Research Articles
Decoding the Ubiquitin Code: Chain Topology, Biological Functions, and Therapeutic Targeting
This article provides a comprehensive overview of the ubiquitin code, focusing on the structural and functional diversity of ubiquitin chain topologies.
The Complete Guide to Selecting Linkage-Specific Ubiquitin Antibodies for Precision Research
This guide provides researchers, scientists, and drug development professionals with a comprehensive framework for selecting and applying linkage-specific ubiquitin antibodies.
Decoding the Ubiquitin Code: A Comprehensive Guide to Interpreting Linkage-Specific Western Blot Results
This article provides a definitive guide for researchers and drug development professionals on interpreting ubiquitin linkage Western blot data.
Decoding the Functions of Atypical Ubiquitin Linkages: K6, K11, K27, K29, and K33 in Cellular Regulation and Disease
This article provides a comprehensive analysis of atypical ubiquitin linkages (K6, K11, K27, K29, K33), exploring their foundational roles in cellular processes like DNA repair and autophagy, methodological advances for...
K48 vs K63 Ubiquitin Chains: Decoding the Proteasomal Degradation Signal
This article synthesizes current understanding of how K48- and K63-linked ubiquitin chains direct proteasomal degradation, moving beyond the classical K48-degradation paradigm.
Linkage-Specific Ubiquitin Antibodies: A Definitive Guide for Researchers and Drug Developers
This article provides a comprehensive overview of linkage-specific ubiquitin antibodies, essential tools for deciphering the complex ubiquitin code.
K27-Linked Ubiquitination: A Critical Checkpoint Balancing IRF3 and NF-κB in Antiviral Immunity
This review synthesizes current knowledge on the distinct and opposing roles of K27-linked ubiquitin chains in regulating the IRF3-driven type I interferon response and the NF-κB-mediated inflammatory pathway.
K6 vs. K27 vs. K63: Decoding Parkin's Ubiquitin Chain Specificity in Parkinson's Disease Pathways
This article provides a comprehensive analysis of the E3 ubiquitin ligase Parkin's specificity for K6-, K27-, and K63-linked ubiquitin chains, a critical determinant in Parkinson's disease pathogenesis.
BCAAs and NF-κB Signaling: Unveiling Dual Roles in Inflammation and Disease Pathogenesis
This article synthesizes current evidence on the intricate relationship between Branched-Chain Amino Acids (BCAAs) and NF-κB signaling, a central pathway in inflammation.
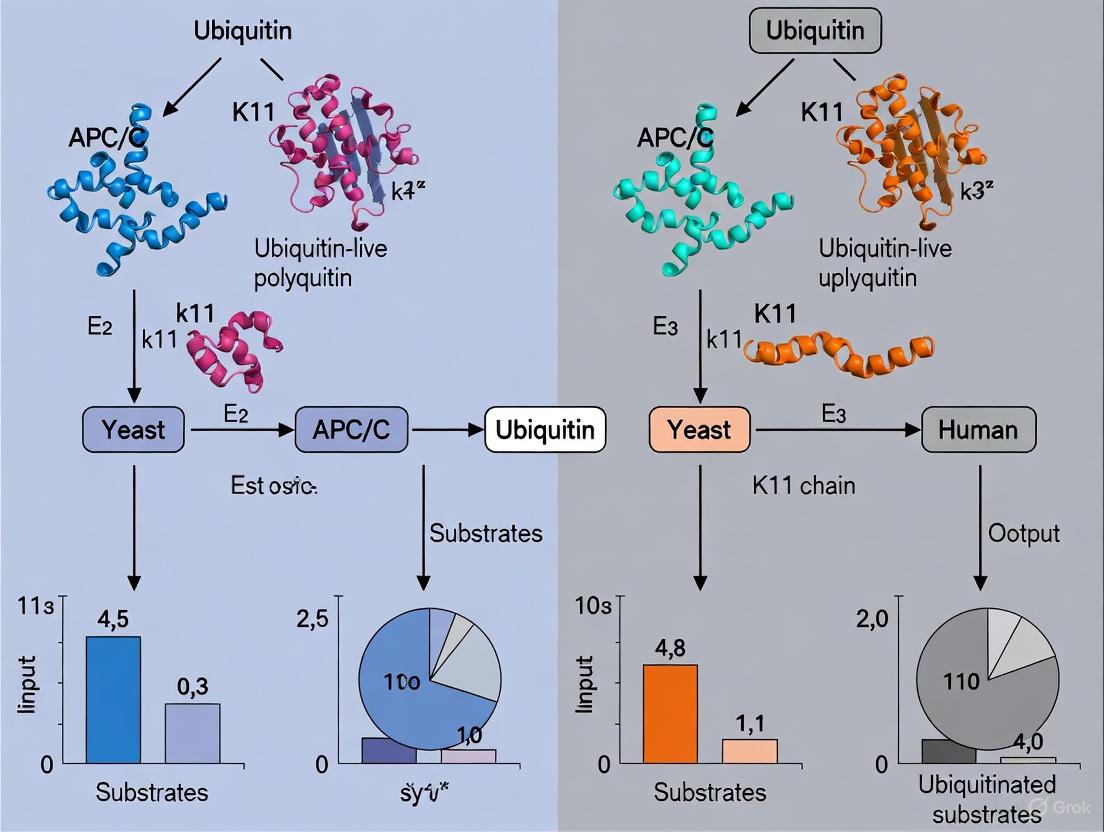
Evolution of Destruction Signals: Comparing K11 Ubiquitin Chain Usage in Yeast and Human APC/C
This article provides a comprehensive comparison of K11-linked ubiquitin chain utilization by the Anaphase-Promoting Complex/Cyclosome (APC/C) in yeast versus humans.