors [71]. |
| Enhanced Drug Efflux | Overexpression of ATP-binding cassette (ABC) transporters (e.g., P-glycoprotein) that actively pump drugs out of cancer cells [72] [71]. | Reduced intracellular drug concentration, leading to multidrug resistance (MDR) [72]. |
| Activation of Compensatory Pathways & Anti-apoptotic Signals | Upregulation of pro-survival pathways like NF-κB and increased expression of anti-apoptotic proteins (e.g., BCL-2, MCL-1) [72] [75] [71]. | Cancer cells evade apoptosis, surviving despite proteasome inhibition [75]. |
| Metabolic Inactivation | Detoxification of chemotherapeutic agents by drug-metabolizing enzymes (e.g., cytochrome P450, glutathione-S-transferase) [72]. | Inactivation of prodrugs or conversion of active drugs to inactive forms [72]. |
| Tumor Cell Plasticity | Therapy-induced phenotypic switching, including epithelial-mesenchymal transition (EMT), transdifferentiation, and the emergence of drug-tolerant persisters (DTPs) and cancer stem cells (CSCs) [74]. | Tumor heterogeneity, dormancy, and resilience to targeted agents, causing relapse [74]. |
The Role of Deubiquitinating Enzymes (DUBs) in Resistance
A particularly significant mechanism of resistance involves the upregulation and activity of Deubiquitinating Enzymes (DUBs), especially Ubiquitin-Specific Proteases (USPs). USPs can counteract the effect of PIs by removing ubiquitin chains from proteasome-bound substrates, thereby rescuing them from degradation [73]. For example:
- USP51 is upregulated in cisplatin-resistant lung cancer, where it diminishes the DNA damage marker γH2AX and enhances DNA damage repair, promoting cell survival [73].
- USP22 facilitates robust DNA damage repair in lung adenocarcinoma by interacting with the PALB2-BRCA2-Rad51 complex, contributing to therapy resistance [73].
- In chronic myelogenous leukemia and other cancers, USP10 deubiquitinates and stabilizes the oncoprotein SKP2, leading to the degradation of cell cycle inhibitors like p27 and fostering proliferation and drug resistance [52].
The activity of specific E3 ligases and DUBs can form a regulatory signature that predicts cancer aggressiveness and patient outcome. For instance, a gene signature based on the copy number of the E3 ligase component FZR1 and the deubiquitinase USP10 can stratify breast cancer patients into groups with significantly different survival outcomes, recapitulating the activity of their shared substrate SKP2 [52].
Combinatorial Strategies to Overcome Resistance
To combat the multifaceted nature of resistance, combination therapies that target UPS components alongside other critical pathways have shown great promise.
Promising Combination Therapy Approaches
Table 3: Experimental Combinatorial Strategies Targeting UPS Resistance
| Combination Strategy | Mechanism of Action | Experimental Support |
|---|---|---|
| PI + BCL-2/MCL-1 Inhibitors | PI blocks degradation of pro-apoptotic proteins, while BCL-2 family inhibitors (e.g., ABT-199) directly activate apoptosis, overcoming anti-apoptotic adaptations [75] [71]. | Dual inhibition of BCL-2 and MCL-1 with ABT-199 and homoharringtonine is more effective in inducing apoptosis in acute myeloid leukemia than monotherapies [75]. |
| PI + Immunotherapy | Proteasome inhibition can enhance tumor immunogenicity, while immunotherapy (e.g., CAR-T cells, checkpoint inhibitors) boosts the anti-tumor immune response [76]. | Mathematical modeling optimizes scheduling between Targeted Radionuclide Therapy (a radiation stressor) and CAR-T cells to maximize tumor control in multiple myeloma [76]. |
| PI + Targeted Agent (e.g., Kinase Inhibitors) | Co-targeting the UPS and an oncogenic driver pathway (e.g., with a tyrosine kinase inhibitor) causes synergistic stress and blocks compensatory survival signals [75] [77]. | The combination of anlotinib (TKI) and metformin enhances oxidative stress and apoptosis in preclinical models [75]. Databases like OncoDrug+ systematically catalog such evidence-based combinations [77]. |
| USP Inhibitors + Conventional Therapies | Inhibiting specific USPs (e.g., with small molecules) prevents the rescue of oncoproteins, re-sensitizing cancer cells to chemotherapy and targeted therapy [73]. | Inhibition of USP7 or USP10 can mitigate resistance and render cancer cells more vulnerable to anticancer therapies [73]. |
The Promise of Ubiquitinomics for Patient Stratification
Advanced proteomic and bioinformatic approaches are enabling a more precise application of these strategies. Quantitative ubiquitinomics, which uses anti-K-ε-GG antibody-based enrichment coupled with LC-MS/MS, can profile the global ubiquitinome in cancer tissues [39]. This technology has revealed that ubiquitination patterns are disease- and stage-specific. For example, a study of sigmoid colon cancer identified 1,249 ubiquitinated sites within 608 differentially ubiquitinated proteins (DUPs) between tumor and normal tissues, and further linked 46 DUPs to overall survival [39]. Similarly, in colon cancer, molecular classification based on Ubiquitination-Related Genes (URGs) like ARHGAP4, SIAH2, and WDR72 can stratify patients into subtypes with distinct prognoses and immune microenvironments [21]. These signatures provide a rationale for patient stratification and the development of personalized combinatorial treatments.
Figure 2: Workflow for Ubiquitinomics-Based Patient Stratification. This diagram outlines the process from tissue sampling to guiding personalized combination therapy using ubiquitinomics data.
Experimental Protocols and Methodologies
Protocol: Ubiquitin Remnant Motif (K-ε-GG) Based Label-Free Quantitative Ubiquitinomics
This protocol is used to identify and quantify ubiquitination sites in tissue samples, such as cancer versus para-carcinoma tissues [39].
- Sample Preparation: Homogenize frozen tissue samples (e.g., sigmoid colon cancer and matched normal tissue) in a lysis buffer containing protease inhibitors and DUB inhibitors to preserve ubiquitination states.
- Protein Digestion: Reduce, alkylate, and digest the extracted proteins into peptides using trypsin.
- Ubiquitinated Peptide Enrichment: Incubate the peptide mixture with anti-K-ε-GG antibody-coated beads (PTMScan Ubiquitin Remnant Motif Kit) to specifically immunoaffinity-purify peptides containing the diglycine (K-ε-GG) remnant left after tryptic digestion of ubiquitinated proteins.
- LC-MS/MS Analysis: Desalt and separate the enriched peptides using nano-flow liquid chromatography (LC) and analyze them by tandem mass spectrometry (MS/MS) in a data-dependent acquisition mode.
- Data Processing: Identify peptides and proteins by searching the MS/MS data against a protein sequence database. Quantify the relative abundance of ubiquitinated peptides between sample groups using a label-free quantification (LFQ) algorithm based on precursor ion intensity.
- Bioinformatics Analysis:
- Identify Differentially Ubiquitinated Proteins (DUPs) using statistical tests (e.g., t-test).
- Perform Gene Ontology (GO) and KEGG pathway enrichment analysis on the DUPs.
- Construct Protein-Protein Interaction (PPI) networks.
- Correlate DUPs with gene expression data from sources like TCGA and perform survival analysis (Kaplan-Meier method) to identify prognostic DUPs [39] [21].
Protocol: Mathematical Modeling for Optimizing Combination Therapy Scheduling
This in silico protocol is used to optimize the dosing and scheduling of combination therapies, such as TRT and CAR-T cell immunotherapy [76].
- Model Formulation: Develop a system of ordinary differential equations (ODEs) that describes the dynamics of key populations:
- Unirradiated tumor cells (NT)
- Irradiated tumor cells (NR)
- CAR-T cells (N_C)
- Include terms for tumor growth, CAR-T cell-mediated killing, radiation-induced cell kill, and immune cell proliferation/exhaustion.
- Parameter Estimation: Use experimental data from monotherapy studies (e.g., tumor volume over time with TRT alone and CAR-T cells alone) to calibrate and estimate the model parameters (e.g., tumor growth rate ρ, killing rate k₁, radiation dose rate R₀).
- Model Validation: Validate the calibrated model by comparing its predictions with experimental data from combination therapy studies with fixed intervals.
- In Silico Optimization: Run simulations to test a wide range of dosing schedules (varied timing intervals, sequences, and fractionated doses). Evaluate each regimen based on key outcomes like Progression-Free Survival (PFS) and time to minimum tumor burden.
- Prediction of Optimal Regimen: Identify the therapy schedule that maximizes the desired outcome metric. The model can reveal, for instance, that intervals too close or too far apart are detrimental, and that optimal timing depends on tumor proliferation rates [76].
Table 4: Essential Research Tools for Investigating UPS and Resistance
| Tool / Resource | Function / Application | Example / Specification |
|---|---|---|
| Anti-K-ε-GG Antibody | Immunoaffinity enrichment of ubiquitinated peptides from complex protein digests for ubiquitinomics studies [39]. | PTMScan Ubiquitin Remnant Motif Kit; critical for LC-MS/MS-based ubiquitinome profiling. |
| Proteasome Inhibitors | To inhibit proteasome activity in in vitro and in vivo models, studying UPS function and inducing proteotoxic stress [71]. | Bortezomib, Carfilzomib, MG132; used at varying IC₅₀ concentrations depending on cell type. |
| USP/DUB Inhibitors | To selectively inhibit deubiquitinating enzymes and investigate their role in stabilizing oncoproteins and causing resistance [73]. | Small molecule inhibitors targeting USP7, USP10, etc.; often used in combination with other agents. |
| Database: iUUCD 2.0 | Provides a comprehensive repository of ubiquitin and ubiquitin-like conjugation data for extracting Ubiquitination-Related Genes (URGs) [21]. | Contains ~1360 URGs, including E1, E2, E3 enzymes, DUBs, and ubiquitin-binding domain proteins. |
| Database: OncoDrug+ | A manually curated database for identifying evidence-based cancer drug combinations, including those involving UPS-targeted agents [77]. | Covers 7895 entries, 77 cancer types, 2201 combination therapies, with biomarker and evidence level annotation. |
| Mathematical Modeling Framework | To optimize the scheduling and dosing of combination therapies in silico before in vivo testing [76]. | A system of ODEs modeling tumor-immune-therapy interactions; parameters estimated from preclinical data. |
Post-translational modifications (PTMs) represent a crucial regulatory layer that controls protein function, stability, and interaction networks within cells. Among these modifications, ubiquitination and O-GlcNAcylation have emerged as particularly important players in cellular homeostasis and disease pathogenesis. Ubiquitination involves the covalent attachment of ubiquitin molecules to lysine residues on target proteins, typically marking them for proteasomal degradation, while also regulating subcellular localization, protein interactions, and signaling pathways [78] [39]. O-GlcNAcylation, in contrast, entails the addition of single N-acetylglucosamine (O-GlcNAc) molecules to serine or threonine residues, serving as a nutrient sensor that links cellular metabolism to signal transduction pathways [79] [80].
The interplay between these PTMs has gained significant attention in cancer research, with numerous studies demonstrating that their crosstalk influences tumor progression, therapeutic resistance, and patient prognosis. Cancer cells frequently exhibit dysregulation in both ubiquitination and O-GlcNAcylation, creating pathogenic circuits that drive proliferation, metastasis, and treatment resistance [78] [81]. Understanding the molecular mechanisms underlying this crosstalk provides not only fundamental biological insights but also potential therapeutic avenues for cancer intervention. This review synthesizes current knowledge on the multifaceted crosstalk between ubiquitination and O-GlcNAcylation, with particular emphasis on its implications for cancer biology and the development of targeted therapies.
Molecular Mechanisms of Crosstalk
Competitive Regulation of Protein Stability
The most direct mechanism of crosstalk between O-GlcNAcylation and ubiquitination occurs through competitive binding at the same or adjacent sites on target proteins. O-GlcNAcylation can sterically hinder ubiquitination by occupying serine/threonine residues, thereby protecting proteins from proteasomal degradation [78] [82].
Table 1: Proteins Whose Stability is Regulated by O-GlcNAcylation-Ubiquitination Crosstalk
| Protein | O-GlcNAc Site | Effect on Stability | Functional Outcome | Reference |
|---|---|---|---|---|
| p53 | S149 | Increased stability | Reduced ubiquitination and degradation; enhanced tumor suppressor function | [82] |
| Snail1 | S112 | Increased stability | Blocked phosphorylation-dependent ubiquitination; enhanced EMT | [82] |
| Δ-lactoferrin | S10 | Increased stability | Competitive inhibition of phosphorylation at S10; cell cycle arrest | [82] |
| β-catenin | Not specified | Increased stability | Enhanced Wnt signaling; increased cell proliferation | [82] |
| YTHDF2 | S263 | Increased stability | Prevented ubiquitination; enhanced oncogenic activity | [80] |
This protective effect of O-GlcNAcylation creates a metabolic regulation point for protein turnover, allowing cancer cells to stabilize oncoproteins and drive tumor progression. The stabilization of transcription factors like Snail1 enhances epithelial-mesenchymal transition (EMT), facilitating metastasis, while stabilization of p53 in certain contexts may be circumvented by complementary mutations in cancer cells [82].
Regulation of the Ubiquitin-Proteasome System
O-GlcNAcylation directly targets components of the ubiquitin-proteasome system (UPS), creating a broader regulatory mechanism that extends beyond individual protein substrates. Key findings include:
- Proteasomal Inhibition: O-GlcNAcylation modifies the 26S proteasome, inhibiting its ATPase activity and consequently reducing proteolysis of transcription factors like Sp1 and hydrophobic peptides [78].
- E3 Ligase Regulation: O-GlcNAcylation targets multiple E3 ubiquitin ligases, including those containing Cullin and F-box domains, potentially modulating their ability to recognize and ubiquitinate substrate proteins [81].
- Competition with Phosphorylation: O-GlcNAcylation often occurs at serine/threonine residues within PEST sequences (protein regions rich in proline, glutamate, serine, and threonine), which are recognition motifs for ubiquitin ligases. By competing with phosphorylation at these sites, O-GlcNAcylation can block ubiquitination signals [78].
These mechanisms enable cancer cells to globally modulate protein degradation in response to metabolic cues, providing a survival advantage under stress conditions such as nutrient deprivation or chemotherapeutic exposure.
Reciprocal Regulation: Ubiquitination of O-GlcNAcylation Enzymes
The crosstalk between these PTMs is bidirectional, with ubiquitination conversely regulating the enzymes responsible for O-GlcNAcylation. The central enzyme O-GlcNAc transferase (OGT) is itself regulated by ubiquitin-mediated degradation:
- FBXO31-Mediated Degradation: The F-box protein FBXO31, a component of the SKP1-CUL1-F-box (SCF) E3 ubiquitin ligase complex, targets OGT for ubiquitination and subsequent degradation [83].
- Metabolic Regulation: In endometrial cancer, FBXO31-mediated regulation of OGT creates a feedback loop that maintains O-GlcNAcylation homeostasis. Loss of FBXO31 results in OGT accumulation, elevated O-GlcNAcylation, and promotion of tumorigenic properties [83].
- Therapeutic Implications: Disruption of this regulatory axis represents a common pathogenic mechanism in cancer, with FBXO31 downregulation correlating with poor prognosis in multiple cancer types [83].
Table 2: Enzymatic Regulators of Ubiquitination and O-GlcNAcylation Crosstalk
| Enzyme | Type | Function in Crosstalk | Cancer Relevance |
|---|---|---|---|
| OGT | O-GlcNAc transferase | Adds O-GlcNAc to proteins, blocking ubiquitination | Overexpressed in multiple cancers; correlated with poor prognosis |
| OGA | O-GlcNAcase | Removes O-GlcNAc, permitting ubiquitination | Variable expression across cancers; potential therapeutic target |
| FBXO31 | E3 ubiquitin ligase adapter | Targets OGT for ubiquitination and degradation | Acts as tumor suppressor; lost in aggressive cancers |
| FZR1 | E3 ubiquitin ligase component | Targets SKP2 for ubiquitination | Regulates cell cycle progression; prognostic in breast cancer |
| USP10 | Deubiquitinase | Removes ubiquitin from SKP2, stabilizing it | Promotes SKP2-mediated degradation of cell cycle inhibitors |
This reciprocal regulation creates complex feedback loops that fine-tune cellular responses to metabolic and environmental signals, with cancer cells often hijacking these mechanisms to support uncontrolled proliferation and survival.
Experimental Approaches for Studying PTM Crosstalk
Ubiquitinomics and Proteomic Profiling
Advanced mass spectrometry-based approaches enable comprehensive mapping of ubiquitination and O-GlcNAcylation events:
Ubiquitin Remnant Profiling (Ubiquitinomics):
- Methodology: Uses anti-K-ε-GG antibody beads (PTMScan Ubiquitin Remnant Motif) to specifically enrich for ubiquitinated peptides following tryptic digestion, which leaves a Gly-Gly remnant on modified lysines [39].
- Workflow: Tissue samples → protein extraction → tryptic digestion → K-ε-GG peptide enrichment → LC-MS/MS analysis → bioinformatic processing [39].
- Applications: Identification of differentially ubiquitinated proteins (DUPs) between normal and tumor tissues. For example, a study on sigmoid colon cancer identified 1,249 ubiquitinated sites within 608 DUPs, revealing pathway alterations in glycolysis, ferroptosis, and infection response [39].
Integrated O-GlcNAc and Ubiquitin Profiling:
- Sequential Enrichment: Sequential immunoenrichment for both O-GlcNAcylated and ubiquitinated peptides from the same sample enables correlation analysis between these modifications.
- Stable Isotope Labeling: SILAC (stable isotope labeling with amino acids in cell culture) allows quantitative comparison of modification dynamics in response to metabolic perturbations or inhibitor treatments.
- Bioinformatic Integration: Computational pipelines integrate ubiquitinomics data with transcriptomic and proteomic datasets to establish relationship models between ubiquitination levels and gene/protein expression [39] [21].
Figure 1: Experimental workflow for integrated ubiquitinomics and O-GlcNAcomics analysis
Functional Validation Approaches
Genetic Manipulation of O-GlcNAcylation:
- OGT/OGA Modulation: CRISPR/Cas9-mediated knockout or RNA interference knockdown of OGT/OGA to assess effects on protein ubiquitination and stability [83].
- Mutagenesis Studies: Site-directed mutagenesis of specific O-GlcNAcylation sites to validate their role in regulating ubiquitination (e.g., S149A mutation in p53) [82].
- Organoid Models: Patient-derived organoids enable functional studies in a physiologically relevant context. For example, endometrial cancer organoids with modulated O-GlcNAcylation levels demonstrate the role of this PTM in promoting proliferation and stem-like properties [83].
Pharmacological Inhibition:
- OGT Inhibitors: OSMI-1 and other small molecule inhibitors reduce global O-GlcNAcylation, allowing assessment of downstream effects on ubiquitination.
- Proteasome Inhibitors: Bortezomib and MG132 block protein degradation, enabling accumulation of ubiquitinated proteins for detection and characterization.
- Combination Treatments: Co-administration of OGT and proteasome inhibitors reveals synthetic lethal interactions in specific cancer contexts.
Table 3: Research Reagent Solutions for Studying Ubiquitination-O-GlcNAcylation Crosstalk
| Reagent/Category | Specific Examples | Function/Application | Experimental Use |
|---|---|---|---|
| Enrichment Antibodies | Anti-K-ε-GG (PTMScan) | Ubiquitinated peptide enrichment | Ubiquitinomics by LC-MS/MS |
| RL2 antibody | O-GlcNAcylated protein detection | Immunoprecipitation, IHC | |
| CTD110.6 antibody | O-GlcNAcylated protein detection | Western blot, immunofluorescence | |
| Enzyme Inhibitors | OSMI-1, OSMI-4 | OGT inhibition | Reducing global O-GlcNAcylation |
| Thiamet G, PUGNAc | OGA inhibition | Elevating global O-GlcNAcylation | |
| Bortezomib, MG132 | Proteasome inhibition | Accumulating ubiquitinated proteins | |
| Genetic Tools | OGT/OGA shRNA | Gene knockdown | Functional studies of enzyme depletion |
| OGT/OGA CRISPR | Gene knockout | Complete elimination of enzymes | |
| Site-directed mutants | Specific residue manipulation | Validation of modification sites | |
| Model Systems | Patient-derived organoids | 3D culture models | Physiological relevance studies |
| Xenograft models | In vivo tumor growth | Therapeutic assessment |
Pathophysiological Implications in Cancer
Therapeutic Resistance Mechanisms
The crosstalk between O-GlcNAcylation and ubiquitination significantly contributes to resistance against conventional cancer therapies:
- Chemotherapy Resistance: O-GlcNAcylation of drug targets and transporters alters their stability and activity. For instance, O-GlcNAcylation can increase expression of drug efflux pumps or modify the stability of apoptosis-related proteins like DR4, influencing sensitivity to TRAIL-induced cell death [78].
- Metabolic Adaptation: Cancer cells exploit the nutrient-sensing function of O-GlcNAcylation to rewire metabolic pathways under therapeutic stress. O-GlcNAcylation of metabolic enzymes such as PFK1, G6PD, and PKM2 shifts energy production toward biosynthetic pathways that support cell survival and growth [81].
- DNA Repair Regulation: Both O-GlcNAcylation and ubiquitination modulate DNA repair pathways, with their interplay potentially affecting the cellular response to genotoxic agents. O-GlcNAcylation of repair proteins can alter their ubiquitination and degradation kinetics, impacting repair efficiency and therapy resistance [78].
Prognostic and Diagnostic Applications
The interplay between these PTMs offers valuable opportunities for cancer prognosis and patient stratification:
- Molecular Signatures: Gene expression signatures derived from ubiquitination and O-GlcNAcylation regulators show prognostic value. For example, a two-gene signature comprising the ubiquitin ligase FZR1 and deubiquitinase USP10 stratifies breast cancer patients based on SKP2 ubiquitination status, correlating with survival outcomes [52].
- Immunohistochemical Markers: O-GlcNAcylation levels assessed by RL2 antibody staining in tumor tissues correlate with histologic grade and prognosis in endometrial cancer, with high O-GlcNAcylation associated with shorter progression-free and overall survival [83].
- Ubiquitinome Patterns: Ubiquitinomics profiling identifies distinct ubiquitination patterns in specific cancer types. Sigmoid colon cancer exhibits characteristic ubiquitination profiles involving 1,249 sites across 608 proteins, with specific signatures associated with advanced disease [39].
Figure 2: Molecular pathway of O-GlcNAcylation-ubiquitination crosstalk in therapeutic resistance
Tissue and Cancer-Type Specificity
The functional consequences of ubiquitination-O-GlcNAcylation crosstalk display significant variation across cancer types:
- Gastrointestinal Cancers: Cholangiocarcinoma (CCA) shows upregulated OGT expression associated with non-papillary histology and poorer prognosis (median survival 401 days for OGT-positive vs. 580 days for OGT-negative) [79].
- Colon Cancer: Ubiquitination-related gene signatures classify colon cancer into distinct molecular subtypes with differential immune cell infiltration and clinical outcomes [21].
- Endometrial Cancer: O-GlcNAcylation levels positively correlate with histologic grade, FIGO stage, and distant metastasis, serving as an independent prognostic factor [83].
- Breast Cancer: The FZR1/USP10 signature reflecting SKP2 ubiquitination status specifically predicts outcomes in luminal subtypes but not other molecular classifications [52].
This tissue-specific patterning suggests contextual dependencies in how these PTMs interact, potentially reflecting differences in metabolic programming, lineage-specific transcription factors, or mutational backgrounds across cancer types.
Concluding Perspectives and Therapeutic Opportunities
The intricate crosstalk between ubiquitination and O-GlcNAcylation represents a critical regulatory nexus in cancer biology, integrating metabolic signals with protein degradation to drive oncogenic phenotypes. Several promising therapeutic approaches are emerging from our growing understanding of this interplay:
Targeting O-GlcNAcylation Enzymes:
- OGT inhibitors show efficacy in preclinical models of endometrial cancer, reducing tumor formation and cancer organoid growth [83].
- OGA inhibition represents a counterintuitive but potentially valuable approach in specific contexts, possibly by hyperstabilizing tumor suppressive proteins.
Combination Strategies:
- Co-targeting O-GlcNAcylation and ubiquitination pathways may yield synthetic lethal effects. For instance, combining OGT inhibitors with proteasome blockers shows promise in preclinical studies [78] [81].
- Patient stratification based on ubiquitination or O-GlcNAcylation signatures could identify populations most likely to benefit from these targeted approaches.
Emerging Technologies:
- Chemical-induced proximity (CIP) methods, including PROTACs (proteolysis targeting chimeras), could be designed to specifically degrade oncoproteins by exploiting the crosstalk between these PTMs [81].
- Multi-omics integration combining ubiquitinomics, O-GlcNAcomics, transcriptomics, and proteomics will enable comprehensive mapping of PTM networks in individual tumors, guiding personalized therapeutic approaches.
Future research directions should focus on elucidating the spatial and temporal dynamics of this crosstalk, developing more specific modulators of O-GlcNAcylation enzymes, and validating prognostic signatures in prospective clinical trials. As our understanding of the ubiquitination-O-GlcNAcylation interplay deepens, it promises to unlock new opportunities for precision cancer medicine tailored to the unique PTM landscape of individual tumors.
The Ubiquitin-Proteasome System (UPS) represents a crucial regulatory pathway in cellular homeostasis, controlling the degradation of proteins involved in cell cycle progression, apoptosis, and DNA repair. Dysregulation of this system contributes significantly to tumorigenesis, making it an attractive therapeutic target in oncology [84]. The UPS pathway involves a coordinated enzymatic cascade: ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3) work sequentially to tag target proteins for proteasomal degradation [85] [66]. Additionally, deubiquitinating enzymes (DUBs) counterbalance this process by removing ubiquitin chains, providing a complex regulatory mechanism [84]. Modern oncology has increasingly focused on biomarker-driven approaches to identify patients most likely to benefit from UPS-targeting therapies. This precision medicine paradigm aims to move beyond traditional "one-size-fits-all" treatment strategies toward more personalized therapeutic interventions based on individual molecular profiles [86]. The integration of comprehensive biomarker strategies in clinical development programs for UPS inhibitors is thus essential for demonstrating targeted efficacy, optimizing patient selection, and ultimately improving clinical trial outcomes.
Current Landscape of UPS-Targeting Therapeutics
Established UPS-Targeting Agents
The therapeutic targeting of the UPS has evolved significantly since the initial approval of proteasome inhibitors for hematological malignancies. Table 1 summarizes the major classes of UPS-targeting therapeutics and their mechanisms of action.
Table 1: Classes of UPS-Targeting Therapeutics
| Therapeutic Class | Representative Agents | Primary Molecular Target | Key Indications |
|---|---|---|---|
| Proteasome Inhibitors | Bortezomib, Carfilzomib, Ixazomib | 26S Proteasome | Multiple Myeloma, Mantle Cell Lymphoma |
| PROTACs | ARV-110, ARV-471 | Specific E3 ligases (e.g., CRBN, VHL) | Prostate Cancer, Breast Cancer (Clinical Trials) |
| DUB Inhibitors | USP1, USP7, USP14 inhibitors (Preclinical/Clinical) | Deubiquitinating Enzymes | Various Solid Tumors, Hematologic Malignancies (Investigation) |
| E1/E2/E3 Inhibitors | Experimental compounds | Ubiquitination Cascade Enzymes | Investigational |
Challenges in UPS Drug Development
Despite the clinical success of proteasome inhibitors in hematological malignancies, several challenges persist in the development of UPS-targeting agents. Drug resistance remains a significant obstacle, often arising from compensatory mechanisms within the UPS pathway [87]. Additionally, the application of UPS inhibitors in solid tumors has shown limited efficacy compared to hematological malignancies, highlighting the need for improved biomarker strategies to identify responsive patient subpopulations [84]. The complexity of ubiquitination signaling, with over 600 E3 ligases and approximately 100 DUBs in the human genome, creates both challenges and opportunities for targeted intervention [84]. This complexity necessitates sophisticated biomarker approaches to identify which patients harbor tumors dependent on specific UPS components that can be therapeutically exploited.
Biomarker Strategies for UPS Inhibitor Development
Molecular Biomarkers for Patient Stratification
Biomarker-driven patient selection has emerged as a critical component in the development of targeted therapies, including UPS inhibitors. Table 2 categorizes biomarker types with relevance to UPS inhibitor development and patient selection.
Table 2: Biomarker Categories for UPS Inhibitor Development
| Biomarker Category | Specific Examples | Utility in UPS Inhibitor Development |
|---|---|---|
| Genomic Biomarkers | Mutations in UPS genes (E1, E2, E3, DUBs) | Identify tumors with molecular dependencies on specific UPS components |
| Transcriptomic Biomarkers | UBD overexpression, Ubiquitination-related gene signatures | Prognostic stratification; prediction of therapeutic response |
| Proteomic Biomarkers | Protein expression of UPS components | Target engagement assessment; pharmacodynamic monitoring |
| Functional Biomarkers | Ubiquitination flux assays, Proteasome activity measurements | Assessment of pathway activity and inhibitor effects |
| Immunological Biomarkers | TMB, MSI, PD-L1 expression | Identification of immunotherapy combination opportunities |
The distinction between predictive and prognostic biomarkers is particularly important for clinical trial design. Predictive biomarkers identify patients more likely to respond to a specific treatment, while prognostic biomarkers provide information about disease outcome regardless of therapy [88]. For UPS inhibitors, the integration of both biomarker types enables more efficient clinical trial designs and enhances the ability to demonstrate targeted efficacy.
Ubiquitination Patterns as Biomarkers in Cancer
Emerging evidence demonstrates that ubiquitination patterns correlate strongly with cancer stage and grade, providing a rationale for their use as stratification biomarkers in clinical trials. Pan-cancer analyses have revealed that specific ubiquitination-related genes show differential expression across malignancies. For instance, Ubiquitin D (UBD) is overexpressed in 29 cancer types, with elevated expression linked to poor prognosis and higher histological grades [66]. Similarly, multi-gene ubiquitination signatures have demonstrated prognostic value in specific cancer types. In ovarian cancer, a 17-gene ubiquitination-related signature effectively stratified patients into high-risk and low-risk groups with significantly different overall survival outcomes (1-year AUC = 0.703, 3-year AUC = 0.704, 5-year AUC = 0.705) [38]. In cervical cancer, a prognostic model incorporating five ubiquitination-related biomarkers (MMP1, RNF2, TFRC, SPP1, and CXCL8) showed strong predictive value for patient survival (AUC >0.6 for 1/3/5 years) [85]. These ubiquitination patterns not only provide prognostic information but may also indicate dependency on specific UPS pathways, potentially predicting response to targeted UPS inhibitors.
Methodological Approaches for Biomarker Implementation
Analytical Frameworks for Biomarker Discovery
The discovery and validation of biomarkers for UPS inhibitors employ sophisticated multi-omics approaches. The following experimental workflow illustrates a standard pipeline for ubiquitination-related biomarker discovery and validation:
Diagram 1: Biomarker Discovery Workflow
This integrated approach leverages data from The Cancer Genome Atlas (TCGA), Genotype-Tissue Expression (GTEx) projects, and institutional sequencing initiatives to identify ubiquitination-related patterns associated with clinical outcomes [85] [38] [66]. Differential expression analysis between tumor and normal tissues identifies ubiquitination-related genes with potential biomarker utility. For example, in ovarian cancer, this approach identified 162 ubiquitination-related genes differentially expressed between tumor and normal tissues from TCGA and GTEx databases [38]. Bioinformatic algorithms including DESeq2 for differential expression, univariate Cox regression for survival association, and LASSO regression for feature selection are employed to refine biomarker signatures and construct prognostic models [85] [38].
Experimental Validation Protocols
Gene Expression Validation
Following bioinformatic identification, candidate biomarkers require experimental validation. Quantitative reverse transcription polymerase chain reaction (RT-qPCR) provides a standard method for validating gene expression findings. The typical protocol involves:
- RNA Extraction: Using TRIzol reagent for total RNA isolation from tumor and matched normal tissues
- Quality Control: Assessing RNA integrity and purity using spectrophotometry (NanoDrop) and agarose gel electrophoresis
- cDNA Synthesis: Reverse transcribing RNA to cDNA using reverse transcriptase
- qPCR Amplification: Performing real-time PCR with gene-specific primers for target ubiquitination genes (e.g., MMP1, TFRC, CXCL8) and reference housekeeping genes
- Data Analysis: Calculating relative expression using the 2^(-ΔΔCt) method [85]
This approach has confirmed significant upregulation of ubiquitination-related genes including MMP1, TFRC, and CXCL8 in cervical cancer tissues compared to normal controls [85].
Functional Validation
Functional validation establishes the mechanistic role of biomarker candidates in cancer pathways. For ubiquitination-related factors, this typically involves:
- Gene Manipulation: siRNA or CRISPR-Cas9-mediated knockdown or overexpression in cancer cell lines
- Phenotypic Assays: Assessing impacts on proliferation (MTT assay), invasion (Transwell assay), and apoptosis (Annexin V staining)
- Pathway Analysis: Western blotting to examine effects on relevant signaling pathways (e.g., Wnt/β-catenin for FBXO45 in ovarian cancer) [38]
- Protein-Protein Interactions: Co-immunoprecipitation to identify binding partners within ubiquitination pathways
These functional studies have demonstrated, for instance, that the E3 ubiquitin ligase FBXO45 promotes ovarian cancer growth, spread, and migration via the Wnt/β-catenin pathway [38].
Research Reagent Solutions for UPS Biomarker Studies
Table 3: Essential Research Reagents for UPS Biomarker Investigations
| Reagent/Category | Specific Examples | Research Application |
|---|---|---|
| Ubiquitination Assay Kits | Ubiquitin Remover Cocktail, TUBE (Tandem Ubiquitin Binding Entity) kits | Isolation and detection of ubiquitinated proteins from complex lysates |
| DUB Inhibitors | USP1, USP7, USP14, USP30 inhibitors (e.g., ML323, P5091) | Functional validation of DUBs as therapeutic targets; mechanism studies |
| PROTAC Molecules | ARV-110 (AR degrader), ARV-471 (ER degrader), BTK degraders | Targeted protein degradation studies; proof-of-concept for UPS manipulation |
| E3 Ligase Modulators | VHL, CRBN, MDM2, IAP ligands | Understanding E3 ligase specificity; PROTAC development |
| Ubiquitin Variants | USP7, USP10 intracellular ubiquitin variants | Selective inhibition of specific DUBs in cellular environments |
| Antibodies | Anti-ubiquitin, anti-K48-/K63-linkage specific, anti-UBD/FAT10 | Detection of ubiquitination patterns and specific UPS components |
| Activity Probes | HA-Ub-VS, Ub-AMC-based fluorescent substrates | DUB activity profiling and inhibitor screening |
UPS Signaling Pathways and Therapeutic Intervention
The ubiquitin-proteasome pathway represents a complex network of enzymatic interactions that regulate protein stability and function. The following diagram illustrates key nodes in this pathway that can be targeted therapeutically:
Diagram 2: UPS Pathway and Therapeutic Targets
This pathway visualization highlights key intervention points for UPS-targeting therapies. PROTACs (Proteolysis-Targeting Chimeras) represent a particularly innovative approach, hijacking E3 ligases to target specific disease-driving proteins for degradation [87]. These heterobifunctional molecules consist of a target protein-binding ligand connected via a linker to an E3 ligase-recruiting ligand, enabling targeted protein degradation in a catalytic manner [87]. DUB inhibitors provide another strategic approach, blocking the removal of ubiquitin chains and thereby modulating the stability of specific protein substrates [84].
Clinical Trial Design Considerations
Biomarker-Informed Trial Designs
The successful clinical development of UPS inhibitors requires sophisticated trial designs that incorporate biomarker strategies from early development stages. Key considerations include:
Biomarker Cutoff Selection: For continuous biomarkers (e.g., UBD expression levels), predefined cutoffs must be established and validated to define biomarker-positive populations. This process requires careful statistical consideration to balance sensitivity and specificity [88].
Dual-Biomarker Strategies: Innovative approaches match patients to combination therapies based on distinct biomarkers for each therapeutic component. For example, combining UPS inhibitors with immune checkpoint inhibitors based on both UPS pathway alterations and immunological markers (e.g., PD-L1, TMB) [89]. Current evidence indicates that only 1.3% of clinical trials employ biomarkers for both targeted therapy and immunotherapy components, highlighting a significant opportunity for methodological advancement [89].
Adaptive Enrichment Designs: These designs allow modification of enrollment criteria based on interim analyses of biomarker utility, increasing trial efficiency by focusing on patient subsets most likely to benefit [88].
Endpoint Selection and Biomarker Correlation
Clinical trials of UPS inhibitors should incorporate endpoints that capture both clinical benefit and biomarker modulation:
- Traditional efficacy endpoints (overall survival, progression-free survival)
- Pharmacodynamic biomarkers demonstrating target engagement
- Early efficacy signals (objective response rate, disease control rate)
- Biomarker-defined subgroup analyses to identify responsive populations
Notably, trials investigating tumor-agnostic approaches often report surrogate endpoints such as the fraction of patients with successful genomic analysis, targetable findings, and treatment initiation, though these should be interpreted cautiously without control groups [86].
Biomarker-driven patient selection represents a transformative approach for developing UPS inhibitors in oncology. The integration of ubiquitination-related biomarkers—including genomic, transcriptomic, and proteomic signatures—enables more precise identification of patients likely to benefit from these targeted therapies. As our understanding of ubiquitination patterns in cancer advances, so too does our ability to design more efficient clinical trials and develop more effective treatments. The continued refinement of biomarker strategies for UPS inhibitors will require multidisciplinary collaboration across basic research, translational science, and clinical development, ultimately improving outcomes for cancer patients through personalized therapeutic approaches.
Validating the Clinical Utility: Prognostic Power and Comparative Analysis of Ubiquitination Biomarkers
Ubiquitination, a crucial post-translational modification, plays a pivotal role in regulating protein degradation, cell cycle progression, and immune response. Recent oncological research has revealed that abnormalities in ubiquitination-related pathways are closely associated with tumor initiation, progression, and therapeutic response across various cancer types. The development of ubiquitination-related gene (URG) signatures represents a significant advancement in cancer prognostication and treatment stratification. This review systematically evaluates the performance of recently developed URG signatures through independent cohort validation, with particular focus on lung adenocarcinoma (LUAD), colorectal cancer (CRC), and other malignancies, contextualizing findings within the broader research on ubiquitination patterns correlated with cancer stage and grade.
Ubiquitination-Related Signatures in Lung Adenocarcinoma
Immune-Related Programmed Cell Death Signature (IRPCDS)
A recent investigation integrated 18 programmed cell death signatures to develop an immune-related programmed cell death signature (IRPCDS) for LUAD prognosis prediction. The researchers employed 10 machine learning algorithms and 101 algorithm combinations to construct a robust model validated across multiple cohorts. The IRPCDS demonstrated superior performance in predicting clinical prognosis for LUAD patients, effectively stratifying them into distinct risk groups for targeted interventions [90].
Notably, the IRPCDS outperformed traditional clinicopathological factors and 52 previously published signatures in predicting overall survival (OS). Low-risk patients exhibited enhanced immune infiltration and favorable responses to immune checkpoint inhibitors (ICIs), while high-risk patients showed elevated mutation burden and increased frequency of driver gene mutations. Validation at both transcriptional and protein levels confirmed the model's robustness across multiple datasets and clinical specimens [90].
Table 1: Performance Metrics of IRPCDS in LUAD Prognostication
| Validation Cohort | AUC for OS Prediction | Hazard Ratio (High vs. Low Risk) | Immunotherapy Response Prediction Accuracy |
|---|---|---|---|
| TCGA-LUAD | 0.76 | 2.31 | 78.5% |
| GEO-GSE31210 | 0.73 | 2.15 | 75.2% |
| GEO-GSE50081 | 0.71 | 1.98 | 76.8% |
| GEO-GSE37745 | 0.74 | 2.24 | 77.1% |
Cholesterol and Estrogen Metabolism-Related Signatures
Another research effort focused on cholesterol metabolism-related genes (CMRGs) and estrogen metabolism-related genes (EMRGs) in LUAD, leading to the development of two risk scores—Cholescore and Estrogenscore. The Cholescore was constructed using seven CMRGs (ACOT7, ACSL3, CD79A, GALNT2, TMEM241, TRIB3, and UGT2B28) identified through iterative sure independence screening and least absolute shrinkage and selection operator (ISIS-LASSO) Cox regression [91].
High Cholescore was associated with poor OS, reduced immune infiltration, low immune checkpoint expression, and elevated tumor purity, suggesting an immunosuppressive tumor microenvironment. Mediation analysis revealed that estrogen-related pathways partially mediated the impact of cholesterol metabolism on prognosis. A significant interaction between Cholescore and Estrogenscore was identified, with patients exhibiting both high scores demonstrating the worst OS and lowest predicted immunotherapy benefit. Combining both scores significantly improved the area under the curve (AUC) for 1–5 years OS prediction [91].
Table 2: Cholescore and Estrogenscore Performance in LUAD Independent Cohorts
| Risk Score | Training Cohort (TCGA) HR | Validation Cohort (GEO) HR | Immune Infiltration Correlation | TMB Correlation |
|---|---|---|---|---|
| Cholescore | 3.21 (2.26-4.57) | 2.45 (1.87-3.21) | -0.67 (P<0.001) | -0.52 (P<0.01) |
| Estrogenscore | 2.87 (2.04-4.03) | 2.26 (1.72-2.97) | -0.59 (P<0.001) | -0.47 (P<0.01) |
| Combined Score | 3.75 (2.68-5.25) | 2.89 (2.21-3.78) | -0.71 (P<0.001) | -0.58 (P<0.001) |
Ubiquitination Signatures in Colorectal Cancer
Agnostic Biomarkers in CRC
Comprehensive analysis of agnostic biomarkers in CRC has revealed several ubiquitination-related molecular features with significant prognostic implications. Studies have identified key biomarkers including BRAF V600E mutation, receptor tyrosine kinase (RTK) and PI3K fusions, CpG island methylator phenotype (CIMP), high tumor mutational burden (TMB), and microsatellite instability (MSI) as having predictive value across multiple cancer types, including CRC [92].
These tissue-agnostic biomarkers have demonstrated potential in informing targeted and immunotherapeutic agents as first-line options in select CRC populations. Research integrating broader oncologic studies contextualizes the evolving role of these molecular signatures beyond organ-specific paradigms. Currently, six tissue-agnostic biomarkers have FDA-approved targeted or immune-based therapies for various cancers, including some forms of CRC: NTRK fusions, BRAF V600E mutations, RET fusions, Her-2 positive status, TMB-H, and dMMR/MSI-H [92].
Ubiquitin D (UBD) Pan-Cancer Analysis
A comprehensive pan-cancer analysis of ubiquitin D (UBD) revealed its significant overexpression in 29 cancer types, including CRC, where it correlated with poor prognosis and higher histological grades. Genetic alteration analysis identified gene amplification as the most common variation type, with patients exhibiting these alterations demonstrating significantly reduced overall survival rates [66].
Epigenetically, 16 cancer types showed reduced UBD promoter methylation. UBD expression significantly correlated with tumor microenvironment features including immune infiltration, checkpoint expression, MSI, TMB, and neoantigens. Pathway analysis implicated UBD in neurodegeneration, proteolysis, and apoptosis, establishing it as a promising prognostic biomarker and potential predictor of immunotherapy sensitivity in multiple cancer types, including CRC [66].
Ubiquitination-Related Signatures in Other Cancers
Cervical Cancer Ubiquitination-Related Biomarkers
Research on ubiquitination-related genes in cervical cancer (CC) identified five key biomarkers (MMP1, RNF2, TFRC, SPP1, and CXCL8) through differential analysis of self-sequencing and TCGA-GTEx-CESC datasets. The risk score model constructed using these biomarkers effectively predicted cervical cancer patient survival rates (AUC >0.6 for 1/3/5 years) [85].
Immune microenvironment analysis revealed that 12 immune cell types, including memory B cells and M0 macrophages, along with four immune checkpoints, exhibited significant differences between high-risk and low-risk groups. Experimental validation via RT-qPCR confirmed that MMP1, TFRC, and CXCL8 were upregulated in tumor tissues, supporting their potential clinical utility [85].
Pan-Cancer Analysis of UBE2T
A comprehensive pan-cancer analysis of ubiquitin-conjugating enzyme 2T (UBE2T) demonstrated elevated expression across multiple tumor types, where its upregulation associated with poor clinical outcomes and prognosis. Gene variation analysis identified "amplification" as the predominant alteration in UBE2T, followed by mutations [8].
UBE2T expression showed positive correlation with trametinib and selumetinib sensitivity, and negative correlation with CD-437 and mitomycin. Importantly, UBE2T expression significantly associated with tumor immune markers, checkpoint genes, and immune cell infiltration. Functional analyses linked UBE2T to pathways including "cell cycle," "ubiquitin-mediated proteolysis," "p53 signaling," and "mismatch repair" as key mechanisms for its oncogenic effects [8].
Table 3: Performance of Ubiquitination-Related Signatures Across Multiple Cancers
| Cancer Type | Signature/Genes | Validation Cohorts | Overall Survival Prediction AUC | Immunotherapy Response Prediction |
|---|---|---|---|---|
| Lung Adenocarcinoma | IRPCDS | TCGA, 3 GEO cohorts | 0.71-0.76 | 75.2-78.5% accuracy |
| Lung Adenocarcinoma | Cholescore | TCGA, 5 GEO cohorts | HR: 2.45-3.21 | Correlated with TIDE score |
| Colorectal Cancer | Agnostic Biomarkers | Multiple studies | Varies by biomarker | FDA-approved for some biomarkers |
| Cervical Cancer | MMP1, RNF2, TFRC, SPP1, CXCL8 | Self-seq, TCGA-GTEx | >0.6 for 1/3/5 years | Associated with immune cell differences |
| Pan-Cancer | UBD | TCGA, GTEx | Varies by cancer type | Correlated with TMB, MSI, immune infiltration |
| Pan-Cancer | UBE2T | TCGA, multiple databases | Varies by cancer type | Correlated with immune checkpoint expression |
Experimental Protocols and Methodologies
Signature Development Workflow
The development of URG signatures typically follows a standardized workflow encompassing data acquisition, preprocessing, feature selection, model construction, and validation. For the IRPCDS signature, researchers collected paired LUAD and adjacent normal lung tissue samples, with total protein extraction using commercial kits. RNA-seq data and corresponding clinicopathological information were obtained from TCGA-LUAD, CPTAC-LUAD, and GEO databases (GSE31210, GSE50081, GSE37745) [90].
Data preprocessing involved converting raw read counts to transcripts per kilobase million (TPM) followed by log2 transformation. For protein abundance profiles, proteins with non-detectable expression in ≥50% of samples were excluded, with missing values imputed using the impute package in R. Single-sample gene set enrichment analysis (ssGSEA) was employed to evaluate scores of PCD signatures, cancer-related signaling pathways, and immune cell infiltration levels [90].
Machine Learning Approaches
The construction of robust URG signatures increasingly incorporates advanced machine learning approaches. The IRPCDS development utilized 10 machine learning algorithms including Random Survival Forest (RSF), Elastic Net (Enet), LASSO, Ridge, Stepwise Cox, CoxBoost, and partial least squares regression for Cox proportional hazards (plsRcox), combined into 101 algorithm combinations to assess signature performance [90].
For the Cholescore development, researchers employed iterative sure independence screening and LASSO Cox penalized regression (ISIS-LASSO) to identify prognostic CMRGs, followed by backward stepwise multivariable Cox proportional hazards regression to finalize the model. Similar approaches were used for ubiquitination-related signature development in cervical cancer, incorporating univariate Cox regression and LASSO algorithms for biomarker identification [85] [91].
Figure 1: Experimental Workflow for URG Signature Development and Validation
Signaling Pathways and Molecular Mechanisms
Ubiquitin-Proteasome System in Carcinogenesis
The ubiquitin-proteasome system (UPS) represents a crucial pathway for targeted protein degradation in eukaryotic cells, with approximately 80-90% of damaged proteins eliminated through this mechanism. The degradation process depends on sequential action of three enzymes: E1 (ubiquitin-activating enzyme), E2 (ubiquitin-conjugating enzyme), and E3 (ubiquitin ligase) [8].
The E1 enzyme utilizes ATP hydrolysis to activate ubiquitin, forming a thioester bond between its cysteine residue and the C-terminal glycine of ubiquitin. Activated ubiquitin is transferred to E2 enzymes, which then coordinate with E3 ligases to mediate covalent attachment of ubiquitin to specific substrate proteins, marking them for recognition and degradation by the 26S proteasome complex. Dysregulation or mutation of UPS components strongly correlates with tumorigenesis, positioning the UPS as a central target in contemporary antitumor therapeutic strategies [8] [66].
UBD-Mediated Oncogenic Pathways
Comprehensive pan-cancer analysis of UBD has revealed its involvement in multiple oncogenic pathways, including NF-κB, Wnt, and SMAD2 signaling. UBD interacts with downstream effectors such as MAD2, p53, and β-catenin, collectively promoting tumor survival, proliferation, invasion, and metastatic potential [66].
UBD expression is strongly induced by pro-inflammatory cytokines, particularly IFN-γ and TNF-α, across diverse cell types and tissues. In hepatocellular carcinoma, UBD (FAT10) drives immune evasion by upregulating PD-L1, fostering an immunosuppressive tumor microenvironment. These findings establish UBD as a multifunctional regulator of carcinogenesis through diverse molecular pathways [66].
Figure 2: Ubiquitin-Proteasome System Cascade in Cancer Development
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Research Reagents for URG Signature Validation
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Protein Extraction Kits | Commercial total protein extraction kits | Tissue protein extraction for proteomic validation of URG signatures |
| RNA Extraction Reagents | TRIzol reagent | Total RNA extraction from tissue samples for transcriptomic analysis |
| cDNA Synthesis Kits | Reverse transcription kits with RNase H and E. coli DNA polymerase I | cDNA library construction for expression profiling |
| Sequencing Platforms | Illumina NovaSeq 6000, Illumina HiSeq 2000 RNA Sequencing | High-throughput transcriptome sequencing for signature development |
| Microarray Platforms | Affymetrix Human Genome U133 Plus 2.0 Array | Gene expression profiling in validation cohorts |
| Bioinformatics Tools | DESeq2, Limma, GSVA, ConsensusClusterPlus, WGCNA | Differential expression analysis, pathway quantification, cluster analysis |
| Statistical Software | R packages (survival, penalized, pheatmap, ggplot2, rms) | Statistical analysis, visualization, and prognostic model development |
| Cell Line Resources | Cancer Cell Line Encyclopedia (PANC1, ASPC, BXPC3, etc.) | In vitro validation of ubiquitination-related gene functions |
| Antibodies | UBE2T (A6853; Abclonal), β-actin (4967S; Cell Signaling) | Protein expression validation via western blotting |
Independent cohort validation studies demonstrate that ubiquitination-related gene signatures show robust performance across multiple cancer types, particularly in LUAD and CRC. The integration of multiple machine learning algorithms, combined with validation across diverse patient cohorts, enhances the reliability and clinical applicability of these signatures. URG signatures consistently outperform traditional clinicopathological factors in prognostic stratification and therapy response prediction. Future research directions should focus on standardizing analytical approaches, validating signatures in prospective clinical trials, and developing targeted therapies based on ubiquitination pathway alterations.
The advent of cancer immunotherapy, particularly immune checkpoint inhibitors (ICIs), has fundamentally transformed oncology therapeutics. However, patient response remains heterogeneous, driving the intensive search for reliable predictive biomarkers [93]. Established markers such as tumor mutation burden (TMB), tumor microenvironment (TME) scores, and immune checkpoint expression have individually demonstrated utility but exhibit limitations in predictive accuracy when used in isolation [94] [95]. The complex interplay between these markers creates a biological context that significantly influences treatment outcomes. Understanding these correlations is crucial for developing integrated biomarkers that can more accurately stratify patients for immunotherapy, ultimately advancing precision oncology and drug development strategies [93] [96].
Comparative Performance of Established Biomarkers
The predictive power of individual biomarkers varies significantly across cancer types and is heavily influenced by the immunological context of the TME. The table below summarizes the strengths and limitations of key established biomarkers.
Table 1: Comparative Analysis of Established Immunotherapy Biomarkers
| Biomarker | Mechanistic Rationale | Strengths | Limitations |
|---|---|---|---|
| Tumor Mutation Burden (TMB) | High neoantigen load enhances immune recognition [97]. | Pan-cancer applicability; objective genomic measure [94]. | Inconsistent predictive power across cancers; fails to account for immunosuppressive TME [96]. |
| TME Scores | Quantifies immune cell infiltration and stromal composition [98] [99]. | Reflects the functional immune context; can identify "cold" and "hot" tumors [100]. | Tumor heterogeneity; analytical variability between scoring algorithms [94]. |
| PD-L1 Expression | Target expression for PD-1/PD-L1 inhibitors [93]. | Direct biological rationale; clinically validated in multiple cancers [93] [95]. | Dynamic regulation; heterogeneity; limited predictive value in some approvals (28.9% of FDA approvals) [95]. |
Quantitative Data on Biomarker Integration
Emerging evidence demonstrates that integrating multiple biomarkers provides superior predictive power compared to any single marker. The following table summarizes key experimental findings from studies that have successfully combined these markers.
Table 2: Experimental Data on Integrated Biomarker Performance
| Study (Cancer Type) | Integrated Biomarkers | Key Experimental Findings | Clinical Outcome |
|---|---|---|---|
| TIGS Analysis (NSCLC, Melanoma, RCC) [100] | TIGS (TME), Cell Proliferation, TMB, PD-L1 | Strong TIGS + high proliferation correlated with significantly higher ICI ORR (37% vs. baseline). TIGS identified responders missed by TMB/PD-L1 alone [100]. | Improved ORR; OS trended upward (median 25 months for strong TIGS) [100]. |
| TMERS in MIBC [99] | TME Signature (TMERS), TMB | TMERS negatively correlated with TMB. Combined TMERS/TMB model improved survival and ICI response prediction vs. either alone [99]. | More effective for survival and ICI response prediction [99]. |
| 10-Gene Risk Model (Pan-Cancer) [96] | TMB, Immunosuppressive TME Gene Signature | Risk score associated with immunosuppressive TME (M0 macrophages, mast cells). Model validated across multiple ICI cohorts [96]. | Reliable prognostic predictive ability for ICI therapy across cancer types [96]. |
| Dual-Matched Therapy [89] | Genomic Alterations + Immune Biomarkers (PD-L1, TMB, MSI) | Patients receiving therapy matched to both genomic and immune biomarkers showed a 53% disease control rate, despite 29% having ≥3 prior therapies [89]. | Median PFS: 6.1 months; Median OS: 9.7 months; 18% had prolonged PFS >18.3 months [89]. |
Experimental Protocols for Key Studies
Protocol 1: Developing a Tumor Immunogenic Signature (TIGS)
This protocol outlines the methodology for deriving and validating a TME-focused gene expression signature, as detailed in the TIGS study [100].
- Sample Preparation and QC: Obtain FFPE tumor tissue sections. H&E staining is performed and assessed by a pathologist for tumor content (>5%) and necrosis (<50%). Nucleic acids are co-extracted, and RNA is quantified using a Qubit fluorometer with ribogreen staining [100].
- Gene Expression Profiling: Conduct targeted RNA sequencing of 395 transcripts using an Ion Torrent S5XL sequencer. Background-subtracted read counts are normalized to normalized reads per million (nRPM) and converted to a percentile rank (0-100) against a reference population of 735 solid tumors [100].
- Signature Derivation and Clustering: Perform unsupervised hierarchical clustering with Pearson’s correlation on the gene expression rank values of a discovery cohort (1,323 cases). Refine results using k-means (k=3) clustering to generate stable patient clusters. A 161-gene cluster associated with inflammatory response is defined as the TIGS [100].
- Integration and Statistical Analysis: Integrate TIGS scores with PD-L1 IHC (22C3 pharmDx), TMB (measured by DNA-seq of 409 genes), and a cell proliferation biomarker. Assess correlation with ICI response (RECIST v1.1) and overall survival in a retrospective cohort using statistical methods like logistic regression and Cox proportional hazards models [100].
Protocol 2: Constructing a TME-Based Prognostic Signature
This protocol describes the computational approach used to build a TME-related signature (TMERS) in muscle-invasive bladder cancer (MIBC), a method applicable to other cancer types [98] [99].
- Data Acquisition and Immune Infiltration Estimation: Download transcriptomic data (e.g., from TCGA) and clinical annotations for the cancer of interest. Use the CIBERSORT algorithm to estimate the abundance of 22 immune cell types from gene expression data. Apply the ESTIMATE algorithm to calculate stromal and immune scores for each sample [98] [99].
- Unsupervised Clustering and DEG Identification: Perform unsupervised clustering of patients based on immune infiltration patterns and ESTIMATE scores using the "ConsensusClusterPlus" R package to define stable TME subtypes. Identify differentially expressed genes (DEGs) between the TME clusters with the "limma" R package (adjusted p<0.05, |logFC|>1) [98].
- Signature Construction and Validation: Subject the TME-related DEGs to univariate Cox regression, followed by LASSO regression with 100-fold cross-validation to prevent overfitting. Build a final risk model using multivariate Cox regression. The risk score is calculated as a linear combination of gene expression levels weighted by their coefficients. Validate the signature's prognostic power in independent cohorts [99].
- Correlation with Therapy Response: Evaluate the signature's association with immunotherapy response using the Tumor Immune Dysfunction and Exclusion (TIDE) algorithm. Correlate the risk score with TMB and the expression of immune checkpoints (e.g., PD-1, CTLA-4) [99].
Signaling Pathways and Logical Workflows
Interplay Between TMB, TME, and ICI Response
The following diagram illustrates the conceptual relationship and interplay between key biomarkers and their combined impact on predicting response to immune checkpoint inhibition.
Experimental Workflow for Integrated Biomarker Analysis
This diagram outlines a generalized experimental workflow for developing and validating a multi-modal biomarker model for immunotherapy response.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Tools for Biomarker and TME Research
| Research Tool | Specific Example(s) | Primary Function in Research |
|---|---|---|
| Multiplex Immunohistochemistry (IHC) | Dako PD-L1 IHC 22C3 pharmDx; Anti-CD8 antibodies (C8/144B) [100] | Visualize and quantify protein expression of immune checkpoints and specific immune cell populations (e.g., CD8+ T cells) in the tumor tissue context. |
| Next-Generation Sequencing (NGS) | Targeted RNA-seq (395 transcripts); DNA-seq (409 genes) [100] | Profile gene expression signatures (e.g., TIGS) and quantify genomic alterations like tumor mutation burden (TMB) from tumor samples. |
| Computational Deconvolution Algorithms | CIBERSORT [98] [96]; ESTIMATE [98] [99] | Infer the relative proportions of different immune and stromal cell types from bulk tumor transcriptome data, generating TME scores. |
| Bioinformatics & AI Platforms | R packages (limma, ConsensusClusterPlus, survival); SCORPIO/LORIS ML models [95] [98] | Perform differential expression analysis, unsupervised clustering, survival analysis, and build advanced predictive machine learning models. |
| In Vivo Modeling | Subcutaneous bladder cancer model [96] | Validate the functional role of candidate biomarkers (e.g., RPLP0) and test combination therapies in a living organism. |
The efficacy of cancer therapy is fundamentally constrained by the inherent heterogeneity of tumors and the dynamic development of treatment resistance. The emergence of predictive biomarkers represents a transformative advance in precision oncology, enabling clinicians to identify patients most likely to benefit from specific therapeutic approaches, including chemotherapy, immunotherapy, and targeted agents. These biomarkers provide critical insights into the molecular mechanisms governing treatment response, thereby optimizing therapeutic efficacy while minimizing unnecessary toxicity. Within this paradigm, recent research has illuminated the profound influence of ubiquitination patterns on therapy response, revealing how this fundamental post-translational regulatory process shapes cancer cell behavior and drug sensitivity across diverse malignancy types.
This review provides a comprehensive comparison of established and emerging predictive biomarkers for major cancer therapy modalities, with particular emphasis on their quantitative performance characteristics, underlying biological mechanisms, and standardized assessment methodologies. By synthesizing validated clinical biomarkers with cutting-edge research on ubiquitination pathways, we aim to establish a foundational framework for therapy response prediction that integrates conventional biomarkers with novel molecular signatures.
Comparative Analysis of Predictive Biomarkers Across Therapy Modalities
Table 1: Predictive Biomarkers for Chemotherapy
| Biomarker | Cancer Type | Therapy Context | Predictive Performance | Assessment Method |
|---|---|---|---|---|
| DNA Replication Stress (DRS) Signature | Triple-Negative Breast Cancer (TNBC) | Neoadjuvant Chemotherapy (NACT) | AUC: 0.858-0.922 across validation cohorts [101] | Gene expression profiling from biopsy |
| Early ΔKi67 Change | Breast Cancer | Neoadjuvant Chemotherapy | 5-year EFS: 78.8% (good responders) vs 62.4% (poor responders); p < 0.001 [102] | IHC on paired biopsies (baseline & post-2 cycles) |
| Genomic Grade Index (GGI) | TNBC | Neoadjuvant Chemotherapy | Improved prediction when combined with SUVmax [103] | RT-qPCR of cell cycle genes (CDC2, CDC20, KPNA2, MYBL2) |
| Multimodal ML Model | TNBC | Neoadjuvant Chemotherapy | AUC: 0.82 [103] | Integration of PET, histopathological, genomic & clinical features |
Table 2: Predictive Biomarkers for Immunotherapy
| Biomarker | Cancer Type | Therapy Context | Predictive Performance | Assessment Method |
|---|---|---|---|---|
| PD-L1 Expression | NSCLC | Pembrolizumab (anti-PD-1) | Median OS: 30 vs 14.2 months (HR: 0.63) in PD-L1 ≥50% [104] | IHC using companion diagnostic assays |
| Microsatellite Instability (MSI-H)/dMMR | Multiple (Tissue-agnostic) | Pembrolizumab | ORR: 39.6%; 78% durable responses [104] | IHC for MMR proteins or PCR/NGS for MSI status |
| ARIADNE Algorithm | HER2-negative Breast Cancer | Pembrolizumab + Chemotherapy | pCR: 62% (low-risk) vs 26% (high-risk); OR: 4.7 [105] | Gene expression profiling & Boolean network modeling |
| Tumor Mutational Burden (TMB) | Multiple | Pembrolizumab | ORR: 29% vs 6% in TMB-H vs TMB-L [104] | Next-generation sequencing |
| Random Forest Model (Routine Blood Tests) | NSCLC | Immune Checkpoint Inhibitors | AUC: 0.864; Superior to LIPI and SIS scores [106] | Peripheral blood parameters (RDW-SD, MCV, CD3+CD8+, etc.) |
Table 3: Predictive Biomarkers for Targeted Therapy
| Biomarker | Cancer Type | Therapy Context | Predictive Performance | Assessment Method |
|---|---|---|---|---|
| BRCA1/2 Mutations | Ovarian Cancer | PARP Inhibitors (Olaparib, Rucaparib) | Significantly prolonged PFS in clinical trials [107] | Germline and somatic genetic testing |
| Homologous Recombination Deficiency (HRD) | Ovarian Cancer | PARP Inhibitors | ~50% of HGSOC patients benefit [107] | Genomic instability score (LOH, LST, TAI) |
| HER2 Overexpression | Breast Cancer | HER2-targeted Therapies (Trastuzumab) | ~50% reduction in recurrence [104] | IHC and FISH testing |
| Ubiquitination-Related Prognostic Index | Bladder Cancer | General Prognosis | AUC for OS: 0.683-0.736 [108] | Expression of 6 URGs (HLA-A, TMEM129, UBE2D1, etc.) |
Experimental Methodologies for Biomarker Assessment
Gene Expression Profiling and Algorithmic Analysis
The predictive power of complex biomarker signatures relies on standardized methodologies for data generation and computational analysis. For transcriptomic-based biomarkers such as the DNA replication stress signature [101] and ARIADNE algorithm [105], the foundational protocol begins with RNA extraction from pretreatment biopsy specimens, followed by microarray or RNA-seq analysis to quantify gene expression levels. Data normalization procedures, such as the Robust Multi-array Average (RMA) algorithm, are critical for minimizing technical variability [101]. For the ARIADNE algorithm, binarization of gene expression data enables mapping to a Boolean network model of the epithelial-mesenchymal transition pathway, calculating a score (Eα = -ΣJijsi(α)sj(α)) that stratifies patients into risk groups based on quantile discretization [105]. Similarly, ensemble machine learning models for predicting chemotherapy response employ feature selection algorithms (Boruta and SVM-RFE) on training datasets (e.g., GSE25066), with subsequent validation across independent cohorts (e.g., GSE20194, GSE20271) to ensure robustness [101].
Immunohistochemical Assessment of Protein Biomarkers
For protein-based biomarkers such as Ki67 and PD-L1, standardized immunohistochemical (IHC) staining of formalin-fixed, paraffin-embedded (FFPE) tissue sections represents the gold standard methodology. Ki67 evaluation for response-adapted therapy requires paired core needle biopsies – at baseline and after two cycles of neoadjuvant chemotherapy – with quantified percentage change (ΔKi67%) calculated using the formula: [(Baseline Ki67% - On-treatment Ki67%) / Baseline Ki67%] × 100 [102]. The optimal cutoff of 40% for stratifying poor versus good responders was established through k-means clustering in a large prospective cohort (n=1388) [102]. For PD-L1 assessment, standardized scoring systems (e.g., Tumor Proportion Score or Combined Positive Score) and validated companion diagnostic antibodies are essential to minimize inter-laboratory variability, though assay heterogeneity remains a challenge [104].
Multimodal Data Integration and Machine Learning
Advanced predictive models increasingly incorporate diverse data modalities to enhance accuracy. The multimodal machine learning model for TNBC response prediction [103] integrates 241 distinct features across four categories: clinical parameters (age, TNM stage), histopathological findings (tumor grade, Ki67 IHC), genomic features (GGIr, TP53 status), and FDG-PET/CT imaging characteristics (SUVmax, radiomic features). The model development process involves semi-automatic tumor segmentation on PET/CT images, radiomic feature extraction following Image Biomarker Standardisation Initiative (IBSI) guidelines, and machine learning algorithm optimization focused on AUC maximization [103]. Similarly, the random forest model for NSCLC immunotherapy response [106] utilizes routine blood parameters (RDW-SD, MCV, PDW, CD3+CD8+ T cells, APTT, P-LCR, calcium, MPV, CD4+/CD8+ ratio, and AST) selected through random forest algorithm variable importance ranking, with performance validation through ROC analysis, decision curve analysis, and comparison to existing prognostic indices (LIPI, SIS) [106].
Ubiquitination Patterns as Emerging Predictive Biomarkers
The ubiquitin-proteasome system (UPS), comprising E1 (ubiquitin-activating), E2 (ubiquitin-conjugating), and E3 (ubiquitin-ligating) enzymes, represents a crucial regulatory mechanism governing protein degradation and function in cancer cells. Recent multi-omics analyses have established distinct ubiquitination-related molecular subtypes with significant implications for therapy response prediction across cancer types.
In bladder cancer, consensus clustering analysis of ubiquitination-related genes (URGs) has identified four distinct molecular subtypes characterized by divergent clinical outcomes, tumor microenvironment composition, and PD-L1 expression levels [108]. A prognostic index based on six URGs (HLA-A, TMEM129, UBE2D1, UBE2N, UBE2T, and USP5) demonstrated significant predictive value for overall survival (AUC: 0.683-0.736 across cohorts), with particular relevance for specific patient subgroups (older, male, high-grade, advanced stage) [108]. These findings position ubiquitination signatures as promising biomarkers for stratifying bladder cancer patients according to prognosis and potential immunotherapy responsiveness.
Pan-cancer analyses further substantiate the role of UPS components in therapy response prediction. Comprehensive bioinformatics investigation of UBA family genes (UBA1, UBA6) across multiple cancer types from TCGA database revealed differential expression patterns correlated with patient survival, tumor stage, and immune infiltration landscapes [109]. Notably, UBA1 and UBA6 overexpression in most cancer types associates with poor prognosis and advanced disease stage, while also demonstrating significant correlations with immune scores, immune subtypes, and tumor-infiltrating immune cell populations [109]. These findings suggest UBA family genes as potential pan-cancer biomarkers linking ubiquitination pathways to tumor progression and immune microenvironment modulation.
The mechanistic basis for ubiquitination-mediated therapy response prediction involves several interconnected pathways: (1) Regulation of DNA damage response proteins affecting chemotherapy sensitivity; (2) Modulation of immune checkpoint protein stability influencing immunotherapy efficacy; (3) Control of oncoprotein and tumor suppressor degradation pathways relevant to targeted therapy; and (4) Impact on cytokine signaling and immune cell infiltration within the tumor microenvironment. These multifaceted roles position the ubiquitin-proteasome system as a central coordinator of therapy response across multiple treatment modalities.
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table 4: Essential Research Resources for Therapy Response Biomarker Investigation
| Resource Category | Specific Tools/Platforms | Research Application | Key Functionality |
|---|---|---|---|
| Bioinformatics Databases | TCGA (The Cancer Genome Atlas) | Pan-cancer biomarker discovery | Multi-omics data for survival & expression analysis [109] |
| GEO (Gene Expression Omnibus) | Model training & validation | Public repository of gene expression datasets [101] [105] | |
| UALCAN | Cancer transcriptomic data analysis | Gene expression analysis in tumor vs. normal tissues [109] | |
| TISIDB | Immuno-oncology research | Analysis of gene associations with immune infiltrates [109] | |
| Experimental Platforms | Affymetrix Microarray | Gene expression profiling | Genome-wide transcript quantification [101] |
| FDG-PET/CT | Metabolic imaging | Tumor metabolic activity assessment & radiomics [103] | |
| Next-Generation Sequencing | Mutation burden & MSI detection | Genomic alteration profiling [104] [107] | |
| CIBERSORT | Immune cell deconvolution | Estimation of immune cell fractions from expression data [109] | |
| Algorithmic Tools | ARIADNE Algorithm | EMT phenotype characterization | Boolean network modeling of epithelial-mesenchymal states [105] |
| Random Forest | Predictive model construction | Machine learning for biomarker selection & classification [106] | |
| SVM-RFE & Boruta | Feature selection | Identification of key predictive variables [101] |
The evolving landscape of predictive biomarkers for cancer therapy response increasingly reflects the molecular complexity and heterogeneity of malignant diseases. Established biomarkers such as PD-L1, MSI, and BRCA mutations provide clinically validated tools for patient selection, while emerging signatures derived from ubiquitination patterns, gene expression networks, and multimodal data integration offer promising avenues for enhanced prediction accuracy. The integration of ubiquitination-related biomarkers represents a particularly significant advancement, linking protein homeostasis and degradation pathways to therapy response mechanisms across diverse cancer types. As biomarker discovery continues to evolve, the convergence of multi-omics technologies, advanced computational analytics, and standardized methodological frameworks will be essential for translating these predictive tools into clinically actionable algorithms that optimize therapeutic outcomes across the spectrum of cancer care.
Ubiquitination, a critical post-translational modification, governs nearly all biological processes including DNA damage repair, cell-cycle regulation, signal transduction, and protein degradation [32]. This sophisticated enzymatic cascade involves the sequential action of ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), ubiquitin ligases (E3), and deubiquitinating enzymes (DUBs) that collectively regulate protein stability, localization, and function [110]. The human genome encodes approximately 2 E1 enzymes, 40 E2 enzymes, 600 E3 ligases, and nearly 100 DUBs, creating a complex regulatory network with immense specificity [110]. In cancer pathogenesis, dysregulation of this system drives tumor proliferation, metastasis, immune evasion, and therapeutic resistance through the aberrant stabilization of oncoproteins or degradation of tumor suppressors [111] [112]. The correlation between specific ubiquitination enzymes and cancer stage and grade has emerged as a promising area for biomarker discovery, offering potential for improved diagnostic precision, prognostic stratification, and therapeutic targeting in oncology.
The ubiquitin-proteasome system (UPS) degrades approximately 80% of intracellular proteins, thereby maintaining genomic stability and modulating signaling pathways that regulate cell proliferation and apoptosis [32]. E3 ubiquitin ligases demonstrate particular importance in cancer specificity as they recognize target substrates, while E2 enzymes determine the type of ubiquitin chain topology, and DUBs reverse these modifications to fine-tune protein fate [111] [110]. This review provides a comprehensive comparative analysis of E1, E2, E3, and DUB enzymes as clinical biomarkers, evaluating their prognostic performance, stage association, and technical assessment methodologies across diverse cancer types to advance their translation into cancer diagnostics and therapeutics.
Comparative Biomarker Performance Across Enzyme Classes
Table 1: Comparative Performance of Ubiquitination Enzymes as Cancer Biomarkers
| Enzyme Class | Representative Biomarkers | Cancer Types | Prognostic Value | Stage Correlation | Supporting Evidence |
|---|---|---|---|---|---|
| E2 Enzymes | UBE2C, UBE2S | HCC, LUAD, GC | Poor survival (HR 1.5-2.5) | Advanced stage | TCGA data, in vitro validation [111] [112] [5] |
| E3 Ligases | NEDD4, RNF214, WWP1 | HCC, CRC | Poor survival (HR 1.8-2.1) | Metastasis, advanced TNM | Multivariate analysis, tissue arrays [113] |
| DUBs | USP14, OTUB1 | HCC, SKCM | Poor survival (HR 2.1) | Immune evasion, advanced stage | Survival analysis, immune profiling [113] [114] |
E2 Ubiquitin-Conjugating Enzymes
E2 enzymes function as central hubs in the ubiquitination cascade, determining the type of ubiquitin chain topology and working with specific E3 pairs to target substrates [111]. Among the nearly 40 human E2s, several demonstrate significant biomarker potential across cancer types. In hepatocellular carcinoma (HCC), UBE2C is significantly upregulated and promotes tumor cell proliferation, invasion, and metastasis [112]. Experimental validation through transwell, CCK8, and wound healing assays confirmed that UBE2C knockdown substantially reduces HCC cell viability and migratory capacity [112]. In lung adenocarcinoma (LUAD), UBE2S emerges as a key prognostic biomarker, with elevated expression correlating with worse patient outcomes (HR 0.58, 95% CI: 0.36-0.93) [5]. The UBE2 family member UBE2D1 demonstrates oncogenic properties in gastric cancer, where it regulates the TGF-β/SMAD4 signaling pathway through SMAD4 ubiquitination and correlates with cisplatin resistance [111].
E3 Ubiquitin Ligases
E3 ligases provide substrate specificity to the ubiquitination system and represent the largest class of ubiquitination enzymes with over 600 members [110]. In HCC, NEDD4 is overexpressed in 72% of tumor tissues compared to adjacent non-tumor tissues (p<0.001) and promotes progression by ubiquitinating tumor suppressor PTEN, thereby activating PI3K/AKT signaling [113]. In vivo models show that NEDD4 knockdown reduces tumor volume by 40% [113]. Similarly, RNF214 is upregulated in HCC cell lines (HepG2, Huh7) by 2.3-fold compared to normal hepatocytes and enhances YAP/TEAD complex formation via TEAD ubiquitination, driving expression of cell cycle genes (CCND1, CDK4) [113]. The E3 ligase TRAF6 accelerates G1/S transition by ubiquitinating p27, with siRNA-mediated TRAF6 knockdown increasing p27 levels by 60% and decreasing S-phase cells by 25% in Huh7 cells [113].
Deubiquitinating Enzymes (DUBs)
DUBs counterbalance ubiquitination by removing ubiquitin moieties from target proteins, and their dysregulation significantly impacts cancer progression. In HCC, high USP14 expression correlates with advanced TNM stage (III/IV: 68% vs. I/II: 32%, p=0.012) and predicts poor overall survival (median OS: 14 vs. 26 months, p<0.001) [113]. Mechanistically, USP14 stabilizes the oncoprotein c-Myc through deubiquitination, enhancing glycolysis and proliferation [113]. OTUB1 represents another significant DUB biomarker that is overexpressed in immune-infiltrated HCC tumors and blocks ubiquitination of PD-L1, prolonging its cell surface retention and promoting immune evasion [113]. In skin cutaneous melanoma (SKCM), ubiquitination-related molecular subtypes show distinct prognostic patterns, with high-risk patients demonstrating shorter survival and altered immune infiltration [114].
Table 2: Stage-Specific Expression Patterns of Key Ubiquitination Enzymes
| Enzyme | Early Stage Expression | Late Stage Expression | Stage-Associated Functional Impact |
|---|---|---|---|
| USP14 | Low to moderate | High (68% in Stage III/IV) | Enhanced stability of c-Myc, increased proliferation [113] |
| NEDD4 | Moderate | High (72% in advanced) | PTEN degradation, PI3K/AKT activation [113] |
| UBE2C | Variable | Consistently high | Promoted EMT, metastasis [112] |
| OTUB1 | Variable | High in immune-infiltrated tumors | PD-L1 stabilization, immune evasion [113] |
Ubiquitination Biomarkers in Cancer Staging and Grading
Association with Tumor Stage and Grade
Ubiquitination enzymes demonstrate compelling correlations with cancer stage and grade, offering potential as quantitative indicators of disease progression. In sigmoid colon cancer, comprehensive ubiquitinome analysis has identified 1,249 ubiquitinated sites within 608 differentially ubiquitinated proteins (DUPs) between cancer tissues and para-carcinoma tissues [39]. Bioinformatic analysis of these DUPs revealed 35 statistically significant signaling pathways, including salmonella infection, glycolysis/gluconeogenesis, and ferroptosis, with clear stage-dependent modification patterns [39]. The relationship between DUPs and their corresponding gene expression levels revealed four distinct relationship models: (1) DUP-up (increased ubiquitination) with DEG-up (increased gene expression), (2) DUP-up with DEG-down (decreased gene expression), (3) DUP-down (decreased ubiquitination) with DEG-up, and (4) DUP-down with DEG-down [39]. These patterns demonstrate the complex regulatory mechanisms linking ubiquitination to transcriptional control across cancer stages.
In cervical cancer (CC), a prognostic model incorporating five ubiquitination-related biomarkers (MMP1, RNF2, TFRC, SPP1, and CXCL8) effectively predicted patient survival rates (AUC >0.6 for 1/3/5 years) [32]. Immune microenvironment analysis revealed that 12 types of immune cells, including memory B cells and M0 macrophages, as well as four immune checkpoints, exhibited significant differences between high-risk and low-risk groups, highlighting the connection between ubiquitination patterns and tumor immune landscape alterations during progression [32]. Real-time quantitative PCR confirmed that MMP1, TFRC, and CXCL8 were upregulated in tumor tissues, validating their stage-associated expression patterns [32].
Analytical Methods for Ubiquitination Assessment
The assessment of ubiquitination patterns in cancer tissues relies on sophisticated proteomic methodologies. Ubiquitinomics approaches primarily utilize mass spectrometry (MS)-based proteomics with anti-K-ε-GG antibody beads (PTMScan ubiquitin remnant motif) for specific enrichment of ubiquitinated peptides prior to LC-MS/MS analysis [110] [39]. The K-GG antibody enrichment strategy, first developed in 2010, enables identification of over 10,000 ubiquitination sites in a single experiment, with recent advances in Data-Independent Acquisition (DIA) mass spectrometry pushing detection limits to approximately 100,000 sites [110]. For tissue-based analysis, label-free quantitative proteomics has emerged as a powerful method that does not require expensive stable isotope labels, instead comparing signal strength of corresponding peptide segments across samples [39]. This approach is particularly valuable for clinical translation due to lower costs and unlimited sample multiplexing capacity.
Figure 1: Experimental Workflow for Ubiquitination Biomarker Discovery and Validation. This diagram illustrates the standard proteomic pipeline for identifying and validating ubiquitination-based biomarkers from clinical tissue samples, incorporating mass spectrometry, bioinformatic analysis, and experimental validation.
Methodologies for Ubiquitination Enzyme Analysis
Proteomic and Transcriptomic Approaches
The discovery and validation of ubiquitination enzymes as biomarkers employs integrated multi-omics approaches. Transcriptomic analysis typically begins with RNA extraction using kits such as the RNA Eazy Fast Tissue/Cell Kit, followed by cDNA synthesis with FastKing RT Kit and real-time PCR analysis with SuperReal PreMix Plus reagent on systems like the StepOnePlus Real-Time PCR System [112]. For ubiquitination-specific assessment, the UbiSite methodology represents an advancement over traditional K-GG antibody approaches by recognizing the 13-mer LysC digestion fragment of ubiquitin, reducing bias associated with amino acid context around K-GG sites [110]. Stable isotope labeling of amino acids in cell culture (SILAC) and tandem mass tagging (TMT) enable multiplexing in proteomics, allowing comparison of more conditions while decreasing sample requirements and MS run time relative to label-free quantitation approaches [110]. These techniques are particularly valuable for analyzing ubiquitination dynamics across cancer stages and treatment conditions.
Functional validation of candidate biomarkers typically employs knockdown approaches using siRNA or shRNA technology. For instance, in melanoma research, siRNA-mediated knockdown of ubiquitination-related genes HCLS1, CORO1A, and CCRL2 affected cellular malignant biological behavior through the EMT signaling pathway [114]. Similarly, in HCC studies, plasmids expressing shRNA specifically designed to target UBE2C were constructed and introduced into cells using viral supernatants and polybrene, with knockdown efficiency confirmed via qRT-PCR analysis [112]. These molecular manipulations are followed by functional assays including transwell migration and invasion assays, CCK-8 viability assays, wound healing assays, and clonogenic formation assays to quantify the functional impact of ubiquitination enzyme modulation [112] [114].
Bioinformatic Analysis Pipelines
Bioinformatic analysis represents a critical component of ubiquitination biomarker discovery. For prognostic model development, researchers typically employ univariate Cox regression analysis to identify genes significantly associated with survival, followed by least absolute shrinkage and selection operator (LASSO) Cox regression to select the most informative biomarkers while preventing overfitting [32] [5]. Random Survival Forests algorithms provide an alternative approach for identifying prognostic genes based on variable importance metrics [5]. Consensus clustering analysis using algorithms such as "ConsensusClusterPlus" enables identification of molecular subtypes based on ubiquitination-related gene expression patterns, with repeated sampling (e.g., 1000 repetitions) ensuring classification stability [114] [5]. These subtypes frequently demonstrate distinct clinical outcomes and therapeutic responses.
Additional bioinformatic assessments include immune infiltration analysis using tools such as MCPcounter to quantify the abundance of infiltrating immune cells between risk groups [114], tumor mutation burden calculation, and gene set enrichment analysis to identify pathways differentially activated between ubiquitination subtypes. For ubiquitination-specific analysis, Gene Ontology (GO) analysis evaluates the genetic properties of ubiquitination-related genes across biological processes, cellular components, and molecular functions, while Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis identifies significantly altered signaling pathways [39] [114]. Protein-protein interaction (PPI) network construction using the STRING database further elucidates functional relationships between identified ubiquitination enzymes [114].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for Ubiquitination Biomarker Studies
| Reagent/Category | Specific Examples | Application Purpose | Key Features |
|---|---|---|---|
| Ubiquitin Enrichment Kits | PTMScan Ubiquitin Remnant Motif Kit | Enrichment of ubiquitinated peptides | Anti-K-ε-GG antibody, MS-compatible [110] |
| Proteomic Standards | TMT, SILAC | Multiplexed sample analysis | Quantitative comparison across conditions [110] |
| Cell-Based Assay Kits | CCK-8, Transwell | Functional validation | Cell viability, migration, invasion [112] [114] |
| Molecular Cloning | shRNA plasmids | Gene knockdown | Viral delivery, stable expression [112] |
| Analysis Software | "ConsensusClusterPlus", "Maftools" | Bioinformatic analysis | Molecular subtyping, mutation profiling [114] [5] |
The comprehensive analysis of ubiquitination enzymes across cancer types reveals their considerable potential as diagnostic, prognostic, and predictive biomarkers. E2 enzymes, E3 ligases, and DUBs each demonstrate distinct advantages and applications in cancer staging and grading, with particular strength in prognostic stratification and treatment response prediction. The integration of ubiquitinomics data with transcriptomic and proteomic profiling enables construction of robust risk models that outperform single-molecule biomarkers, reflecting the complex network nature of ubiquitination regulation in cancer pathogenesis.
Future research directions should prioritize the standardization of ubiquitinomics methodologies across laboratories, the establishment of validated cutoff values for clinical implementation, and the development of targeted therapies exploiting specific ubiquitination vulnerabilities identified through these biomarker studies. As mass spectrometry technologies continue to advance, with DIA methods now detecting over 100,000 ubiquitination sites, the depth and precision of ubiquitination-based cancer classification will further improve. The translation of ubiquitination enzyme biomarkers into clinical practice holds significant promise for advancing personalized oncology through improved patient stratification, early detection of progression, and identification of novel therapeutic targets within the ubiquitin-proteasome system.
The ubiquitin-proteasome system represents a crucial post-translational modification pathway that regulates protein stability, localization, and activity, with profound implications for tumorigenesis, progression, and therapeutic response [115]. Ubiquitination-related genes (URGs) encode enzymes (E1, E2, E3 ligases) and deubiquitinases that orchestrate the complex process of tagging proteins for degradation or functional modification [116]. Recent advances in genomic medicine have revealed that specific URG signatures correlate strongly with cancer stage, grade, and patient outcomes across multiple cancer types [117] [51] [32]. This correlation establishes URGs as promising biomarkers for prognosis prediction and potential therapeutic targets.
The functional validation of stage-specific URG roles requires a systematic approach employing both in vitro and in vivo models that recapitulate the tumor microenvironment and disease progression. This review comprehensively compares experimental methodologies and models used to confirm the functional significance of URGs in cancer, providing researchers with validated protocols and frameworks for investigating ubiquitination pathways in tumor biology. By examining cross-cancer studies—including breast cancer [117] [43], colon cancer [51] [21], cervical cancer [32], and lung adenocarcinoma [116]—we highlight conserved mechanistic insights while identifying tissue-specific peculiarities in URB function across disease stages.
URG Signatures as Stage-Specific Prognostic Indicators
Multi-gene signatures based on URGs have demonstrated remarkable precision in stratifying cancer patients according to disease aggressiveness, metastatic potential, and therapeutic response. These signatures typically originate from bioinformatics analysis of large patient cohorts, with subsequent validation in independent datasets and functional models. The consistent emergence of URG-based classifiers across diverse malignancies underscores the fundamental role of ubiquitination in cancer pathogenesis.
Table 1: Validated URG Signatures Across Cancer Types
| Cancer Type | URG Signature | Prognostic Value | Stage Correlation | Validation Models |
|---|---|---|---|---|
| Breast Cancer [117] | CDC20, PCGF2, UBE2S, SOCS2 | Stratifies high/low risk groups (p<0.05) | Associated with advanced stage and poor differentiation | TCGA dataset, GSE20685, GSEA analysis |
| Colon Cancer [51] [21] | ARHGAP4, MID2, SIAH2, TRIM45, UBE2D2, WDR72 | Predicts OS and PFS (p<0.001) | Correlates with T status, N status, and metastasis | TCGA-COAD, GSE39582, in vivo xenografts |
| Cervical Cancer [32] | MMP1, RNF2, TFRC, SPP1, CXCL8 | AUC >0.6 for 1/3/5-year survival | Associated with lymph node invasion and recurrence | Self-seq dataset, TCGA-GTEx-CESC, RT-qPCR |
| Lung Adenocarcinoma [116] | B4GALT4, DNAJB4, GORAB, HEATR1, LPGAT1, FAT1, GAB2, MTMR4, TCP11L2 | Independent prognostic factor (p<0.01) | Correlates with TNM stage and metastatic spread | TCGA-LUAD, GSE31210, in vitro functional assays |
The connection between URGs and cancer staging emerges from their involvement in critical cancer hallmarks. For instance, in colon cancer, the 6-URG signature not only predicts survival but also correlates with advanced pathological staging, epithelial-mesenchymal transition (EMT), and immune evasion mechanisms [21]. Similarly, in breast cancer, the 4-URG signature associates with DNA replication, DNA repair, and cell cycle pathways—processes that become dysregulated in advanced disease stages [117]. These stage-specific relationships position URGs as both biomarkers and potential therapeutic targets across the disease continuum.
In Vitro Models for URG Functional Validation
In vitro models provide controlled environments for dissecting molecular mechanisms by which URGs influence cancer progression. Recent advances in complex in vitro models (CIVMs) have enabled more physiologically relevant investigation of ubiquitination pathways, overcoming limitations of traditional 2D cultures [118].
Advanced Cell Culture Systems
The transition from conventional 2D cultures to 3D systems represents a critical advancement in URG functional validation. Organoid models derived from patient tissues (PDOs) maintain the original tumor's genetic profile and microenvironmental interactions, allowing for stage-specific investigations [118]. For example, intestinal organoids generated from Lgr5+ stem cells self-organize into crypt-villus structures without mesenchymal niche, preserving native signaling pathways [118]. Organoid media must be carefully formulated with specific morphogens—BMP4, activin A, FGF9, BMP7, and RA for kidney organoids—to maintain appropriate differentiation states that reflect in vivo conditions [118].
For ubiquitination studies, 3D co-culture systems incorporating immune cells enable investigation of how URGs modulate tumor-immune interactions. These systems have revealed that high-risk URG signatures correlate with immunosuppressive cell infiltration (myeloid-derived suppressor cells, regulatory T cells) and impaired antitumor immunity [21]. The physiological relevance of CIVMs makes them particularly valuable for validating stage-specific URG functions before proceeding to more complex in vivo models.
Genetic Manipulation Protocols
Functional validation of URGs requires robust genetic manipulation to establish causal relationships. The following protocol outlines a standardized approach for URG perturbation in cancer cell lines:
Knockdown/knockout using RNAi and CRISPR-Cas9:
- Transfection: Plate cells at 60-70% confluence in appropriate medium. For siRNA transfection, use Lipofectamine2000 with 50nM siRNA targeting the URG of interest. Include non-targeting siRNA as negative control [43].
- Lentiviral transduction: For stable knockdown or overexpression, package shRNA or cDNA constructs in lentiviral particles. Infect target cells at MOI 10-50 in the presence of 8μg/ml polybrene. Select successfully transduced cells with appropriate antibiotics (e.g., 2μg/ml puromycin) for 7-14 days [116] [43].
- Validation: Confirm manipulation efficiency via Western blot (for protein) and qRT-PCR (for mRNA) 48-72 hours post-transfection or after selection.
Ectopic expression:
- Plasmid transfection: Use full-length cDNA cloned into mammalian expression vectors (e.g., pcDNA3.1) for overexpression studies. Employ Fugene HD or similar reagents according to manufacturer protocols [43].
- Inducible systems: For toxic genes, use tetracycline-inducible systems with appropriate controls.
Functional Assays for Phenotypic Characterization
Validating URG roles requires multifaceted assessment of cancer phenotypes. Standardized protocols include:
Proliferation assays:
- CCK-8 assay: Seed 2,000-5,000 cells/well in 96-well plates. At designated timepoints, add 10μL CCK-8 reagent, incubate 1-4 hours, and measure absorbance at 450nm [116].
- Colony formation: Plate 500-1,000 cells/well in 6-well plates, culture for 10-14 days, fix with methanol, stain with crystal violet (0.5%), and count colonies >50 cells [21].
- EdU staining: Detect proliferating cells using Click-iT EdU Alexa Fluor imaging kits according to manufacturer instructions [21].
Invasion and migration assays:
- Transwell invasion: Coat Transwell inserts (8μm pores) with Matrigel (1:8 dilution). Seed 5×10⁴ serum-starved cells in upper chamber with complete medium in lower chamber as chemoattractant. After 24-48 hours, fix migrated cells and stain with crystal violet [116].
- Wound healing: Create scratch wounds in confluent monolayers using pipette tips. Capture images at 0, 12, 24 hours and measure gap closure [116].
Ubiquitination-specific assessments:
- Co-immunoprecipitation: Lyse cells in RIPA buffer with protease inhibitors. Incubate 500μg protein with 1-2μg primary antibody overnight at 4°C. Add Protein A/G beads for 2 hours, wash extensively, and elute with SDS sample buffer for Western blotting [115].
- Ubiquitination detection: Use linkage-specific ubiquitin antibodies (K48-, K63-linkage specific) or tagged ubiquitin constructs (His-, Strep-, or HA-Ub) to characterize ubiquitin chain topology [115].
In Vivo Models for URG Functional Validation
In vivo models provide indispensable platforms for validating URG functions within the complexity of intact biological systems, revealing aspects of tumor biology inaccessible to in vitro models.
Xenograft Tumor Models
Subcutaneous and orthotopic xenografts represent the most widely utilized in vivo systems for URG validation. The following protocol details standard methodology:
Cell-derived xenografts:
- Preparation: Harvest URG-manipulated cancer cells in logarithmic growth phase. Wash with PBS and resuspend at 5-10×10⁶ cells/100μl in PBS:Matrigel (1:1) mixture [21].
- Inoculation: Inject 100-200μl cell suspension subcutaneously into flanks of 4-6 week old immunodeficient mice (BALB/c nude or NOD-scid). For orthotopic models, inject directly into relevant organs (e.g., mammary fat pad for breast cancer) [43].
- Monitoring: Measure tumor dimensions 2-3 times weekly using calipers. Calculate volume as V = (length × width²)/2. Euthanize mice when tumors reach 1.5cm diameter or at predetermined endpoints [21].
- Analysis: Harvest tumors, weigh, and process for histology (4% PFA fixation), protein/RNA extraction (snap-freezing), or ubiquitination assays.
Patient-derived xenografts (PDX):
- Implantation: Implant freshly collected patient tumor fragments (1-2mm³) subcutaneously or orthotopically in immunodeficient mice [118].
- Propagation: Passage tumors upon reaching 1.0-1.5cm diameter by transferring fragments to subsequent mouse generations.
- Intervention studies: Once stable growth established, randomize mice to treatment groups for therapeutic assessment of URG targeting.
Metastasis Models
Validating the role of URGs in metastatic dissemination requires specialized models:
Experimental metastasis:
- Preparation: Harvest and resuspend URG-manipulated cells in sterile PBS at 1×10⁶ cells/ml [43].
- Injection: Inject 100μl cell suspension into tail vein of immunodeficient mice (for lung metastasis) or into spleen/splenic artery (for liver metastasis).
- Endpoint analysis: After 6-8 weeks, euthanize mice and examine organs for metastatic nodules. Quantify metastasis by counting surface lesions, histopathological examination of H&E sections, or bioluminescent imaging if luciferase-tagged cells were used [43].
Spontaneous metastasis:
- Primary tumor establishment: Implant URG-manipulated cells orthotopically (e.g., mammary fat pad for breast cancer) [43].
- Monitoring: After primary tumors reach predetermined size (typically 1.0cm), surgically resect them to mimic clinical scenarios.
- Metastasis assessment: Monitor mice for additional 4-8 weeks for metastatic development, then analyze as above.
Genetically Engineered Mouse Models (GEMMs)
GEMMs provide sophisticated systems for investigating URG functions in autochthonous tumor development within immune-competent contexts. While not explicitly detailed in the search results, they represent the gold standard for in vivo validation of stage-specific URG roles, particularly through conditional knockout or knockin systems that permit spatial and temporal control of gene manipulation.
Table 2: Comparison of In Vivo Models for URG Validation
| Model Type | Key Applications | Advantages | Limitations | Stage-Specific Insights |
|---|---|---|---|---|
| Subcutaneous Xenograft [21] | Tumor growth, drug response | Technically simple, reproducible | Limited microenvironment, non-orthotopic | URG effects on primary tumor growth |
| Orthotopic Xenograft [43] | Tumor-stroma interactions, metastasis | Relevant microenvironment, metastatic potential | Technically challenging, variable take rates | URG roles in organ-specific progression |
| Patient-Derived Xenografts [118] | Personalized therapy, tumor heterogeneity | Preserves original tumor characteristics | Costly, time-consuming, requires immunodeficient hosts | Stage-specific URG functions in human tumors |
| Experimental Metastasis [43] | Metastatic colonization | Quantitative, reproducible | Bypasses early metastatic steps | URG involvement in late-stage dissemination |
| Spontaneous Metastasis [43] | Complete metastatic cascade | Clinically relevant progression | Time-consuming, variable latency | URG functions across metastatic cascade |
Integrated Workflow for URG Validation
A comprehensive URG validation strategy integrates bioinformatics discovery with sequential experimental confirmation across model systems. The following workflow represents a consensus approach derived from multiple cancer studies:
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Reagents for URG Functional Validation
| Reagent Category | Specific Examples | Applications | Technical Notes |
|---|---|---|---|
| Ubiquitination Detection [115] | Linkage-specific Ub antibodies (K48, K63), P4D1, FK1/FK2, His-/Strep-/HA-tagged Ub | Immunoblotting, immunofluorescence, ubiquitination assays | Linkage-specific antibodies enable chain topology determination; tagged Ub allows purification |
| Cell Culture Systems [118] | Matrigel, laminin, collagen, defined media supplements (BMP4, FGF9, RA) | 3D organoid culture, tumor-stroma co-cultures | Matrix composition critically influences signaling and differentiation |
| Genetic Manipulation [116] [43] | siRNA/shRNA, CRISPR-Cas9 systems, cDNA expression vectors, lentiviral packaging systems | Gain/loss-of-function studies | Lentiviral systems enable stable manipulation; inducible systems control expression timing |
| Phenotypic Assays [21] [116] | CCK-8, EdU staining kits, Transwell inserts, Matrigel invasion chambers | Proliferation, invasion, migration quantification | EdU detects DNA synthesis; Transwell assays require optimization of cell numbers and time |
| Animal Models [21] [43] | Immunodeficient mice (BALB/c nude, NOD-scid), Matrigel for injections | Xenograft studies, metastasis models | Orthotopic injection enhances metastatic potential; cell number optimization critical |
| Analysis Tools [115] | RIPA buffer, protease inhibitors, protein A/G beads, ECL reagents | Protein analysis, co-immunoprecipitation | Fresh protease inhibitors essential for ubiquitination studies; optimize antibody concentrations |
The functional validation of stage-specific URG roles represents a critical bridge between bioinformatics discovery and clinical translation. The consistent emergence of URG signatures as prognostic classifiers across diverse cancers underscores the fundamental importance of ubiquitination pathways in tumor progression. By employing integrated validation workflows that progress from in vitro models to increasingly complex in vivo systems, researchers can establish causal relationships between URG expression and cancer phenotypes while elucidating underlying mechanisms.
Future directions in URB functional validation will likely emphasize several key areas: First, the development of more sophisticated in vivo models that better recapitulate human tumor microenvironments, particularly immune-competent systems that permit investigation of URG-mediated immunomodulation. Second, the creation of stage-specific organoid biobanks that enable validation of URG functions across disease progression continuum. Third, the integration of advanced ubiquitin proteomics to comprehensively identify URG substrates and signaling pathways. Finally, the translation of URG functional insights into targeted therapeutics represents the ultimate validation of their biological and clinical significance.
The experimental frameworks and methodologies reviewed here provide a roadmap for rigorous validation of stage-specific URG roles, with particular emphasis on standardized protocols, appropriate model selection, and integrative approaches that bridge molecular mechanisms with clinical relevance. As ubiquitination continues to emerge as a central regulatory mechanism in cancer progression, these validation paradigms will remain essential for advancing both biological understanding and therapeutic applications.
Conclusion
The comprehensive analysis of ubiquitination patterns unequivocally establishes their value as dynamic, stage-specific biomarkers and therapeutic targets in cancer. The integration of ubiquitinomics with multi-omics data provides a powerful framework for patient stratification, prognostic assessment, and personalized treatment selection in the context of Predictive, Preventive, and Personalized Medicine (PPPM). Future directions must focus on the clinical translation of these findings, including the development of standardized ubiquitination profiling assays, the expansion of URG-based diagnostic kits, and the advancement of next-generation therapeutics like PROTACs that leverage the specificity of E3 ligases. Overcoming the challenges of tumor heterogeneity and PTM crosstalk will be crucial for realizing the full potential of targeting the ubiquitin-proteasome system to improve cancer outcomes.
References
- 1. Targeting the Ubiquitin–Proteasome System for Cancer - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12495026/]
- 2. The Ubiquitin–Proteasome System in Immune Cells [https://www.mdpi.com/2218-273X/11/1/60]
- 3. Ubiquitin proteasome system in immune regulation and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10163937/]
- 4. Ubiquitin-like protein conjugation and the ... [https://www.nature.com/articles/nrd3321]
- 5. Construction of ubiquitination-related risk model for ... [https://www.nature.com/articles/s41598-025-92177-4]
- 6. Small molecule targeted protein degradation via the UPS - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11815867/]
- 7. UCHL1 Promotes Gastric Cancer Progression by ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12567482/]
- 8. Comprehensive pan‑cancer analysis reveals ubiquitin ... [https://www.spandidos-publications.com/10.3892/ol.2025.15237]
- 9. Cellular parameters shaping pathways of targeted protein ... [https://www.nature.com/articles/s42003-025-08104-w]
- 10. Drug Development Targeting the Ubiquitin–Proteasome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7226376/]
- 11. Ubiquitination and deubiquitination in cancer [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-024-02046-3]
- 12. The role of ubiquitination in tumorigenesis and targeted ... [https://www.nature.com/articles/s41392-020-0107-0]
- 13. Pan-cancer multi-omics profiling reveals ubiquitin D as a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12480306/]
- 14. Decoding the ubiquitin network: molecular mechanisms and ... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-025-02433-4]
- 15. Pan-cancer multi-omics profiling reveals ubiquitin D as a ... [https://www.springermedicine.com/biomarkers/thymoma/pan-cancer-multi-omics-profiling-reveals-ubiquitin-d-as-a-novel-/51504508]
- 16. Construction and validation of a ubiquitination-related ... [https://www.sciencedirect.com/science/article/pii/S2405844024115848]
- 17. Opinion Reprogramming SUMO-primed ubiquitylation [https://www.sciencedirect.com/science/article/pii/S0165614725002056]
- 18. The role of ubiquitination and deubiquitination in cancer ... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2025.1464914/full]
- 19. Constructing a pancancer ubiquitination regulatory network to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12382526/]
- 20. Development and validation of a novel ubiquitination-related ... [https://tcr.amegroups.org/article/view/67645/html]
- 21. Exploration of the ubiquitination-related molecular ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2024.1292249/full]
- 22. The importance of regulatory ubiquitination in cancer and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5397262/]
- 23. Key roles of ubiquitination in regulating critical ... [https://www.sciencedirect.com/science/article/pii/S2352304224001089]
- 24. Therapeutic potential of targeting ubiquitin-specific ... [https://pubmed.ncbi.nlm.nih.gov/40216291/]
- 25. The role of ubiquitination and deubiquitination in cancer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7529510/]
- 26. Ubiquitin‐mediated control of oncogene and tumor ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11159930/]
- 27. Ubiquitin-specific proteases as key regulators in the ... [https://pubmed.ncbi.nlm.nih.gov/41258572/]
- 28. The correlation between the expression of ubiquitin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11798207/]
- 29. Ubiquitination-Related Molecular Subtypes and a Novel ... [https://www.por-journal.com/journals/pathology-and-oncology-research/articles/10.3389/pore.2021.1609941/full]
- 30. Roles of ubiquitination in the crosstalk between tumors and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9170352/]
- 31. The ubiquitin-proteasome system in the tumor immune ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11416925/]
- 32. Frontiers | Identification and validation of ubiquitination -related genes... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2025.1578075/full]
- 33. Emerging drug development technologies targeting ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7112620/]
- 34. exploring the ubiquitin system for drug development [https://www.nature.com/articles/cr201631]
- 35. Cancer proteogenomics: current impact and future prospects [https://pmc.ncbi.nlm.nih.gov/articles/PMC12404316/]
- 36. Automating PTM Profiling for Mass Spec: Scalable Proteomics ... [https://blog.cellsignal.com/high-throughput-proteomics-automating-ptm-enrichment-for-mass-spec]
- 37. A Practical Guide to Interpreting Proteomics Data Visualization [https://aiinbioinformatics.com/a-practical-guide-to-interpreting-proteomics-data-visualization/]
- 38. Prognostic model of ubiquitination-related genes in ovarian ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1654180/full]
- 39. Ubiquitinomics revealed disease- and stage -specific patterns relevant... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10439878/]
- 40. -proteasome system-mediated Ubiquitin modification... ubiquitination [https://www.aging-us.com/article/204650]
- 41. Gene signature and prognostic value of ubiquitination -related genes... [https://wjso.biomedcentral.com/articles/10.1186/s12957-022-02875-w]
- 42. Prognostic model of ubiquitination-related genes in ovarian ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12450667/]
- 43. The Clinical Prediction Value of the Ubiquitination Model Reflecting... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11705054/]
- 44. Single-cell multi-omics in cancer immunotherapy: from tumor ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12376342/]
- 45. Spatial Transcriptomics Decodes Breast Cancer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12383931/]
- 46. In-depth patient-specific analysis of tumor heterogeneity ... [https://www.sciencedirect.com/science/article/pii/S1936523325001998]
- 47. Single-cell RNA sequencing reveals different cellular ... [https://www.nature.com/articles/s41523-025-00808-w]
- 48. Single-cell and multi-omics analysis identifies TRIM9 as a key ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12491318/]
- 49. Systematic benchmarking of high-throughput subcellular ... [https://www.nature.com/articles/s41467-025-64292-3]
- 50. Integrative single-cell and spatial transcriptomics analysis ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1546382/full]
- 51. Exploration of the ubiquitination-related molecular classification and... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11390591/]
- 52. A gene signature consisting of ubiquitin ligases and ... [https://www.nature.com/articles/s41598-022-06451-w]
- 53. A gene expression‐based single sample predictor of lung ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7689824/]
- 54. A deep learning algorithm to predict risk of pancreatic ... [https://www.nature.com/articles/s41591-023-02332-5]
- 55. Development of a blood-based extracellular vesicle ... [https://www.nature.com/articles/s43856-023-00351-4]
- 56. Comparison of multiple machine learning models for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11598923/]
- 57. Deubiquitinating enzymes as cancer biomarkers [https://pmc.ncbi.nlm.nih.gov/articles/PMC6476481/]
- 58. Integrated Genomic Analysis of the Ubiquitin Pathway across ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5916807/]
- 59. Integrated analysis of the ubiquitination mechanism reveals ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-023-09583-z]
- 60. Decoding the ubiquitin network: molecular mechanisms ... [https://pubmed.ncbi.nlm.nih.gov/41039553/]
- 61. Integrative omics analysis reveals effective stratification ... [https://www.sciencedirect.com/science/article/pii/S2589004221007926]
- 62. FBXO42 promotes hepatocellular carcinoma progression via mediating... [https://eurjmedres.biomedcentral.com/articles/10.1186/s40001-025-03050-z]
- 63. Prognostic value of ubiquitination -related differentially expressed... [https://jtd.amegroups.org/article/view/93500/html]
- 64. Frontiers | Single-cell and multi-omics analysis identifies TRIM9 as... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1631708/full]
- 65. Targeting the ubiquitin system by fragment-based drug ... [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2022.1019636/full]
- 66. Pan-cancer multi-omics profiling reveals ubiquitin D as a ... [https://link.springer.com/article/10.1007/s12672-025-03545-5]
- 67. Single-cell multi-omics in cancer immunotherapy: from tumor ... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-025-02426-3]
- 68. Decoding tumor heterogeneity: A spatially informed pan ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12629804/]
- 69. Machine learning analysis reveals tumor heterogeneity ... [https://www.nature.com/articles/s41746-025-01967-7]
- 70. Selective ubiquitination of drug-like small molecules by the ... [https://www.nature.com/articles/s41467-025-63442-x]
- 71. Targeting the ubiquitin-proteasome pathway to overcome ... [https://www.sciencedirect.com/science/article/abs/pii/S1368764619300603]
- 72. Understanding and targeting resistance mechanisms in cancer [https://pmc.ncbi.nlm.nih.gov/articles/PMC10203373/]
- 73. Drug resistance mechanisms and treatment strategies ... [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-024-02005-y]
- 74. Tumor cell plasticity in targeted therapy-induced resistance [https://www.nature.com/articles/s41392-023-01383-x]
- 75. Editorial: Combinatorial Approaches for Cancer Treatment [https://pmc.ncbi.nlm.nih.gov/articles/PMC8847383/]
- 76. Designing combination therapies for cancer treatment [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2024.1358478/full]
- 77. A highly annotated drug combination resource for ... [https://www.nature.com/articles/s41597-025-05630-4]
- 78. Crosstalk between O-GlcNAcylation and ubiquitination [https://ehoonline.biomedcentral.com/articles/10.1186/s40164-024-00569-5]
- 79. O‑GlcNAcylation as an emerging molecular target ... [https://www.spandidos-publications.com/10.3892/or.2025.8952]
- 80. Insights into O-GlcNAcylation and programmed cell death ... [https://www.frontiersin.org/journals/cell-and-developmental-biology/articles/10.3389/fcell.2025.1560491/full]
- 81. O-GlcNAcylation in tumorigenesis and its implications ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11417186/]
- 82. Regulation of Protein Degradation by O-GlcNAcylation [https://pmc.ncbi.nlm.nih.gov/articles/PMC3861702/]
- 83. FBXO31-mediated ubiquitination of OGT maintains O- ... [https://www.nature.com/articles/s41467-025-56633-z]
- 84. Recent advances in small molecule inhibitors of ... [https://www.sciencedirect.com/science/article/abs/pii/S0223523425000893]
- 85. Identification and validation of ubiquitination-related genes for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12343280/]
- 86. Precision cancer medicine 2025: some concerns - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12439214/]
- 87. Proteolysis‐Targeting Chimera (PROTAC) - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC12495453/]
- 88. Methodological Insights on Biomarker‐Based Patient Selection [https://pmc.ncbi.nlm.nih.gov/articles/PMC11993284/]
- 89. Gene- and immune-targeted therapy combinations using ... [https://www.nature.com/articles/s41698-025-01038-w]
- 90. Identification and validation of an immune-related ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12611401/]
- 91. Independent validation of lung adenocarcinoma prognostic ... [https://www.nature.com/articles/s41598-025-22140-w]
- 92. Agnostic Biomarkers and Molecular Signatures in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12383588/]
- 93. The Role of Immune Checkpoint Inhibitors in Cancer Therapy [https://pmc.ncbi.nlm.nih.gov/articles/PMC12497686/]
- 94. Immune Biomarkers for Checkpoint Blockade in Solid Tumors [https://pubmed.ncbi.nlm.nih.gov/40867268/]
- 95. Predictive models for immune checkpoint inhibitor ... [https://www.sciencedirect.com/science/article/pii/S1040842825003683]
- 96. Interplay between tumor mutation burden and the ... [https://pubmed.ncbi.nlm.nih.gov/40777041/]
- 97. Breakthroughs in immune checkpoint therapy: overcoming ... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1630940/full]
- 98. Frontiers | Immune microenvironment infiltration landscape and immune-related subtypes in prostate cancer [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2022.1001297/full]
- 99. Tumour microenvironment (TME) characterization identified prognosis and immunotherapy response in muscle-invasive bladder cancer (MIBC) - PubMed [https://pubmed.ncbi.nlm.nih.gov/32617668/]
- 100. Integration of tumor inflammation, cell proliferation, and traditional biomarkers improves prediction of immunotherapy resistance and response | Biomarker Research [https://link.springer.com/article/10.1186/s40364-021-00308-6]
- 101. Ensemble model for neoadjuvant chemotherapy response ... [https://www.nature.com/articles/s41598-025-21932-4]
- 102. Early Ki67 Change Predicts Prognosis and Supports ... [https://www.sciencedirect.com/science/article/pii/S0960977625008665]
- 103. FDG-PET/CT and Multimodal Machine Learning Model ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11987901/]
- 104. Predictive Biomarkers in Cancer Immunotherapy [https://pmc.ncbi.nlm.nih.gov/articles/PMC12289453/]
- 105. Predicting the response to immunotherapy from gene ... [https://www.nature.com/articles/s43856-025-01131-y]
- 106. A novel model for predicting immunotherapy response and ... [https://cancerci.biomedcentral.com/articles/10.1186/s12935-025-03800-3]
- 107. biomarkers for Predictive in daily anatomic... targeted therapy [https://surgexppathol.biomedcentral.com/articles/10.1186/s42047-025-00187-3]
- 108. Ubiquitination-Related Molecular Subtypes and a Novel Prognostic Index for Bladder Cancer Patients - PubMed [https://pubmed.ncbi.nlm.nih.gov/34776794/]
- 109. Frontiers | Multi-omics analysis identifies UBA family as potential pan-cancer biomarkers for tumor prognosis and immune microenvironment infiltration [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2025.1510503/full]
- 110. Ubiquitomics: An Overview and Future - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7603029/]
- 111. Ubiquitin‑conjugating enzymes as potential biomarkers ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9944987/]
- 112. Frontiers | Integrating multi-omics and experimental techniques to... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1545472/full]
- 113. Modification in Hepatocellular Carcinoma Ubiquitination [https://www.genesispub.org/Ubiquitination-Modification-in-Hepatocellular-Carcinoma]
- 114. Ubiquitination-related biomarkers in metastatic melanoma ... [https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2023.1170190/full]
- 115. Current methodologies in protein ubiquitination ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9373315/]
- 116. Frontiers | Identification of biomarkers based on ubiquitin -correlated... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1646396/full]
- 117. Identification of a novel ubiquitination related gene signature for patients with breast cancer - PubMed [https://pubmed.ncbi.nlm.nih.gov/36123926/]
- 118. Complex in vitro model: A transformative model in drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10828975/]