Validating Ubiquitination Sites: A Comprehensive Guide to Mutagenesis and Immunoblotting for Researchers
This article provides a detailed methodological guide for researchers and drug development professionals on validating protein ubiquitination sites through the combined power of site-directed mutagenesis and immunoblotting.
Validating Ubiquitination Sites: A Comprehensive Guide to Mutagenesis and Immunoblotting for Researchers
Abstract
This article provides a detailed methodological guide for researchers and drug development professionals on validating protein ubiquitination sites through the combined power of site-directed mutagenesis and immunoblotting. Covering the entire workflow from foundational principles to advanced validation techniques, we explore how to confirm putative ubiquitination sites by mutating candidate lysine residues to arginine and detecting molecular weight shifts via immunoblotting. The content includes practical troubleshooting advice, optimization strategies for enhancing specificity, and comparative analysis with mass spectrometry and computational prediction methods. This resource aims to equip scientists with robust experimental frameworks for elucidating ubiquitination-mediated regulatory mechanisms in health and disease.
Ubiquitination Fundamentals: From Molecular Mechanisms to Functional Consequences
The Ubiquitin-Proteasome System (UPS) is the primary pathway for targeted protein degradation in eukaryotic cells, representing a crucial regulatory mechanism that governs virtually all cellular processes, from cell cycle progression to DNA repair and immune responses [1] [2]. This system functions through a sophisticated enzymatic cascade that tags proteins for degradation with the small, highly conserved protein ubiquitin [3]. The specificity and precision of this system are conferred by its multi-enzyme architecture, involving E1 activating enzymes, E2 conjugating enzymes, and E3 ligases working in concert [1] [4]. Countering this process is a family of specialized proteases known as deubiquitinases (DUBs) that reverse ubiquitination by cleaving ubiquitin from substrate proteins, creating a dynamic equilibrium essential for cellular homeostasis [5] [6]. Dysregulation of UPS components is implicated in numerous pathologies, particularly cancer and neurodegenerative disorders, making this system an attractive target for therapeutic development [7] [2]. This review explores the enzymatic cascade, regulatory components, and experimental methodologies for studying ubiquitination, with particular focus on validation through mutagenesis and immunoblotting.
The Ubiquitination Enzymatic Cascade
The Three-Step Enzymatic Reaction
Ubiquitination involves a sequential cascade of three enzyme families that ultimately conjugate ubiquitin to specific substrate proteins. The process begins with E1 ubiquitin-activating enzymes, which initiate the pathway in an ATP-dependent manner. E1 enzymes catalyze the formation of a high-energy thioester bond between their active-site cysteine residue and the C-terminal glycine of ubiquitin [1] [2]. Humans possess only two E1 enzymes, highlighting their fundamental role as entry points to the ubiquitination pathway [7] [2].
The activated ubiquitin is then transferred to E2 ubiquitin-conjugating enzymes (also called ubiquitin-carrier enzymes) through a trans-thioesterification reaction, forming an E2~Ub thioester intermediate [1] [4]. The human genome encodes approximately 30-40 E2 enzymes, which begin to impart specificity to the system [3] [7].
The final step involves E3 ubiquitin ligases, which function as matchmakers that specifically recognize both the E2~Ub complex and the target substrate protein. E3 ligases facilitate the direct or indirect transfer of ubiquitin from the E2 to a lysine residue on the substrate protein, forming an isopeptide bond [1] [4]. With over 600 members in humans, E3 ligases provide the remarkable substrate specificity of the ubiquitination system [1] [7].
Table 1: Core Enzymes in the Ubiquitination Cascade
| Enzyme Class | Human Genes | Core Function | Key Features |
|---|---|---|---|
| E1 (Activating) | 2 [7] [2] | Ubiquitin activation via ATP hydrolysis | ATP-dependent, forms E1-Ub thioester |
| E2 (Conjugating) | ~30-40 [3] [7] | Ubiquitin carrier | Determines ubiquitin chain topology |
| E3 (Ligating) | ~600 [1] [7] | Substrate recognition | Provides specificity to UPS |
E3 Ligase Classifications and Mechanisms
E3 ubiquitin ligases are categorized into three major families based on their structural features and catalytic mechanisms. RING (Really Interesting New Gene) E3 ligases represent the largest family, characterized by a RING finger domain that binds both the E2~Ub complex and the substrate. Rather than forming a covalent intermediate, RING E3s function as scaffolds that position the E2~Ub close to the substrate to enable direct ubiquitin transfer [1] [8]. A prominent subgroup includes the multi-subunit Cullin-RING ligases (CRLs), which utilize cullin proteins as scaffolds to bring together substrate-recognition modules and RING components [1].
HECT (Homologous to E6AP C-terminus) E3 ligases employ a distinct catalytic mechanism involving a conserved HECT domain with an active-site cysteine residue. These enzymes form a transient thioester intermediate with ubiquitin before transferring it to the substrate [1]. The HECT family includes the well-characterized Nedd4 subfamily, which typically contains WW domains for substrate recognition and C2 domains for membrane localization [1].
RBR (RING-Between-RING) E3 ligases represent a hybrid class that combines features of both RING and HECT mechanisms. While they contain RING domains that bind E2~Ub complexes, they also feature a catalytic cysteine residue that forms a thioester intermediate with ubiquitin before final transfer to substrates, similar to HECT E3s [1] [2]. PARKIN, mutations in which cause familial Parkinson's disease, is a notable RBR E3 ligase [2].
Diagram 1: Ubiquitination Enzymatic Cascade
Deubiquitinases (DUBs): Reversal of Ubiquitination
DUB Classification and Functions
Deubiquitinases (DUBs) constitute a superfamily of proteases that counterbalance ubiquitination by cleaving ubiquitin from modified substrates. These enzymes perform several essential functions: they process inactive ubiquitin precursors to generate mature ubiquitin, remove ubiquitin chains from substrates to reverse their fate, and edit or disassemble unanchored ubiquitin chains for recycling [5] [6] [9]. The human genome encodes approximately 100 DUBs, which are classified into two major classes based on their catalytic mechanisms [5] [6].
Cysteine protease DUBs represent the largest group and are further subdivided into four main families: Ubiquitin-Specific Proteases (USPs), Ovarian Tumor Proteases (OTUs), Ubiquitin C-terminal Hydrolases (UCHs), and Machado-Josephin Domain Proteases (MJDs) [6] [9]. These enzymes employ a catalytic triad or dyad containing a nucleophilic cysteine residue that attacks the isopeptide bond between ubiquitin and the substrate [6].
Metalloprotease DUBs belong to the JAMM/MPN+ family and utilize a coordinated zinc ion to activate a water molecule for nucleophilic attack on the isopeptide bond [6] [9]. Unlike cysteine proteases, JAMM/MPN+ DUBs do not form covalent intermediates during catalysis [6].
Table 2: Major Deubiquitinase (DUB) Families
| DUB Family | Catalytic Type | Human Members | Representative Examples |
|---|---|---|---|
| USP | Cysteine protease | ~58 [9] | USP7, USP9X, USP28 |
| OTU | Cysteine protease | 14 [9] | OTUB1, A20 |
| UCH | Cysteine protease | 4 [9] | UCH-L1, UCH-L3, BAP1 |
| MJD | Cysteine protease | 5 [9] | Ataxin-3, Josephin-1 |
| JAMM/MPN+ | Zinc metalloprotease | 14 [9] | RPN11, BRCC36 |
Regulation of DUB Activity and Function
DUBs are subject to multiple layers of regulation to ensure precise spatiotemporal control of deubiquitination. Many DUBs feature modular architectures with regulatory domains that influence their catalytic activity, subcellular localization, and substrate specificity [6]. For instance, USP7 contains five C-terminal ubiquitin-like (UBL) domains that undergo intramolecular interactions to regulate its enzymatic activity [6]. Similarly, the tumor suppressor BAP1 forms a stable complex between its N-terminal catalytic domain and C-terminal domain, which is essential for cofactor-mediated activation [6].
DUBs are also regulated through obligate and facultative protein complexes that control their stability, activity, and substrate targeting. For example, the proteasomal DUB RPN11 (PSMD14) is only active when incorporated into the 26S proteasome, while UCHL5 (UCH37) requires binding to either the proteasome or the INO80 chromatin-remodeling complex for full activation [6].
Post-translational modifications represent another crucial regulatory mechanism for DUB function. Phosphorylation, acetylation, and ubiquitination can modulate DUB stability, catalytic activity, protein-protein interactions, and subcellular localization [6]. These multiple regulatory layers ensure that DUBs maintain ubiquitin homeostasis and execute precise substrate deubiquitination without promiscuous activity.
Experimental Validation of Ubiquitination
Methodologies for Detecting Protein Ubiquitination
The experimental validation of protein ubiquitination employs various methodologies, each with specific applications, advantages, and limitations. Traditional immunoblotting approaches remain widely used for initial detection and validation of ubiquitination. In this method, proteins are separated by SDS-PAGE and transferred to membranes, which are then probed with ubiquitin-specific antibodies. A characteristic shift to higher molecular weights suggests ubiquitination, which can be further confirmed by mutating putative lysine residues to arginine to abolish ubiquitination sites [3].
Ubiquitin tagging-based approaches enable higher-throughput profiling of ubiquitinated substrates. These methods involve expressing epitope-tagged ubiquitin (e.g., His, HA, or FLAG tags) in cells, followed by affinity purification under denaturing conditions to capture ubiquitinated proteins. After tryptic digestion, ubiquitination sites are identified by mass spectrometry through the detection of a 114.04 Da mass shift on modified lysine residues [3]. While this approach is relatively accessible and cost-effective, potential artifacts may arise because tagged ubiquitin may not completely mimic endogenous ubiquitin.
Ubiquitin antibody-based approaches allow enrichment of endogenously ubiquitinated proteins without genetic manipulation. Pan-specific ubiquitin antibodies (e.g., P4D1, FK1/FK2) can enrich various ubiquitinated proteins, while linkage-specific antibodies (e.g., K48-, K63-specific) enable the study of specific chain types [3]. This approach is particularly valuable for clinical samples where genetic manipulation is infeasible, though antibody cost and potential non-specific binding represent limitations.
Ubiquitin-binding domain (UBD)-based approaches utilize natural ubiquitin receptors to enrich ubiquitinated proteins. Single UBDs typically exhibit low affinity, necessitating tandem-repeated UBDs for efficient purification [3]. This method provides a more physiological approach to studying endogenous ubiquitination.
Table 3: Methods for Ubiquitination Detection and Validation
| Method | Principle | Applications | Advantages | Limitations |
|---|---|---|---|---|
| Immunoblotting + Mutagenesis | Lysine mutation and antibody detection | Target validation | Accessible, specific | Low-throughput, candidate-based |
| Ubiquitin Tagging | Affinity purification of tagged ubiquitin conjugates | Ubiquitinome profiling | Relatively easy, low-cost | Potential artifacts from tagged Ub |
| Ub Antibody Enrichment | Immunoaffinity with ubiquitin antibodies | Endogenous ubiquitination | Works with clinical samples | Antibody cost, potential non-specificity |
| UBD-based Enrichment | Affinity purification with ubiquitin-binding domains | Physiological ubiquitination | Mimics natural recognition | May require tandem domains for affinity |
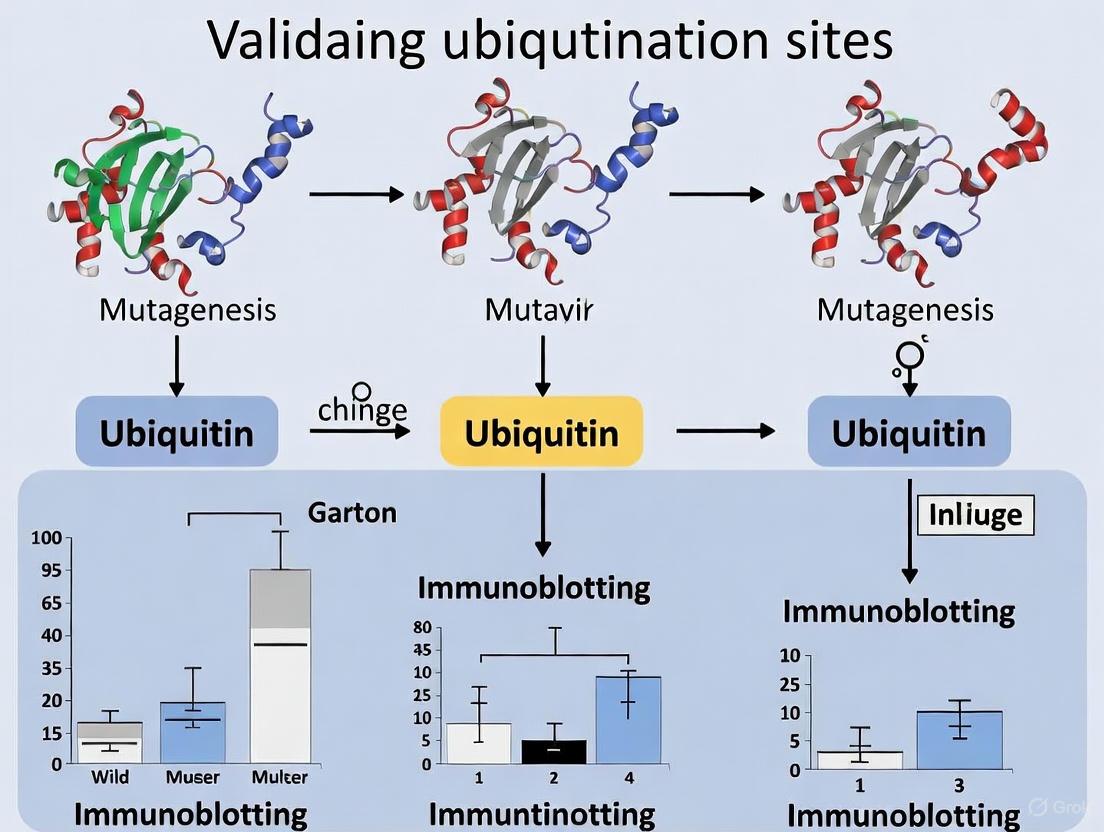
Validating Ubiquitination Sites by Mutagenesis and Immunoblotting
The combination of site-directed mutagenesis and immunoblotting represents a cornerstone method for validating specific ubiquitination sites. This approach follows a systematic workflow that typically includes target selection based on prior experimental evidence or predictive algorithms, followed by design and generation of lysine-to-arginine (K-to-R) mutants that prevent ubiquitin conjugation while maintaining the positive charge of lysine [3].
The experimental protocol involves co-transfecting cells with vectors expressing your protein of interest (wild-type or mutants) along with tagged ubiquitin. After treatment with proteasome inhibitors (e.g., MG132) to enhance detection by preventing degradation of ubiquitinated proteins, cells are lysed under denaturing conditions (e.g., with 1% SDS) to disrupt non-covalent interactions while preserving ubiquitin conjugates [3]. The protein of interest is then immunoprecipitated using specific antibodies or tags, followed by immunoblotting with anti-ubiquitin antibodies to detect ubiquitination.
Key controls for these experiments include monitoring protein expression levels to ensure mutants are properly expressed, verifying that mutations do not alter protein stability or function unrelated to ubiquitination, and confirming that observed higher molecular weight species represent authentic ubiquitin conjugates through ubiquitin immunoblotting [3]. A successful validation demonstrates reduced or abolished ubiquitination signals in K-to-R mutants compared to wild-type protein, as exemplified by a study of Merkel cell polyomavirus large tumor antigen where mutation of K585 to R585 significantly reduced ubiquitination [3].
Diagram 2: Ubiquitination Site Validation Workflow
UPS-Targeted Therapeutic Development
Targeting UPS Components in Cancer Therapy
The ubiquitin-proteasome system has emerged as a promising therapeutic target, particularly in oncology, where dysregulated protein degradation drives tumor progression. Several strategic approaches have been developed to target specific nodes within the UPS. Proteasome inhibitors represent the first clinically successful class of UPS-targeting drugs, with bortezomib and carfilzomib approved for treating multiple myeloma and other hematological malignancies [7] [2]. These compounds directly inhibit the proteolytic activity of the 20S proteasome core particle, leading to accumulation of polyubiquitinated proteins and ultimately apoptosis in malignant cells [7].
E1 enzyme inhibitors target the apex of the ubiquitination cascade. The NEDD8-activating enzyme (NAE) inhibitor MLN4924 (pevonedistat) has shown promise in clinical trials by blocking neddylation of cullins, thereby disrupting the activity of Cullin-RING ligases (CRLs) – the largest class of E3 ubiquitin ligases [2]. This approach induces cell death through uncontrolled DNA synthesis, DNA damage, and apoptosis, with particular efficacy against proliferating tumor cells [2].
E2 enzyme inhibitors offer potentially greater specificity than E1 targeting. Compounds such as CC0651 allosterically inhibit CDC34, while NSC697923 and BAY 11-7082 target the UBE2N-UBE2V1 heterodimer that synthesizes K63-linked ubiquitin chains [2]. Although development challenges have limited clinical translation of E2 inhibitors, they remain an active area of investigation.
E3 ligase modulators represent perhaps the most specific approach, leveraging the substrate specificity of E3s. Small molecules targeting MDM2 (e.g., nutlins) activate p53 by disrupting its interaction with this E3 ligase [2]. Similarly, compounds targeting SCFSKP2 promote accumulation of cell cycle inhibitors p27KIP1 and p21CIP1, exerting anti-proliferative effects in cancer cells [2].
Emerging Technologies and Therapeutic Strategies
Recent technological advances have expanded the toolbox for targeting the UPS. PROTACs (Proteolysis-Targeting Chimeras) represent a revolutionary approach that hijacks the ubiquitin system to degrade specific target proteins. These bifunctional molecules consist of one moiety that binds the target protein connected to another that recruits an E3 ubiquitin ligase, thereby inducing target ubiquitination and degradation [1] [7]. PROTACs have shown remarkable efficacy in degrading previously "undruggable" targets, including transcription factors and scaffold proteins [7] [2].
DUB inhibitors have emerged as another promising therapeutic strategy. The DUB USP9X inhibitor WP1130 has demonstrated synergistic effects with cisplatin in triple-negative breast cancer models by shifting the balance toward E3 ligase WWP1-mediated degradation of the m6A reader IGF2BP2, thereby suppressing oncogenic signaling [10]. This approach highlights how modulating DUB activity can alter the stability of key regulatory proteins in cancer cells.
Advanced screening technologies have accelerated the discovery of UPS-targeting compounds. DNA-encoded compound libraries (DELs) enable ultra-high-throughput screening of vast chemical spaces, while fragment-based screening provides more cost-effective sampling of chemical diversity [7]. Protein engineering approaches, including phage display of ubiquitin variants (UbVs), have generated highly specific inhibitors targeting E3 ligases like NEDD4L and DUBs such as USP7 and USP8 [7].
Table 4: Selected UPS-Targeting Therapeutic Approaches
| Target | Compound/Approach | Mechanism of Action | Development Status |
|---|---|---|---|
| Proteasome | Bortezomib, Carfilzomib | Inhibits 20S proteolytic activity | FDA-approved |
| NEDD8 E1 | MLN4924 (Pevonedistat) | Inhibits cullin neddylation | Phase II trials |
| E3 Ligases | Nutlins, PROTACs | Modulates substrate degradation | Preclinical/Clinical |
| DUBs | WP1130, FT671 | Inhibits deubiquitination | Preclinical development |
The Scientist's Toolkit: Essential Research Reagents
Table 5: Key Research Reagents for Ubiquitination Studies
| Reagent/Category | Function/Application | Examples |
|---|---|---|
| Ubiquitin Tags | Affinity purification of ubiquitinated proteins | His-Ub, HA-Ub, FLAG-Ub |
| Ubiquitin Antibodies | Detection and enrichment of ubiquitinated proteins | P4D1, FK1, FK2, linkage-specific antibodies |
| Proteasome Inhibitors | Stabilize ubiquitinated proteins for detection | MG132, Bortezomib, Carfilzomib |
| DUB Inhibitors | Investigate DUB functions and therapeutic potential | WP1130 (USP9X), P5091 (USP7) |
| E1/E2/E3 Modulators | Dissect specific pathway components | MLN4924 (NAE inhibitor), CC0651 (CDC34 inhibitor) |
| Mutagenesis Kits | Generate lysine mutants for validation | Site-directed mutagenesis systems |
| Ubiquitin Binding Domains | Enrich endogenous ubiquitinated proteins | Tandem UBDs, UIM, UBA domains |
The Ubiquitin-Proteasome System represents a sophisticated regulatory network that maintains protein homeostasis through a delicate balance between ubiquitination and deubiquitination. The E1-E2-E3 enzymatic cascade provides remarkable specificity in targeting proteins for degradation or functional modification, while deubiquitinases offer precise counter-regulation. Methodologies for validating ubiquitination sites, particularly through mutagenesis and immunoblotting, remain fundamental tools for elucidating the physiological and pathological roles of specific ubiquitination events. Continuing advances in targeting UPS components, especially with emerging modalities like PROTACs and DUB inhibitors, highlight the therapeutic potential of manipulating this system. For researchers and drug development professionals, a comprehensive understanding of both the fundamental biology and experimental approaches for studying the UPS is essential for advancing both basic science and therapeutic development in this rapidly evolving field.
Ubiquitination represents a crucial post-translational modification that regulates virtually all aspects of eukaryotic cell biology. The diversity of ubiquitin signals—ranging from single mono-ubiquitination events to complex polyubiquitin chains of specific linkages—generates a sophisticated "ubiquitin code" that determines distinct functional outcomes for modified proteins. This guide provides a comprehensive comparison between mono-ubiquitination and various polyubiquitin chain architectures, with a specific focus on methodological approaches for validating ubiquitination sites through mutagenesis and immunoblotting. We synthesize current experimental data and protocols to equip researchers with practical tools for deciphering ubiquitin signaling in drug development contexts.
Ubiquitin is a small, 76-amino acid protein that is covalently attached to target substrates through a sequential enzymatic cascade involving E1 activating, E2 conjugating, and E3 ligase enzymes [11] [12]. What makes ubiquitin signaling remarkably diverse is the variety of modifications it can form: single ubiquitin molecules (mono-ubiquitination), multiple single ubiquitins on different lysines (multi-mono-ubiquitination), or polyubiquitin chains connected through specific lysine residues within ubiquitin itself [12] [13]. The human genome encodes approximately 40 E2 enzymes and over 600 E3 ligases that confer substrate specificity and determine chain linkage type [13], while around 100 deubiquitinases (DUBs) counter these modifications by cleaving ubiquitin chains [13].
The structural basis for ubiquitin's versatility lies in its compact β-grasp fold, which provides remarkable stability, and the presence of seven lysine residues (K6, K11, K27, K29, K33, K48, K63) plus an N-terminal methionine (M1) that serve as potential linkage sites for chain formation [12]. Each linkage type adopts a distinct three-dimensional structure that is recognized by specific effector proteins containing ubiquitin-binding domains (UBDs), enabling the transduction of diverse cellular signals [14]. This review systematically compares the architectures, functional consequences, and detection methodologies for major ubiquitin modifications, with particular emphasis on experimental validation approaches relevant to pharmaceutical research.
Comparative Analysis of Ubiquitin Modifications
Table 1: Functional and Structural Characteristics of Major Ubiquitin Modifications
| Modification Type | Structural Features | Primary Functions | Key Readers/Effectors | Experimental Detection Approaches |
|---|---|---|---|---|
| Mono-ubiquitination | Single ubiquitin attached to substrate lysine | Endocytosis, DNA repair, histone regulation, protein activity modulation | UBD-containing proteins with preference for mono-Ub | Immunoblotting showing discrete band shifts |
| K48-linked Chains | Compact structures with hydrophobic patches | Proteasomal degradation targeting | Proteasome receptors (Rpn10, Rpn13) | TUBE-based pulldown [15]; characteristic smears in western blot |
| K63-linked Chains | Extended, open conformations | NF-κB activation, kinase signaling, DNA repair, endocytosis | TAB2/3, RAP80, ESCRT components | Linkage-specific TUBEs [15]; K63-selective DUBs [13] |
| M1-linked (Linear) Chains | Rigid, straight chain conformation | NF-κB activation, inflammatory signaling | NEMO/IKKγ | K63-TUBEs (cross-reactivity) [15] |
| Branched/Heterotypic Chains | Complex 3D architectures with multiple linkage types | Signal integration, fine-tuning of degradation kinetics | Specific UBD combinations | Sequential immunoprecipitation, advanced mass spectrometry |
Table 2: Quantitative Degradation Dynamics of Polyubiquitin Chain Types
| Chain Type | Minimum Degradation Unit | Relative Degradation Rate | Chain Disassembly Dynamics | Cellular Half-life of Modified Protein |
|---|---|---|---|---|
| K48-linked | ≥3 ubiquitin molecules [16] | High (primary degradation signal) | Subject to DUB-mediated disassembly [16] | ~1 minute for K48-GFP constructs [16] |
| K63-linked | Not a primary degradation signal | Low (rapidly deubiquitinated) [16] | Rapid DUB-mediated disassembly [16] | Stable (non-degradative function) [16] |
| K48/K63 Branched | Hierarchy with substrate-proximal chain dominant [16] | Intermediate (context-dependent) | Complex regulation by multiple DUB classes | Determined by proximal chain type [16] |
The functional specificity of different ubiquitin modifications is exemplified by the distinct roles of K48 versus K63 linkages. K48-linked polyubiquitin chains, which constitute approximately 72% of chains identified on KCNQ1 ion channels, primarily target substrates for proteasomal degradation [13]. In contrast, K63-linked chains (comprising about 24% of KCNQ1 chains) function in regulatory processes such as inflammatory signaling and protein trafficking [15] [13]. Recent research using the UbiREAD system has demonstrated that K48-linked chains require at least three ubiquitin molecules to efficiently target substrates for degradation, with a half-life of approximately one minute, while K63 chains are rapidly deubiquitinated and do not promote degradation [16].
Branched ubiquitin chains containing multiple linkage types introduce additional complexity to the ubiquitin code. Studies have revealed that in chains with both K48 and K63 linkages, the ubiquitin chain directly conjugated to the substrate protein overrides the influence of the branching chain in determining the substrate's fate [16]. This hierarchical organization enables sophisticated regulation of protein stability and function that is only beginning to be understood.
Experimental Methodologies for Ubiquitination Analysis
Chain-Specific TUBE-Based Affinity Enrichment
Tandem Ubiquitin Binding Entities (TUBEs) are engineered affinity reagents with nanomolar affinities for polyubiquitin chains that can be utilized in high-throughput screening assays to investigate ubiquitination dynamics [15]. The protocol involves several key steps:
Cell Lysis and Preparation: Cells are lysed in optimized buffer systems that preserve polyubiquitination states. For RIPK2 analysis in THP1 cells, lysis is performed under conditions that maintain chain integrity [15].
Affinity Capture: Chain-specific TUBEs (K48-selective, K63-selective, or pan-selective) coated on 96-well plates are incubated with cell lysates. For endogenous RIPK2 studies, TUBEs faithfully capture stimulus-dependent ubiquitination: K63-TUBEs specifically capture L18-MDP-induced ubiquitination, while K48-TUBEs capture PROTAC-induced ubiquitination [15].
Detection and Analysis: Captured ubiquitinated proteins are detected through immunoblotting with target-specific antibodies. This approach enables quantitative analysis of linkage-specific ubiquitination in response to various stimuli or therapeutic agents [15].
The application of TUBE-based technologies has been demonstrated in the characterization of PROTACs (Proteolysis Targeting Chimeras), where K48-selective TUBEs specifically capture PROTAC-induced ubiquitination, contributing to drug development efforts targeting the ubiquitin-proteasome system [15].
Linkage-Selective Engineered DUBs (enDUBs)
Engineered deubiquitinases (enDUBs) represent a innovative approach for investigating the functions of specific polyubiquitin linkages on target proteins in live cells [13]. The methodology involves:
enDUB Construction: Catalytic domains of DUBs with known linkage preferences (OTUD1 for K63, OTUD4 for K48, Cezanne for K11, TRABID for K29/K33) are fused to a GFP-targeted nanobody [13].
Cellular Application: enDUBs are co-expressed with the GFP/YFP-tagged protein of interest, enabling selective hydrolysis of specific polyubiquitin linkages from the target protein [13].
Functional Assessment: Following enDUB-mediated cleavage, changes in protein abundance, localization, and function are measured. For KCNQ1 ion channels, this approach revealed that different linkages regulate distinct trafficking steps: K11 and K29/K33 promote ER retention, K63 enhances endocytosis and reduces recycling, and K48 is necessary for forward trafficking [13].
This technology enables researchers to move beyond correlation to causation in defining the functional consequences of specific ubiquitin linkages on target proteins.
Ubiquitin-Specific Proximity-Dependent Labeling (Ub-POD)
Ub-POD is a proximity labeling technique that enables selective biotinylation of substrates of a specific E3 ligase, facilitating the identification of novel ubiquitination substrates [17]. The protocol includes:
Construct Design: The candidate E3 ligase is fused to the biotin ligase BirA, while ubiquitin is fused to a modified biotin acceptor peptide (-2)AP [17].
Cell Transfection and Biotin Exposure: Cells are co-transfected with these constructs and exposed to biotin, resulting in BirA-catalyzed biotinylation of (-2)AP-Ub when in complex with E2~Ub [17].
Substrate Capture and Identification: Biotinylated substrates are enriched under denaturing conditions using streptavidin pulldown and identified through mass spectrometry or immunoblotting [17].
This method is particularly valuable for capturing transient E3 ligase-substrate interactions that are difficult to detect with conventional co-immunoprecipitation approaches.
Ubiquitination Pathway Diagrams
Ubiquitin Enzymatic Cascade and Modification Outcomes
Experimental Workflow for Ubiquitin Modification Analysis
Research Reagent Solutions Toolkit
Table 3: Essential Research Reagents for Ubiquitination Studies
| Reagent Category | Specific Examples | Key Applications | Advantages/Limitations |
|---|---|---|---|
| Linkage-Specific Affinity Reagents | K48-TUBEs, K63-TUBEs, Pan-TUBEs [15] | Enrichment of linkage-specific polyubiquitinated proteins from cell lysates | High affinity (nM range), applicable to HTS; may require optimization for different targets |
| Engineered DUBs (enDUBs) | OTUD1-nanobody (K63-selective), OTUD4-nanobody (K48-selective) [13] | Selective cleavage of specific ubiquitin linkages from target proteins in live cells | Enables functional assessment of specific linkages; requires GFP-tagged target proteins |
| Ubiquitin Variants (UbVs) | K48-linkage specific UbVs, K63-linkage specific UbVs [12] | Modulation of HECT E3 ligases, detection of specific chain types | High specificity; limited commercial availability for all linkage types |
| Linkage-Selective DUBs | Catalytic domains of OTUD1, OTUD4, Cezanne, TRABID [13] [14] | Analytical cleavage of specific chain types for validation | Well-characterized specificity; requires experimental optimization for cellular use |
| Mutagenesis Tools | Ubiquitin K-to-R mutants, Substrate K-to-R mutants [13] | Identification of specific ubiquitination sites by immunoblotting | Direct assessment of site functionality; may disrupt protein structure/function |
| Activity-Based Probes | DUB substrates, E1/E2/E3 inhibitors [14] | Monitoring enzyme activities in ubiquitination cascade | Functional readouts; may lack specificity for individual enzymes |
The sophisticated diversity of ubiquitin modifications—from single mono-ubiquitination events to complex homotypic and branched polyubiquitin chains—constitutes a complex regulatory language that controls protein fate and function in eukaryotic cells. The experimental toolbox for deciphering this ubiquitin code has expanded significantly, with linkage-specific TUBEs, engineered DUBs, proximity labeling techniques, and traditional mutagenesis approaches providing complementary insights into ubiquitin signaling pathways. For researchers in drug development, understanding these distinct ubiquitin architectures and their functional consequences is particularly relevant given the emergence of therapeutic strategies that target the ubiquitin-proteasome system, such as PROTACs and molecular glues. As our methodological capabilities continue to advance, so too will our ability to decipher the complex ubiquitin code and develop novel therapeutic interventions for human diseases characterized by ubiquitin pathway dysregulation.
Ubiquitination is a crucial post-translational modification that regulates virtually all aspects of cellular physiology in eukaryotic cells. This process involves the covalent attachment of ubiquitin, a 76-amino acid protein, to target substrates through a sequential enzymatic cascade [18] [19]. The functional consequences of ubiquitination extend far beyond its canonical role in targeting proteins for proteasomal degradation, encompassing diverse non-proteolytic functions in signal transduction, subcellular localization, DNA repair, and inflammatory responses [20] [21] [18]. The specificity of ubiquitin signaling is determined by the architecture of ubiquitin chains formed on substrate proteins, with different linkage types encoding distinct cellular outcomes [20] [19]. Understanding these functional outcomes is essential for researchers investigating cellular signaling pathways, protein homeostasis, and developing targeted therapeutic strategies. This guide provides a comprehensive comparison of ubiquitination functions, supported by experimental data and methodologies relevant for validating ubiquitination sites through mutagenesis and immunoblotting.
The Ubiquitin Code: Linkage Types and Functional Consequences
The versatility of ubiquitin signaling arises from its capacity to form polymeric chains through different linkage types. Ubiquitin contains seven internal lysine residues (K6, K11, K27, K29, K33, K48, and K63) and an N-terminal methionine (M1) that can serve as linkage points for polyubiquitin chain formation [19]. The type of ubiquitin linkage determines the functional outcome for the modified protein, creating a complex "ubiquitin code" that cells decipher to regulate physiological processes.
Table 1: Ubiquitin Linkage Types and Their Primary Functional Outcomes
| Linkage Type | Primary Function | Key Biological Processes | Experimental Detection Methods |
|---|---|---|---|
| K48-linked chains | Proteasomal degradation [20] [21] | Cell cycle regulation, protein quality control [19] | K48-TUBE pulldown + immunoblotting [21] |
| K63-linked chains | Signal activation, endocytic trafficking [20] [21] | DNA repair, NF-κB signaling, inflammation [20] [21] [18] | K63-TUBE pulldown + immunoblotting [21] |
| M1-linked (linear) chains | Inflammation regulation [18] | NF-κB activation, immune responses [18] | Linear ubiquitin-specific antibodies [18] |
| K11-linked chains | ER-associated degradation, cell cycle regulation [19] | Mitotic progression, protein quality control [19] | Linkage-specific antibodies, mass spectrometry |
| K27/K29/K33-linked chains | Atypical functions, lysosomal degradation [19] | Immune signaling, stress responses [18] [19] | Ubiquitinomics, mutagenesis studies |
The functional diversity of ubiquitin linkages enables precise control over cellular processes. For example, K48-linked polyubiquitination represents the canonical signal for proteasomal degradation, ensuring the removal of damaged, misfolded, or regulatory proteins [20] [19]. In contrast, K63-linked chains are primarily associated with non-proteolytic functions, including activation of kinase pathways, DNA damage response, and endocytic trafficking [20] [21]. M1-linked linear chains play specialized roles in regulating inflammatory responses through the NF-κB pathway [18].
Diagram 1: The ubiquitination enzymatic cascade. This diagram illustrates the three-step enzymatic process of ubiquitination, highlighting the E3 ligase role in substrate recognition.
Proteasomal Degradation: The K48-Linked Pathway
The K48-linked ubiquitin pathway serves as the primary route for targeted protein degradation in eukaryotic cells. This pathway regulates the abundance of key regulatory proteins, including cyclins, transcription factors, and damaged proteins, thereby controlling essential cellular processes.
Molecular Mechanism of K48-Linked Degradation
The K48-linked ubiquitination pathway begins with the E1-mediated activation of ubiquitin, followed by transfer to an E2 conjugating enzyme. An E3 ubiquitin ligase then recruits both the E2~Ub complex and the specific substrate protein, facilitating the transfer of ubiquitin to form K48-linked chains [19]. These chains are recognized by the 26S proteasome, which unfolds and degrades the target protein while recycling ubiquitin molecules for reuse [19]. The 26S proteasome consists of a 20S core particle (CP) that contains the proteolytic active sites and one or two 19S regulatory particles (RP) that recognize ubiquitinated substrates, remove ubiquitin chains, and unfold target proteins [19].
Experimental Analysis of K48-Linked Ubiquitination
Researchers can investigate K48-linked ubiquitination using various experimental approaches. Tandem Ubiquitin Binding Entities (TUBEs) specific for K48 linkages provide a powerful tool for enriching and detecting proteins modified with K48-linked chains [21]. In a study investigating RIPK2 ubiquitination, K48-TUBEs successfully captured endogenous RIPK2 only when treated with a specific PROTAC molecule that induces K48-linked ubiquitination, but not when cells were stimulated with L18-MDP, which induces K63-linked chains [21]. This demonstrates the linkage specificity of these reagents.
Table 2: Experimental Models of K48-Linked Ubiquitination
| Experimental System | Inducer/Stimulus | Target Protein | Detection Method | Key Findings |
|---|---|---|---|---|
| THP-1 human monocytic cells | RIPK2 PROTAC (degrader-2) [21] | RIPK2 [21] | K48-TUBE pulldown + immunoblotting [21] | Specific capture of K48-ubiquitinated RIPK2 in PROTAC-treated cells [21] |
| Reconstituted in vitro system | E1, E2, E3 enzymes, ATP [22] | Small molecule inhibitors (BI8622/BI8626) [22] | Mass spectrometry, SDS-PAGE [22] | HUWE1 catalyzes ubiquitination of primary amine-containing compounds [22] |
| Cancer cell lines | Various cellular stresses | p53, NF-κB, β-catenin | Immunoblotting, ubiquitination assays | Multiple oncoproteins and tumor suppressors regulated by K48 ubiquitination |
Immunoblotting remains a fundamental technique for detecting ubiquitinated proteins, though researchers must use validated antibodies and determine the linear dynamic range for accurate quantification [23]. For studying endogenous protein ubiquitination, protocols typically involve protein extraction under denaturing conditions to preserve ubiquitination, enrichment of ubiquitinated proteins using TUBEs or immunoprecipitation, followed by immunoblotting with target-specific antibodies [21] [23]. Site-directed mutagenesis of acceptor lysines in substrate proteins, particularly mutation to arginine (K→R), provides a critical approach for validating specific ubiquitination sites and their functional consequences [22].
Non-Proteolytic Functions of Ubiquitination
Beyond targeting proteins for degradation, ubiquitination serves diverse non-proteolytic functions that regulate cellular signaling, protein interactions, and subcellular localization. These functions are primarily mediated through K63-linked and linear (M1-linked) ubiquitin chains.
K63-Linked Ubiquitination in Signal Transduction
K63-linked ubiquitination plays a crucial role in inflammatory signaling pathways, particularly in the activation of NF-κB. Upon stimulation of nucleotide-binding oligomerization domain (NOD)-like receptors by bacterial components such as muramyldipeptide (MDP), the NOD2 receptor oligomerizes and recruits RIPK2 and E3 ligases including XIAP, leading to K63-linked ubiquitination of RIPK2 [21]. These K63 ubiquitin chains serve as a signaling scaffold that recruits and activates the TAK1/TAB1/TAB2/IKK kinase complexes, ultimately driving NF-κB activation and production of proinflammatory cytokines [21].
The functional role of K63 ubiquitination in inflammatory signaling has been demonstrated using chain-specific TUBEs. In THP-1 cells stimulated with L18-MDP, K63-TUBEs specifically captured ubiquitinated RIPK2, while K48-TUBEs showed minimal binding [21]. This linkage-specific ubiquitination was time-dependent, with higher levels observed at 30 minutes compared to 60 minutes post-stimulation [21]. Furthermore, pretreatment with the RIPK2 inhibitor Ponatinib completely abrogated L18-MDP-induced RIPK2 ubiquitination, confirming the specificity of the response [21].
Diagram 2: K63-linked ubiquitination in inflammatory signaling. This pathway shows how bacterial stimulation leads to NOD2-RIPK2-XIAP complex formation, K63 ubiquitination, and downstream NF-κB activation.
Linear (M1-Linked) Ubiquitination in Inflammation
Linear ubiquitin chains, formed through linkage via the N-terminal methionine of ubiquitin, play specialized roles in regulating inflammatory and cell death pathways. The linear ubiquitin assembly complex (LUBAC), composed of HOIP, HOIL-1L, and SHARPIN, is the primary E3 ligase responsible for generating M1-linked chains [18]. During tumor necrosis factor (TNF) signaling, LUBAC catalyzes linear ubiquitination of components in the TNFR signaling complex, including RIPK1 and NEMO (IKKγ), facilitating optimal NF-κB activation [18]. This modification creates a signaling platform that recruits additional effector proteins through ubiquitin-binding domains.
The functional importance of linear ubiquitination is highlighted by studies in sepsis models, where LUBAC-mediated linear ubiquitination of RIPK1 helps regulate the balance between cell survival and necroptosis [18]. Furthermore, the deubiquitinating enzyme USP5 has been shown to remove K63-linked polyubiquitin chains from RIPK1, thereby inhibiting necroptosis and protecting cardiomyocytes during sepsis [18]. These findings demonstrate how different ubiquitin linkages and their removal by specific DUBs precisely control inflammatory signaling outcomes.
Ubiquitination in Subcellular Localization and Trafficking
Ubiquitination also regulates protein localization and trafficking within cells. Monoubiquitination or multi-monoubiquitination of membrane proteins serves as a signal for endocytosis and subsequent sorting to lysosomes for degradation [20]. This process is distinct from K48-linked proteasomal targeting and involves different sets of ubiquitin-binding adaptor proteins. Additionally, ubiquitination regulates the assembly and disassembly of protein complexes, chromatin remodeling, and DNA repair through both proteasome-independent and dependent mechanisms [20] [19].
Experimental Approaches for Studying Ubiquitination
Detecting Ubiquitination: Methodological Comparisons
Researchers have multiple methodological options for detecting and characterizing protein ubiquitination. The choice of method depends on the specific research question, available resources, and whether the focus is on global ubiquitination changes or specific target proteins.
Table 3: Comparison of Ubiquitination Detection Methods
| Method | Sensitivity | Throughput | Linkage Specificity | Key Applications | Limitations |
|---|---|---|---|---|---|
| Immunoblotting [23] | Moderate | Low to moderate | Limited (depends on antibody quality) | Target protein validation, modification detection [23] | Semiquantitative, antibody-dependent variability [23] |
| TUBE-Based Assays [21] | High | High (96-well format) [21] | High (chain-specific TUBEs) [21] | High-throughput screening, linkage-specific analysis [21] | Specialized reagents required |
| Mass Spectrometry | High | Low | High with enrichment | Proteome-wide ubiquitinome mapping | Technically challenging, expensive instrumentation |
| Reporter Gene Assays | Variable | High | Limited | PROTAC screening, degradation kinetics | Potential artifacts from reporter tags [21] |
Recommended Protocol for Ubiquitination Detection
Based on current methodologies, the following optimized protocol can be used for detecting ubiquitination of endogenous proteins:
Cell Lysis: Lyse cells in denaturing buffer (e.g., containing SDS) to preserve ubiquitination signatures and prevent deubiquitination by DUBs during processing [21] [23].
Enrichment of Ubiquitinated Proteins: Use chain-specific TUBEs (K48, K63, or pan-selective) immobilized on magnetic beads or microplate wells to capture ubiquitinated proteins. Incubate lysates with TUBEs for 2-4 hours at 4°C with gentle agitation [21].
Washing and Elution: Wash beads thoroughly with wash buffer to remove non-specifically bound proteins. Elute bound proteins with SDS-PAGE sample buffer containing DTT or β-mercaptoethanol [21].
Immunoblotting: Separate proteins by SDS-PAGE, transfer to nitrocellulose membrane, and probe with target-specific antibodies. Include controls for equal loading and specificity [23].
Validation: Confirm ubiquitination dependence by treating cells with proteasome inhibitors (e.g., MG132) for K48-linked chains or using mutagenesis of putative ubiquitination sites (K→R mutations) [22].
The Scientist's Toolkit: Essential Research Reagents
Table 4: Key Research Reagents for Ubiquitination Studies
| Reagent/Tool | Function | Example Applications | Commercial Sources |
|---|---|---|---|
| Chain-specific TUBEs [21] | High-affinity capture of linkage-specific polyubiquitin chains | Differentiation between K48 and K63 ubiquitination in inflammatory signaling [21] | LifeSensors [21] |
| Proteasome Inhibitors (MG132, bortezomib) | Block proteasomal degradation, accumulate ubiquitinated proteins | Stabilization of K48-ubiquitinated proteins for detection | Multiple vendors |
| Linkage-Specific Antibodies | Detect specific ubiquitin linkages in immunoblotting | Verification of chain type in ubiquitination assays | Multiple vendors |
| HA-Ubiquitin, MYC-Ubiquitin Plasmids | Expression of tagged ubiquitin in cells | Pull-down experiments, identification of ubiquitinated proteins | Addgene, commercial vendors |
| E1/E2/E3 Enzyme Systems | Reconstitute ubiquitination in vitro | Biochemical characterization of ubiquitination mechanisms [22] | Multiple vendors |
The functional outcomes of ubiquitination extend far beyond the canonical role in proteasomal degradation to include sophisticated regulation of signaling pathways, protein localization, and complex assembly. The specific biological consequence of ubiquitination is determined by the type of ubiquitin linkage, with K48-linked chains primarily targeting proteins for degradation, while K63-linked and linear chains regulate signal transduction and inflammatory responses. Advanced research tools including chain-specific TUBEs, validated antibodies, and optimized immunoblotting protocols enable researchers to dissect these diverse functions with increasing precision. A comprehensive understanding of ubiquitination outcomes, combined with robust experimental methodologies for validation, provides the foundation for targeting the ubiquitin system in therapeutic development, particularly in the rapidly advancing field of targeted protein degradation.
The Critical Need for Site-Specific Validation in Understanding Disease Mechanisms
In the pursuit of understanding complex disease mechanisms, site-specific validation of protein modifications has emerged as a non-negotiable requirement for rigorous biological research. While high-throughput omics technologies can identify potential molecular targets, functional validation through precise experimental approaches remains the critical gateway to mechanistic understanding. This is particularly evident in the study of ubiquitination—a versatile post-translational modification where abnormalities are closely linked to various pathologies, including cancer and neurodegenerative diseases [24] [3]. The versatility of ubiquitination, which can range from single ubiquitin monomers to polymers with different lengths and linkage types, creates a complex signaling landscape that demands precise dissection [3]. This guide objectively compares the performance of current methodologies for ubiquitination site validation, providing researchers with experimental frameworks to bridge the gap between putative site identification and mechanistic understanding of disease pathways.
The Ubiquitination Landscape and Disease Implications
Ubiquitination is a sophisticated post-translational modification process involving a cascade of E1 (activating), E2 (conjugating), and E3 (ligase) enzymes that covalently attach ubiquitin to target proteins [3] [25]. This modification can result in diverse outcomes depending on the ubiquitin chain topology, with K48-linked chains typically targeting substrates for proteasomal degradation, while K63-linked chains are primarily involved in signal transduction and protein trafficking [15]. The critical role of ubiquitination in cellular homeostasis becomes evident in its dysregulation, which is implicated in numerous diseases. For instance, recent research has identified specific ubiquitination-related genes—MMP1, RNF2, TFRC, SPP1, and CXCL8—as significantly associated with cervical cancer outcomes, demonstrating the clinical relevance of understanding site-specific ubiquitination patterns [24].
Table 1: Ubiquitin Chain Linkages and Their Functional Consequences
| Linkage Type | Primary Function | Associated Cellular Processes | Disease Implications |
|---|---|---|---|
| K48-linked chains | Proteasomal degradation | Cell cycle regulation, protein turnover | Cancer, neurodegenerative disorders |
| K63-linked chains | Signal transduction | NF-κB activation, DNA repair, inflammation | Inflammatory diseases, immune disorders |
| K6-linked chains | Mitochondrial homeostasis, DNA damage response | Mitophagy, genomic stability | Parkinson's disease, cancer |
| K11-linked chains | ER-associated degradation, cell cycle regulation | Protein quality control, division | Cancer, developmental disorders |
| K27-linked chains | Immune signaling, kinase activation | Inflammatory pathways, autophagy | Autoimmune diseases, infection |
| K29-linked chains | Proteasomal degradation, Wnt signaling | Protein turnover, development | Neurodegeneration, cancer |
| K33-linked chains | Trafficking, kinase regulation | Intracellular transport, signaling | Metabolic disorders |
| M1-linear chains | NF-κB signaling, inflammation | Immune response, cell death | Autoinflammatory diseases |
Methodological Comparison for Ubiquitination Site Detection
Computational Prediction Methods
Computational approaches provide the first line of screening for potential ubiquitination sites, with machine learning algorithms increasingly outperforming traditional methods. Recent benchmarks demonstrate that deep learning models achieve superior performance with a 0.902 F1-score, 0.8198 accuracy, 0.8786 precision, and 0.9147 recall when utilizing both raw amino acid sequences and hand-crafted features [26]. These tools analyze sequence motifs, evolutionary conservation, and structural features to predict lysine residues likely to be ubiquitinated, offering a cost-effective strategy to prioritize targets for experimental validation.
Mass Spectrometry-Based Approaches
Mass spectrometry (MS) has become the gold standard for ubiquitination site identification, with advanced workflows enabling comprehensive mapping of modification sites. The typical MS workflow involves protein extraction and digestion, ubiquitin enrichment, LC-MS/MS analysis, and data interpretation using software tools like MaxQuant, Proteome Discoverer, and PEAKS [25]. The key advantage of MS-based approaches is their ability to identify modification sites with high precision through the detection of characteristic mass shifts (~8.5 kDa) corresponding to ubiquitin modification [3] [25]. However, challenges remain in detecting low-abundance ubiquitinated peptides and deciphering complex polyubiquitin chain architectures, particularly with multi-ubiquitination events [25].
Immunoblotting and Functional Validation
Traditional immunoblotting remains a cornerstone for ubiquitination validation, particularly when combined with mutagenesis approaches. The conventional protocol involves testing ubiquitination levels of putative substrates using anti-ubiquitin antibodies, followed by systematic mutation of candidate lysine residues to assess whether specific mutations reduce ubiquitination signals [3]. This approach was successfully employed to identify K585 as the ubiquitination site in Merkel cell polyomavirus large tumor antigen, where substitution with arginine significantly reduced ubiquitination levels [3]. When integrated with site-directed mutagenesis, immunoblotting provides a direct method for establishing causal relationships between specific residues and ubiquitination patterns.
Table 2: Performance Comparison of Ubiquitination Detection Methods
| Method | Throughput | Sensitivity | Site Specificity | Key Limitations | Best Use Cases |
|---|---|---|---|---|---|
| Machine Learning Prediction | High | Moderate | Moderate (requires validation) | Limited by training data quality; cannot detect novel motifs | Initial screening and prioritization of candidate sites |
| Mass Spectrometry | Medium-High | High (with enrichment) | High | Low-abundance peptides masked by non-modified counterparts; complex data interpretation | Comprehensive site mapping; discovery workflows |
| Immunoblotting with Mutagenesis | Low | Medium | High (when combined with mutagenesis) | Time-consuming; low-throughput; antibody-dependent | Functional validation of specific candidate sites |
| Linkage-Specific TUBEs | Medium | High for specific linkages | Linkage-level specificity | Requires linkage-specific reagents; may miss rare linkages | Studying chain topology and linkage-specific functions |
| In Vitro Ubiquitination Assays | Medium | High for confirmed substrates | High in controlled systems | May not recapitulate cellular context | Mechanism studies; E3 ligase substrate specificity |
Advanced Techniques for Site-Specific Validation
Tandem Ubiquitin Binding Entities (TUBEs) for Linkage-Specific Analysis
The recent development of chain-specific TUBEs represents a significant advancement for studying ubiquitination in physiological contexts. These specialized affinity matrices with nanomolar affinities for polyubiquitin chains enable precise capture of linkage-specific ubiquitination events on endogenous proteins [15]. This technology has been successfully applied to differentiate context-dependent ubiquitination, as demonstrated in studies of RIPK2, where K63-TUBEs specifically captured inflammatory stimulus-induced ubiquitination, while K48-TUBEs identified PROTAC-induced degradation signals [15]. This approach provides researchers with a tool to dissect the functional consequences of different ubiquitin chain types on specific protein targets.
Integrated Computational-Experimental Frameworks
Machine learning methods that combine evolutionary information with biophysical models offer enhanced ability to identify functionally important sites. Recent models integrate predicted changes in thermodynamic protein stability (ΔΔG), evolutionary sequence information (ΔΔE), hydrophobicity, and weighted contact number to distinguish residues critical for function from those primarily important for structural stability [27]. This approach successfully identifies "stable but inactive" (SBI) variants—mutations that affect function without disrupting protein stability—which are strong indicators of direct functional involvement [27]. This methodology was prospectively validated for HPRT1 variants associated with Lesch-Nyhan syndrome, demonstrating its utility for pinpointing molecular disease mechanisms.
Experimental Protocols for Ubiquitination Validation
Protocol 1: In Vitro Ubiquitination Assay
- Principle: Reconstitute the ubiquitination cascade using recombinant enzymes to test substrate modification under controlled conditions [25].
- Procedure:
- Recombinant Enzyme Preparation: Combine E1, E2, and E3 enzymes with recombinant ubiquitin in reaction buffer containing ATP.
- Substrate Addition: Introduce the recombinant substrate protein (often a truncated version of the target) to the reaction mixture.
- Incubation: Conduct the reaction for 30-60 minutes at 30°C.
- Reaction Termination: Stop the reaction by boiling in SDS-PAGE loading buffer.
- Analysis: Resolve proteins via SDS-PAGE and detect ubiquitin-modified species by Western blotting using anti-ubiquitin or target-specific antibodies [25].
- Applications: Identifying potential ubiquitination sites, investigating E3 ligase specificity, and examining ubiquitin chain formation mechanisms.
Protocol 2: Mass Spectrometry-Based Site Identification
- Principle: Utilize high-resolution mass spectrometry to precisely identify ubiquitinated lysine residues through characteristic mass shifts [3] [25].
- Procedure:
- Protein Extraction and Digestion: Isolate proteins from biological samples and digest with trypsin to generate peptides.
- Ubiquitin Enrichment: Employ immunoprecipitation with anti-ubiquitin antibodies, affinity chromatography with ubiquitin-binding domains, or chemical enrichment strategies to isolate ubiquitinated peptides.
- LC-MS/MS Analysis: Separate enriched peptides using liquid chromatography and analyze with tandem mass spectrometry.
- Data Interpretation: Use specialized software to identify ubiquitination sites based on the diagnostic mass shift (GG remnant, +114.04 Da) on modified lysine residues [3] [25].
- Applications: Large-scale ubiquitinome mapping, identification of specific modification sites, and quantification of ubiquitination dynamics across conditions.
Protocol 3: Mutagenesis Validation Workflow
- Principle: Systematically mutate candidate ubiquitination sites to assess their necessity for ubiquitination [3].
- Procedure:
- Site Identification: Select candidate lysine residues based on computational predictions, MS data, or structural analysis.
- Mutant Generation: Create lysine-to-arginine (K→R) mutants using site-directed mutagenesis to prevent ubiquitination while maintaining similar physicochemical properties.
- Expression and Treatment: Express wild-type and mutant proteins in relevant cell lines, with or without proteasomal inhibition (e.g., MG132) to enhance ubiquitination detection.
- Immunoprecipitation and Immunoblotting: Immunoprecipitate the target protein and detect ubiquitination using anti-ubiquitin antibodies.
- Functional Assays: Assess the functional consequences of ubiquitination-deficient mutations through protein stability, localization, or activity assays [3].
- Applications: Establishing causal relationships between specific residues and ubiquitination, functional characterization of modification sites, and mechanistic studies.
Visualizing Experimental Workflows
Ubiquitination Cascade and Validation Pathway
Experimental Decision Framework for Method Selection
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Ubiquitination Studies
| Reagent Category | Specific Examples | Function/Application | Considerations |
|---|---|---|---|
| Linkage-Specific Antibodies | K48-linkage specific, K63-linkage specific, M1-linear specific | Immunoblotting, immunoprecipitation, immunofluorescence | Specificity validation required; may exhibit cross-reactivity |
| Tandem Ubiquitin Binding Entities (TUBEs) | K48-TUBEs, K63-TUBEs, Pan-TUBEs | Affinity enrichment, linkage-specific capture, proteasome inhibition | Enables study of endogenous proteins without genetic manipulation |
| Recombinant Enzymes | E1, E2 (UBE2L3), E3 ligases (RNF19A/B) | In vitro ubiquitination assays, mechanism studies | Enzyme activity validation critical for assay success |
| Ubiquitin Mutants | K48R, K63R, K0 (all lysines mutated) | Linkage-specific function studies, chain topology mapping | May not fully recapitulate wild-type ubiquitin biology |
| Deubiquitinase Inhibitors | PR-619, P22077, G5 | Stabilize ubiquitinated species, pathway manipulation | Varying specificity across DUB families |
| Proteasome Inhibitors | MG132, Bortezomib, Carfilzomib | Accumulation of ubiquitinated proteins, degradation studies | Can induce cellular stress responses at high concentrations |
| Tagged Ubiquitin | HA-Ub, FLAG-Ub, GFP-Ub, His-Ub | Affinity purification, microscopy, pull-down assays | Tags may alter ubiquitin structure/function |
Site-specific validation remains the critical gateway to transforming correlative observations into mechanistic understanding of disease pathways. The evolving methodology landscape—spanning computational predictions, advanced mass spectrometry, linkage-specific capture technologies, and classical mutagenesis—provides researchers with a powerful toolkit for deconvoluting ubiquitination complexity. The integration of these approaches, each with complementary strengths and limitations, enables the construction of rigorous evidentiary chains linking specific molecular modifications to functional consequences. As research progresses toward therapeutic interventions targeting the ubiquitin system, including PROTACs and molecular glues, the imperative for precise site-specific validation only intensifies. By employing the comparative frameworks and standardized protocols outlined in this guide, researchers can advance our understanding of disease mechanisms with the precision necessary for meaningful biological insight and therapeutic development.
Protein ubiquitination, the covalent attachment of a small regulatory protein (ubiquitin) to substrate proteins, represents one of the most crucial post-translational modifications in eukaryotic cells, governing processes including proteasomal degradation, DNA repair, signal transduction, and cell cycle progression [28] [29]. The traditional experimental identification of ubiquitination sites through methods like mass spectrometry, antibody recognition, and mutagenesis is often time-consuming, expensive, and labor-intensive [30] [31]. Consequently, computational prediction tools have emerged as indispensable resources for preliminary screening, enabling researchers to prioritize candidate sites for subsequent experimental validation. These tools leverage machine learning algorithms to identify patterns in protein sequences that signify potential ubiquitination sites, dramatically accelerating the initial discovery phase [32] [33].
Within the broader thesis context of validating ubiquitination sites via mutagenesis and immunoblotting, computational predictors serve as the critical first step in the research pipeline. They provide specific, testable hypotheses about which lysine residues are most likely to be ubiquitinated, thereby informing the design of mutagenesis experiments and making the subsequent immunoblotting validation more efficient and targeted [28]. This guide objectively compares three established prediction tools—UbiPred, UbiSite, and UbiProber—evaluating their methodologies, performance, and practical applications to equip researchers with the knowledge to select the appropriate tool for their specific research needs.
Tool Comparison: Core Algorithms and Feature Encoding
The predictive performance of any computational tool is fundamentally determined by its underlying machine learning algorithm and the features it extracts from protein sequences. The table below summarizes the core methodologies of UbiPred, UbiSite, and UbiProber.
Table 1: Core Algorithmic Comparison of Ubiquitination Prediction Tools
| Tool | Core Algorithm | Feature Encoding Methods | Species Focus |
|---|---|---|---|
| UbiPred | Support Vector Machine (SVM) [32] [30] | Informative Physicochemical Properties (PCPs) [32] | General [32] |
| UbiSite | Support Vector Machine (SVM) [34] | Amino Acid Composition (AAC), Position Weight Matrix, Amino Acid Pair Composition (AAPC), Position-Specific Scoring Matrix (PSSM) [34] | General [34] |
| UbiProber | Support Vector Machine (SVM) [33] | K-nearest neighbor, Amino Acid Composition, Physicochemical Properties [33] | General & Species-Specific [33] |
UbiPred distinguishes itself by employing an Informative Physicochemical Property (PCP) mining algorithm. This method selects the most relevant features from a large pool of 531 physicochemical properties in the AAindex database, which are then used to train its SVM classifier [32] [30]. In contrast, UbiSite utilizes a broader set of sequence-based features, including evolutionary information derived from the Position-Specific Scoring Matrix (PSSM), to capture the biological context around lysine residues [34]. UbiProber offers a unique flexibility; it is not only a general predictor but can also be trained to create species-specific models for Homo sapiens, Mus musculus, and Saccharomyces cerevisiae, acknowledging the sequence pattern differences that exist between species [33]. This capability is particularly valuable for researchers focusing on model organisms, as species-specific models often outperform general ones [33].
Performance Evaluation and Experimental Data
When selecting a prediction tool, empirical performance on benchmark datasets is a critical deciding factor. The following table summarizes key performance metrics for the three tools as reported in their foundational studies.
Table 2: Reported Performance Metrics of Prediction Tools
| Tool | Accuracy (Acc) | Sensitivity (Sn) | Specificity (Sp) | Area Under Curve (AUC) | Matthews Correlation Coefficient (MCC) |
|---|---|---|---|---|---|
| UbiPred | ~84% [30] | ~83% [30] | ~85% [30] | ~0.85 [30] | Not Reported |
| UbiSite | Not Reported | Not Reported | Not Reported | ~0.87 [34] | Not Reported |
| UbiProber | Not Reported | Not Reported | Not Reported | >0.80 (Species-specific models) [33] | Not Reported |
It is essential to interpret these metrics within their experimental context. The Area Under the Curve (AUC) of the Receiver Operating Characteristic (ROC) curve is a robust indicator of a model's overall classification ability, with values closer to 1.0 indicating better performance. UbiProber's strength lies in its species-specific models, which have been shown to achieve AUCs greater than 0.80, a significant improvement over general models when working with data from specific organisms [33]. Furthermore, a key concept in machine learning is the trade-off between Sensitivity (the ability to correctly identify true ubiquitination sites) and Specificity (the ability to correctly exclude non-ubiquitination sites). UbiPred's balanced performance across these two metrics suggests it is a reliable all-rounder [30].
Integrating Prediction with Experimental Validation
Computational predictions gain their ultimate value when integrated into a robust experimental workflow for validation. The following diagram illustrates the standard pipeline from computational prediction to experimental confirmation, which aligns with the thesis context of mutagenesis and immunoblotting.
Figure 1: Workflow from Prediction to Validation
The initial in silico analysis involves submitting the protein sequence to one or more prediction tools. The resulting scores allow researchers to prioritize lysine residues for experimental testing. Site-directed mutagenesis is then employed to create lysine-to-arginine (K→R) mutants, which prevent ubiquitination at the specific residue. Finally, immunoblotting (Western blotting) is used to compare the ubiquitination status of the wild-type protein versus the mutant. A key principle utilized here is that ubiquitination, particularly polyubiquitination, causes a discernible upward shift in the protein's apparent molecular weight, which can be visualized as a laddering pattern on the blot [28]. A disappearance of the ubiquitination signal in the K→R mutant, as detected by ubiquitin-specific antibodies, provides strong confirmation of the computational prediction [28].
The Scientist's Toolkit: Essential Research Reagents
To execute the experimental validation workflow, researchers require a set of essential reagents. The table below details key materials and their functions in the context of ubiquitination site validation.
Table 3: Essential Reagents for Ubiquitination Site Validation
| Research Reagent | Critical Function in Experimentation |
|---|---|
| Ubiquitin-Specific Antibodies | Core reagent for immunoblotting; detects ubiquitin-protein conjugates via recognition of ubiquitin or specific tags (e.g., HA, MYC, FLAG) [28]. |
| Epitope-Tagged Ubiquitin (e.g., HIS, FLAG, HA) | Enables affinity purification of ubiquitinated proteins under denaturing conditions (e.g., 8M urea) to reduce co-purification of non-specific proteins before MS analysis or immunoblotting [28]. |
| Mutagenesis Kits | Facilitates the creation of lysine-to-arginine (K→R) point mutations in the expression plasmid to confirm the specific ubiquitination site [28]. |
| Ni-NTA Agarose Resin | Critical for immobilised metal affinity chromatography (IMAC) to purify polyhistidine (6xHis)-tagged ubiquitin conjugates from complex cell lysates [28]. |
| Denaturing Lysis Buffer (e.g., with 8M Urea) | Preserves the transient ubiquitination modification during cell lysis by denaturing and inactivating deubiquitinating enzymes (DUBs) [28]. |
The use of epitope-tagged ubiquitin systems, such as 6xHis-myc-ubiquitin, has become a gold standard in the field. This system allows for the highly specific purification of ubiquitinated conjugates from total cell lysates under denaturing conditions, which is crucial for minimizing false positives from non-specifically bound proteins [28]. Furthermore, the choice of ubiquitin-specific antibodies is critical for immunoblotting. These antibodies can either recognize endogenous ubiquitin or the epitope tag (e.g., MYC) on the transfected ubiquitin, with the latter often providing a cleaner signal with reduced background.
The choice among UbiPred, UbiSite, and UbiProber is not a matter of identifying a single "best" tool, but rather of selecting the most appropriate one for a specific research context. UbiPred is a strong, general-purpose predictor with a proven balance of sensitivity and specificity, making it an excellent starting point for most investigations. UbiSite incorporates valuable evolutionary information, which may provide deeper biological insight. For researchers working with specific model organisms, UbiProber offers a distinct advantage through its dedicated species-specific models, which have demonstrated superior performance for their target species.
For a robust research strategy, it is advisable to run multiple prediction tools and look for consensus sites predicted with high confidence by more than one algorithm. These high-probability candidates should then be prioritized for the experimental validation pipeline of mutagenesis and immunoblotting outlined in this guide. This integrated approach of computational prediction followed by rigorous experimental confirmation provides an efficient and powerful methodology for expanding our understanding of the ubiquitin code and its functional roles in health and disease.
Experimental Workflow: From Mutagenesis to Immunoblot Detection
Lysine to arginine (K-to-R) mutagenesis serves as a cornerstone technique in molecular biology for dissecting protein function, particularly in the validation of ubiquitination sites. This guide explores the structural and biochemical rationale behind this mutagenesis approach, provides a comparative analysis of modern molecular cloning strategies for its implementation, and outlines detailed experimental protocols for validating ubiquitination sites through immunoblotting. Designed for researchers and drug development professionals, this resource synthesizes current methodologies with practical experimental workflows to inform strategic decision-making in protein engineering and functional genomics.
Lysine to arginine mutagenesis represents a fundamental protein engineering strategy that preserves positive charge while eliminating the epsilon-amino group targeted for post-translational modifications. This technique proves particularly invaluable in ubiquitination research, where identifying specific modification sites remains methodologically challenging. The approach capitalizes on the biochemical similarities and differences between these two basic amino acids to systematically probe lysine-dependent cellular processes without disrupting overall protein charge or structure.
Within molecular cloning, K-to-R mutagenesis has evolved from traditional restriction enzyme-based methods to sophisticated systems enabling precise, multi-site mutations with high efficiency. This technical evolution, coupled with advanced validation methodologies, has positioned K-to-R mutagenesis as an essential tool for deconvoluting complex ubiquitination signaling networks and understanding their implications in disease pathogenesis and therapeutic development.
Rationale: Biochemical and Structural Basis for K-to-R Substitution
Charge Preservation with Functional Elimination
The strategic value of K-to-R mutagenesis lies in its unique ability to maintain physicochemical properties while disrupting specific biochemical functionality:
- Charge conservation: Both lysine and arginine carry positive charges at physiological pH, minimizing perturbations to electrostatic interactions and protein folding.
- Amino group removal: Arginine lacks the primary epsilon-amino group that serves as the attachment point for ubiquitin molecules, thereby preventing ubiquitination at mutated sites.
- Structural compatibility: The longer, planar guanidinium group of arginine can often fit into spatial regions occupied by lysine without major structural rearrangements.
Enhanced Protein Stability through Structural Interactions
Beyond its application in ubiquitination studies, K-to-R mutagenesis can strategically enhance protein stability under specific conditions. Research using green fluorescent protein (GFP) as a model system demonstrates that surface lysine to arginine mutations can improve stability against chemical denaturants including urea, alkaline pH, and ionic detergents [35] [36].
The structural basis for this stabilization originates from the superior interaction capabilities of arginine's guanidinium group, which enables:
- Multi-directional interactions: Three asymmetrical nitrogen atoms (Nε, Nη1, Nη2) allow interactions in three possible directions versus the single direction available from lysine's amino group [36]
- Enhanced electrostatic networks: Capacity to form more salt bridges and hydrogen bonds compared to lysine
- Higher pKa advantage: The higher pKa of arginine's guanidinium group (∼12.5) versus lysine's amino group (∼10.5) maintains positive charge and ionic interactions under alkaline conditions [36]
Table 1: Comparative Properties of Lysine and Arginine Relevant to Mutagenesis
| Property | Lysine | Arginine | Implications for Mutagenesis |
|---|---|---|---|
| Charge at pH 7 | Positive | Positive | Preserves electrostatic interactions |
| Reactive Group | ε-amino group | Guanidinium group | Eliminates ubiquitination site |
| pKa of Group | ~10.5 | ~12.5 | Better maintains charge at alkaline pH |
| Interaction Directions | One | Three | Enables more stable salt bridges |
| Average H-bonds in GFP | ~2 | ~3 | Enhanced structural stability [36] |
Trade-offs in Protein Folding and Function
Despite the strategic advantages, K-to-R mutagenesis presents important limitations that must be considered in experimental design:
- Folding efficiency challenges: Comprehensive surface lysine replacement (e.g., 19 mutations in GFP) can severely compromise protein folding, yielding misfolded, non-functional proteins [36]
- Context-dependent effects: Residues critical for folding pathways or catalytic functions may not tolerate substitution despite surface location
- Productivity reduction: Altered electrostatic interactions can decrease functional protein yield even when folding occurs correctly [35]
These considerations highlight the importance of strategic, rather than comprehensive, K-to-R mutagenesis approaches focused on specific residues of interest while preserving structurally or functionally critical lysines.
Molecular Cloning Strategies for K-to-R Mutagenesis
Modern molecular cloning offers multiple pathways for implementing K-to-R mutations, each with distinct advantages in efficiency, scalability, and technical requirements. The following section compares mainstream methodologies, with performance data summarized in Table 2.
Site-Directed Mutagenesis (SDM) Methods
Site-directed mutagenesis enables precise, targeted nucleotide substitutions to create specific amino acid changes. Contemporary SDM systems have largely moved beyond early methods that required unique bacterial strains or uracil-containing DNA templates [37].
Q5 Site-Directed Mutagenesis System employs a "back-to-back" primer design that enables exponential amplification while producing non-nicked circular plasmids for higher transformation efficiency [37]. This system supports:
- Single amino acid substitutions through codon changes
- Multi-site mutagenesis (typically 1-3 sites simultaneously)
- Insertions (limited primarily by primer synthesis constraints)
- Deletions of various sizes
GeneArt System utilizes a methylase-based approach combined with high-fidelity DNA polymerase and McrBC endonuclease digestion to achieve mutagenesis efficiencies exceeding 90% [38]. This system demonstrates robust performance across various mutation types:
- 12-base substitutions, insertions, and deletions with >90% efficiency
- Simultaneous 3-site mutagenesis in plasmids up to 14kb
- Typical time-to-results of under 3 hours for standard plasmids
Random Mutagenesis Approaches
While SDM creates predetermined mutations, random mutagenesis generates diverse mutation libraries useful for exploratory studies or directed evolution:
Error-Prone PCR artificially increases nucleotide misincorporation during amplification by altering buffer conditions (e.g., unbalanced nucleotide ratios, elevated magnesium concentrations) [39]. This method benefits from adjustable mutation frequency through template concentration control but requires subsequent cloning steps.
XL1-Red Bacterial Strains utilize Escherichia coli deficient in DNA repair pathways to accumulate random mutations during plasmid replication [39]. This approach offers technical simplicity (no subcloning required) but lower mutation frequency (~1 mutation per 2000 base pairs).
Table 2: Performance Comparison of Molecular Cloning Methods for Mutagenesis
| Method | Mutation Efficiency | Key Advantages | Limitations | Best Applications |
|---|---|---|---|---|
| Q5 SDM | >90% (1-3 sites) | High efficiency; non-nicked products; exponential amplification [37] | Primer design constraints; cost | Targeted single/multi-site mutations |
| GeneArt SDM | >90% (even for 12-base changes) | Rapid (<3 hours); handles large plasmids [38] | Proprietary system; cost | Complex mutations in large constructs |
| Error-Prone PCR | Adjustable via template concentration | Controllable mutation rate; no specialized strains [39] | Requires subcloning; random mutations | Diverse mutant library generation |
| XL1-Red Cells | ~1/2000 bases | Technically simple; no in vitro manipulation [39] | Low frequency; unpredictable mutations | Low-frequency random mutagenesis |
Advanced DNA Assembly Methods
For complex mutagenesis projects involving multiple dispersed K-to-R substitutions or combination with other genetic elements, advanced DNA assembly methods provide powerful alternatives:
NEBuilder HiFi DNA Assembly enables seamless construction of large DNA molecules from multiple fragments, facilitating simultaneous introduction of numerous mutations across a gene [37].
Gibson Assembly allows one-step isothermal assembly of overlapping DNA fragments, suitable for combining multiple mutagenized regions into a complete expression construct [40].
These assembly methods are particularly valuable for comprehensive ubiquitination studies requiring systematic mutation of multiple putative modification sites or combination of K-to-R mutations with other genetic modifications.
Experimental Framework: Validating Ubiquitination Sites by K-to-R Mutagenesis and Immunoblotting
The following section provides detailed protocols for implementing K-to-R mutagenesis within ubiquitination validation workflows, from molecular cloning to functional confirmation.
Molecular Cloning Workflow for K-to-R Mutations
The experimental pathway for creating and validating K-to-R mutants follows a systematic process from design to protein characterization:
Diagram 1: K-to-R Mutagenesis Experimental Workflow
Immunoblotting Protocol for Ubiquitination Validation
After successful creation and expression of K-to-R mutants, immunoblotting provides definitive validation of ubiquitination status through these key steps:
Sample Preparation and Denaturation:
- Lyse cells in denaturing buffer (e.g., containing 8M urea) to preserve ubiquitination status and prevent deubiquitinase activity [28] [36]
- Include protease inhibitors and N-ethylmaleimide (NEM) to inhibit deubiquitinating enzymes
- Denature samples at 95°C for 5 minutes in SDS-containing buffer
Gel Electrophoresis and Transfer:
- Resolve proteins using 6-12% gradient SDS-PAGE for optimal separation of ubiquitinated species [28]
- Transfer to PVDF or nitrocellulose membranes using standard western blot protocols
Immunodetection and Analysis:
- Probe with primary antibodies against:
- Target protein (to confirm equal loading)
- Ubiquitin (e.g., P4D1, FK1, FK2) or epitope tags if using tagged ubiquitin systems [41]
- Use appropriate HRP-conjugated secondary antibodies
- Develop with enhanced chemiluminescence substrate
- Interpret results by comparing wild-type and mutant migration patterns
Key Validation Criteria:
- Molecular weight shifts: Ubiquitination causes characteristic upward shifts (~8 kDa for mono-ubiquitination, larger for polyubiquitination) [28]
- Signal elimination: Successful K-to-R mutation at ubiquitination sites eliminates or reduces ubiquitin signal compared to wild-type
- Ladder patterns: Polyubiquitinated species often appear as characteristic ladder patterns on immunoblots
Advanced Validation: Mass Spectrometry Corroboration
While immunoblotting provides initial validation, mass spectrometry offers definitive site-specific confirmation:
- Di-glycine remnant detection: Trypsin digestion leaves a 114.043 Da mass signature on ubiquitinated lysines [28] [41]
- Virtual Western blots: Computational reconstruction of molecular weight from MS data validates ubiquitination status [28]
- Multiple reaction monitoring: Targeted MS assays quantitatively confirm absence of ubiquitination at specific sites in K-to-R mutants
Essential Research Tools for K-to-R Mutagenesis Studies
Successful implementation of K-to-R mutagenesis workflows requires specific reagents and tools optimized for ubiquitination research:
Table 3: Essential Research Reagents for K-to-R Mutagenesis and Ubiquitination Validation
| Reagent/Tool | Function | Key Considerations |
|---|---|---|
| High-Fidelity DNA Polymerase | Amplifies mutant sequences with minimal errors | Critical for multi-site mutagenesis; examples: Q5, Pfu |
| Site-Directed Mutagenesis Kits | Streamlined systems for creating specific mutations | Consider efficiency, number of sites, plasmid size [37] [38] |
| Tagged Ubiquitin Constructs | Enables affinity purification of ubiquitinated proteins | His-tag, Strep-tag, or HA-tag systems available [41] |
| Ubiquitin Antibodies | Detects ubiquitinated species in immunoblots | Linkage-specific (e.g., K48, K63) or pan-specific options [41] |
| Tandem Ubiquitin-Binding Entities (TUBEs) | Enriches endogenous ubiquitinated proteins without genetic tags | Higher affinity than single UBDs; preserves native ubiquitination [41] |
| Deubiquitinase Inhibitors | Preserves ubiquitination status during lysis | N-ethylmaleimide (NEM) prevents DUB activity [42] |
| Denaturing Lysis Buffers | Maintains ubiquitination during protein extraction | 8M urea or similar denaturants prevent deubiquitination [28] |
Lysine to arginine mutagenesis represents a powerful, precise approach for interrogating protein ubiquitination and engineering enhanced protein stability. The strategic selection of molecular cloning methods—from high-efficiency site-directed mutagenesis for targeted studies to random mutagenesis for exploratory work—enables researchers to effectively implement this technique across diverse experimental contexts. When coupled with robust immunoblotting validation protocols and mass spectrometry confirmation, K-to-R mutagenesis provides unequivocal evidence for ubiquitination site identification, advancing our understanding of ubiquitin signaling in both physiological and pathological processes. As mutagenesis technologies continue evolving toward higher efficiency and greater multiplexing capacity, this fundamental protein engineering strategy will remain indispensable for deconvoluting complex post-translational regulatory networks in biomedical research and therapeutic development.
Protein ubiquitination is a crucial post-translational modification that regulates diverse cellular functions, including targeted degradation, cell signaling, and immune responses [21] [43]. Validation of ubiquitination sites through mutagenesis and immunoblotting remains a cornerstone of ubiquitination research. This guide objectively compares core methodologies for cell-based ubiquitination assays, providing researchers with a framework to select the optimal approach for their specific experimental needs, from initial detection to high-throughput inhibitor screening.
Key Assay Methodologies: A Comparative Analysis
The choice of ubiquitination detection method significantly impacts the specificity, throughput, and biological relevance of the data obtained. The table below summarizes the core characteristics of prevalent techniques.
Table 1: Comparison of Core Ubiquitination Assay Methodologies
| Methodology | Key Principle | Linkage Specificity | Throughput | Key Advantages | Primary Limitations |
|---|---|---|---|---|---|
| Immunoblotting/ Immunoprecipitation (IP) [44] [3] | Uses ubiquitin-or linkage-specific antibodies to detect or enrich target proteins. | Variable (Pan- or linkage-specific antibodies available) | Low | Accessible, works for endogenous proteins, can be linkage-specific. | Semi-quantitative, low-throughput, antibody quality is critical. |
| Tandem Ubiquitin Binding Entities (TUBEs) [21] [3] | High-affinity matrices with tandem ubiquitin-binding domains enrich polyubiquitinated proteins. | High (Pan-selective or chain-specific TUBEs available) | Medium-High (adaptable to 96-well format) | Protects ubiquitin chains from deubiquitinases (DUBs), enables high-throughput application. | Requires specialized TUBE reagents. |
| Denaturing Lysis & Affinity Purification [3] [45] | Cells lysed under denaturing conditions (e.g., SDS, guanidinium-HCl) to preserve ubiquitination, followed by enrichment (e.g., His-purification). | Depends on enrichment tag (e.g., His-tagged ubiquitin) | Low | Preserves unstable modifications, minimizes post-lysis deubiquitination and protein-protein interactions. | Requires genetic manipulation (tagged ubiquitin), labor-intensive. |
| Fluorescence Polarization (UbiReal Assay) [46] | Monitors real-time ubiquitin transfer and conjugation using fluorescently-labeled ubiquitin. | Can be designed for specificity | High | Real-time kinetic data, excellent for inhibitor screening and mechanistic studies. | Typically in vitro or semi-in vivo, may not reflect full cellular complexity. |
Detailed Experimental Protocols
Conventional Immunoprecipitation and Immunoblotting for Endogenous Proteins
This protocol is ideal for validating ubiquitination of a specific endogenous protein, such as mitochondrial antiviral signaling protein (MAVS), and is foundational for mutagenesis studies [44].
- Cell Lysis: Lyse cells in a modified RIPA or cell lysis buffer for Western/IP, supplemented with protease and deubiquitinase (DUB) inhibitors (e.g., 10 μM PR-619). Note: For validation via mutagenesis, this step follows transfection with plasmid encoding your protein of interest with lysine-to-arginine (K-to-R) mutations [3].
- Immunoprecipitation: Incubate the cleared lysate with a target-specific antibody (e.g., anti-MAVS antibody [44]) and Protein G PLUS-Agarose beads for several hours at 4°C.
- Washing and Elution: Wash beads stringently to remove non-specifically bound proteins. Elute bound proteins by boiling in 1X SDS-PAGE sample buffer.
- Immunoblotting: Resolve proteins by SDS-PAGE, transfer to a membrane, and probe with a linkage-specific ubiquitin antibody (e.g., anti-Ub-K27 [44] or anti-Ub-K63 [21]) to confirm the ubiquitin chain topology. Reprobing with the target protein antibody confirms successful IP.
TUBE-Based High-Throughput Ubiquitination Assay
This method, demonstrated for monitoring endogenous RIPK2 ubiquitination, leverages TUBEs for sensitive, linkage-specific detection in a format suitable for screening [21].
- Cell Treatment and Lysis: Treat cells (e.g., THP-1) with your stimulus (e.g., L18-MDP for K63-linked ubiquitination) or PROTAC (e.g., RIPK2 degrader for K48-linked ubiquitination) in a 96-well plate. Lyse cells using a buffer optimized to preserve polyubiquitination.
- Capture with TUBEs: Incubate cell lysates in wells pre-coated with chain-specific TUBEs (e.g., K63-TUBE, K48-TUBE, or Pan-TUBE).
- Detection: After washing, detect captured ubiquitinated proteins using an antibody against your target protein (e.g., anti-RIPK2). Quantification can be achieved via colorimetric, chemiluminescent, or fluorescent readouts.
Denaturing Lysis and Affinity Purification for Tagged Ubiquitin
This protocol is critical for reducing false negatives caused by deubiquitination during processing and is widely used for proteomic studies [3] [45].
- Cell Transfection and Harvest: Transfect cells with a plasmid encoding 6xHis-tagged ubiquitin. Harvest cells and pellet them by centrifugation.
- Denaturing Lysis: Lyse the cell pellet in a denaturing buffer (e.g., 1% SDS, 8 M Urea, or 6 M Guanidinium-HCl) with Benzonase to degrade nucleic acids and reduce viscosity. Boiling for 10 minutes ensures complete denaturation.
- Affinity Purification: Dilute the lysate to reduce denaturant concentration. Incubate with Ni-NTA agarose resin for 2-3 hours at room temperature with end-over-end rotation. Critical: Avoid EDTA or strong reducing agents (DTT, BME) in the buffer, as they will disrupt the nickel-resin interaction [45].
- Washing and Elution: Wash the resin sequentially with wash buffers of decreasing denaturant strength and increasing imidazole concentration. Elute the bound His-tagged ubiquitinated proteins with elution buffer containing high-concentration imidazole (e.g., 250-300 mM).
Diagram 1: A workflow to guide researchers in selecting the most appropriate ubiquitination assay based on their experimental goals, such as confirming endogenous protein modification, high-throughput screening, or kinetic studies.
The Scientist's Toolkit: Key Reagent Solutions
Successful execution of ubiquitination assays relies on a suite of specialized reagents. The following table details essential tools and their functions.
Table 2: Essential Research Reagents for Ubiquitination Assays
| Reagent Category | Specific Examples | Function in Assay |
|---|---|---|
| Linkage-Specific Antibodies | Anti-Ubiquitin (K48-linkage specific), Anti-Ubiquitin (K63-linkage specific), Anti-Ubiquitin (K27-linkage specific) [44] [3] | Detect specific polyubiquitin chain topologies in immunoblotting or IP. Critical for determining functional outcome. |
| Affinity Enrichment Tools | Tandem Ubiquitin Binding Entities (TUBEs - K48, K63, Pan) [21], Ni-NTA Agarose (for His-tagged Ub) [3] [45], Anti-FLAG M2 Affinity Gel [47] | High-affinity capture and purification of ubiquitinated proteins from complex lysates. TUBEs offer protection from DUBs. |
| Activity Probes & Substrates | Fluorescein-Ubiquitin (F-Ub) [46], Ub-AMC [46], Ub-Rhodamine [46] | Fluorescent substrates for real-time activity monitoring of E1/E2/E3 enzymes (F-Ub) or DUBs (Ub-AMC) in high-throughput screens. |
| Pharmacologic Inhibitors/Activators | Bortezomib/Epoxomicin (Proteasome) [48] [47], PYR-41 (E1 inhibitor) [46], PROTACs (e.g., RIPK2 degrader-2) [21] | Tools to perturb the ubiquitin-proteasome system. PROTACs induce targeted K48-linked ubiquitination and degradation. |
| Critical Buffers & Kits | Denaturing Lysis Buffer (Guanidinium-HCl/Urea) [45], Protease Inhibitor Cocktail (without DUB inhibitors), DUB Inhibitor (e.g., PR-619) | Maintain integrity of ubiquitin signals during cell lysis and processing. |
Advanced Applications and Integrated Workflow
Understanding the ubiquitin-proteasome system (UPS) as a whole is vital for contextualizing assay data. Assays can be designed to probe specific stages of this pathway, from ubiquitin conjugation to proteasomal degradation.
Diagram 2: The Ubiquitin-Proteasome System Pathway. This diagram illustrates the sequential action of E1, E2, and E3 enzymes in attaching ubiquitin (Ub) to a substrate protein. The fate of the ubiquitinated substrate—either degradation by the proteasome (typically K48-linked chains) or another functional outcome (e.g., signaling via K63-linked chains)—is counterbalanced by the activity of deubiquitinating enzymes (DUBs). Assays can target any node in this pathway.
The integration of these methodologies forms a powerful, iterative research cycle. Initial discovery via denaturing purification or TUBE-based assays can identify candidate ubiquitinated proteins or specific linkages. These findings are then validated and pinpointed through mutagenesis and immunoblotting, where putative ubiquitination sites are mutated to arginine to confirm their necessity for the modification [3]. Finally, high-throughput fluorescence polarization or TUBE assays enable the screening for small-molecule modulators or detailed kinetic studies of the ubiquitination process [21] [46]. This systematic approach ensures robust and translatable findings in the complex landscape of ubiquitin signaling.
Immunoprecipitation (IP) under denaturing conditions represents a critical methodological approach for studying labile post-translational modifications (PTMs), particularly ubiquitination, SUMOylation, acetylation, and tyrosine phosphorylation. This technique enables researchers to preserve transient modification states by inhibiting endogenous hydrolases and disrupting protein complexes that might otherwise obscure PTM detection. As research increasingly reveals extensive crosstalk between different PTMs operating on single protein targets, the development of unified denaturing systems capable of investigating multiple modifications simultaneously has become essential for accurate pathway elucidation. This guide objectively compares current methodologies, buffer systems, and affinity reagents for studying covalent modifications, with particular emphasis on experimental workflows for validating ubiquitination sites through mutagenesis and immunoblotting.
Post-translational modifications represent dynamic, reversible alterations that dramatically expand protein functional diversity, increasing proteoforms from approximately 30,000 gene products to over a million distinct entities [49]. Among these, ubiquitination stands as a particularly versatile regulator of protein stability, activity, and localization, with dysregulation leading to cancer, neurodegenerative diseases, and other pathologies [3]. The inherent challenges in studying ubiquitination and other PTMs include their transient nature, low stoichiometry under physiological conditions, and the liability of modifications when native cellular environments are preserved [49].
Traditional immunoprecipitation under native conditions often proves inadequate for PTM investigation due to several factors:
- PTM Lability: Native buffers fail to inhibit endogenous enzymes like deubiquitinases and deSUMOylases that rapidly remove modifications during processing
- Protein Complex Interference: Non-denaturing buffers preserve protein interactions that can mask PTM sites or generate false positives through indirect associations
- Compartmental Limitations: Gentle lysis methods inefficiently extract proteins from all cellular compartments, particularly nuclear proteins [49]
Denaturing IP conditions address these limitations by employing agents that disrupt protein structure and inhibit enzymatic activity, thereby "freezing" PTM states at the moment of cell lysis. This approach provides a more accurate snapshot of modification status, though it introduces unique technical challenges including genomic DNA contamination, buffer incompatibility issues, and potential antibody epitope disruption [49].
Comparative Methodologies for PTM Investigation
Systematic Comparison of IP Approaches
Table 1: Comparison of immunoprecipitation methodologies for PTM analysis
| Method | Conditions | Primary Applications | Advantages | Limitations |
|---|---|---|---|---|
| Native IP/Co-IP | Non-denaturing buffers, mild detergents | Protein-protein interaction mapping, intact complex isolation | Preserves native interactions, maintains protein function | PTM loss from endogenous enzymes, false positives from indirect interactions [50] [51] |
| Denaturing IP | Strong denaturants (SDS, urea), reducing agents | Labile PTM preservation (ubiquitination, SUMOylation), covalent modification studies | Inhibits modifying enzymes, dissociates protein complexes, improves specificity | Potential antibody incompatibility, genomic DNA contamination, epitope disruption [49] [52] |
| Tag-Based Affinity Purification | Variable (native or denaturing) | High-throughput PTM screening, recombinant protein studies | High affinity capture, well-characterized reagents, consistent performance | Tag may alter protein function/ localization, requires genetic manipulation [3] [51] |
Quantitative Performance Metrics
Table 2: Performance comparison of denaturing IP methodologies for ubiquitination analysis
| Enrichment Method | Sensitivity | Specificity | Ubiquitination Sites Identified | Compatibility with Tissue Samples |
|---|---|---|---|---|
| Ub Antibody-Based | Moderate | High | 96 sites (MCF-7 breast cancer cells) [3] | Excellent (works with clinical samples) [3] |
| His-Tag Ub Exchange | Moderate | Moderate | 277 sites (HeLa cells) [3] | Limited (requires genetic manipulation) [3] |
| Strep-Tag Ub System | High | High | 753 sites (U2OS/HEK293T cells) [3] | Limited (requires genetic manipulation) [3] |
| UBD-Based Approaches | Variable | High | Dependent on UBD affinity and specificity [3] | Excellent (works with endogenous proteins) [3] |
Integrated Denaturing System for Multiple PTM Analysis
Recent methodological advances have addressed the historical challenge of investigating multiple PTMs using different buffer systems. A comprehensive denaturing lysis system has been developed that effectively preserves and enables investigation of four key PTMs—ubiquitination, tyrosine phosphorylation, SUMOylation 2/3, and acetylation—within a single experimental framework [49]. This system incorporates several innovations:
- Unified Denaturing Lysis Buffer: A unique formulation that maintains compatibility with PTM affinity reagents while effectively denaturing proteins
- Genomic DNA Removal: A specialized filter system that eliminates contaminating genomic DNA, a common problem with denaturing buffers that traditionally requires sonication or syringe lysis
- Comprehensive Inhibitor Cocktails: Simultaneous inhibition of tyrosine phosphatases, deubiquitinases, deSUMOylases, histone deacetylases, and proteases [49]
This integrated approach streamlines the investigation of potential PTM crosstalk, which has emerged as a critical regulatory mechanism for fine-tuning protein function [49].
Experimental Protocols for Ubiquitination Analysis
Denaturing Lysis and Immunoprecipitation Protocol
Sample Preparation and Lysis
- Grow cells to appropriate confluency (e.g., A431 cells to 50% confluency) and apply relevant treatments
- Place cells on ice, wash twice with 10 mL of cold 1x PBS, removing as much PBS as possible
- Add denaturing lysis buffer (600 µL per 150 mm plate) containing comprehensive inhibitors:
- Tyrosine phosphatase inhibitor
- Deubiquitinase and deSUMOylase inhibitor
- Histone deacetylase (HDAC) inhibitor
- Protease inhibitor cocktail [49]
- Lyse cells using a cell scraper; lysate will become viscous due to nuclear lysis
Lysate Clarification and DNA Removal
- Transfer crude lysate using a snipped pipette tip to a specialized DNA removal filter placed in a 15 mL collection tube
- Use supplied filter plunger to compress the filter and collect clarified lysate flow-through
- Optional: Centrifuge clarified lysate at 10,000 × g for 1 minute at 4°C to remove any remaining particulates
- Transfer supernatant to a new tube and dilute with appropriate dilution buffer to achieve final buffer composition compatible with IP reactions [49]
Immunoprecipitation Procedure
- Quantitate protein concentration using compatible assay (e.g., precision red advanced protein assay)
- Pre-clear lysate with protein A/G beads (optional for magnetic beads) [53]
- Incubate lysate with primary antibody or pre-immobilized antibody-bead complexes
- For ubiquitination studies: Use linkage-specific Ub antibodies or general Ub antibodies (FK2, P4D1) [3]
- Antibody amount: Typically 1-5 µg per IP, optimize based on abundance
- Incubate with gentle agitation for 1-4 hours at 4°C or overnight for low-abundance targets
- Wash beads 3-4 times with appropriate wash buffer (increasing stringency if needed)
- Elute proteins by boiling in SDS-PAGE sample buffer or using specific elution buffers [49] [54]
Ubiquitination Validation by Mutagenesis and Immunoblotting
Site-Directed Mutagenesis Approach
- Identify putative ubiquitination sites through sequence analysis or mass spectrometry data
- Design primers to mutate target lysine residues to arginine (conservative mutation that prevents ubiquitination)
- Perform site-directed mutagenesis and verify mutations by sequencing
- Express wild-type and mutant proteins in relevant cell lines [3]
Immunoblotting Validation
- Perform denaturing IP as described above from both wild-type and mutant expressing cells
- Separate proteins by SDS-PAGE and transfer to nitrocellulose or PVDF membrane
- Probe with ubiquitin-specific antibodies and target protein antibodies
- Compare ubiquitination signals between wild-type and mutant proteins
- A significant reduction in ubiquitination signal for the mutant compared to wild-type provides validation of the specific ubiquitination site [3]
This conventional mutagenesis approach, while time-consuming, remains widely used for validating ubiquitination sites identified through proteomic screening [3].
Research Reagent Solutions
Table 3: Essential reagents for denaturing immunoprecipitation experiments
| Reagent Category | Specific Examples | Function | Considerations |
|---|---|---|---|
| Denaturing Agents | SDS, urea | Disrupt protein interactions, inhibit modifying enzymes | Concentration critical; must be diluted for antibody compatibility [49] [52] |
| Bead Matrices | Magnetic protein A/G beads, agarose resin | Solid support for antibody immobilization | Magnetic beads offer easier handling; agarose may have higher capacity [53] |
| Ubiquitin Antibodies | P4D1, FK1/FK2 (pan-specific), linkage-specific (K48, K63, etc.) | Detect and enrich ubiquitinated proteins | Linkage-specific antibodies enable chain type determination [3] |
| Protease Inhibitors | PMSF, complete protease inhibitor cocktails | Prevent protein degradation during processing | Essential in all steps before denaturation [49] |
| Deubiquitinase Inhibitors | PR-619, N-ethylmaleimide | Preserve ubiquitination state | Critical for accurate ubiquitination assessment [49] |
| Tag Systems | His, Strep, FLAG, HA | Alternative enrichment strategy | High affinity, consistent performance; may alter protein behavior [3] [51] |
| Lysis Buffers | blastR buffer, RIPA, SDS-containing buffers | Extract proteins while preserving PTMs | Denaturing buffers better for nuclear proteins and disrupting interactions [49] |
Visualization of Experimental Workflows
Denaturing IP Workflow for Ubiquitination Analysis
Ubiquitination Validation by Mutagenesis
Technical Considerations and Optimization
Antibody Selection and Validation
Choosing appropriate antibodies represents one of the most critical factors in successful denaturing IP experiments. Key considerations include:
- IP Validation: Select antibodies specifically validated for immunoprecipitation under denaturing conditions, as antibodies working in native IP may fail when proteins are denatured [54]
- Epitope Accessibility: Ensure the antibody recognizes an epitope that remains accessible after denaturation; linear epitopes often perform better in denaturing conditions
- Species Compatibility: Match antibody host species with appropriate protein A/G beads (protein A for rabbit IgG, protein G for mouse IgG) [54] [55]
- Control Antibodies: Include species-matched non-specific IgG controls to distinguish specific binding from non-specific interactions [55]
For ubiquitination studies, antibody selection expands to include:
- Pan-ubiquitin Antibodies: Recognize ubiquitin regardless of linkage type (e.g., P4D1, FK1/FK2)
- Linkage-Specific Antibodies: Target specific ubiquitin chain linkages (K48, K63, M1, etc.), enabling functional insights [3]
- Tag-Specific Antibodies: When using tagged ubiquitin systems (His, Strep, HA, FLAG), select high-affinity antibodies against the tag [3] [51]
Bead Selection and Handling
The solid support matrix significantly impacts IP efficiency and specificity:
- Magnetic vs. Agarose Beads: Magnetic beads (1-4 μm diameter) offer easier handling, better reproducibility, and reduced background; agarose beads (50-150 μm) may provide higher theoretical capacity but require centrifugation [53]
- Binding Capacity: Despite lower theoretical capacity, magnetic beads often yield equal or better antigen recovery due to complete antibody accessibility on their smooth surface [53]
- Handling Precautions: Always remove supernatant by pipetting, not vacuum aspiration, to prevent bead loss [54] [53]
- Wash Stringency: Optimize wash buffer composition and number of washes to balance signal retention with background reduction [55]
Troubleshooting Common Issues
- High Background: Increase wash stringency (salt concentration, add mild detergents), pre-clear lysate, optimize antibody concentration, and ensure thorough washing [54] [55]
- Low Signal: Verify antibody compatibility with denaturing conditions, increase input material, extend incubation times, and check inhibitor efficacy [49] [54]
- Genomic DNA Contamination: Implement specialized filter systems or brief sonication to shear DNA when using strongly denaturing buffers [49]
- Antibody Interference in Detection: Use covalently immobilized antibodies or ensure detection antibodies recognize different species than IP antibody to avoid heavy/light chain detection [55]
Immunoprecipitation under denaturing conditions provides an essential toolset for investigating labile post-translational modifications, particularly ubiquitination. The development of unified denaturing systems that preserve multiple PTMs simultaneously represents a significant advancement, enabling more accurate investigation of potential crosstalk between different modification types. When combined with mutagenesis and immunoblotting validation approaches, these methods form a robust framework for elucidating the complex regulatory networks governed by covalent protein modifications. As mass spectrometry sensitivity continues to improve and more specific affinity reagents become available, denaturing IP methodologies will remain foundational for precise mapping of ubiquitination sites and understanding their functional consequences in both physiological and pathological contexts.
In the study of protein ubiquitination, immunoblotting (Western blotting) remains an indispensable analytical technique for researchers seeking to validate post-translational modifications. The detection of characteristic molecular weight shifts and heterogeneous smearing patterns serves as a hallmark signature of ubiquitination events, providing critical evidence beyond what mass spectrometry alone can offer [28]. This guide examines how these distinctive immunoblotting patterns enable the identification and validation of ubiquitinated proteins, comparing traditional and automated methodologies while placing these techniques within the broader context of ubiquitination research.
The fundamental principle underlying this application stems from the nature of ubiquitination itself. As a library, NLM provides access to scientific literature. Inclusion in an NLM database does not imply endorsement of, or agreement with, the contents by NLM or the National Institutes of Health. [28] Each ubiquitin modification adds approximately 8 kDa to a protein's molecular weight, with polyubiquitination creating even larger shifts [28]. Furthermore, the variable number and linkage types of ubiquitin chains frequently generate heterogeneous modified substrates that manifest as the characteristic laddering or smearing patterns observed on Western blots [28]. These visual signatures provide researchers with immediate, accessible evidence of ubiquitination status while guiding subsequent validation experiments.
The Molecular Basis of Ubiquitination Detection
Ubiquitination Machinery and Detection Implications
Protein ubiquitination occurs through a sequential enzymatic cascade involving E1 activating enzymes, E2 conjugating enzymes, and E3 ligases, resulting in the covalent attachment of ubiquitin to substrate proteins primarily through lysine ε-amino groups [28]. Recent research has revealed that this process can extend beyond protein substrates, with evidence showing direct ubiquitination of small molecules like BRD1732, which contains an azetidine secondary amine that forms an amide bond with the ubiquitin C-terminus [56]. This unexpected finding demonstrates the versatility of ubiquitination machinery while underscoring the importance of immunoblotting for detecting novel modification types.
The detection of ubiquitination relies on recognizing two interconnected phenomena:
- Molecular weight increases: Monoubiquitination adds approximately 8 kDa, while polyubiquitination creates even larger shifts [28]
- Band patterns: Heterogeneous modification generates smears or ladders rather than discrete bands [28]
These features remain consistent across various ubiquitination contexts, whether studying traditional protein substrates, regulatory factors like Midnolin [57], or disease-associated mutants such as NFIX frameshift proteins in Malan syndrome [58].
Ubiquitination Site Validation Through Mutagenesis
A critical application of immunoblotting in ubiquitination research involves validating modification sites through targeted mutagenesis. As illustrated in Figure 1, this process creates an experimental workflow where potential ubiquitination sites are systematically mutated and the effects analyzed through immunoblotting.
Figure 1. Experimental workflow for validating ubiquitination sites through mutagenesis and immunoblotting. The process begins with identification of potential ubiquitination sites, followed by site-directed mutagenesis, protein expression, immunoblotting analysis, and confirmation through characteristic banding pattern changes.
This approach was effectively demonstrated in midnolin research, where systematic mutation of six lysine residues (K76, K84, K264, K354, K372, and K402) to arginine progressively reduced ubiquitination levels, with the 6KR mutant showing significantly diminished ubiquitination compared to single mutants [57]. Similarly, studies of the small molecule BRD1732 showed that methylation of the azetidine nitrogen (the ubiquitination site) completely prevented ubiquitin conjugate accumulation [56]. These mutagenesis experiments, coupled with immunoblotting analysis, provide compelling evidence for specific ubiquitination sites.
Comparative Methodologies in Ubiquitination Detection
Traditional vs. Automated Immunoblotting Platforms
The detection of ubiquitination signatures can be accomplished through various immunoblotting platforms, each with distinct advantages and limitations. Table 1 provides a direct comparison of traditional Western blotting with two automated systems that offer different degrees of automation.
Table 1. Comparison of Traditional and Automated Western Blotting Methods
| Parameter | Traditional Western Blotting | iBind Flex (Semi-Automated) | JESS Simple Western (Fully Automated) |
|---|---|---|---|
| Automation Level | Manual | Semi-automated (immunodetection only) | Fully automated (separation to analysis) |
| Hands-on Time | 1-3 days [23] [59] | Reduced for immunodetection steps | Minimal (sample preparation and loading only) |
| Sample Requirement | Larger amounts (10-50 μg) [59] | Similar to traditional | Significantly less (limited sample beneficial) [59] |
| Sensitivity | Standard | Comparable to traditional | Enhanced sensitivity [59] |
| Reproducibility | Variable due to multiple manual steps [59] | Improved for immunodetection | High (automation of critical steps) [59] |
| Cost Considerations | Lower equipment cost | Moderate (device and reagents) | Higher (device and reagent costs) [59] |
| Ubiquitination Detection Capability | Excellent for smears and shifts | Suitable for established targets | Excellent sensitivity for low-abundance targets |
| Method Flexibility | High (easily adjustable protocols) | Moderate | Lower (standardized protocols) |
Experimental Protocols for Ubiquitination Detection
Traditional Immunoblotting for Ubiquitination Analysis
The following protocol outlines the standard approach for detecting ubiquitination through traditional Western blotting, with specific considerations for identifying characteristic molecular weight shifts and smearing patterns:
Sample Preparation: Lyse cells in RIPA buffer (4°C, 30 min) with proteasome inhibitors (e.g., MG132) to stabilize ubiquitinated species [57] [59]. Clear lysates by centrifugation (2000× g, 5 min) and quantify protein concentration using BCA assay [59].
Gel Electrophoresis: Resolve 10-50 μg total protein on 4-20% gradient SDS-PAGE gels [59]. Gradient gels are particularly valuable for ubiquitination studies as they maximize resolution across a broad molecular weight range, essential for detecting ubiquitin ladders [28].
Protein Transfer: Transfer to nitrocellulose membranes (0.2 μm) using standard wet or semi-dry transfer systems [59].
Immunodetection:
Data Interpretation: Analyze membranes for characteristic upward shifts (∼8 kDa increments) and smearing patterns indicative of heterogeneous ubiquitination [28].
Virtual Western Blot Alternative
For large-scale ubiquitination studies, researchers have developed "virtual Western blot" approaches that combine gel electrophoresis, mass spectrometry, and computational analysis. This method computes experimental molecular weights from the distribution of spectral counts in gel bands using Gaussian curve fitting, allowing systematic differentiation of ubiquitinated species from co-purified unmodified components [28]. This approach has demonstrated that approximately 95% of proteins with defined ubiquitination sites show convincing molecular weight increases on virtual blots [28].
Research Reagent Solutions for Ubiquitination Studies
Table 2. Essential Research Reagents for Ubiquitination Immunoblotting
| Reagent/Category | Specific Examples | Function in Ubiquitination Research |
|---|---|---|
| Ubiquitination Inhibitors | MG132 [57] | Proteasome inhibitor that stabilizes ubiquitinated proteins for detection |
| Lysis Buffers | RIPA Buffer [59], Denaturing Buffer (8M urea) [28] | Extract proteins while preserving ubiquitination modifications |
| Ubiquitin Detection Antibodies | Anti-K-ε-GG antibodies [57] | Specifically detect di-glycine remnant on ubiquitinated lysines |
| Secondary Antibodies | HRP-conjugated goat anti-rabbit IgG [59] | Enable chemiluminescent detection of target proteins |
| Detection Substrates | ECL Plus Western Blotting Substrate [59] | Generate light signal for imaging ubiquitinated species |
| Protein Standards | Pre-stained MW markers (10-220 kDa) [28] | Reference for identifying ubiquitination-induced molecular weight shifts |
| Proteasome Components | RNF19A/B E3 ligases, UBE2L3 E2 enzyme [56] | Key ubiquitination machinery for functional validation studies |
Applications in Disease Research and Drug Development
The integration of immunoblotting-based ubiquitination detection with mutagenesis has yielded significant insights into disease mechanisms and therapeutic development. In cancer research, the discovery that small molecules like BRD1732 undergo direct stereospecific ubiquitination via RNF19A/B E3 ligases and UBE2L3 E2 enzyme reveals novel targeting opportunities for proteasome inhibition [56]. Immunoblotting confirmed both monoubiquitin and diubiquitin conjugates of BRD1732, with accumulation dependent on specific ubiquitination machinery [56].
In genetic disorders such as Malan syndrome, immunoblotting demonstrated that NFIX frameshift mutations (c.164delC, p.Ala55Glyfs*2) cause significantly diminished protein expression despite normal mRNA levels, with proteasome inhibitor (MG132) experiments confirming ubiquitin-proteasome pathway-mediated degradation of the truncated protein [58]. Similarly, studies of midnolin have revealed a complex relationship between ubiquitination and function, where a 6KR mutant (simultaneous mutation of six ubiquitination sites) displayed impaired substrate degradation despite normal proteasome binding [57].
These applications highlight how molecular weight shifts and smearing patterns on immunoblots provide critical functional insights, enabling researchers to connect ubiquitination status with biological outcomes in both basic research and therapeutic development contexts.
Characteristic molecular weight shifts and heterogeneous smearing patterns remain definitive hallmarks of protein ubiquitination detectable through immunoblotting. When combined with site-directed mutagenesis, these detection methods form a powerful validation framework for identifying modification sites and understanding functional consequences. While traditional Western blotting offers flexibility and accessibility, automated systems provide enhanced reproducibility and sensitivity for challenging targets. As research continues to expand beyond traditional protein substrates to include small molecules and novel modification types, immunoblotting detection of ubiquitination signatures will remain essential for advancing both basic science and therapeutic development.
The post-translational modification of proteins by ubiquitination is a critical regulatory mechanism controlling numerous cellular processes, including protein degradation, signaling, and localization. This case study focuses on the experimental validation of a specific ubiquitination site, lysine 222 (K222), on Peroxisome Proliferator-activated receptor γ1 (PPARγ1). PPARγ1 is a nuclear receptor transcription factor that serves as a master regulator of adipogenesis, glucose homeostasis, and cell growth [60]. Its expression and activity are tightly controlled by various post-translational modifications, with ubiquitination playing a pivotal role in determining its protein stability and transcriptional activity [60] [61]. The identification of specific ubiquitination sites is essential for understanding the molecular mechanisms governing PPARγ1 function in both physiological and pathological contexts.
This guide objectively compares the methodological approaches and presents the experimental data that established K222 as the key site for MuRF2-mediated ubiquitination, providing researchers with a framework for similar ubiquitination site validation projects.
Experimental Findings and Data Comparison
Key Experimental Results Validating K222 Ubiquitination
Table 1: Summary of Key Experimental Findings for PPARγ1-K222 Ubiquitination
| Experimental Approach | Key Finding | Biological Implication |
|---|---|---|
| Computational Site Prediction | K222 identified among top candidate sites (with K68, K228, K242, K356) [60] | K222 is a high-probability ubiquitination acceptor site |
| In Vitro Ubiquitination Assay | K222R mutation significantly decreased ubiquitination levels [60] | K222 is a critical residue for MuRF2-mediated ubiquitination in a purified system |
| In Vivo Ubiquitination Assay | Smeared PPARγ1 K222R protein almost disappeared in immunoprecipitation [60] | K222 is the primary acceptor for polyubiquitination chains in cellular contexts |
| Cycloheximide Chase Assay | PPARγ1 K222R protein had significantly longer half-life than wild-type [60] | Ubiquitination at K222 targets PPARγ1 for proteasomal degradation |
| RT-qPCR Target Genes | mRNA levels of PLIN2 and CPT1b increased dramatically in PPARγ1 K222R group [60] | K222 ubiquitination inhibits PPARγ1 transcriptional activity for specific target genes |
Research Reagent Solutions for Ubiquitination Studies
Table 2: Essential Research Reagents for Ubiquitination Site Validation
| Research Reagent | Specific Example | Experimental Function |
|---|---|---|
| PPARγ Mutant Plasmids | K222R (Lysine to Arginine mutation) [60] | Site-specific validation of ubiquitination acceptor residues |
| E3 Ligase Expression Vector | His-MuRF2 [60] | Source of ubiquitin ligase activity for pathway manipulation |
| Ubiquitin Tag Plasmids | HA-Ub [60] | Affinity tag for purification and detection of ubiquitinated species |
| Proteasome Inhibitor | MG132 (20 µM) [60] | Blocks degradation of ubiquitinated proteins to enhance detection |
| Protein Synthesis Inhibitor | Cycloheximide (CHX) [60] | Enables measurement of protein half-life and stability |
| Affinity Purification Resins | Ni-NTA beads (for His-tag), Anti-GFP affinity beads [60] | Isolation of specific protein complexes for ubiquitination analysis |
| Ubiquitination Pathway Antibodies | Anti-His, Anti-HA, Anti-PPARγ [60] | Detection and confirmation of ubiquitinated proteins in assays |
Detailed Experimental Protocols
Computational Prediction of Ubiquitination Sites
The initial identification of candidate ubiquitination sites on PPARγ1 employed computational prediction tools based on acceptable sensitivity and specificity parameters [60]. Researchers utilized:
- UbiSite and UbiProber prediction tools: Sites with SVM score >0.8 in UbiProber or SVM score >0.9 in UbiSite were considered candidates [60]
- Candidate site visualization: Predicted sites were visualized in a 3D PPARγ1 molecular model established by PyMOL software [60]
- Site selection criteria: This computational approach identified K68, K222, K228, K242, and K356 as the leading candidate ubiquitination sites on PPARγ1 [60]
Site-Directed Mutagenesis and Plasmid Construction
To experimentally test the candidate sites, lysine residues were systematically mutated to arginine (K to R), which preserves the positive charge but prevents ubiquitin conjugation [60]. The experimental workflow included:
- Mutant generation: Creation of K68R, K222R, K228R, K242R, and K356R PPARγ1 mutants [60]
- Expression vector construction: Cloning of wild-type and mutant PPARγ1 into appropriate expression vectors [60]
- Cell line selection: Utilization of HEK293T cells for transfection studies due to high transfection efficiency [60]
In Vitro Ubiquitination Assay
The in vitro ubiquitination assay provided a controlled system to evaluate direct ubiquitination effects [60]:
- Protein purification: PPARγ1 (wild-type and mutants) were transfected into HEK293T cells, then total protein was extracted and purified using anti-GFP affinity beads [60]
- Reaction system setup: An in vitro ubiquitination reaction system was constructed with purified components [60]
- Ubiquitination detection: Immunoblot analysis demonstrated that PPARγ1 was poly-ubiquitinated by MuRF2, with K222R and K242R mutants showing significantly decreased ubiquitination levels [60]
In Vivo Ubiquitination Assay
Cellular ubiquitination assays confirmed the physiological relevance of findings [60]:
- Cell transfection: HEK293T cells were cotransfected with plasmids for His-MuRF2, HA-Ub, and PPARγ1 (wild-type or mutants) [60]
- Proteasome inhibition: MG132 (20 µM) was administered 6 hours before harvest to prevent degradation of ubiquitinated proteins [60]
- Immunoprecipitation and immunoblotting: Proteins were precipitated with anti-His antibody (targeting MuRF2) and ubiquitination was detected by immunoblotting [60]
- Key result: The smeared PPARγ1 K222R protein pattern (indicative of polyubiquitination) almost completely disappeared compared to wild-type and other mutants [60]
Protein Stability Assessment
The functional consequence of K222 ubiquitination on PPARγ1 stability was assessed using:
- Cycloheximide chase assay: HEK293T cells cotransfected with His-MuRF2 and either PPARγ1 or PPARγ1 K222R were treated with cycloheximide to inhibit new protein synthesis [60]
- Time-course sampling: Cells were harvested at 3 h and 6 h post-CHX treatment [60]
- Half-life determination: Immunoblot analysis showed PPARγ1 K222R protein had significantly longer half-life than wild-type PPARγ1, confirming that ubiquitination at K222 promotes proteasomal degradation [60]
Functional Assessment of Transcriptional Activity
The impact on PPARγ1 function was evaluated by measuring downstream target gene expression:
- Gene expression analysis: RT-qPCR was performed to measure mRNA levels of PPARγ1-regulated genes involved in cardiac glycolipid metabolism (ACOX1, PLIN2, CPT1b) [60]
- Comparative analysis: mRNA levels of PLIN2 and CPT1b increased dramatically in the PPARγ1 K222R group compared to wild-type PPARγ1 group [60]
- Functional interpretation: This demonstrated that MuRF2-mediated ubiquitination at K222 selectively inhibits PPARγ1 transcriptional activity for specific metabolic target genes [60]
Signaling Pathway and Experimental Workflow
This case study demonstrates a comprehensive methodological framework for validating specific ubiquitination sites, using PPARγ1-K222 as a model system. The convergent evidence from computational prediction, in vitro and in vivo ubiquitination assays, protein stability measurements, and functional transcriptional analyses established K222 as the critical residue for MuRF2-mediated ubiquitination. This modification directly regulates PPARγ1 protein stability and selectively modulates its transcriptional activity on metabolic target genes.
The experimental approaches detailed here provide researchers with a validated toolkit for similar ubiquitination site validation projects, emphasizing the importance of multiple orthogonal methods to establish both the molecular mechanism and functional consequences of specific ubiquitination events. These findings have significant implications for understanding the regulation of PPARγ1 in metabolic diseases and potentially for developing targeted therapeutic strategies.
Optimizing Assay Specificity: Overcoming Common Technical Challenges
In the study of protein ubiquitination, deubiquitinating enzymes (DUBs) present a significant experimental challenge. These enzymes rapidly reverse ubiquitination events, potentially obscuring research findings. To address this, scientists employ specific inhibitors to preserve ubiquitin signals during experiments. This guide compares two fundamental approaches to DUB inhibition: the broad-spectrum cysteine protease inhibitor N-Ethylmaleimide (NEM) and the more targeted Ubiquitin Aldehyde (Ub-aldehyde). Understanding their distinct mechanisms, applications, and limitations is essential for researchers validating ubiquitination sites through mutagenesis and immunoblotting, as the choice of inhibitor directly impacts data reliability and experimental interpretation.
The ubiquitin-proteasome system (UPS) regulates countless cellular processes, from protein degradation to DNA repair and signal transduction [62]. Deubiquitinases, comprising over 100 enzymes in humans, counterbalance this system by removing ubiquitin marks [63] [62]. Their activity, if unchecked during cell lysis and protein extraction, can swiftly erase the ubiquitination landscape an experiment aims to capture. Therefore, effective DUB inhibition is not merely a technical step but a prerequisite for accurate analysis.
Mechanism of Action: How DUB Inhibitors Function
N-Ethylmaleimide (NEM): A Broad-Spectrum Alkylating Agent
NEM is a cell-permeable small molecule that acts as an irreversible covalent inhibitor. Its primary mechanism involves alkylating sulfhydryl groups (-SH) on cysteine residues. This activity is not selective for DUBs and affects any cysteine-dependent enzyme, including E1 ubiquitin-activating enzymes and E2 ubiquitin-conjugating enzymes critical for the ubiquitination cascade [64] [17]. This broad reactivity is both its strength and its primary limitation, as it can disrupt the very system researchers seek to study.
Ubiquitin Aldehyde (Ub-aldehyde): A Transition-State Analogue
Ub-aldehyde is a mechanism-based inhibitor that acts as a potent, reversible transition-state analogue for many cysteine protease DUBs. It mimics the tetrahedral intermediate formed during ubiquitin C-terminal hydrolysis, competitively occupying the active site of DUBs with high affinity [65]. Unlike NEM, Ub-aldehyde specifically targets ubiquitin-binding sites, offering greater selectivity. However, its effectiveness varies across DUB subfamilies due to structural differences in their catalytic domains.
Table 1: Fundamental Properties and Mechanisms of DUB Inhibitors
| Property | N-Ethylmaleimide (NEM) | Ubiquitin Aldehyde (Ub-aldehyde) |
|---|---|---|
| Chemical Nature | Small molecule (125.13 g/mol) | Modified protein (~8.6 kDa) |
| Inhibition Mechanism | Irreversible cysteine alkylation | Reversible transition-state analogue |
| Primary Target | Cysteine residues globally | Active sites of many cysteine protease DUBs |
| Selectivity | Low (non-selective) | Medium (DUB-family selective) |
| Cell Permeability | Yes | No |
| Stability in Assays | Stable during lysis and processing | Sensitive to reducing conditions |
Comparative Performance Analysis in Experimental Settings
Effectiveness in Preserving Ubiquitin Signals
In ubiquitination pull-down assays and immunoblotting experiments, Ub-aldehyde generally provides superior protection against deubiquitination during cell lysis and subsequent steps. Its targeted action preserves ubiquitin conjugates without disrupting the enzymatic machinery required for ubiquitin transfer. Research shows that targeted DUB inhibition can significantly enhance the detection of specific ubiquitinated proteins, such as TAU aggregates in neurodegenerative disease models [66].
NEM, while effective at inactivating cysteine-dependent DUBs, can be problematic. Its non-specific alkylation of E1 and E2 enzymes may partially inhibit the ubiquitination cascade itself, potentially creating experimental artifacts. Furthermore, its action is "all-or-nothing," providing no gradation of inhibition across different DUB classes.
Application in Modern Proteomic Studies
Advanced proteomic techniques like tandem ubiquitin enrichment workflows benefit from a combination approach. Protocols often recommend using NEM in initial lysis buffers to provide immediate, broad DUB inhibition, followed by more specific inhibitors like Ub-aldehyde in subsequent steps to maintain suppression of deubiquitination during longer processing times [67]. This layered strategy leverages the strengths of both inhibitors while mitigating their individual limitations.
For specialized techniques such as Ub-POD (Ubiquitin-specific Proximity-Dependent Labeling), which identifies E3 ligase substrates, NEM is routinely included in lysis buffers at concentrations of 10-20 mM to prevent deubiquitination during the critical substrate capture step [17].
Table 2: Experimental Performance Comparison
| Experimental Parameter | N-Ethylmaleimide (NEM) | Ubiquitin Aldehyde (Ub-aldehyde) |
|---|---|---|
| Usage Concentration | 1-20 mM | 0.1-10 µM |
| Optimal Application | Cell lysis buffer, initial fixation | Lysis buffer, immunoprecipitation buffers |
| Impact on Ubiquitination Cascade | High (disrupts E1/E2 enzymes) | Low (primarily affects DUBs) |
| Compatibility with MS | Can alkylate cysteine peptides | No direct interference |
| Cost per Experiment | Low | High |
| Ease of Use | Simple addition to buffers | Requires aliquoting and careful storage |
Detailed Experimental Protocols
Protocol 1: Ubiquitination Site Validation via Mutagenesis and Immunoblotting with DUB Inhibitors
This protocol is designed for validating putative ubiquitination sites identified through mass spectrometry or other screening methods.
Reagents and Solutions:
- Lysis Buffer: 20 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% SDS, 1 mM EDTA
- Complete Protease Inhibitor Cocktail
- 1M N-Ethylmaleimide (NEM) stock in ethanol (freshly prepared)
- 1 mM Ubiquitin Aldehyde stock in DMSO (store at -80°C)
- N-Ethylmaleimide (NEM)
- PBS (Phosphate Buffered Saline)
- Protein A/G Agarose Beads
- Anti-ubiquitin Antibody (e.g., P4D1 or FK2)
- Anti-target Protein Antibody
- Laemmli Sample Buffer with 355 mM 2-mercaptoethanol [17]
Procedure:
- Cell Transfection and Treatment:
- Culture HEK-293 or relevant cell line in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% FBS.
- Transfect cells with plasmids encoding: (a) wild-type target protein, (b) lysine-to-arginine (K→R) mutant target protein, and (c) empty vector control using polyethyleneimine (PEI) or Lipofectamine.
- Treat cells with proteasome inhibitor MG132 (10-20 µM) for 4-6 hours before harvesting to stabilize ubiquitinated proteins.
Cell Lysis with DUB Inhibition:
- Prepare ice-cold lysis buffer supplemented with 1× protease inhibitor cocktail, 10 mM NEM, and 1 µM Ubiquitin Aldehyde.
- Lyse cells directly in culture dishes by adding appropriate volume of lysis buffer (e.g., 500 µL for 10 cm dish).
- Scrape cells and transfer lysates to microcentrifuge tubes.
- Sonicate lysates briefly (3× 5-second pulses) to shear DNA and reduce viscosity.
- Incubate lysates on ice for 30 minutes with occasional vortexing.
- Clarify lysates by centrifugation at 16,000 × g for 15 minutes at 4°C.
- Transfer supernatant to new tubes. [17]
Immunoprecipitation:
- Pre-clear lysates with Protein A/G Agarose beads for 30 minutes at 4°C.
- Incubate pre-cleared lysates with anti-target protein antibody (1-2 µg) overnight at 4°C with gentle rotation.
- Add Protein A/G Agarose beads and incubate for additional 2-4 hours.
- Pellet beads by gentle centrifugation (2,500 × g for 5 minutes) and wash 3× with wash buffer (20 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.5% NP-40, 10 mM NEM).
Immunoblotting Analysis:
- Elute proteins from beads by boiling in 2× Laemmli sample buffer with 355 mM 2-mercaptoethanol for 10 minutes.
- Separate proteins by SDS-PAGE and transfer to PVDF membrane.
- Probe membrane with anti-ubiquitin antibody to detect ubiquitinated species.
- Strip and re-probe membrane with anti-target protein antibody to confirm equal loading and immunoprecipitation efficiency.
- Compare ubiquitination patterns between wild-type and K→R mutant proteins to validate specific ubiquitination sites. [64]
Protocol 2: Tandem Enrichment of Ubiquitinated Peptides for Mass Spectrometry
This advanced protocol enables simultaneous enrichment of multiple post-translational modifications from a single sample.
Key Steps with DUB Inhibition:
- Protein Extraction and Digestion:
- Lyse cells or tissues in SDS-containing buffer with 10 mM NEM present throughout the extraction process.
- Process samples using the SCASP (SDS-cyclodextrin-assisted sample preparation) method.
- Digest proteins with trypsin/Lys-C mixture.
- Tandem Peptide Enrichment:
- Subject digested peptides to ubiquitinated peptide enrichment without desalting.
- Utilize the flowthrough for subsequent phosphorylated or glycosylated peptide enrichment.
- Desalt enriched peptides prior to mass spectrometric analysis.
- Analyze data using DIA (Data-Independent Acquisition) MS methods. [67]
Diagram 1: Experimental workflow for ubiquitination site validation using DUB inhibitors. The combined use of NEM and Ub-aldehyde during lysis is critical for preserving ubiquitin signals.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for DUB Inhibition Studies
| Reagent | Function/Application | Key Considerations |
|---|---|---|
| N-Ethylmaleimide (NEM) | Broad DUB inhibition during cell lysis | Prepare fresh stock solutions; concentration optimization required |
| Ubiquitin Aldehyde | Specific DUB inhibition in enzymatic assays | Sensitive to reducing agents; expensive for large-scale use |
| MG132 (Proteasome Inhibitor) | Stabilizes polyubiquitinated proteins | Use 4-6 hour treatment before harvesting; dissolved in DMSO |
| Anti-Ubiquitin Antibodies (P4D1, FK2) | Detection of ubiquitinated proteins in immunoblots | P4D1 recognizes polyubiquitin chains; FK2 detects mono/polyubiquitin |
| Anti-K48- or K63-linkage Specific Antibodies | Detection of specific ubiquitin linkages | Essential for determining ubiquitin chain topology and function |
| Biotin-Ub-VME/Biotin-Ub-PA | Activity-based probes for DUB profiling | Used in ABPP (Activity-Based Protein Profiling) screens [65] |
| Streptavidin Agarose | Enrichment of biotinylated ubiquitinated proteins | Used in pull-down assays under denaturing conditions [17] |
| N-Ethylmaleimide (NEM) | Alkylating free thiols to prevent disulfide rearrangement | Also included in sample buffers for proteomic studies |
The choice between NEM and Ubiquitin Aldehyde is not merely technical but strategic, dictated by experimental goals and resources. NEM offers a cost-effective, broad-spectrum solution for initial DUB suppression during cell lysis, while Ubiquitin Aldehyde provides targeted inhibition valuable for specific applications and mechanistic studies. For the most robust preservation of ubiquitination states—particularly in site validation studies—a combination approach often yields superior results.
The field continues to evolve with emerging technologies. Advanced chemical biology approaches, such as activity-based protein profiling (ABPP) with purpose-designed covalent libraries, are accelerating the discovery of selective DUB inhibitors [65]. Furthermore, new techniques like Ub-POD (Ubiquitin-specific Proximity-Dependent Labeling) are enhancing our ability to identify E3 ligase substrates with unprecedented specificity [17]. These innovations, combined with the foundational use of classical inhibitors like NEM and Ub-aldehyde, provide researchers with an expanding toolkit to decipher the complex ubiquitin code and its profound implications for cellular function and disease.
In molecular biology research, particularly in the study of post-translational modifications like ubiquitination, the reliability of experimental results is paramount. False positive signals can compromise data interpretation, leading to incorrect conclusions about protein function, interaction, and regulation. This guide objectively compares key methodological approaches for controlling false positives, with a specific focus on techniques applicable to ubiquitination site validation through mutagenesis and immunoblotting. We evaluate experimental protocols based on their stringency, specificity, and practical implementation, providing researchers with a framework for selecting appropriate methods for their specific applications.
Comparative Analysis of False Positive Control Methods
The table below summarizes three distinct methodological approaches for controlling false positives, each employing different principles of stringency and denaturation.
Table 1: Comparison of False Positive Control Methodologies
| Method Name | Core Principle | Key Applications | Stringency Controls | Experimental Evidence |
|---|---|---|---|---|
| Sequential Denaturation and Protein Precipitation (SDPP) | Sequential application of multiple denaturation mechanisms (thermal, solvent, pH) to progressively precipitate proteins and amplify solubility shifts [68] | Ligand target identification; protein interaction studies | Thermal denaturation (hydrophobic core exposure), organic solvent (reduced dielectric solution), low pH (altered charge distribution) [68] | Identified 54%, 38%, 48%, and 21% more kinase targets than single denaturation methods (thermal, solvent, pH) and IPSSA, respectively [68] |
| TurboID-Matched Expression Controls | Expression-level matching of TurboID control to bait protein to standardize background biotinylation in proximity labeling [69] | Proximity labeling proteomics; protein-protein interaction mapping | Control TurboID expression matched to bait-TurboID fusion expression levels; normalization strategies [69] | Discordant expression levels caused high-frequency false negatives/positives; matched expression reduced background interference [69] |
| Ubiquitination Site-Directed Mutagenesis | Chemical mutagenesis of potential ubiquitination sites to confirm modification specificity [56] | Ubiquitination site validation; E3 ligase substrate confirmation | Methylation of secondary amine on azetidine nitrogen (BRD1732) prevents ubiquitin conjugation [56] | BRD-NMe and BRD-NOMe analogs showed complete prevention of ubiquitination vs. BRD-OMe (no effect); IC50 increased to >30µM [56] |
Detailed Experimental Protocols
Protocol 1: Sequential Denaturation and Protein Precipitation (SDPP)
The SDPP method employs sequential denaturation treatments to individual samples to enable stepwise protein precipitation without sample pooling, preserving and accumulating solubility shifts [68].
- Cell Lysis: Suspend frozen cell pellets in ice-cold PBS lysis buffer with 1% EDTA-free protease inhibitor cocktail [68].
- Ligand Incubation: Treat lysates with ligand or vehicle control for 30 minutes at room temperature [68].
- First Denaturation (Thermal): Heat samples at 55°C for 3 minutes, then centrifuge at 16,000×g for 10 minutes at room temperature [68].
- Second Denaturation (Solvent): Transfer supernatant to fresh tube, add acetonitrile to 30% final concentration, incubate 10 minutes at room temperature, then centrifuge as above [68].
- Protein Precipitation: Transfer supernatant to new tube, add 4 volumes of acetone, precipitate at -20°C overnight [68].
- Protein Digestion: Centrifuge at 16,000×g for 10 minutes, wash pellet with 90% acetone, then digest for LC-MS/MS analysis [68].
Protocol 2: Ubiquitination Validation via Site-Directed Mutagenesis
This protocol confirms direct small-molecule ubiquitination through strategic chemical mutagenesis [56].
- Analog Synthesis: Synthesize methylated analogs of the target molecule:
- Cell Treatment: Treat RNF19A-overexpressing Expi293F cells with 10µM BRD1732 or methylated analogs for 6 hours [56].
- Ubiquitin Purification: Harvest cells and purify endogenous ubiquitin using cation exchange followed by size-exclusion chromatography [56].
- Mass Spectrometry Analysis: Analyze ubiquitin samples by LC-MS to detect mass shifts indicating ubiquitin conjugates [56].
- Trypsin Digestion: Perform trypsin digestion under nondenaturing conditions and analyze fragments for Gly-Gly-adduct formation [56].
- Viability Assay: Conduct proliferation assays with each analog to determine IC50 values and stereospecificity [56].
Visualization of Key Methodological Pathways
Ubiquitination Validation Workflow
Research Reagent Solutions for Ubiquitination Studies
Table 2: Essential Research Reagents for Controlling False Positives
| Reagent/Condition | Function | Application Example |
|---|---|---|
| sAB-K29 Antibody | Highly specific antibody for K29-linked ubiquitin chains; minimal cross-reactivity with other linkage types [70] | Chromatin profiling of K29-linked ubiquitination via CUT&Tag; specific detection in immunofluorescence [70] |
| Stringent Wash Buffers | Reduce non-specific antibody binding and background signal in immunoblotting | Washes with high-salt (e.g., 300-500mM NaCl) and/or detergent-containing buffers after primary/secondary antibody incubation |
| Methylated Compound Analogs | Chemically inactivated controls for ubiquitination site validation [56] | BRD-NMe analog methylation prevents ubiquitination on azetidine nitrogen, confirming site specificity [56] |
| Expression-Matched TurboID Controls | Control for background biotinylation in proximity labeling experiments [69] | TurboID-GFP expressed at levels matching TurboID-bait fusion to normalize background signal [69] |
| Multi-Mechanism Denaturation Buffers | Sequential application of different denaturation conditions to amplify ligand-induced solubility shifts [68] | Thermal denaturation (hydrophobic exposure) followed by organic solvent (hydration membrane disruption) [68] |
Discussion and Performance Comparison
The methodological approaches compared herein demonstrate that controlling false positives requires strategic application of stringency at multiple experimental levels. SDPP achieves enhanced sensitivity through accumulating denaturation effects without signal dilution [68], while matched expression controls address a frequently overlooked variable in proximity labeling studies [69]. Most notably for ubiquitination researchers, site-directed mutagenesis provides the most direct evidence for modification specificity, as demonstrated by the complete abolition of BRD1732 ubiquitination upon methylation of the azetidine nitrogen [56].
These methods highlight that effective false positive control often requires orthogonal approaches—combining chemical, genetic, and physical separation techniques to validate findings. Researchers should select methods based on their specific experimental system, with ubiquitination studies particularly benefiting from the mutagenesis approach combined with stringent immunoblotting conditions.
Controlling false positives remains a critical challenge in protein biochemistry and ubiquitination research. The methods compared in this guide—SDPP, expression-matched controls, and site-directed mutagenesis—each offer distinct advantages for different experimental contexts. By implementing these stringent approaches and carefully selecting research reagents, scientists can significantly enhance the reliability of their ubiquitination validation studies, leading to more robust conclusions about protein function and regulation in health and disease.
Ubiquitination is a critical post-translational modification (PTM) that regulates diverse cellular functions, including protein degradation, signal transduction, and DNA repair [3]. This modification involves the covalent attachment of ubiquitin—a small, highly conserved 76-residue protein—to substrate proteins via a three-enzyme cascade involving E1 (activating), E2 (conjugating), and E3 (ligase) enzymes [3]. The complexity of ubiquitin signaling arises from the ability of ubiquitin itself to form polymers through any of its seven lysine residues (K6, K11, K27, K29, K33, K48, K63) or its N-terminal methionine (M1), creating a diverse "ubiquitin code" with distinct functional consequences [3] [71]. For instance, K48-linked polyubiquitin chains primarily target substrates for proteasomal degradation, while K63-linked chains typically regulate non-proteolytic functions such as kinase activation and inflammatory signaling [3].
Detecting and characterizing ubiquitination events is fundamental to understanding cellular homeostasis and the pathogenesis of numerous diseases, including cancer and neurodegenerative disorders [3]. Researchers have developed various antibody-based tools to study ubiquitination, which primarily fall into two categories: pan-ubiquitin antibodies that recognize ubiquitin regardless of linkage type, and linkage-specific antibodies that detect particular polyubiquitin chain topologies [3] [72]. This guide objectively compares these antibody classes within the experimental context of validating ubiquitination sites through mutagenesis and immunoblotting, providing researchers with a framework for appropriate reagent selection.
Technical Comparison of Antibody Types
Pan-Ubiquitin Antibodies
Pan-ubiquitin antibodies, also known as broad-spectrum ubiquitin detection reagents, recognize ubiquitin in monomeric, polymeric, or substrate-conjugated states without preference for specific linkage types [72]. These antibodies target conserved regions of ubiquitin itself, making them valuable for initial detection of ubiquitination events.
Key Characteristics:
- Epitope Recognition: Target invariant regions of ubiquitin, such as the core ubiquitin fold or C-terminal sequence
- Detection Scope: Identify monoubiquitination, multi-monoubiquitination, and all polyubiquitin chain types
- Common Clones: Include widely used antibodies such as P4D1, FK1, and FK2 [3]
- Applications: Initial ubiquitination screening, total ubiquitin quantification, and detection of atypical ubiquitination forms
The primary advantage of pan-ubiquitin antibodies lies in their broad detection capability, making them suitable for initial experiments when the specific chain topology is unknown. However, this lack of specificity can also be a limitation, as these antibodies cannot distinguish between functionally distinct polyubiquitin chains [3].
Linkage-Specific Antibodies
Linkage-specific antibodies are engineered to recognize particular polyubiquitin chain architectures, allowing researchers to decipher the functional ubiquitin code. These antibodies detect the unique structural features presented by specific lysine linkages in polyubiquitin chains.
Key Characteristics:
- Epitope Recognition: Target unique conformational epitopes formed by specific ubiquitin-ubiquitin linkages
- Detection Scope: Identify homotypic chains with particular linkages (e.g., K48, K63, K11, etc.)
- Common Targets: Include K48-linked chains (degradation signaling) and K63-linked chains (non-degradative signaling) [73] [3]
- Applications: Functional ubiquitination studies, pathway-specific signaling analysis, and mechanistic investigations
These antibodies provide functional insights into ubiquitination events by revealing which specific signaling pathways are activated. For example, the anti-ubiquitin (linkage-specific K48) antibody [EP8589] specifically recognizes K48-linked polyubiquitin chains without cross-reacting with other linkage types [73]. This specificity enables researchers to correlate K48-linked ubiquitination with proteasomal degradation of specific substrates.
Table 1: Comparison of Pan-Ubiquitin and Linkage-Specific Antibodies
| Feature | Pan-Ubiquitin Antibodies | Linkage-Specific Antibodies |
|---|---|---|
| Detection Scope | All ubiquitin forms | Specific polyubiquitin linkages |
| Primary Applications | Initial screening, total ubiquitin detection | Functional studies, pathway analysis |
| Advantages | Broad reactivity, comprehensive detection | Functional correlation, precise signaling insights |
| Limitations | Cannot distinguish chain types | May miss alternative ubiquitination forms |
| Common Validation | Detect ubiquitin protein standard | Verify against specific linkage standards [73] |
| Typical Use Cases | Ubiquitination confirmation | Degradation vs. signaling discrimination |
Performance Data and Experimental Validation
Robust validation is essential for both antibody types to ensure reliability and specificity in experimental outcomes. Linkage-specific antibodies require rigorous testing against various ubiquitin chain types to confirm specificity.
Table 2: Experimental Performance Characteristics of Ubiquitin Antibodies
| Parameter | Pan-Ubiquitin Antibodies | Linkage-Specific Antibodies |
|---|---|---|
| Specificity Validation | Reactivity with monoubiquitin and polyubiquitin | Testing against competing linkage types [73] |
| Detection Sensitivity | High for total ubiquitin | Variable depending on linkage abundance |
| Common Applications | WB, IHC, ICC, IP, Flow Cytometry [73] | WB, ICC/IF, IHC-P, Flow Cytometry (Intra) [73] |
| Cross-Reactivity Concerns | Minimal (targets conserved epitopes) | Significant (requires validation against multiple linkages) |
| Typical Dilutions | 1:1000 (WB) [73] | 1:100-1:2000 (application-dependent) [73] |
| Quantitative Capability | Semi-quantitative for total ubiquitin | Semi-quantitative for specific linkage abundance |
For linkage-specific antibodies, validation should include testing against recombinant di-ubiquitin or polyubiquitin chains with exact linkage configurations to ensure specificity [72]. As demonstrated in commercial antibody development, specificity is confirmed through ELISA, Western blotting, and dot blot assays against competing linkages [72]. For example, a well-validated K48-linkage-specific antibody should show strong signal with K48-linked ubiquitin chains but minimal reactivity with K63-linked, linear, or other chain types [73].
Experimental Protocols for Ubiquitination Validation
Immunoblotting Workflow for Ubiquitination Detection
The following diagram illustrates the core experimental workflow for validating protein ubiquitination using mutagenesis and immunoblotting:
Step-by-Step Immunoblotting Protocol
Sample Preparation:
- Cell Treatment: Treat cells with proteasomal inhibitors (e.g., 10-20 μM MG132 for 4-6 hours) to prevent degradation of ubiquitinated proteins and enhance detection sensitivity [71].
- Cell Lysis: Use denaturing lysis buffers (e.g., RIPA buffer with 1% SDS) to preserve ubiquitination status and prevent deubiquitination. Include protease inhibitors and specific deubiquitinase inhibitors (e.g., N-ethylmaleimide) to maintain ubiquitin conjugates.
- Protein Quantification: Determine protein concentration using BCA or Bradford assay to ensure equal loading.
Immunoprecipitation:
- Pre-clear Lysate: Incubate cell lysate with protein A/G beads for 30-60 minutes at 4°C to reduce non-specific binding.
- Antibody Incubation: Add specific antibody against the protein of interest (1-5 μg per 500 μg lysate) and incubate for 2-4 hours at 4°C with gentle rotation.
- Bead Capture: Add protein A/G beads and incubate for an additional 1-2 hours to capture antibody-protein complexes.
- Washing: Wash beads 3-4 times with lysis buffer (reduced SDS concentration) to remove non-specifically bound proteins.
Immunoblotting:
- Gel Electrophoresis: Separate immunoprecipitated proteins by SDS-PAGE (8-10% gels) to resolve ubiquitinated species, which typically appear as higher molecular weight smears or discrete bands.
- Protein Transfer: Transfer proteins to PVDF or nitrocellulose membrane using standard wet or semi-dry transfer methods.
- Blocking: Block membrane with 5% non-fat dry milk or BSA in TBST for 1 hour at room temperature to prevent non-specific antibody binding.
- Antibody Incubation:
- Incubate with primary antibody (pan-ubiquitin or linkage-specific) diluted in blocking buffer overnight at 4°C.
- Common dilutions: 1:1000 for pan-ubiquitin, 1:500-1:2000 for linkage-specific antibodies [73].
- Detection: Incubate with appropriate HRP-conjugated secondary antibody (1:2000-1:5000 dilution) for 1 hour at room temperature, followed by chemiluminescent detection.
Mutagenesis Validation Protocol
Site-directed mutagenesis of putative ubiquitination sites provides critical validation of immunoblotting results:
Identification of Putative Sites:
- Bioinformatic Analysis: Use prediction tools like Ubigo-X, which employs machine learning algorithms combining sequence-based, structure-based, and function-based features to identify potential ubiquitination sites [74].
- Conserved Lysine Mapping: Identify conserved lysine residues across homologs and structural domains.
Mutagenesis and Validation:
- Site-Directed Mutagenesis: Mutate candidate lysine (K) residues to arginine (R) using PCR-based mutagenesis kits to create non-ubiquitinatable mutants while maintaining similar physicochemical properties.
- Transfection: Express wild-type and mutant proteins in appropriate cell lines (e.g., HEK293T for high transfection efficiency).
- Comparative Analysis: Process wild-type and mutant samples in parallel following the immunoblotting protocol above.
- Interpretation: Reduced or absent ubiquitination signal in mutants compared to wild-type confirms the identification of bona fide ubiquitination sites.
Advanced Detection Methodologies
Alternative Ubiquitination Detection Platforms
Beyond traditional immunoblotting, several advanced methodologies have emerged for ubiquitination detection:
Tandem Ubiquitin-Binding Entity (TUBE) Systems:
- Utilize engineered ubiquitin-binding domains with high affinity for polyubiquitinated proteins
- Can be coated on 96-well plates for high-throughput screening [75]
- Less biased than antibodies for different ubiquitin chain types
Thioesterase-Based Ubiquitin Fusion (ThUBD) Technology:
- Coats high-density 96-well plates with ThUBD for unbiased capture of polyubiquitin chains
- Exhibits no bias toward any ubiquitin linkage type [75]
- Enables specific binding to approximately 5 pmol of polyubiquitin chains [75]
Engineered Deubiquitinases (enDUBs):
- Fusion proteins combining linkage-selective DUB catalytic domains with target-specific nanobodies
- Allow selective hydrolysis of specific polyubiquitin chains from target proteins in live cells [71]
- Useful for investigating functional roles of distinct polyubiquitin linkages
Ubi-Tagging Approach:
- Repurposes the ubiquitination machinery for site-specific protein conjugation
- Enables controlled multimerization through specific ubiquitin linkages [76] [77]
- Achieves conjugation efficiencies of 93-96% within 30 minutes [76]
The Ubiquitination Signaling Network
The following diagram illustrates the complexity of ubiquitin signaling, highlighting how different chain types regulate distinct cellular outcomes:
Research Reagent Solutions
Table 3: Essential Research Reagents for Ubiquitination Studies
| Reagent Category | Specific Examples | Function & Application |
|---|---|---|
| Pan-Ubiquitin Antibodies | P4D1, FK1, FK2 clones [3] | Broad detection of ubiquitinated proteins in various applications |
| Linkage-Specific Antibodies | Anti-K48 (e.g., EP8589) [73], Anti-K63 | Detection of specific polyubiquitin chain types for functional studies |
| Proteasome Inhibitors | MG132, Bortezomib [71] | Enhance detection by preventing degradation of ubiquitinated proteins |
| Deubiquitinase Inhibitors | N-ethylmaleimide, PR-619 | Preserve ubiquitin conjugates during sample preparation |
| Ubiquitin-Activating Enzyme Inhibitor | PYR-41 | Blocks E1 enzyme for functional studies of ubiquitination dynamics |
| Recombinant Ubiquitin Variants | K48-only, K63-only ubiquitin mutants [76] | Positive controls for antibody validation and linkage specificity |
| Engineered DUBs | enDUBs with linkage selectivity [71] | Targeted hydrolysis of specific ubiquitin chains in live cells |
| Ubiquitin Binding Domains | TUBEs, ThUBD [75] | Affinity enrichment of polyubiquitinated proteins with reduced linkage bias |
The selection between pan-ubiquitin and linkage-specific antibodies should be guided by research objectives and experimental context. Pan-ubiquitin antibodies serve as excellent tools for initial detection and confirmation of ubiquitination events, offering broad reactivity across various ubiquitin forms. In contrast, linkage-specific antibodies provide functional insights by connecting specific ubiquitin chain topologies to biological outcomes, such as proteasomal targeting versus signaling functions.
For comprehensive ubiquitination validation, we recommend a sequential approach: begin with pan-ubiquitin antibodies to confirm overall ubiquitination status, then employ linkage-specific reagents to decipher functional implications. This strategy should be combined with mutagenesis studies to validate modification sites and advanced methodologies like TUBEs or ThUBD when unbiased ubiquitin chain capture is required. As the ubiquitin field continues to evolve, emerging technologies such as enDUBs and ubi-tagging offer increasingly sophisticated tools for precise manipulation and detection of ubiquitin signals, enabling researchers to better understand this complex post-translational regulatory system.
Within the framework of validating ubiquitination sites through mutagenesis and immunoblotting, a central challenge is the reliable and specific enrichment of ubiquitinated proteins. Traditional antibodies, such as the commonly used P4D1 and FK1/FK2, are powerful tools but can be hampered by non-specific binding and high cost [3]. This guide objectively compares the performance of epitope-tagged ubiquitin systems as alternatives to conventional antibody-based methods for studying protein ubiquitination, providing researchers with data to inform their experimental design.
Comparative Analysis of Ubiquitin Enrichment Methodologies
The table below summarizes the core characteristics, advantages, and limitations of the primary methods used for ubiquitination studies.
| Methodology | Key Feature | Advantages | Limitations & Non-Specific Binding Risks |
|---|---|---|---|
| Epitope-Tagged Ubiquitin [3] | Genetic fusion of Ub with tags like 6×His, Strep, or HA. | Relatively low-cost; easy to use; enables high-throughput profiling in living cells [3]. | Co-purification of histidine-rich or endogenously biotinylated proteins; potential artifacts as tagged Ub may not perfectly mimic endogenous Ub [3]. |
| Ub Antibody-Based Enrichment [3] | Use of anti-Ub antibodies (e.g., P4D1, FK2) or linkage-specific antibodies. | Applicable to animal tissues and clinical samples without genetic manipulation; provides linkage-specific information [3]. | High cost; potential for non-specific binding of non-ubiquitinated proteins [3]. |
| UBD-Based Enrichment [3] | Use of Ub-Binding Domains (UBDs) from E3 ligases, DUBs, or Ub receptors. | Can recognize Ub linkages generally or selectively; enriches endogenous proteins. | Low affinity of single UBDs limits purification efficiency, though tandem-repeated UBDs can mitigate this [3]. |
Experimental Protocols for Key Methodologies
Epitope-Tagged Ubiquitin System Workflow
The foundational protocol for using epitope-tagged ubiquitin involves expressing the tagged ubiquitin in cells, enriching the conjugated proteins, and identifying the ubiquitination sites via mass spectrometry [3].
- Cell Line Construction: Generate a cell line (e.g., HEK293T, U2OS) stably expressing ubiquitin tagged at its N-terminus with an affinity tag such as 6×His or Strep-II [3]. The correct conjugation and functionality of the tagged ubiquitin must be validated [78] [79].
- Enrichment of Ubiquitinated Conjugates: Lyse cells and pass the whole-cell lysate over an affinity resin corresponding to the tag—for example, Ni-NTA agarose for 6×His tags or Strep-Tactin for Strep-tags [3].
- Washing and Elution: Wash the resin with buffer to remove non-specifically bound proteins. Elute the bound ubiquitinated proteins, often by competition (e.g., imidazole for His-tags, biotin for Strep-tags) or by low-pH conditions [3].
- Downstream Analysis: The eluted proteins can be analyzed by immunoblotting to confirm ubiquitination or digested with trypsin for MS analysis. Ubiquitination sites are identified by searching for a diagnostic mass shift of 114.04 Da on modified lysine residues [3] [80].
Conventional Validation by Mutagenesis and Immunoblotting
This classical biochemistry approach serves as a critical validation step within the broader research context.
- Putative Site Identification: Based on MS data, bioinformatic prediction (e.g., using tools like UbPred [80]), or structural cues, identify specific lysine residues suspected of being ubiquitinated.
- Site-Directed Mutagenesis: Generate mutant constructs of the protein of interest where the candidate lysine(s) are substituted, typically with arginine (K→R) [3].
- Ubiquitination Assay: Co-express the wild-type or mutant protein with ubiquitin in cells. Immunoprecipitate the protein of interest under denaturing conditions (e.g., using SDS lysis buffers) to preserve labile ubiquitin conjugates.
- Immunoblotting Detection: Analyze the immunoprecipitated proteins by SDS-PAGE and immunoblotting using an anti-ubiquitin antibody (e.g., P4D1) [3]. A reduction in the ubiquitination signal for the K→R mutant compared to the wild-type protein confirms the specific lysine as a ubiquitination site [3].
Visualization of Ubiquitin Signaling and Experimental Workflow
Ubiquitin Conjugation Pathway
Epitope-Tagged Ubiquitin Enrichment Workflow
The Scientist's Toolkit: Key Research Reagent Solutions
| Reagent / Tool | Primary Function | Key Features & Considerations |
|---|---|---|
| 6×His-Tagged Ubiquitin [3] | Affinity purification of ubiquitinated conjugates using Ni-NTA resin. | Cost-effective; potential for co-purification of endogenous histidine-rich proteins. |
| Strep-Tagged Ubiquitin [3] | Affinity purification using Strep-Tactin resin. | High binding specificity and affinity; potential co-purification of endogenously biotinylated proteins. |
| Linkage-Specific Ub Antibodies [3] | Immunoblotting or enrichment of polyUb chains with specific linkages (K48, K63, etc.). | High specificity for chain architecture; valuable for functional studies; high cost. |
| Tandem Ub-Binding Domains (TUBEs) [3] | High-affinity enrichment of endogenous ubiquitinated proteins. | Protects ubiquitin chains from deubiquitinases (DUBs) during purification; no genetic manipulation required. |
| UbPred Algorithm [80] | In silico prediction of ubiquitination sites on lysine residues. | Random forest-based predictor; helps prioritize lysine residues for experimental validation. |
Epitope-tagged ubiquitin systems offer a practical and cost-effective alternative to antibody-based methods, directly addressing the issue of non-specific binding in ubiquitination research. While considerations regarding their ability to fully mimic endogenous ubiquitin exist, their utility for high-throughput profiling in live cells is well-established. The optimal choice of method—be it epitope-tagging, antibody-based enrichment, or UBD-based approaches—depends on the specific research question, experimental model, and available resources. Integrating these enrichment techniques with the gold-standard validation of site-directed mutagenesis and immunoblotting provides a robust framework for conclusively identifying and validating protein ubiquitination sites.
Western blotting remains one of the most utilized analytical techniques in biological research, particularly for detecting specific proteins from complex samples. In the context of post-translational modification (PTM) studies, such as validating ubiquitination sites by mutagenesis, Western blotting provides a crucial methodology for researchers to confirm modifications through characteristic molecular weight shifts. Traditional Western blotting, invented over four decades ago, involves separating proteins using gel electrophoresis, transferring them to a membrane, and probing target proteins with specific antibodies followed by imaging of antibody binding [81].
The detection of ubiquitination through Western blotting typically reveals distinctive patterns: unmodified proteins appear at their expected molecular weights, while ubiquitinated species manifest as higher molecular weight smears or discrete bands, depending on whether mono-ubiquitination or poly-ubiquitination has occurred. This molecular weight shift methodology serves as a valuable secondary validation technique alongside primary confirmation methods like mass spectrometry, especially when validating ubiquitination sites through mutagenesis experiments where specific lysine residues are altered to prevent ubiquitin attachment.
Comparative Analysis of Western Blotting Methodologies
Traditional Western Blotting
Traditional Western blotting represents the conventional approach that has been widely used for decades. This methodology requires multiple manual steps performed over an extended period.
Experimental Protocol for Traditional Western Blotting:
- Protein Separation: Proteins are separated using SDS-PAGE protocol, typically utilizing 4-20% gradient pre-cast gels. A total of 10 µg, 2.5 µg, and 1 µg of total protein from each sample is loaded onto gels in 1x Laemmli sample buffer [81].
- Membrane Transfer: Following electrophoresis, separated proteins are transferred to 0.2 µm nitrocellulose membranes using transfer systems such as the Trans-Blot Turbo Transfer System [81].
- Blocking: Nonspecific antibody binding is blocked using 5% bovine serum albumin (BSA) in Tris-buffered solution with 0.05% Tween (TBST) [81].
- Antibody Incubation: Primary antibodies (e.g., rabbit IgG anti-target protein) are diluted in 5% BSA in TBST and incubated overnight at 4°C on a shaker. Membranes are washed four times using TBST solution before adding secondary antibodies (e.g., HRP-conjugated goat anti-rabbit IgG) diluted in TBST and allowed to bind at room temperature for 2 hours [81].
- Detection: Chemiluminescent substrate (e.g., Pierce ECL Plus Western Blotting substrate) is added according to manufacturer's instructions and imaged using systems like Bio-Rad ChemiDoc Imaging System [81].
- Normalization: For loading controls, membranes are stripped and reblotted with antibodies for housekeeping proteins like GAPDH to normalize protein amounts in lysates [81].
Semi-Automated Systems: iBind Flex
The iBind Flex system represents a semi-automated approach that streamlines specific components of the Western blotting process while still requiring substantial manual intervention.
Experimental Protocol for iBind Flex:
- Initial Steps: Protein separation via SDS-PAGE and membrane transfer are performed identically to traditional Western blotting [81].
- Automated Immunodetection: The iBind Flex device and iBind Flex cards are used for immunodetection according to manufacturer's protocol. Primary antibodies (2 mL) and secondary antibodies (2 mL) are applied using the automated system [81].
- Detection and Analysis: Membranes are imaged using standard systems like Bio-Rad ChemiDoc Imaging System. The instrument utilizes sequential lateral flow to process blocking, antibodies, and washing solutions in a controlled fashion [81].
Fully Automated Systems: JESS Simple Western
JESS Simple Western represents a fully automated, capillary-based system that performs all steps downstream of sample preparation, fundamentally changing the Western blotting workflow.
Experimental Protocol for JESS Simple Western:
- Sample Preparation: Lysates are diluted to total protein concentrations of 100 ng/µL, 50 ng/µL, 25 ng/µL, and 12.5 ng/µL in 1x fluorescent master mix, and 3 µL is added per well [81].
- Automated Processing: The system automatically performs protein size separation, immunoblotting, imaging, and image analysis within capillaries that replace traditional gels and membranes [81].
- Antibody Incubation: Primary antibodies are used at specific dilutions (e.g., anti-target protein at 1:50) diluted in appropriate buffers [81].
Quantitative Comparison of Western Blot Methods
Table 1: Performance Comparison of Western Blotting Methodologies
| Parameter | Traditional Western | iBind Flex (Semi-Automated) | JESS Simple Western (Fully Automated) |
|---|---|---|---|
| Total Hands-on Time | 1-3 days [81] | Reduced hands-on time (approximately 3 hours for immunodetection) [81] | Minimal hands-on time post-sample preparation [81] |
| Total Protein Required | 1-10 µg per sample [81] | Similar to traditional Western | Significantly less - 0.3 µg per sample at 100 ng/µL concentration [81] |
| Sensitivity | Standard sensitivity | Similar to traditional | Enhanced sensitivity, particularly beneficial for limited sample amounts [81] |
| Reproducibility | Subject to variability due to multiple manual steps [81] | Improved reproducibility for immunodetection steps | High reproducibility through automation of all critical steps [81] |
| Antibody Consumption | Standard volumes | Higher concentration but smaller volumes [81] | Optimized volumes |
| Equipment Cost | Low | Moderate | High [81] |
| Throughput | Low to moderate | Moderate | High |
Table 2: Data Quality and Analytical Performance Comparison
| Analysis Feature | Traditional Western | iBind Flex | JESS Simple Western |
|---|---|---|---|
| Molecular Weight Resolution | High with proper gel concentration | Equivalent to traditional | High with capillary separation |
| Quantitative Capability | Requires careful normalization [82] | Similar to traditional | Built-in quantification software |
| Linear Detection Range | Must be empirically determined [82] | Similar to traditional | Automated determination |
| Multi-target Detection | Possible with stripping/reprobing | Similar to traditional | Limited by capillary capacity |
| Data Documentation | Manual | Manual | Automated digital record |
Application to Ubiquitination Site Validation
Molecular Weight Shift Analysis for Ubiquitination
The core principle of using Western blotting for ubiquitination validation relies on the significant molecular weight shift that occurs when ubiquitin (approximately 8.6 kDa) attaches to target proteins. This shift manifests differently depending on the ubiquitination type:
- Mono-ubiquitination: Discrete bands shifting approximately 8.6 kDa higher than the unmodified protein
- Poly-ubiquitination: Characteristic smears or ladder patterns representing multiple ubiquitin attachments
- Lysine Mutagenesis Validation: Comparison between wild-type and mutant proteins (where putative ubiquitination sites are mutated) showing absence of higher molecular weight species in mutants
Experimental Design for Ubiquitination Validation
Critical Controls for Ubiquitination Experiments:
- Wild-type vs. Mutant Proteins: Express both wild-type and lysine-to-arginine mutant proteins to confirm specific ubiquitination sites
- Proteasome Inhibition: Treat cells with MG132 or other proteasome inhibitors to accumulate ubiquitinated species
- Ubiquitin Co-expression: Co-express tagged ubiquitin (HA-Ub, Myc-Ub, or FLAG-Ub) for enhanced detection
- Loading Controls: Use consistent housekeeping proteins (GAPDH, actin, tubulin) validated for stable expression under experimental conditions [82]
Methodological Considerations for Ubiquitination Detection:
- Gel Concentration: Use appropriate acrylamide concentrations to resolve expected molecular weight ranges
- Transfer Conditions: Optimize transfer for high molecular weight proteins
- Antibody Selection: Choose antibodies specific for target protein and ubiquitin (or tags if using tagged ubiquitin)
- Lysate Preparation: Include deubiquitinase inhibitors in lysis buffers to preserve ubiquitinated species
Virtual Western Blot Workflow for Ubiquitination Validation
The following workflow diagram illustrates the integrated process of using molecular weight shifts in Western blotting for validating ubiquitination sites through mutagenesis:
Method Selection for Ubiquitination Studies
Choosing the appropriate Western blotting methodology depends on several factors specific to ubiquitination research:
Traditional Western Blotting is preferable when:
- Resources are limited
- Multiple reprobing steps are anticipated
- Large molecular weight ranges need examination
- Method development and optimization are ongoing
Semi-Automated Systems (iBind Flex) are advantageous for:
- Standardized ubiquitination assays
- Laboratories with moderate sample throughput
- Reducing variability in antibody incubation steps
- Experiments requiring consistent immunodetection
Fully Automated Systems (JESS Simple Western) excel in:
- High-throughput ubiquitination screening
- Studies with limited protein availability
- Quantitative comparisons across multiple samples
- Environments prioritizing reproducibility and documentation
Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Ubiquitination Validation Studies
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Pre-cast Gels (4-20% gradient) | Protein size separation | Optimal for resolving ubiquitinated species with different molecular weights [81] |
| Nitrocellulose Membranes (0.2 µm) | Protein immobilization | Standard support for immunoblotting [81] |
| Primary Antibodies (Target-specific) | Target protein detection | Should be validated for Western blotting; concentration needs optimization [81] |
| Primary Antibodies (Ubiquitin-specific) | Ubiquitin detection | For direct ubiquitin detection (e.g., P4D1) or tag-specific if using tagged ubiquitin |
| HRP-conjugated Secondary Antibodies | Signal generation | Species-specific conjugates for chemiluminescent detection [81] |
| Chemiluminescent Substrate | Signal detection | ECL-based reagents for sensitive detection [81] |
| Housekeeping Protein Antibodies | Loading control | GAPDH, actin, or tubulin for normalization [81] [82] |
| Proteasome Inhibitors | Stabilize ubiquitinated proteins | MG132, lactacystin to prevent degradation of polyubiquitinated proteins |
| Deubiquitinase Inhibitors | Preserve ubiquitin conjugates | Include in lysis buffers (e.g., PR-619, N-ethylmaleimide) |
| Site-Directed Mutagenesis Kits | Create ubiquitination-deficient mutants | For lysine-to-arginine mutations at putative sites |
| Protein Ladders | Molecular weight reference | Broad range ladders essential for shift identification |
| Blocking Reagents | Reduce nonspecific binding | BSA or non-fat milk in TBST [81] |
Best Practices and Methodological Considerations
Image Processing and Data Integrity
When processing Western blot images for publication, maintaining data integrity is paramount:
- Minimal Processing: Apply only simple adjustments (brightness/contrast) uniformly to entire images [83]
- Avoid Selective Modifications: Do not enhance or obscure specific features within images; use arrows for emphasis instead [83]
- Retain Original Data: Always preserve unprocessed image files for verification [83]
- Consistent Cropping: Retain approximately five bandwidths of background above and below bands when cropping [83]
- Clear Splicing Indication: If splicing is necessary, clearly indicate with white spaces or black lines and describe in figure legends [83]
Quantitative Analysis Considerations
For accurate quantification of ubiquitination signals:
- Ensure Linear Range: Empirical determination of the amount of protein to load onto a gel is critical; ensure this amount is within the linear range for accurate quantitation [82]
- Validate Housekeeping Proteins: Housekeeping proteins used as internal loading controls must be validated first to ensure no variability in expression that can introduce error [82]
- Appropriate Normalization: Use normalization techniques to correct for unavoidable small variations, such as sample concentration or cell number [82]
- Replication: Replicate results to confirm reproducibility and distinguish actual differences from experimental variability [82]
Troubleshooting Molecular Weight Shift Detection
Common challenges in detecting ubiquitination through molecular weight shifts include:
- Smear Visualization: Polyubiquitinated smears may require longer exposure times or more sensitive detection methods
- Specificity Confirmation: Use ubiquitin mutants (e.g., lysine-less ubiquitin) to confirm chain topology
- Membrane Transfer Efficiency: Optimize transfer conditions for high molecular weight complexes
- Antibody Specificity: Validate antibodies for specific ubiquitin linkages if studying chain-type specific functions
Virtual Western blot analysis utilizing molecular weight shifts provides a robust secondary validation method for ubiquitination site identification when combined with mutagenesis approaches. While traditional Western blotting offers flexibility and established protocols, semi-automated and fully automated systems present advantages in reproducibility, sensitivity, and throughput that may benefit large-scale ubiquitination studies. The selection of appropriate methodology should be guided by experimental needs, resource availability, and required throughput. When properly implemented with appropriate controls and validation steps, molecular weight shift analysis serves as a reliable technique confirming ubiquitination events in the broader context of PTM validation.
Integrative Validation: Correlating Mutagenesis with Complementary Methodologies
Within the framework of research focused on validating ubiquitination sites by mutagenesis and immunoblotting, the analysis of protein half-life serves as a critical functional correlate. Protein ubiquitination, a versatile post-translational modification, is a primary signal targeting proteins for degradation by the 26S proteasome [3]. Consequently, the targeted disruption of a ubiquitination site often results in a measurable stabilization of the protein. The Cycloheximide (CHX) Chase Assay is a widely recognized biochemical method used to observe intracellular protein degradation and determine the half-life of a given protein in eukaryotes [84]. This guide provides an objective comparison of the CHX chase assay against alternative methodologies for analyzing protein turnover, framing them within the context of ubiquitination research.
The Cycloheximide Chase Assay: Principle and Workflow
Core Principle
Cycloheximide (CHX) is a small molecule derived from Streptomyces griseus that acts as a global inhibitor of eukaryotic translation elongation. By restricting the ribosome translocation process, CHX halts new protein synthesis [84]. In a CHX chase assay, cells are treated with a suitable concentration of CHX, and the subsequent decay of a pre-existing protein pool is monitored over time. A short half-life protein will decrease in abundance over time, while a long half-life protein will exhibit little change. This assay efficiently visualizes protein degradation kinetics without radioactive isotopes [84] [85].
Detailed Experimental Protocol
A standardized protocol for the CHX chase assay is as follows [84] [85]:
- Cell Preparation: Seed cells (e.g., approximately 6 × 10^5 cells in a 35-mm dish) and incubate overnight to achieve appropriate confluence.
- CHX Treatment: Remove the medium and add fresh complete medium containing a predetermined concentration of CHX (typically ranging from 50–300 µg/mL, dissolved in DMSO). A cell line test is recommended to determine a concentration that inhibits translation without inducing apoptosis within the experimental timeframe.
- Time-Course Harvesting: Harvest cells at predetermined time points (e.g., t = 0, 1, 2, 3, 6 hours). For an unfamiliar protein, a time course spanning up to 24 hours is advised, noting that CHX cytotoxicity can become significant beyond 20–24 hours.
- Lyse cells at each time point with a suitable protein lysis buffer containing fresh protease inhibitors.
- Clarify lysates by centrifugation and determine protein concentrations.
- Target Protein Detection: Analyze protein levels by SDS-PAGE and Western blotting using target-specific antibodies (e.g., anti-Flag) and a loading control antibody (e.g., β-actin or tubulin).
- Quantification and Half-Life Calculation: Quantify band intensities using software such as MetaMorph or ImageJ. Calculate the ratio of the target protein to the loading control at each time point. Express the protein level as a percentage of the level at t=0. Plot these percentages over time and fit the data to an exponential decay curve to determine the half-life.
The workflow and the primary signaling pathway connecting ubiquitination to degradation that can be investigated with this assay are summarized in the diagram below.
Figure 1: CHX Chase Assay Workflow and Associated Ubiquitin-Proteasome Pathway. The experimental steps (red-to-green) are used to investigate protein degradation typically mediated by the ubiquitin-proteasome pathway (blue).
Comparative Analysis of Protein Turnover Methodologies
While the CHX chase assay is a cornerstone technique, several other methods exist for studying protein turnover, each with distinct strengths and limitations. A comparative analysis is essential for selecting the appropriate tool.
Table 1: Comparison of Key Protein Turnover Analysis Methodologies
| Method | Core Principle | Key Advantages | Key Limitations / Cytotoxicity | Throughput | Suitability for Ubiquitination Studies |
|---|---|---|---|---|---|
| CHX Chase Assay [84] [85] | Global translation inhibition to monitor pre-existing protein decay. | Easy to operate; no need for specialized equipment or isotopes; direct assessment of degradation. | High cytotoxicity, unsuitable for long-lived proteins; blocks all new synthesis. | Medium (single protein focus) | Excellent: Direct functional readout for ubiquitination-mediated degradation. |
| Stable Isotope Labeling (SILAC, Heavy Water) [86] [87] | Metabolic incorporation of heavy isotopes to track newly synthesized proteins. | In vivo measurement; proteome-wide coverage; can measure synthesis and degradation. | High cost; complex data analysis; requires specialized instrumentation (LC-MS/MS). | High (proteome-wide) | Good: Identifies proteins with altered turnover; can be combined with ubiquitin enrichment. |
| Pulse-Chase Assay [84] | Incorporation of radiolabeled amino acids (e.g., ³⁵S-Methionine) to track a cohort of proteins. | Considered a "gold standard"; allows simultaneous monitoring of synthesis and degradation. | Use of radioactive materials; complex operation; health and safety concerns. | Low | Good: Directly measures degradation but less commonly used due to radioactivity. |
The following diagram illustrates the logical decision-making process for selecting the most appropriate methodology based on research goals.
Figure 2: Methodology Selection Workflow for Protein Turnover Analysis. The decision tree guides researchers to the most suitable technique based on the scope of their study and specific measurement objectives.
The Scientist's Toolkit: Essential Reagents for CHX Chase Assays
Successful execution of a CHX chase assay relies on a set of core reagents. The following table details these essential components and their functions.
Table 2: Key Research Reagent Solutions for CHX Chase Assays [84] [85]
| Reagent / Solution | Function in the Assay | Example Specifications / Considerations |
|---|---|---|
| Cycloheximide (CHX) | Core inhibitor of eukaryotic translation elongation; halts new protein synthesis to isolate degradation. | Soluble in DMSO; working concentration typically 50-300 µg/mL; requires cytotoxicity testing for each cell line. |
| Cell Culture Medium | Supports cell health during the assay period. | e.g., DMEM or RPMI-1640, supplemented with serum (e.g., 10% FBS) and antibiotics. |
| Lysis Buffer | Extracts proteins from cells at each time point while preserving protein integrity and modifications. | Contains detergent (e.g., 1% IGEPAL CA-630), salts, and protease inhibitor cocktail to prevent post-lysis degradation. |
| Protease Inhibitor Cocktail | Prevents protein degradation by cellular proteases after cell lysis, crucial for accurate quantification. | Often includes inhibitors of serine, cysteine, and metalloproteases; some formulations include EDTA. |
| Target Protein Antibody | Primary antibody for detection and quantification of the protein of interest via Western blot. | Specificity and titer are critical; validation for use in Western blot is required (e.g., Anti-Flag, Anti-Tyk2). |
| Loading Control Antibody | Primary antibody for detecting a stable internal control protein to normalize for loading variance. | Detects a constitutively expressed protein with a long half-life (e.g., Anti-Tubulin, Anti-β-actin). |
| HRP-Conjugated Secondary Antibody | Enables chemiluminescent detection of the primary antibodies bound to the target and loading control. | Species-specific (e.g., Goat anti-rabbit IgG-HRP); dilution typically 1:10,000. |
Within the investigative pipeline for validating ubiquitination sites, the CHX chase assay stands as an accessible, direct, and powerful method for functionally confirming the consequences of ubiquitination—accelerated protein degradation. Its utility in demonstrating the stabilization of a protein upon mutation of a putative ubiquitination site is unmatched for focused, single-protein studies. However, researchers must be mindful of its limitations, particularly its cytotoxicity and inability to resolve proteome-wide dynamics. The choice between the CHX chase assay, pulse-chase, and modern mass spectrometry-based methods should be guided by the specific research question, available resources, and desired throughput. Used judiciously, the CHX chase assay remains an indispensable tool in the molecular biologist's arsenal for elucidating the functional correlates of protein ubiquitination.
Protein ubiquitination is a fundamental post-translational modification (PTM) that regulates diverse cellular functions, including protein degradation, cell cycle progression, DNA damage repair, and numerous signaling pathways [3] [43]. This modification involves the covalent attachment of ubiquitin, a small 76-amino acid protein, to substrate proteins via a three-enzyme cascade comprising E1 (activating), E2 (conjugating), and E3 (ligating) enzymes [43]. The versatility of ubiquitination stems from the complexity of ubiquitin conjugates, which can range from single ubiquitin monomers to polymers of different lengths and linkage types [3]. Dysregulation of ubiquitination pathways leads to many pathologies, including cancer and neurodegenerative diseases, making accurate detection and validation of ubiquitination sites crucial for both basic research and drug development [3] [43].
Traditional methods for validating ubiquitination sites have relied heavily on mutagenesis and immunoblotting, where putative ubiquitinated lysines are substituted and analyzed via Western blotting with anti-ubiquitin antibodies [3]. While these approaches remain valuable for studying individual proteins, they are time-consuming, low-throughput, and provide limited information about specific modification sites [3]. The emergence of mass spectrometry (MS)-based methods, particularly those focusing on the direct detection of di-glycine remnants on modified lysines, has revolutionized the field by enabling high-throughput, site-specific mapping of ubiquitination events across the entire proteome [88] [89] [90].
This guide objectively compares the performance of leading MS-based methodologies for di-glycine remnant detection against traditional validation techniques, providing researchers with experimental data and protocols to inform their study design.
The Biochemical Basis of Di-glycine Remnant Detection
The Trypsin Digestion Principle
The di-glycine remnant strategy capitalizes on a fundamental aspect of ubiquitin biochemistry. When ubiquitinated proteins are digested with trypsin, the protease cleaves after arginine and lysine residues but leaves a distinctive signature on ubiquitinated lysines [88] [89]. Specifically, trypsin digestion cleaves off all but the two C-terminal glycine residues of ubiquitin, leaving these two glycine (GG) residues linked via an isopeptide bond to the epsilon-amino group of the modified lysine in the substrate peptide [88]. This K-ε-GG moiety adds a characteristic mass shift of 114.04 Da to the modified lysine residue and prevents tryptic cleavage at this site, resulting in an internal modified lysine within the tryptic peptide [88]. This tryptic remnant serves as a universal marker for ubiquitination sites, enabling the development of enrichment and detection strategies that have transformed the ubiquitin field.
It is important to note that modification by ubiquitin-like proteins Nedd8 and ISG15 also result in a GG remnant being retained on modified lysine residues, making these modifications indistinguishable from ubiquitination based solely on the tryptic remnant [88]. However, control experiments have demonstrated that >94% of K-ε-GG sites in HCT116 cells result from ubiquitination rather than Nedd8ylation or ISG15ylation [88].
Table 1: Key Characteristics of Di-glycine Remnant Detection
| Aspect | Specification | Biological Significance |
|---|---|---|
| Origin | C-terminal glycine residues of ubiquitin (G75-G76) | Ubiquitin-specific signature with high conservation across eukaryotes |
| Mass Shift | +114.04 Da on modified lysine | Detectable by high-resolution mass spectrometry |
| Trypsin Effect | Prevents cleavage at modified lysine | Creates longer peptides with internal modified lysines |
| Specificity | Also generated by Nedd8 and ISG15 | ~94% of sites originate from ubiquitination in human cells |
| Stability | Stable under standard MS conditions | Suitable for various enrichment and separation techniques |
Visualizing the Di-glycine Remnant Generation Pathway
The following diagram illustrates the biochemical process that generates the detectable di-glycine remnant during mass spectrometry sample preparation:
Comparative Analysis of Ubiquitination Detection Methodologies
Methodological Approaches and Their Performance Characteristics
Researchers have developed multiple strategies for detecting and validating ubiquitination sites, each with distinct advantages and limitations. The table below provides a comprehensive comparison of the primary methodologies:
Table 2: Performance Comparison of Ubiquitination Detection Methods
| Method | Throughput | Sensitivity | Site Resolution | Key Limitations | Typical Applications |
|---|---|---|---|---|---|
| Mutagenesis + Immunoblotting | Low (single protein) | Moderate | Indirect inference | Cannot distinguish specific sites on same protein; antibody specificity issues | Validation of putative ubiquitination sites on individual proteins |
| Ubiquitin Tagging | Medium (hundreds of sites) | High with enrichment | Direct site identification | Tag may alter ubiquitin function; requires genetic manipulation | Proteome-wide screening in engineered cell lines |
| Ubiquitin Antibody Enrichment | Medium (hundreds of sites) | High with enrichment | Direct site identification | Linkage-specific antibodies limited; non-specific binding | Physiological conditions without genetic manipulation |
| K-ε-GG Antibody MS | High (>10,000 sites) | Very high | Direct site identification | Cannot distinguish ubiquitination from Nedd8/ISG15; requires specific antibody | Large-scale ubiquitinome profiling; quantitative studies |
| UbiSite Antibody MS | Very high (>63,000 sites) | Very high | Direct site identification | Requires LysC digestion; specialized antibody | Comprehensive ubiquitinome mapping; N-terminal ubiquitination studies |
Traditional Validation: Mutagenesis and Immunoblotting
The conventional approach for validating ubiquitination involves immunoblotting with anti-ubiquitin antibodies combined with site-directed mutagenesis [3]. In this methodology, putative ubiquitinated lysine residues are substituted with non-modifiable residues (typically arginine), and the ubiquitination status is analyzed by Western blotting [3]. For example, Ortiz et al. demonstrated that ubiquitination of Merkel cell polyomavirus large tumor (LT) antigen was significantly reduced when K585 was mutated to R585, identifying K585 as a ubiquitination site [3].
Key Protocol Steps:
- Generate lysine-to-arginine mutants of the protein of interest
- Express wild-type and mutant proteins in appropriate cell lines
- Treat cells with proteasome inhibitors (e.g., MG132) to accumulate ubiquitinated species if necessary
- Perform immunoprecipitation of the target protein
- Analyze by Western blotting with anti-ubiquitin antibodies (e.g., P4D1, FK1, FK2)
- Compare ubiquitination signals between wild-type and mutant proteins
While this approach is widely used and accessible, it suffers from several limitations: it is time-consuming, provides low-throughput analysis, relies on antibody specificity, and cannot distinguish between multiple ubiquitination sites on the same protein [3]. Additionally, mutagenesis approaches only provide indirect evidence of ubiquitination sites rather than direct identification.
Mass Spectrometry-Based Di-glycine Remnant Detection Methods
Anti-K-ε-GG Antibody Enrichment Methodology
The development of antibodies specifically recognizing the tryptic K-ε-GG remnant has dramatically improved the sensitivity and scale of ubiquitination site mapping [88] [89] [90]. This approach enables specific enrichment of formerly ubiquitinated peptides from complex protein digests, significantly reducing sample complexity and enhancing detection of low-abundance ubiquitination events [88].
Detailed Experimental Protocol [88]:
Sample Preparation
- Lyse cells or tissue in urea lysis buffer (8 M urea, 50 mM Tris HCl pH 8.0, 150 mM NaCl) supplemented with protease and deubiquitinase inhibitors (e.g., PR-619)
- Reduce disulfide bonds with dithiothreitol (DTT) and alkylate with iodoacetamide or chloroacetamide
- Digest proteins first with LysC (Wako) followed by trypsin (Promega)
- Desalt peptides using solid-phase extraction (SPE)
Fractionation (Critical for High Coverage)
- Fractionate peptides by basic pH reversed-phase chromatography (bRP) using ammonium formate pH 10
- Collect 8-12 fractions across acetonitrile gradient (increases identifications 2-3 fold)
Antibody Enrichment
- Cross-link anti-K-ε-GG antibody (Cell Signaling Technology) to protein A beads using dimethyl pimelimidate (DMP) to prevent antibody leakage
- Incubate peptide fractions with cross-linked antibody beads for 2 hours at 4°C
- Wash beads extensively with PBS and water
- Elute K-ε-GG peptides with 0.15% trifluoroacetic acid
Mass Spectrometric Analysis
- Analyze enriched peptides by LC-MS/MS on high-resolution instruments (Orbitrap platforms)
- Identify K-ε-GG sites using database search algorithms (e.g., MaxQuant, Spectrum Mill) with 114.04292 Da modification on lysine
This methodology routinely identifies >10,000 distinct ubiquitination sites in single experiments from moderate protein input amounts (5-15 mg) [88] [90]. When combined with SILAC (Stable Isotope Labeling by Amino Acids in Cell Culture), it enables quantitative assessment of ubiquitination dynamics in response to cellular perturbations [88].
UbiSite Method for Comprehensive Ubiquitination Mapping
The UbiSite approach represents an advancement in ubiquitination site mapping by using an antibody that recognizes the C-terminal 13 amino acids of ubiquitin, which remain attached to modified peptides after digestion with the endoproteinase LysC [91]. This method offers two significant advantages: exceptional specificity for ubiquitin (excluding other ubiquitin-like modifiers), and the ability to detect N-terminal ubiquitination in addition to lysine ubiquitination [91].
Key Differentiating Features:
- Employes LysC instead of trypsin for proteolytic digestion
- Detects both lysine and protein N-terminal ubiquitination events
- Identified >63,000 unique ubiquitination sites on 9,200 proteins in human cell lines
- Revealed inverse association between protein N-terminal ubiquitination and acetylation
The UbiSite methodology has uncovered widespread involvement of ubiquitination in all cellular aspects and demonstrated a complete lack of correlation between changes in protein abundance and alterations in ubiquitination sites upon proteasome inhibition, highlighting the complex regulation of ubiquitin signaling [91].
Visualizing the K-ε-GG Enrichment Workflow
The following flowchart details the complete experimental workflow for anti-K-ε-GG antibody-based ubiquitinome profiling:
Quantitative Performance Comparison
Method Efficacy and Throughput Assessment
The table below summarizes the quantitative performance of different ubiquitination detection methods based on published data:
Table 3: Quantitative Performance of Ubiquitination Detection Methods
| Method | Typical Sites Identified | Sample Requirements | Quantification Capability | Specialized Detection |
|---|---|---|---|---|
| Mutagenesis + Immunoblotting | Individual sites | Variable; typically transfected cells | Semi-quantitative | Linkage-specific antibodies available |
| His-Ub Tagging (Peng et al.) | 110 sites (72 proteins) | Engineered yeast cells | Relative quantification | Specific chain linkages with linkage-specific antibodies |
| Strep-Ub Tagging (Danielsen et al.) | 753 sites (471 proteins) | Engineered mammalian cells | Relative quantification | Specific chain linkages with linkage-specific antibodies |
| Anti-K-ε-GG (Standard) | 1,000-5,000 sites | 5-15 mg protein input | SILAC quantification | Mixed linkage detection |
| Anti-K-ε-GG (Optimized) | ~20,000 sites | 15-30 mg protein input | SILAC/TMT quantification | Mixed linkage detection |
| UbiSite (LysC-based) | >63,000 sites (9,200 proteins) | Large-scale input | Label-free quantification | N-terminal ubiquitination detection |
Technical Considerations and Limitations
Each methodology presents distinct technical considerations that influence their application:
Anti-K-ε-GG Antibody Limitations:
- Cannot distinguish ubiquitination from Nedd8ylation or ISG15ylation (though ubiquitination represents >94% of modifications)
- Requires high-quality, specific antibodies with careful cross-linking to prevent contamination
- Dependent on efficient trypsin digestion and sample preparation
- Lower abundance sites may require extensive fractionation and high sample input
UbiSite Method Limitations:
- Requires specialized antibody not commercially widespread
- LysC digestion may produce different peptide populations compared to trypsin
- Protocol optimization may be needed for different sample types
Traditional Methods Limitations:
- Poor suitability for proteome-wide studies
- Limited quantitative capabilities
- Inability to detect exact modification sites
- Antibody specificity concerns
Essential Research Reagents and Tools
The Scientist's Toolkit for Di-glycine Remnant Detection
Successful implementation of di-glycine remnant detection methods requires specific reagents and tools:
Table 4: Essential Research Reagents for Di-glycine Remnant Detection
| Reagent/Tool | Function | Examples/Specifications |
|---|---|---|
| Anti-K-ε-GG Antibody | Enrichment of ubiquitinated peptides | Cell Signaling Technology #5562; cross-linked to protein A beads |
| Ubiquitin Tagging System | Expression of tagged ubiquitin | His-Ub, Strep-Ub, HA-Ub for affinity purification |
| Linkage-specific Antibodies | Detection of specific ubiquitin linkages | K48-, K63-, M1-linkage specific antibodies available |
| Protease Inhibitors | Preservation of ubiquitination | PR-619 (DUB inhibitor), PMSF, Aprotinin, Leupeptin |
| Digestion Enzymes | Protein cleavage | Trypsin (Promega), LysC (Wako) |
| Fractionation Materials | Sample complexity reduction | High-pH reversed-phase chromatography |
| Mass Spectrometers | Peptide identification | High-resolution instruments (Orbitrap platforms) |
| Database Search Software | Site identification | MaxQuant, Spectrum Mill with K-ε-GG modification (114.04292 Da) |
The direct detection of di-glycine remnants by mass spectrometry has fundamentally transformed our ability to map and validate ubiquitination sites at a proteome-wide scale. The anti-K-ε-GG antibody enrichment method represents a robust, well-established approach capable of identifying >20,000 ubiquitination sites in a single experiment, providing unprecedented coverage of the ubiquitinome [88] [90]. The newer UbiSite methodology offers complementary advantages, including exceptional specificity for ubiquitin and the ability to detect N-terminal ubiquitination events typically missed by traditional approaches [91].
For researchers validating specific ubiquitination sites initially identified through mutagenesis and immunoblotting, mass spectrometry-based di-glycine remnant detection provides orthogonal validation with site-specific resolution. The choice between methods should be guided by research goals: traditional mutagenesis and immunoblotting remain valuable for focused studies of individual proteins, while MS-based approaches are indispensable for comprehensive ubiquitinome profiling and systems-level understanding of ubiquitin signaling.
As mass spectrometry technology continues to advance with improved sensitivity, speed, and quantification capabilities, di-glycine remnant detection will undoubtedly remain a cornerstone methodology for elucidating the complex roles of ubiquitination in health and disease, ultimately facilitating the development of targeted therapeutics for cancer, neurodegenerative disorders, and other ubiquitination-related diseases.
The validation of protein ubiquitination sites is a critical step in understanding a vast array of cellular processes, including protein degradation, DNA repair, and cell signaling. Dysregulation of ubiquitination is implicated in numerous pathologies, such as cancer and neurodegenerative diseases [3] [92]. For researchers and drug development professionals, selecting the appropriate methodological pathway is paramount. This guide provides an objective comparison of two foundational approaches: the conventional method of mutagenesis coupled with immunoblotting and the increasingly prevalent mass spectrometry (MS)-based proteomics. We will dissect their strengths, limitations, and ideal applications, supported by experimental data and detailed protocols, to inform your experimental design within the broader context of ubiquitination research.
Mutagenesis and Immunoblotting
This traditional, hypothesis-driven approach is used to confirm both the ubiquitination of a specific protein and the identity of the modified lysine residue(s).
Core Protocol:
- Generate Mutants: Create point mutants of the protein of interest, substituting suspected lysine (Lys/K) residues with arginine (Arg/R).
- Express Proteins: Transfect cells with plasmids encoding both the wild-type (WT) and mutant proteins.
- Immunoprecipitation (IP): Harvest cell lysates and immunoprecipitate the target protein using a specific antibody or an affinity tag (e.g., Flag, HA).
- Immunoblotting (IB): Analyze the immunoprecipitated proteins by Western blot.
The logical relationship and workflow of this method is outlined in the diagram below.
Mass Spectrometry-Based Proteomics
This is a discovery-oriented, high-throughput approach that enables the unbiased identification of ubiquitination sites across thousands of proteins in a single experiment.
Core Protocol: The most common and effective strategy is the ubiquitin remnant profiling technique [93] [92].
- Digest Proteins: Lyse cells and digest the proteome into peptides with trypsin.
- Enrich Ubiquitinated Peptides: Use monoclonal antibodies (e.g., di-Gly lysine antibodies) to specifically immunoprecipitate peptides that contain the di-glycine remnant left on modified lysines after trypsin digestion.
- LC-MS/MS Analysis: Separate the enriched peptides by liquid chromatography (LC) and analyze them by tandem mass spectrometry (MS/MS).
- Data Analysis: Use bioinformatics tools to search MS/MS spectra against protein databases. The identification of a peptide with a di-glycine modification (a 114.04 Da mass shift on a lysine) localizes the ubiquitination site [3] [92].
The workflow for this method is detailed in the following diagram.
Comparative Analysis of Technical Parameters
The choice between mutagenesis and MS-based approaches depends heavily on project goals, scale, and resource constraints. The table below summarizes the core characteristics of each method.
Table 1: Core Characteristics of Ubiquitination Site Validation Methods
| Feature | Mutagenesis + Immunoblotting | MS-Based Proteomics |
|---|---|---|
| Primary Application | Targeted validation of a single protein | Proteome-wide discovery |
| Throughput | Low-throughput (one protein at a time) | High-throughput (100s-1000s of sites) |
| Key Strength | Directly tests functional necessity of a specific lysine | Unbiased, systems-level view of the ubiquitinome |
| Major Limitation | Prone to false negatives from compensatory mutagenesis | May miss low-abundance or transient modifications |
| Quantitative Accuracy | Semi-quantitative (based on band intensity) | Good to high (using SILAC, TMT, or label-free methods) [94] |
| Typical Workflow Duration | 1-2 weeks | Several days to weeks (including analysis) |
Detailed Comparison of Strengths and Limitations
Mutagenesis and Immunoblotting
Strengths:
- Functional Insight: It directly links a specific lysine residue to the ubiquitination of the protein. Demonstrating that mutation alters protein stability or function provides powerful mechanistic evidence [92].
- Accessibility: The protocol utilizes standard molecular biology and biochemistry equipment found in most labs, requiring no specialized MS instrumentation [92].
- Cost-Effectiveness for Single Targets: For validating ubiquitination of one or a few proteins, it is more economical than large-scale MS projects.
Limitations:
- Low-Throughput: The process is slow and laborious, making it unsuitable for discovering novel ubiquitination events or profiling entire pathways [3].
- Indirect Detection: It infers ubiquitination based on antibody signal and mobility shifts; it does not directly identify the modified site [92].
- Compensatory Mutagenesis: When a major ubiquitination site is mutated, nearby lysines may become secondarily modified, leading to false negatives or misinterpretation of the primary site [92].
- Artifact Potential: Extensive lysine-to-arginine mutations can disrupt protein folding, interaction with E3 ligases, or subcellular localization, confounding results [92].
MS-Based Proteomics
Strengths:
- Comprehensive and Unbiased: Capable of identifying thousands of ubiquitination sites from a single experiment without prior knowledge of targeted proteins, enabling discovery of novel regulatory events [3] [93].
- Direct and Definitive Site Mapping: MS/MS data provides direct evidence by localizing the di-glycine modification to a specific lysine residue within a peptide sequence, offering high confidence in identification [92].
- Capability for Quantification: Can be coupled with isotopic labeling (e.g., SILAC, TMT) or label-free methods to quantitatively track dynamic changes in ubiquitination in response to stimuli or drug treatments [94] [93].
Limitations:
- Technical Complexity and Cost: Requires sophisticated, expensive instrumentation and specialized expertise in proteomics and bioinformatics for data analysis [94] [95].
- Challenges with Low-Abundance Proteins: The dynamic range of the proteome is a major hurdle. High-abundance proteins can dominate the analysis, masking the detection of low-abundance, but biologically critical, ubiquitinated substrates [94].
- Sample Preparation Artifacts: Multi-step preparation (enrichment, fractionation) can introduce variability and loss of material, affecting reproducibility and quantitative accuracy [94].
Table 2: Summary of Key Advantages and Disadvantages
| Aspect | Mutagenesis + Immunoblotting | MS-Based Proteomics |
|---|---|---|
| Throughput & Scope | - Low-throughput, focused | + High-throughput, global |
| Site Identification | - Indirect, inferential | + Direct, precise |
| Functional Link | + Strong (via mutation) | - Weak (correlative) |
| Technical Barrier | + Low | - High |
| Cost for Single Target | + Lower | - Higher |
| Risk of Artifacts | - High (protein misfolding) | - Medium (sample handling) |
| Handling Complex Samples | - Poor | + Excellent |
Research Reagent Solutions
Successful execution of these methodologies relies on a suite of specific reagents. The following table details essential materials and their functions.
Table 3: Key Research Reagents for Ubiquitination Studies
| Reagent / Tool | Function / Application | Method |
|---|---|---|
| K-to-R Mutant Plasmids | Substitutes lysine with arginine to prevent ubiquitination, used for functional validation. | Mutagenesis |
| Anti-Ubiquitin Antibodies (e.g., P4D1, FK2) | Detect polyubiquitinated proteins in Western blots or enrich ubiquitinated proteins. | Mutagenesis, MS Sample Prep |
| di-Gly (K-ε-GG) Remnant Antibodies | Immunoaffinity enrichment of tryptic peptides derived from ubiquitinated proteins for MS analysis. | MS-Based Proteomics |
| Tagged-Ubiquitin (e.g., His-, HA-, Strep-Ub) | Expression in cells allows purification of ubiquitinated conjugates under denaturing conditions. | MS Sample Prep |
| Tandem Mass Tag (TMT) | Isobaric labels for multiplexed relative quantification of ubiquitinated peptides across multiple samples. | MS-Based Proteomics |
| Stable Isotope Labeling with Amino Acids in Cell Culture (SILAC) | Metabolic labeling for quantitative comparison of ubiquitination changes between two or three cell states. | MS-Based Proteomics |
The decision between mutagenesis and MS-based approaches is not a matter of which is superior, but which is most appropriate for the specific research question.
- Employ mutagenesis with immunoblotting when the goal is to definitively validate the ubiquitination of a specific, pre-identified protein and to understand the functional consequence of modifying a particular lysine residue.
- Employ MS-based proteomics when the objective is to discover novel ubiquitination events, profile global changes in the ubiquitinome in response to a perturbation, or understand system-wide network regulation.
The most powerful strategies in modern ubiquitination research often involve an integrated approach: using MS-based discovery to identify a landscape of potential targets and sites, followed by targeted mutagenesis and functional validation to establish mechanistic causality and biological relevance.
In the intricate world of post-translational modifications, the ubiquitin-proteasome system (UPS) stands as a crucial regulatory mechanism governing diverse cellular processes from protein degradation to signal transduction. At the heart of this system lie E3 ubiquitin ligases, the primary determinants of specificity that recognize and facilitate ubiquitin transfer to particular substrate proteins. With over 600 putative E3 ligases encoded in the human genome but only a fraction characterized, the challenge of definitively establishing E3-substrate relationships remains a central focus in biochemical research [96].
While in vivo methods like co-immunoprecipitation can identify potential E3 interactors, they often fail to distinguish genuine ubiquitination targets from mere complex associations. In vitro reconstitution with purified components provides an unparalleled approach for direct validation, free from confounding cellular factors. This guide systematically compares established and emerging methodologies for validating E3 ligase specificity, providing researchers with experimental frameworks to advance our understanding of the ubiquitin code.
Established Methodologies for E3-Substrate Validation
Classical In Vitro Reconstitution Assays
The foundational approach for validating E3 ligase activity involves reconstituting the ubiquitination cascade using purified components. This method provides direct evidence of enzymatic capability without interference from cellular regulators or competing enzymes.
Core Protocol Components:
The minimal system requires several purified components: E1 activating enzyme, E2 conjugating enzyme, E3 ligase, ubiquitin, substrate protein, and ATP regeneration system. The reaction typically proceeds at 30°C for 1-2 hours before termination with SDS-PAGE loading buffer [97].
Key Technical Considerations:
Enzyme Titration: Careful titration of E1 and E2 enzymes is critical, as their abundance can mask the enhancing effect of E3 ligases. Reduced E1/E2 concentrations often better reveal genuine E3 activity [97].
Detection Methods: Substrate ubiquitination is typically detected via immunoblotting using substrate-specific antibodies to visualize upward mobility shifts, or ubiquitin-specific antibodies to confirm modification.
Specificity Controls: Essential controls include omitting individual components (E1, E2, E3, or ATP) to confirm requirement of all cascade elements, and using catalytically inactive E3 mutants to verify enzymatic dependence.
Table 1: Core Components for In Vitro Ubiquitination Assays
| Component | Function | Example Sources | Key Considerations |
|---|---|---|---|
| E1 Activating Enzyme | Activates ubiquitin via ATP-dependent thioester bond formation | Commercial recombinant proteins, insect cell expression | Single E1 (UBA1) in humans; titration critical to avoid masking E3 effects |
| E2 Conjugating Enzyme | Accepts ubiquitin from E1, conjugates to substrate | Commercial recombinant proteins, bacterial expression | ~40 human E2s; choice influences linkage specificity |
| E3 Ubiquitin Ligase | Confers substrate specificity, promotes ubiquitin transfer | Varies by study (bacterial, insect cell, or mammalian expression) | >600 human E3s; catalytic activity must be validated |
| Ubiquitin | Protein modifier conjugated to substrates | Commercial recombinant ubiquitin | Wild-type or mutant forms for specific chain types |
| Substrate Protein | Target of ubiquitination | Dependent on research focus | Purification must maintain native conformation |
| ATP Regeneration System | Provides energy for E1 activation | Commercial systems or laboratory-prepared | Essential for reaction progression |
Advantages and Limitations:
This approach provides direct biochemical evidence of E3 activity under controlled conditions. However, it may not recapitulate cellular regulation, and false negatives can occur if reaction conditions are suboptimal or post-translational modifications necessary for recognition are absent.
Mutagenesis Approaches for Degron Mapping
Once E3-mediated ubiquitination is established, identifying specific degrons—the molecular motifs recognized by E3 ligases—becomes essential. Lysine-to-arginine mutagenesis of putative acceptor sites in substrates represents the gold standard for mapping ubiquitination sites [3].
Experimental Framework:
Systematic Mutagenesis: Individual lysine residues or degron motifs are mutated, and ubiquitination assays repeated to identify essential sites.
Domain Mapping: Truncation mutants help identify protein regions necessary for E3 recognition.
Functional Validation: Mutated substrates are tested in cellular degradation assays to confirm functional consequences of disrupted ubiquitination.
Case Example:
A compelling example comes from the study of the Merkel cell polyomavirus large tumor (LT) antigen, where substitution of K585 with arginine significantly reduced ubiquitination levels, identifying K585 as a critical ubiquitination site [3].
Emerging High-Throughput Validation Technologies
COMET: Combinatorial Mapping of E3 Targets
The COMET framework represents a significant advancement for testing E3-substrate interactions at scale. This approach enables systematic assessment of thousands of E3-substrate combinations within single experiments, moving beyond the one-by-one validation of traditional methods [98].
Methodological Principles:
COMET utilizes pooled expression libraries encoding numerous E3 ligases and potential substrates. Through sophisticated barcoding and sequencing strategies, it tracks degradation relationships en masse, revealing complex E3-substrate networks that often diverge from simple one-to-one associations.
Applications and Insights:
Application of COMET to SCF ubiquitin ligase subunits screened 6,716 F-box-ORF combinations, while screening of E3s degrading short-lived transcription factors assessed 26,028 E3-TF combinations. The data revealed that many E3-substrate relationships are complex rather than simple binary associations, with multiple E3s often targeting the same substrate and individual E3s modifying multiple substrates [98].
Multiplex CRISPR Screening Platforms
Recent advances in CRISPR technology have enabled the development of multiplex screening platforms that simultaneously map E3 ligases to hundreds of substrates in parallel. This approach combines the Global Protein Stability (GPS) expression system with CRISPR-mediated E3 knockout [99].
Workflow Integration:
The method employs a dual vector system encoding both GFP-tagged substrates and sgRNAs targeting E3 ubiquitin ligases. When expressed in Cas9-containing cells, disruption of a cognate E3 stabilizes its substrate, increasing GFP fluorescence. FACS isolation of stabilized populations followed by paired-end sequencing identifies both the substrate and its regulating E3 [99].
Validation and Performance:
A proof-of-principle screen successfully performed approximately 100 CRISPR screens in a single experiment, refining known C-degron pathways and identifying novel relationships, such as Cul2-FEM1B targeting of C-terminal proline degrons. The system compatibly works with pools of full-length protein substrates of varying stabilities and, when combined with site-saturation mutagenesis, can assign E3 ligases to their cognate degron motifs [99].
BioE3: Proximity-Dependent Substrate Identification
The BioE3 system represents an innovative strategy combining proximity-dependent biotinylation with ubiquitin tagging to identify bona fide E3 substrates. This method addresses the critical challenge of distinguishing direct ubiquitination targets from mere interactors [100].
Technical Innovation:
BioE3 utilizes BirA-E3 fusion proteins combined with a bioUbL (biotinylatable ubiquitin-like protein) strategy. An engineered AviTag variant with lower BirA affinity (bioGEF) minimizes non-specific labeling, enabling precise capture of substrates as they are being ubiquitinated by the BirA-E3 fusion [100].
Experimental Workflow:
- Stable cell lines expressing bioGEFUb are generated
- BirA-E3 fusions are introduced under controlled expression
- Limited biotin pulses allow proximity-dependent labeling
- Streptavidin capture isolates biotinylated substrates
- LC-MS/MS identifies specific E3 targets
Validation and Applications:
BioE3 has successfully identified both known and novel targets for RING-type E3s (RNF4, MIB1, MARCH5, RNF214) and HECT-type E3s (NEDD4). The system can detect altered specificity in response to chemical treatments, opening avenues for targeted protein degradation research [100].
BioE3 Experimental Workflow
Comparative Analysis of Methodologies
Table 2: Method Comparison for E3-Substrate Validation
| Method | Throughput | Key Advantages | Limitations | Ideal Use Cases |
|---|---|---|---|---|
| Classical In Vitro Reconstitution | Low (1:1 testing) | Direct biochemical evidence; controlled conditions; minimal cellular confounding factors | May not recapitulate cellular context; requires protein purification | Initial validation of suspected E3-substrate pairs; degron mapping |
| Mutagenesis + Immunoblotting | Low to medium | Definitive site identification; functional validation possible | Labor-intensive; low-throughput | Mapping precise ubiquitination sites; validating degron motifs |
| COMET Framework | High (thousands of combinations) | Unbiased discovery; reveals complex E3 networks | Computational complexity; validation required | Systematic E3-substrate mapping; network biology studies |
| Multiplex CRISPR Screening | High (hundreds parallel) | Functional assessment in cellular context; identifies endogenous regulators | False positives from indirect effects; technical complexity | Functional genomics; identification of degron-E3 relationships |
| BioE3 System | Medium (E3-specific) | Distinguishes targets from interactors; subcellular resolution | Proximity labeling artifacts; optimization required | Specific E3 substrate identification; organelle-specific ubiquitination |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for E3-Substrate Validation
| Reagent/Tool | Function | Application Notes |
|---|---|---|
| BirA-E3 Fusion Constructs | Proximity-dependent labeling of E3 substrates | N-terminal fusions recommended to avoid RING domain disruption [100] |
| bioGEFUb (AviTag Mutant) | Biotinylatable ubiquitin with reduced non-specific labeling | Superior to wild-type AviTag for proximity labeling; minimal background [100] |
| GPS (Global Protein Stability) Vector | Lentiviral platform for stability profiling | Enables pooled screening of substrate libraries; GFP-DsRed reporting [99] |
| COMET Framework Software | Analysis of combinatorial E3-substrate data | Requires bioinformatics support; identifies complex relationships [98] |
| Linkage-Specific Ub Antibodies | Immunoblot detection of specific ubiquitin chains | K48-linked most abundant; K63-linked regulates signaling [3] |
| Catalytically Inactive E3 Mutants | Essential controls for E3 activity dependence | Typically cysteine mutations in HECT domains; RING domain disruptions |
Integrated Experimental Design for Comprehensive Validation
Based on methodological comparisons, a robust framework for E3-substrate validation emerges:
Phase 1: Discovery
- Implement high-throughput methods (COMET or multiplex CRISPR) for unbiased identification of potential E3-substrate relationships
- Utilize BioE3 for specific E3 ligases of interest
Phase 2: Reductionist Validation
- Employ classical in vitro reconstitution with purified components to confirm direct ubiquitination
- Conduct mutagenesis studies to map critical degron motifs and ubiquitination sites
Phase 3: Functional Confirmation
- Validate findings in cellular contexts through degradation assays
- Assess biological consequences through phenotypic studies
Comprehensive E3 Validation Strategy
This integrated approach leverages the complementary strengths of each methodology while mitigating their individual limitations. The sequential framework progresses from high-throughput discovery to reductionist biochemical validation and culminates in functional confirmation within biological systems.
The evolving methodological landscape for validating E3 ligase specificity offers researchers powerful tools to decipher the complex ubiquitination code. While classical in vitro approaches remain essential for biochemical proof, emerging technologies enable unprecedented scale and precision in defining E3-substrate relationships. The integration of these complementary approaches—from multiplexed CRISPR screens to proximity-dependent labeling strategies—accelerates our understanding of ubiquitin signaling networks and creates new opportunities for therapeutic intervention in ubiquitination-related diseases. As these methodologies continue to mature, they promise to illuminate the vast landscape of uncharacterized E3 ligases, advancing both fundamental knowledge and drug discovery efforts targeting the ubiquitin-proteasome system.
Ubiquitination is a versatile post-translational modification (PTM) that involves the covalent attachment of a small, 76-amino-acid protein, ubiquitin, to target proteins [101] [102]. This modification is orchestrated by a sequential enzymatic cascade involving ubiquitin-activating (E1), ubiquitin-conjugating (E2), and ubiquitin-ligating (E3) enzymes, which together confer substrate specificity [103] [102]. The versatility of ubiquitination stems from its ability to form diverse structures—including monoubiquitination, multiple monoubiquitination, and polyubiquitin chains linked through different lysine residues (K6, K11, K27, K29, K33, K48, K63) or the N-terminal methionine (M1)—each encoding distinct functional consequences [103] [102] [41]. This complexity, often referred to as the "ubiquitin code," regulates virtually all cellular processes, from protein degradation and DNA repair to immune signaling and cell death [102] [104].
Dysregulation of the ubiquitin system contributes significantly to disease pathogenesis, particularly in cancer, neurodegenerative disorders, and immune dysfunctions [101] [103] [102]. Mutations in E3 ligases or deubiquitinases (DUBs) can lead to aberrant stabilization of oncoproteins or accelerated degradation of tumor suppressors, driving tumorigenesis [103]. For instance, in von Hippel-Lindau (VHL) disease, loss-of-function mutations in the VHL E3 ligase result in accumulation of hypoxia-inducible factor-alpha (HIF-α), promoting angiogenesis and renal cell carcinoma [101]. The clinical significance of targeting ubiquitination is exemplified by the proteasome inhibitor bortezomib, which has achieved tangible success in treating multiple myeloma [101] [103]. However, emerging therapeutic strategies are moving beyond proteasome inhibition to target specific E3 ligases, DUBs, and other components of the ubiquitin system with greater precision [103] [102].
Methodologies for Detecting and Validating Site-Specific Ubiquitination
Understanding the biological roles and clinical implications of specific ubiquitination events relies on robust methods for detection and validation. These techniques range from conventional approaches to advanced proteomic strategies, each with distinct advantages and limitations.
Conventional Immunoblotting and Mutagenesis Approaches
The conventional workflow for validating site-specific ubiquitination typically involves immunoblotting combined with lysine mutagenesis [41]. This method tests whether substituting a specific lysine residue with a non-ubiquitinatable amino acid (e.g., arginine) reduces the ubiquitination signal of the substrate protein [41]. The process generally involves:
- Plasmid Preparation: Generating expression plasmids for the protein of interest (POI), both wild-type and with lysine-to-arginine (K-to-R) mutations at suspected ubiquitination sites [105].
- Cell Transfection and Treatment: Transfecting cells (e.g., HEK293T) with these plasmids and often treating with proteasome inhibitors (e.g., MG-132) to stabilize ubiquitinated species [105].
- Immunoprecipitation: Isolating the POI or the ubiquitin conjugate using specific antibodies or affinity tags (e.g., His-tagged ubiquitin with Ni-NTA agarose) [105] [41].
- Immunoblotting: Detecting ubiquitinated proteins with anti-ubiquitin antibodies (e.g., P4D1, FK1/FK2) or tags (e.g., anti-HA for HA-tagged ubiquitin) [105] [41] [42].
A significant challenge in these assays is distinguishing between direct ubiquitination of the POI and ubiquitination of associated proteins in a complex. The lysine mutagenesis approach is critical for confirming the specific lysine residue responsible for the modification [41].
Advanced Proteomic and Chemical Biology Tools
While conventional methods are invaluable for validating specific proteins, advanced proteomic approaches enable system-wide profiling of ubiquitination sites:
- Ubiquitin Tagging-Based Proteomics: Cells are engineered to express affinity-tagged ubiquitin (e.g., His-, Strep-, or FLAG-tags). Ubiquitinated proteins are enriched en masse using corresponding resins (Ni-NTA, Strep-Tactin) and identified via mass spectrometry (MS). The signature Gly-Gly (GG) remnant left on modified lysines after trypsin digestion allows precise site localization [41].
- Antibody-Based Enrichment: Endogenous ubiquitinated proteins are captured using general anti-ubiquitin antibodies (e.g., FK2) or linkage-specific antibodies (e.g., for K48 or K63 chains) [41]. This is particularly useful for clinical samples where genetic manipulation is infeasible.
- UBD-Based Approaches: Tandem-repeated Ub-binding entities (TUBEs) exhibit high affinity for polyubiquitin chains and protect them from deubiquitinase (DUB) activity during lysis, enhancing recovery [41] [42].
- Site-Specific Ubiquitin Antibodies: Generating antibodies that recognize ubiquitination at a specific lysine on a specific protein is challenging but highly valuable. This typically requires chemical synthesis of stable ubiquitin-peptide conjugates using non-hydrolyzable linkages (e.g., triazole isosteres) for immunization [106].
Table 1: Comparison of Major Methodologies for Ubiquitination Detection
| Methodology | Key Features | Advantages | Limitations | Best Suited For |
|---|---|---|---|---|
| Immunoblotting & Mutagenesis [105] [41] | - Lysine-to-arginine mutations- Proteasome inhibition (MG-132)- IP with tag-specific beads | - Direct causal validation of sites- Accessible, widely used- Quantitative with chemiluminescence | - Low throughput- Cannot distinguish chain linkages- Antibody specificity issues | Validating putative ubiquitination sites on individual proteins |
| Tag-Based Proteomics [41] | - Expression of tagged-Ub (e.g., His-Ub)- Ni-NTA/Strep-Tactin enrichment- MS detection of GG remnant | - High-throughput, system-wide- Identifies thousands of sites- Unbiased discovery | - Tag may alter Ub function- High false-positive rate from contaminants- Not suitable for tissues | Global discovery of ubiquitination sites in cell lines |
| Linkage-Specific Antibodies [41] | - Antibodies specific to chain types (K48, K63, etc.)- IP or enrichment before WB/MS | - Provides linkage information- Applicable to clinical samples- High specificity when available | - Limited antibody availability- High cost- Potential cross-reactivity | Studying specific ubiquitin signaling pathways in disease contexts |
| TUBEs [41] [42] | - Recombinant proteins with multiple UBDs- High-affinity Ub binding- DUB protection during lysis | - Protects labile ubiquitination- Enriches diverse chain types- Higher yield than single UBDs | - Protein expression and purification required- May not distinguish chain types | Stabilizing and enriching unstable ubiquitination events |
Figure 1: Experimental Workflow for Validating Site-Specific Ubiquitination by Mutagenesis and Immunoblotting.
Quantitative Landscape of Ubiquitination and its Biological Implications
Recent systems-scale studies have quantified the occupancy and turnover of ubiquitination sites, providing fundamental insights into its regulatory logic. The median occupancy of a ubiquitination site is remarkably low—approximately 0.0081%—which is over three orders of magnitude lower than the median phosphorylation occupancy (28%) [107]. This low occupancy indicates that the ubiquitin system operates on a principle of fast turnover and low abundance, allowing for rapid and dynamic control of protein fate without accumulating high levels of modified proteins [107].
Quantitative profiling reveals that ubiquitination sites can be broadly divided into two classes:
- The Low-Occupancy Majority (~80% of sites): These sites have very low occupancy (median ~0.0015%) and faster half-lives. They are often found in unstructured protein regions and are less responsive to proteasome inhibition, suggesting roles in non-proteolytic signaling [107].
- The High-Occupancy Minority (~20% of sites): These sites have higher occupancy (median ~0.12%), longer half-lives, and are strongly upregulated by proteasome inhibitors. They are frequently located in structured protein domains and are enriched in proteins involved in proteasomal degradation, such as solute carriers (SLCs) [107].
This quantitative framework is crucial for clinical translation. It suggests that targeting low-abundance, fast-turnover ubiquitination events (e.g., in signaling pathways) requires different therapeutic strategies than targeting high-occupancy sites involved in protein degradation.
Connecting Site-Specific Ubiquitination to Disease Pathogenesis
Dysregulation of specific ubiquitination events is a hallmark of numerous diseases. The following examples illustrate how connecting specific ubiquitination events to disease mechanisms reveals potential therapeutic targets.
Cancer
- Von Hippel-Lindau (VHL) Disease: Loss-of-function mutations in the VHL E3 ligase prevent the ubiquitination and proteasomal degradation of HIF-1α. This leads to constitutive activation of HIF-1α target genes (e.g., VEGF, EPO), promoting angiogenesis and tumor development in multiple organs [101] [103].
- Colorectal Cancer: Mutations in the adenomatous polyposis coli (APC) protein, part of a multi-subunit E3 complex, disrupt the ubiquitination of β-catenin. Consequently, β-catenin accumulates and drives uncontrolled proliferation of colorectal epithelial cells [101].
- Diffuse Large B-Cell Lymphoma (DLBCL): A ubiquitination-based prognostic signature comprising CDC34 (an E2 enzyme), FZR1 (an activator of the APC/C E3 complex), and OTULIN (a Met1-linkage-specific DUB) predicts patient outcomes. Elevated CDC34 and FZR1 with low OTULIN expression correlates with poor prognosis [108].
Neurodegenerative Disorders
- Alzheimer's Disease: K48-linked polyubiquitination of tau protein is abnormally accumulated in the brains of Alzheimer's patients, suggesting a failure in the clearance of pathological proteins [41]. Impaired ubiquitin-dependent degradation of other aggregate-prone proteins, such as α-synuclein and huntingtin, is a common theme in neurodegenerative diseases [102] [41].
- Angelman Syndrome: This neurodevelopmental disorder is caused by mutations in the gene encoding the E3 ubiquitin ligase UBE3A. This disrupts the ubiquitination of key neuronal substrates, leading to severe cognitive and motor deficits [101].
Genetic and Immune Disorders
- 3-M Syndrome: This inherited growth retardation disorder results from mutations in the CUL7 gene. CUL7 is essential for assembling an E3 ubiquitin ligase complex, and its disruption impairs normal ubiquitination-dependent growth regulation [101].
- Inflammatory and Autoimmune Diseases: Dysregulated Met1-linked (linear) ubiquitination in the NF-κB signaling pathway is implicated in immune disorders. For example, the DUB OTULIN, which specifically cleaves Met1 chains, is critical for terminating inflammatory signaling; its dysregulation can lead to autoinflammation [102] [104].
Table 2: Disease-Associated Ubiquitination Events and Clinical Implications
| Disease | Ubiquitination Component | Molecular Consequence | Clinical/Translational Implication |
|---|---|---|---|
| VHL Syndrome [101] [103] | VHL E3 Ligase (Loss of function) | HIF-1α stabilization → Angiogenesis | Renal cell carcinoma, hemangioblastomas; HIF inhibitors in therapy |
| Colorectal Cancer [101] | APC/E3 Complex (Mutation) | β-catenin stabilization → Proliferation | Tumorigenesis; Targeting Wnt/β-catenin pathway |
| DLBCL [108] | CDC34 (E2), FZR1, OTULIN (DUB) | Altered cell cycle & signaling | Prognostic biomarker signature; potential for targeted E2/DUB therapies |
| Alzheimer's Disease [41] | K48-linked Tau Ubiquitination | Impaired tau degradation → Aggregates | Proteasome dysfunction; Investigational therapies to boost clearance |
| Angelman Syndrome [101] | UBE3A E3 Ligase (Mutation) | Altered neuronal substrate degradation | Neurodevelopmental disorder; Gene therapy approaches under investigation |
| 3-M Syndrome [101] | CUL7 (E3 Complex subunit) | Disrupted growth regulation | Prenatal and postnatal growth retardation |
| Autoinflammation [102] [104] | OTULIN DUB (Dysregulation) | Excessive Met1-Ub → NF-κB hyperactivation | Autoimmune pathology; Small molecule DUB modulators in development |
Figure 2: The Ubiquitination Enzyme Cascade and its Connection to Disease. A three-enzyme cascade (E1, E2, E3) conjugates ubiquitin to specific protein substrates, forming a polyubiquitin chain that alters the protein's fate. Dysregulation at any point in this pathway can lead to disease.
Targeted Drug Discovery in the Ubiquitin System
The ubiquitin system offers a rich landscape for therapeutic intervention. Drug discovery efforts have evolved from broad proteasome inhibitors to highly specific agents targeting individual components of the ubiquitin machinery.
Established and Emerging Therapeutic Strategies
- Proteasome Inhibitors: Drugs like bortezomib, carfilzomib, and ixazomib inhibit the 26S proteasome, preventing the degradation of proteins marked for destruction. They are approved for hematological malignancies like multiple myeloma but can have off-target effects due to their broad mechanism [101] [103].
- E1 Enzyme Inhibitors: MLN7243 (TAK-243) inhibits the ubiquitin-activating enzyme UBA1, blocking the entire ubiquitin cascade. It has shown anti-tumor activity in preclinical models but may face toxicity challenges due to its global effect [103].
- E2 Enzyme Inhibitors: Compounds like CC0651 target specific E2 enzymes (e.g., CDC34), offering more selectivity than E1 inhibition. They are primarily in preclinical development [103].
- E3 Ligase Modulators: This is a rapidly growing area due to the high substrate specificity of E3s.
- Deubiquitinase (DUB) Inhibitors: Targeting DUBs (e.g., with compounds G5 or F6) can rebalance ubiquitination to promote the degradation of oncoproteins. DUB inhibitors are considered high-potential but challenging targets, with several in early-stage development [103].
Targeted Protein Degradation (TPD)
A revolutionary approach that hijacks the ubiquitin system is TPD, using bifunctional molecules like PROTACs (Proteolysis-Targeting Chimeras) and molecular glues [104]. These molecules recruit a specific E3 ligase to a target protein of interest, inducing its ubiquitination and degradation. This allows direct targeting of proteins previously considered "undruggable," such as transcription factors [104].
Table 3: Targeted Therapies in the Ubiquitin-Proteasome System
| Therapeutic Class | Target | Example Agent | Mechanism of Action | Development Stage |
|---|---|---|---|---|
| Proteasome Inhibitor [101] [103] | 26S Proteasome | Bortezomib, Carfilzomib | Blocks protein degradation, inducing ER stress & apoptosis | Approved (Multiple Myeloma) |
| E1 Inhibitor [103] | UBA1 (E1) | MLN7243 (TAK-243) | Global inhibition of ubiquitin activation | Preclinical/Phase I |
| E2 Inhibitor [103] | Specific E2 (e.g., CDC34) | CC0651, Leucettamol A | Inhibits activity of a subset of E2 enzymes | Preclinical |
| E3 Ligase Modulator [101] [103] | MDM2 (E3) | Nutlin, MI-219 | Blocks MDM2-p53 interaction, stabilizing p53 | Advanced Clinical Trials |
| DUB Inhibitor [103] | Specific Deubiquitinases | Compounds G5, F6 | Inhibits DUB activity, promoting degradation of its substrates | Preclinical/Early Clinical |
| Targeted Protein Degradation [104] | E3 Ligase + POI | PROTACs, Molecular Glues | Recruits E3 ligase to neo-substrate for degradation | Early Clinical (e.g., ARV-471) |
The Scientist's Toolkit: Key Reagents and Methodologies
This section provides a curated list of essential tools and reagents for ubiquitination research, based on methodologies cited in this review.
Table 4: Essential Research Reagent Solutions for Ubiquitination Studies
| Reagent / Tool | Category | Primary Function | Example Use Case |
|---|---|---|---|
| Tagged Ubiquitin (His-, HA-, Strep-) [105] [41] | Affinity Tag | High-affinity purification of ubiquitinated proteome | Global ubiquitome profiling by MS; in vivo ubiquitination assays |
| Linkage-Specific Ub Antibodies [41] | Antibody | Immunodetection of specific chain types (K48, K63, M1) | Assessing degradation vs. signaling ubiquitination by WB/IP |
| Tandem UBDs (TUBEs) [41] [42] | Affinity Tool | High-affinity enrichment, protects ubiquitination from DUBs | Stabilizing and detecting labile ubiquitination events |
| Site-Specific Ubiquitin Antibodies [106] | Antibody | Detect ubiquitination at a specific lysine on a specific protein | Monitoring endogenous H2B ubiquitination in ChIP assays |
| Proteasome Inhibitors (MG-132, Bortezomib) [105] | Small Molecule | Blocks 26S proteasome, stabilizes ubiquitinated proteins | Enhancing detection of ubiquitinated species in cell assays |
| DUB Inhibitors (NEM, PR-619) [42] | Small Molecule | Inhibits deubiquitinating enzymes during lysis | Preserving the native ubiquitome during sample preparation |
| QuikChange Mutagenesis Kit [105] | Molecular Biology | Rapid introduction of point mutations (K-to-R) | Validating specific ubiquitination sites by mutagenesis |
| Ni-NTA Agarose / Strep-Tactin Beads [105] [41] | Chromatography Resin | Immobilized affinity matrix for purifying tagged proteins | Isolating His-Ub or Strep-Ub conjugated proteins from lysates |
The journey from mapping site-specific ubiquitination to clinical translation is accelerating. The integration of conventional validation methods like mutagenesis with advanced quantitative proteomics is providing an unprecedented view of the ubiquitin code's complexity and dynamics. As the quantitative principles of ubiquitination—such as its characteristically low occupancy and fast turnover—become clearer, and as the disease-specific roles of E3 ligases and DUBs are further elucidated, the pipeline for targeted therapies will continue to expand. The emergence of revolutionary strategies like targeted protein degradation underscores the immense clinical potential of deliberately manipulating the ubiquitin system. For researchers and drug developers, the future lies in combining deep mechanistic knowledge of specific ubiquitination pathways with innovative chemical and biological tools to develop the next generation of precise and effective therapeutics.
Conclusion
The integration of lysine mutagenesis with immunoblotting remains a cornerstone methodology for validating ubiquitination sites, providing a accessible yet powerful approach to establish causal relationships between specific lysine residues and ubiquitin modification. When combined with computational predictions, mass spectrometry, and functional assays, this approach forms a robust validation framework essential for deciphering the ubiquitin code. As research progresses, the refinement of linkage-specific detection methods and the development of small-molecule inhibitors targeting specific E3 ligases promise to open new therapeutic avenues for cancers, neurodegenerative diseases, and other conditions driven by ubiquitination dysregulation. The continued optimization of these validation strategies will be crucial for advancing both basic science and targeted drug development in the ubiquitin field.
References
- 1. E3 ubiquitin ligases: styles, structures and functions - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC8607428/]
- 2. exploring the ubiquitin system for drug development [https://www.nature.com/articles/cr201631]
- 3. Current methodologies in protein ubiquitination ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9373315/]
- 4. Functional reconstruction of a eukaryotic-like E1/E2/(RING) ... [https://www.nature.com/articles/s41467-017-01162-7]
- 5. Deubiquitinases: From mechanisms to their inhibition by ... [https://www.sciencedirect.com/science/article/pii/S1097276521009473]
- 6. Mechanisms orchestrating the enzymatic activity and cellular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9356280/]
- 7. Emerging drug development technologies targeting ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7112620/]
- 8. Structural insights into the biochemical mechanism of ... [https://www.sciencedirect.com/science/article/abs/pii/S0969212624005379]
- 9. Deubiquitinating enzyme [https://en.wikipedia.org/wiki/Deubiquitinating_enzyme]
- 10. The deubiquitinase USP9X and E3 ligase WWP1 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12500958/]
- 11. Chain reactions: molecular mechanisms of RBR ubiquitin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7458406/]
- 12. Review Ubiquitin—A structural perspective [https://www.sciencedirect.com/science/article/abs/pii/S1097276524010104]
- 13. Decoding polyubiquitin regulation of KV7. 1 (KCNQ1) ... [https://www.nature.com/articles/s41467-025-60893-0]
- 14. The Molecular Toolbox for Linkage Type-Specific Analysis ... [https://pubmed.ncbi.nlm.nih.gov/40192223/]
- 15. Unraveling chain specific ubiquitination in cells using tandem ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12217038/]
- 16. Decoding ubiquitin signals inside cells [https://www.nature.com/articles/s41580-025-00919-z]
- 17. Ub-POD: A Ubiquitin-Specific Proximity-Dependent ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12222633/]
- 18. The Role of Protein Ubiquitination in the Onset and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12248740/]
- 19. Protein Degradation Through Ubiquitination: Insights into ... [https://www.preprints.org/manuscript/202504.2585]
- 20. Unraveling Chain specific Ubiquitination in Cells Using ... [https://lifesensors.com/unraveling-chain-specific-ubiquitination-in-cells-using-tandem-ubiquitin-binding-entities-in-a-high-throughput-assay/]
- 21. Unraveling chain specific ubiquitination in cells using ... [https://www.nature.com/articles/s41598-025-07242-9]
- 22. Selective ubiquitination of drug-like small molecules by the ... [https://www.nature.com/articles/s41467-025-63442-x]
- 23. Western Blotting (Immunoblotting): History, Theory, Uses ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12303220/]
- 24. Identification and validation of ubiquitination-related genes ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2025.1578075/full]
- 25. Protein Ubiquitination: Assays, Sites & Proteomics Methods [https://www.creative-proteomics.com/resource/protein-ubiquitination-detection-assays-site-identification-proteomics.htm]
- 26. Machine learning-based approaches for ubiquitination site ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-023-05581-w]
- 27. Discovering functionally important sites in proteins [https://www.nature.com/articles/s41467-023-39909-0]
- 28. Systematic approach for validating the ubiquitinated proteome [https://pmc.ncbi.nlm.nih.gov/articles/PMC2673951/]
- 29. DeepUbi: a deep learning framework for prediction of ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-2677-9]
- 30. Computational methods for ubiquitination site prediction using ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-016-0959-z]
- 31. A Caps-Ubi Model for Protein Ubiquitination Site Prediction [https://pmc.ncbi.nlm.nih.gov/articles/PMC9175003/]
- 32. UbiSitePred: A novel method for improving the accuracy of ubiquitination sites prediction by using LASSO to select the optimal Chou's pseudo components - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S0169743918305227]
- 33. DeepTL-Ubi: A novel deep transfer learning method for effectively predicting ubiquitination sites of multiple species - ScienceDirect [https://www.sciencedirect.com/science/article/abs/pii/S1046202320301560]
- 34. Species-specific model based on sequence and structural ... [https://www.sciencedirect.com/science/article/abs/pii/S0022283624004017]
- 35. A study on the effect of surface lysine to arginine ... [https://pubmed.ncbi.nlm.nih.gov/22792305/]
- 36. A Study on the Effect of Surface Lysine to Arginine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3392243/]
- 37. Site Directed Mutagenesis [https://www.neb.com/en-us/applications/cloning-and-synthetic-biology/site-directed-mutagenesis?srsltid=AfmBOoqwnW0LOoPvKOOneTjWnfPXUbWJWeW4aWHX8Zl9wQr2OfARyTFc]
- 38. Site-Directed Mutagenesis | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/cloning/mutagenesis.html]
- 39. Random mutagenesis strategies for construction of large ... [https://pubmed.ncbi.nlm.nih.gov/15153637/]
- 40. Introduction to Molecular Cloning Methods [https://www.geneious.com/guides/molecular-cloning-methods]
- 41. Current methodologies in protein ubiquitination characterization [https://cellandbioscience.biomedcentral.com/articles/10.1186/s13578-022-00870-y]
- 42. Optimising methods for the preservation, capture and ... [https://www.sciencedirect.com/science/article/pii/S0006291X15305015]
- 43. Ubiquitination detection techniques - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC10625345/]
- 44. An optimized protocol to detect ubiquitination modification ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10594630/]
- 45. Denaturing in vivo ubiquitination assay [https://bio-protocol.org/exchange/preprintdetail?id=2303&type=3]
- 46. A High-Throughput Assay for Monitoring Ubiquitination in ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2019.00816/full]
- 47. Inhibition of the ubiquitin-proteasome system by an NQO1- ... [https://www.nature.com/articles/s41419-021-04191-9]
- 48. Measuring activity in the ubiquitin-proteasome system [https://pmc.ncbi.nlm.nih.gov/articles/PMC3758771/]
- 49. Utilizing a Comprehensive Immunoprecipitation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5908451/]
- 50. Co-immunoprecipitation (Co-IP): The Complete Guide [https://www.antibodies.com/applications/co-immunoprecipitation]
- 51. Immunoprecipitation (IP): The Complete Guide [https://www.antibodies.com/applications/immunoprecipitation]
- 52. Short note A reducing and denaturing step maximizes the ... [https://www.sciencedirect.com/science/article/abs/pii/S0165022X06000649]
- 53. Overview of the Immunoprecipitation (IP) Technique [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/immunoprecipitation-ip.html]
- 54. Immunoprecipitation Experimental Design Tips [https://www.cellsignal.com/applications/immunoprecipitation/ip-experimental-design-tips?srsltid=AfmBOooksdZFYi4jgP05TIhG0l5-MyRVRKpS_NDOnQTxYPlrvfqg5l25]
- 55. Co-immunoprecipitation Protocol: 9 Easy Steps To Co-IPs [https://bitesizebio.com/19773/co-immunoprecipitation-protocol-guide/]
- 56. Highly specific intracellular ubiquitination of a small molecule [https://www.nature.com/articles/s41589-025-02011-1]
- 57. Midnolin Ubiquitination is Required for its Proteasome‐ ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12022415/]
- 58. Mechanistic insights into NFIX frameshift mutations in ... [https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2025.1648420/full]
- 59. Comparison of Automated and Traditional Western Blotting ... [https://www.mdpi.com/2409-9279/6/2/43]
- 60. Lysine 222 in PPAR γ1 functions as the key site of MuRF2- ... [https://www.nature.com/articles/s41598-023-28905-5]
- 61. Suppression of PPARγ through MKRN1-mediated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3950322/]
- 62. Therapeutic targeting of ubiquitin-specific protease 7 (USP7) [https://www.sciencedirect.com/science/article/abs/pii/S0223523425006373]
- 63. Recent advances in small molecule inhibitors of ... [https://pubmed.ncbi.nlm.nih.gov/39908798/]
- 64. Ubiquitin-interacting Motifs Confer Full Catalytic Activity, ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3900984/]
- 65. Accelerating inhibitor discovery for deubiquitinating enzymes [https://www.nature.com/articles/s41467-023-36246-0]
- 66. Deubiquitinating Enzymes Ubiquitin-Specific Proteases 7 ... [https://www.mdpi.com/1422-0067/26/22/11062]
- 67. Protocol for tandem enrichment of ubiquitinated, ... [https://www.sciencedirect.com/science/article/pii/S2666166725003508]
- 68. Sequential Denaturation and Protein Precipitation assay ... [https://www.sciencedirect.com/science/article/abs/pii/S000326702501116X]
- 69. Employing Expression-Matched Controls Enables High- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12226361/]
- 70. K29‐Linked Ubiquitination of Transcription Regulators ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12622412/]
- 71. Decoding polyubiquitin regulation of KV7. 1 (KCNQ1) ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12218124/]
- 72. Ubiquitination Specific Antibody Discovery Service [https://www.creative-biolabs.com/Ubiquitination-Specific-Antibody-Production-Service.html]
- 73. Anti-Ubiquitin (linkage-specific K48) antibody [EP8589] [https://www.abcam.com/en-us/products/primary-antibodies/ubiquitin-linkage-specific-k48-antibody-ep8589-ab140601]
- 74. Ubigo-X: Protein ubiquitination site prediction using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12303043/]
- 75. A High-Throughput Method for Specific, Rapid, Precise and ... [https://www.sciencedirect.com/science/article/abs/pii/S0039914025015681]
- 76. Site-directed multivalent conjugation of antibodies to ... [https://www.nature.com/articles/s41551-024-01342-z]
- 77. Advancing Antibody Conjugates with Ubi-Tagging [https://communities.springernature.com/posts/behind-the-paper-advancing-antibody-conjugates-with-ubi-tagging]
- 78. Epitope-tagged ubiquitin. A new probe for analyzing ... [https://www.sciencedirect.com/science/article/pii/S002192581854833X]
- 79. Epitope-tagged ubiquitin. A new probe for analyzing ... - PubMed [https://pubmed.ncbi.nlm.nih.gov/1718971/]
- 80. Identification, Analysis and Prediction of Protein Ubiquitination ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3006176/]
- 81. Comparison of Automated and Traditional Western Blotting ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10142486/]
- 82. How to Achieve Quantitative Western Blot Data: a Step-by- ... [https://www.licorbio.com/blog/step-by-step-guide-to-achieving-quantitative-western-blot-data]
- 83. Biomedical images – how much processing is too much? [https://www.internationalscienceediting.com/biomedical-images-how-much-processing-is-too-much/]
- 84. Cycloheximide (CHX) Chase Assay to Examine Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10262206/]
- 85. Analysis of Protein Stability by the Cycloheximide Chase ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5659619/]
- 86. Determining and interpreting protein lifetimes in mammalian ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9868050/]
- 87. An atlas of protein turnover rates in mouse tissues [https://www.nature.com/articles/s41467-021-26842-3]
- 88. Large-Scale Identification of Ubiquitination Sites by Mass ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4725055/]
- 89. Characterizing Ubiquitination Sites by Peptide-based ... [https://www.sciencedirect.com/science/article/pii/S1535947620334502]
- 90. Refined preparation and use of anti-diglycine remnant ... [https://pubmed.ncbi.nlm.nih.gov/23266961/]
- 91. UbiSite approach for comprehensive mapping of lysine ... [https://pubmed.ncbi.nlm.nih.gov/29967540/]
- 92. Proteomic Identification of Protein Ubiquitination Events - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3937853/]
- 93. Proteomic approaches for the profiling of ubiquitylation ... [https://www.sciencedirect.com/science/article/abs/pii/S187439192030364X]
- 94. Current Status and Advances in Quantitative Proteomic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3606794/]
- 95. Advantages and Disadvantages of MS-Based Relative ... [https://www.mtoz-biolabs.com/advantages-and-disadvantages-of-ms-based-relative-protein-quantification.html]
- 96. Systematic approaches to identify E3 ligase substrates - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5103871/]
- 97. In Vitro SUMOylation Assay to Study SUMO E3 Ligase Activity - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5912215/]
- 98. Combinatorial mapping of E3 ubiquitin ligases to their ... [https://www.sciencedirect.com/science/article/pii/S1097276525000516]
- 99. Defining E3 ligase–substrate relationships through ... [https://www.nature.com/articles/s41556-023-01229-2]
- 100. BioE3 identifies specific substrates of ubiquitin E3 ligases | Nature Communications [https://www.nature.com/articles/s41467-023-43326-8]
- 101. Biochemistry, Ubiquitination - StatPearls - NCBI Bookshelf - NIH [https://www.ncbi.nlm.nih.gov/books/NBK556052/]
- 102. The ubiquitin system: from cell signalling to disease ... [https://www.nature.com/articles/s41418-020-00703-w]
- 103. The role of ubiquitination in tumorigenesis and targeted ... [https://www.nature.com/articles/s41392-020-0107-0]
- 104. The Ubiquitin Code in Disease Pathogenesis and ... [https://pubmed.ncbi.nlm.nih.gov/39973547/?utm_source=FeedFetcher&utm_medium=rss&utm_campaign=None&utm_content=14EXV6AaFU8htrIOCjRMdpDNiKtgASVCScQXcx3qOBm2jl8k8j&fc=None&ff=20250221011334&v=2.18.0.post9+e462414]
- 105. An optimized protocol to detect protein ubiquitination and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10050764/]
- 106. Strategy for Development of Site-Specific Ubiquitin ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2020.00111/full]
- 107. Global, site-resolved analysis of ubiquitylation occupancy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11136510/]
- 108. A novel ubiquitination-based molecular signature predicts ... [https://pubmed.ncbi.nlm.nih.gov/41134336/]