Ubiquitin-Like Proteins: From Ancient Origins to Modern Drug Discovery
This article provides a comprehensive synthesis of ubiquitin-like proteins (UBLs), exploring their conserved β-grasp fold structure, intricate enzymatic conjugation cascades, and profound evolutionary lineage tracing back to prokaryotic sulfur-transfer systems.
Ubiquitin-Like Proteins: From Ancient Origins to Modern Drug Discovery
Abstract
This article provides a comprehensive synthesis of ubiquitin-like proteins (UBLs), exploring their conserved β-grasp fold structure, intricate enzymatic conjugation cascades, and profound evolutionary lineage tracing back to prokaryotic sulfur-transfer systems. Tailored for researchers and drug development professionals, it details the critical functions of UBLs—such as SUMO, NEDD8, and ATG8—in regulating protein degradation, autophagy, DNA repair, and immune response. The content further examines contemporary methodological approaches for studying UBL pathways, analyzes challenges and emerging strategies in therapeutic targeting, including E1, E2, and E3 enzyme inhibitors, and validates these targets through a multiomics and comparative disease biology lens. By integrating foundational knowledge with cutting-edge therapeutic applications, this review serves as a vital resource for navigating the complexity of UBL systems and exploiting their vast potential in biomedical research and clinical intervention.
The Architecture and Evolutionary Legacy of Ubiquitin-Like Proteins
Ubiquitin-like proteins (UBLs) constitute a family of small proteins involved in the post-translational modification of other proteins, thereby regulating a vast array of cellular functions [1]. The family derives its name from its first discovered and most well-known member, ubiquitin (Ub), which is renowned for its central role in targeting proteins for degradation by the proteasome [1] [2]. Following ubiquitin's discovery, many structurally and evolutionarily related proteins were identified, leading to the recognition of a larger protein family with parallel regulatory processes and similar chemical mechanisms [1] [3]. These UBLs are involved in a widely varying array of cellular functions including autophagy, protein trafficking, inflammation and immune responses, transcription, DNA repair, RNA splicing, and cellular differentiation [1].
Structurally, UBLs are defined by a characteristic three-dimensional structure known as the β-grasp fold [1] [4]. This fold consists of a five-strand antiparallel beta sheet surrounding a central alpha helix [1] [3]. While this structural motif is shared, UBLs are functionally diverse and are primarily classified into two categories based on their ability to form covalent conjugates with other molecules: Type I (conjugated) and Type II (non-conjugated) [1] [3] [5].
Core Structural Characteristics: The β-Grasp Fold
The defining structural feature of ubiquitin and ubiquitin-like proteins is the β-grasp fold [1] [4]. This compact protein fold is highly versatile and is found in a wide range of proteins beyond the UBL family, including those with catalytic roles, iron-sulfur cluster scaffolds, and RNA-binding proteins [4].
- Ubiquitin Prototype: Ubiquitin itself is a 76-amino acid polypeptide whose structure reveals a distinctive fold dominated by a beta-sheet with five anti-parallel beta-strands and a single helical segment [4]. The beta-sheet appears to "grasp" the helical segment, giving the fold its name [4].
- Structural Conservation in UBLs: Most UBLs share this core β-grasp architecture, though variations exist [3]. For instance, some UBLs, like ISG15 and FAT10, are composed of tandem β-grasp domains [3]. Members of the ATG8 family possess two additional α-helices near their N-terminus, while ATG12, UFM1, and SUMO proteins often have N-terminal extensions that can be disordered [3].
Despite shared structure, critical surface residues of ubiquitin are not always conserved in other UBLs, leading to distinct interaction surfaces and functions [6]. For example, UBL5/Hub1, an atypical UBL, adopts the β-grasp fold but has a highly charged electrostatic surface with no large hydrophobic patches, unlike ubiquitin, and it lacks the characteristic C-terminal diglycine motif [5] [6].
Classification of UBLs: Type I vs. Type II
The ubiquitin-like protein family is fundamentally divided into two categories based on their mechanism of action and covalent conjugation capabilities.
Table 1: Core Characteristics of Type I and Type II Ubiquitin-Like Proteins
| Feature | Type I UBLs (Conjugated) | Type II UBLs (Non-Conjugated) |
|---|---|---|
| Defining Action | Covalently conjugated to target proteins or lipids [1] [3]. | Not covalently conjugated; function as protein-protein interaction domains or integral domains within larger proteins [1] [5]. |
| C-Terminal Motif | Characteristic glycine residue(s) exposed after proteolytic processing [1] [3]. | Lack the C-terminal glycine motif required for conjugation [5] [6]. |
| Expression & Processing | Often expressed as inactive precursors requiring C-terminal proteolysis for activation [1]. | May occur as protein domains genetically fused in a single polypeptide [1]. |
| Representative Members | Ubiquitin, SUMO, NEDD8, ATG8, ATG12, URM1, UFM1, FAT10, ISG15 [1] [3]. | Ubiquilins (e.g., UBQLN1), Hub1/UBL5, Ubiquitin-associated (UBX) domain proteins, Rad23 [7] [5] [8]. |
| Primary Function | Post-translational modification to alter substrate activity, stability, localization, or interactions [1] [2]. | Act as adaptors, scaffolds, or modifiers in processes like proteasomal targeting, splicing, and DNA repair [7] [5] [8]. |
Type I UBLs: Covalent Protein Modifiers
Type I UBLs are characterized by their ability to be activated and covalently ligated to target proteins (or, in one case, a lipid) through an enzymatic cascade [1] [3]. This process is analogous to ubiquitination. A key hallmark of Type I UBLs is a C-terminal sequence that ends with one or two glycine residues [1]. These UBLs are typically expressed as inactive precursors and must be activated by proteases that cleave the C-terminus to expose the reactive glycine [1] [3]. The conjugation cascade involves E1 (activating), E2 (conjugating), and E3 (ligating) enzymes, which are often specific to each UBL family [1] [3] [2].
Type II UBLs: Non-Covalent Regulators
Type II UBLs contain domains that share the ubiquitin-like β-grasp fold but are not covalently conjugated to other proteins [1] [5]. They generally lack the C-terminal glycine motif [5]. These UBL domains can be found within larger multidomain proteins and often function as protein-protein interaction modules [1] [8]. For example:
- Ubiquilins contain an N-terminal UBL domain and a C-terminal ubiquitin-associated (UBA) domain. They function as ubiquitin receptors, shuttling polyubiquitinated proteins to the proteasome by using their UBL domain to bind the proteasome and their UBA domain to bind ubiquitin [7] [8].
- Hub1/UBL5 lacks the diglycine motif and binds its protein partners, such as the spliceosomal protein Snu66, non-covalently to modulate pre-mRNA splicing [5] [6].
- UBX domain proteins contain a domain that structurally mimics ubiquitin but functions as a binding module within larger proteins involved in ubiquitin-related processes [1] [8].
The Conjugation Machinery and Mechanism for Type I UBLs
The conjugation of Type I UBLs to their targets is a tightly regulated, multi-step process that consumes ATP. The well-characterized ubiquitination pathway serves as the paradigm.
The Enzymatic Cascade
The conjugation mechanism involves a cascade of three enzymes:
- Activation (E1): The UBL is first activated by an E1 activating enzyme in an ATP-dependent reaction. The E1 forms a high-energy thioester bond between its active-site cysteine and the C-terminal glycine of the UBL [3] [2].
- Conjugation (E2): The activated UBL is then transferred from the E1 to the active-site cysteine of an E2 conjugating enzyme, forming a similar E2~UBL thioester intermediate [3] [2].
- Ligation (E3): Finally, an E3 ligase facilitates the transfer of the UBL from the E2 to a primary amine (typically the ε-amino group of a lysine) on the target protein, resulting in an isopeptide bond [3] [2]. E3s are primarily responsible for substrate recognition and specificity.
Table 2: Human Type I UBLs and Their Cognate Enzymes
| UBL Family | Representative UBLs in Humans | E1 Activating Enzyme | E2 Conjugating Enzyme(s) |
|---|---|---|---|
| Ubiquitin | Ubiquitin | UBA1, UBA6 | Many [2] |
| SUMO | SUMO1, SUMO2, SUMO3 | UBA2/SAE1 | UBC9 [3] |
| NEDD8 | NEDD8 | UBA3/NAE1 | UBC12, UBE2F [3] |
| ATG8 | LC3A, LC3B, GABARAP | ATG7 | ATG3 [3] |
| ATG12 | ATG12 | ATG7 | ATG10 [3] |
| URM1 | URM1 | UBA4 | – [3] |
| UFM1 | UFM1 | UBA5 | UFC1 [3] |
| FAT10 | FAT10 | UBA6 | UBE2Z [3] |
| ISG15 | ISG15 | UBA7 | UBCH8 [3] |
Deconjugation and Regulation
UBL modifications are dynamic and reversible. A class of enzymes known as deubiquitinating enzymes (DUBs) or UBL-specific proteases (ULPs) cleave the isopeptide bonds, releasing the UBL from the substrate and allowing for recycling of the UBL tag and termination of the signal [1] [2]. Furthermore, many UBLs, including ubiquitin and SUMO, can themselves be modified by other UBLs, creating a complex network of cross-talk that integrates different cellular signals [1].
Experimental Analysis of UBL Pathways
Research into UBL pathways relies on a suite of molecular and biochemical techniques to dissect the conjugation cascades, identify substrates, and understand functional consequences.
Key Methodologies
- Trapping Reaction Intermediates: A critical strategy for studying the mechanism of E1 and E2 enzymes involves trapping thioester-linked intermediates. This is often achieved by using ATP analogues like ATPγS, which leads to the formation of a stable UBL~adenylate, or by employing E1/E2 active-site mutants (e.g., cysteine to alanine) that prevent thioester transfer, allowing for the isolation and structural characterization of complexes [3].
- In Vitro Reconstitution Assays: Purified components of the UBL pathway (E1, E2, E3, UBL, and substrate) are combined in a test tube to reconstitute the conjugation reaction. This allows for direct manipulation of reaction conditions and identification of minimal essential components [3].
- Mass Spectrometry (MS): MS is indispensable for identifying sites of UBL modification on target proteins. Tandem MS (MS/MS) following immunoprecipitation of UBL-conjugated proteins can precisely map the specific lysine residue modified by the UBL [3].
- Yeast Two-Hybrid (Y2H) Screening: This technique is used to identify novel protein-protein interactions. For UBLs, it has been successfully used to find interacting partners of Type II UBLs like Hub1/UBL5, revealing connections to spliceosomal components and other factors [5] [6].
- Structural Biology Techniques: X-ray crystallography and NMR spectroscopy have been pivotal in determining the three-dimensional structures of UBLs (e.g., UBL5) and their complexes with binding partners (e.g., the Hub1-Snu66 complex), providing atomic-level insights into recognition and mechanism [6] [8].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for UBL Research
| Research Reagent | Function in UBL Research |
|---|---|
| ATPγS (Adenosine 5'-O-[γ-thio]triphosphate) | A non-hydrolyzable ATP analogue used to trap the UBL~adenylate intermediate on the E1 enzyme for mechanistic studies [3]. |
| Active-Site Mutant E1/E2 Enzymes | E1 or E2 enzymes with catalytic cysteine mutated to alanine (Cys→Ala) are used to block thioester transfer and stabilize complexes for structural analysis [3]. |
| UBL-Specific Proteases (ULPs) | Enzymes like SENP/ULP for SUMO are used to confirm the identity of a UBL modification by cleaving it from substrates in control experiments [1]. |
| Tandem Affinity Purification (TAP) Tags | Used to purify UBL-conjugated protein complexes from cell lysates under native conditions for subsequent identification by mass spectrometry [3]. |
| NMR Isotope Labeling (¹⁵N, ¹³C) | Production of isotopically labeled UBLs (e.g., UBL5) for NMR studies to determine solution structure and map binding interfaces with partners [6]. |
Diagram of UBL Classification and Function
The following diagram summarizes the classification and primary functional roles of the Ubiquitin-Like Protein family.
The ubiquitin-like protein family, defined by the conserved β-grasp fold, is a cornerstone of eukaryotic cellular regulation. The fundamental classification into Type I (conjugated) and Type II (non-conjugated) UBLs reflects a fundamental divergence in their mechanisms of action. Type I UBLs act as reversible covalent modifiers in a manner analogous to ubiquitin, while Type II UBLs function primarily as non-covalent interaction modules within larger proteins. Understanding the distinct characteristics, conjugation machinery, and cellular functions of these two types is essential for comprehending their vast regulatory potential. Continued research into their specific pathways, substrates, and the cross-talk between them remains a vibrant and critical area of cell biology, with direct implications for understanding disease mechanisms and developing novel therapeutics.
The β-grasp fold (β-GF) represents a remarkable evolutionary solution in structural biology—a compact and versatile protein architecture that has been recruited for a staggering array of biochemical functions across all domains of life. Prototyped by ubiquitin (Ub), this fold has been extensively studied for its central role in eukaryotic protein regulation through the ubiquitin-conjugation system [9] [1]. However, its functional repertoire extends far beyond this celebrated role, encompassing catalytic functions, iron-sulfur cluster scaffolding, RNA binding, sulfur transfer, co-factor binding, and adaptor functions in signaling complexes [9] [10].
This whitepaper examines the β-grasp fold through the lens of evolutionary structural biology, exploring how a single structural blueprint has been adapted to serve diverse physiological necessities. The persistence of this fold from the last universal common ancestor (LUCA) to modern organisms underscores its fundamental utility in molecular evolution, while its diversification in eukaryotes highlights its critical role in the development of complex cellular regulation [9] [11]. Understanding the structural principles and evolutionary history of the β-grasp fold provides valuable insights for drug development professionals seeking to target ubiquitin-like pathways in disease states, particularly in cancer, neurodegenerative disorders, and infectious diseases.
Structural Characteristics of the β-Grasp Fold
Core Architectural Features
The β-grasp fold is defined by a highly conserved core structure consisting of a β-sheet with four to five antiparallel strands that partially envelop or "grasp" a single α-helix [9] [1]. This arrangement creates a stable structural scaffold that can withstand significant sequence variation while maintaining structural integrity. The core structural elements typically include:
- A mixed β-sheet with strands arranged in antiparallel fashion
- A single α-helix positioned adjacent to the β-sheet
- Conservative hydrophobic core that stabilizes the fold despite low sequence conservation
- Variable loop regions that confer functional specificity
The fold's stability arises from a conserved hydrophobic core that packs the α-helix against the β-sheet, creating a robust platform that can tolerate extensive surface modifications for specialized functions [9]. This structural robustness has allowed the β-grasp fold to evolve into numerous specialized variants while maintaining its core architectural identity.
Structural Elaborations and Variations
Despite conservation of the core fold, numerous elaborations have evolved to support functional diversity. These include:
- Insertions between core elements: Additional secondary structures such as β-hairpins (e.g., in transcobalamin) or additional helices that extend functional surfaces [12]
- Terminal extensions: N- or C-terminal elongations that add interaction interfaces
- Surface residue specialization: Strategic placement of charged, polar, or hydrophobic residues on the solvent-exposed surface to facilitate specific interactions
- Oligomerization interfaces: Modifications that enable self-association or hetero-complex formation
The prominent β-sheet provides an exposed surface for diverse interactions, and in some cases, these sheets curve to form open barrel-like structures that can accommodate ligands or protein partners [9]. This structural plasticity explains how the fold can support functions as diverse as small molecule binding (e.g., vitamin B12 in transcobalamin) and protein-protein interactions (e.g., in ubiquitin signaling) [12].
Table 1: Major Structural Variations of the β-Grasp Fold
| Structural Variant | Key Features | Representative Proteins | Primary Functions |
|---|---|---|---|
| Classical Ubiquitin-like | Minimal insertions, compact structure | Ubiquitin, NEDD8, SUMO | Protein modification, signaling |
| SLBB Superfamily | β-hairpin insert after helix | Transcobalamin, Nqo1 subunit | Soluble ligand binding |
| Enzyme-associated | Active site integration | NUDIX hydrolases, Staphylokinases | Catalysis, enzymatic activity |
| Fe-S Cluster Binding | Cysteine motifs for cluster coordination | 2Fe-2S Ferredoxins | Electron transport |
| Sulfur Carrier | C-terminal glycines for thiocarboxylate | ThiS, MoaD | Sulfur transfer in cofactor biosynthesis |
Evolutionary Origins and Diversification
Deep Evolutionary Roots
Evolutionary reconstruction indicates that the β-grasp fold had already differentiated into at least seven distinct lineages by the time of the last universal common ancestor (LUCA) of all extant organisms [9] [10]. This early diversification encompassed much of the structural diversity observed in modern versions of the fold, suggesting rapid adaptation to various functional niches in primordial life forms.
The earliest β-grasp members were likely involved in RNA metabolism, with subsequent radiation into various functional niches [9] [10]. The fold's initial recruitment probably exploited its stable platform for RNA interactions, as seen in contemporary TGS domains of aminoacyl tRNA synthetases and other translation regulators [9]. This deep evolutionary history demonstrates the fold's fundamental utility in core cellular processes that date back to the dawn of cellular life.
Prokaryotic Radiation and Functional Innovation
Most of the structural diversification of the β-grasp fold occurred in prokaryotes, where it was adapted for numerous biochemical functions [9] [10]. The fold provided an ideal scaffold for the evolution of various enzymatic activities (e.g., NUDIX phosphohydrolases) and the binding of diverse co-factors (e.g., molybdopterin), each independently evolving on at least three occasions [9]. Similarly, iron-sulfur-cluster-binding emerged at least twice independently within the fold [9].
Comparative genomic analyses reveal that precursors of the eukaryotic ubiquitin system were already present in prokaryotes [13]. These include:
- Metabolic sulfur transfer systems: Combining a Ubl and an E1-like enzyme in pathways for metallopterin, thiamine, cysteine, siderophore, and modified base biosynthesis [13]
- Primitive protein-tagging systems: Including Sampylation in archaea and potential conjugation systems in bacteria with Ubls of the YukD family [13]
- Complete ubiquitin-like pathways: Some prokaryotic systems developed additional elements that closely resemble the eukaryotic state, possessing an E2, a RING-type E3, or both [13]
Eukaryotic Expansion and Specialization
The eukaryotic phase of β-grasp evolution was characterized primarily by a specific expansion of ubiquitin-like (Ubl) members [9] [10]. The eukaryotic UB superfamily diversified into at least 67 distinct families, with approximately 19-20 families already present in the eukaryotic common ancestor [9] [10]. This expansion included:
- Multiple protein-conjugated forms (e.g., ubiquitin, SUMO, NEDD8, ATG12)
- One lipid-conjugated form (ATG8)
- Versions functioning as adaptor domains in multi-module polypeptides
This diversification played a major role in the emergence of characteristic eukaryotic cellular systems, including:
- Nucleo-cytoplasmic compartmentalization
- Vesicular trafficking networks
- Lysosomal targeting pathways
- Protein processing in the endoplasmic reticulum
- Chromatin dynamics and regulation
A key aspect of the eukaryotic phase was the dramatic increase in domain architectural complexity, with Ubl domains incorporated into numerous proteins as adaptors or regulatory modules [9]. This expansion facilitated the evolution of complex regulatory networks that underlie eukaryotic cellular complexity.
Figure 1: Evolutionary diversification of the β-grasp fold from LUCA to modern functional systems in prokaryotes and eukaryotes.
Functional Diversity and Classification
Ubiquitin and Ubiquitin-like Proteins (UBLs)
Ubiquitin and ubiquitin-like proteins represent the most extensively studied class of β-grasp fold proteins. These small proteins are involved in post-translational modification of target proteins through covalent conjugation, typically via their C-terminal glycine residues [1]. The human genome encodes at least eight families of UBLs capable of covalent conjugation: SUMO, NEDD8, ATG8, ATG12, URM1, UFM1, FAT10, and ISG15 [1].
These UBLs regulate an enormous range of physiological processes, primarily by controlling protein interactions with other macromolecules [11]. Their functions include:
- Targeting proteins for degradation (ubiquitin-proteasome system)
- Autophagy (ATG8 and ATG12)
- Transcription regulation (SUMO)
- DNA repair (ubiquitin and SUMO)
- Immune response (ISG15)
- Cell signaling and trafficking (ubiquitin)
The regulatory apparatus for UBL conjugation involves conserved enzyme cascades: E1 (activating), E2 (conjugating), and E3 (ligating) enzymes, with specific sets of enzymes dedicated to each UBL family [1].
Enzymatic and Catalytic Functions
The β-grasp fold has been independently recruited for enzymatic activities on multiple occasions throughout evolution [9]. These include:
- NUDIX phosphohydrolases: Enzymes that hydrolyze nucleoside diphosphates
- Staphylokinases and streptokinases: Fibrinolytic enzymes in bacteria
- Various other catalytic activities that have evolved at least three times independently
In these proteins, the β-grasp fold provides a stable scaffold that positions catalytic residues and binding pockets for substrates, demonstrating how a structural domain can be adapted for chemical catalysis through strategic placement of active site residues.
Soluble Ligand Binding
A novel superfamily within the β-grasp fold, termed the Soluble-Ligand-Binding β-grasp (SLBB) superfamily, has been identified through sensitive sequence and structure similarity searches [12]. This superfamily includes:
- Transcobalamin and intrinsic factor: Vitamin B12 uptake proteins in animals
- Bacterial polysaccharide export proteins
- Competence DNA receptor ComEA in bacteria
- Cobalamin reductase PduS
- Nqo1 subunit of respiratory electron transport chain
These proteins are characterized by two major clades: the transcobalamin-like clade with a β-hairpin insert after the helix, and the Nqo1-like clade with an insert between strands 4 and 5 of the core fold [12]. Members of both clades interact with ligands in a similar spatial location, with their specific inserts playing important roles in ligand binding.
Iron-Sulfur Cluster Binding
The β-grasp fold serves as a scaffold for iron-sulfur cluster binding in proteins such as 2Fe-2S ferredoxins, which are involved in electron transport [9] [12]. This function has evolved on at least two independent occasions within the fold [9], demonstrating convergent evolution for cofactor binding. The fold provides a stable platform with cysteine residues appropriately positioned for cluster coordination, enabling electron transfer capabilities.
Sulfur Transfer in Cofactor Biosynthesis
The evolutionary link between UBLs and sulfur transfer systems is exemplified by proteins like ThiS and MoaD, which function as sulfur carriers in thiamine and molybdopterin biosynthesis, respectively [9] [11]. These proteins share not only the β-grasp fold with ubiquitin but also similar activation mechanisms involving C-terminal thiocarboxylate formation catalyzed by E1-like enzymes (ThiF and MoeB) [9]. The eukaryotic protein URM1 represents a molecular fossil that functions both as a UBL and a sulfur carrier, bridging these functional classes [1].
Adaptor Functions in Protein Complexes
The β-grasp fold frequently serves as an adaptor domain in larger multidomain proteins, mediating specific protein-protein interactions in various contexts [9]. Examples include:
- RA, PB1, and DCX domains: Adaptors in animal signaling proteins
- FERM N-terminal domains: Mediators of membrane-cytoskeleton interactions
- UBX domains: Ubiquitin-regulatory domains in larger polypeptides
These adaptor functions leverage the fold's stable platform and customizable interaction surfaces to facilitate the assembly of macromolecular complexes.
Table 2: Functional Classification of β-Grasp Fold Proteins
| Functional Category | Key Examples | Organismic Distribution | Central Biochemical Role |
|---|---|---|---|
| Protein Modification | Ubiquitin, SUMO, NEDD8 | Eukaryotes | Post-translational modification, signaling |
| Sulfur Transfer | ThiS, MoaD, URM1 | All domains | Cofactor biosynthesis |
| Electron Transfer | 2Fe-2S Ferredoxins | Bacteria, Eukaryotes | Electron transport |
| Soluble Ligand Binding | Transcobalamin, ComEA | Bacteria, Animals | Vitamin uptake, metabolite binding |
| RNA Binding | TGS domain, IF3 | All domains | Translation regulation |
| Enzymatic Catalysis | NUDIX hydrolases, Staphylokinases | Primarily bacteria | Hydrolysis, fibrinolysis |
| Adaptor Functions | RA, PB1, FERM domains | Primarily eukaryotes | Protein complex assembly |
Research Methodologies and Experimental Approaches
Identification and Characterization Techniques
Comprehensive identification of β-grasp fold members requires specialized bioinformatic approaches due to the fold's small size and high sequence divergence. Successful strategies include:
- Multi-pronged sequence-structure searches: Iterative sequence profile searches using PSI-BLAST combined with structural comparisons [9]
- Hidden Markov Model (HMM) searches: Profile-based detection of distant homologs [12]
- Structural similarity searches: Using programs like DALI to identify fold similarities despite sequence divergence [12]
- Topological analysis: Recognition of characteristic β-grasp structural motifs
These approaches have revealed previously unrecognized members of the fold, including domains in bacterial flagellar and fimbrial assembly components, and five new UB-like domains in eukaryotes [9].
Structural Determination and Analysis
Structural characterization of β-grasp proteins employs multiple complementary techniques:
- X-ray crystallography: Provides high-resolution structures of fold variants
- NMR spectroscopy: Reveals dynamic properties and solution structures
- Computational modeling: Rosetta-based protein design to explore fold compatibility [14]
- Chemical shift analysis: CS-Rosetta for structure determination from NMR data [14]
These methods have been instrumental in identifying core distinguishing features of the fold and numerous elaborations, including several previously unrecognized variants [9].
Functional Assay Systems
Functional characterization of β-grasp proteins employs specialized assay systems tailored to their diverse activities:
- Binding assays: For protein-protein, protein-ligand, and protein-RNA interactions
- Enzymatic activity assays: For catalytic members like NUDIX hydrolases
- Conjugation assays: For UBLs, measuring transfer to target proteins
- Inhibition studies: For functional characterization of engineered variants [14]
For example, inhibition constants (Kᵢ) for engineered protease inhibitors based on the S6 ribosomal protein fold can be measured using engineered subtilisin proteases and fluorescent substrates [14].
Figure 2: Integrated research methodology for studying β-grasp fold proteins, combining bioinformatic, structural, and functional approaches.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for β-Grasp Fold Studies
| Reagent/Material | Specific Function | Application Examples |
|---|---|---|
| Ubiquitin Activating Enzyme (E1) | Activates UBLs through ATP-dependent adenylation | UBL conjugation assays, mechanistic studies |
| Ubiquitin Conjugating Enzymes (E2) | Transfers activated UBL to E3 or substrate | Enzyme cascade reconstitution, specificity studies |
| Ubiquitin Ligases (E3) | Mediates target specificity in UBL transfer | Substrate identification, functional characterization |
| Deubiquitinating Enzymes (DUBs) | Cleaves UBL conjugates | Conjugation dynamics, signaling regulation studies |
| Proteasome Complexes | Recognizes and degrades ubiquitinated substrates | Functional validation of ubiquitination |
| NMR Isotope-labeled Media | Production of ¹⁵N/¹³C-labeled proteins for NMR | Structural studies, dynamics characterization |
| Subtilisin Protease Columns | Affinity purification of engineered inhibitors | Functional analysis of engineered β-grasp variants [14] |
| Cross-linking Reagents | Stabilizes transient protein complexes | Interaction mapping, structural characterization |
| Fluorescent Ubiquitin Probes | Monitoring ubiquitination in cellular contexts | Live-cell imaging, kinetic studies |
| Computational Design Software | Rosetta-based protein design | Engineering fold-switching variants [14] |
Engineering and Therapeutic Applications
Protein Engineering and Design
Recent advances have demonstrated the potential for engineering β-grasp proteins with novel properties and functions. Notably, researchers have successfully designed mutational pathways connecting three common folds (3α, β-grasp, and α/β-plait) [14]. This engineering approach involves:
- Threading sequences of smaller folds through larger fold structures
- Identifying alignments that minimize catastrophic interactions
- Computational design using tools like Rosetta to resolve unfavorable interactions
- Experimental validation of designed proteins using NMR and functional assays
These efforts have created protein pairs where a single sequence can adopt either a 3α or β-grasp fold in smaller forms but an α/β-plait fold in larger forms [14]. The ability to design such fold-switching proteins highlights the underlying ambiguity in the protein folding code and demonstrates how new protein structures can evolve via abrupt fold switching.
Therapeutic Targeting Opportunities
The ubiquity and functional importance of β-grasp proteins, particularly in ubiquitin signaling pathways, make them attractive therapeutic targets. Key targeting strategies include:
- Proteasome inhibitors (e.g., bortezomib) for cancer treatment
- DUB inhibitors for modulating ubiquitin signaling
- E1-E2-E3 cascade inhibitors for specific pathway modulation
- SUMOylation modifiers for cancer and neurological disorders
- ISG15 pathway modulators for antiviral applications
Understanding the structural principles and evolutionary relationships of β-grasp proteins provides valuable insights for drug development, particularly in designing specific inhibitors that exploit conserved structural features while targeting functionally divergent family members.
The β-grasp fold represents a remarkable example of evolutionary economy—a single structural blueprint adapted through billions of years of evolution to serve diverse functional roles. From its origins in primordial RNA metabolism to its expansion in eukaryotic regulatory systems, this versatile fold has been continuously reinvented while maintaining its core architectural identity.
The fold's functional versatility stems from its stable structural core, which can tolerate extensive surface modifications and insertions that create specialized functional sites. This plasticity has allowed the evolution of functions ranging from enzymatic catalysis to signal transduction, with different functional classes emerging independently on multiple occasions throughout evolutionary history.
For researchers and drug development professionals, understanding the structural principles and evolutionary relationships of β-grasp proteins provides valuable insights for manipulating these systems therapeutically. The continued discovery of new β-grasp variants and functions underscores the importance of this fold in cellular physiology and highlights potential opportunities for therapeutic intervention in human diseases.
The conjugation of ubiquitin (Ub) and ubiquitin-like proteins (Ubls) represents a crucial post-translational modification mechanism that regulates a vast array of cellular processes in eukaryotic cells. This modification involves the covalent attachment of these small protein modifiers to target substrates, thereby influencing their activity, stability, subcellular localization, and macromolecular interactions [15] [3]. The fundamental importance of these pathways is evidenced by their involvement in essential physiological processes including cell division, immune responses, embryonic development, and protein quality control [16]. Defects in these conjugation systems are associated with numerous diseases, particularly cancer, neurodegenerative disorders, and inflammatory conditions [17] [18].
The conjugation of Ub and Ubls is achieved through a conserved enzymatic cascade comprising activating (E1), conjugating (E2), and ligase (E3) enzymes [15] [3]. These multienzyme cascades utilize labile thioester intermediates, extensive conformational changes, and vast combinatorial diversity of short-lived protein-protein complexes to conjugate Ub/Ubls to various substrates in a tightly regulated manner [19]. This whitepaper provides a comprehensive technical overview of the E1-E2-E3 conjugation cascade, examining the structural mechanisms, evolutionary origins, and experimental approaches that define this essential biological system.
The Enzymatic Cascade: E1, E2, and E3 Mechanisms
E1 Activating Enzymes: The Apex of the Cascade
E1 enzymes serve at the apex of each Ubl conjugation cascade, initiating the activation process and directing the Ubl to downstream pathways [16]. These enzymes employ a conserved mechanism to activate the C-terminus of Ub/Ubls in an ATP-dependent process. The human genome encodes eight E1 enzymes that initiate conjugation for Ub and various Ubls, each exhibiting specificity for their cognate modifiers [16].
The activation mechanism proceeds through two distinct steps [16] [18]. First, the E1 enzyme binds MgATP and ubiquitin, catalyzing ubiquitin C-terminal acyl-adenylation. In the second step, the catalytic cysteine residue in the E1 active site attacks the ubiquitin~adenylate to form an activated ubiquitin~E1 thioester complex (the tilde "~" represents a high-energy thioester bond). Throughout this mechanism, the E1 enzyme binds two ubiquitin molecules simultaneously, with the secondary ubiquitin facilitating conformational changes during the subsequent transthioesterification process [18].
Table 1: Human E1 Enzymes and Their Cognate Ubls
| E1 Enzyme | Composition | Cognate Ubl(s) | Cellular Functions |
|---|---|---|---|
| UBA1 | Homodimeric | Ubiquitin | Protein degradation, signaling |
| UBA6 | Homodimeric | Ubiquitin, FAT10 | Immune regulation |
| UBA2/SAE1 heterodimer | Heterodimeric | SUMO1, SUMO2, SUMO3 | Nuclear transport, transcription |
| UBA3/NAE1 heterodimer | Heterodimeric | NEDD8 | Cullin-RING ligase activation |
| ATG7 | Homodimeric | ATG8, ATG12 | Autophagy |
| UBA4 | Homodimeric | URM1 | Sulfur transfer, oxidative stress |
| UBA5 | Homodimeric | UFM1 | Endoplasmic reticulum stress |
| UBA7 | Homodimeric | ISG15 | Immune response, antiviral defense |
E2 Conjugating Enzymes: Central Hubs in Specificity
E2 enzymes (ubiquitin-conjugating enzymes or UBCs) function as central hubs in the conjugation cascade, receiving the activated Ubl from E1 and cooperating with E3 ligases to modify specific substrates [20]. The human genome encodes at least 38 E2s, which can be classified into 17 phylogenetically distinct subfamilies [20]. These enzymes contain a conserved core ubiquitin-conjugating (UBC) domain that houses the active-site cysteine residue responsible for thioester linkage with the Ubl.
E2 enzymes have emerged as key mediators of ubiquitin chain assembly, governing the switch from chain initiation to elongation, regulating the processivity of chain formation, and establishing the topology of assembled chains [20]. After being charged with ubiquitin, E2s engage specific E3s to catalyze substrate ubiquitylation. A single E2 can interact with multiple different E3s, with members of the UBE2D family exhibiting particularly broad E3 specificity [20].
Table 2: Selected Human E2 Enzymes and Their Characteristics
| E2 Enzyme | Alternative Names | Cognate Ubl | Key Functions |
|---|---|---|---|
| UBE2D1 | UbcH5a | Ubiquitin | Works with multiple RING E3s |
| UBE2N | Ubc13 | Ubiquitin | Forms K63-linked chains with UBE2V1 |
| UBE2C | UbcH10 | Ubiquitin | Cell cycle regulation |
| UBE2L3 | UbcH7 | Ubiquitin | Works with HECT E3s |
| UBC9 | - | SUMO | Sole SUMO E2 |
| UBE2M | Ubc12 | NEDD8 | Neddylation of cullins |
| UBE2Z | - | FAT10 | Immune regulation |
| UBCH8 | - | ISG15, Ubiquitin | Antiviral response |
E3 Ligases: Determinants of Substrate Specificity
E3 ubiquitin ligases function as the final enzymatic components in the conjugation cascade, providing substrate specificity through direct recognition of target proteins [15]. The human genome encodes an estimated 600-1,000 E3s, which can be broadly categorized into three major classes based on their structural features and mechanisms of action: Really Interesting New Gene (RING), U-box, and Homologous to E6AP C-Terminus (HECT) domain-containing ligases [20].
RING and U-box E3s function primarily as scaffolds that simultaneously bind charged E2s and substrate proteins, facilitating direct transfer of ubiquitin from the E2 to the substrate without forming a covalent E3-ubiquitin intermediate [21]. In contrast, HECT E3s employ a two-step mechanism where ubiquitin is first transferred from the E2 to an active-site cysteine within the HECT domain, forming a thioester intermediate, before final conjugation to the substrate [21]. This mechanistic diversity allows for precise temporal and spatial control over protein ubiquitylation, enabling specific regulation of countless cellular processes.
Evolutionary Origins and Conservation
The Ub/Ubl conjugation systems have their origins in prokaryotic biosynthetic pathways, with structural and mechanistic parallels observed in bacterial sulfur transfer systems [16] [22]. The bacterial proteins MoaD and ThiS, which share the characteristic Ubl fold, function in sulfur incorporation for the biosynthesis of molybdopterin and thiamine, respectively [16]. These proteins are activated by C-terminal acyl-adenylation through the bacterial enzymes MoeB and ThiF, which share significant sequence homology and structural similarity with the adenylation domain of eukaryotic E1s [16] [2].
The evolutionary relationship between these prokaryotic biosynthetic systems and eukaryotic Ubl conjugation pathways highlights the exaptation of ancient enzymatic mechanisms for novel regulatory functions in eukaryotic cells [22] [2]. The core β-grasp fold characteristic of Ub and Ubls appears in these prokaryotic antecedents, with the eukaryotic system expanding through gene duplication and functional diversification [3]. Surprisingly, proteins similar to Ubl-conjugating and Ubl-deconjugating enzymes appear to have already become widespread by the time of the last universal common ancestor, suggesting that Ubl-protein conjugation is not exclusively a eukaryotic invention [2].
The conservation of key mechanistic features across evolution is remarkable. The core adenylation chemistry, thioester transfer, and ultimate isopeptide bond formation remain fundamentally similar from prokaryotic sulfur transfer to eukaryotic protein modification [16]. This conservation underscores the functional efficiency of this enzymatic strategy and explains its adaptation for diverse regulatory purposes throughout eukaryotic evolution.
Experimental Approaches and Methodologies
Chemical Tools for Probing Conjugation Mechanisms
Recent advances in chemical biology have yielded sophisticated tools for studying the transient intermediates and complex dynamics of the Ub/Ubl conjugation cascades [19]. These approaches are essential because the E1-E2-E3 enzymatic cascades utilize labile thioester intermediates and short-lived protein-protein complexes that are often too elusive for direct observation under physiological conditions [19].
One powerful strategy involves the development of mechanism-based probes that trap specific intermediates in the activation and transfer process. These include ATP analogs, ubiquitin variants with modified C-termini, and oxime/hydrazide-based probes that form stable adducts with thioester intermediates [19]. Additionally, disulfide-trapping approaches using engineered E1 and E2 variants containing additional cysteine residues can stabilize otherwise transient E1-E2 complexes for structural characterization [19].
Phage display has emerged as a particularly valuable methodology for profiling the specificity of E1 enzymes toward the C-terminal sequences of Ub/Ubls [21]. This approach involves creating libraries of Ub variants with randomized C-terminal sequences displayed on phage surfaces, followed by selection based on the catalytic formation of thioester conjugates with E1 enzymes [21]. This method has revealed that while certain residues (particularly Arg72) are absolutely required for E1 recognition, other positions in the Ub C-terminus can accommodate substantial variation while maintaining E1 reactivity [21].
Structural Biology Techniques
Structural biology has been instrumental in elucidating the molecular mechanisms of the conjugation cascade. X-ray crystallography has provided high-resolution snapshots of various states in the E1 catalytic cycle, including the E1-UB/ATP complex, the ubiquitin-adenylate intermediate, and the E1-E2 transthioesterification complex [16]. These structures have revealed the dramatic conformational changes that occur during ubiquitin activation and transfer.
Cryo-electron microscopy (cryo-EM) has enabled visualization of larger complexes, including full-length E3 ligases bound to their cognate E2s and substrates. Nuclear magnetic resonance (NMR) spectroscopy has provided insights into the dynamics of these processes, particularly for transient complexes that are difficult to crystallize. Together, these techniques have revealed how E1s serve as molecular switches that undergo substantial conformational rearrangements to coordinate the different steps of Ubl activation and transfer to E2s [16].
Research Reagents and Tools
Table 3: Essential Research Reagents for Studying the Conjugation Cascade
| Reagent/Tool | Function/Application | Key Features |
|---|---|---|
| Phage-displayed UB library [21] | Profiling E1 specificity toward UB C-terminal sequences | Randomized residues 71-75 of UB; library size ~10^8 clones |
| Biotin-CoA conjugate [21] | Labeling PCP-E1 fusions via Sfp phosphopantetheinyl transferase | Enables immobilization of E1 enzymes for phage selection |
| ATP analogs (e.g., ATPγS) [19] | Trapping adenylate intermediates | Non-hydrolyzable or slowly hydrolyzable ATP variants |
| Ubiquitin C-terminal mutants [21] | Studying sequence requirements for E1 recognition and DUB resistance | Includes L73F, L73Y, G75S, G75D, G75N mutants |
| Disulfide-trapping E1/E2 variants [19] | Stabilizing transient E1-E2 complexes for structural studies | Engineered cysteine residues for covalent crosslinking |
| Activity-based E1 probes [19] | Monitoring E1 activation states and inhibitor screening | Covalently modify active site cysteine |
Therapeutic Targeting and Diagnostic Applications
The Ub/Ubl conjugation pathways represent promising therapeutic targets for numerous diseases, particularly cancer, neurodegenerative disorders, and viral infections [17]. The clinical success of the proteasome inhibitor bortezomib for multiple myeloma validation this approach and has stimulated intensive drug discovery efforts targeting various components of the conjugation machinery [17].
E1 enzymes have emerged as particularly attractive targets due to their position at the apex of the cascades. The NEDD8 E1 inhibitor MLN4924 (pevonedistat) has shown promising clinical activity by blocking cullin neddylation and thereby modulating the activity of cullin-RING E3 ligases [17]. Similarly, inhibitors targeting the SUMO E1 and ubiquitin E1 enzymes are under investigation for cancer therapy [17].
E2 enzymes offer another attractive point for therapeutic intervention, with their central role in determining ubiquitin chain topology and their more limited diversity compared to E3s [20]. E3 ligases present the greatest challenge for drug discovery due to their extensive diversity, but also offer the potential for exquisite specificity [17]. Small molecules targeting specific protein-protein interactions between E3s and their substrates are currently in development, with MDM2 inhibitors representing a prominent example [17].
Visualizing the Conjugation Cascade
The following diagram illustrates the core E1-E2-E3 enzymatic cascade and the key conformational changes that drive ubiquitin activation and transfer.
Diagram 1: The E1-E2-E3 Ubiquitin Conjugation Cascade. This diagram illustrates the sequential steps of ubiquitin activation and transfer, highlighting the key intermediates and conformational changes involved.
The subsequent diagram details an experimental workflow for phage display selection of ubiquitin variants based on their reactivity with E1 enzymes, a key methodology for profiling E1 specificity.
Diagram 2: Phage Display Workflow for Profiling E1 Specificity. This experimental methodology enables identification of ubiquitin C-terminal sequences that are reactive with E1 enzymes through iterative selection rounds.
The E1-E2-E3 conjugation cascade represents a sophisticated enzymatic system that has evolved from prokaryotic antecedents to become a central regulatory mechanism in eukaryotic cell biology. The structural and mechanistic insights gained from biochemical, structural, and chemical biology approaches have revolutionized our understanding of these pathways and created new opportunities for therapeutic intervention. Continued investigation of these systems will undoubtedly yield further insights into their biological functions and enhance our ability to target them for treating human disease.
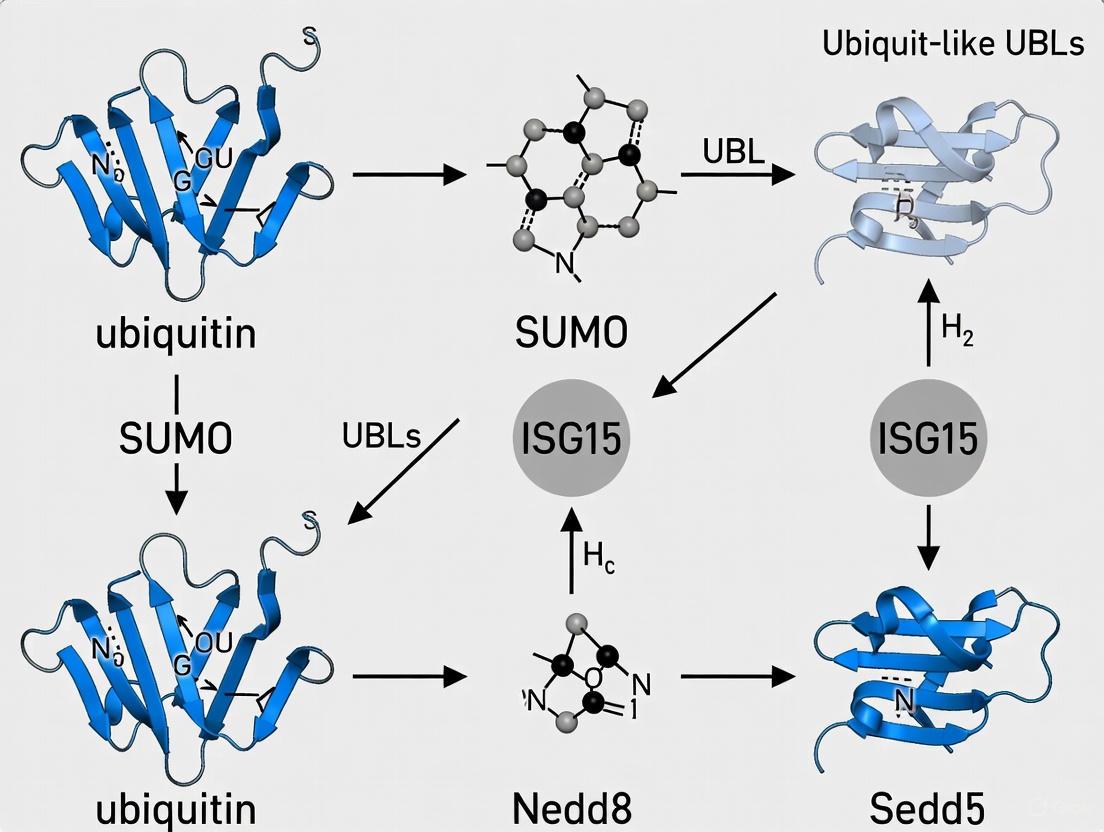
Ubiquitin-like proteins (UBLs) are a family of protein modifiers that share a characteristic three-dimensional structure known as the β-grasp fold and are conjugated to target substrates via a conserved enzymatic cascade [23] [24]. This family includes prominent members such as SUMO, NEDD8, ISG15, ATG8, and ATG12, which regulate a diverse array of cellular processes including transcription, cell cycle progression, DNA repair, autophagy, and immune responses [23] [24] [25]. The attachment of UBLs to cellular proteins represents a crucial post-translational modification mechanism that expands the functional diversity of the proteome, allowing cells to rapidly fine-tune protein activity, stability, localization, and interaction networks in response to genetic and environmental changes [2] [25].
The mechanistic conservation across UBL pathways is remarkable. Typically, UBL conjugation involves a three-step enzymatic cascade: an E1 activating enzyme catalyzes UBL activation in an ATP-dependent manner, an E2 conjugating enzyme accepts the activated UBL, and an E3 ligase facilitates the transfer of the UBL to specific target proteins [24] [26]. This modification is dynamic and reversible, with dedicated proteases responsible for UBL deconjugation and recycling [27]. While these pathways share mechanistic similarities, each UBL system maintains specificity through dedicated enzymatic machinery [24].
Table: Core Components of Major UBL Pathways
| UBL | E1 Activating Enzyme | E2 Conjugating Enzyme | E3 Ligase Examples | Primary Functions |
|---|---|---|---|---|
| SUMO | SAE1/SAE2 heterodimer | Ubc9 | PIAS family, RanBP2 | Nuclear transport, transcription, DNA repair |
| NEDD8 | UBA3-NAE1 heterodimer | Ubc12 | RBX1/2 | Cullin activation, ubiquitin ligase regulation |
| ISG15 | UBE1L | UbcH8 | HERC5, TRIM25 | Antiviral response, immune modulation |
| ATG12 | ATG7 | ATG10 | ATG5-ATG16 complex (E3-like) | Autophagosome formation |
| ATG8 | ATG7 | ATG3 | ATG12-ATG5-ATG16 complex | Autophagosome membrane expansion |
The UBL Conjugation Machinery
Structural and Mechanistic Conservation
The conjugation of UBLs to target proteins follows a conserved enzymatic pathway that begins with UBL activation. Structural studies reveal that E1 activating enzymes share a conserved domain architecture consisting of two pseudosymmetric adenylation domains that form a composite active site for ATP•Mg²⁺ and UBL binding, a catalytic cysteine domain that harbors the active-site cysteine needed for E1~UBL thioester bond formation, and the Ub-fold domain (UFD) that interacts with E2 proteins [24]. The E1 enzyme catalyzes adenylation of the UBL C-terminal glycine, followed by nucleophilic attack by its conserved active-site cysteine, forming an E1~UBL thioester intermediate [24]. A second UBL adenylation completes formation of the E1 ternary complex, which then catalyzes thioester transfer of the UBL to the conserved active-site cysteine of the E2 conjugating enzyme [24].
Significant conformational changes enable this process. During E1 activation, the CYS domain rotates approximately 130°, transiting the catalytic cysteine to a position proximal to the UBL C-terminal adenylate [24]. Similarly, a ~25° rotation of the UFD brings the E2 from a distal position to a proximal position suitable for thioester transfer [24]. These coordinated movements and disassembly of the adenylation active site help drive the reaction forward.
UBL-Specific Enzymatic Cascades
Each UBL employs a dedicated set of E1, E2, and E3 enzymes that confer pathway specificity:
SUMOylation: The heterodimeric E1 enzyme (SAE1/SAE2) activates SUMO, which is transferred to the sole E2 enzyme Ubc9 [28]. Ubc9 can directly recognize substrates through non-covalent interactions with SUMO molecules and may bypass E3 ligases under specific conditions, though E3s like the PIAS family and RanBP2 enhance specificity and efficiency [28].
NEDD8ylation: The NEDD8 E1 (UBA3-NAE1 heterodimer) activates NEDD8, which is transferred primarily to the E2 Ubc12 [24] [25]. The best-characterized NEDD8 targets are cullin proteins, which require neddylation for activation of their E3 ubiquitin ligase activity [25].
ISG15ylation: UBE1L serves as the E1 for ISG15, with UbcH8 as its primary E2 [2] [28]. ISG15 E3 ligases include HERC5 and TRIM25, which facilitate ISG15 conjugation to target proteins involved in antiviral defense [28].
ATG8 and ATG12 Conjugation: Both UBLs share the same E1 enzyme (ATG7) but have distinct E2s—ATG10 for ATG12 and ATG3 for ATG8 [23] [25]. The ATG12-ATG5 conjugate then functions as an E3-like complex to promote ATG8 lipidation [25].
Major UBL Families: Structures and Functions
SUMO (Small Ubiquitin-like Modifier)
SUMO proteins possess a molecular weight of approximately 11 kDa and contain a characteristic βββαβαβ fold with a C-terminal diglycine motif [28]. Mammals encode five SUMO paralogs (SUMO1-5) that exhibit both complementary and antagonistic roles in cellular regulation [28]. For instance, SUMO-1 enhances transcriptional activity by promoting the interaction between specificity protein-1 (Sp1) and histone acetyltransferase p300, while SUMO-2 disrupts this complex and destabilizes Sp1 [28].
SUMOylation influences numerous cellular processes through several mechanisms. It modulates protein-protein interactions via recognition by SUMO-interacting motifs (SIMs), alters subcellular localization, affects protein stability, and regulates transcriptional activity [28]. Unlike ubiquitination, SUMOylation does not typically target proteins for degradation but rather acts as a molecular switch that controls protein function, complex formation, and trafficking between nuclear and cytoplasmic compartments [28].
NEDD8 (Neural Precursor Cell Expressed, Developmentally Down-regulated 8)
NEDD8 shares approximately 55% sequence identity with ubiquitin and possesses a nearly identical three-dimensional structure, with the exception of two surface regions that mediate specialized interactions with target proteins [29]. The primary function of NEDD8 is the regulation of cullin-RING ubiquitin ligases (CRLs), which constitute the largest family of E3 ubiquitin ligases [25] [29].
Neddylation of cullin proteins induces a conformational change that activates CRLs by promoting their assembly and facilitating ubiquitin transfer to substrates [29]. Through this mechanism, NEDD8 indirectly regulates the ubiquitination and degradation of numerous proteins involved in cell cycle progression, signaling transduction, and transcription [25] [29]. The NEDD8 pathway is therefore intimately connected to the ubiquitin-proteasome system, serving as a critical regulator of its activity.
ISG15 (Interferon-Stimulated Gene 15)
ISG15 is a unique UBL consisting of two ubiquitin-like domains in tandem, with 32% and 37% identity to ubiquitin at its N- and C-termini, respectively [28] [25]. It is strongly induced by type I interferons and functions as a key mediator of antiviral immunity [2] [28]. ISG15 can exist as both a free molecule and a conjugated form, each with distinct biological activities.
As a covalent modifier, ISG15 conjugates to numerous target proteins in infected cells, modulating their function to establish an antiviral state [28]. Free ISG15 can act as a cytokine with immunomodulatory properties [28]. The conjugation of ISG15 to viral and host proteins can interfere with viral replication through various mechanisms, including blocking viral assembly, counteracting host restriction factors, and modulating immune signaling pathways [28].
ATG8 and ATG12 (Autophagy-related Proteins 8 and 12)
ATG8 and ATG12 are central regulators of autophagy, an intracellular degradation system that delivers cytoplasmic components to lysosomes for breakdown and recycling [23] [25]. ATG12 is conjugated to ATG5 in a ubiquitin-like reaction that requires the E1 enzyme ATG7 and the E2 enzyme ATG10 [25]. The ATG12-ATG5 conjugate then forms a complex with ATG16L1, which functions as an E3-like enzyme to promote the lipidation of ATG8 [25].
ATG8 is activated by ATG7 (E1) and transferred to ATG3 (E2) before being conjugated to the lipid phosphatidylethanolamine (PE) [25]. This lipidated form of ATG8 localizes to autophagosomal membranes, where it facilitates membrane expansion and curvature and serves as a docking site for receptors that recruit cargo destined for degradation [25]. The ATG8/ATG12 conjugation systems are essential for autophagosome biogenesis and selective autophagy, playing critical roles in cellular homeostasis, adaptation to stress, and elimination of intracellular pathogens [23].
Table: Functional Roles and Disease Associations of Major UBLs
| UBL | Key Biological Functions | Cellular Localization | Disease Associations |
|---|---|---|---|
| SUMO | Transcriptional regulation, genome stability, stress response, protein localization | Predominantly nuclear | Cancer, neurodegenerative disorders, heart diseases |
| NEDD8 | Activation of cullin-RING ligases, cell cycle regulation, transcription | Nuclear and cytoplasmic | Cancer (often overexpressed), targeted by therapeutic inhibitor MLN4924 |
| ISG15 | Antiviral defense, immune modulation, interferon signaling | Cytoplasmic, secreted form | Infectious diseases, autoimmune disorders |
| ATG12 | Autophagosome formation, mitochondrial homeostasis | Cytoplasmic, autophagosomal membranes | Neurodegenerative diseases, cancer, inflammatory disorders |
| ATG8/LC3 | Autophagosome membrane expansion, cargo recognition | Autophagosomal membranes, lipidated form | Neurodegenerative diseases, cancer, metabolic disorders |
Experimental Methods for UBL Research
The bioUbL Platform for Studying UBL Modifications
The bioUbL system represents a comprehensive set of tools for studying UBL modifications, based on multicistronic expression and in vivo biotinylation using the E. coli biotin protein ligase BirA [30]. This approach addresses several challenges in UBL research, including the cost and specificity of reagents, removal of UBLs by proteases, distinguishing UBL conjugates from interactors, and the low abundance of modified substrates [30].
The bioUbL methodology involves:
- Multicistronic vector design encoding three open reading frames separated by viral 2A sequences
- In vivo biotinylation of UBLs containing an N-terminal biotinylation-target peptide (Bio tag)
- Purification under denaturing conditions to inactivate deconjugating enzymes and preserve UBL modifications
- Stringent washes using the high-affinity biotin-streptavidin interaction to remove UBL interactors and non-specific background [30]
This system has been successfully applied in Drosophila cells, transgenic flies, and mammalian cells, identifying extensive sets of putative SUMOylated proteins and novel potential substrates for UFM1 [30]. The flexibility of this platform makes it a powerful complement to existing strategies for studying UBL modifications.
Proteomic Approaches for UBL Substrate Identification
Advanced proteomic techniques have been developed to detect and quantify UBL modifications:
- Enrichment strategies using epitope-tagged UBLs (e.g., 6xHIS, FLAG, HA) or specific antibodies enable isolation of UBL-conjugated proteins [30].
- Mass spectrometry analysis of tryptic peptides containing the characteristic Gly-Gly remnant left after digestion of UBL-modified substrates helps identify modification sites [23].
- The PRISM method (for SUMO) involves chemical blockade of all free lysines, treatment with SUMO-specific proteases, biotin-tagging of freed lysines, and identification by high-resolution mass spectrometry [30].
- Activity-based probes and inhibitors targeting UBL-specific enzymatic machinery facilitate capture, identification, and characterization of UBL pathway components [23].
Diagram: The Ubiquitin-Like Protein Conjugation Cascade. UBLs are first processed to expose their C-terminal glycine, then undergo E1-mediated activation, E2-mediated conjugation, and E3-mediated ligation to specific substrates, ultimately triggering diverse cellular responses.
Research Reagent Solutions for UBL Studies
Table: Essential Research Tools for UBL Pathway Investigation
| Reagent Type | Specific Examples | Primary Applications | Key Features |
|---|---|---|---|
| Activity-based Probes | Suicide substrates, Ub/Ubl-adenylate analogs | Enzyme mechanism studies, inhibitor screening | Covalently trap intermediate states, enable structural studies |
| Biotinylation Tools | bioUbL system, BirA ligase, AviTag | Purification under denaturing conditions, MS analysis | High-affinity streptavidin binding, minimal background |
| Specific Inhibitors | MLN4924 (NEDD8 E1), PYR-41 (Ub E1) | Pathway validation, therapeutic development | Mechanism-based inhibition, target specificity |
| Protease-resistant Mutants | SUMO2-T90R (MS optimization) | Proteomic identification of modification sites | Enhanced peptide detection, reduced ambiguity |
| Chain Formation Mutants | SUMO2-K11R (chain formation study) | Elucidating functions of poly-Ubl chains | Disrupted specific lysine for chain formation |
| Tagged UBL Variants | 6xHIS-FLAG, 6xHIS-HA, FLAG-TEV | Affinity purification, interactome studies | Compatibility with multiple detection methods |
Therapeutic Targeting of UBL Pathways
Dysregulation of UBL pathways is implicated in various human diseases, making them attractive therapeutic targets. In cancer, abnormal UBL activity contributes to uncontrolled proliferation, evasion of apoptosis, and metastasis [25] [29]. Notable examples include:
NEDD8 pathway inhibition: MLN4924 (Pevonedistat) inhibits the NEDD8 E1 activating enzyme, preventing cullin neddylation and subsequent ubiquitin ligase activation [29]. This agent has demonstrated anticancer activity in clinical trials by inducing cell cycle arrest and apoptosis [29].
SUMOylation in cancer: SUMO pathway components are frequently overexpressed in malignancies, and SUMO inhibition sensitizes cancer cells to chemotherapy and radiation [25].
NUB1 as a tumor suppressor: The protein NEDD8 ultimate buster 1 (NUB1) negatively regulates the NEDD8 conjugation system and recruits both NEDD8- and FAT10-conjugated proteins to the proteasome for degradation [29]. NUB1 exhibits growth-inhibitory properties and induces apoptosis in cancer cells, particularly in renal cell carcinoma [29].
Beyond oncology, UBL pathways are being investigated as therapeutic targets for neurodegenerative disorders and cardiac diseases [25] [26]. In cardiovascular pathology, insufficient ubiquitin-mediated proteolysis contributes to the accumulation of toxic protein aggregates in cardiomyocytes, while modulation of SUMOylation and NEDD8 pathways affects cardiac hypertrophy and ischemic injury [26].
Diagram: Therapeutic Strategies for Targeting Dysregulated UBL Pathways. Multiple intervention points exist for correcting aberrant UBL signaling, including E1 inhibition, disruption of E2-E3 interactions, modulation of deconjugating enzymes, and enhancement of natural negative regulators like NUB1.
The major UBL families—SUMO, NEDD8, ISG15, ATG8, and ATG12—represent crucial regulatory systems that expand the functional capacity of the proteome through reversible protein modification. Each UBL pathway employs a conserved enzymatic cascade while maintaining specificity through dedicated enzymes and regulatory mechanisms. Continued advances in research tools, particularly proteomic platforms like the bioUbL system and highly specific inhibitors, are accelerating our understanding of these complex pathways. The therapeutic targeting of UBL systems in cancer and other diseases highlights the translational importance of fundamental research in this field. As our knowledge of UBL structure, function, and regulation expands, so too will opportunities for manipulating these pathways for therapeutic benefit.
The ubiquitin-like protein (UBL) system, a hallmark of eukaryotic cellular regulation, did not originate de novo but evolved from ancient prokaryotic precursors. This whitepaper synthesizes current evidence demonstrating that UBLs and their conjugation machinery originated from sulfur carrier systems and enzyme cofactor biosynthesis pathways present in the last universal common ancestor (LUCA). Structural and phylogenetic analyses reveal that modern UBLs, including ubiquitin itself, evolved from the β-grasp fold (β-GF) that initially functioned in RNA binding and sulfur transfer in prokaryotic metabolic pathways. The conservation of this structural architecture over billions of years, coupled with its functional diversification from basic metabolic roles to sophisticated regulatory signaling, represents a remarkable evolutionary trajectory. Understanding these origins provides critical insights for drug development targeting UBL pathways in cancer, neurodegenerative diseases, and autoimmune disorders.
Ubiquitin-like proteins constitute a family of protein modifiers that regulate virtually every aspect of eukaryotic cellular function through post-translational modification of target proteins. The canonical ubiquitin protein comprises 76 amino acids and adopts a compact globular structure characterized by a central α-helix embraced by five β-strands, forming the distinctive β-grasp domain [31] [32]. This structural motif is shared across UBLs, despite considerable sequence divergence among family members.
The UBL conjugation system operates through a conserved enzymatic cascade involving E1 (activating), E2 (conjugating), and E3 (ligating) enzymes [32] [33]. This system facilitates the covalent attachment of UBLs to target proteins, modulating their activity, stability, localization, and interactions. The human genome encodes numerous UBLs, including SUMO, NEDD8, ATG8, ATG12, URM1, UFM1, Ubl5/Hub1, FUB1, FAT10, and ISG15, each with specialized cellular functions [31]. The evolutionary transformation of this system from prokaryotic metabolic functions to eukaryotic regulatory roles represents one of the most significant developments in cellular evolution.
Structural Conservation and the β-Grasp Fold
The Ubiquitin Superfold
The β-grasp fold represents an ancient structural motif that predates the divergence of prokaryotes and eukaryotes. Comprehensive structural analyses indicate that the β-GF differentiated into at least seven distinct clades by the time of LUCA, encompassing much of the structural diversity observed in modern versions [13]. This fold is characterized by a five-strand β-sheet (five antiparallel β-strands) and a single α-helix at the apex, forming a compact globular domain that has been recruited for strikingly diverse biochemical functions [31] [13].
Table 1: Diversity of β-Grasp Fold Functions Across Evolution
| Organismal Domain | Representative Functions | Example Proteins/Domains |
|---|---|---|
| Prokaryotes | Catalytic roles, RNA binding, sulfur transfer, co-factor binding | NUDIX hydrolases, MoaD, ThiS, Molybdopterin synthases |
| Eukaryotes | Protein modification, signaling, autophagy, immune response | Ubiquitin, SUMO, NEDD8, ATG8, ATG12 |
| Universal | Scaffolding iron-sulfur clusters, mediation of protein interactions | Various UDPs (Ubiquitin-like Domain Proteins) |
Structural Phylogeny of UBLs
The structural conservation between prokaryotic sulfur carriers and eukaryotic UBLs provides compelling evidence for their evolutionary relationship. Solution NMR structure of Urm1 from Saccharomyces cerevisiae has revealed it as a unique "molecular fossil" that preserves the most conserved structural and sequence features of the common ancestor of the entire ubiquitin superfamily [34]. Striking similarities in 3D structure and hydrophobic and electrostatic surface features exist between Urm1 and MoaD (molybdopterin synthase small subunit), suggesting they interact with partners in analogous manners [34].
The structural resemblance extends beyond individual proteins to functional complexes. Notably, similarities between the Urm1-Uba4 and MoaD-MoeB pairs establish an evolutionary link between ATP-dependent protein conjugation in eukaryotes and ATP-dependent cofactor sulfuration in prokaryotes [34]. This connection reveals how the eukaryotic UBL system co-opted existing structural frameworks from prokaryotic metabolic pathways.
Evolutionary Trajectory from Prokaryotic Systems
Origins in Sulfur Transfer and Cofactor Biosynthesis
The evolutionary origin of UBL conjugation systems can be traced to prokaryotic pathways involved in sulfur transfer and enzyme cofactor biosynthesis. Comparative genomic analyses indicate that precursors of the eukaryotic Ub-system were already present in prokaryotes, with the most basic versions combining a Ubl and an E1-like enzyme involved in metabolic pathways related to metallopterin, thiamine, cysteine, siderophore, and modified base biosynthesis [13]. These primordial systems primarily functioned in the biosynthesis of essential enzyme cofactors through sulfur transfer mechanisms.
The Urm1 pathway represents a particularly significant evolutionary link, as it shares characteristics with both prokaryotic sulfur carrier systems and modern eukaryotic UBL pathways. Urm1 is involved in both thiolation of transfer RNAs and protein conjugation, serving as a functional bridge between these distinct biochemical processes [34]. This dual functionality provides a plausible evolutionary pathway for the transition from metabolic to regulatory functions.
Development of Conjugation Machinery
The evolution of the complete UBL conjugation machinery (E1-E2-E3) occurred through a process of gene duplication, fusion, and functional specialization. Prokaryotic systems containing Ubls of the YukD and other families, including some very close to Ub itself, developed additional elements that more closely resemble the eukaryotic state in possessing an E2 conjugating enzyme, a RING-type E3 ligase, or both [13]. These findings demonstrate that the fundamental components of the UBL system were already emerging in prokaryotic lineages.
Table 2: Evolutionary Development of UBL Conjugation Machinery
| Evolutionary Stage | Key Components | Functional Capabilities |
|---|---|---|
| Primordial Prokaryotic | Ubl + E1-like enzyme | Sulfur transfer for cofactor biosynthesis |
| Intermediate Prokaryotic | Ubl + E1 + E2/E3 elements | Limited protein modification |
| Early Eukaryotic | Multiple UBL families + E1-E2-E3 cascade | Diverse protein tagging functions |
| Modern Eukaryotic | Expanded UBL families + Complex E3 ligases | Sophisticated regulatory networks |
The evolutionary trajectory involved both the conservation of a universal structural fold of UBLs and the rise in complexity of the superfamily of ligases that conjugate UBLs to substrates. This complexity increased in terms of the number of enzyme variants, structural organization diversity, and diversification of catalytic domains [31]. The E2 ubiquitin-conjugating enzymes of metazoan phyla are highly conservative, whereas the homology of E3 ubiquitin ligases with human orthologues gradually decreases depending on evolutionary distance and "molecular clock" timing [31].
Experimental Approaches for Studying UBL Evolution
Structural Biology Techniques
Solution NMR Spectroscopy has been instrumental in elucidating the evolutionary relationships between UBLs and prokaryotic sulfur carriers. The protocol for determining the solution structure of Urm1 involved:
- Protein Expression and Purification: Recombinant Urm1 was expressed in E. coli and purified using affinity and size-exclusion chromatography.
- NMR Data Collection: A comprehensive set of NMR experiments including (^1\text{H})-(^{15}\text{N}) HSQC, (^1\text{H})-(^{13}\text{C}) HSQC, HNCA, HNCOCA, HNCACB, and CBCACONH were performed to obtain backbone and side-chain assignments.
- Structure Calculation: Distance constraints from NOESY spectra combined with torsion angle constraints were used to calculate the three-dimensional structure using simulated annealing protocols in programs such as X-PLOR or CYANA.
- Structural Comparison: The solved structure was compared with known structures of prokaryotic sulfur carriers (e.g., MoaD) and other UBLs using structural alignment algorithms like DALI or VAST.
X-ray Crystallography has provided additional insights into the structural conservation of the β-grasp fold across evolution. Recent structural studies of E1 enzymes have revealed their conserved architecture and the dynamic changes that accompany UBL activation and transfer [32]. These structures build upon foundational biochemical research to elucidate the determinants of activity, specificity, and novel regulatory mechanisms governing UBL conjugation.
Phylogenomic Analysis
Comparative Genomics approaches have been essential for tracing the evolutionary history of UBL systems:
- Sequence Database Mining: Iterative BLAST searches using known UBLs and conjugation enzymes as queries against diverse taxonomic groups.
- Profile-based Methods: Hidden Markov Models (HMMs) and PSI-BLAST to detect distant homologs that may have diverged significantly in sequence while retaining structural and functional similarity.
- Phylogenetic Tree Construction: Multiple sequence alignment of identified homologs followed by maximum likelihood or Bayesian inference methods to reconstruct evolutionary relationships.
- Synteny Analysis: Examination of genomic context and conservation of gene neighborhoods to identify functional associations and evolutionary events.
These methods have revealed that the β-grasp fold first emerged in the context of translation-related RNA-interactions and subsequently diversified to occupy various functional niches [13]. Most biochemical diversification of the fold occurred in prokaryotes, with the eukaryotic phase of its evolution mainly marked by the expansion of the Ubl clade of the β-GF [13].
Biochemical and Functional Assays
Enzyme Kinetics Studies have been crucial for understanding the mechanistic evolution of UBL activation. Foundational biochemical research revealed that activation of ubiquitin follows a pseudo-ordered substrate binding mechanism, where ATP-Mg(^{2+}) preferentially binds first, followed by ubiquitin [32]. Kinetic analysis of thioester formation, combined with site-directed mutagenesis of conserved residues like Asp576 in human UBA1, has demonstrated this preferential binding and provided insights into the evolution of enzyme specificity.
In Vitro Reconstitution Assays have allowed researchers to test the functional capabilities of putative ancestral UBL systems. These experiments typically involve:
- Purified Component Incubation: Combining recombinant UBL, E1, E2, and E3 enzymes with ATP and potential substrate proteins.
- Reaction Monitoring: Using techniques like western blotting, mass spectrometry, or fluorescence-based assays to detect UBL conjugation.
- Cross-species Complementation: Testing whether components from evolutionarily distant organisms can function together, providing evidence for conserved mechanisms.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Investigating UBL Evolution
| Reagent/Category | Specific Examples | Research Application | Evolutionary Insight Provided |
|---|---|---|---|
| Recombinant Proteins | Urm1, MoaD, ThiS, MoeB | Structural comparison, biochemical assays | Reveals conserved structural features and functional mechanisms |
| Antibodies | Anti-ubiquitin, anti-SUMO, anti-Urm1 | Detection of UBL conjugates in diverse organisms | Identifies conservation of modification patterns across species |
| NMR Isotope Labels | (^{15}\text{NH}_4\text{Cl}), (^{13}\text{C})-glucose | Protein structure determination | Enables atomic-level structural comparisons between modern and ancestral forms |
| Phylogenetic Software | MEGA, PhyML, RAxML | Evolutionary tree construction | Reconstructs evolutionary relationships and divergence times |
| Structural Alignment Tools | DALI, VAST, PyMOL | 3D structure comparison | Quantifies structural conservation despite sequence divergence |
| Activity-Based Probes | HA-Ub-VS, SUMO-AMC | Enzyme activity profiling | Traces functional conservation of conjugation machinery |
Implications for Human Disease and Therapeutic Development
The evolutionary perspective on UBL systems provides valuable insights for drug development targeting these pathways in human disease. The high conservation of UBL components from prokaryotes to humans validates their fundamental importance in cellular function and supports their relevance as therapeutic targets. Dysregulation of UBL pathways has been implicated in diverse pathological conditions, including cancer, neurodegenerative diseases, autoimmune disorders, and developmental syndromes [32] [33].
The E1 enzymes that initiate UBL activation have emerged as particularly attractive therapeutic targets. As the gatekeepers of UBL cascades, E1s represent potential choke points for pharmacological intervention. Current research is exploring E1 inhibitors as potential anticancer therapies, leveraging the evolutionary conservation of their catalytic mechanisms while exploiting structural differences for specificity [32]. Understanding the evolutionary origins of these enzymes provides a framework for predicting potential side effects and assessing conservation across therapeutic models.
Neurodegenerative diseases exemplify the pathological consequences of UBL system dysfunction. Reduced expression of UBA1, the major E1 enzyme for ubiquitin, has been reported in Huntington's disease, while rare missense mutations that impair UBA1 function cause X-linked infantile spinal muscular atrophy [32]. The essential nature of UBA1 is evidenced by the embryonic lethality of complete knockout in mice, highlighting the critical role of this evolutionarily ancient enzyme in neuronal health [32].
Future Directions and Research Opportunities
Several promising research directions emerge from our current understanding of UBL evolution:
- Ancestral Protein Reconstruction: Resurrecting putative ancestral UBLs and conjugation enzymes to experimentally test evolutionary hypotheses about their structure and function.
- Metagenomic Mining: Exploring the diversity of UBL systems in uncultured prokaryotes and microbial dark matter to fill gaps in the evolutionary record.
- Comparative Mechanistic Studies: Detailed biochemical characterization of UBL conjugation across diverse taxonomic groups to identify universally conserved versus lineage-specific mechanisms.
- Integrative Modeling: Combining evolutionary analysis with structural biology and systems biology to develop comprehensive models of UBL system evolution.
These approaches will continue to illuminate how simple prokaryotic sulfur carriers transformed into the sophisticated regulatory systems that govern eukaryotic cell biology, with important implications for understanding disease mechanisms and developing novel therapeutic strategies.
The evolutionary journey from prokaryotic sulfur carriers to eukaryotic UBL regulatory systems represents a remarkable example of functional diversification built upon conserved structural foundations. The β-grasp fold, originating in LUCA for basic metabolic functions, was progressively elaborated through evolution to create the sophisticated UBL conjugation machinery that regulates virtually every aspect of eukaryotic biology. The Urm1 protein stands as a pivotal "molecular fossil" that bridges the evolutionary gap between sulfur metabolism in prokaryotes and protein modification in eukaryotes. This evolutionary perspective not only deepens our understanding of cell biology but also provides valuable insights for therapeutic intervention in the numerous diseases associated with UBL pathway dysfunction. As research continues to unravel the complexities of UBL evolution, we can anticipate new opportunities for targeting these ancient systems in modern medicine.
Ubiquitin-like proteins (UBLs) constitute a family of small proteins involved in the post-translational modification of other cellular proteins, thereby playing crucial regulatory roles in virtually all eukaryotic cellular processes. The UBL protein family derives its name from its first discovered member, ubiquitin (Ub), which is best known for its central role in regulating protein degradation via the proteasome. Following ubiquitin's discovery, numerous structurally and evolutionarily related proteins have been identified, all sharing a characteristic three-dimensional core structure known as the β-grasp fold [1] [35]. This fold consists of a five-strand antiparallel beta sheet surrounding a central alpha helix, providing a stable platform for interactions with various enzymatic machineries and target proteins [1].
UBLs are fundamentally categorized into two types based on their capacity for covalent conjugation. Type I UBLs can be covalently attached to target proteins (or lipids) through an enzymatic cascade analogous to the ubiquitin pathway. This process typically requires proteolytic activation of the UBL to expose a C-terminal glycine residue, followed by sequential action of E1 (activating), E2 (conjugating), and E3 (ligating) enzymes specific to each UBL family [1] [3]. In contrast, Type II UBLs are not conjugated but often exist as domains within larger multidomain proteins, where they frequently mediate protein-protein interactions [1]. The functional spectrum of UBL modifications is remarkably diverse, encompassing roles in autophagy, protein trafficking, inflammation and immune responses, transcription, DNA repair, RNA splicing, and cellular differentiation [1] [36].
The evolutionary history of UBLs reveals a fascinating journey from prokaryotic precursor systems to the elaborate eukaryotic regulatory networks observed today. mounting evidence indicates that UBL-protein modification evolved from prokaryotic sulfurtransferase systems or related enzymes involved in cofactor biosynthesis [35]. Surprisingly, proteins structurally and mechanistically similar to UBL-conjugating and deconjugating enzymes appear to have been widespread by the time of the last universal common ancestor, challenging the long-held notion that UBL-protein conjugation is exclusively a eukaryotic invention [35]. This review provides a comprehensive analysis of the genomic distribution of UBL systems across humans, plants, and prokaryotes, highlighting both conserved features and lineage-specific adaptations.
Comparative Genomic Analysis of UBL Families
UBL Distribution in Humans
The human genome encodes a sophisticated and diversified array of UBL conjugation systems. current genomic analyses identify at least eight distinct families of Type I UBLs in humans, in addition to ubiquitin itself, that are experimentally confirmed to form covalent modifications on target proteins [1] [3]. These include SUMO (Small Ubiquitin-like Modifier), NEDD8 (Neural precursor cell-expressed, Developmentally down-regulated 8), ATG8 and ATG12 (both involved in autophagy), URM1 (Ubiquitin Related Modifier 1), UFM1 (Ubiquitin Fold Modifier 1), FAT10 (HLA-F Adjacent Transcript 10), and ISG15 (Interferon-Stimulated Gene 15) [1].
Table 1: Major UBL Families in Homo sapiens
| UBL Family | Representative Members | E1 Activating Enzyme | E2 Conjugating Enzyme | Primary Cellular Functions |
|---|---|---|---|---|
| Ubiquitin | Ubiquitin | UBA1, UBA6 | ~40 different E2s | Protein degradation, signaling, trafficking |
| SUMO | SUMO1, SUMO2, SUMO3, SUMO4 | UBA2/SAE1 | UBC9 | Transcription, DNA repair, nuclear transport |
| NEDD8 | NEDD8 | UBA3/NAE1 | UBC12, UBE2F | Regulation of cullin-RING ligases |
| ATG8 | LC3A, LC3B, GABARAP, etc. | ATG7 | ATG3 | Autophagosome formation |
| ATG12 | ATG12 | ATG7 | ATG10 | Autophagy regulation |
| URM1 | URM1 | UBA4 | - | Thiolation, oxidative stress response |
| UFM1 | UFM1 | UBA5 | UFC1 | ER homeostasis, erythroid development |
| FAT10 | FAT10 | UBA6 | UBE2Z | Immune response, proteasomal degradation |
| ISG15 | ISG15 | UBA7 | UBCH8 | Antiviral response, interferon signaling |
Notably, some human UBL families have undergone significant gene expansion. while a single SUMO gene exists in lower eukaryotes like yeast, humans possess at least four functional SUMO genes (SUMO1-4) with partially overlapping functions [1] [35]. similarly, the ATG8 family in humans has expanded to include seven members categorized as LC3A, LC3B, LC3B2, LC3C, GABARAP, GABARAPL1, and GATE-16, each with potentially specialized roles in different forms of autophagy [3]. This diversification enables nuanced regulation of cellular processes through tissue-specific, developmental stage-specific, or stress-responsive expression of different UBL paralogs.
The genes encoding UBLs exhibit varied genomic organizations. some, like those for ubiquitin, are often arranged as head-to-tail tandem repeats or fused with ribosomal proteins, requiring proteolytic processing to generate the mature UBL [3]. others, such as UFM1, ISG15, NEDD8, SUMO, and ATG8, are translated as preproteins that must be C-terminally processed by specific proteases to expose the active glycine residue essential for conjugation [3]. in contrast, ATG12, FAT10, and URM1 are genetically encoded in their mature forms, ready for activation [3].
UBL Distribution in Plants
Plant genomes harbor a complex repertoire of UBL systems that reflect both conserved eukaryotic features and plant-specific adaptations. comprehensive genomic surveys have identified at least seven major families of Type I UBLs in plants in addition to ubiquitin: SUMO, RUB (the plant homolog of NEDD8), ATG8, ATG12, MUB (Membrane-anchored UBL), UFM1, and HUB1 [1]. interestingly, some UBL families in plants have undergone dramatic expansion, likely due to both whole-genome duplication events and other forms of gene duplication [1].
Table 2: UBL Distribution Across Evolutionary Lineages
| UBL Family | Human | Plants (e.g., Arabidopsis) | Prokaryotes |
|---|---|---|---|
| Ubiquitin | Yes (multiple genes) | Yes (multiple genes) | Limited (certain archaea) |
| SUMO | Yes (4 isoforms) | Yes (≥8 in Arabidopsis) | No |
| NEDD8/RUB | Yes | Yes | No |
| ATG8 | Yes (7 isoforms) | Yes (expanded family) | No |
| ATG12 | Yes | Yes | No |
| URM1 | Yes | Not reported | Related proteins (ThiS, MoaD) |
| UFM1 | Yes | Yes | No |
| FAT10 | Yes | No | No |
| ISG15 | Yes | No | No |
| SAMPs/Pup | No | No | Yes (various forms) |
Genomic analyses reveal that the ubiquitin, SUMO, ATG8, and MUB families collectively account for nearly 90% of all UBL genes in plant genomes [1]. for instance, the model plant Arabidopsis thaliana possesses at least eight SUMO genes, significantly more than the four found in humans, suggesting substantial diversification of SUMO-mediated regulatory pathways in plants [1]. this expansion may reflect the need for sophisticated regulation of plant-specific processes such as complex developmental patterning, specialized stress responses, and symbiotic interactions.
proteins associated with both ubiquitin and SUMO signaling pathways are highly enriched in the genomes of embryophytes (land plants), indicating their crucial roles in plant adaptation to terrestrial environments [1]. the MUB UBL family, with its C-terminal prenylation motif, represents a plant-specific adaptation for membrane association, highlighting how UBL families can evolve distinct functional specializations in different phylogenetic lineages [1].
UBL Distribution in Prokaryotes
while traditionally considered a eukaryotic innovation, UBL-related proteins and conjugation systems do exist in prokaryotes, though they are phylogenetically restricted and generally less complex than their eukaryotic counterparts [1] [35]. the most widespread prokaryotic UBL is Pup (prokaryotic ubiquitin-like protein), found in certain actinobacteria, which serves a function analogous to ubiquitin in tagging proteins for proteasomal degradation [1]. however, unlike ubiquitin, Pup is intrinsically disordered, and its evolutionary relationship to eukaryotic UBLs remains unclear [1].
more recently, a related protein termed UBact has been identified in some gram-negative bacterial lineages [1]. in contrast to Pup, the protein TtuB in Thermus thermophilus shares the characteristic β-grasp fold with eukaryotic UBLs and has been reported to serve dual functions as both a sulfur carrier and a covalently conjugated protein modifier [1]. in archaea, SAMPs (small archaeal modifier proteins) possess the β-grasp fold and play ubiquitin-like roles in protein modification and degradation [1] [3].
perhaps most remarkably, seemingly complete sets of genes corresponding to a eukaryote-like ubiquitin pathway have been identified in certain archaeal lineages, including "Euryarchaeota," Thermoproteota (formerly Crenarchaeota), and "Aigarchaeota" [1]. the discovery of an operon-like cluster in Candidatus Caldiarchaeum subterraneum encoding a UBL alongside proteins structurally related to eukaryotic E1, E2, and E3 enzymes provides compelling evidence that the basic components of UBL conjugation systems predate the emergence of eukaryotes [3].
the evolutionary link between eukaryotic UBLs and prokaryotic systems is further strengthened by the structural and mechanistic similarities between ubiquitin-activating enzymes (E1s) and bacterial enzymes involved in sulfur mobilization, such as ThiF and MoeB, which participate in the biosynthesis of the cofactors thiamine and molybdopterin [1] [35]. the eukaryotic UBL URM1, which functions both as a protein modifier and a sulfur carrier, has been described as a "molecular fossil" that bridges these evolutionary pathways [1].
Evolutionary Perspectives on UBL Genomic Distribution
the comparative genomic analysis of UBL systems across the tree of life reveals several key evolutionary patterns. first, eukaryotic UBLs are monophyletic, indicating a shared evolutionary origin from a common progenitor [1]. second, while the core conjugation machinery was established early in cellular evolution, different lineages have experienced both gene loss and gene family expansion, resulting in the specialized UBL repertoires observed in modern organisms [1] [3].
the evolutionary trajectory of UBL systems demonstrates a clear trend toward increased complexity in eukaryotic lineages, particularly in multicellular organisms. this is evident in the expansion of UBL gene families (e.g., SUMO and ATG8 in humans, SUMO in plants) and the emergence of entirely new UBL families dedicated to specialized functions, such as the immune-related UBLs FAT10 and ISG15 in vertebrates [1] [3]. the absence of these immune-specific UBLs from lower eukaryotes and prokaryotes underscores how UBL systems have been adapted to meet lineage-specific physiological requirements.
the presence of simplified UBL conjugation systems in archaea suggests that the evolutionary transition from prokaryotic to eukaryotic UBL systems involved both vertical inheritance and extensive elaboration. the endosymbiotic events that gave rise to mitochondria and chloroplasts may have facilitated the transfer and integration of UBL-related genes between evolutionary lineages, contributing to the diversification of UBL functions in eukaryotes [1].
Experimental Approaches for Studying UBL Genomics and Function
Genomic Mining and Phylogenetic Analysis
Protocol 1: Identification and Annotation of UBL Genes in Genomic Sequences
- Sequence Retrieval: Obtain complete genomic sequences from databases such as NCBI Genome, Ensembl, or Phytozome.
- Homology Searching:
- Perform BLASTP or TBLASTN searches using known UBL protein sequences as queries.
- Use profile hidden Markov models (HMMs) from databases like Pfam (PF00240 for ubiquitin family) to identify UBL domains.
- Gene Prediction: Employ ab initio gene prediction algorithms (e.g., AUGUSTUS, GeneMark) combined with homology-based methods to identify complete coding sequences.
- Domain Architecture Analysis: Use tools like InterProScan to identify conserved domains and classify UBL types.
- Phylogenetic Reconstruction:
- Perform multiple sequence alignment using MAFFT or Clustal Omega.
- Construct phylogenetic trees with maximum likelihood (RAxML) or Bayesian (MrBayes) methods.
- Calibrate evolutionary timescales using fossil records or molecular dating approaches.
Comprehensive Platform for UBL Modification Analysis
Protocol 2: In Vivo Biotinylation System for UBL Conjugate Isolation [37]
This protocol enables efficient isolation and identification of UBL conjugates under denaturing conditions, which inactivates deconjugating enzymes and preserves modification states.
Vector Construction:
- Generate multicistronic expression vectors encoding:
- ORF1: Biotinylation target peptide (Bio tag) followed by the UBL of interest
- ORF2: E. coli biotin protein ligase (BirA)
- ORF3: Fluorescent marker (e.g., GFP) for transfection monitoring
- Separate ORFs using viral 2A self-cleaving peptide sequences.
- Generate multicistronic expression vectors encoding:
Cell Transfection and Biotinylation:
- Transfect mammalian cells (e.g., HEK293T) or Drosophila cells using appropriate methods.
- Culture cells for 24-48 hours to allow expression and in vivo biotinylation.
- Verify biotinylation efficiency by streptavidin blotting.
Denaturing Lysis and Affinity Purification:
- Lyse cells in 8M urea or 1% SDS buffer to denature proteins and inhibit UBL proteases.
- Incubate lysates with streptavidin-coated beads for 2-4 hours.
- Wash beads stringently with urea-containing buffers to remove non-specific interactors.
Analysis of UBL Conjugates:
- Elute bound proteins with Laemmli buffer containing 2mM biotin.
- Identify conjugates by immunoblotting with UBL-specific antibodies.
- For proteomic identification, perform on-bead tryptic digestion and analyze by LC-MS/MS.
Diagram 1: Experimental workflow for bioUbL conjugate analysis showing key steps from vector design to conjugate characterization.
Chemical Biology Approaches for UBL Study
Protocol 3: Chemical Synthesis of UBLs and Their Conjugates [38]
Chemical synthesis enables precise control over UBL structure and incorporation of specific modifications, overcoming limitations of enzymatic methods.
Solid-Phase Peptide Synthesis (SPPS):
- Synthesize UBL fragments (typically 30-50 amino acids) using Fmoc/t-Bu chemistry.
- Incorporate non-hydrolyzable isopeptide linkage mimics (e.g., thioether, triazole) for stable conjugate analogs.
- Include specially modified amino acids (phosphorylated, acetylated) at desired positions.
Native Chemical Ligation (NCL):
- Combine peptide fragments containing C-terminal thioester and N-terminal cysteine via NCL.
- Perform ligation in 6M guanidinium HCl, 0.1M phosphate buffer, pH 7.0, with 2% thiophenol.
- Incubate at 25-37°C for 12-24 hours.
Desulfurization/Deselenization:
- For cysteine-free UBLs, perform metal-free desulfurization using VA-044 radical initiator.
- Use glutathione and TCEP in 6M guanidinium HCl, pH 6.8-7.2, at 37°C for 2-4 hours.
Folding and Purification:
- Refold synthesized UBLs by dialysis from 6M guanidinium HCl to physiological buffer.
- Purify by reverse-phase HPLC using C4 or C8 columns.
- Verify correct folding by NMR spectroscopy or circular dichroism.
Table 3: Key Research Reagent Solutions for UBL Studies
| Reagent/Resource | Type | Primary Function | Examples/Specifics |
|---|---|---|---|
| BioUbL System | Multicistronic vector platform | In vivo biotinylation and isolation of UBL conjugates | bioSUMO, bioNEDD8, bioUFM1; enables purification under denaturing conditions [37] |
| Chemical Synthesis Tools | Synthetic peptides and proteins | Generation of site-specifically modified UBLs and conjugates | NCL, KAHA ligation, SPPS; allows incorporation of non-hydrolyzable linkages, PTMs [38] |
| Activity-Based Probes (ABPs) | Chemical probes with electrophilic traps | Profiling UBL-specific protease activity and identification of DUBs/ULPs | Ub-/SUMO-vinyl sulfones, -aldehydes; covalent modification of active site cysteines [38] [27] |
| ULP/DUB Inhibitors | Small molecule inhibitors | Specific inhibition of deconjugating enzymes to stabilize UBL modifications | MLN4924 (NEDD8 activation inhibitor), ginkgolic acid (SUMOylation inhibitor) [39] |
| Chain-Specific Antibodies | Monoclonal antibodies | Detection of specific polyUb/UBL chain types | K48- and K63-linkage specific Ub antibodies; SUMO2/3-specific antibodies [37] |
| Tandem Ubiquitin/Ubl Binding Entities (TUBEs) | Engineered binding proteins | Affinity enrichment of Ub/UBL conjugates from native lysates | Multi-domain constructs with high affinity for Ub/UBLs; preserve labile modifications [37] |
Diagram 2: Decision framework for selecting UBL research reagents based on experimental goals and applications.
the genomic distribution of UBL systems across humans, plants, and prokaryotes reveals a fascinating evolutionary narrative of conservation, diversification, and lineage-specific adaptation. while the core β-grasp fold and fundamental conjugation mechanisms remain conserved across eukaryotes, different lineages have expanded and specialized their UBL repertoires to meet specific physiological needs. the presence of simplified UBL-like systems in prokaryotes confirms that the evolutionary roots of protein modification by UBLs predate the emergence of eukaryotes, with the eukaryotic lineage representing an explosion of complexity from these primordial systems.
current experimental approaches, including genomic analyses, sophisticated biochemical tools like the bioUbL platform, and chemical biology methods for UBL synthesis, continue to reveal new dimensions of UBL biology. these technical advances are particularly important for understanding the nuanced functional specialization of UBL family members in different organisms and the potential for targeting specific UBL pathways for therapeutic intervention in human disease or for agricultural improvement in plants. as genomic databases continue to expand and experimental methodologies become increasingly sophisticated, our understanding of the genomic distribution and functional evolution of UBL systems will undoubtedly continue to deepen.
Decoding UBL Pathways: Research Techniques and Therapeutic Targeting
The conjugation of ubiquitin and ubiquitin-like proteins (Ubls) represents a crucial regulatory mechanism in eukaryotic cell biology, governing protein degradation, signaling, and homeostasis. This whitepaper elucidates the complex enzymatic cascades and structural mechanisms underpinning Ubl conjugation, framed within their evolutionary context. We detail cutting-edge biochemical and structural biology methodologies enabling researchers to capture transient intermediates and delineate reaction pathways. The content specifically addresses the information needs of research scientists and drug development professionals, providing technical protocols, analytical frameworks, and visual tools to advance both fundamental knowledge and therapeutic applications in this rapidly evolving field.
Ubiquitin (Ub) and ubiquitin-like proteins (Ubls) constitute a family of post-translational modifiers that orchestrate a vast array of cellular processes through covalent attachment to target substrates. The Ub/Ubl conjugation pathway employs multi-step reactions orchestrated by specific E1 (activating), E2 (conjugating), and E3 (ligating) enzymes [40]. This system regulates essential cellular functions including protein degradation, DNA repair, cell signaling, and autophagy [41]. Despite shared structural features—particularly the characteristic ubiquitin superfold comprising two α-helices and five β-strands (ββαββαβ)—Ubls demonstrate remarkable functional diversity [41].
The conjugation process typically culminates in an isopeptide bond between the Ubl C-terminal carboxyl group and the ε-amino group of a lysine residue in the target protein [40]. Recent research has revealed alternative linkages, including ester bonds and modifications of non-protein substrates such as lipids and sugars [40]. The specificity and outcomes of ubiquitylation are determined by combinatorial E2-E3 pairing, with hundreds of such combinations enabling precise substrate selection [41]. Furthermore, the system's complexity is amplified by the capacity to form polymeric chains through different lysine linkages, with Lys48-linked chains typically targeting proteins for proteasomal degradation, while Lys63-linked chains function in signaling pathways [41].
Ubl Conjugation Cascade: Mechanism and Evolution
The Enzymatic Cascade
The conjugation of Ubls proceeds through a conserved three-step enzymatic mechanism:
- E1-Mediated Activation: E1 activating enzymes initiate the cascade through an ATP-dependent process. The E1 first catalyzes the formation of a Ubl-adenylate (Ubl-AMP) intermediate, which then undergoes nucleophilic attack by the E1 active site cysteine to form a reactive E1~Ubl thioester bond (denoted as ~) [40] [42].
- E2-Mediated Conjugation: The activated Ubl is transferred from E1~Ubl to the active site cysteine of an E2 conjugating enzyme via a transthiolation reaction, generating an E2~Ubl thioester intermediate [40].
- E3-Mediated Ligation: E3 ligases facilitate the final transfer of Ubl to the target substrate. RING-type E3s function as scaffolds that position the E2~Ubl complex optimally for direct substrate modification, while HECT, RBR, and RCR E3s employ a catalytic cysteine to form a transient E3~Ubl thioester intermediate before substrate modification [40].
Table 1: Major E1 Enzymes in Human Ubl Pathways
| E1 Enzyme | Ubl Substrate | Type | Key Features |
|---|---|---|---|
| UBA1, UBA6 | Ubiquitin | Canonical | Domain structure: adenylation, catalytic Cys, C-terminal UFD [42] |
| SAE1-UBA2 | SUMO | Canonical | Heterodimeric; related domain structure to ubiquitin E1 [42] |
| NAE1-UBA3 | NEDD8 | Canonical | Activates NEDD8 for cullin modification [42] |
| UBA7 | ISG15 | Canonical | Induced by interferon signaling [42] |
| ATG7 | ATG8, ATG12 | Non-canonical | Critical for autophagy; structurally distinct from canonical E1s [42] |
| UBA5 | UFM1 | Non-canonical | Divergent mechanism [42] |
| UBA4 (MOCS3) | URM1 | Non-canonical | Initiates sulfur transfer pathway; related to bacterial ancestors [42] |
This enzymatic cascade utilizes labile thioester intermediates, extensive conformational changes, and vast combinatorial diversity of short-lived protein complexes to achieve regulated substrate modification [40]. The system's dynamics are further modulated by deubiquitylating enzymes (DUBs) and Ubl-specific proteases (ULPs) that reverse modifications, making Ub/Ubl signaling both diverse and highly dynamic [40] [43].
Evolutionary Origins and Relationships
Ubl proteins and their conjugation machinery have evolutionary origins in prokaryotic biosynthetic pathways [42]. Structural and mechanistic parallels exist between eukaryotic E1 enzymes and bacterial enzyme systems like MoeB-MoaD (involved in molybdenum cofactor biosynthesis) and ThiF-ThiS (involved in thiamine biosynthesis) [42]. These ancestral systems utilize similar adenylation mechanisms but for sulfur transfer rather than protein modification, suggesting that the eukaryotic Ubl system evolved from these fundamental metabolic pathways through gene duplication and functional diversification.
The human genome encodes eight known E1 enzymes that initiate distinct Ubl conjugation pathways, classified as canonical or non-canonical based on their domain architecture and mechanistic features [42]. Canonical E1s (for ubiquitin, SUMO, NEDD8, and ISG15) share related domain structures, while non-canonical E1s (for URM1, UFM1, ATG12, and ATG8) exhibit greater divergence [42]. This evolutionary diversity enables specific pathway regulation while conserving fundamental activation mechanics.
Diagram 1: E1 enzyme evolutionary origins and functional diversification from prokaryotic ancestors to specialized eukaryotic Ubl pathways.
Key Methodological Approaches in Ubl Research
Structural Biology Techniques
Structural biology has been instrumental in elucidating the mechanisms of Ubl conjugation, particularly through X-ray crystallography and cryo-electron microscopy (cryo-EM). These techniques have revealed:
- E1 Mechanism: Structures of E1 enzymes in complex with Ubls and E2s have illuminated the sequential adenylation, thioester transfer, and E2 charging steps [42]. Canonical E1 structures consistently show an adenylation domain resembling prokaryotic ancestors, a catalytic Cys domain containing the active site cysteine, and a C-terminal ubiquitin-fold domain (UFD) that recruits cognate E2s [42].
- E2 Specificity: Structural analyses have identified the molecular determinants of E1-E2 specificity, demonstrating how distinct E1 surfaces recognize only their cognate E2s to ensure fidelity in downstream pathway selection [42].
- E3 Mechanisms: Structures of various E3 types (RING, HECT, RBR) have revealed how they position E2~Ubl for direct transfer (RING) or form catalytic intermediates (HECT, RBR) [40].
Activity-Based Probes (ABPs) and Chemical Tools
Activity-based probes (ABPs) represent powerful chemical tools for studying the transient intermediates and enzymatic activities in Ubl cascades. These probes typically feature three modular components [40]:
- Reactive Group ("Warhead"): An electrophilic moiety that forms covalent bonds with nucleophilic active site cysteines in E1, E2, or E3 enzymes, mimicking the natural thioester chemistry.
- Recognition Element: Typically the Ubl protein itself or specific domains that confer binding specificity to target enzymes.
- Reporter Group: A detectable tag (e.g., fluorophore, affinity handle) for visualization and isolation of probe-labeled enzymes.
ABPs have been designed to trap and characterize each step of the conjugation cascade, including E1~Ubl thioesters, E2~Ubl intermediates, and E3~Ubl complexes [40]. These tools enable investigation of enzymatic mechanisms in complex biological systems and facilitate screening for selective inhibitors.
Table 2: Quantitative Features and Applications of Major Ubl Research Tools
| Tool Category | Key Examples | Target Enzymes/Processes | Detection Method | Throughput Capacity |
|---|---|---|---|---|
| Activity-Based Probes | Ub/Ubl-based ABPs with electrophilic warheads | E1, E2, E3 active sites | Fluorescence, affinity purification-mass spectrometry | Medium to High [40] |
| Fluorescence-Based Assays | UbL-Enterokinase, UbL-Granzyme B, FRET-based assays | DUBs/ULPs, proteolytic activity | Fluorescence intensity, resonance energy transfer | High (adaptable to 384-well) [43] [44] |
| Binding Assays | Surface Plasmon Resonance (SPR) | Protein-protein interactions, inhibitor binding | Refractive index changes | Medium [44] |
| Enzymatic Assays | Direct and coupled assays with chromogenic/fluorogenic substrates | E1, E2, E3 catalytic activity | Spectrophotometry, fluorescence | High [45] |
| Cellular Assays | Reporter gene assays, mammalian two-hybrid, ToxTracker | Pathway activity, protein interactions, toxicity | Luminescence, fluorescence, cell viability | Medium to High [44] |
Biochemical Assay Development
Biochemical assays form the foundation for quantifying enzymatic activities and interactions in Ubl pathways. Essential assay formats include:
- Direct Enzymatic Assays: Measure substrate consumption or product formation using spectrophotometric, fluorometric, or luminescent detection [45]. These include coupled systems that link Ubl activation to detectable signals.
- Binding Assays: Techniques like surface plasmon resonance (SPR) quantify interactions between conjugation enzymes, Ubls, and substrates without covalent modification [44].
- High-Throughput Screening (HTS) Assays: Automated platforms using fluorescence polarization, FRET, or other homogeneous formats enable rapid screening of compound libraries for Ubl pathway modulators [43] [44].
Assay validation requires careful optimization of temperature, pH, ion concentration, and reagent stability to minimize false positives/negatives, with quality typically assessed using the Z' factor (values >0.4 indicate robust assays) [44].
Diagram 2: Modular design and application workflow of activity-based probes for Ubl research.
Detailed Experimental Protocols
Protocol: Using Activity-Based Probes to Capture E1~Ubl Thioester Intermediates
Purpose: To trap and detect the transient thioester intermediate formed between E1 enzymes and Ubls.
Principle: ABPs containing Ubl domains C-terminally fused to electrophilic warheads (e.g., vinyl sulfones, acyl phosphates) mimic the adenylated Ubl intermediate and form stable covalent adducts with E1 active site cysteines.
Materials:
- Purified E1 enzyme
- Ubl-based ABP with appropriate reactive group and reporter tag
- Reaction buffer (e.g., 50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 10 mM MgCl₂, 1 mM TCEP)
- ATP (optional, depending on probe design)
- SDS-PAGE equipment
- Fluorescence scanner or Western blot system
Procedure:
- Reaction Setup: Incubate E1 enzyme (0.5-5 µM) with ABP (1-10 µM) in reaction buffer at 25-30°C for 15-60 minutes. Include controls without enzyme or without probe.
- Reaction Termination: Stop reactions by adding SDS-PAGE loading buffer with or without reducing agent (note: thioester bonds are labile under reducing conditions).
- Analysis:
- Fluorescence Detection: Resolve proteins by SDS-PAGE, then scan gels for fluorescence signal using appropriate wavelengths for the probe's fluorophore.
- Western Blot: Transfer proteins to membranes and detect using antibodies against the tag (e.g., His-tag, FLAG) or the E1 enzyme.
- Mass Spectrometry Confirmation: For structural validation, excise labeled bands and analyze by mass spectrometry to confirm the modification site.
Troubleshooting:
- Low Labeling Efficiency: Optimize enzyme:probe ratio, incubation time, and temperature. Verify enzyme activity with conventional assays.
- Non-specific Labeling: Include competition experiments with native Ubl or specific inhibitors. Titrate probe concentration to minimize off-target effects.
Protocol: Fluorescence-Based Multiplex Assay for DUB/ULP Inhibition Profiling
Purpose: To simultaneously evaluate inhibitor selectivity across multiple deubiquitylating enzymes (DUBs) and Ubl-specific proteases (ULPs) in a high-throughput format.
Principle: This multiplex assay combines distinct Ubl protease substrates with different cleavage specificities in a single reaction, each producing unique fluorescent signals upon proteolysis.
Materials:
- Recombinant DUBs/ULPs (e.g., USP7, UCH-L1, SENP1)
- Ubl protease substrates (e.g., UbL-Enterokinase, UbL-Granzyme B) with different fluorescent reporters
- Test compounds/inhibitors
- Assay buffer optimized for multiple enzymes
- Multiwell plate reader capable of detecting multiple fluorescence wavelengths
- Black 384-well plates
Procedure:
- Assay Setup: Prepare master mixes containing each Ubl protease substrate at predetermined concentrations in assay buffer.
- Inhibitor Addition: Dispense test compounds (e.g., P022077 for USP7 inhibition) or controls into assay plates using serial dilutions.
- Enzyme Addition: Add enzyme mixture to initiate reactions, ensuring linear reaction conditions.
- Incubation and Detection: Incubate at 25-30°C while monitoring fluorescence development at multiple wavelengths over time (30-120 minutes).
- Data Analysis: Calculate inhibition values for each enzyme by comparing initial velocities of substrate cleavage in treated versus control wells.
Validation:
- Specificity Assessment: Confirm selective inhibition patterns across different protease classes.
- Quality Control: Determine Z' factor for each detection channel (>0.4 indicates robust assay).
- Counter-Screening: Include unrelated proteases to exclude non-specific inhibition.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Research Reagents for Ubl Conjugation Studies
| Reagent Category | Specific Examples | Primary Function | Key Applications |
|---|---|---|---|
| Activity-Based Probes | Ub~VS, SUMO~acyl phosphate, NEDD8~vinyl sulfone | Covalent trapping of active site nucleophiles in E1, E2, E3 enzymes | Target identification, mechanistic studies, enzyme profiling [40] |
| Fluorescent Substrates | Ub-AMC, SUMO-MCA, UbL-EK, UbL-GrB | Report proteolytic activity through fluorescence dequenching | DUB/ULP activity assays, high-throughput inhibitor screening [43] |
| Recombinant Enzymes | E1 (UBA1, NAE1-UBA3), E2 (UbcH5, Ubc12), E3 (RING, HECT types) | Catalytic components for in vitro reconstitution | Enzyme kinetics, structural studies, cascade reconstitution [40] [42] |
| Chemical Inhibitors | MLN4924 (NAE1 inhibitor), P022077 (USP7 inhibitor), TAK-243 (UBA1 inhibitor) | Selective pathway inhibition through ATP-competitive or active site targeting | Mechanistic studies, target validation, therapeutic development [43] [42] |
| Non-hydrolyzable Analogs | Ub-adenylate analogs, Ub~SR (thioester mimics) | Stable mimics of transient reaction intermediates | Structural studies, enzyme mechanism elucidation [40] |
| Antibody Reagents | Anti-ubiquitin, anti-SUMO, anti-NEDD8, linkage-specific antibodies | Detection and purification of modified proteins | Western blot, immunoprecipitation, localization studies [45] |
Disease Relevance and Therapeutic Applications
Dysregulation of Ubl conjugation pathways underlies numerous human diseases, making them promising therapeutic targets:
- Neurodegenerative Disorders: Ubiquilin-1 and ubiquilin-2, human homologs of yeast Dsk2, localize to pathological inclusions in Alzheimer's disease and amyotrophic lateral sclerosis [41]. Genetic variations in ubiquilin-1 increase Alzheimer's risk, potentially through disrupted presenilin handling and GABA receptor trafficking [41].
- Cancer: Numerous E3 ligases and DUBs function as oncogenes or tumor suppressors. The UBL/UBA domain proteins facilitate degradation of cell cycle regulators and oncoproteins, making them attractive targets for cancer therapy [41].
- Therapeutic Development: E1 enzymes represent particularly attractive drug targets as they are the only components of Ubl cascades that consume ATP, enabling development of selective ATP-competitive inhibitors [42]. The NEDD8 E1 inhibitor MLN4924 (pevonedistat) has advanced to clinical trials, validating this approach [42].
The emergence of proteolysis-targeting chimeras (PROTACs) and other induced proximity therapies further highlights the therapeutic potential of harnessing Ubl conjugation machinery for targeted protein degradation [46] [47].
Concluding Perspectives
The integration of structural biology, chemical biology, and biochemical assays has dramatically advanced our understanding of Ubl conjugation mechanisms. Future progress will depend on developing more sophisticated tools to capture the dynamic nature of these pathways in native cellular environments. Key challenges include elucidating the structural basis of E3 substrate specificity, understanding the regulation of branched ubiquitin chains, and developing isoform-selective inhibitors for therapeutic applications. As chemical tools and structural methods continue to evolve, they will undoubtedly reveal new biological insights and therapeutic opportunities in the complex landscape of ubiquitin and ubiquitin-like signaling.
Ubiquitin-like proteins (UBLs) constitute a family of evolutionarily conserved, small proteins that share structural similarities with ubiquitin and are involved in the post-translational modification (PTM) of cellular proteins [1]. These modifications typically serve crucial regulatory functions, enabling cells to rapidly respond to environmental changes, manage protein degradation, coordinate DNA repair mechanisms, and mount effective immune responses [1]. The UBL family derives its name from its founding member, ubiquitin (Ub), which remains the best-characterized member known primarily for its role in targeting proteins for proteasomal degradation. Following ubiquitin's discovery, numerous structurally and mechanistically related UBLs have been identified, including SUMO (Small Ubiquitin-like Modifier), NEDD8, ATG8, ATG12, ISG15, and UFM1, among others [1] [48].
These UBLs participate in parallel regulatory processes utilizing similar enzymatic cascades while governing widely varying cellular functions including autophagy, protein trafficking, inflammation and immune responses, transcription, DNA repair, RNA splicing, and cellular differentiation [1]. From an evolutionary perspective, UBL regulatory systems share common origins with prokaryotic biosynthesis pathways for cofactors such as thiamine and molybdopterin, with bacterial sulfur transfer proteins ThiS and MoaD sharing the characteristic beta-grasp fold of UBLs [1]. The eukaryotic UBL URM1 notably functions as both a ubiquitin-like modifier and a sulfur-carrier protein, representing a "molecular fossil" that establishes this evolutionary connection [1]. This whitepaper provides a comprehensive technical guide to contemporary proteomic and multiomic strategies for mapping UBL interaction networks, with emphasis on methodological considerations, experimental protocols, and data integration approaches relevant to researchers and drug development professionals.
UBL Biology and Technical Challenges
Structural and Functional Classification
UBLs are characterized by their small size and a common three-dimensional structure known as the "beta-grasp" fold, which consists of a five-strand antiparallel beta sheet surrounding a central alpha helix [1]. UBLs are typically classified into two primary categories based on their conjugation capabilities. Type I UBLs can be covalently conjugated to target proteins or other molecules through a characteristic C-terminal glycine residue [1]. These UBLs are initially expressed as inactive precursors that require proteolytic processing to expose the active glycine residue. The human genome encodes at least eight families of Type I UBLs: SUMO, NEDD8, ATG8, ATG12, URM1, UFM1, FAT10, and ISG15 [1]. Type II UBLs typically function as protein-protein interaction domains within larger polypeptide chains and do not form covalent conjugates, though they may be proteolytically processed to release free UBL domains [1].
The enzymatic cascades governing UBL conjugation parallel that of ubiquitin, typically involving three sequential steps: (1) activation by E1 activating enzymes, (2) conjugation by E2 conjugating enzymes, and (3) ligation by E3 ligases to specific substrates [1]. This process results in a covalent isopeptide bond between the C-terminus of the UBL and a lysine residue on the target protein, though some UBLs like ATG8 conjugate to phospholipids instead of proteins [1]. Deconjugation is equally important and is mediated by specialized proteases including deubiquitinating enzymes (DUBs) and UBL-specific proteases (ULPs) that reverse these modifications [1].
Technical Challenges in UBL Interaction Mapping
Mapping UBL interactions presents several significant technical challenges that distinguish it from conventional proteomic studies. Unlike ubiquitination sites, modification sites for most UBLs cannot be reliably identified using standard mass spectrometry methods without introducing mutations to the UBL protein [48]. The transient nature of many UBL-substrate interactions, low stoichiometry of modifications, and rapid dynamics of conjugation and deconjugation further complicate comprehensive mapping. Additionally, the high degree of sequence similarity between different UBLs, particularly in their active sites, creates challenges for specific enrichment and detection. Another layer of complexity arises from the ability of some UBLs (including SUMO, NEDD8, and URM1) to form polymeric chains, and the existence of complex crosstalk between different UBL modifications, where one UBL can modify another [1]. Recent research has also revealed that UBLs can modify unexpected non-protein substrates, such as the conjugation of spermidine to fission yeast SUMO Pmt3, expanding the potential landscape of UBL interactions beyond traditional protein substrates [48].
Proteomic Strategies for UBL Interaction Mapping
Advanced Mass Spectrometry Approaches
Mass spectrometry (MS)-based proteomics has emerged as the cornerstone technology for mapping UBL interactions, though it requires specialized methodologies to address the unique challenges of UBL biology. A significant advancement in this field is the development of pLink-UBL, a dedicated search engine built upon the pLink software framework originally designed for identifying cross-linked peptide pairs [48]. This specialized tool has demonstrated superior precision, sensitivity, and speed compared to "make-do" search engines such as MaxQuant, pFind, and standard pLink. In benchmark evaluations, pLink-UBL increased the number of identified SUMOylation sites by 50-300% from the same datasets compared to MaxQuant [48].
The fundamental workflow for UBL interaction mapping typically involves multiple critical steps: (1) stabilization of UBL-substrate interactions, (2) specific enrichment of UBL-modified peptides or proteins, (3) high-resolution LC-MS/MS analysis, and (4) specialized computational analysis using tools like pLink-UBL. For enrichment, antibody-based approaches remain widely used, particularly for UBLs like SUMO and ISG15 where high-quality antibodies are available. However, the development of novel enrichment strategies continues to evolve, including the use of engineered UBL variants with affinity tags and the exploitation of specific interacting domains that recognize particular UBLs.
Workflow for Concurrent PTM Profiling
The integration of UBL interaction mapping with other PTM analyses provides a more comprehensive view of cellular regulatory networks. A recently developed workflow enables simultaneous analysis of protein thiol oxidation and phosphorylation alongside UBL modifications from the same sample [49]. This integrated approach consists of several key steps: cell lysis under controlled conditions, acetone precipitation, tryptic digestion, isobaric labeling, and subsequent parallel enrichment of thiol-containing peptides utilizing resin-assisted capture (RAC) and phosphopeptides using immobilized metal affinity chromatography (IMAC) [49]. Critical to this workflow is the immediate alkylation of samples to prevent artificial oxidation of nascent free thiols and preserve phosphorylation sites, ensuring accurate identification and quantification of both PTM types [49].
Table 1: Quantitative Performance of pLink-UBL Compared to Conventional Search Engines
| Search Engine | Identified SUMOylation Sites | Precision | Sensitivity | Processing Speed |
|---|---|---|---|---|
| pLink-UBL | 50-300% increase | Superior | Superior | Superior |
| MaxQuant | Baseline | Moderate | Moderate | Moderate |
| pFind | Below baseline | Moderate | Moderate | Moderate |
| Standard pLink | Below baseline | Moderate | Moderate | Moderate |
The following diagram illustrates the core proteomic workflow for UBL interaction mapping, integrating both specific UBL profiling and concurrent multi-PTM analysis:
Figure 1: Proteomic Workflow for UBL and Multi-PTM Profiling
Small-Molecule Substrate Identification
Beyond traditional protein substrates, recent research has revealed that UBLs can also modify small molecules, expanding the functional scope of UBL-mediated regulation. A method for identifying these non-protein substrates involves antibody enrichment of a UBL C-terminal peptide following initial UBL protein enrichment, subsequent LC-MS/MS analysis, and a pFind 3 blind search to identify unexpected modifications [48]. Using this approach, researchers discovered that spermidine serves as a major non-protein substrate for fission yeast SUMO Pmt3 [48]. Spermidine conjugates to the C-terminal carboxylate group of Pmt3 through its N1 or N8 amino group in the presence of SUMO E1, E2, and ATP, in a process that doesn't require E3 enzymes and can be reversed by SUMO isopeptidase Ulp1 [48]. This conjugation has been observed in mice and humans, and spermidine can also be conjugated to ubiquitin in vitro by E1 and E2 enzymes in the presence of ATP, suggesting it may be a common small molecule substrate for SUMO and possibly ubiquitin across eukaryotic species [48].
Multiomics Integration for UBL Network Analysis
Multiomics Technologies and Platforms
Multiomics approaches integrate data from multiple molecular layers—including genomics, transcriptomics, proteomics, epigenomics, and metabolomics—to provide a comprehensive, systems-level understanding of biological processes [50]. For UBL research, multiomics integration is particularly valuable because UBL pathways involve complex regulatory networks that span multiple molecular layers. The emergence of single-cell multiomics enables researchers to correlate specific genomic, transcriptomic, and epigenomic changes within individual cells, providing unprecedented resolution for understanding cellular heterogeneity in UBL responses [51] [50]. Recent technological innovations have significantly advanced multiomic data collection, moving beyond traditional approaches that required separate sample processing for different molecular classes.
Several integrated platforms now enable more efficient multiomic profiling from limited samples. The Multi-Omic Single-Shot Technology (MOST) integrates proteome and lipidome analysis in a single LC-MS run using one reverse-phase column and a binary mobile phase system [50]. The Bead-enabled Accelerated Monophasic Multi-omics (BAMM) method combines n-butanol-based monophasic extraction with magnetic beads and accelerated protein digestion for separate analysis of metabolites, lipids, and proteins [50]. One of the most comprehensive platforms is Omni-MS, a multiomic assay that simultaneously profiles proteins, lipids, electrolytes, metabolites, and other small molecules in a single preparation and single LC-MS analysis [50]. These integrated approaches significantly reduce sample requirements, processing time, and technical variation while improving correlation analysis across different molecular classes.
Table 2: Integrated Multiomics Platforms for Comprehensive UBL Studies
| Platform/Method | Molecular Classes Analyzed | Key Features | Applications in UBL Research |
|---|---|---|---|
| Omni-MS | Proteins, lipids, metabolites, electrolytes | Single preparation, single LC-MS analysis | Biomarker discovery across molecular classes |
| MOST | Proteome, lipidome | Single LC-MS run, one reverse-phase column | Correlation of UBL changes with lipid profiles |
| BAMM | Metabolites, lipids, proteins | n-butanol extraction, magnetic beads | High-throughput UBL substrate identification |
| Sequential Extraction | DNA, RNA, proteins, metabolites, lipids | TRIzol-based sequential isolation | Integrated transcriptional and UBL profiling |
Data Integration and Computational Strategies
The complexity and volume of multiomics data necessitate advanced computational approaches for meaningful integration and interpretation. Machine learning and artificial intelligence have become indispensable tools for extracting insights from multiomics datasets, enabling the detection of intricate patterns and interdependencies that would be impossible to derive from single-analyte studies [51] [50]. A particularly powerful approach is network integration, where multiple omics datasets are mapped onto shared biochemical networks to improve mechanistic understanding [51]. In this framework, analytes (genes, transcripts, proteins, and metabolites) are connected based on known interactions—for example, transcription factors mapped to the transcripts they regulate, or metabolic enzymes mapped to their associated metabolite substrates and products [51].
The RGCCA (Regularized Generalized Canonical Correlation Analysis) package implements a unified and flexible statistical framework for heterogeneous data integration that enables the identification of putative biomarkers across omics datasets [50]. Similarly, the mixOmics project provides a suite of multivariate methods, including sparse Partial Least Squares regression for feature selection [50]. These tools are particularly valuable for UBL research because they can help identify how perturbations in UBL pathways affect multiple molecular layers and contribute to phenotypic outcomes. The integration of both extracellular and intracellular protein measurements, including cell signaling activity, provides another critical layer for understanding the functional consequences of UBL modifications in specific biological contexts [51].
The following diagram illustrates the multiomics data integration process for UBL network analysis:
Figure 2: Multiomics Data Integration Workflow
Experimental Protocols for UBL Interaction Mapping
Protocol for Global UBL Substrate Identification
This protocol outlines a comprehensive approach for identifying protein and small-molecule substrates of UBLs, adapted from recently published methodologies [48].
Materials and Reagents:
- Lysis buffer (e.g., RIPA buffer with protease inhibitors)
- UBL-specific antibodies (commercial or custom)
- Protein A/G magnetic beads
- Cross-linker (e.g., DSS or BS3)
- Trypsin/Lys-C mix for digestion
- StageTips for desalting
- LC-MS/MS system (high-resolution mass spectrometer)
Procedure:
- Cell Culture and Treatment: Culture cells under appropriate conditions. Include treatments with relevant stimuli (e.g., interferon for ISG15 studies, proteasome inhibitors for ubiquitin studies) and appropriate controls.
- Cell Lysis and Protein Extraction: Lyse cells in lysis buffer supplemented with N-ethylmaleimide (NEM) to preserve UBL conjugates. Maintain samples at 4°C throughout preparation to prevent artificial deconjugation.
- Cross-linking: Add cross-linker to final concentration of 1-2 mM and incubate for 30 minutes at room temperature to stabilize transient UBL-substrate interactions.
- Immunoaffinity Enrichment: Incubate lysates with UBL-specific antibodies overnight at 4°C. Add protein A/G magnetic beads and incubate for 2 hours. Wash beads extensively with lysis buffer followed by PBS.
- On-bead Digestion: Reduce and alkylate proteins on beads. Digest with trypsin/Lys-C mix overnight at 37°C.
- Peptide Cleanup: Desalt peptides using StageTips or similar solid-phase extraction method.
- LC-MS/MS Analysis: Analyze peptides using a high-resolution LC-MS/MS system with a gradient optimized for peptide separation.
- Data Analysis: Process raw data using pLink-UBL with appropriate database and search parameters. Use a false discovery rate (FDR) threshold of 1% for identifications.
Protocol for Small-Molecule Substrate Identification
This specialized protocol focuses on identifying non-protein substrates of UBLs, such as the spermidine conjugation recently discovered [48].
Materials and Reagents:
- UBL-specific antibodies
- Strong cation exchange (SCX) material
- C18 StageTips
- LC-MS/MS system
- pFind 3 software for blind searches
Procedure:
- UBL Enrichment: Perform initial UBL enrichment as described in steps 1-5 of the previous protocol.
- Antibody Enrichment of C-terminal Peptides: Following UBL protein enrichment, use antibodies specific to the C-terminal peptide of the UBL to further enrich for conjugation sites.
- LC-MS/MS Analysis with Extended Mass Range: Analyze samples using LC-MS/MS with an extended mass range to detect potential small-molecule adducts.
- Blind Search Analysis: Process data using pFind 3 with blind search mode enabled to identify unexpected modifications.
- Validation Experiments: Confirm identifications using synthetic standards and in vitro reconstitution assays with purified E1 and E2 enzymes.
Table 3: Essential Research Reagents and Computational Tools for UBL Studies
| Resource | Type | Function/Application | Examples/Sources |
|---|---|---|---|
| pLink-UBL | Software | Dedicated search engine for UBL modification sites | [48] |
| UBL-specific Antibodies | Reagent | Immunoaffinity enrichment of UBLs and their conjugates | Commercial suppliers (multiple) |
| Cross-linkers | Chemical Reagent | Stabilization of transient UBL-substrate interactions | DSS, BS3 |
| Tandem Ubiquitin Binding Entities (TUBEs) | Reagent | Recognition and protection of poly-Ub chains | Commercial kits |
| Deubiquitinase Inhibitors | Chemical Reagent | Preservation of Ub/UBL conjugates during processing | PR-619, etc. |
| Ubiquitin-activating Enzyme (E1) Inhibitors | Chemical Reagent | Inhibition of specific UBL conjugation pathways | PYR-41, TAK-243 |
| mixOmics/RGCCA | Software | Multivariate statistical analysis of multiomics data | Bioconductor/CRAN [50] |
| Network Analysis Tools | Software | Integration of multiomics data onto biological networks | Cytoscape with appropriate plugins |
| MultiAssayExperiment | Software | Bioconductor interface for multiomics data with overlapping samples | Bioconductor [50] |
Evolutionary and Structural Perspectives on UBL Interactions
From an evolutionary standpoint, UBL regulatory systems share a common origin with prokaryotic biosynthesis pathways for the cofactors thiamine and molybdopterin [1]. The bacterial sulfur transfer proteins ThiS and MoaD from these pathways share the characteristic beta-grasp fold with eukaryotic UBLs, while sequence similarity and a common catalytic mechanism link the pathway members ThiF and MoeB to ubiquitin-activating enzymes [1]. This evolutionary conservation underscores the fundamental importance of UBL-mediated regulatory mechanisms across domains of life.
Structural studies have revealed that despite shared fold architecture, UBLs have evolved distinct surface features that determine their specific interactions. For example, research on avian OASL has revealed that its tandem Ubl domain shares structural characteristics with mammalian ISG15, including the LRLRGG motif required for conjugation [52]. This structural similarity has functional implications, as viral deubiquitinases and deISGylases from pathogens like nairoviruses and coronavirases can sometimes recognize and cleave UBL modifications in a species-specific manner [52]. Several ovarian tumor domain proteases (OTUs) that lack deubiquitinase and deISGylase activity were found to have moderate "deOASLylase" activity, with this activity mirroring viral host preferences [52]. Understanding these evolutionary relationships and structural features provides valuable insights for interpreting interaction mapping data and designing functional experiments.
The field of UBL interaction mapping continues to evolve rapidly, driven by advances in mass spectrometry instrumentation, computational tools, and multiomics integration frameworks. The development of specialized search engines like pLink-UBL has already significantly improved the sensitivity and precision of UBL modification site identification [48]. Looking forward, several emerging trends are poised to further transform UBL research. The increasing adoption of single-cell multiomics approaches will enable the characterization of UBL regulatory networks with unprecedented resolution, revealing cell-to-cell heterogeneity in UBL responses that are obscured in bulk analyses [51] [50]. The integration of spatial omics technologies will add another dimension, allowing researchers to situate UBL networks within their tissue context.
From a clinical perspective, UBL interaction mapping holds significant promise for drug discovery and therapeutic development. Many disease processes, including cancer, neurodegenerative disorders, and infectious diseases, involve dysregulation of UBL pathways [1] [52]. The ability to comprehensively map these networks in patient samples using multiomics approaches will facilitate the identification of novel therapeutic targets and biomarkers for patient stratification. As these technologies continue to mature and become more accessible, they will undoubtedly yield new insights into the fundamental biological processes governed by ubiquitin-like proteins and their relevance to human health and disease.
The ubiquitin-proteasome system (UPS) is a quintessential regulator of eukaryotic biology, controlling protein stability, function, and localization. Within this system, ubiquitin-like proteins (UBLs) constitute a family of small proteins that share structural homology with ubiquitin and mediate critical post-translational modifications. UBLs are involved in a widely varying array of cellular functions including autophagy, protein trafficking, inflammation, immune responses, transcription, DNA repair, RNA splicing, and cellular differentiation [1]. These proteins are characterized by the beta-grasp fold, a three-dimensional structure consisting of a five-strand antiparallel beta sheet surrounding an alpha helix [1] [13]. From a therapeutic perspective, the UBL family represents a promising class of drug targets, as evidenced by the clinical success of proteasome inhibitors and the emerging potential of NEDD8 activation blockade.
UBLs can be functionally divided into two primary categories. Type I UBLs are capable of covalent conjugation to target proteins or lipids through a characteristic C-terminal glycine motif. This class includes well-characterized members such as ubiquitin itself, SUMO (Small Ubiquitin-like MOdifier), NEDD8 (NEural precursor cell-expressed Developmentally Down-regulated 8), ATG8, ATG12, and ISG15 [1]. The conjugation process for these UBLs typically follows a three-step enzymatic cascade involving E1 (activating), E2 (conjugating), and E3 (ligating) enzymes, analogous to the ubiquitination pathway. In contrast, Type II UBLs do not form covalent conjugates but instead exist as protein domains within larger polypeptides, often functioning as protein-protein interaction modules [1]. The evolutionary conservation of UBLs from prokaryotes to humans, alongside their frequent dysregulation in human diseases—particularly cancers—has positioned them as attractive targets for therapeutic intervention [1] [13] [53].
Proteasome Inhibitors: Pioneering UBL-Targeted Therapeutics
Mechanism of Action and Clinical Development
Proteasome inhibitors represent the first clinically validated class of pharmaceuticals targeting the ubiquitin-proteasome pathway. These compounds function by blocking the proteolytic activity of the 26S proteasome, a multi-subunit complex responsible for the degradation of ubiquitin-tagged proteins. The 26S proteasome consists of a 20S catalytic core and one or two 19S regulatory subunits. The 20S core is a barrel-shaped structure composed of four heptameric rings, with the two inner beta rings containing three primary proteolytic active sites: chymotrypsin-like (β5), trypsin-like (β2), and caspase-like (β1) activities [54]. Proteasome inhibitors primarily target the chymotrypsin-like site, though at higher concentrations they can inhibit all three catalytic activities [54].
The mechanism by which proteasome inhibitors induce cell death is multifactorial. By preventing proteasomal degradation, these agents cause accumulation of polyubiquitinated proteins, creating endoplasmic reticulum (ER) stress and triggering the unfolded protein response (UPR) [54]. Additionally, proteasome inhibition prevents degradation of pro-apoptotic factors such as NOXA, Bim, and p53, while simultaneously inhibiting the NF-κB pro-survival pathway by stabilizing its endogenous inhibitor IκBα [54]. The net result is a shift in cellular equilibrium toward apoptosis, which is particularly pronounced in malignant cells with high protein turnover rates, such as multiple myeloma cells producing abundant immunoglobulins [54].
Table 1: Clinically Approved Proteasome Inhibitors
| Name | Kinetics | Active Moiety | Key Indications | Administration | Common Toxicities |
|---|---|---|---|---|---|
| Bortezomib (Velcade) | Slowly reversible | Boronate | First-line, relapsed/refractory MM and MCL | IV/SC | Peripheral neuropathy, nausea, diarrhea, cytopenias |
| Carfilzomib (Kyprolis) | Irreversible | Epoxyketone | Relapsed/refractory MM | IV | Dyspnea, cytopenias, fatigue, hypertension |
| Ixazomib (Ninlaro) | Reversible | Boronate | MM after one prior therapy | Oral | Diarrhea, constipation, thrombocytopenia, rash |
Experimental Protocols for Proteasome Inhibition Studies
In vitro assessment of proteasome inhibitor activity typically involves cell viability assays, proteasome activity measurements, and analysis of protein accumulation. For cell viability testing, malignant cells are plated in 96-well plates (e.g., 5,000 cells/well) and exposed to serial dilutions of the proteasome inhibitor for 48 hours. Viability is then quantified using tetrazolium-based colorimetric assays such as MTS (CellTiter Aqueous One Solution Cell Proliferation Assay) [55]. To directly measure proteasome inhibition, cells can be lysed and incubated with fluorogenic substrates specific for each proteasomal catalytic activity (e.g., Suc-LLVY-aminoluciferin for chymotrypsin-like activity) [54] [56]. Accumulation of polyubiquitinated proteins and specific proteasome substrates such as p27, IκBα, and NOXA can be detected by Western blot analysis of cell lysates prepared under denaturing conditions [54] [55].
For in vivo evaluation, mouse models of human cancers (e.g., subcutaneous xenograft models) are commonly employed. Animals are randomized when tumors reach a predetermined volume (typically 100-150 mm³) and treated with vehicle control or the proteasome inhibitor via appropriate routes (intravenous, subcutaneous, or oral) according to established schedules. Tumor measurements are performed regularly with calipers, and volume is calculated using the formula: (length × width²)/2. At endpoint, tumors are harvested for immunohistochemical analysis of ubiquitin-protein conjugates, apoptosis markers (e.g., cleaved caspase-3), and proliferation indices (e.g., Ki-67) [54] [55].
NEDD8 Activation Enzyme (NAE) Inhibition: MLN4924/Pevonedistat
Molecular Mechanism and Anticancer Activity
Neddylation, the process of conjugating NEDD8 to target proteins, plays a crucial regulatory role in cellular homeostasis, with its overactivation observed in multiple human cancers [57] [58]. The primary substrates of neddylation are cullin family proteins, which form the scaffold of Cullin-RING ligases (CRLs)—the largest family of E3 ubiquitin ligases that control approximately 20% of proteasome-mediated protein degradation [57] [58]. Neddylation activates CRLs by inducing conformational changes that facilitate ubiquitin transfer to substrate proteins, thereby regulating the turnover of key regulatory proteins involved in cell cycle progression, DNA damage response, and signal transduction [57].
MLN4924 (pevonedistat) is a first-in-class NEDD8-activating enzyme (NAE) inhibitor that functions through a unique mechanism. Structurally, MLN4924 forms a covalent adduct with NEDD8 that mimics the NEDD8-AMP intermediate, effectively blocking NAE activity and subsequent transfer of NEDD8 to E2 conjugating enzymes [57] [53]. This inhibition leads to suppression of cullin neddylation, resulting in inactivation of CRLs and subsequent accumulation of their substrate proteins [57] [58]. The anticancer effects of MLN4924 are primarily mediated through induction of cell cycle arrest, apoptosis, senescence, and autophagy in a cell-type dependent manner [58]. Notably, the compound demonstrates selectivity for NAE over other E1 enzymes, with IC50 values of 4 nM for NAE compared to 1.8 μM for UBA6, 8.2 μM for SAE, and 1.5 μM for UAE [57].
Immunomodulatory Effects in the Tumor Microenvironment
Recent research has revealed that the antitumor activity of MLN4924 extends beyond direct cancer cell cytotoxicity to include significant immunomodulatory effects within the tumor microenvironment (TME). Pevonedistat treatment has been shown to enhance CD8+ T-cell function by increasing production of TNFα and IFNγ while promoting T-cell cytotoxicity [55]. In syngeneic lymphoma models, pevonedistat induced a CD8+ T-cell inflamed microenvironment and delayed tumor progression in a manner dependent on MHC class I interactions, as demonstrated by the diminished antitumor effect following CD8+ T-cell depletion or B2M knockout [55].
Mechanistically, NAE inhibition appears to modulate T-cell function in a HIF-1α-dependent manner, as shRNA-mediated knockdown of this CRL substrate abrogated the effects of pevonedistat in vitro [55]. Furthermore, combination therapy with PD-1 blockade synergistically enhanced tumor immune infiltration, T-cell activation, and chemokine expression, resulting in superior tumor growth control compared to either agent alone [55]. Single-cell RNA sequencing analysis of clinical samples from lymphoma patients receiving pevonedistat therapy confirmed upregulation of interferon response signatures in immune cells, providing translational validation of these immunomodulatory mechanisms [55].
Table 2: Cellular Responses to NAE Inhibition by MLN4924
| Biological Process | Key Molecular Events | Functional Outcome |
|---|---|---|
| Cell Cycle Regulation | Accumulation of CRL substrates (p21, p27, WEE1) | S-phase arrest, DNA re-replication, and checkpoint activation |
| Apoptosis Induction | Accumulation of NOXA, BIM, and other pro-apoptotic factors | Caspase activation and mitochondrial apoptosis |
| Senescence | DNA damage response activation (γH2AX) | Irreversible growth arrest with senescence-associated phenotype |
| Autophagy | Disruption of mTOR signaling and ULK1 stabilization | Enhanced autophagic flux as protective response |
| Immune Modulation | HIF-1α accumulation, enhanced cytokine production | CD8+ T-cell activation and enhanced antitumor immunity |
Comparative Analysis of Therapeutic Approaches
Target Specificity and Therapeutic Windows
The therapeutic targeting of UBL pathways represents a paradigm shift in cancer treatment, with proteasome inhibitors and NAE inhibitors offering distinct mechanisms of action and clinical applications. Proteasome inhibitors target the final common pathway of protein degradation, affecting a broad spectrum of cellular processes and exhibiting particular efficacy in hematologic malignancies characterized by high protein turnover, such as multiple myeloma and mantle cell lymphoma [54] [59]. In contrast, NAE inhibitors act further upstream, specifically disrupting the neddylation pathway and consequently inhibiting a defined subset of E3 ubiquitin ligases (CRLs) while sparing other ubiquitination processes [57] [53]. This more selective mechanism theoretically offers an improved therapeutic window, though the clinical validation of this hypothesis remains ongoing.
The differential effects of these targeting strategies extend to their immunomodulatory properties. While proteasome inhibitors have demonstrated some immunosuppressive effects—partially explaining their utility in graft-versus-host disease prevention—NAE inhibition appears to exert immunostimulatory effects, particularly on CD8+ T-cell function [55]. This distinction has important implications for combination therapy strategies, with NAE inhibition showing particular promise in combination with immune checkpoint inhibitors [55]. Additionally, the targeting of different nodes within the UPS/UBL system results in distinct resistance mechanisms, with proteasome resistance often involving mutations in proteasome subunits or upregulation of alternative protein clearance pathways, while resistance to NAE inhibition may emerge through alterations in the neddylation pathway components or compensatory ubiquitination mechanisms.
Table 3: Comparison of UBL-Targeting Therapeutic Approaches
| Parameter | Proteasome Inhibitors | NAE Inhibitors |
|---|---|---|
| Molecular Target | 20S proteasome catalytic core | NEDD8-activating enzyme (NAE) |
| Primary Mechanism | Block protein degradation | Inhibit cullin neddylation and CRL activity |
| Cellular Effects | Accumulation of polyubiquitinated proteins, ER stress | Accumulation of specific CRL substrates |
| Clinical Status | FDA-approved (bortezomib, carfilzomib, ixazomib) | Phase II/III clinical trials |
| Key Toxicities | Peripheral neuropathy, cytopenias, fatigue | Fatigue, nausea, rash, cytopenias |
| Immunomodulation | Generally immunosuppressive | T-cell activation and enhanced antitumor immunity |
Signaling Pathways and Molecular Interactions
The following diagram illustrates the key molecular pathways targeted by proteasome inhibitors and NAE inhibitors, highlighting points of therapeutic intervention and downstream consequences:
The Scientist's Toolkit: Essential Research Reagents and Methodologies
Advancing research in UBL biology and therapeutics requires specialized experimental tools and methodologies. The following table summarizes key research reagents and their applications in studying UBL pathways and inhibitor mechanisms:
Table 4: Essential Research Reagents for UBL-Targeted Drug Development
| Reagent/Method | Function/Application | Key Features |
|---|---|---|
| BioUbL Platform [37] | Comprehensive analysis of UBL modifications using in vivo biotinylation | Enables purification under denaturing conditions; identifies covalent UBL conjugates; minimal background |
| In vitro NEDD8 conjugation assay [57] | Screening for NAE inhibitors | Measures NEDD8 activation and transfer; uses purified NAE, E2s, NEDD8, and ATP |
| Cellular thermal shift assay (CETSA) [57] | Target engagement assessment | Detects drug-induced thermal stabilization of target proteins in cells |
| AlphaScreen technology [57] | High-throughput screening for NAE inhibitors | Homogeneous bead-based proximity assay for protein-protein interactions |
| TUBEs (Tandem Ubiquitin Binding Entities) [37] | Enrichment of ubiquitinated proteins | High-affinity capture of polyubiquitinated substrates; protects from DUBs |
| PRISM (Protein Interaction Screen on MST) [37] | Identification of SUMOylation sites | Chemical blockade of free lysines followed by SUMO protease treatment and biotin-tagging |
| scRNA-Seq of patient samples [55] | Analysis of drug effects on tumor microenvironment | Identifies cell-type specific responses; reveals immune modulation mechanisms |
Experimental Protocol for NAE Inhibitor Evaluation
Comprehensive assessment of NAE inhibitors involves a multi-tiered experimental approach. Initial enzymatic assays utilize purified NAE complex (APPBP1/UBA3 heterodimer) incubated with NEDD8, ATP, and the test compound in reaction buffer (e.g., 50 mM Tris-HCl pH 7.5, 5 mM MgCl₂, 2 mM ATP, 0.5 mM DTT). Reactions proceed for 60-90 minutes at 30°C, followed by quantification of NEDD8-NAE thioester formation via Western blotting under non-reducing conditions or using homogenous time-resolved fluorescence (HTRF) assays [57].
For cellular mechanistic studies, tumor cells are treated with varying concentrations of the NAE inhibitor (typically 10 nM to 1 μM) for 6-48 hours. Cells are then lysed in RIPA buffer supplemented with protease and phosphatase inhibitors, and neddylation status is assessed by immunoblotting for key substrates such as cullin-1 (showing decreased neddylation) and accumulation of CRL substrates including p21, p27, and HIF-1α [58] [55]. Functional consequences are evaluated through cell cycle analysis (PI staining and flow cytometry), apoptosis assays (Annexin V/7-AAD staining), and senescence-associated β-galactosidase staining [58].
In vivo efficacy studies employ appropriate xenograft or syngeneic models, with animals randomized when tumors reach 100-150 mm³. Pevonedistat is typically administered intravenously at 30-60 mg/kg on varying schedules (e.g., once daily for 5 days every 2 weeks). Tumor volume is monitored regularly, and at study endpoint, tumors are harvested for immunohistochemical analysis of neddylation markers (e.g., NEDD8-conjugates), proliferation (Ki-67), apoptosis (cleaved caspase-3), and immune cell infiltration (CD4, CD8, CD11b) [55]. For combination studies with immune checkpoint inhibitors, anti-PD-1 antibodies are administered intraperitoneally at 200 μg/dose 2-3 times per week [55].
The targeting of ubiquitin-like protein pathways represents a promising frontier in cancer therapeutics, with proteasome inhibitors establishing clinical proof-of-concept and NAE inhibitors demonstrating the potential for more selective intervention. The continued development of UBL-targeted therapies will likely focus on enhancing specificity, identifying predictive biomarkers for patient selection, and developing rational combination strategies that leverage the unique immunomodulatory properties of these agents. As our understanding of the complex interplay between different UBL pathways deepens, opportunities for multi-node targeting within the system may emerge, offering new avenues for overcoming resistance mechanisms and improving therapeutic outcomes across a broader spectrum of malignancies.
The integration of advanced research tools—including the BioUbL platform for comprehensive UBL modification analysis, single-cell technologies for dissecting tumor microenvironment effects, and structural biology approaches for rational drug design—will accelerate the development of next-generation UBL-targeted therapeutics. Furthermore, the exploration of UBL pathways beyond ubiquitin, NEDD8, and SUMO may uncover additional druggable nodes with unique biological functions and therapeutic potential. As this field evolves, the precedent established by proteasome inhibitors and NAE inhibitors will continue to guide strategic approaches for targeting the complex yet pharmacologically rewarding landscape of ubiquitin-like protein modifications.
The ubiquitin-proteasome system (UPS) and ubiquitin-like protein (UBL) conjugation pathways represent a sophisticated enzymatic hierarchy that maintains cellular protein homeostasis. This intricate system, comprising E1 activating, E2 conjugating, and E3 ligating enzymes, has emerged as a promising therapeutic target for various diseases, including cancer, neurodegenerative disorders, and viral infections. While the proteasome inhibitor bortezomib validated the UPS as a drug target, current research focuses on developing specific inhibitors targeting individual components of the enzymatic cascade to achieve greater therapeutic precision. This technical review comprehensively analyzes the development status of E1, E2, and E3-specific inhibitors, provides detailed experimental methodologies for studying these pathways, and examines the evolutionary context of UBL systems. The growing understanding of ubiquitin-like protein structure and function, coupled with advances in targeted protein degradation technologies like PROTACs, promises to unlock new therapeutic opportunities for previously undruggable targets.
The ubiquitin-proteasome system represents a crucial regulatory mechanism for controlling protein stability and function in eukaryotic cells. This system employs a sequential enzymatic cascade involving E1 (activating), E2 (conjugating), and E3 (ligating) enzymes that conjugate ubiquitin or ubiquitin-like proteins (UBLs) to target substrates [53]. Similar to kinases, components of the ubiquitin system are frequently dysregulated in various diseases, making them attractive targets for therapeutic intervention [53].
Ubiquitin itself is a highly conserved 76-amino acid protein that can be covalently attached to substrate proteins via an isopeptide bond between its C-terminal glycine and lysine residues on target proteins [53] [60]. The ubiquitination process involves three sequential enzymatic steps: (1) E1 activating enzyme utilizes ATP to adenylate the ubiquitin C-terminus; (2) E2 conjugating enzyme receives the activated ubiquitin via transthioesterification; and (3) E3 ligase facilitates the transfer of ubiquitin from E2 to the target substrate [60]. This hierarchical system offers multiple points for therapeutic intervention, with varying levels of specificity.
The complexity of ubiquitin signaling is magnified by the existence of ubiquitin-like proteins (UBLs), including NEDD8, SUMO, ISG15, ATG8, ATG12, and UFM1, which share structural similarities with ubiquitin but have distinct biological functions [17] [61]. These UBLs utilize parallel E1/E2/E3 cascades dedicated to their specific conjugation pathways. Recent research has revealed that bacterial antiviral defense pathways encode eukaryotic-like ubiquitination systems, suggesting that these pathways arose first in bacteria [62]. This evolutionary conservation underscores the fundamental importance of protein modification systems across domains of life.
Enzymatic Hierarchy and Biological Significance
The Ubiquitin/UBL Conjugation Cascade
The ubiquitin and UBL conjugation cascades follow a conserved mechanism while maintaining distinct biological functions. The process begins with E1 activating enzymes, which initiate the conjugation cascade in an ATP-dependent manner. E1 enzymes catalyze the formation of a thioester bond between the C-terminal carboxyl group of ubiquitin/UBL and a catalytic cysteine residue within the E1 [18]. Humans possess two ubiquitin E1 enzymes (UBA1 and UBA6), while specific UBLs have their own dedicated E1s, which can be single proteins or heterodimers [53] [63].
Following activation, ubiquitin/UBL is transferred to an E2 conjugating enzyme (approximately 38 in humans) through a transthioesterification reaction [53]. E2 enzymes not only serve as ubiquitin carriers but also dictate ubiquitin chain linkage specificity and length, playing a crucial role in determining the functional outcome of ubiquitination [53].
The final step involves E3 ligases (approximately 600-700 in humans), which are responsible for substrate recognition and facilitating ubiquitin transfer from E2 to the target protein [53] [60]. E3 ligases are categorized into three main families: RING (Really Interesting New Gene), HECT (Homologous to E6-AP Carboxyl Terminus), and RBR (RING-Between-RING) [64]. RING E3s act as scaffolds to bring E2 and substrate into proximity, while HECT and RBR E3s form catalytic intermediates with ubiquitin before transferring it to substrates [64].
Table 1: Major Enzyme Classes in the Ubiquitin/UBL Conjugation Cascade
| Enzyme Class | Number in Humans | Primary Function | Key Characteristics |
|---|---|---|---|
| E1 Activating Enzymes | 2 for ubiquitin (UBA1, UBA6); multiple for UBLs | ATP-dependent activation of ubiquitin/UBL | Catalytic cysteine forms thioester with ubiquitin/UBL; some function as heterodimers for UBLs |
| E2 Conjugating Enzymes | ~38 | Accepts activated ubiquitin/UBL from E1; determines chain topology | Contains catalytic cysteine; works with E3 to transfer ubiquitin to substrate |
| E3 Ligases | ~600-700 | Substrate recognition and ubiquitin ligation | Three main classes: RING, HECT, RBR; provides substrate specificity |
| Deubiquitinases (DUBs) | ~100 | Removal of ubiquitin from substrates | Counteracts ubiquitination; regulates ubiquitin homeostasis |
Evolutionary Conservation of UBL Pathways
The evolutionary origins of ubiquitin and UBL pathways extend deep into prokaryotic systems. The β-grasp fold (β-GF), characteristic of Ub/Ubls, had already differentiated into at least seven clades by the time of the last universal common ancestor, encompassing much of the structural diversity observed in extant versions [13]. Most biochemical diversification of the fold occurred in prokaryotes, with the eukaryotic phase of its evolution mainly marked by the expansion of the Ubl clade [13].
Recent research has demonstrated that bacterial antiviral defense systems encode complete ubiquitination pathways with striking architectural parallels to eukaryotic ubiquitination machinery [62]. Structural studies of bacterial E1:E2:Ubl complexes reveal similar mechanisms for ubiquitin activation and transfer, suggesting convergent evolution or conservation of an ancient protein modification system [62]. These bacterial systems include not only E1 and Ubl components but also E2 enzymes and RING-type E3 elements, closely resembling the eukaryotic state [13].
The NEDD8 pathway illustrates the evolutionary adaptations in UBL activation. While the E1 for NEDD8 is a heterodimer (APPBP1-UBA3), research has shown that UBA3 alone can activate NEDD8, with APPBP1 primarily functioning as a scaffold to enhance reaction rates [63]. This contrasts with SUMO activation, which requires both subunits of its E1 heterodimer (AOS1-Uba2), suggesting distinct evolutionary pathways for different UBL activation mechanisms [63].
E1 Inhibitors: Targeting the Apex of the Cascade
Mechanism and Therapeutic Approaches
E1 enzymes occupy the apex of the ubiquitination cascade, making them attractive but challenging therapeutic targets. Inhibition of E1 enzymes would theoretically block entire ubiquitination pathways, offering broad biological effects but potentially increasing toxicity concerns. The two ubiquitin E1 enzymes in humans, UBA1 and UBA6, control ubiquitination of all downstream targets, while specific UBLs like NEDD8 and SUMO have their own dedicated E1 enzymes [53].
The most clinically advanced E1 inhibitor is MLN4924 (Pevonedistat), which targets the NEDD8 activating enzyme (NAE) [53]. MLN4924 interacts with the nucleotide-binding site within NAE and forms a covalent adduct that mimics NEDD8-AMP, blocking NAE function and subsequent neddylation of cullins [53]. This disruption of cullin RING ligase (CRL) activity leads to accumulation of cell cycle regulators and DNA damage response proteins, ultimately inducing apoptosis in proliferating tumor cells [53].
For ubiquitin E1 inhibition, PYR-41 was the first identified cell-permeable inhibitor of UBA1 [53]. This compound is believed to irreversibly inhibit UBA1 by covalently modifying the active cysteine (Cys632), while PYZD-4409, based on a pyrazolidine pharmacophore, also shows preferential induction of cell death in malignant cells [53]. However, the precise mechanisms and off-target profiles of these compounds remain incompletely characterized.
Table 2: E1 Inhibitors in Development
| Compound | Target | Mechanism of Action | Development Status | Key Findings |
|---|---|---|---|---|
| MLN4924 (Pevonedistat) | NEDD8 E1 (NAE) | Forms covalent adduct with NEDD8-AMP, blocking enzyme function | Phase II clinical trials | Induces DNA damage, apoptosis; accumulates cell cycle regulators (p27, IκB) |
| PYR-41 | Ubiquitin E1 (UBA1) | Irreversible inhibitor; covalently modifies active cysteine | Preclinical | Preferentially induces cell death in malignant cells; mechanism not fully characterized |
| PYZD-4409 | Ubiquitin E1 (UBA1) | Pyrazolidine pharmacophore | Preclinical | Similar activity to PYR-41; limited off-target characterization |
Experimental Protocols for E1 Inhibition Studies
FRET-Based E1 Activity Assay: Quantitative Förster Resonance Energy Transfer (FRET) technology enables real-time monitoring of E1 activation dynamics. The protocol involves constructing fluorescent protein fusions (CyPet and YPet) with E1 enzymes and their corresponding UBLs [63]. Upon E1-UBL interaction, FRET efficiency changes can be measured to determine binding affinities, reaction intermediates, and enzymatic kinetics. This approach has been instrumental in dissecting distinct roles of heterodimeric E1 subunits, revealing that APPBP1 is dispensable for NEDD8 activation while UBA3 alone suffices for the reaction [63].
Electrostatic Similarity Analysis: The Analysis of Electrostatic Similarities Of Proteins (AESOP) computational framework performs computational alanine scans to quantitatively assess the impact of mutations on protein-protein interaction stability [63]. Using the Poisson-Boltzmann method, AESOP calculates electrostatic potentials on a grid surrounding protein complexes, enabling hierarchical clustering based on electrostatic potential similarities and calculation of electrostatic free energies of association [63]. This approach is particularly valuable for understanding the role of charged amino acids in the formation of complexes between highly charged E1 enzymes and their binding partners.
E2 Inhibitors: Intermediate Specificity Targets
Strategic Approaches to E2 Inhibition
E2 conjugating enzymes represent an intermediate specificity target in the ubiquitination cascade, with approximately 38 E2s in mammals offering greater selectivity than E1 enzymes but less specificity than E3 ligases [53] [60]. E2 enzymes not only serve as ubiquitin carriers but also play a crucial role in determining ubiquitin chain linkage specificity and length [53]. This functional diversity makes specific E2 enzymes attractive targets for therapeutic intervention.
The compound CC0651 was identified as an allosteric inhibitor of the E2 enzyme CDC34 [53]. Unlike active-site inhibitors, CC0651 inserts into a cryptic binding pocket distant from the catalytic site, causing conformational rearrangement that interferes with ubiquitin discharge to acceptor lysine residues [53]. Although promising in vitro, development of CC0651 has faced challenges in optimization for clinical application.
For UBE2N-UBE2V1, an E2 heterodimer that catalyzes synthesis of K63-specific polyubiquitin chains, NSC697923 inhibits formation of the UBE2N~Ub thioester conjugate, thereby blocking ubiquitin transfer to substrates [53]. Another compound, BAY 11-7082, initially thought to inhibit IKK, was later found to exert its effects by covalently modifying reactive cysteine residues in UBE2N and possibly other E2 enzymes [53].
Experimental Approaches for E2 Functional Characterization
E2~Ub Thioester Formation Assay: This fundamental assay monitors the formation of the E2~Ub thioester intermediate, essential for E2 function. The protocol involves incubating E2 enzyme with E1, ubiquitin, and ATP in reaction buffer at 30°C, followed by non-reducing SDS-PAGE analysis to detect the covalently linked E2~Ub intermediate, which migrates at a higher molecular weight than unmodified E2 [53]. Inhibition of this process can be quantified by measuring decreased E2~Ub formation in the presence of candidate inhibitors.
Ubiquitin Chain Typing Assay: To characterize the role of specific E2 enzymes in determining ubiquitin chain topology, in vitro ubiquitination reactions are performed with specific E2 enzymes in the absence of E3 ligases [17]. The resulting ubiquitin chains are analyzed by mass spectrometry or using linkage-specific antibodies to determine lysine preference. This approach has demonstrated that E2 enzymes are capable of directing chain formation to distinct subsets of ubiquitin lysines independently of E3 enzymes [17].
E3 Inhibitors: Maximizing Therapeutic Specificity
Diverse E3 Ligase Families as Drug Targets
E3 ubiquitin ligases represent the most promising and specific targets in the ubiquitination cascade, with approximately 600-700 members in humans offering the potential for highly selective therapeutic intervention [53] [60]. The three main E3 families - RING, HECT, and RBR - employ distinct catalytic mechanisms, providing multiple avenues for inhibitor development [64].
The SCF (SKP1-CUL1-F-box) complex, a multimeric RING E3 ligase, represents a well-characterized target. SCF complexes utilize F-box proteins (approximately 70 in humans) as substrate receptors, with different F-box proteins conferring specificity toward distinct substrates [53]. The F-box protein SKP2, which targets cell cycle regulators p27KIP1 and p21CIP1 for degradation, is frequently overexpressed in human cancers, making it an attractive therapeutic target [53].
MDM2 (HDM2 in humans), another RING E3 ligase, regulates the tumor suppressor p53 by targeting it for degradation [60]. MDM2 is overexpressed in various cancers, often associated with poor prognosis, and inhibition of MDM2 offers a strategy to reactivate p53-mediated tumor suppression [60]. Additional E3 ligases with validated roles in disease include BRCA1 (breast and ovarian cancer), PARKIN (Parkinson's disease), and various CRL family members [64] [60].
Challenges in E3 Ligase Drug Development
Despite their therapeutic potential, E3 ligases present significant challenges for drug discovery. Most E3 ligases lack easily targetable active sites, function through complex protein-protein interactions, and have proven difficult to assay in high-throughput screening formats [64] [60]. Additionally, existing small molecule chemical libraries have not been rich sources of E3 ligase modulators, necessitating novel screening approaches and compound collections [60].
The emergence of PROTAC (Proteolysis Targeting Chimera) technology has transformed the E3 ligase field by hijacking E3 ligases to induce targeted protein degradation [65]. PROTACs are heterobifunctional molecules consisting of an E3 ligase-binding moiety connected to a target-binding warhead via a chemical linker. By bringing the E3 ligase into proximity with the target protein, PROTACs induce ubiquitination and subsequent degradation of the target [65]. This catalytic mode of action allows for lower dosing and targeting of previously undruggable proteins.
Table 3: E3 Ligase-Targeting Therapeutic Approaches
| Target/E3 Ligase | Therapeutic Approach | Mechanism | Development Status |
|---|---|---|---|
| MDM2 | Small molecule inhibitors | Block MDM2-p53 interaction, stabilizing p53 | Multiple candidates in clinical trials |
| SKP2 | Small molecule inhibitors | Inhibit SCFSKP2 complex formation or activity | Preclinical development |
| CRBN | PROTAC technology | Hijack CRBN E3 ligase for targeted protein degradation | ARV-110 and ARV-471 in clinical trials |
| VHL | PROTAC technology | Recruit VHL E3 ligase to degrade target proteins | Multiple candidates in preclinical and clinical development |
Visualization of Key Pathways and Relationships
Ubiquitin/UBL Conjugation Cascade
Ubiquitin/UBL Conjugation Cascade - This diagram illustrates the sequential enzymatic steps in ubiquitin/UBL conjugation, from E1-mediated activation to E3-mediated substrate modification.
PROTAC Mechanism of Action
PROTAC Mechanism of Action - This diagram illustrates how heterobifunctional PROTAC molecules bring E3 ligases and target proteins together to induce targeted protein degradation.
The Scientist's Toolkit: Essential Research Reagents
Table 4: Essential Research Reagents for Ubiquitin/UBL Studies
| Reagent Category | Specific Examples | Function/Application | Key Features |
|---|---|---|---|
| Recombinant Enzymes | E1 (UBA1, UBA6, NAE), E2 (CDC34, UBE2N, Ubc12), E3 (MDM2, PARKIN, SCF complexes) | In vitro ubiquitination assays, enzyme kinetics, inhibitor screening | Active enzymes with proper folding; availability of mutant forms for mechanistic studies |
| UBL Proteins | Ubiquitin, NEDD8, SUMO, ISG15, ATG8, UFM1 | Substrates for conjugation assays; protein interaction studies | Wild-type and mutant forms; fluorescently labeled variants for FRET-based assays |
| Activity Assay Systems | FRET-based E1/E2/E3 assays, thioester formation assays, ubiquitin chain typing platforms | Quantitative measurement of enzyme activities; inhibitor characterization | High sensitivity; compatibility with high-throughput screening; real-time monitoring capability |
| Linkage-Specific Antibodies | K48-linkage, K63-linkage, linear ubiquitin chain antibodies | Detection of specific ubiquitin chain types in cells and tissues | High specificity; minimal cross-reactivity with other chain types |
| Cell-Based Reporter Systems | Ubiquitin reference technique reporters, GFP-based degradation reporters | Monitoring ubiquitination and degradation in cellular contexts | Physiological relevance; compatibility with live-cell imaging |
| Proteasome Inhibitors | Bortezomib, MG132, Lactacystin | Control compounds for validating ubiquitin-proteasome pathway dependence | Well-characterized specificity; appropriate potency for cellular studies |
The development of inhibitors targeting the enzymatic hierarchy of the ubiquitin-proteasome system represents a rapidly advancing frontier in therapeutic discovery. While E1 inhibitors offer broad pathway modulation and E2 inhibitors provide intermediate specificity, E3 ligase inhibitors and hijacking technologies like PROTACs promise unprecedented target selectivity. The growing understanding of ubiquitin-like protein structure and evolutionary conservation, including the recent discovery of bacterial ubiquitination systems, continues to inform drug design strategies.
Technical advances in screening methodologies, including quantitative FRET assays, structural biology approaches, and computational modeling, are overcoming previous barriers to drug discovery in this pathway. The successful clinical validation of PROTAC technology, with multiple candidates now in clinical trials, demonstrates the therapeutic potential of targeting the ubiquitin system beyond proteasome inhibition. As our understanding of ubiquitin and UBL biology expands, particularly regarding chain specificity and non-proteolytic functions, new opportunities will emerge for developing increasingly sophisticated therapeutics targeting this essential regulatory system.
MLN4924 (Pevonedistat) represents a pioneering first-in-class inhibitor of the NEDD8-activating enzyme (NAE), a key regulator of the ubiquitin-like protein (UBL) modification pathway. This case study details its mechanism-based inhibition of NAE, which disrupts the activity of Cullin-RING Ligases (CRLs), leading to the accumulation of CRL substrates that drive DNA damage, cell cycle arrest, and apoptosis in cancer cells. We summarize the compound's core biochemical properties, its preclinical and clinical development, and its established synergies with standard chemotherapies. Framed within the broader context of UBL structure and evolutionary relevance, MLN4924 serves as a paradigm for the therapeutic targeting of a specific UBL pathway. The document provides structured quantitative data, experimental protocols, pathway visualizations, and a reagent toolkit to support ongoing research and development efforts.
Neuronal precursor cell-expressed developmentally down-regulated protein 8 (NEDD8) is a 9 kDa ubiquitin-like protein (UBL) that shares approximately 60% amino acid sequence identity with ubiquitin [66] [57]. Like other UBLs, NEDD8 is conjugated to target proteins via a sequential enzymatic cascade in a process known as NEDDylation. The primary substrates of NEDDylation are the Cullin family members (CUL1-5), which form the core scaffolding of Cullin-RING Ligases (CRLs) [57]. NEDD8 modification of a Cullin protein induces a conformational change that activates the CRL complex, enabling the ubiquitination of a wide array of substrate proteins that regulate fundamental cellular processes, including cell cycle progression, signal transduction, and DNA damage response [66] [67]. It is estimated that CRLs control the turnover of up to 20% of the cellular proteome, positioning the NEDD8 pathway as a master regulator of cellular homeostasis [66].
Molecular Mechanism of Action of MLN4924
Inhibition of NEDD8-Activating Enzyme (NAE)
MLN4924 is a potent, selective, and mechanism-based inhibitor of NAE [68] [57]. NAE is a heterodimeric E1 enzyme composed of APPBP1 and UBA3 subunits, responsible for the initial, ATP-dependent activation of NEDD8. The enzyme catalyzes the formation of a high-energy NEDD8-adenylate (NEDD8-AMP) intermediate, which then forms a thioester bond with the catalytic cysteine of UBA3 [69] [67].
MLN4924, an AMP mimetic, exploits this catalytic mechanism. The inhibitor is processed by NAE in the presence of NEDD8 and ATP to form a covalent NEDD8-MLN4924 adduct that closely resembles the NEDD8-AMP intermediate [68]. However, unlike the labile native intermediate, the NEDD8-MLN4924 adduct is stable and remains tightly bound within the enzyme's active site. This effectively blocks the subsequent catalytic steps, including the transfer of NEDD8 to the E2 conjugating enzymes (UBC12 and UBE2F), thereby halting the entire NEDDylation cascade [68] [69] [67].
Cellular Consequences and Induction of Cell Death
Inhibition of NAE by MLN4924 leads to the rapid inactivation of CRLs. This results in the stabilization and accumulation of numerous CRL substrates, which drives multiple pro-death cellular outcomes [66] [70].
- DNA Re-Replication and Damage: The accumulation of the DNA replication licensing factor CDT1 upon CRL inactivation leads to unscheduled DNA re-replication in the S-phase. This causes replication stress and DNA double-strand breaks, triggering a robust DNA damage response characterized by the phosphorylation of CHK1 and the accumulation of γH2AX [66] [70].
- Activation of Apoptotic Pathways: MLN4924 treatment induces cell death through both intrinsic and extrinsic apoptotic pathways. Research in colorectal cancer (CRC) models has shown that the apoptotic response is enhanced in p53 wild-type cells and involves the upregulation of TRAIL-R2/DR5 and activation of caspase-8. The anti-apoptotic protein c-FLIP has been identified as a novel mediator of resistance to MLN4924, and its depletion enhances apoptosis [66].
- Senescence and Cell Cycle Arrest: Beyond apoptosis, MLN4924 can induce senescence, an irreversible cell cycle arrest. This is associated with an enlarged cellular morphology, persistent accumulation of p21, and sustained DNA damage response signaling [70].
Table 1: Key Biochemical and Cellular Properties of MLN4924
| Property | Description | Reference |
|---|---|---|
| Target | NEDD8-Activating Enzyme (NAE) | [71] |
| IC₅₀ (Cell-Free) | 4 - 4.7 nM | [72] [70] |
| Mechanism | Forms a covalent NEDD8-MLN4924 adduct, acting as a NEDD8-AMP mimetic | [68] |
| Selectivity | >300-fold selective for NAE over SAE (SUMO E1) and UAE (Ubiquitin E1) | [57] |
| Key Effect | Inactivation of Cullin-RING Ligases (CRLs) | [66] [67] |
| Cellular Outcomes | DNA damage, cell cycle arrest, senescence, and apoptosis | [66] [70] |
Preclinical and Clinical Development
Preclinical Synergy with Chemotherapeutic Agents
MLN4924 has demonstrated significant synergistic effects when combined with various standard-of-care chemotherapies, enhancing its anti-tumor potential.
- Synergy with Irinotecan (SN38) in Colorectal Cancer: MLN4924 synergized with the irinotecan metabolite SN38 in CRC models. The cell death induced by this combination was dependent on BAX/BAK-mediated mitochondrial activation but occurred in a p53-independent manner, a crucial finding for treating advanced CRC with a high frequency of TP53 mutations [66].
- Synergy with Cytarabine (ara-C) in Acute Myeloid Leukemia (AML): MLN4924 was found to disrupt nucleotide metabolism and deplete intracellular nucleotide pools in AML cells. This disruption promoted the increased incorporation of ara-C into DNA, leading to synergistic levels of DNA damage and apoptosis [73].
Table 2: Demonstrated Synergistic Combinations of MLN4924 in Preclinical Models
| Combination Therapy | Cancer Type | Proposed Mechanism of Synergy | Reference |
|---|---|---|---|
| Cytarabine (ara-C) | Acute Myeloid Leukemia (AML) | MLN4924 disrupts nucleotide metabolism, increasing incorporation of ara-C into DNA. | [73] |
| Irinotecan (SN38) | Colorectal Cancer (CRC) | Induces BAX/BAK-dependent apoptosis, bypassing p53 requirement. | [66] |
| TNF-α | Various | Synergistically activates apoptotic cell death pathways. | [67] |
| Rituximab | Mantle Cell Lymphoma | Enhances anti-tumor activity of the therapeutic antibody in vivo. | [67] |
MLN4924 has been evaluated in numerous Phase I and II clinical trials, primarily as a combination therapy. Early phase I trials in patients with acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS) showed modest clinical activity as a single agent [67]. Subsequent trials in relapsed/refractory multiple myeloma, lymphoma, metastatic melanoma, and advanced solid tumors demonstrated a more promising therapeutic effect [67].
Based on encouraging data, the U.S. FDA granted Breakthrough Therapy Designation for Pevonedistat in 2020 for the treatment of patients with higher-risk MDS (HR-MDS) [67]. Current clinical research is focused on investigating MLN4924 in combination therapies rather than as a single agent [67] [57].
Mechanisms of Resistance
A key mechanism of acquired resistance to MLN4924 involves mutations in the UBA3 subunit of NAE. In vitro, selection of resistant leukemic cell lines revealed mutations such as I310N and Y352H in UBA3. These mutations increase the enzyme's affinity for ATP while decreasing its affinity for NEDD8, thereby reducing the potency of MLN4924 without completely abrogating essential NAE function for cell survival [69]. This insight is critical for the development of next-generation NAE inhibitors capable of overcoming such resistance.
Experimental Protocols for Key Assays
In Vitro NAE Activity Assay (Time-Resolved Fluorescence Energy Transfer)
This protocol is used to measure the inhibitory activity of compounds like MLN4924 against NAE in a cell-free system [70].
- Procedure:
- Prepare a 50 µL reaction mixture containing: 50 mM HEPES (pH 7.5), 0.05% BSA, 5 mM MgCl₂, 20 µM ATP, 250 µM glutathione, 10 nM Ubc12-GST, 75 nM NEDD8-Flag, and 0.3 nM recombinant human NAE.
- Incubate the reaction at 24°C for 90 minutes in a 384-well plate.
- Terminate the reaction by adding 25 µL of stop/detection buffer containing: 0.1 M HEPES (pH 7.5), 0.05% Tween-20, 20 mM EDTA, 410 mM KF, 0.53 nM Europium-Cryptate-labelled anti-Flag antibody, and 8.125 µg/mL allophycocyanin (APC)-labelled anti-GST antibody.
- Incubate the plate for 2 hours at 24°C.
- Read the plate using a time-resolved fluorescence method on a compatible plate reader (e.g., LJL Analyst HT). The signal is generated by the energy transfer between the antibody pairs upon successful E2 (Ubc12~NEDD8) conjugate formation.
Analysis of Cell Viability and Apoptosis
This method is used to determine the cytotoxic effects of MLN4924 and its combinations on cancer cells [66] [73].
- Procedure:
- Seed cell suspensions (e.g., 3,000–8,000 cells per well) in 96-well culture plates and incubate overnight.
- Treat cells with MLN4924, other chemotherapeutics, or their combinations for a defined period (e.g., 72 hours).
- Quantify cell viability using ATP-based assays like ATPlite. Luminescence is proportional to the number of viable cells.
- For apoptosis analysis, treat cells as above, then harvest and fix with ethanol.
- Stain cellular DNA with a solution containing Propidium Iodide (PI) and RNase A.
- Analyze the cells by flow cytometry to quantify the sub-G0/G1 population (hypodiploid cells), which represents apoptotic cells.
Immunoblotting for Pharmacodynamic Markers
This protocol assesses the on-target effects of MLN4924 in cells and tissues.
- Procedure:
- Treat cells with MLN4924 and lyse them in an appropriate buffer (e.g., Triton X-100 lysis buffer with protease inhibitors).
- Separate proteins by SDS-PAGE and transfer to a nitrocellulose membrane.
- Probe the membrane with specific antibodies to monitor:
- Inhibition of NEDDylation: Loss of NEDD8-Cullin conjugates.
- Accumulation of CRL Substrates: Increased levels of CDT1, p21, p27, NRF2.
- DNA Damage Response: Phosphorylation of CHK1 (Ser317) and accumulation of γH2AX.
- Apoptosis: Cleavage of PARP and Caspases.
- Detect bands using enhanced chemiluminescence (ECL). β-tubulin or similar proteins should be used as loading controls [66] [73].
Visualization of Signaling Pathways and Workflows
NEDD8 Activation Pathway and MLN4924 Inhibition
The following diagram illustrates the multistep mechanism of NEDD8 activation by NAE and the precise point of inhibition by MLN4924.
MLN4924-Induced Apoptotic Signaling
This diagram summarizes the key apoptotic pathways activated by MLN4924 treatment, as identified in colorectal cancer models.
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents for Investigating MLN4924 and the NEDD8 Pathway
| Reagent / Material | Function / Application | Example Specification / Source |
|---|---|---|
| MLN4924 (Pevonedistat) | Selective NAE inhibitor for mechanistic studies in vitro and in vivo. | >98% purity; reconstitute in DMSO for 5-10 mM stock solutions [71] [70]. |
| Anti-NEDD8 Antibody | Immunoblotting to monitor global neddylation status and inhibition efficacy. | Monoclonal antibody for detecting NEDD8-Cullin conjugates. |
| Anti-CDT1 Antibody | Immunoblotting to confirm on-target NAE inhibition via accumulation of CRL substrate. | Polyclonal or monoclonal antibody [73] [70]. |
| Anti-γH2AX Antibody | Immunofluorescence or immunoblotting to quantify DNA damage response. | Phospho-specific antibody [73]. |
| Anti-Cleaved Caspase-3 Antibody | Immunoblotting or flow cytometry to detect and quantify apoptosis. | Antibody recognizing the activated, cleaved fragment [73]. |
| Recombinant NAE Enzyme | For in vitro biochemical assays to measure direct enzyme inhibition (IC₅₀). | Heterodimeric APPBP1/UBA3, purified [70]. |
| Recombinant NEDD8 & E2 (UBC12) | Essential components for in vitro NAE activity assays. | NEDD8-Flag, Ubc12-GST for TR-FRET assays [70]. |
| Cellular Thermal Shift Assay (CETSA) | Used to study drug-target engagement in a cellular context [57]. | Validates binding of MLN4924 to NAE within cells. |
Targeted protein degradation (TPD) represents a paradigm shift in therapeutic intervention and biological research, moving beyond traditional inhibition to the complete elimination of pathogenic proteins. This whitepaper provides an in-depth technical analysis of two principal TPD modalities—molecular glues and proteolysis-targeting chimeras (PROTACs). Framed within the evolutionary context of ubiquitin-like proteins (UBLs), we examine their mechanistic foundations, design principles, and experimental applications. The content includes structured quantitative data, detailed protocols for emerging technologies like the Guided Protein Labeling and Degradation (GPlad) system, and essential reagent toolkits to equip researchers for advanced work in this rapidly evolving field.
The ubiquitin-proteasome system (UPS), the central executioner of targeted protein degradation in eukaryotes, has its origins deeply rooted in the evolutionary history of the β-grasp fold (β-GF). Ubiquitin (Ub) and ubiquitin-like proteins (Ubls) are defined by this fold, which diversified into at least seven distinct clades by the time of the last universal common ancestor [13] [74]. This fold was initially recruited for translation-related RNA interactions before expanding into a stunning array of biochemical functions, from catalytic roles to sulfur transfer and scaffolding [13].
The eukaryotic phase of β-GF evolution is characterized by a dramatic expansion of the Ubl clade, with at least 70 distinct Ubl families identified across eukaryotes [13] [74]. Nearly 20 of these families were present in the last eukaryotic common ancestor, enabling the emergence of sophisticated cellular systems such as vesicular trafficking, nuclear transport, and chromatin regulation [13]. The core mechanism of protein conjugation—the covalent attachment of Ub/Ubls to target proteins or lipids—represents a molecular logic co-opted for TPD technologies. Modern molecular glues and PROTACs are therefore not mere inventions but sophisticated applications of an ancient evolutionary template, hijacking a natural protein-tagging system that dates back to prokaryotic predecessors [75] [74].
Mechanistic Principles of Molecular Glues and PROTACs
The Ubiquitin-Proteasome System (UPS)
The UPS is the primary cellular pathway for regulated protein degradation in eukaryotes. It involves a cascade of three enzymes:
- E1 (Ubiquitin-activating enzyme): Activates ubiquitin in an ATP-dependent manner.
- E2 (Ubiquitin-conjugating enzyme): Accepts the activated ubiquitin from E1.
- E3 (Ubiquitin ligase): Recognizes specific protein substrates and facilitates the transfer of ubiquitin from E2 to the target protein [76].
A protein tagged with a chain of at least four ubiquitin molecules is recognized and degraded by the 26S proteasome, a multi-subunit complex comprising a 20S catalytic core and 19S regulatory caps [76]. This process is central to the mechanism of action for both molecular glues and PROTACs.
Molecular Glues: Monovalent Inducers of Proximity
Molecular glues are small, monovalent compounds (typically <500 Da) that induce or stabilize interactions between two proteins that would not otherwise associate productively. In TPD, they function by binding to an E3 ubiquitin ligase, creating a new molecular surface that can recruit a target protein of interest (POI), or vice versa [77] [76]. This induced proximity leads to the polyubiquitination and subsequent proteasomal degradation of the POI.
A key advantage of molecular glues is their catalytic mode of action; a single molecule can facilitate the degradation of multiple copies of a target protein [77]. Their small size often confers favorable drug-like properties, including better membrane permeability and potential to cross the blood-brain barrier compared to larger degraders [77]. However, their discovery has historically been serendipitous, as rationally designing a small molecule to create a novel, productive protein-protein interface remains challenging [76].
PROTACs: Bivalent Catalytic Degraders
PROteolysis TArgeting Chimeras (PROTACs) are heterobifunctional molecules consisting of three key elements:
- A ligand that binds to a target protein of interest (POI).
- A ligand that recruits an E3 ubiquitin ligase.
- A chemical linker connecting these two moieties [78].
PROTACs act as catalytic "matchmakers," physically bridging the E3 ligase and the POI to form a productive ternary complex. This complex enables the transfer of ubiquitin from the E2 enzyme to lysine residues on the POI, marking it for proteasomal degradation [78] [76]. The table below summarizes a quantitative comparison of these two modalities.
Table 1: Quantitative Comparison of Molecular Glues and PROTACs
| Property | Molecular Glues | PROTACs |
|---|---|---|
| Molecular Weight | Typically <500 Da [76] | Larger, often >700 Da [78] |
| Structure | Monovalent [76] | Heterobifunctional (Two ligands + linker) [78] |
| Mechanism | Induce novel binding interface between E3 and POI [77] [76] | Form a ternary complex by linking E3 and POI [78] |
| Discovery Paradigm | Largely serendipitous; high-throughput screening [76] | More amenable to rational design [78] |
| Key Advantage | Drug-like properties, catalytic action [77] | Modularity; ability to target numerous proteins [78] |
The following diagram illustrates the core mechanistic pathway shared by molecular glues and PROTACs, leading to target protein degradation via the ubiquitin-proteasome system.
Diagram 1: TPD mechanism via the ubiquitin-proteasome system.
Research Landscape and Quantitative Data
The TPD field is experiencing explosive growth, with research and development focused on a wide range of diseases. The following tables consolidate quantitative data from the CAS Content Collection to illustrate current trends and focus areas.
Table 2: Prevalence of E3 Ligases in TPD Publications and Associated Diseases
| E3 Ligase | Prevalence in Publications | Primary Disease Associations |
|---|---|---|
| CRBN (Cereblon) | Highest [77] | Cancer (Multiple Myeloma, Leukemia), Autoimmune Diseases [77] |
| VHL (von Hippel-Lindau) | High [77] | Cancer, Inflammation [77] |
| MDM2 | High [77] | Cancer [77] |
Table 3: Disease Areas Targeted by Protein Degraders (Based on Publication Analysis)
| Disease Area | Proportion of Publications |
|---|---|
| Cancer | 44% |
| Infectious Diseases | 11% |
| Neurodegenerative Diseases | 10% |
| Inflammatory Diseases | 10% |
| Autoimmune Diseases | 8% |
| Metabolic Diseases | 6% |
| Cardiovascular Diseases | 5% |
Experimental Protocols and Novel Systems
The GPlad System for Bacterial Protein Degradation
Most TPD systems function in eukaryotes. The Guided Protein Labeling and Degradation (GPlad) system is a novel, de novo designed technology for precise protein degradation in E. coli, offering a plug-and-play strategy without the need for pre-fused degrons or chemical degraders [79].
System Components:
- Marking Protein (MP): McsB, an arginine kinase from B. subtilis that phosphorylates arginine residues on proximal proteins, marking them for degradation.
- Guide Protein (GP): A de novo designed protein that binds specifically to a target protein.
- Protease: The ClpCP protease complex, which recognizes and degrades the McsB-marked proteins [79].
Detailed Workflow:
- System Construction: Co-express the three components on plasmids in E. coli: the GP-McsB fusion, the ClpCP protease, and the target protein [79].
- Spatial Proximity: The de novo designed GP binds with high affinity to a specific surface on the target protein, bringing McsB into close proximity.
- Target Labeling: McsB phosphorylates arginine residues on the surface of the target protein.
- Protein Degradation: The ClpCP protease complex recognizes the phosphorylated arginine marks, unfolds the target protein, and degrades it into small peptides [79].
The GPlad system has been successfully used to degrade various proteins in E. coli, including fluorescent proteins (e.g., mKate2), metabolic enzymes, and human proteins. The system's efficiency was demonstrated by a 49.5% reduction in fluorescence and a 56.7% decrease in mKate2 protein abundance within 6 hours of induction [79].
The following diagram outlines the key stages of the GPlad experimental workflow.
Diagram 2: GPlad system workflow for targeted protein degradation.
Screening Method for Molecular Glues
Identifying molecular glues remains a significant challenge. The following protocol outlines a yeast two-hybrid screening approach, adapted from a method used to discover modulators of plant hormone receptors [76].
Objective: To identify small molecules that induce or stabilize the interaction between an E3 ubiquitin ligase and a target protein.
Procedure:
- Strain Generation: Engineer a yeast reporter strain where:
- The "bait" is your E3 ubiquitin ligase (or a key subunit, e.g., an F-box protein) fused to a DNA-binding domain.
- The "prey" is your target protein fused to a transcriptional activation domain.
- Library Screening: Treat the yeast strain with a library of small molecules (e.g., ~22,500 compounds).
- Selection and Identification: Plate the yeast on selective media that lacks a key nutrient (e.g., histidine). Only yeast cells where the small molecule has successfully "glued" the bait and prey together will activate the reporter gene (e.g., HIS3), allowing them to grow.
- Hit Validation: Isolate the growing colonies and validate the hits through secondary assays, such as measuring target protein degradation in mammalian cells [76].
The Scientist's Toolkit: Key Research Reagents
The table below details essential reagents and tools used in the featured GPlad experiment and broader TPD research.
Table 4: Key Research Reagent Solutions for TPD Experiments
| Reagent / Tool | Function in Research | Example from GPlad Protocol |
|---|---|---|
| De Novo Designed Binders | Custom-designed proteins that bind with high specificity to a target protein of interest. | GP12959 (GPmkate2) guide protein designed to bind mKate2 [79]. |
| Marking Enzyme | An enzyme that catalytically labels a target protein for recognition by a degradation machinery. | McsB, an arginine kinase that phosphorylates target proteins [79]. |
| Orthogonal Protease | A protease system that does not interfere with the host's native degradation machinery. | The ClpCP protease complex from B. subtilis used in E. coli [79]. |
| High-Affinity Heterodimers | Protein pairs that bind tightly and specifically, used as modular proximity scaffolds. | 1/1′ (37ABXBA and 37ABXBB) used for initial system validation [79]. |
| Reporter Proteins | Proteins (e.g., fluorescent) used to easily quantify degradation efficiency. | mKate2 (red fluorescent protein) used as a degradation target [79]. |
| Bacterial Two-Hybrid System | A tool to detect and quantify protein-protein interactions in vivo. | Used for the initial screening of designed guide proteins [79]. |
Molecular glues and PROTACs have fundamentally expanded the druggable proteome, enabling the targeting of proteins that defy conventional inhibition. Their development is a testament to the power of leveraging deep evolutionary biology—specifically the versatile UBL architecture—for transformative therapeutic applications.
The future of TPD is focused on overcoming current challenges and expanding its boundaries. Key areas include achieving greater spatiotemporal precision in degradation, increasing the throughput of degrader synthesis, and optimizing the cooperativity of chemically induced complexes [80]. The integration of nanotechnology is also emerging as a powerful strategy to improve the solubility, delivery, and efficacy of TPD agents [81]. Furthermore, the clinical landscape is poised for validation, with several PROTACs and molecular glues in advanced trials, and partnerships within the pharmaceutical industry exceeding $200 million in 2025 alone [82]. As the field matures from serendipitous discovery to rational design, these modalities are set to reshape the treatment of cancer, neurodegenerative disorders, and many other diseases with high unmet need.
Navigating Complexity: Challenges and Strategies in UBL Drug Discovery
Ubiquitin-like proteins (UBLs) constitute a critical family of post-translational modifiers that regulate virtually all cellular processes through parallel, yet distinct, signaling pathways. Despite sharing a conserved β-grasp fold, UBLs including ubiquitin, SUMO, NEDD8, and ISG15 orchestrate diverse physiological outcomes—from protein degradation to immune response—by maintaining exquisite pathway specificity. This technical analysis examines the molecular mechanisms that enforce specificity throughout UBL activation, conjugation, and deconjugation cycles. We integrate structural insights with quantitative kinetic data to elucidate how cognate enzyme-substrate pairs achieve selective recognition while minimizing aberrant cross-talk. The emerging principles not only clarify fundamental ubiquiton biology but also reveal strategic vulnerabilities for therapeutic intervention in cancer, neurodegenerative diseases, and viral pathogenesis.
The ubiquitin-like protein family encompasses at least eight covalently conjugatable modifiers (Type I UBLs) in humans, including ubiquitin, SUMO, NEDD8, ATG8, ATG12, URM1, UFM1, FAT10, and ISG15 [1]. Each UBL modifies specific cellular targets through a dedicated enzymatic cascade comprising E1 activating, E2 conjugating, and E3 ligating enzymes, while deconjugating enzymes (DUBs and ULPs) reverse these modifications [83] [84]. The central challenge in UBL biology lies in understanding how parallel pathways maintain specificity despite using chemically identical isopeptide bond formation and structurally similar protein folds.
Evolutionary analyses reveal that UBL systems originated from prokaryotic sulfurtransferase systems, with UBL-conjugating and deconjugating enzymes already widespread in the last common eukaryotic ancestor [83]. This deep conservation underscores the fundamental importance of regulated protein modification throughout eukaryotic evolution. The human genome encodes approximately 100 deubiquitinating enzymes (DUBs) and UBL-specific proteases (ULPs) that must distinguish between highly similar UBLs and their polymeric forms [84]. Pathway fidelity is further complicated by the formation of hybrid chains, such as SUMO-ubiquitin hybrids, which create unique signaling platforms [84].
This technical guide examines the molecular principles governing specificity in UBL pathways, with emphasis on structural recognition features, kinetic partitioning mechanisms, and experimental approaches for probing selective interactions. The insights presented herein frame UBL specificity within the broader context of evolutionary adaptation and therapeutic development.
Molecular Basis of UBL Recognition and Specificity
Structural Determinants of UBL Identity
UBLs share a common β-grasp fold consisting of a five-strand antiparallel β sheet surrounding a central α helix [1]. Despite this structural conservation, each UBL displays distinct surface features that enable specific recognition by cognate enzymes. Key recognition patches include:
- Ile44 patch (comprising Ile44, Leu8, Val70, and His68 in ubiquitin)
- Ile36 patch (Ile36, Leu71, and Leu73 in ubiquitin)
- TEK box (Lys6, Lys11, Thr12, Thr14, and Glu34 in ubiquitin)
- Flexible C-terminal tail (residues P6-P1) [84]
NEDD8 shares significant sequence similarity with ubiquitin in these patches, particularly the Ile44 and Ile36 patches, which directly bind to the deneddylase Den1/SENP8 [84]. In contrast, SUMO and ISG15 exhibit minimal conservation in these regions, contributing to their discrimination [84]. The C-terminal tail represents perhaps the most critical specificity determinant, with single amino acid substitutions sufficient to alter protease affinity [84].
Table 1: Key Recognition Patches in Human UBLs
| UBL | Ile44 Patch Conservation | Ile36 Patch Conservation | C-terminal Tail Sequence | Primary Cognate Proteases |
|---|---|---|---|---|
| Ubiquitin | Ile44, Leu8, Val70, His68 | Ile36, Leu71, Leu73 | LRLRGG | Multiple DUB families |
| NEDD8 | High conservation | High conservation | LRLRGG | Den1/SENP8 |
| SUMO-1 | Low conservation | Low conservation | QQQTGG | SENP/ULP family |
| ISG15 | No conservation | No conservation | LRLRGG | USP21, SARS-CoV-2 PLpro |
Mechanisms of Deconjugating Enzyme Specificity
Deubiquitinating enzymes (DUBs) and Ubl-specific proteases (ULPs) employ multiple strategies to distinguish among different UBLs:
Topographical specificity: DUBs and ULPs contain binding pockets that complement the surface features of specific UBLs. For example, the ubiquitin-specific protease CYLD recognizes distal ubiquitin in Met1- or Lys63-linked diubiquitin without engaging the Ile44 patch [84]. Similarly, the SUMO-specific protease SENP1 shows minimal interaction with the β1-β2 loop that is critical for ubiquitin recognition by UCH and USP family DUBs [84].
Active site complementarity: The catalytic clefts of DUBs and ULPs are shaped to accommodate specific UBL C-terminal sequences. USP21, which processes both ubiquitin and ISG15, contains an invariant glutamate that forms a salt bridge with Arg72 (P5) present in both ubiquitin and ISG15 but absent in NEDD8 [84]. This explains USP21's discrimination against NEDD8 despite its dual specificity.
Allosteric regulation: UBL domains within deubiquitinating enzymes themselves can modulate specificity. In USP4, a UBL domain mimics ubiquitin binding to the catalytic domain, autoinhibiting non-cognate interactions [85]. Conversely, in USP7, C-terminal UBL domains promote catalytic activity by inducing favorable conformational changes rather than inhibiting it [85].
Linkage and chain-type selectivity: Many DUBs display marked preferences for specific ubiquitin chain linkages. OTU family DUBs frequently specialize in cleaving particular linkage types, while JAMM/MPN+ metalloproteases show strong preferences for Lys63-linked chains [84].
Quantitative Analysis of UBL Protease Specificity
Kinetic parameters provide crucial insights into the efficiency and selectivity of UBL proteases. Recent studies on viral and human proteases reveal how specificity is achieved through differential recognition.
Table 2: Kinetic Parameters of SARS-CoV-2 PLpro Wild-Type (WT) vs. Truncated (TR) Variant [86]
| Enzyme | Substrate | kcat (s-1) | Km (μM) | kcat/Km (M-1s-1) | Specificity Relative to Z-RLRGG-AMC |
|---|---|---|---|---|---|
| PLpro WT | Z-RLRGG-AMC | 0.47 ± 0.03 | 182.3 ± 21.5 | (2.58 ± 0.33) × 103 | 1.0 |
| PLpro TR | Z-RLRGG-AMC | 0.19 ± 0.01 | 245.6 ± 25.1 | (7.74 ± 0.89) × 102 | 0.3 |
| PLpro WT | Ub-AMC | 2.85 ± 0.12 | 0.16 ± 0.03 | (1.78 ± 0.35) × 107 | 6.9 × 103 |
| PLpro TR | Ub-AMC | 0.66 ± 0.04 | 0.22 ± 0.04 | (3.00 ± 0.59) × 106 | 3.9 × 103 |
| PLpro WT | ISG15-AMC | 1.72 ± 0.08 | 0.11 ± 0.02 | (1.56 ± 0.29) × 107 | 6.0 × 103 |
| PLpro TR | ISG15-AMC | 0.52 ± 0.03 | 0.18 ± 0.03 | (2.89 ± 0.51) × 106 | 3.7 × 103 |
The quantitative data reveal several key principles:
- Catalytic efficiency maintenance: Removal of the Ubl domain in PLpro (TR variant) reduces kcat/Km for all substrates by 4-6 fold, demonstrating the Ubl domain's role in optimizing catalytic efficiency without completely abolishing activity [86].
- Substrate discrimination: Both WT and TR PLpro show 1000-fold higher efficiency toward natural substrates (Ub-AMC, ISG15-AMC) versus the peptide substrate Z-RLRGG-AMC, highlighting their adaptation to recognize full UBL structures [86].
- Dual specificity mechanism: The similar efficiency toward Ub-AMC and ISG15-AMC explains PLpro's dual deubiquitinating and deISGylating activity, a key viral immune evasion strategy [86].
Experimental Approaches for Profiling UBL Specificity
Functional Proteomic Profiling
Comprehensive understanding of UBL pathway specificity requires systematic analysis of modification patterns across cellular states. The experimental workflow below outlines a robust approach for UBL substrate profiling:
Diagram 1: UBL Substrate Profiling Workflow. This approach enables identification of 80-200 exclusive targets per UBL across cellular states [87].
This methodology has revealed that UBL networks are non-random, with most protein targets mapping to a single UBL, and has uncovered previously underappreciated roles for UBLs like FAT10 in mitotic regulation [87]. The technique leverages active mammalian cell extracts to maintain physiological enzyme activities and protein microarrays for high-throughput target identification.
Structural and Biophysical Characterization
Molecular dynamics simulations and X-ray crystallography provide atomic-level insights into specificity mechanisms. For example, all-atom molecular dynamics simulations of Parkin activation revealed how phosphorylation of Ser65 in its UBL domain initiates a sequential unfolding pathway that releases autoinhibition [88]. Similar approaches applied to SARS-CoV-2 PLpro demonstrate how its Ubl domain stabilizes critical structural elements like the ridge helix in the thumb subdomain [86].
Differential scanning fluorimetry (DSF) assays enable quantitative assessment of structural stability by measuring protein melting temperatures under different conditions. In PLpro studies, DSF revealed that the Ubl domain contributes to structural integrity, with the truncated variant showing altered thermal stability [86].
Enzyme Inhibition and Specificity Profiling
Competitive inhibition assays using natural UBLs reveal unexpected regulatory relationships. Free ubiquitin enhances PLpro enzymatic activity, likely through non-canonical binding sites distinct from the SUb1 and SUb2 sites [86]. This allosteric modulation represents an additional layer of regulatory complexity in UBL pathway specificity.
Table 3: Research Reagent Solutions for UBL Specificity Studies
| Reagent/Category | Specific Examples | Function/Application | Key Insights Enabled |
|---|---|---|---|
| Fluorogenic Substrates | Ub-AMC, ISG15-AMC, Z-RLRGG-AMC | Steady-state kinetic analysis | Quantification of catalytic efficiency (kcat/Km) and specificity constants |
| Activity-Based Probes | Ubiquitin vinyl sulfone, SUMO-1 vinyl methyl ester | Active-site labeling and profiling | Identification of active deubiquitinases in complex proteomes |
| Recombinant UBLs | Wild-type and mutant UBLs (e.g., NEDD8 A5R) | Competition assays, structural studies | Determination of specificity-determining residues in UBLs |
| Specialized Inhibitors | GRL0617 (PLpro inhibitor), Ubiquitin variants (UbVs) | Mechanistic and functional studies | Allosteric regulation analysis and therapeutic development |
| Cellular Extraction Kits | Active cell extract preparation kits | Functional proteomic profiling | Maintenance of physiological enzyme activities for pathway analysis |
Regulatory Plasticity and Allosteric Modulation
UBL pathways exhibit remarkable regulatory plasticity, with UBL domains often serving as allosteric regulators of catalytic activity. Several illustrative examples demonstrate this principle:
SARS-CoV-2 PLpro: The N-terminal Ubl domain stabilizes the ridge helix in the thumb subdomain, and its removal alters substrate processing and reduces catalytic efficiency [86]. Molecular dynamics simulations show that the Ubl domain stabilizes critical structural elements, with its removal increasing flexibility in the thumb and palm subdomains [86].
USP Family Regulation: UBL domains within ubiquitin-specific proteases employ diverse regulatory mechanisms. In USP14, the UBL domain mediates proteasome association, enhancing catalysis [85]. In USP4, the UBL domain acts as an autoinhibitory element by competing with ubiquitin binding [85]. USP7 contains five UBL domains that promote catalytic activity through allosteric activation rather than inhibition [85].
Parkin Activation: The E3 ubiquitin ligase Parkin maintains an autoinhibited conformation through intra-molecular interactions. Phosphorylation of Ser65 in its UBL domain by PINK1 initiates conformational opening that releases this autoinhibition, enabling Parkin's catalytic activity [88]. Molecular dynamics simulations show that phosphorylation-induced UBL domain dissociation propagates through further opening conformations that allow E2-Ub binding [88].
Implications for Therapeutic Development
The molecular principles governing UBL pathway specificity present attractive therapeutic opportunities. Several strategic approaches have emerged:
Targeted inhibition of viral UBL proteases: SARS-CoV-2 PLpro represents a promising antiviral target due to its essential roles in viral replication and immune evasion [86]. Its dual deubiquitinating and deISGylating activity makes it particularly vulnerable to selective inhibition that preserves host DUB functions.
Allosteric modulation of E3 ligases: The conformational plasticity of Parkin and other E3 ligases suggests opportunities for small molecule activators that stabilize active conformations [88]. Such approaches could mitigate pathological Parkin mutations associated with Parkinson's disease.
Exploitation of structural differences: Despite shared folds, UBLs contain sufficient structural variation to enable selective targeting. Engineered ubiquitin variants (UbVs) that bind allosteric sites on DUBs can achieve remarkable specificity, as demonstrated by potent inhibition of PLpro and reduction of viral replication in cell culture [86].
The continuing evolution of structural, biochemical, and proteomic methodologies will undoubtedly uncover additional specificity mechanisms and therapeutic opportunities within the intricate networks of UBL signaling pathways.
The ubiquitin-proteasome system (UPS) represents a master regulatory network controlling protein degradation and function in eukaryotic cells. Central to this system are ubiquitin ligases (E3s), which confer substrate specificity by recruiting target proteins for ubiquitination. The Really Interesting New Gene (RING) E3 ligases constitute the largest class of E3s, characterized by a RING finger domain that typically coordinates two Zinc ions in a cross-braced arrangement and functions as a scaffold that directly facilitates ubiquitin transfer from an E2 conjugating enzyme to a substrate [89]. The human genome encodes over 600 RING E3 ligases, which are implicated in virtually all cellular processes and are frequently dysregulated in human diseases, particularly cancer [89] [90]. For instance, BRCA1 (involved in DNA repair), Mdm2 (a key regulator of p53), and components of the FANC ligase are all RING-type E3s with established roles in cancer pathogenesis [89].
Despite their profound therapeutic potential, RING E3 ligases have largely been considered "undruggable" due to several intrinsic challenges. Their enzymatic activity primarily depends on protein-protein interactions (PPIs) rather than deep catalytic pockets, their surfaces are often large and flat, and they frequently function within multi-subunit complexes with dynamic conformational changes [91]. This whitepaper delineates the current strategies to overcome these challenges, focusing on the mechanistic structural biology of RING E3 ligases and the innovative approaches being developed to target their protein-protein interfaces.
Mechanistic and Structural Basis of RING E3 Ligase Function
The Catalytic Core and E2~Ub Engagement
RING E3 ligases function by binding both a substrate and a ubiquitin-loaded E2 conjugating enzyme (E2~Ub), facilitating the direct transfer of ubiquitin from the E2 to a lysine residue on the substrate. This distinguishes them from HECT-type E3s, which form a covalent thioester intermediate with ubiquitin [89] [90]. The RING domain itself, typically comprising 40–60 residues that coordinate two Zn²⁺ ions, creates the primary platform for E2 binding [89]. A landmark structural study of the RING E3 RNF4 in complex with an E2~Ub conjugate revealed the mechanistic details of this process. The structure showed that while the E2 (UbcH5A) contacts a single protomer of the RING dimer, the ubiquitin molecule is folded back onto the E2, making contacts with both RING protomers. This intricate network of interactions locks the C-terminal tail of ubiquitin into an active site groove on the E2, priming the complex for catalysis by deprotonating the incoming substrate lysine and stabilizing the consequent tetrahedral transition-state intermediate [92].
The Critical Role of Dimerization and Complex Assembly
A defining feature of many RING E3 ligases is their propensity to form homodimers and heterodimers, a key regulatory mechanism for their activity. Prominent examples include cIAP, RNF4, and the heterodimers BRCA1-BARD1 and Mdm2-MdmX [89]. Dimerization can occur either through the RING domains themselves or via flanking sequences. In structures of RING dimers, the E2 binding surfaces face away from each other, suggesting that the two RING-bound E2s within a dimer do not cooperate directly [89]. Dimerization can serve as a critical control point; for example, the RING homodimerization interface of cIAP1 is sequestered in an inactive conformation until activated by IAP antagonists like SMAC/DIABLO, which stabilizes an "open" conformation permitting RING dimerization and subsequent E2 binding [89].
Table 1: Characterized RING E3 Ligase Dimers and Their Functional Roles
| E3 Ligase Complex | Dimer Type | Functional Significance |
|---|---|---|
| RNF4 | Homodimer | Forms via interleaved C-termini; involved in DNA damage response [89] [92]. |
| BRCA1-BARD1 | Heterodimer | BARD1 stabilizes BRCA1 and enhances its E3 ligase activity [89]. |
| Mdm2-MdmX | Heterodimer | Critical for regulation of p53 levels; MdmX alone is largely inactive [89]. |
| cIAP1 | Homodimer | Dimerization and activity activated by SMAC/DIABLO binding [89]. |
Substrate Recognition: The Degron Code
The specificity of RING E3 ligases is governed by their ability to recognize short linear motifs, structural elements, or post-translational modifications on their substrates, collectively known as degrons [90]. Key degron types include:
- N-degrons: The identity of the N-terminal amino acid, according to the N-end rule, can dictate a protein's half-life [90].
- Phosphodegrons: Phosphorylation of specific serine, threonine, or tyrosine residues can create a high-affinity binding site for an E3. The F-box protein FBW7, part of an SCF E3 complex, specifically recognizes phosphorylated substrates through arginine residues that form hydrogen bonds with the phosphate group [90].
- Oxygen-dependent degrons: The VHL E3 ligase complex recognizes HIF-α only when a specific proline residue is hydroxylated under normal oxygen conditions [90].
- Misfolding and Glycan degrons: E3s like San1 in yeast can recognize hydrophobic patches exposed on misfolded proteins, while Fbs1/2 recognize high-mannose glycans on misfolded glycoproteins [90].
Therapeutic Strategies for Targeting RING E3 Ligases
Targeting E3-Substrate Interactions
A primary strategy involves developing small molecules that disrupt the interaction between an E3 and its specific oncogenic substrate. This approach is exemplified by efforts targeting Mdm2 to reactivate the p53 tumor suppressor pathway. Similarly, the VHL complex can be targeted to stabilize HIF-α, which may have therapeutic implications. The main challenge lies in the often extensive and flat binding interfaces involved in these PPIs [91].
Modulation of E3 Ligase Activity
An alternative is to target allosteric sites to modulate the E3's intrinsic activity. As described earlier, the activity of cIAP1 is allosterically regulated by SMAC mimetics, which promote its RING dimerization and activation [89]. The discovery of molecular glues that induce or stabilize specific E3 conformations or complex assemblies represents a powerful, albeit serendipitous, approach in this category. The immunomodulatory drug thalidomide and its analogs (lenalidomide, pomalidomide) are classic examples that redirect the CRL4CRBN E3 ligase to target specific transcription factors for degradation [91].
The PROTAC Platform: Inducing Targeted Protein Degradation
Proteolysis-Targeting Chimeras (PROTACs) are bifunctional molecules that represent a paradigm shift in drug discovery. A PROTAC consists of one ligand that binds a target protein of interest, another ligand that recruits a specific E3 ubiquitin ligase, and a linker connecting them [91] [93]. By bringing the E3 ligase into proximity with the target protein, the PROTAC induces ubiquitination and subsequent proteasomal degradation of the target. This technology effectively hijacks the cell's natural degradation machinery to eliminate pathogenic proteins, including those previously considered "undruggable" by conventional small-molecule inhibitors. The versatility of this platform is being explored with various RING E3 ligases, including CRBN, VHL, and others, as the recruitment module [91].
Figure 1: Mechanism of Action of PROTACs. A heterobifunctional PROTAC molecule simultaneously binds a target protein and an E3 ubiquitin ligase, forming a ternary complex that leads to target ubiquitination and proteasomal degradation.
Inhibiting E3 Complex Assembly and Neddylation
Cullin-RING ligases (CRLs), a major subclass of RING E3s, require the ubiquitin-like protein NEDD8 to be conjugated (neddylated) to the cullin subunit for full activation [1]. This makes the neddylation pathway a compelling target for indirect inhibition of a large family of E3s. MLN4924 (Pevonedistat) is a first-in-class small molecule that inhibits the NEDD8-activating enzyme (E1), thereby blocking the neddylation of all cullins and the activity of CRLs [91]. This approach has validated CRLs as promising anti-cancer targets. Furthermore, specific protein-protein interactions within E3 complexes can be targeted, such as the interaction between UBE2M and DCN1, which is required for the neddylation and activation of CUL1 and CUL3 [91].
Table 2: Representative Small-Molecule Inhibitors and Modulators of RING E3 Pathways
| Compound/Approach | Target | Mechanism of Action | Therapeutic Stage |
|---|---|---|---|
| MLN4924 (Pevonedistat) | NAE (NEDD8 E1) | Inhibits cullin neddylation, blocking CRL activity [91]. | Clinical Trials |
| SMAC Mimetics | IAPs (e.g., cIAP1) | Promote RING dimerization and auto-ubiquitination or activate apoptosis [89]. | Clinical Trials |
| Nutlins | Mdm2-p53 interaction | Blocks Mdm2 from binding and ubiquitinating p53 [89]. | Preclinical/Clinical |
| PROTACs | Various targets via E3 recruitment | Induces targeted protein degradation [91] [93]. | Early Clinical/Preclinical |
| UBE2M-DCN1 inhibitors | UBE2M-DCN1 PPI | Blocks neddylation of CUL1/3, inhibiting specific CRLs [91]. | Preclinical |
The Scientist's Toolkit: Experimental Methods for Elucidating RING E3 Mechanisms
Structural Biology Techniques
- X-ray Crystallography: Essential for determining high-resolution structures of RING domains, E2~Ub-RING complexes, and full-length multi-subunit E3s. The structure of the RNF4~UbcH5A~Ub complex was solved using this method [92].
- NMR Spectroscopy: Provides dynamic information on conformational changes, binding interfaces, and allosteric regulation that may be missed in static crystal structures [89].
- Cryo-Electron Microscopy (Cryo-EM): Increasingly vital for solving structures of large, flexible multi-subunit E3 complexes like the anaphase-promoting complex/cyclosome (APC/C) [90] that are difficult to crystallize.
Biochemical and Cellular Assays
- In Vitro Ubiquitination Assays: Reconstitute the ubiquitination reaction using purified E1, E2, E3, ubiquitin, and ATP to study enzyme activity and specificity directly [92] [94].
- Tandem Ubiquitin-Binding Entities (TUBEs): Used in proteomics to capture and identify ubiquitinated proteins, helping to map E3 substrates [90].
- Proximity-Dependent Biotin Identification (BioID): Identifies proximal and interacting proteins in live cells, useful for mapping E3 interactomes and identifying novel substrates or regulators [90].
Table 3: Key Research Reagent Solutions for E3 Ligase Studies
| Reagent / Tool | Function and Application |
|---|---|
| Active E2~Ub Thioester Complexes | Pre-formed, reactive intermediates for structural and biochemical studies of RING mechanism [92]. |
| NEDD8/WT-Ubiquitin and Mutants | Wild-type and lysine-mutant ubiquitins (e.g., K48-only, K63-only) to study chain linkage specificity [90] [1]. |
| E3-Substrate Trapping Mutants | Engineered E3s (e.g., with inactive RING) used with crosslinkers to identify transient substrate interactions [90]. |
| Deubiquitinase (DUB) Inhibitors | Preserve cellular ubiquitin signals during lysis for more accurate analysis of ubiquitination states [1]. |
| Phospho-specific Substrate Antibodies | Detect post-translational modifications that create degrons, enabling study of phosphodegron-dependent recognition [90]. |
Figure 2: Integrated Workflow for RING E3 Ligase Mechanistic Studies. A multi-pronged experimental approach combining structural, biochemical, and cellular/proteomic methods is required to fully elucidate the function and regulation of a RING E3 ligase.
The field of RING E3 ligase drug discovery is at a pivotal juncture. The historical challenges posed by their extensive, flat PPI interfaces are being systematically addressed through a combination of deep mechanistic understanding and technological innovation. The elucidation of RING-E2~Ub complex structures has provided a blueprint for catalysis, while the recognition of dimerization and allosteric regulation has revealed new avenues for therapeutic intervention. The most promising developments, such as the PROTAC platform and molecular glues, have reframed the challenge from one of inhibiting an "undruggable" enzyme to one of repurposing it. By hijacking the cell's natural degradation machinery, these modalities transform RING E3 ligases from targets into tools, opening a vast new frontier for targeting oncoproteins, pathological aggregates, and other disease-causing proteins.
Future progress will depend on continued structural biology efforts to visualize more E3-ligase complexes in active states, the expansion of the repertoire of E3 ligases that can be productively engaged by small molecules, and the development of strategies to achieve tissue and cell-type specificity. As our knowledge of the intricate structure and evolutionary conservation of the ubiquitin and ubiquitin-like systems deepens [1], so too will our ability to rationally design the next generation of therapeutics targeting this fundamental regulatory network.
The ubiquitin-proteasome system (UPS) is a crucial regulatory mechanism for intracellular protein degradation, influencing essential processes such as cell cycle progression, signal transduction, and apoptosis. Central to this system are E2 ubiquitin-conjugating enzymes, which serve as pivotal mediators in the transfer of ubiquitin to target protein substrates. The human E2 enzyme Cdc34 (hCdc34) is particularly notable for its role in conjugating ubiquitin to numerous protein substrates in collaboration with the cullin-RING (CRL) superfamily of E3 enzymes, thereby controlling protein stability and interactions [95]. Traditional drug discovery efforts have primarily focused on orthosteric inhibitors that compete with natural substrates at the enzyme's active site. However, these approaches often face challenges related to specificity and off-target effects. In contrast, allosteric inhibition—modulating enzyme activity by binding at a site distinct from the catalytic center—offers a promising alternative. The discovery of CC0651, a small molecule that selectively inhibits hCdc34 through an allosteric mechanism, validates E2 enzymes as a viable class of drug targets within the UPS and opens new avenues for therapeutic intervention [95] [96].
The Ubiquitin-Conjugation Cascade: A Primer on E2 Enzyme Function
E2 enzymes operate within a well-defined enzymatic cascade. The process initiates with an E1 activating enzyme, which activates ubiquitin in an ATP-dependent reaction, forming a thioester bond between its active site cysteine and the C-terminal glycine of ubiquitin [97]. The activated ubiquitin is then transferred to the active site cysteine of an E2 conjugating enzyme, forming a E2~Ub thioester intermediate [98] [99]. Finally, often with the assistance of an E3 ubiquitin ligase which aids in substrate recognition, the ubiquitin is conjugated via an isopeptide bond to a lysine residue on the target protein [98]. E2 enzymes are not merely passive carriers; they are main determinants for the specific lysine linkage within ubiquitin chains, a feature that directly influences the cellular fate of the substrate protein [98].
Table 1: Key Enzymes in the Ubiquitin/UBL Conjugation Pathway
| Enzyme | Role in Conjugation Cascade | Key Functional Features |
|---|---|---|
| E1 Activating Enzyme | Activates Ub/UBL and transfers it to E2 | Adenylation domain, catalytic cysteine domain, C-terminal Ub-fold domain (UFD) for E2 recruitment [97]. |
| E2 Conjugating Enzyme | Accepts Ub/UBL from E1 and catalyzes its transfer to substrate | Active site cysteine for thioester linkage, determines lysine linkage specificity in Ub/UBL chains [98]. |
| E3 Ligase | Facilitates substrate selection and final transfer | Recognizes specific protein substrates; works with E2 to catalyze isopeptide bond formation [98]. |
CC0651: A Case Study in Allosteric Inhibition of hCdc34
Discovery and Biological Impact
The small molecule CC0651 was identified as a selective inhibitor of the human Cdc34 (hCdc34) E2 enzyme. Its cellular activity underscores its therapeutic potential: treatment with analogs of CC0651 inhibited the proliferation of human cancer cell lines and led to the accumulation of the SCF(Skp2) substrate p27(Kip1, a key cell cycle regulator [95]. This evidence confirms that CC0651 effectively disrupts the normal function of the UPS in a cellular context.
Unique Allosteric Mechanism of Action
Structural biology, specifically X-ray crystallography (PDB ID: 3RZ3), was instrumental in deciphering CC0651's novel mechanism. The structure revealed that CC0651 does not bind to the enzyme's active site. Instead, it inserts into a cryptic binding pocket on hCdc34 that is distant from the catalytic cysteine residue [95] [96]. The binding of CC0651 to this allosteric site induces subtle but wholesale displacement of E2 secondary structural elements [95]. Functionally, this structural perturbation has a precise effect: CC0651 does not interfere with hCdc34's interactions with E1 or E3 enzymes, nor does it prevent the initial formation of the ubiquitin thioester intermediate on the E2 active site. The sole mechanism of inhibition is the blockage of the final discharge of ubiquitin to the acceptor lysine residues on the target protein [95]. This makes CC0651 a pure allosteric inhibitor of the catalytic step of ubiquitin transfer.
Figure 1: Mechanism of Allosteric Inhibition of hCdc34 by CC0651. CC0651 binds a cryptic allosteric pocket, blocking ubiquitin discharge without affecting earlier steps.
Methodologies for Studying Allosteric Inhibition in E2 Enzymes
Structural Biology and Biophysical Techniques
Determining the high-resolution three-dimensional structure of the E2-inhibitor complex is paramount. The X-ray crystal structure of hCdc34 in complex with CC0651 (PDB: 3RZ3) was solved at 2.30 Å resolution, revealing the cryptic nature of the binding pocket and the subtle conformational changes induced by inhibitor binding [96]. Key steps include:
- Protein Purification: Recombinant expression of hCdc34 in E. coli and purification to homogeneity.
- Crystallization: Formation of diffraction-quality crystals of the E2-inhibitor complex.
- Data Collection and Refinement: Using synchrotron radiation for data collection and computational tools like REFMAC for structural refinement [96].
Functional and Mechanistic Assays
To pinpoint the exact step inhibited, a series of in vitro and cellular assays are employed:
- Thioester Formation Assay: Confirms that CC0651 does not affect the E1-mediated charging of hCdc34 with ubiquitin. This is assessed by detecting the E2~Ub thioester intermediate via non-reducing SDS-PAGE [95].
- Ubiquitin Discharge Assay: Directly measures the inability of CC0651-bound hCdc34 to transfer ubiquitin to a target lysine residue, either in a substrate protein or to form polyubiquitin chains [95].
- Cellular Proliferation and Substrate Accumulation Assays: Validates the functional consequence of inhibition in a physiological context. For CC0651, this involved demonstrating reduced cancer cell proliferation and accumulation of the Cdc34 substrate p27(Kip1 [95].
Table 2: Experimental Profile of CC0651's Effects on hCdc34 Function
| Experimental Assay | Observed Outcome with CC0651 | Biological Interpretation |
|---|---|---|
| E1-E2 Thioester Formation | No effect [95] | Inhibitor does not prevent E2 activation. |
| E3 Binding | No effect [95] | Inhibitor does not disrupt E2-E3 complex formation. |
| Ubiquitin Discharge to Lysine | Inhibited [95] | Allosteric mechanism blocks the catalytic step of ubiquitin transfer. |
| Cancer Cell Proliferation | Inhibited [95] | Validates on-target effect and therapeutic potential. |
| p27 |
Accumulated [95] | Confirms disruption of specific Cdc34-dependent degradation in cellulo. |
Computational and Theoretical Modeling
Computational approaches are increasingly vital for understanding and predicting allosteric regulation. Methods such as the Structure-Based Statistical Mechanical Model of Allostery (SBSMMA) quantify the energetics and cooperativity of allosteric communication, helping to predict novel allosteric sites [100]. Molecular Dynamics (MD) simulations can capture the dynamic motion of proteins, revealing how allosteric binding at one site (like the CC0651 pocket) propagates through the protein structure to influence the active site [101] [100].
The Evolutionary Context of UBLs and Their Regulatory Mechanisms
The ubiquitin-like protein family is evolutionarily ancient. Ubiquitin and UBLs share a common structural fold, the "beta-grasp" fold, which consists of a five-strand antiparallel beta sheet surrounding a central alpha helix [1]. Phylogenetic studies suggest that eukaryotic UBLs are monophyletic, sharing a common evolutionary origin with prokaryotic biosynthesis pathways for cofactors like thiamine and molybdopterin [1]. The bacterial sulfur transfer proteins ThiS and MoaD share the beta-grasp fold with UBLs, and their associated enzymes, ThiF and MoeB, share sequence similarity and a common catalytic mechanism with ubiquitin-activating enzymes (E1) [1]. This deep evolutionary connection is further exemplified by the human UBL URM1, which functions both as a protein modifier and a sulfur-carrier, acting as a "molecular fossil" [1]. The conservation of this structural and mechanistic framework across billions of years of evolution underscores the fundamental importance of UBL-based regulation in cellular life and provides a rich context for understanding the function and inhibition of modern E2 enzymes.
The Scientist's Toolkit: Essential Reagents for E2 Allosteric Research
Table 3: Key Research Reagents for Investigating E2 Allosteric Inhibition
| Research Reagent / Tool | Function and Application in E2 Research |
|---|---|
| Recombinant E2 Enzymes (e.g., hCdc34) | Essential substrate for in vitro mechanistic studies including structural biology (crystallography), biochemical activity assays, and inhibitor screening [96]. |
| Allosteric Inhibitors (e.g., CC0651) | Chemical probes to dissect E2 mechanism, validate targets, and serve as lead compounds for therapeutic development [95] [96]. |
| E1 Activating Enzymes | Required component in in vitro ubiquitination assays to activate ubiquitin/UBL and transfer it to the E2 enzyme under investigation [99]. |
| E3 Ligases (e.g., CRL Complexes) | Used in reconstituted ubiquitination assays to study the functional consequence of E2 inhibition on specific substrate ubiquitination [95]. |
| Ubiquitin & UBL Proteins | The core modifiers whose conjugation and transfer are monitored to measure E2 enzyme activity and the efficacy of inhibition [1]. |
| Activity Assays (Thioester/Discharge) | Biochemical kits or established protocols to specifically monitor the formation and breakdown of the E2~Ub thioester intermediate, pinpointing the step of inhibition [95]. |
The discovery and characterization of CC0651 as an allosteric inhibitor of hCdc34 represents a paradigm shift, demonstrating that E2 enzymes are druggable targets with promising therapeutic applications, particularly in oncology. Allosteric inhibitors offer distinct advantages over traditional orthosteric compounds, including higher selectivity and the potential to fine-tune enzyme activity rather than completely ablating it [101]. However, the hunt for allosteric drugs presents unique challenges. Allosteric sites are often cryptic (not present in the unliganded structure) and can be highly dynamic, making them difficult to detect with conventional screening methods [102] [101]. The future of allosteric drug discovery for E2 enzymes and other UPS targets will rely on integrated strategies combining advanced computational modeling to predict allosteric sites and communication pathways, high-throughput screening adapted for allosteric mechanisms, and high-resolution structural biology to visualize inhibitor binding and guide rational drug design [102] [101] [100]. As these techniques mature, allosteric inhibition is poised to unlock the full potential of the ubiquitin-proteasome system as a source of novel therapeutics for cancer and other diseases.
Managing System Redundancy and Compensatory Mechanisms
The concept of system redundancy, a cornerstone of reliable engineering design, finds a profound parallel in the intricate workings of cellular biology. In engineering, redundancy involves the deliberate inclusion of backup components or processes that ensure continuous operation despite component failures [103]. This principle is quantitatively measured by metrics such as Mean Time Between Failures (MTBF) and system Availability, which gauge reliability and operational uptime respectively [103]. Biological systems have evolved analogous, yet far more complex, compensatory mechanisms to maintain homeostasis and functionality despite genetic perturbations, environmental stresses, or internal failures. At the molecular level, the ubiquitin-proteasome system (UPS) and its extended family of ubiquitin-like proteins (Ubls) represent a quintessential example of such biological redundancy, enabling precise control over protein stability, function, and localization [13] [75]. This article explores the management of system redundancy and compensatory mechanisms, framing the discussion within the context of Ubl structure and evolutionary biology, with specific relevance for research and therapeutic development.
Core Principles of System Redundancy
In system design, redundancy is a strategic approach to enhance reliability and fault tolerance. It is not merely duplication but a structured methodology for ensuring service continuity.
Types and Strategies of Redundancy
Engineering systems implement several distinct forms of redundancy, each applicable to different components and failure modes [103].
- Hardware Redundancy: Involves replicating critical physical components. A common example is a RAID (Redundant Array of Independent Disks) configuration, where data is distributed across multiple hard drives, allowing recovery if one fails [103].
- Software Redundancy: Relies on multiple instances of an application or service running concurrently. Load balancers distribute traffic across these instances; if one fails, traffic is rerouted to healthy instances, ensuring continuous operation [103].
- Data Redundancy: Ensures data availability by storing copies in multiple locations or through replication techniques like database replication across several servers [103].
- Geographic Redundancy: Protects against regional disasters by distributing redundant systems or data centers across different geographical locations [103].
These redundancy types can be deployed in either an active or passive mode. Active Redundancy involves multiple entities performing the same task simultaneously, with immediate takeover if one fails. Passive Redundancy maintains an inactive backup that only engages when a primary component fails [103].
Quantitative Metrics for Redundancy
The effectiveness of redundancy is measured using key reliability engineering metrics, which can also be adapted to quantify biological robustness [103].
Table 1: Key Metrics for Measuring Redundancy Effectiveness
| Metric | Formula | Interpretation |
|---|---|---|
| Mean Time Between Failures (MTBF) | Total Operating Time / Number of Failures |
Measures the average time a system or component operates before a failure occurs. A higher MTBF indicates greater reliability. |
| Mean Time To Recovery (MTTR) | Total Downtime / Number of Failures |
Measures the average time required to repair a failed system and restore it to full functionality. A lower MTTR is desirable. |
| Availability | (Total Uptime / (Total Uptime + Total Downtime)) * 100% |
Represents the percentage of time a system is operational and available for use. High availability (e.g., 99.999% or "five nines") is a common goal for critical systems. |
Ubiquitin-like Proteins: Compensatory Mechanisms in Molecular Evolution
The ubiquitin system and its Ubl counterparts are a master regulatory network in eukaryotes, embodying nature's approach to managing cellular function through redundant and compensatory pathways.
Structural Commonalities and Functional Diversification
Ubiquitin and Ubls share a common structural fold, the β-grasp fold (β-GF), despite often having low sequence similarity [13] [74]. This fold was already highly diversified in the last universal common ancestor (LUCA), giving rise to at least seven distinct clades with functions ranging from catalysis to RNA binding and sulfur transfer [13]. In eukaryotes, this fold was co-opted for the Ubl clade, which expanded dramatically. Comparative genomics indicates at least 70 distinct Ubl families across eukaryotes, with nearly 20 already present in the last eukaryotic common ancestor (LECA) [13] [74]. This structural redundancy—where a single fold supports numerous functions—provided the raw material for the evolution of complex compensatory systems. The primary function of many Ubls is to act as protein or lipid modifiers, covalently conjugating to target molecules to drastically alter their biochemistry, stability, interaction partners, or localization [13] [75].
Evolutionary Origins and Redundant Pathways
Precursors of the eukaryotic ubiquitin system are found in prokaryotes, hinting at its deep evolutionary origins [13] [74]. Simple versions combining a Ubl and an E1-like enzyme exist in metabolic pathways. Some of these evolved into protein-tagging systems like Sampylation in archaea and Urmylation in eukaryotes, the latter being considered a molecular fossil of the evolutionary link between sulfur transfer and protein modification [13] [75]. Other prokaryotic systems developed more complex elements, including E2-like enzymes and RING-type E3 ligases, closely mirroring the eukaryotic state [13]. This evolutionary history has created a network with built-in redundancy and cross-talk. For example, the loss of one Ubl modification pathway can sometimes be partially compensated for by the activity of another, and the UPS itself can be seen as a redundant protein degradation backup to lysosomal pathways [13] [75]. The early diversification of Ubls was critical for the emergence of key eukaryotic features, including vesicular trafficking, chromatin dynamics, and protein processing in the endoplasmic reticulum [13] [74].
Experimental Analysis of UBL Systems
Studying the redundant and compensatory functions of Ubls requires a suite of well-defined experimental methodologies.
Key Research Reagents and Materials
Table 2: Essential Research Reagent Solutions for Ubl Studies
| Research Reagent | Function and Application |
|---|---|
| E1, E2, and E3 Enzymes | Essential components for the in vitro reconstitution of Ubl conjugation cascades. Used to study enzyme kinetics, specificity, and the biochemical requirements for target modification. |
| Mutant Ubls (e.g., GG-deletion, point mutants) | Used to dissect the functional requirements of Ubl conjugation. The GG-motif is often critical for the conjugation cascade. These mutants serve as critical negative controls. |
| Proteasome Inhibitors (e.g., MG132, Bortezomib) | Used to block proteasomal degradation, allowing for the accumulation and study of ubiquitinated proteins and to investigate the role of the UPS in specific cellular processes. |
| Specific Antibodies (against Ubls, conjugates, targets) | Crucial for detecting protein expression, post-translational modification (e.g., via western blot), and assessing subcellular localization (e.g., via immunofluorescence). |
| Expression Plasmids (for Ubls, enzymes, targets) | Enable the overexpression or knock-down of system components in cell culture models to study gain-of-function or loss-of-function phenotypes and compensatory mechanisms. |
Core Methodological Workflows
A standard workflow for investigating Ubl conjugation and function involves both in vitro biochemical reconstitution and in vivo cellular assays.
Protocol 1: In Vitro Reconstitution of Ubl Conjugation
- Purification: Express and purify recombinant Ubl, E1 activating enzyme, E2 conjugating enzyme, E3 ligase, and the target protein substrate.
- Reaction Setup: Combine the purified components in a reaction buffer containing ATP and an energy-regenerating system to sustain the conjugation cascade.
- Incubation: Allow the reaction to proceed at a physiological temperature (e.g., 30-37°C) for a defined period.
- Analysis: Terminate the reaction and analyze the products via SDS-PAGE and western blotting, using specific antibodies against the Ubl to detect conjugated species. Mass spectrometry can be used for precise identification of modification sites.
Protocol 2: Assessing Functional Compensation in Cellulae
- Perturbation: Use siRNA, CRISPR/Cas9, or small-molecule inhibitors to knock down or inhibit a specific Ubl pathway component.
- Phenotypic Analysis: Monitor cellular phenotypes using assays for proliferation, viability, stress response, or subcellular morphology.
- Global Proteomic Analysis: Employ quantitative mass spectrometry (e.g., SILAC, TMT) to profile changes in the global ubiquitinome or proteome upon perturbation.
- Validation: Identify and validate potential compensatory pathways that are upregulated or activated in the knockdown background, often involving related Ubls or alternative degradation mechanisms.
The following diagram illustrates the logical workflow and key decision points in a functional analysis of a Ubl system, from genetic perturbation to the identification of compensatory mechanisms.
Diagram 1: Functional analysis workflow for UBL systems.
A Network View of UBL Conjugation and Compensation
The process of Ubl modification is a canonical enzymatic cascade that can be represented as a biological circuit. The following diagram details this core conjugation machinery and hints at the potential for compensatory interactions between different Ubl pathways, a key feature of biological redundancy.
Diagram 2: UBL conjugation cascade and compensation.
Implications for Drug Discovery and Therapeutic Development
The redundant and compensatory nature of the Ubl network presents both challenges and opportunities for pharmaceutical intervention. Targeting specific E3 ligases or Ubl pathways offers a strategy for precise manipulation of disease-driving proteins, such as oncoproteins. However, functional redundancy can lead to drug resistance, as inhibition of one pathway may be compensated by the activity of another [13] [75]. A key approach is to develop combination therapies that simultaneously inhibit a primary target and its compensatory backup pathways. Furthermore, targeting the upstream, shared components of the conjugation cascade (e.g., specific E1 enzymes for a Ubl) could overcome redundancy but requires careful management of toxicity due to broader system effects. Understanding the evolutionary history and network properties of Ubls is therefore not merely an academic exercise but a critical component of designing robust and effective therapeutic regimens.
The ubiquitin-proteasome system (UPS) and ubiquitin-like protein (UBL) conjugation pathways represent master regulators of cellular homeostasis, governing essential processes from protein degradation to DNA repair, cell signaling, and immune responses [104]. These pathways employ an elegant hierarchical enzymatic cascade, beginning with E1 activating enzymes, proceeding through E2 conjugating enzymes, and culminating with E3 ligases that provide substrate specificity [105]. The dynamic and reversible nature of these modifications is maintained by deubiquitinating enzymes (DUBs) and UBL-specific proteases (ULPs) [106]. The fundamental importance of these systems in normal physiology is underscored by their frequent dysregulation in human diseases, including cancer, neurodegenerative disorders, and autoimmune conditions [104] [105]. This nexus of biological significance and disease relevance has positioned components of the UPS and UBL pathways as attractive targets for therapeutic intervention.
The journey toward clinical validation of this target class began with the proteasome inhibitor bortezomib, which demonstrated that targeting the UPS could yield transformative cancer therapeutics [104]. However, the development of inhibitors against more specific components, particularly those in UBL pathways, presents unique challenges and opportunities for optimizing drug-like properties. Early-stage inhibitors in this domain have revealed critical lessons about balancing potency, selectivity, and pharmacological properties. This review synthesizes these lessons within the structural and evolutionary context of UBLs, providing a technical framework for advancing therapeutic candidates from concept to clinic.
Structural and Evolutionary Framework of UBLs
The UBL Family: Structural Commonalities and Variations
Ubiquitin-like proteins constitute a family of small proteins characterized by a conserved three-dimensional core structure known as the β-grasp fold [1] [107]. This structural motif consists of a five-strand antiparallel β sheet wrapped around a central α helix, creating a compact, stable globular domain [1]. Despite limited sequence conservation, this shared architectural principle unifies UBLs including SUMO (Small Ubiquitin-like Modifier), NEDD8 (Neural precursor cell-expressed, developmentally down-regulated 8), ISG15 (Interferon-Stimulated Gene 15), UFM1 (Ubiquitin-fold modifier 1), and ATG8/12 (Autophagy-related protein 8/12) [1] [107]. UBLs are typically expressed as inactive precursors that require proteolytic processing to expose a C-terminal glycine or diglycine motif, which is essential for their conjugation to target proteins or other molecules [1].
Functionally, UBLs are divided into two principal categories. Type I UBLs can be covalently conjugated to target proteins through an enzymatic cascade analogous to ubiquitination, thereby altering the target's activity, stability, localization, or interactions [1]. Type II UBLs often exist as domains within larger proteins and typically function as protein-protein interaction modules without covalent conjugation [1]. The human genome encodes at least eight families of Type I UBLs, including SUMO, NEDD8, ATG8, ATG12, URM1, UFM1, FAT10, and ISG15, each with distinct biological functions and target spectra [1].
Evolutionary Origins and Conservation
Mounting evidence indicates that UBL-protein modification systems evolved from prokaryotic sulfurtransferase pathways involved in cofactor biosynthesis [1] [2]. The bacterial sulfur carrier proteins ThiS and MoaD, essential for thiamine and molybdopterin synthesis respectively, share the characteristic β-grasp fold with eukaryotic UBLs [1] [2]. Remarkably, the eukaryotic UBL URM1 functions as both a ubiquitin-like modifier and a sulfur carrier, representing a molecular "living fossil" that bridges these ancient and modern systems [1]. The conservation of the β-grasp fold across domains of life, coupled with mechanistic parallels in activation enzymes, suggests that UBL conjugation was not a eukaryotic innovation but rather was cobbled together from pre-existing prokaryotic components [2].
Ubiquitin itself stands as one of the most evolutionarily conserved proteins known, with plant ubiquitin differing from human ubiquitin by only three amino acids [107]. This extraordinary sequence conservation across eukaryotic lineages highlights the structural and functional constraints on this protein family. Such evolutionary insights are not merely academic; they inform drug discovery by identifying conserved functional surfaces and structural motifs that may represent privileged targeting sites for therapeutic intervention.
UBL Pathways: Key Targets for Therapeutic Intervention
The Enzymatic Conjugation Cascade
UBL conjugation follows a conserved three-step enzymatic mechanism. First, a dedicated E1 activating enzyme consumes ATP to adenylate the UBL C-terminus, forming a high-energy thioester bond between the E1 catalytic cysteine and the UBL [104] [105]. The activated UBL is then transferred to the catalytic cysteine of an E2 conjugating enzyme via a transthiolation reaction [105]. Finally, an E3 ligase facilitates the transfer of the UBL from the E2 to a specific substrate protein, typically forming an isopeptide bond between the UBL C-terminus and a lysine ε-amino group on the target [105]. Each UBL family possesses its own dedicated E1 and E2 enzymes, though E3 ligases may exhibit broader specificity [1] [104].
Deconjugation of UBLs is equally important for pathway regulation and is mediated by specialized proteases. Deubiquitinating enzymes (DUBs) remove ubiquitin, while ULPs (UBL-specific proteases) reverse the modification of specific UBLs [106]. These enzymes demonstrate remarkable specificity for their cognate UBLs, often recognizing subtle surface features distinct from ubiquitin despite structural similarities [106].
Table 1: Major Ubiquitin-like Protein Families and Their Key Characteristics
| UBL | Identity with Ubiquitin | E1 Activating Enzyme | Key Biological Functions |
|---|---|---|---|
| Ubiquitin | 100% | UBA1, UBA6 | Protein degradation, signaling, trafficking, DNA repair |
| NEDD8 | ~55% | NAE (APPB1-UBA3) | Cullin activation, cell cycle regulation |
| SUMO | ~18% | SAE (SAE1-UBA2) | Transcription, DNA repair, stress response |
| ISG15 | ~32-37% | UBA7 | Antiviral response, immune regulation |
| ATG8 | Limited | ATG7 | Autophagosome formation, autophagy |
| UFM1 | Limited | UBA5 | Endoplasmic reticulum homeostasis, development |
Disease Relevance and Target Validation
Dysregulation of UBL pathways has been implicated in a wide spectrum of human diseases, validating their therapeutic potential. In cancer, aberrant ubiquitination and NEDDylation drive uncontrolled proliferation and evade apoptosis, while defective SUMOylation contributes to genomic instability [104]. Neurodegenerative diseases such as Alzheimer's, Parkinson's, and Huntington's often feature accumulation of misfolded proteins, highlighting failures in UPS function [104] [105]. Autoimmune and inflammatory disorders involve dysregulated immune signaling controlled by ubiquitin and ISG15, while viral pathogens frequently hijack UBL pathways to facilitate infection [104].
The clinical success of bortezomib, a proteasome inhibitor approved for multiple myeloma and mantle cell lymphoma, provided definitive proof-of-concept that targeting the UPS could yield effective therapeutics [104] [105]. This breakthrough demonstrated that despite the UPS's central role in cellular homeostasis, selective inhibition could produce manageable toxicity while delivering therapeutic effects, encouraging drug development efforts against more specific components of UBL pathways.
Case Studies: Early-Stage UBL Pathway Inhibitors
Proteasome Inhibitors: Pioneering the Therapeutic Class
Bortezomib (Velcade) represents the foundational success in UPS-targeted therapy. This peptide boronic acid analog selectively and reversibly inhibits the chymotrypsin-like activity of the proteasome's β5 subunit, with IC₅₀ values of 2.4-7.9 nM for the β5 subunit compared to 24-74 nM for the β1 subunit and 590-4,200 nM for the β2 subunit [104]. Bortezomib's clinical validation created a roadmap for targeting previously considered "undruggable" components of the UPS and UBL systems.
Second-generation proteasome inhibitors have built upon this foundation. Carfilzomib, an epoxyketone-based irreversible inhibitor, demonstrates improved selectivity for the β5 subunit (IC₅₀ = 6 nM) and reduced neurotoxicity compared to bortezomib [104]. MLN9708 (ixazomib), a boronic acid derivative like bortezomib, offers the advantage of oral bioavailability while maintaining potent proteasome inhibition [104]. These compounds collectively highlight the importance of subunit selectivity, binding kinetics, and administration route in optimizing the therapeutic profile of UPS-targeting agents.
NEDD8-Activating Enzyme (NAE) Inhibition: MLN4924
The investigational compound MLN4924 (pevonedistat) represents a paradigm-shifting approach in UBL pathway inhibition. Rather than directly inhibiting a protease or ligase, MLN4924 targets the NEDD8 E1 activating enzyme (NAE), forming a covalent adduct with NEDD8 that blocks its activation and subsequent conjugation to cullin proteins [104] [105]. This mechanism disrupts the function of cullin-RING ligases (CRLs), a major class of E3 ubiquitin ligases that require neddylation for activity.
MLN4924's action ultimately prevents the ubiquitination and degradation of CRL substrates, leading to accumulation of tumor suppressive proteins and cell cycle arrest. This compound demonstrates the therapeutic potential of targeting UBL-specific E1 enzymes and has shown promising activity in clinical trials for various malignancies [105]. Its mechanism underscores the value of understanding pathway interdependencies—by inhibiting neddylation, MLN4924 indirectly modulates a broad but specific subset of ubiquitination events.
E2 Conjugating Enzyme Inhibitors: CC0651
The discovery of CC0651, an allosteric inhibitor of the E2 enzyme Cdc34, illustrated that E2 enzymes represent viable drug targets despite initial skepticism about their selectivity [105]. CC0651 does not impair Cdc34's interactions with E1 or E3 enzymes, nor does it affect the initial formation of the E2~ubiquitin thioester intermediate [105]. Instead, it specifically blocks the discharge of ubiquitin to acceptor lysine residues by inducing a conformational change that mispositions the active site cysteine relative to the target lysine.
This allosteric mechanism provides important insights for inhibitor design. First, it demonstrates that enzymes beyond the canonical active site can be effectively targeted. Second, it shows that blocking specific steps in the catalytic cycle rather than complete enzyme inhibition can achieve sufficient pathway modulation for therapeutic effect. Third, it highlights that targeting E2 enzymes can provide greater specificity than E1 inhibition while maintaining broader effects than single E3 ligase inhibition.
E3 Ligase and DUB Inhibitors
The development of HDM2 inhibitors such as Nutlin-3, MI-219, and RG7388 exemplifies the pursuit of specificity through targeting E3 ligases [105]. These compounds block the interaction between HDM2 (an E3 ubiquitin ligase) and p53 (a critical tumor suppressor), preventing p53 ubiquitination and degradation [105]. This leads to p53 stabilization and activation of apoptosis in cancer cells retaining wild-type p53. The progression from initial tool compounds to clinical candidates has required extensive optimization of binding affinity, selectivity over related proteins, and pharmaceutical properties.
Similarly, DUB inhibitors represent an emerging class with significant therapeutic potential. Compounds targeting USP7 indirectly modulate p53 levels by stabilizing its negative regulator HDM2, while inhibitors of UCHL1 are being explored for neurodegenerative diseases and cancer [105]. The development of these agents faces unique challenges, including achieving selectivity among the approximately 100 human DUBs and understanding the complex biological consequences of modulating deubiquitination.
Table 2: Representative Early-Stage Inhibitors of UBL Pathways
| Inhibitor | Target | Mechanism | Development Stage | Key Insights |
|---|---|---|---|---|
| Bortezomib | Proteasome β5 subunit | Reversible inhibition | Approved (multiple myeloma) | Demonstrated clinical viability of UPS targeting |
| Carfilzomib | Proteasome β5 subunit | Irreversible inhibition | Approved (multiple myeloma) | Improved selectivity and reduced neurotoxicity |
| MLN4924 | NEDD8 E1 (NAE) | Covalent NEDD8 adduct | Clinical trials | Validated E1 enzymes as druggable targets |
| CC0651 | E2 (Cdc34) | Allosteric inhibition | Preclinical | Demonstrated E2 targetability and allosteric mechanisms |
| Nutlin-3/RG7388 | HDM2-p53 interaction | Protein-protein interaction inhibitor | Clinical trials | Achieved specificity through E3 targeting |
| USP7 inhibitors | Deubiquitinase | Active site inhibition | Preclinical/early clinical | Highlighted complex consequences of DUB inhibition |
Experimental Approaches for UBL Inhibitor Characterization
Biochemical Assays for Enzyme Inhibition
Characterizing UBL pathway inhibitors requires a hierarchy of biochemical and cellular assays. Primary enzyme activity assays typically employ purified E1, E2, or DUB/ULP enzymes with fluorogenic or luminescent substrates to quantify inhibition potency (IC₅₀ values) and mechanism (reversible/irreversible, competitive/non-competitive) [106]. For E1 enzymes, assays often monitor ATP consumption, ubiquitin/UBL adenylation, or thioester formation. E2 charging assays measure transfer from E1 to E2, while E3 assays typically monitor substrate ubiquitination using gel electrophoresis or immunoassays.
Mechanistic studies employ techniques such as surface plasmon resonance (SPR) or isothermal titration calorimetry (ITC) to determine binding kinetics and thermodynamics. X-ray crystallography of inhibitor-enzyme complexes provides atomic-level insights for structure-based optimization, revealing key interactions with active site residues or allosteric pockets [106]. These structural insights are particularly valuable for understanding specificity determinants between highly related enzymes in the UPS and UBL pathways.
Cellular Target Engagement and Pathway Modulation
Demonstrating cellular activity requires assays that confirm target engagement and downstream pathway effects. Cellular thermal shift assays (CETSA) can verify compound binding to intended targets in cells. Immunoblotting techniques monitor changes in substrate ubiquitination/UBLylation, such as accumulation of high-molecular-weight ubiquitin conjugates after proteasome inhibition or reduced cullin neddylation after NAE inhibition.
More sophisticated reporter systems employ engineered constructs where luciferase or fluorescent protein stability is controlled by specific ubiquitination events. For DUB inhibitors, activity-based probes can visualize target engagement in cells through gel-based or microscopy readouts. These cellular assays bridge the gap between biochemical potency and functional activity, informing structure-activity relationship (SAR) campaigns.
Specific Methodologies for UBL Research
Advanced methodologies have been developed specifically for studying UBL modifications. The bioUBL system utilizes in vivo biotinylation with the E. coli BirA ligase to tag UBLs, enabling purification under denaturing conditions that preserve labile UBL conjugates by inactivating deconjugating enzymes [37]. This approach facilitates the identification of novel UBL substrates and monitoring of UBL conjugation dynamics in response to inhibitor treatment.
Proteomic techniques employing diGly remnant antibodies (for ubiquitin) or analogous approaches for other UBLs enable system-wide monitoring of UBL modifications [107] [37]. These "ubiquitylome" studies can reveal both specific substrates and global changes in UBL signaling in response to inhibitor treatment, providing comprehensive insights into mechanism of action and potential biomarkers.
Diagram 1: UBL Inhibitor Characterization Workflow - This integrated experimental approach bridges from biochemical screening to mechanistic understanding and cellular validation.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for UBL Inhibitor Development
| Reagent Category | Specific Examples | Research Applications | Technical Considerations |
|---|---|---|---|
| Activity Assay Kits | Ubiquitin E1/E2/E3 kits, DUB/ULP profiling panels | High-throughput screening, IC₅₀ determination | Select appropriate substrate (fluorogenic, luminescent) based on detection platform |
| Active Enzymes | Recombinant E1 (UBA1, NAE), E2 (UbcH5, Ubc12), E3 (HDM2, SCF) | Mechanistic studies, crystallography | Verify activity and purity; consider post-translational modifications needed for function |
| UBL Modification Probes | BioUbL tags, TUBEs (Tandem Ubiquitin Binding Entities), diGly antibodies | Enrichment and detection of UBL conjugates | Use denaturing conditions to preserve labile modifications; include protease inhibitors |
| Cellular Reporters | Ubiquitination-dependent degrons, UBL pathway sensors | Cellular target engagement, functional activity | Optimize expression levels; include controls for non-specific effects |
| Structural Biology Tools | Crystallization screens, cryo-EM grids, NMR isotope labeling | Structure-based drug design | Consider protein engineering (mutations, tags) to improve crystallization behavior |
| Activity-Based Probes | HA-Ub-VS, SUMO-AMC, NEDD8 fluorogenic substrates | Monitoring enzyme activity in complex mixtures | Validate specificity with control enzymes; optimize concentration and incubation time |
Optimization Strategies for Drug-Like Properties
Balancing Potency, Selectivity, and Physicochemical Properties
Early-stage UBL pathway inhibitors often face challenges related to suboptimal physicochemical properties, inadequate pharmacokinetics, or insufficient selectivity. Potency optimization typically begins with structural biology to understand key ligand-protein interactions, followed by systematic SAR exploration. However, potency gains must be balanced against potential impacts on other properties, particularly selectivity.
Selectivity profiling is especially critical for UBL pathway inhibitors due to the structural similarities between enzyme families. Comprehensive screening against related E1/E2/E3 enzymes, DUBs/ULPs, and off-targets ensures that observed cellular phenotypes derive from intended target engagement. For example, E1 inhibitors should be counterscreened against other E1s (both ubiquitin and UBL-specific), while DUB inhibitors require profiling across the approximately 100 human DUBs [106] [105].
Physicochemical optimization focuses on improving solubility, metabolic stability, and membrane permeability while maintaining potency. Strategies include modulating lipophilicity, introducing solubilizing groups, and reducing hydrogen bond donors/acceptors. For compounds targeting protein-protein interactions (such as HDM2-p53 inhibitors), addressing the typically high molecular weight and lipophilicity presents particular challenges that may require innovative scaffold design.
Addressing Challenges Specific to UBL-Targeted Therapeutics
UBL pathway inhibitors face several unique development challenges. Mechanistic toxicity concerns arise from the fundamental biological roles of UBL pathways, requiring careful therapeutic index determination. Biomarker development is essential for demonstrating target engagement and pathway modulation in clinical settings. Combination strategies may be necessary given the network buffering and redundancy in UBL pathways.
The lessons from early-stage inhibitors provide guiding principles for addressing these challenges. First, crystallographic guidance enables rational optimization rather than random exploration. Second, cellular target engagement assays bridge biochemical potency to functional activity. Third, comprehensive selectivity profiling prevents unexpected off-target effects. Fourth, pharmacodynamic biomarkers facilitate dose selection and regimen optimization.
Diagram 2: UBL Inhibitor Optimization Strategy - This multidimensional approach balances multiple compound properties to identify development candidates with the highest probability of success.
The development of therapeutics targeting ubiquitin-like protein pathways has progressed from initial skepticism to clinical validation, with multiple compounds now in advanced development. The lessons from these early-stage inhibitors highlight both the challenges and opportunities in this target space. Success requires deep understanding of UBL biology, sophisticated biochemical and cellular tools for compound characterization, and multidimensional optimization strategies that balance potency with drug-like properties.
Future directions in this field include developing bifunctional molecules such as PROTACs (Proteolysis-Targeting Chimeras) that harness E3 ubiquitin ligases to target specific proteins for degradation, allosteric inhibitors that provide greater specificity than active-site directed compounds, and tissue-specific targeting approaches to minimize systemic toxicity. As structural and mechanistic understanding of UBL pathways continues to advance, so too will opportunities for therapeutic intervention in diseases ranging from cancer to neurodegeneration.
The extraordinary evolutionary conservation of UBL pathways underscores their fundamental biological importance, while their dysregulation in disease highlights their therapeutic relevance. By applying the lessons from early-stage inhibitors—rigorous selectivity profiling, comprehensive mechanistic understanding, and balanced optimization of drug-like properties—the next generation of UBL-targeted therapeutics holds promise for addressing unmet medical needs across diverse disease areas.
From Bench to Bedside: Validating UBL Targets in Disease and Therapy
Functional validation represents a critical bridge between genetic association and causality in biomedical research. This process is indispensable for confirming the pathological role of specific genes, proteins, or genetic variants identified through high-throughput methodologies. Within the context of ubiquitin-like proteins (UBLs)—a family of post-translational modifiers with profound regulatory functions—functional validation provides essential insights into their roles in health and disease [25]. UBLs, characterized by their structural similarity to ubiquitin and their modification via E1-E2-E3 enzymatic cascades, orchestrate diverse cellular processes including protein degradation, DNA repair, autophagy, and immune responses [1] [108]. The evolutionary conservation of UBL systems from prokaryotes to humans, alongside their mechanistic links to ancient biosynthetic pathways for cofactors like thiamine and molybdopterin, underscores their fundamental biological importance [1]. This guide details the experimental paradigms and methodological frameworks for generating genetic and pharmacological evidence to validate UBL functions across disease models, providing technical protocols and resources for the research community.
Methodological Frameworks for Functional Validation
Genetic Manipulation in Model Organisms
Model organisms serve as foundational platforms for in vivo functional validation, enabling researchers to prove causality for genes and genetic variants associated with disease [109]. The selection of an appropriate model depends on genetic conservation, physiological similarities to humans, and practical experimental considerations.
Table 1: Model Organisms for Functional Validation of UBL Pathways
| Organism | Key Advantages | Limitations | Representative UBL Research Applications |
|---|---|---|---|
| Mouse | High genetic, anatomical, and physiological similarity to human kidney and other organs; well-established genetic tools [109] | Time-consuming and expensive breeding; ethical considerations | Validation of STUB1/CHIP mutations in cerebellar ataxia and hypogonadism (Gordon Holmes Syndrome) [110] |
| Zebrafish | Rapid external development; optical transparency for visualization; high fecundity; amenability to high-throughput screening [109] | Lesser anatomical similarity for some organs | |
| Fruit Fly (Drosophila) | Powerful genetic toolbox; short generation time; low maintenance cost [109] | Limited relevance for some complex human diseases | |
| Frog (Xenopus) | Large embryos for micromanipulation; suitable for embryogenesis and large-scale screens [109] | Tetrapod with some physiological differences |
The application of these models is exemplified by studies on STUB1, the gene encoding the CHIP protein, which possesses both chaperone and ubiquitin ligase activities. Exome sequencing of patients with autosomal recessive cerebellar ataxia (ARCA) identified multiple pathogenic STUB1 mutations [110]. Functional validation in CHIP-/- mouse models recapitulated key patient phenotypes, including motor impairments, cerebellar atrophy, and hypogonadism, thereby confirming the causal role of CHIP loss-of-function in disease progression [110].
Pharmacological Inhibition and Targeted Therapeutics
Pharmacological approaches provide complementary evidence to genetic studies by enabling reversible, acute disruption of target proteins. These strategies are particularly valuable for validating druggable targets within the ubiquitin-proteasome system (UPS) and UBL pathways.
Proteasome Inhibitors: Bortezomib, a reversible 26S proteasome inhibitor, received FDA approval in 2003 for relapsed multiple myeloma. Its mechanism of action involves accumulating regulatory proteins, ultimately triggering apoptosis in malignant cells [111]. This clinical success validates the UPS as a therapeutic target, though drug resistance and side effects have spurred interest in more specific agents.
UBL-Specific Inhibitors: The development of compounds targeting specific UBL pathways has provided both therapeutic leads and powerful research tools.
- TAK-981 (Subasumstat): This inhibitor targets the SUMO-activating enzyme (SAE), thereby disrupting the global SUMOylation process. TAK-981 enhances type I interferon signaling and has been investigated primarily for cancer, though its utility extends to basic research on SUMO function [108].
- E1 Enzyme Inhibitors: Research has identified inhibitors for ubiquitin-activating enzymes (UBA1, UBA6), which are essential for cellular stress responses in cancer cells [25].
Experimental Protocols for UBL Functional Analysis
Protocol: Global Identification of UBL Modification Sites Using pLink-UBL
The identification of specific modification sites on substrate proteins has been a historical challenge for most UBLs. This protocol describes a mass spectrometry-based method for the global analysis of UBL modification sites, particularly SUMOylation, without requiring mutation of the UBL itself [48].
Workflow Overview:
- Sample Preparation: Extract proteins from cells or tissues of interest under denaturing conditions to preserve post-translational modifications.
- UBL Enrichment: Perform immunoprecipitation using antibodies specific to the UBL of interest (e.g., anti-SUMO) to enrich for UBL-conjugated proteins.
- Proteolytic Digestion: Digest enriched protein complexes with trypsin or another suitable protease to generate peptides for mass spectrometry analysis.
- Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS): Separate peptides via liquid chromatography and analyze using a tandem mass spectrometer.
- Data Analysis with pLink-UBL: Utilize the dedicated search engine pLink-UBL, which is based on cross-linked peptide identification algorithms, to identify UBL modification sites from the MS/MS data. This software has demonstrated superior precision, sensitivity, and speed compared to "make-do" search engines like MaxQuant, pFind, and standard pLink, increasing the identification of SUMOylation sites by 50-300% from the same datasets [48].
Applications: This protocol can be adapted to identify not only protein substrates but also small-molecule modifications of UBLs. For example, researchers used an analogous method involving antibody enrichment and a pFind 3 blind search to discover that spermidine is a major non-protein substrate for fission yeast SUMO (Pmt3) and is also conjugated to ubiquitin in vitro [48].
Protocol: Functional Validation of a UBL in Cancer ModelsIn VitroandIn Vivo
This protocol outlines a consolidated workflow for validating the tumor-suppressive or oncogenic functions of a UBL, as exemplified by studies on Ubiquilin4 (Ubqln4) in gastric cancer [112].
Part A: In Vitro Functional Characterization
- Genetic Manipulation in Cell Lines:
- Generate stable overexpression or knockdown/knockout cell lines using lentiviral transduction of the UBL gene (e.g., Ubqln4) in relevant cancer cells (e.g., MKN45, BGC-823 gastric cancer cells).
- Validate expression changes via western blotting.
- Phenotypic Assays:
- Cell Proliferation: Perform MTT assays and colony-formation assays to assess growth and clonogenic potential.
- Cell Cycle Analysis: Use flow cytometry with propidium iodide staining to determine cell cycle distribution. Overexpression of Ubqln4, for instance, was shown to increase the proportion of cells in G1 phase and decrease those in S phase [112].
- Cellular Senescence: Conduct Senescence-Associated β-Galactosidase (SA-β-Gal) staining. Ubqln4 overexpression induced a senescent morphology and increased SA-β-Gal positive cells [112].
Part B: In Vivo Tumorigenesis Assay
- Xenograft Models: Subcutaneously inject control and UBL-modified cancer cells into immunodeficient mice.
- Tumor Monitoring: Measure tumor volume regularly over several weeks.
- Endpoint Analysis: Excise and weigh tumors at the end of the study. Ubqln4 overexpression resulted in smaller tumor volume and weight compared to controls, confirming its tumor-suppressive role in vivo [112].
Protocol: Exome Sequencing for Identification of Disease-Causing UBL Mutations
Exome sequencing has proven highly effective for identifying novel mutations in UBL-related genes in patients with rare diseases, such as cerebellar ataxias [110].
- Patient Selection: Identify affected individuals and families with the disease phenotype, ideally including multiple affected and unaffected family members for comparison.
- Exome Sequencing: Perform exome capture and high-throughput sequencing on DNA from selected participants.
- Bioinformatic Analysis:
- Map sequences to a reference genome and call variants.
- Filter variants against public databases (e.g., dbSNP, 1000 Genomes Project) to exclude common polymorphisms.
- Prioritize variants based on predicted functional impact (e.g., nonsense, missense, splice-site), segregation with the disease phenotype in families, and occurrence in genes of interest (e.g., UBL genes, components of the UPS).
- Functional Validation: Confirm the pathogenicity of candidate mutations using in vitro assays (e.g., assessing the impact of a STUB1 mutation on CHIP's ubiquitin ligase activity) and animal models [110].
Figure 1: Integrated workflow for identifying and validating disease-causing UBL mutations using exome sequencing and functional studies.
Signaling Pathways and Molecular Mechanisms
UBLs regulate a vast network of cellular signaling pathways. Functional validation often requires delineating these pathways and the specific molecular mechanisms by which a UBL exerts its effect.
The Ubqln4-p21 Tumor Suppressor Pathway in Gastric Cancer: Research has established that Ubqln4 functions as a tumor suppressor in gastric cancer by stabilizing the cell cycle inhibitor p21. The mechanism involves a dual regulatory approach:
- p53-Dependent Regulation: Ubqln4 overexpression activates the p53/p21 signaling axis.
- p53-Independent Regulation: Ubqln4 interacts with the E3 ubiquitin ligase RNF114 and negatively regulates its expression levels. Since RNF114 targets p21 for proteasomal degradation, its downregulation by Ubqln4 leads to p21 stabilization, inducing G1-S cell cycle arrest and cellular senescence [112]. This mechanism was validated through experiments showing that the effects of Ubqln4 were partially abrogated upon silencing of p21 [112].
UBL Cross-Talk in Immune Regulation: The interplay between different UBLs adds a layer of complexity to their functional validation. For instance, SUMOylation can have both activating and suppressive effects on immune responses. SUMO1 can protect IκBα from degradation, inhibiting NF-κB translocation, while SUMO2/3 can activate NEMO, leading to prolonged NF-κB activation [108]. Furthermore, viral proteases, such as papain-like proteases (PLpros) from coronaviruses, can cleave multiple UBLs (ubiquitin, ISG15, NEDD8) from host proteins, impairing immune functions—a finding that underscores the need to study UBL pathways in the context of host-pathogen interactions [111].
Figure 2: Molecular mechanism of Ubqln4 tumor suppressor function via p21 stabilization.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for UBL Functional Validation Studies
| Reagent / Resource | Function / Application | Examples / Key Characteristics |
|---|---|---|
| pLink-UBL Software | Identification of UBL modification sites from MS/MS data; superior to generic search engines [48] | Increased SUMOylation site identification by 50-300% [48] |
| UBL-Specific Antibodies | Immunoprecipitation and immunohistochemistry for UBL localization and expression analysis | Used to score Ubqln4 expression in gastric cancer tissues [112] |
| Lentiviral Vectors | Stable overexpression or knockdown of UBL genes in cell lines; enables in vitro and in vivo studies | Generation of stable Ubqln4-expressing gastric cancer cell lines [112] |
| SAE Inhibitors | Pharmacological disruption of SUMOylation cascade; research tool and therapeutic candidate | TAK-981 (Subasumstat) [108] |
| Proteasome Inhibitors | Inhibition of protein degradation via the proteasome; validates UPS involvement | Bortezomib (FDA-approved for multiple myeloma) [111] |
| Defined Model Organisms | In vivo functional validation of UBL gene causality | Mouse (STUB1/CHIP), Zebrafish, Fruit Fly, Frog [109] [110] |
Functional validation through genetic and pharmacological approaches remains the cornerstone of translational research, providing the necessary evidence to move from observational association to mechanistic understanding. Within the complex and evolutionarily conserved systems governed by UBLs, these methodologies are particularly vital for deciphering disease mechanisms and identifying novel therapeutic targets. The continuous refinement of model systems, mass spectrometry techniques, and bioinformatic tools, coupled with the development of specific pharmacological agents, promises to accelerate the validation of UBL-related pathways in human disease. This progress will undoubtedly enhance both our fundamental biological knowledge and our ability to develop targeted interventions for cancer, neurodegenerative disorders, autoimmune diseases, and other conditions linked to UBL dysregulation.
Ubiquitin-like proteins (UBLs) represent a family of critical post-translational modifiers that govern virtually all cellular processes in eukaryotes. These small proteins, which share a characteristic β-grasp fold structure, are conjugated to target proteins through dedicated enzymatic cascades, thereby modulating their stability, activity, localization, and interactions. This whitepaper examines the pathological consequences of UBL system dysregulation across three major disease domains: cancer, neurodegenerative disorders, and viral infections. Drawing upon recent clinical and experimental evidence, we detail how disrupted UBL signaling contributes to disease pathogenesis through mechanisms including impaired protein degradation, aberrant DNA repair, defective stress responses, and altered immune signaling. The analysis extends to emerging therapeutic strategies that target UBL pathways, highlighting both current clinical applications and future directions for targeted drug development. This comprehensive review underscores the central role of UBL homeostasis in human health and disease, positioning UBL pathways as promising targets for next-generation therapeutics.
Ubiquitin-like proteins (UBLs) constitute an evolutionarily conserved family of small proteins involved in the post-translational modification of cellular targets. The paradigm for this family is ubiquitin (Ub) itself, a 76-amino acid polypeptide first discovered in the 1970s and best known for its role in targeting proteins for proteasomal degradation [1]. Subsequent research has identified numerous UBLs, including SUMO (Small Ubiquitin-like Modifier), NEDD8, ISG15, ATG8, ATG12, UFM1, FAT10, and UBL5, among others [1] [113]. These modifiers share a common three-dimensional structure known as the β-grasp fold, characterized by a mixed β-sheet structure surrounding a central α-helix [1] [114].
UBLs are broadly classified into two functional categories:
- Type I UBLs (e.g., Ubiquitin, SUMO, NEDD8, ISG15) are capable of covalent conjugation to target proteins or lipids through a characteristic C-terminal glycine residue. This conjugation typically involves a dedicated enzymatic cascade comprising E1 (activating), E2 (conjugating), and E3 (ligating) enzymes [1] [115].
- Type II UBLs (e.g., UBL5/Hub1) lack the C-terminal diglycine motif and typically function as non-covalent modifiers, often acting as protein interaction domains or structural components within macromolecular complexes [113].
The enzymatic cascades responsible for UBL conjugation are highly specific, with distinct E1, E2, and E3 enzymes dedicated to each UBL family. This specificity ensures precise regulation of diverse cellular processes, including protein degradation, DNA repair, transcription, autophagy, and immune responses [1] [115]. The modification of cellular proteins by UBLs is a dynamic and reversible process, with dedicated deconjugating enzymes (e.g., deubiquitinases/DUBs for ubiquitin, ULPs for SUMO) providing an additional layer of regulation [114] [115].
Dysregulation of UBL signaling pathways—whether through mutation of UBLs themselves, their associated enzymes, or protein targets—underlies the pathogenesis of numerous human diseases. The following sections explore these connections in cancer, neurodegeneration, and viral infection, highlighting shared mechanisms and disease-specific alterations.
UBL Dysregulation in Cancer Pathogenesis
The role of UBL dysregulation in cancer is multifaceted, impacting oncogene stability, tumor suppressor function, DNA damage response, and cell cycle progression. Malignant cells often exploit UBL pathways to promote proliferation, survival, and metastasis [116].
Key UBL Pathways in Carcinogenesis
Ubiquitin-Proteasome System (UPS): The UPS controls the degradation of key regulators of cell proliferation and apoptosis. Notably, the E3 ubiquitin ligase MDM2 frequently undergoes amplification in cancers, leading to excessive ubiquitination and degradation of the tumor suppressor p53 [116]. Conversely, mutations in the deubiquitinase BAP1 (BRCA1-associated protein 1), a tumor suppressor, are associated with various malignancies, including mesothelioma and uveal melanoma [116].
SUMOylation: SUMO modification regulates the activity, stability, and subcellular localization of numerous cancer-relevant proteins. SUMOylation of the transcription factor NF-κB can either activate or inhibit its transcriptional activity depending on cellular context, thereby influencing expression of pro-survival genes [116]. Altered SUMO pathway components are observed in several cancers; for example, the SUMO E2 enzyme UBC9 is overexpressed in lung adenocarcinoma and melanoma [116].
NEDDylation: The conjugation of NEDD8 to cullin proteins activates cullin-RING ligases (CRLs), a major class of E3 ubiquitin ligases. Hyperactivation of NEDDylation can drive oncogenesis by enhancing the degradation of tumor suppressors. The NEDD8-activating enzyme (NAE) inhibitor pevonedistat is currently under clinical investigation for several cancer types [116].
Cross-talk Between UBL Pathways in Cancer
UBL pathways do not function in isolation but engage in complex cross-talk. A prominent example is the coordinated regulation of p53: while MDM2-mediated ubiquitination targets p53 for proteasomal degradation, SUMOylation can both positively and negatively regulate p53 activity and stability. Additionally, NEDDylation of MDM2 itself can enhance its E3 ligase activity toward p53, creating a multi-layered regulatory network that is frequently disrupted in cancer [116].
Table 1: UBL Pathways Dysregulated in Human Cancers
| UBL Pathway | Dysregulated Components | Cancer Types | Oncogenic Consequences |
|---|---|---|---|
| Ubiquitin | MDM2 amplification; BAP1 mutation | Sarcoma, Mesothelioma, Uveal Melanoma | p53 degradation; Genomic instability |
| SUMO | UBC9 overexpression | Lung Adenocarcinoma, Melanoma | Altered transcription factor activity |
| NEDD8 | CRL hyperactivation | Multiple Solid and Hematologic Cancers | Enhanced degradation of tumor suppressors |
| ISG15 | ISG15 overexpression | Breast Cancer | Altered DNA damage response |
UBL Dysregulation in Neurodegenerative Diseases
Neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson's disease (PD), and Amyotrophic Lateral Sclerosis (ALS), are characterized by the accumulation of misfolded, ubiquitinated proteins in affected neurons [117]. UBL pathways are central to the clearance of these toxic aggregates, and their dysfunction is a hallmark of these disorders.
Ubiquilins and Protein Quality Control
The ubiquilin (UBQLN) family of proteins, which function as ubiquitin receptors, play a particularly critical role in neuronal proteostasis. Ubiquilins contain an N-terminal ubiquitin-like (UBL) domain that interacts with the proteasome and a C-terminal ubiquitin-associated (UBA) domain that binds polyubiquitinated proteins, thereby shuttling substrates to the proteasome for degradation [117] [7].
- Alzheimer's Disease: UBQLN1 interacts with presenilins, components of the γ-secretase complex that processes amyloid precursor protein (APP). Polymorphisms in UBQLN1 are associated with increased AD risk, and reduced UBQLN1 levels are observed in the brain cortex of AD patients [117] [7].
- Amyotrophic Lateral Sclerosis and Frontotemporal Dementia: Dominant, missense mutations in UBQLN2 cause X-linked ALS, often associated with frontotemporal dementia. These mutations lead to the formation of UBQLN2-containing inclusions in spinal motor neurons, which are observed even in sporadic ALS patients without UBQLN2 mutations [117] [7]. UBQLN1 and UBQLN2 also interact with TDP-43, a DNA/RNA binding protein that forms pathological aggregates in the majority of ALS cases [117].
Autophagy-Related UBLs in Neurodegeneration
ATG8 and ATG12 are UBLs essential for the process of autophagy, a key pathway for clearing protein aggregates and damaged organelles. Impaired autophagy is implicated in several neurodegenerative diseases. ATG8 (LC3 in mammals) is uniquely conjugated to the lipid phosphatidylethanolamine, enabling its association with autophagosomal membranes and facilitating the engulfment of cytoplasmic cargo [1]. Dysregulation of this conjugation system disrupts the efficient clearance of aggregation-prone proteins like α-synuclein (in PD) and mutant huntingtin (in Huntington's disease), contributing to disease pathogenesis [1] [117].
Table 2: UBL Dysregulation in Major Neurodegenerative Diseases
| Disease | Dysregulated UBL/Pathway | Genetic/Molecular Evidence | Functional Consequence |
|---|---|---|---|
| Alzheimer's Disease (AD) | Ubiquilin (UBQLN1) | Polymorphisms increase AD risk; Reduced protein levels in cortex | Impaired presenilin handling & proteasomal degradation |
| Amyotrophic Lateral Sclerosis (ALS) | Ubiquilin (UBQLN2) | Missense mutations cause X-linked ALS; Protein aggregates | Defective substrate shuttling; TDP-43 pathology |
| Parkinson's Disease (PD) | ATG8/LC3 (Autophagy) | Impaired autophagy observed in models | Accumulation of α-synuclein aggregates |
| Huntington's Disease (HD) | Ubiquitin; ATG8/LC3 | Ubiquitinated huntingtin aggregates; Impaired autophagy | Neuronal inclusion formation |
UBLs in Viral Infection and Immune Evasion
Viruses have evolved sophisticated mechanisms to manipulate host UBL pathways to promote viral replication and evade immune responses [113] [114]. The host-virus interface involves multiple UBL systems, including ISG15, SUMO, and ubiquitin itself.
Antiviral Roles of UBLs: ISG15
ISG15 is an interferon-stimulated UBL that functions as a critical broad-spectrum antiviral molecule. It is conjugated to host and viral proteins in a process called ISGylation, which can inhibit the replication of viruses such as influenza, herpes, and Sindbis viruses [1] [114]. For example, ISG15 inhibits the budding of Ebola virus particles by blocking the activity of the viral E3 ligase adaptor, VP40 [114]. Interestingly, several viruses encode proteases that specifically deconjugate ISG15 to counteract its antiviral effects [114].
Viral Subversion of UBL Pathways
Viruses actively hijack or disrupt host UBL signaling:
- Herpesviruses and Adenoviruses: Encode viral E3 ubiquitin ligases that target host defense proteins for degradation. They also encode deconjugases (DUBs) that remove ubiquitin and UBLs from host proteins to prevent immune activation [114].
- Influenza B Virus: Its NS1 protein directly binds to and inhibits the conjugation of ISG15, thereby suppressing the host's interferon-induced antiviral state [114].
- Rice Stripe Virus: The host UBL5 protein binds to the viral non-structural protein 3 (NS3) and mediates its degradation, representing a direct antiviral role for UBL5. Conversely, UBL5 is also exploited by some viruses to support processes like pre-mRNA splicing of viral transcripts [113].
The diagram below illustrates the complex interplay between host UBL pathways and viral factors during infection.
Figure 1: Host-Virus Interactions in UBL Signaling. Host UBL pathways (yellow) activate antiviral immunity (green), but viral factors (red) subvert these pathways to promote replication.
Experimental Methodologies for UBL Research
Studying UBL dynamics requires specialized methodologies to detect UBL conjugation, identify substrates, and characterize enzymatic activities.
Detecting UBL Conjugation and Chain Topology
Mass Spectrometry (MS)-Based Proteomics: This is the primary method for comprehensive identification of UBL modification sites and polyubiquitin/UBL chain topology. The workflow typically involves:
- Protein Digestion: Proteolytic cleavage of protein mixtures (e.g., with trypsin).
- Ubiquitin Affinity Enrichment: Use of antibodies specific for ubiquitin or UBLs (e.g., anti-diGly antibodies for ubiquitin) to enrich modified peptides from complex lysates.
- Liquid Chromatography-Tandem MS (LC-MS/MS): Separation and sequencing of peptides to identify modification sites and the specific lysine residues within Ub/UBLs used for chain linkage [116] [115].
Yeast Two-Hybrid (Y2H) Screening: Used to identify novel protein-protein interactions involving UBLs, their enzymes, and potential substrates. For instance, Y2H screens identified the spliceosomal protein Snu66 and the ferulic acid decarboxylase Fdc1 as interactors of UBL5/Hub1 [113].
Structural Analysis of UBLs and Complexes
Nuclear Magnetic Resonance (NMR) Spectroscopy: Particularly valuable for determining the 3D solution structures of small UBLs like UBL5 and for mapping interaction surfaces by monitoring chemical shift perturbations upon binding to partners like the HIND domain of Snu66 [113].
X-ray Crystallography: Provides high-resolution atomic structures of UBLs alone or in complex with their binding partners, such as E1/E2/E3 enzymes or UBL-recognition domains (e.g., UBA, UIM, SIM domains) [113].
Table 3: Essential Research Reagents and Methods for UBL Studies
| Reagent / Method | Function/Application | Key Utility in UBL Research |
|---|---|---|
| Anti-diGly Antibodies | Immunoaffinity enrichment of ubiquitinated peptides | Identification of endogenous ubiquitination sites by MS |
| E1/E2/E3 Recombinant Enzymes | Reconstitution of conjugation cascades in vitro | Biochemical characterization of UBL transfer mechanisms |
| Active-Site Mutant UBLs (E.g., C-terminal Gly to Ala) | Acts as a substrate trap; inhibits conjugation | Identifying and validating specific UBL substrates |
| Tandem Ubiquitin Binding Entities (TUBEs) | High-affinity probes for polyubiquitin chains | Isolate and analyze endogenous polyubiquitinated proteins |
| NMR Spectroscopy | Determine 3D protein structure and dynamics | Characterize UBL fold and protein interaction interfaces |
| Yeast Two-Hybrid Screening | Identify protein-protein interactions | Discover novel UBL-binding partners and substrates |
The centrality of UBL pathways in human disease makes them attractive targets for therapeutic intervention. Several strategies are being actively pursued.
Proteasome Inhibitors: Bortezomib (Velcade), a reversible inhibitor of the proteasome's chymotrypsin-like activity, was the first Ub-system drug approved for cancer (multiple myeloma and mantle cell lymphoma) [116] [115]. It capitalizes on the dependence of cancer cells on the UPS to clear misfolded proteins and regulate cell cycle proteins.
Targeting E1, E2, and E3 Enzymes: Developing specific inhibitors for components of the UBL conjugation cascade is a major focus. The NEDD8 E1 inhibitor pevonedistat is in clinical trials for solid and hematologic cancers [116]. MDM2-p53 interaction antagonists are being developed to reactivate p53 in cancers with wild-type p53 [116] [115].
Targeting Deconjugating Enzymes: Inhibiting viral or host deconjugases (DUBs/ULPs) represents another therapeutic avenue, particularly for antiviral applications and cancer [114] [115].
In conclusion, UBLs are master regulators of cellular homeostasis, and their dysregulation is a common denominator in the pathogenesis of cancer, neurodegeneration, and viral infections. Continued research into the intricate mechanisms of UBL signaling will undoubtedly unveil new pathological connections and yield novel, targeted therapies for a wide spectrum of human diseases. The integration of basic structural biology, proteomic profiling, and disease modeling will be essential to fully exploit the therapeutic potential of the UBL universe.
Ubiquitin-like proteins (UBLs) constitute a family of small, evolutionarily conserved proteins that are covalently attached to target substrates in a process reminiscent of ubiquitination [1]. This reversible post-translational modification is a fundamental regulatory mechanism that governs virtually every aspect of cellular function. The UBL family derives its name from its founding member, ubiquitin, which was first discovered in the 1970s and originally named "ubiquitous immunopoietic polypeptide" [1]. Following the discovery of ubiquitin, many additional evolutionarily related members were described, including ISG15 (the first shown to share the key feature of covalent protein modification), SUMO, NEDD8, and Apg12 [1].
UBLs share a common three-dimensional structure known as the "beta-grasp" protein fold, consisting of a five-strand antiparallel beta sheet surrounding an alpha helix [1]. Despite this structural conservation, UBLs have evolved distinct biological functions through the development of dedicated enzymatic cascades and recognition systems. These UBL modifications act as sophisticated molecular switches that can alter the activity, stability, localization, or interaction partners of their target proteins [104] [39].
This review provides a comprehensive analysis of the two primary functional paradigms of UBL signaling: the classical degradative pathway and the diverse non-degradative regulatory mechanisms. Understanding the intricate balance between these pathways is essential for elucidating their roles in cellular homeostasis and disease pathogenesis, particularly in the context of developing targeted therapeutic interventions.
Classification and Structural Characteristics
UBLs can be systematically classified into two primary categories based on their conjugation capabilities:
- Type I UBLs: Capable of covalent conjugation to target molecules through a characteristic C-terminal glycine motif. These are expressed as inactive precursors that require proteolytic processing to expose the active glycine residue [1]. This category includes ubiquitin itself and well-characterized UBLs such as SUMO, NEDD8, ATG8, ATG12, URM1, UFM1, FAT10, and ISG15 [1].
- Type II UBLs: Not covalently conjugated but often exist as protein domains fused to other domains within a single polypeptide chain. These may be proteolytically processed to release the UBL domain or function as protein-protein interaction modules [1].
The structural unity of the UBL family stems from the conserved beta-grasp fold, yet each member possesses distinct surface properties that dictate specific interactions with their cognate enzymes and effector proteins [1] [118]. For instance, NEDD8, the closest relative of ubiquitin with 58% sequence identity and 80% similarity, contains subtle but critical structural differences that ensure its discrimination from ubiquitin within dedicated conjugation pathways [118].
Evolutionary Conservation and Distribution
UBLs exhibit remarkable evolutionary conservation across eukaryotes, with phylogenetic studies suggesting a monophyletic origin for eukaryotic UBLs [1]. The UBL regulatory systems share common evolutionary origins with prokaryotic biosynthesis pathways for cofactors such as thiamine and molybdopterin [1]. The eukaryotic protein URM1, which functions as both a UBL and a sulfur-carrier protein, represents a molecular fossil establishing this evolutionary link [1].
The distribution of UBL families varies significantly across evolutionary lineages:
- Humans: Encode at least eight families of Type I UBLs in addition to ubiquitin itself, including SUMO, NEDD8, ATG8, ATG12, URM1, UFM1, FAT10, and ISG15 [1].
- Plants: Contain at least seven UBL families including SUMO, RUB (the plant homolog of NEDD8), ATG8, ATG12, MUB, UFM1, and HUB1 [1].
- Prokaryotes: Exhibit phylogenetically restricted UBL-related proteins, such as Pup in actinobacteria, which functions analogously to ubiquitin but is intrinsically disordered and evolutionarily distinct [1].
Table 1: Major UBL Families and Their Primary Characteristics
| UBL | Size (aa) | E1 Activating Enzyme | Primary Functions | Conjugation Type |
|---|---|---|---|---|
| Ubiquitin | 76 | UAE (UBA1) | Protein degradation, trafficking, signaling [119] | Type I |
| SUMO | ~100 | SAE (SAE1-SA2) | Transcription, DNA repair, stress response [1] | Type I |
| NEDD8 | 81 | NAE (APPB1-UBA3) | Cullin activation, regulation of CRL complexes [118] | Type I |
| ATG8 | 117-124 | ATG7 | Autophagosome formation, autophagy [1] | Type I (to lipids) |
| ATG12 | 186 | ATG7 | Autophagy initiation [1] | Type I |
| ISG15 | 165 | UBA7 | Immune response, antiviral defense [1] | Type I |
The UBL Conjugation Machinery
The Enzymatic Cascade
UBL conjugation follows a conserved three-step enzymatic cascade that is parallel for each UBL family member:
Activation: UBLs are activated in an ATP-dependent manner by specific E1 activating enzymes, forming a high-energy thioester bond between the UBL's C-terminal glycine and a catalytic cysteine residue in the E1 [119] [104]. This step is highly specific, with each UBL typically having dedicated E1 enzymes [104].
Conjugation: The activated UBL is transferred to the catalytic cysteine of an E2 conjugating enzyme through a trans-thiolation reaction [119]. The human genome encodes approximately 40 E2 enzymes that play central roles in determining aspects of UBL signaling, including the efficiency of chain assembly and specific configurations of different chains [39].
Ligation: E3 ligases facilitate the final transfer of the UBL to specific substrate proteins, typically forming an isopeptide bond with a lysine ε-amino group [119]. E3 ligases provide substrate specificity and are classified into several families, including RING, HECT, and RBR types [119].
The specificity of UBL pathways is maintained through precise molecular discrimination at each enzymatic step. For example, in the neddylation pathway, structural studies have revealed that the interface between NEDD8 and its E1 enzyme (APPBP1-UBA3 heterodimer) involves three distinct contact sites that collectively ensure pathway specificity [118].
Deconjugation and Regulation
UBL modifications are reversible through the action of specialized proteases:
- Deubiquitinating enzymes (DUBs): Approximately 100 in humans, responsible for removing ubiquitin from modified substrates [119] [39].
- ULPs (UBL-specific proteases): Family of enzymes that reverse UBL modifications, such as SENP proteases for de-SUMOylation [1] [39].
These regulatory enzymes provide dynamic control over UBL signaling and play crucial roles in maintaining cellular homeostasis.
Degradative UBL Signaling
The Ubiquitin-Proteasome System
The most characterized degradative UBL pathway is the ubiquitin-proteasome system (UPS), where polyubiquitin chains linked through lysine 48 (K48) of ubiquitin target substrate proteins for degradation by the 26S proteasome [119] [120]. This process requires a minimum of four ubiquitin molecules attached in a chain to effectively trigger degradation [120]. The UPS is responsible for the regulated proteolysis of numerous cellular proteins and is essential for maintaining protein homeostasis, with implications for various human diseases including cancer, cardiovascular disease, and neurodegenerative disorders [104].
Structural Basis for Degradative Signaling
The specificity of K48-linked polyubiquitin chains for proteasomal recognition lies in the structural properties of the chain itself and the presence of ubiquitin receptors on the proteasome. The proteasome contains multiple ubiquitin receptors that recognize and bind to polyubiquitin chains, initiating the unfolding and degradation of the attached substrate while recycling ubiquitin molecules for reuse [104].
Table 2: UBL Chains with Predominantly Degradative Functions
| Chain Type | Structural Features | E2/E3 Enzymes | Cellular Functions |
|---|---|---|---|
| K48-linked ubiquitin | Compact structure, proteasome recognition [119] | Various E2s and E3s | Primary signal for proteasomal degradation [119] [120] |
| K11-linked ubiquitin | Mixed extended/compact conformation [119] | UBE2S, APC/C | Cell cycle regulation, ER-associated degradation [119] |
| K29-linked ubiquitin | Less characterized | UBE2A, UBE2B, UBE2D1 | Wnt/β-catenin signaling, neurodegenerative disorders [119] |
Non-degradative UBL Signaling
Diversity of Non-proteolytic Functions
Non-degradative UBL signaling encompasses a wide array of regulatory mechanisms that control protein function without targeting substrates for destruction. These non-proteolytic functions are mediated by various types of UBL modifications, including monoubiquitination, multi-monoubiquitination, and atypical polyubiquitin chains linked through lysine residues other than K48 [119].
Key Non-degradative UBL Modifications
K63-linked ubiquitin chains represent the best-characterized non-degradative ubiquitin signal and play crucial roles in:
- Inflammatory signaling: Activation of NF-κB pathway through modification of signaling components such as RIP kinases [119] [120].
- DNA damage repair: Recruitment of repair factors to sites of DNA damage through histone ubiquitination [119].
- Endocytic trafficking: Regulation of membrane protein internalization and sorting [119].
M1-linked (linear) ubiquitin chains are generated by the LUBAC complex and function in:
- Immune signaling: Regulation of TNF and IL-1β signaling pathways [119].
- Cell death regulation: Modulation of necroptosis and apoptosis [119].
SUMO modification controls diverse cellular processes including:
- Transcriptional regulation: Modification of transcription factors and co-regulators [1].
- DNA repair: Coordination of repair complex assembly and disassembly [1] [119].
- Protein stability and subcellular localization: Regulation of nuclear-cytoplasmic trafficking [104].
NEDD8 conjugation (neddylation) primarily regulates:
- CRL complex activation: Modification of cullin scaffolds stimulates the ubiquitin ligase activity of CRL complexes, thereby indirectly regulating protein degradation [118].
- Non-cullin substrates: Emerging evidence indicates neddylation of additional substrates with non-degradative outcomes [118].
Table 3: Non-degradative UBL Chains and Their Functions
| Chain Type | Primary Functions | Representative Enzymes | Cellular Processes |
|---|---|---|---|
| K63-linked ubiquitin | Recruitment platform, activation | UBC13, TRAF6, RNF8 [119] | NF-κB signaling, DNA repair, endocytosis [119] [120] |
| M1-linked ubiquitin | Inflammation, cell death | LUBAC complex, HOIP [119] | TNF signaling, immunity, protein quality control [119] |
| K6-linked ubiquitin | Mitophagy, protein stabilization | PARKIN, UBE3A | Mitochondrial quality control [119] |
| K27-linked ubiquitin | Innate immunity | RNF168 | Histone modification, DNA damage response [119] |
| K33-linked ubiquitin | Protein trafficking | Unknown | Kinase regulation, intracellular trafficking [119] |
Experimental Approaches for UBL Research
Methodologies for Identifying UBL Modifications
Advanced mass spectrometry-based techniques have revolutionized the identification and quantification of UBL modifications. Recent methodological developments include:
pLink-UBL Search Engine: A dedicated computational tool for identifying UBL modification sites without requiring mutation of the UBL protein. This approach has demonstrated superior precision, sensitivity, and speed compared to conventional search engines, increasing the identification of SUMOylation sites by 50-300% from the same datasets [48].
Small-Molecule Substrate Identification: A novel method involving antibody enrichment of UBL C-terminal peptides followed by LC-MS/MS analysis and blind searching to identify unexpected modifications. Using this approach, researchers discovered that spermidine can be conjugated to the C-terminus of SUMO in fission yeast, mice, and humans, suggesting that small molecules may represent a novel class of UBL substrates [48].
Experimental Workflow for UBL Modification Analysis
The following diagram illustrates a comprehensive workflow for the identification and functional characterization of UBL modifications:
Diagram 1: Workflow for UBL modification analysis. This comprehensive pipeline encompasses sample preparation, mass spectrometric identification, and functional validation of UBL modifications.
Research Reagent Solutions
Table 4: Essential Research Reagents for UBL Studies
| Reagent Category | Specific Examples | Applications and Functions |
|---|---|---|
| E1 Inhibitors | MLN4924 (NAE inhibitor) [104] | Selective inhibition of NEDD8 activation, blocks cullin neddylation |
| Proteasome Inhibitors | Bortezomib, Carfilzomib [104] | Inhibit proteasomal degradation, allow accumulation of ubiquitinated proteins |
| DUB/ULP Inhibitors | PR-619 (pan-DUB inhibitor) | Broad inhibition of deubiquitinating enzymes, stabilize ubiquitination |
| UBL-Specific Antibodies | Anti-K48 ubiquitin, Anti-K63 ubiquitin, Anti-SUMO, Anti-NEDD8 [48] | Enrichment and detection of specific UBL modifications |
| Expression Plasmids | E1/E2/E3 constructs, UBL mutants, Substrate plasmids | Functional studies of UBL pathways in cellular systems |
| Activity Assays | E1-E2-E3 cascade assays, Deconjugation assays | In vitro analysis of UBL conjugation and deconjugation kinetics |
Structural Mechanisms of Signal Discrimination
Molecular Basis of UBL Chain Recognition
The functional diversity of UBL modifications is encoded in the structural properties of the chains themselves, which are interpreted by specialized receptor proteins containing UBL-binding domains. The concept of the "ubiquitin code" proposes that different UBL chain types create distinct signals that are recognized by specific receptors to initiate appropriate cellular responses [119] [39].
Structural studies have revealed that ubiquitin-binding domains (UBDs) exhibit remarkable specificity for distinct chain types. For example:
- Proteasomal receptors (e.g., Rpn10, Rpn13) preferentially recognize K48-linked ubiquitin chains through specific interaction surfaces that accommodate the compact conformation of these chains [119].
- K63-linkage specific receptors (e.g., TAB2, TAB3) contain specialized UBDs that recognize the unique extended conformation of K63-linked chains [119].
- SUMO-interaction motifs (SIMs) recognize the hydrophobic surfaces of SUMO proteins and facilitate the assembly of SUMO-dependent protein complexes [1].
Structural Basis for NEDD8 Specificity
NEDD8 presents a particularly interesting case for understanding signal discrimination, as it shares high sequence similarity with ubiquitin yet maintains functional specificity. The molecular mechanisms ensuring discrimination between ubiquitin and NEDD8 include:
E1-E2 recognition: The NEDD8 E1 enzyme (APPBP1-UBA3) contains unique structural elements that specifically recognize NEDD8 through three distinct contact sites, while rejecting ubiquitin [118].
E2-substrate interactions: NEDD8-specific E2 enzymes (UBC12, UBE2F) contain N-terminal extensions that interact specifically with the NEDD8 E1, maintaining pathway fidelity [118].
Receptor discrimination: Recent identification of novel NEDD8-binding domains in KHNYN and N4BP1 proteins has shed light on how NEDD8-specific signals are interpreted in cells, despite the ability of many ubiquitin-binding domains to interact with both ubiquitin and NEDD8 with varying affinities [118].
The following diagram illustrates the specific molecular interactions that maintain discrimination between ubiquitin and NEDD8 conjugation pathways:
Diagram 2: Specificity and cross-talk in ubiquitin and NEDD8 pathways. While maintaining distinct conjugation cascades, neddylation of cullins activates CRL complexes, enhancing their ubiquitin ligase activity and creating functional cross-talk.
Evolutionary Perspectives on UBL Functions
Conservation and Diversification of UBL Pathways
UBL systems display complex evolutionary patterns that reflect both functional conservation and lineage-specific adaptations:
Ubiquilin gene family: Evolutionary analysis of ubiquilins, which function as ubiquitin receptors, reveals that many organisms (e.g., fungi, many animals) have single ubiquilin genes, but duplications have occurred in animal, plant, alveolate, and excavate species [7]. Vertebrates possess up to seven different ubiquilins, with mammals exhibiting the most complex family composition of up to six genes resulting from recent evolutionary expansions [7].
Testis-specific expansions: Mammals have experienced exceptional mammalian-specific expansion of ubiquilin genes, with three newly emerged genes (UBQLN3, UBQLN5, and UBQLNL) showing precise testis-specific expression patterns, suggesting roles in postmeiotic spermatogenesis [7]. A similar independent expansion has occurred in Drosophila species, indicating convergent evolution of reproductive function specialization [7].
Positive selection signatures: Detection of positive selection acting on some mammalian ubiquilins suggests adaptive evolution and functional diversification within this protein family [7].
Prokaryotic UBL Systems
The presence of UBL-related systems in prokaryotes provides insights into the evolutionary origins of eukaryotic UBL pathways:
Pup in actinobacteria: Functions analogously to ubiquitin in labeling proteins for proteasomal degradation but is intrinsically disordered and evolutionarily distinct from eukaryotic UBLs [1].
TtuB in Thermus bacteria: Shares the beta-grasp fold with eukaryotic UBLs and has dual functions as both a sulfur carrier protein and a covalently conjugated protein modification [1].
Archaeal systems: Some archaea possess seemingly complete sets of genes corresponding to eukaryote-like ubiquitin pathways, identified in "Euryarchaeota", Thermoproteota, and "Aigarchaeota" [1].
These evolutionary patterns highlight how UBL systems have been adapted and specialized throughout evolution to meet specific cellular requirements in different organisms.
Therapeutic Targeting of UBL Pathways
Clinical Implications of UBL Signaling
Dysregulation of UBL pathways is implicated in numerous human diseases, making them attractive therapeutic targets:
Cancer: Aberrant ubiquitination and neddylation are hallmarks of many cancers, with mutations in UPS components and altered degradation of tumor suppressors or oncoproteins contributing to tumorigenesis [104].
Neurodegenerative disorders: Protein aggregation in diseases such as Alzheimer's, Parkinson's, and amyotrophic lateral sclerosis (ALS) is closely linked to impaired UBL signaling. Mutations in ubiquilin-encoding genes have been directly linked to neurodegenerative diseases, with UBQLN2 mutations causing familial ALS [7].
Inflammatory and immune disorders: Non-degradative ubiquitination plays crucial roles in immune signaling, and dysregulation of these pathways can lead to autoimmune and inflammatory conditions [119] [120].
Current and Emerging Therapeutic Strategies
Several therapeutic approaches targeting UBL pathways have been developed:
Proteasome inhibitors: Bortezomib was the first FDA-approved drug targeting the UPS, demonstrating the clinical validity of this approach for multiple myeloma treatment [104]. Second-generation proteasome inhibitors such as carfilzomib have further improved therapeutic options [104].
NEDD8 activation inhibitor: MLN4924 (Pevonedistat) inhibits the NEDD8 E1 enzyme (NAE), blocking cullin neddylation and subsequent CRL-mediated ubiquitination [104]. This compound is under investigation in clinical trials for various malignancies.
DUB inhibitors: Developing selective inhibitors for specific DUBs represents an emerging therapeutic strategy, with several compounds in preclinical and early clinical development [104].
The following table summarizes key therapeutic targets in UBL pathways:
Table 5: Therapeutic Targeting of UBL Pathways in Human Disease
| Therapeutic Target | Compound Examples | Development Stage | Primary Indications |
|---|---|---|---|
| 20S Proteasome | Bortezomib, Carfilzomib | FDA-approved, Phase III [104] | Multiple myeloma, lymphoma |
| NEDD8 E1 (NAE) | MLN4924 (Pevonedistat) | Phase I/II trials [104] | Acute myeloid leukemia, solid tumors |
| Ubiquitin E1 (UAE) | TAK-243 | Phase I trials | Advanced solid tumors |
| DUBs | Multiple candidates | Preclinical/early clinical | Cancer, neurodegenerative diseases |
| E3 Ligases | MDM2 inhibitors, IAP antagonists | Preclinical/clinical development | Cancer, immune disorders |
The comparative analysis of degradative versus non-degradative UBL signaling reveals a sophisticated regulatory network that extends far beyond the initial paradigm of protein degradation. While degradative UBL signaling, primarily through K48-linked ubiquitin chains, remains a fundamental mechanism for controlling protein half-life, non-degradative UBL modifications have emerged as equally crucial regulators of diverse cellular processes.
The functional specificity of UBL signaling is encoded at multiple levels, including the specificity of E1-E2-E3 enzymatic cascades, the structural diversity of UBL chains, and the selective recognition of these chains by specialized effector proteins. The evolutionary conservation of UBL systems, alongside lineage-specific expansions and adaptations, underscores their fundamental importance in cellular regulation.
Future research directions should focus on elucidating the complete repertoire of UBL modifications, including the recently discovered conjugation to small molecules such as spermidine [48], and developing more sophisticated tools for probing the dynamics and functional consequences of these modifications in physiological and pathological contexts. The continued development of therapeutic agents targeting specific components of UBL pathways holds significant promise for treating a wide range of human diseases, particularly cancer and neurodegenerative disorders.
Understanding the intricate balance between degradative and non-degradative UBL signaling will continue to provide fundamental insights into cellular regulation and open new avenues for therapeutic intervention across a spectrum of human diseases.
The ubiquitin-like protein (UBL) system, comprising ubiquitin, SUMO, and NEDD8, constitutes a crucial regulatory network governing virtually all eukaryotic cellular processes. While each UBL modifies distinct substrates through dedicated enzymatic cascades, they do not operate in isolation. Emerging evidence reveals sophisticated cross-pathway communication where these modifiers reciprocally regulate one another, generating a complex signaling language that coordinates cellular responses to physiological and pathological stimuli. This review delineates the structural, functional, and evolutionary underpinnings of this interplay, with particular focus on hybrid chain formation, shared recognition domains, and bacterial exploitation of these pathways. We further provide experimental methodologies for investigating UBL cross-talk and discuss the therapeutic implications of targeting this network in human diseases, including cancer and infection.
Ubiquitin-like proteins (UBLs) are a family of small proteins involved in the post-translational modification of cellular targets, thereby drastically altering their fate, activity, localization, or interactions [1]. The family derives its name from its founding member, ubiquitin, and includes well-characterized modifiers such as SUMO (Small Ubiquitin-like Modifier) and NEDD8 (Neural precursor cell Expressed Developmentally Down-regulated protein 8) [121]. These UBLs share a common structural fold, the β-grasp fold, characterized by a five-strand antiparallel beta sheet surrounding a central alpha helix [13] [1].
The conjugation of UBLs to target proteins is a hallmark of their function, typically occurring through a conserved three-step enzymatic cascade:
- Activation: A UBL-specific E1 activating enzyme catalyzes UBL adenylation in an ATP-dependent manner.
- Conjugation: The activated UBL is transferred to a catalytic cysteine residue on a UBL-specific E2 conjugating enzyme.
- Ligation: A UBL-specific E3 ligase facilitates the transfer of the UBL from the E2 to a lysine residue on the target protein, forming an isopeptide bond [122] [123] [1].
This reversible process is counteracted by specific deconjugating enzymes (e.g., deubiquitinases for ubiquitin, ULPs for SUMO), allowing for dynamic regulation [1]. The system's complexity is magnified by the ability of UBLs like ubiquitin and SUMO to form polymeric chains of various linkages and topologies, which encode distinct functional outcomes [122]. The interplay between ubiquitin, SUMO, and NEDD8 represents a sophisticated layer of regulation, enabling the cell to integrate diverse signals and mount appropriate responses. This cross-pathway communication is not only fundamental to cellular homeostasis but is also a key target in pathological contexts, including bacterial infection and cancer [124] [125].
Structural and Evolutionary Foundations
The functional synergy between ubiquitin, SUMO, and NEDD8 is deeply rooted in their shared evolutionary history and conserved structural architecture.
A Common Structural Fold
Ubiquitin, SUMO, and NEDD8 all adopt the β-grasp fold (β-GF), a structural motif widely distributed in proteins across all domains of life [13] [74]. This fold is typified by a β-sheet with five antiparallel β-strands that "grasp" a central α-helix [1]. Despite relatively low sequence identity—NEDD8 shares approximately 60% sequence identity with ubiquitin, while SUMO is more distantly related—this structural conservation is paramount [123]. It allows for analogous interactions with enzymatic machineries and binding domains, while sequence variations confer specificity.
Evolutionary Origins and Diversification
Evolutionary analyses indicate that the β-grasp fold first emerged in prokaryotes in the context of translation-related RNA interactions [13] [74]. The eukaryotic phase of its evolution was marked by a significant expansion of the UBL clade. It is believed that precursors of the eukaryotic ubiquitin system were present in prokaryotes, with UBLs and E1-like enzymes involved in metabolic pathways for cofactors like thiamine and molybdopterin [13] [74] [1]. Proteins like ThiS and MoaD in these biosynthetic pathways share the β-grasp fold and are considered evolutionary progenitors of modern UBLs [1]. The eukaryotic protein URM1, which functions both as a UBL and a sulfur carrier, is often described as a "molecular fossil" bridging these ancient sulfur-transfer systems and protein-modification systems [1].
By the time of the last eukaryotic common ancestor, nearly 20 distinct UBL families were already present, playing crucial roles in the emergence of characteristic eukaryotic cellular structures and systems, including vesicular trafficking, nuclear compartmentalization, and chromatin dynamics [13] [74]. The high degree of structural similarity, particularly between ubiquitin and NEDD8, has direct functional consequences, as it complicates the specific recognition of these modifiers by cellular machinery, a challenge that nature has solved through the evolution of specialized domains [123].
Table 1: Structural and Evolutionary Features of Ubiquitin, SUMO, and NEDD8
| Feature | Ubiquitin | SUMO | NEDD8 |
|---|---|---|---|
| Length (aa) | 76 | ~100 (varies by isoform) | 81 |
| Sequence Identity vs. Ubiquitin | 100% | ~20% | ~60% |
| Core Structural Fold | β-grasp | β-grasp | β-grasp |
| Key Conjugation Site | C-terminal Gly76 | C-terminal Gly | C-terminal Gly |
| Evolutionary Link | Derived from prokaryotic sulfur-carrier proteins (ThiS/MoaD) | Derived from prokaryotic sulfur-carrier proteins (ThiS/MoaD) | Derived from prokaryotic sulfur-carrier proteins (ThiS/MoaD) |
| Prokaryotic Analogues | Pup, UBact (functionally analogous) | Limited evidence | Limited evidence |
Recognition and Specificity: The Challenge of Similarity
The high sequence and structural identity between ubiquitin and NEDD8 creates a problem of promiscuity in recognition by ubiquitin-binding domains (UBDs) [123]. However, cells have evolved specific solutions to ensure faithful signal decoding. A prime example is the evolution of the CUBAN (Cullin-Binding domain Associating with NEDD8) domain, which binds monomeric NEDD8 and neddylated cullins with high specificity. Intriguingly, its evolutionary relative, the CoCUN (Cousin of CUBAN) domain, binds only ubiquitin [123]. This divergence from a common three-helix bundle ancestor illustrates how nature achieved specificity in recognizing these highly related molecules, employing distinct interaction modes—electrostatic for NEDD8 (recalling RBX1/2 E3 ligase interactions) and hydrophobic for ubiquitin (similar to CUE domains) [123].
Mechanisms of Functional Interplay
The crosstalk between ubiquitin, SUMO, and NEDD8 is not merely parallel operation; it involves direct, reciprocal regulation that amplifies the coding potential of the ubiquitin code.
Hybrid Chain Formation
A primary mechanism of cross-talk is the formation of hybrid chains, where one UBL modifies another [122]. This creates branched structures that can alter the original message encoded by a single UBL type or introduce a completely new one.
- SUMO-UBiquitin Hybrids: Proteomic studies have identified ubiquitination on multiple lysine residues of SUMO-1, -2, and -3 [122]. While SUMO-2/3 can form polymeric chains, SUMO-1 cannot, but it can be ubiquitinated on at least six different lysines [122]. These hybrid chains are predominantly formed under stress conditions and are recognized by specific receptors containing combined SUMO-interacting motifs (SIMs) and ubiquitin-binding domains (UBDs), conferring high affinity and specificity [122].
- NEDD8-UBiquitin Hybrids: Although less characterized than SUMO-ubiquitin hybrids, NEDD8 can also be modified by ubiquitin, and vice versa [123]. The CUBAN domain itself can bind di-ubiquitin chains in addition to NEDD8, highlighting the inherent cross-reactivity and potential for hybrid signaling at the level of recognition [123].
The formation of these hybrid chains introduces a "collaboration" between UBL pathways, allowing for the integration of signals. For instance, a SUMO modification can be "read" not only by SUMO readers but also by ubiquitin readers if it becomes ubiquitinated, potentially targeting a SUMOylated substrate for proteasomal degradation [122].
Sequential and Competitive Modification
Another layer of interplay involves the sequential or competitive modification of a shared substrate. The most characterized example is the SUMO-targeted ubiquitin ligase (STUbL) pathway. STUbLs, such as RNF4, recognize poly-SUMO chains via multiple SIMs and catalyze the ubiquitination of the same substrate, leading to its proteasomal degradation [125]. This pathway is critical in processes like the arsenic-induced degradation of the PML protein [125].
Conversely, NEDD8's most defined role is the neddylation of cullin proteins, which activates the cullin-RING ligase (CRL) family of E3 ubiquitin ligases [125]. Thus, NEDD8 directly controls the activity of a major class of the ubiquitin system, creating a hierarchical relationship where neddylation governs global ubiquitination patterns [121] [125].
Table 2: Types of UBL Cross-Talk and Their Functional Consequences
| Type of Interplay | Molecular Mechanism | Example | Functional Outcome |
|---|---|---|---|
| Hybrid Chain Formation | Covalent modification of one UBL by another on lysine residues. | Ubiquitination of SUMO-1/2/3 [122] | Generates a novel composite signal; can recruit combined UBD-SIM readers; may target SUMOylated proteins for degradation. |
| Sequential Modification | One UBL modification primes the substrate for modification by another. | STUbL (RNF4) ubiquitination of poly-SUMOylated substrates [125] | Targets SUMOylated proteins for proteasomal degradation. |
| Regulatory Hierarchy | One UBL directly regulates the enzymatic machinery of another. | Neddylation of cullins activates CRL E3 ubiquitin ligases [125] | NEDD8 controls a major branch of the ubiquitin-proteasome system, affecting stability of thousands of proteins. |
| Competitive Modification | Different UBLs modify the same lysine residue on a substrate. | SUMOylation vs. Ubiquitination on identical lysines (e.g., IkBα, PCNA) | Creates a molecular switch to control protein activity, stability, or interactions. |
The following diagram illustrates the key mechanisms of cross-pathway communication, including hybrid chain formation and sequential modification.
Diagram 1: UBL Cross-Pathway Communication. This diagram illustrates key mechanisms of interplay: 1) SUMO-ubiquitin hybrid chain formation via STUbLs, targeting substrates for degradation; 2) NEDD8-mediated activation of CRL ubiquitin ligases, creating a regulatory hierarchy.
Pathogenic Exploitation of UBL Interplay
Bacterial pathogens, despite lacking eukaryotic-like UBL systems, have evolved a remarkable arsenal of effectors that precisely manipulate host ubiquitin, SUMO, and NEDD8 pathways to promote infection [124] [121]. This exploitation highlights the critical importance of these pathways in host defense.
Targeting the NF-κB Pathway
A common theme is the disruption of the NF-κB inflammatory signaling pathway, which is heavily regulated by ubiquitination. For example:
- Shigella flexneri delivers effector proteins like IpaH1.4, IpaH2.5, and IpaH9.8 (NEL-family E3 ubiquitin ligases) that ubiquitinate and degrade components of the NF-κB pathway, including LUBAC, TRAF2, and NEMO [121].
- Shigella OspI is a deamidase that inactivates the E2 ubiquitin-conjugating enzyme UBE2N/UBC13, thereby shutting down NF-κB signaling [121].
- Legionella pneumophila uses the effector SdeA, which possesses a non-canonical ubiquitin ligase activity. SdeA can modify host RAB GTPases and RTN4 with ubiquitin in an E1/E2-independent manner, disrupting cellular trafficking and signaling [121].
Direct Targeting of UBLs and Their Machinery
Pathogens also directly target SUMO and NEDD8 conjugation. Bacterial effectors can mimic host E3 ligases, DUBs, or SUMO proteases to rewire the host post-translational modification landscape [124]. The interplay between pathways is exploited; for instance, by simultaneously inhibiting ubiquitin-dependent NF-κB activation and manipulating SUMOylation of host transcription factors, bacteria can effectively suppress the host immune response [124]. The fact that bacterial effectors have evolved to target multiple nodes in the UBL network underscores the functional integration of these pathways and their collective importance in maintaining cellular homeostasis during infection.
Experimental and Therapeutic Applications
A Toolkit for Studying UBL Cross-Talk
Deciphering the complex language of UBL interplay requires a specialized set of reagents and methodologies.
Table 3: Research Reagent Solutions for Investigating UBL Interplay
| Research Tool / Reagent | Function / Description | Application in UBL Cross-Talk Research |
|---|---|---|
| Chain-Specific UBD Probes | Recombinant proteins or domains (e.g., CUBAN, CoCUN, UIM, SIM) with defined binding specificity for UBL chains or hybrids [123]. | Used in pull-down assays to enrich and identify specific hybrid chains from cell lysates. |
| Activity-Based Probes (ABPs) | Chemical probes that covalently bind to active-site residues of E1, E2, E3, or DUBs/ULPs [122]. | Profiling enzyme activity in different cellular states; identifying enzymes that process hybrid chains. |
| SUMO-Trap / TUBE | Tandem-repeated binding motifs (e.g., SIMs for SUMO-Trap; UBDs for Tandem Ubiquitin Binding Entities) immobilized on beads [122]. | High-affinity enrichment of SUMOylated or ubiquitinated proteins, including those with hybrid modifications, for proteomics. |
| In Vitro Reconstitution System | Purified components: E1, E2, E3 enzymes for one or multiple UBLs; UBLs (wild-type and mutant); potential substrates [122]. | Biochemical dissection of the hierarchy and kinetics of hybrid chain formation (e.g., SUMOylation before ubiquitination). |
| Linkage-Specific Antibodies | Antibodies recognizing specific ubiquitin or SUMO linkages (e.g., K48-Ub, K63-Ub, SUMO-2/3 chains) [122]. | Detection and quantification of specific chain types and hybrid chains in Western blotting or immunofluorescence. |
| Mass Spectrometry (PTM Proteomics) | Proteomic techniques, often using di-glycine (K-ε-GG) remnant antibodies for ubiquitin/NEDD8 and specific SUMO proteomics workflows [122] [125]. | Global, site-specific mapping of UBL modification sites, including identification of UBLs modified by other UBLs. |
Detailed Experimental Protocol: Analyzing SUMO-Ubiquitin Hybrid Chains
The following workflow provides a methodology for the identification and validation of SUMO-ubiquitin hybrid chains, a key feature of UBL cross-talk.
Objective: To isolate and identify proteins modified by SUMO-Ubiquitin hybrid chains under proteotoxic stress (e.g., heat shock).
Materials:
- Cell Line: HEK293T or U2OS cells.
- Lysis Buffer: RIPA buffer supplemented with 1% SDS, 50mM N-Ethylmaleimide (NEM) to inhibit deubiquitinases and SUMO proteases, and complete EDTA-free protease inhibitor cocktail.
- Enrichment Reagents: Anti-SUMO2/3 antibody (or SIM-based affinity resin) for initial enrichment. Anti-Ubiquitin remnant motif (K-ε-GG) antibody for subsequent enrichment.
- Elution Buffer: Low-pH glycine buffer (100mM, pH 2.5) or 2x Laemmli buffer containing 100mM DTT.
- Mass Spectrometry: Trypsin/Lys-C mix, C18 StageTips for desalting, LC-MS/MS system.
Procedure:
- Induction and Harvest: Subject cells to heat shock (e.g., 43°C for 60 minutes). Immediately wash cells with ice-cold PBS and lyse using the pre-heated (95°C) SDS-containing lysis buffer to rapidly denature proteins and inactivate enzymes.
- Immunoaffinity Purification:
- a. SUMO-Enrichment: Dilute the cleared lysates 10-fold with RIPA buffer (without SDS) to reduce SDS concentration. Incubate with anti-SUMO2/3 antibody-conjugated beads overnight at 4°C.
- b. Washing: Wash beads stringently with high-salt buffer (e.g., 1M NaCl in RIPA) and then with standard RIPA buffer to remove non-specifically bound proteins.
- c. Elution: Elute the bound SUMOylated proteins using the low-pH glycine buffer, and immediately neutralize with Tris-base.
- Post-Translational Modification (PTM) Cross-Enrichment:
- a. Denaturation and Digestion: Denature the eluate, reduce with DTT, alkylate with iodoacetamide, and digest with trypsin/Lys-C.
- b. Ubiquitin Remnant Enrichment: Desalt the digested peptides and subject them to immunoaffinity purification using the anti-K-ε-GG antibody to specifically isolate peptides that carry a ubiquitin remnant (di-glycine) on lysine. This step will capture ubiquitinated peptides derived from the initial SUMO-enriched pool, i.e., the hybrid chains [122].
- Mass Spectrometric Analysis:
- a. Analyze the enriched peptides by LC-MS/MS.
- b. Use database search engines (e.g., MaxQuant, Proteome Discoverer) configured to search for the following variable modifications: GlyGly (K) for ubiquitination, and the specific SUMO-2/3 remnant peptides (e.g., TGG, QTGG) on lysine.
- c. Data interpretation should focus on peptides that contain both the SUMO-2/3 remnant and the ubiquitin (di-glycine) modification, providing direct evidence for hybrid chain formation [122].
Validation: Confirm key hits by western blotting of purified material using antibodies against SUMO-2/3 and ubiquitin sequentially. Genetic validation can be achieved by siRNA knockdown of suspected hybrid chain readers or enzymes like STUbLs and assessing the stability of the hybrid-modified proteins.
Therapeutic Targeting in Cancer
The dysregulation of ubiquitin, SUMO, and NEDD8 pathways is a hallmark of many cancers, making them attractive therapeutic targets [125]. The interplay between these pathways offers unique opportunities for combination therapy.
- Neddylation Inhibition: The NEDD8-activating enzyme (NAE) inhibitor Pevonedistat (MLN4924) blocks cullin neddylation, thereby inactivating CRLs. This leads to the accumulation of CRL substrates, many of which are oncoproteins or cell-cycle regulators, causing cell cycle arrest and apoptosis [125].
- SUMO Pathway Inhibition: Developing SUMO E1 inhibitors and STUbL activators is an active area of research. Since SUMOylation often stabilizes oncogenic transcription factors, its inhibition can have anti-tumor effects. Furthermore, because SUMO can antagonize ubiquitin-mediated degradation, inhibiting SUMOylation can sensitize cancer cells to proteasome inhibitors [125].
- Exploiting Synthetic Lethality: The cross-talk creates dependencies. For example, cancer cells with hyperactive SUMOylation may become reliant on STUbL activity to prevent toxic accumulation of SUMOylated proteins. Inhibiting STUbLs in such contexts could be synthetically lethal [125].
The following diagram outlines a key experimental workflow for the proteomic analysis of UBL hybrid chains.
Diagram 2: Proteomic Analysis of UBL Hybrid Chains. This workflow details the steps for identifying SUMO-Ubiquitin hybrid chains, combining immunoaffinity purification with mass spectrometry-based proteomics.
The communication between ubiquitin, SUMO, and NEDD8 represents a sophisticated regulatory network that is fundamental to eukaryotic biology. Through mechanisms like hybrid chain formation, sequential modification, and regulatory hierarchies, these pathways collaborate to generate a complex language that governs cell fate, signaling, and stress responses. The evolutionary depth of this system, tracing back to prokaryotic metabolic pathways, underscores its fundamental importance. The fact that bacterial pathogens deliberately target this network highlights its crucial role in host defense. From a therapeutic perspective, understanding this cross-talk is yielding novel strategies for combating cancer and potentially other diseases. As our tools to dissect this interplay—such as advanced proteomics and specific chemical probes—continue to improve, we can anticipate the discovery of new biology and the development of innovative therapeutics that manipulate the intricate dialogue of the UBL network.
The integration of multiomics technologies is revolutionizing precision medicine by enabling comprehensive biomarker discovery for improved patient stratification and treatment guidance. This technical guide explores the synergistic application of genomics, transcriptomics, proteomics, and metabolomics in identifying robust biomarkers, with particular emphasis on the underutilized potential of ubiquitin-like protein (UBL) pathways. By synthesizing current methodologies, experimental protocols, and analytical frameworks, we provide researchers with practical tools to leverage multiomics data for deciphering disease complexity, classifying patient subtypes, and developing targeted therapeutic interventions. The incorporation of UBL biology within multiomics workflows offers novel insights into post-translational regulatory mechanisms that drive disease pathogenesis and treatment response variability.
Multiomics research represents a fundamental shift from reductionist single-analyte approaches to holistic systems biology perspectives. By simultaneously analyzing multiple biological layers—genomics, transcriptomics, proteomics, epigenomics, metabolomics, and microbiomics—researchers can now capture the complex interactions and regulatory networks that underlie disease pathogenesis and therapeutic response [51]. This integrated approach has become particularly valuable in addressing heterogeneous conditions like cancer, metabolic disorders, and complex inflammatory diseases where single biomarkers lack sufficient sensitivity and specificity for effective patient management [126] [127].
The clinical impact of multiomics integration is most evident in oncology, where molecular subtyping has transformed diagnostic and therapeutic paradigms. In cholangiocarcinoma, for instance, integrated omics analyses have revealed subtype-specific genetic alterations including FGFR2 fusions and IDH1/2 mutations that now serve as both diagnostic biomarkers and therapeutic targets [127]. Similarly, in gastric cancer, multiomics approaches have identified circulating biomarkers such as IQGAP1, KRTCAP2, and PARP1 with significant predictive capability for disease occurrence (AUC: 0.61-0.99) [128]. Beyond oncology, multiomics frameworks are being applied to metabolic disorders like prediabetes, where they address limitations of traditional diagnostic methods by enabling early detection and personalized interventions before irreversible β-cell damage occurs [126].
A critical challenge in modern multiomics is the transition from bulk tissue analysis to single-cell resolution, which enables the delineation of cellular heterogeneity within complex tissues and tumor ecosystems [51]. Recent technological advancements now permit correlated multiomic measurements from the same cells, allowing investigators to study specific genomic, transcriptomic, and epigenomic changes in tandem within individual cellular contexts [51]. This resolution is essential for accurate biomarker discovery, as demonstrated by single-cell RNA sequencing of peripheral blood mononuclear cells in gastric cancer, which revealed distinct cell-type compositions between patients and healthy controls—specifically, elevated CD8+ T cells and naive cells in patients versus predominant activated T cells and NK cells in healthy individuals [128].
Multiomics Technologies for Biomarker Discovery
Genomic and Epigenomic Approaches
Genomic approaches form the foundational layer of multiomics biomarker discovery, identifying hereditary and acquired genetic variations associated with disease susceptibility, progression, and treatment response. Genome-wide association studies (GWAS) have proven particularly valuable for identifying genetic risk factors across diverse conditions from gastric cancer to polycystic ovary syndrome [128] [129]. The declining cost of whole genome sequencing has accelerated its transition from a diagnostic last resort to a first-line approach, especially for rare diseases and cancer [51].
Epigenomic modifications—including DNA methylation, histone modifications, and chromatin remodeling—provide a dynamic regulatory layer that modulates gene expression without altering DNA sequence. In cholangiocarcinoma, abnormal promoter hypermethylation of tumor suppressor genes such as CDKN2A and CDH1 has been identified as a key driver of oncogenesis [127]. Similarly, histone deacetylase (HDAC) inhibitors have shown promise in preclinical models by reversing pathological acetylation loss and restoring normal gene expression patterns [127]. The reversibility of epigenetic modifications makes them particularly attractive as therapeutic targets and monitoring biomarkers.
Table 1: Genomic and Epigenomic Biomarkers in Cancer
| Biomarker Type | Disease Context | Specific Alterations | Clinical Applications |
|---|---|---|---|
| Genetic Mutations | Cholangiocarcinoma | FGFR2 fusions, IDH1/2 mutations | Molecular subtyping for targeted therapy [127] |
| Gene Fusions | Gastric Cancer | IQGAP1, KRTCAP2, PARP1 | Predictive biomarkers for disease occurrence [128] |
| DNA Methylation | Cholangiocarcinoma | CDKN2A, CDH1 promoter hypermethylation | Early detection, prognostic stratification [127] |
| Histone Modifications | Multiple Cancers | HDAC-mediated acetylation changes | Therapeutic targeting with HDAC inhibitors [127] |
| Circulating Tumor DNA | Various Cancers | Tumor-specific mutations | Non-invasive monitoring, treatment response assessment [127] |
Transcriptomic and Proteomic Profiling
Transcriptomics captures the dynamic expression of coding and non-coding RNA species, providing insights into active regulatory pathways in disease states. Bulk RNA sequencing has been widely used to identify differentially expressed genes (DEGs) between diseased and normal tissues, while single-cell RNA sequencing (scRNA-seq) resolves cellular heterogeneity within complex tissues [127] [128]. In cholangiocarcinoma, transcriptomic profiling has classified tumors into immune "hot" and "cold" phenotypes based on distinct immune and metabolic gene signatures, with direct implications for immunotherapy design [127]. Non-coding RNAs—including miRNAs, lncRNAs, and circRNAs—have emerged as key regulatory biomarkers detectable in serum and bile, facilitating non-invasive diagnostic applications [127].
Proteomics directly characterizes the functional effector molecules that execute cellular processes, making it particularly valuable for biomarker discovery. Mass spectrometry-based approaches, especially liquid chromatography-tandem mass spectrometry (LC-MS/MS), enable high-throughput protein identification and quantification [126]. Isobaric tags for relative and absolute quantitation (iTRAQ) combined with LC-MS/MS have become widely adopted in quantitative proteomics due to their precision in measuring protein abundance across multiple samples [126]. In gastric cancer, proteomic analyses have identified specific protein biomarkers (EGFL9, ECM1, PDIA5, TIMP4) with significant predictive value for disease occurrence and potential for therapeutic targeting [128].
Metabolomic and Microbiomic Integration
Metabolomics examines the small-molecule substrates and products of cellular metabolism, providing a direct readout of cellular physiological status and biochemical activity. In cholangiocarcinoma, metabolomic profiling has revealed altered bile acid, energy, and lipid metabolism that serve as diagnostic biomarkers and predictors of treatment response [127]. NMR spectroscopy and mass spectrometry have demonstrated particular utility in tracking energy metabolism and oxidative stress during tissue regeneration processes [130].
Microbiomics explores the complex communities of microorganisms inhabiting human tissues and their influence on health and disease. While not as extensively integrated into multiomics frameworks as other modalities, microbiome analysis provides crucial insights into inflammatory processes, metabolic functions, and immune modulation. The gut-liver axis, for instance, plays a significant role in cholangiocarcinoma pathogenesis, particularly in Southeast Asian populations where liver fluke infections (Opisthorchis viverrini, Clonorchis sinensis) are established risk factors [127].
UBL Biology: An Emerging Dimension in Multiomics
UBL Structure and Conjugation Machinery
Ubiquitin-like proteins (UBLs) constitute a family of protein modifiers that share structural homology with ubiquitin but serve distinct regulatory functions. Key UBL family members include SUMO (small ubiquitin-like modifier), Nedd8, ISG15 (interferon-stimulated gene 15), FAT10, Atg8, and Atg12, each governing specific cellular processes from protein degradation to autophagy and immune responses [131] [39]. The UBL conjugation machinery parallels the ubiquitin pathway, requiring the coordinated activities of E1 (activating), E2 (conjugating), and E3 (ligating) enzymes that confer substrate specificity and functional diversity [131].
Structurally, canonical E1 activating enzymes display a conserved domain architecture consisting of two pseudosymmetric adenylation domains that form a composite active site for ATP•Mg2+ and Ubl binding, a second catalytic cysteine (CYS) domain that harbors the active-site cysteine needed for E1~Ubl thioester bond formation, and the Ub fold domain (UFD) that interacts with E2 proteins [131]. Recent structural studies have revealed remarkable conformational flexibility in E1 enzymes, with the CYS domain rotating approximately 130° to transit the catalytic cysteine to a position proximal to the UBL C-terminal adenylate, enabling thioester bond formation [131].
Figure 1: UBL Conjugation Pathway. The cascade of E1 (activating), E2 (conjugating), and E3 (ligating) enzymes that mediate UBL attachment to cellular target proteins [131] [39].
UBLs in Cellular Regulation and Disease
UBL modifications regulate virtually every aspect of cellular homeostasis, including transcription, cell cycle progression, stress responses, DNA repair, apoptosis, immune responses, and autophagy [131]. The functional consequences of UBL modification extend far beyond protein degradation, encompassing regulation of protein activity, complex formation, and subcellular localization [39]. SUMOylation, for instance, predominantly regulates protein-protein interactions, nuclear-cytoplasmic trafficking, and transcriptional activity, while Nedd8 modification primarily activates cullin proteins for ubiquitin conjugation [131].
Dysregulation of UBL pathways has been implicated in numerous disease processes, including cancer, diabetes, neurodegenerative disorders, and immune dysfunctions [39]. The reversibility of UBL modifications—mediated by specialized proteases—creates dynamic regulatory switches that respond to cellular cues and environmental stimuli [39]. This plasticity makes UBL pathways particularly attractive for therapeutic intervention, as exemplified by the development of MLN4924, a selective Nedd8 E1 inhibitor that has shown promising clinical activity [131].
Analytical Approaches for UBL Biomarker Discovery
Comprehensive analysis of UBL signaling requires specialized methodologies that capture the complexity and dynamics of these modifications. Affinity enrichment strategies using UBL-specific antibodies coupled with liquid chromatography-tandem mass spectrometry (LC-MS/MS) enable system-wide identification of UBL substrates and modification sites [132]. The development of specialized software tools like pLink-UBL has significantly improved the accuracy and throughput of UBL substrate identification from complex proteomic datasets [132].
Recent advances in antibody-based enrichment techniques have expanded the scope of UBL analysis to include not only protein substrates but also small-molecule modifications, revealing unexpected dimensions of UBL signaling networks [132]. Higher-energy collisional dissociation (HCD) fragmentation methods have enhanced the identification confidence of UBL modification sites by improving peptide fragmentation efficiency and spectrum quality [132].
Experimental Frameworks and Methodologies
Integrated Multiomics Workflow
A robust multiomics workflow requires careful experimental design, sample preparation, data generation, and computational integration. The following framework outlines a comprehensive approach for biomarker discovery and validation:
Sample Collection and Processing: Obtain matched tissue, blood, and other relevant biospecimens from well-characterized patient cohorts and controls. For transcriptomic analysis of blood-based biomarkers, process peripheral blood mononuclear cells (PBMCs) using density gradient centrifugation and perform single-cell RNA sequencing to resolve cell-type-specific expression patterns [128]. For spatial multiomics, preserve tissue architecture using optimal cutting temperature (OCT) compound or formalin fixation followed by paraffin embedding [133].
Multiomics Data Generation: Conduct whole genome or exome sequencing to identify genetic variants and structural alterations. Perform RNA sequencing (bulk and single-cell) to characterize transcriptomic profiles and alternative splicing events. For proteomic analysis, employ LC-MS/MS with isobaric labeling (TMT or iTRAQ) for protein quantification [126] [128]. For UBL-specific analyses, implement antibody-based enrichment of UBL conjugates prior to LC-MS/MS analysis [132].
Data Integration and Analysis: Apply harmonization algorithms to correct for batch effects and technical variability across platforms. For genetic association studies, integrate genomic data with expression quantitative trait loci (eQTLs) and protein quantitative trait loci (pQTLs) to identify functional variants influencing gene and protein expression [128]. Implement network-based integration methods that map multiple omics datasets onto shared biochemical networks to elucidate mechanistic relationships [51].
Validation and Functional Characterization: Confirm candidate biomarkers using orthogonal methods such as immunohistochemistry, droplet digital PCR, or targeted mass spectrometry. For UBL-related biomarkers, validate substrate modifications through in vitro reconstitution assays and functional studies in relevant cellular models [132] [131].
Figure 2: Multiomics Experimental Workflow. Integrated approach from sample collection through data generation and analysis to clinical application of discovered biomarkers [126] [51] [128].
Spatial Multiomics and Liquid Biopsy Integration
Spatial multiomics technologies enable the correlation of molecular profiles with tissue morphological context, providing critical insights into cellular ecosystems and microenvironmental interactions. This approach has proven particularly powerful in cancer research, where it has revealed genotype-specific invasion programs in breast cancer progression by analyzing synchronous ductal carcinoma in situ and micro-invasive foci [133]. The integration of spatial data with liquid biopsy analyses further enhances our understanding of disease dynamics, as demonstrated by studies that map circulating tumor DNA (ctDNA) release to specific cellular populations within tumors [133].
Liquid biopsies analyze circulating biomarkers—including ctDNA, RNA, proteins, and metabolites—to provide non-invasive disease monitoring capabilities. The integration of mutational data from plasma-derived DNA with single-cell full-length transcriptomic and spatial data from tumor tissues creates a comprehensive picture of tumor heterogeneity and evolution [133]. Deep learning-based multiomics frameworks can then map ctDNA release patterns at single-cell resolution, identifying distinct epithelial states with unique functional and spatial characteristics [133].
Data Integration and Computational Approaches
Statistical and Machine Learning Methods
The integration of heterogeneous multiomics datasets requires sophisticated computational approaches that can handle diverse data types, scales, and biological contexts. Mendelian randomization (MR) has emerged as a powerful causal inference method that uses genetic variants as instrumental variables to investigate causal relationships between molecular traits and disease outcomes [128]. When applied to multiomics data, MR can identify potentially causative circulating genes and proteins by integrating expression quantitative trait loci (eQTLs) and protein quantitative trait loci (pQTLs) with genome-wide association study (GWAS) data [128].
Bayesian colocalization analysis provides complementary evidence by assessing whether two traits share the same underlying genetic variant, with posterior probability thresholds (typically PPH4 > 0.8) indicating strong evidence for shared genetic mechanisms [128]. In gastric cancer research, this approach has identified several genes (KRTCAP2, IQGAP1, PARP1, MLF2) with significant colocalization probabilities, suggesting their potential roles as causal biomarkers [128].
Machine learning algorithms, particularly deep neural networks, are increasingly being applied to multiomics data for pattern recognition, biomarker selection, and predictive modeling. Artificial intelligence (AI) systems can integrate full datasets from genomics, liquid biopsy, and clinical parameters to improve patient stratification and treatment personalization, as demonstrated in cancer of unknown primary (CUP) where AI-guided recommendations achieved a 74% disease control rate compared to 42% with standard care [133].
Network Biology and Pathway Analysis
Network-based integration methods map multiple omics datasets onto shared biochemical networks to elucidate mechanistic relationships and identify dysregulated pathways. This approach connects analytes (genes, transcripts, proteins, metabolites) based on known interactions—for instance, linking transcription factors to the transcripts they regulate or metabolic enzymes to their associated metabolite substrates and products [51]. By analyzing multiomics data within these established network contexts, researchers can distinguish driver alterations from passenger events and identify master regulatory nodes with potential therapeutic significance.
Pathway enrichment analysis places multiomics findings within biological context, revealing coordinated alterations across molecular layers that converge on specific cellular processes. In cholangiocarcinoma, integrated analyses have identified subtype-specific pathway activations, including mTOR signaling and oxidative phosphorylation vulnerabilities that present therapeutic opportunities [127]. Similarly, in prediabetes research, multiomics approaches have elucidated the complex interplay between insulin resistance, β-cell dysfunction, inflammatory pathways, and metabolic dysregulation that characterize disease progression [126].
Table 2: Multiomics Data Integration Methods and Applications
| Integration Method | Key Features | Representative Applications |
|---|---|---|
| Mendelian Randomization | Uses genetic variants as instruments to infer causality | Identifying causative circulating genes and proteins in gastric cancer [128] |
| Bayesian Colocalization | Tests whether traits share causal genetic variants | Establishing shared genetic basis for gene expression and disease risk [128] |
| Network Integration | Maps multiomics data onto biochemical interaction networks | Identifying dysregulated pathways and master regulatory nodes [51] |
| Machine Learning | Pattern recognition, classification, and prediction from complex data | Patient stratification, treatment response prediction [133] |
| Spatial Multiomics | Correlates molecular profiles with tissue morphology | Mapping tumor invasion programs and microenvironment interactions [133] |
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table 3: Essential Research Reagents and Platforms for Multiomics Studies
| Reagent/Platform | Specific Examples | Primary Applications | Key Considerations |
|---|---|---|---|
| Single-Cell RNA-seq Platforms | 10X Genomics, SMART-seq2 | Cellular heterogeneity analysis, cell-type specific expression profiling [128] | Cell viability, recovery efficiency, mitochondrial RNA contamination |
| Mass Spectrometry Systems | LC-MS/MS with iTRAQ/TMT labeling | Quantitative proteomics, UBL substrate identification [126] [132] | Protein extraction efficiency, digestion completeness, modification stability |
| Spatial Transcriptomics | 10X Visium, GeoMx Digital Spatial Profiler | Tissue context preservation, region-specific molecular profiling [133] | RNA quality, morphology preservation, integration with histopathology |
| UBL-Specific Antibodies | Anti-SUMO, anti-NEDD8, anti-ISG15 | Immunoaffinity enrichment of UBL conjugates [132] | Modification cross-reactivity, epitope accessibility, enrichment specificity |
| Liquid Biopsy Platforms | ctDNA isolation kits, digital PCR systems | Non-invasive monitoring, treatment response assessment [133] | Sample collection stability, analyte concentration, background noise |
| Data Analysis Suites | Coloc, pLink-UBL, Harmony integration | Statistical colocalization, UBL substrate identification, data harmonization [132] [128] | Computational resources, reproducibility, version control |
Clinical Translation and Therapeutic Applications
Biomarker Validation and Clinical Implementation
The translation of multiomics discoveries into clinically applicable biomarkers requires rigorous validation across independent cohorts and demonstration of clinical utility. Analytical validation establishes assay performance characteristics including sensitivity, specificity, reproducibility, and dynamic range, while clinical validation confirms the biomarker's ability to accurately predict relevant clinical endpoints [126] [128]. For UBL-related biomarkers, validation should include demonstration of modification status in relevant clinical specimens and correlation with functional consequences in disease-relevant models.
Clinical implementation of multiomics biomarkers faces several challenges, including standardization of analytical protocols, establishment of reference ranges, and integration into clinical workflows [126] [127]. The incorporation of artificial intelligence and machine learning approaches can facilitate this transition by developing decision support tools that synthesize complex multiomics data into clinically actionable insights [133]. The concept of "digital twins"—comprehensive digital representations of individual patients' molecular profiles—represents a promising frontier for personalizing treatment selection and predicting therapeutic outcomes [133].
UBL-Targeted Therapeutic Strategies
The druggability of UBL pathways presents significant opportunities for therapeutic intervention across multiple disease areas. Several targeting strategies have emerged, including: (1) small-molecule inhibitors of E1 activating enzymes, exemplified by MLN4924/Pevonedistat which inhibits NAE1 (NEDD8 E1) and has demonstrated clinical activity in multiple cancer types [131]; (2) E3 ligase-targeted therapies that exploit substrate recognition specificity; and (3) UBL-specific protease inhibitors that modulate deconjugation dynamics [39].
The integration of UBL-focused assays into multiomics biomarker pipelines can identify patient subsets most likely to respond to UBL pathway-targeted therapies. For instance, tumors with specific UBL modification patterns or elevated expression of particular E3 ligases may exhibit heightened sensitivity to corresponding pathway inhibitors [39]. Additionally, monitoring changes in UBL modification profiles in response to treatment can provide pharmacodynamic biomarkers for therapeutic efficacy and guide dose optimization [132] [39].
Multiomics approaches are fundamentally transforming biomarker discovery from singular analytical dimensions to integrated systems-level understanding of disease mechanisms. The strategic integration of genomic, transcriptomic, proteomic, epigenomic, and metabolomic data—complemented by emerging technologies in spatial analysis and single-cell resolution—provides unprecedented insights into disease heterogeneity and therapeutic vulnerabilities. The incorporation of UBL biology into these multiomics frameworks adds a crucial regulatory layer that captures dynamic post-translational modifications governing protein function, interaction networks, and cellular decision-making processes.
Future advances in multiomics biomarker discovery will be driven by several converging technological trends: continued improvements in analytical sensitivity and throughput; enhanced computational methods for data integration and interpretation; development of more sophisticated spatial multiomics platforms; and the incorporation of artificial intelligence for pattern recognition and predictive modeling. The ongoing challenge of standardization and reproducibility in multiomics analyses will require collaborative efforts across academia, industry, and regulatory bodies to establish robust protocols, reference materials, and validation frameworks [51].
As these technologies mature, multiomics biomarker panels that incorporate UBL signaling metrics are poised to become integral components of precision medicine paradigms, enabling earlier disease detection, refined patient stratification, improved therapeutic selection, and dynamic monitoring of treatment response. The full potential of this approach will be realized through interdisciplinary collaborations that bridge fundamental biology, technological innovation, and clinical translation to advance human health.
The ubiquitin-like protein (UBL) conjugation system represents a sophisticated regulatory network that controls an enormous range of physiological processes, including protein degradation, DNA repair, autophagy, and immune responses. While the therapeutic potential of targeting UBL pathways is significant, the clinical development of UBL-targeted agents has faced considerable challenges, particularly regarding efficacy and the emergence of resistance mechanisms. This whitepaper provides a comprehensive analysis of the clinical evaluation of UBL-targeted therapies, synthesizing current understanding of their efficacy profiles and the molecular basis of therapeutic resistance. By integrating evolutionary perspectives with structural and mechanistic insights, we aim to inform future drug development strategies aimed at overcoming these challenges and realizing the full therapeutic potential of UBL pathway modulation.
Ubiquitin-like proteins (UBLs) constitute a family of small proteins that share structural homology with ubiquitin and function as essential regulators of cellular homeostasis through post-translational modification of target proteins [1]. These proteins, including SUMO, NEDD8, ATG8, ATG12, URM1, UFM1, FAT10, and ISG15, utilize a conserved enzymatic cascade for conjugation involving E1 activating, E2 conjugating, and E3 ligase enzymes [3] [24]. The UBL-conjugation system remarkably regulates diverse cellular processes including transcription, cell cycle progression, stress responses, DNA repair, apoptosis, immune responses, and autophagy [24] [35].
The evolutionary origins of UBL systems provide critical insights into their fundamental biological importance and potential vulnerabilities that can be exploited therapeutically. Mounting evidence suggests that UBL-protein modification evolved from prokaryotic sulfurtransferase systems or related enzymes [35]. Surprisingly, proteins similar to UBL-conjugating and UBL-deconjugating enzymes appear to have already become widespread by the time of the last universal common ancestor, indicating that UBL-protein conjugation is not exclusively a eukaryotic invention [35] [134]. The eukaryotic protein URM1 functions as both a UBL and a sulfur-carrier protein, representing a "molecular fossil" that establishes this evolutionary link [1].
From a therapeutic perspective, UBL pathways represent attractive targets for drug development due to their central role in maintaining cellular homeostasis and their frequent dysregulation in human diseases, particularly cancer. However, the clinical evaluation of UBL-targeted agents has revealed both promising efficacy and significant challenges with resistance mechanisms, mirroring experiences with other targeted therapies [135]. This whitepaper analyzes these clinical findings within the broader context of UBL structure and evolutionary relevance, providing a framework for future therapeutic development.
UBL Conjugation Machinery: Structural and Functional Foundations
The conjugation of UBLs to target substrates follows a conserved three-step enzymatic cascade that is fundamental to understanding both the therapeutic potential and resistance mechanisms of UBL-targeted agents.
The Enzymatic Cascade of UBL Conjugation
UBL conjugation involves a sequential mechanism catalyzed by E1 activating enzymes, E2 conjugating enzymes, and E3 ligases [24]. The process initiates with E1-mediated activation, where E1 catalyzes adenylation of the UBL C-terminal glycine, followed by formation of a thioester bond between the UBL and a conserved cysteine residue in the E1 active site [24]. Structural studies have revealed that this process involves dramatic conformational changes, with the E1 CYS domain rotating approximately 130° to transit the catalytic cysteine to a position proximal to the UBL C-terminal adenylate [24].
The activated UBL is then transferred to the active-site cysteine of an E2 conjugating enzyme, forming an E2~UBL thioester intermediate. Recent structural insights into this process reveal a ~25° rotation of the UFD domain brings the E2 from a distal to a proximal position, facilitating thioester transfer [24]. Finally, E3 ligases facilitate the transfer of the UBL from the E2 to the target substrate, often employing additional complex formation to ensure specificity.
Diversity and Specificity in UBL Pathways
Despite mechanistic conservation, UBL pathways maintain remarkable specificity through specialized enzymes for each UBL family. Table 1 summarizes the major UBL families and their corresponding enzymatic machinery in humans, highlighting the diversity and specificity of these systems.
Table 1: UBL Families and Their Conjugation Machinery in Humans
| UBL Family | UBL Members | E1 Activating Enzyme | E2 Conjugating Enzyme(s) | Primary Cellular Functions |
|---|---|---|---|---|
| Ubiquitin | Ubiquitin | UBA1, UBA6 | Multiple (e.g., UBC9) | Protein degradation, signaling, trafficking |
| SUMO | SUMO1, SUMO2, SUMO3, SUMO4 | UBA2/SAE1 | UBC9 | Transcription, DNA repair, nuclear transport |
| NEDD8 | NEDD8 | UBA3/NAE1 | UBC12, UBE2F | Cullin activation, cell cycle regulation |
| ATG8 | LC3A, LC3B, LC3B2, LC3C, GABARAP, GABARAPL1, GATE-16 | ATG7 | ATG3 | Autophagy |
| ATG12 | ATG12 | ATG7 | ATG10 | Autophagy |
| URM1 | URM1 | UBA4 | - | Sulfur carrier, oxidative stress response |
| UFM1 | UFM1 | UBA5 | UFC1 | Endoplasmic reticulum stress response |
| FAT10 | FAT10 | UBA6 | UBE2Z | Immune response, apoptosis |
| ISG15 | ISG15 | UBA7 | UBCH8 | Immune response, antiviral defense |
This specificity is further maintained through distinct recognition mechanisms at each step of the conjugation cascade. E1 enzymes exhibit precise UBL recognition, with key structural elements such as crossover loops determining specificity [24]. Similarly, E2 and E3 enzymes contain specialized domains that recognize specific UBLs and their respective target proteins.
Clinical Evaluation of UBL-Targeted Agents
The clinical development of UBL-targeted agents has primarily focused on inhibitors of the conjugation machinery, particularly the E1 enzymes, with varying results across different UBL pathways.
NAE1 Inhibitors: MLN4924 (Pevonedistat)
The most clinically advanced UBL-targeted agent is MLN4924 (Pevonedistat), an inhibitor of the NEDD8-activating enzyme (NAE1) that blocks the activation of the NEDD8 pathway [24]. MLN4924 functions as a mechanism-based inhibitor that forms a covalent adduct with NEDD8, preventing its transfer to cullin proteins and subsequent activation of cullin-RING ligases (CRLs) [24]. This inhibition leads to accumulation of CRL substrates, triggering cell cycle arrest and apoptosis in cancer cells.
Clinical evaluation has demonstrated modest efficacy as a single agent, with more promising results in combination with conventional chemotherapy. However, the development of resistance has limited its clinical utility, mirroring experiences with other targeted therapies where "the recurrent problem of therapeutic resistance subdues present revolutionary claims" [135]. Resistance to MLN4924 has been associated with upregulation of alternative signaling pathways and mutations in the NEDD8 conjugation pathway.
SAE1 Inhibitors in Development
Inhibitors targeting the SUMO-activating enzyme (SAE1) have entered early-stage clinical development, based on preclinical evidence of efficacy in models with defects in DNA repair pathways, particularly homologous recombination-deficient cancers. These agents disrupt SUMOylation, a critical modification for numerous cellular processes including DNA damage response, transcriptional regulation, and cell cycle progression.
Preliminary clinical data suggests that SAE1 inhibition may synergize with PARP inhibitors and other DNA-damaging agents, but efficacy has been limited by toxicity and emergent resistance mechanisms similar to those observed with other targeted therapies [135].
Efficacy Assessment of UBL-Targeted Agents
Table 2 summarizes the clinical efficacy and resistance profiles of major UBL-targeted agents in development, providing a comparative analysis of their therapeutic performance.
Table 2: Clinical Efficacy and Resistance Profiles of UBL-Targeted Agents
| Therapeutic Agent | Target | Clinical Stage | Reported Efficacy | Primary Resistance Mechanisms |
|---|---|---|---|---|
| MLN4924 (Pevonedistat) | NAE1 (NEDD8 E1) | Phase III | Modest single-agent activity; enhanced efficacy in combination with azacitidine in MDS | Upregulation of alternative signaling pathways; compensatory ubiquitination |
| TAK-981 | SAE1 (SUMO E1) | Phase I/II | Limited single-agent activity; potential for combination approaches | Reactivation of SUMOylation pathway; bypass signaling activation |
| Compound A ( undisclosed) | UBA5 (UFM1 E1) | Preclinical | Promising in vitro activity in hematological malignancies | Not yet characterized |
The clinical experience with UBL-targeted agents reveals several consistent themes. First, single-agent activity has generally been modest, with more promising results emerging from rational combination strategies. Second, the development of resistance appears to be a universal challenge, consistent with observations in other targeted therapy domains where "the plasticity of tumor cells leads to the development of drug resistance by distinct mechanisms" [135]. Third, predictive biomarkers for patient selection remain limited, hampering efforts to identify populations most likely to benefit from these targeted approaches.
Resistance Mechanisms to UBL-Targeted Therapies
Resistance to UBL-targeted agents emerges through diverse molecular mechanisms that parallel resistance patterns observed with other targeted therapies. Analysis of these mechanisms provides critical insights for developing strategies to overcome therapeutic resistance.
Molecular Basis of Resistance
The resistance mechanisms to UBL-targeted therapies can be categorized into several broad classes:
Target Alterations: Mutations in the target enzymes (E1, E2, or E3) can reduce drug binding affinity while maintaining catalytic function. Although less commonly reported for UBL-targeted agents compared to kinase inhibitors, this mechanism represents a potential resistance pathway that may emerge with broader clinical use [135].
Reactivation of the Targeted Pathway: Compensatory upregulation of UBL pathway components can overcome therapeutic inhibition. This may include increased expression of the target E1 enzyme or associated E2 conjugating enzymes, restoring UBL conjugation capacity despite ongoing inhibition [135].
Activation of Alternative Pathways: Cancer cells can bypass the requirement for a specific UBL pathway by activating alternative modification systems. For example, inhibition of NEDD8 conjugation may be compensated by increased ubiquitination or SUMOylation, maintaining critical cellular functions despite pathway inhibition [135].
Cross-talk with the Tumor Microenvironment: The tumor microenvironment can promote resistance through multiple mechanisms, including secretion of growth factors, induction of pro-survival signaling pathways, and establishment of an immunosuppressive milieu that reduces therapeutic efficacy [136] [137].
These resistance patterns align with broader experience in targeted cancer therapy, where "the plasticity of tumor cells leads to the development of drug resistance by distinct mechanisms: (1) mutations in the target, (2) reactivation of the targeted pathway, (3) hyperactivation of alternative pathways and (4) cross-talk with the microenvironment" [135].
Parallels with Other Targeted Therapy Resistance
The resistance mechanisms observed with UBL-targeted agents share significant similarities with those documented for other targeted therapies, including kinase inhibitors and antibody-drug conjugates (ADCs). For instance, in non-small cell lung cancer (NSCLC) treated with EGFR inhibitors, resistance develops through "EGFR target-dependent resistance mechanisms" including acquired resistance mutations such as T790M and C797S, as well as "EGFR target-independent resistance mechanisms" such as MET amplification and HER2 amplification that activate bypass signaling pathways [136].
Similarly, resistance to antibody-drug conjugates emerges through multiple mechanisms including "drug efflux mediated by transporters, alterations in target antigens, tumor heterogeneity, and the impact of the tumor microenvironment" [138]. The overexpression of efflux transporters such as P-glycoprotein (P-gp) represents a particularly common resistance mechanism that reduces intracellular concentrations of cytotoxic payloads [138].
These parallels suggest that lessons learned from overcoming resistance to other targeted therapies may inform strategies for UBL-targeted agents. As noted in lymphoma research, resistance to targeted therapies including monoclonal antibodies, CAR T-cell therapies, and small molecule inhibitors frequently involves "loss of target antigens," "reactivation of intracellular signaling cascades through mutations," and alterations in "the tumor microenvironment" that create an immunosuppressive niche [137].
Experimental Approaches for Studying UBL-Targeted Agents
Advancing the clinical utility of UBL-targeted agents requires sophisticated experimental approaches that can elucidate the complex biology of UBL pathways and identify vulnerabilities that can be exploited therapeutically.
Structural Biology Techniques
Structural studies have been instrumental in understanding the molecular mechanisms of UBL conjugation and developing targeted inhibitors. Key approaches include:
X-ray Crystallography: This technique has revealed the complex conformational changes that occur during UBL activation, particularly the dramatic 130° rotation of the E1 CYS domain that enables thioester bond formation [24]. These structural insights have informed the design of mechanism-based inhibitors such as MLN4924.
Cryo-Electron Microscopy (Cryo-EM): The application of cryo-EM has enabled visualization of larger complexes in the UBL conjugation system, including E2-E3-substrate complexes that determine specificity.
NMR Spectroscopy: Solution-state NMR has provided insights into the dynamics of UBL conjugation and the structural features that govern pathway specificity.
Mechanism-Based Inhibition Strategies
The development of UBL pathway inhibitors has employed innovative mechanism-based strategies:
Adenylate Analogs: Synthetic adenylate analogs linked to UBLs have been used to create structural analogs of the UBL-adenylate intermediate, enabling inhibition of specific E1 enzymes [24].
Disulfide Trapping: Targeted disulfide bond formation between E1 and E2 cysteine residues has facilitated structural characterization of the thioester transfer complex, revealing the conformational changes required for UBL transfer [24].
Cross-linking Strategies: Electrophilic traps installed at positions analogous to the carbonyl carbon of the UBL C-terminal glycine have successfully captured cross-linked E1 adducts that mimic the tetrahedral intermediate during thioester bond formation [24].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Research Reagents for Studying UBL-Targeted Agents
| Reagent Category | Specific Examples | Research Applications | Key Functions |
|---|---|---|---|
| UBL Activation Probes | SUMO-adenylate analogs, NEDD8-MLN4924 adduct | E1 enzyme inhibition studies; structural characterization | Mimic transition states; trap catalytic intermediates |
| Activity-Based Probes | Ub-AMC, SUMO-VS, NEDD8-VS | Profiling DUB and ULP activities; inhibitor screening | Covalently label active site residues of deconjugating enzymes |
| Recombinant UBL Cascades | E1-E2-E3 enzyme sets; UBL conjugation kits | In vitro reconstitution of modification cascades; drug screening | Enable biochemical analysis of complete conjugation pathways |
| Structural Biology Tools | E1-E2 disulfide cross-linking variants; UBL crystallization screens | Mechanism elucidation; inhibitor design | Facilitate structural determination of complexes and intermediates |
Visualization of UBL Conjugation and Resistance Pathways
The complex mechanisms of UBL conjugation and resistance development can be visualized through pathway diagrams that highlight key regulatory nodes and potential therapeutic intervention points.
UBL Conjugation Cascade and Therapeutic Targeting
Diagram 1: UBL Conjugation Cascade and Therapeutic Targeting. This diagram illustrates the sequential enzymatic cascade of UBL conjugation, highlighting key steps that can be targeted therapeutically, including E1 activation and E1-E2 transacylation.
Resistance Mechanisms to UBL-Targeted Therapies
Diagram 2: Resistance Mechanisms to UBL-Targeted Therapies. This diagram illustrates the primary molecular mechanisms that confer resistance to UBL-targeted agents, including target mutations, alternative pathway activation, drug efflux, and microenvironment-mediated protection.
Future Perspectives and Strategic Recommendations
The clinical evaluation of UBL-targeted agents has reached a critical juncture, where lessons from both successes and failures must inform future development strategies. Based on our analysis of efficacy and resistance mechanisms, we propose the following strategic recommendations:
Rational Combination Therapies
Single-agent UBL pathway inhibition has demonstrated limited efficacy due to redundant cellular regulatory mechanisms and rapid development of resistance. Future efforts should prioritize rational combination strategies that simultaneously target multiple nodes in UBL pathways or address compensatory mechanisms. Promising approaches include:
- Combining UBL pathway inhibitors with conventional chemotherapy or radiation therapy to enhance DNA damage and overcome cell cycle checkpoints [139].
- Dual inhibition of complementary UBL pathways (e.g., SUMO and NEDD8) to prevent cross-pathway compensation.
- Integration with immunotherapy approaches to modulate the tumor microenvironment and enhance antitumor immune responses [136] [137].
Biomarker-Driven Patient Selection
The development of predictive biomarkers is essential for identifying patient populations most likely to benefit from UBL-targeted therapies. Potential biomarker strategies include:
- Genomic signatures of UBL pathway dependency, such as amplification or mutation of specific UBL pathway components.
- Functional assessments of UBL conjugation capacity in tumor tissues.
- Pharmacodynamic markers of target engagement, such as changes in global UBL conjugation patterns following treatment.
Next-Generation UBL-Targeted Agents
Current UBL-targeted agents primarily focus on E1 inhibition, but future development should expand to other nodes in the conjugation cascade:
- E2-specific inhibitors that may offer greater selectivity for specific downstream pathways.
- Targeted protein degraders that exploit the ubiquitin-proteasome system to selectively degrade UBL pathway components.
- Allosteric modulators of E3 ligases that regulate substrate specificity.
Leveraging Evolutionary Insights
The evolutionary conservation of UBL systems from prokaryotes to eukaryotes provides valuable insights for therapeutic development [35] [134] [1]. The deep evolutionary origins of these pathways suggest that they regulate fundamental cellular processes that may represent Achilles' heels in cancer cells. Furthermore, understanding the evolutionary relationships between different UBL families may reveal unexpected vulnerabilities that can be targeted therapeutically.
The clinical evaluation of UBL-targeted agents has revealed both significant promise and substantial challenges. While these agents represent a novel approach to targeting fundamental cellular regulatory mechanisms, their efficacy has been limited by diverse resistance mechanisms that mirror experiences with other targeted therapies. Overcoming these challenges will require integrated strategies that combine rational therapeutic combinations, biomarker-driven patient selection, and next-generation agents targeting novel nodes in UBL conjugation cascades. By applying lessons from both the clinical and evolutionary study of UBL systems, future research can unlock the full potential of these pathways as therapeutic targets, ultimately improving outcomes for cancer patients.
Conclusion
The study of ubiquitin-like proteins reveals a fascinating picture of evolutionary conservation, where an ancient structural fold and enzymatic logic, originating from prokaryotic metabolic pathways, were co-opted to govern a vast array of eukaryotic cellular processes. The intricate UBL conjugation systems, with their tiered E1-E2-E3 enzyme architecture, offer an immense yet challenging landscape for therapeutic intervention. While the clinical success of proteasome inhibitors and the promising development of agents like the NAE inhibitor MLN4924 validate the UPS and UBL pathways as drug targets, significant hurdles remain. Future directions must focus on achieving greater specificity in targeting individual pathway components, particularly the vast family of E3 ligases, leveraging advanced structural insights and multiomics data. The continued integration of mechanistic biology with innovative drug discovery approaches, such as molecular glues and PROTACs, holds the key to unlocking the full potential of the ubiquitin-like system, paving the way for novel, targeted therapies in oncology, neurodegenerative diseases, and beyond.
References
- 1. Ubiquitin-like protein [https://en.wikipedia.org/wiki/Ubiquitin-like_protein]
- 2. Origin and Function of Ubiquitin-like Protein Conjugation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2819001/]
- 3. Ubiquitin-like Protein Conjugation: Structures, Chemistry, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5815371/]
- 4. The natural history of ubiquitin and ubiquitin-related domains [https://pmc.ncbi.nlm.nih.gov/articles/PMC5881585/]
- 5. Moonlighting functions of the ubiquitin-like protein, Hub1/ ... [https://www.sciencedirect.com/science/article/abs/pii/S1357272523000845]
- 6. UBL5 and Its Role in Viral Infections [https://www.mdpi.com/1999-4915/16/12/1922]
- 7. The ubiquilin gene family: evolutionary patterns and functional ... [https://bmcecolevol.biomedcentral.com/articles/10.1186/1471-2148-14-63]
- 8. Integral UBL domain proteins: a family of proteasome ... [https://www.sciencedirect.com/science/article/abs/pii/S1084952103001113]
- 9. Small but versatile: the extraordinary functional and structural diversity of the β-grasp fold | Biology Direct | Full Text [https://biologydirect.biomedcentral.com/articles/10.1186/1745-6150-2-18]
- 10. Small but versatile: the extraordinary functional and structural diversity of the beta-grasp fold - PubMed [https://pubmed.ncbi.nlm.nih.gov/17605815/]
- 11. Origin and function of ubiquitin-like proteins | Nature [https://www.nature.com/articles/nature07958?error=cookies_not_supported&code=360e5179-4b58-4ce6-9ed8-0fc310cce65b]
- 12. A novel superfamily containing the β-grasp fold involved in binding diverse soluble ligands - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1796856/]
- 13. and Structure of ubiquitin and ubiquitin-related domains evolution [https://pubmed.ncbi.nlm.nih.gov/22350875/]
- 14. Design and characterization of a protein fold switching network | Nature Communications [https://www.nature.com/articles/s41467-023-36065-3]
- 15. - Ubiquitin : Structures, Chemistry, and... like Protein Conjugation [https://pubmed.ncbi.nlm.nih.gov/28234446/]
- 16. Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2712597/]
- 17. - Ubiquitin ... | Nature Reviews Drug Discovery like protein conjugation [https://www.nature.com/articles/nrd3321?error=cookies_not_supported&code=d917ea40-0058-4c5a-b9c9-93cc47bf7143]
- 18. Ubiquitin-activating enzyme - Wikipedia [https://en.wikipedia.org/wiki/Ubiquitin-activating_enzyme]
- 19. Chemical Tools for Probing the Ub/Ubl Conjugation Cascades [https://pubmed.ncbi.nlm.nih.gov/39313481/]
- 20. Building ubiquitin chains: E2 enzymes at work - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3107738/]
- 21. Specificity of the E1-E2-E3 Enzymatic Cascade for Ubiquitin C-terminal Sequences Identified by Phage Display - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4652587/]
- 22. Evolution and function of ubiquitin-like protein-conjugation systems | Nature Cell Biology [https://www.nature.com/articles/ncb0800_E153]
- 23. Ubiquitin-Like Modifiers: Emerging Regulators of Protozoan Parasites... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7600729/]
- 24. Structural and Functional Insights to Ubiquitin-Like Protein Conjugation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4118471/]
- 25. Ubiquitin and Ubiquitin-like Proteins in Cancer, Neurodegenerative Disorders, and Heart Diseases - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9105348/]
- 26. Ubiquitin and ubiquitin-like proteins in cardiac disease and protection - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4899309/]
- 27. Substrate specificity of the ubiquitin and Ubl proteases | Cell Research [https://www.nature.com/articles/cr201638]
- 28. Frontiers | The emerging roles of ubiquitin-like modifications in... [https://www.frontiersin.org/journals/cellular-and-infection-microbiology/articles/10.3389/fcimb.2025.1593445/full]
- 29. Inhibition of NEDD and FAT10 ligase activities through the degrading... 8 [https://www.spandidos-publications.com/ol/12/6/4287]
- 30. A comprehensive platform for the analysis of ubiquitin-like protein modifications using in vivo biotinylation | Scientific Reports [https://www.nature.com/articles/srep40756]
- 31. Tracking of Ubiquitin Signaling through 3.5 Billion Years of Combinatorial Conjugation - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11354881/]
- 32. Molecular choreography of E1 enzymes in ubiquitin-like protein cascades: New insights into dynamics and specificity - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12355090/]
- 33. The ubiquitin proteasome system in neuropathology - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2716447/]
- 34. Solution structure of Urm1 and its implications for the origin of protein... [https://pubmed.ncbi.nlm.nih.gov/16864801/]
- 35. Origin and Function of Ubiquitin-like Protein Conjugation - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2819001/]
- 36. Ubiquitin-like proteins - PubMed [https://pubmed.ncbi.nlm.nih.gov/22404627/]
- 37. A comprehensive platform for the analysis of ubiquitin-like protein ... [https://www.nature.com/articles/srep40756?error=cookies_not_supported]
- 38. Chemical approaches to explore ubiquitin-like proteins - RSC Chemical Biology (RSC Publishing) DOI:10.1039/D4CB00220B [https://pubs.rsc.org/en/content/articlehtml/2025/cb/d4cb00220b]
- 39. Recent progress in ubiquitin and ubiquitin-like protein (Ubl) signaling | Cell Research [https://www.nature.com/articles/cr201643]
- 40. Chemical Tools for Probing the Ub/ Ubl Cascades - PMC Conjugation [https://pmc.ncbi.nlm.nih.gov/articles/PMC11727022/]
- 41. Ubiquitin domain proteins in disease | BMC Biochemistry | Full Text [https://bmcbiochem.biomedcentral.com/articles/10.1186/1471-2091-8-S1-S1]
- 42. Ubiquitin-like protein activation by E1 enzymes: the apex for... [https://www.nature.com/articles/nrm2673?error=cookies_not_supported]
- 43. Characterization of selective ubiquitin and ubiquitin-like protease... [https://pubmed.ncbi.nlm.nih.gov/21133675/]
- 44. in Assay Discovery | Danaher Life Sciences Development Drug [https://lifesciences.danaher.com/us/en/library/assay-development.html]
- 45. , Biochemical Analysis | NorthEast... Assay Development Biochemical [https://www.nebiolab.com/biochemical-assays-development/]
- 46. Ubiquitin and ubiquitin-like proteins in health and disease [https://meetings.embo.org/event/24-ubiquitin]
- 47. The Ubiquitin Family in Biology and Disease | Keystone Symposia [https://www.keystonesymposia.org/conferences/conference-listing/meeting/k52026]
- 48. Global analysis of protein and small-molecule substrates of... [https://pubmed.ncbi.nlm.nih.gov/40254064/]
- 49. A Mass -Based Spectrometry Workflow for... | PNNL Proteomics [https://www.pnnl.gov/publications/mass-spectrometry-based-proteomics-workflow-concurrent-profiling-protein-thiol]
- 50. - Wikipedia Multiomics [https://en.wikipedia.org/wiki/Multiomics]
- 51. Trends: 2025 Multiomics [https://www.genengnews.com/topics/omics/2025-trends-multiomics/]
- 52. Frontiers | The Structure and Immune Regulatory Implications of the... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2021.794664/full]
- 53. Drugging the undruggables: exploring the ubiquitin system for drug development | Cell Research [https://www.nature.com/articles/cr201631]
- 54. : Structure and Function - PMC Proteasome Inhibitors [https://pmc.ncbi.nlm.nih.gov/articles/PMC6020165/]
- 55. Pharmacologic targeting of Nedd8-activating enzyme reinvigorates T-cell responses in lymphoid neoplasia | Leukemia [https://www.nature.com/articles/s41375-023-01889-x]
- 56. ; source for design of newer... Proteasome inhibitors mechanism [https://www.nature.com/articles/ja201184?error=cookies_not_supported&code=718bec74-8eac-4fb9-9d85-47c08ddba68e]
- 57. Targeting NEDD8-activating enzyme for cancer therapy: developments, clinical trials, challenges and future research directions | Journal of Hematology & Oncology | Full Text [https://jhoonline.biomedcentral.com/articles/10.1186/s13045-023-01485-7]
- 58. Neddylation: a novel modulator of the tumor microenvironment | Molecular Cancer | Full Text [https://molecular-cancer.biomedcentral.com/articles/10.1186/s12943-019-0979-1]
- 59. - Wikipedia Proteasome inhibitor [https://en.wikipedia.org/wiki/Proteasome_inhibitor]
- 60. Ubiquitination, E3 Ligases and Drug Discovery - Novel technologies for a challenging pathway - Drug Discovery World (DDW) [https://www.ddw-online.com/ubiquitination-e3-ligases-and-drug-discovery-novel-technologies-for-a-challenging-pathway-757-201208/]
- 61. Frontiers | Editorial: Structure, Function, and Evolution of E3 Ligases... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2021.767281/full]
- 62. Bacterial antiviral defense pathways encode eukaryotic- like ... [https://pubmed.ncbi.nlm.nih.gov/37808811/]
- 63. Dissecting Distinct Roles of NEDDylation E1 Ligase Heterodimer... [https://www.nature.com/articles/s41598-018-28214-2?error=cookies_not_supported&code=f19bef81-f777-4579-87be-bd0f2ee0bf71]
- 64. Screening for E3-Ubiquitin ligase inhibitors: challenges and opportunities | Oncotarget [https://www.oncotarget.com/article/2431/text/]
- 65. Proteolysis targeting chimera - Wikipedia [https://en.wikipedia.org/wiki/Proteolysis_targeting_chimera]
- 66. Pevonedistat ( MLN ): 4924 of cell death induction and... mechanism [https://www.nature.com/articles/s41420-020-00296-w?error=cookies_not_supported&code=19d0b0cc-2896-4873-ac70-2fcf68e6ac72]
- 67. Pevonedistat - Wikipedia [https://en.wikipedia.org/wiki/Pevonedistat]
- 68. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ - PubMed [https://pubmed.ncbi.nlm.nih.gov/20129059/]
- 69. Mutations in UBA3 Confer Resistance to the NEDD8-Activating Enzyme Inhibitor MLN4924 in Human Leukemic Cells | PLOS One [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0093530]
- 70. Pevonedistat ( MLN ) Datasheet 4924 [https://www.selleckchem.com/datasheet/mln-4924-S710906-DataSheet.html]
- 71. MLN4924 (Pevonedistat) | Cell Signaling Technology [https://www.cellsignal.com/products/activators-inhibitors/mln4924-pevonedistat/85923]
- 72. MLN4924 - Inhibitors - Products [https://www.ubpbio.com/index.php/product/proteasome-deubiquitinase-inhibitors/mln4924-1.html]
- 73. The NEDD8-activating enzyme inhibitor MLN disrupts nucleotide... 4924 [https://pmc.ncbi.nlm.nih.gov/articles/PMC4297545/]
- 74. and Structure of Evolution and... | SpringerLink Ubiquitin [https://link.springer.com/protocol/10.1007/978-1-61779-474-2_2]
- 75. and function of Evolution - ubiquitin -conjugation systems like protein [https://www.nature.com/articles/ncb0800_E153?error=cookies_not_supported]
- 76. : enhanced Molecular - glues interactions and cell... protein protein [https://link.springer.com/article/10.1007/s00044-022-02882-2]
- 77. tackle undruggable targets | CAS Molecular glues [https://www.cas.org/ja/resources/cas-insights/molecular-glues-protein-degraders]
- 78. and PROTACs in targeted Molecular ... glues protein degradation [https://pubmed.ncbi.nlm.nih.gov/40907798/]
- 79. De novo designed protein guiding targeted protein degradation | Nature Communications [https://www.nature.com/articles/s41467-025-62050-z]
- 80. Next steps for targeted protein degradation - PubMed [https://pubmed.ncbi.nlm.nih.gov/39500325/]
- 81. Targeted protein degradation technologies: Emerging mechanisms and nano-based innovations [https://www.sciopen.com/article/10.26599/NR.2025.94907983]
- 82. 8th Annual TPD & Induced Proximity Summit [https://proteindegradation.com/]
- 83. Origin and function of ubiquitin-like proteins | Nature [https://www.nature.com/articles/nature07958?error=cookies_not_supported&code=9a3bc035-0e5f-47df-952d-c67f84f8034f]
- 84. Substrate specificity of the ubiquitin and Ubl proteases - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4822132/]
- 85. The role of UBL in ubiquitin-specific proteases domains [https://pubmed.ncbi.nlm.nih.gov/22616864/]
- 86. Structural and functional insights into Ubl -mediated regulation... domain [https://biologydirect.biomedcentral.com/articles/10.1186/s13062-025-00690-3]
- 87. Profiling of ubiquitin-like modifications reveals features of mitotic control - PubMed [https://pubmed.ncbi.nlm.nih.gov/23452859/]
- 88. Phosphorylation by PINK1 Releases the UBL and Initializes... Domain [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1003935]
- 89. -type RING : Master manipulators of... E 3 ligases [https://pmc.ncbi.nlm.nih.gov/articles/PMC4109693/]
- 90. Ubiquitin ligase - Wikipedia [https://en.wikipedia.org/wiki/Ubiquitin_ligase]
- 91. [ Targeting Cullin- RING for anti-cancer therapy: efforts on...] E 3 ligases [https://pubmed.ncbi.nlm.nih.gov/32621419/]
- 92. of a Structure and RING -loaded... | Nature E 3 ligase ubiquitin [https://www.nature.com/articles/nature11376?error=cookies_not_supported&code=99bc95ae-9145-41d7-a23a-bf4f5dc37021]
- 93. sciencedirect.com/topics/neuroscience/ ubiquitin - protein - ligase - e 3 [https://www.sciencedirect.com/topics/neuroscience/ubiquitin-protein-ligase-e3]
- 94. Structure of a RING trapped in action reveals ligation mechanism... E 3 [https://pubmed.ncbi.nlm.nih.gov/24949976/]
- 95. An allosteric of the human inhibitor ubiquitin-conjugating... Cdc 34 [https://pubmed.ncbi.nlm.nih.gov/21683433/]
- 96. RCSB PDB - 3RZ3: Human Cdc in complex with 34 E 2 CC 0651 inhibitor [https://www.rcsb.org/structure/3RZ3]
- 97. E1 Ubiquitin/Ubl Activating Enzymes Research Areas: R&D Systems [https://www.rndsystems.com/research-area/e1-ubiquitin-ubl-activating-enzymes]
- 98. E2 Ubiquitin/Ubl Conjugating Enzymes Research Areas: R&D Systems [https://www.rndsystems.com/research-area/e2-ubiquitin-ubl-conjugating-enzymes]
- 99. Structural insights into E1-catalyzed ubiquitin activation and transfer to conjugating enzymes - PubMed [https://pubmed.ncbi.nlm.nih.gov/18662542/]
- 100. Allosteric inhibition explained through conformational ensembles sampling distinct “mixed” states - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7720024/]
- 101. Frontiers | Recent applications of computational methods to allosteric drug discovery [https://www.frontiersin.org/journals/molecular-biosciences/articles/10.3389/fmolb.2022.1070328/full]
- 102. Allosteric drugs: New principles and design approaches - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S0959440X23002324]
- 103. in Redundancy Design - GeeksforGeeks System [https://www.geeksforgeeks.org/system-design/redundancy-system-design/]
- 104. Ubiquitin- like protein conjugation and the ubiquitin–proteasome system... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7097807/]
- 105. Targeting the ubiquitin-proteasome system [https://www.drugtargetreview.com/article/34078/targeting-the-ubiquitin-proteasome-system/]
- 106. Substrate specificity of the ubiquitin and Ubl proteases | Cell Research [https://www.nature.com/articles/cr201638?error=cookies_not_supported&code=87109cbd-96b5-4448-8825-4c40f96d7f55]
- 107. The Ubiquitin Tale: Current Strategies and Future Challenges - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11406696/]
- 108. Ubiquitin-like Proteins in Autoimmune Diseases: Current Evidence and Therapeutic Opportunities [https://immunenetwork.org/DOIx.php?id=10.4110/in.2025.25.e21]
- 109. organisms for Model in functional renal validation genetic disease [https://pubmed.ncbi.nlm.nih.gov/38836086/]
- 110. Emerging evidence of coding mutations in the ubiquitin–proteasome system associated with cerebellar ataxias | Human Genome Variation [https://www.nature.com/articles/hgv201418]
- 111. Targeting the Ubiquitin–Proteasome System and Recent Advances in Cancer Therapy - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10778545/]
- 112. The UbL-UBA Ubiquilin4 protein functions as a tumor suppressor in gastric cancer by p53-dependent and p53-independent regulation of p21 | Cell Death & Differentiation [https://www.nature.com/articles/s41418-018-0141-4]
- 113. UBL5 and Its Role in Viral Infections - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11680113/]
- 114. Viral Ubiquitin and Ubiquitin-Like Deconjugases—Swiss Army Knives for Infection - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7464072/]
- 115. Recent progress in ubiquitin and ubiquitin-like protein (Ubl) signaling - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4822135/]
- 116. Ubiquitin and ubiquitin-like proteins in cancer ... | Nature Reviews Cancer [https://www.nature.com/articles/nrc1994?error=cookies_not_supported&code=19b9ac49-c520-4932-b48c-cf53017961e1]
- 117. Non-Proteasomal UbL-UbA Family of Proteins in Neurodegeneration - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6514573/]
- 118. New Insights into the Mechanisms Underlying NEDD8 Structural and... [https://www.intechopen.com/chapters/66145]
- 119. Non-proteolytic ubiquitylation in cellular signaling and human disease | Communications Biology [https://www.nature.com/articles/s42003-022-03060-1]
- 120. Regulation of Signaling by Non-degradative Ubiquitination - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2659175/]
- 121. Ubiquitin, SUMO, and NEDD8: Key Targets of Bacterial Pathogens - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7028394/]
- 122. Frontiers | Hybrid Chains: A Collaboration of Ubiquitin and... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2019.00931/full]
- 123. Old and New Concepts in Ubiquitin and NEDD8 Recognition - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7226360/]
- 124. , Ubiquitin , and SUMO : Key Targets of Bacterial Pathogens NEDD 8 [https://pubmed.ncbi.nlm.nih.gov/30107971/]
- 125. , Ubiquitin , and SUMO as Therapeutic Targets in... | SpringerLink Nedd 8 [https://link.springer.com/chapter/10.1007/978-3-030-38266-7_2]
- 126. Frontiers | Multi-omics approaches for biomarker discovery and precision diagnosis of prediabetes [https://www.frontiersin.org/journals/endocrinology/articles/10.3389/fendo.2025.1520436/full]
- 127. Approaches for the Multiomics of Identification and... Biomarkers [https://www.intechopen.com/online-first/1227653]
- 128. Multi-omics analysis identifies diagnostic circulating biomarkers and potential therapeutic targets, revealing IQGAP1 as an oncogene in gastric cancer | npj Precision Oncology [https://www.nature.com/articles/s41698-025-00895-9]
- 129. Multiomics Analysis-Based Biomarkers in Diagnosis of Polycystic Ovary Syndrome - PubMed [https://pubmed.ncbi.nlm.nih.gov/35084716/]
- 130. The role of multi-omics in biomarker discovery, diagnosis, prognosis, and therapeutic monitoring of tissue repair and regeneration processes - ScienceDirect [https://www.sciencedirect.com/science/article/pii/S2214031X25001196]
- 131. Structural and Functional Insights to Ubiquitin-Like Protein Conjugation - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4118471/]
- 132. Global Analysis of Protein and Small-Molecule Substrates of Ubiquitin-Like Proteins - ScienceDirect [https://www.sciencedirect.com/science/article/pii/S1535947625000738]
- 133. spatial profiling & ctDNA to understand cancer Multiomic [https://dailyreporter.esmo.org/molecular-analysis-for-precision-oncology-congress-map-2025/integrating-multiomic-spatial-profiling-and-ctdna-for-a-deeper-understanding-of-cancer-complexity]
- 134. Evolution and function of ubiquitin-like protein-conjugation systems - PubMed [https://pubmed.ncbi.nlm.nih.gov/10934491/]
- 135. -based cancer therapy: Mechanism to therapy, therapy for... resistance [https://pubmed.ncbi.nlm.nih.gov/25263438/]
- 136. Frontiers | Mechanisms of resistance to targeted therapy and... [https://www.frontiersin.org/journals/immunology/articles/10.3389/fimmu.2024.1366260/full]
- 137. Overcoming Resistance in Lymphoma: Advances and Future... [https://bioengineer.org/overcoming-resistance-in-lymphoma-advances-and-future-directions-in-targeted-therapy/]
- 138. Overcoming resistance to antibody-drug conjugates: from... [https://jhoonline.biomedcentral.com/articles/10.1186/s13045-025-01752-9]
- 139. Selenium targets biomarkers enhancing resistance while... efficacy [https://www.oncotarget.com/article/24297/text/]